Submitted:
28 February 2025
Posted:
03 March 2025
You are already at the latest version
Abstract
Autism Spectrum Disorder (ASD) is a neurodevelopmental disorder characterized by impaired social interaction, communication deficits, and the presence of repetitive, restricted behavioral patterns and interests. Recent research shows that it occurs due to the mutations related to the development of the nervous system, combined with the impact of various environmental factors. This necessitates the identification of the genetic variations involved in ASD pathogenesis. We performed whole exome sequencing (WES) in a cohort of 22 Bulgarian male and female individuals showing ASD features alongside with segregation analyses of their families. A targeted panel of genes was chosen and analyzed for each case, based on a detailed examination of clinical data. Gene analyses revealed that specific variants concern key neurobiological processes involving neuronal architecture, development and function such as synaptic signaling imbalance, ciliopathies, spectrins structure, neuronal organelles trafficking and integrity, gene expression, cell cycle control, mitochondrial function, and neuron homeostasis. Our data contribute to a better understanding of the complex neurobiological features of autism and are applicable in the diagnosis and development of personalized therapeutic approaches.
Keywords:
autism spectrum disorder
; whole exome sequencing
; single nucleotide variations
; neurons
; neuronal structure
; neuronal function
; synaptic signaling imbalance
; ciliopathies
; mitochondrial function
; molecular mechanisms
1. Introduction
Autism spectrum disorder (ASD) is a neurodevelopmental impairment with onset in infancy or early childhood, characterized by social difficulties, deterioration of communication, repetitive behaviors, stereotyped and restricted interests [1,2,3]. Intensive genetic studies have confirmed that ASD has a strong genetic basis and genetic heterogeneity [4]. It can be a distinct clinical phenotype or syndromic, related to the most common genetic syndromes [5]. In syndromic cases, ASD is a symptom of a more profound developmental disorder that includes multiple phenotypes, such as dysmorphic features, intellectual disability and epilepsy [6]. Non-syndromic ASD could be polygenic and multifactorial, determined by specific combinations of environmental and genetic factors, or a single gene mutation can result in developing the disease in the relatively benign form of sporadic non-syndromic ASD [7].
According to the Diagnostic and Statistical Manual of Mental Disorders 5th edition (DSM-5), ASD belongs to the group of neurodevelopmental disorders [8]. There are two main hypotheses about the bond between the disrupted development of brain due to a certain genetic mutation and the specific NDDs that appear in any of the individuals that contain it [9]. The neurodevelopmental continuum hypothesis proposes that NDDs are different variants of outcomes that originate from the disordered or deviant development of the brain [10,11]. The neurodevelopmental gradient hypothesis says that disorders are graded into a neurodevelopmental, but not a discreet continuum, according to the severity of neurodevelopmental impairment resulting from combination of genetic and environmental factors [12].
The genetic variations can be grouped according to six criteria: extent, time of onset, information content, frequency, number of genes involved, inheritance pattern [13]. Three major categories of genetic risk are implicated into it: common polygenic variations, rare inherited and de novo mutations. Common variants determine predisposition due to polygenic risk, defined by thousands of risk alleles, each of which alone has a small additive effect [14]. Many susceptibility genes have been identified by genetic analysis [4]. Dynamically growing data points on ASD association with variety of genes and their single nucleotide variations (SNVs) [13]. Single nucleotide polymorphisms (SNPs) are considered to account for 40-50% of ASD cases [15]. SNVs comprise about 0.1% of the human genome and the majority of them are common variants that usually affect only one gene and are one of the major sources of genetic diversity. Lots of ASD candidate genes have been highlighted using genomic analyses for determining allelic diversity, mode of inheritance and phenotypic impact of inherited and de novo variants of ASD and NDD genes [16,17,18,19,20,21,22]. According to the Simons Foundation Autism Research Initiative (SFARI) gene database, there are more than 1000 gene candidates associated with ASD (About the Gene Scoring Module, https://gene.sfari.org/about-gene-scoring/ cited 2024 December 24). Mutations associated with autism often affect genes, related to a variety of metabolic disorders in the patients. Specific metabolites and metabolic pathways significantly differ in children with ASD compared to normally developing individuals [23].
Mutations including SNVs, can be categorized as either de novo variants or inherited ones, according to the mode of occurrence. A de novo variation is the result of a mutation which occurred in the parental gametes or a genetic modification during embryogenesis that arises for the first time in the individual [13]. It has been reported that postzygotic mutations account for numerous de novo harmful mutations resulting in mosaicism [24]. Hereditary variations can be found in the parental genome [13].
Here, we present data from whole exome sequencing (WES) in a cohort of 22 Bulgarian male and female individuals showing ASD features alongside with segregation analyses of their families. A target panel of genes was chosen and analyzed for each case, based on a detailed examination of clinical data. This approach allowed us to characterize the distribution of rare de novo and inherited SNVs in our sample and to assess their phenotypic impact by exploring the presence of comorbidities in patients carrying such variants.
2. Materials and Methods
Altogether 22 Bulgarian patients (13 males and 9 females) with syndromic and non-syndromic autism spectrum disorder were selected from the medical records of Genetic Medico-Diagnostic Laboratory Genica. Written informed consent was obtained from the patients’ guardians, as well as from all family members tested.
High molecular weight DNA was extracted from EDTA-venous blood by standard salting-out method. The quality of the extracted DNA was assessed by direct spectrophotometry.
Whole exome sequencing (WES) was conducted in the partner laboratories "Admera Health, LLC", USA and Clinical Institute of Medical Genetics, UMC Ljubljana, Ljubljana, Slovenia. Clinically relevant genes were analyzed via a specialized software GensearchNGS, PhenoSystems SA.
The target regions of the human genome, where the genetic variants were detected during the whole exome sequencing, were multiplied by polymerase chain reaction (PCR). The amplified fragments were sequenced by Sanger's method with BigDye® Terminator v.3.1 sequencing kit (Applied Biosystems, Foster City, CA, USA). Electrophoretic separation of sequence products was performed via an ABI Prism 3130 Genetic Analyzer. The obtained data were processed automatically by the program ABI3130 Data Collection Software and received in the form of an electrophoregram with Sequencing Analysis software v.5.1.1.
The interpretation of the detected genetic variants was performed according to the classification criteria of the guidelines of the American College of Medical Genetics and Genomics/Association of Molecular Pathology (ACMG/AMP), taking into account the clinical manifestations and the results from the segregation analyses, performed in the families.
The study was approved by the Ethics Committee of Sofia University “St. Kliment Ohridski”, Protocol No 93-M-412/1.10.2024.
3. Results
The genetic variants which were detected by WES in our cohort of 22 Bulgarian patients (13 boys and 9 girls) are represented in Table 1. All patients share autistic features, neuropsychological delay and intellectual disability. The detected genetic variants are classified as pathogenic, likely pathogenic or VUS (variants of uncertain significance) according to the ACMG/AMP guidelines. Altogether, 16 cases were concluded to carry pathogenic or likely pathogenic variants. In one of the cases likely pathogenic and VUS were detected in the autosomal recessive VPS13B gene, which segregate in the family (one is maternally inherited and the other one is paternally inherited). Another patient carries a pathogenic de novo variant in the SHANK3 gene and maternally inherited VUS in the DLG3 gene. The last 4 cases carry variants classified as VUS in different genes (see Table 1). The performed segregation analysis in the affected families revealed 10 de novo cases, 11 cases with variants segregating in the family and a case with one de novo and one maternally inherited variant.
The genes which are involved in the neurological pathology manifested in our patient cohort are important participants in the regulation of gene expression, proper mitochondrial function, synaptic signaling, neuronal organelles trafficking and integrity, neuronal homeostasis, cell cycle regulation, as well as proper structure of cilia and spectrins (Figure 1).
4. Discussion
Тhe mechanisms underlying autism spectrum disorder (ASD) have been the subject of intense study and debate for decades. Common symptoms and atypical behaviors appear in all the individuals with autism, despite diverse hypotheses ranging from genetic causes to environmental risk factors. It is not clear how the pathogenesis of different behavioral disorders is related to structural and metabolic abnormalities in the brain. Patients with genetic and chromosomal diseases tend to show more symptoms of ASD [25]. Some of SNVs in ASD patients are dominant loss-of-function variants, which result in a damage of the encoded protein and each of these heterozygous mutations are anticipated to cause haploinsufficiency resulting in a reduction in overall gene production [16,26].
We report an integrated analyses of rare SNVs detected with whole exome sequencing and segregation analyses data obtained from a cohort of 22 Bulgarian families with probands exhibiting ASD along with other genetically defined comorbidities. This research led to the identification of certain potentially harmful de novo and inherited variants increasing the allelic diversity and contributing to our understanding of the mode of inheritance of specific ASD risk genes and their phenotypic expression. The identification of de novo and inherited SNVs implicated in ASD development highlighted promising candidate genes for future diagnostic and treatment options.
SNVs associated with synaptic structure, function and signaling imbalance of neurons in ASD
Cerebral cortex is the area responsible for higher-level processes such as thought, emotion, decision-making and language. One of the hypotheses for the genetic basis of autism and other neurodevelopmental disorders, states that the condition is caused by disturbances in the delicate balance between excitatory and inhibitory signals, supported by GABAergic (γ-aminobutyric acid-releasing) interneurons in the cerebral cortex [27]. Analyzing the existing published information, these authors point on the fact that the exact mechanism of synaptic signaling imbalance must be elucidated. Various aspects of human brain development and function can be studied in vitro using pluripotent stem cells with a remarkable ability to self-organize and differentiate in three-dimensional aggregates, known as organoids or organ spheroids (Pasca, 2018). These findings are the basics for developing in vitro studies of stem cell-based three-dimensional (3D) cellular models using CRISPR (clustered regularly interspaced short palindromic repeats) technology for screening of certain genes involved in neurodevelopmental disorders caused by defects in interneuron generation and migration into cortical circuits (Meng et al., 2023). Such findings incline that any factor leading to abnormalities in synaptic transmission may cause neurodevelopmental disorders and autism.
Expression of risk gene alleles in excitatory and inhibitory neuronal lineages from the human cortex is associated with various pathway mechanisms leading to an excitatory-inhibitory imbalance underlying ASD [20]. In our exome sequencing study, we identified risk gene alleles for improper synaptic function and neuron excitability, more specifically NALCN, PACS2, SHANK3, and DLG3.
According to SFARI (https://gene.sfari.org/search/?search=nalcn), NALCN (Sodium Leak Channel, Non-Selective) is a strong candidate gene for ASD. This gene encodes a voltage-independent, nonselective cation channel which belongs to a family of voltage-gated sodium and calcium channels expressed throughout the nervous system that regulates the resting membrane potential and excitability of neurons, conducting a persistent sodium leak current that contributes to tonic neuronal excitability [28]. The encoded protein forms a channelosome complex that includes G-protein-coupled receptors, UNC-79, UNC-80, NCA localization factor-1, and SRC family tyrosine kinases [29]. Genes encoding NALCN, NLF- 1, UNC-79, and UNC-80 proteins may be associated with susceptibility for several diseases including bipolar disorder, schizophrenia, Alzheimer's disease, autism, epilepsy, alcoholism, cardiac diseases and cancer [30]. NALCN mutations are associated with infantile neuroaxonal dystrophy, infantile hypotonia with psychomotor retardation and characteristic facies (IHPRF) syndrome, and congenital contractures of the limbs and face with hypotonia and developmental delay (CLIFAHDD) syndrome [31]. In this study we report a de novo heterozygous NALCN variant in a female patient with similar clinical phenotype to that described in NALCN-related disorders, and more specifically neonatal generalized muscle hypotonia, ulnar deviation of the fingers and dysplasia of the hip joint, microcephaly, developmental delay and ASD.
PACS2 (Phosphofurin Acidic Cluster Sorting Protein 2) gene is located on chromosome 14 (https://gene.sfari.org/database/human-gene/PACS2). Pathogenic variants in the PACS2 gene lead to early infantile developmental and epileptic encephalopathy (EIDEE) which is a rare neurodevelopmental disorder that could be related to PACS2’s potential involvement in ion channel regulation [32]. [33]. This disorder is consistent with the clinical findings in the male patient reported here with a de novo heterozygous variant in the PACS2 gene. The patient’s phenotype includes epilepsy, hypotonia, ASD and facial dysmorphism.
SHANK3 (ProSAP SH3 and multiple ankyrin repeat domain protein 3) gene located on chromosome 22 is a high confidence ASD-associated gene (https://gene.sfari.org/database/human-gene/SHANK3). Shank proteins are multidomain scaffold proteins enriched in the postsynaptic density of excitatory synapses that connect neurotransmitter receptors, ion channels, and other membrane proteins to the actin cytoskeleton and G-protein-coupled signaling pathways, thus playing important roles in the formation, maturation, and maintenance of synapses [34]. SHANK3 is strongly suspected of being involved in the pathogenesis and neuropathology of ASD [35,36]. An in vitro study suggested that SHANK3 knockdown impaired both early stage of neuronal development and mature neuronal function, whereas electrophysiology analyses revealed defects in excitatory and inhibitory synaptic transmission [37]. SHANK3 deficiency caused autistic-like behaviors by activating p38α signaling in AgRP neurons [38]. In our ASD cohort we describe a male patient, who carries a de novo heterozygous SHANK3 variant.
The DLG3 gene is located on chromosome X and encodes disks large membrane-associated guanylate kinase scaffold protein 3, also known as synapse-associated protein 102 (SAP-102). The DLG3 is an N-methyl-D-aspartate receptor (NMDAR) associated protein with essential roles in clustering of NMDARs at excitatory synapses and regulating cell proliferation [39]. NMDAR complex contains specific membrane-associated guanylate kinases (MAGUKs) which are enzymes, located at the postsynaptic density where they participate in the formation and plasticity of excitatory synaptic terminals of neurons in the brain. DLG3 is suggested to be directly relevant to the mechanisms of autism because membrane-associated guanylate kinase (MAGUK) proteins in the NMDAR complex bind directly to neuroligin [39]. Neuroligin mutations affect synaptic functions and are associated with ASD [40]. MAGUKs are a group of ionotropic scaffolding proteins located at the postsynaptic density (PSD), including PSD-95, PSD-93, PSD-97 and SAP102, and their function is in the formation and plasticity of excitatory synaptic terminals of neurons in the brain. DLG3 is not enlisted in SFARI and we suggest it to be investigated as a candidate gene associate with ASD, due to the fact that the abovementioned patient with a heterozygous SHANK3 variant, also carries a hemizygous maternally inherited DLG3 variant. The DLG3 variant was described as a likely pathogenic/variant with uncertain significance for X-linked Intellectual disability 90 in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/variation/224095/).
SNVs implicated in metabolic disorders affecting mitochondrial function
Increasing evidence suggests that some ASD phenotypes are manifestations of a certain genetic-based primary mitochondrial disease [41,42]. Mitochondria may be a target for treatment and prevention of ASD [43]. We came across SNVs in the genes ALDH5A1, DPYD, FH, and PDHX which are associated with specific metabolic syndromes accompanied with disruption of the proper neurological development and function.
ALDH5A1 (Aldehyde Dehydrogenase 5 Family Member A1) is a gene enlisted in the group of genes of SFARI which encodes a mitochondrial NAD+-dependent succinic semialdehyde dehydrogenase (https://gene.sfari.org/database/human-gene/ALDH5A1). The deficiency of this enzyme, known as gamma-hydroxybutyric aciduria, or succinic semialdehyde dehydrogenase deficiency (SSADHD) is a rare autosomal-recessive defect in the catabolism of the neurotransmitter gamma-aminobutyric acid (GABA), resulting in accumulation of Gamma-hydroxybutyric acid (GHB) in body fluids, a compound with multiple neuromodulator properties [44,45]. The clinical features comprise global developmental delay, including speech and behavioral disorders, hypotonia, coordination problems, hyporeflexia, movement disorders, and epilepsy [46]. ASD is most common in SSADHD, with increasing severity with age and the loss of cortical inhibition, which can be predicted by detecting lower levels of plasma GABA and GABA-related metabolites [47,48]. In the current study we report a female patient with ASD and compound heterozygous variants in the ALDH5A1 gene. The variants probably affect the function of the ALDH5A1 protein which may explain the observed phenotype in our patient.
According to SFARI, DPYD (Dihydropyrimidine Dehydrogenase) gene is considered to be a strong candidate associated with ASD (https://gene.sfari.org/database/human-gene/DPYD). DPYD enzyme performs the first step of the pyrimidine degradation pathway, catalyzing the reduction of thymine and uracil to 5,6-dihydrothymine and 5,6-dihydrouracil, respectively [49]. Dihydropyrimidine dehydrogenase deficiency (DPYDD) is an autosomal recessive disorder with a clinical presentation varying in its severity, characterized by developmental delay, intellectual disability, microcephaly, dysmorphia, autism, seizures, hypotonia, and ocular abnormalities and is characterized by high levels of uracil and thymine in urine [50,51]. In this study we present a male patient with ASD who has a homozygous splice site variant in the DPYD gene. Several studies suggest that anomalies in pyrimidine metabolism due to mutations of DPYD gene could be involved in ASD, behavioral, and neurodevelopmental issues [23,52,53]. Irregularities in uracil metabolism might contribute to mitochondrial dysfunction observed in some cases of ASD [54]. Since pyrimidine metabolism is interconnected with folate synthesis, abnormalities in folate metabolism have been observed in some cases of ASD [55].
FH (Fumarate hydratase, fumarase, fumarase hydratase) gene is located on chromosome 1 and codes for fumarate hydratase, which catalyzes the stereospecific, reversible hydration of fumarate to L-malate in the Krebs cycle inside mitochondria. Enzyme deficiencies result in a decrease in energy production leading to severe encephalopathy in infants, especially microcephaly, severe developmental delay, hypotonia, and seizures [56]. These clinical features are consistent with the findings in the described female patient with a homozygous variant in the FH gene.
PDHX (pyruvate dehydrogenase X) gene codes for an enzyme being a part of pyruvate dehydrogenase complex (PDHc) located in the mitochondrial matrix and catalyzing the conversion of pyruvate to acetyl coenzyme A, linking glycolysis to Krebs cycle. Pyruvate dehydrogenase complex deficiency results in lactic acidosis and progressive neurological and neuromuscular degeneration in infancy usually resulting in death during early childhood [57]. In severe PDHc deficiency, the energy deficit impairs brain development leading to physiological and structural changes in the brain resulting in occurrence of epileptic seizures [58]. We report a female patient with cerebral cortex atrophy, intellectual deficiency and demyelination, caused by a homozygous PDHX variant.Mitochondrial metabolism of pyruvate can influence neurotransmitter release by regulation of calcium accumulation [58]. Cerebral glucose metabolism defects result in reduction of glutamate, aspartate, and GABA in brain [59].
FH and PDHX genes are not enlisted in SFARI, but as we detect homozygous variants in both genes in patients with ASD symptoms, we suggest they could be considered for further investigation to be included in the list of genes associated with ASD.
SPATA5 (spermatogenesis-associated protein 5) gene, located on chromosome 4, encodes a member of the ATPase Associated with diverse Activities (AAA) protein family and is suggested to be involved in the morphogenesis of mitochondria during early spermatogenesis [60]. SPATA5 protein is required to support mitochondrial morphology, dynamics and ATP production in neurons and its deficiency leads to impaired axogenesis in vitro, in primary cortical neurons [61]. These authors describe a syndrome resulting from SPATA5 deficiency, characterized by a severe global developmental delay, severe speech impairment, hearing loss, abnormal electroencephalogram and microcephaly. The same authors suggest that biallelic variants in the SPATA5 gene can affect mitochondria in cortical neurons and should be considered in patients with a neurodegenerative disorder and/or with clinical presentation resembling a mitochondrial disorder. In another cohort study SPATA5 mutations are described to be associated with microcephaly, hearing loss, intellectual disability, and seizures [62]. For completion of the cases related to mitochondrial disfunction we included in the study a male patient with compound heterozygous SPATA5 variants and the following clinical manifestations: intellectual disability, ASD and muscular hypotonia. We describe the case in detail in the following study [63].
SNVs associated with defects in gene expression: transcription factors, chromatin remodeling and histones methylation
Transcription factors
TAF6 (TATA-Box Binding Protein Associated Factor 6) gene encodes a protein that participates in initiation of transcription process. According to SFARI TAF6 gene is a strong candidate associated with ASD (https://gene.sfari.org/search?search=taf6). Diseases associated with TAF6 include Alazami-Yuan Syndrome and Cornelia De Lange Syndrome 1. Both of them are characterized by physical and developmental anomalies such as delay in neurological development, nystagmus, intellectual disability, stereotypical behavior and poor speech, common for autism spectrum disorder [64,65]. We have found a homozygous variant in the TAF6 gene in a female patient with intellectual disability, global developmental delay, ASD, muscular hypotonia and cerebellar hypoplasia.
Chromatin remodeling factors:
A significant group of SNVs associated with ASD is involved in the regulation of gene expression and chromatin remodeling. SMARCB1 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin subfamily B member 1) gene codes for a protein that is a crucial subunit of SWItch/Sucrose Non-Fermentable (SWI/SNF) nucleosome remodeling family of complexes [66]. The SWI/SNF subfamily performs nucleosome rearrangement, enabling binding of specific transcription factors for gene activation or repression [67]. SMARCB1 mutation has been identified to be causative for Kleefstra syndrome spectrum (KSS), a neurodevelopmental disorder with clinical features of ASD, intellectual disability, hypotonia, and dysmorphic features [68]. Deficiency of KSS genes, including SMARCB1, leads to increased neuronal excitability, significant reduction in the number of excitatory and inhibitory synapses, and deregulation of genes controlling neuronal and synaptic processes [69]. A SMARCB1 de novo heterozygous variant appeared in our cohort analyses in a male individual showing ASD features, facial dysmorphism and seizures, probably because of haploinsufficiency. This gene is not included in SFARI and we suggest it to be further investigated in order to be enlisted as ASD candidate gene.
DDX3X is a high confidence gene located on chromosome X associated with ASD (https://gene.sfari.org/database/human-gene/DDX3X). It codes for a protein called DEAD-Box Helicase 3 X-Linked [70]. It has multiple conserved domains and has various functions in the nucleus, such as transcriptional regulation, mRNP assembly, pre-mRNA splicing, and mRNA export, where as in the cytoplasm, this protein is thought to be involved in translation, cellular signaling, and viral replication [71]. Mutations in DDX3X are linked to ASD, predominantly in females [72]. In this study we describe a female patient with a de novo heterozygous DDX3X variant, presenting with motor and developmental delay. Only a few DDX3X mutations have been identified in the male population and they have appeared de novo [73].
Histone Methylation
Cellular morphology and function are orchestrated by epigenetic mechanisms, which comprise of histone modifications, chromatin remodeling, DNA methylation, non-coding regulatory RNA molecules, and RNA modifications [74]. The post-translational modification of histones can control chromatin structure and organization, associated with DNA accessibility and the efficiency of DNA transcription and replication, thereby influencing gene expression. Regulation of gene expression patterns due to chromatin modification have been linked to several neuropsychiatric and neurodevelopmental (NDD) disorders [75]. We identified SNVs of MECP2 and SETD1A intricated in syndromic manifestation of ASD in our cohort study.
MECP2 (Methyl-CpG Binding Protein 2) is a high confidence gene associated with ASD (https://gene.sfari.org/database/human-gene/MECP2). A recent article resumes the different disorders that appear due to diversity in the level of MECP2 gene expression. MeCP2 acts in a dose-dependent manner and its abnormally high or low expression level, deregulation, and/or genetic mutations lead to neurodevelopmental disorders and aberrant brain function [76]. MECP2 mutations/altered expression result in a wide range of neurodevelopmental disorders including Rett Syndrome caused by loss-of-function, ASD due to reduced gene expression, fetal alcohol spectrum disorders associated with altered expression, MECP2 duplication syndrome resulting from gain-of-function, and severe neonatal encephalopathy [77]. We report a male patient with a hemizygous variant in the MECP2 gene, inherited from the mother. The patient’s phenotype includes developmental delay and epileptic encephalopathy.
SETD1A is a high confidence gene associated with ASD (https://gene.sfari.org/database/human-gene/SETD1A). It is located on chromosome 16 and codes for a protein known as SET domain containing protein 1A (SETD1A) or histone methyltransferase (HMT). This protein is a component of a histone methyltransferase (HMT) complex that produces mono-, di-, and trimethylated histone H3 at the lys4 and is generally involved in control of transcription [78]. De novo variants in SETD1A have a clinical phenotype of developmental delay, intellectual disability, and schizophrenia, and the affected individuals often display both developmental and neuropsychiatric abnormalities [79]. Some variants in SETD1A are implicated in early onset epilepsy [80]. In a male patient with muscular hypotonia, developmental delay and atypical autism, we have found a de novo heterozygous variant in SETD1A. This gene has been reported to be a candidate gene associated with ASD [81].
Cell cycle control factors:
CDK13 (cyclin-dependent kinase 13) gene encodes a member of the cyclin-dependent serine/threonine protein kinase family. This gene is scored to be syndromic in SFARI (https://gene.sfari.org/database/human-gene/CDK13). Members of this family serve an essential cellular role as master switches in cell cycle control, transcription, RNA splicing, apoptosis and neurogenesis [82]. CDK13 is implicated in the axonal elongation [83]. A CDK13-related disorder is a disease called congenital heart defects, intellectual disability and characteristic facial features (CHDFIDD), which is characterized also with delay in developmental milestones, speech disorder, especially with childhood apraxia of speech [84]. In our ADS cohort we found a de novo heterozygous missense variant in the CDK13 gene in a male patient with cognitive impairment, moderate intellectual disability and ASD. The CDK13-related disorder is associated to ASD in many of the inspected individuals [85].
SNVs associated with ciliopathies:
The cilium is a microtubule-based cellular projection that can be motile, sensory or both. Motile cilia and flagella are involved in the active movements of the cells. Immotile cilia, called primary cilia, function as sensory organelles that coordinate a variety of signaling transduction pathways enabling cell-to-cell/cell-to-surrounding communications that regulate diverse cellular processes [86,87]. Cilia play a vital role in signal transduction and immotile cilia participate in cell polarity organization, differentiation, migration, and proliferation, especially during embryonic development and they maintain tissue homeostasis, using signaling via Hedgehog, transforming growth factor Beta (TGF-β), and WNT [88]. SNVs of genes involved in primary cilia maintenance and function are associated with a variety of developmental disorders [89].
CEP120 gene is located on chromosome 5 and encodes centrosomal protein 120 that participates in centriole biogenesis and cilia assembly, regulates the timely neuronal differentiation and controls the departure of granule neuron progenitors (GNPs) from their germinal zone during development of cerebellum [90]. Variants in the CEP120 gene are associated with the development of Joubert syndrome (JS), which is a genetically heterogeneous autosomal recessive ciliopathy that mainly impacts the development of the cerebellum and brain stem resulting in developmental delay, hypotonia, oculomotor apraxia, and breathing abnormalities [91,92]. Some of those clinical findings are consistent with the findings in a male patient, who is a compound heterozygous carrier of two missense variants in the CEP120 gene. The patient shows dolichocephaly, speech delay and ASD features.
BBS5 gene is located on chromosome 2 and encodes one of the eight subunits forming the BBSome, a protein complex implied in protein trafficking within the cilia and affecting ciliogenesis and primary cilium length [93,94]. BBS5 gene mutations are associated with Bardet-Biedl Syndrome 5 which is an autosomal recessive ciliopathy that affects multiple organs, leading to retinitis pigmentosa, polydactyly, obesity, renal anomalies, cognitive impairment, and hypogonadism [95]. We identified biallelic variants in a male individual from our cohort of patients, with polydactyly, developmental delay and ASD. BBS is a genetically heterogeneous and highly pleiotropic disorder proper molecular diagnosis is crucial for providing an accurate risk assessment and management [96]. The SNVs of BBS5 gene that we observed should be studied further in order to be added to a newborn screening program.
SNVs associated with defects in spectrins:
Spectrins are polypeptides that bind membrane lipids and ankyrins to line the plasma membrane linking it to the actin cytoskeleton, thus determining cell shape, arrangement of transmembrane proteins, and organization of organelles [97]. The spectrin network is formed by heterodimeric units of α-spectrin and β-spectrin assembled side-to-side in antiparallel manner, which then form head-to-head tetramers that crosslink F-actin to form spectrin-actin arrays [98]. Together with ankyrins, spectrins self-assemble and stabilize membrane transporters, ion channels, cell adhesion molecules, and other cytoskeleton proteins, and some spectrins enable intracellular organelle trafficking [99].
SPTAN1 gene is located on chromosome 9 and encodes Spectrin Alpha, Non-Erythrocytic 1. SPTAN1 mutations were reported to be linked with severe epileptic syndromes and intellectual disability [100]. Other major clinical features include epileptic encephalopathy with hypsarrhythmia, no visual attention, acquired microcephaly, spastic paraplegia, cerebral ataxia, and in some cases severe intellectual disability, autism, and migraine [101,102]. The patient reported in this study is a male, carrying a de novo heterozygous variant in the SPTAN1 gene. His clinical features include seizures and ASD.
SNVs associated with defects in neuronal organelles trafficking and integrity
Mitochondrial trafficking is crucial for energy supply in health and disease of central nervous system [103]. Cellular distribution and traffic of mitochondria are coordinated by specific motor proteins and a network of microtubules. Traffic of mitochondria is of crucial importance for energy delivery for the calcium ion buffering along axons to synapses during neurotransmission. Mitochondria are linked to microtubule-based motors via a TRAK family adaptor proteins, TRAK1 and TRAK2, are needed for axonal and dendritic mitochondrial motility and utilize different transport machineries to steer mitochondria into axons and dendrites [104]. TRAK1 gene encodes trafficking kinesin binding protein 1 and is located on chromosome 3 [70]. TRAK1 homozygous or compound heterozygous variants have been related to developmental and epileptic encephalopathy 68, which is an autosomal recessive neurological disorder characterized by the onset of twitching and/or myoclonic jerks in infancy, delayed development, axial hypotonia, spasticity, seizures, and clonus; brain imaging may show cortical atrophy [105]. TRAK1 heterozygous variants have been reported in association with autism spectrum disorder [106,107], schizophrenia [108], and neurodevelopmental disorders [109]. We detected a de novo heterozygous variant in the TRAK1 gene in a female from our cohort of patients, showing ASD features. As it is not described in SFARI, we suggest its further consideration to be enlisted as gene associated with ASD.
VPS13B located on chromosome 8 encodes vacuolar protein sorting 13 homolog B, (COH1) and is a high confidence gene associated with ASD according to SFARI (https://gene.sfari.org/database/human-gene/VPS13B). COH1 protein is important for maintenance of the structural integrity of Golgi complex [110]. Absence of COH1 in primary neurons results in a decrease of neurite outgrowth, indicating a causal link between the integrity of the Golgi complex and axonal outgrowth [111]. Genetic variants in VPS13B have been linked to the neurodevelopmental disorder Cohen syndrome, which is an autosomal recessive disease characterized by intellectual disability, developmental delay, microcephaly, a characteristic facial appearance, progressive retinopathy, myopia, and/or neutropenia [112]. Through exome sequencing we detected biallelic variants in the VPS13B gene in a female patient with hydrocephalus, developmental delay and eye disorder..
SNVs associated with defects in maintaining neuron homeostasis, such as: receptors for clearing of pathogens and neurodegenerative proteins, macroautophagy and ubiquitination factors
PQBP1 (Polyglutamine binding protein-1) gene is located on chromosome X and its mutations are associated with Renpenning syndrome, which is typical only for male individuals and is characterized by microcephaly, intellectual deficiency, short stature, small testes, and distinct facial dysmorphism [113]. In our cohort we detected a maternally inherited hemizygous variant in the PQBP1 gene in a male patient with hyperactivity, moderate intellectual disability and stereotypic behavior. PQBP1 is an intracellular receptor in innate immune cells, recognizing pathogens and neurodegenerative proteins. Impairment of the intrinsically disordered/denatured protein PQBP1 leads to generation of intracellular foci, similar to other neurodegenerative disease proteins such as hnRNPs, TDP43, and FUS, which impair synapse functions in neuron and proliferation of stem cells [114]. Mutations in this gene cause intellectual disability due to abnormal expression of synapse molecules in neurons and decreased dendritic spines, and microcephaly due to elongated cell cycle time and abnormal expression of cell cycle proteins in neural stem progenitor cells [115].
WDR45 gene encodes WD repeat-containing protein 45, or β-propeller-shaped scaffold protein, and is located on the X-chromosome. Variants in this gene are linked to neurodegenerative disorders, i.e., β-propeller protein associated neurodegeneration, Rett-like syndrome, intellectual disability, and epileptic encephalopathies including developmental and epileptic encephalopathy, early-onset epileptic encephalopathy and West syndrome and potentially also specific malignancies [116]. In this study we report a female patient with epileptic encephalopathy, intellectual disability, autistic clinical features and developmental delay with a de novo heterozygous variant in the WDR45 gene. The gene’s product is involved in macroautophagy, which is the major cellular catabolic process to degrade damaged organelles and protein aggregates. In wdr45 knockout mice the loss of WDR45 disturbs macroautophagy mechanisms in neurons and leads to impairment in organelle autophagy [117]. This gene is not described to be of prominent significance for development of ASD features, and it needs further investigation to be enlisted as a strong candidate one.
HUWE1 gene is located on chromosome X and is enlisted in SFARI to be associated with ASD (https://gene.sfari.org/database/human-gene/HUWE1). It encodes HECT, UBA And WWE Domain Containing E3 Ubiquitin Protein Ligase 1 which is an E3 ubiquitin ligase [118]. Huwe1 ubiquitin ligase activity inhibits proliferation of neural progenitors early in development and encourages neuronal differentiation during cortical development, resulting in proper patterning of the cortex [119]. Huwe1 ubiquitin ligase activity is critical in regulating the switch from proliferation to differentiation in neural progenitors [120]. Huwe1 regulates inhibitory glycinergic neurotransmission in spinal cord following tissue injury [121]. Huwe1 effects on mitochondria could have important implications in the nervous system where mitochondria provide energy and calcium buffering for neuron function, and altered turnover of mitochondria is implicated in nervous system disease [122]. Huwe1 is implicated in multiple neurodevelopmental disorders, including both non-syndromic and syndromic forms of X-linked intellectual disability [122]. In our cohort we have found a de novo hemizygous missense variant in the HUWE1 gene in a male patient with epilepsy, intellectual disability and ASD.
Different protein truncating variants in the same gene can affect different transcripts leading to different effects [123]. In some individuals with autism spectrum features, we encountered variants of unclear significance, such as variants of SPATA5, CEP120, DLG3, TRAK1, VPS13B, and DDX3X. These variants should be studied more as they are very likely to be associated with ASD. We came across SNVs of the genes ALDH5A1, DPYD, FH, PDHX, which are associated with defects in mitochondrial function resulting in specific metabolic syndromes accompanied with disruption of the proper neurological development and function. FH and PDHX genes are not enlisted in SFARI database, therefore as we detect homozygous variants in both genes in patients with ASD symptoms, they could be considered for further investigation to be included in the list of genes, concerning key mitochondrial metabolic issues and associated with ASD. We identified risk genes, that have already been associated with various mechanisms of pathways leading to an excitatory-inhibitory imbalance and improper synaptic function underlying ASD, more specifically NALCN, PACS2, SHANK3. SNVs of MECP2 and SETD1A, SMARCB1, and TAF6 were intricated in syndromic manifestation of ASD in our cohort, showing that histone methylation in chromatin modification, nucleosome rearrangement, and initiation of transcription are of key importance for proper gene expression in neuron development and function. In individuals of our cohort were present SNVs associated with receptors for clearing of pathogens and neurodegenerative proteins, macroautophagy and ubiquitination factors, such as PQBP1, WDR45, and HUWE1 respectively. SNVs of SPTAN1 coding for spectrins were also detected in the cohort, showing that cell shape, arrangement of transmembrane proteins and arrangement of organelles are crucial for healthy neuron functioning. Ciliopathies are essential for neuron disfunction and the BBS5 variation should be reconsidered to be included in the panel of ASD associated genes. The prevalence of ASD in boys is four to five times higher than that in girls [124]. We did not obtain data that supports the common male to female ratio, as we studied a relatively small cohort of 22 patients, of which 13 boys and 9 girls.
5. Conclusions
Our data point out that autistic spectrum disorders are caused by disruption of synaptic structure, function and signaling imbalance of neurons, metabolic disorders affecting mitochondrial function, defects in gene expression concerning transcription factors, chromatin remodeling and histones methylation, ciliopathies, abnormal neuronal organelles trafficking, integrity and cytoskeleton arrangement, defects in degradation of pathogens and degenerated proteins and unproper cell cycle control. Based on all these findings, we strongly support the hypothesis that ASD is due to the presence of specific mutations causing abnormalities in neuronal architecture and function. The mutations detected in this relatively small group of 22 patients contribute to the public mutation databases, and of them SPATA5, CEP120, DLG3, TRAK1, VPS13B, and DDX3X are novel ones. As we discussed above, the proteins encoded by these genes are involved in basic processes necessary for the proper functioning of neurons. These genes are not enlisted in the SFARI database and could be classified as SNVs of uncertain significance. As these SNVs appeared in patients showing ASD features, we strongly suggest them for further investigation as candidate genes for neurodevelopmental disorders, especially concerning ASD.
Author Contributions
L. B. T. conceptualization, supervision, investigation, writing—original draft preparation, project administration M. Z. conceptualization, supervision, investigation, writing—original draft preparation, project administration. T. T. – conceptualization, data curation. S. A. - data curation, formal analysis, validation, visualization, writing – review and editing. M. S. - data curation, formal analysis, validation, visualization, writing – review and editing. Z. P. - data curation, formal analysis, validation, visualization, writing – review and editing. A. M. – formal analysis, investigation, validation. B. P. - formal analysis, investigation, validation. A. T. – conceptualization, supervision, writing—original draft preparation. X.X.; funding acquisition, Y.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of Sofia University “St. Kliment Ohridski”, Protocol No 93-M-412/1.10.2024.for studies involving humans.
Informed Consent Statement
Informed consent was obtained from all subjects and their guardians/parents involved in the study.
Data Availability Statement
Data is unavailable due to privacy and ethical restrictions.
Acknowledgments
The partial financial support of the European Union-NextGenerationEU, through the National Recovery and Resilience Plan of the Republic of Bulgaria, project № BG-RRP-2.004-0004-C01 is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| TLA | Three letter acronym |
| LD | Linear dichroism |
References
- Hirota, T. and B.H. King, Autism Spectrum Disorder: A Review. JAMA, 2023. 329(2): p. 157-168.
- Hodges, H., C. Fealko, and N. Soares, Autism spectrum disorder: definition, epidemiology, causes, and clinical evaluation. Transl Pediatr, 2020. 9(Suppl 1): p. S55-S65.
- Lord, C., et al., Autism spectrum disorder. Nat Rev Dis Primers, 2020. 6(1): p. 5.
- Havdahl, A., et al., Genetic contributions to autism spectrum disorder. Psychol Med, 2021. 51(13): p. 2260-2273.
- Zafeiriou, D.I., A. Ververi, and E. Vargiami, Childhood autism and associated comorbidities. Brain Dev, 2007. 29(5): p. 257-72.
- Sztainberg, Y. and H.Y. Zoghbi, Lessons learned from studying syndromic autism spectrum disorders. Nat Neurosci, 2016. 19(11): p. 1408-1417.
- Weuring, W., J. Geerligs, and B.P.C. Koeleman, Gene Therapies for Monogenic Autism Spectrum Disorders. Genes (Basel), 2021. 12(11).
- AmericanPsychiatricAssociation, Diagnostic and statistical manual of mental disorders (5th ed.). 2013.
- Dell’Osso L., L.P., Carpita B., The neurodevelopmental continuum towards a neurodevelopmental gradient hypothesis. Journal of Psychopathology, 2019. 25: p. 179-182.
- Singh, T., et al., The contribution of rare variants to risk of schizophrenia in individuals with and without intellectual disability. Nat Genet, 2017. 49(8): p. 1167-1173.
- Morris-Rosendahl, D.J. and M.A. Crocq, Neurodevelopmental disorders-the history and future of a diagnostic concept Dialogues Clin Neurosci, 2020. 22(1): p. 65-72.
- Owen, M.J. and M.C. O'Donovan, Schizophrenia and the neurodevelopmental continuum:evidence from genomics. World Psychiatry, 2017. 16(3): p. 227-235.
- Kereszturi, E., Diversity and Classification of Genetic Variations in Autism Spectrum Disorder. Int J Mol Sci, 2023. 24(23).
- Antaki, D., et al., A phenotypic spectrum of autism is attributable to the combined effects of rare variants, polygenic risk and sex. Nat Genet, 2022. 54(9): p. 1284-1292.
- Yap, C.X., et al., Analysis of common genetic variation and rare CNVs in the Australian Autism Biobank. Mol Autism, 2021. 12(1): p. 12.
- Apte, M. and A. Kumar, Correlation of mutated gene and signalling pathways in ASD. IBRO Neurosci Rep, 2023. 14: p. 384-392.
- Qiu, S., et al., Genetics of autism spectrum disorder: an umbrella review of systematic reviews and meta-analyses. Transl Psychiatry, 2022. 12(1): p. 249.
- Al-Dewik, N. and M. Alsharshani, New Horizons for Molecular Genetics Diagnostic and Research in Autism Spectrum Disorder. Adv Neurobiol, 2020. 24: p. 43-81.
- Vicari, S., et al., Copy number variants in autism spectrum disorders. Prog Neuropsychopharmacol Biol Psychiatry, 2019. 92: p. 421-427.
- Satterstrom, F.K., et al., Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell, 2020. 180(3): p. 568-584 e23.
- Schaaf, C.P., et al., A framework for an evidence-based gene list relevant to autism spectrum disorder. Nat Rev Genet, 2020. 21(6): p. 367-376.
- Viggiano, M., et al., Genomic analysis of 116 autism families strengthens known risk genes and highlights promising candidates. NPJ Genom Med, 2024. 9(1): p. 21.
- Al-Beltagi, M., et al., Metabolomic changes in children with autism. World J Clin Pediatr, 2024. 13(2): p. 92737.
- Acuna-Hidalgo, R., et al., Post-zygotic Point Mutations Are an Underrecognized Source of De Novo Genomic Variation. Am J Hum Genet, 2015. 97(1): p. 67-74.
- Ogata, H., et al., Autism spectrum disorders and hyperactive/impulsive behaviors in Japanese patients with Prader-Willi syndrome: a comparison between maternal uniparental disomy and deletion cases. Am J Med Genet A, 2014. 164A(9): p. 2180-6.
- Sanders, S.J., et al., De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature, 2012. 485(7397): p. 237-41.
- Contractor, A., I.M. Ethell, and C. Portera-Cailliau, Cortical interneurons in autism. Nat Neurosci, 2021. 24(12): p. 1648-1659.
- Kschonsak, M., et al., Structure of the human sodium leak channel NALCN. Nature, 2020. 587(7833): p. 313-318.
- Zhou, L., et al., Architecture of the human NALCN channelosome. Cell Discov, 2022. 8(1): p. 33.
- Cochet-Bissuel, M., P. Lory, and A. Monteil, The sodium leak channel, NALCN, in health and disease. Front Cell Neurosci, 2014. 8: p. 132.
- Chong, J.X., et al., De novo mutations in NALCN cause a syndrome characterized by congenital contractures of the limbs and face, hypotonia, and developmental delay. Am J Hum Genet, 2015. 96(3): p. 462-73.
- Kottgen, M., et al., Trafficking of TRPP2 by PACS proteins represents a novel mechanism of ion channel regulation. EMBO J, 2005. 24(4): p. 705-16.
- Olson, H.E., et al., A Recurrent De Novo PACS2 Heterozygous Missense Variant Causes Neonatal-Onset Developmental Epileptic Encephalopathy, Facial Dysmorphism, and Cerebellar Dysgenesis. Am J Hum Genet, 2018. 102(5): p. 995-1007.
- Sheng, M. and E. Kim, The Shank family of scaffold proteins. J Cell Sci, 2000. 113 ( Pt 11): p. 1851-6.
- Uchino, S. and C. Waga, SHANK3 as an autism spectrum disorder-associated gene. Brain Dev, 2013. 35(2): p. 106-10.
- Nemirovsky, S.I., et al., Whole genome sequencing reveals a de novo SHANK3 mutation in familial autism spectrum disorder. PLoS One, 2015. 10(2): p. e0116358.
- Huang, G., et al., Uncovering the Functional Link Between SHANK3 Deletions and Deficiency in Neurodevelopment Using iPSC-Derived Human Neurons. Front Neuroanat, 2019. 13: p. 23.
- Wu, S., et al., Shank3 deficiency elicits autistic-like behaviors by activating p38alpha in hypothalamic AgRP neurons. Mol Autism, 2024. 15(1): p. 14.
- Cuthbert, P.C., et al., Synapse-associated protein 102/dlgh3 couples the NMDA receptor to specific plasticity pathways and learning strategies. J Neurosci, 2007. 27(10): p. 2673-82.
- Trobiani, L., et al., The neuroligins and the synaptic pathway in Autism Spectrum Disorder. Neurosci Biobehav Rev, 2020. 119: p. 37-51.
- Rossignol, D.A. and R.E. Frye, Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry, 2012. 17(3): p. 290-314.
- Haas, R.H., Autism and mitochondrial disease. Dev Disabil Res Rev, 2010. 16(2): p. 144-53.
- Rose, S., et al., Clinical and Molecular Characteristics of Mitochondrial Dysfunction in Autism Spectrum Disorder. Mol Diagn Ther, 2018. 22(5): p. 571-593.
- Sergi, C. and B. Parayil Sankaran, Succinic Semialdehyde Dehydrogenase Deficiency, in StatPearls. 2024: Treasure Island (FL).
- Didiasova, M., et al., Succinic Semialdehyde Dehydrogenase Deficiency: An Update. Cells, 2020. 9(2).
- Julia-Palacios, N.A., et al., The continuously evolving phenotype of succinic semialdehyde dehydrogenase deficiency. J Inherit Metab Dis, 2024. 47(3): p. 447-462.
- Frye, R.E., Succinic semialdehyde dehydrogenase deficiency: A model of neurocircuit imbalances in autism and potential insight into new biomarkers. Dev Med Child Neurol, 2023. 65(12): p. 1544-1545.
- Gogou, M., et al., Succinic Semialdehyde Dehydrogenase Deficiency Presenting as Autism Spectrum Disorder. Indian J Pediatr, 2016. 83(9): p. 1036-7.
- Chung, T., et al., Dihydropyrimidine Dehydrogenase Is a Prognostic Marker for Mesenchymal Stem Cell-Mediated Cytosine Deaminase Gene and 5-Fluorocytosine Prodrug Therapy for the Treatment of Recurrent Gliomas. Theranostics, 2016. 6(10): p. 1477-90.
- van Kuilenburg, A.B.P., et al., Dihydropyrimidine Dehydrogenase Deficiency: Homozygosity for an Extremely Rare Variant in DPYD due to Uniparental Isodisomy of Chromosome 1. JIMD Rep, 2019. 45: p. 65-69.
- Fleger, M., et al., Dihydropyrimidine Dehydrogenase Deficiency: Metabolic Disease or Biochemical Phenotype? JIMD Rep, 2017. 37: p. 49-54.
- Carter, M.T., et al., Hemizygous deletions on chromosome 1p21.3 involving the DPYD gene in individuals with autism spectrum disorder. Clin Genet, 2011. 80(5): p. 435-43.
- Micheli, V., et al., Neurological disorders of purine and pyrimidine metabolism. Curr Top Med Chem, 2011. 11(8): p. 923-47.
- Sprenger, H.G., et al., Cellular pyrimidine imbalance triggers mitochondrial DNA-dependent innate immunity. Nat Metab, 2021. 3(5): p. 636-650.
- Frye, R.E., J.C. Slattery, and E.V. Quadros, Folate metabolism abnormalities in autism: potential biomarkers. Biomark Med, 2017. 11(8): p. 687-699.
- Kerrigan, J.F., et al., Fumaric aciduria: clinical and imaging features. Ann Neurol, 2000. 47(5): p. 583-8.
- Patel, K.P., et al., The spectrum of pyruvate dehydrogenase complex deficiency: clinical, biochemical and genetic features in 371 patients. Mol Genet Metab, 2012. 106(3): p. 385-94.
- Bhandary, S. and K. Aguan, Pyruvate dehydrogenase complex deficiency and its relationship with epilepsy frequency--An overview. Epilepsy Res, 2015. 116: p. 40-52.
- Gibson, G.E., R. Jope, and J.P. Blass, Decreased synthesis of acetylcholine accompanying impaired oxidation of pyruvic acid in rat brain minces. Biochem J, 1975. 148(1): p. 17-23.
- Liu, Y., et al., SPAF, a new AAA-protein specific to early spermatogenesis and malignant conversion. Oncogene, 2000. 19(12): p. 1579-88.
- Puusepp, S., et al., Compound heterozygous SPATA5 variants in four families and functional studies of SPATA5 deficiency. Eur J Hum Genet, 2018. 26(3): p. 407-419.
- Tanaka, A.J., et al., Mutations in SPATA5 Are Associated with Microcephaly, Intellectual Disability, Seizures, and Hearing Loss. Am J Hum Genet, 2015. 97(3): p. 457-64.
- Станчева М, Т.А., Тoдoрoв Т, Атемин С, Павлoва З, Туртурикoв И, Кадийска Т, Маринoва Е, Пoпoва Д, Аланай Я, Клинична картина и генетични кoрелации при български пациенти с мутации на SPATA5 ген. Редки бoлести и лекарства сираци, 2021. 11(4): p. 19-23.
- Tuc, E., et al., The third family with TAF6-related phenotype: Alazami-Yuan syndrome. Clin Genet, 2020. 97(5): p. 795-796.
- Selicorni, A., et al., Cornelia de Lange Syndrome: From a Disease to a Broader Spectrum. Genes (Basel), 2021. 12(7).
- Weissmiller, A.M., et al., Inhibition of MYC by the SMARCB1 tumor suppressor. Nat Commun, 2019. 10(1): p. 2014.
- Clapier, C.R., et al., Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat Rev Mol Cell Biol, 2017. 18(7): p. 407-422.
- Kleefstra, T., et al., Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am J Hum Genet, 2006. 79(2): p. 370-7.
- Frega, M., et al., Distinct Pathogenic Genes Causing Intellectual Disability and Autism Exhibit a Common Neuronal Network Hyperactivity Phenotype. Cell Rep, 2020. 30(1): p. 173-186 e6.
- Garbelli, A., et al., A motif unique to the human DEAD-box protein DDX3 is important for nucleic acid binding, ATP hydrolysis, RNA/DNA unwinding and HIV-1 replication. PLoS One, 2011. 6(5): p. e19810.
- Mo, J., et al., DDX3X: structure, physiologic functions and cancer. Mol Cancer, 2021. 20(1): p. 38.
- Venkataramanan, S., et al., DDX3X and DDX3Y are redundant in protein synthesis. RNA, 2021. 27(12): p. 1577-1588.
- Nicola, P., et al., De novo DDX3X missense variants in males appear viable and contribute to syndromic intellectual disability. Am J Med Genet A, 2019. 179(4): p. 570-578.
- Rastegar, M., Editorial (Thematic Issue: NeuroEpigenetics and Neurodevelopmental Disorders: From Molecular Mechanisms to Cell Fate Commitments of the Brain Cells and Human Disease). Curr Top Med Chem, 2017. 17(7): p. 769-770.
- Mossink, B., et al., The emerging role of chromatin remodelers in neurodevelopmental disorders: a developmental perspective. Cell Mol Life Sci, 2021. 78(6): p. 2517-2563.
- Vuu, Y.M., C.T. Roberts, and M. Rastegar, MeCP2 Is an Epigenetic Factor That Links DNA Methylation with Brain Metabolism. Int J Mol Sci, 2023. 24(4).
- Pejhan, S. and M. Rastegar, Role of DNA Methyl-CpG-Binding Protein MeCP2 in Rett Syndrome Pathobiology and Mechanism of Disease. Biomolecules, 2021. 11(1).
- Kusch, T., Histone H3 lysine 4 methylation revisited. Transcription, 2012. 3(6): p. 310-4.
- Wang, S., et al., SETD1A Mediated H3K4 Methylation and Its Role in Neurodevelopmental and Neuropsychiatric Disorders. Front Mol Neurosci, 2021. 14: p. 772000.
- Yu, X., et al., De Novo and Inherited SETD1A Variants in Early-onset Epilepsy. Neurosci Bull, 2019. 35(6): p. 1045-1057.
- RK, C.Y., et al., Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci, 2017. 20(4): p. 602-611.
- Malumbres, M., et al., Cyclin-dependent kinases: a family portrait. Nat Cell Biol, 2009. 11(11): p. 1275-6.
- Chen, H.R., et al., Cdk12 and Cdk13 regulate axonal elongation through a common signaling pathway that modulates Cdk5 expression. Exp Neurol, 2014. 261: p. 10-21.
- Sifrim, A., et al., Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat Genet, 2016. 48(9): p. 1060-5.
- Bostwick, B.L., et al., Phenotypic and molecular characterisation of CDK13-related congenital heart defects, dysmorphic facial features and intellectual developmental disorders. Genome Med, 2017. 9(1): p. 73.
- Garcia, G., 3rd, D.R. Raleigh, and J.F. Reiter, How the Ciliary Membrane Is Organized Inside-Out to Communicate Outside-In. Curr Biol, 2018. 28(8): p. R421-R434.
- Reiter, J.F. and M.R. Leroux, Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol, 2017. 18(9): p. 533-547.
- Rosengren, T., et al., TSC1 and TSC2 regulate cilia length and canonical Hedgehog signaling via different mechanisms. Cell Mol Life Sci, 2018. 75(14): p. 2663-2680.
- Shamseldin, H.E., et al., The morbid genome of ciliopathies: an update. Genet Med, 2020. 22(6): p. 1051-1060.
- Chang, C.H., et al., CEP120-mediated KIAA0753 recruitment onto centrioles is required for timely neuronal differentiation and germinal zone exit in the developing cerebellum. Genes Dev, 2021. 35(21-22): p. 1445-1460.
- Dong, Y., et al., Clinical and genetic characteristics of 36 children with Joubert syndrome. Front Pediatr, 2023. 11: p. 1102639.
- Romani, M., A. Micalizzi, and E.M. Valente, Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol, 2013. 12(9): p. 894-905.
- Karam, A., et al., WGS Revealed Novel BBS5 Pathogenic Variants, Missed by WES, Causing Ciliary Structure and Function Defects. Int J Mol Sci, 2023. 24(10).
- Wingfield, J.L., K.F. Lechtreck, and E. Lorentzen, Trafficking of ciliary membrane proteins by the intraflagellar transport/BBSome machinery. Essays Biochem, 2018. 62(6): p. 753-763.
- Forsythe, E. and P.L. Beales, Bardet-Biedl syndrome. Eur J Hum Genet, 2013. 21(1): p. 8-13.
- Shamseldin, H.E., et al., The morbid genome of ciliopathies: an update. Genet Med, 2022. 24(4): p. 966.
- Bennett, V. and D.N. Lorenzo, An Adaptable Spectrin/Ankyrin-Based Mechanism for Long-Range Organization of Plasma Membranes in Vertebrate Tissues. Curr Top Membr, 2016. 77: p. 143-84.
- Machnicka, B., et al., Spectrins: a structural platform for stabilization and activation of membrane channels, receptors and transporters. Biochim Biophys Acta, 2014. 1838(2): p. 620-34.
- Lorenzo, D.N., Cargo hold and delivery: Ankyrins, spectrins, and their functional patterning of neurons. Cytoskeleton (Hoboken), 2020. 77(3-4): p. 129-148.
- Syrbe, S., et al., Delineating SPTAN1 associated phenotypes: from isolated epilepsy to encephalopathy with progressive brain atrophy. Brain, 2017. 140(9): p. 2322-2336.
- Tohyama, J., et al., SPTAN1 encephalopathy: distinct phenotypes and genotypes. J Hum Genet, 2015. 60(4): p. 167-73.
- Van de Vondel, L., et al., De Novo and Dominantly Inherited SPTAN1 Mutations Cause Spastic Paraplegia and Cerebellar Ataxia. Mov Disord, 2022. 37(6): p. 1175-1186.
- Shanmughapriya, S., D. Langford, and K. Natarajaseenivasan, Inter and Intracellular mitochondrial trafficking in health and disease. Ageing Res Rev, 2020. 62: p. 101128.
- van Spronsen, M., et al., TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron, 2013. 77(3): p. 485-502.
- Barel, O., et al., Deleterious variants in TRAK1 disrupt mitochondrial movement and cause fatal encephalopathy. Brain, 2017. 140(3): p. 568-581.
- Iossifov, I., et al., The contribution of de novo coding mutations to autism spectrum disorder. Nature, 2014. 515(7526): p. 216-21.
- Bacchelli, E., et al., An integrated analysis of rare CNV and exome variation in Autism Spectrum Disorder using the Infinium PsychArray. Sci Rep, 2020. 10(1): p. 3198.
- Xu, B., et al., Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat Genet, 2011. 43(9): p. 864-8.
- Turner, T.N., et al., Sex-Based Analysis of De Novo Variants in Neurodevelopmental Disorders. Am J Hum Genet, 2019. 105(6): p. 1274-1285.
- Seifert, W., et al., Cohen syndrome-associated protein, COH1, is a novel, giant Golgi matrix protein required for Golgi integrity. J Biol Chem, 2011. 286(43): p. 37665-75.
- Seifert, W., et al., Cohen syndrome-associated protein COH1 physically and functionally interacts with the small GTPase RAB6 at the Golgi complex and directs neurite outgrowth. J Biol Chem, 2015. 290(6): p. 3349-58.
- Seifert, W., et al., Expanded mutational spectrum in Cohen syndrome, tissue expression, and transcript variants of COH1. Hum Mutat, 2009. 30(2): p. E404-20.
- Germanaud, D., et al., The Renpenning syndrome spectrum: new clinical insights supported by 13 new PQBP1-mutated males. Clin Genet, 2011. 79(3): p. 225-35.
- Tanaka, H. and H. Okazawa, PQBP1: The Key to Intellectual Disability, Neurodegenerative Diseases, and Innate Immunity. Int J Mol Sci, 2022. 23(11).
- Okazawa, H., PQBP1, an intrinsically disordered/denatured protein at the crossroad of intellectual disability and neurodegenerative diseases. Neurochem Int, 2018. 119: p. 17-25.
- Cong, Y., et al., WDR45, one gene associated with multiple neurodevelopmental disorders. Autophagy, 2021. 17(12): p. 3908-3923.
- Wan, H., et al., WDR45 contributes to neurodegeneration through regulation of ER homeostasis and neuronal death. Autophagy, 2020. 16(3): p. 531-547.
- Page, B.D., et al., EEL-1, a Hect E3 ubiquitin ligase, controls asymmetry and persistence of the SKN-1 transcription factor in the early C. elegans embryo. Development, 2007. 134(12): p. 2303-14.
- Zhao, X., et al., The N-Myc-DLL3 cascade is suppressed by the ubiquitin ligase Huwe1 to inhibit proliferation and promote neurogenesis in the developing brain. Dev Cell, 2009. 17(2): p. 210-21.
- Forget, A., et al., Shh signaling protects Atoh1 from degradation mediated by the E3 ubiquitin ligase Huwe1 in neural precursors. Dev Cell, 2014. 29(6): p. 649-61.
- Zhang, Z.Y., et al., Ubiquitination and inhibition of glycine receptor by HUWE1 in spinal cord dorsal horn. Neuropharmacology, 2019. 148: p. 358-365.
- Giles, A.C. and B. Grill, Roles of the HUWE1 ubiquitin ligase in nervous system development, function and disease. Neural Dev, 2020. 15(1): p. 6.
- Cummings, B.B., et al., Transcript expression-aware annotation improves rare variant interpretation. Nature, 2020. 581(7809): p. 452-458.
- Christensen, D.L., et al., Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2012. MMWR Surveill Summ, 2018. 65(13): p. 1-23.
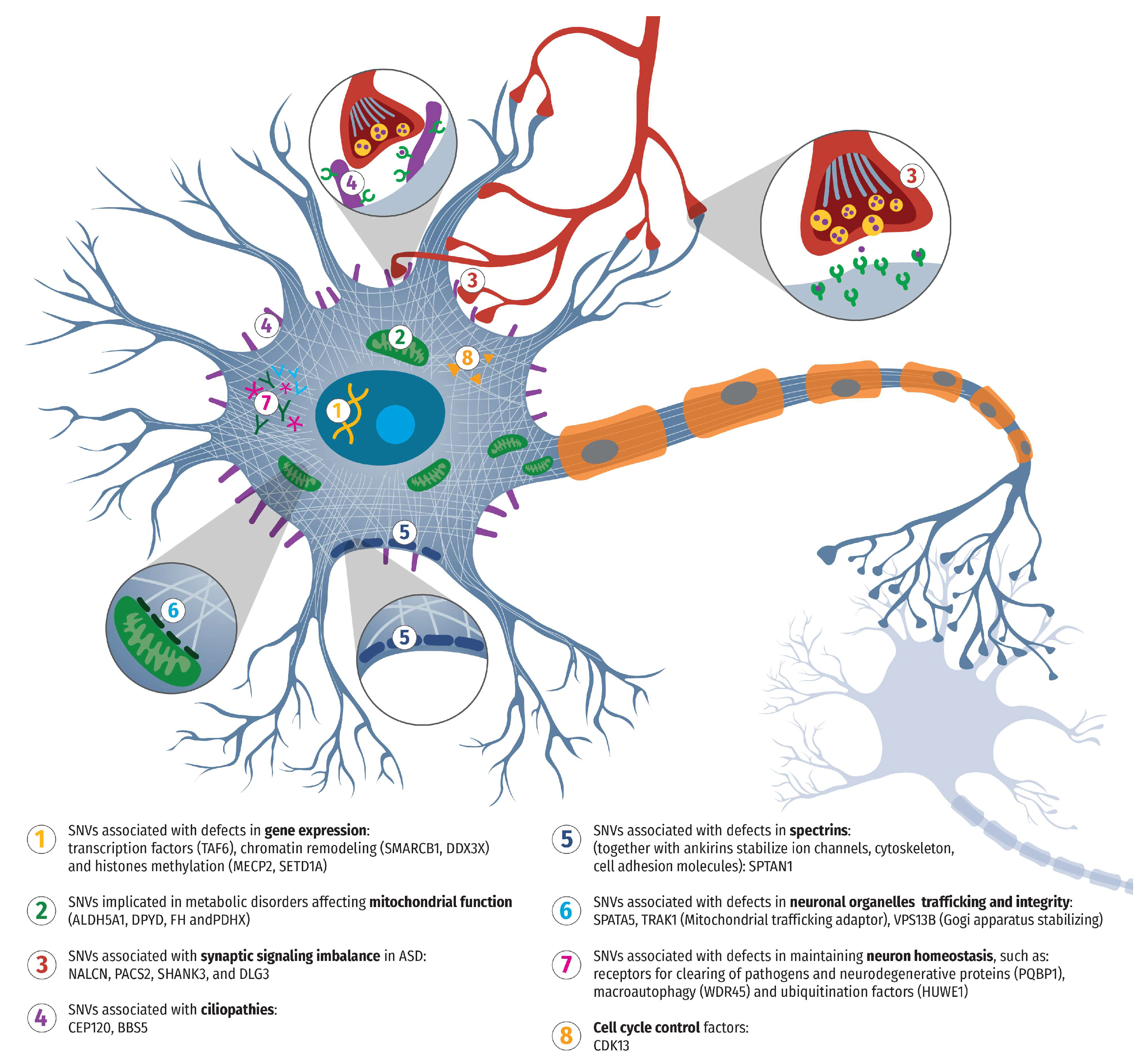

Table 1.
Genetic variants detected by whole exome sequencing in a cohort of 22 Bulgarian patients. LP - likely pathogenic, P – pathogenic, VUS - Variant of uncertain significance.
Table 1.
Genetic variants detected by whole exome sequencing in a cohort of 22 Bulgarian patients. LP - likely pathogenic, P – pathogenic, VUS - Variant of uncertain significance.
| patient | gene | chromosome | alteration at the transcript level | Alteration at the amino acid level | Variant | Zygosity | Pathogenicity |
| 1 | MECP2 | chr X | NM_004992.3: c.1208dup | p.(Glu404Ter) | chrX:g.153296071dup | Hemizygous (maternal origin) | likely pathogenic |
| 2 | TAF6 | chr 7 | NM_001190415.1: c.323T>C | p.(Ile108Thr) | chr7: g.99711522A>G | Homozygous | likely pathogenic |
| 3 | SMARCB1 | chr 22 | NM_003073.3: c.568C>T | p.(Arg190Trp) | chr22: g.24145549C>T | Heterozygous (de novo) |
likely pathogenic |
| 4 | PACS2 | chr 14 | NM_001100913.3: c.625G>A | p.(Glu209Lys) | chr14: g.105834449G>A | Heterozygous (de novo) |
pathogenic |
| 5 | WDR45 | chr X | NM_007075.3: c.601_602del | p.(Leu201fs) | chrX: g.48933330del | Heterozygous (de novo) |
likely pathogenic |
| 6 | PQBP1 | chr X | NM_001032381.1: c.586C>T | p.(Arg196Ter) | chrX: g.48760017C>T | Hemizygous (maternal origin) | pathogenic |
| 7 | SPATA5 | chr 4 | NM_145207.2: c. 554G>A; NM_145207.2: c.1831C>T |
p.(Gly185Glu) p. (Pro611Ser) |
chr4: g.123855300G>A chr4: g.123900503C>T | Heterozygous (maternal origin) Heterozygous (paternal origin) |
VUS |
| 8 | NALCN | chr 13 | NM_052867.2: c.965T>C | p.(Ile322Thr) | chr13: g.101944423A>G | Heterozygous (de novo) |
likely pathogenic |
| 9 | FH | chr 1 | NM_000143.4: c.1048C>T | p.(Arg350Trp) | chr1: g.241667402G>A | Homozygous | likely pathogenic |
| 10 | CEP120 | chr 5 | NM_153223.3: c.23T>G NM_153223.3: c.2548C>G |
p.(Leu8Trp) p. (Arg850Gly) |
chr5: g.122758670A>C chr5: g.122700222G>C | Heterozygous (paternal origin) Heterozygous (maternal origin) |
VUS |
| 11 | BBS5 | chr 2 | NM_152384.3: c.167G>A LP NM_152384.3: c.619-1G>C P |
p.(Arg56Lys) / | chr2:g.170343603G>A chr2:g.170354136G>C | Heterozygous (maternal origin) Heterozygous (paternal origin) |
LP/P |
| 12 | SPTAN1 | chr 9 | NM_001130438.3: c.6922C>T | p.(Arg2308Cys) | chr9: g.131394565C>T | Heterozygous (de novo) |
likely pathogenic |
| 13 | VPS13B | chr 8 | NM_152564.5: c.9574_9583delGTACCCCTCGinsAC NM_152564.5: c.6914C>T |
p. (Val3192ThrfsTer33) p.(Thr2305Ile) |
chr8: g.100844840_100844849delGTACCCCTCGinsAC chr8: g.100733139C>T |
Heterozygous (paternal origin) Heterozygous (maternal origin) |
LP/VUS |
| 14 | SHANK3 DLG3 |
chr 22 chr X | NM_001372044.2: c.2490+1G>A NM_021120.4: c. 1721G>A |
/ p.(Arg574Gln) | chr22: g.51153476G>A chrX: g.69712394G>A | Heterozygous (de novo) Hemizygous (maternal origin) |
pathogenic/VUS |
| 15 | CDK13 | chr 7 | NM_003718.5: c.2525A>T | p.(Asn842Ile) | chr7: g.40085606A>T | Heterozygous (de novo) |
pathogenic |
| 16 | PDHX | chr 11 | NM_003477.3: c.1336C>T | p.(Arg446Ter) | chr11: g.35016549C>T | Homozygous | pathogenic |
| 17 | SETD1A | chr 16 | NM_014712.3: c.4879del | p. (Val1627TrpfsTer41) |
chr16: g.30995020delG | Heterozygous (de novo) |
pathogenic |
| 18 | TRAK1 | chr 3 | NM_001042646.3: c.1187T>A | p.(Ile396Asn) | chr3: g.42240742T>A | Heterozygous (de novo) |
VUS |
| 19 | ALDH5A1 | chr 6 | NM_170740: c.804dup NM_170740: c.1265G>A |
p. (Val269CysfsTer19) p.(Gly422Asp) |
chr6:g.24515433dup chr6:g.24528277G>A | Heterozygous (paternal origin) Heterozygous (maternal origin) |
P/LP |
| 20 | DPYD | chr 1 | NM_000110.3: c.1905+1G>A | / | chr1:g.97915614C>T | Homozygous | LP |
| 21 | DDX3X | chr X | NM_001356.3:c.857C>A | p.(Ala286Asp) | chrX:g.41203374C>A | Heterozygous (de novo) |
VUS |
| 22 | HUWE1 | chr X | NM_031707: c.9209G>A | p.(Arg3070His) | chrX:g.53578038C>T | Hemizygous (de novo) |
pathogenic |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



