
Submitted:
02 December 2024
Posted:
04 December 2024
You are already at the latest version
Abstract
Background and Objectives: Neurodevelopmental disorders (NDDs), including developmental delay (DD), autism spectrum disorder (ASD), intellectual disability (ID), attention-deficit/hyperactivity disorder (ADHD), and specific learning disorders, affect 15% of children and adolescents worldwide. Advances in next-generation sequencing, particularly whole exome sequencing (WES), have improved the understanding of NDD genetics. Methodology: This study analyzed 3244 patients undergoing WES (single, duo, trio analyses), with 1028 meeting inclusion criteria (67% male; age 0–50 years). Results: Pathogenic (P) or likely pathogenic (LP) variants were identified in 190 patients, achieving a diagnostic yield of 13.4% (singleton), 14% (duo), and 21.2% (trio). A total of 207 P/LP variants were identified in NDD-associated genes: 38% were missense (48 de novo), 29% frameshift (26 de novo), 21% nonsense (14 de novo), 11% splicing site (14 de novo), and 1% inframe (1 de novo). De novo variants accounted for 49.8% of cases, with 87 novel de novo variants and 27 novel non-de novo variants unreported in databases like ClinVar or scientific literature. Conclusions: This is the largest study on WES in Colombian children with NDDs and one of the largest in Latino populations. It highlights WES as a cost-effective first-tier diagnostic tool in low-income settings, reducing diagnostic timelines and improving clinical care. These findings underscore the feasibility of implementing WES in underserved populations and contribute significantly to understanding NDD genetics, identifying novel variants with potential for further research and clinical applications.
Keywords:
neurodevelopmental disorders
; whole exome sequencing
; genetic testing
1. Introduction
Neurodevelopmental disorders (NDDs) are a heterogeneous group of conditions that affect brain development. These disorders, including developmental delay (DD), autism spectrum disorder (ASD), intellectual disability (ID), attention-deficit/hyperactivity disorder (ADHD), and specific learning disorders, affect approximately 15% of children and adolescents worldwide [1,2]. The etiology of NDDs is complex, involving both genetic and environmental factors. Recent advances in genetic research, particularly next-generation sequencing technologies, have significantly enhanced our understanding of the genetic landscape underlying these disorders, enlightening us about the intricate mechanisms at play [2,3,4].
The genetic architecture of NDDs exhibits extensive locus heterogeneity and a spectrum of variant types, including single nucleotide variants (SNVs), copy number variations (CNVs), and structural variations. Large-scale exome sequencing studies have revealed that de novo and inherited rare variants contribute substantially to individual risk for NDDs [4,5]. For instance, studies have identified over 100 high-confidence ASD-associated genes enriched with likely deleterious de novo variants. However, the genetic landscape is incomplete, with estimates suggesting that up to 1,000 genes may harbour de novo variants in ASD alone [5].
Recent meta-analyses and large-scale studies have provided important insights into the heritability and genetic overlap of various NDDs [2]. Family-based studies indicate that approximately two-thirds of the variation in NDDs can be attributed to genetic differences between individuals [3]. Moreover, there is significant genetic overlap between ASD, ID, ADHD and other neurodevelopmental conditions such as epilepsy [2,3]. For example, substantial genetic correlations have been observed between ASD and ADHD and between communication disorders and specific learning disorders [3,6].
As mentioned before, whole exome sequencing (WES) has emerged as a powerful first-tier diagnostic tool. In a previous study of our group, we reported 30-43 % diagnostic yields for unexplained NDDs, representing a significant improvement over traditional diagnostic methods such as chromosomal microarray analysis [7]. Molecular diagnosis follow-up of the clinical progression and associated phenotypes could have profound implications for clinical management, including initiating targeted surveillance and providing accurate genetic counselling for families [2,7]. As our understanding of the genetic basis of NDDs continues to evolve, it is essential to identify overlapping phenotypes because it could help develop a more personalized therapeutic approach in the future, offering hope for more effective treatments [1,5,7].
2. Materials and Methods
2.1. Patient Recruitment and Sampling
Patients who attended consultation for medical genetics at Clínica Colsanitas between January 2021 and October 2023. The inclusion criteria were the following: 1. patients with suspected or non-genetic clinical diagnosis of neurodevelopmental disorders (NDD), recorded in terms of HPO (Human Phenotype Oncology) or terms of the Diagnostic and Statistical Manual of Mental Disorders, DSM-5; 2. patients with request for Whole Exome Sequencing (WES) in trio (WES patient and both parents), duo (WES patient and one of his parents) or singleton (WES patient). The following patients were excluded from the study: 1. patients with identification of NDD caused by an extrinsic event such as perinatal noxa, dystocic delivery, severe childhood or adolescent trauma with clear evidence of structural injury, complicated infection and neurological symptomatic such as localized or generalised meningoencephalitis, clear history of perinatal or childhood and adolescent anoxia or hypoxia due to traumatic event; 2. patients with clear and defined metabolopathy of the perinatal stage with severe neonatal or perinatal distress leading to neurological deterioration; 3. patients with known syndromes, defined by aneuploidies identified in the cytogenetic analysis.
2.2. Review of Medical Records
The research group extracted patients' information from electronic medical records, and results were recorded in the information system of the Specialized Laboratory of Clínica Colsanitas. The information collected consisted of perinatal history, detailed developmental milestones, clinical suspicion or diagnosis of the patient, sex, age, personal history, imaging and electrophysiological test results, family history, clinical laboratory results and genetic tests. Based on the clinical histories, the patients were categorized into the following phenotypes associated with NDD: Neurodevelopmental Delay, Autism Spectrum Disorder, Cognitive impairment and Neurodevelopmental Delay + Epilepsy.
2.3. Sampling, Sequencing, Bioinformatics Processing and Variant Filtering
WES (clinical exome or trio-exome sequencing) was performed in the Specialized Laboratory of Clinica Colsanitas from DNA extracted from peripheral blood by next-generation sequencing (NGS), using Capture Probes targeting exomic regions based on Illumina DNA prep with enrichment® and MGIEasy Exome Capture V5 Probe Set®. Sequencing was performed on NextSeq 2000 (Illumina, San Diego, CA, USA), NovaSeq 6000 Sequencing System (Illumina, San Diego, CA, USA) or G-400 (MGI Tech Co, San Jose, CA, USA). Sequence reads were aligned with ConsortiumHuman Build 37, and visualisation and variant identification were done with the SOPHiA DDM and VarSome Clinical platforms. This methodology allows detection of single nucleotide variants (SNV), insertions/deletions (Indel), copy number changes (CNV) and structural variants (SV). The genetic variants found, were classified as pathogenic (P), likely pathogenic (LP), of uncertain clinical significance (VUS), likely benign (LB) or benign (B) according to the guidelines of the American College of Medical Genetics and Genomics (ACMG), supported by different clinical databases such as ClinVar from NCBI (https://www.ncbi.nlm.nih.gov/clinvar/), ClinGen (https://clinicalgenome.org/), GnomAD (https://gnomad.broadinstitute.org/), Franklin (https://franklin.genoox.com/clinical-db/home), UniProt (https://uniprot.org/) and OMIM (https://omim.org/). Variant analyses in inpatients with NDD were performed as follows: single or panel (patient only analysed), duo (patient and one parent analysed), and trio (patient and both parents analysed).
2.4. Classification and Ontology Analysis of Associated Genes
The identified genes associated with NDD were classified into four groups based on patient history and clinical diagnosis. Genes and patients without specific phenotypes were classified into neurodevelopmental delay. To demonstrate the biological significance of the genes associated with the eight phenotypic groups, gene ontology network analysis was performed using Cytoscape software (v.3.10.2) with the ClueGo plug-in (v.2.5.10). With ClueGo, enrichment of the studied genes with GO terms linked to the kappa score is performed. Only GO terms with p-values < 0.05 were adjusted to the Bonferroni step-down.
2.5. Statistical Analysis
Qualitative variables are presented as absolute and relative frequencies. Quantitative variables are described with measures of central tendency and dispersion. To calculate the diagnostic yield, a positive result was considered to be any result that had a variant classified as pathogenic (P) or likely pathogenic (LP) in a gene associated with a phenotype that correlates clinically with the phenotype of the patient under study (gene-disease association was performed according to the OMIM database). All results in which no variants related to the patient's phenotype were reported were considered harmful. The diagnostic yield of the test was calculated considering the total number of patients included in the study, which corresponds to the percentage of patients with a positive result.
2.6. Ethical Considerations
The present study was developed within the framework of the macro research entitled “Development of a comprehensive care model based on personalised medicine for the diagnosis, treatment and follow-up of pediatric patients with NDD in the Colombian population”, which was reviewed and approved by the Research Ethics Committee of the Fundación Universitaria Sanitas (CEIFUS code 1419-21, date of approval, July 2, 2021), following the principles of the Declaration of Helsinki. Clinica Colsanitas have approved the informed consent form for this study, and the participants, or their parents/legal representatives, have signed the corresponding informed consent form to perform WES.
The information associated with the patients above was handled exclusively by the principal investigators, who always respected the provisions of law 1581 of Colombian legislation regarding the handling of personal data.
3. Results
Between January 2021 and October 2023, a total of 3244 patients with single, duo and trio exome analyses were sequenced by NGS. 1028 (31.7%) met the inclusion criteria, 67% (688) were male and 33% (340) were female. The age range of patients associated with NDD was between 0 and 50 years of age (mean X̅: 5 SD: 4.7). Of the 1028 patients who met NDD criteria, 695 patients had clinically suspected DD (68% of patients), 199 with ASD (19% of patients), 79 with DD and epilepsy (8% of patients), and finally, 55 (5% of patients) with CI.
3.1. Diagnostic Yield
Pathogenic (P) or likely pathogenic (LP) variants were identified in 190 patients, corresponding to 18.5% of patients with NDD admissions and 5.6% of the total patients analysed in this period. The diagnostic yield varied depending on the type of analysis performed: Clinical exome 13.4% (299 admissions/ 40 positive), duo WES 14% (63 admissions/ 9 positive), and trio WES 21.2% (666 admissions /141 positive) (Figure 1).
A total of 139 genes associated with NDD were identified in 190 patients with P/LP variants, of which 116 genes were identified in 151 patients with DD, 16 genes in 22 patients with DD and Epilepsy, 13 genes in 12 patients with ASD and seven genes in 5 patients with CI (Table 1). Some genes were associated with more than one of the above phenotypes (Figure 2). 186 P/LP variants were identified in the 190 NDD-positive patients (Table S1). The majority of patients (78%, 146/186) presented SNV-type variants associated with genes with autosomal dominant (AD) inheritance patterns, followed by 9% (17/186) of patients with genes with autosomal recessive (AR) inheritance patterns. Finally, genes with X-linked dominant (XLD), X-linked recessive (XLR) and X-linked (XL) inheritance patterns were identified in 8% (14/186), 3% (6/186) and 2% (3/186) of patients, respectively. 82% per cent (156/190) of patients had variants in a heterozygous state, 8% (15/190) homozygous, 6% (11/190) compound heterozygous and 4% (8/190) hemizygous.
3.2. Characterization of Novel Variants
A total of 207 P/LP SNV variants were identified in NDD-associated genes, and missense variants were the most represented with 78 (38%), 48 were de novo, 61 (29%) Frameshift, 26 de novo; 44 (21%) Nonsense, 14 de novo; 22 (11%) Splicing site, 14 de novo; 2 (1%) Inframe, one de novo. Finally, 49.8% (103/207) of the variants were identified de novo in 100 patients with NDD. Additionally, we report 87 new variants de novo that have not been listed in ClinVar nor reported previously in any of the databases consulted or in scientific articles (marked with * in Table 2). In addition, 27 non-de novo SNVs had not been previously reported in scientific articles or databases (marked without * in Table 2). However, it was possible to classify them as P or LP according to ACMG, in silico analysis using relevant databases and tools (Table 2).
Genes with the highest number of variants were MECP2 (n=6) and CUL3 (n=4). Two Nonsense and two frameshifts were identified in this gene, three were de novo (c.764C>G, p.Ser255*;c.769delG, p.Glu257Lysfs*5; c.494dup, p.Leu166Ilefs*37), two were not previously reported (.769delG, p.Glu257Lysfs*5; c.494dup, p.Leu166Ilefs*37).
3.3. Gene Ontology Analysis
GO network analysis of biological processes, based on the four categories in which the NDD genes were classified, the genes associated with ASD, CI and DD presented different patterns among them. In the case of the genes related to DD and Epilepsy, they did not show any pattern of association. Three ASD genes and four CI genes presented significant association (false discovery rate and p-value < 0.05). IC genes showed a single association with the term GO protein demethylation and ASD genes with social behavior (Figure 3A,B). As for the DD genes, 113 genes formed a complex network without a solid or consistent association with each other. Some fragmented associations were observed, such as protein methylation (12 genes), telencephalon development (12 genes), associative learning (9 genes), regulation of cell cycle G1/S phase transition (8 genes), positive regulation of type I interferon production (6 genes), glial cell migration (5 genes), face development (5 genes), negative regulation of cell-matrix adhesion (5 genes), negative regulation of cell-matrix adhesion (5 genes), negative regulation of cell-substrate adhesion (5 genes), DNA duplex unwinding (5 genes), axo-dendritic transport (5 genes), head morphogenesis (4 genes), voltage-gated sodium channel activity (4 genes) and protein destabilization (4 genes) (Figure 3c).
4. Discussion
Despite the genomic analyses, understanding the etiology of NDDs remains challenging due to their broad genetic and phenotypic heterogeneity. WES has played an essential role in identifying causatives and has proven to be a valuable diagnostic tool. This study achieved a molecular diagnosis for 190/1028 patients with NDD spectrum in Clínica Colsanitas between January 2021 and October 2023.
In patients with NDD and the complete spectrum of ASD, ID, and DD, we applied a retrospective chart review of the probands' and their relatives' medical records before continuing to the molecular analyses. The epilepsy phenotype was excluded. It was taken into account for the results but omitted from the analyses as it is an extensive phenotype and will be considered for future group publications.
Clinical studies should address the effectiveness of genetic studies targeting the aetiological diagnosis. The NDD spectrum is highly heritable and heterogeneous and affects a significant proportion of the population. The etiology of such disorders arises from environmental factors such as malnutrition, perinatal infections, drug misuse or pollution, which may contribute to the risk for these disorders through epigenetic dysregulation and mutations; synaptopathies are also a significant cause of NDD in the context of structural as corticogenesis and functional as synaptic disruption, affecting the plasticity, signalling and disrupting cerebral connectivity characterized by an imbalance between excitatory and inhibitory transmissions. It is also known that differentially expressed gene networks enriched neurotransmitter and synapse activity, immune processes, and cortical development (6). Achieving a molecular diagnosis of NDD has an economic impact not only on the patient’s healthcare system but also on their families.
The array comparative genomic hybridization (aCGH) has supported greater diagnostic effectiveness concerning historical cytogenetic techniques (3 vs. 10%, respectively) (8). Development of next-generation sequencing (NGS) techniques, which allow genome sequencing (GS), or WES, have shown a high diagnostic capacity, leading to an exponential drop in costs and expanded sequencing coverage of the genome, as well as the ability to capture high read depth to detect low-level mutations. In our study, the diagnostic yield of trio-exome sequencing was significantly high (21.2%) and like reported yields in other studies (7,8), supporting its utility in molecular diagnosis of NDD spectrum patients. The strengths of this study are the sizeable Colombian cohort, which included all patients assessed by a clinical geneticist, and the extensive clinical and phenotypic data that were available, which were used for the stratification of the central overlapping NDD spectrum phenotype.
The most common clinical symptom shared by all NDDs was cognitive dysfunction and Autism. The classification of patients in the diagnostic spectrum categories might have also affected the proportion of ASD; patients with ASD features were diagnosed as DD due to either not being old enough to be diagnosed as ASD or not being assessed with a specific ASD-standardized scale to provide an accurate diagnosis, indeed. Statistical significance between molecular findings on each phenotype ID/NDD, ASD/NDD and DD/NDD.
Evidence of diagnostic yield supports NGS-based genetic testing for diagnosing NDD (2,8,9, 10). Current evidence suggests that a diagnostic yield of 35% can be obtained for WES in patients with intellectual disability and global developmental delay and 15% for patients with ASD (11). The overall WES yield for NDD was 36%, 31% for isolated DD, and 53% for NDD with associated conditions. In the present study, an overall diagnostic rate of 18.5% was observed for WES, within the range reported in the literature (12). Additionally, when evaluating only patients with clinical suspicion of DD, the diagnostic yield was 22% (151/695), ASD was 6% (12/199), DD and epilepsy was 28% (22/79) and IC with 5% (5/55).
The diagnostic yield was significantly different comparing clinical exome with 13.4% and trio, analyzing the parents (diagnostic rates of 21.2%), conditioning the modification of the genetic algorithms in the diagnosis of different NDD, positioning the use of WES as an initial analysis in this type of patients, and going above directed molecular studies such as the triplet expansion study for the FMR1 gene as well as aCGH (12, 13,23).
Mutations in various genes have been shown to interfere with the proliferation and migration of neurons. Genetic advances resulting from new sequencing techniques have also expanded our understanding of neuronal migration disorders that affect each stage of neurodevelopment. From the early stages of gestation, the development of the neuroepithelial progenitors that cover the wall of the ventricles begins. This process requires different stages, such as proliferation, differentiation, and migration. Enhancing corticogenesis is characterized by several unique features, including a unique germinal zone and the outer subventricular zone, increasing size from week 20, and making an integration circuit (14, 21). Pathogenic variants that could modify cortical development and the timing and complexity of cortical neurogenesis or synaptogenesis could be linked to neurodevelopmental disorders, providing evidence for their physiological relevance (17, 18).
We report the mutational spectrum of MECP2, CUL3 and SHANK3 genes. The MECP2 (MIM*300005) binds methylated CpGs, is a chromatin-associated protein required to mature neurons and is developmentally regulated. The MECP2 syndrome is a neurodevelopmental disorder that occurs almost exclusively in females and has an XLD inheritance pattern. It is characterized by arrested development between 6 and 18 months of age, regression of acquired skills, loss of speech, stereotypic movements (classically of the hands), microcephaly, seizures and mental retardation. Also, it is associated with susceptibility to X-linked autism 3, with an XL inheritance pattern. Pathogenic and probably pathogenic variants in homozygous or compound heterozygous state in the MECP2 gene have also been associated with severe neonatal encephalopathy, syndromic X-linked intellectual development disorder 13 and syndromic X-linked intellectual development disorder, Lubs type, phenotypes with XLR inheritance pattern (19). Two Nonsense and three Missense variants were identified (one present in two patients).
The CUL3 gene (MIM *603136) encodes a scaffolding component of the Cullin-RING ligase (CRL) complex, essential for mitotic division. CUL3 transcript levels are relatively high during early embryonic development, playing an important role during fetal development and maturation. CUL3 neurodevelopmental disorder with or without autism or seizures, with autosomal dominant inheritance pattern, is characterized by global developmental delay evident in childhood, impaired intellectual development and speech delay. Some patients develop seizures and may show regression after the onset of seizures. Others present with autistic features or behavioural abnormalities. Additional variable systemic features such as cardiac defects, growth retardation or brain imaging abnormalities may also be present. Also, it is associated with pseudohypoaldosteronism type IIE, which is related to an AD inheritance pattern (20, 25).
SHANK3 gene (MIM*606230), expressed predominantly in the cerebral cortex and cerebellum, encodes a scaffolding protein that is enriched in postsynaptic densities of excitatory synapses; it has been shown to bind to neuroligins, which, together with the neurexins, form a complex at glutamatergic synapses. SHANK3 was shown to coincide with the most severe cases of autism and Phelan-McDermid Syndrome (22q13.3) deletion syndrome when including the gene); because it affects the development and morphology of dendritic spines and reduces synaptic transmission in mature neurons, contributing to an imbalance of inhibition to excitation (26).
In positive Trio Exomes, 100 LP/P variants were identified as de novo (absent in the parents). De novo variants gain value and increase the diagnostic performance of the test when there is no family history of similar conditions or reports of consanguinity (21). However, it is essential to highlight that in cases where variants inherited from the parents are identified, the inheritance pattern of the phenotype should be considered since there are cases where the variants can be heterozygous in the mother, being carriers, and hemizygous in male children, who would express the phenotype for diseases with recessive patterns linked to the X chromosome. Another possibility is that there are diseases for which incomplete penetrance and variable expressivity have been reported due to differences in genetic background, environmental factors or a combination of both, so it is possible that within the same family, there are affected individuals with different levels of involvement, or even that they are asymptomatic despite having the same variant (22, 24).
The present study has several limitations, such as its retrospective design, the possible overestimation of the clinical exome sequencing diagnostic yield due to a bias in the selection of samples for NGS, and the fact that aCGH is not being compared with WES. To confirm our results, a prospective study comparing the diagnostic yield of aCGH and clinical exome sequencing in an unbiased sample would be desirable, and for those recently described new variants, further mechanistic and phenotypic characterization of additional patients could confirm their roles in human neurodevelopment disease and to delineate their associated phenotypic spectrums.
To our knowledge, this is the first study to evaluate the clinical utility of WES for children with NDD spectrum in Colombia in a large cohort of patients; we show the importance of reducing the expenses in genetic testing for NDD disorders with a cost-saving first-tier diagnostic test that would serve for developing countries, and to define an aetiological diagnosis in less time.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: List of P/LP variants identified in patients with NDD.
Author Contributions
J. L., T. A. C. C., and M. A. B. performed the conceptualization, writing, review and article preparation; D.A.R.G, J.L., L.C.R.P., P.R.G., Y.N.S.R, and J.J.L.R performed patient data analysis and reports. T.C., M. A. B., C.A., O. L., C. R., and J.J.L.R. participated in the clinical discussion of the variants analysis results. J.J.L.R and M.I.R. did the funding acquisition. J.J.L.R. takes responsibility for the data's integrity and the data analysis's accuracy. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Research Ethics Committee of the Fundación Universitaria Sanitas (CEIFUS code 1419-21, date of approval, July 2, 2021).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Acknowledgments
We thank all the patients and families, and all the laboratory and medical staff involved in this process.
References
- Gidziela, A., Ahmadzadeh, Y. I., Michelini, G., Allegrini, A. G., Agnew-Blais, J., Lau, L. Y., Duret, M., Procopio, F., Daly, E., Ronald, A., Rimfeld, K., & Malanchini, M. A meta-analysis of genetic effects associated with neurodevelopmental disorders and co-occurring conditions. Nature Human Behaviour. 2023; 7(4), 642–656. [CrossRef]
- Srivastava, S., Love-Nichols, J. A., Dies, K. A., Ledbetter, D. H., Martin, C. L., Chung, W. K., Firth, H. v, Frazier, T., Hansen, R. L., Prock, L., Brunner, H., Hoang, N., Scherer, S. W., Sahin, M., & Miller, D. T. Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders and the NDD Exome Scoping Review Work Group. Genetics in Medicine. 2019; 21, 2413–2421. [CrossRef]
- Hu, W. F., Chahrour, M. H., & Walsh, C. A. The diverse genetic landscape of neurodevelopmental disorders. Annual Review of Genomics and Human Genetics. 2014; 15, 195–213. [CrossRef]
- Wayhelova, M., Vallova, V., Broz, P. et al. Exome sequencing improves the molecular diagnostics of paediatric unexplained neurodevelopmental disorders. Orphanet J Rare Dis. 2024; 19, 41. [CrossRef]
- Satterstrom, F. K., Kosmicki, J. A., Wang, J., Breen, M. S., de Rubeis, S., An, J. Y., Peng, M., Collins, R., Grove, J., Klei, L., Stevens, C., Reichert, J., Mulhern, M. S., Artomov, M., Gerges, S., Sheppard, B., Xu, X., Bhaduri, A., Norman, U., … Buxbaum, J. D. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell. 2020; 180(3), 568-584.e23. [CrossRef]
- Griffin A, Mahesh A, Tiwari VK. Disruption of the gene regulatory programme in neurodevelopmental disorders. Biochim Biophys Acta Gene Regul Mech. 2022 Oct;1865(7):194860. [CrossRef]
- Ballesta-Martínez, M.J., Pérez-Fernández, V., López-González, V. et al. Validation of clinical exome sequencing in the diagnostic procedure of patients with intellectual disability in clinical practice. Orphanet J Rare Dis. 2023; 18, 201. [CrossRef]
- Martinez-Granero, F., Blanco-Kelly, F., Sanchez-Jimeno, C. et al. Comparison of the diagnostic yield of aCGH and genome-wide sequencing across different neurodevelopmental disorders. npj Genom. Med. 2021; 6, 25. [CrossRef]
- Monroe GR, Frederix GW, Savelberg SM, de Vries TI, Duran KJ, et al. Effectiveness of whole-exome sequencing and costs of the traditional diagnostic trajectory in children with intellectual disability. Genet Med. 2016; 18:949–56. [CrossRef]
- Vrijenhoek T, Middelburg EM, Monroe GR, van Gassen KL, Geenen JW, Hövels AM, Knoers NV, van Amstel HK, Frederix GW. Whole-exome sequencing in intellectual disability; cost before and after a diagnosis. European Journal of Human Genetics. 2018; Nov;26(11):1566-71.
- Savatt JM, Myers SM. Genetic testing in neurodevelopmental disorders. Frontiers in Pediatrics. 2021; 19;9:526779.
- Zhou, X., Feliciano, P., Shu, C., Wang, T., Astrovskaya, I., Hall, J. B., Obiajulu, J. U., Wright, J. R., Murali, S. C., Xu, S. X., Brueggeman, L., Thomas, T. R., Marchenko, O., Fleisch, C., Barns, S. D., Snyder, L. A. G., Han, B., Chang, T. S., Turner, T. N., … Chung, W. K. Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nature Genetics. 2022; 54(9), 1305–1319. [CrossRef]
- Servetti M, Pisciotta L, Tassano E, Cerminara M, Nobili L, Boeri S, Rosti G, Lerone M, Divizia MT, Ronchetto P, Puliti A. Neurodevelopmental disorders in patients with complex phenotypes and potential complex genetic basis involving non-coding genes, and double CNVs. Frontiers in Genetics. 2021; 21;12:732002. [CrossRef]
- Leite AJ, Pinto IP, Leijsten N, Ruiterkamp-Versteeg M, Pfundt R, de Leeuw N, da Cruz AD, Minasi LB. Diagnostic yield of patients with undiagnosed intellectual disability, global developmental delay and multiples congenital anomalies using karyotype, microarray analysis, whole exome sequencing from Central Brazil. PLoS One. 2022; 7;17(4):e0266493.
- Charouf, D., Miller, D., Haddad, L., White, F. A., Boustany, R.-M., & Obeid, M. High Diagnostic Yield and Clinical Utility of Next-Generation Sequencing in Children with Epilepsy and Neurodevelopmental Delays: A Retrospective Study. International Journal of Molecular Sciences. 2024; 25(17), 9645. [CrossRef]
- Vissers, L., Gilissen, C. & Veltman, J. Genetic studies in intellectual disability and related disorders. Nat Rev Genet. 2016; 17, 9–18. [CrossRef]
- Wilfert, A.B., Sulovari, A., Turner, T.N. et al. Recurrent de novo mutations in neurodevelopmental disorders: properties and clinical implications. Genome Med. 2017; 9, 101. [CrossRef]
- Vanderhaeghen, P., Polleux, F. Developmental mechanisms underlying the evolution of human cortical circuits. Nat Rev Neurosci. 2023; 24, 213–232. [CrossRef]
- Swanberg SE, Nagarajan RP, Peddada S, Yasui DH, LaSalle JM. Reciprocal co-regulation of EGR2 and MECP2 is disrupted in Rett syndrome and autism. Human Molecular Genetics. 2009; 1;18(3):525-34. [CrossRef]
- Fischer S, Schlotthauer I, Kizner V, Macartney T, Dorner-Ciossek C, Gillardon F. Loss-of-function Mutations of CUL3, a High Confidence Gene for Psychiatric Disorders, Lead to Aberrant Neurodevelopment In Human Induced Pluripotent Stem Cells. Neuroscience. 2020; 10;448:234-254. [CrossRef]
- Bruno, L. P., Doddato, G., Valentino, F., Baldassarri, M., Tita, R., Fallerini, C., Bruttini, M., Rizzo, C. lo, Mencarelli, M. A., Mari, F., Pinto, A. M., Fava, F., Fabbiani, A., Lamacchia, V., Carrer, A., Caputo, V., Granata, S., Benetti, E., Zguro, K., … Ariani, F.. New candidates for autism/intellectual disability identified by whole-exome sequencing. International Journal of Molecular Sciences. 2021; 22(24). [CrossRef]
- Heiman P, Drewes S, Ghaloul-Gonzalez L. A familial case of CAMK2B mutation with variable expressivity. SAGE Open Medical Case Reports. 2021 Feb;9:2050313X21990982.
- Jin C, Zhang X, Lei Q, Chen P, Hu H, Shen S, Liu J and Ye S. Case report: genetic analysis of a novel frameshift mutation in FMR1 gene in a Chinese family. Front. Genet. 2023; 14:1228682. [CrossRef]
- Pande, S., Majethia, P., Nair, K. et al. De novo variants underlying monogenic syndromes with intellectual disability in a neurodevelopmental cohort from India. Eur J Hum Genet. 2024; 32, 1291–1298. [CrossRef]
- Lin P, Yang J, Wu S, Ye T, Zhuang W, Wang W, Tan T. Current trends of high-risk gene Cul3 in neurodevelopmental disorders. Front Psychiatry. 2023; 28;14:1215110. [CrossRef]
- Uchino S, Waga C. SHANK3 as an autism spectrum disorder-associated gene. Brain Dev. 2013; 35(2):106-10. [CrossRef]
Figure 1.
Summary of the sample of patients according to 1) the presence of pathogenic (P) of likely pathogenic (LP) variants in agreement with the ACMG criteria, and 2) if the exome sequencing was applied to a singleton, duo, or trio unit of analysis.
Figure 1.
Summary of the sample of patients according to 1) the presence of pathogenic (P) of likely pathogenic (LP) variants in agreement with the ACMG criteria, and 2) if the exome sequencing was applied to a singleton, duo, or trio unit of analysis.
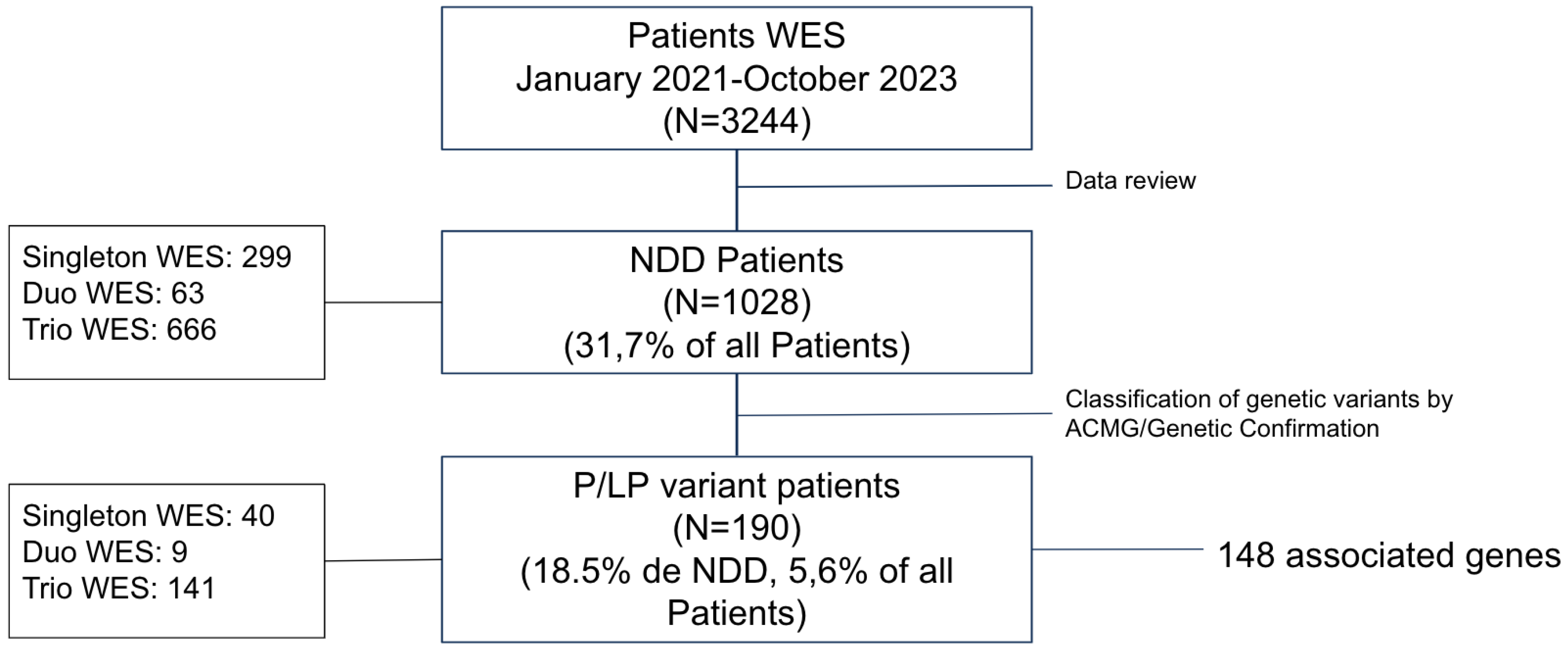
Figure 2.
Genes with LP/P variants overlapped between phenotypes. DD, ASD and CI variants in the MECP2, CUL3 and SHANK3 genes were described. ASD and CI variants were described on DD and CI in KDM4B and KDM3B variants.
Figure 2.
Genes with LP/P variants overlapped between phenotypes. DD, ASD and CI variants in the MECP2, CUL3 and SHANK3 genes were described. ASD and CI variants were described on DD and CI in KDM4B and KDM3B variants.
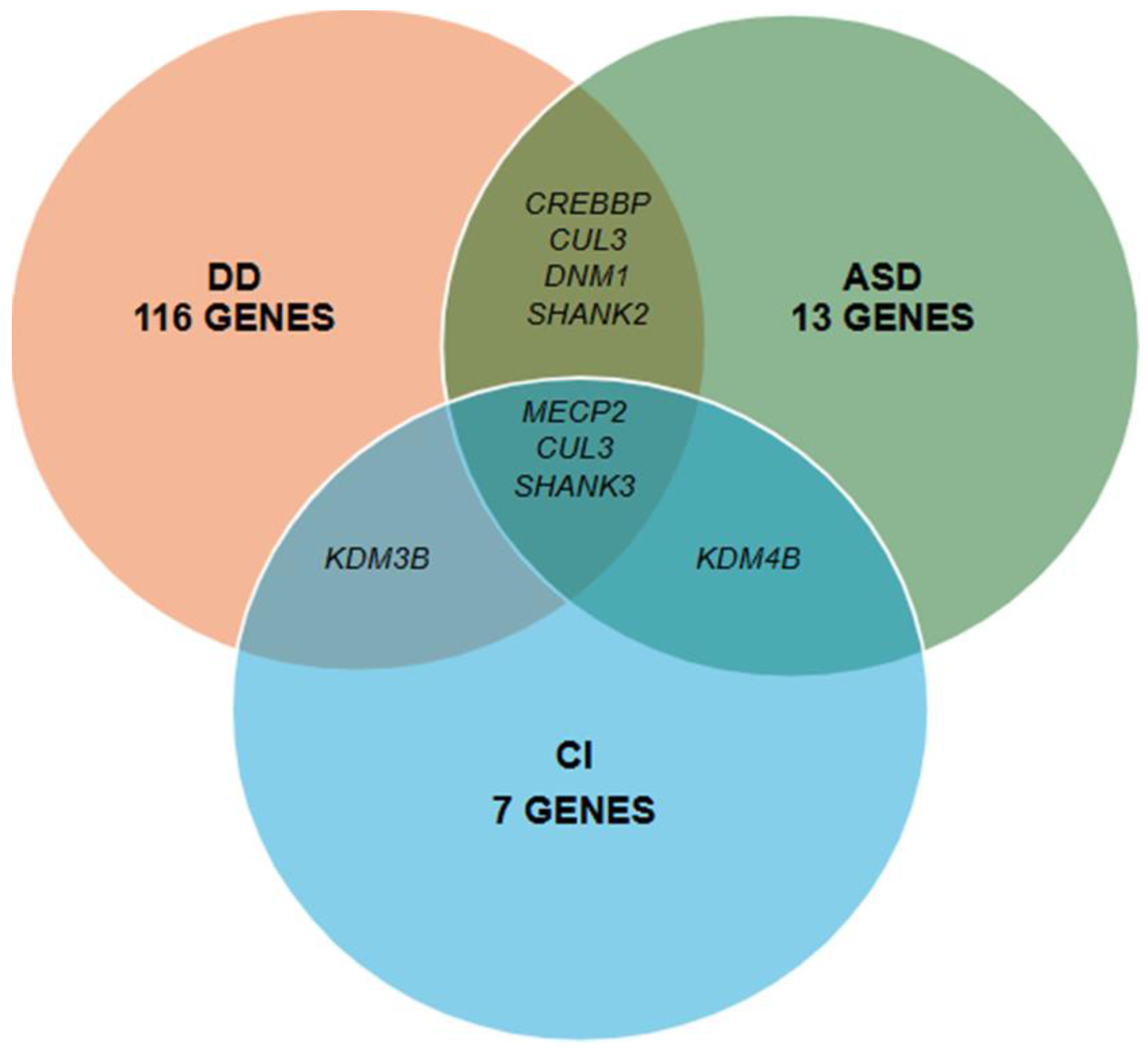
Figure 3.
Functionally cluster gene networks for the CI (A), ASD (B) and DD (C) phenotypes. Networks were obtained from ClueGo enrichment analysis. Gene ontology terms and associated genes are in the same color. The node size of each term corresponds to its importance in enrichment.
Figure 3.
Functionally cluster gene networks for the CI (A), ASD (B) and DD (C) phenotypes. Networks were obtained from ClueGo enrichment analysis. Gene ontology terms and associated genes are in the same color. The node size of each term corresponds to its importance in enrichment.
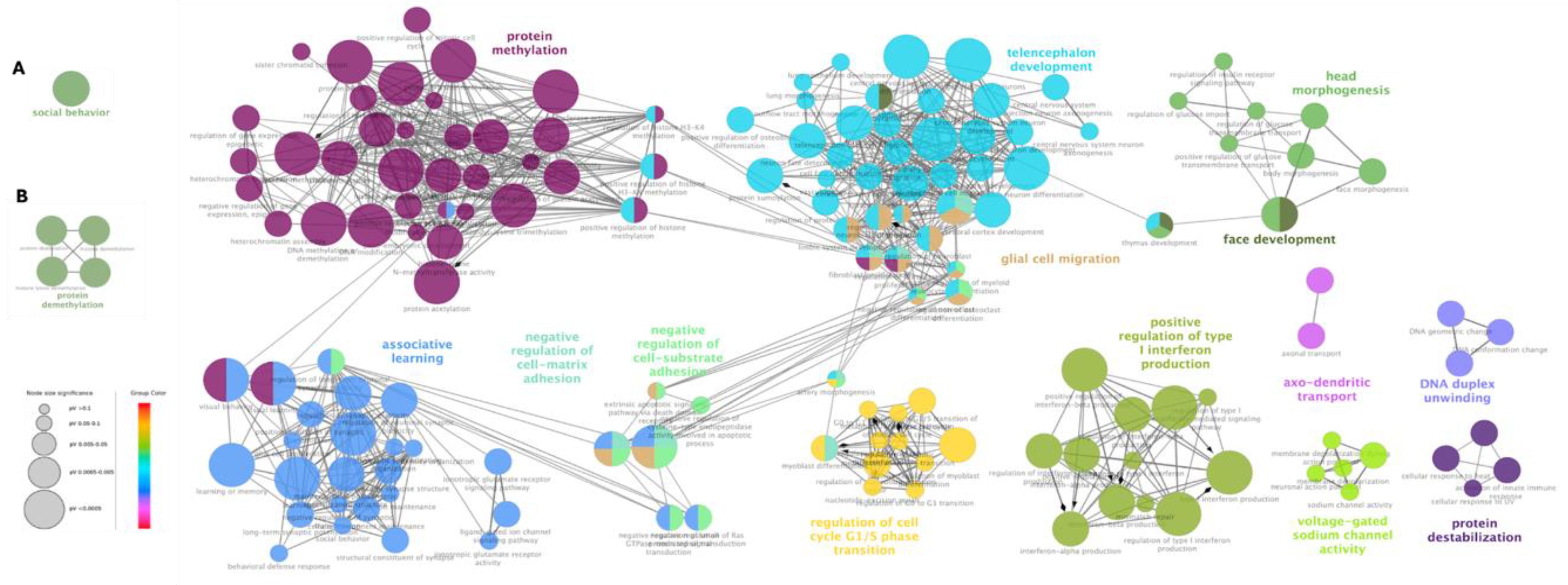

Table 1.
Genes in which LP/P variants were identified by phenotype.
| Phenotype | Gene |
|---|---|
| Developmental Delay | ACTL6B, AHDC1, ANKRD11, ARID2, ATRX, BCL11A, BCL11B, BRAF, CHD3, CREBBP, CTNNB1, CUL3, DDX3X, DNM1, DNMT3A, DPYD, DYNC1H1, FBXO11, FLNB, FOXG1, GLUD2, GNB1, GRIA2, GRIN1, H1-4, IFIH1, INTS1, IVD, JAG1, KCNT2, KDM6B, KIAA1109, KIF1A, KMT2A, KMT2C, LARP7, LARS2, LMAN2L, LZTR1, MECP2, MN1, MSTO1, NACC1, NALCN, NARS, NEXMIF, NF1, NFIB, PHF8, PIK3R1, PPM1D, PTPN11, PURA, QRICH1, RAF1, RAI1, RBMX, RPS6KA3, SATB2, SCN1A, SCN2A, SCN8A, SETD2, SETD5, SHANK2, SHANK3, SIX3, SLC13A5, SPEN, STAG2, SYNGAP1, TAOK1, TBK1, TBR1, TCF20, TCF4, TOE1, TREX1, TRIP12, TSC2, TSPAN7, TUBB, UBTF, USP7, WAC, ZMIZ1, AP1S2, ARSA, CAPN3, CDC42, COL6A2, COL6A3, CTBP1, DCX, DHTKD1, EP300, FBN2, GCDH, PMM2, POLR3B, PUF60, RTN2, RYR1, SPG11, SPG7, SPTBN2, TNPO3, TRPV4, TRIO, RNASEH2B, RFX7, KMT2E, ATP7B, ARID1B, INPP5E, KDM3B, CSNK2A1 |
| Developmental Delay & Epilepsy | ABCC8, AFF3, ANKRD17, CDKL5, CHD2, GNAO1, GRIN2B, HNRNPU, KAT6A, KCNB1, KCNMA1, PTCH1, SETD1A, SLC2A1, STXBP1, MN1 |
| Autism Spectrum Disorder | CACNA1E, CAMK2A, CHD8, CPT2, CREBBP, CSF1R, CUL3, DNM1, EBF3, KDM4B, PTEN, SHANK2, SHANK3 |
| Cognitive impairment | KDM4B, KDM5C, KDM6A, PAX8, NAA15, KDM3B, SHANK3 |
Table 2.
Description of variants without previous reports in literature or databases.
| Gene* | DNA | Protein | Variant Type | ACMG Classification | Transcripts | OMIM code |
|---|---|---|---|---|---|---|
| ACTL6B | c.521_522insA | p.Thr175Hisfs*7 | Frameshift | LP | NM_016188.4 | 612458 |
| ACTL6B* | c.991G>A | p.Gly331Ser | Missense | LP | NM_016188.5 | 612458 |
| AHDC1 | c.4294del | p.Ala1432Profs*13 | Frameshift | LP | NM_001371928.1 | 615790 |
| ANKRD11 | c.741C>A | p.Tyr247* | Nonsense | LP | NM_013275.6 | 611192 |
| ANKRD11* | c.7124_7152del | p.Glu2375Alafs*147 | Frameshift | P | NM_013275.5 | 611192 |
| ANKRD17* | c.4453_4457del | p.Lys1485Glufs*17 | Frameshift | P | NM_032217.4 | 615929 |
| AP1S2 | c.180-1G>C | Splicing site | LP | NM_001272071.2 | 300629 | |
| BCL11B* | c.2345dup | p.Gly785Argfs*100 | Frameshift | P | NM_138576.3 | 606558 |
| BRAF* | c.2030A>G | p.Asp677Gly | Missense | P | NM_004333.6 | 164757 |
| CACNA1E* | c.5365-2A>G | - | Splicing site | LP | NM_001205293.1 | 601013 |
| CHD2* | c.2822A>T | p.Gln941Leu | Missense | LP | NM_001271.4 | 602119 |
| CHD3 | c.3481C>T | p.His1161Tyr | Missense | LP | NM_001005273.3 | 7806365 |
| CHD8* | c.4987_5003del | p.Val1663Glnfs*59 | Frameshift | P | NM_020920.4 | 610528 |
| COL6A3* | c.6156+2T>C | - | Splicing site | P | NM_004369.4 | 120250 |
| COL9A3 | c.1021C>T | p.Arg341* | Nonsense | LP | NM_001853.4 | 120270 |
| CUL3* | c.769delG | p.Glu257Lysfs*5 | Frameshift | P | NM_003590 | 603136 |
| CUL3* | c.494dup | p.Leu166Ilefs*37 | Frameshift | P | NM_003590.4 | 603136 |
| DCX | c.166C>G | p.Arg56Gly | Missense | LP | NM_001195553.1 | 300121 |
| DDX3X | c.1646A>T | p.Asn549Ile | Missense | LP | NM_001356.4 | 300160 |
| DNM1* | c.1751A>T | p.His584Leu | Missense | LP | NM_004408.4 | 602377 |
| DNM1* | c.2318+2T>C | - | Splicing site | P | NM_004408.4 | 602377 |
| DYNC1H1* | c.11632C>G | p.Gln3878Glu | Missense | LP | NM_001376.5 | 600112 |
| EP300* | c.4779+1G>A | - | Splicing site | P | NM_001429.3 | 602700 |
| FBXO11* | c.1685A>G | p.Tyr562Cys | Missense | LP | NM_025133.4 | 607871 |
| GNB1* | c.310G>C | p.Ala104Pro | Missense | LP | NM_002074.5 | 139380 |
| GRIN1* | c.2248G>A | p.Gly750Arg | Missense | LP | NM_000832.7 | 138249 |
| GRIN1* | c.1824G>C | p.Trp608Cys | Missense | P | NM_007327.3 | 138249 |
| GRIN2B* | c.1990T>C | p.Ser664Pro | Missense | LP | NM_000834.3 | 138252 |
| HNRNPU | c.1484_1487del | p.Lys495Ilefs*5 | Frameshift | LP | NM_031844.3 | 602869 |
| HNRNPU* | c.1576_1580del | p.Asn526Serfs*9 | Frameshift | P | NM_031844.2 | 602869 |
| IFIH1 | c.2863C>T | p.Gln955* | Nonsense | LP | NM_022168.3 | 606951 |
| JAG1 | c.1725_1726dupTG | p.Asp576Valfs*168 | Frameshift | LP | NM_000214.3 | 601920 |
| KAT6A* | c.1019del | p.Asn340Thrfs*3 | Frameshift | P | NM_006766.4 | 601408 |
| KAT6A* | c.4140_4141insA | p.Asp1381Argfs*13 | Frameshift | P | NM_006766.4 | 601408 |
| KCNB1* | c.1223C>T | p.Pro408Leu | Missense | LP | NM_004975.3 | 600397 |
| KCNB1* | c.1202G>T | p.Gly401Val | Missense | P | NM_004975.4 | 600397 |
| KCNMA1 | c.2095A>T | p.Lys699* | Nonsense | LP | NM_001161352.1 | 600150 |
| KCNT2 | c.3118C>T | p.Arg1040* | Nonsense | LP | NM_198503 | 610044 |
| KDM3B* | c.3638dup | p.(Asp1214Ter | Nonsense | LP | NM_016604.4 | 609373 |
| KDM4B | c.2147del | p.Leu716Tyrfs*42 | Frameshift | LP | NM_015015.2 | 609765 |
| KDM5C* | c.1571A>T | p.Asn524Ile | Missense | LP | NM_004187.5 | 314690 |
| KIAA1109 | c.4118_4119del | p.Ser1373* | Nonsense | LP | NM_015312.3 | 611565 |
| KIAA1109* | c.18T>A | p.Asn6Lys | Missense | LP | NM_015312.3 | 611565 |
| KIAA1109* | c.4118_4119del | p.Ser1373* | Nonsense | LP | NM_015312.3 | 611565 |
| KMT2A* | c.7187_7188del | p.Pro2396Argfs*2 | Frameshift | P | NM_001197104.2 | 159555 |
| KMT2C* | c.5551C>T | p.Gln1851* | Nonsense | P | NM_170606.2 | 606833 |
| KMT2E | c.1944_1948del | p.Lys649GlufsTer8 | Frameshift | LP | NM_182931.3 | 608444 |
| LARP7 | c.1118_1130del | p.Val373Glufs*11 | Frameshift | LP | NM_016648.4 | 612026 |
| LARS2* | c.1420del | p.Leu474Trpfs*6 | Frameshift | P | NM_015340.4 | 604544 |
| MN1 | c.97del | p.His33ThrfsTer20 | Frameshift | LP | NM_002430.3 | 156100 |
| NAA15* | c.2214del | p.Met738IlefsTer18 | Frameshift | P | NM_057175.5 | 608000 |
| NACC1* | c.166C>T | p.Arg56Trp | Missense | LP | NM_052876.3 | 610672 |
| NEXMIF | c.1998del | p.Glu667Lysfs*5 | Frameshift | LP | NM_001008537.2 | 300524 |
| NEXMIF | c.1998del | p.Glu667Lysfs*5 | Frameshift | LP | NM_001008537.3 | 300524 |
| NF1 | c.2056A>T | p.Lys686* | Nonsense | LP | NM_001042492.2 | 613113 |
| NFIB* | c.626A>G | p.Glu209Gly | Missense | LP | NM_001190737.3 | 600728 |
| NFIX* | c.442del | p.Ile148Serfs*71 | Frameshift | P | NM_001271043.2 | 164005 |
| LPM1D* | c.1245dupT | p.Thr416Tyrfs*18 | Frameshift | P | NM_003620.4 | 605100 |
| PUF60* | c.1334C>T | p.Thr445Ile | Missense | LP | NM_014281.5 | 604819 |
| RFX7* | c.2236C>T | p.Gln746* | Nonsense | P | NM_022841.7 | 612660 |
| RPS6KA3* | c.383C>T | p.Pro128Leu | Missense | LP | NM_004586.3 | 300075 |
| SATB2 | c.1165C>A | p.Arg389Ser | Missense | LP | NM_001172509.2 | 608148 |
| SCN1A* | c.4987G>T | p.Gly1663Cys | Missense | P | NM_006920.6 | 182389 |
| SCN1A* | c.4582-1_4583del | - | Splicing site | P | NM_001165963.4 | 182389 |
| SCN1A* | c.4987G>T | p.Gly1663Cys | Missense | P | NM_006920.6 | 182389 |
| SCN2A | c.641C>A | p.Ser214* | Nonsense | LP | NM_001040143.2 | 182390 |
| SCN8A* | c.5235C>A | p.Phe1745Leu | Missense | P | NM_001330260 | 600702 |
| SETD1A* | c.5116C>G | p.Leu1706Val | Missense | LP | NM_014712.3 | 611052 |
| SHANK3* | c.352dup | p.Leu118ProfsTer28 | Frameshift | P | NM_001372044.2 | 606230 |
| SIX3* | c.221del | p.Pro74Argfs*177 | Frameshift | P | NM_005413.4 | 603714 |
| SPEN* | c.5485_5486insTTTGAAC | p.Gln1829Leufs*2 | Frameshift | P | NM_015001.4 | 613484 |
| STAG2 | c.1018-2_1018-1delinsTT | - | Splicing site | LP | NM_001042750.2 | 300826 |
| STXBP1* | c.903-1G>C | - | Splicing site | P | NM_001032221.3 | 602926 |
| SYNGAP1* | c.1713_1714delinsAC | p.Trp572Arg | Missense | LP | NM_006772.2 | 603384 |
| SYNGAP1* | c.1216_1218delins | p.Tyr406Asnfs*4 | Frameshift | P | NM_006772 | 603384 |
| TAOK1 | c.1721dupA | p.Ser575Glufs*28 | Frameshift | LP | NM_020791 | 610266 |
| TAOK1 | c.1489_1492del | p.Asp497Lysfs*42 | Frameshift | LP | NM_020791.4 | 610266 |
| TBR1* | c.893dup | p.His298Glnfs*23 | Frameshift | P | NM_006593.3 | 604616 |
| TCF20* | c.5047_5054del | p.Pro1683Valfs*34 | Frameshift | P | NM_005650.3 | 603107 |
| TCF4 | c.-20-184_72+815del | - | Splicing site | LP | NM_001083962.2 | 602272 |
| TNPO3 | c.120+2T>G | - | Splicing site | LP | NM_012470.4 | 610032 |
| TRIP12 | c.3206+1G>T | - | Splicing site | LP | NM_001348323.3 | 604506 |
| TUBB* | c.1145C>T | p.Ser382Leu | Missense | LP | NM_178014.4 | 191130 |
| TUBB* | c.1017C>G | p.Ser339Arg | Missense | LP | NM_178014.4 | 191130 |
| USP7* | c.502T>C | p.Ser168Pro | Missense | LP | NM_003470.2 | 602519 |
| WAC* | c.620del | p.Lys207Serfs*124 | Frameshift | P | NM_016628.5 | 615049 |
| ZMIZ1* | c.1413+4A>G | - | Splicing site | LP | NM_020338.3 | 607159 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



