
Submitted:
03 July 2025
Posted:
04 July 2025
You are already at the latest version
Abstract
Multidrug-resistant (MDR) strains of Klebsiella pneumoniae present an acute threat as they continue to disseminate globally. Phage therapy has shown promise as a powerful approach to combat MDR infections, but narrow phage host ranges make development of broad acting therapeutics more challenging. The goal of this effort was to use in vitro directed evolution (the “Appelmans protocol”) to isolate K. pneumoniae phages with broader host ranges for improved therapeutic cocktails. Five myophages in the genus Jiaodavirus (family Straboviridae) with complementary activity were mixed and passaged against a panel of 11 bacterial strains including a permissive host and phage-resistant clinical isolates. Following multiple rounds of training, we collected phage variants displaying altered specificity and/or expanded host ranges compared with parental phages when tested against a 100-strain diversity panel of K. pneumoniae. Some phage variants gained the ability to lyse previously phage-resistant strains but lost activity towards previously phage-susceptible strains, while several variants had expanded activity. Whole genome sequencing identified mutations and recombination events impacting genes associated with host tropism including tail fiber genes that most likely underlie the observed changes in host ranges. Evolved phages with broader activity are promising candidates for improved K. pneumoniae therapeutic phage cocktails.
Keywords:
Klebsiella pneumoniae
; phage training
; appelmans protocol
; jiaodavirus
; host range expansion
; recombinant events
; phage cocktail improvement
1. Introduction
Klebsiella pneumoniae is an important human pathogen that tends to acquire and spread drug resistance and causes various hospital-acquired infections including lung, urinary tract, bloodstream, brain, wound, and surgical site infections. Carbapenem-resistant K. pneumoniae is categorized as an urgent public health threat by the Centers for Disease Control and Prevention [1,2,3]. Multidrug-resistant (MDR) infections caused by K. pneumoniae strains have been reported from multiple hospitals on all continents, from developing and developed countries alike and thus are a global challenge [4,5,6,7,8,9,10,11]. Some high-risk MDR K. pneumoniae lineages, especially hypervirulent strains, cause infections with high mortality that can reach 50% [12,13,14,15,16,17].
The need for effective alternative antibacterials is great, and bacteriophage (phage) therapy presents a promising avenue for the treatment of MDR K. pneumoniae infections, often in combination with standard-of-care antibiotics. Phages have been employed to successfully treat K. pneumoniae infections both in animal models and in human compassionate use cases [18,19,20,21,22,23,24]. Our team is focused on the development of broadly active, durable phage cocktails for utilization in multiple clinical cases against diverse strains. This proves to be a difficult task for K. pneumoniae because of the high diversity of its surface factors, particularly capsule types [25] and the narrow host ranges of Klebsiella phages, which typically vary from 1% to 31%, even when tested against relatively small strain panels or those with limited or unspecified diversity [26,27,28,29,30]. We screen Klebsiella phages against a 100-strain diversity panel of mostly MDR K. pneumoniae clinical isolates that includes 94 different sequence types, 54 KL serotypes, and 11 OL serotypes [31]. Given this high diversity, even a library of >150 Klebsiella phages harvested on four continents and belonging to 25 genera was unable to cover >63% of the K. pneumoniae panel.
We therefore employed an in vitro directed evolution strategy (also known as phage training or host adaptation) using a general method called the Appelmans protocol [23,32,33], to generate phage variants with expanded coverage against the highly diverse panel of clinical K. pneumoniae isolates. This approach yielded nine Jiaodavirus phage variants with altered specificity and genome sequence, including three isolates with expanded host ranges, one of which provided dramatically expanded activity in iterative phage cocktail development. The findings of this study demonstrate the potential of phage training in the rational design of therapeutic phage cocktails to overcome the limitations of narrow lytic spectra of cocktail components within a target bacterial species, particularly with species like K. pneumoniae that typically have phages with narrower host ranges.
2. Results
2.1. Training Led to Phage Variants with Altered Lytic Spectra
Eleven K. pneumoniae strains used for phage training (Table 1) were selected based on their phage resistance, antibiotic resistance and diversity, representing 11 sequence types, 10 capsule (KL) serotypes, and five O-antigen (OL) serotypes. A phage-susceptible strain, MRSN 414780, was used for general propagation of training lysates and maintaining phage titers during training. Five training phages (Table 2) included vB_Kpn11382-KEN22 (KEN22), vB_Kpn529046-KEN25-1 (KEN25-1), vB_Kpn529046-KEN25-2 (KEN25-2), vB_Kpn529046-KEN37 (KEN37), and vB_Kpn529046-KEN39 (KEN39). Their genomes were previously published [34]. They belong to the genus Jiaodavirus in the myophage family Straboviridae, whose members are reported to have relatively broad host ranges, quick lysis time, and efficacy at lower multiplicities of infection [35]. These phages were selected based on their genetic similarity, which is important to help facilitate recombination, a likely major driver of change in training experiments [33]. Another criterion for selection was complementarity in host range coverage to achieve expansion of activity.
Table displays antibiotic and phage susceptibility of strains used in the training. Strains were assessed by reviewing antibiotic MIC data [31] and phage susceptibility data. 1 XDR, extensively drug-resistant. 2 XPR, extensively phage-resistant; PPR, pan-phage-resistant; PS, phage-susceptible.
Kp, K. pneumoniae; Kq, Klebsiella quasipneumoniae. Phages KEN22, KEN25-1, KEN25-2, KEN37, and KEN39 [34] were used for training. Other phages were employed later for cocktail development. The genome of K. quasipneumoniae phage EKq1 was published [36]. All other phages have also been sequenced. The GenBank accession numbers for genome sequences of phages vB_Kpn3619-KEN42 (KEN42), vB_Kpn529046-KEN1821 (KEN1821), vB_Kpn3619-AFR4 (AFR4), and EKq2 are listed in the “Data Availability Statement” below. *Unclassified phage EKq1 formerly belonged to the family Siphoviridae that is now excluded from the phage classification scheme.
Following 10 rounds of phage training (see Materials and Methods), we were able to isolate multiple phage variants infecting highly phage-resistant strains that were initially resistant to the parental phages. Phage plaques were collected from and purified on K. pneumoniae strains MRSN 15687, MRSN 15882, MRSN 27989, and MRSN 681054, but some of these variants were seemingly unstable and were either lost before being purified or recalcitrant to propagation to workable phage titers. Nine relatively stable phage variants were isolated on highly resistant strains MRSN 15882 and MRSN 27989; six trained variants were isolated and purified on MRSN 15882 and three on MRSN 27989 (Figure 1).
After three rounds of plaque isolation, nine evolved phages were tested for host ranges against the panel of 100 highly diverse K. pneumoniae clinical isolates [31]. Additionally, parental phages KEN22, KEN25-1, KEN25-2, KEN37, and KEN39 were plated on the panel at the same time. All these new phage variants displayed altered lytic spectra and three of them, 15882-3, 15882-6, and 15882-7, demonstrated expanded host ranges of 44%, 40%, and 38%, respectively (Table 3 and Figure 2).
Compared with the best performing parental phage, KEN39 that was active against 36% of strains (Table 3), this represented an expansion of 8%, 4%, or 2%, respectively. No other trained phages revealed expanded host range. Isolates 15882-1 and 15882-5 showed 33% and 36% activity (roughly equivalent to the parental phages). Finally, phage variants 15882-8, 27989-4, 27989-11 and 27989-12 showed host ranges narrowed by 14%, 11%, 9%, and 10%, respectively (see Table 3 and Figure 2). All phages with expanded host ranges showed productive infection and plaque formation on previously resistant strains, but the quality of lysis was limited, at score 2+ (Table 4). Even evolved phages with narrowed or equivalent host ranges were capable of lysing highly phage-resistant strains MRSN 15882 and MRSN 27989.
2.2. Serial Propagation on a Phage-susceptible K. pneumoniae Strain Resulted in Host Range Changes
Purified evolved phages were assessed for the stability of the acquired host range changes by passaging against the phage-susceptible strain MRSN 414780. Following five serial propagations on MRSN 414780, progeny phage clones were collected, and their host ranges were determined and compared to their parental trained phage variants (Figure 3). Three clones were collected from each lysate, 15882-3, 15882-6, and 15882-7. All of the progeny clones displayed some narrowing of overall host range due to tapered 2+ activity shown in yellow. However, the proportion of strains lysed with full activity (scores 3+ and 4+: see Table 4 and Figure 3) increased in all the progeny clones. For example, the originally evolved isolates lysed with 4+ activity only one strain, MRSN 414780, in contrast to four, five or four strains for the 15882-3, 15882-6, and 15882-7 progeny clones, respectively (Figure 3). The strains lysed with higher level activity primarily expressed O-antigen types O3b and O1v1. Therefore, to maintain stability, we intend to store phage stocks with expanded host ranges propagated on the phage-resistant strain MRSN 15882 and to use the same strain for propagation of the evolved phages in the future.

Table 4.
Scoring system for phage plaque assay results.
| Score | Observation |
|---|---|
 |
No activity, no lysis (negative result). |
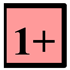 |
Lysis from without: very faint, turbid spots or clear spots in first dilutions, no plaque formation and no negative dynamics of lysis: lysis, lysis, then nothing (negative result). |
 |
Clear or turbid spots, tiny plaques, countable or uncountable, or lack of visible isolated plaques but clear negative dynamics of lysis intensity from lower to higher dilution (slightly positive result). |
 |
Clear spots, clear plaques of medium or small size (strictly positive result). |
 |
Totally clear spots, there are isolated large clear plaques in the highest phage dilutions (highly positive result). |
2.3. A Broad-host-range Cocktail Containing an Evolved Phage
The ultimate goal of this study was to isolate phage variants with expanded host range that could be incorporated in a therapeutic phage cocktail for improved formulations. Our previous best-performing cocktail targeting K. pneumoniae, WRAIR_KPM1 (KPM1), included five phages, KEN39, EKq1, EKq2, AFR4, and KEN42, which collectively lyse 50% of the diversity panel strains. Addition of the trained phage 15882-3 to KPM1 expanded its activity by 21%. We developed an improved version of KPM1, designated WRAIR_KPM2 (KPM2) (Table 5), comprising the core (KPM1) phages along with the evolved phage 15882-3 and recently isolated natural phage KEN1821. This cocktail covered 81% of the diversity panel (Table 5, Figure 4a). KPM2 also outperformed KPM1 in quality of lysis with more robust plaque formation (Figure 4b) and more strains lysed by two or more phages (Figure 4c).
2.4. Sequence Analysis of Trained Phages Reveals Multiple Recombination Events and Accumulation of Mutations
Four evolved phages were selected for variant and recombination analysis: 15882-1, 15882-3, 15882-5, and 15882-6. Since the five input parental phages shared a nucleotide identity of ~90-94%, recombination was expected. All four isolates were found to be recombinant phages, primarily derived from parental phages KEN22 and KEN25-2 (~94% identity, see Table 6). Multiple recombination events were detected (Figure 5) in genes that encode for RNA and nucleotide metabolism, baseplate, and tail proteins. Genes that are likely involved in host range expansion and phage-host interactions with significant recombination events and/or polymorphisms resulting in unique variation from parents are described below.
2.5. Recombinant Events Resulted in Few Nonsynonymous Mutations Compared to Parental Phages
Although one or more recombination events were identified in genes involved in phage-host interaction, these recombination events did not frequently result in amino acid substitutions unique from parental genomes (Table S1). Genes that housed amino acid substitutions unique from parents include those encoding an anti-sigma factor gene and the hinge connector protein of the long tail fiber. The anti-sigma factor gene is homologous to gp49 of T4, a recombination endonuclease VII, which is expressed at early and late stages of T4 infection and is involved in mismatch repair, resolution of branched DNA, and DNA packaging [37]. The hinge connector protein is homologous to gp34 of phage T4, which is a homo-trimer that forms the proximal half-fiber, with the N-terminal end binding to the baseplate, and the C-terminal end being involved in the hinge between the proximal and distal half fibers [38].
Figure 4.
Comparative activity of phage cocktails WRAIR_KPM1 and WRAIR_KPM2. (a) KPM1 covers 50% of strains in the 100-strain K. pneumoniae diversity panel, while KPM2 is active against 81% of the strains; (b) overall higher lytic activity of KPM2 phages; (c) greater numbers of strains lysed by two or more KPM2 components compared to KPM1.
Figure 4.
Comparative activity of phage cocktails WRAIR_KPM1 and WRAIR_KPM2. (a) KPM1 covers 50% of strains in the 100-strain K. pneumoniae diversity panel, while KPM2 is active against 81% of the strains; (b) overall higher lytic activity of KPM2 phages; (c) greater numbers of strains lysed by two or more KPM2 components compared to KPM1.
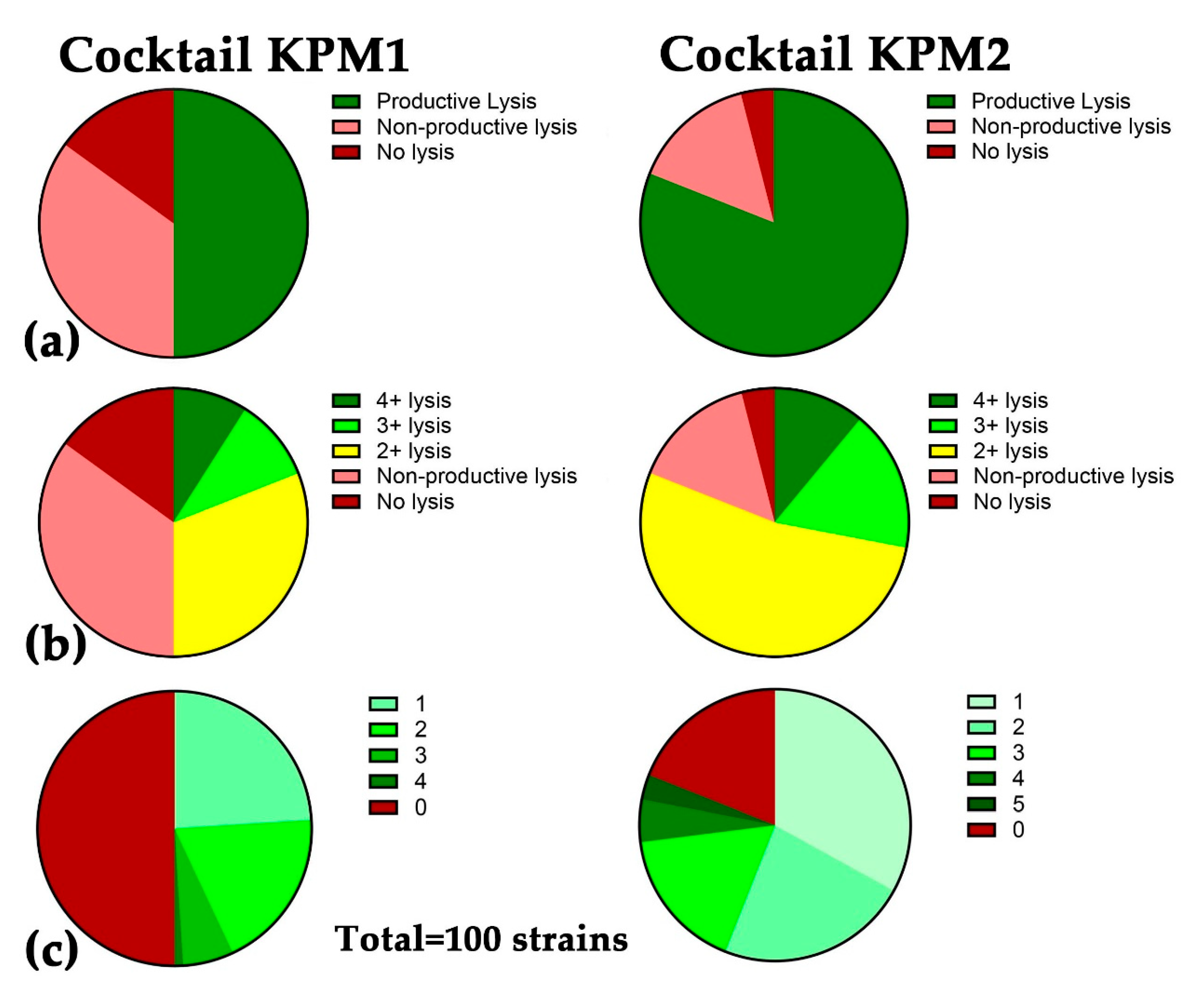
Table 6.
Whole genome nucleotide distance matrix based on multiple sequence alignment of five Jiaodavirus phages and four recombinant phages isolated from phage training.
Table 6.
Whole genome nucleotide distance matrix based on multiple sequence alignment of five Jiaodavirus phages and four recombinant phages isolated from phage training.
| KEN22 | KEN25-1 | KEN25-2 | KEN37 | KEN39 | 15882-1 | 15882-3 | 15882-5 | 15882-6 | |
|---|---|---|---|---|---|---|---|---|---|
| KEN22 | 92.94 | 90.04 | 94.03 | 90.72 | 94.38 | 94.21 | 94.77 | 94.40 | |
| KEN25-1 | 92.94 | 87.70 | 92.12 | 89.44 | 90.17 | 90.23 | 90.52 | 90.19 | |
| KEN25-2 | 90.04 | 87.70 | 91.72 | 88.94 | 94.43 | 94.18 | 93.89 | 94.44 | |
| KEN37 | 94.03 | 92.12 | 91.72 | 91.33 | 92.94 | 93.07 | 92.54 | 92.97 | |
| KEN39 | 90.72 | 89.44 | 88.94 | 91.33 | 89.77 | 89.92 | 90.21 | 89.71 | |
| 15882-1 | 94.38 | 90.17 | 94.43 | 92.94 | 89.77 | 99.34 | 99.15 | 99.65 | |
| 15882-3 | 94.21 | 90.23 | 94.18 | 93.07 | 89.92 | 99.34 | 99.15 | 99.45 | |
| 15882-5 | 94.77 | 90.52 | 93.89 | 92.54 | 90.21 | 99.15 | 99.15 | 99.27 | |
| 15882-6 | 94.40 | 90.19 | 94.44 | 92.97 | 89.71 | 99.65 | 99.45 | 99.27 |
Figure 5.
Genome maps of four recombinant Jiaodavirus phages isolated from phage training experiments. Segments derived from each parental phage are shown in different colors. Variant analysis was conducted using NucDiff; figure was generated using gggenomes in RStudio.
Figure 5.
Genome maps of four recombinant Jiaodavirus phages isolated from phage training experiments. Segments derived from each parental phage are shown in different colors. Variant analysis was conducted using NucDiff; figure was generated using gggenomes in RStudio.
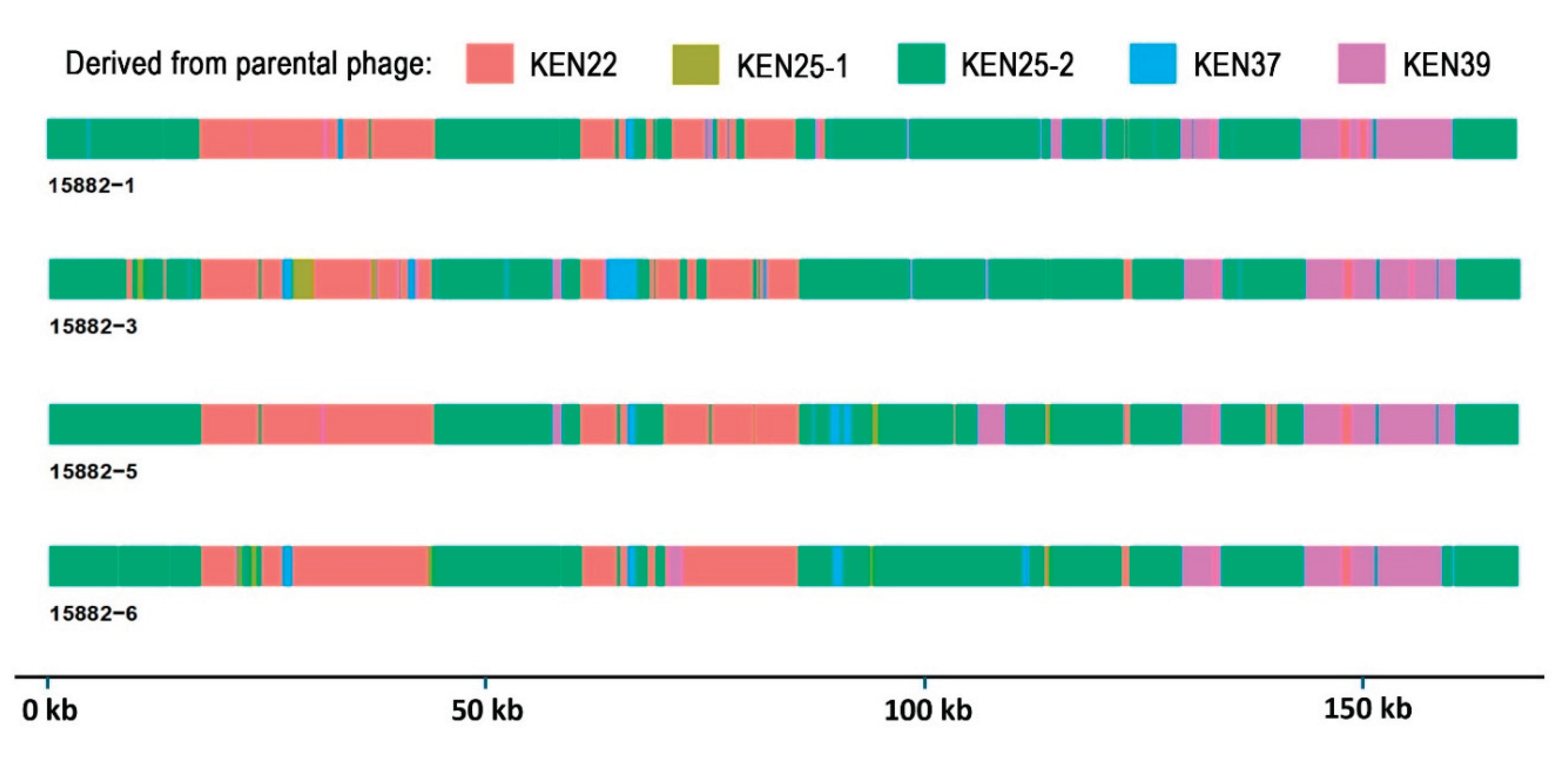
Recombination events were detected in a baseplate hub subunit and tail length protein gene (reference genome KEN22; MBLPYHJN_CDS_0225) in three evolved phages (except 15882-1 that shared 100% nucleotide identity in this gene to KEN22), with segments derived from KEN22 and KEN25-2. Variant 15882-6 had multiple recombination events in this gene, but, surprisingly, had no amino acid changes compared to KEN25-2. 15882-3 and 15882-5 shared 100% identity in this gene and had two amino acid changes in the C-terminal compared to KEN22 (residues 555 His-> Tyr, and 559 Met->Thr); these amino acid substitutions reflected those found in parental phage KEN25-2. This gene showed weak homology to the tape measure protein of phage DT57C [39]. Preliminary structural predictions and analysis using AlphaFold 3 [40] and the RCSB pairwise structure alignment tool [41] for phages KEN22, KEN25-2 and 15882-3 suggest that these structures may differ in their length.
The tail fiber protein (MBLPYHJN_CDS_0281) showed recombination between KEN22 and KEN25-2 in all four evolved phage variants. 15882-1 and 15882-6 shared 100% identity. 15882-3 and 15882-5 shared 100% identity that also included a recombination region from KEN39 at the N-terminal. Despite recombination events detected in this gene, no unique amino acid changes were identified compared to the parental phages. This gene had regions of homology to the putative tail fiber protein of Ralstonia phage GP4 [42] (probability 98.63%, e-value 2.6e-6), the L-shaped tail fiber assembly of phage T5 [43] (probability 98.12-98.35%, e-value 2.1e-5 - 1.2e-6), and the phage T4 proximal long tail fiber gp34 [44] (probability 91.96%, e-value 7.4). Preliminary structural predictions and analysis for KEN22, KEN25-2, KEN39, 15882-3, and 15882-6 tail fibers showed that the recombinant tail fiber proteins from trained phages 15882-3 and 15882-6 have predicted structures unique from parents.
Recombination was detected between KEN22 and KEN39 in a gene annotated as baseplate hub subunit and tail lysozyme for isolate 15882-1 (MBLPYHJN_CDS_0184), which is homologous to gp5 of the T4 phage [45]. This results in two amino acid changes compared to KEN22 at residues 494 (Asn->Asp) and 516 (Ser->Asp) which corresponds to the residues present in KEN39, however, no unique amino acid substitutions were identified.
Genetic analysis of phage clones collected after serial propagation on the permissive host strain K. pneumoniae MRSN 414780 showed that only two clones had additional mutations compared to the original trained phage they were derived from. 15882-3-2 had two mutations, one was a SNP in an intergenic region between genes that encode for lysis inhibition protein and a head morphogenesis protein (see Table S1) and a nonsynonymous mutation in a thioredoxin domain contain protein. Phage clone 15882-6-4 showed one additional mutation in a gene that encodes a tail collar fiber protein. This gene showed homology (probability 100%, e-value 2.1e-32) to the phage T4 gene encoding gp12, a short tail fiber protein, which is part of the tail fiber network forming a spring-like mechanism that extends upon interaction with a suitable host cell, with the N-terminal of domain of gp12 orienting towards the host cell surface to bind to a receptor [46].
3. Discussion
K. pneumoniae is a challenging target species for the development of broad-range phage cocktail therapeutics. In large part, this is because of the tremendous diversity of cell surface targets, especially capsule and lipopolysaccharide (LPS) types, which are known to serve as phage receptors or otherwise influence phage susceptibility [25,47,48]. For example, the 100-strain diversity panel of K. pneumoniae clinical isolates used in this study was created based on multilocus sequence typing (MLST), includes 94 sequence types (STs), 54 capsule types and 11 O-antigen serotypes [31]. The strains in this panel were isolated between 2003 and 2020 in multiple military hospitals in North and South America, Europe, Asia, and Africa, from blood, wound, urine, respiratory, perianal specimens, and environmental swabs.
Klebsiella phage host range has been correlated to ST and capsule types, which themselves are correlated, with the 92% probability of a phage lytic against one strain of certain capsule type to be lytic against another strain of the same capsule type [49]. Phages need to overcome the thick capsule layer to reach the cell surface. This often requires the phage to encode depolymerase enzymes that can degrade the capsule layer. Klebsiella phages encoding one or a few depolymerases are more likely to have a narrow host range in comparison to phages that encode multiple or divergent depolymerases [50,51,52,53]. This makes a broadly active Klebsiella phage a relatively rare phenomenon as it requires a phage encoding many copies of divergently active depolymerases. One such rare K. pneumoniae phage possessed 11 distinct depolymerases, enabling lysis of 10 distinct capsule types [54]. Interestingly, phages isolated on capsule-deficient mutants of K. pneumoniae have shown relatively broad host ranges [55].
The diversity in capsule and LPS types results in narrow specificity of most Klebsiella phages that usually cover only 1-31% of the tested K. pneumoniae strains [26,27,28,29,30]. Moreover, the panels of strains used for host range testing in these published studies were small and/or limited in diversity, or diversity was not specified. Only a few reported Klebsiella phages have shown broader activity, and there was no evidence for any of them to be effective against diversity strain panels. For example, phages KP34 [56] and vB_KpnM_M1 [23] covered 42/101 (42%) and 76/121 (63%) of K. pneumoniae strains, respectively, but the diversity of the strains was not specified. Phage Kpp95 was lytic against 47% of 108 K. pneumoniae isolates from the same hospital [57]. Phages vB_Klp_3 and vB_Klp_4 were active against 76% of 73 Klebsiella spp. isolates from University Hospitals of Leicester (UK), but only 26% of 50 Georgian isolates [58]. Finally, phages P545 and P546 showed as broad lytic spectrum as 96%, but they were evaluated against 54 K. pneumoniae strains isolated from the same hospital, 48 of which (89%) belonged to the same sequence type, ST11 [59]. Given that the 100-strain K. pneumoniae panel used in this work is highly diverse [31], it should not be surprising that the majority in our >150-phage library had host ranges varying between 1% and 7% (data not shown), with the broadest activity of 36% in phages KEN39 and KEN1821 (Table 2). Therefore, the goal of this work was to use directed evolution to obtain K. pneumoniae phages with broader host ranges and employ them for improving therapeutic phage cocktails.
Phage directed evolution (training, host adaptation) can result in host range expansion [60]. Several researchers have successfully used for host range expansion the Appelmans protocol – incubation of serial phage dilutions with bacterial cultures and lysis monitoring in liquid media first described in 1921 [61]. Using both permissive and phage-resistant bacterial strains, this method has been employed to expand the lytic spectra of phages specific for Staphylococcus aureus [62,63,64,65], Pseudomonas aeruginosa [33,66,67], Enterococcus faecium [68], Acinetobacter baumannii [69], Listeria monocytogenes [70], as well as Escherichia coli [71,72], Streptococcus spp. and Enterococcus spp. isolates [72] from urinary tract infections (UTI). Klebsiella phage vB_KpnM_M1 that belongs to the genus Slopekvirus was adapted by the Appelmans protocol to pandrug-resistant K. pneumoniae isolates from a polytrauma patient; this led to a reduced incidence of phage resistance and higher efficacy of phage treatment [23]. Compassionate use therapy with trained phages was also effective in 6/9 patients with UTIs caused by E. coli, Streptococcus spp. and Enterococcus spp. [72].
Using two phages that belong to the same genus resulted in recombinant phages with expanded activity against P. aeruginosa [33] and L. monocytogenes [70]. In this work, we utilized five myophages of the genus Jiaodavirus (Table 2). This group of phages is characterized by broad host range, quick lysis time, and efficacy at lower multiplicities of infection [35]. These five phages were trained against 11 diverse MDR and XDR K. pneumoniae clinical isolates, including one phage-susceptible and 10 phage-resistant strains (Table 1 and Table 2). Nine evolved phage variants with altered host ranges were isolated that were capable of lysing previously phage-resistant strains. Four of these variants demonstrated host range expansion of up to 8% compared to the parental phage KEN39 with the broadest activity (Table 3, Figure 2). Finding few unique substitutions (Table S1) and multiple recombination events (Figure 5) in the evolved phages suggests that recombination plays a significant role in host range alteration and expansion in Jiaodavirus phages, likely due to the generation of chimeric proteins involved in phage-host interactions, such as those observed in the tail fiber protein of trained phages 15882-3 and 15882-6. However, unique mutations in the gene encoding hinge connector protein of long tail fiber were identified in three of the four genomically characterized evolved phages, thus this gene may also be significant for host range expansion.
Several groups of scientists recently reported that the Appelmans procedures were thwarted by prophage induction: expanded phage activity occurred not because of mutations or recombination events in input phages but because of induced prophages. For example, an attempt to train lytic A. baumannii phages P115, P711 and P577 resulted in isolation of four phages with broader host ranges that were recombinant derivatives of prophages from development bacterial strains [73]. A study with the Appelmans method employing two Yuavirus and one Detrevirus phages detected a Casadabanvirus prophage that was induced from the P. aeruginosa chromosome and caused host range expansion [74]. The use of three lytic Przondovirus phages in a training experiment on K. pneumoniae enabled the isolation of temperate phage vB_KpnS-KpLi5, which expanded the activity of the input phages [75]. Such temperate phages derived from training procedures cannot be used for phage therapy. This kind of prophage induction that expands lytic spectra of input phages was not observed in the work described herein.
Phage training is currently recommended to use for improvement of therapeutic phage cocktails [32,76]. Our team is developing durable fixed phage cocktails against MDR ESKAPE pathogens, including K. pneumoniae [77]. The first iteration of K. pneumoniae phage cocktail, KPM1 (Table 5), consisted of five natural phage isolates and covered 50/100 (50%) of strains in the high diversity panel [31], including 48 different STs, 32 KL serotypes and 9 OL serotypes. Most of the STs are global epidemic, MDR and XDR lineages, e.g., ST11, ST14, ST15, ST20, ST37, ST45, ST101, ST107, ST147, ST258, ST322, ST336, ST340, ST394, and ST512. However, 50% coverage is not enough for an off-the-shelf phage cocktail. Addition of trained phage 15882-3 to this cocktail expanded its activity by 21%. After incorporation of recently isolated wild-type phage KEN1821, the 7-phage cocktail KPM2 had a broad host range of 81% (Table 5, Figure 4a). KPM2 covered 78/94 STs, 45/54 KL serotypes and representatives of all 11 OL serotypes in the diversity strain panel. This improved cocktail also outperformed KPM1 in quality of lysis with more robust plaque formation (Figure 4b) and higher numbers of strains lysed by two or more phages (Figure 4c).
To conclude, the use of five Jiaodavirus phages in the Appelmans procedure against broadly phage-resistant strains allowed for the isolation of progeny phage variants with altered and expanded host ranges. Genome recombination played a significant role in the alteration and expansion of phage lytic spectra. The incorporation of a trained phage into iterative phage cocktail design dramatically expanded host range against K. pneumoniae global diversity represented in a 100-strain panel comprising 94 sequence types.
4. Materials and Methods
4.1. Bacterial strains, Phages, Growth and Storage Conditions
In addition to 11 K. pneumoniae strains used for phage training (Table 1), strains MRSN 3619, MRSN 11382 and MRSN 529046, and Klebsiella quasipneumoniae strain MRSN 829456 were utilized for propagation of phages before training (Table 2). Bacterial cultures were grown in Heart Infusion Broth (HIB, Becton, Dickinson and Company, Franklin Lakes, New Jersey, USA) at 37°C, with shaking at 120 rpm, or on HIB agar. Fresh overnight cultures were prepared for each experiment. Phages used in this work are listed in Table 2. They included five training phages (KEN22, KEN25-1, KEN25-2, KEN37, and KEN39) and five phages used for the development of phage cocktails broadly active against MDR K. pneumoniae isolates (KEN42, KEN1821, AFR4, EKq1, and EKq2). For propagation, a concentrated phage stock was added to bacterial culture at mid-log phase in HIB with 2 mM CaCl2 and 10 mM MgSO4 (HIB-CM) at a multiplicity of infection of ca. 0.01, allowed to incubate for 4-6 h, until the culture was cleared. Debris was pelleted, and phage lysate was sterilized by passing through a 0.22-µm syringe filter. Filtered phage lysates were used as the initial inocula for phage training. Phage lysates were stored at +4°C, protected from light.
4.2. The Appelmans Training
Phage training using the Appelmans protocol was conducted as described previously [33], with modifications. The initial phage inoculum consisted of a 1:1:1:1:1 mix of KEN22, KEN25-1, KEN25-2, KEN37, and KEN39 at a final concentration of 1×1010 PFU/mL (2×109 PFU/mL per each phage). One hundred microliters of the mix was titrated with 100 µL of SM buffer (Alpha Teknova, Inc., Hollister, California, USA) down the columns of 96-well plate, and 100 µL of 2×HIB-CM was added to each well. Titrated phage mixes were inoculated with 2 µL of overnight cultures of K. pneumoniae phage-resistant clinical isolates (Table 1), one strain per column. An uninfected bacterial control was maintained for all strains used. The assembled plate was incubated overnight at 37°C with shaking, and the OD at 600 nm was visualized in a microplate reader (SpectraMax Plus 384, Molecular Devices, San Jose, California, USA). Wells that showed reduction in OD relative to the uninfected control were tracked. The two most diluted lysates showing OD reduction were collected to select for the best performing possible recombinant/mutant phages. If no reduction in OD was observed, the two least diluted samples were collected. All collected lysates were pooled together, filter sterilized and stored at 4°C for subsequent use. Filtered lysates from Round 1 of phage training were titrated in SM buffer, diluted with 2×HIB-CM and inoculated with K. pneumoniae cultures as described above (Round 2), and the procedure was repeated for all subsequent rounds of training. To assess for activity on phage-resistant strains, pooled lysates were titrated in SM buffer to 10–7 and plated on the strains used in the phage training. Plaque formation was assessed, and plaques collected for follow up analysis.
4.3. Purification of Collected Trained Phage Variants
Phages isolated on previously resistant strains were collected by picking plaques on double-layer HIB agar (1.5%/0.7%) and transferring to 500 µL of sterile SM buffer. Fifty microliters of chloroform was added to assist in destroying any remaining cells and releasing phage still inside. After a 15-min incubation at room temperature, the plaque suspension was centrifuged at 5,000×g for 5 min. The aqueous phase was collected, and filter sterilized. The process of phage plating, single plaque isolation and filter sterilization was repeated three times, and phage purity was then assessed by confirming uniform plaque morphology.
4.4. Host Range Determination
Host range was determined by assessing lysis against the 100-strain K. pneumoniae diversity panel [31] of mostly MDR clinical isolates as described previously [64], with minor modifications. Briefly, overnight cultures of bacterial strains were grown in HIB. Ten-fold serial dilutions of the tested phages were prepared in a sterile round bottom 96-well plate. An aliquot (2 μL) of each phage dilution, ranging from 10−1 to 10−8, was spotted using a multichannel pipette on 0.7% HIB agar overlay infused with K. pneumoniae culture and incubated overnight at 37°C. The following day, the quality of lysis was assessed.
4.5. Assessment of Stability of Host Range Expansion
The stability of observed changes in lytic spectra was assessed by conducting serial propagations on a single phage-susceptible bacterial strain. This was followed by re-isolation and purification of clones and re-assessment of host range, which was compared with the originally isolated trained phage clones.
4.6. DNA Isolation, Library Preparation, Sequencing and Genome Assembly
Phages were propagated on a corresponding host strain, and their DNA was extracted as described [78] using the QIAamp DNA Mini Kit (Qiagen, Germantown, Maryland, USA). Sequencing libraries were constructed using the KAPA HyperPlus Kit (Roche Diagnostics, Indianapolis, Indiana, USA) and sequenced on an Illumina MiSeq (Illumina, Inc., San Diego, California, USA) with a 600 cycle MiSeq Reagent Kit v3 that produced 300-bp paired-end reads. The quality of reads was assessed, and then trimmed using Fastp [79] v0.22.0. Genomes were then de novo assembled from the trimmed reads using Unicycler [80] v0.5.0 using both paired and unpaired reads. Where necessary, trimmed read datasets were subsampled using seqtk (https://github.com/lh3/seqtk) v1.4 to achieve ~100× of expected genome prior to genome assembly. Assembly statistics and average coverage of assembled genomes were determined using BBmap (https://sourceforge.net/projects/bbmap) v38.9 and SAMtools [81] v1.13.
4.7. Genome Annotation
The termini of each assembled phage genome were identified using PhageTerm [82] v1.0.12. Phage protein coding sequences (CDSs) were annotated using the Pharokka pipeline [83,84,85,86,87,88,89,90,91,92,93,94]. Pharokka [83] integrates predicted coding sequences (CDS) from PHANOTATE [84] with functional annotations generated by matching each CDS to the PHROG [85], VFDB [86] and CARD [87] databases using MMseqs2 [88] and PyHMMER [89]. tRNAs and tmRNAs were predicted with tRNAscan-SE 2.0 [90] and ARAGORN [91], respectively, and CRISPRs were predicted with CRT [92]. Phage contigs were also matched to their closest hit in the INPHARED database [93] using mash [94].
4.8. Genome Variation Analysis
Structural variations, single nucleotide polymorphisms (SNPs), insertions and deletions (indels) were identified between phage mutant genomes and their respective parental strains using NucDiff [95] v2.0.3. NucDiff aligns input genomes and detects variants including substitutions, insertions, deletions, inversions, and translocations. The R package gggenome [96] was used to visualize genomic variations in RStudio (2024.09.0+375). Aligned genomes were provided as input to generate figures depicting SNPs, indels, and structural rearrangements. Default gggenome plotting parameters were used, except for varying figure size and color schemes. Protein homology searches were performed using HHpred [97] and Phyre2 [98].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Predicted mutations in trained phages and their clones isolated from stability testing experiments.
Author Contributions
Conceptualization, K.A.B. and A.A.F.; methodology, K.A.B., T.L.P., O.A.K., C.D.U., B.D.W., J.T.B., N.M., and M.O.G.; software, K.A.B. and T.L.P.; validation, K.A.B., T.L.P., J.T.B., N.M., P.L.; L.A.M., M.P.N., and A.A.F.; formal analysis, K.A.B., T.L.P., O.A.K., C.D.U., J.T.B., N.M., M.O.G., and A.A.F.; investigation, K.A.B., T.L.P., O.A.K., C.D.U., B.D.W., J.T.B., N.M., M.O.G., and A.A.F.; resources, K.A.B., T.L.P., P.L., L.A.M., M.P.N., and A.A.F.; data curation, K.A.B., T.L.P., J.T.B., N.M., P.L.; L.A.M., M.P.N., and A.A.F.; writing—original draft preparation, K.A.B. and A.A.F.; writing—review and editing, K.A.B., T.L.P., O.A.K., C.D.U., B.D.W., J.T.B., N.M., M.O.G., P.L., L.A.M., M.P.N., and A.A.F.; visualization, K.A.B., T.L.P., C.D.U., and A.A.F.; supervision, P.L., L.A.M., M.P.N., and A.A.F.; project administration, M.P.N. and A.A.F.; funding acquisition, M.P.N. and A.A.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by DoD Congressionally Directed Medical Research Programs, Peer Reviewed Medical Research Program, Focused Program Award PR182667, and Military Infectious Diseases Research Program, grant MI210045.
Data Availability Statement
The BioProject number for this study is PRJNA1173206. GenBank accession numbers for complete genome sequences of phages AFR4, EKq2, KEN1821, and KEN42 are PV240279, PV240280, PV240281, and PV240282, respectively. GenBank accession numbers for genomes of evolved phages 15882-1, 15882-3, 15882-5, and 15882-6 are PQ537369, PQ537375, PQ537376, and PQ537382. GenBank accession numbers for genomes of phage clones 15882-3-1 through 15882-3-5 and 15882-6-1 through 1588-6-5 from stability testing are PQ537370-PQ537374 and PQ537377-PQ537381, respectively.
Acknowledgments
Material has been reviewed by the Walter Reed Army Institute of Research. There is no objection to its presentation and/or publication. The opinions or assertions contained herein are the private views of the authors and are not to be construed as official, or as reflecting true views of the Department of the Army or the Department of Defense. Multidrug-Resistant Organism Repository and Surveillance Network (MRSN) at the Walter Reed Army Institute of Research provided strains of K. pneumoniae and performed DNA sequencing. Generative artificial intelligence (GenAI) has not been used in this paper.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| CDS | Protein coding sequence |
| HIB | Heart Infusion Broth with calcium chloride and magnesium sulfate |
| HIB-CM | Heart Infusion Broth with |
| KPM | Klebsiella phage mix |
| LPS | Lipopolysaccharide |
| MDPI | Multidisciplinary Digital Publishing Institute |
| MDR | Multidrug-resistant |
| MIC | Minimum inhibitory concentration |
| MLST | Multilocus sequence typing |
| MRSN | Multidrug-Resistant Organism Repository and Surveillance Network |
| PPR | Pan-phage-resistant |
| PS | Phage-susceptible |
| SNP | Single nucleotide polymorphism |
| ST | Sequence type |
| UTI | Urinary tract infections |
| WRAIR | Walter Reed Army Institute of Research |
| XDR | Extensively drug-resistant |
| XPR | Extensively phage-resistant |
References
- Chang, D.; Sharma, L.; Dela Cruz, C.S.; Zhang, D. Clinical epidemiology, risk factors, and control strategies of Klebsiella pneumoniae infection. Front. Microbiol. 2021, 12, 750662. [Google Scholar] [CrossRef]
- Kochan, T.J.; Nozick, S.H.; Medernach, R.L.; Cheung, B.H.; Gatesy, S.W.M.; Lebrun-Corbin, M.; Mitra, S.D.; Khalatyan, N.; Krapp, F.; Qi, C.; Ozer, E.A.; Hauser, A.R. Genomic surveillance for multidrug-resistant or hypervirulent Klebsiella pneumoniae among United States bloodstream isolates. BMC Infect. Dis. 2022, 22, 603. [Google Scholar] [CrossRef]
- Ullah, S.R.; Jamal, M.; Rahman, A.; Andleeb, S. Comprehensive insights into Klebsiella pneumoniae: unravelling clinical impact, epidemiological trends and antibiotic-resistance challenges. J. Antimicrob. Chemother. 2024, 79, 1484–1492. [Google Scholar] [CrossRef]
- Shoma, S.; Kamruzzaman, M.; Ginn, A.N.; Iredell, J.R.; Partridge, S.R. Characterization of multidrug-resistant Klebsiella pneumoniae from Australia carrying blaNDM-1. Diagn. Microbiol. Infect. Dis. 2014, 78, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Long, S.W.; Olsen, R.J.; Eagar, T.N.; Beres, S.B.; Zhao, P.; Davis, J.J.; Brettin, T.; Xia, F.; Musser, J.M. Population genomic analysis of 1,777 extended-spectrum beta-lactamase-producing Klebsiella pneumoniae isolates, Houston, Texas: unexpected abundance of clonal group 307. mBio 2017, 8, e00489–17. [Google Scholar] [CrossRef] [PubMed]
- Rojas, R.; Macesic, N.; Tolari, G.; Guzman, A.; Uhlemann, A.-C. Multidrug-resistant Klebsiella pneumoniae ST307 in traveler returning from Puerto Rico to Dominican Republic. Emerg. Infect. Dis. 2019, 25, 1583–1585. [Google Scholar] [CrossRef]
- Heiden, S.E.; Hübner, N.O.; Bohnert, J.A.; Heidecke, C.-D.; Kramer, A.; Balau, V.; Gierer, W.; Schaefer, S.; Eckmanns, T.; Gatermann, S.; et al. A Klebsiella pneumoniae ST307 outbreak clone from Germany demonstrates features of extensive drug resistance, hypermucoviscosity, and enhanced iron acquisition. Genome Med. 2020, 12, 113. [Google Scholar] [CrossRef]
- Mbelle, N.M.; Feldman, C.; Sekyere, J.O.; Maningi, N.E.; Modipane, L.; Essack, S.Y. Pathogenomics and evolutionary epidemiology of multi-drug resistant clinical Klebsiella pneumoniae isolated from Pretoria, South Africa. Sci. Rep. 2020, 10, 1232. [Google Scholar] [CrossRef]
- Martin, M.J.; Corey, B.W.; Sannio, F.; Hall, L.R.; MacDonald, U.; Jones, B.T.; Mills, E.G.; Harless, C.; Stam, J.; Maybank, R.; et al. Anatomy of an extensively drug-resistant Klebsiella pneumoniae outbreak in Tuscany, Italy. Proc. Natl. Acad. Sci. USA 2021, 118, e2110227118. [Google Scholar] [CrossRef]
- Salazar, C.; Antelo, V.; Vieytes, M.; Dávila, C.; Grill, F.; Galiana, A.; Iraola, G. First detection and origin of multi-drug resistant Klebsiella pneumoniae ST15 harboring OXA-48 in South America. J. Glob. Antimicrob. Resist. 2022, 30, 480–484. [Google Scholar] [CrossRef]
- Pham, M.H.; Hoi, L.T.; Beale, M.A.; Khokhar, F.A.; Hoa, N.T.; Musicha, P.; Blackwell, G.A.; Long, H.B.; Huong, D.T.; Binh, N.G.; et al. Evidence of widespread endemic populations of highly multidrug resistant Klebsiella pneumoniae in hospital settings in Hanoi, Vietnam: a prospective cohort study. Lancet Microbe 2023, 4, e255–e263. [Google Scholar] [CrossRef]
- Zarkotou, O.; Pournaras, S.; Tselioti, P.; Dragoumanos, V.; Pitiriga, V.; Ranellou, K.; Prekates, A.; Themeli-Digalaki, K.; Tsakris, A. Predictors of mortality in patients with bloodstream infections caused by KPC-producing Klebsiella pneumoniae and impact of appropriate antimicrobial treatment. Clin. Microbiol. Infect. 2011, 17, 1798–1803. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Sun, X.; Ma, X. Systematic review and meta-analysis of mortality of patients infected with carbapenem-resistant Klebsiella pneumoniae. Ann. Clin. Microbiol. Antimicrob. 2017, 16, 18. [Google Scholar] [CrossRef]
- Karlsson, M.; Stanton, R.A.; Ansari, U.; McAllister, G.; Chan, M.Y.; Grass, S.E.; Duffy, N.; Anacker, M.L.; Witwer, M.L.; Rasheed, J.K.; et al. Identification of a carbapenemase-producing hypervirulent Klebsiella pneumoniae isolate in the United States. Antimicrob. Agents Chemother. 2019, 63, e00519–19. [Google Scholar] [CrossRef]
- Choby, J.E.; Howard-Anderson, J.; Weiss, D.S. Hypervirulent Klebsiella pneumoniae - clinical and molecular perspectives. J. Intern. Med. 2020, 287, 283–300. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ding, Y.; Xu, Y.; Li, Z.; Zeng, Z.; Liu, J. An outbreak of extensively drug-resistant and hypervirulent Klebsiella pneumoniae in an intensive care unit of a teaching hospital in Southwest China. Front. Cell. Infect. Microbiol. 2022, 12, 979219. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, H.; Liao, X. Hypervirulent Klebsiella pneumoniae. Infect. Drug Resist. 2023, 16, 5243–5249. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, S.; Ruth, M.M.; Mientjes, M.; de Sévaux, R.G.L.; van Ingen, J. A Dutch case report of successful treatment of chronic relapsing urinary tract infection with bacteriophages in a renal transplant patient. Antimicrob. Agents Chemother. 2019, 64, e01281–19. [Google Scholar] [CrossRef]
- Anand, T.; Virmani, N.; Kumar, S.; Mohanty, A.K.; Pavulraj, S.; Bera, B.C.; Vaid, R.K.; Ahlawat, U.; Tripathi, B.N. Phage therapy for treatment of virulent Klebsiella pneumoniae infection in a mouse model. J. Glob. Antimicrob. Resist. 2020, 21, 34–41. [Google Scholar] [CrossRef]
- Bao, J.; Wu, N.; Zeng, Y.; Chen, L.; Li, L.; Yang, L.; Zhang, Y.; Guo, M.; Li, L.; Li, J.; et al. Non-active antibiotic and bacteriophage synergism to successfully treat recurrent urinary tract infection caused by extensively drug-resistant Klebsiella pneumoniae. Emerg. Microbes Infect. 2020, 9, 771–774. [Google Scholar] [CrossRef]
- Cano, E.J.; Caflisch, K.M.; Bollyky, P.L.; Van Belleghem, J.D.; Patel, R.; Fackler, J.; Brownstein, M.J.; Horne, B.; Biswas, B.; Henry, M.; et al. Phage therapy for limb-threatening prosthetic knee Klebsiella pneumoniae infection: case report and in vitro characterization of anti-biofilm activity. Clin. Infect. Dis. 2021, 73, e144–e151. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Fu, H.; Tian, Z.; Cui, J.; Yan, C.; Xue, G.; Fan, Z.; Du, B.; Feng, J.; Zhao, H.; et al. Bacteriophage effectively rescues pneumonia caused by prevalent multidrug-resistant Klebsiella pneumoniae in the early stage. Microbiol. Spectr. 2022, 10, e0235822. [Google Scholar] [CrossRef]
- Eskenazi, A.; Lood, C.; Wubbolts, J.; Hites, M.; Balarjishvili, N.; Leshkasheli, L.; Askilashvili, L.; Kvachadze, L.; van Noort, V.; Wagemans, J.; et al. Combination of pre-adapted bacteriophage therapy and antibiotics for treatment of fracture-related infection due to pandrug-resistant Klebsiella pneumoniae. Nat. Commun. 2022, 13, 302. [Google Scholar] [CrossRef]
- Ichikawa, M.; Nakamoto, N.; Kredo-Russo, S.; Weinstock, E.; Weiner, I.N.; Khabra, E.; Ben-Ishai, N.; Inbar, D.; Kowalsman, N.; Mordoch, R.; et al. Bacteriophage therapy against pathological Klebsiella pneumoniae ameliorates the course of primary sclerosing cholangitis. Nat. Commun. 2023, 14, 3261. [Google Scholar] [CrossRef]
- Wyres, K.L.; Holt, K.E. Klebsiella pneumoniae population genomics and antimicrobial-resistant clones. Trends Microbiol. 2016, 24, 944–956. [Google Scholar] [CrossRef]
- Kęsik-Szeloch, A.; Drulis-Kawa, Z.; Weber-Dąbrowska, B.; Kassner, J.; Majkowska-Skrobek, G.; Augustyniak, D.; Lusiak-Szelachowska, M.; Zaczek, M.; Górski, A.; Kropinski, A.M. Characterising the biology of novel lytic bacteriophages infecting multidrug resistant Klebsiella pneumoniae. Virol. J. 2013, 10, 100. [Google Scholar] [CrossRef]
- Lin, T.-L.; Hsieh, P.-F.; Huang, Y.-T.; Lee, W.-C.; Tsai, Y.-T.; Su, P.-A.; Pan, Y.-J.; Hsu, C.-R.; Wu, M.-C.; Wang, J.-T. Isolation of a bacteriophage and its depolymerase specific for K1 capsule of Klebsiella pneumoniae: implication in typing and treatment. J. Infect. Dis. 2014, 210, 1734–1744. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, R.; Xu, M.; Liu, Y.; Zhu, X.; Qiu, J.; Liu, Q.; He, P.; Li, Q. A novel polysaccharide depolymerase encoded by the phage SH-KP152226 confers specific activity against multidrug-resistant Klebsiella pneumoniae via biofilm degradation. Front. Microbiol. 2019, 10, 2768. [Google Scholar] [CrossRef]
- Zhang, C.; Yuan, J.; Guo, C.; Ge, C.; Wang, X.; Wei, D.; Li, X.; Si, H.; Hu, C. Identification and complete genome of lytic "Kp34likevirus" phage vB_KpnP_Bp5 and therapeutic potency in the treatment of lethal Klebsiella pneumoniae infections in mice. Virus Res. 2021, 297, 198348. [Google Scholar] [CrossRef]
- Ferriol-González, C.; Concha-Eloko, R.; Bernabéu-Gimeno, M.; Fernández-Cuenca, F.; Cañada-García, J.E.; García-Cobos, S.; Sanjuán, R.; Domingo-Calap, P. Targeted phage hunting to specific Klebsiella pneumoniae clinical isolates is an efficient antibiotic resistance and infection control strategy. Microbiol. Spectr. 2024, 12, e00254–24. [Google Scholar] [CrossRef]
- Martin, M.J.; Stribling, W.; Ong, A.C.; Maybank, R.; Kwak, Y.I.; Rosado-Mendez, J.A.; Preston, L.N.; Lane, K.F.; Julius, M.; Jones, A.R.; et al. A panel of diverse Klebsiella pneumoniae clinical isolates for research and development. Microb. Genom. 2023, 9, mgen000967. [Google Scholar] [CrossRef] [PubMed]
- Merabishvili, M.; Pirnay, J.P.; De Vos, D. Guidelines to compose an ideal bacteriophage cocktail. Methods Mol. Biol. 2018, 1693, 99–110. [Google Scholar] [CrossRef]
- Burrowes, B.H.; Molineux, I.J.; Fralick, J.A. Directed in vitro evolution of therapeutic bacteriophages: the Appelmans protocol. Viruses 2019, 11, 241. [Google Scholar] [CrossRef]
- Peters, T.L.; Urick, C.D.; Georges, M.; Burke, K.A.; Kirillina, O.A.; Mzhavia, N.; Musila, L.; Filippov, A.A.; Nikolich, M.P. Genome sequences of five Klebsiella bacteriophages that belong to the genus Jiaodavirus. Microbiol. Resour. Announc. 2024, 13, e0105624. [Google Scholar] [CrossRef]
- Townsend, E.M.; Kelly, L.; Gannon, L.; Muscatt, G.; Dunstan, R.; Michniewski, S.; Sapkota, H.; Kiljunen, S.J.; Kolsi, A.; Skurnik, M.; et al. Isolation and characterization of Klebsiella phages for phage therapy. Phage (New Rochelle) 2021, 2, 26–42. [Google Scholar] [CrossRef]
- Bird, J.T.; Burke, K.A.; Urick, C.D.; Braverman, J.L.; Mzhavia, N.; Ellison, D.W.; Nikolich, M.P.; Filippov, A.A. Genome sequence of the Klebsiella quasipneumoniae bacteriophage EKq1 with activity against Klebsiella pneumoniae. Microbiol. Resour. Announc. 2024, 13, e0095423. [Google Scholar] [CrossRef]
- Raaijmakers, H.; Törö, I.; Birkenbihl, R.; Kemper, B.; Suck, D. Conformational flexibility in T4 endonuclease VII revealed by crystallography: implications for substrate binding and cleavage. J. Mol. Biol. 2001, 302, 2. [Google Scholar] [CrossRef]
- Granel,l M. ; Namura, M.; Alvira, S.; Kanamaru, S.; Van Raaij, M.J. Crystal structure of the carboxy-terminal region of the bacteriophage T4 proximal long tail fiber protein Gp34. Viruses 2017, 9, 168. [Google Scholar] [CrossRef]
- Ayala, R.; Moiseenko, A.V.; Chen, T.H.; Eugene, E.E.; Golomidova, A.K.; Orekhov, P.S.; Street, M.A.; Sokolova, O.S.; Letarov, A.V.; Wolf, M. Nearly complete structure of bacteriophage DT57C reveals architecture of head-to-tail interface and lateral tail fibers. Nat. Commun. 2023, 14, 8205. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Bittrich, S.; Segura, J.; Duarte, J.M.; Burley, S.K.; Rose, Y. RCSB protein Data Bank: exploring protein 3D similarities via comprehensive structural alignments. Bioinformatics 2024, 40, btae370. [Google Scholar] [CrossRef]
- Zheng, J.; Chen, W.; Xiao, H.; Yang, F.; Song, J; Cheng, L. ; Liu, H. Asymmetric structure of podophage GP4 reveals a novel architecture of three types of tail fibers. J. Mol. Biol. 2023, 435, 168258. [Google Scholar] [CrossRef]
- Garcia-Doval, C.; Castón, J.R.; Luque, D.; Granell, M.; Otero, J.M.; Llamas-Saiz, A.L.; Renouard, M.; Boulanger, P.; Van Raaij, M.J. Structure of the receptor-binding carboxy-terminal domain of the bacteriophage T5 L-shaped tail fibre with and without its intra-molecular chaperone. Viruses 2015, 7, 6424–6440. [Google Scholar] [CrossRef]
- Granell, M.; Namura, M.; Alvira, S.; Kanamaru, S.; Van Raaij, M.J. Crystal structure of the carboxy-terminal region of the bacteriophage T4 proximal long tail fiber protein gp34. Viruses 2017, 9, 168. [Google Scholar] [CrossRef] [PubMed]
- Kanamaru, S.; Ishiwata, Y.; Suzuki, T.; Rossmann, M.G.; Arisaka, F. Control of bacteriophage T4 tail lysozyme activity during the infection process. J. Mol. Biol. 2005, 346, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.M.; Prokhorov, N.S.; Guerrero-Ferreira, R.C.; Shneider, M.M.; Browning, C.; Goldie, K.N.; Stahlberg, H.; Leiman, P.G. Structure of the T4 baseplate and its function in triggering sheath contraction. Nature 2016, 533, 346–352. [Google Scholar] [CrossRef]
- Follador, R.; Heinz, E.; Wyres, K.L.; Ellington, M.J.; Kowarik, M.; Holt, K.E.; Thomson, N.R. The diversity of Klebsiella pneumoniae surface polysaccharides. Microb. Genom. 2016, 2, e000073. [Google Scholar] [CrossRef]
- Hesse, S.; Rajaure, M.; Wall, E.; Johnson, J.; Bliskovsky, V.; Gottesman, S.; Adhya, S. Phage resistance in multidrug-resistant Klebsiella pneumoniae ST258 evolves via diverse mutations that culminate in impaired adsorption. mBio 2020, 11, e02530–19. [Google Scholar] [CrossRef]
- Beamud, B.; García-González, N.; Gómez-Ortega, M.; González-Candelas, F.; Domingo-Calap, P.; Sanjuan, R. Genetic determinants of host tropism in Klebsiella phages. Cell Rep. 2023, 42, 112048. [Google Scholar] [CrossRef]
- Domingo-Calap, P.; Beamud, B.; Mora-Quilis, L.; González-Candelas, F.; Sanjuán, R. Isolation and characterization of two Klebsiella pneumoniae phages encoding divergent depolymerases. Int. J. Mol. Sci. 2020, 21, 3160. [Google Scholar] [CrossRef]
- Majkowska-Skrobek, G.; Latka, A.; Berisio, R.; Squeglia, F.; Maciejewska, B.; Briers, Y.; Drulis-Kawa, Z. Phage-borne depolymerases decrease Klebsiella pneumoniae resistance to innate defense mechanisms. Front. Microbiol. 2018, 9, 2517. [Google Scholar] [CrossRef]
- Wu, J.W.; Wang, J.-T.; Lin, T.-L.; Liu, Y.-Z.; Wu, L.-T.; Pan, Y.-J. Identification of three capsule depolymerases in a bacteriophage infecting Klebsiella pneumoniae capsular types K7, K20, and K27 and therapeutic application. J. Biomed. Sci. 2023, 30, 31. [Google Scholar] [CrossRef]
- Li, M.; Wang, H.; Chen, L.; Guo, G.; Li, P.; Ma, J.; Chen, R.; Du, H.; Liu, Y.; Zhang, W. Identification of a phage-derived depolymerase specific for KL47 capsule of Klebsiella pneumoniae and its therapeutic potential in mice. Virol. Sin. 2022, m37, 538–546. [Google Scholar] [CrossRef]
- Pan, Y.-J.; Lin, T.-L.; Chen, C.-C.; Tsai, Y.-T.; Cheng, Y.-H.; Chen, Y.-Y.; Hsieh, P.-F.; Lin, Y.-T.; Wang, J.-T. Klebsiella phage ΦK64-1 encodes multiple depolymerases for multiple host capsular types. J. Virol. 2017, 91, e02457–16. [Google Scholar] [CrossRef]
- Lourenco, M.; Osbelt, L.; Passet, V.; Gravey, F.; Megrian, D.; Strowig, T.; Rodrigues, C.; Brisse, S. Phages against noncapsulated Klebsiella pneumoniae: broader host range, slower resistance. Microbiol. Spectr. 2023, 11, e0481222. [Google Scholar] [CrossRef]
- Drulis-Kawa, Z.; Mackiewicz, P.; Kęsik-Szeloch, A.; Maciaszczyk-Dziubinska, E.; Weber-Dąbrowska, B.; Dorotkiewicz-Jach, A.; Augustyniak, D.; Majkowska-Skrobek, G.; Bocer, T.; Empel, J.; et al. Isolation and characterisation of KP34 – a novel ɸKMV-like bacteriophage for Klebsiella pneumoniae. Appl. Microbiol. Biotechnol. 2011, 90, 1333–1345. [Google Scholar] [CrossRef]
- Wu, L.-T.; Chang, S.-Y.; Yen, M.-R.; Yang, T.-C.; Tseng, Y.-H. Characterization of extended-host-range pseudo-T-even bacteriophage Kpp95 isolated on Klebsiella pneumoniae. Appl. Environ. Microbiol. 2007, 73, 2532–2540. [Google Scholar] [CrossRef]
- Karumidze, N.; Kusradze, I.; Rigvava, S.; Goderdzishvili, M.; Rajakumar, K.; Alavidze, Z. Isolation and characterisation of lytic bacteriophages of Klebsiella pneumoniae and Klebsiella oxytoca. Curr. Microbiol. 2013, 66, 251–258. [Google Scholar] [CrossRef]
- Li, M.; Guo, M.; Chen, L.; Zhu, C.; Xiao, Y.; Li, P.; Guo, H.; Chen, L.; Zhang, W.; Du, H. Isolation and characterization of novel lytic bacteriophages infecting epidemic carbapenem-resistant Klebsiella pneumoniae strains. Front. Microbiol. 2020, 1, 1554. [Google Scholar] [CrossRef]
- Hall, J.P.J.; Harrison, E.; Brockhurst, M.A. Viral host-adaptation: insights from evolution experiments with phages. Curr. Opin. Virol. 2013, 3, 572–577. [Google Scholar] [CrossRef]
- Appelmans, R. Le dosage du bacteriophage. Compt. Rend. Soc. Biol. 1921, 85, 1098–1099. [Google Scholar]
- O'Flaherty, S.; Ross, R.P.; Meaney, W.; Fitzgerald, G.F.; Elbreki, M.F.; Coffey, A. Potential of the polyvalent anti-Staphylococcus bacteriophage K for control of antibiotic-resistant staphylococci from hospitals. Appl. Environ. Microbiol. 2005, 71, 1836–1842. [Google Scholar] [CrossRef] [PubMed]
- Kvachadze, L.; Balarjishvili, N.; Meskhi, T.; Tevdoradze, E.; Skhirtladze, N.; Pataridze, T.; Adamia, R.; Topuria, T.; Kutter, E.; Rohde, C.; et al. Evaluation of lytic activity of staphylococcal bacteriophage Sb-1 against freshly isolated clinical pathogens. Microb. Biotechnol. 2011, 4, 643–650. [Google Scholar] [CrossRef]
- Sergueev, K.V.; Filippov, A.A.; Farlow, J.; Su, W.; Kvachadze, L.; Balarjishvili, N.; Kutateladze, M.; Nikolich, M.P. Correlation of host range expansion of therapeutic bacteriophage Sb-1 with allele state at a hypervariable repeat locus. Appl. Environ. Microbiol. 2019, 85, e01209–19. [Google Scholar] [CrossRef] [PubMed]
- Kolenda, C.; Bonhomme, M.; Medina, M.; Pouilly, M.; Rousseau, C.; Troesch, E.; Martins-Simoes, P.; Stegger, M.; Verhoeven, P.O.; Laumay, F.; et al. Potential of training of anti-Staphylococcus aureus therapeutic phages against Staphylococcus epidermidis multidrug-resistant isolates is restricted by inter- and intra-sequence type specificity. mSystems 2024, 9, e0085024. [Google Scholar] [CrossRef] [PubMed]
- Burrowes, B. Analysis of the Appelmans protocol for the generation of therapeutic bacteriophages. Ph.D. thesis, Texas Tech Univeristy Health Sciences Center, Lubbock, Texas, USA, 2011.
- Mapes, A.C.; Trautner, B.W.; Liao, K.S.; Ramig, R.F. Development of expanded host range phage active on biofilms of multi-drug resistant Pseudomonas aeruginosa. Bacteriophage 2016, 6, e1096995. [Google Scholar] [CrossRef]
- Lossouarn, J.; Beurrier, E.; Bouteau, A.; Moncaut, E.; Sir Silmane, M.; Portalier, H.; Zouari, A.; Cattoir, V.; Serror, P.; Petit, M.-A. The virtue of training: extending phage host spectra against vancomycin-resistant Enterococcus faecium strains using the Appelmans method. Antimicrob. Agents Chemother. 2024, 68, e0143923. [Google Scholar] [CrossRef]
- Blasco, L.; Bleriot, I.; González de Aledo, M.; Fernández-García, L.; Pacios, O.; Oliveira, H.; López, M.; Ortiz-Cartagena, C.; Fernández-Cuenca, F.; Pascual, Á.; et al. Development of an anti-Acinetobacter baumannii biofilm phage cocktail: genomic adaptation to the host. Antimicrob. Agents Chemother. 2022, 66, e0192321. [Google Scholar] [CrossRef]
- Peters, T.L.; Song, Y.; Bryan, D.W.; Hudson, L.K.; Denes, T.G. Mutant and recombinant phages selected from in vitro coevolution conditions overcome phage-resistant Listeria monocytogenes. Appl. Environ. Microbiol. 2020, 86, e02138–20. [Google Scholar] [CrossRef]
- Sybesma, W.; Zbinden, R.; Chanishvili, N.; Kutateladze, M.; Chkhotua, A.; Ujmajuridze, A.; Mehnert, U.; Kessler, T.M. Bacteriophages as potential treatment for urinary tract infections. Front. Microbiol. 2016, 7, 465. [Google Scholar] [CrossRef]
- Ujmajuridze, A.; Chanishvili, N.; Goderdzishvili, M.; Leitner, L.; Mehnert, U.; Chkhotua, A.; Kessler, T.M.; Sybesma, W. Adapted bacteriophages for treating urinary tract infections. Front. Microbiol. 2018, 9, 1832. [Google Scholar] [CrossRef]
- Vu, T.N.; Clark, J.R.; Jang, E.; D'Souza, R.; Nguyen, L.P.; Pinto, N.A.; Yoo, S.; Abadie, R.; Maresso, A.W.; Yong, D. Appelmans protocol - a directed in vitro evolution enables induction and recombination of prophages with expanded host range. Virus Res. 2024, 339, 199272. [Google Scholar] [CrossRef]
- Peters, T.L.; Schow, J.; Spencer, E.; Van Leuven, J.T.; Wichman, H.; Miller, C. Directed evolution of bacteriophages: thwarted by prolific prophage. Appl. Environ. Microbiol. 2024, 90, e0088424. [Google Scholar] [CrossRef]
- Jakob, N.; Hammerl, J.A.; Swierczewski, B.E.; Würstle, S.; Bugert, J.J. Appelmans protocol for in vitro Klebsiella pneumoniae phage host range expansion leads to induction of the novel temperate linear plasmid prophage vB_KpnS-KpLi5. Virus Genes 2025, 61, 132–135. [Google Scholar] [CrossRef]
- Merabishvili, M.; Pirnay, J.P.; De Vos, D. Guidelines to compose an ideal bacteriophage cocktail. Methods Mol. Biol. 2024, 2734, 49–66. [Google Scholar] [CrossRef]
- Nikolich, M.P.; Filippov, A.A. Bacteriophage therapy: developments and directions. Antibiotics (Basel) 2020, 9, 135. [Google Scholar] [CrossRef]
- Mencke, J.L.; He, Y.; Filippov, A.A.; Nikolich, M.P.; Belew, A.T.; Fouts, D.E.; McGann, P.T.; Swierczewski, B.E.; Getnet, D.; Ellison, D.W.; et al. Identification and characterization of vB_PreP_EPr2, a lytic bacteriophage of pan-drug resistant Providencia rettgeri. Viruses 2022, 14, 708. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLOS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Garneau, J.R.; Depardieu, F.; Fortier, L.-C.; Bikard, D.; Monot, M. PhageTerm: a tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017; 7, 8292. [CrossRef]
- Bouras, G.; Nepal, R.; Houtak, G.; Psaltis, A.J.; Wormald, P.-J.; Vreugde, S. Pharokka: a fast scalable bacteriophage annotation tool. Bioinformatics 2023, 39, btac776. [Google Scholar] [CrossRef]
- McNair, K.; Zhou, C.; Dinsdale, E.A.; Souza, B.; Edwards, R.A. PHANOTATE: a novel approach to gene identification in phage genomes. Bioinformatics 2019, 35, 4537–4542. [Google Scholar] [CrossRef] [PubMed]
- Terzian, P.; Olo Ndela, E.; Galiez, C.; Lossouarn, J.; Pérez Bucio, R.E.; Mom, R.; Toussaint, A.; Petit, M.-A.; Enault, F. PHROG: families of prokaryotic virus proteins clustered using remote homology. NAR Genomics and Bioinformatics 2021, 3, 1–12. [Google Scholar] [CrossRef]
- Chen, L. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 2004, 33, D325–D328. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.; Cheng, A.A.; Liu, S.; et al. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2019, 48, D517–D525. [Google Scholar] [CrossRef]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef]
- Larralde, M.; Zeller, G. PyHMMER: a Python library binding to HMMER for efficient sequence analysis. Bioinformatics 2023, 39, btad214. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Bland, C.; Ramsey, T.L.; Sabree, F.; Lowe, M.; Brown, K.; Kyrpides, N.C.; Hugenholtz, P. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 2007, 8, 209. [Google Scholar] [CrossRef]
- Cook, R.; Brown, N.; Redgwell, T.; Rihtman, B.; Barnes, M.; Clokie, M.; Stekel, D.J.; Hobman, J.; Jones, M.A.; Millard, A. INfrastructure for a PHAge REference Database: identification of large-scale biases in the current collection of cultured phage genomes. Phage (New Rochelle) 2021, 2, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef]
- Khelik, K.; Lagesen, K.; Sandve, G.K.; Rognes, T.; Nederbragt, A.J. NucDiff: in-depth characterization and annotation of differences between two sets of DNA sequences. BMC Bioinformatics 2017, 18, 338. [Google Scholar] [CrossRef]
- Hackl, T.; Ankenbrand, M.; van Adrichem, B.; Wilkins, D.; Haslinger, K. gggenomes: effective and versatile visualizations for comparative genomics. arXiv:2411.13556. https://thackl.github.io/gggenomes/authors.html.
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucl. Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
Figure 1.
Plaque morphology of phage isolates following 10 rounds of training on phage-resistant strains MRSN 15882 and MRSN 27989.
Figure 1.
Plaque morphology of phage isolates following 10 rounds of training on phage-resistant strains MRSN 15882 and MRSN 27989.
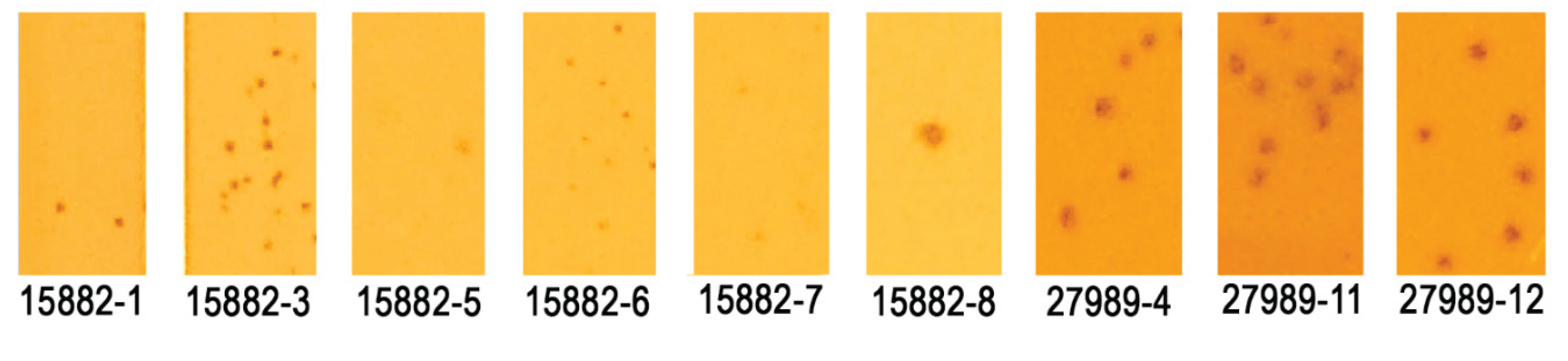
Figure 2.
Host ranges of trained and parental phages on the 100-strain K. pneumoniae diversity panel following 10 rounds of training. Lysis scoring is derived from a qualitative scoring system based on plaque formation and quality (see Table 4).
Figure 2.
Host ranges of trained and parental phages on the 100-strain K. pneumoniae diversity panel following 10 rounds of training. Lysis scoring is derived from a qualitative scoring system based on plaque formation and quality (see Table 4).
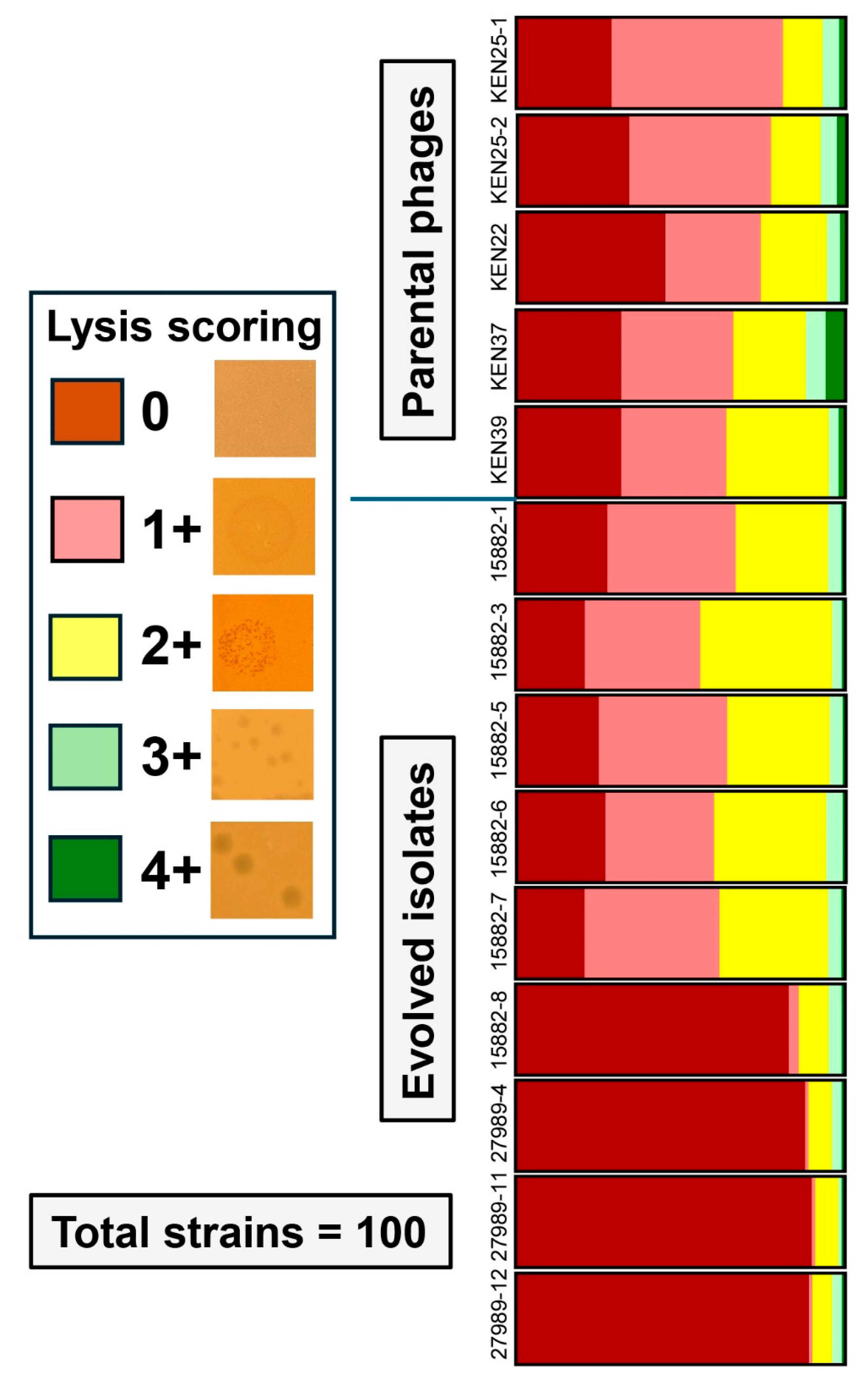
Figure 3.
Host ranges of phage clones collected after serial propagation on the permissive host strain K. pneumoniae MRSN 414780. These clones were compared to the originally evolved trained phages by plating on the 100-strain diversity panel.
Figure 3.
Host ranges of phage clones collected after serial propagation on the permissive host strain K. pneumoniae MRSN 414780. These clones were compared to the originally evolved trained phages by plating on the 100-strain diversity panel.
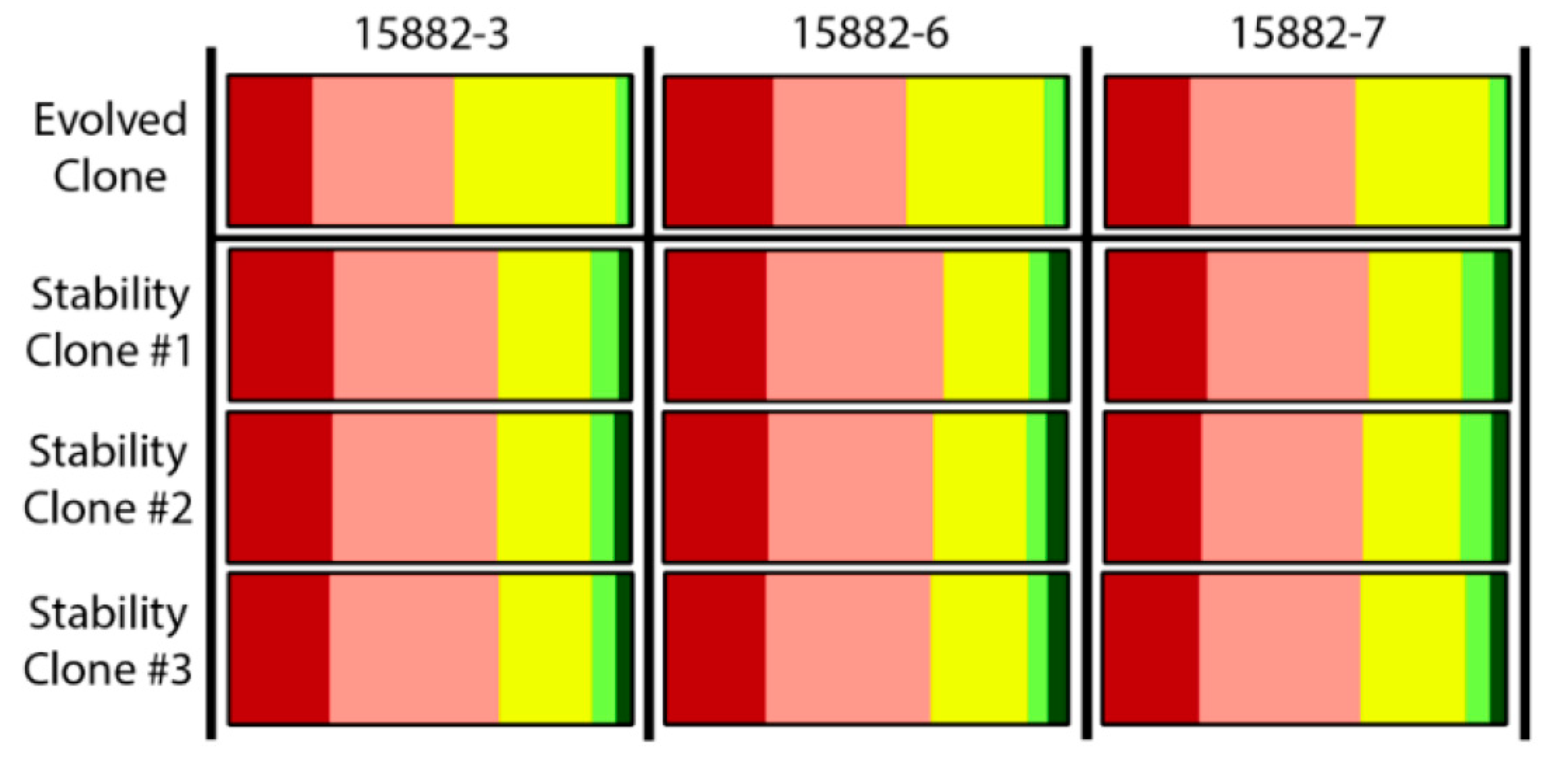
Table 1.
Clinical isolates of K. pneumoniae used for phage training.
| Strain | Sample type | Antibiotic susceptibility 1 | Phage susceptibility 2 | Sequence type | KL serotype | OL serotype |
|---|---|---|---|---|---|---|
| MRSN 4759 | Urine | MDR | XPR | ST37 | KL38 | O3b |
| MRSN 6778 | Urine | MDR | XPR | ST1842 | KL3 | O2v2 |
| MRSN 15687 | Urine | MDR | XPR | ST5446 | KL62 | O2v1 |
| MRSN 15882 | Perianal | MDR | XPR | ST1686 | KL113 | O1v1 |
| MRSN 22232 | Respiratory | XDR | XPR | ST405 | KL151 | O4 |
| MRSN 27989 | Wound | MDR | XPR | ST2279 | KL3 | O2v2 |
| MRSN 479404 | Wound | XDR | XPR | ST16 | KL51 | O3b |
| MRSN 511348 | Unknown | XDR | XPR | ST14 | KL2 | O1v1 |
| MRSN 614201 | Environ. | MDR | PPR | ST1838 | KL14 | O3b |
| MRSN 681054 | Urine | XDR | PPR | ST340 | KL15 | O4 |
| MRSN 414780 | Urine | XDR | PS | ST323 | KL21 | O3b |
Table 2.
Phages used in this work.
| Phage | Propagation strain | Genome length, bp | Family | Genus | Host range, % |
|---|---|---|---|---|---|
| KEN22 | Kp. MRSN 11382 | 166,645 | Straboviridae | Jiaodavirus | 26 |
| KEN25-1 | Kp MRSN 529046 | 169,768 | Straboviridae | Jiaodavirus | 19 |
| KEN25-2 | Kp MRSN 529046 | 165,574 | Straboviridae | Jiaodavirus | 23 |
| KEN37 | Kp MRSN 529046 | 166,503 | Straboviridae | Jiaodavirus | 34 |
| KEN39 | Kp MRSN 529046 | 166,254 | Straboviridae | Jiaodavirus | 36 |
| KEN42 | Kp MRSN 3619 | 38,200 | Autographiviridae | Teetrevirus | 18 |
| KEN1821 | Kp MRSN 529046 | 168,619 | Straboviridae | Jiaodavirus | 36 |
| AFR4 | Kp MRSN 3619 | 48,962 | Drexlerviridae | Webervirus | 17 |
| EKq1 | Kq MRSN 829456 | 48,244 | Fmr. Siphoviridae* | Unclass. | 15 |
| EKq2 | Kq MRSN 829456 | 51,496 | Drexlerviridae | Webervirus | 7 |
Table 3.
Evolved K. pneumoniae phages with altered or expanded host ranges.
| Phage | Host range | Expansion? |
|---|---|---|
| KEN22 | 26 | Parental phage |
| KEN25-1 | 19 | Parental phage |
| KEN25-2 | 23 | Parental phage |
| KEN37 | 34 | Parental phage |
| KEN39 | 36 | Parental phage |
| 15882-1 | 33 | No |
| 15882-3 | 44 | Yes |
| 15882-5 | 36 | No |
| 15882-6 | 40 | Yes |
| 15882-7 | 38 | Yes |
| 15882-8 | 14 | No |
| 27989-4 | 11 | No |
| 27989-11 | 9 | No |
| 27989-12 | 10 | No |
Table 5.
Comparison of K. pneumoniae phage cocktails WRAIR_KPM1 and WRAIR_KPM2.
| Cocktail | Phage ID | Genome size, bp | Family | Genus | Host Range | Mix host range |
|---|---|---|---|---|---|---|
| KPM1 | AFR4 | 48,962 | Drexlerviridae | Webervirus | 17% | 50% |
| KEN39 | 166,254 | Straboviridae | Jiaodavirus | 36% | ||
| KEN42 | 38,200 | Autographiviridae | Teetrevirus | 18% | ||
| EKq1 | 48,244 | Fmr. Siphoviridae* | Unclass. | 15% | ||
| EKq2 | 51,496 | Drexlerviridae | Webervirus | 7% | ||
| KPM2 | AFR4 | 48,962 | Drexlerviridae | Webervirus | 17% | 81% |
| KEN39 | 166,254 | Straboviridae | Jiaodavirus | 36% | ||
| KEN42 | 38,200 | Autographiviridae | Teetrevirus | 18% | ||
| EKq1 | 48,244 | Fmr. Siphoviridae* | Unclass. | 15% | ||
| EKq2 | 51,496 | Drexlerviridae | Webervirus | 7% | ||
| KEN1821 | 168,619 | Straboviridae | Jiaodavirus | 36% | ||
| 15882-3 | 167,537 | Straboviridae | Jiaodavirus | 44% |
*Unclassified phage EKq1 [36] formerly belonged to the family Siphoviridae now excluded from the phage classification scheme.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



