
Submitted:
01 July 2025
Posted:
02 July 2025
You are already at the latest version
Abstract
Corynebacterium ulcerans is an emerging zoonotic pathogen that can cause diphtheria-like infections in humans. In this study, we report the novel detection and comprehensive phenotypic and genomic characterization of three atoxigenic C. ulcerans strains isolated from domestic animals in Brazil. Notably, all isolates belonged to the multilocus sequence type ST-339, which has been previously identified in both human and animal hosts from geographically distant regions, suggesting the potential for international dissemination. Whole-genome sequencing confirmed species identity and revealed high genetic simila-rity among isolates, although distinct phylogenetic subclades were observed. Genomic analyses identified conserved virulence-associated determinants, including incomplete pilus gene clusters, iron acquisition systems, and the pld gene encoding phospholipase D. In contrast, the tox gene was absent in all strains. Notably, one isolate exhibited ciproflo-xacin resistance associated with double mutations (S89L and D93G) in the quinolone- resistance-determining region of GyrA. Molecular modeling and dynamics simulations demonstrated that these mutations impair key interactions within the ciprofloxacin– magnesium–water complex, thereby compromising the stability of drug binding. Additionally, the presence of diverse mobile genetic elements, prophages, and CRISPR-Cas systems highlighted the genomic plasticity of these isolates. Our findings provide new insights into the zoonotic potential, antimicrobial resistance mechanisms, and genomic diversity of C. ulcerans, underscoring the need for strengthened surveillance and mole-cular monitoring of this emerging pathogen in both veterinary and public health contexts.
Keywords:
Corynebacterium ulcerans
; non-toxigenic
; virulence factors
; resistance genes
; CRISPR-Cas system
; quinolone resistance
1. Introduction
The genus Corynebacterium includes 166 valid species, some of them pathogenic to humans and animals [1]. Corynebacterium diphtheriae, the type species, is the leading cause of diphtheria, a potentially fatal disease characterized by local and systemic effects due to diphtheria toxin (DT), an exotoxin produced by the microorganism when lysogenized by phages carrying the tox gene [2,3]. Despite the global decline in cases following widespread vaccination, diphtheria remains endemic in some regions and is still reported among vaccinated individuals [4,5,6], with concerns heightened by decreased vaccination coverage during the COVID-19 pandemic [7]. Consequently, World Health Organization and the Pan American Health Organization (WHO/PAHO) have issued alerts stressing the need for preventive actions.
Beyond C. diphtheriae, other species in the C. diphtheriae complex, including Corynebacterium belfantii, Corynebacterium pseudotuberculosis, Corynebacterium ramonii, Coryneba-cterium rouxii, Corynebacterium silvaticum, and Corynebacterium ulcerans, may also carry the tox gene and cause diphtheria-like infections in humans and animals [8,9,10,11,12,13,14,15].
C. ulcerans is an emerging zoonotic pathogen that colonizes domestic [16,17] and wild animals [18,19], with human infections primarily linked to close contact with pets [20]. Both toxigenic and atoxigenic strains are increasingly reported worldwide, with the former often causing diphtheria-like disease [21]. While early treatment with erythromycin or penicillin is recommended [22], resistance is emerging [4], prompting consideration of alternatives such as quinolones.
However, limited genomic data constrain understanding of C. ulcerans transmission and the emergence of toxigenic clones. A recent genomic study of 582 isolates from diverse hosts and regions applied a novel core genome genotyping approach, revealing major sublineages and diverse mobile elements, including five tox prophage families and a novel tox-carrying element, indicating interspecies prophage transfer within the C. diphtheriae complex [23].
As potentially toxigenic corynebacteria from animals are not subject to mandatory reporting in most countries, epidemiological data remain scarce, limiting public health responses and knowledge on zoonotic transmission risks. This study aimed to phenoty-pically and molecularly characterize three atoxigenic C. ulcerans strains isolated from domestic animals in Brazil, identifying potential virulence factors and antimicrobial resistance genes, with a focus on the structural implications of mutations conferring quinolone resistance.
2. Results
2.1. Phenotypic Identification and Antimicrobial Susceptibility Profile
After a 48 h incubation period, the aspect of all grown colonies was dry, white and opaque on the surface of blood agar. The Gram stain from these colonies revealed Gram-positive bacillary forms arranged in pallid shapes with angular formations between cells. In all cultures, MALDI-TOF MS (Matrix-Assisted Laser Desorption Ionization Time-of-Flight) analysis identified isolates as C. ulcerans (99% probability), which were named as C. ulcerans IHP37393, IHP103889 and IHP106492, respectively. In the first culture of ear injury in a dog from the State of Paraná, another colony was also identified as Staphylo-coccus aureus (99% probability). In the second culture of neck abscess in a cat from the State of Pernambuco, Enterococcus spp. was also identified with 99% probability. Finally, in the third culture of ear injury in a dog from the State of São Paulo, Morganella morganii was also identified with 99% probability. There is no information about monitoring or researching careers at home.
According to the halos obtained by disk diffusion method, the clinical isolates were categorized as sensitive to the following antimicrobials tested: clindamycin (IHP106492 = 15mm; cut-off ≥ 15mm), erythromycin (IHP37393 = 38mm; IHP103889 = 38mm; IHP106492 = 34mm; cut-off ≥ 24mm), linezolid (IHP37393 = 38mm; IHP103889 = 36mm; IHP106492 = 32mm; cut-off ≥ 25mm), rifampicin (IHP37393 = 36mm; IHP103889 = 36mm; IHP106492 = 36mm; cut-off ≥ 24mm), trimethoprim-sulfamethoxazole (IHP37393 = 31mm; IHP103889 = 35mm; IHP106492 = 33mm; cut-off ≥ 23mm ) and tetracycline (IHP37393 = 33mm; IHP103889 = 36mm; IHP106492 = 35mm; cut-off ≥ 24mm). Susceptibility with increased exposure was observed to: benzylpenicillin (IHP37393 = 24mm; IHP103889 = 20mm; IHP106492 = 19mm; cut-off 12-49mm) and ciprofloxacin (IHP103889 = 35mm; IHP106492 = 36mm; cut-off 24-49mm). Resistance to clindamycin (IHP37393 = 14mm; IHP103889 = 14mm; cut-off < 15mm) and ciprofloxacin (IHP37393 = 0mm; cut-off < 24mm ) were also observed.
2.2. Genome Features and Taxonomy
The average genomes size is approximately 2.5 Mb. The genomes had predicted GC content of 53.3%. Relevant information about the assemblies of IHP37393, IHP103889 and IHP106492 strains, as well as the median coverage, N50, number of CDS, rRNA and tRNA are shown in Table 1. No chimerism was found.
The taxonomy position of the strains was confirmed as C. ulcerans by TYGS and GTDB-Tk. ANI values obtained from the comparison between our strains and close refe-rence genomes identified by TYGS are shown in a heatmap (Supplementary Materials Figure S1). The results showed ANI values of 99.9% for our strains in comparison to C. ulcerans NCTC 7910T. Moreover, as expected, the results of DDH in silico comparing our genomes with the C. ulcerans NCTC 7910T revealed values of 88.5%, 88.1% and 88.4% to IHP37393, IHP103889 and IHP106492, respectively (Supplementary Materials Table S1).
2.3. Multilocus Sequence Typing (MLST) Characterization and Phylogenetic Analysis
Analysis of the 7 housekeeping genes showed that all strains included in this study belong to ST-339. The allelic profile found was 41 (atpA), 35 (dnaE), 79 (dnaK), 49 (fusA), 52 (leuA), 48 (odhA) and 39 (rpoB).
In the phylogenetic analysis, it was possible to observe that all strains belong to ST-339 formed a well-defined clade with strong bootstrap support (100%), highlighted in Figure 1 by a yellow square. This clade is divided into two subclades which are highlighted in Figure 1 by orange and green squares. The first one contained one of our strains (IHP103889) and two Austrian strains (04-15 and 06-19). The last ones had more phylogenetic proximity to each other than to IHP103889, which was evidenced by another subclade with good bootstrap support (100%) between them, and probably because these strains were isolated from humans, while IHP103889 was isolated from a cat. The other subclade was composed of two strains from this study (IHP37393 and IHP106492) and another Brazilian strain (BR-AD 22), all of them isolated from dogs. Moreover, it was possible to observe that ST-339 clade was separated from C. ulcerans 4724 strain (ST-327) and isolated from a dog in Switzerland by a distinct clade having a common ancestor among them and having a strong bootstrap support (100%).
2.4. Prediction of Mobile Genetic Elements and CRISPR-Cas Systems
No plasmids were detected in the genomes of our strains using PlasmidFinder. Besides, IntegronFinder did not identify any integrons in the genomes. Detailed analysis of the ISs annotated by ISEScan identified three different IS families (IS110, IS21, IS256) and the same number of insertion sequences in the genome of each strain. In all of them, IS110 (n = 2 in each strain) was the most abundant IS, followed by IS21 and IS256 (n = 1 in each strain). The copy number of the annotated ISs was stable in all strains. All information about the IS copies in each strain, besides the transposase gene into the complete IS are in Supplementary Materials Table S2.
Using PHASTEST, we predicted only one intact prophage in the genome of IHP37389 strain (region length: 47.9 Kb; completeness: 130; total number of CDS: 28; region position: 201090-249006; most common phage: Rhodococcus phage Jace - GenBank accession number: NC047974; GC content: 55.58%). In the genome of IHP103889 strain, two prophages were predicted, one intact prophage (region length: 38.2 Kb; completeness: 130; total number of CDS: 29; region position: 322879-361082; most common phage: Gordonia phage Nyceirae - GenBank accession number: NC031004; GC content: 55.29%) and one questionable prophage (region length: 14.3 Kb; completeness: 80; total number of CDS: 14; region position: 61225-75524; most common phage: Gordonia phage GMA5 - GenBank accession number: NC030907; GC content: 57.32%). Lastly, in the genome of IHP106492 strain, two prophages were also detected, one intact prophage (region length: 42.5 Kb; completeness: 100; total number of CDS: 20; region position: 205082-247623; most common phage: Corynebacterium phage Adelaide - GenBank accession number: NC048791; GC content: 54.21%) and one questionable prophage (region length: 14.1 Kb; completeness: 90; total number of CDS: 13; region position: 59635-73774; most common phage: Gordonia phage GMA5 - GenBank accession number: NC030907; GC content: 55.11%). All predicted phage genes predicted in intact prophages of each strains are shown in Figure 2.
CRISPRCasFinder identified the type I-E CRISPR-Cas system, lacking the cse1 gene in all strains. IHP106492 strain harbored the addition type IU CRISPR-Cas system, which contained cas2, cas1, cas3, csb2 and csb1genes. Three CRISPR arrays with evidence levels equal to 4 were found in each one of the strains. Through the analysis of the spacer diversity among all CRISPR arrays, we found a total of 192 spacer sequences, in which IHP103889 strain carried the largest number of them, 82 spacers, followed by IHP37393 and IHP106492 with 55 spacers each one. The CRISPRTarget database identified 86 spa-cers, 24 spacers in IHP37393, 37 spacers in IHP103889 and 25 spacers in IHP106492. The identity of spacers with the highest matches values is shown in Supplementary Materials Table S3. Besides, linear genome maps including the location of the cas genes and some CRISPR arrays are shown in Supplementary Materials Figure S2.
2.5. Identification of Genes Encoding Antimicrobial Resistance and Virulence Factors
As shown in Figure 3, compared to reference C. ulcerans NCTC 7910T, incomplete pilus clusters spaABC (srtA and spaC), spaDEF (srtB and srtC) and spaGHI (spaI) were predicted in all strains, encoding the pilus SpaA-, SpaD- and SpaH-type, respectively. Moreover, all strains harbored one gene that encodes surface-anchored pili proteins (sapD). Genes involved in ABC transporter (fagABC operon and fagD gene), ABC-type heme transporter (hmuTUV cluster), Ciu iron uptake and siderophore biosynthesis system (ciuABCDE cluster) and iron-dependent regulator of diphtheria toxin production (dtxR) were also identified in all strains.
The embC, mptC and aftB genes associated with the biosynthesis of lipoarabinomannan-like lipoglycan (CdiLAM) were also found in all isolates, as well as some urease- encoding genes, ureB and ureG, and ctpV gene, a putative copper exporter. As expected, the pld gene encoding the sphingomyelin-degrading phospholipase D was found in all strains. Other genes were predicted by PanViTa, including leuD, mprA, ndk, regX3, sigE, sigH e tufA, however they presented low percentage of identity. The tox gene was not found in any of our strains. The antimicrobial resistance genes including rpoB2, rbpA, gyrA and gyrB were predicted in all strains.
2.6. Mutation Analysis
The DNA gyrase subunit A (GyrA) of the ciprofloxacin resistant strain IHP37393 exhibited two mutations compared to susceptible strains (Figure 4; Supplementary Materials Figure S3). The mutant protein has a leucine and a glycine at positions 89 and 93, respectively, whereas susceptible strains possess a serine and an aspartate at these positions [24]. In contrast, the DNA gyrase subunit B (GyrB) of the resistant strain showed no mutations relative to susceptible strains.
The gyr89L-93G model, after undergoing a 5000-step energy minimization using the steepest descent algorithm, exhibited 95.2% of amino acid residues in favored regions, while the remaining 4.8% were in allowed regions (Supplementary Materials Figure S4A). Additionally, a 100-nanosecond simulation of the model in its apo conformation confirmed that the protein maintains a stable structure (Supplementary Figure S4B, S4C). These results suggest that the model is structurally stable and functionally viable, making it suitable for subsequent in silico analyses.
Molecular docking analysis revealed that the 5BTC-ciprofloxacin complex had a binding score of -6.204, which is comparable to the gyr89L-93G-ciprofloxacin complex, with a score of -6.246. However, in the 5BTC-ciprofloxacin complex, ciprofloxacin inte-racts with Ser91 and Asp94 from chain A via a water/magnesium ion bridge and forms hydrophobic interactions with arginine and glycine at positions 60 and 61 of chain B (Figures 5A, 5B). In contrast, in the gyr89L-93G-ciprofloxacin complex, the interaction at position 89 is weakened due to the serine-to-leucine substitution, resulting in a hydrophobic interaction in the region (Figures 5C, 5D). Additionally, the aspartate-to-glycine substitution at position 93 leads to a loss of interaction of the mutated protein with the water/magnesium ion bridge complex. It is worth noting that serine 91 and aspartate 94 in the 5BTC protein occupies the same positions as serine 89 and aspartate 93 in the gyr89L-93G model. Thus, despite the similar binding affinity energies obtained from molecular docking, our findings indicate a significant loss of key interactions in the cipro-floxacin-bound mutated DNA gyrase, which impacts its binding stability and function.
The 100-nanosecond molecular dynamics simulation of the 5BTC-ciprofloxacin complex demonstrated that ciprofloxacin remained bound to the DNA gyrase protein throughout the entire simulation (Figure 6).
Like the other ciprofloxacin-susceptible C. ulcerans strains, the 5BTC DNA gyrase contains serine and aspartate at positions 89 and 93 in the GyrA subunit, highlighting these residues as key determinants of ciprofloxacin inhibitory activity (Figure 7).
In contrast, the molecular simulation of the gyr89L-93G-ciprofloxacin complex revealed a highly unstable interaction, with the ligand dissociating from the enzyme's active site within the first 10 nanoseconds of the simulation (Figure 6). The final frame of the 5BTC-ciprofloxacin simulation (A,B) showed that, despite some positional flu-ctuations, ciprofloxacin maintained interactions with the key amino acid residues throughout the simulation. Conversely, in the gyr89L-93G-ciprofloxacin complex, cipro-floxacin completely lost interaction with the mutated protein almost immediately, with no interaction observed beyond 0.04 nanoseconds (Figures 8C, 8D).
3. Discussion
In the present study, we characterized three C. ulcerans strains (IHP37393, IHP103889, IHP106492) isolated from domestic animals in Brazil. Identified by MALDI-TOF, all showed ANI values above the boundary proposed to species definition (95–96%) [25] and dDDH values above the recommended cut-off point of 70% [26], confirming their classification. Given the limited genomic data available for this species, we conducted molecular typing and phylogenetic analysis.
Classical MLST remains a valuable tool for epidemiological studies, supporting the surveillance of C. diphtheriae complex species and contributing to outbreak investigations [27]. In the present study, all isolates belonged to sequence type ST-339, a lineage pre-viously reported in atoxigenic C. ulcerans strains isolated from Austrian patients with skin infections [28], in a nasal sample from an asymptomatic dog in Brazil [29], and in a dog with an ulcerative lesion in Italy [30]. Additionally, ST-339 strains have been identified in Canada, France, and Germany according to Institut Pasteur MLST database.
The detection of ST-339 across such geographically distant regions and diverse hosts raises the hypothesis of a potential international dissemination of this lineage, possibly facilitated by animal-human-environment interactions. The repeated identification of ST-339 in both human and animal samples suggests that certain clones may possess adaptive advantages favoring their persistence and spread across different ecological niches. This finding underscores the importance of integrating molecular typing data with global surveillance systems to better understand transmission dynamics and the potential for transboundary circulation of C. ulcerans strains.
In our study, the phylogenetic analysis using the core genome, although it showed that our strains grouped with those that had the same ST, also evidenced the formation of distinct clades among them, characterizing genetic diversity among the isolates. Corro-borating our findings, genetic diversity among C. ulcerans species has already been observed in a previously published study, in which the formation of two distinct clades was evidenced [31]. Interestingly, it was also observed that regardless of the formation of the clades, the genetic variability within both clades allows isolates from humans and animals to be distributed heterogeneously in the study carried out, once again showing the tremendous genetic variation among the isolates, thus suggesting a significant adaptive evolution across the different hosts. The observation of our results also allows us to reaffirm, mainly from an epidemiological point of view, that C. ulcerans is geographically distributed, including the isolates that present significant genetic variations.
Horizontal gene transfer is key to bacterial genome evolution, promoting survival, adaptability, and acquisition of virulence and antimicrobial resistance genes, with mobile genetic elements (MGEs) playing a central role [32]. Thus, we analyzed our bacterial genomes for MGEs, including plasmids, integrons, insertion sequences, and prophages.
Plasmids and integrons contribute to the development of antimicrobial resistance and to the spread of virulence genes conferring selective advantage to bacteria [33,34]. None were detected in our strains, consistent with other C. ulcerans studies. In contrast, C. diphtheriae harbors plasmids such as pNG2 (erythromycin resistance) and pLRPD (multidrug resistance) [35,36], as well as integrons carrying resistance genes [37].
Insertion sequences (IS), the smallest and most common MGEs, significantly in- fluence bacterial genome structure, plasticity and function [38,39]. They vary in copy number and can mobilize within genomes and through other MGEs like plasmids and phages [40]. We identified three IS families (IS110, IS21, IS256) with identical copy numbers in our strains, a finding previously reported in C. silvaticum [9]. These IS families were originally isolated from Streptomyces coelicolor, Pseudomonas aeruginosa, and S. aureus, respectively [41], and have been associated with antimicrobial resistance in Corynebacterium striatum [42] and Corynebacterium jeikeium [43].
Prophages, common in C. diphtheriae complex genomes, contribute to their genomic plasticity [44]. We identified one intact prophage per strain, all from the Siphoviridae family, with GC contents exceeding the typical 53.3% of C. ulcerans genomes. In strain IHP37393, Rhodococcus phage Jace carries integrase and repressor genes, suggesting it is a temperate, circularly permuted phage [45]. In IHP103889, Gordonia phage Nyceirae is linked to prophage-mediated viral defense [46], while for IHP106492, Corynebacterium phage Adelaide's lifestyle remains uncertain (https://phagesdb.org/phages/).
CRISPR-Cas is an adaptive defense system widely found in many bacteria and archaea that helps them to recognize and destroy invading genetic elements, including viruses, phages and plasmids [47]. This system is composed of a repeated short array separated by spacers, a leader sequence containing the promoter and located upstream of the CRISPR array and, a set of CRISPR-associated genes (cas) which encode Cas proteins with endonuclease activity [48]. In this study, we found the type I-E CRISPR-Cas system in all strains, which was also detected in other C. diphtheriae complex species, including C. diphtheriae [49] and C. rouxii [13]. However, in our strains it was observed a lack of cse1 gene, which is required for Cas3 to recognize the DNA target site and position itself adjacently to the protospacer adjacent motif to ensure cleavage [50]. This lack might indicate an incomplete or modified system, although no study with the absence of this gene in C. diphtheriae complex species has been published. The high diversity of targets for the spacer sequences found in this study, like corynebacterial (C. ulcerans and C. diphtheriae), Phage Rhodococcus and Phage Streptococcus phiSASD1, highlights the effectiveness of the CRISPR-Cas in providing protection against phage infections. Moreover, IHP106492 strain harbored an additional type IU CRISPR-Cas system, containing cas2, cas1, cas3, csb2 and csb1 genes. This type of CRISPR-Cas system has also been found in Bifidobacterium spp. and other components of the intestinal microbiota of humans and animals [51,52,53]. However, even in these species, type IU CRISPR-Cas system has not yet been well cha-racterized. Additional studies should be performed to characterize this type of CRISPR-Cas system in C. ulcerans.
Several virulence factors related to adhesion, invasion, and iron acquisition were identified, supporting the pathogenic potential of this species. All strains harbored three incomplete pilus clusters - spaABC (srtA, spaC), spaDEF (srtB, srtC), and spaGHI (spaI) - indicating a pilus gene variation possibly linked to altered adherence mechanisms. These clusters are essential for epithelial adherence and colonization [54,55]. The spaD gene, encoding a surface-anchored pilus protein, was also present in all strains. Iron acquisition systems, critical for C. diphtheriae complex survival, were consistently identified. All strains carried the putative fagABC operon with fagD. An in vitro study with C. pseudotuberculosis fagB(C) mutant demonstrated that the fag genes expression in the host contribute to virulence when compared to wild type in a goat model of caseous lymphadenitis [56]. Similarly, the ciu cluster (ciuABCDE), important for survival under iron limitation, was detected in all isolates [57]. While C. diphtheriae possesses both the hmuTUV hemin transporter and HtaA-C proteins for hemin uptake [58,59], only the hmuTUV cluster was fully present in our C. ulcerans strains, suggesting limited hemin utilization, as also observed in C. rouxii [14].
The diphtheria toxin (DT) production, encoded by tox, has been described in C. diphtheriae, C. ulcerans, and C. pseudotuberculosis [60]. In this study, tox gene was absent in all C. ulcerans strains, but the dtxR gene, encoding the DtxR regulator of DT and side-rophore synthesis, oxidative stress response, and other promoters, was detected [61]. Additionally, all strains possessed the pld gene, encoding phospholipase D (PLD), a key virulence factor shared with C. pseudotuberculosis, involved in host invasion, persistence, and lesion formation [62,63,64,65]. All strains also carried embC, mptC, and aftB genes linked to CdiLAM, contributing to epithelial adherence and cell wall biosynthesis [66,67]. As expected, urease-related genes, including ureB and ureG, were present in all isolates, distinguishing C. ulcerans and C. pseudotuberculosis from C. diphtheriae [68,69].
As shown in Figure 3, the rpoB2 and rbpA genes, previously associated with rifampin resistance [36,70], were predicted in all strains. Nevertheless, further studies are required to demonstrate how this resistance occurs in Corynebacterium species. Moreover, gyrA and gyrB genes, which encode DNA gyrase, were also detected in all strains.
Fluoroquinolones, like ciprofloxacin, inhibit bacterial growth by targeting DNA gyrase. They prevent the resealing of double-stranded DNA breaks that normally occur after DNA strand passage, resulting in the formation of stable covalent enzymes–DNA adducts, known as cleaved complexes [71]. At higher concentrations, fluoroquinolones cause the release of these DNA breaks from the complexes, leading to chromosome fragmentation and, ultimately, cell death [72]. Ciprofloxacin primarily interacts with the α4 helix of the DNA gyrase A subunit, with additional non-specific contacts involving amino acid residues near the ciprofloxacin C7 group in the GyrB subunit. A key residue implicated in fluoroquinolone resistance is the second position of the α4 helix in GyrA, which is typically serine in fluoroquinolone-sensitive DNA gyrases [73]. This serine residue has been observed to participate in a water/magnesium ion bridge that stabilizes enzyme–drug interactions [74]. Also, acquired fluoroquinolone resistance is commonly associated with mutations in the α4 helix region, particularly at residues 87–94 in GyrA Mycobacterium tuberculosis and Corynebacterium spp. [75,76,77].
Previous studies on Corynebacterium spp. have shown that amino acid substitutions within the QRDR, particularly the replacement of a polar amino acid with a hydrophobic one at positions 87 and 91, correlate with increased minimum inhibitory concentration (MIC) values and resistance to quinolones, including ciprofloxacin [75,77]. In C. ulcerans susceptible to fluoroquinolones, these positions correspond to serine and aspartate at residues 89 and 93, respectively. Both are polar amino acids capable of forming hydrogen bonds with water molecules via their hydroxyl and carboxyl side chains [78], making them crucial for ciprofloxacin binding and overall quinolone efficacy.
Here, we provide significant insights into the antimicrobial resistance and pathogenic potencial of C. ulcerans, particularly concerning fluoroquinolone resistance. In this study, the mutated gyrA sequence (gyr89L-93G) contains leucine and glycine substitutions at positions 89 and 93, respectively. As aliphatic and nonpolar residues, they lack functional groups necessary for hydrogen bonding with water molecules [79], disrupting the interaction between the ciprofloxacin–water/magnesium ion bridge complex and the mutated enzyme. These observations underscore the critical role of this bridge in fluoroquinolone action and may contribute to the development of novel antibacterial agents to combat resistance.
Zoonotically acquired C. ulcerans infections are a matter of concern worldwide [23] and highlight the need for surveillance and expansion of knowledge about this pathogen. The present study provided a comprehensive genomic analysis of three C. ulcerans strains isolates from domestic animals in Brazil. Although additional studies are still necessary, the data obtained in the present work can contribute to understanding the dissemination and evolution of virulent and antimicrobial resistant strains of this zoonotic pathogen and, also to the establishment of monitoring, prevention and treatment measures for C. ulcerans infections in humans and animals.
4. Materials and Methods
4.1. Origin of Bacterial Strains
Three animals from different Brazilian states with signs of discomfort were treated at veterinary clinics. The first one, with an ear injury, was a 14-year-old dog from the State of Paraná. The second one, with a neck abscess, was an 8-year-old cat from the State of Pernambuco. The last one, with an ear injury, was a 3-year-old dog from the State of São Paulo. For analysis, three swabs were collected and sent to Hermes Pardini Institute (Fleury Group), Minas Gerais, Brazil. Bacterial cultures were performed on 5% sheep’s blood agar (Plastlabor®, Brazil) and incubated at 37°C for 48 h. The semi-automated system VITEK® MS (bioMérieux®, France) was used to identify isolated strains through MALDI-TOF MS analysis. Bacterial spots of 1 to 3 colonies were placed on the target slide. Then, 1 μL α-cyano-4-hydroxycyanic acid matrix - VITEK MS-CHCA (bioMérieux®, Brazil) was applied over the samples and air dried until the matrix and samples co- crystallized. The slide was loaded into the VITEK® MS system to acquire protein mass spectra, mainly composed of ribosomal protein. The obtained mass spectra were compared with the MYLA® software version 4.7.1 database (bioMérieux®, France).
4.2. Antimicrobial Susceptibility Testing In Vitro
Antimicrobial susceptibility profiles of all isolates were performed using disk diffusion method according to the guideline provided by the Brazilian Committee on Antimicrobial Susceptibility Testing (BrCAST) version valid from April 2024 (https://brcast.org.br/). Bacterial suspensions were prepared in saline to the density of a 0.5 McFarland turbidity standard and seeded on a Mueller-Hinton agar supplemented with 5% defibrinated horse's blood and 20 mg/L β-NAD (Plastlabor®️, Brazil). Then, the following antimicrobials (Oxoid®, Brazil) were placed on the surface of the seeded plates: benzylpenicillin (1 U), ciprofloxacin (5 μg), clindamycin (2 µg), erythromycin (15 µg), linezolid (10 µg), rifampicin (5 µg), trimethoprim-sulfamethoxazole (23,75-1,25 µg) and tetracycline (30 μg). Test plates were incubated at 35 ± 1°C in a 5% CO2 atmosphere for 40-44 h. The interpretation of susceptibility was performed according to the breakpoints established by the BrCAST guideline as well as the quality control, which was carried out using Streptococcus pneumoniae ATCC 49619.
4.3. Genome Sequencing, Assembling and Annotation
Bacterial genomic DNA was extracted using the QIAamp® DNA Blood Mini Kit (QIAGEN®, Germany) according to the manufacturer’s instructions. Whole-genome sequencing (WGS) was performed using Illumina® NextSeq 550 platform (California, USA). Library was constructed with the DNA Prep Library Preparation Kit (Illumina Inc.). Sequences quality analysis was performed using FastQC v.0.12.1 (https://github.com/s-andrews/FastQC). All genomes were assembled de novo using Unicycler v.0.5.1 [80] and contigs with less than 200 bp were trimmed. To evaluate the guanine and cytosine (GC) content, size, and fragmentation of the genomes, we used QUAST v.5.2.0 [81]. Completeness and contamination levels were estimated using CheckM2 v.1.0.2 [82] and the location of ribosomal RNA genes in genomes was predicted using Barrnap v.0.9 (https://github.com/tseemann/barrnap). Chimerism was checked using GUNC v.1.0.6 [83]. Genomes were annotated using Prokka v.1.14.6 [84] and deposited in GenBank.
4.4. Genomic Taxonomy
Taxonomy classification of the strains was performed using Type Strain Genome Server (TYGS) [85] and Genome Taxonomy Database Toolkit (GTDB-Tk) v.2.4.0 [86]. Average Nucleotide Identity (ANI) values among our strains and close reference genomes identified by TYGS were calculated using PyANI v.0.2.12 [87]. DNA-DNA hybridization (DDH) was determined in silico comparing our genomes with the type strains of the closely related species: C. diphtheriae NCTC 11397T, C. belfanti FRC0043T, C. ramonii FRC0011T, C. rouxii FRC0190T, C. pseudotuberculosis ATCC 19410T, C. silvaticum KL0182T, and C. ulcerans NCTC 7910T using the Genome-to-Genome Distance Calculator (GGDC) v.3.0 with BLAST+ [88]. The results were based on the recommended formula 2 (identities/HSP length) [89].
4.5. Multilocus Sequence Typing (MLST) Characterization and Phylogenetic Analysis
Considering the seven housekeeping genes, atpA, dnaE, dnaK, fusA, leuA, odhA, and rpoB obtained from whole genome, sequence type (ST) was determined in silico using the Institute Pauster MLST database (https://bigsdb.pasteur.fr/diphtheria/). The sequences of these seven housekeeping genes were also deposited in this database which provides access to genotyping data for C. diphtheriae complex isolates worldwide.
We used the Genbank database from National Center of Biotechnology Information (NCBI - https://www.ncbi.nlm.nih.gov) to retrieve some genomic sequences of C. ulce-rans that we consider important and related to our study. Before downloading the genomes in nucleotide FASTA format, we checked completeness and contamination levels using CheckM2 v.1.0.2 [82]. The genome of C. silvaticum KL0182T was added as an outgroup. For multiple sequence alignment of the core genome, we used PPanGGOLiN v.2.2.0 [90] with MAFFT with default option to perform the alignment [91]. SNP-sites v.2.5.1 [92] was chosen to extract single nucleotide polymorphisms (SNPs). The evolutio-nary model and phylogenetic inference were estimated by the IQ-TREE2 v.2.0.7, with the maximum likelihood method. The support values were calculated using 1000 bootstrap replications. The tree was visualized using iTOL v.6 [93].
4.6. Prediction of Mobile Genetic Elements and CRISPR-Cas Systems
PlasmidFinder v.2.1.6 was used for in silico detection of plasmids [94], and IntegronFinder v.2.0 was used for identifying and analyzing integrons across the genomes [95]. Insertion sequences (IS) were identified using ISEScan v.1.7.2.3 [96]. Prophage sequences were identified and annotated with Phage Search Tool with Enhanced Sequence Translation (PHASTEST) [97]. Intact prophage regions and their predicted phage genes were visualized using Circular Genome View Server in Proksee [98].
CRISPRCasFinder v.4.2.30 was used to analyze the presence of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs) and Cas proteins [99]. We only included CRISPR arrays with evidence levels equal to 3 or 4 [99], and the type of CRISPR-Cas cassette was determined according to the previously described nomenclature and classification [100]. Spacer sequences were analyzed for their identity in the CRISPRTarget database [101]. Spacer hits were selected from the CRISPRTarget with a cut-off identity cover (IC) score above 0.80 [102].
4.7. Identification of Genes Encoding Antimicrobial Resistance and Virulence Factors
VFanalyzer was implemented to screen potential virulence factors using the VFDB (Virulence Factor Database) database [103]. Furthermore, PanViTa v.1.1.3 [104] to search for antimicrobial resistance genes and virulence genes using CARD (Comprehensive Antibiotic Resistance Database) and VFDB databases, respectively. For more accurate results, we also used the BlastKOALA server [105,106]. Circular genome map comparisons were built with C. ulcerans NCTC 7910T as reference using BRIG (Blast Ring Image Generator) software v.0.95 [106], to show positions for virulence factors and antimicrobial resistance genes.
4.8. Mutation Analysis
For the identification of mutations in the quinolone resistance-determining region (QRDR) in our strains, we used the susceptible DNA gyrase sequence obtained from C. ulcerans strain 809 (GenBank accession number: GCA_000215645.1) [24]. This sequence was aligned against IHP37393 strain (resistant to ciprofloxacin), and with IHP103889 and IHP103492 strains (susceptible to increased exposure to ciprofloxacin). The alignment was performed using MUSCLE in MEGA software v.11.0.13 with default parameters [89] and analyzed using Jalview v.2.4.11.1 [107].
The 3D structure of IHP37393 DNA gyrase protein (gyr89L-93G) was modeled using Swiss-Model [108], based on the DNA gyrase complex structure deposited in the Protein Data Bank (https://doi.org/10.2210/pdb5bta/pdb) resolved by X-ray diffraction (resolution 2.55 Angstroms), which shares 78.11% sequence identity and coverage of 0.48. The gene-rated model was protonated in pH 7.0 in Charmm-gui web server [109] and then, su-bjected to energy minimization with 5000 steps using the steepest descent algorithm in GROMACS – 2023 [110] and it was submitted for quality analysis using the MolProbity web server [111].
Molecular docking was performed using DockThor [112] with the M. tuberculosis DNA gyrase complexed with ciprofloxacin (redocking) (https://doi.org/10.2210/pdb5btc/pdb), which shares 76.0% sequence identity with gyr89L-93G. During the docking process we included the magnesium ion which is described as involved in protein-ligand interaction along with its associated water molecules [73]. Si-milarly, molecular docking was performed with ciprofloxacin and the gyr89L-93G stru-cture, also accounting for the magnesium ions and their coordinating water molecules.
The molecular dynamics simulations were conducted using GROMACS - 2023, employing the CHARMM36 force field [113]. Initially, the system was solvated with TIP3P water within the simulation box and then neutralized. The system underwent energy mi-nimization using the steepest descent method until the maximum force was reduced to less than 1000 kJ/mol/nm. Following minimization, the system was gradually relaxed and heated to 310 K while maintaining the protein in a fixed position. The system was equilibrated at 310 K before performing 100 nanoseconds of molecular dynamics simulations under standard in vitro conditions, including physiological temperature (25ºC), pH 7.0, and 1 atm pressure. GROMACS routines were used to calculate root mean square deviation (RMSD), root mean square fluctuation (RMSF) for the protein and ligand backbone, and the radius of gyration (Rg) of the protein.
Ciprofloxacin parameterization was performed using the CGenFF web server [114], following the GROMACS protein-ligand complex simulation tutorial [115]. All simulation plots were generated using Xmgrace (Grace-5.1.25). The 2D-dimensional interaction maps were produced with LigPlot+ v.2.2.9 [116], while 3D-structural representations of the protein-ligand complexes and docking analyses were visualized using PyMOL v.2.5.0 (The PyMOL Molecular Graphics System, v.1.2r3pre, Schrödinger, LLC).
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Heatmap representing the ANI percentage nu- cleotide identity of all matching regions between IHP37393, IHP103889, IHP106492 isolates and the closest related type strains; Table S1: DDH in silico results obtained by GGDC v.3.0 for C. ulcerans strains compared to the closest related type strains; Figure S2: Linear genome map visualized using Circular Genome View Server in Proksee showing the location of the cas genes and some CRISPR arrays; Table S2: Number of predicted IS family in each one of C. ulcerans strain using ISEScan v.1.7.2.3; Figure S3: Multiple sequence alignment of C. ulcerans DNA gyrase; Table S3: Hits found to spacer sequences in the CRISPRTarget databases; Figure S4: Quality assessment of the gyr89L-93G model.
Author Contributions
Conceptualization; data curation; formal analysis; investigation; methodo-logy; visualization; writing – original draft preparation; writing – review & editing: F.D.P and M.R.B.A. Data curation; formal analysis; investigation; methodology; visualization: J.S.S and L.S.S. Formal analysis; investigation; methodology; visualization: L.O.S and B.S.A. Formal analysis; vi-sualization: T.C.S.C, A.C.C.S and S.C.S. Conceptualization; data curation; formal analysis; investigation; methodology project administration; visualization; writing – original draft preparation; writing – review & editing: V.A.C.A.
Funding
L.S.S acknowledges the support from the Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro – FAPERJ (E-26/202.088/2020; E-26/205.900/2022; E26/211.554/2019), the Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq (311233/2022-8), and Fundação de Amparo à Pesquisa do Estado de Minas Gerais – FAPEMIG (APQ-00214-20). S.C.S acknowledges the support from Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq (311249/2024-0), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (no grant number) and Fundação de Amparo à Pesquisa do Estado de Minas Gerais (APQ-01323-15). B.S.A acknowledges the support from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior through a Visiting Senior Professor Scholarship (88887.023263/2024-00).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data generated or analyzed during this study are included in this published article. The whole genome sequences of Corynebacterium ulcerans IHP37393, IHP103889 and IHP106492 strains were uploaded in NCBI with accession number of JBGNWQ000000000, JBGNWN000000000 and JBGNWM000000000, respectively.
Acknowledgments
We thank the Hermes Pardini Institute (Fleury Group), especially Júnia Pérez and Vanessa Oliveira.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Parte, A.C.; Sardà Carbasse, J.; Meier-Kolthoff, J.P.; Reimer, L.C.; Göker, M. List of Prokaryotic Names with Standing in Nomenclature (LPSN) Moves to the DSMZ. Int J Syst Evol Microbiol 2020, 70, 5607–5612. [Google Scholar] [CrossRef]
- Sharma, N.C.; Efstratiou, A.; Mokrousov, I.; Mutreja, A.; Das, B.; Ramamurthy, T. Diphtheria. Nat Rev Dis Primers 2019, 5, 81. [Google Scholar] [CrossRef] [PubMed]
- Sugiman-Marangos, S.N.; Gill, S.K.; Mansfield, M.J.; Orrell, K.E.; Doxey, A.C.; Melnyk, R.A. Structures of Distant Diphtheria Toxin Homologs Reveal Functional Determinants of an Evolutionarily Conserved Toxin Scaffold. Commun Biol 2022, 5, 375. [Google Scholar] [CrossRef] [PubMed]
- Batista Araújo, M.R.; Bernardes Sousa, M.Â.; Seabra, L.F.; Caldeira, L.A.; Faria, C.D.; Bokermann, S.; Sant’Anna, L.O.; dos Santos, L.S.; Mattos-Guaraldi, A.L. Cutaneous Infection by Non-Diphtheria-Toxin Producing and Penicillin-Resistant Corynebacterium Diphtheriae Strain in a Patient with Diabetes Mellitus. Access Microbiol 2021, 3. [Google Scholar] [CrossRef]
- Araújo, M.R.B.; Ramos, J.N.; de Oliveira Sant’Anna, L.; Bokermann, S.; Santos, M.B.N.; Mattos-Guaraldi, A.L.; Azevedo, V.; Prates, F.D.; Rodrigues, D.L.N.; Aburjaile, F.F.; et al. Phenotypic and Molecular Characterization and Complete Genome Sequence of a Corynebacterium Diphtheriae Strain Isolated from Cutaneous Infection in an Immunized Individual. Brazilian Journal of Microbiology 2023, 54, 1325–1334. [Google Scholar] [CrossRef]
- Ramos, J.N.; Araújo, M.R.B.; Sant’Anna, L.O.; Bokermann, S.; Camargo, C.H.; Prates, F.D.; Sacchi, C.T.; Vieira, V.V.; Campos, K.R.; Santos, M.B.N.; et al. Molecular Characterization and Whole-Genome Sequencing of Corynebacterium Diphtheriae Causing Skin Lesion. European Journal of Clinical Microbiology & Infectious Diseases 2024, 43, 203–208. [Google Scholar] [CrossRef]
- Moura, C.; Truche, P.; Sousa Salgado, L.; Meireles, T.; Santana, V.; Buda, A.; Bentes, A.; Botelho, F.; Mooney, D. The Impact of COVID-19 on Routine Pediatric Vaccination Delivery in Brazil. Vaccine 2022, 40, 2292–2298. [Google Scholar] [CrossRef] [PubMed]
- Dangel, A.; Berger, A.; Rau, J.; Eisenberg, T.; Kämpfer, P.; Margos, G.; Contzen, M.; Busse, H.-J.; Konrad, R.; Peters, M.; et al. Corynebacterium Silvaticum Sp. Nov., a Unique Group of NTTB Corynebacteria in Wild Boar and Roe Deer. Int J Syst Evol Microbiol 2020, 70, 3614–3624. [Google Scholar] [CrossRef]
- Viana, M.V.C.; Galdino, J.H.; Profeta, R.; Oliveira, M.; Tavares, L.; de Castro Soares, S.; Carneiro, P.; Wattam, A.R.; Azevedo, V. Analysis of Corynebacterium Silvaticum Genomes from Portugal Reveals a Single Cluster and a Clade Suggested to Produce Diphtheria Toxin. PeerJ 2023, 11, e14895. [Google Scholar] [CrossRef]
- Dazas, M.; Badell, E.; Carmi-Leroy, A.; Criscuolo, A.; Brisse, S. Taxonomic Status of Corynebacterium Diphtheriae Biovar Belfanti and Proposal of Corynebacterium Belfantii Sp. Nov. Int J Syst Evol Microbiol 2018, 68, 3826–3831. [Google Scholar] [CrossRef]
- Hall, A.J.; Cassiday, P.K.; Bernard, K.A.; Bolt, F.; Steigerwalt, A.G.; Bixler, D.; Pawloski, L.C.; Whitney, A.M.; Iwaki, M.; Baldwin, A.; et al. Novel Diphtheriae in Domestic Cats. Emerg Infect Dis 2010, 16, 688–691. [Google Scholar] [CrossRef]
- Crestani, C.; Arcari, G.; Landier, A.; Passet, V.; Garnier, D.; Brémont, S.; Armatys, N.; Carmi-Leroy, A.; Toubiana, J.; Badell, E.; et al. Corynebacterium Ramonii Sp. Nov., a Novel Toxigenic Member of the Corynebacterium Diphtheriae Species Complex. Res Microbiol 2023, 174, 104113. [Google Scholar] [CrossRef]
- Ramos, J.N.; Araújo, M.R.B.; Baio, P.V.P.; Sant’Anna, L.O.; Veras, J.F.C.; Vieira, É.M.D.; Sousa, M.Â.B.; Camargo, C.H.; Sacchi, C.T.; Campos, K.R.; et al. Molecular Characterization and Phylogenetic Analysis of the First Corynebacterium Rouxii Strains Isolated in Brazil: A Recent Member of Corynebacterium Diphtheriae Complex. BMC Genom Data 2023, 24, 65. [Google Scholar] [CrossRef]
- Prates, F.D.; Araújo, M.R.B.; Sousa, E.G.; Ramos, J.N.; Viana, M.V.C.; Soares, S. de C.; dos Santos, L.S.; Azevedo, V.A. de C. First Pangenome of Corynebacterium Rouxii, a Potentially Toxigenic Species of Corynebacterium Diphtheriae Complex. Bacteria 2024, 3, 99–117. [Google Scholar] [CrossRef]
- Araújo, M.R.B.; Prates, F.D.; Viana, M.V.C.; Santos, L.S.; Mattos-Guaraldi, A.L.; Camargo, C.H.; Sacchi, C.T.; Campos, K.R.; Vieira, V.V.; Santos, M.B.N.; et al. Genomic Analysis of Two Penicillin- and Rifampin-Resistant Corynebacterium Rouxii Strains Isolated from Cutaneous Infections in Dogs. Res Vet Sci 2024, 179, 105396. [Google Scholar] [CrossRef]
- Katsukawa, C.; Komiya, T.; Yamagishi, H.; Ishii, A.; Nishino, S.; Nagahama, S.; Iwaki, M.; Yamamoto, A.; Takahashi, M. Prevalence of Corynebacterium Ulcerans in Dogs in Osaka, Japan. J Med Microbiol 2012, 61, 266–273. [Google Scholar] [CrossRef]
- Hoefer, A.; Herrera-León, S.; Domínguez, L.; Gavín, M.O.; Romero, B.; Piedra, X.B.A.; Calzada, C.S.; Uría González, M.J.; Herrera-León, L. Zoonotic Transmission of Diphtheria from Domestic Animal Reservoir, Spain. Emerg Infect Dis 2022, 28. [Google Scholar] [CrossRef]
- Contzen, M.; Sting, R.; Blazey, B.; Rau, J. Corynebacterium Ulcerans from Diseased Wild Boars. Zoonoses Public Health 2011, 58, 479–488. [Google Scholar] [CrossRef]
- Sting, R.; Pölzelbauer, C.; Eisenberg, T.; Bonke, R.; Blazey, B.; Peters, M.; Riße, K.; Sing, A.; Berger, A.; Dangel, A.; et al. Corynebacterium Ulcerans Infections in Eurasian Beavers (Castor Fiber). Pathogens 2023, 12, 979. [Google Scholar] [CrossRef]
- De Zoysa, A.; Hawkey, P.M.; Engler, K.; George, R.; Mann, G.; Reilly, W.; Taylor, D.; Efstratiou, A. Characterization of Toxigenic Corynebacterium Ulcerans Strains Isolated from Humans and Domestic Cats in the United Kingdom. J Clin Microbiol 2005, 43, 4377–4381. [Google Scholar] [CrossRef]
- Fuursted, K.; Søes, L.M.; Crewe, B.T.; Stegger, M.; Andersen, P.S.; Christensen, J.J. Non-Toxigenic Tox Gene-Bearing Corynebacterium Ulcerans in a Traumatic Ulcer from a Human Case and His Asymptomatic Dog. Microbes Infect 2015, 17, 717–719. [Google Scholar] [CrossRef]
- Kneen, R.; Giao, P.N.; Solomon, T.; Van, T.T.M.; Hoa, N.T.T.; Long, T.B.; Wain, J.; Day, N.P.J.; Hien, T.T.; Parry, C.M.; et al. Penicillin vs. Erythromycin in the Treatment of Diphtheria. Clinical Infectious Diseases 1998, 27, 845–850. [Google Scholar] [CrossRef]
- Crestani, C.; Passet, V.; Rethoret-Pasty, M.; Zidane, N.; Brémont, S.; Badell, E.; Criscuolo, A.; Brisse, S. Microevolution and Genomic Epidemiology of the Diphtheria-Causing Zoonotic Pathogen Corynebacterium Ulcerans. Nat Commun 2025, 16, 4843. [Google Scholar] [CrossRef]
- Mattos-Guaraldi, A.; Sampaio, J.; Santos, C.; Pimenta, F.; Pereira, G.; Pacheco, L.; Miyoshi, A.; Azevedo, V.; Moreira, L.; Gutierrez, F.; et al. First Detection of Corynebacterium Ulcerans Producing a Diphtheria-like Toxin in a Case of Human with Pulmonary Infection in the Rio de Janeiro Metropolitan Area, Brazil. Mem Inst Oswaldo Cruz 2008, 103, 396–400. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R. Shifting the Genomic Gold Standard for the Prokaryotic Species Definition. Proceedings of the National Academy of Sciences 2009, 106, 19126–19131. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA–DNA Hybridization Values and Their Relationship to Whole-Genome Sequence Similarities. Int J Syst Evol Microbiol 2007, 57, 81–91. [Google Scholar] [CrossRef]
- Vieira, V.V.; Ramos, J.N.; dos Santos, L.S.; Mattos-Guaraldi, A.L. Corynebacterium: Molecular Typing and Pathogenesis of Corynebacterium Diphtheriae and Zoonotic Diphtheria Toxin-Producing Corynebacterium Species. In Molecular Typing in Bacterial Infections, Volume I; Springer International Publishing: Cham, 2022; pp. 3–35. [Google Scholar]
- Schaeffer, J.; Huhulescu, S.; Stoeger, A.; Allerberger, F.; Ruppitsch, W. Draft Genome Sequences of Six Corynebacterium Ulcerans Strains Isolated from Humans and Animals in Austria, 2013 to 2019. Microbiol Resour Announc 2020, 9. [Google Scholar] [CrossRef]
- Trost, E.; Al-Dilaimi, A.; Papavasiliou, P.; Schneider, J.; Viehoever, P.; Burkovski, A.; Soares, S.C.; Almeida, S.S.; Dorella, F.A.; Miyoshi, A.; et al. Comparative Analysis of Two Complete Corynebacterium Ulcerans Genomes and Detection of Candidate Virulence Factors. BMC Genomics 2011, 12, 383. [Google Scholar] [CrossRef]
- Carfora, V.; Scarampella, F.; Iurescia, M.; Donati, V.; Stravino, F.; Lorenzetti, S.; Menichini, E.; Franco, A.; Caprioli, A.; Battisti, A. Non-Toxigenic Corynebacterium Ulcerans Sequence Types 325 and 339 Isolated from Two Dogs with Ulcerative Lesions in Italy. Journal of Veterinary Diagnostic Investigation 2018, 30, 447–450. [Google Scholar] [CrossRef]
- Subedi, R.; Kolodkina, V.; Sutcliffe, I.C.; Simpson-Louredo, L.; Hirata, R.; Titov, L.; Mattos-Guaraldi, A.L.; Burkovski, A.; Sangal, V. Genomic Analyses Reveal Two Distinct Lineages of Corynebacterium Ulcerans Strains. New Microbes New Infect 2018, 25, 7–13. [Google Scholar] [CrossRef]
- Arnold, B.J.; Huang, I.-T.; Hanage, W.P. Horizontal Gene Transfer and Adaptive Evolution in Bacteria. Nat Rev Microbiol 2022, 20, 206–218. [Google Scholar] [CrossRef]
- San Millan, A. Evolution of Plasmid-Mediated Antibiotic Resistance in the Clinical Context. Trends Microbiol 2018, 26, 978–985. [Google Scholar] [CrossRef]
- Bhat, B.A.; Mir, R.A.; Qadri, H.; Dhiman, R.; Almilaibary, A.; Alkhanani, M.; Mir, M.A. Integrons in the Development of Antimicrobial Resistance: Critical Review and Perspectives. Front Microbiol 2023, 14. [Google Scholar] [CrossRef]
- Tauch, A.; Bischoff, N.; Brune, I.; Kalinowski, J. Insights into the Genetic Organization of the Corynebacterium Diphtheriae Erythromycin Resistance Plasmid PNG2 Deduced from Its Complete Nucleotide Sequence. Plasmid 2003, 49, 63–74. [Google Scholar] [CrossRef]
- Hennart, M.; Panunzi, L.G.; Rodrigues, C.; Gaday, Q.; Baines, S.L.; Barros-Pinkelnig, M.; Carmi-Leroy, A.; Dazas, M.; Wehenkel, A.M.; Didelot, X.; et al. Population Genomics and Antimicrobial Resistance in Corynebacterium Diphtheriae. Genome Med 2020, 12, 107. [Google Scholar] [CrossRef]
- Arcari, G.; Hennart, M.; Badell, E.; Brisse, S. Multidrug-Resistant Toxigenic Corynebacterium Diphtheriae Sublineage 453 with Two Novel Resistance Genomic Islands. Microb Genom 2023, 9. [Google Scholar] [CrossRef]
- Ngan, W.Y.; Parab, L.; Bertels, F.; Gallie, J. A More Significant Role for Insertion Sequences in Large-Scale Rearrangements in Bacterial Genomes. mBio 2025, 16. [Google Scholar] [CrossRef]
- Vandecraen, J.; Chandler, M.; Aertsen, A.; Van Houdt, R. The Impact of Insertion Sequences on Bacterial Genome Plasticity and Adaptability. Crit Rev Microbiol 2017, 43, 709–730. [Google Scholar] [CrossRef]
- Udaondo, Z.; Abram, K.Z.; Kothari, A.; Jun, S.-R. Insertion Sequences and Other Mobile Elements Associated with Antibiotic Resistance Genes in Enterococcus Isolates from an Inpatient with Prolonged Bacteraemia. Microb Genom 2022, 8. [Google Scholar] [CrossRef]
- Mahillon, J.; Chandler, M. Insertion Sequences. Microbiology and Molecular Biology Reviews 1998, 62, 725–774. [Google Scholar] [CrossRef] [PubMed]
- Leyton-Carcaman, B.; Abanto, M. Beyond to the Stable: Role of the Insertion Sequences as Epidemiological Descriptors in Corynebacterium Striatum. Front Microbiol 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Leyton, B.; Ramos, J.N.; Baio, P.V.P.; Veras, J.F.C.; Souza, C.; Burkovski, A.; Mattos-Guaraldi, A.L.; Vieira, V.V.; Abanto Marin, M. Treat Me Well or Will Resist: Uptake of Mobile Genetic Elements Determine the Resistome of Corynebacterium Striatum. Int J Mol Sci 2021, 22, 7499. [Google Scholar] [CrossRef]
- Sekizuka, T.; Yamamoto, A.; Komiya, T.; Kenri, T.; Takeuchi, F.; Shibayama, K.; Takahashi, M.; Kuroda, M.; Iwaki, M. Corynebacterium Ulcerans 0102 Carries the Gene Encoding Diphtheria Toxin on a Prophage Different from the C. Diphtheriae NCTC 13129 Prophage. BMC Microbiol 2012, 12, 72. [Google Scholar] [CrossRef]
- Garza, D.R.; Di Blasi, D.; Bruns, J.A.; Empson, B.; Light, I.; Ghannam, M.; Castillo, S.; Quijada, B.; Zorawik, M.; Garcia-Vedrenne, A.E.; et al. Characterization of Genomic Diversity In Bacteriophages Infecting Rhodococcus 2022.
- Pope, W.H.; Mavrich, T.N.; Garlena, R.A.; Guerrero-Bustamante, C.A.; Jacobs-Sera, D.; Montgomery, M.T.; Russell, D.A.; Warner, M.H.; Hatfull, G.F. Bacteriophages of Gordonia Spp. Display a Spectrum of Diversity and Genetic Relationships. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Alkhnbashi, O.S.; Meier, T.; Mitrofanov, A.; Backofen, R.; Voß, B. CRISPR-Cas Bioinformatics. Methods 2020, 172, 3–11. [Google Scholar] [CrossRef]
- Karginov, F. V.; Hannon, G.J. The CRISPR System: Small RNA-Guided Defense in Bacteria and Archaea. Mol Cell 2010, 37, 7–19. [Google Scholar] [CrossRef]
- Hong, K.-W.; Asmah Hani, A.W.; Nurul Aina Murni, C.A.; Pusparani, R.R.; Chong, C.K.; Verasahib, K.; Yusoff, W.N.W.; Noordin, N.M.; Tee, K.K.; Yin, W.-F.; et al. Comparative Genomic and Phylogenetic Analysis of a Toxigenic Clinical Isolate of Corynebacterium Diphtheriae Strain B-D-16-78 from Malaysia. Infection, Genetics and Evolution 2017, 54, 263–270. [Google Scholar] [CrossRef]
- Rath, D.; Amlinger, L.; Rath, A.; Lundgren, M. The CRISPR-Cas Immune System: Biology, Mechanisms and Applications. Biochimie 2015, 117, 119–128. [Google Scholar] [CrossRef]
- Ou, L.; Long, J.; Teng, Y.; Yang, H.; Xi, Y.; Duan, G.; Chen, S. Diversity of the Type I-U CRISPR-Cas System in Bifidobacterium. Arch Microbiol 2021, 203, 3235–3243. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhou, X.; Pei, Z.; Stanton, C.; Ross, R.P.; Zhao, J.; Zhang, H.; Yang, B.; Chen, W. Characterization of CRISPR-Cas Systems in Bifidobacterium Breve. Microb Genom 2022, 8. [Google Scholar] [CrossRef] [PubMed]
- Kahraman Ilıkkan, Ö. Analysis of Probiotic Bacteria Genomes: Comparison of CRISPR/Cas Systems and Spacer Acquisition Diversity. Indian J Microbiol 2022, 62, 40–46. [Google Scholar] [CrossRef]
- Broadway, M.M.; Rogers, E.A.; Chang, C.; Huang, I.-H.; Dwivedi, P.; Yildirim, S.; Schmitt, M.P.; Das, A.; Ton-That, H. Pilus Gene Pool Variation and the Virulence of Corynebacterium Diphtheriae Clinical Isolates during Infection of a Nematode. J Bacteriol 2013, 195, 3774–3783. [Google Scholar] [CrossRef]
- Mandlik, A.; Swierczynski, A.; Das, A.; Ton-That, H. Corynebacterium Diphtheriae Employs Specific Minor Pilins to Target Human Pharyngeal Epithelial Cells. Mol Microbiol 2007, 64, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Billington, S.J.; Esmay, P.A.; Songer, J.G.; Jost, B.H. Identification and Role in Virulence of Putative Iron Acquisition Genes from Corynebacterium Pseudotuberculosis. FEMS Microbiol Lett 2002, 208, 41–45. [Google Scholar] [CrossRef]
- Kunkle, C.A.; Schmitt, M.P. Analysis of a DtxR-Regulated Iron Transport and Siderophore Biosynthesis Gene Cluster in Corynebacterium Diphtheriae. J Bacteriol 2005, 187, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Lyman, L.R.; Peng, E.D.; Schmitt, M.P. Corynebacterium Diphtheriae Iron-Regulated Surface Protein HbpA Is Involved in the Utilization of the Hemoglobin-Haptoglobin Complex as an Iron Source. J Bacteriol 2018, 200. [Google Scholar] [CrossRef]
- Drazek, E.S.; Hammack, C.A.; Schmitt, M.P. Corynebacterium Diphtheriae Genes Required for Acquisition of Iron from Haemin and Haemoglobin Are Homologous to ABC Haemin Transporters. Mol Microbiol 2000, 36, 68–84. [Google Scholar] [CrossRef]
- Burkovski, A. Proteomics of Toxigenic Corynebacteria. Proteomes 2023, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Yellaboina, S.; Ranjan, S.; Chakhaiyar, P.; Hasnain, S.E.; Ranjan, A. Prediction of DtxR Regulon: Identification of Binding Sites and Operons Controlled by Diphtheria Toxin Repressor in Corynebacterium Diphtheriae. BMC Microbiol 2004, 4, 38. [Google Scholar] [CrossRef]
- McNamara, P.J.; Cuevas, W.A.; Songer, J.G. Toxic Phospholipases D of Corynebacterium Pseudotuberculosis, C. Ulcerans and Arcanobacterium Haemolyticum: Cloning and Sequence Homology. Gene 1995, 156, 113–118. [Google Scholar] [CrossRef]
- McNamara, P.J.; Bradley, G.A.; Songer, J.G. Targeted Mutagenesis of the Phospholipase D Gene Results in Decreased Virulence of Corynebacterium Pseudotuberculosis. Mol Microbiol 1994, 12, 921–930. [Google Scholar] [CrossRef]
- McKean, S.C.; Davies, J.K.; Moore, R.J. Expression of Phospholipase D, the Major Virulence Factor of Corynebacterium Pseudotuberculosis, Is Regulated by Multiple Environmental Factors and Plays a Role in Macrophage Death. Microbiology (N Y) 2007, 153, 2203–2211. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Kutzer, P.; Peters, M.; Sing, A.; Contzen, M.; Rau, J. Nontoxigenic Tox -Bearing Corynebacterium Ulcerans Infection among Game Animals, Germany. Emerg Infect Dis 2014, 20. [Google Scholar] [CrossRef] [PubMed]
- Moreira, L.O.; Mattos-Guaraldi, A.L.; Andrade, A.F.B. Novel Lipoarabinomannan-like Lipoglycan (CdiLAM) Contributes to the Adherence of Corynebacterium Diphtheriae to Epithelial Cells. Arch Microbiol 2008, 190, 521–530. [Google Scholar] [CrossRef]
- Jankute, M.; Alderwick, L.J.; Moorey, A.R.; Joe, M.; Gurcha, S.S.; Eggeling, L.; Lowary, T.L.; Dell, A.; Pang, P.-C.; Yang, T.; et al. The Singular Corynebacterium Glutamicum Emb Arabinofuranosyltransferase Polymerises the α(1 → 5) Arabinan Backbone in the Early Stages of Cell Wall Arabinan Biosynthesis. The Cell Surface 2018, 2, 38–53. [Google Scholar] [CrossRef]
- Park, J.-U.; Song, J.-Y.; Kwon, Y.-C.; Chung, M.-J.; Jun, J.-S.; Park, J.-W.; Park, S.-G.; Hwang, H.-R.; Choi, S.-H.; Baik, S.-C.; et al. Effect of the Urease Accessory Genes on Activation of the Helicobacter Pylori Urease Apoprotein. Mol Cells 2005, 20, 371–377. [Google Scholar] [CrossRef]
- Simpson-Louredo, L.; Ramos, J.N.; Peixoto, R.S.; Santos, L.S.; Antunes, C.A.; Ladeira, E.M.; Santos, C.S.; Vieira, V.V.; Bôas, M.H.S.V.; Hirata, R.; et al. Corynebacterium Ulcerans Isolates from Humans and Dogs: Fibrinogen, Fibronectin and Collagen-Binding, Antimicrobial and PFGE Profiles. Antonie Van Leeuwenhoek 2014, 105, 343–352. [Google Scholar] [CrossRef]
- Araújo, M.R.B.; Prates, F.D.; Ramos, J.N.; Sousa, E.G.; Bokermann, S.; Sacchi, C.T.; de Mattos-Guaraldi, A.L.; Campos, K.R.; Sousa, M.Â.B.; Vieira, V.V.; et al. Infection by a Multidrug-Resistant Corynebacterium Diphtheriae Strain: Prediction of Virulence Factors, CRISPR-Cas System Analysis, and Structural Implications of Mutations Conferring Rifampin Resistance. Funct Integr Genomics 2024, 24, 145. [Google Scholar] [CrossRef]
- Aldred, K.J.; Kerns, R.J.; Osheroff, N. Mechanism of Quinolone Action and Resistance. Biochemistry 2014, 53, 1565–1574. [Google Scholar] [CrossRef]
- Drlica, K.; Malik, M.; Kerns, R.J.; Zhao, X. Quinolone-Mediated Bacterial Death. Antimicrob Agents Chemother 2008, 52, 385–392. [Google Scholar] [CrossRef]
- Blower, T.R.; Williamson, B.H.; Kerns, R.J.; Berger, J.M. Crystal Structure and Stability of Gyrase–Fluoroquinolone Cleaved Complexes from Mycobacterium Tuberculosis. Proceedings of the National Academy of Sciences 2016, 113, 1706–1713. [Google Scholar] [CrossRef] [PubMed]
- Wohlkonig, A.; Chan, P.F.; Fosberry, A.P.; Homes, P.; Huang, J.; Kranz, M.; Leydon, V.R.; Miles, T.J.; Pearson, N.D.; Perera, R.L.; et al. Structural Basis of Quinolone Inhibition of Type IIA Topoisomerases and Target-Mediated Resistance. Nat Struct Mol Biol 2010, 17, 1152–1153. [Google Scholar] [CrossRef] [PubMed]
- Ramos, J.N.; Valadão, T.B.; Baio, P.V.P.; Mattos-Guaraldi, A.L.; Vieira, V.V. Novel Mutations in the QRDR Region GyrA Gene in Multidrug-Resistance Corynebacterium Spp. Isolates from Intravenous Sites. Antonie Van Leeuwenhoek 2020, 113, 589–592. [Google Scholar] [CrossRef]
- Maruri, F.; Sterling, T.R.; Kaiga, A.W.; Blackman, A.; van der Heijden, Y.F.; Mayer, C.; Cambau, E.; Aubry, A. A Systematic Review of Gyrase Mutations Associated with Fluoroquinolone-Resistant Mycobacterium Tuberculosis and a Proposed Gyrase Numbering System. Journal of Antimicrobial Chemotherapy 2012, 67, 819–831. [Google Scholar] [CrossRef]
- Sierra, J.M.; Martinez-Martinez, L.; Vázquez, F.; Giralt, E.; Vila, J. Relationship between Mutations in the GyrA Gene and Quinolone Resistance in Clinical Isolates of Corynebacterium Striatum and Corynebacterium Amycolatum. Antimicrob Agents Chemother 2005, 49, 1714–1719. [Google Scholar] [CrossRef]
- Morris, A.S.; Thanki, N.; Goodfellow, J.M. Hydration of Amino Acid Side Chains: Dependence on Secondary Structure. “Protein Engineering, Design and Selection” 1992, 5, 717–728. [Google Scholar] [CrossRef]
- Ide, M.; Maeda, Y.; Kitano, H. Effect of Hydrophobicity of Amino Acids on the Structure of Water. J Phys Chem B 1997, 101, 7022–7026. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving Bacterial Genome Assemblies from Short and Long Sequencing Reads. PLoS Comput Biol 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Chklovski, A.; Parks, D.H.; Woodcroft, B.J.; Tyson, G.W. CheckM2: A Rapid, Scalable and Accurate Tool for Assessing Microbial Genome Quality Using Machine Learning. Nat Methods 2023, 20, 1203–1212. [Google Scholar] [CrossRef]
- Orakov, A.; Fullam, A.; Coelho, L.P.; Khedkar, S.; Szklarczyk, D.; Mende, D.R.; Schmidt, T.S.B.; Bork, P. GUNC: Detection of Chimerism and Contamination in Prokaryotic Genomes. Genome Biol 2021, 22, 178. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS Is an Automated High-Throughput Platform for State-of-the-Art Genome-Based Taxonomy. Nat Commun 2019, 10, 2182. [Google Scholar] [CrossRef]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A Toolkit to Classify Genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and Taxonomy in Diagnostics for Food Security: Soft-Rotting Enterobacterial Plant Pathogens. Analytical Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A Database Tandem for Fast and Reliable Genome-Based Classification and Nomenclature of Prokaryotes. Nucleic Acids Res 2022, 50, D801–D807. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol Biol Evol 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Gautreau, G.; Bazin, A.; Gachet, M.; Planel, R.; Burlot, L.; Dubois, M.; Perrin, A.; Médigue, C.; Calteau, A.; Cruveiller, S.; et al. PPanGGOLiN: Depicting Microbial Diversity via a Partitioned Pangenome Graph. PLoS Comput Biol 2020, 16, e1007732. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol Biol Evol 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Page, A.J.; Taylor, B.; Delaney, A.J.; Soares, J.; Seemann, T.; Keane, J.A.; Harris, S.R. SNP-Sites: Rapid Efficient Extraction of SNPs from Multi-FASTA Alignments. Microb Genom 2016, 2. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v6: Recent Updates to the Phylogenetic Tree Display and Annotation Tool. Nucleic Acids Res 2024, 52, W78–W82. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Møller Aarestrup, F.; Hasman, H. In Silico Detection and Typing of Plasmids Using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob Agents Chemother 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [PubMed]
- Néron, B.; Littner, E.; Haudiquet, M.; Perrin, A.; Cury, J.; Rocha, E. IntegronFinder 2.0: Identification and Analysis of Integrons across Bacteria, with a Focus on Antibiotic Resistance in Klebsiella. Microorganisms 2022, 10, 700. [Google Scholar] [CrossRef]
- Xie, Z.; Tang, H. ISEScan: Automated Identification of Insertion Sequence Elements in Prokaryotic Genomes. Bioinformatics 2017, 33, 3340–3347. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Han, S.; Saha, S.; Oler, E.; Peters, H.; Grant, J.R.; Stothard, P.; Gautam, V. PHASTEST: Faster than PHASTER, Better than PHAST. Nucleic Acids Res 2023, 51, W443–W450. [Google Scholar] [CrossRef]
- Grant, J.R.; Enns, E.; Marinier, E.; Mandal, A.; Herman, E.K.; Chen, C.; Graham, M.; Van Domselaar, G.; Stothard, P. Proksee: In-Depth Characterization and Visualization of Bacterial Genomes. Nucleic Acids Res 2023, 51, W484–W492. [Google Scholar] [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an Update of CRISRFinder, Includes a Portable Version, Enhanced Performance and Integrates Search for Cas Proteins. Nucleic Acids Res 2018, 46, W246–W251. [Google Scholar] [CrossRef]
- Makarova, K.S.; Koonin, E. V. Annotation and Classification of CRISPR-Cas Systems. In; 2015; pp. 47–75.
- Biswas, A.; Gagnon, J.N.; Brouns, S.J.J.; Fineran, P.C.; Brown, C.M. CRISPRTarget. RNA Biol 2013, 10, 817–827. [Google Scholar] [CrossRef]
- Sangal, V.; Fineran, P.C.; Hoskisson, P.A. Novel Configurations of Type I and II CRISPR–Cas Systems in Corynebacterium Diphtheriae. Microbiology (N Y) 2013, 159, 2118–2126. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Zhou, S.; Chen, L.; Yang, J. VFDB 2022: A General Classification Scheme for Bacterial Virulence Factors. Nucleic Acids Res 2022, 50, D912–D917. [Google Scholar] [CrossRef]
- Rodrigues, D.L.N.; Ariute, J.C.; Rodrigues da Costa, F.M.; Benko-Iseppon, A.M.; Barh, D.; Azevedo, V.; Aburjaile, F. PanViTa: Pan Virulence and ResisTance Analysis. Frontiers in Bioinformatics 2023, 3. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J Mol Biol 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Alikhan, N.-F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple Prokaryote Genome Comparisons. BMC Genomics 2011, 12, 402. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—a Multiple Sequence Alignment Editor and Analysis Workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-based Graphical User Interface for CHARMM. J Comput Chem 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and Better Reference Data for Improved All-atom Structure Validation. Protein Science 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Guedes, I.A.; Pereira da Silva, M.M.; Galheigo, M.; Krempser, E.; de Magalhães, C.S.; Correa Barbosa, H.J.; Dardenne, L.E. DockThor-VS: A Free Platform for Receptor-Ligand Virtual Screening. J Mol Biol 2024, 436, 168548. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-atom Additive Biological Force Fields. J Comput Chem 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Lemkul, J. From Proteins to Perturbed Hamiltonians: A Suite of Tutorials for the GROMACS-2018 Molecular Simulation Package [Article v1.0]. Living J Comput Mol Sci 2019, 1. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J Chem Inf Model 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Phylogenetic tree based on single nucleotide polymorphism of the core genome. The tree was built using IQ-TREE2 v.2.0.7 and distance was inferred with the maximum likelihood method. Bootstrap values were calculated using 1000 replicates. The main cluster containing our strains is highlighted in a yellow square, while the subclusters are highlighted in an orange and green square.
Figure 1.
Phylogenetic tree based on single nucleotide polymorphism of the core genome. The tree was built using IQ-TREE2 v.2.0.7 and distance was inferred with the maximum likelihood method. Bootstrap values were calculated using 1000 replicates. The main cluster containing our strains is highlighted in a yellow square, while the subclusters are highlighted in an orange and green square.
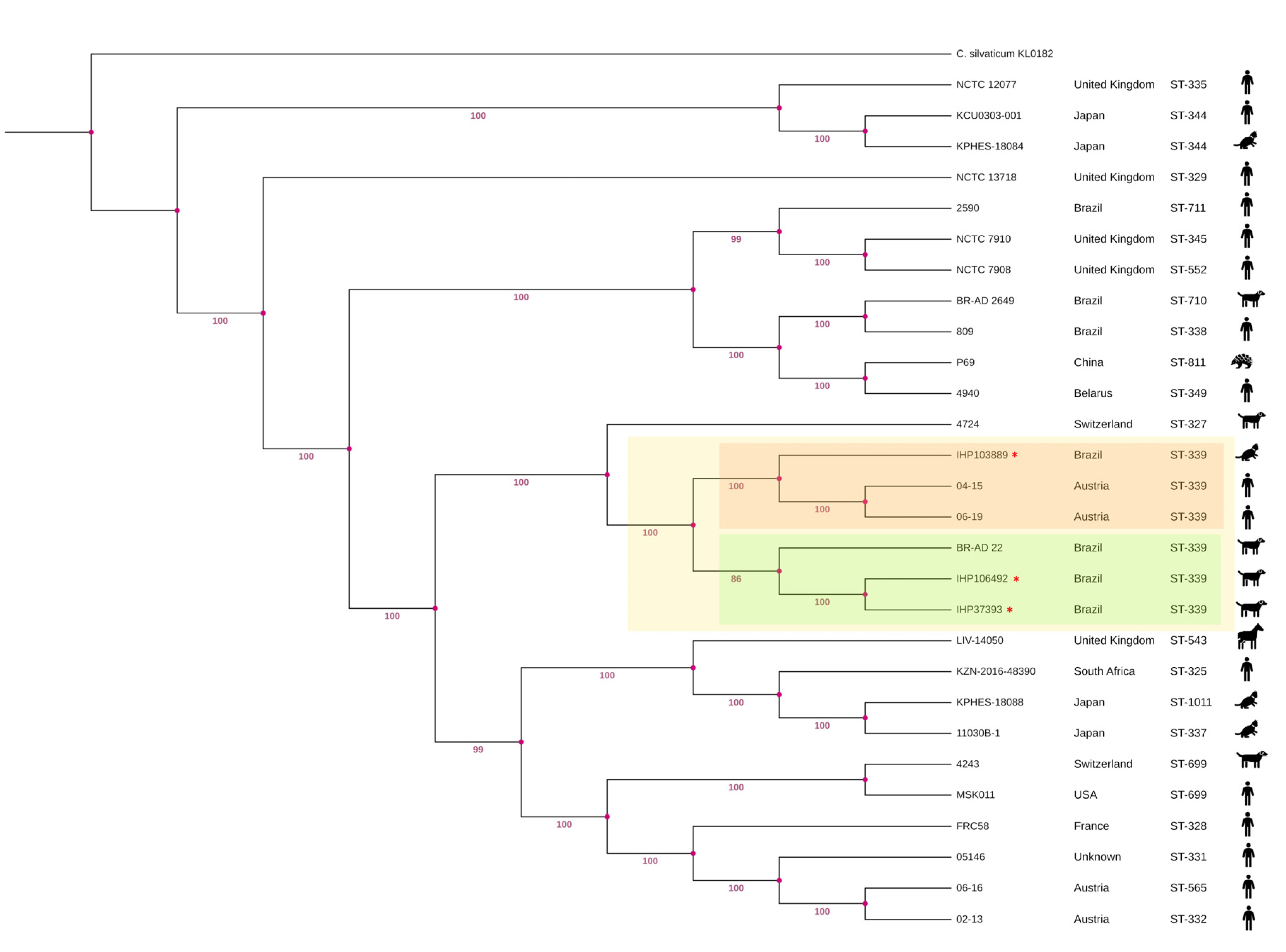
Figure 2.
Linear genome map visualized using Circular Genome View Server in Proksee showing the location of the intact prophage regions and predicted phage genes in the IHP37393 (A), IHP103889 (B) and IHP106492 (C) strains.
Figure 2.
Linear genome map visualized using Circular Genome View Server in Proksee showing the location of the intact prophage regions and predicted phage genes in the IHP37393 (A), IHP103889 (B) and IHP106492 (C) strains.
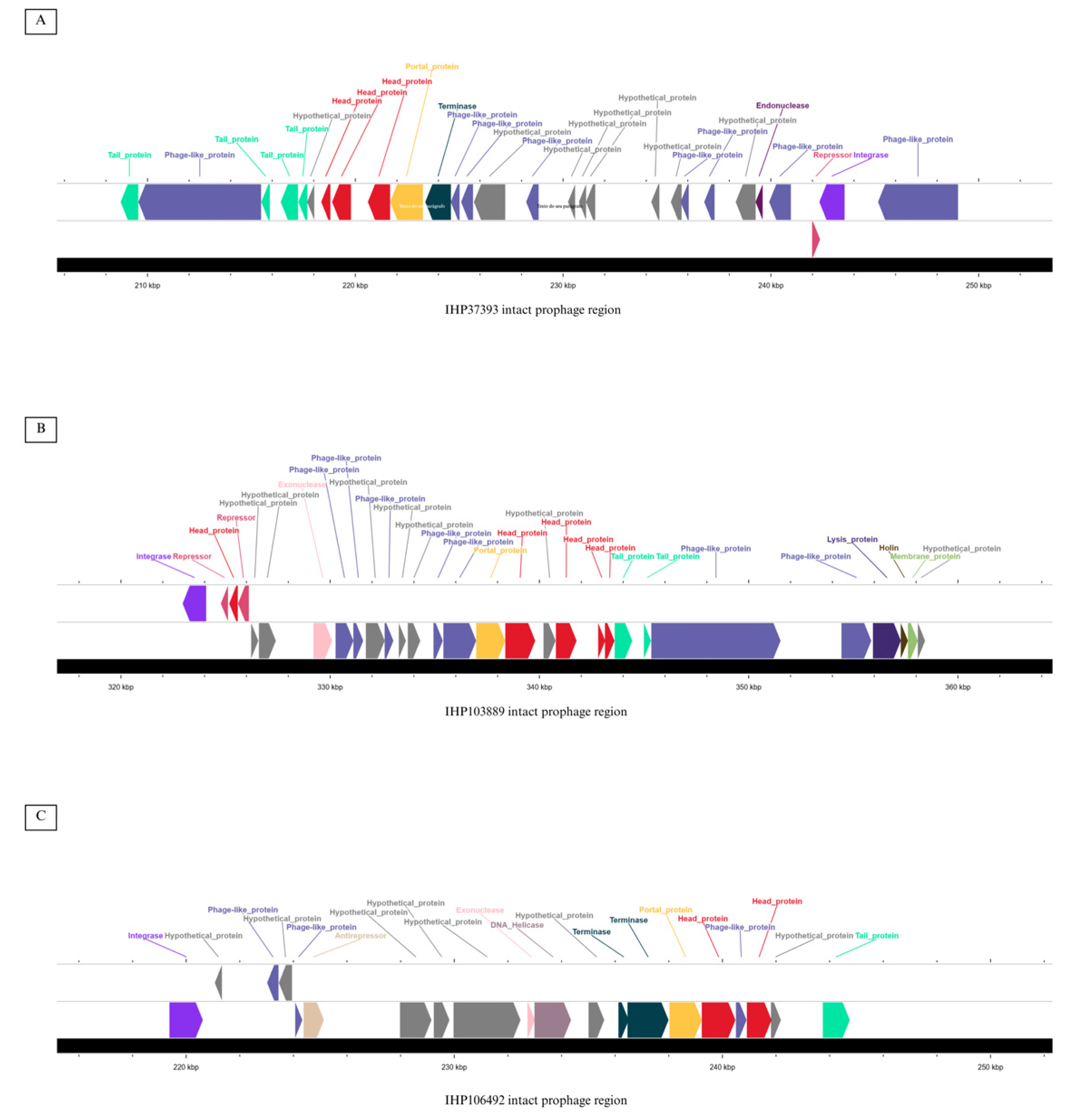
Figure 3.
Linear genome map visualized using Circular Genome View Server in Proksee showing the location of the intact prophage regions and predicted phage genes in the IHP37393 (A), IHP103889 (B) and IHP106492 (C) strains.
Figure 3.
Linear genome map visualized using Circular Genome View Server in Proksee showing the location of the intact prophage regions and predicted phage genes in the IHP37393 (A), IHP103889 (B) and IHP106492 (C) strains.
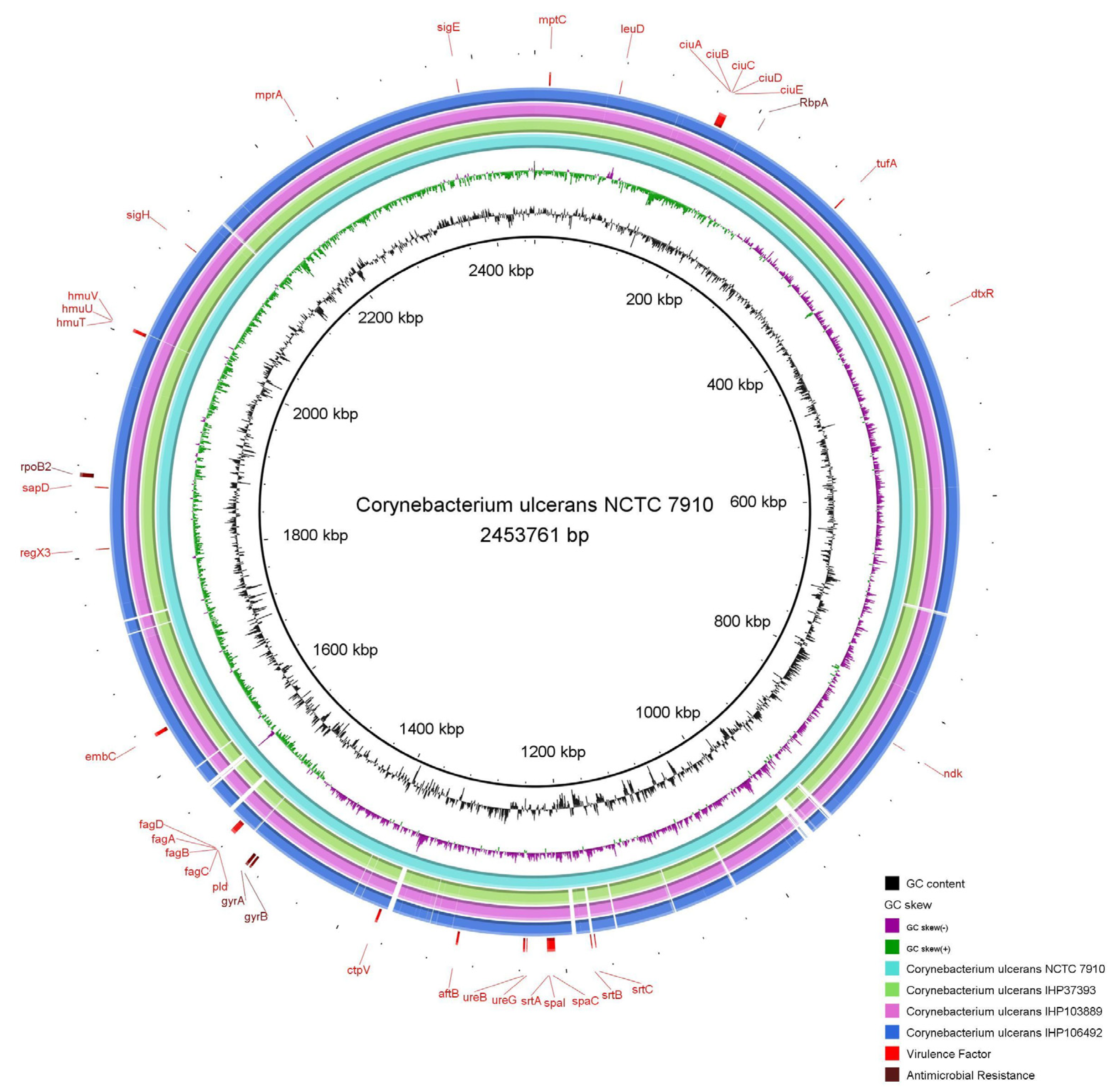
Figure 4.
Multiple sequence alignment of the QRDR regions of Corynebacterium ulcerans DNA gyrase subunit A, highlighting mutations present in the ciprofloxacin resistant IHP37393 strain.
Figure 4.
Multiple sequence alignment of the QRDR regions of Corynebacterium ulcerans DNA gyrase subunit A, highlighting mutations present in the ciprofloxacin resistant IHP37393 strain.
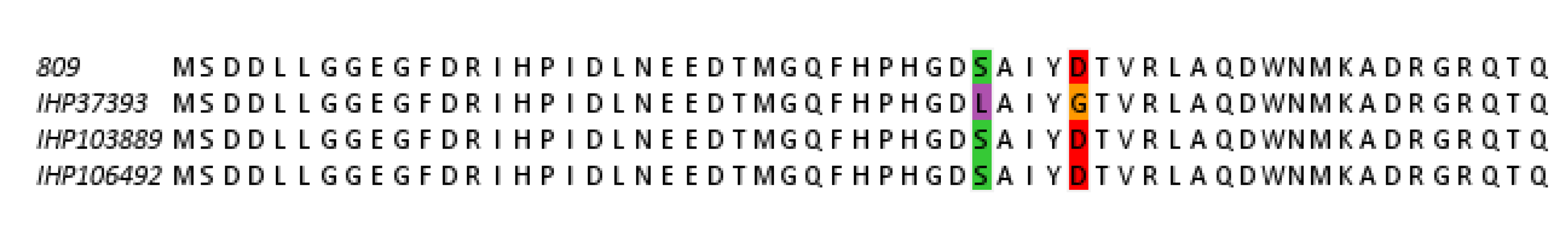
Figure 5.
Docking simulations. (A, B) Redocking of the 5BTC-ciprofloxacin (CPF) complex, showing CPF interactions with magnesium ions (green spheres) and water molecules (blue and red spheres in A and B, respectively). (B) Comparison of CPF poses with the original pose in yellow and the redocked pose in green. (C, D) CPF interactions with the mutated DNA gyrase (gyr89L-93G).
Figure 5.
Docking simulations. (A, B) Redocking of the 5BTC-ciprofloxacin (CPF) complex, showing CPF interactions with magnesium ions (green spheres) and water molecules (blue and red spheres in A and B, respectively). (B) Comparison of CPF poses with the original pose in yellow and the redocked pose in green. (C, D) CPF interactions with the mutated DNA gyrase (gyr89L-93G).
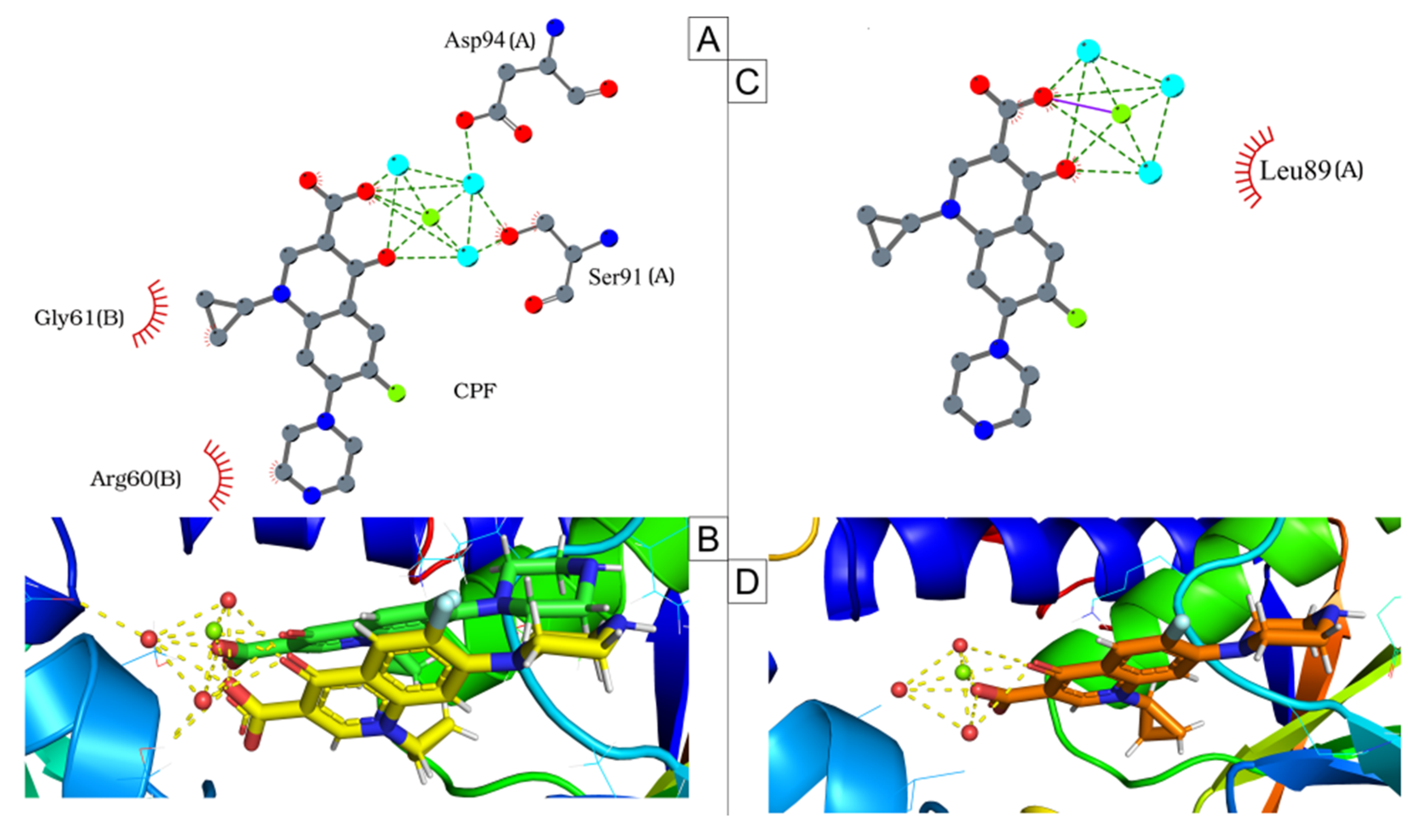
Figure 6.
Root Mean Square Deviation (RMSD) of ciprofloxacin following least-squares fitting to the complex backbone over a 100-nanosecond simulation.
Figure 6.
Root Mean Square Deviation (RMSD) of ciprofloxacin following least-squares fitting to the complex backbone over a 100-nanosecond simulation.
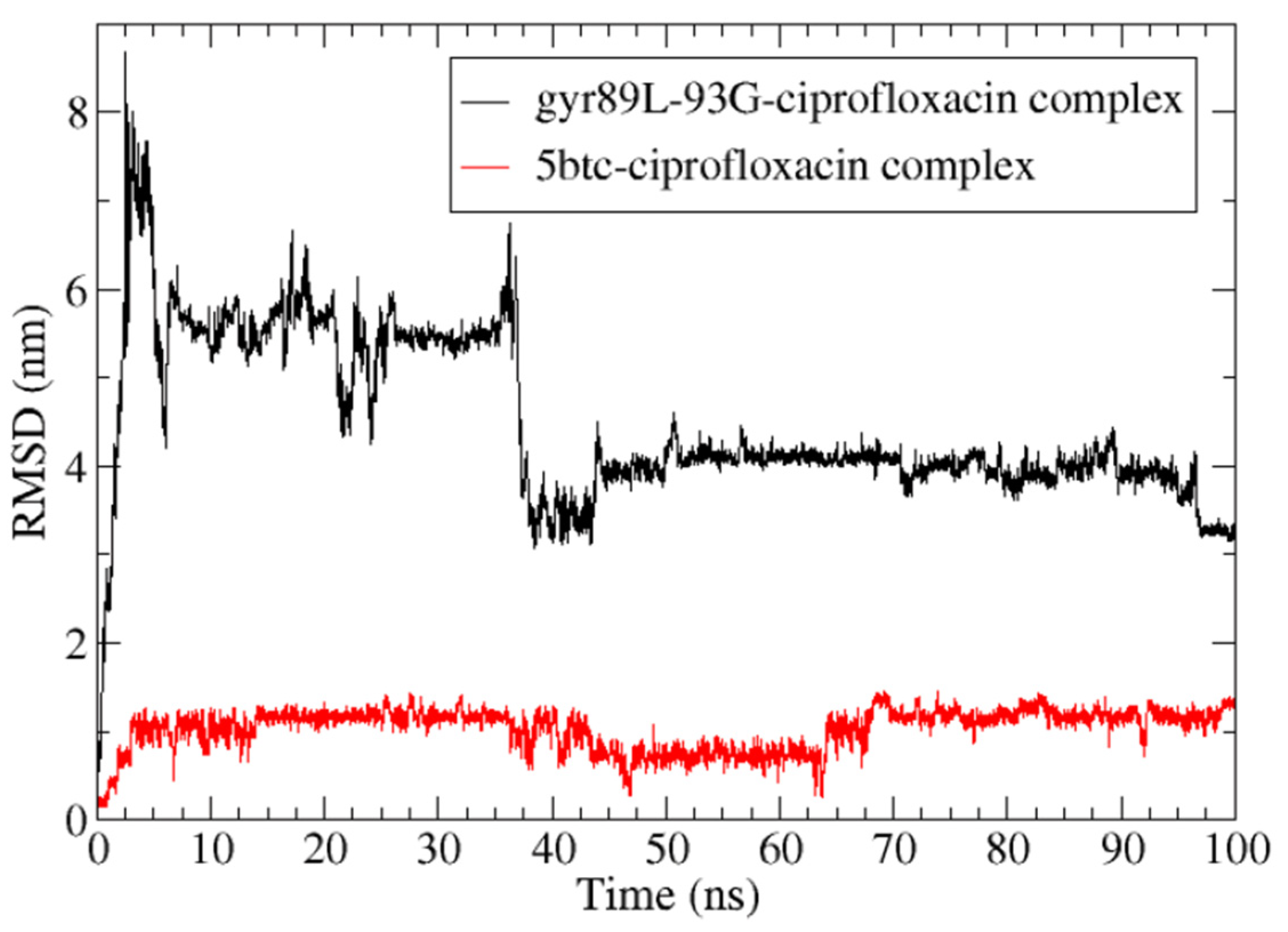
Figure 7.
Root Mean Square Fluctuation (RMSF) of the protein, illustrating amino acid movement over a 100-nanosecond simulation. The red arrow highlights amino acids in the QRDR region of the GyrA subunit, while the blue arrow points to amino acids in the GyrB subunit that interact with ciprofloxacin.
Figure 7.
Root Mean Square Fluctuation (RMSF) of the protein, illustrating amino acid movement over a 100-nanosecond simulation. The red arrow highlights amino acids in the QRDR region of the GyrA subunit, while the blue arrow points to amino acids in the GyrB subunit that interact with ciprofloxacin.
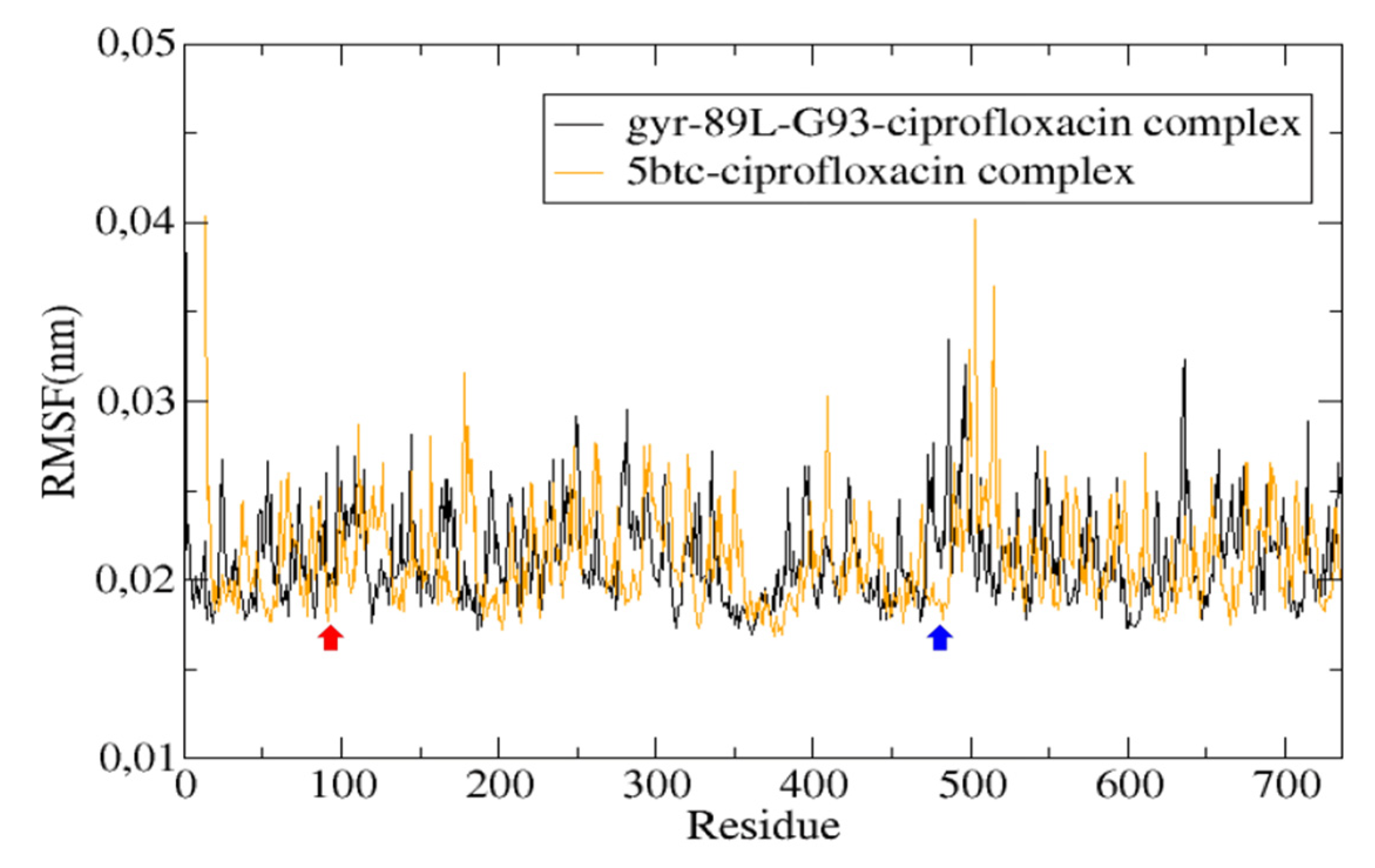
Figure 8.
Protein-ligand interactions during molecular dynamics simulations. (A, C) Last frame of the 5BTC-ciprofloxacin complex. (C, D) 5th frame (0.04 nanoseconds) of the gyr89L-93G-ciprofloxacin complex.
Figure 8.
Protein-ligand interactions during molecular dynamics simulations. (A, C) Last frame of the 5BTC-ciprofloxacin complex. (C, D) 5th frame (0.04 nanoseconds) of the gyr89L-93G-ciprofloxacin complex.
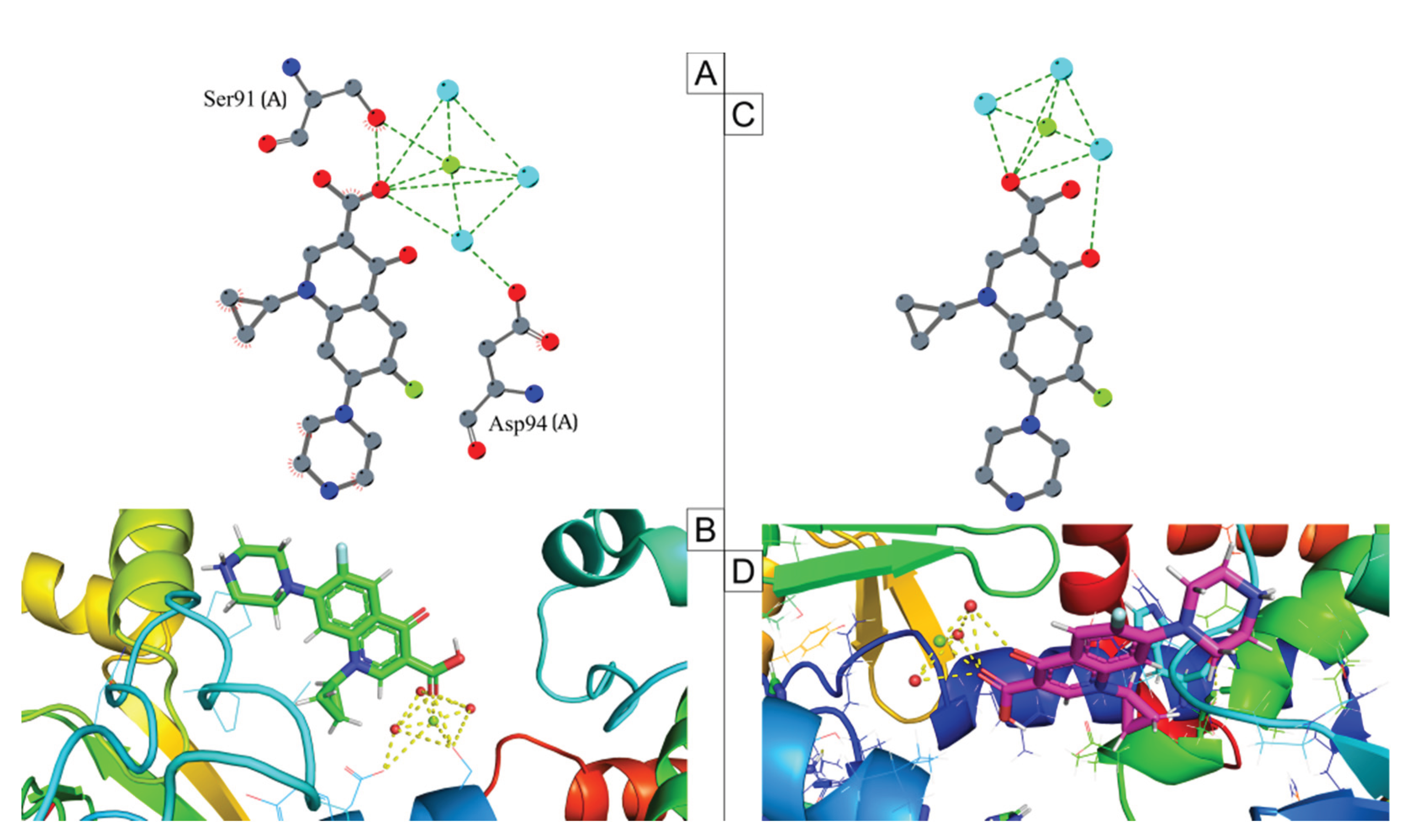

Table 1.
Genome features of Corynebacterium ulcerans IHP37393, IHP103889 and IHP106492 strains.
| Feature | Strain | ||
|---|---|---|---|
| IHP37393 | IHP103889 | IHP106492 | |
| Accession number | JBGNWQ000000000 | JBGNWN000000000 | JBGNWM000000000 |
| Platform | Illumina® NextSeq 550 | Illumina® NextSeq 550 | Illumina® NextSeq 550 |
| Completeness (%) | 99.99 | 99.99 | 99.99 |
| Contamination (%) | 0,74 | 0,29 | 0,60 |
| Coverage | 251x | 293x | 261x |
| Chimerism | No | No | No |
| Total length (bp) | 2,489,063 | 2,496,757 | 2,521,744 |
| GC (%) | 53.3 | 53.3 | 53.3 |
| Contigs | 6 | 6 | 5 |
| N50 | 815040 | 808602 | 828787 |
| L50 | 2 | 2 | 2 |
| CDS | 2242 | 2252 | 2293 |
| rRNAs | 3 | 3 | 3 |
| tRNAs | 51 | 51 | 51 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



