
Submitted:
19 June 2025
Posted:
20 June 2025
You are already at the latest version
Abstract
Aurora kinase B (AURKB), a serine/threonine protein kinase, is essential for accurate chromosome segregation and cytokinesis during mitosis. Dysregulation of AURKB, often characterized by overexpression, has been implicated in various malignancies, including breast cancer. However, the mechanisms governing its dysregulation remain incompletely understood. Here, we identify a pivotal role of the MOF/MSL complex, which includes the histone acetyltransferase MOF (KAT8), in modulating AURKB stability through acetylation at lysine 215 (K215). This post-translational modification prevents AURKB ubiquitination, thereby stabilizing its expression. MOF/MSL-mediated AURKB stabilization promotes the proper assembly of the chromosomal passenger complex (CPC), ensuring mitotic fidelity. Notably, inhibition of MOF reduces AURKB K215 acetylation, leading to a decline in both AURKB expression and activity. Consequently, this downregulation suppresses the expression of the downstream target c-MYC, ultimately attenuating the malignant proliferation of breast cancer cells. Collectively, our findings reveal a novel mechanism by which lysine acetylation governs AURKB stability, highlight the significance of the MOF-AURKB-c-MYC axis in breast cancer progression, and propose potential therapeutic strategies targeting this pathway in clinical settings.
Keywords:
AURKB
; MOF
; histone acetyltransferase
; acetylation
; cell proliferation
1. Introduction
The Mitosis is a fundamental biological process orchestrated by a complex network of proteins, among which Aurora kinase B (AURKB) plays a pivotal role in mitotic regulation and cell cycle progression [1]. Aurora kinases, a family of three highly homologous serine/threonine kinases—Aurora A, Aurora B, and Aurora C—execute distinct yet interconnected functions in mitotic control. AURKB, also known by various aliases, including AIK2, AIM1, ARK2, AurB, IPL1, and STK5, was first identified as a cell cycle-dependent kinase in NIH3T3 cells, highlighting its role in cell proliferation [2].
AURKB is a core component of the chromosome passenger complex (CPC) [3], functioning in concert with the scaffolding protein inner centromere protein (INCENP) and the non-enzymatic subunits Survivin (BIRC5) and Borealin (CDCA8). The CPC ensures faithful chromosome segregation by dynamically localizing to specific mitotic structures at different stages [4,5]. Precise regulation of AURKB-mediated substrate phosphorylation is essential for mitotic progression [6], particularly during early mitosis, where AURKB phosphorylates kinetochore substrates to correct erroneous microtubule attachments [7,8]. As mitosis progresses, the CPC targets additional substrates, coordinating distinct mitotic events. AURKB translocates from metaphase kinetochores to the central spindle during anaphase and subsequently to the contractile ring during telophase [9,10,11], ensuring successful cytokinesis. Aberrant AURKB expression is frequently observed in various cancers, where it is associated with chromosomal instability and heightened malignancy [12]. Dysregulated AURKB is thought to confer a proliferative advantage to cancer cells [13].
MOF (males absent on the first), a histone acetyltransferase (HAT) of the MYST family, was initially identified as part of the Drosophila dosage compensation complex [14,15]. In humans, MOF shares structural features with its Drosophila homolog dMOF, including a MYST catalytic domain, a chromatin domain, and a C2HC-type zinc finger [16]. MOF forms two distinct complexes: the male-specific lethal (MSL) complex, which primary acetylates histone H4K16, and the non-specific lethal (NSL) complex, which acetylates histones H4K5, H4K8, and H4K16 [17,18]. Beyond histone modification, human MOF plays critical roles in transcriptional regulation, chromatin dynamics, cell proliferation, differentiation, and DNA damage repair [19,20,21]. Increasing evidence suggests that MOF is implicated in tumorigenesis, influencing cancer cell proliferation, apoptosis, and stemness [22]. For instance, MOF overexpression in non-small cell lung cancer (NSCLC) promotes tumor progression by acetylating Nrf2, thereby contributing to poor prognosis and therapeutic resistance [23]. Additionally, MOF-mediated acetylation of MDM2 has been linked to cisplatin resistance in ovarian cancer [24].
Post-translational modifications (PTMs) fine-tune AURKB activity and localization during mitosis, with ubiquitin-mediated degradation being a well-established regulatory mechanism [25,26]. Emerging studies suggest that HATs and histone deacetylases (HDACs) modulate AURKB in cancer. In oesophageal cancer, BRD4, a histone acetylation reader, is recruited to the promoters of AURKA and AURKB, while its inhibition by JQ1 induces senescence [27]. In lymphoma cells, AURKB and HDACs cooperatively regulate proliferation, with inhibition of either trigging cell cycle arrest and apoptosis [28].
However, direct acetylation of AURKB remains poorly understood. A 2016 study reported TIP60 (KAT5)-dependent AURKB acetylation enhances its kinase activity, ensuring proper mitotic procession and genomic stability [29]. Similarly, the MOF-containing MSL complex modulates YY1 stability and transcriptional activity via acetylation [30]. Whether MOF directly acetylates AURKB and how this modification influences AURKB stability and cancer progression remain unknown. Here, we demonstrate that MOF-mediated acetylation of AURKB enhances its stability and activity. Specifically, MOF-driven AURKB acetylation promotes c-MYC accumulation, thereby facilitating malignant proliferation in breast cancer cells.
2. Materials and Methods
2.1. Antibodies and Reagents
The following antibodies were used in this study: Anti-MSL1 (mouse monoclonal, 24373-1-AP) from Proteintech (Wuhan, China); anti-MOF (rabbit polyclonal, A3390) from ABclonal Technology (Wuhan, China); anti-MSL2 (rabbit polyclonal, ab83911) from Abcam (Shanghai, China); anti-AURKB (rabbit monoclonal, AF1930) from Beyotime (Shanghai, China); anti-c-MYC (9E10), anti-INCENP (sc-376514), anti-CDCA8 (sc-376635) and anti-BIRC5 (sc-17779), anti-Akt (sc-81434), anti-mTOR (sc-517464), anti-ERK (sc-135900), anti-C-jun (sc-166540), anti-MAPK (sc-7972) (mouse monoclonal antibodies), as well as anti-CyclinB1 (sc-752), anti-E-Cadherin (sc-59778) and anti-N-Cadherin (sc-393933) (rabbit polyclonal antibodies), all from Santa Cruz Biotechnology (Dallas, TX, USA). Anti-HA (RLM3003) and anti-H3 (RLM3038) mouse monoclonal antibodies were obtained from Ruiying Biological (Suzhou, China). Anti-H3S10Ph (mouse monoclonal, #26436) was sourced from Upstate (New York USA). Anti-Flag (M2) (A2220), anti-Myc (M2)-agarose (A7470), anti-Flag M2 (mouse monoclonal, F3165), and anti-H4K16ac (H9164) (rabbit polyclonal) antibodies were obtained from Sigma (St. Louis, MO, USA). Anti-c-MYC-T58Ph (rabbit polyclonal) was from Bioss (Beijing, China). Pan-acetylation (Pan-ac, PTM0105RM) (rabbit polyclonal) antibody was from Jingjie Biotechnology (Hangzhou, China). Anti-β-Tubulin (M30109), anti-PanPh (M210030F) (mouse monoclonal), and anti-Ki67 (TW0001F) (rabbit monoclonal) antibodies were purchased from Abmart (Shanghai China). Anti-His (GB151251-100) (mouse monoclonal) antibody was from Servicebio (Wuhan, China). Anti-MSL3L1, and anti-GAPDH (rabbit polyclonal) antibodies were raised against bacterially expressed proteins at Jilin University.
The following reagents were used: cycloheximide (CHX, DH466-1) from Beijing Dingguo Changsheng Biotechnology Co., Ltd. (Beijing, China); the histone acetyltraferase (HAT) inhibitor MG149 (S7476) from Selleck Chemicals (Shanghai, China); the AURKB inhibitor AZD1152-HQPA from Abmole Bioscience (Beijing, China); and hydroxyurea (HU, H8267), Nocodazole (M1404), and MG132 (Z-Leu-Leu-al) from Sigma (St. Louis, MO, USA).
2.2. Cell Culture
HEK293T and HeLa cells were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The breast cancer lines MCF-7 and MDA-MB-231 were purchased from the Shanghai Biotechnology Co., Ltd. (Shanghai, China). All cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Meilunbio®, Dalian, China) supplemented with 10% fetal bovine serum ((FBS, Procell, Wuhan, China) and 1% penicillin-streptomycin (P/S, Thermo Fisher Scientific, Waltham, MA, USA). Cells were maintained at 37◦C in a humidified incubator with 5% CO2. Each cell line was authenticated by short tandem repeat (STR) profiling within the past three years. All experiments were conducted using mycoplasma-free cells.
2.3. Plasmid Construction and Transfection
The coding region of full-length AURKB (NM_001313950.2), MSL1 (NM_001012241), MOF (NM_032188), INCENP (NM_020238.3), and MYC (AH002906.2), along with various truncations—including MOF (1-157aa, 1-216aa and 158-458aa)—were subcloned into a pcDNA3.1(–) vectors with Flag, Myc, or HA tags. Additionally, point mutants, including AURKB (K215Q, K215R) and MOF (G327E), were generated. Plasmids were transiently transfected into cells using polyethyleneimine (PEI, 23966, PolySciences, Beijing, China) according to the manufacturer’s instructions.
2.4. Expression of Recombinant Proteins in Escherichia Coli
Full-length AURKB and INCENP were subcloned into pET41a vector. His-GST-tagged AURKB and INCENP proteins that were expressed from pET41a vector in BL21 (DE3) Codon Plus Escherichia coli. Cells.
2.5. siRNA/shRNA Knockdown
293T, MCF-7, and MDA-MB-231 breast cancer cells were transfected with non-targeting (NT) siRNA (D-001206), AURKB siRNA (#1, 5’-CCUGCGUCUCUACAACUAUtt-3’, #2, 5’-UCGUCAAGGUGGACCUAAAtt-3’), and an siRNA SMART pool (Dharmacon, Shanghai, China), using Lipofectamine RNAi MAX (13778150, Invitrogen) according to the manufacturer’s instructions. Seventy-two hours post-transfection, cells were subjected to subsequent experiments. For stable knockdown, the pLVX-shRNA system was used to express shRNA targeting AURKB, MSL1, and MOF in 293T, MCF-7, and MDA-MB-231 cells. The specific shRNA target sequences were: shAURKB (CCUGCGUCUCUACAACUAU), shMSL1 (GCACCGGACGTGTAGGAAAT), and shMOF (CGAAATTGATGCCTGGTAT).
2.6. Immunoprecipitation (IP)
MCF7, MDA-MB-231, and HEK293T cells were cultured in 10 cm tissue culture plates and transiently transfected with Flag- or Myc-tagged plasmids. Forty-eight hours post-transfection, cells were collected and lysed using RIPA buffer containing: 1% NP-40, 150 mM NaCl, 50 mM Tris-HCl, 10% glycerol, 1 mM dithiothreitol (DTT), and a complete protease inhibitor cocktail. Whole-cell lysates were incubated overnight at 4 ◦C with anti-Flag (M2) or anti-Myc-agarose beads. The immunoprecipitated proteins were then eluted using 4 × SDS loading buffer and analyzed by western blot with anti-Flag or anti-Myc antibodies.
2.7. Immunofluorescence Staining
Hela and MCF7 cells were cultured in 24-well plates containing coverslips (Nest, 8D1007) and grown to approximately 30% confluence. Cells were then transfected with plasmids and incubated for 48 hours. Cells were then fixed and immunostained with primary antibodies, followed by FITC/TRITC-conjugated secondary antibodies (1:300, Santa Cruz sc-2012). Nuclei were counterstained with Vectashield containing DAPI (H-1200, Vector Laboratories, Inc., Burlingame, CA, USA). Fluorescent images were acquired using an Olympus BX40F microscope equipped with a 40× silicon immersion objective (Olympus Corporation, Miyazaki, Japan).
2.8. Reverse Transcription PCR
Total RNA was extracted using RNAiso Plus (9109; Takara,Tokyo, Japan). 1 μg of total RNA from each sample was reverse transcribed into cDNA using the PrimeScript 1st Strand synthesis Kit (6110A, Takara, Tokyo, Japan). Relative mRNA levels were quantified using TB Green® Premix Ex Taq™ II (RR820A, Takara, Tokyo, Japan) on the Eco Real-Time PCR System (Illumina, Gene Company Limited, Hong Kong, China). The qPCR primers were as follows: AURKB, 5ʹ-CAGTGGGACACCCGACATC -3ʹ (forward) and 5ʹ-GTACACGTTTCCAAACTTGCC -3ʹ (reverse); MSL1, 5ʹ- CAAGACTCTCCACTCCCCAAAA -3ʹ (forward) and 5ʹ- CCTCCAAGAAGGAATTGCTACAG -3ʹ (reverse); MOF, 5ʹ- CCCAAACCAGTCAGACCAGC -3ʹ (forward) and 5ʹ- GGGCCACCAGAACTGACTTT -3ʹ (reverse); GAPDH, 5ʹ-ATCACTGCCACCCAGAAGAC-3ʹ (forward) and 5ʹ-ATGAGGTCCACCACCCTGTT-3ʹ (reverse).
2.9. In Vitro KAT Assay
Mix the following reactants in a total volume of 20 μl: cold acetyl coenzyme A (12.5 μM), recombinant AURKB proteins expressed in Escherichia coli,anti-Flag MOF beads,HAT reaction buffer to reach 20 μl. The HAT reaction buffer contains 50 mM Tris-HCl (pH 8.0), 50 mM KCl, 0.1 mM EDTA, 1 mM dithiothreitol, 5% (v/v) glycerol, 1 mM phenylmethylsulfonyl fluoride, 10 mM sodium butyrate. Incubate the reaction mixture at 30°C for 30 to 60 min. Finally, add 4× SDS loading buffer to terminate the reaction, and heat the sample at 95°C for western blot analysis.
2.10. Flow Cytometry Analysis
MCF7 and MDA-MB-231 cells were cultured in DMEM supplemented with 10% FBS. For fixation, cells were harvested and resuspended as single-cell in 70% ethanol at 20ºC for at least 4 hours. Following ethanol fixation, cells were centrifuged at 300×g for 5 min, and the supernatant was discarded. The cell pellets were washed and resuspended in 300 μL PBS containing 0.1% (v/v) Triton X-100 (Sigma, Cat. T8787), 0.3 mg/mL DNase-free RNase A (Sigma, Cat. R5500), and 50 μg/mL propidium iodide (CF0031, Beijing Dingguo, China), followed by incubation at 37 C for 1 hour. Flow cytometry was performed using an EPICS XL™ cytometers (Beckman Coulter), and data were analyzed with ModFit LT software (Verity Software House, USA).
2.11. EdU Assay
The EdU incorporation assay was performed using the BeyoCleck™ EdU Cell Proliferation Kit and the Alexa Fluor 488 in vitro Imaging Kit (Beyotime, C0071s, Shanghai, China). MCF7 and MDA-MB-231 cells were incubated with 10 µM EdU (5-ethyl-2´-deoxyuridine) at 37◦C for 2 hours. Cells were then fixed with 4% paraformaldehyde for 15 minutes and washed with PBS containing 0.5% Triton-X-100. Nuclei were counterstained with Hoechst 33342 (GC10939, GLPBIO). The proliferation rate was determined according to the manufacturer's instructions. Fluorescent images were aquired using a fluorescence microscope, with three randomly selected fields captured per group.
2.12. Cell Viability Assay
MCF7 and MDA-MB-231 cells (1,000 cells/well) were seeded in 96-well plates, and cell viability was assessed using the CellTiter 96® Aqueous One Solution Cell Proliferation Assay Kit (G3580, Promega Corporation, Madison, WI, USA) at 24, 48, 72, and 96 hours. Absorbance was measured at 490 nm using a microplate reader (Infinite F200 Pro, TECAN, Shanghai, China).
2.13. Colony Formation Assay
Stably transferred MCF7 and MDA-MB-231 cell lines (3000-4000 cells/well) were seeded into 6-well plates and cultured for 10–14 days at 37 C. Colonies were then fixed with 4% paraformaldehyde for 15 minutes and stained with 0.1% crystal violet for 20 min. The number and size of colonies were recorded for comparison and imaged using a digital camera.
2.14. In Vivo Tumor Metastasis Experiments
All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of Jilin University. Male Balb/c mice (7–8 weeks old, weighing18–20 g) were purchased from Vital River Biotechnology Company (Beijing, China). The animal experiment protocol was approved under the ethics review number (2024) YNPZSY No. (0512). Mice were housed under a 12-hour light/dark cycle with ad libitum access to food and water. Randomization was performed based on body weight, and sample sizes were determined according to the “Resource Equation” method. MDA-MB-231 cells infected with lentiviral-mediated pLVX-Flag-AURKB-WT (n=5) and pLVX-Flag-AURKB K215R (n=5) were subcutaneously injected into mice. Thirth days post-injection, mice were euthanized, and tumor tissues were collected for further analysis.
2.15. Statistical Analysis
Statistical analyses were conducted using data from at least three independent experiments. Data were processed with SPSS software, version 26 (IBM Corp., Armonk, NY, USA). Results are presented as mean ± SD. Differences between two groups were evaluated using an unpaired Student´s t-test, while comparisons among multiple groups were analyzed using one-way analysis of variance (ANOVA). A P-value < 0.05 was considered statistically significant.
3. Results
3.1. A Reciprocal Interaction Between the MOF/MSL Complex and CPC in 293T Cells
Previously, we generated an MSL1-knockout (MSL1-KO) HEK293T cell line using CRISPR/Cas9 gene editing [31]. To explore the regulatory effects of the MSL complex, we performed SILAC-based mass spectrometry on MSL1-KO cells. Among 4,449 differentially expressed proteins (Figure 1A), we identified several CPC subunits—AURKB, INCENP, and BIRC5—among the significantly downregulated proteins (fold change > 1.2, p < 0.05) (Figure 1B). This observation suggested a regulatory link between the MOF/MSL complex and the CPC. To validate this hypothesis, we overexpressed Flag-tagged MOF or MSL1 in 293T cells and observed a dose-dependent increase in endogenous AURKB protein levels, accompanied by an increase in its substrate phosphorylation at H3S10 (Figure 1C). Conversely, shRNA-mediated knockdown of MOF significantly reduced AURKB expression and H3S10 phosphorylation (Figure 1D). Notably, quantitative PCR (qPCR) analysis confirmed that neither MOF nor MSL1 overexpression or knockdown affected AURKB mRNA levels (Figure 1E,F), suggesting that the MOF/MSL complex regulates CPC components at the post-transcriptional level. Consistent with this, in MSL1-KO cells exhibited reduced expression of all CPC subunits, as well as diminished H3S10 phosphorylation (Figure 1G, lane 2 vs. lane 1). Importantly, reintroduction of Flag-MSL1 into MSL1-KO cells restored the expression of CPC components, including AURKB, BIRC5, CDCA8, and INCENP (Figure 1G, lane 4 vs. lane 3). To further assess the role of the MOF/MSL1 complex in AURKB stability, we conducted a cycloheximide (CHX) chase assay. AURKB degradation was markedly acceleratewd in 293T cells treated with shMOF (Figure 1H,I, lanes 7-12) or shMSL1 (Figure 1J,K, lanes 7-12) compared to cells expressing non-targeting shRNA (shNT), confirming that MOF/MSL1 depletion shortens AURKB’s half-life. Finally, an in vitro phosphorylation assay demonstrated that AURKB catalytic activity toward INCENP (Figure 1L, lane 4) was significantly enhanced in the presence of MSL1 (Figure 1M, lane 3). Collectively, these findings establish a reciprocal interaction between the MOF/MSL complex and the CPC, wherein the MOF/MSL complex functions as a stabilizer and enhancer of CPC activity by regulating AURKB stability and function.
3.2. MOF, MSL1, and AURKB May Collaborate During Early Mitosis
Given the regulatory effects of the MOF/MSL complex on AURKB kinase activity and CPC subunit expression, we hypothesized that MOF and MSL1 physically interact with AURKB. Flag immunoprecipitation (IP) assays confirmed this interaction (Figure 2A–C), demonstrating that endogenous MOF, MSL1, and AURKB co-precipitated with Flag-AURKB, Flag-MOF, and Flag-MSL1, indicating a direct binding affinity. This interaction was further validated by co-transfection of MOF and AURKB, followed by co-IP experiments, which confirmed their association (Figure 2D,E, lane 3). To delineate the minimal binding region of MOF required for AURKB interaction, we co-transfected Flag-AURKB with the full-length Myc-MOF (1-458 aa) and its truncated mutants—Myc-MOF-F1 (1-157 aa), HA-MOF-L1 (1-216 aa), and HA-MOF-L2 (158-458 aa) (Figure 2F, upper)—into 293T cells. Flag IP followed by Western blot analysis revealed that the C-terminal region of MOF (216-458 aa) was essential for AURKB binding (Figure 2G, lower), suggesting that the MOF HAT domain is required for their interaction.This section may be divided by subheadings. It should provide a concise and precise description of the experimental results, their interpretation, as well as the experimental conclusions that can be drawn.
As CPC localization dynamically changes throughout the cell cycle, with AURKB exhibiting distinct catalytic activities at different mitotic phases [5], we investigated the subcellular co-localization of MOF, MSL1, and AURKB during mitosis via immunofluorescence (IF) staining in HeLa cells. β-tubulin (red) was used to distinguish prophase, metaphase, anaphase, and telophase. AURKB, MOF, and MSL1 (stained green) were observed on chromosomes during prophase and metaphase. However, in anaphase, while MOF and MSL1 remained chromosomally associated (Figure 2H–J), AURKB relocated to the equatorial plate, facilitating cytokinesis as cells progressed into telophase (Figure 2H). These findings suggest that the MOF/MSL complex plays a role in mediating AURKB function during early mitosis.
To further validate the interaction between the MOF/MSL and CPC complexes, we co-transfected 293T cells with Flag-INCENP, a CPC scaffolding protein, alongside MOF or MSL1. Co-IP assays confirmed that both MOF and MSL1 interacted with Flag-INCENP, with endogenous AURKB detected in Flag IP (Figure 2L, lane 4 vs. lane 2; Figure 2L, lane 3 vs. lane 1). Overexpression of MOF also enhanced phospho-INCENP levels compared to Flag-INCENP alone (Figure 2K, lane 4). In contrast, MSL1-knockdown (KD) cells exhibited markedly reduced co-IP of CPC subunits with Flag-INCENP (Figure 2M, lane 4 vs. lane 2), further supporting the role of the MOF/MSL complex in stabilizing CPC.
3.3. MOF/MSL Complex Mediates AURKB Acetylation, Stabilizing CPC Integrity in 293T Cells
The observed interaction between MOF, MSL1, and AURKB prompted us to investigate whether the MOF/MSL complex acetylates AURKB and how this modification affects its stability. To address this, Flag-AURKB was co-transfected with Myc-MOF or Myc-MSL1 in 293T cells, and AURKB acetylation and ubiquitin-mediated degradation were analyzed via Flag IP in the presence of HA-ubiquitin and the proteasome inhibitor MG132. As expected, both MOF (Figure 3A) and MSL1 (Figure 3B) bound to and acetylated AURKB (Flag IP/Pan-ac). Notably, this acetylation inhibited AURKB ubiquitination (Flag IP/HA), suggesting that the MOF/MSL complex stabilizes AURKB by preventing its proteasomal degradation. This conclusion was further supported by the reduced acetylation and enhanced degradation of Flag-AURKB following MOF knockdown (Figure 3C, Flag IP/Pan-ac and HA). Additionally, co-transfection with the enzymatically inactive MOF G327E mutant [32], which lacks acetyltransferase activity, significantly diminished AURKB binding and acetylation (Figure 3D, lane 6 vs. lane 4). These findings were corroborated by an in vitro lysine acetyltransferase (KAT) assay (upper panel) using His-AURKB purified from E. coli (Figure 3E, lane 5).
To further elucidate the role of the MOF/MSL complex in regulating AURKB acetylation, protein stability, and kinase activity, we performed a series of functional assays. As shown in Figure 3F and 3G, co-transfection of Flag-AURKB and MOF into 293T cells, with either MSL1 overexpression or knockdown, demonstrated that MSL1 enhances AURKB acetylation (Flag IP/Pan-ac), stabilizes AURKB (Flag IP/HA), and promotes phosphorylation of its substrate H3S10 (Input/H3S10p). Conversely, MSL1 knockdown resulted in the opposite effect, indicating that MSL1 positively regulates AURKB in a MOF-dependent manner. Furthermore, co-transfection of Myc-AURKB and Flag-INCENP into MSL1-KO 293T cells revealed that MSL1 deletion not only impaired AURKB binding but also significantly reduced its acetylation and stability (Figure 3H, lane 2 vs. lane 1), suggesting that MOF-dependent CPC regulation requires MSL1. Collectively, our results suggest that MOF/MSL complex-mediated acetylation of AURKB is critical for maintaining CPC stability.
3.4. MOF/MSL Complex-Mediated Acetylation of AURKB at K215 Maintains CPC Integrity and Kinase Activity
Our findings demonstrate that MOF/MSL complex-mediated acetylation positively regulates AURKB kinase activity and CPC stability. To identify the specific acetylation site on AURKB, we analyzed its predicted structure alongside MOF using data retrieved from the RCSB Protein Data Bank (https://www.rcsb.org/) and performed protein docking analysis via the HDOCK Server. This analysis identified lysine 215 (K215) of AURKB as a key interaction site, forming hydrogen bond with aspartic acid 254 (D254) in MOF, with a bond length of 3.2Å and a confidence score of 0.9239, suggesting a stable interaction (Figure 4A). Consistent with this prediction, high-throughput mass spectrometry (https://www.phosphosite.org) identified K215 as a potential acetylation site (Figure 4B). To validate K215 as the primary MOF-targeted acetylation site on AURKB, we performed Flag IP in 293T cells co-transfected with Myc-MOF and Flag-tagged AURKB (wild-type or K215R mutant). Western blot analysis revealed that K215 mutation abolished AURKB acetylation by MOF, confirming K215 as the major acetylation site (Figure 4C, Flag IP/Pan-ac). A CHX chase assay further demonstrated that the half-life of AURKB was markedly reduced upon K215 mutation (Figure 4D; lanes 7-12 vs. lanes 1-6, quantification shown in lower panel). Additionally, co-transfection with HA-ubiquitin and MG132 showed that the K215R mutant underwent significantly enhanced ubiquitin-mediated degradation compared to wild type AURKB and the acetylation-mimic AURKB-K215Q mutant (Figure 4E, lane 4 vs. lane 2 and 3).
To assess the role of MOF-mediated acetylation in AURKB stability, we transfected Flag-tagged wild type or K215R AURKB into 293T cells, with or without Myc-MOF. Flag IP results indicated that mutating K215 accelerated AURKB degradation via the proteasome pathway (Figure 4F, Flag IP/HA, lane 3 & 5). Furthermore, the K215R mutation reduced INCENP phosphorylation (Figure 4G, lane 4 vs. lane 3), whereas the acetylation-mimic K215Q mutant exhibited increased phospho-INCENP levels (Lane 5 vs. Lane 3). Beyond CPC stability, the K215 mutation impaired AURKB’s ability to maintain CPC integrity, as evidenced by reduced expression of INCENP, CDCA8 and BIRC5 (Figure 4H, Flag IP/ lane 3 vs. lane 2 & 4). It also attenuated AURKB kinase activity, as indicated by decreased H3S10 phosphorylation (Input/H3S10p). Notably, K215 mutation reduced c-MYC protein levels while increasing c-MYC phosphorylation at T58 (Input/c-MYC, Input/c-MYC-T58p), a modification linked with c-MYC degradation. In summary, MOF-mediated acetylation of AURKB at K215 (Figure 4I) is crucial for stabilizing CPC integrity, regulating AURKB kinase activity, and modulating its downstream signaling effects.
3.5. MOF/MSL1 Complex-Mediated Acetylation of AURKB at K215 Regulates G2/M Phase Progression in HeLa and MCF-7 Cells
Our findings indicate that the MOF/MSL complex plays a crucial role in mitotic progression by acetylating AURKB at K215. This modification is essential for maintaining the integrity and stability of the CPC, thereby ensuring proper spindle formation and chromosome segregation. To investigate the role of MOF and MSL1 in mitosis, we performed IF staining in HeLa cells following the knockdown of MOF or MSL1. Compared to non-targeting shRNA (shNT) controls, cells transfected with shMOF or shMSL1 exhibited a significant increase in spindle multipolarity—an indicator of mitotic disruption (Figure 5A,B). Notably, this phenotype was rescued by AURKB overexpression (Figure 5C, shMOF + AURKB vs. shMOF groups). A similar phenotype was observed in cells expressing the AURKB K215R mutant (Figure 5D), suggesting that MOF/MSL1 regulates mitotic progression through AURKB K215 acetylation. Quantification of spindle multipolarity (Figure 5E–H) further corroborated these findings. Western blot analysis confirmed efficient knockdown of MOF and MSL1 (Figure 5I, lane 2 & 4; Figure 5J, lane 2–4), correlating their depletion with mitotic abnormalities.
To further assess the impact of AURKB K215 acetylation on cell cycle progression, HeLa cells were transiently transfected with either wild-type or K215R-mutant AURKB, synchronized at G1/S using 1 mM hydroxyurea, and subsequently released. Flow cytometry analysis confirmed that cells expressing the K215R mutant progressed more slowly through the G2/M phase transition compared to those expressing wild-type AURKB (Figure 5K). Western blot analysis of samples collected at 0, 6, 9, and 12 hours post-release showed distinct expression kinetics: wild-type AURKB peaked at 9 hours, whereas the K215R mutant exhibited a delayed peak at 12 hours (Figure 5L), further supporting the role of K215 acetylation in timely mitotic progression. In both MCF-7 and MDA-MB-231 breast cancer cells, AURKB depletion led to G2/M phase arrest. Reintroduction of wild type (WT) or K215Q AURKB restored normal cell cycle progression, whereas the K215R mutant failed to do so (Figure 5M). Phase-specific analysis of the cell cycle (G1, S, G2/M, and M phases) revealed that MOF, MSL1, and H4K16ac levels were highest during G2/M and M phases (Figure 5N, lane 3), aligning with AURKB expression and reinforcing their role in CPC regulation. Collectively, these findings provide compelling evidence that the MOF/MSL complex acetylates AURKB at K215, stabilizing the CPC, enhancing AURKB kinase activity, and ensuring proper spindle assembly and chromosomal alignment during early mitosis. The delayed mitotic progression observed in K215R-expressing cells underscores the critical role of this acetylation event in maintaining mitotic fidelity.
3.6. MOF-Mediated Acetylation of AURKB at K215 Is Essential for Breast Cancer Cell Proliferation
Given that MOF-mediated acetylation of AURKB at K215 is implicated in early mitosis, we hypothesized that MOF regulates cancer cell proliferation through AURKB. As expected, EdU incorporation assays revealed that MOF depletion significantly reduced DNA replication, indicating impaired cell proliferation. Notably, cells expressing WT or K215Q AURKB exhibited near-normal EdU incorporation, whereas cells expressing the K215R mutant displayed markedly reduced incorporation (Figure 6A,B). Consistent with these findings, CCK-8 cell viability assays demonstrated a significant decrease in viability following MOF depletion, which was rescued by WT or K215Q AURKB overexpression, but not by the K215R mutant (Figure 6C). Similarly, clonogenic assays revealed a significant reduction in colony-forming ability upon MOF knockdown in both MCF-7 and MDA-MB-231 cells (Figure 6D,E). Overexpression of WT or K215Q AURKB restored clonogenic potential, whereas the K215R mutant failed to rescue this defect. Collectively, these results suggest that MOF regulates the proliferation of MCF-7 and MDA-MB-231 breast cancer cells via AURKB K215 acetylation.
To further examine the role of AURKB K215 acetylation in tumorigenesis, we established stable MDA-MB-231 cell lines expressing either Flag-AURKB or Flag-AURKB K215R mutants (Figure 6F). To assess the in vivo relevance of MOF-mediated AURKB K215 acetylation, we employed a xenograft model, in which BALB/c nude mice were subcutaneously injected with these stable cell lines (5 mice per group) (Figure 6G). Over a 30-day period, body weight remained unchanged between groups; however, a marked difference in tumor growth was observed. Tumors in the AURKB K215R group exhibited significantly slower growth than those in the AURKB WT group (Figure 6H), suggesting that MOF promotes MDA-MB-231 proliferation by mediating AURKB K215 acetylation. At the end of the study, excised tumors were photographed (Figure 6I) and weighted, confirming a significantly smaller tumor size in the AURKB K215R group (Figure 6J). Western blot analysis of whole-cell lysates from excised tumors further validated these findings, revealing decreased Ki67 and H3S10p expression in the AURKB K215R group (Figure 6K), consistent with impaired proliferation. Interestingly, we also observed reduced c-MYC expression and increased phosphorylation at c-MYC-T58 in the AURKB K215R group, suggesting a potential regulatory link between the MOF-AURKB axis and c-MYC. This finding warrants further investigation into the broader oncogenic implications of MOF-mediated AURKB acetylation.
3.7. Acetylation of AURKB at K215 Promotes Breast Cancer Cell Proliferation by Stabilizing c-MYC
Xenograft studies suggest that AURKB plays a regulatory role in c-MYC expression and phosphorylation. To elucidate the molecular mechanism by which MOF-mediated acetylation of AURKB at K215 promotes breast cancer cell proliferation, we first examined the expression levels of key oncogenic proteins, particularly c-MYC, following AURKB knockdown in MCF-7 and MDA-MB-231 cells. Notably, depletion of AURKB resulted in a marked reduction in c-JUN, ERK, c-MYC, and H3S10p levels in both cell lines (Figure 7A,B, lane 2). To further explore the regulatory network of AURKB, we performed an integrated Venn analysis incorporating multiple datasets: CRISPR screen data from the BioGRID ORCS database (https://orcs.thebiogrid.org/Gen) for MCF7 and MDA-MB-231 cell proliferation, protein interaction data from the STRING database (https://cn.string-db.org/), and genes positively associated with AURKB in breast cancer from the TCGA database (https://ualcan.path.uab.edu/analysis). This analysis identified four overlapping proteins—c-MYC, BIRC5, CDC20 and NUF2 (Figure 7C,D)—suggesting that AURKB positively regulates these proliferation-associated factors.
c-MYC is a well-established oncogene transcription factor that governs tumor cell proliferation, apoptosis, and metabolic reprogramming [33]. AURKB-mediated phosphorylation of c-MYC has been reported to inhibit its ubiquitination, thereby stabilizing the protein and promoting tumorigenesis [34]. In line with this, inhibition of AURKB with the AURKB kinase inhibitor AZD1152 at concentration of 10, 20, and 40 nM in MCF-7 cells led to a significant reduction in Ki67 and H3S10p levels, accompanied by a dose-dependent decrease in c-MYC protein levels (Figure 7E). To determine whether MOF is implicated in AURKB-mediated c-MYC accumulation, we treated MCF-7 cells with the MOF enzymatic activity inhibitor MG149 (20, 30, and 40 μM) for 24 hours, followed by Western blot analysis. MG149 treatment led to a dose-dependent reduction in AURKB and c-MYC expression, as well as the proliferation marker Ki67, suggesting that MOF activity is linked to breast cancer cells proliferation (Figure 7F). To further confirm the specific role of MOF-mediated AURKB K215 acetylation in regulating c-MYC protein levels, we transfected MCF-7 and MDA-MB-231 cells with WT AURKB, acetylation-deficient AURKB-K215R, or acetylation-mimic AURKB-K215Q, following AURKB knockdown. Consistent with previous observations, AURKB depletion significantly decreased in c-MYC protein levels (Figure 7G,I, lanes 2-3), as quantified in Fig. 7H,J. This effect was reversed by overexpression of WT AURKB or AURKB-K215Q, but not by AURKB-K215R, indicating that acetylation at K215 is essential for c-MYC stabilization. To further validate the role of the MOF-AURKB axis in regulating c-MYC, we overexpressed WT, K215R, and K215Q AURKB mutants in MOF-knockdown MCF-7 and MDA-MB-231 cells. Overexpression of WT or K215Q AURKB increased c-MYC levels, whereas K215R AURKB overexpression failed to restore c-MYC expression (Figure 7K–N). In summary, these findings demonstrate that acetylation of AURKB at K215 stabilizes c-MYC, which may present a key mechanism by which MOF-mediated AURKBK215 acetylation promotes breast cancer cell proliferation.
4. Discussion
RNAi screening identified MOF as a critical regulator of cancer cell survival. MOF has been previously implicated in the regulation of the G2/M cell cycle checkpoint, underscoring its essential role in cancer biology and highlighting its potential as a therapeutic target [35]. Another key oncogene, AURKB, is widely recognized as a target in multiple cancer types, with AURKB inhibitors showing enhanced efficacy in combination with osimertinib for non-small-cell lung cancer (NSCLC) [36]. Our study provides novel insights into the molecular interplay between the MOF/MSL complex and the chromosomal passenger complex (CPC). We demonstrate, for the first time, that MOF directly acetylates AURKB, reducing its ubiquitination and enhancing its kinase activity. Furthermore, the MSL1 subunit strengthens the interaction between MOF and AURKB, stabilizing the complex during the G2/M phase. Notably, acetylation of AURKB at K215 is crucial for mitotic progression. In breast cancer cells, MOF-mediated AURKB acetylation enhances AURKB protein levels and promotes c-MYC phosphorylation, leading to its accumulation and subsequent tumor cell proliferation.
The CPC plays a crucial role in mitotic regulation, with AURKB serving as its key catalytic component. AURKB is essential for assembling functional kinetochores, aligning spindle microtubules, and correcting improper microtubule-kinetochore attachments [37, 38]. Our proteomic analysis of MSL1-deficient cells identified differential expression of NUF2, a subunit of the NDC80 (nuclear division cycle 80) complex, which is a well-established AURKB substrate [39]. AURKB modulates the microtubules-binding affinity of the NDC80 complex by phosphorylating NDC80 at multiple sites, thereby influencing kinetochore-microtubule interactions [40]. Given that AURKB activity is dynamically regulated by post-translational modifications, our findings suggest that MOF-mediated acetylation serves as an additional regulatory layer that modulates both AURKB stability and kinase activity. Interestingly, we observed co-localization of the MOF/MSL complex and the CPC at the equatorial plate during mid-mitosis, raising the possibility that MOF/MSL-mediated acetylation dynamically regulates AURKB function during chromosome segregation. Further structural studies, including high-resolution cryo-electron microscopy [41], will be necessary to delineate the precise molecular mechanisms governing MOF/MSL-AURKB interactions during mitosis.
AURKB has garnered significant attention in cancer research due to its elevated expression in multiple malignancies, including colorectal adenocarcinoma, thyroid follicular carcinoma, laryngeal carcinoma, and lung cancer [42]. In breast cancer, AURKB expression is markedly upregulated and correlates with increased tumor cell proliferation and resistance to therapy [43]. Furthermore, AURKB overexpression has been implicated in paclitaxel resistance in NSCLC [44] and is associated with poor prognosis in hematological malignancies, such as acute lymphoblastic leukemia and acute myeloid leukemia [45]. Similarly, in hepatocellular carcinoma, AURKB mRNA levels are significantly elevated in tumor tissues and serve as an independent prognostic marker for disease aggressiveness [46]. The c-MYC oncogenes family (MYC, MYCN, MYCL) plays pivotal role in tumorigenesis, particularly in cancers characterized by MYC overexpression or amplification [47]. Deregulated MYC activity is a hallmark of various malignancies, including breast cancer, where it is linked to aggressive disease progression [48]. Clinical evidence indicates that c-MYC is frequently overexpressed in breast tumors and contributes to oncogenic transformation and tumor maintenance [49, 50].
5. Conclusions
Our study demonstrates that MOF-mediated acetylation of AURKB at K215 enhances AURKB stability and kinase activity, subsequently promoting c-MYC accumulation. Given the central role of this pathway in breast cancer progression, targeting MOF and AURKB presents a promising therapeutic strategy. Pharmacological inhibition of MOF or AURKB could disrupt this oncogenic axis, thereby suppressing tumor growth and providing a novel avenue for therapeutic intervention. Future studies should explore the development of specific inhibitors targeting MOF-AURKB interactions, as well as their potential synergy with existing anti-cancer therapies.
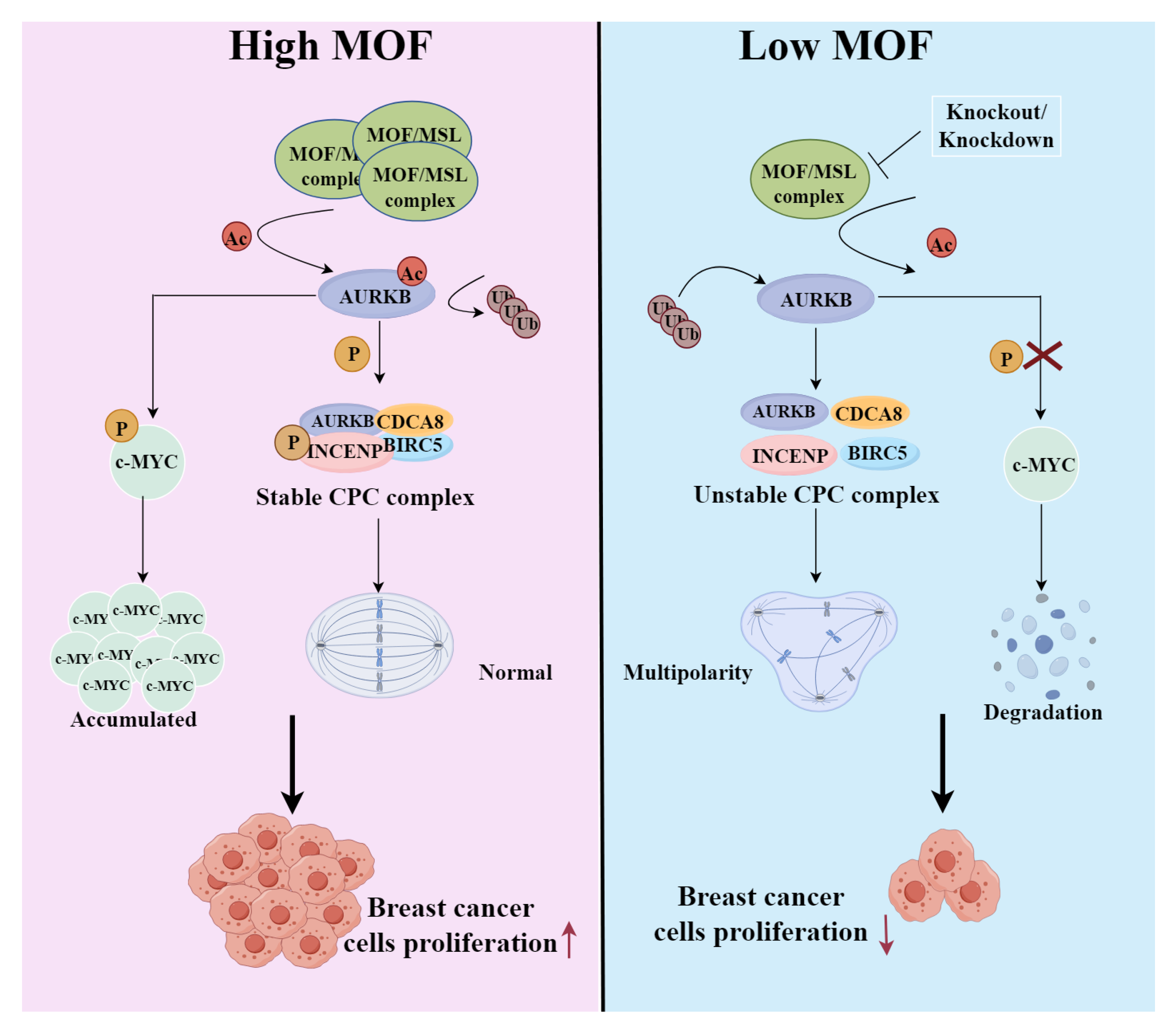
Figure 8.
The MOF/MSL complex acetylates AURKB, enhancing its protein stability and kinase activity, thereby stabilizing the CPC and ensuring proper mitotic progression. In breast cancer cells, this acetylation upregulates AURKB expression and activity, leading to robust phosphorylation of c MYC, which promotes its accumulation and drives malignant proliferation. Conversely, MOF/MSL depletion reduces AURKB acetylation, resulting in decreased AURKB stability and kinase activity. This destabilization of the CPC induces mitotic defects, attenuates c-MYC phosphorylation, and ultimately suppresses malignant proliferation or triggers cell death. (Figure 8, created with FigDraw).
Figure 8.
The MOF/MSL complex acetylates AURKB, enhancing its protein stability and kinase activity, thereby stabilizing the CPC and ensuring proper mitotic progression. In breast cancer cells, this acetylation upregulates AURKB expression and activity, leading to robust phosphorylation of c MYC, which promotes its accumulation and drives malignant proliferation. Conversely, MOF/MSL depletion reduces AURKB acetylation, resulting in decreased AURKB stability and kinase activity. This destabilization of the CPC induces mitotic defects, attenuates c-MYC phosphorylation, and ultimately suppresses malignant proliferation or triggers cell death. (Figure 8, created with FigDraw).
Author Contributions
Y.M, Y.C. and J.J. conceived and coordinated the project and designed the experiments. Y.M., Y.C and J.J. interpreted the data and wrote and edited the manuscript. Y.M. performed the most of the experiments. N.Z., F.L. F.W., Y-Y.C., F- Q.L., X.C., and Q.Z. assisted with the experiments and helped to analyze the data. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 32170599). The funders had no role in study design, data collection and analysis, publication decisions, or manuscript preparation.
Institutional Review Board Statement
All animal studies were approved by the Animal Care and Use Committee (IACUC) of Jilin University. An ethics review form for animal experiments has been approved, with an approval number: IACUC Issue No.: (2024) Shen YNPZSY No (0512).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
We declare that, all authors have declared that no competing interests exist.
Abbreviations
The following abbreviations are used in this manuscript:
| AURKB | Aurora kinase B |
| INCENP | Inner centromere protein |
| KATs | lysine acetyltransferases |
| CPC H3S19pho MOF MSL H4K16ac CHX |
chromosome passenger complex phosphorylation of histone H3S10 males absent on the first male-specific lethal acetylation of histone H4 at lysine16 cycloheximide |
| PTMs HATs HDACs |
post-translational modifications histone acetyltransferases histone deacetylases |
References
- Bolanos-Garcia, V.M. Aurora kinases. Int. J. Biochem. Cell Biol. 2005, 37, 1572–1527. [Google Scholar] [CrossRef] [PubMed]
- Niwa, H.; Abe, K.; Kunisada, T.; Yamamura, K. Cell-cycle-dependent expression of the STK-1 gene encoding a novel murine putative protein kinase. Gene 1996, 169, 197–201. [Google Scholar] [CrossRef]
- van der Horst, A.; Lens, SM. Cell division: control of the chromosomal passenger complex in time and space. Chromosoma 2014, 123, 25–42. [Google Scholar] [CrossRef]
- Carmena, M.; Wheelock, M.; Funabiki, H.; Earnshaw, W.C. The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat. Rev. Mol. Cell Biol. 2012, 13, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Lampson, M.A.; Cheeseman, I.M. Sensing centromere tension: Aurora B and the regulation of kinetochore function. Trends. Cell Biol. 2011, 21, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Welburn, J.P.; Vleugel, M.; Liu, D.; Yates, J.R., 3rd; Lampson, M.A.; Fukagawa, T.; Cheeseman, I.M. Aurora B phosphorylates spatially distinct targets to differentially regulate the kinetochore-microtubule interface. Mol. Cell. 2010, 38, 383–392. [Google Scholar] [CrossRef]
- Liu, D.; Vleugel, M.; Backer, C.B.; Hori, T.; Fukagawa, T.; Cheeseman, I.M.; Lampson, M.A. Regulated targeting of protein phosphatase 1 to the outer kinetochore by KNL1 opposes Aurora B kinase. J. Cell Biol. 2010, 188, 809–820. [Google Scholar] [CrossRef]
- DeLuca, K.F.; Lens, S.M.; DeLuca, J.G. Temporal changes in Hec1 phosphorylation control kinetochore-microtubule attachment stability during mitosis. J. Cell Sci. 2011, 124, 622–34. [Google Scholar] [CrossRef]
- Sumara, I.; Quadroni, M.; Frei, C.; Olma, M.H.; Sumara, G.; Ricci, R.; Peter, M. A Cul3-based E3 ligase removes Aurora B from mitotic chromosomes, regulating mitotic progression and completion of cytokinesis in human cells. Dev. Cell. 2007, 12, 887–900. [Google Scholar] [CrossRef]
- Adams, R.R.; Maiato, H.; Earnshaw, W.C.; Carmena, M. Essential roles of Drosophila inner centromere protein (INCENP) and aurora B in histone H3 phosphorylation, metaphase chromosome alignment, kinetochore disjunction, and chromosome segregation. J. Cell Biol. 2001, 153, 865–880. [Google Scholar] [CrossRef]
- Monje-Casas, F.; Prabhu, V.R.; Lee, B.H.; Boselli, M.; Amon, A. Kinetochore orientation during meiosis is controlled by Aurora B and the monopolin complex. Cell 2007, 128, 477–490. [Google Scholar] [CrossRef]
- Borah, N.A.; Reddy, M.M. Aurora Kinase B Inhibition: A Potential Therapeutic Strategy for Cancer. Molecules. 2021, 26, 1981. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.R.; Eckley, D.M.; Vagnarelli, P.; Wheatley, S.P.; Gerloff, D.L.; Mackay, A.M.; Svingen, P.A.; Kaufmann, S.H.; Earnshaw, W.C. Human INCENP colocalizes with the Aurora-B/AIRK2 kinase on chromosomes and is overexpressed in tumour cells. Chromosoma. 2001, 110, 65–74. [Google Scholar] [CrossRef]
- Belote, J.M.; Lucchesi, J.C. Male-specific lethal mutations of Drosophila melanogaster. Genetics. 1980, 96, 165–186. [Google Scholar] [CrossRef] [PubMed]
- Rea, S.; Xouri, G.; Akhtar, A. Males absent on the first (MOF): from flies to humans. Oncogene. 2007, 26, 5385–5394. [Google Scholar] [CrossRef] [PubMed]
- Neal, K.C.; Pannuti, A.; Smith, E.R.; Lucchesi, J.C. A new human member of the MYST family of histone acetyl transferases with high sequence similarity to Drosophila MOF. Biochim; Biophys; Acta. 2000, 149, 170–174. [Google Scholar] [CrossRef]
- Smith, E.R.; Cayrou, C.; Huang, R.; Lane, W.S.; Côté, J.; Lucchesi, J.C. A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol. Cell Biol. 2005, 25, 9175–9188. [Google Scholar] [CrossRef]
- Zhao, X.; Su, J.; Wang, F.; Liu, D.; Ding, J.; Yang, Y.; Conaway, J.W.; Conaway, R.C.; Cao, L.; Wu, D.; Wu, M.; Cai, Y.; Jin, J. Crosstalk between NSL histone acetyltransferase and MLL/SET complexes: NSL complex functions in promoting histone H3K4 di-methylation activity by MLL/SET complexes. PLoS Genet. 2013, 9, e1003940. [Google Scholar] [CrossRef]
- Kapoor-Vazirani, P.; Kagey, J.D.; Powell, D.R.; Vertino, P.M. Role of hMOF-dependent histone H4 lysine 16 acetylation in the maintenance of TMS1/ASC gene activity. Cancer Res. 2008, 68, 6810–6821. [Google Scholar] [CrossRef]
- Singh, M.; Bacolla, A.; Chaudhary, S.; Hunt, C.R.; Pandita, S.; Chauhan, R.; Gupta, A.; Tainer, J.A.; Pandita, T.K. Histone Acetyltransferase MOF Orchestrates Outcomes at the Crossroad of Oncogenesis, DNA Damage Response, Proliferation, and Stem Cell Development. Mol. Cell Biol. 2020, 40, e00232–20 (). [Google Scholar] [CrossRef]
- Zhong, J.; Li, X.; Cai, W.; Wang, Y.; Dong, S.; Yang, J.; Zhang, J.; Wu, N.; Li, Y.; Mao, F.; Zeng, C.; Wu, J.; Xu, X.; Sun, Z.S. TET1 modulates H4K16 acetylation by controlling auto-acetylation of hMOF to affect gene regulation and DNA repair function. Nucleic Acids Res. 2017, 45, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, L.; Pandey, R.; Byun, J.S.; Gardner, K.; Qin, Z.; Dou, Y. The histone acetyltransferase MOF is a key regulator of the embryonic stem cell core transcriptional network. Cell Stem Cell. 2012, 11, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ye, X.; Tang, N.; Shen, S.; Li, Z.; Niu, X.; Lu, S.; Xu, L. The histone acetylranseferase hMOF acetylates Nrf2 and regulates anti-drug responses in human non-small cell lung cancer. Br. J. Pharmacol. 2014, 171, 3196–3211. [Google Scholar] [CrossRef]
- Cai, M.; Xu, S.; Jin, Y.; Yu, J.; Dai, S.; Shi, X.J.; Guo, R. hMOF induces cisplatin resistance of ovarian cancer by regulating the stability and expression of MDM2. Cell Death Discov. 2023, 9, 179. [Google Scholar] [CrossRef]
- Chen, B.B.; Glasser, J.R.; Coon, T.A.; Mallampalli, R.K. Skp-cullin-F box E3 ligase component FBXL2 ubiquitinates Aurora B to inhibit tumorigenesis. Cell Death Dis. 2013, 4, e759. [Google Scholar] [CrossRef]
- Krupina, K.; Kleiss, C.; Metzger, T.; Fournane, S.; Schmucker, S.; Hofmann, K.; Fischer, B.; Paul, N.; Porter, I.M.; Raffelsberger, W.; Poch, O.; Swedlow, J.R.; Brino, L.; Sumara, I. Ubiquitin Receptor Protein UBASH3B Drives Aurora B Recruitment to Mitotic Microtubules. Dev. Cell. 2016, 36, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.L.; Yuan, Y.J.; Lv, J.; Qi, D.; Wu, M.D.; Lan, J.; Liu, S.N.; Yang, Y.; Zhai, J.; Jiang, H.M. Inhibition of BRD4 triggers cellular senescence through suppressing aurora kinases in oesophageal cancer cells. J. Cell Mol. Med. 2020, 24, 13036–13045. [Google Scholar] [CrossRef]
- Wang, C.; Chen, J.; Cao, W.; Sun, L.; Sun, H.; Liu, Y. Aurora-B and HDAC synergistically regulate survival and proliferation of lymphoma cell via AKT, mTOR and Notch pathways. Eur. J. Pharmacol. 2016, 779, 1–7. [Google Scholar] [CrossRef]
- Mo, F.; Zhuang, X.; Liu, X.; Yao, P.Y.; Qin, B.; Su, Z.; Zang, J.; Wang, Z.; Zhang, J.; Dou, Z.; Tian, C.; Teng, M.; Niu, L.; Hill, D.L.; Fang, G.; Ding, X.; Fu, C.; Yao, X. Acetylation of Aurora B by TIP60 ensures accurate chromosomal segregation. Nat. Chem. Biol. 2016, 12, 226–223. [Google Scholar] [CrossRef]
- Wu, T.; Zhao, B.; Cai, C.; Chen, Y.; Miao, Y.; Chu, J.; Sui, Y.; Li, F.; Chen, W.; Cai, Y.; Wang, F.; Jin, J. The Males Absent on the First (MOF) Mediated Acetylation Alters the Protein Stability and Transcriptional Activity of YY1 in HCT116 Cells. Int. J. Mol. Sci. 2023, 24, 8719. [Google Scholar] [CrossRef]
- Wei, T.; Liu, H.; Zhu, H.; Chen, W.; Wu, T.; Bai, Y.; Zhang, X.; Miao, Y.; Wang, F.; Cai, Y.; Jin, J. Two distinct males absent on the first (MOF)-containing histone acetyltransferases are involved in the epithelial-mesenchymal transition in different ways in human cells. Cell Mol. Life Sci. 2022, 79, 238. [Google Scholar] [CrossRef] [PubMed]
- Valerio, D.G.; Xu, H.; Eisold, M.E.; Woolthuis, C.M.; Pandita, T.K.; Armstrong, S.A. Histone acetyltransferase activity of MOF is required for adult but not early fetal hematopoiesis in mice. Blood. 2017, 129, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Rohrberg, J.; Van de Mark, D.; Amouzgar, M.; Lee, J.V.; Taileb, M.; Corella, A.; Kilinc, S.; Williams, J.; Jokisch, M.L.; Camarda, R.; Balakrishnan, S.; Shankar, R.; Zhou, A.; Chang, A.N.; Chen, B.; Rugo, H.S.; Dumont, S.; Goga, A. MYC Dysregulates Mitosis, Revealing Cancer Vulnerabilities. Cell Rep. 2020, 30, 3368–3382. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, J.; Yue, M.; Cai, X.; Wang, T.; Wu, C.; Su, H.; Wang, Y.; Han, M.; Zhang, Y.; Zhu, X.; Jiang, P.; Li, P.; Sun, Y.; Xiao, W.; Feng, H.; Qing, G.; Liu, H. Direct Phosphorylation and Stabilization of MYC by Aurora B Kinase Promote T-cell Leukemogenesis. Cancer Cell. 2020, 37, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, X.; Zhang, Y.; Cheng, Y.; Li, Y. RNAi screening identifies KAT8 as a key molecule important for cancer cell survival. Int. J. Clin. Exp. Pathol. 2013, 6, 870–877. [Google Scholar]
- Tanaka, K.; Helena, A.Y.; Yang, S.; Han, S.; Selcuklu, S.D.; Kim, K.; Ramani, S.; Ganesan, Y.T.; Moyer, A.; Sinha, S.; Xie, Y.; Ishizawa, K.; Osmanbeyoglu, H.U.; Lyu, Y.; Roper, N.; Guha, U.; Rudin, C.M.; Kris, M.G.; Hsieh, J.J.; Cheng, E.H. Targeting Aurora B kinase prevents and overcomes resistance to EGFR inhibitors in lung cancer by enhancing BIM- and PUMA-mediated apoptosis. Cancer Cell. 2021, 39, 1245–1261. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, F.; Ward, T.; Yan, F.; Wu, Q.; Wang, Z.; McGlothen, T.; Peng, W.; You, T.; Sun, M.; Cui, T.; Hu, R.; Dou, Z.; Zhu, J.; Xie, W.; Rao, Z.; Ding, X.; Yao, X. Phosphorylation of HsMis13 by Aurora B kinase is essential for assembly of functional kinetochore. J. Biol. Chem. 2008, 283, 26726–26736. [Google Scholar] [CrossRef]
- Cimini, D.; Wan, X.; Hirel, C.B.; Salmon, E.D. Aurora kinase promotes turnover of kinetochore microtubules to reduce chromosome segregation errors. Curr. Biol. 2006, 16, 1711–1718. [Google Scholar] [CrossRef]
- Alushin, G.M.; Musinipally, V.; Matson, D.; Tooley, J.; Stukenberg, P.T.; Nogales, E. Multimodal microtubule binding by the Ndc80 kinetochore complex. Nat. Struct. Mol. Biol. 2012, 19, 1161–1167. [Google Scholar] [CrossRef]
- Umbreit, N.T.; Gestaut, D.R.; Tien, J.F.; Vollmar, B.S.; Gonen, T.; Asbury, C.L.; Davis, T.N. The Ndc80 kinetochore complex directly modulates microtubule dynamics. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 16113–1611. [Google Scholar] [CrossRef]
- Muir, K.W.; Batters, C.; Dendooven, T.; Yang, J.; Zhang, Z.; Burt, A.; Barford, D. Structural mechanism of outer kinetochore Dam1-Ndc80 complex assembly on microtubules. Science. 2023, 382, 1184–1119. [Google Scholar] [CrossRef]
- Ahmed, A.; Shamsi, A.; Mohammad, T.; Hasan, G.M.; Islam, A.; Hassan, M.I. Aurora B kinase: a potential drug target for cancer therapy. J. Cancer Res. Clin. Oncol. 2021, 147, 2187–2198. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, Y.; Zhang, C.; Zhang, Q. Role of aurora kinase B in regulating resistance to paclitaxel in breast cancer cells. Hum. Cell. 2022, 35, 678–693. [Google Scholar] [CrossRef]
- Al-Khafaji, A.S.; Davies, M.P.; Risk, J.M.; Marcus, M.W.; Koffa, M.; Gosney, J.R.; Shaw, R.J.; Field, J.K.; Liloglou, T. Aurora B expression modulates paclitaxel response in non-small cell lung cancer. Br. J. Cancer. 2017, 116, 592–599. [Google Scholar] [CrossRef]
- Hartsink-Segers, S.A.; Zwaan, C.M.; Exalto, C.; Luijendijk, M.W.; Calvert, V.S.; Petricoin, E.F.; Evans, W.E.; Reinhardt, D.; de Haas, V.; Hedtjärn, M.; Hansen, B.R.; Koch, T.; Caron, H.N.; Pieters, R.; Den Boer, M.L. Aurora kinases in childhood acute leukemia: the promise of aurora B as therapeutic target. Leukemia. 2013, 27, 560–568. [Google Scholar] [CrossRef]
- Lin, Z.Z.; Jeng, Y.M.; Hu, F.C.; Pan, H.W.; Tsao, H.W.; Lai, P.L.; Lee, P.H.; Cheng, A.L.; Hsu, H.C. Significance of Aurora B overexpression in hepatocellular carcinoma. Aurora B Overexpression in HCC. BMC. Cancer. 2010, 10, 461. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC on the path to cancer. Cell. 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, C.; Kalkat, M.; Houlahan, K.E.; De Melo, J.; Longo, J.; Done, S.J.; Boutros, P.C.; Penn, L.Z. Modelling the MYC-driven normal-to-tumour switch in breast cancer. Dis. Model. Mech. 2019, 12, dmm038083. [Google Scholar] [CrossRef]
- Li, M.; Li, A.; Zhou, S.; Lv, H.; Yang, W. SPAG5 upregulation contributes to enhanced c-MYC transcriptional activity via interaction with c-MYC binding protein in triple-negative breast cancer. J. Hematol. Oncol. 2019, 12, 14. [Google Scholar] [CrossRef]
- Gao, F.Y.; Li, X.T.; Xu, K.; Wang, R.T.; Guan, X.X. c-MYC mediates the crosstalk between breast cancer cells and tumor microenvironment. Cell Commun. Signal. 2023, 21, 28. [Google Scholar] [CrossRef]
Figure 1.
Crosstalk between the MOF/MSL complex and CPC in 293T cells. (A) Summary of MSL1-KO-based mass spectrometry data. (B) Volcano plot of the MSL1-KO protein expression profile, highlighting the downregulation of AURKB, INCENP, and BIRC5. (C) MOF and MSL1 Overexpression increases endogenous AURKB protein levels and kinase activity. 293T cells were transfected with Flag-tagged MOF or MSL1 for 48 hours, and AURKB levels and H3S10 phosphorylation (H3S10p) were detected by Western blot. (D) MOF Knockdown reduces AURKB protein and H3S10p levels. (E,F) Relative AURKB mRNA levels, measured by RT-qPCR, remain unchanged following MOF/MSL1 overexpression or knockdown. GAPDH was used for normalization. (G) MSL1 overexpression restores AURKB, BIRC5, CDCA8, and INCENP protein levels in MSL1-KO cells. (H–K) AURKB protein Half-life assessment. 293T cells transfected with shNT, shMOF, or shMSL1 were treated with 20 μg/mL CHX, and AURKB protein levels were analyzed at 0, 1, 2, 3, 4, and 5 hours. GAPDH and H3 were used as internal controls. (L) In vitro phosphorylation assay: Flag IP was performed in cells overexpressing Flag-AURKB, followed by an in vitro phosphorylation assay using Flag IP eluates, E. coli-expressed/purified His-INCENP, and ATP. Phosphorylated INCENP was detected using an anti-Pan-phospho antibody. (M) Myc-MSL1 overexpression enhances INCENP phosphorylation. GAPDH, β-tubulin, and H3 were used as internal controls throughout.
Figure 1.
Crosstalk between the MOF/MSL complex and CPC in 293T cells. (A) Summary of MSL1-KO-based mass spectrometry data. (B) Volcano plot of the MSL1-KO protein expression profile, highlighting the downregulation of AURKB, INCENP, and BIRC5. (C) MOF and MSL1 Overexpression increases endogenous AURKB protein levels and kinase activity. 293T cells were transfected with Flag-tagged MOF or MSL1 for 48 hours, and AURKB levels and H3S10 phosphorylation (H3S10p) were detected by Western blot. (D) MOF Knockdown reduces AURKB protein and H3S10p levels. (E,F) Relative AURKB mRNA levels, measured by RT-qPCR, remain unchanged following MOF/MSL1 overexpression or knockdown. GAPDH was used for normalization. (G) MSL1 overexpression restores AURKB, BIRC5, CDCA8, and INCENP protein levels in MSL1-KO cells. (H–K) AURKB protein Half-life assessment. 293T cells transfected with shNT, shMOF, or shMSL1 were treated with 20 μg/mL CHX, and AURKB protein levels were analyzed at 0, 1, 2, 3, 4, and 5 hours. GAPDH and H3 were used as internal controls. (L) In vitro phosphorylation assay: Flag IP was performed in cells overexpressing Flag-AURKB, followed by an in vitro phosphorylation assay using Flag IP eluates, E. coli-expressed/purified His-INCENP, and ATP. Phosphorylated INCENP was detected using an anti-Pan-phospho antibody. (M) Myc-MSL1 overexpression enhances INCENP phosphorylation. GAPDH, β-tubulin, and H3 were used as internal controls throughout.
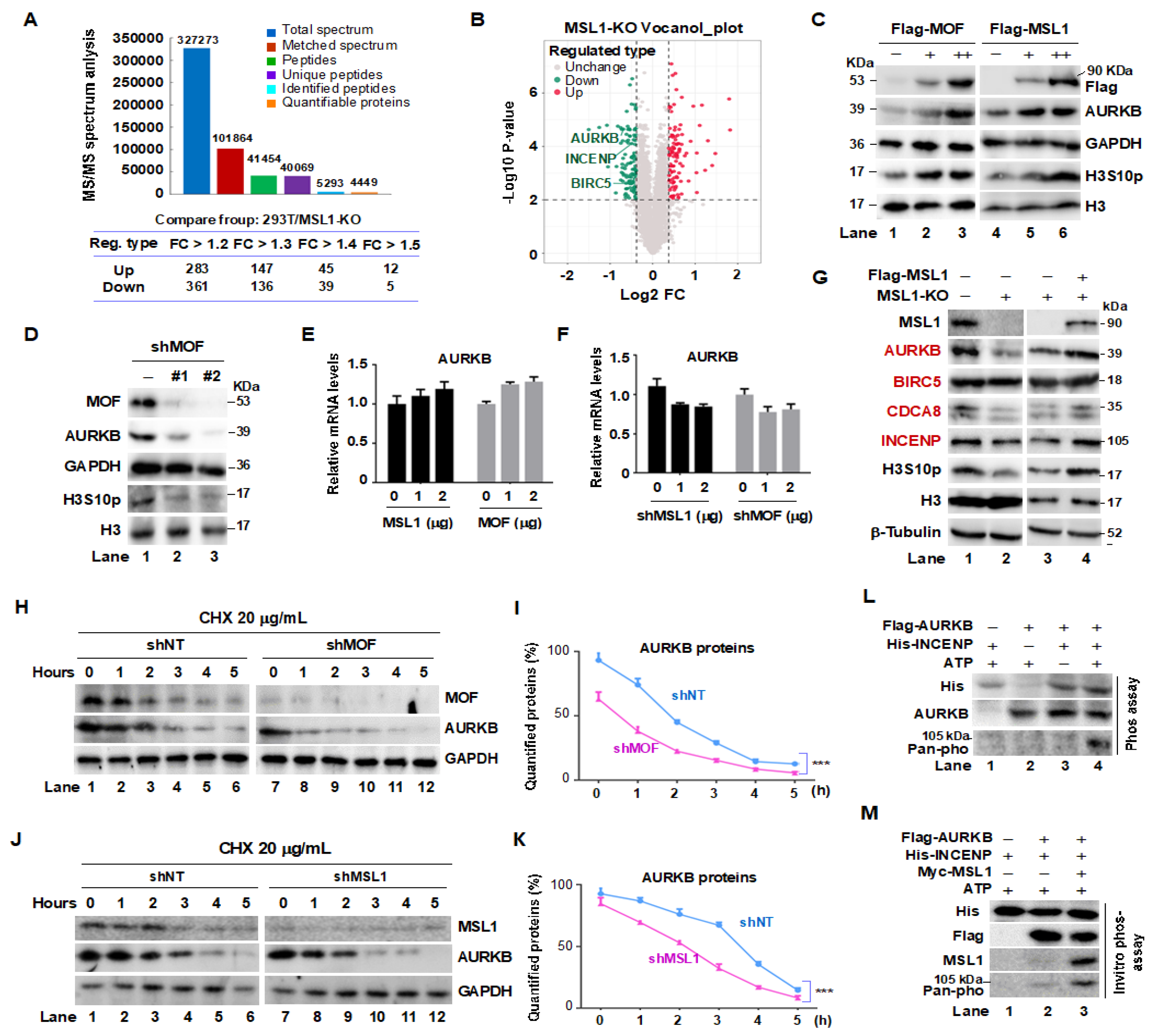
Figure 2.
Interaction and co-distribution of MOF/MSL1 and AURKB during mitosis in HeLa cells. (A–C) Interaction between the MOF/MSL complex and AURKB. Endogenous MOF, MSL1, and AURKB proteins bound to Flag-AURKB, Flag-MOF, or Flag-MSL1 were identified through Flag IP in 293T cells. (D, E) Co-transfection and Co-IP experiments further validate the interaction between AURKB and the MOF/MSL complex in 293T cells. (F, G) Domain mapping analysis. MOF deletion mutants were generated based on distinct structural domains (upper panel). Full-length Flag-AURKB and MOF truncations were transfected into 293T cells. After 48 hours, Flag IP was conducted, and bound proteins were analyzed by Western blot. (H–J) IF staining showing the subcellular localization of AURKB, MOF, and MSL1 (green) at different mitotic phases. β-tubulin (red) marks the mitotic phases, and DAPI (blue) stains nuclei. Scale bar: 20 μm. Images were captured with a 40× objective. (K,L) Co-IP assays confirm interactions between the MOF/MSL1 complex and the CPC. 293T cells were co-transfected with Flag-INCENP and Myc-tagged MOF or MSL1. 48 hours later, Flag IP was performed, and bound proteins, including phospho-INCENP, were detected by Western blot. (M) MSL1 Knockdown destabilizes the CPC. Flag IP was carried out in cells overexpressing Flag-INCENP with or without shMSL1. Input and IP eluates were analyzed for Western blot for the indicated proteins.
Figure 2.
Interaction and co-distribution of MOF/MSL1 and AURKB during mitosis in HeLa cells. (A–C) Interaction between the MOF/MSL complex and AURKB. Endogenous MOF, MSL1, and AURKB proteins bound to Flag-AURKB, Flag-MOF, or Flag-MSL1 were identified through Flag IP in 293T cells. (D, E) Co-transfection and Co-IP experiments further validate the interaction between AURKB and the MOF/MSL complex in 293T cells. (F, G) Domain mapping analysis. MOF deletion mutants were generated based on distinct structural domains (upper panel). Full-length Flag-AURKB and MOF truncations were transfected into 293T cells. After 48 hours, Flag IP was conducted, and bound proteins were analyzed by Western blot. (H–J) IF staining showing the subcellular localization of AURKB, MOF, and MSL1 (green) at different mitotic phases. β-tubulin (red) marks the mitotic phases, and DAPI (blue) stains nuclei. Scale bar: 20 μm. Images were captured with a 40× objective. (K,L) Co-IP assays confirm interactions between the MOF/MSL1 complex and the CPC. 293T cells were co-transfected with Flag-INCENP and Myc-tagged MOF or MSL1. 48 hours later, Flag IP was performed, and bound proteins, including phospho-INCENP, were detected by Western blot. (M) MSL1 Knockdown destabilizes the CPC. Flag IP was carried out in cells overexpressing Flag-INCENP with or without shMSL1. Input and IP eluates were analyzed for Western blot for the indicated proteins.
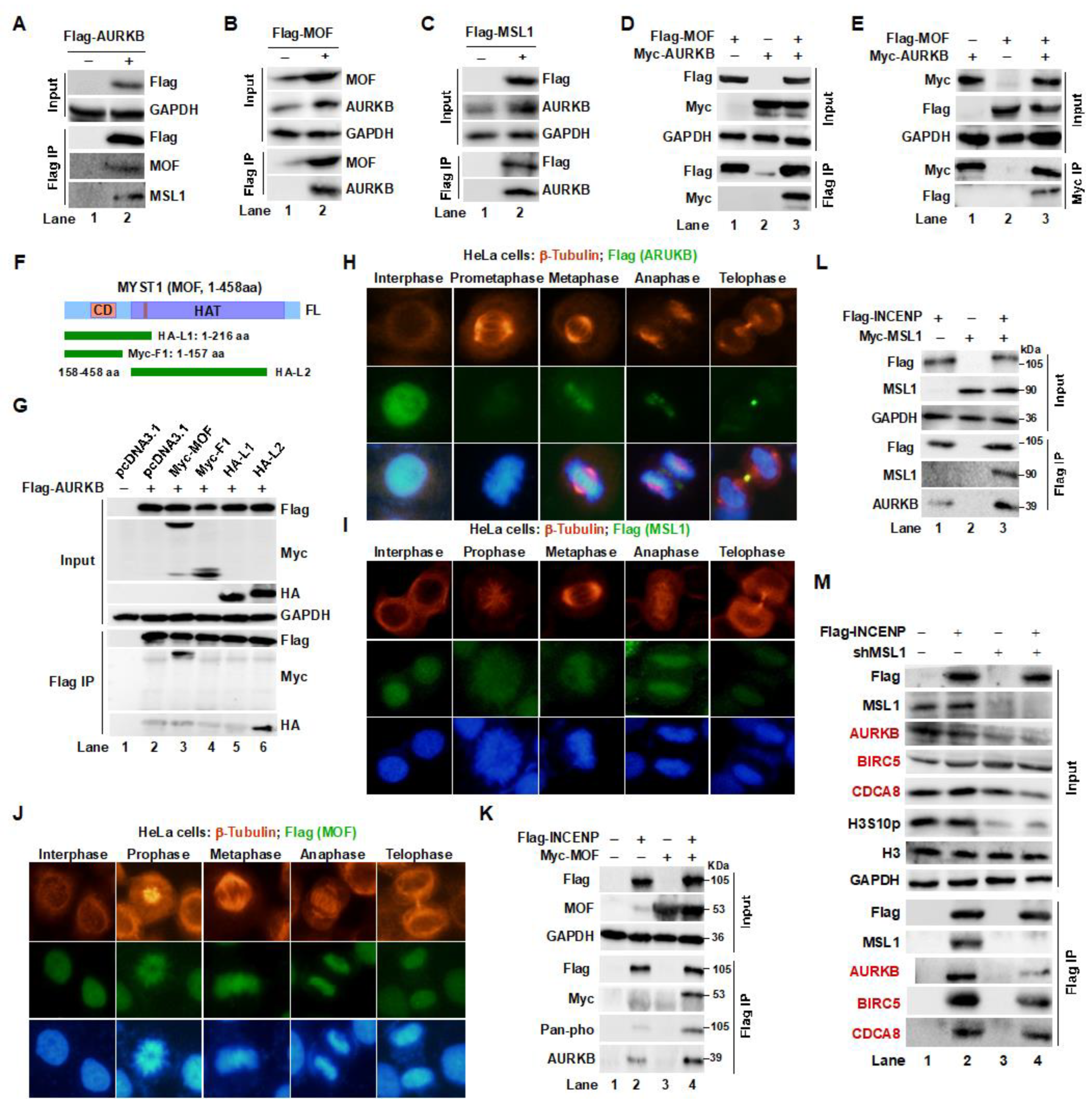
Figure 3.
Acetylation of AURKB by the MOF/MSL1 complex stabilizes the CPC in 293T cells. (A, B) MOF/MSL1 complex stabilizes AURKB. Acetylation and stability of AURKB were assessed following co-transfection of Flag-AURKB with Myc-tagged MOF or MSL1, in the presence of HA-Ubiquitin and the proteasome inhibitor MG132, followed by Flag IP. (C) AURKB instability in 293T cells upon MOF knockdown (shMOF). (D) The MOF G327E mutant, which lacks enzymatic activity, failed to acetylate AURKB. (E) In vitro lysine acetyltransferase (KAT) assay. Recombinant His-AURKB protein was expressed and purified from E. coli. The KAT assay was performed by incubating recombinant His-AURKB with Flag IP eluates from MOF-overexpressing 293T cells and acetyl coenzyme-A (AcCoA) (upper panel). Acetylation of His-AURKB was assessed by Western blot using an anti-Pan-ac antibody (lower panel, lane 4). (F, G) Positive regulation of AURKB by MOF/MSL1. The effects of MOF on Flag-AURKB were analyzed in 293T cells with MSL1 overexpression or knockdown. Protein levels in the input and Flag IP eluates were detected by Western blot using specific antibodies. (H) Role of MSL1 in CPC complex integrity. Myc-AURKB and Flag-INCENP were co-transfected into MSL1-KO 293T cells. The interaction between AURKB and INCENP, as well as the acetylation, phosphorylation, and ubiquitination levels of AURKB and H3S10p, were analyzed by Western blot using specific antibodies.
Figure 3.
Acetylation of AURKB by the MOF/MSL1 complex stabilizes the CPC in 293T cells. (A, B) MOF/MSL1 complex stabilizes AURKB. Acetylation and stability of AURKB were assessed following co-transfection of Flag-AURKB with Myc-tagged MOF or MSL1, in the presence of HA-Ubiquitin and the proteasome inhibitor MG132, followed by Flag IP. (C) AURKB instability in 293T cells upon MOF knockdown (shMOF). (D) The MOF G327E mutant, which lacks enzymatic activity, failed to acetylate AURKB. (E) In vitro lysine acetyltransferase (KAT) assay. Recombinant His-AURKB protein was expressed and purified from E. coli. The KAT assay was performed by incubating recombinant His-AURKB with Flag IP eluates from MOF-overexpressing 293T cells and acetyl coenzyme-A (AcCoA) (upper panel). Acetylation of His-AURKB was assessed by Western blot using an anti-Pan-ac antibody (lower panel, lane 4). (F, G) Positive regulation of AURKB by MOF/MSL1. The effects of MOF on Flag-AURKB were analyzed in 293T cells with MSL1 overexpression or knockdown. Protein levels in the input and Flag IP eluates were detected by Western blot using specific antibodies. (H) Role of MSL1 in CPC complex integrity. Myc-AURKB and Flag-INCENP were co-transfected into MSL1-KO 293T cells. The interaction between AURKB and INCENP, as well as the acetylation, phosphorylation, and ubiquitination levels of AURKB and H3S10p, were analyzed by Western blot using specific antibodies.
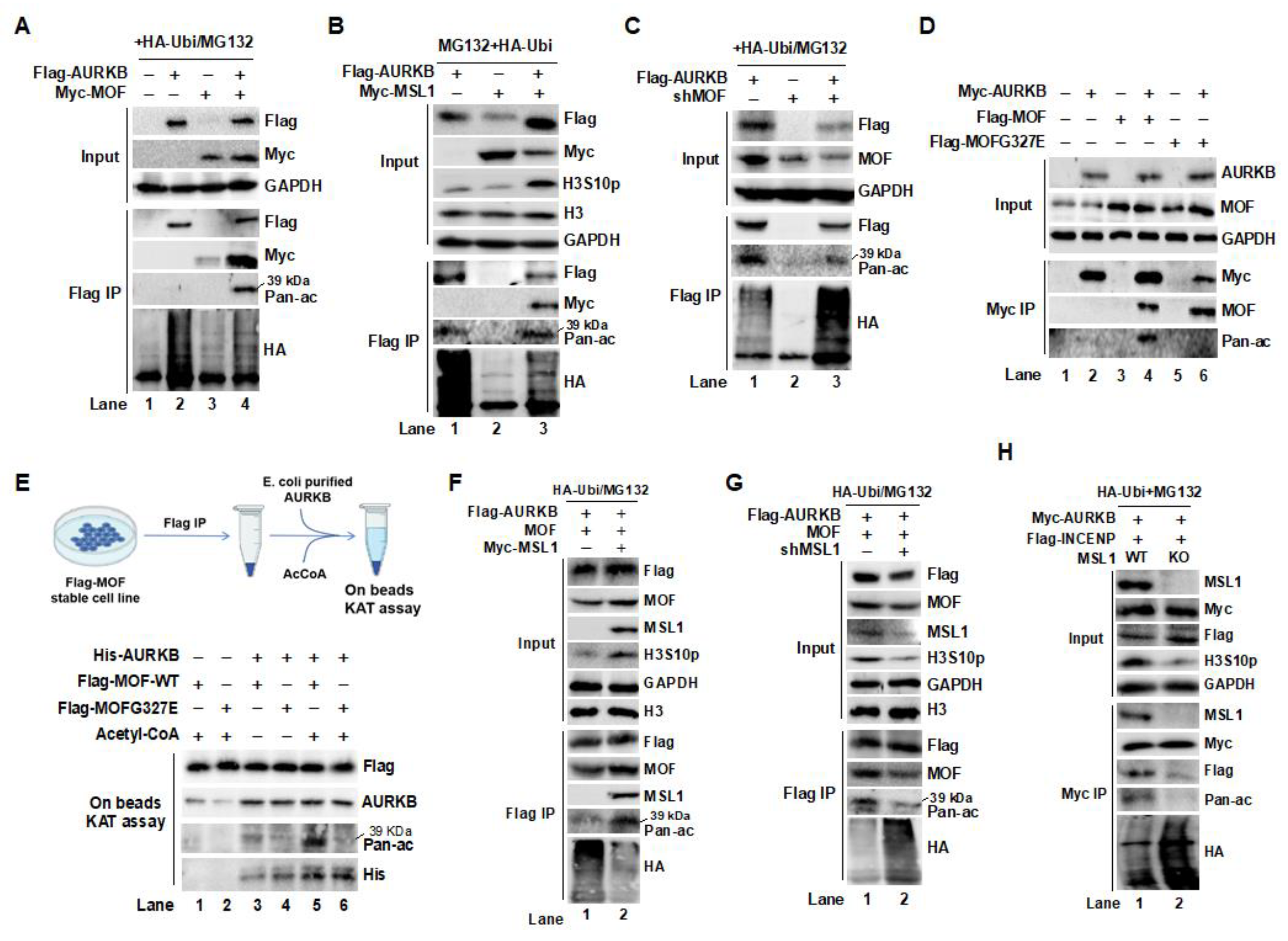
Figure 4.
Lysine 215 is the primary acetylation site targeted by the MOF/MSL complex on AURKB in 293T cells. (A) 3D docking analysis of MOF (pink) and AURKB (green) reveals binding sites, with a zoomed-in view in the right panel. Hydrogen bonds between the two proteins are indicated by yellow dashed lines. (B) Prediction of AURKB K215 acetylation based on the PhosphoSitePlus® database. (C) Flag IP and Western blot analysis confirm AURKB K215 as the primary acetylation site targeted by the MOF/MSL complex. (D) Mutation of the K215 site significantly reduces AURKB protein half-life. (E) Acetylation of AURKB at K215 is essential for maintaining its stability and kinase activity. (F) MOF stabilizes AURKB by acetylating the K215 site, preventing its degradation. (G) The AURKB K215R (non-acetylatable) mutant retains kinase activity on its substrate INCENP, compared to the WT and acetylation-mimic mutant AURKB (K215Q). (H) The K215R mutation disrupts AURKB’s ability to form the CPC and reduces the expression of key complex components, including INCENP, CDCA8, and BIR5. Additionally, the K215R mutation decreases c-MYC expression while increasing phosphorylation of c-MYC at T58, a modification associated with c-MYC degradation. GAPDH and H3 were used as internal controls. (I) The MSL/MOF complex acetylates AURKB at K215, which is crucial for maintaining CPC integrity and AURKB activity.
Figure 4.
Lysine 215 is the primary acetylation site targeted by the MOF/MSL complex on AURKB in 293T cells. (A) 3D docking analysis of MOF (pink) and AURKB (green) reveals binding sites, with a zoomed-in view in the right panel. Hydrogen bonds between the two proteins are indicated by yellow dashed lines. (B) Prediction of AURKB K215 acetylation based on the PhosphoSitePlus® database. (C) Flag IP and Western blot analysis confirm AURKB K215 as the primary acetylation site targeted by the MOF/MSL complex. (D) Mutation of the K215 site significantly reduces AURKB protein half-life. (E) Acetylation of AURKB at K215 is essential for maintaining its stability and kinase activity. (F) MOF stabilizes AURKB by acetylating the K215 site, preventing its degradation. (G) The AURKB K215R (non-acetylatable) mutant retains kinase activity on its substrate INCENP, compared to the WT and acetylation-mimic mutant AURKB (K215Q). (H) The K215R mutation disrupts AURKB’s ability to form the CPC and reduces the expression of key complex components, including INCENP, CDCA8, and BIR5. Additionally, the K215R mutation decreases c-MYC expression while increasing phosphorylation of c-MYC at T58, a modification associated with c-MYC degradation. GAPDH and H3 were used as internal controls. (I) The MSL/MOF complex acetylates AURKB at K215, which is crucial for maintaining CPC integrity and AURKB activity.
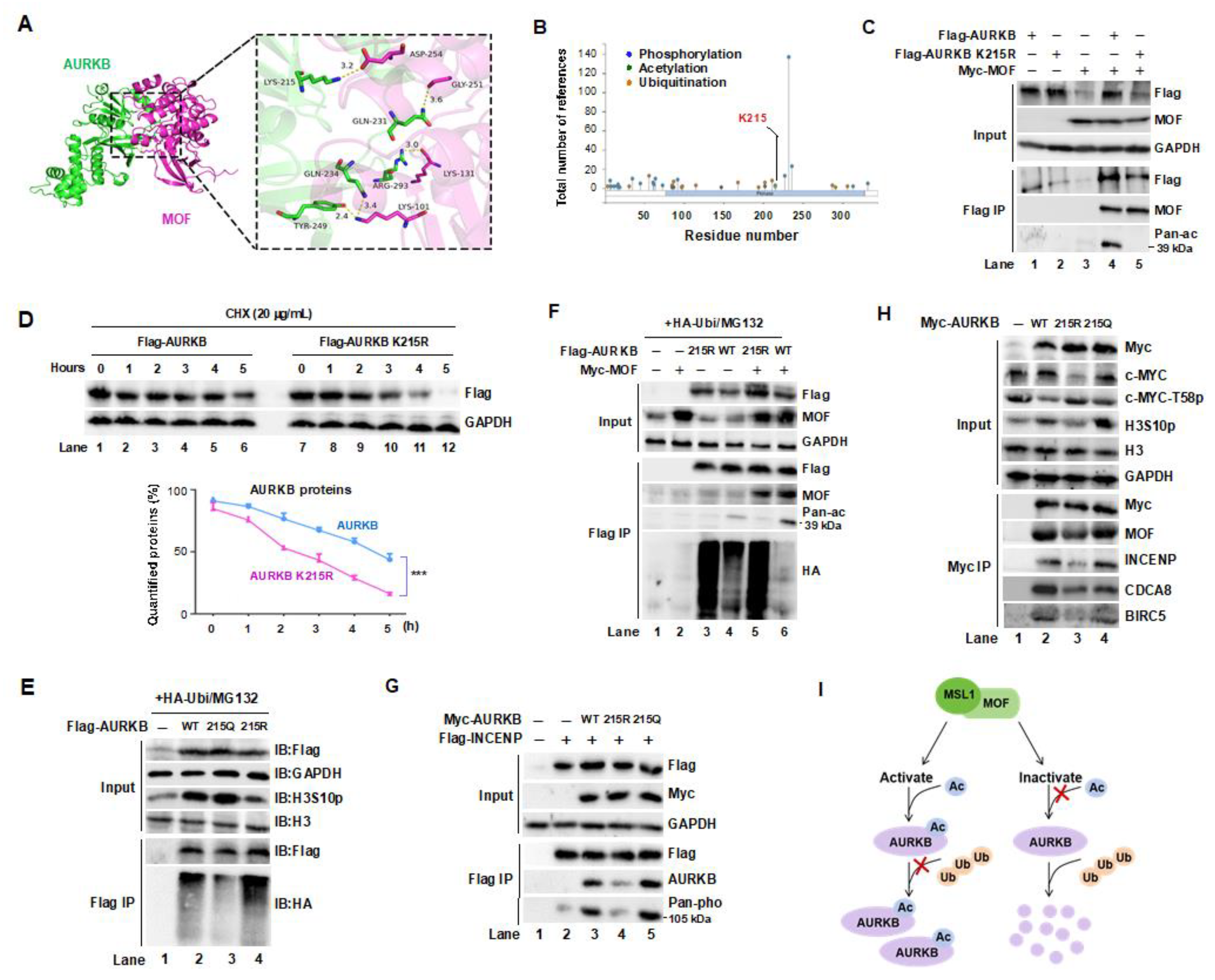
Figure 5.
MOF/MSL complex-mediated acetylation of AURKB at K215 regulates G2/M phase progression. (A,B) Immunofluorescence (IF) staining of β-Tubulin (red or green), AURKB (red), H3S10p (green) in HeLa cells treated with shMOF (A) or shMSL1 (B). Nuclei were stained with DAPI (blue). Scale bars: 200 μm. (C) IF staining of β-Tubulin (red) and AURKB (green) in MCF-7 cells treated with shMOF. Nuclei were stained with DAPI (blue). Scale bars:100 μm. (D) IF staining of β-Tubulin (red) in HeLa cells overexpressing wild-type (WT) or K215R mutant AURKB Nuclei were stained with DAPI (blue). Scale bars:20 μm. (E–H) Quantification of spindle multipolar cells corresponding to panels A D, respectively. (I,J) Validation of MOF and MSL1 knockdown efficiency in the experiments shown in panels A C. (K) Transient transfection of the AURKB K215R mutant in HeLa cells delayed cell cycle progression compared to the WT AURKB. Effect of MOF overexpression on AURKB protein levels in MCF7 and MDA-MB-231 cells. (L) Subcellular fractionation of HeLa cells arrested in the S and M phases using hydroxyurea (HU) and nocodazole, respectively. Cytoplasmic and nuclear fractions were separated by centrifugation and analyzed by Western blot. GAPDH was used as a cytoplasmic marker. (M) Flow cytometry analysis of cell cycle progression. MCF-7 and MDA_MB-231 cells were transfected with wild-type or mutant AURKB were analyzed in AURKB knockdown backgrounds. (N) Co-localization of MOF, MSL1, and AURKB across different cell cycle phases. HeLa cells were synchronized at specific phases: (1) G1 phase by serum starvation (24 hours); (2) S phase by HU treatment (1 mM, 24 hours); (3) G2/M phase by nocodazole treatment (500 ng/mL, 16 hours); (4) M phase by release from nocodazole arrest (1 hour).
Figure 5.
MOF/MSL complex-mediated acetylation of AURKB at K215 regulates G2/M phase progression. (A,B) Immunofluorescence (IF) staining of β-Tubulin (red or green), AURKB (red), H3S10p (green) in HeLa cells treated with shMOF (A) or shMSL1 (B). Nuclei were stained with DAPI (blue). Scale bars: 200 μm. (C) IF staining of β-Tubulin (red) and AURKB (green) in MCF-7 cells treated with shMOF. Nuclei were stained with DAPI (blue). Scale bars:100 μm. (D) IF staining of β-Tubulin (red) in HeLa cells overexpressing wild-type (WT) or K215R mutant AURKB Nuclei were stained with DAPI (blue). Scale bars:20 μm. (E–H) Quantification of spindle multipolar cells corresponding to panels A D, respectively. (I,J) Validation of MOF and MSL1 knockdown efficiency in the experiments shown in panels A C. (K) Transient transfection of the AURKB K215R mutant in HeLa cells delayed cell cycle progression compared to the WT AURKB. Effect of MOF overexpression on AURKB protein levels in MCF7 and MDA-MB-231 cells. (L) Subcellular fractionation of HeLa cells arrested in the S and M phases using hydroxyurea (HU) and nocodazole, respectively. Cytoplasmic and nuclear fractions were separated by centrifugation and analyzed by Western blot. GAPDH was used as a cytoplasmic marker. (M) Flow cytometry analysis of cell cycle progression. MCF-7 and MDA_MB-231 cells were transfected with wild-type or mutant AURKB were analyzed in AURKB knockdown backgrounds. (N) Co-localization of MOF, MSL1, and AURKB across different cell cycle phases. HeLa cells were synchronized at specific phases: (1) G1 phase by serum starvation (24 hours); (2) S phase by HU treatment (1 mM, 24 hours); (3) G2/M phase by nocodazole treatment (500 ng/mL, 16 hours); (4) M phase by release from nocodazole arrest (1 hour).
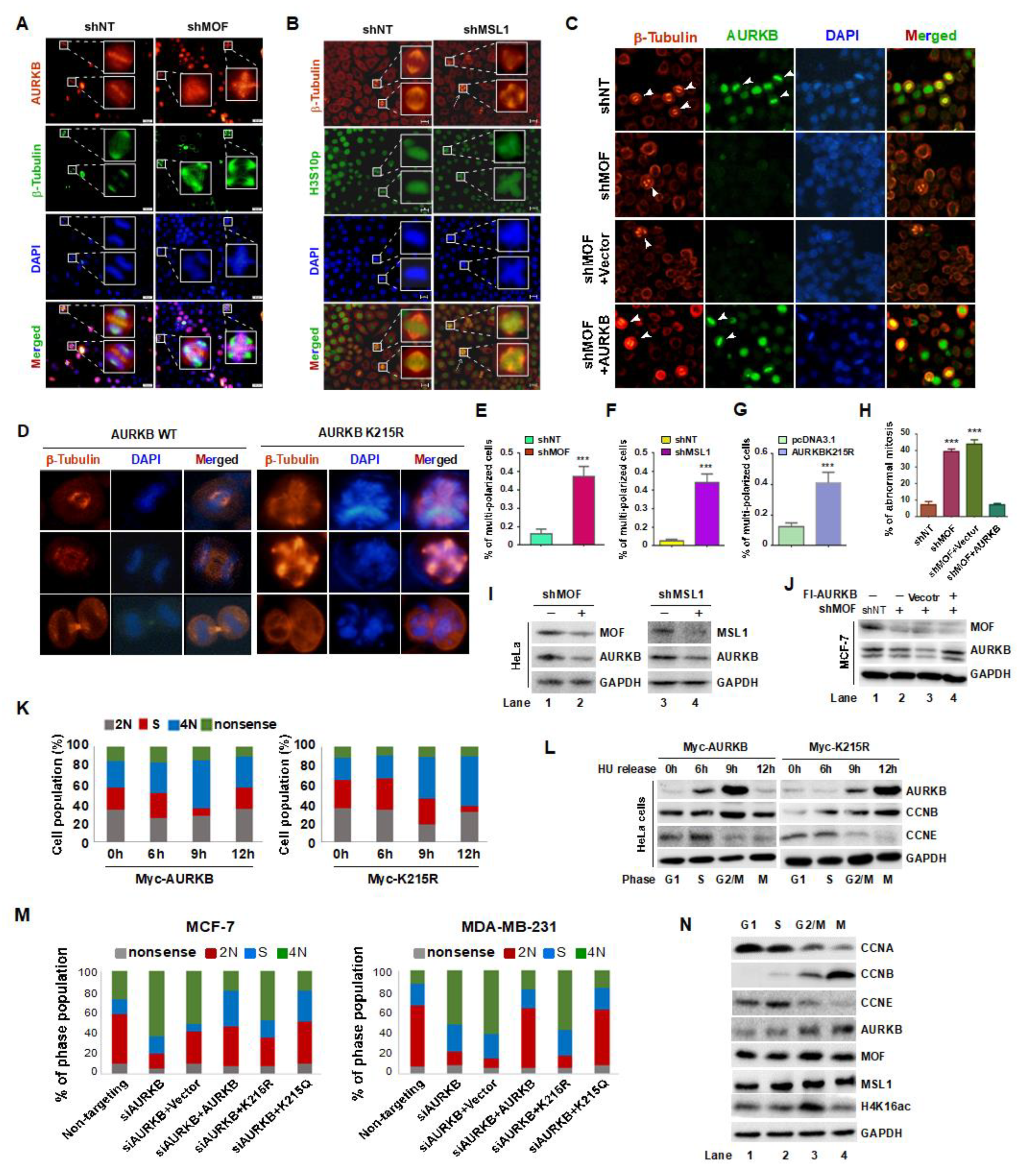
Figure 6.
MOF-mediated acetylation of AURKB at K215 regulates the proliferation of MCF-7 and MDA-MB-231 cells. (A) EdU incorporation assay to assess cell proliferation. Cells were incubated with EdU for 2 hours and analyzed using the BeyoCleck™ EdU Cell Proliferation Kit (C0071s). Scale bar: 50 μm. (B) Quantification of EdU-positive cells. (C) CCK-8 assay to evaluate cell viability. (D) Colony formation assays, with colonies stained using crystal violet. (E) Quantification of colony formation efficiency. (F) Generation of stable MDA-MB-231 cell lines expressing Flag-tagged AURKB or the AKRKB K215R mutant. (G) Schematic representation of the animal experiment design. (H) Tumor growth progression over time. (I) Representative images of tumors at the end of the experiment (n = 5 per group). (J) Tumor weights at the experiments termination in mice injected with AURKB or AURKB K215R-expressing cells. ***p < 0.001 compared to the AURKB group. (K) Western blot analysis of protein expression in whole-cell lysates from four tumors per group.
Figure 6.
MOF-mediated acetylation of AURKB at K215 regulates the proliferation of MCF-7 and MDA-MB-231 cells. (A) EdU incorporation assay to assess cell proliferation. Cells were incubated with EdU for 2 hours and analyzed using the BeyoCleck™ EdU Cell Proliferation Kit (C0071s). Scale bar: 50 μm. (B) Quantification of EdU-positive cells. (C) CCK-8 assay to evaluate cell viability. (D) Colony formation assays, with colonies stained using crystal violet. (E) Quantification of colony formation efficiency. (F) Generation of stable MDA-MB-231 cell lines expressing Flag-tagged AURKB or the AKRKB K215R mutant. (G) Schematic representation of the animal experiment design. (H) Tumor growth progression over time. (I) Representative images of tumors at the end of the experiment (n = 5 per group). (J) Tumor weights at the experiments termination in mice injected with AURKB or AURKB K215R-expressing cells. ***p < 0.001 compared to the AURKB group. (K) Western blot analysis of protein expression in whole-cell lysates from four tumors per group.
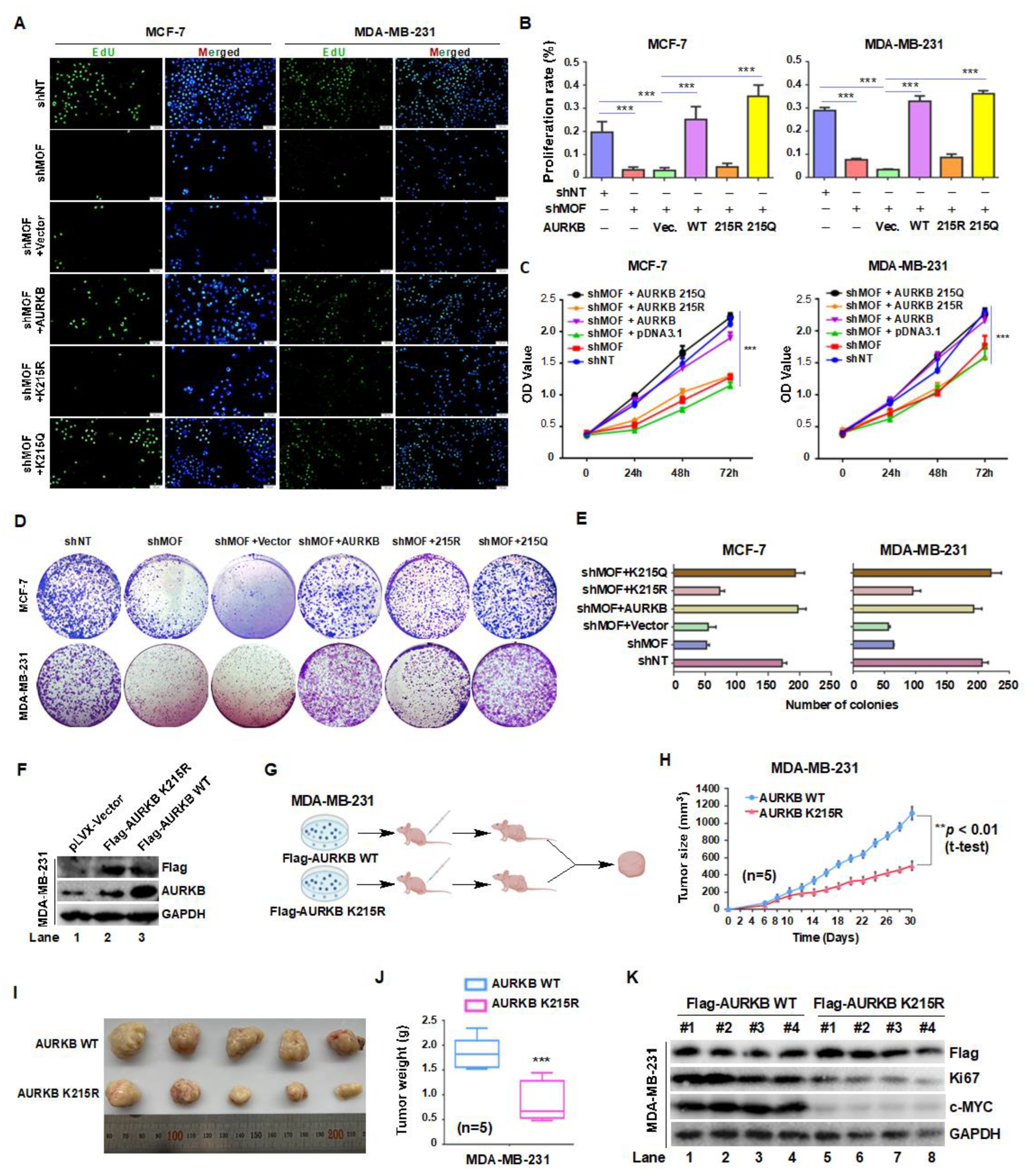
Figure 7.
The MOF-AURKB K215ac axis promotes breast cancer cell proliferation by stabilizing c-MYC protein. (A,B) Western blot analysis of the indicated protein expression levels in MCF-7 and MDA-MB-231 cells transfected with either shNT or shAURKB. (C,D) Venn diagram showing the intersection of data from BioGRID ORCS, STRING and TCGA Databases, identifying AURKB downstream targets involved in breast cancer proliferation. (E) Treatment of MCF-7 cells with the AURKB kinase inhibitor AZD1152 (10, 20, and 40 nM for 24 hours) resulted in decreased in c-MYC- levels. (F) Inhibition of MOF activity using the enzyme inhibitor MG149 reduced the expression of AURKB, c-MYC, and Ki67 in MCF7 cells. (G,H) Effects of overexpressing WT, K215R, and K215Q AURKB in MCF-7 and MDA-MB-231 cells on c-MYC levels following AURKB knockdown. GAPDH and histone H3 were used as internal controls. (I,J) Quantification of c-MYC protein levels. ***P < 0.001 compared to non-targeting siRNA group. (K,L) Effects of overexpressing WT, K215R, and K215Q AURKB on c-MYC, H3S10p, and AURKB levels following MOF knockdown. (M,N) Quantification of c-MYC protein levels. ***P < 0.001 compared to non-targeting shRNA group.
Figure 7.
The MOF-AURKB K215ac axis promotes breast cancer cell proliferation by stabilizing c-MYC protein. (A,B) Western blot analysis of the indicated protein expression levels in MCF-7 and MDA-MB-231 cells transfected with either shNT or shAURKB. (C,D) Venn diagram showing the intersection of data from BioGRID ORCS, STRING and TCGA Databases, identifying AURKB downstream targets involved in breast cancer proliferation. (E) Treatment of MCF-7 cells with the AURKB kinase inhibitor AZD1152 (10, 20, and 40 nM for 24 hours) resulted in decreased in c-MYC- levels. (F) Inhibition of MOF activity using the enzyme inhibitor MG149 reduced the expression of AURKB, c-MYC, and Ki67 in MCF7 cells. (G,H) Effects of overexpressing WT, K215R, and K215Q AURKB in MCF-7 and MDA-MB-231 cells on c-MYC levels following AURKB knockdown. GAPDH and histone H3 were used as internal controls. (I,J) Quantification of c-MYC protein levels. ***P < 0.001 compared to non-targeting siRNA group. (K,L) Effects of overexpressing WT, K215R, and K215Q AURKB on c-MYC, H3S10p, and AURKB levels following MOF knockdown. (M,N) Quantification of c-MYC protein levels. ***P < 0.001 compared to non-targeting shRNA group.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



