Submitted:
10 June 2025
Posted:
12 June 2025
Read the latest preprint version here
Abstract
ERCC6, also known as CSB (Cockayne Syndrome B), is a key protein involved in transcription-coupled nucleotide excision repair (TC-NER), a DNA repair process that removes lesions blocking RNA polymerase. ERCC6’s multifaceted roles include chromatin remodeling, transcription regulation, oxidative stress response, and coordination with other DNA repair proteins. Mutations in ERCC6 lead to Cockayne Syndrome and other neurodegenerative disorders, but some variants, such as M1097V, have been associated with cancer risk, particularly in African American (AA) populations. Recent studies have explored the functional impact of ERCC6 variants in prostate cancer (PCa), especially among AAs, who face higher incidence and more aggressive forms of the disease. A notable finding is that the M1097V variant increases cellular tolerance to UV damage, suggesting a possible evolutionary benefit but also a potential risk for mutagenesis when exposed to complex environmental carcinogens. Other ERCC6 mutations, such as S636N, located near regulatory regions, may alter repair activity, though their effects remain unclear. Given the high mutation burden in mismatch repair (MMR) and NER genes observed in AA PCa patients, a synthetic lethality strategy targeting both TC-NER and homologous recombination repair (HRR) pathways could be effective. This includes combining agents like CPT (cisplatin) with inhibitors of RAD54, such as J54. These approaches may offer alternatives to androgen deprivation therapy (ADT), which is often ineffective in advanced or treatment-resistant PCa common among AA men. This work underscores the importance of integrating genetic, environmental, and therapeutic insights to address PCa disparities.
Keywords:
ERCC6
; DNA damage
; UV
; TC-NER
; Prostate Cancer
Introduction
Prostate cancer (PCa) is the most commonly diagnosed non-cutaneous malignancy and the second leading cause of cancer-related death among men in the United States. Notably, African American (AA) men bear a disproportionate burden of this disease compared to men of European ancestry (Caucasians - CC), exhibiting higher incidence rates, earlier onset, and increased mortality from disease that is refractory to treatment [1]. This is to some extent explained by genetic differences in some cases attributed to alterations of the “repairome” [2]. While socioeconomic, environmental, and healthcare access disparities contribute to these differences, accumulating evidence suggests that biological factors, including genomic and molecular alterations, may play a critical role in driving the aggressive phenotype observed in AA patients.
The DNA damage response and repair pathways (DDRR), collectively known as the "repairome," are essential for maintaining genomic stability and preventing malignant transformation. Defects in DNA repair mechanisms are a well-established hallmark of cancer, contributing to increased mutational burden, chromosomal instability, and therapeutic resistance. In prostate cancer, alterations in key DNA repair genes—such as BRCA1, BRCA2, ATM, and MLH1—have been associated with tumor progression, poor prognosis, and sensitivity to targeted therapies, including PARP inhibitors and platinum-based agents. Inflicting DNA damage and enhancing apoptosis of cancer cells are the mechanistic strategies of all radiotherapeutic (e.g., external beam radiation therapy, brachytherapy, radium- 223) and some chemotherapeutic (PARP inhibitors; topoisomerase inhibitors, platinum-based therapy [3], and DNA crosslinking agents) [4,5]. Relative success of PARPi for mCRPC patients with and without homologous recombination repair (HRR) mutations (PROfound [6] and TRITON2 [7]) is defining new biology and more effective treatment options for previously unmanageable PCa; and while cisplatin (CPT)-based therapy is currently not the treatment of choice for PCa, partly due to significant renal toxicity (which can be faithfully recapitulated in mice [8]), the current trend suggests that lower dosing in combination with inhibitors of DNA Damage Repair, encompassing TC-NER, could be quite effective [3].
Helicases are central to maintaining genomic integrity, mediating processes such as DNA replication, repair, transcription, and chromatin remodeling. Dysregulation of helicase activity has been implicated in various cancers, yet their specific contributions to prostate cancer remain underexplored. Preliminary studies suggest that certain helicases are overexpressed or mutated in prostate cancer and may influence key oncogenic pathways, including androgen receptor signaling, genomic instability, and cellular stress responses. Furthermore, therapies targeting key aspects of DNA Repair mechanisms, including NER, BER, and HRR with PARPis, particularly for PCa cases manifesting BRCAness, have proven to be the greatest clinical advancement since the use of ARSI. ERCC6 is a helicase whose primary function is mediating dislodging of Transcription Elongation Complexes (EC) stuck at DNA-distorting lesions. Mutations in this protein reduce the rate of rRNA synthesis, and cancer cells with functional knockout display detrimental growth effects. This protein also acts as an anti-apoptotic factor, tipping the cell towards proliferation and survival, while loss of function results in cell cycle arrest and senescence via its interaction with p53, and in the rare Cockayne syndrome, in Progeria.
Emerging studies have identified race-specific differences in the frequency, type, and functional consequences of repairome alterations in prostate tumors. African American men may harbor unique germline and somatic variants in DDR genes, as well as distinct patterns of gene expression and epigenetic regulation within DNA repair networks. Among these molecular differences that may underlie the observed disparities in tumor biology and response to therapy, yet remain incompletely characterized due to the historical underrepresentation of AA men in genomic studies, is a specific polymorphism in ERCC6 (M1097V) [9] . Although this haplotype was reported for greater prevalence of this mutation in AA-PCa (21% frequency vs 1% for CC [9]), this was determined from only a small number of patients [9]. However, this same polymorphism was identified as a significant risk for ontogeny and worse outcome in meta-analyses of other types of cancer worldwide [10,11,12]
In this study, we introduced this genomic mutation via CRISPR/Cas in a panel of common PCa cells lines, including PCa2 cells derived from a AA patient, and we investigate the consequences for repair of lesions requiring TC-NER (UV and cisplatin resistance) as a first assessment of the possible altered interaction of this mutant protein with the environment that these individuals may be exposed to. For instance, in Louisiana, PCa disparity is far more prevalent, with higher incidence and worse overall survival (OS) that can be attributed to both genetic components and dietary habits [13], compounded by much greater health risk from a regional toxic environment that was built to disproportionally impact the AA population [14].
Crispr Site-Directed Mutagenesis Experiment
The guide RNA and donor DNA were designed using the Thermofisher Invitrogen TrueDesign Genome Editor. The cells were seeded to 70% confluency. After 24 hours, in tube 1, TrueCut Cas9 protein, TrueGuide sgRNA, and donor DNA were diluted with Cas9 Plus Reagent in Opti-MEM solutions and in another tube, CRISPRMAX reagent was diluted in Opti-MEM medium. The reagents were mixed, incubated for 10 minutes, and added to the cells. After 48 hours, clones were created and screened.
Gel Electrophoresis
Agarose gels (1%) were prepared with 1x TAE buffer (from a 50x stock solution; EDTA disodium salt 372.24 g/mol, Tris 121.14 g/mol, glacial/acetic acid 60.05), after which fresh EtBr was added before use. Electrophoresis was performed with a constant 80 V. After electrophoresis, the gel image was captured by the BIORAD ChemiDoc Imaging System
PCR- Restriction Fragment Length Polymorphism(RFLP)
Hin1II (NlaIII) cleavage sites were identified in our expected amplicons using the New England Biolabs NEBcutter tool. following PCR amplifications of the ERCC6 M1097V region, 20%(10ul of 50ul reaction) of the reaction volume was incubated with the enzyme and run on a gel electrophoresis as described in the gel electrophoresis method
Dot Blot (DNA Southwestern Blot)
50, 000 cells were plated and incubated for 24 hours, and then exposed to UV for 30 seconds. They were then allowed to recover for different time points, after which the cells were lysed using the X-Amp DNA reagent, Cat. No IB47441 and dot blotted on Milipore INYC00010 IMMOBILON via a dot blot apparatus (Bio-rad). Baked at 80 degrees for 30 minutes. The membrane was dipped in water and then 0.1% methylene blue and incubated with primary antibody, mouse monoclonal Anti-Cyclobutane pyrimidine dimer [C3B6] – Absolute antibody diluted 1:1000 in 0.5% BSA overnight. The membrane was washed twice gently in 0.02% TBST for 10 min and incubated in secondary antibody, Anti-mouse IgG, HRP-linked Antibody – Cell Signaling technology 1:5000 in 0.5% BSA in 0.02% TBST, and then washed three times and visualized using the BIORAD ChemiDoc Imaging System
Proliferation Assay
Cells were seeded into Greiner Cell culture Microplate, 96 Well, Ps, F-Bottom 655180 at 50% confluency(10,000 cells). After 24 hours, the cells were exposed to different doses of UV and placed into IncuCyte S3. Cell growth was monitored using the Incucyte® Live-Cell Analysis System to capture phase contrast images every four hours and analyzed using the integrated confluence algorithm.
Statistical Analysis
Graphpad prism 9 was used to perform statistical analysis, and also Microsoft Excel software (Version 16.88) was used for data handling. Results are viewed as mean ± standard error of the mean (SEM). Statistical significance was calculated by a Student’s t-test when comparing the mean between two groups. P-values < 0.05 were considered significant(P value: P < 0.05*, P < 0.01**, and P < 0.001٭٭٭)
Results
ERCC6 is an essential, highly conserved gene from yeast to mammals (Figure 1). Given its essentiality, mutations are rare and almost absent from the PCa TCGA-500 database. Therefore, the unusual frequency of the M1097V variant, more so in AA, may be a peculiarity of the Louisiana population. Starting from the genomic SDM work, we have introduced the M1097V mutations in the indicated cell lines (Figure 2) via CRISPR-mediated recombination and obtained hetero- and homozygous (bi-allelic) SDM. We have begun assessing the UV and CPT sensitivity-dependence on the ERCC6 mutation in these derivate clones, and surprisingly (against our initial hypothesis) we found that the M1097V mutation conferred somewhat greater resistance to UV doses (Figure 4) and faster resolution of UV-induced CPDs (Figure 3). Interestingly, the PCa2 line from an AA patient, which surprisingly carries an ERCC6 mutation (Y776C), showed also remarkable activity in CPD removal when compared to all other lines (Figure 3). This clearly depends largely on ERCC6, as siRNA-KD for it drastically reduced their survival from UV (Figure 5). For control, we also used our NT1-Nek1-KO cells [15], where we know that Nek1 (a key substrate of TLK1) phosphorylates and activates ERCC6 (not published - although ERCC6 was previously reported as a target of Nek4 [16]), and these showed almost no repair (removal of CPDs) even after 1day. The faster CPD repair activity in the ERCC6-M1097V mutants and in PCa2 cells forced to rethink our initial hypothesis of the greater sensitivity to bulky/distorting-lesions damage (mainly CPDs) in AA subpopulations exposed to the Louisiana toxic racial disparity environment, but only by a changing a bit our point of view. In practice, an ‘overactive’ TC-NER mechanism can be more mutagenic (under the right conditions) than an underactive, deficient one. During NER strand replacement/polymerization at the incision site, there is a significant chance of introducing mismatches if the bulky lesions are elevated, particularly in presence of 8OG. It may not be so surprising that MMR can be more mutagenic than NER-mediated correction of bulky-lesions due to the lack of precise ‘mutated strand’ discrimination during MMR [17]; hence, the ERCC6 variants found in AA-PCa can still be targeted by NER and HRR combination strategy, as these are expected to yield more DSBs during CPT-ISLs incision/processing. In short, UV-sensitivity (orchestrated via CPDs removal) and CPT sensitivity (via complex combination pathways that include NER and HRR) are not overlapping as one may think, and CSB may be beneficial in one and not the other damage
As there are no available cell lines carrying the M1097V mutation, we have introduced this via CRISPR-mediated editing/recombination. After the generation of a panel of clones per each cell line, we proceeded with their analysis, as shown in the example in Figure 2. The introduced mutation removes one of the two Hinf3 cleavage sites, so that the wt sequence gets cleaved by Hinf3 into 2 products of the ~500 bp PCR product (lanes 1, 2 top gel) but if it is mutated, it does not. Note that in the well from clone D3(C42B) and A8(PC3), there is an intact band +the 2 cleavage products(at ~300 bp and 100bp PCR product), which indicates a heterozygous SDM change. In contrast, G3 is a genuine (bi-allelic) homozygous SDM.
We have begun assessing the UV sensitivity-dependence on the ERCC6 mutation in these derivate clones, and surprisingly (against our initial hypothesis) we found that the M1097V mutation conferred greater resistance to UV doses (Figure 4) and faster resolution of UV-induced CPDs (Figure 3). Interestingly, the PCa2 line from an AA patient, which surprisingly carries an ERCC6 mutation (Y776C), showed remarkable activity in CPD removal when compared to all other lines. For control, we used our NT1-Nek1-KO cells [15], where we know that Nek1 (a key substrate of TLK1) phosphorylates and activates ERCC6 (unpublished; although ERCC6 was reported as a target of Nek4 [16]), and these showed almost no repair (removal of CPDs) even after 1day. The faster NER activity in the ERCC6-M1079V mutants and in PCa2 cells made us rethink our initial hypothesis for the greater sensitivity to bulky-lesions (e.g., CPDs) damage in AA subpopulations exposed to the Louisiana toxic, historically racial, disparity environment [13,14], but only by a changing a bit our point of view (see Discussion).
To establish if the rate of clearance of the CPD translates to differences in survival/fitness, we performed a proliferation assay following exposure to UV using the prostate cancer cells and their ERCC6 m1097v counterparts. Without UV, exposure, the ERCC6 m1097v mutant achieved confluency faster than the WT variant and at 10S UV exposure, there is a clear difference between both PC3 and DU145 and their m1097v counterpart’s survival rates (Figure 4). Since UV exposure results in a cell cycle arrest, there is an expected recovery lag before proliferation resumes, but more importantly, when it does, the slope of proliferation rate gives a key estimate of how good the repair was, as an indication of the overall ‘fitness’’ of the cells population after recovery from a highly mutagenic event. When we compare the slopes in the plot, at 10S UV exposure, G3 is growing at roughly 2X the rate of the WT variant once past the recovery lad, and similarly for A8. In conclusion, the ERCC6-M1079V variant, introduced by SDM replacement at the natural genomic location with its expression regulated at the normal level, confers better TC-NER activity on UV-induced CPDs and greater survival/repair fitness.
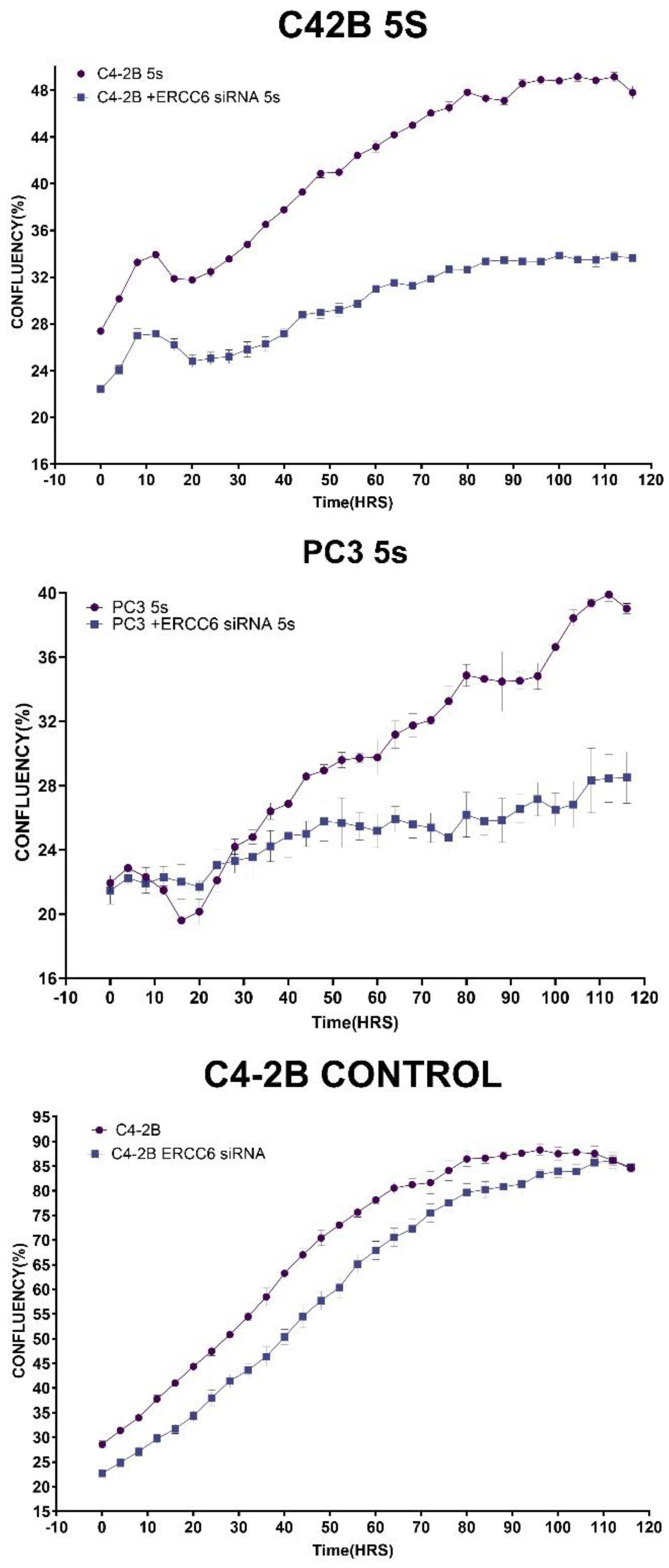
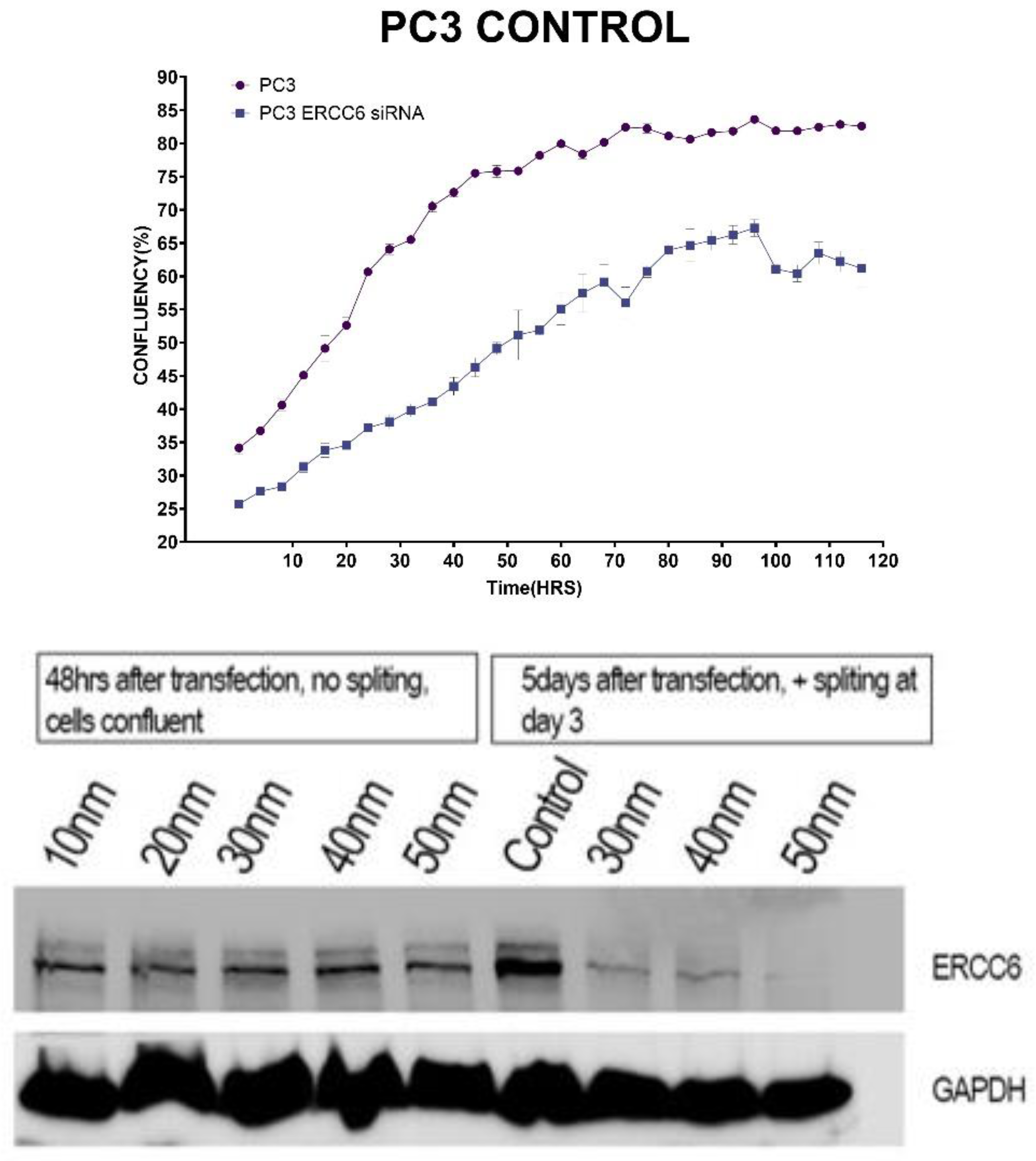
Since it is critical to establish that these differences in recovery capacity are attributable to directly to ERCC6 repair activity, that inherntly differ between the wt and M0197V variant, we performed a viability assay in cells in which ERCC6 was knocked down and exposed to a low dose of UV. This experiment demonstrated that although there are other mechanisms to deal with UV damage (CPDs), including GG-NER, ERCC6-mediated TC-NER is still the most critical repair pathway (Figure 5).
Discussion
ERCC6 (also known as CSB – Cockayne Syndrome B protein) is a gene encoding a critical protein involved in DNA repair, specifically in transcription-coupled nucleotide excision repair (TC-NER). Helix-distorting DNA lesions that are dealt with in this pathway include CPDs, bulky adducts (including cisplatin), and in some instances modified or oxidized bases like 5OG. Many more roles have been ascribed to this large protein that belongs to the class of helicases/translocases. In fact, ERCC6/CSB is a multifunctional protein that couples transcription to DNA repair, remodels chromatin, regulates gene expression, and helps maintain genomic and cellular integrity, especially in the face of damage that disrupts transcription of PolI and PolII (https://www.genecards.org/cgi-bin/carddisp.pl?gene=ERCC6).
- General Activities of ERCC6/CSB:
-
Transcription-Coupled DNA Repair (TC-NER):
- ○
- ERCC6 plays a central role in detecting and initiating the repair of DNA lesions that block transcription.
- ○
- When RNA polymerase II stalls at DNA damage (such as UV-induced cyclobutane pyrimidine dimers), ERCC6 helps recruit repair factors to remove the lesion and resume transcription.
-
Chromatin Remodeling:
- ○
- ERCC6 possesses ATP-dependent chromatin remodeling activity, allowing it to alter nucleosome positioning.
- ○
- This activity is crucial for providing repair machinery access to DNA in compact chromatin regions.
-
Transcription Regulation:
- ○
- In addition to DNA repair, ERCC6 can regulate gene expression by interacting with transcription machinery.
- ○
- It influences RNA polymerase 1 and RNA polymerase II pausing and restart, ensuring proper transcription resumption after repair.
-
Interaction with Other Repair Proteins:
- ○
- ERCC6 interacts with other TC-NER factors such as CSA (ERCC8), XPG, TFIIH, and UVSSA to coordinate the repair process.
-
Response to Oxidative Stress:
- ○
- ERCC6 has been implicated in the repair of oxidative DNA damage, not just UV-induced lesions.
- ○
- It helps maintain mitochondrial function and cellular redox balance under stress conditions.
-
Role in Disease:
- ○
- Mutations in ERCC6 cause Cockayne Syndrome, a rare autosomal recessive disorder characterized by growth failure, neurodegeneration, and premature aging.
- ○
- It is also linked to other neurodevelopmental and progeroid syndromes.
While LOF mutations result in severe syndromes, the function of most missense mutations (or variants) have not been well studied nor are understood. The M1097V polymorphism was identified as a significant risk for ontogeny and worse outcome in meta-analyses of several types of cancer, particularly kidney, worldwide [10,11,12], and more recently noted as a significant incidence in a study of possible racial disequilibrium in Louisiana AA PCa patients. But the functional significance of this ERCC6 variant and its activity in PCa development or progression was unknown. The fact that there are very few established PCa cell lines from AA that can be used as representative of their typical Repairome forced to approach this problem by introducing this mutation, at the correct genomic location and functional regulation, in a panel of the most common PCa cell lines, including PCa2 derived from a AA patient, and that interestingly already carries a different mutation (Y776C) in ERCC6 of unknown significance. We should add that in our limited exome study of PCa patient from the more northern region of Louisiana, we did not identify the M1097V mutation, although our collection included a S636N missense variant that resides very closely to SYSY-624-628 ‘hydroxy patch’ that we have found to be highly phosphorylated by Nek1 and strongly regulates the activity of ERCC6 after UV damage (unpublished work). The close proximity of the S636N on the same loop fold suggests that the variant may affect the activity of the protein, although it is hard to assess in which direction. But we did study the activity of the M1097V variant, and perhaps unexpectedly, it increased the tolerance to UV of all the cell lines in which we carried out the appropriate gene replacement. The potential implications for this are significant, considering some segments of the Louisiana population have remained local for generations. If we should think about the AA slaves working in the sun-drenched cotton fields and having greater protection from UV damage from a protective germ-line mutation might have been advantageous over several generations. Getting PCa or other types later in life was certainly not a main concern then, whereas it could be a significant issue in modern days if one also considers the regional toxic environment of Louisiana, dubbed ‘cancer alley’. In this respect, it is important to emphasize that an advantageous mutation against UV utilizing TC-NER does not necessarily protect against more complex lesions partially utilizing this repair pathway, like those caused by petrochemically derived aromatic alkylating agents, or in the case we have studied, CPT that also causes ICLs that require the action of several pathways in addition to NER for completion of repair. In fact, the opposite could happen from an overzealous TC-NER that can lead to the accumulation of mismatch mutations during replacement of the damaged ‘flap’ of DNA. In practice, an ‘overactive’ TC-NER mechanism can be more mutagenic (under the right conditions) than an underactive/deficient one. During NER strand replacement/polymerization at the incision site, there is a significant chance of introducing mismatches when bulky lesions are numerous, particularly in the presence of 8OG. It should not be so surprising that MMR can be more mutagenic than NER-mediated correction of bulky-lesions due to the lack of precise ‘mutated strand’ discrimination during MMR [17]; In this regard, we should note that it was remarkable how many LOF mutations were found among a panel of MMR genes in our Repairome study of PCa patients (unpublished), both in AA and CC, clearly supporting the concept of the "mutator phenotype" in cancer [18] championed by Louis A. Loeb. Hence, the ERCC6 variants found in AA-PCa can still be successfully targeted by NER and HRR combination strategy (e.g., with a synthetic lethal combination of CPT and J54), as these are expected to yield more DSBs during CPT-ISLs processing (see Fig?).
African Americans (AA) are at higher risk for developing PCa than Caucasians (CC) and have more aggressive disease that is refractory to treatment [1], to some extent explained by genetic differences in some cases attributed to alterations of the “repairome” [2]. Since androgen receptor (AR) signaling regulates the growth, survival, and proliferation of prostate tumors, the majority of PCa therapies are focused on either inhibition of androgen synthesis or blockade of androgen receptor transactivation. However, the drug effect does not last long and the tumor relapses within 18-24 months with a more aggressive phenotype known as metastatic castration resistant prostate cancer (mCRPC). Androgen ablation, radiotherapy and chemotherapy are commonly employed for the treatment of both localized PCa and mCRPC. Inflicting DNA damage and enhancing apoptosis of cancer cells are the mechanistic strategies of all radiotherapeutic (e.g., external beam radiation therapy, brachytherapy, radium- 223) and some chemotherapeutic (PARP inhibitors; topoisomerase inhibitors, platinum-based therapy [3], and DNA crosslinking agents) [4,5]. Relative success of PARPi for mCRPC patients with and without homologous recombination repair (HRR) mutations (PROfound [6] and TRITON2 [7]) is defining new biology and more effective treatment options for previously unmanageable PCa; and while cisplatin (CPT)-based therapy is currently not the treatment of choice for PCa, partly due to significant renal toxicity (which can be faithfully recapitulated in mice [8]), the current trend suggests that lower dosing in combination with inhibitors of DNA Damage Repair could be quite effective [3]. In Louisiana, PCa disparity is far more prevalent, with higher incidence and worse overall survival (OS) that can be attributed to both genetic components and dietary habits [13], compounded by much greater health risk from a regional toxic environment that was built to disproportionally impact the AA population [14]. We see many of these patients presenting to LSU Health Shreveport with advanced stage disease both in the North and from a central region sadly named Cancer Alley. Perhaps paradoxically, the more frequent germline and somatic mutations in NER genes identified in AA [9] that may be negatively impacted from this toxic environment and that are likely germane to higher mutation-induced cancer, are also key to a therapeutic strategy that could be largely beneficial to AA PCa patients. Alterations of key genes involved in NER could result in incomplete or aberrant repair of the bulky lesions (or ISLs) and result in potentially lethal effects for the cancer cells. This relies on a synthetic lethality approach simultaneously targeting the Transcription-Coupled-Nucleotide-Excision-Repair (TC-NER) and Homologous-Recombination-Repair (HRR) pathways. In particular, we propose one could target a function TLK1 as a regulator of RAD54 activity and the availability of some novel specific inhibitors like J54 [19,20,21] as a potential synthetic lethal approach we have recently employed [22]. In contrast, in this work we have focused our efforts to direct a future synthetic lethal therapy designed for the more active TC-NER activity of one specific haplotype variant of the gene ERCC6 that is frequently found in mCRPC of AA patients [9], reportedly a germline. Such combination therapy approaches may bypass altogether the need for ADT (i.e., castration) and its significant side-effects that few men would readily choose, or clearly for those who wouldn’t respond to ADT/ARSI from the start, as epitomized by NEPC cases, which is also a more common occurrence for AA-PCa patients, in addition to the already mentioned higher incidence of mutations in their repairome [23].
Conclusions
Our findings underscore the critical and multifaceted role of ERCC6/CSB in DNA repair and transcription regulation, particularly through the TC-NER pathway. The M1097V variant of ERCC6 enhances UV resistance in prostate cancer cells, suggesting a possible evolutionary adaptation with modern therapeutic implications. This variant, along with other ERCC6 alterations found predominantly in African American (AA) prostate cancer patients, could be exploited using synthetic lethality strategies targeting TC-NER and HRR. Such precision therapies may be especially beneficial in regions like Louisiana, where environmental exposures and inherited variants converge to heighten cancer risk and severity.
Author Contributions
Conceptualization: O.O., A.D.B., experimental analysis and interpretation and A.D.B.
Funding
LSUHS Chancellor Award
Institutional Review Board Statement
N/A.
Acknowledgments
We like to thank the INLET facility of LSU Health Shreveport, especially Ana Maria Dragoi and Brian Latimer for their assistance in working with the IncuCyte machines.
Conflicts of Interest
Authors declare no conflicts of interest.
References
- CHANG C-H, CHIU C-F, WANG H-C, WU H-C, TSAI R-Y, TSAI C-W, WANG R-F, WANG C-H, TSOU Y-A, BAU D-T: Significant Association of ERCC6 Single Nucleotide Polymorphisms with Bladder Cancer Susceptibility in Taiwan. Anticancer Research 2009, 29(12):5121-5124.
- Forrest IS, Chaudhary K, Vy HMT, Bafna S, Kim S, Won HH, Loos RJF, Cho J, Pasquale LR, Nadkarni GN et al: Genetic pleiotropy of ERCC6 loss-of-function and deleterious missense variants links retinal dystrophy, arrhythmia, and immunodeficiency in diverse ancestries. Hum Mutat 2021, 42(8):969-977. [CrossRef]
- Lin X, Wu Y, Li Q, Yu H, Li X, Li X, Zheng J: Genetic Association of ERCC6 rs2228526 Polymorphism with the Risk of Cancer: Evidence from a Meta-Analysis. Biomed Res Int 2022, 2022:2662666. [CrossRef]
- Putnam CD: Strand discrimination in DNA mismatch repair. DNA Repair (Amst) 2021, 105:103161. [CrossRef]
- Loeb LA: A mutator phenotype in cancer. Cancer Res 2001, 61(8):3230-3239.
- Kakarla M, ChallaSivaKanaka S, Hayward SW, Franco OE: Race as a Contributor to Stromal Modulation of Tumor Progression. Cancers 2021, 13(11):2656. [CrossRef]
- Teng PC, Huang SP, Liu CH, Lin TY, Cho YC, Lai YL, Wang SC, Yeh HC, Chuu CP, Chen DN et al: Identification of DNA Damage Repair-Associated Prognostic Biomarkers for Prostate Cancer Using Transcriptomic Data Analysis. Int J Mol Sci 2021, 22(21). [CrossRef]
- Schmid S, Omlin A, Higano C, Sweeney C, Martinez Chanza N, Mehra N, Kuppen MCP, Beltran H, Conteduca V, Vargas Pivato de Almeida D et al: Activity of Platinum-Based Chemotherapy in Patients With Advanced Prostate Cancer With and Without DNA Repair Gene Aberrations. JAMA network open 2020, 3(10):e2021692-e2021692. 2021. [CrossRef]
- Zhang W, Van Gent DC, Incrocci L, Van Weerden WM, Nonnekens J: Role of the DNA damage response in prostate cancer formation, progression and treatment. Prostate cancer and prostatic diseases 2020, 23(1):24-37. [CrossRef]
- Banks P, Xu W, Murphy D, James P, Sandhu S: Relevance of DNA damage repair in the management of prostate cancer. Current problems in cancer 2017, 41(4):287-301. [CrossRef]
- de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, Chi KN, Sartor O, Agarwal N, Olmos D et al: Olaparib for Metastatic Castration-Resistant Prostate Cancer. New England Journal of Medicine 2020, 382(22):2091-2102. [CrossRef]
- Abida W, Campbell D, Patnaik A, Shapiro JD, Sautois B, Vogelzang NJ, Voog EG, Bryce AH, McDermott R, Ricci F et al: Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis From the Phase II TRITON2 Study. Clin Cancer Res 2020, 26(11):2487-2496. [CrossRef]
- Perše M: Cisplatin Mouse Models: Treatment, Toxicity and Translatability. Biomedicines 2021, 9(10):1406. [CrossRef]
- Su LJ, Arab L, Steck SE, Fontham ET, Schroeder JC, Bensen JT, Mohler JL: Obesity and prostate cancer aggressiveness among African and Caucasian Americans in a population-based study. Cancer Epidemiol Biomarkers Prev 2011, 20(5):844-853. [CrossRef]
- Baurick T, Younes L, Meiners J: Welcome to "Cancer Alley" where toxic air is about to get worse. In: Propublica. 2019.
- Yadav S, Anbalagan M, Baddoo M, Chellamuthu VK, Mukhopadhyay S, Woods C, Jiang W, Moroz K, Flemington EK, Makridakis N: Somatic mutations in the DNA repairome in prostate cancers in African Americans and Caucasians. Oncogene 2020, 39(21):4299-4311. [CrossRef]
- Singh V, Bhoir S, Chikhale RV, Hussain J, Dwyer D, Bryce RA, Kirubakaran S, De Benedetti A: Generation of phenothiazine with potent anti-TLK1 activity for prostate cancer therapy. Iscience 2020, 23(9):101474. [CrossRef]
- Singh V, Jaiswal PK, Ghosh I, Koul HK, Yu X, De Benedetti A: Targeting the TLK1/NEK1 DDR axis with Thioridazine suppresses outgrowth of androgen independent prostate tumors. International journal of cancer 2019, 145(4):1055-1067. [CrossRef]
- Singh V, Jaiswal PK, Ghosh I, Koul HK, Yu X, De Benedetti A: The TLK1-Nek1 axis promotes prostate cancer progression. Cancer letters 2019, 453:131-141. [CrossRef]
- Bhoir S, Ogundepo O, Yu X, Shi R, De Benedetti A: Exploiting TLK1 and Cisplatin Synergy for Synthetic Lethality in Androgen-Insensitive Prostate Cancer. Biomedicines 2023, 11(11):2987. [CrossRef]
- Hinata N, Fujisawa M: Racial Differences in Prostate Cancer Characteristics and Cancer-Specific Mortality: An Overview. World J Mens Health 2022, 40(2):217-227. [CrossRef]
- Kakarla M, ChallaSivaKanaka S, Hayward SW, Franco OE: Race as a Contributor to Stromal Modulation of Tumor Progression. Cancers 2021, 13(11):2656. [CrossRef]
- Teng PC, Huang SP, Liu CH, Lin TY, Cho YC, Lai YL, Wang SC, Yeh HC, Chuu CP, Chen DN et al: Identification of DNA Damage Repair-Associated Prognostic Biomarkers for Prostate Cancer Using Transcriptomic Data Analysis. Int J Mol Sci 2021, 22(21). [CrossRef]
- Schmid S, Omlin A, Higano C, Sweeney C, Martinez Chanza N, Mehra N, Kuppen MCP, Beltran H, Conteduca V, Vargas Pivato de Almeida D et al: Activity of Platinum-Based Chemotherapy in Patients With Advanced Prostate Cancer With and Without DNA Repair Gene Aberrations. JAMA network open 2020, 3(10):e2021692-e2021692. [CrossRef]
- Zhang W, Van Gent DC, Incrocci L, Van Weerden WM, Nonnekens J: Role of the DNA damage response in prostate cancer formation, progression and treatment. Prostate cancer and prostatic diseases 2020, 23(1):24-37. [CrossRef]
- Banks P, Xu W, Murphy D, James P, Sandhu S: Relevance of DNA damage repair in the management of prostate cancer. Current problems in cancer 2017, 41(4):287-301. [CrossRef]
- de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, Chi KN, Sartor O, Agarwal N, Olmos D et al: Olaparib for Metastatic Castration-Resistant Prostate Cancer. New England Journal of Medicine 2020, 382(22):2091-2102. [CrossRef]
- Abida W, Campbell D, Patnaik A, Shapiro JD, Sautois B, Vogelzang NJ, Voog EG, Bryce AH, McDermott R, Ricci F et al: Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis From the Phase II TRITON2 Study. Clin Cancer Res 2020, 26(11):2487-2496. [CrossRef]
- Perše M: Cisplatin Mouse Models: Treatment, Toxicity and Translatability. Biomedicines 2021, 9(10):1406. [CrossRef]
- Yadav S, Anbalagan M, Baddoo M, Chellamuthu VK, Mukhopadhyay S, Woods C, Jiang W, Moroz K, Flemington EK, Makridakis N: Somatic mutations in the DNA repairome in prostate cancers in African Americans and Caucasians. Oncogene 2020, 39(21):4299-4311. [CrossRef]
- CHANG C-H, CHIU C-F, WANG H-C, WU H-C, TSAI R-Y, TSAI C-W, WANG R-F, WANG C-H, TSOU Y-A, BAU D-T: Significant Association of ERCC6 Single Nucleotide Polymorphisms with Bladder Cancer Susceptibility in Taiwan. Anticancer Research 2009, 29(12):5121-5124.
- Forrest IS, Chaudhary K, Vy HMT, Bafna S, Kim S, Won HH, Loos RJF, Cho J, Pasquale LR, Nadkarni GN et al: Genetic pleiotropy of ERCC6 loss-of-function and deleterious missense variants links retinal dystrophy, arrhythmia, and immunodeficiency in diverse ancestries. Hum Mutat 2021, 42(8):969-977. [CrossRef]
- Lin X, Wu Y, Li Q, Yu H, Li X, Li X, Zheng J: Genetic Association of ERCC6 rs2228526 Polymorphism with the Risk of Cancer: Evidence from a Meta-Analysis. Biomed Res Int 2022, 2022:2662666. [CrossRef]
- Su LJ, Arab L, Steck SE, Fontham ET, Schroeder JC, Bensen JT, Mohler JL: Obesity and prostate cancer aggressiveness among African and Caucasian Americans in a population-based study. Cancer Epidemiol Biomarkers Prev 2011, 20(5):844-853. [CrossRef]
- Baurick T, Younes L, Meiners J: Welcome to "Cancer Alley" where toxic air is about to get worse. In: Propublica. 2019.
- Khalil MI, Ghosh I, Singh V, Chen J, Zhu H, De Benedetti A: NEK1 Phosphorylation of YAP Promotes Its Stabilization and Transcriptional Output. Cancers 2020, 12(12):3666. [CrossRef]
- Basei FL, Meirelles GV, Righetto GL, Dos Santos Migueleti DL, Smetana JH, Kobarg J: New interaction partners for Nek4.1 and Nek4.2 isoforms: from the DNA damage response to RNA splicing. Proteome Sci 2015, 13:11. [CrossRef]
- Putnam CD: Strand discrimination in DNA mismatch repair. DNA Repair (Amst) 2021, 105:103161. [CrossRef]
- Loeb LA: A mutator phenotype in cancer. Cancer Res 2001, 61(8):3230-3239.
- Singh V, Bhoir S, Chikhale RV, Hussain J, Dwyer D, Bryce RA, Kirubakaran S, De Benedetti A: Generation of phenothiazine with potent anti-TLK1 activity for prostate cancer therapy. Iscience 2020, 23(9):101474. [CrossRef]
- Singh V, Jaiswal PK, Ghosh I, Koul HK, Yu X, De Benedetti A: Targeting the TLK1/NEK1 DDR axis with Thioridazine suppresses outgrowth of androgen independent prostate tumors. International journal of cancer 2019, 145(4):1055-1067. [CrossRef]
- Singh V, Jaiswal PK, Ghosh I, Koul HK, Yu X, De Benedetti A: The TLK1-Nek1 axis promotes prostate cancer progression. Cancer letters 2019, 453:131-141. [CrossRef]
- Bhoir S, Ogundepo O, Yu X, Shi R, De Benedetti A: Exploiting TLK1 and Cisplatin Synergy for Synthetic Lethality in Androgen-Insensitive Prostate Cancer. Biomedicines 2023, 11(11):2987. [CrossRef]
- Hinata N, Fujisawa M: Racial Differences in Prostate Cancer Characteristics and Cancer-Specific Mortality: An Overview. World J Mens Health 2022, 40(2):217-227. [CrossRef]
Figure 1.
ERCC6 is a conserved protein in Humans, Mouse, Plants, and Yeast. A. A table summarizing and comparing the conserved regions in ERCC6 and similar proteins taken from … B. Phylogenetic relationship between ERCC6 and related protein, including CHR8/24 in plant and RAD26 in yeast, taken from Tianyi Fan et al., The Plant Cell, 2024 C. ERCC6 schematic structure taken from Spyropoulou, Z et al., MDPI Cells 2021.
Figure 1.
ERCC6 is a conserved protein in Humans, Mouse, Plants, and Yeast. A. A table summarizing and comparing the conserved regions in ERCC6 and similar proteins taken from … B. Phylogenetic relationship between ERCC6 and related protein, including CHR8/24 in plant and RAD26 in yeast, taken from Tianyi Fan et al., The Plant Cell, 2024 C. ERCC6 schematic structure taken from Spyropoulou, Z et al., MDPI Cells 2021.
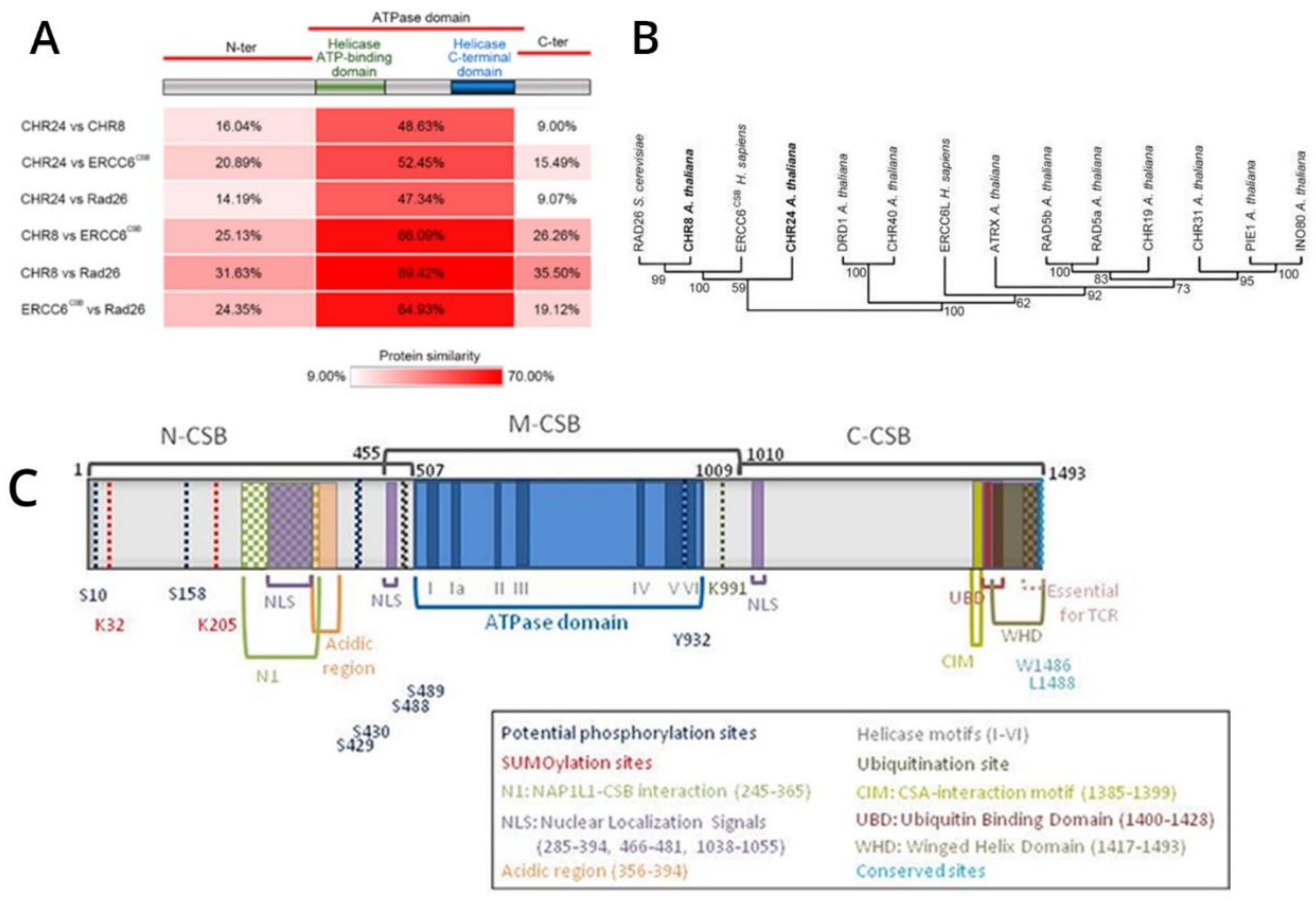
Figure 2.
Generation and Validation of the M1097V knock-in in prostate cancer cells with the CRISPR/Cas9 system. A. a schematic diagram of the recurring ERCC6 M1097V mutation, made with Biorender B. a schematic diagram of the CRISPR/Cas9 editing system made with biorender C. PCR amplification of the M1097V region and cleavage with HIN1II D. confirmation of cleavage site using ERCC6 plasmid from Origene E. Clone confirmation after generation of the M1097V variants using the CRISPR/Cas9 system via PCR-RFLP.
Figure 2.
Generation and Validation of the M1097V knock-in in prostate cancer cells with the CRISPR/Cas9 system. A. a schematic diagram of the recurring ERCC6 M1097V mutation, made with Biorender B. a schematic diagram of the CRISPR/Cas9 editing system made with biorender C. PCR amplification of the M1097V region and cleavage with HIN1II D. confirmation of cleavage site using ERCC6 plasmid from Origene E. Clone confirmation after generation of the M1097V variants using the CRISPR/Cas9 system via PCR-RFLP.
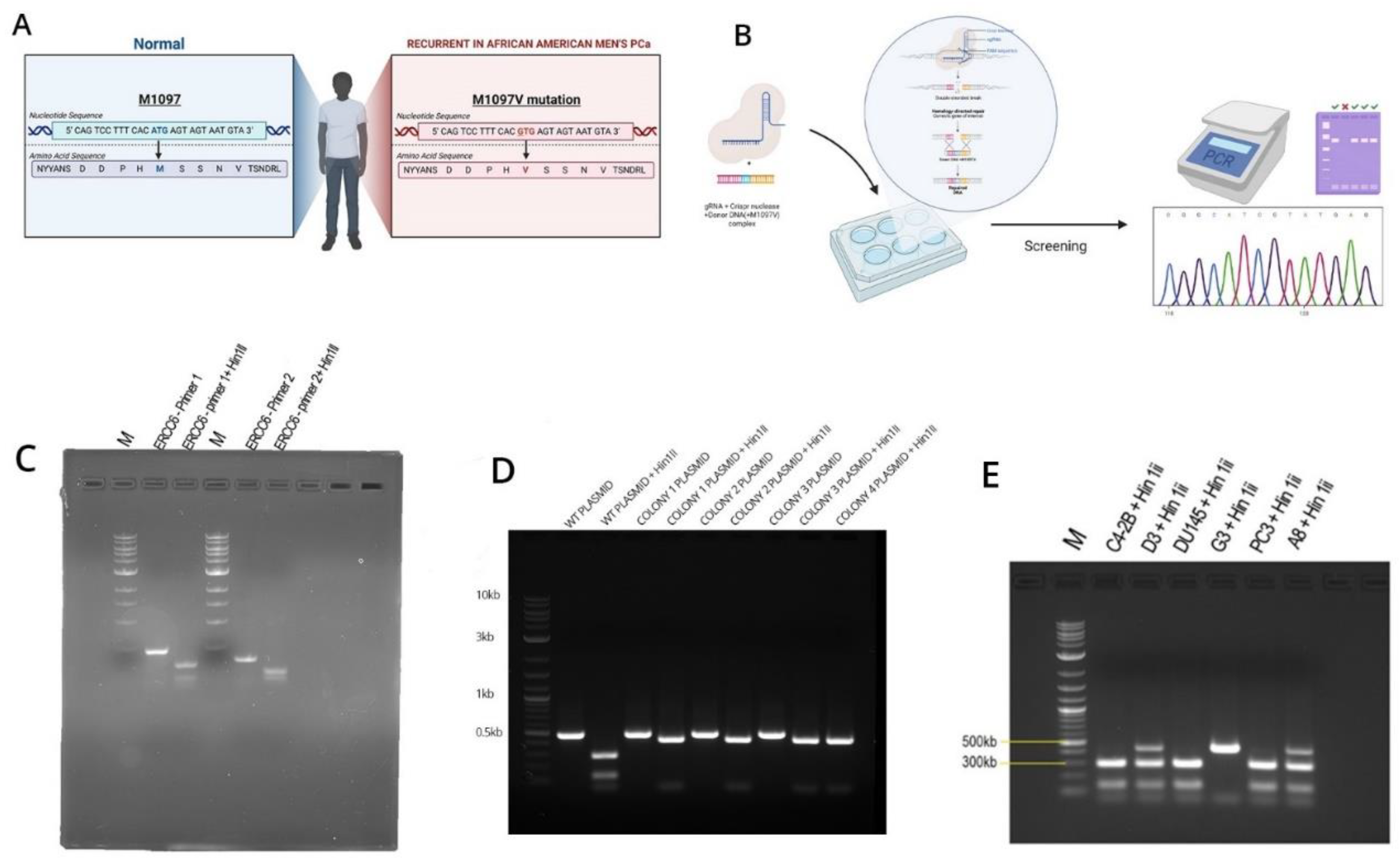
Figure 3.
Kinetics of CPD removal. Cells were irradiated with UV (200 mJ/cm2) and allowed to recover for the indicated times. DNA was isolated and affixed to Hi-Bond via a manifold. Following brief staining with MB to ensure even DNA application, the blot is probed with CPD antiserum.
Figure 3.
Kinetics of CPD removal. Cells were irradiated with UV (200 mJ/cm2) and allowed to recover for the indicated times. DNA was isolated and affixed to Hi-Bond via a manifold. Following brief staining with MB to ensure even DNA application, the blot is probed with CPD antiserum.

Figure 4.
A-B. Proliferation assay of the wild type and M1097V mutants following exposure to UV at different time points and recovery for 4 days. C-D. Rate of change in cell number confluency(%) to time(HRS) P value: P < 0.05*, P < 0.01**, and P < 0.001٭٭٭.
Figure 4.
A-B. Proliferation assay of the wild type and M1097V mutants following exposure to UV at different time points and recovery for 4 days. C-D. Rate of change in cell number confluency(%) to time(HRS) P value: P < 0.05*, P < 0.01**, and P < 0.001٭٭٭.
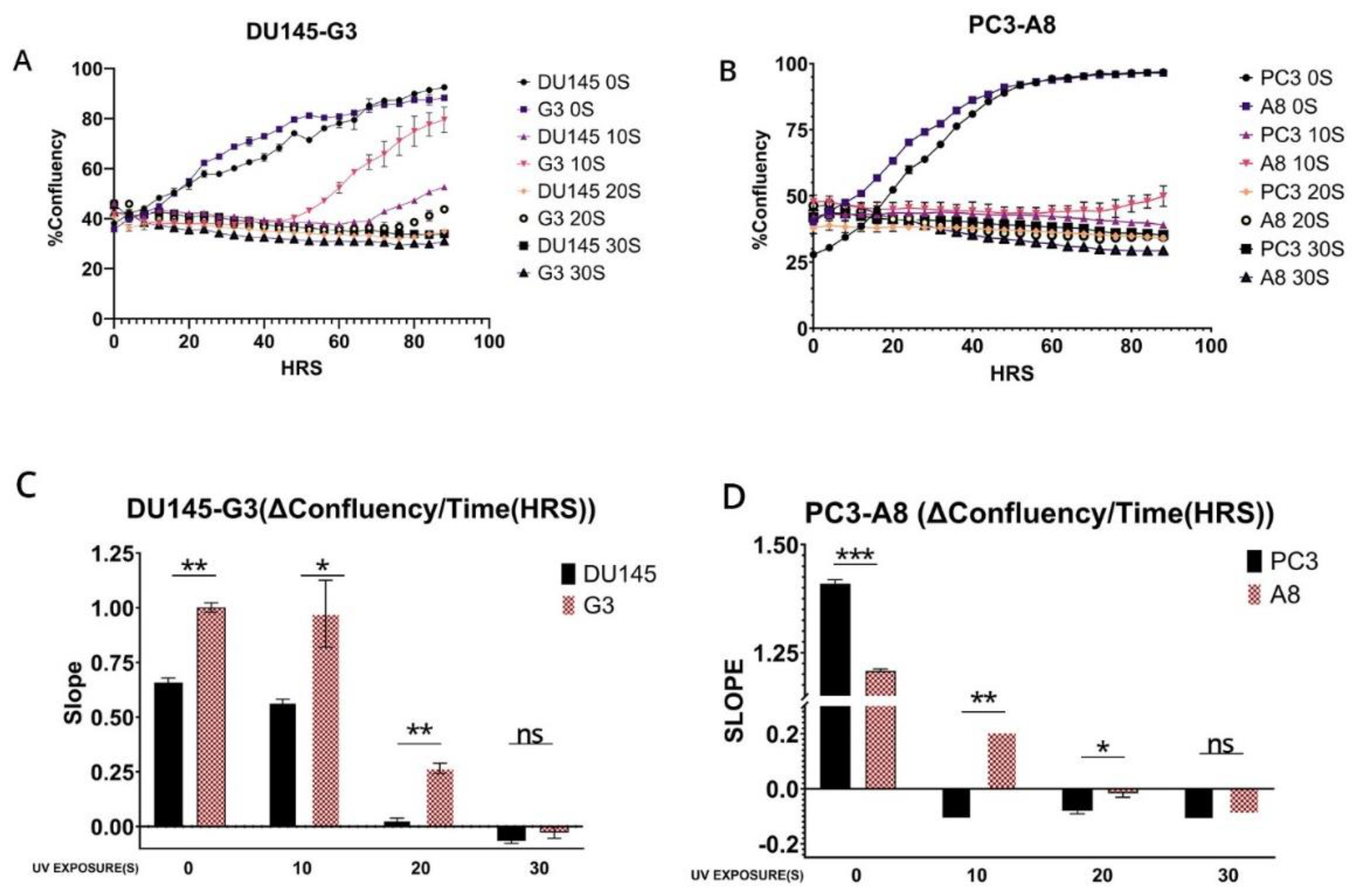
Figure 5.
Proliferation assays in parental cells treated with siRNA and exposed to 5s UV.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



