
Submitted:
05 June 2025
Posted:
11 June 2025
You are already at the latest version
Abstract
Autoimmune diseases such as systemic lupus erythematosus and Sjögren’s syndrome show pronounced sex disparities in prevalence, severity, and clinical outcomes, with females disproportionately affected. Emerging evidence highlights sex-based differences in immune and inflammatory responses as key contributors to this bias. Genetic factors—including sex chromosomes, skewed X chromosome inactivation, and sex-biased microRNAs—as well as sex hormones and pregnancy modulate gene expression and immune cell function in a sex-specific manner. Additionally, sex hormone-dependent epigenetic modifications influence the transcription of critical immune regulators. These genetic and hormonal factors collectively shape the activation, differentiation, and effector functions of diverse immune cell types. Environmental factors—including infections, gut microbiota, environmental chemicals and pollutants, and lifestyle behaviors such as diet, smoking, UV exposure, alcohol and caffeine intake, physical activity, and circadian rhythms—further modulate immune function and autoimmune disease pathogenesis in a sex-dependent manner. Together, these mechanisms contribute to the heightened risk and distinct clinical features of autoimmunity in females. A deeper understanding of sex-biased immune regulation will facilitate the identification of novel biomarkers, enable patient stratification, and inform the development of sex-specific diagnostic and therapeutic strategies for autoimmune diseases.
Keywords:
autoimmune diseases
; epigenetic regulation
; estrogens
; immune responses
; inflammation
; Sjögren's syndrome
; systemic lupus erythematosus
; X chromosome inactivation
1. Introduction
The immune system defends the host against harmful pathogens through tightly regulated inflammatory responses essential for tissue repair and homeostasis maintenance. However, dysregulation of these pathways can result in persistent inflammation and the breakdown of self-tolerance, ultimately leading to autoimmune diseases [1]. These conditions emerge when the immune system mistakenly targets self-antigens in various tissues—including skin, joints, endocrine organs, and the nervous system—resulting in diverse and often debilitating clinical manifestations [2].
Over 100 autoimmune diseases have been identified, including systemic lupus erythematosus (SLE), Sjögren’s syndrome (SS), rheumatoid arthritis (RA), scleroderma (systemic sclerosis, SD/SSc), multiple sclerosis (MS), type 1 diabetes mellitus, and inflammatory bowel disease. These diseases are chronic, often lifelong, and significantly impair quality of life, increase morbidity, and pose substantial socioeconomic burdens. Their global incidence and prevalence continue to rise, with an estimated 23.5 million individuals affected in the United States alone—approximately 10% of the global population. Prevalence varies according to genetic background, geographic location, environmental exposures, and coexisting conditions, emphasizing the need for nuanced epidemiological insights to inform public health interventions and precision medicine approaches [3].
A defining feature of autoimmune diseases is their marked sex disparity [4,5]. Female-to-male incidence and prevalence ratios vary across diseases and populations, with the greatest skew observed in SLE and SS [3,6,7]. SLE shows incidence and prevalence rate ratios of 5.8 and 8.5, respectively, while SS exhibits even higher ratios of 9.2 and 10.7 (Figure 1a and 1b). Given these pronounced differences, this review highlights SLE and SS as representative models of female-biased autoimmunity.
SLE is a prototypic systemic autoimmune disorder characterized by multisystem involvement— including the skin, joints, kidneys, central nervous system, lungs, and vasculature. Its clinical presentation is heterogeneous, with common symptoms such as fatigue, fever, cytopenia, arthritis, malar rash, and proteinuria [16]. Lupus nephritis (LN), a major complication, occurs in ~ 40% of patients and progresses to end-stage renal disease in approximately 10% within a decade [17,18,19]. LN is more prevalent in females and peaks in incidence between the ages 30–39, then declines after age 60 [20].
SS is a chronic autoimmune disease that predominantly affects the exocrine glands, particularly the lacrimal and salivary glands, resulting in dryness of the eyes (xerophthalmia) and mouth (xerostomia). It can occur as primary SS (pSS) or as secondary SS in association with other autoimmune diseases such as SLE, RA, or SD. SS is characterized by lymphocytic infiltration of the glands, reduced secretory capacity, and systemic complications, including interstitial lung disease, cardiovascular manifestations, and renal dysfunction [21,22].
Biological sex and sex hormones are critical determinants of immune responses to pathogens and self-antigens [23,24]. Females generally exhibit stronger innate and adaptive immune responses than males, enhancing pathogen defense but increasing susceptibility to autoimmunity [2,25]. Section 2 of this review outlines immunological disparities between sexes, while Section 3 explores how these differences contribute to sex-biased susceptibility, onset, and progression of SLE and SS. The heightened autoimmune risk observed in females is attributed to X chromosome-linked gene dosage effects and epigenetic regulation, sex hormone-mediated modulation of immune pathways, and environmental factors [26]. Section 4 explores these genetic and hormonal mechanisms and environmental insults underlying sex bias in autoimmunity. Understanding these mechanisms is essential to advance our knowledge of autoimmune pathogenesis and to guide the development of sex-informed, personalized therapeutic strategies.
2. Sex-Specific Disparities in Immune Responses
Sex-based differences in immune function profoundly influence susceptibility to infections, vaccine efficacy, and autoimmune disease prevalence. Females and males display distinct innate and adaptive immune profiles, which drive divergent immune and inflammatory responses and disease outcomes. Understanding these mechanisms is crucial for advancing personalized immunotherapies and clinical care of autoimmune diseases.
2.1. Innate Immune Responses
The innate immune system provides rapid, nonspecific defense through physical barriers, innate immune cells—including dendritic cells (DCs), macrophages, natural killer (NK) cells, and invariant NK T (iNKT) cells—and soluble mediators such as cytokines and chemokines (Figure 2a). These responses enhance antigen presentation by promoting the generation of antigen-presenting cells (APCs) and driving adaptive and inflammatory responses.
Toll-like receptors (TLRs) play a central role in recognizing pathogen-associated and damage-associated molecular patterns and in mediating cell-based immunity [27]. Surface TLRs (e.g., TLR1/2, TLR4-6, TLR10) detect microbial components, while endosomal TLRs (e.g., TLR3, TLR7-9) recognize nucleic acids from viruses, bacteria, or self-origin [28]. Upon ligand binding, TLRs assemble the Myddosome complex, comprising myeloid differentiation factor 88 (MyD88) and interleukin 1 receptor-associated kinases (IRAKs) [29]. This complex activates transforming growth factor- β-activated kinase 1 (TAK1), nuclear factor kappa B (NF-κB), mitogen-activated protein kinase (MAPK), and interferon regulatory factor (IRF) pathways [28] (Figure 2b). TLR3/7/8/9 are trafficked from the endoplasmic reticulum to endosomes by UNC93B1, an endoplasmic transmembrane protein highly expressed in DCs, macrophages, monocytes, and B cells. Endosomal TLR7-9 activate IRF5 via TASL, a TLR adaptor that interacts with the endolysosomal solute carrier family 15 member 4 (SLC15A4) [30,31]. This signaling induces proinflammatory cytokines, including interleukins (IL-1β, IL-6), tumor necrosis factor-alpha (TNF-α), and type I interferons (IFN-Is), which are crucial for pathogen clearance but contribute to autoimmunity when dysregulated [32,33].
Sex-specific differences shape these immune responses [24,34]. Females exhibit higher phagocyte numbers, TLR expression, and production of IL-6 and TNF-α, promoting stronger innate activation. Although females have a lower percentage of NK cells, males exhibit greater NK cell-mediated cytotoxicity and more robust anti-inflammatory response. These differences may underlie the higher autoimmune susceptibility observed in females.
2.2. Adaptive Immune Responses
Adaptive immunity is mediated by antigen-specific responses of T and B cells. Upon antigen presentation via major histocompatibility complex class II (MHC-II) on APCs, naïve CD4⁺ T cells differentiate into specialized helper T cell subsets (e.g., Th1, Th2, Th17, Treg, Tfh, Th9, Th22) to orchestrate immune responses (Figure 3). These subsets exhibit distinct transcriptional programs, cytokine secretion profiles, and immunological functions. Notably, Th1, Th9, Th17, and Th22 cells promote inflammation and contribute to autoimmunity [35].
B cells are activated either independently or with Th cell help, undergoing class switching and affinity maturation in germinal centers [36]. These processes generate short-lived IgM-secreting plasma cells, long-lived IgG-secreting plasma cells, and memory B cells (Figure 4a). B cell survival and differentiation are regulated by B cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL) via signaling through BAFF receptor (BAFFR), transmembrane activator and calcium moderator and cyclophilin ligand interactor (TACI), and B cell maturation antigen (BCMA) [37].
CD8+ cytotoxic T cell (Tctx), activated by MHC-I–presented antigens and costimulatory signals (e.g., CD28–CD80/CD86), eliminate infected or abnormal cells through perforin/granzyme release and proinflammatory cytokine production (Figure 4b). Their cytotoxic activity is tightly regulated by inhibitory checkpoints, including Tctx-associated protein 4 (CTLA-4), PD-1/PD-L1, TIM-3, and VISTA [38], as well as Treg-mediated suppression. iNKT cells further support memory T cell formation and cross-priming [39].
During immune maturation, self-reactive immune cells are eliminated through central tolerance [40]. However, this process is incomplete and reinforced by peripheral tolerance mechanisms, including the conversion of self-reactive Th cells into Tregs [41]. In autoimmune diseases, Treg cells are often reduced in number, exhibit impaired suppressive function, or both. The presence of autoantibodies and autoreactive T cells, along with decreased Treg populations, is a hallmark of autoimmunity [42,43].
Sex-specific differences further shape adaptive immune responses. In the humoral compartment, females exhibit higher B cell counts and a greater propensity for differentiation into autoantibody-producing plasma cells compared with males [34]. These autoantibodies activate the classical complement pathway, perpetuating inflammation and tissue damage [44]. Elevated BAFF/APRIL activity in females may further reinforce these responses, contributing to the higher severity and mortality of autoimmune diseases [34].
Females also display heightened lymphocyte activation, increased CD4⁺ Th cell counts, elevated cytokine production (e.g., ILs, IFNs), and a higher CD4⁺/CD8⁺ T cell ratio, resulting in stronger—yet often pathological—immune responses [34,45]. In contrast, males exhibit a predominance of Treg cells, contributing to weaker immune responses [46]. Th17-skewed inflammation is more prominent in females and is closely associated with autoimmune diseases such as SLE and RA. Additionally, sex-specific differences in APC function, including MHC-II usage, modulate TCR signaling thresholds and immune tolerance [47]. Overexpression of X-linked immune genes (e.g., CD40L, CXCR3, OGT) in females further amplifies immune responses.
3. Alterations of Immune and Inflammatory Responses in SLE and SS
Despite distinct clinical presentations, SLE and SS share core immunopathological features, including loss of self-tolerance, hyperactivation of innate and adaptive immune cells, and persistent production of proinflammatory cytokines and autoantibodies. These dysregulated responses contribute tissue damage and disease progression. The following sections outline the specific immune and inflammatory alterations characteristic of each disease.
3.1. Immune and Inflammatory Responses Manifested in SLE
SLE pathogenesis involves innate and adaptive immune activation, cytokine dysregulation, autoantibody production, and immune complex deposition, ultimately driving chronic inflammation and organ damage [19,48] (Figure 5). A hallmark feature is the production of antinuclear antibodies (ANAs) against double-stranded DNA (anti-dsDNA) and small nuclear ribonucleoproteins (snRNPs), such as Smith protein (Sm) and Sjogren’s autoantibodies (SSA, and SSB) [33]. However, the data regarding their effectiveness as a predictive marker for LN is not fully established [18]. Upregulation of IFN-stimulated genes (ISGs), transcription factors (e.g., STAT3, TASL), and proinflammatory cytokines (e.g., IL-6, TNF-α) links innate activation to adaptive immune dysregulation, promoting B cell activation, autoantibody production, and tissue inflammation [49].
Enhanced TLR7 signaling is central to SLE pathogenesis through (1) increased TLR7 expression via gene duplication or stabilizing SNPs, (2) sustained activation by endosomal ligand accumulation, and (3) gain-of-function mutations lowering activation thresholds [28,33,50,51]. In plasmacytoid and myeloid DCs (pDCs and mDCs), TLR7 drives IFN-I and cytokine production, and chemokines migration to inflammatory sites, promoting autoimmunity and tissue damage [52]. RNA-containing immune complexes further activate TLR7/8, sustaining inflammation via the JAK–STAT pathway and promoting autoreactive B cell differentiation into autoantibody-producing cells through Bruton’s tyrosine kinase (BTK), BAFFR, TACI, CD19, and CD20 [53].
B–T cell interactions amplify adaptive immune dysregulation. CD40–CD40L engagement promotes B cell activation, class switching, and production of IgA, IgG, and IgM autoantibodies, forming immune complexes that deposit in tissues—particularly the kidneys [17,18,54]. B cells also act as APCs, activating autoreactive CD4⁺ Th cells and CD8⁺ Tctx, which further sustain inflammation through IFN-I and IFN-γ production [55]. Altered TCR signaling and endocytic recycling in Th cells promote proinflammatory polarization and impair Treg function, contributing to systemic inflammation [43,56]. Genetic, hormonal, and environmental factors exacerbate these immune disturbances.
CD8⁺ Tctx cells in SLE show impaired cytotoxic function despite elevated activation markers (CD38, HLA-DR), contributing to both defective pathogen clearance and autoimmune tissue damage [57]. γδ T cells and IL-15–driven CD4⁺ CD28⁻ T cells also promote tissue injury in LN through antigen presentation and proinflammatory cytokine secretion [58]. Tctx cells targeting modified self-antigens contribute directly to tissue damage [59], while altered immunometabolism, marked by increased glycolysis and oxidative stress, exacerbates inflammation in both T and B cells [60]. Autoreactive B cells further sustain inflammation by functioning as APCs and producing cytokines. Loss of B cell tolerance is driven by BCR and IFN-I signaling from pDCs, while Tfh cells promote B cell activation through IL-4, IL-17, IL-21, and IFN-γ [61].
Sex-based immune differences play a crucial role in the pathogenesis of SLE. Females exhibit broader organ involvement and higher autoantibody titers, while males often present later with more severe renal disease and higher mortality. Effective treatment of SLE requires a multifaceted approach targeting these interconnected pathways.
3.2. Immune and Inflammatory Responses Manifested in SS
SS is a multifactorial autoimmune disease characterized by immune dysregulation, with elevated proinflammatory cytokines and autoantibodies driving chronic immune activation and glandular damage [62]. Disease onset is primarily mediated by innate immune overactivation, particularly through IFN-I signaling. Activated pDCs produce IFN-I, BAFF, and APRIL via the JAK–STAT pathway (Figure 6), further stimulating macrophages, NK cells, and CD8⁺ Tctx cells [63]. Tissue damage activates TLRs, rapidly inducing IFN-I and initiating proinflammatory cascades [64].
As the disease progresses, adaptive immunity sustains chronic inflammation through (1) autoreactive B cell activation and autoantibody production, (2) B cell–mediated T cell activation and cytokine release, and (3) lymphocyte infiltration of exocrine glands. IFN-I promotes BAFF production, enhancing B cell survival and activation. Stimulated by BCR, BAFF/APRIL-TACI, and TLR signaling, B cells produce pathogenic autoantibodies, notably anti-SSA/Ro and anti-SSB/La, contributing to glandular dysfunction [65].
Autoreactive B cells also act as APCs, engaging CD4⁺ Th cells via MHC-II and CD40–CD40L, promoting proinflammatory cytokine release (e.g., IFN-γ, IL-1, IL-6, TNF-α) and skewing T cell polarization toward Th1 and Th17 phenotypes while reducing Th2 and Treg subsets [66]. IFN-λ further reinforces Th1 and CD8⁺ Tctx responses, perpetuating chronic inflammation and reactivating innate pathways through cytokines, BAFF, and immune complexes, establishing a self-sustaining inflammatory loop and characteristic IFN signature [67]. Damaged epithelial cells also contribute as nonprofessional APCs, maintaining immune activation.
SS shows a pronounced female predominance (female-to-male ratio 9:1 to 14:1) [9]. Women typically exhibit stronger humoral responses and higher autoantibody levels, while men present with more severe systemic complications, including vasculitis and pulmonary involvement. Musculoskeletal symptoms (e.g., arthralgia, myalgia) are more frequent in women, whereas men show greater CD8⁺ Tctx infiltration in glandular tissues. The complex interplay between innate and adaptive immunity, along with sex-specific immune variations, presents challenge in SS management. Therapeutic strategies targeting IFN signaling, B-T cell interactions, and cytokine pathways hold promise. Understanding sex-specific immunological differences will be critical for developing more effective, personalized treatments.
4. Sex-Specific Immune Mechanisms in Autoimmune Diseases
Understanding sex-specific differences in immune and inflammatory responses is crucial for improving the diagnosis, treatment, and long-term management of autoimmune diseases, particularly through the development of personalized therapies. This section examines the key contributors to sex bias in autoimmunity, focusing on the roles of sex chromosome–linked genes, sex hormones, and their interaction with environmental factors in modulating immune cell function and disease susceptibility
4.1. Sex-Linked Genetic Factors
4.1.1. Escape from X Chromosome Inactivation
Sex-based disparities in autoimmune disease prevalence persist even in hormone-independent contexts, such as juvenile rheumatic diseases and postmenopausal women, underscoring a critical role for sex chromosomes in disease pathogenesis [45,68]. Comparable hormone levels in prepubescent boys and girls with SLE or SS further support the contribution of X chromosome–linked genetic susceptibility [69].
Females possess two X chromosomes, while males have one. To maintain dosage compensation, one X chromosome undergoes random inactivation during early embryogenesis in females [70]. However, 20–30% of X-linked genes escape X chromosome inactivation (XCI), leading to functional gene dosage imbalances [69,71]. As the X chromosome carries significantly more (11-fold) immune-related genes than the Y chromosome, this escape amplifies immune-regulatory gene expression, thereby increasing autoimmune susceptibility in females [45,72,73]. Clinical observations support this: SLE is rare in Turner syndrome (45,X) but markedly increased (14-fold) in Klinefelter syndrome (47,XXY) and 47,XXX females [74]. Single-cell analyses show biallelic expression of TLR7 in pDCs, B cells, and monocytes from 46 XX women and 47 XXY Klinefelter males due to skewed XCI [50].
XCI is regulated by the long noncoding RNA Xist, which recruits silencing complexes to epigenetically repress one X chromosome [75]. Disruptions in Xist expression or associated protein complexes, observed in thymocytes and peripheral T cells of SLE patients, result in incomplete XCI and reactivation of X-linked immune genes [76]. Together, skewed XCI, gene dosage imbalance, and epigenetic dysregulation contribute significantly to the female predominance in autoimmune diseases [45,77,78].
4.1.2. Immune-Associated Genes Escaping XCI
A subset of immune-related genes on the X chromosome escapes XCI, resulting in biallelic expression in females and individuals with Klinefelter syndrome [30,51,69,79]. Key genes include TLR7, TLR8, CD40L, CXCR3, IRAK1, BTK, FoxP3, CXorf21, and CYBB (Table 1).
Overexpression of TLR7 and TLR8, encoding endosomal pattern recognition receptors, enhances IFN-I signaling and proinflammatory cytokine production, particularly in pDCs, driving upregulation of IFN-stimulated genes and predisposing females to SLE and related autoimmune diseases [50,77,80]. These receptors also promote B cell activation, class-switching, and autoantibody production, central to SLE pathogenesis [81]. Notably, TLR7-mediated IFN-I production is amplified by estrogen signaling, highlighting a convergence of genetic and hormonal effects.
CD40L, expressed on activated T cells, promotes pathogenic Th cell responses and B cell activation. Elevated CXCR3 and IRAK1 enhances T cell trafficking and innate immune signaling, while BTK escape supports autoreactive B cell survival. Dysregulation of FoxP3, critical for Treg development and function, impairs peripheral tolerance. CXorf21 (TASL) amplifies IFN-I responses in pDCs and monocytes. CYBB encodes a NOX2 subunit essential for ROS production in phagocytes.
X-linked cytokine receptors (IL13RA1/2, IL2RG, IL9R) contribute to lymphocyte development and sex-biased immune regulation. Epigenetic regulators KDM6A (UTX) and KDM5C (JARID1C), which modulate histone marks, are active in females and associated with increased autoimmune risk [69,82]. KDM6A, in particular, enhances NK cell function, as evidenced by reduced IFN-γ production in male and KDM6A-deficient female NK cells [83].
Collectively, these findings underscore the critical role of X–linked gene dosage in shaping immune responses and driving female-biased autoimmunity [84]. Further research into cell type–specific expression and the functional impact of XCI escape genes is essential for advancing precision medicine in autoimmune diseases.
4.1.3. Genetic Variations Across the Genome
Sex differences in immune responses are shaped by a complex interplay of X-linked gene expression, XCI, and genome-wide genetic variations. Immune regulatory gene variants and HLA alleles interact with sex to influence disease susceptibility and progression [26]. Recent genome-wide association studies (GWAS) have identified over 300 loci associated with sex-biased immune responses, underscoring the intricate genetic contributions to autoimmunity [85]. Notably, most autoimmune-associated GWAS variants lie in non-coding regions and are believed to regulate gene expression through long non-coding RNAs (lncRNAs), which may interact with sex hormones and environmental triggers to modulate immune responses [86]. However, the mechanisms linking these variants to sex-biased autoimmunity remain poorly understood, highlighting the need for functional validation and integrative genomic studies.
X-linked variants play a particularly significant role in sex-biased autoimmunity. Variants in genes such as TLR7, FoxP3, IRAK1, and MECP2 are strongly associated with increased SLE risk [87]. A notable gain-of-function variant of TLR7 (Y264H), localized in its ligand-binding domain, enhances its affinity for guanosine-rich ligands, promoting aberrant activation of innate and adaptive immunity by single-stranded RNAs and leading to spontaneous lupus-like disease in kika model mice [88]. This disease phenotype is reversed by MyD88 deletion, underscoring the pathogenic role of the TLR7–MyD88 axis [51]. Additionally, TLR7 polymorphisms such as rs3853839 and rs179019 have been linked to increased transcript levels and heightened SLE susceptibility, although results remain inconsistent across studies [89]. In contrast, TLR9 appears to exert a protective effect, as its deficiency exacerbates disease severity in animal models [90].
Variants affecting the regulation of TLR7 signaling further contribute to disease risk. Risk variants in SLC29A3, which modulates TLR7 ligand export from endosomes, lead to nucleoside accumulation and enhanced TLR7 activation. Specifically, rs780669 has been associated with reduced SLC29A3a expression in monocytes from Asian SLE patients [91]. Mutations in FoxP3 are implicated in X-linked immune dysregulation syndromes and increase SLE susceptibility [92]. An X-linked SNP in CCDC22 (rs2294020) has also been associated with enhanced NF-κB activation and increased SLE risk [93].
In addition to X-linked factors, autosomal variants and somatic mutations contribute to autoimmune susceptibility. Gain-of-function variants in the IFN-I pathway, a central pathogenic axis in SLE, have been linked to increased disease risk [94]. Sex hormone receptor gene polymorphisms also modulate disease onset and severity. Notably, HLA class II alleles, particularly HLA-DR, HLA-DQA1, and HLA-DQB1, represent some of the most consistent and robust risk factors for autoimmune diseases, including SLE and SS [22].
4.1.4. Sex-Biased microRNAs and Gene Expression
MicroRNAs (miRs) are small noncoding RNAs (19–24 nucleotides) that regulate gene expression post-transcriptionally. Aberrant miR expression has been reported in immune cells, including peripheral blood mononuclear cells (PBMCs) and T cells, from autoimmune disease patients [95,96]. Notably, sex-specific differences in miR profiles, observed both intracellularly and in circulating extracellular vesicles, modulate T and B cell functions and contribute to sex-biased immune regulation [26,97]. Dysregulated, sex-biased miRs interact with genetic susceptibility loci, transcription factors, and epigenetic modifiers, playing critical roles in the pathogenesis of sex-specific autoimmunity [98]. Consequently, miRs are emerging as potential diagnostic biomarkers, prognostic indicators, and therapeutic targets in autoimmune diseases [95,96,99].
The X chromosome encodes approximately 118 miRs, compared to only 4 on the Y chromosome, contributing to female-biased miR expression in autoimmunity [100]. Skewed XCI further enhances the expression of X-linked miRs. In diseases such as SLE and SS, miRs including miR-20b, miR-23b, miR-98, and miR-222 are frequently downregulated, while miR-106a, miR-223, miR-224, and others are upregulated, supporting their relevance as disease biomarkers and therapeutic targets [101] (Table 2).
In SLE, reduced miR-23b and miR-98 activate NF-κB and STAT3 pathways, promoting proinflammatory cytokine production and autoimmunity [102]. Restoration of these miRs suppresses inflammation and ameliorate disease phenotypes in experimental models [103]. Estrogen may exacerbate disease by repressing anti-inflammatory miR-98 in B cells. Additionally, miR-548m is upregulated in SLE PBMCs, suppressing PTEN and activating the PI3K–AKT pathway, thereby enhancing immune cell survival [104]. Inhibition of miR-548m restores PTEN and attenuates disease progression. In LN, downregulation of miR-222 correlates with increased CFHR5 expression and complement activation, contributing to tissue damage [105]. This mechanism is further supported by evidence that lncRNA MIAT exacerbates inflammation by sponging miR-222, leading to upregulation of CFHR5 [106].
In pSS, downregulation of miR-125b relieves repression on PRDM1, promoting plasma cell differentiation and autoantibody production. Restoring miR-125b via exosomes-mediated delivery suppresses PRDM1 and reduces plasma cell expansion in experiments [107]. Similarly, decreased miR-506 increases NFATC1 expression, enhancing CD4⁺ T cell activation and proliferation. Pharmacologic upregulation of miR-506 using fangchinoline mitigates T cell–mediated inflammation [108].
miR-223 shows disease-specific expression pattern—upregulated in CD4⁺ T cells and glandular tissues in pSS, but downregulated in active LN [109]. It regulates T cell migration and suppresses proinflammatory chemokines such as CXCL2 and CCL3 by targeting S1PR1. In lupus model mice, miR-223 deficiency worsens nephritis, while in SS, its dysregulation promotes epithelial inflammation and cell death [110]. Elevated miR-223 also correlates with altered B cell subset distributions, implicating X chromosome demethylation in the female-biased lupus susceptibility to lupus [96].
Although X-linked miRs play a prominent role, autosomal miRs also contribute to sex-biased autoimmunity. Some function independently of sex hormones, while others are hormonally regulated. For example, miR-21 (Chr17) enhances proinflammatory cytokine production via activation of X-linked TLR8 [111]. Additionally, dysregulation of miR-145 (Chr5) and miR-224 (X-linked) modulates T cell apoptosis through the STAT1 and API5 pathways, particularly in LN [112].
Collectively, both X-linked and autosomal miRs shape sex differences in autoimmune diseases through their complex regulation of immune cell function and inflammatory pathways. Further research into sex-specific miR expression and function will be critical for advancing precision medicine strategies in the diagnosis, prognosis, and treatment of autoimmune diseases.
4.2. Sex Hormones, Pregnancy, and Autoimmunity
4.2.1. Sex Hormones and Autoimmunity
The pronounced female predominance in autoimmune diseases such as SLE and pSS is most evident during reproductive years, implicating sex hormones as critical modulators of disease risk [25,113]. For instance, SLE incidence increases nearly ninefold in females after puberty (ages 15–45), coinciding with elevated estrogen levels [4,114]. In affected women, fluctuations in estrogen and progesterone during menstrual cycles and pregnancy correlate with disease activity, while androgen levels, including testosterone, are often lower [115].
Estrogen promotes immune activation by enhancing B cell responses, increasing autoantibody production, and skewing cytokine profiles toward Th2 dominance [77,116]. It also inhibits activation-induced T cell apoptosis by downregulating Fas ligand (FasL) and prolongs the survival of activated peripheral T cells. While these effects may support pathogen clearance, they also heighten the risk of autoreactivity. In contrast, androgens exert immunosuppressive effects, suppressing B cell responses, promoting Th1/Th17 immune profiles, and limiting pathogenic autoantibody production [23,117]. This hormonal environment contributes to the lower prevalence and severity of autoimmune diseases in males [73]. Thus, the post-pubertal hormonal milieu in females predisposes them to heightened immune activation and autoimmunity [118].
Sex hormones also influence disease onset and severity. In pSS, elevated estrogen levels in middle-aged women are associated with disease development, whereas higher testosterone levels reduce disease severity in animal models [21]. Conversely, the decline of estrogen after menopause promotes glandular apoptosis, increases autoantibody production, and contributes to disease onset. These findings underscore the critical importance of the estrogen-to-testosterone balance in shaping autoimmune disease risk and clinical outcomes.
4.2.2. Pregnancy and Autoimmunity
During pregnancy, rising estrogen and progesterone levels significantly modulate immune responses. Early gestation is characterized by a shift from Th1- to Th2-dominant immunity, increased Treg cells, and a higher Treg/Th17 ratio, promoting to maternal–fetal tolerance and reducing inflammation [5]. Progesterone further suppresses Th1/Th17 responses and B cell activity, potentially mitigating autoimmune flares during pregnancy, particularly in SLE. However, impaired Treg function or pregnancy-related metabolic changes can trigger or exacerbate autoimmunity. Some of these immunological adaptations persist for up to a year postpartum.
Pregnancy also introduces microchimerism—the bidirectional exchange of fetal and maternal cells. Fetal cells can persist in maternal circulation for years, potentially interacting with the maternal immune system and contributing to autoimmune disease development or exacerbation [119]. This may partially explain the higher prevalence of autoimmune diseases in reproductive-aged women. However, pregnancy’s effects on disease course vary: some conditions, such as RA, often improve during gestation, while others, including SLE, may worsen or flare [120].
4.3. Sex Hormone-Dependent Mechanisms of Immune Regulation
Sex hormones regulate immune responses through four key mechanisms: (1) modulation of transcription factor activity, (2) amplification of cytokine signaling pathways, (3) induction of epigenetic modifications, and (4) interaction with environmental factors [73,121]. These processes collectively influence immune cell survival, differentiation, and apoptosis, shaping the immune landscape. Understanding these mechanisms is critical for explaining the female predominance in autoimmune diseases and advancing hormone-based therapeutic strategies for SLE, SS, and other immune-mediated conditions.
4.3.1. Modulation of Transcription Factors
Sex hormones regulate immune function primarily through transcriptional control of key immune-related genes. Estrogen receptors (ERα and ERβ, which exert opposing effects on immune responses) are expressed in most immune cells [122], act as nuclear transcription factors, binding estrogen response element (ERE) to regulate genes such as TLRs, IRF5, IFN-I, ILs, BAFF, UNC93B1, S1PR2, AIRE, AID, and SLC15A4 [53,77,123,124] (Table 3). ERs also participate in membrane-initiated signaling, further enhancing immune cell activation [125].
Estrogens upregulate IRF5, a key risk factor for SLE and SS, promoting IFN-α and proinflammatory cytokine production. In contrast, ERα deficiency reduces IRF5 expression and impairs pDC function [25,113]. Moreover, estrogens decrease AIRE expression, thereby promoting the survival of autoreactive T cells [126]. Conversely, androgen increases its expression, lowering susceptibility of males to develop autoreactive T cells [24]. Estrogens reduces Treg cell numbers by downregulating FoxP3 expression [127]. Progesterone exhibits dose-dependent immunomodulatory effects. At physiological levels, it supports Treg function via FoxP3 and Ikzf2 (Helios) upregulation, promoting maternal–fetal tolerance. PR deficiency in lupus-prone mice leads to reduced Treg and increased Tfh cells [115]. At higher concentrations, progesterone may activate glucocorticoid receptors (GRs) or, under certain conditions, cooperate with estrogen to promote RORγt expression and Th17 differentiation, contributing to inflammation [128].
Sex hormones also regulate other transcription factors central to autoimmunity. Estrogen-ER complexes activate STAT1 and NF-κB, promoting proinflammatory cytokine production and sustaining the IFN signature in SLE [115,129]. Estrogen additionally induces HoxC4, facilitating immunoglobulin class-switch recombination and autoantibody production [129]. In contrast, testosterone and progesterone tend to suppress STAT1, NF-κB, and HoxC4 activity, reducing inflammatory responses and autoimmunity [125,129,130].
Collectively, sex hormones shape immune responses through complex regulation of transcription factors—including IRF5, FoxP3, STAT1, NF-κB, RORγt, and HoxC4—contributing to heightened inflammation and reduced immune tolerance in female-predominant autoimmune diseases such as SLE and pSS.
4.3.2. Amplification of Cytokine Signaling
Aberrant IFN-I signaling is central to the pathogenesis of SLE and SS, with sex hormones serving as critical modulators. In female SLE patients, elevated estradiol correlates with increased expression of IFN-stimulated genes and cytokines such as IL-21 [123]. Estrogen amplifies IFN-I responses by upregulating TLR7, TLR8, and TLR9 expression on B cells and DCs, enhancing sensitivity to nucleic acid ligands—particularly during high-estrogen states such as late menstrual phases and pregnancy [115,123]. This effect is mediated through IRF5 and STAT1 signaling, establishing a feed-forward loop that sustains chronic inflammation [25]. In contrast, testosterone suppresses IFN-I production; androgen depletion in lupus-prone mice leads to increased IFN-α secretion and autoantibody production, effects that are further exacerbated by exogenous estrogen administration [25,115].
Sex hormones also exert differential regulation of the NF-κB pathway, a key mediator of inflammatory responses. Estrogen promotes NF-κB activation by suppressing miR-145 and enhancing IKKε activity, thereby increasing proinflammatory cytokine production. In contrast, testosterone and progesterone inhibit NF-κB signaling, reducing the production of TNF-α, IL-6, and Th1/Th17 cytokines [125,130]. Progesterone, particularly at levels observed during pregnancy, acts through GRs to suppress NF-κB–mediated C-C chemokine ligand 2 (CCL2) expression, supporting immune tolerance [131]. Additionally, progesterone receptor (PR) signaling appears protective in SLE, as PR deficiency exacerbates NF-κB–driven inflammation and worsens disease severity [115].
Beyond IFN-I and NF-κB pathways, sex hormones modulate TLR and BCR signaling, shaping both innate and humoral immune responses. X-linked TLR7 and TLR9 escape XCI and are more highly expressed in females [25]. Estrogen further enhances TLR7/8/9 expression [115] and promotes pDC-driven IFN-α production, reinforcing the IFN signature characteristic of SLE [123]. This involves upregulation of UNC93B1 and MyD88, and direct ERα binding to ERE near the TLR8 locus, further amplifying proinflammatory cytokine production [77,123,132].
In contrast, testosterone downregulates TLR7 expression and dampens IFN-I responses, providing a protective effect against autoimmunity in males. Experimental evidence shows that castration of lupus-prone mice followed by TLR stimulation induces lupus-like pathology, underscoring the protective role of androgens [113,115]. Sex-specific TLR responses also contribute to distinct autoantibody profiles: estrogen promotes anti-RNP/Sm autoantibodies via TLR7, whereas males predominantly produce anti-dsDNA antibodies through TLR9 activation [25].
In SS, estrogen initially protects glandular epithelial cells but sustained exposure maintains chronic TLR activation and inflammation [113]. Estrogen also promotes autoreactive B cell survival by upregulating CD22, SHP-1, and Bcl-2 expression [25]. Conversely, testosterone reduces B cell development and BAFF levels, limiting autoreactive B cell expansion [115]. Although less well characterized, progesterone appears to promote Th2 responses and enhance Treg activity, particularly during pregnancy, by increasing IL-10 production and suppressing TLR-induced inflammation [113].
4.3.3. Induction of Epigenetic Changes
Sex hormones critically influence epigenetic modifications—including DNA methylation, histone modifications, and non-coding RNA regulation—that shape immune gene expression and contribute to sex-based differences in autoimmune susceptibility. In SLE and SS, estrogens promote DNA hypomethylation by inhibiting DNMT1, leading to overexpression of inflammatory genes in CD4⁺ T cells [25,115]. The ESR1 gene (encoding ERα) itself becomes demethylated in SLE T cells, creating a self-amplifying loop of heightened estrogen sensitivity. In contrast, testosterone enhances DNA methylation and silences proinflammatory genes, partly by promoting the development of FoxP3⁺ Treg cells [115]. Progesterone similarly supports Treg expansion and limits Tfh cell differentiation. Supporting this, studies in transgender individuals show that estrogen and anti-androgen therapy reduce DNA methylation at proinflammatory loci, whereas testosterone increases it [133]. These hormone-driven epigenetic changes may underlie long-term, sex-specific immune programming [134,135].
Epigenetic dysregulation also directly contributes to autoimmune disease pathogenesis. In SLE, demethylation of X-linked genes such as CD40L, CXCR3, and OGT in CD4⁺ T cells promotes their overexpression, contributing to the female-biased disease phenotype [82,136]. Similarly, in diseases such as Takayasu arteritis and psoriatic arthritis, altered DNA methylation in CD8⁺ and γδ T cells affects the expression of key inflammatory genes (IL1RN, IL10, IL27, IL32) and components of the TCR signaling cascade [137].
Non-coding RNAs also play a vital role in sex-biased epigenetic regulation. LncRNAs, including Xist, regulate DNA and histone modifications to maintain XCI. Dysregulated Xist expression in female SLE T cells may result in reactivation of X-linked immune genes [138]. Additionally, m6A RNA methylation influences T cell development, RNA stability, and alternative splicing, playing an important role in immune homeostasis [139].
Estrogen-bound ERα further promotes epigenetic reprogramming by recruiting histone acetyltransferases (HATs) such as p300/CBP, increasing histone acetylation at cytokine gene loci and enhancing inflammatory gene expression [129]. Estrogen also enhances AICDA expression, sustaining activation-induced cytidine deaminase (AID) activity and promoting autoantibody production [125,140]. Moreover, estrogen inhibits histone deacetylases (HDACs), further sustaining AID expression and autoantibody production [129]. AID interacts with UBN1, a component of the HIRA histone chaperon complex that regulates chromatin structure [124]. In contrast, HDAC inhibitors reduce B cell differentiation and disease activity in lupus-prone mice, indicating their therapeutic potential [141].
Conversely, androgens promote immune tolerance by recruiting co-repressors to condense chromatin and suppress inflammatory genes. AR binding at the FoxP3 locus alters histone acetylation, enhancing Treg differentiation [115]. AR deficiency leads to increased BAFF levels and B cell hyperactivation, further highlighting its immunoregulatory role.
Incomplete XCI of histone demethylases such as KDM6A (UTX) and KDM5C amplifies immune gene expression in females. Depletion of KDM6A reduces inflammatory cytokine production and tissue damage, underscoring its role in female-biased autoimmunity and its potential as a therapeutic target [25,141].
Finally, environmental exposures—including infections, dietary factors, and xenobiotic agents—interact with sex hormones to modulate epigenetic regulation and influence autoimmune risk [26]. Understanding these complex interactions is critical for identifying sex-specific biomarkers and developing targeted therapies for autoimmune diseases.
4.3.4. Regulation of miR Expression
Sex hormones regulate immune gene expression at the post-transcriptional level by modulating miR expression through receptor-mediated mechanisms, thereby shaping immune cell function [25,129,142]. In SLE, estrogen increases disease-associated miRs in castrated mice, suggesting a pathogenic role for estrogen-regulated miRs in female-biased autoimmunity [77]. Notably, many of these miRs are encoded on autosomes rather than the X chromosome.
Estrogen promotes immune activation by suppressing anti-inflammatory miRs in B cells, including let-7e-5p, miR-98-5p, and miR-145a-5p, leading to increased IKKε expression and enhanced IFN-I signaling [115,130]. Estrogen also downregulates miR-26a, a negative regulator of AICDA (a gene of activation-induced cytidine deaminase, AID), thereby promoting class-switch recombination, somatic hypermutation, and autoantibody production by autoreactive B cells [129]. In T cells, estrogen upregulates miR-10b-5p, inhibiting SRSF1 and skewing cytokine expression toward pro-inflammatory profiles [115].
In contrast, androgens promote immune tolerance by enhancing IL-10 production and expanding FoxP3⁺ Tregs [130]. Dihydrotestosterone (DHT) upregulates miR-26a, reducing AICDA expression and limiting B cell activation [129]. These effects contribute to the immunosuppressive influence of androgens and the lower incidence of autoimmunity in males.
Both estradiol and progesterone also promote IL-17A production by inhibiting let-7f and increasing IL-23R expression, facilitating Th17 differentiation and inflammation [128]. These hormone-regulated miR networks add an important layer of control over immune responses, linking hormonal signaling to key effector pathways in autoimmunity. Table 4 summarizes the chromosomal locations, hormone-regulated expression changes, target genes, and immunological functions of key miRs implicated in SLE and related diseases.
4.4. Interplay with Environmental Factors
Emerging evidence highlights the contribution of environmental factors such as infections, gut microbiota, environmental chemicals and pollutants, and lifestyle behaviors such as diet, smoking, UV exposure, alcohol and caffeine intake, physical activity, and circadian habits to immune dysregulation and the pathogenesis of autoimmune diseases. Major autoimmune risk may be attributable to gene-environment interactions. Sex hormones further modulate these effects through sex-specific interactions with environmental insults. These interactions alter hormone-responsive immune gene expression and epigenetic landscapes, increasing disease susceptibility and severity in females.
4.4.1. Infections
Infectious agents—including viruses (e.g., EBV, CMV, parvovirus B19, HIV, influenza, and SARS-CoV-2) and bacterial components—are major environmental triggers of SLE and pSS [1,143,144]. These pathogens contribute to disease onset and flares by activating innate immunity, inducing IFN-I, recruiting autoreactive lymphocytes, and promoting epitope spreading, molecular mimicry, and bystander activation [145,146]. Any robust immune stimulus could theoretically tip the balance in susceptible individuals. Particularly, intercurrent infections often stimulate the IFN-I pathway, T cell activation, and the formation of neutrophil extracellular traps (NETs), precipitating SLE flares [143,147].
EBV shows the strongest association with SLE and pSS [1,148,149]. SLE patients exhibit higher EBV seropositivity and antibody titers, with elevated (up to 100-fold) latent membrane protein (LMP1) expression in B cells, indicating latent viral reactivation [1,150]. EBV drives autoimmunity by activating TLRs via noncoding RNAs and by molecular mimicry—e.g., EBNA-1 cross-reacts with Sm autoantigens in lupus, and EBNA-2 shares homology with Ro60 in pSS [149]. EBV DNA is frequently detected in salivary glands of SS patients, with a meta-analysis showing strong serological association with pSS [148]. EBV miRNAs may impair glandular function by targeting calcium signaling molecules, while its viral IL-10 homolog fosters local immune tolerance and chronic infection in pSS [149]. Although EBV exposure is nearly universal, only individuals with underlying genetic susceptibility—often associated with HLA-DR variants and high viral loads—develop autoimmune diseases [151]. Taken together, microbial exposure is an important environmental insult that can trigger nucleic-acid sensing pathways and loss of tolerance in genetically susceptible hosts, potentially initiating or exacerbating SLE and pSS in females.
Sex hormones modulate antiviral responses and may contribute to sex-biased autoimmunity. Estrogen enhances TLR7-IFN-α signaling, amplifying responses to viral stimuli in females [123], whereas testosterone dampens inflammation, reducing SLE flares in males [25].
Despite their immunosuppressive treatment burden, preventive measures like vaccination remain underutilized in SLE, though vaccine-associated autoimmunity appears rare and unsupported at the population level [1,152]. Persistent viral infections serve as chronic immune stimuli in genetically predisposed hosts, driving IFN-I production, immune dysregulation, and loss of tolerance in both SLE and pSS [153].
4.4.2. Gut Microbiota
The gut microbiota is a key modulator of immune homeostasis and mediates host–environment interactions. Gut dysbiosis contributes to autoimmunity through immune activation, gut barrier disruption, and microbial mimicry [146,154,155]. In lupus-prone models, microbiota depletion reduces inflammation predominantly in females, highlighting sex-dependent microbial influences [156]. Estrogen fosters a proinflammatory microbiome, while androgens promote protective profiles [146]. Microbiota transfer studies show male-derived microbiota can protect against T1D and lupus in female mice [113]. Gut microbes also regulate sex hormone levels, influencing disease trajectories [157]. Microbial metabolites interacting with ERs and PPARs further modulate immune responses.
In SLE, female patients exhibit reduced microbial diversity and enrichment of pathobionts like Ruminococcus (Blautia) gnavus (RG), which correlates with disease activity and LN [158]. RG strains from SLE patients increase gut permeability and translocate to lymphoid tissues, inducing systemic inflammation, whereas Lactobacillus exerts protective effects [159]. Some RG antigens cross-react with anti-dsDNA antibodies, exemplifying molecular mimicry [155]. Other gut microbes, such as Bacteroides and Odoribacter, express autoantigen-mimicking peptides (e.g., Ro60, Sm) that activate IFN-γ/IL-17–producing T cells or autoantibody responses in murine models [155,160]. Therapeutic strategies targeting gut integrity or composition (e.g., probiotics, zonulin inhibitors) reduce microbial translocation and lupus activity in animal models [155,158]. Thus, dysbiosis acts as an environmental amplifier of SLE by disrupting mucosal tolerance and promoting systemic autoimmunity.
Emerging data indicate similar microbial disturbances in pSS. Patients show decreased microbial richness and lower levels of beneficial commensals (e.g., Bifidobacterium, Agathobacter), alongside increased Prevotella, which is linked to Th17-driven inflammation and dry eye severity [161]. Gut microbiota from pSS patients can reduce Tregs and enhance Th17 responses in recipient mice [162], implicating dysbiosis in systemic immune dysregulation. Additionally, gut-derived metabolites and translocated microbial products may trigger innate immune pathways and cytokine release that affect glandular function.
Although mechanistic studies are ongoing, molecular mimicry involving glandular autoantigens (e.g., Ro/La, muscarinic receptors) is a plausible link. Collectively, these findings suggest that gut microbiota imbalances in SLE and pSS contribute to immune dysfunction and symptom exacerbation, representing a promising target for therapeutic intervention of sex-biased autoimmune diseases.
4.4.3. Environmental Chemicals and Pollutants
Environmental exposures are critical contributors to systemic autoimmunity. Low concordance rates for SLE among monozygotic twins (~24%), geographic clustering near polluted areas, and urban–rural differences underscore the role of external factors [134,163]. Chemical agents—including pesticides, bisphenol A (BPA), silica, air pollutants, and heavy metals—are linked to increased SLE risk [164,165]. In particular, BPA acts as an endocrine disruptor, promoting autoimmunity via estrogenic signaling [166], while silica and asbestos induce oxidative stress, Treg depletion, and proinflammatory cytokine production [167]. Exposure to other xenobiotics, such as drugs, cosmetics, food additives, plant constituents, and environmental pollutants induces oxidative stress and mitochondrial DNA release, activating innate immune pathways [26]. Sex hormones modulate susceptibility to these environmental insults. Estrogen amplifies immune responses to oxidative and apoptotic stress, lowering the threshold for autoimmunity, while testosterone offers partial protection unless exposure is severe [25,26].
Silica exposure, particularly in occupational settings (e.g. miners, sandblasters), is a well-established SLE risk factor [165]. Inhaled silica particles trigger cell death and innate immune activation, promoting IFN-I responses and ectopic lymphoid formation in lungs of lupus-prone mice, accelerating loss of tolerance and autoantibody production [1,168]. Another class of harmful exposures is heavy metals. Mercury, in particular, is a pro-inflammatory immunotoxicant that can modify host proteins (creating neoantigens), disrupt redox homeostasis, and activate autoreactive B cells, contributing to lupus-like features in both humans and animal models.
Agricultural exposures are particularly relevant. High cumulative pesticide exposure significantly increases the risk of SLE or pSS overtime, with herbicides like metribuzin linked to more than fivefold elevated risk in older individuals [165]. These findings suggest certain pesticides can act as triggers, potentially via immune-adjuvant effects or direct toxicity to lymphocytes. However, not all chemicals have the same impact and that some correlations may reflect complex behavioral or exposure patterns. Notably, early-life farm residence has been associated with reduced risk, supporting the hygiene hypothesis.
Organic solvents and pollutants are another concern. Occupational contact with solvents (such as trichloroethylene or benzene derivatives) has been associated with SLE development, possibly through mechanisms of oxidative stress and epigenetic changes in immune cells [165]. Air pollutants, including particulate matter 2.5 (PM₂.₅), nitrogen dioxide (NO₂), and polycyclic aromatic hydrocarbons (PAHs), have been associated with increased lupus incidence and disease flares [169,170]. Mechanistically, inhaled PM particles alter DNA methylation in immune cells, upregulate IFN-responsive genes, and promote ongoing disease by epigenetic reprogramming of immune responses [171]. NO₂ exposure is linked to higher hospitalization rates and mortality in SLE patients, likely through oxidative stress–induced immune dysregulation [169]. While NO₂ affects both sexes, sex-specific susceptibilities remain to be fully clarified. PAH, a component of smoke and smog, can activate aryl hydrocarbon receptors on immune cells, skewing T cell differentiation and enhancing autoreactive B cell survival.
While fewer studies have examined chemical exposures in pSS, shared environmental risk factors are likely [165]. Gut and lung immune activation by xenobiotics, including silica and pesticides, may promote glandular autoimmunity. Dysregulated clearance of cellular debris, cytokine production (e.g., IL-1, TNFα), and IFN-I activation underlie a common immunopathogenic axis in both SLE and pSS.
Taken together, environmental chemicals and air pollutants act as potent triggers or amplifiers of autoimmunity, especially in genetically predisposed individuals. Avoidance strategies—such as limiting pesticide use, minimizing air pollution exposure, and using protective equipment—may reduce disease risk and progression.
4.4.4. Lifestyle Behaviors
Lifestyle behaviors—such as diet, smoking, UV exposure, alcohol and caffeine intake, physical activity, and circadian habits—profoundly shape autoimmune disease risk and severity [163]. Diet and nutritional status influence immune responses via modulation of inflammation, gut microbiota, and hormone interactions [172]. In lupus, high-fat and high-sugar diets that lead to obesity may worsen SLE outcomes through proinflammatory adipokines (e.g., IL-6, TNFα), while fiber-rich, low-carbohydrate diet or Mediterranean diets rich in antioxidants and polyunsaturated fats may attenuate systemic inflammation and disease activity [1]. In pSS, dietary research is limited, but adherence to anti-inflammatory diets correlates with lower disease scores and improved hydration supports symptomatic relief [173]. Adequate hydration and avoidance of diuretic substances (like caffeine or alcohol) are often recommended in pSS to help manage dryness, though these are symptomatic measures. These dietary factors modulate inflammation, gut microbiota composition, and interact with sex hormones [172].
Cigarette smoking is a prominent lifestyle factor impacting systemic autoimmunity, though its effects differ between SLE and pSS. In SLE, current smoking exacerbates disease risk and morbidity by inducing DNA adducts, oxidative DNA damage, upregulation of BAFF/BLyS/TNFα/IL-6, activation of IFN-I, increased anti-dsDNA autoantibody production, and NETosis, wherein neutrophils release NETs to trap and neutralize pathogens [1,174]. Smokers also show reduced IL-10 and increased disease activity and organ damage. Therefore, smoking cessation is strongly recommended for those at risk of or living with SLE, as it removes a significant pro-inflammatory stimulus to the immune system [175]. In contrast, some studies suggest current smoking may correlate with lower pSS risk [173], possibly due to nicotine’s immunomodulatory effects or disease-related aversion to smoking. Nonetheless, smoking cessation is universally recommended for immune and general health.
UV exposure, particularly UVB, is a well-established environmental trigger in flares and cutaneous lupus lesions. UVB induces keratinocyte apoptosis, exposing nuclear antigens and promoting IFN-I signaling, IFN-regulated gene expression, and T cell activation [1,176]. UV-damaged keratinocytes can externalize autoantigens like Ro/SSA, which then incite autoantibody responses. Photosensitivity affects up to 70% of patients, and UV exposure often precedes flares. Females are more susceptible to UV radiation due to hormonal factors. Sunscreen use has been shown to reduce cutaneous and systemic activity [1]. However, UV also contributes to vitamin D synthesis—often deficient in SLE—necessitating a balance between sun protection and supplementation [177]. In pSS, UV associations are weaker as the primary target organs (glands) are internal. However, may trigger symptoms in Ro/SSA-positive or lupus-overlap patients. Photoprotection is generally advised if patients have such symptoms.
Circadian disruption (e.g., shift work, insufficient sleep) is linked to autoimmunity through hormonal rhythm disturbance and immune imbalance [178]. Sleep deprivation increases IL-6 and TNFα levels and reduces Treg function, leading to a pro-inflammatory cytokine milieu and loss of self-tolerance, and promoting autoreactivity [179]. Estrogen fluctuations during menstrual cycles correlate with autoimmune flares in SLE and SS [115]. Nighttime light exposure suppresses melatonin, potentially amplifying estrogen-driven inflammation. Chronotherapy, aligning treatment with circadian and hormonal cycles, holds promise for improving disease management.
Moderate alcohol and caffeine consumption may exert anti-inflammatory effects, while excessive intake promotes immune dysregulation and autoimmunity [180]. Regular physical activity is protective, likely through anti-inflammatory and metabolic benefits. Collectively, lifestyle factors interact with genetic and hormonal contexts to modulate immune tolerance. In both SLE and pSS, interventions targeting modifiable behaviors—such as diet, smoking, UV protection, and sleep hygiene—offer practical strategies to reduce disease onset and flares. While the precise biological mechanisms are still being explored, maintaining healthy lifestyle habits – no smoking, balanced diet, regular exercise, and sufficient sleep– is generally thought to support immune tolerance and reduce the chances of autoimmune disease onset or flare. One notable nuance is sun exposure: UV light is a lifestyle factor (related to outdoor activity) that can trigger lupus flares (see below), yet moderate sun exposure is also needed for vitamin D synthesis, which has protective immunoregulatory effects. Patients are encouraged to find a balance (using sunscreens and vitamin D supplements as needed) [1].
4.4.5. Psychological and Physical Stressors
Psychological stress is a potent environmental insult that modulates immune responses via the hypothalamic–pituitary–adrenal (HPA) axis and neuroimmune circuits. Chronic stress initially elevates cortisol, but immune cells can become resistant over time, leading to unchecked sympathetic activation and increased pro-inflammatory cytokines [181]. In both SLE and pSS, patients frequently report disease flares following emotional stress or trauma. Post-traumatic stress disorder (PTSD) and major life events are associated with significantly elevated autoimmune risk—doubling the odds of SLE and increasing the risk of pSS [173,182].
Stress alters regulatory immune functions by disrupting Treg activity and promoting inflammatory mediators such as substance P and catecholamines. Neuroendocrine-immune crosstalk may also affect glandular inflammation in pSS by altering salivary gland blood flow and immune cell infiltration. These mechanisms underscore that stress is not merely a secondary factor but an active contributor to disease pathogenesis.
Physical trauma and environmental factors such as extreme cold or injury may act as "second hits" that expose sequestered antigens and initiate autoimmune responses in genetically predisposed individuals. Raynaud’s phenomenon, common in lupus and sometimes in pSS, is often triggered by cold exposure. Infections or surgical trauma have also been reported as preceding events in disease onset.
Air pollutants (e.g., particulate matter) can damage lung tissue, increase antigen exposure, and activate innate immune sensors, thereby promoting systemic autoimmunity. These environmental insults often interact with hormonal and genetic susceptibilities, shaping sex-specific disease trajectories.
Together, stress, trauma, and pollution contribute to the multifactorial nature of SLE and pSS. They act through diverse mechanisms—oxidative stress, antigen exposure, neuroendocrine imbalance, and IFN-I pathway activation—lowering the threshold for immune tolerance breakdown. Understanding these interactions is critical for advancing sex-informed, preventive, and therapeutic strategies against autoimmunity.
5. Conclusions
Autoimmune diseases arise from inappropriate innate and adaptive immune responses to self-antigens, leading to loss of self-tolerance and chronic tissue damage. Growing evidence underscores the profound impact of biological sex on immune function and autoimmune pathogenesis. Females not only have a higher prevalence of autoimmune diseases but also experience more severe clinical manifestations and frequent disease flares compared to males. These disparities highlight the need to consider sex as a fundamental biological variable in immunological research, diagnostics, and clinical trial design.
Sex-specific immune differences result from complex interactions between intrinsic factors—including X chromosome-linked immune gene dosage, genetic variants, miRs, and sex hormone–dependent regulation of transcription, cytokine signaling, epigenetic modifications, and miR expression. These mechanisms shape immune gene expression, immune cell function, cytokine production, self-tolerance, and activation thresholds of inflammatory pathways (Figure 7). Extrinsic environmental factors—including infections, gut microbiota, chemical exposures, lifestyle behaviors, psychological stress, and circadian disruption—further modulate immune responses in a sex-dependent manner.
Together, these factors drive distinct immune gene expression profiles and contribute to sex-specific disease trajectories. Advancing our understanding of these complex mechanisms will improve risk prediction, enable earlier and more accurate diagnoses, support patient stratification into immunologically homogeneous subgroups, and inform the development of personalized, more effective immunotherapies.
6. Future Directions
Future research should focus on elucidating the precise molecular mechanisms underlying the complex interplay among genetic, hormonal, and environmental factors in immune regulation, ideally through integrated multi-omics approaches. Such studies will enhance risk prediction and therapeutic strategies. The development of sex-specific immunotherapies and the consistent inclusion of sex as a biological variable in experimental design, data analysis, and reporting are crucial for advancing personalized prevention and treatment strategies in autoimmunity. Longitudinal, sex-stratified cohort studies and clinical trials are also essential to clarify the influence of sex and hormonal fluctuations on autoimmune disease progression and treatment response
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) with grants given to I.K. and Y.J.J. from the Korean government (MIST) (No. NRF-2018R1A6A1A03025124 and 2021R1A5A2030333, respectively).
Author Contributions
Conceptualization, E.-J.Y., and I.K.; original draft preparation and illustration, E.J.J., Y.R.K, and E.-J.Y.; supervision and edition, E.J.J. and E.-J.Y.; funding acquisition, I.K. and E.J.J. All authors have read and agreed to the published version of the manuscript
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
ABC, age-associated B cell; APCs, antigen-presenting cells; API5, apoptosis inhibitor 5; APRIL, a proliferation-inducing ligand; BAFF, B cell activating factor; BAFFR, B cell activating factor receptor; BCR, B cell receptor; BTK, Bruton’s tyrosine kinase; CXCR, C-X-C motif chemokine receptor; DCs, dendritic cells; ERE, estrogen response element; FoxP3, forkhead box P3; IFN-I; type I interferon; IL, interleukin; iNKT cell, invariant natural killer T cell; IRAK, interleukin-1 receptor-associated kinase; IRF, interferon regulatory factor; JAK-STAT, Janus kinase-signal transducer and activator of transcription; LN, lupus nephritis; MAPK, mitogen-activated protein kinase; MHC, major histocompatibility complex; miR, microRNA; MS, multiple sclerosis; mTOR, mechanistic target of rapamycin; MyD88, myeloid differentiation factor 88; NF-κB, nuclear factor kappa B; NK cell, natural killer cell; PBMCs, peripheral blood mononuclear cells; pDC, plasmacytoid dendritic cell; PDCD4, a selective protein translation inhibitor; pSS, primary Sjögren's syndrome; RA, rheumatoid arthritis; ROR, retinoid-acid receptor related orphan receptor; ROS, reactive oxygen species; SD, scleroderma; SLC15A4, the endolysosomal solute carrier family 15 member 4; SLE, systemic lupus erythematosus; Sm, Smith protein; SOCS1, suppressor of cytokine signaling 1 (a negative regulator of the JAK/STAT pathway); TACI, transmembrane activator and calcium moderator and cyclophilin ligand interactor; SS, Sjögren’s syndrome; SSc, systemic sclerosis; TAK1, transforming growth factor-β-activated kinase 1; TASL, TLR adaptor interacting with SLC15A4; TCR, T cell receptor; Tctx cells, cytotoxic T cells; Th cells, helper T cells; TGF-β, transforming growth factor-beta; TLR, toll-like receptor; TNF-α, tumor necrosis factor-alpha; Treg cells, regulatory T cells; UTX, ubiquitously transcribed tetratricopeptide repeat, X chromosome.
References
- Dai, X.; Fan, Y.; and Zhao, X. Systemic lupus erythematosus: Updated insights on the pathogenesis, diagnosis, prevention and therapeutics. Signal Transduct Target Ther, 2025, 10. 102. [CrossRef]
- Peckham, H.; Webb, K.; Rosser, E.C.; Butler, G., et al. Gender-diverse inclusion in immunological research: Benefits to science and health. Front Med (Lausanne), 2022, 9. 909789. [CrossRef]
- Conrad, N.; Misra, S.; Verbakel, J.Y.; Verbeke, G., et al. Incidence, prevalence, and co-occurrence of autoimmune disorders over time and by age, sex, and socioeconomic status: A population-based cohort study of 22 million individuals in the uk. Lancet, 2023. [CrossRef]
- Desai, M.K.; Brinton, R.D. Autoimmune disease in women: Endocrine transition and risk across the lifespan. Front Endocrinol (Lausanne), 2019, 10. 265. [CrossRef]
- Angum, F.; Khan, T.; Kaler, J.; Siddiqui, L., et al. The prevalence of autoimmune disorders in women: A narrative review. Cureus, 2020, 12. e8094. [CrossRef]
- Izmirly, P.M.; Buyon, J.P.; Wan, I.; Belmont, H.M., et al. The incidence and prevalence of adult primary sjogren's syndrome in new york county. Arthritis Care Res (Hoboken), 2019, 71. 949-960. [CrossRef]
- Izmirly, P.M.; Parton, H.; Wang, L.; McCune, W.J., et al. Prevalence of systemic lupus erythematosus in the united states: Estimates from a meta-analysis of the centers for disease control and prevention national lupus registries. Arthritis Rheumatol, 2021, 73. 991-996. DOI: 10.1002/art.41632,Tian, J.; Zhang, D.; Yao, X.; Huang, Y., et al. Global epidemiology of systemic lupus erythematosus: A comprehensive systematic analysis and modelling study. Ann Rheum Dis, 2023, 82. 351-356. DOI: 10.1136/ard-2022-223035.
- Barber, M.R.W.; Drenkard, C.; Falasinnu, T.; Hoi, A., et al. Global epidemiology of systemic lupus erythematosus. Nat Rev Rheumatol, 2021, 17. 515-532. [CrossRef]
- Seror, R.; Chiche, L.; Beydon, M.; Desjeux, G., et al. Estimated prevalence, incidence and healthcare costs of sjogren's syndrome in france: A national claims-based study. RMD Open, 2024, 10. [CrossRef]
- Royle, J.G.; Lanyon, P.C.; Grainge, M.J.; Abhishek, A., et al. The incidence, prevalence, and survival of systemic sclerosis in the uk clinical practice research datalink. Clin Rheumatol, 2018, 37. 2103-2111. DOI: 10.1007/s10067-018-4182-3,Bairkdar, M.; Rossides, M.; Westerlind, H.; Hesselstrand, R., et al. Incidence and prevalence of systemic sclerosis globally: A comprehensive systematic review and meta-analysis. Rheumatology (Oxford), 2021, 60. 3121-3133. DOI: 10.1093/rheumatology/keab190.
- Myasoedova, E.; Davis, J.; Matteson, E.L.; and Crowson, C.S. Is the epidemiology of rheumatoid arthritis changing? Results from a population-based incidence study, 1985-2014. Ann Rheum Dis, 2020, 79. 440-444. DOI: 10.1136/annrheumdis-2019-216694,Venetsanopoulou, A.I.; Alamanos, Y.; Voulgari, P.V.; and Drosos, A.A. Epidemiology and risk factors for rheumatoid arthritis development. Mediterr J Rheumatol, 2023, 34. 404-413. DOI: 10.31138/mjr.301223.eaf,Collaborators, G.B.D.R.A. Global, regional, and national burden of rheumatoid arthritis, 1990-2020, and projections to 2050: A systematic analysis of the global burden of disease study 2021. Lancet Rheumatol, 2023, 5. e594-e610. DOI: 10.1016/S2665-9913(23)00211-4.
- Calissendorff, J.; Cramon, P.K.; Hallengren, B.; Khamisi, S., et al. Long-term outcome of graves' disease: A gender perspective. Womens Health Rep (New Rochelle), 2023, 4. 487-496. [CrossRef]
- Hu, X.; Chen, Y.; Shen, Y.; Tian, R., et al. Global prevalence and epidemiological trends of hashimoto's thyroiditis in adults: A systematic review and meta-analysis. Front Public Health, 2022, 10. 1020709. [CrossRef]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W., et al. Rising prevalence of multiple sclerosis worldwide: Insights from the atlas of ms, third edition. Mult Scler, 2020, 26. 1816-1821. DOI: 10.1177/1352458520970841,Coyle, P.K. What can we learn from sex differences in ms? J Pers Med, 2021, 11. DOI: 10.3390/jpm11101006.
- Caio, G.; Volta, U.; Sapone, A.; Leffler, D.A., et al. Celiac disease: A comprehensive current review. BMC Med, 2019, 17. 142. DOI: 10.1186/s12916-019-1380-z,King, J.A.; Jeong, J.; Underwood, F.E.; Quan, J., et al. Incidence of celiac disease is increasing over time: A systematic review and meta-analysis. Am J Gastroenterol, 2020, 115. 507-525. DOI: 10.14309/ajg.0000000000000523.
- Fava, A.; Petri, M. Systemic lupus erythematosus: Diagnosis and clinical management. J Autoimmun, 2019, 96. 1-13. [CrossRef]
- Roveta, A.; Parodi, E.L.; Brezzi, B.; Tunesi, F., et al. Lupus nephritis from pathogenesis to new therapies: An update. Int J Mol Sci, 2024, 25. [CrossRef]
- Saleem, A.; Zeeshan, B.; Dissanayake, G.; Zergaw, M., et al. Anti-smith antibodies as a predictive factor for developing lupus nephritis in systemic lupus erythematosus patients: A systematic review. Cureus, 2024, 16. e66270. [CrossRef]
- Siegel, C.H.; Sammaritano, L.R. Systemic lupus erythematosus: A review. JAMA, 2024, 331. 1480-1491. [CrossRef]
- Pryor, K.P.; Barbhaiya, M.; Costenbader, K.H.; and Feldman, C.H. Disparities in lupus and lupus nephritis care and outcomes among us medicaid beneficiaries. Rheum Dis Clin North Am, 2021, 47. 41-53. DOI: 10.1016/j.rdc.2020.09.004,Wang, H.; Ren, Y.L.; Chang, J.; Gu, L., et al. A systematic review and meta-analysis of prevalence of biopsy-proven lupus nephritis. Arch Rheumatol, 2018, 33. 17-25. DOI: 10.5606/ArchRheumatol.2017.6127.
- Morthen, M.K.; Tellefsen, S.; Richards, S.M.; Lieberman, S.M., et al. Testosterone influence on gene expression in lacrimal glands of mouse models of sjogren syndrome. Invest Ophthalmol Vis Sci, 2019, 60. 2181-2197. [CrossRef]
- Zhan, Q.; Zhang, J.; Lin, Y.; Chen, W., et al. Pathogenesis and treatment of sjogren's syndrome: Review and update. Front Immunol, 2023, 14. 1127417. [CrossRef]
- Sciarra, F.; Campolo, F.; Franceschini, E.; Carlomagno, F., et al. Gender-specific impact of sex hormones on the immune system. Int J Mol Sci, 2023, 24. [CrossRef]
- Forsyth, K.S.; Jiwrajka, N.; Lovell, C.D.; Toothacre, N.E., et al. The connexion between sex and immune responses. Nat Rev Immunol, 2024, 24. 487-502. [CrossRef]
- Bose, M.; Jefferies, C. Sex bias in systemic lupus erythematosus: A molecular insight. Immunometabolism (Cobham), 2022, 4. e00004. [CrossRef]
- Fairweather, D.; Beetler, D.J.; McCabe, E.J.; and Lieberman, S.M. Mechanisms underlying sex differences in autoimmunity. J Clin Invest, 2024, 134. [CrossRef]
- Duan, T.; Du, Y.; Xing, C.; Wang, H.Y., et al. Toll-like receptor signaling and its role in cell-mediated immunity. Front Immunol, 2022, 13. 812774. [CrossRef]
- Lind, N.A.; Rael, V.E.; Pestal, K.; Liu, B., et al. Regulation of the nucleic acid-sensing toll-like receptors. Nat Rev Immunol, 2022, 22. 224-235. [CrossRef]
- Balka, K.R.; De Nardo, D. Understanding early tlr signaling through the myddosome. J Leukoc Biol, 2019, 105. 339-351. [CrossRef]
- Heinz, L.X.; Lee, J.; Kapoor, U.; Kartnig, F., et al. Tasl is the slc15a4-associated adaptor for irf5 activation by tlr7-9. Nature, 2020, 581. 316-322. [CrossRef]
- Kobayashi, T.; Nguyen-Tien, D.; Sorimachi, Y.; Sugiura, Y., et al. Slc15a4 mediates m1-prone metabolic shifts in macrophages and guards immune cells from metabolic stress. Proc Natl Acad Sci U S A, 2021, 118. [CrossRef]
- Arleevskaya, M.I.; Larionova, R.V.; Brooks, W.H.; Bettacchioli, E., et al. Toll-like receptors, infections, and rheumatoid arthritis. Clin Rev Allergy Immunol, 2020, 58. 172-181. DOI: 10.1007/s12016-019-08742-z,Frasca, L.; Lande, R. Toll-like receptors in mediating pathogenesis in systemic sclerosis. Clin Exp Immunol, 2020, 201. 14-24. DOI: 10.1111/cei.13426.
- Caielli, S.; Wan, Z.; and Pascual, V. Systemic lupus erythematosus pathogenesis: Interferon and beyond. Annu Rev Immunol, 2023, 41. 533-560. [CrossRef]
- Huang, Z.; Chen, B.; Liu, X.; Li, H., et al. Effects of sex and aging on the immune cell landscape as assessed by single-cell transcriptomic analysis. Proc Natl Acad Sci U S A, 2021, 118. [CrossRef]
- Taniuchi, I. Cd4 helper and cd8 cytotoxic t cell differentiation. Annu Rev Immunol, 2018, 36. 579-601. DOI: 10.1146/annurev-immunol-042617-053411,Khantakova, J.N.; Sennikov, S.V. T-helper cells flexibility: The possibility of reprogramming t cells fate. Front Immunol, 2023, 14. 1284178. DOI: 10.3389/fimmu.2023.1284178.
- Cyster, J.G.; Allen, C.D.C. B cell responses: Cell interaction dynamics and decisions. Cell, 2019, 177. 524-540. DOI: 10.1016/j.cell.2019.03.016,Curley, S.M.; Putnam, D. Biological nanoparticles in vaccine development. Front Bioeng Biotechnol, 2022, 10. 867119. DOI: 10.3389/fbioe.2022.867119.
- Chamberlain, C.; Colman, P.J.; Ranger, A.M.; Burkly, L.C., et al. Repeated administration of dapirolizumab pegol in a randomised phase i study is well tolerated and accompanied by improvements in several composite measures of systemic lupus erythematosus disease activity and changes in whole blood transcriptomic profiles. Ann Rheum Dis, 2017, 76. 1837-1844. [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S., et al. Novel immune checkpoint targets: Moving beyond pd-1 and ctla-4. Mol Cancer, 2019, 18. 155. DOI: 10.1186/s12943-019-1091-2,Nishizaki, D.; Kurzrock, R.; Miyashita, H.; Adashek, J.J., et al. Viewing the immune checkpoint vista: Landscape and outcomes across cancers. ESMO Open, 2024, 9. 102942. DOI: 10.1016/j.esmoop.2024.102942.
- Qin, Y.; Bao, X.; and Zheng, M. Cd8(+) t-cell immunity orchestrated by inkt cells. Front Immunol, 2022, 13. 1109347. [CrossRef]
- Sogkas, G.; Atschekzei, F.; Adriawan, I.R.; Dubrowinskaja, N., et al. Cellular and molecular mechanisms breaking immune tolerance in inborn errors of immunity. Cell Mol Immunol, 2021, 18. 1122-1140. [CrossRef]
- Mohr, A.; Atif, M.; Balderas, R.; Gorochov, G., et al. The role of foxp3(+) regulatory t cells in human autoimmune and inflammatory diseases. Clin Exp Immunol, 2019, 197. 24-35. [CrossRef]
- Burbelo, P.D.; Iadarola, M.J.; Keller, J.M.; and Warner, B.M. Autoantibodies targeting intracellular and extracellular proteins in autoimmunity. Front Immunol, 2021, 12. 548469. [CrossRef]
- Dominguez-Villar, M.; Hafler, D.A. Regulatory t cells in autoimmune disease. Nat Immunol, 2018, 19. 665-673. [CrossRef]
- Moghaddam, B.; Marozoff, S.; Li, L.; Sayre, E.C., et al. All-cause and cause-specific mortality in systemic lupus erythematosus: A population-based study. Rheumatology (Oxford), 2021, 61. 367-376. [CrossRef]
- Jiwrajka, N.; Anguera, M.C. The x in sex-biased immunity and autoimmune rheumatic disease. J Exp Med, 2022, 219. [CrossRef]
- Dodd, K.C.; Menon, M. Sex bias in lymphocytes: Implications for autoimmune diseases. Front Immunol, 2022, 13. 945762. [CrossRef]
- Schneider-Hohendorf, T.; Gorlich, D.; Savola, P.; Kelkka, T., et al. Sex bias in mhc i-associated shaping of the adaptive immune system. Proc Natl Acad Sci U S A, 2018, 115. 2168-2173. [CrossRef]
- Herrada, A.A.; Escobedo, N.; Iruretagoyena, M.; Valenzuela, R.A., et al. Innate immune cells' contribution to systemic lupus erythematosus. Front Immunol, 2019, 10. 772. [CrossRef]
- Tanaka, Y.; Kubo, S.; Iwata, S.; Yoshikawa, M., et al. B cell phenotypes, signaling and their roles in secretion of antibodies in systemic lupus erythematosus. Clin Immunol, 2018, 186. 21-25. DOI: 10.1016/j.clim.2017.07.010,Chen, K.; Wu, T.; Wang, D.; Li, R., et al. Transcriptomics and quantitative proteomics reveal changes after second stimulation of bone marrow-derived macrophages from lupus-prone mrl/lpr mice. Front Immunol, 2022, 13. 1004232. DOI: 10.3389/fimmu.2022.1004232,Xu, J.W.; Wang, M.Y.; Mao, Y.; Hu, Z.Y., et al. Inhibition of stat3 alleviates lps-induced apoptosis and inflammation in renal tubular epithelial cells by transcriptionally down-regulating tasl. Eur J Med Res, 2024, 29. 34. DOI: 10.1186/s40001-023-01610-9.
- Souyris, M.; Cenac, C.; Azar, P.; Daviaud, D., et al. Tlr7 escapes x chromosome inactivation in immune cells. Sci Immunol, 2018, 3. [CrossRef]
- Brown, G.J.; Canete, P.F.; Wang, H.; Medhavy, A., et al. Tlr7 gain-of-function genetic variation causes human lupus. Nature, 2022, 605. 349-356. [CrossRef]
- Kalliolias, G.D.; Basdra, E.K.; and Papavassiliou, A.G. Targeting tlr signaling cascades in systemic lupus erythematosus and rheumatoid arthritis: An update. Biomedicines, 2024, 12. [CrossRef]
- Crow, M.K. Pathogenesis of systemic lupus erythematosus: Risks, mechanisms and therapeutic targets. Ann Rheum Dis, 2023, 82. 999-1014. [CrossRef]
- Furie, R.A.; Bruce, I.N.; Dorner, T.; Leon, M.G., et al. Phase 2, randomized, placebo-controlled trial of dapirolizumab pegol in patients with moderate-to-severe active systemic lupus erythematosus. Rheumatology (Oxford), 2021, 60. 5397-5407. [CrossRef]
- Oke, V.; Gunnarsson, I.; Dorschner, J.; Eketjall, S., et al. High levels of circulating interferons type i, type ii and type iii associate with distinct clinical features of active systemic lupus erythematosus. Arthritis Res Ther, 2019, 21. 107. DOI: 10.1186/s13075-019-1878-y,Sun, X.; Pan, H.; Lu, H.; Song, S., et al. Diagnostic significance of combined anti-extractable nuclear antigens antibody, anti-cardiolipin antibody and anti-beta2-glycoprotein 1 in systemic lupus erythematosus patients. Heliyon, 2024, 10. e29230. DOI: 10.1016/j.heliyon.2024.e29230.
- Park, J.S.; Perl, A. Endosome traffic modulates pro-inflammatory signal transduction in cd4(+) t cells-implications for the pathogenesis of systemic lupus erythematosus. Int J Mol Sci, 2023, 24. [CrossRef]
- Yuan, S.; Zeng, Y.; Li, J.; Wang, C., et al. Phenotypical changes and clinical significance of cd4(+)/cd8(+) t cells in sle. Lupus Sci Med, 2022, 9. [CrossRef]
- Zhang, T.; Liu, X.; Zhao, Y.; Xu, X., et al. Excessive il-15 promotes cytotoxic cd4 + cd28- t cell-mediated renal injury in lupus nephritis. Immun Ageing, 2022, 19. 50. [CrossRef]
- Katsuyama, T.; Tsokos, G.C.; and Moulton, V.R. Aberrant t cell signaling and subsets in systemic lupus erythematosus. Front Immunol, 2018, 9. 1088. [CrossRef]
- Morel, L. Immunometabolism in systemic lupus erythematosus. Nat Rev Rheumatol, 2017, 13. 280-290. DOI: 10.1038/nrrheum.2017.43,Psarras, A.; Clarke, A. A cellular overview of immunometabolism in systemic lupus erythematosus. Oxf Open Immunol, 2023, 4. iqad005. DOI: 10.1093/oxfimm/iqad005,Iwata, S.; Hajime Sumikawa, M.; and Tanaka, Y. B cell activation via immunometabolism in systemic lupus erythematosus. Front Immunol, 2023, 14. 1155421. DOI: 10.3389/fimmu.2023.1155421.
- Hamilton, J.A.; Hsu, H.C.; and Mountz, J.D. Autoreactive b cells in sle, villains or innocent bystanders? Immunol Rev, 2019, 292. 120-138. [CrossRef]
- Odani, T.; Chiorini, J.A. Targeting primary sjogren's syndrome. Mod Rheumatol, 2019, 29. 70-86. DOI: 10.1080/14397595.2018.1546268,Seror, R.; Nocturne, G.; and Mariette, X. Current and future therapies for primary sjogren syndrome. Nat Rev Rheumatol, 2021, 17. 475-486. DOI: 10.1038/s41584-021-00634-x.
- Finotti, G.; Tamassia, N.; and Cassatella, M.A. Interferon-lambdas and plasmacytoid dendritic cells: A close relationship. Front Immunol, 2017, 8. 1015. DOI: 10.3389/fimmu.2017.01015,Jiang, J.; Zhao, M.; Chang, C.; Wu, H., et al. Type i interferons in the pathogenesis and treatment of autoimmune diseases. Clin Rev Allergy Immunol, 2020, 59. 248-272. DOI: 10.1007/s12016-020-08798-2.
- Apostolou, E.; Tzioufas, A.G. Type-iii interferons in sjogren's syndrome. Clin Exp Rheumatol, 2020, 38 Suppl 126. 245-252.
- Jonsson, R.; Brokstad, K.A.; Jonsson, M.V.; Delaleu, N., et al. Current concepts on sjogren's syndrome - classification criteria and biomarkers. Eur J Oral Sci, 2018, 126 Suppl 1. 37-48. [CrossRef]
- Lazear, H.M.; Schoggins, J.W.; and Diamond, M.S. Shared and distinct functions of type i and type iii interferons. Immunity, 2019, 50. 907-923. [CrossRef]
- Barrat, F.J.; Crow, M.K.; and Ivashkiv, L.B. Interferon target-gene expression and epigenomic signatures in health and disease. Nat Immunol, 2019, 20. 1574-1583. [CrossRef]
- Cattalini, M.; Soliani, M.; Caparello, M.C.; and Cimaz, R. Sex differences in pediatric rheumatology. Clin Rev Allergy Immunol, 2019, 56. 293-307. DOI: 10.1007/s12016-017-8642-3,Smith, E.M.D.; Lythgoe, H.; Midgley, A.; Beresford, M.W., et al. Juvenile-onset systemic lupus erythematosus: Update on clinical presentation, pathophysiology and treatment options. Clin Immunol, 2019, 209. 108274. DOI: 10.1016/j.clim.2019.108274.
- Miquel, C.H.; Faz-Lopez, B.; and Guery, J.C. Influence of x chromosome in sex-biased autoimmune diseases. J Autoimmun, 2023. 102992. [CrossRef]
- Dossin, F.; Heard, E. The molecular and nuclear dynamics of x-chromosome inactivation. Cold Spring Harb Perspect Biol, 2022, 14. [CrossRef]
- Fang, H.; Disteche, C.M.; and Berletch, J.B. X inactivation and escape: Epigenetic and structural features. Front Cell Dev Biol, 2019, 7. 219. DOI: 10.3389/fcell.2019.00219,Loda, A.; Collombet, S.; and Heard, E. Gene regulation in time and space during x-chromosome inactivation. Nat Rev Mol Cell Biol, 2022, 23. 231-249. DOI: 10.1038/s41580-021-00438-7.
- Oertelt-Prigione, S.; Mariman, E. The impact of sex differences on genomic research. Int J Biochem Cell Biol, 2020, 124. 105774. [CrossRef]
- Gay, L.; Melenotte, C.; Lakbar, I.; Mezouar, S., et al. Sexual dimorphism and gender in infectious diseases. Front Immunol, 2021, 12. 698121. [CrossRef]
- Liu, K.; Kurien, B.T.; Zimmerman, S.L.; Kaufman, K.M., et al. X chromosome dose and sex bias in autoimmune diseases: Increased prevalence of 47,xxx in systemic lupus erythematosus and sjogren's syndrome. Arthritis Rheumatol, 2016, 68. 1290-1300. [CrossRef]
- Jacobson, E.C.; Pandya-Jones, A.; and Plath, K. A lifelong duty: How xist maintains the inactive x chromosome. Curr Opin Genet Dev, 2022, 75. 101927. [CrossRef]
- Syrett, C.M.; Paneru, B.; Sandoval-Heglund, D.; Wang, J., et al. Altered x-chromosome inactivation in t cells may promote sex-biased autoimmune diseases. JCI Insight, 2019, 4. [CrossRef]
- Nusbaum, J.S.; Mirza, I.; Shum, J.; Freilich, R.W., et al. Sex differences in systemic lupus erythematosus: Epidemiology, clinical considerations, and disease pathogenesis. Mayo Clin Proc, 2020, 95. 384-394. [CrossRef]
- Dou, D.R.; Zhao, Y.; Belk, J.A.; Zhao, Y., et al. Xist ribonucleoproteins promote female sex-biased autoimmunity. Cell, 2024, 187. 733-749 e716. [CrossRef]
- Hagen, S.H.; Henseling, F.; Hennesen, J.; Savel, H., et al. Heterogeneous escape from x chromosome inactivation results in sex differences in type i ifn responses at the single human pdc level. Cell Rep, 2020, 33. 108485. DOI: 10.1016/j.celrep.2020.108485,Barhum, L. Why are autoimmune diseases more common in women? 2021,Youness, A.; Miquel, C.H.; and Guery, J.C. Escape from x chromosome inactivation and the female predominance in autoimmune diseases. Int J Mol Sci, 2021, 22. DOI: 10.3390/ijms22031114,Lu, Y.; Xu, M.; Dorrier, C.E.; Zhang, R., et al. Cd40 drives central nervous system autoimmune disease by inducing complementary effector programs via b cells and dendritic cells. J Immunol, 2022, 209. 2083-2092. DOI: 10.4049/jimmunol.2200439,Li, X.; Yue, X.; Sepulveda, H.; Burt, R.A., et al. Ogt controls mammalian cell viability by regulating the proteasome/mtor/ mitochondrial axis. Proc Natl Acad Sci U S A, 2023, 120. e2218332120. DOI: 10.1073/pnas.2218332120,Youness, A.; Cenac, C.; Faz-Lopez, B.; Grunenwald, S., et al. Tlr8 escapes x chromosome inactivation in human monocytes and cd4(+) t cells. Biol Sex Differ, 2023, 14. 60. DOI: 10.1186/s13293-023-00544-5.
- Laffont, S.; Guery, J.C. Deconstructing the sex bias in allergy and autoimmunity: From sex hormones and beyond. Adv Immunol, 2019, 142. 35-64. [CrossRef]
- Souyris, M.; Mejia, J.E.; Chaumeil, J.; and Guery, J.C. Female predisposition to tlr7-driven autoimmunity: Gene dosage and the escape from x chromosome inactivation. Semin Immunopathol, 2019, 41. 153-164. DOI: 10.1007/s00281-018-0712-y,Satterthwaite, A.B. Tlr7 signaling in lupus b cells: New insights into synergizing factors and downstream signals. Curr Rheumatol Rep, 2021, 23. 80. DOI: 10.1007/s11926-021-01047-1.
- Itoh, Y.; Golden, L.C.; Itoh, N.; Matsukawa, M.A., et al. The x-linked histone demethylase kdm6a in cd4+ t lymphocytes modulates autoimmunity. J Clin Invest, 2019, 129. 3852-3863. [CrossRef]
- Cheng, M.I.; Li, J.H.; Riggan, L.; Chen, B., et al. The x-linked epigenetic regulator utx controls nk cell-intrinsic sex differences. Nat Immunol, 2023, 24. 780-791. [CrossRef]
- Oliva, M.; Munoz-Aguirre, M.; Kim-Hellmuth, S.; Wucher, V., et al. The impact of sex on gene expression across human tissues. Science, 2020, 369. [CrossRef]
- Sakkas, L.I.; Chikanza, I.C. Sex bias in immune response: It is time to include the sex variable in studies of autoimmune rheumatic diseases. Rheumatol Int, 2024, 44. 203-209. [CrossRef]
- Aune, T.M.; Crooke, P.S., 3rd; Patrick, A.E.; Tossberg, J.T., et al. Expression of long non-coding rnas in autoimmunity and linkage to enhancer function and autoimmune disease risk genetic variants. J Autoimmun, 2017, 81. 99-109. [CrossRef]
- Boodhoo, K.D.; Liu, S.; and Zuo, X. Impact of sex disparities on the clinical manifestations in patients with systemic lupus erythematosus: A systematic review and meta-analysis. Medicine (Baltimore), 2016, 95. e4272. [CrossRef]
- Jenks, S.A.; Cashman, K.S.; Zumaquero, E.; Marigorta, U.M., et al. Distinct effector b cells induced by unregulated toll-like receptor 7 contribute to pathogenic responses in systemic lupus erythematosus. Immunity, 2018, 49. 725-739 e726. [CrossRef]
- Ranjan, S.; Panda, A.K. Association of toll-like receptor 7 (tlr7) polymorphisms with predisposition to systemic lupus erythematosus (sle): A meta and trial sequential analysis. Biochem Genet, 2024, 62. 3350-3366. [CrossRef]
- Leibler, C.; John, S.; Elsner, R.A.; Thomas, K.B., et al. Genetic dissection of tlr9 reveals complex regulatory and cryptic proinflammatory roles in mouse lupus. Nat Immunol, 2022, 23. 1457-1469. [CrossRef]
- Shibata, T.; Sato, R.; Taoka, M.; Saitoh, S.I., et al. Tlr7/8 stress response drives histiocytosis in slc29a3 disorders. J Exp Med, 2023, 220. DOI: 10.1084/jem.20230054,Wang, Y.F.; Wei, W.; Tangtanatakul, P.; Zheng, L., et al. Identification of shared and asian-specific loci for systemic lupus erythematosus and evidence for roles of type iii interferon signaling and lysosomal function in the disease: A multi-ancestral genome-wide association study. Arthritis Rheumatol, 2022, 74. 840-848. DOI: 10.1002/art.42021.
- Husebye, E.S.; Anderson, M.S.; and Kampe, O. Autoimmune polyendocrine syndromes. N Engl J Med, 2018, 378. 1132-1141. [CrossRef]
- Starokadomskyy, P.; Gluck, N.; Li, H.; Chen, B., et al. Ccdc22 deficiency in humans blunts activation of proinflammatory nf-kappab signaling. J Clin Invest, 2013, 123. 2244-2256. DOI: 10.1172/JCI66466,D'Amico, F.; Skarmoutsou, E.; Lo, L.J.; Granata, M., et al. Association between rs2294020 in x-linked ccdc22 and susceptibility to autoimmune diseases with focus on systemic lupus erythematosus. Immunol Lett, 2017, 181. 58-62. DOI: 10.1016/j.imlet.2016.11.011.
- Postal, M.; Vivaldo, J.F.; Fernandez-Ruiz, R.; Paredes, J.L., et al. Type i interferon in the pathogenesis of systemic lupus erythematosus. Curr Opin Immunol, 2020, 67. 87-94. [CrossRef]
- Wang, Z.; Heid, B.; Dai, R.; and Ahmed, S.A. Similar dysregulation of lupus-associated mirnas in peripheral blood mononuclear cells and splenic lymphocytes in mrl/lpr mice. Lupus Sci Med, 2018, 5. e000290. [CrossRef]
- Chen, J.Q.; Papp, G.; Poliska, S.; Szabo, K., et al. Microrna expression profiles identify disease-specific alterations in systemic lupus erythematosus and primary sjogren's syndrome. PLoS One, 2017, 12. e0174585. [CrossRef]
- Cui, C.; Yang, W.; Shi, J.; Zhou, Y., et al. Identification and analysis of human sex-biased micrornas. Genomics Proteomics Bioinformatics, 2018, 16. 200-211. DOI: 10.1016/j.gpb.2018.03.004,Taneja, V. Sex hormones determine immune response. Front Immunol, 2018, 9. 1931. DOI: 10.3389/fimmu.2018.01931.
- Luo, B.; Zhou, K.; Liufu, Y.; Huang, X., et al. Novel insight into mirna biology and its role in the pathogenesis of systemic lupus erythematosus. Front Immunol, 2022, 13. 1059887. [CrossRef]
- Navarro Quiroz, E.; Navarro Quiroz, R.; Pacheco Lugo, L.; Aroca Martinez, G., et al. Integrated analysis of microrna regulation and its interaction with mechanisms of epigenetic regulation in the etiology of systemic lupus erythematosus. PLoS One, 2019, 14. e0218116. [CrossRef]
- Kozomara, A.; Birgaoanu, M.; and Griffiths-Jones, S. Mirbase: From microrna sequences to function. Nucleic Acids Res, 2019, 47. D155-D162. [CrossRef]
- Zhang, L.; Wu, H.; Zhao, M.; and Lu, Q. Identifying the differentially expressed micrornas in autoimmunity: A systemic review and meta-analysis. Autoimmunity, 2020, 53. 122-136. [CrossRef]
- Yuan, S.; Tang, C.; Chen, D.; Li, F., et al. Mir-98 modulates cytokine production from human pbmcs in systemic lupus erythematosus by targeting il-6 mrna. J Immunol Res, 2019, 2019. 9827574. [CrossRef]
- Xie, L.; Xu, J. Role of mir-98 and its underlying mechanisms in systemic lupus erythematosus. J Rheumatol, 2018, 45. 1397-1405. [CrossRef]
- Yang, X.; Shi, L.; Zheng, X.; Liu, X., et al. Modulation of mir-548m encoded by x chromosome on the pten pathway in systemic lupus erythematosus. Clin Exp Rheumatol, 2022, 40. 56-63. [CrossRef]
- So, B.Y.F.; Yap, D.Y.H.; and Chan, T.M. Micrornas in lupus nephritis-role in disease pathogenesis and clinical applications. Int J Mol Sci, 2021, 22. [CrossRef]
- Zhang, Y.; Xie, L.; Lu, W.; Lv, J., et al. Lncrna miat enhances systemic lupus erythematosus by upregulating cfhr5 expression via mir-222 degradation. Cent Eur J Immunol, 2021, 46. 17-26. [CrossRef]
- Xing, Y.; Li, B.; He, J.; and Hua, H. Labial gland mesenchymal stem cell derived exosomes-mediated mirna-125b attenuates experimental sjogren's syndrome by targeting prdm1 and suppressing plasma cells. Front Immunol, 2022, 13. 871096. [CrossRef]
- Al-Haidose, A.; Hassan, S.; Elhassan, M.; Ahmed, E., et al. Role of ncrnas in the pathogenesis of sjogren's syndrome. Biomedicines, 2024, 12. [CrossRef]
- Hiramatsu-Asano, S.; Sunahori-Watanabe, K.; Zeggar, S.; Katsuyama, E., et al. Deletion of mir223 exacerbates lupus nephritis by targeting s1pr1 in fas(lpr/lpr) mice. Front Immunol, 2020, 11. 616141. [CrossRef]
- Qi, X.; Wang, R.; Jin, L.; Tian, Y., et al. Mir-223-3p aggravates ocular inflammation in sjogren's syndrome. Endocr Metab Immune Disord Drug Targets, 2023, 23. 1087-1095. [CrossRef]
- Young, N.A.; Valiente, G.R.; Hampton, J.M.; Wu, L.C., et al. Estrogen-regulated stat1 activation promotes tlr8 expression to facilitate signaling via microrna-21 in systemic lupus erythematosus. Clin Immunol, 2017, 176. 12-22. [CrossRef]
- Lu, M.C.; Lai, N.S.; Chen, H.C.; Yu, H.C., et al. Decreased microrna(mir)-145 and increased mir-224 expression in t cells from patients with systemic lupus erythematosus involved in lupus immunopathogenesis. Clin Exp Immunol, 2013, 171. 91-99. [CrossRef]
- Punnanitinont, A.; Kramer, J.M. Sex-specific differences in primary sjogren's disease. Front Dent Med, 2023, 4. 1168645. [CrossRef]
- Cornet, A.; Andersen, J.; Myllys, K.; Edwards, A., et al. Living with systemic lupus erythematosus in 2020: A european patient survey. Lupus Sci Med, 2021, 8. [CrossRef]
- Kim, J.W.; Kim, H.A.; Suh, C.H.; and Jung, J.Y. Sex hormones affect the pathogenesis and clinical characteristics of systemic lupus erythematosus. Front Med (Lausanne), 2022, 9. 906475. [CrossRef]
- Taneja, V. Sexual dimorphism, aging and immunity. Vitam Horm, 2021, 115. 367-399. [CrossRef]
- Rey, R.A. The role of androgen signaling in male sexual development at puberty. Endocrinology, 2021, 162. DOI: 10.1210/endocr/bqaa215,Santi, M.; Graf, S.; Zeino, M.; Cools, M., et al. Approach to the virilizing girl at puberty. J Clin Endocrinol Metab, 2021, 106. 1530-1539. DOI: 10.1210/clinem/dgaa948.
- Vom Steeg, L.G.; Klein, S.L. Sex and sex steroids impact influenza pathogenesis across the life course. Semin Immunopathol, 2019, 41. 189-194. DOI: 10.1007/s00281-018-0718-5,Yang, Q.; Kennicott, K.; Zhu, R.; Kim, J., et al. Sex hormone influence on female-biased autoimmune diseases hints at puberty as an important factor in pathogenesis. Front Pediatr, 2023, 11. 1051624. DOI: 10.3389/fped.2023.1051624.
- Keestra, S.M.; Male, V.; and Salali, G.D. Out of balance: The role of evolutionary mismatches in the sex disparity in autoimmune disease. Med Hypotheses, 2021, 151. 110558. [CrossRef]
- Merz, W.M.; Fischer-Betz, R.; Hellwig, K.; Lamprecht, G., et al. Pregnancy and autoimmune disease. Dtsch Arztebl Int, 2022, 119. 145-156. [CrossRef]
- Lipoldova, M.; Demant, P. Gene-specific sex effects on susceptibility to infectious diseases. Front Immunol, 2021, 12. 712688. [CrossRef]
- Mohammad, I.; Starskaia, I.; Nagy, T.; Guo, J., et al. Estrogen receptor alpha contributes to t cell-mediated autoimmune inflammation by promoting t cell activation and proliferation. Sci Signal, 2018, 11. [CrossRef]
- Singh, R.P.; Hahn, B.H.; and Bischoff, D.S. Interferon genes are influenced by 17beta-estradiol in sle. Front Immunol, 2021, 12. 725325. [CrossRef]
- Jaiswal, A.; Roy, R.; Tamrakar, A.; Singh, A.K., et al. Activation-induced cytidine deaminase an antibody diversification enzyme interacts with chromatin modifier ubn1 in b-cells. Sci Rep, 2023, 13. 19615. [CrossRef]
- Shepherd, R.; Cheung, A.S.; Pang, K.; Saffery, R., et al. Sexual dimorphism in innate immunity: The role of sex hormones and epigenetics. Front Immunol, 2020, 11. 604000. [CrossRef]
- Moulton, V.R. Sex hormones in acquired immunity and autoimmune disease. Front Immunol, 2018, 9. 2279. [CrossRef]
- Singh, R.P.; Bischoff, D.S. Sex hormones and gender influence the expression of markers of regulatory t cells in sle patients. Front Immunol, 2021, 12. 619268. [CrossRef]
- Fuseini, H.; Cephus, J.Y.; Wu, P.; Davis, J.B., et al. Eralpha signaling increased il-17a production in th17 cells by upregulating il-23r expression, mitochondrial respiration, and proliferation. Front Immunol, 2019, 10. 2740. [CrossRef]
- Casali, P.; Shen, T.; Xu, Y.; Qiu, Z., et al. Estrogen reverses hdac inhibitor-mediated repression of aicda and class-switching in antibody and autoantibody responses by downregulation of mir-26a. Front Immunol, 2020, 11. 491. [CrossRef]
- Henze, L.; Schwinge, D.; and Schramm, C. The effects of androgens on t cells: Clues to female predominance in autoimmune liver diseases? Front Immunol, 2020, 11. 1567. [CrossRef]
- Harding, A.T.; Heaton, N.S. The impact of estrogens and their receptors on immunity and inflammation during infection. Cancers (Basel), 2022, 14. [CrossRef]
- Ciarambino, T.; Para, O.; and Giordano, M. Immune system and covid-19 by sex differences and age. Womens Health (Lond), 2021, 17. 17455065211022262. [CrossRef]
- Shepherd, R.; Bretherton, I.; Pang, K.; Mansell, T., et al. Gender-affirming hormone therapy induces specific DNA methylation changes in blood. Clin Epigenetics, 2022, 14. 24. [CrossRef]
- Gulati, G.; Brunner, H.I. Environmental triggers in systemic lupus erythematosus. Semin Arthritis Rheum, 2018, 47. 710-717. [CrossRef]
- Brooks, W.H.; Arleevskaya, M.I.; and Renaudineau, Y. Editorial: Epigenetic aspects of autoimmune diseases. Front Cell Dev Biol, 2022, 10. 991693. [CrossRef]
- Golden, L.C.; Itoh, Y.; Itoh, N.; Iyengar, S., et al. Parent-of-origin differences in DNA methylation of x chromosome genes in t lymphocytes. Proc Natl Acad Sci U S A, 2019, 116. 26779-26787. DOI: 10.1073/pnas.1910072116,Marquez, E.J.; Chung, C.H.; Marches, R.; Rossi, R.J., et al. Sexual-dimorphism in human immune system aging. Nat Commun, 2020, 11. 751. DOI: 10.1038/s41467-020-14396-9.
- Charras, A.; Garau, J.; Hofmann, S.R.; Carlsson, E., et al. DNA methylation patterns in cd8(+) t cells discern psoriasis from psoriatic arthritis and correlate with cutaneous disease activity. Front Cell Dev Biol, 2021, 9. 746145. DOI: 10.3389/fcell.2021.746145,Vecellio, M.; Paraboschi, E.M.; Ceribelli, A.; Isailovic, N., et al. DNA methylation signature in monozygotic twins discordant for psoriatic disease. Front Cell Dev Biol, 2021, 9. 778677. DOI: 10.3389/fcell.2021.778677,Kabeerdoss, J.; Danda, D.; Goel, R.; Mohan, H., et al. Genome-wide DNA methylation profiling in cd8 t-cells and gamma delta t-cells of asian indian patients with takayasu arteritis. Front Cell Dev Biol, 2022, 10. 843413. DOI: 10.3389/fcell.2022.843413.
- Yao, Q.; Song, Z.; Wang, B.; Jia, X., et al. Identification of lncrna and mrna expression profile in relapsed graves' disease. Front Cell Dev Biol, 2021, 9. 756560. DOI: 10.3389/fcell.2021.756560,Bost, C.; Arleevskaya, M.I.; Brooks, W.H.; Plaza, S., et al. Long non-coding rna xist contribution in systemic lupus erythematosus and rheumatoid arthritis. Clin Immunol, 2022, 236. 108937. DOI: 10.1016/j.clim.2022.108937.
- Lv, X.; Liu, X.; Zhao, M.; Wu, H., et al. Rna methylation in systemic lupus erythematosus. Front Cell Dev Biol, 2021, 9. 696559. DOI: 10.3389/fcell.2021.696559,Chao, Y.; Li, H.B.; and Zhou, J. Multiple functions of rna methylation in t cells: A review. Front Immunol, 2021, 12. 627455. DOI: 10.3389/fimmu.2021.627455.
- Aye, I.; Gong, S.; Avellino, G.; Barbagallo, R., et al. Placental sex-dependent spermine synthesis regulates trophoblast gene expression through acetyl-coa metabolism and histone acetylation. Commun Biol, 2022, 5. 586. DOI: 10.1038/s42003-022-03530-6,Jiang, Y.; Peng, Z.; Man, Q.; Wang, S., et al. H3k27ac chromatin acetylation and gene expression analysis reveal sex- and situs-related differences in developing chicken gonads. Biol Sex Differ, 2022, 13. 6. DOI: 10.1186/s13293-022-00415-5.
- Bacalao, M.A.; Satterthwaite, A.B. Recent advances in lupus b cell biology: Pi3k, ifngamma, and chromatin. Front Immunol, 2020, 11. 615673. [CrossRef]
- Liu, Y.; Li, C.; Yang, Y.; Li, T., et al. The tgf-beta/mir-31/ceacam1-s axis inhibits cd4(+) cd25(+) treg differentiation in systemic lupus erythematosus. Immunol Cell Biol, 2021, 99. 697-710. DOI: 10.1111/imcb.12449,Pashangzadeh, S.; Motallebnezhad, M.; Vafashoar, F.; Khalvandi, A., et al. Implications the role of mir-155 in the pathogenesis of autoimmune diseases. Front Immunol, 2021, 12. 669382. DOI: 10.3389/fimmu.2021.669382,Li, S.; Wu, Q.; Jiang, Z.; Wu, Y., et al. Mir-31-5p regulates type i interferon by targeting slc15a4 in plasmacytoid dendritic cells of systemic lupus erythematosus. J Inflamm Res, 2022, 15. 6607-6616. DOI: 10.2147/JIR.S383623.
- Illescas-Montes, R.; Corona-Castro, C.C.; Melguizo-Rodriguez, L.; Ruiz, C., et al. Infectious processes and systemic lupus erythematosus. Immunology, 2019, 158. 153-160. [CrossRef]
- Sun, F.; Chen, Y.; Wu, W.; Guo, L., et al. Varicella zoster virus infections increase the risk of disease flares in patients with sle: A matched cohort study. Lupus Sci Med, 2019, 6. e000339. DOI: 10.1136/lupus-2019-000339,Tsai, P.H.; Jang, S.S.; and Liou, L.B. Septicaemia is associated with increased disease activity and mortality in systemic lupus erythematosus: A retrospective analysis from taiwan. Lupus, 2020, 29. 191-198. DOI: 10.1177/0961203319899162,Joo, Y.B.; Kim, K.J.; Park, K.S.; and Park, Y.J. Influenza infection as a trigger for systemic lupus erythematosus flares resulting in hospitalization. Sci Rep, 2021, 11. 4630. DOI: 10.1038/s41598-021-84153-5,Fu, X.L.; Qian, Y.; Jin, X.H.; Yu, H.R., et al. Covid-19 in patients with systemic lupus erythematosus: A systematic review. Lupus, 2022, 31. 684-696. DOI: 10.1177/09612033221093502.
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J., et al. Longitudinal analysis reveals high prevalence of epstein-barr virus associated with multiple sclerosis. Science, 2022, 375. 296-301. DOI: 10.1126/science.abj8222,Sundaresan, B.; Shirafkan, F.; Ripperger, K.; and Rattay, K. The role of viral infections in the onset of autoimmune diseases. Viruses, 2023, 15. DOI: 10.3390/v15030782.
- Martinelli, S.; Nannini, G.; Cianchi, F.; Coratti, F., et al. The impact of microbiota-immunity-hormone interactions on autoimmune diseases and infection. Biomedicines, 2024, 12. [CrossRef]
- Ronnblom, L.; Leonard, D. Interferon pathway in sle: One key to unlocking the mystery of the disease. Lupus Sci Med, 2019, 6. e000270. DOI: 10.1136/lupus-2018-000270,Huang, J.; Hong, W.; Wan, M.; and Zheng, L. Molecular mechanisms and therapeutic target of netosis in diseases. MedComm (2020), 2022, 3. e162. DOI: 10.1002/mco2.162,El Hadiyen, F.; Tsang, A.S.M.W.P.; Lissenberg-Witte, B.I.; Voskuyl, A.E., et al. Intercurrent infection as a risk factor for disease flares in patients with systemic lupus erythematosus. Lupus Sci Med, 2024, 11. DOI: 10.1136/lupus-2023-001131.
- Xuan, J.; Ji, Z.; Wang, B.; Zeng, X., et al. Serological evidence for the association between epstein-barr virus infection and sjogren's syndrome. Front Immunol, 2020, 11. 590444. [CrossRef]
- Liu, Z.; Chu, A. Sjogren's syndrome and viral infections. Rheumatol Ther, 2021, 8. 1051-1059. [CrossRef]
- Lemus, Y.B.; Martinez, G.A.; Lugo, L.P.; Escorcia, L.G., et al. Gene profiling of epstein-barr virus and human endogenous retrovirus in peripheral blood mononuclear cells of sle patients: Immune response implications. Sci Rep, 2024, 14. 20236. [CrossRef]
- Agostini, S.; Mancuso, R.; Guerini, F.R.; D'Alfonso, S., et al. Hla alleles modulate ebv viral load in multiple sclerosis. J Transl Med, 2018, 16. 80. [CrossRef]
- Barber, M.R.W.; Clarke, A.E. Systemic lupus erythematosus and risk of infection. Expert Rev Clin Immunol, 2020, 16. 527-538. [CrossRef]
- Silva, J.M.; Alves, C.E.C.; and Pontes, G.S. Epstein-barr virus: The mastermind of immune chaos. Front Immunol, 2024, 15. 1297994. [CrossRef]
- Kim, Y.S.; Unno, T.; Kim, B.Y.; and Park, M.S. Sex differences in gut microbiota. World J Mens Health, 2020, 38. 48-60. DOI: 10.5534/wjmh.190009,Rio, P.; Caldarelli, M.; Chiantore, M.; Ocarino, F., et al. Immune cells, gut microbiota, and vaccines: A gender perspective. Cells, 2024, 13. DOI: 10.3390/cells13060526.
- Yao, K.; Xie, Y.; Wang, J.; Lin, Y., et al. Gut microbiota: A newly identified environmental factor in systemic lupus erythematosus. Front Immunol, 2023, 14. 1202850. [CrossRef]
- Johnson, B.M.; Gaudreau, M.C.; Gudi, R.; Brown, R., et al. Gut microbiota differently contributes to intestinal immune phenotype and systemic autoimmune progression in female and male lupus-prone mice. J Autoimmun, 2020, 108. 102420. [CrossRef]
- Santos-Marcos, J.A.; Mora-Ortiz, M.; Tena-Sempere, M.; Lopez-Miranda, J., et al. Interaction between gut microbiota and sex hormones and their relation to sexual dimorphism in metabolic diseases. Biol Sex Differ, 2023, 14. 4. [CrossRef]
- Silverman, G.J.; Deng, J.; and Azzouz, D.F. Sex-dependent lupus blautia (ruminococcus) gnavus strain induction of zonulin-mediated intestinal permeability and autoimmunity. Front Immunol, 2022, 13. 897971. [CrossRef]
- Park, H.J.; Choi, J.M. Sex-specific regulation of immune responses by ppars. Exp Mol Med, 2017, 49. e364. [CrossRef]
- Greiling, T.M.; Dehner, C.; Chen, X.; Hughes, K., et al. Commensal orthologs of the human autoantigen ro60 as triggers of autoimmunity in lupus. Sci Transl Med, 2018, 10. DOI: 10.1126/scitranslmed.aan2306,Chen, B.D.; Jia, X.M.; Xu, J.Y.; Zhao, L.D., et al. An autoimmunogenic and proinflammatory profile defined by the gut microbiota of patients with untreated systemic lupus erythematosus. Arthritis Rheumatol, 2021, 73. 232-243. DOI: 10.1002/art.41511.
- Moon, J.; Choi, S.H.; Yoon, C.H.; and Kim, M.K. Gut dysbiosis is prevailing in sjogren's syndrome and is related to dry eye severity. PLoS One, 2020, 15. e0229029. [CrossRef]
- Schaefer, L.; Trujillo-Vargas, C.M.; Midani, F.S.; Pflugfelder, S.C., et al. Gut microbiota from sjogren syndrome patients causes decreased t regulatory cells in the lymphoid organs and desiccation-induced corneal barrier disruption in mice. Front Med (Lausanne), 2022, 9. 852918. [CrossRef]
- Refai, R.H.; Hussein, M.F.; Abdou, M.H.; and Abou-Raya, A.N. Environmental risk factors of systemic lupus erythematosus: A case-control study. Sci Rep, 2023, 13. 10219. [CrossRef]
- Liu, J.L.; Woo, J.M.P.; Parks, C.G.; Costenbader, K.H., et al. Systemic lupus erythematosus risk: The role of environmental factors. Rheum Dis Clin North Am, 2022, 48. 827-843. [CrossRef]
- Parks, C.G.; Costenbader, K.H.; Long, S.; Hofmann, J.N., et al. Pesticide use and risk of systemic autoimmune diseases in the agricultural health study. Environ Res, 2022, 209. 112862. [CrossRef]
- Jochmanova, I.; Lazurova, Z.; Rudnay, M.; Bacova, I., et al. Environmental estrogen bisphenol a and autoimmunity. Lupus, 2015, 24. 392-399. [CrossRef]
- Pollard, K.M. Silica, silicosis, and autoimmunity. Front Immunol, 2016, 7. 97. DOI: 10.3389/fimmu.2016.00097,Lueschow, S.R.; McElroy, S.J. The paneth cell: The curator and defender of the immature small intestine. Front Immunol, 2020, 11. 587. DOI: 10.3389/fimmu.2020.00587.
- Fee, L.; Kumar, A.; Tighe, R.M.; and Foster, M.H. Autoreactive b cells recruited to lungs by silica exposure contribute to local autoantibody production in autoimmune-prone bxsb and b cell receptor transgenic mice. Front Immunol, 2022, 13. 933360. [CrossRef]
- Chen, J.; Qu, W.; Sun, L.; Chen, J., et al. The relationship of polluted air and drinking water sources with the prevalence of systemic lupus erythematosus: A provincial population-based study. Sci Rep, 2021, 11. 18591. [CrossRef]
- Adami, G.; Pontalti, M.; Cattani, G.; Rossini, M., et al. Association between long-term exposure to air pollution and immune-mediated diseases: A population-based cohort study. RMD Open, 2022, 8. DOI: 10.1136/rmdopen-2021-002055,Zhang, M.; Wang, Y.; Hu, S.; and Wu, Y. Causal relationships between air pollution and common autoimmune diseases: A two-sample mendelian randomization study. Sci Rep, 2025, 15. 135. DOI: 10.1038/s41598-024-83880-9.
- Rasking, L.; Roelens, C.; Sprangers, B.; Thienpont, B., et al. Lupus, DNA methylation, and air pollution: A malicious triad. Int J Environ Res Public Health, 2022, 19. [CrossRef]
- Chen, J.; Liao, S.; Pang, W.; Guo, F., et al. Life factors acting on systemic lupus erythematosus. Front Immunol, 2022, 13. 986239. DOI: 10.3389/fimmu.2022.986239,Touil, H.; Mounts, K.; and De Jager, P.L. Differential impact of environmental factors on systemic and localized autoimmunity. Front Immunol, 2023, 14. 1147447. DOI: 10.3389/fimmu.2023.1147447.
- Jin, L.; Dai, M.; Li, C.; Wang, J., et al. Risk factors for primary sjogren's syndrome: A systematic review and meta-analysis. Clin Rheumatol, 2023, 42. 327-338. [CrossRef]
- Cozier, Y.C.; Barbhaiya, M.; Castro-Webb, N.; Conte, C., et al. Relationship of cigarette smoking and alcohol consumption to incidence of systemic lupus erythematosus in a prospective cohort study of black women. Arthritis Care Res (Hoboken), 2019, 71. 671-677. DOI: 10.1002/acr.23703,Hahn, J.; Leatherwood, C.; Malspeis, S.; Liu, X., et al. Associations between smoking and systemic lupus erythematosus-related cytokines and chemokines among us female nurses. Arthritis Care Res (Hoboken), 2021, 73. 1583-1589. DOI: 10.1002/acr.24370.
- Speyer, C.B.; Costenbader, K.H. Cigarette smoking and the pathogenesis of systemic lupus erythematosus. Expert Rev Clin Immunol, 2018, 14. 481-487. [CrossRef]
- Wolf, S.J.; Estadt, S.N.; Theros, J.; Moore, T., et al. Ultraviolet light induces increased t cell activation in lupus-prone mice via type i ifn-dependent inhibition of t regulatory cells. J Autoimmun, 2019, 103. 102291. DOI: 10.1016/j.jaut.2019.06.002,Coss, S.L.; Zhou, D.; Chua, G.T.; Aziz, R.A., et al. The complement system and human autoimmune diseases. J Autoimmun, 2023, 137. 102979. DOI: 10.1016/j.jaut.2022.102979.
- Holick, M.F. Ultraviolet b radiation: The vitamin d connection. Adv Exp Med Biol, 2017, 996. 137-154. [CrossRef]
- Awuah, W.A.; Huang, H.; Kalmanovich, J.; Mehta, A., et al. Circadian rhythm in systemic autoimmune conditions: Potential of chrono-immunology in clinical practice: A narrative review. Medicine (Baltimore), 2023, 102. e34614. [CrossRef]
- Young, K.A.; Munroe, M.E.; Harley, J.B.; Guthridge, J.M., et al. Less than 7 hours of sleep per night is associated with transitioning to systemic lupus erythematosus. Lupus, 2018, 27. 1524-1531. DOI: 10.1177/0961203318778368,Choi, M.Y.; Malspeis, S.; Sparks, J.A.; Cui, J., et al. Association of sleep deprivation and the risk of developing systemic lupus erythematosus among women. Arthritis Care Res (Hoboken), 2023, 75. 1206-1212. DOI: 10.1002/acr.25017.
- Anaya, J.M.; Restrepo-Jimenez, P.; and Ramirez-Santana, C. The autoimmune ecology: An update. Curr Opin Rheumatol, 2018, 30. 350-360. [CrossRef]
- Hsu, T.W.; Bai, Y.M.; Tsai, S.J.; Chen, T.J., et al. Risk of autoimmune diseases after post-traumatic stress disorder: A nationwide cohort study. Eur Arch Psychiatry Clin Neurosci, 2024, 274. 487-495. [CrossRef]
- Mandagere, K.; Stoy, S.; Hammerle, N.; Zapata, I., et al. Systematic review and meta-analysis of post-traumatic stress disorder as a risk factor for multiple autoimmune diseases. Front Psychiatry, 2025, 16. 1523994. [CrossRef]
Figure 1.
The incidence rate ratio (IRR) and prevalence rate ratio (PRR) for females compared to males across various autoimmune diseases. The IRR (a) and PRR (b) are derived from referenced epidemiological data for each disease: systemic lupus erythematosus (SLE) [7,8], Sjögren's syndrome (SS) [6,9], scleroderma (SD) [10], rheumatoid arthritis (RA) [11], Graves' disease (GD) [3,12], Hashimoto's thyroiditis (HT) [3,13], multiple sclerosis (MS) [14], celiac disease (CD) [15]. These ratios highlight the striking sex disparities in disease incidence and prevalence across autoimmune conditions.
Figure 1.
The incidence rate ratio (IRR) and prevalence rate ratio (PRR) for females compared to males across various autoimmune diseases. The IRR (a) and PRR (b) are derived from referenced epidemiological data for each disease: systemic lupus erythematosus (SLE) [7,8], Sjögren's syndrome (SS) [6,9], scleroderma (SD) [10], rheumatoid arthritis (RA) [11], Graves' disease (GD) [3,12], Hashimoto's thyroiditis (HT) [3,13], multiple sclerosis (MS) [14], celiac disease (CD) [15]. These ratios highlight the striking sex disparities in disease incidence and prevalence across autoimmune conditions.
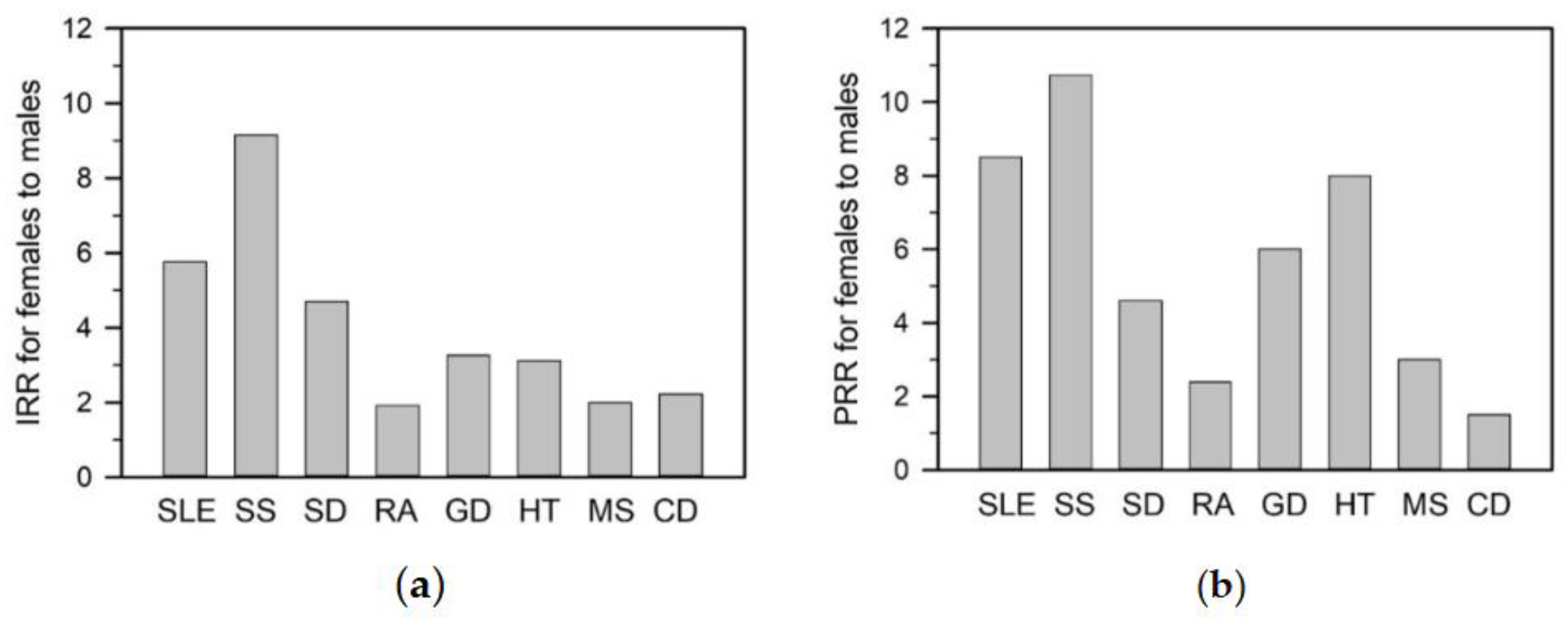
Figure 2.
Innate immune responses. (a) Innate immunity involves epithelial and physiological barriers, complement activation, and recruitment of innate immune cells. TLRs initiate phagocytosis and stimulate the production of IFNs, ILs, and other cytokines and chemokines, facilitating antigen presentation through the generation of APCs. (b) Innate immune responses trigger inflammation primarily through surface TLR-mediated signaling through the Myddosome complex (MyD88 and IRAKs), TAK1, NF-κB, MAPK, and IRF pathways. These cascades promote nuclear translocation of NF-κB, AP-1, and IRF3/5, driving proinflammatory cytokine expression. Endosomal TLR7-9 and TLR4 also activate IRF5 via the SLC15A4-associated adaptor TASL, enhancing IFN-I production. Proinflammatory cytokines amplify inflammation through their receptors, activating the Janus kinase (JAK)-signal transducer and activator of transcription (STAT), NF-κB, and MAPK-AP-1 signaling pathways.
Figure 2.
Innate immune responses. (a) Innate immunity involves epithelial and physiological barriers, complement activation, and recruitment of innate immune cells. TLRs initiate phagocytosis and stimulate the production of IFNs, ILs, and other cytokines and chemokines, facilitating antigen presentation through the generation of APCs. (b) Innate immune responses trigger inflammation primarily through surface TLR-mediated signaling through the Myddosome complex (MyD88 and IRAKs), TAK1, NF-κB, MAPK, and IRF pathways. These cascades promote nuclear translocation of NF-κB, AP-1, and IRF3/5, driving proinflammatory cytokine expression. Endosomal TLR7-9 and TLR4 also activate IRF5 via the SLC15A4-associated adaptor TASL, enhancing IFN-I production. Proinflammatory cytokines amplify inflammation through their receptors, activating the Janus kinase (JAK)-signal transducer and activator of transcription (STAT), NF-κB, and MAPK-AP-1 signaling pathways.
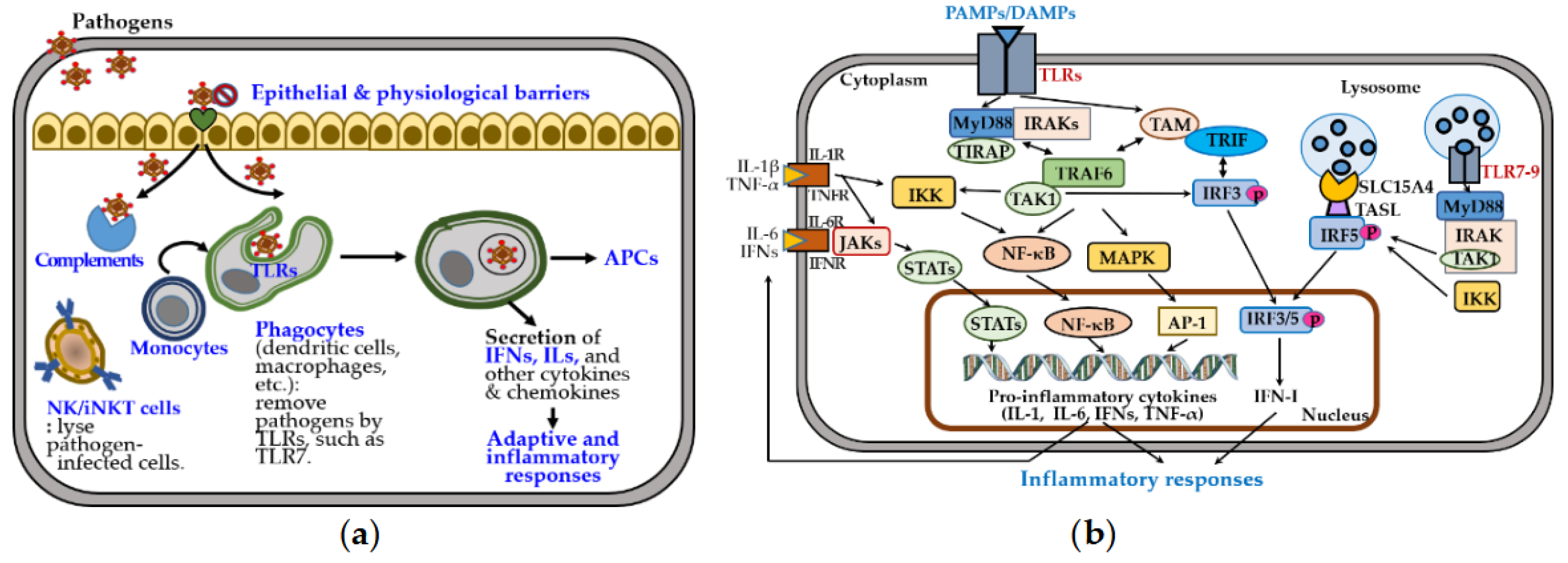
Figure 3.
Activation and differentiation of naïve CD4+ Th cells. Cytokines produced by activated phagocytes and Th cells guide the differentiation of naïve CD4+ Th cells into distinct subsets, including Th1, Th2, Th17, Treg, Tfh, Th9, and Th22. Each subset is characterized by specific phenotypes, transcriptional programs, cytokine profiles, and immune functions. The subsets also exhibit functional plasticity: Th1 and Th2 cells can interconvert, while Th17 and Treg cells are unstable and may transition into other lineages, as indicated by curved arrows in the phenotype panel. In the transcriptional profile panel, activating and inhibitory transcription factors are marked by (+) and (-), respectively, with the master regulator listed first and underlined.
Figure 3.
Activation and differentiation of naïve CD4+ Th cells. Cytokines produced by activated phagocytes and Th cells guide the differentiation of naïve CD4+ Th cells into distinct subsets, including Th1, Th2, Th17, Treg, Tfh, Th9, and Th22. Each subset is characterized by specific phenotypes, transcriptional programs, cytokine profiles, and immune functions. The subsets also exhibit functional plasticity: Th1 and Th2 cells can interconvert, while Th17 and Treg cells are unstable and may transition into other lineages, as indicated by curved arrows in the phenotype panel. In the transcriptional profile panel, activating and inhibitory transcription factors are marked by (+) and (-), respectively, with the master regulator listed first and underlined.
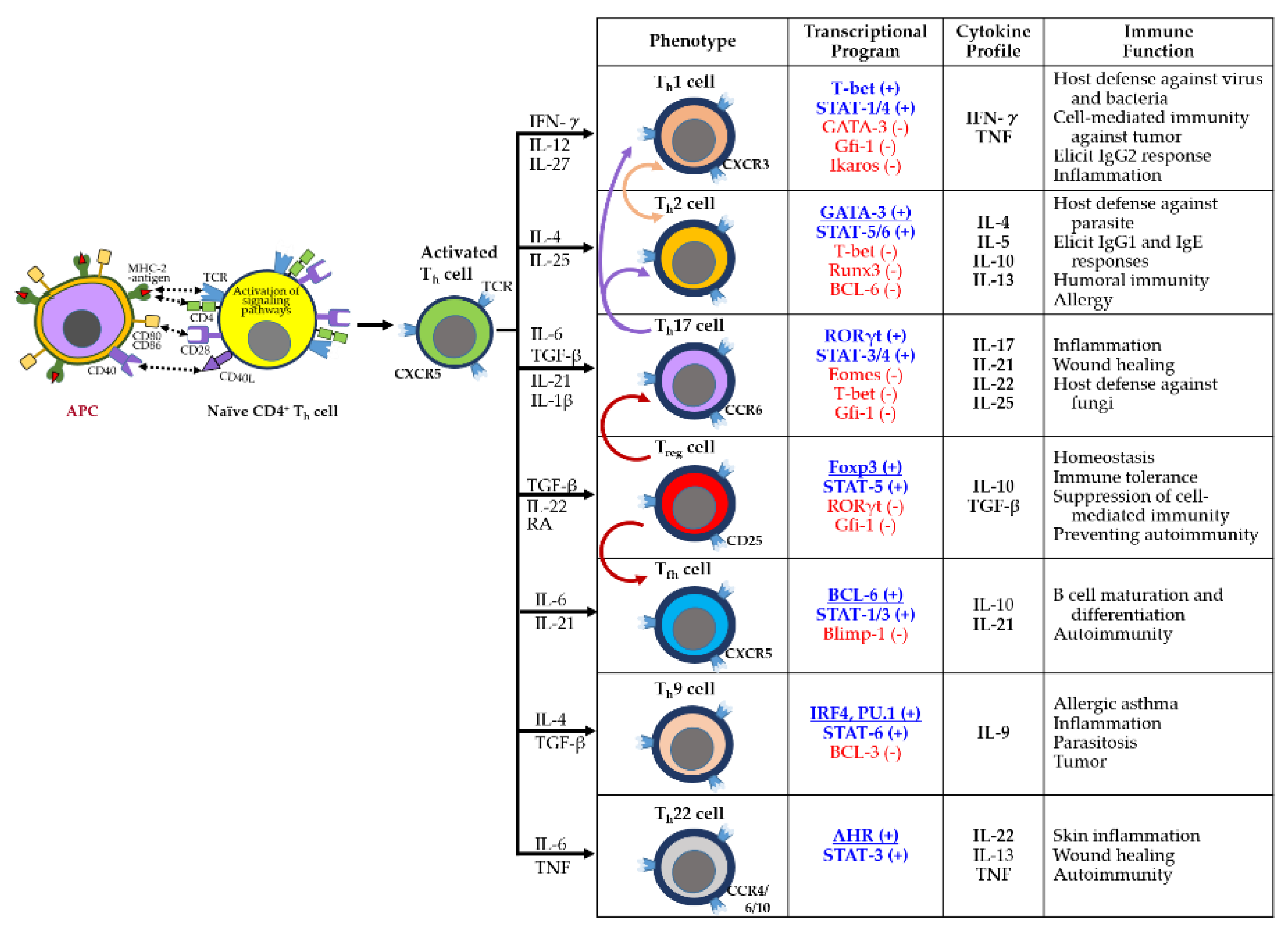
Figure 4.
Activation and differentiation of B cells and CD8+ Tctx cells. (a) B cell activation in humoral immunity. B cells can be activated independently of Th cells by pathogens displaying repetitive epitopes, leading to B cell receptor (BCR) crosslinking and differentiation into short-lived plasma cells that secrete IgM antibodies. Alternatively, BCR-mediated antigen uptake followed by MHC-II presentation to Th cells induces signaling cascades that promote the generation of long-lived plasma cells and memory B cells. This results in high affinity IgG production and robust humoral immunity. (b) CD8+ Tctx cell activation in cell-mediated immunity. CD8+ Tctx cells recognize infected or transformed cells through T cell receptor (TCR)-MHC-I-peptide interactions, along with co-stimulatory signals via CD28 and CD80/CD86. This triggers differentiation into effector and memory Tctx cells. Effector cells secrete cytotoxic molecules such as perforin and granzymes, enabling elimination of target cells. These responses are modulated by Treg and iNKT cells.
Figure 4.
Activation and differentiation of B cells and CD8+ Tctx cells. (a) B cell activation in humoral immunity. B cells can be activated independently of Th cells by pathogens displaying repetitive epitopes, leading to B cell receptor (BCR) crosslinking and differentiation into short-lived plasma cells that secrete IgM antibodies. Alternatively, BCR-mediated antigen uptake followed by MHC-II presentation to Th cells induces signaling cascades that promote the generation of long-lived plasma cells and memory B cells. This results in high affinity IgG production and robust humoral immunity. (b) CD8+ Tctx cell activation in cell-mediated immunity. CD8+ Tctx cells recognize infected or transformed cells through T cell receptor (TCR)-MHC-I-peptide interactions, along with co-stimulatory signals via CD28 and CD80/CD86. This triggers differentiation into effector and memory Tctx cells. Effector cells secrete cytotoxic molecules such as perforin and granzymes, enabling elimination of target cells. These responses are modulated by Treg and iNKT cells.
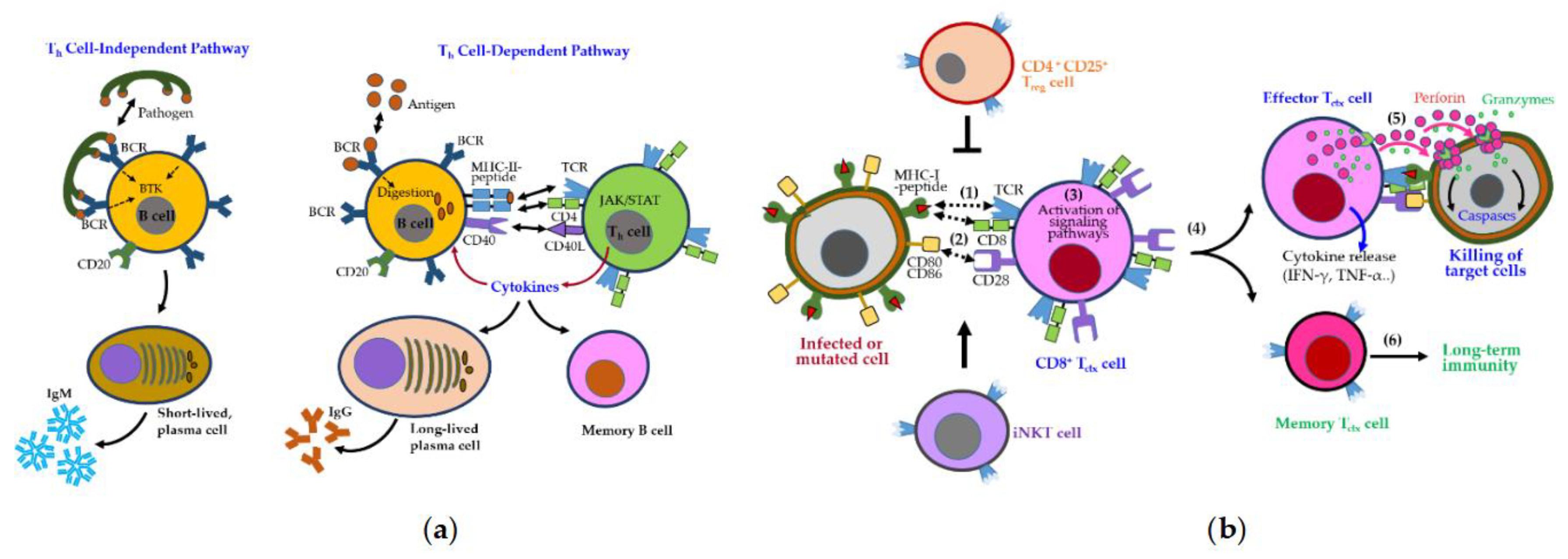
Figure 5.
Key immune and inflammatory responses in SLE. Innate immunity plays a central role in the early stages of SLE through activation of the IFN-I pathway. In SLE, endosomal TLRs recognize single-stranded RNA within immune complexes, inducing IFN-I production by pDCs via the JAK/STAT pathway. TLR7 signaling promotes the differentiation of age-associated B cells (ABCs) into autoantibody-producing cells, while TLR8 activation stimulates TNF-α-producing monocytes that infiltrate the kidney. B cells, activated through BCR engagement and receptors such as BAFFR, TACI, APRIL, and CD19/CD20, interact with T cells via CD40/CD40L signaling. Interleukins (e.g., IL-4/12/23) further promote T cell activation and inflammation. Autoantibodies form immune complexes that perpetuate adaptive immune responses and drive disease progression.
Figure 5.
Key immune and inflammatory responses in SLE. Innate immunity plays a central role in the early stages of SLE through activation of the IFN-I pathway. In SLE, endosomal TLRs recognize single-stranded RNA within immune complexes, inducing IFN-I production by pDCs via the JAK/STAT pathway. TLR7 signaling promotes the differentiation of age-associated B cells (ABCs) into autoantibody-producing cells, while TLR8 activation stimulates TNF-α-producing monocytes that infiltrate the kidney. B cells, activated through BCR engagement and receptors such as BAFFR, TACI, APRIL, and CD19/CD20, interact with T cells via CD40/CD40L signaling. Interleukins (e.g., IL-4/12/23) further promote T cell activation and inflammation. Autoantibodies form immune complexes that perpetuate adaptive immune responses and drive disease progression.
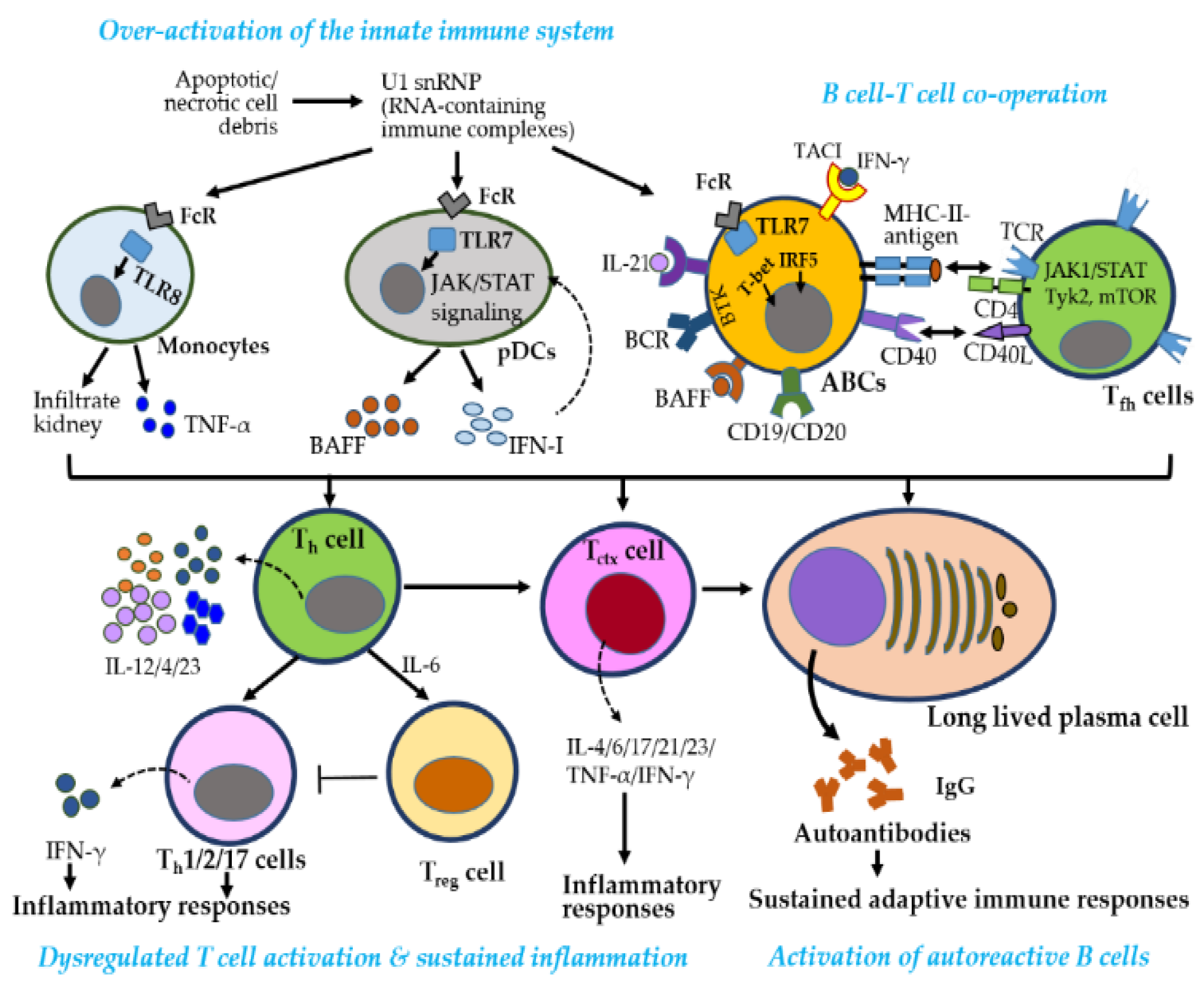
Figure 6.
Figure 6. Key immune and inflammatory responses in pSS. Early activation of the innate immune system, particularly the IFN-I pathway, plays a central role in pSS. Activated pDCs by blood dendritic cell antigen 2 (BDCA2) secrete IFN-I and TNF family cytokines, including BAFF and APRIL, through the JAK/STAT pathway, initiating inflammatory responses. Autoreactive B cells, stimulated by BAFF and APRIL via BAFFR and TACI, and the BTK pathway, produce autoantibodies, with CD19 and CD20 contributing to B cell activation. These autoantibodies form immune complexes that sustain adaptive immune responses. B cells present antigens to Th cells via MHC-II and TCR-CD4 interactions, along with CD40-CD40L signaling. Activated Th cells secrete IL-12, promoting Th1 differentiation and IFN-γ production, which further enhances BAFF expression—creating a pathogenic feedback loop central to pSS.
Figure 6.
Figure 6. Key immune and inflammatory responses in pSS. Early activation of the innate immune system, particularly the IFN-I pathway, plays a central role in pSS. Activated pDCs by blood dendritic cell antigen 2 (BDCA2) secrete IFN-I and TNF family cytokines, including BAFF and APRIL, through the JAK/STAT pathway, initiating inflammatory responses. Autoreactive B cells, stimulated by BAFF and APRIL via BAFFR and TACI, and the BTK pathway, produce autoantibodies, with CD19 and CD20 contributing to B cell activation. These autoantibodies form immune complexes that sustain adaptive immune responses. B cells present antigens to Th cells via MHC-II and TCR-CD4 interactions, along with CD40-CD40L signaling. Activated Th cells secrete IL-12, promoting Th1 differentiation and IFN-γ production, which further enhances BAFF expression—creating a pathogenic feedback loop central to pSS.
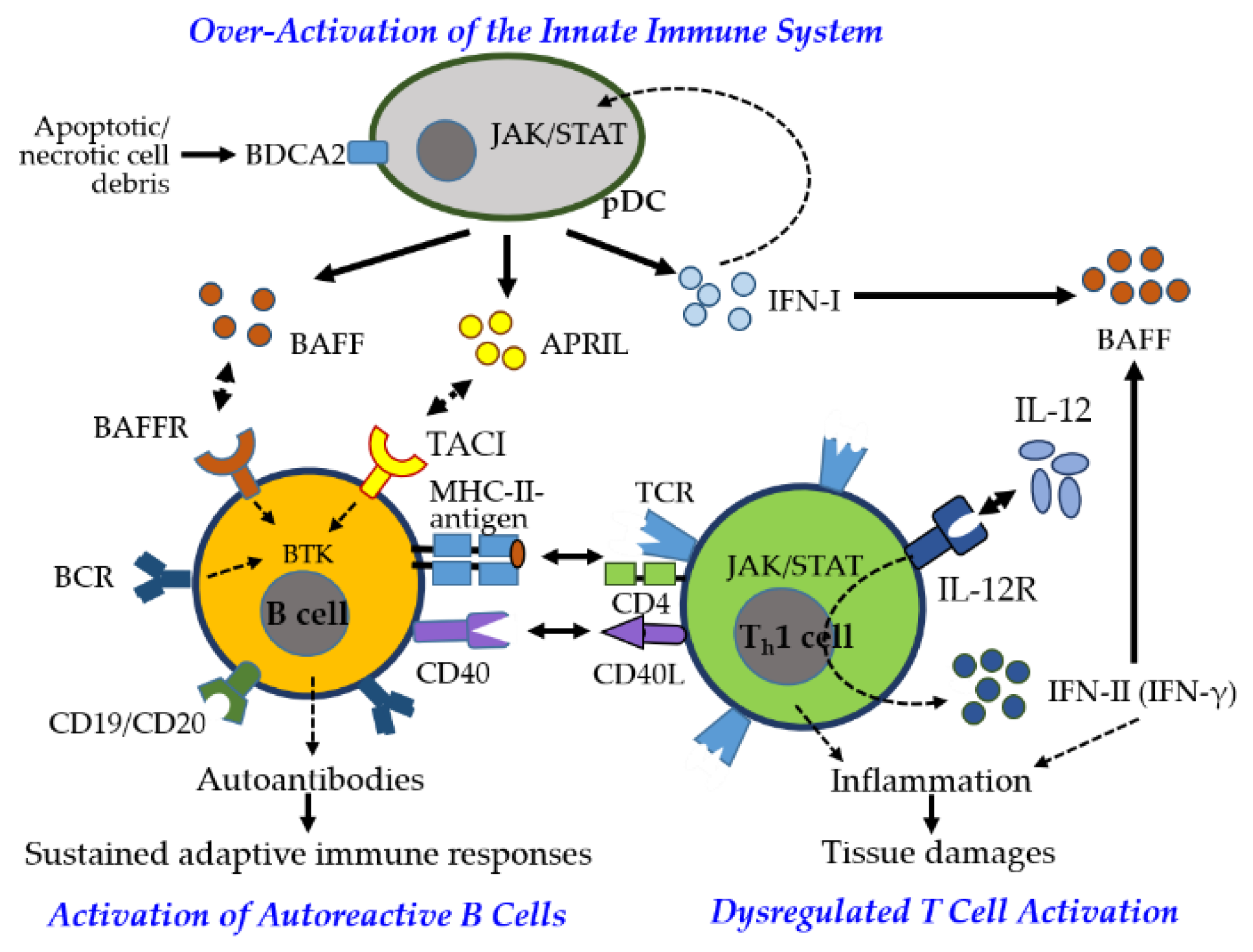
Figure 7.
Factors influencing sex-biased autoimmunity.
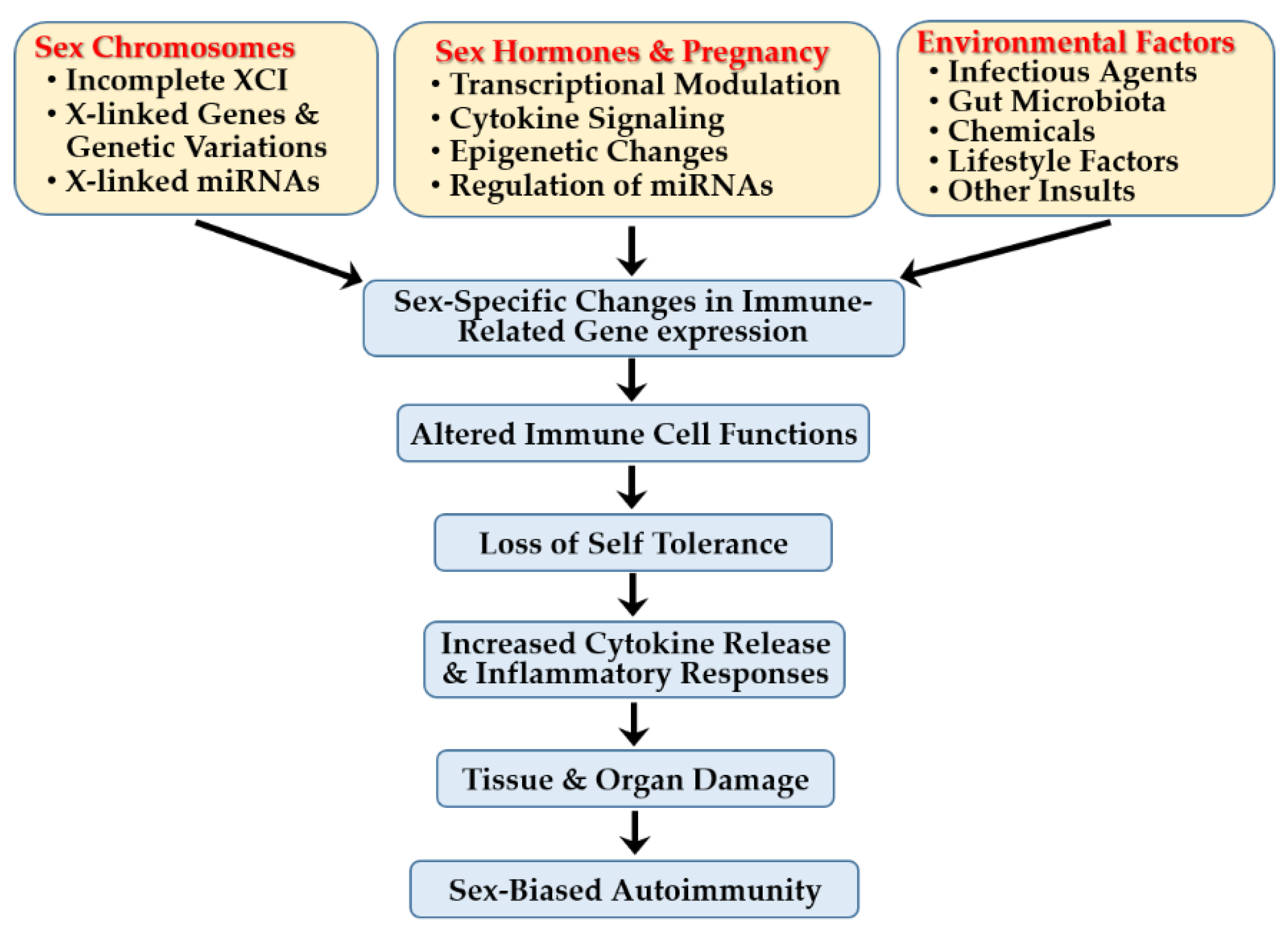

Table 1.
X-linked immune genes escaping XCI and contributing to sex differences in autoimmunity.
| Gene Symbol | Gene Product |
Immunological Function |
|---|---|---|
| BTK | Burton’s tyrosine kinase |
A protein tyrosine kinase that mediates pre-BCR signaling for Ig heavy chain rearrangement, crucial for B cell development and IgE-dependent mast cell activation |
| CD40L | CD40 ligand | A costimulatory molecule on T cells that binds CD40 on APCs, priming pathogenic Th cells, driving immune responses, enabling B-T cell communication and B cell class switching |
| CXCR3 | C-X-C motif chemokine receptor 3 | A chemokine receptor involved in immune cell trafficking, recruiting killer T cells to sites of inflammation |
| CXorf21 | Chromosome X open reading frame 21 | A TLR adaptor that interacts with SLC15A4 on the lysosomal membrane |
| CYBB | Cytochrome b-245 beta chain | A component of the NADPH oxidase complex that generates ROS for microbial killing |
| FoxP3 | Forkhead box P3 | A key regulator of Treg cell development that functions to suppress immune responses |
| IL13RA1/2 | Interleukin 13 receptor subunit alpha 1/2 | A component of the IL-13 receptor complex that mediates immune regulatory functions |
| IL2RG | Interleukin 2 receptor subunit gamma | A component of the IL-2 receptor essential for T cell development and function |
| IL9R | Interleukin 9 receptor | A component of the IL-9 signaling pathway that regulates diverse immune responses |
| IRAK1 | Interleukin 1 receptor associated kinase 1 | A protein kinase that mediates IL-1R and TLR signaling to activate NF-κB and MAPK pathways, promoting innate immune responses and inflammation |
|
KDM6a (UTX) |
Lysine demethylase 6a | An enzyme that demethylates H3K27me3 to regulate gene expression, skewing immunity toward inflammation and enhancing NK cell effector function. |
| OGT | O-linked N-acetylglucosamine transferase | An enzyme involved in protein glycosylation that regulates mTOR activity and influences diverse cellular processes |
| SLC15A4 | Solute carrier family 15 member 4 | A proton-coupled amino acid transporter essential for endolysosomal TLR activation and TLR-mediated IFN-I production in innate immune responses |
| TLR7 | Toll like receptor 7 | A receptor protein that enhances viral RNA sensing and IFN-α production, contributing to female-biased antiviral defense, and promotes ABC accumulation, immune activation, and inflammation |
| TLR8 | Toll like receptor 8 | A receptor protein involved in TLR signaling via MyD88, sufficient to drive B cell tolerance loss, class-switched autoantibody production, enhanced granulopoiesis, and increased IFN-I production |
Table 2.
Changes in X-linked miRs and functional consequences in autoimmune diseases.
| miR | Target Genes | Changes* | Functional Consequences in Autoimmunity |
|---|---|---|---|
| miR-20b | RELA (NF-κB subunit), STAT3 | ↓ | Its downregulation in SLE T cells lifts repression on RELA and STAT3, amplifying NF-κB signaling and Th17 differentiation, thereby promoting inflammation in lupus pathogenesis. |
| miR-23b | TAB2/3 and IKK-α | ↓ | Its downregulation in inflamed tissues of SLE, RA, and MS upregulates NF-κB signaling, promoting proinflammatory cytokine production and autoimmunity, while its ectopic expression suppresses inflammation and reduces disease severity in autoimmune models. |
| miR-92a | KLF2, BCL2L11 (Bim) | ↓ | Its dysregulation in salivary glands and PBMCs of SS patients may disrupt glandular epithelial cell survival and immune cell homeostasis by altering apoptosis and T cell differentiation. |
| miR-98 | IL-6, FAS (CD95), TNF-α | ↓ | Its downregulation in SLE PBMCs lifts suppression of IL-6, increasing proinflammatory cytokines and STAT3 signaling, while FAS upregulation promotes CD4⁺ T cell apoptosis, worsening immune dysregulation and disease activity. |
| miR-106a | IL-10, SOCS5 | ↑ | Its upregulation in CD4⁺ T cells and PBMCs from SLE and pSS patients suppresses IL-10, a regulatory cytokine, and SOCS5, enhancing JAK/STAT signaling and promoting T cell activation and inflammation. |
| miR-125b |
PRDM1 (Blimp-1) |
↓ | Its downregulation in activated CD4⁺ T cells of SS patients lifts repression on PRDM1 (Blimp-1: B-lymphocyte-induced maturation protein 1), enhancing plasma cell differentiation and autoantibody production, while its exosomal delivery from salivary gland-derived mesenchymal stem cells suppresses plasma cell formation and restores secretory function. |
| miR-188 | NFATc2, FOXO1, CBL | ↑ | Its upregulation in PBMCs of SLE, RA, and PA patients suppresses FOXO1, impairing Treg differentiation and immune tolerance, while reducing CBL expression and dampening TCR signaling in lupus CD4+ Th cells, collectively enhancing effector T cell activity. |
| miR-221 /222 cluster |
CDKN1B (p27kip1), ETS1 | ↑ | Its upregulation in SLE PBMCs downregulates CDKN1B and ETS1, driving lymphocyte proliferation and plasma cell differentiation, which enhances autoreactive B cell activity and autoantibody production. |
| miR-222 | CFHR5 | ↓ | Its downregulation in lupus nephritis patients increases CFHR5 expression, overactivating the alternative complement pathway and promoting immune complex–mediated tissue injury. |
| miR-223 |
S1PR1, CXCL2, CCL3 (in SLE) ITPR3 (in SS) |
↑/↓ | Its upregulation in CD4⁺ T cells from SLE patients and in epithelial cells of SS patients suppresses S1PR1 and chemokines, limiting T cell egress and inflammatory cell recruitment, while downregulating ITPR3 to impair Ca²⁺ signaling and activate NF-κB, promoting epithelial inflammation. It is also linked to X chromosome demethylation in female lupus predisposition, whereas its deficiency in LN leads to T cell accumulation and exacerbated renal inflammation. |
| miR-224 | SMAD4, HOXD10, API5 | ↑ | Its upregulation in PBMCs and T cells of SLE, SD/SSc, and RA targets SMAD4, disrupting TGF-β signaling and promoting fibrosis and tissue dysfunction. It also enhances cell proliferation and migration, downregulates apoptosis inhibitor 5 (API5) to facilitate activation-induced cell death in Jurkat and SLE T cells, and upregulates STAT-1, contributing to LN. |
| miR-361-5P | VEGFA, IL-6R | ↑ | Its overexpression in labial salivary glands of SS patients reduces VEGFA and IL-6R expression, potentially compromising vascular integrity and altering cytokine responses in glandular tissues. |
| miR-374a | SOCS1, PTEN, IL-10 | ↑ | Its upregulation in inflamed synovium of SLE and RA downregulates SOCS1 and PTEN, activating JAK/STAT and PI3K pathways, thereby promoting cytokine-driven inflammation and lymphocyte survival, and increased susceptibility to SLE with renal involvement. |
| miR-421 | ATM, E2F1, PDCD4 | ↑ | Its upregulation in LN kidney biopsies and RA synovial tissue impairs the DNA damage response by inhibiting ATM and E2F1 in LN renal tissues, suppresses apoptosis-related genes like PDCD4, promoting fibroblast-like synoviocyte survival and proliferation in RA. |
| miR-424 | CCND1, CDK6 | ↑ | Its upregulation induces cell cycle arrest in SLE PBMCs and SS salivary gland epithelial cells by downregulating CCND1 and CDK6, resulting in tissue atrophy and impaired glandular regeneration. |
| miR-452 | BMI1, RAB11A, CDKN1B | ↓ | Its downregulation derepresses genes that promote T cell proliferation and survival, thereby enhancing autoreactive T cell responses in MS and RA patients. |
| miR-506 | NFATC1 | ↓ | Its underexpression lifts reppression on NFATC1, leading to increased CD4⁺ T cell activation and proliferation in SS patients. |
| miR-548m | PTEN | ↑ | Its overexpression reduces PTEN expression, resulting in hyperactivation of the PI3K–AKT pathway and promoting immune cell survival and activation in SLE patients. |
| hsa-miR-503 | BCL2, CCND1, FGF2 | ↓ | Its downregulation enhances BCL2-mediated survival and CCND1-driven proliferation, leading to synovial hyperplasia and joint inflammation in RA, while its expression is elevated in demethylated CD4+ T cells from women with lupus following 5-azacytidine treatment. |
| hsa-miR-545 | RIG-I, TP53INP1, ZEB2 | ↑ | Its upregulation, observed in some LN datasets, inhibits RIG-I-mediated antiviral responses and regulates p53-dependent apoptosis, potentially shifting immune balance away from effective antiviral surveillance. |
| hsa-let-7f-2 | STAT3, IL-13, TGFBR1 | ↑ | Its upregulation in the plasma and salivary glands of SLE and SS patients modulates Th2 and Th17 differentiation by suppressing STAT3 and IL-13, potentially disrupting effector T cell balance and promoting proinflammatory cytokine production. |
*↑, over-expressed; ↓, under-expressed.
Table 3.
Immune-related gene products regulated by sex hormones.
| Gene Products | Impacts by Sex Hormones |
|---|---|
| TLRs (Toll-like receptors) |
Estrogens and ERα signaling differentially regulate TLR family members, enhancing TLR7/9-mediated IRF5 activation and IFN production in female pDCs, while modulating TLR8 expression independently of IFNs through direct ERα binding to an ERE near the TLR8 locus or indirectly via STAT1-mediated transcriptional activation. |
| IRF5 (Interferon regulatory factor 5) |
Estrogens and ERα signaling upregulate IRF5, a key transcription factor involved in immune responses and a lupus susceptibility factor, leading to IFN-I overproduction and contributing to autoimmune disease progression in SLE. |
| IFNs (Interferons) | Estrogens enhance IFN-α and IFN-γ production via ERα and TLR7/9-mediated IRF5 activation in pDCs, amplifying cytokine output and innate immunity, thereby contributing to the female bias in autoimmunity. IFN-α upregulates MHC-I, while IFN-γ induces MHC-II and alters proteasome composition, facilitating self-peptide presentation to T cells—processes central to SLE pathogenesis. |
| ILs (Interleukins) | Estrogens and ERα signaling promote IL-6 expression and inflammation in both mice and humans, while elevated estradiol in SLE patients enhances the secretion of IL-8, IL-18, and IL-23. Estrogen-regulated cytokines such as IL-4, IL-5, and IL-10 support B cell activation and antibody production. Notably, increased IL-6 and IL-10 levels correlate with higher SLE disease activity index (SLEDAI) scores, linking estrogen-driven cytokine expression to disease activity in SLE. |
| BAFF (B cell activating factor) |
Estrogens enhance BAFF production, which supports B cell survival and maturation, leading to elevated antibody levels and potentially influencing thyroid dysfunction in GD. |
| UNC93B1 (Unc-93 homolog B1) | Estrogens enhance UNC93B1 expression via IFN-α or IFN-γ signaling, with notably higher levels observed in lupus-prone female mice and PBMCs from SLE patients compared to healthy controls. |
| S1PR2 (Sphingosine-1-phosphate receptor 2) | Estrogens regulate S1PR2 expression, a G-protein–coupled receptor, potentially contributing to the female-biased severity of CNS-related autoimmune diseases like MS. |
| AIRE (Autoimmune regulator) | Estrogens increase methylation of CpG sites in the AIRE promoter, inducing epigenetic silencing of AIRE—a central tolerance regulator controlling tissue-specific antigen expression—thereby enhancing autoimmune susceptibility. In contrast, androgens upregulate AIRE, contributing to sex bias in CNS autoimmune diseases. |
| AID (Activation-induced cytidine deaminase) | Estrogens promote AID transcription, enhancing somatic hypermutation and class switch recombination in activated B cells—key processes for antibody diversification—likely through AID’s interaction with the chromatin modifier UBN1, a component of the HIRA chaperone complex. |
| SLC15A4 (Solute carrier family 15 member 4) |
Estrogens upregulate SLC15A4 expression, enhancing IFN-I and pro-inflammatory cytokine production in pDCs, thereby contributing to autoimmune disease progression in SLE and colitis models. |
| Cathepsin S | Estrogens promote inflammation by activating cathepsin S—a lysosomal acidic protease involved in immune regulation—elevated in the lacrimal glands and tears of female SS murine models., whereas testosterone reduces inflammation and enhances glandular function in SS, potentially through cathepsin S suppression. |
Table 4.
Estrogen-dependent autosomal miRs and their functions in autoimmune diseases.
| miR | Location | Change* | Target Gene | Impacts by Sex Hormones |
|---|---|---|---|---|
| miR-10b | Chr2 | ↑ | SRSF1, MAPK7 (TAK1) | Estrogens upregulate miR-10b-5p, which inhibits SRSF1 and MAPK7, enhancing NF-κB signaling, Th17 differentiation, and proinflammatory cytokine expression in T cells. |
| miR-26a | Chr3/ Chr12 |
↓ | AICDA, HMGA2, COX-2, TLR4, MALT1, HMGA1 | Estrogens suppress miR-26a, enhancing class-switch recombination and autoantibody production, while androgens induce miR-26a to restrain AICDA expression and B cell activation. miR-26a downregulation also enhances proinflammatory cytokines via other target genes. |
| miR-31 | Chr9 | ↓ |
RhoA, CEACAM1, IRF5, STAT-1, SLC15A4 |
Estrogens downregulate miR-31 via TGF-β and NF-κB in SLE T cells, impairing IL-2 production by disrupting NFAT, NF-κB, and AP-1 activity, while increasing CREM and dysregulating CaMK-IV and PP2A. This leads to defective IL-2 signaling, impaired Treg differentiation, and T cell dysfunction. |
| miR-96 | Chr7 | ↑ | FoxP3, RHOA, FCGR1, IL-2, CD138, CEACAM1 | Estrogen-induced miR-96 upregulates immune genes such as FoxP3, RHOA, FCGR1, IL-2, CD138, and CEACAM1, influencing SLE susceptibility, onset, clinical heterogeneity, and progression. |
| miR-127 | Chr14 | ↑ | FoxP3, RHOA, FCGR1, IL-2, CD138, CEACAM1 | Estrogen-induced miR-127 promote immune and inflammatory responses, contributing to SLE susceptibility, onset, clinical heterogeneity, and progression. |
| miR-145 | Chr5 | ↓ | STAT1, OPG | Estrogen-mediated miR-145-5p downregulation elevates osteoprotegerin, reducing osteoclast activity and bone resorption, and contributing to joint damage in RA. |
| miR-145a | Chr5 | ↓ | ADAM17, KLF4, SIRT1 | miR-145a targets inflammation- and stress-related genes to suppress immune activation, but estrogen downregulates miR-145a in B cells, promoting immune activation. |
| miR-146a | Chr5 | ↑↓ | IRAK-1, TRAF-6, IRF5, STAT-1, SLC15A4 | miR-146a suppresses IRAK-1 and TRAF-6 translation, serving as a negative regulator of immune activation. Estrogens dysregulate miR-146a in PBMCs and splenocytes of MRL/lpr mice, linking it to epigenetic changes, B cell hyperactivity, and autoantibody production in autoimmunity. |
| miR-148a | Chr7 | ↑ | DNA methytransferase 1(DNMT1) | Upregulated miR-148a promotes DNA hypomethylation, contributing to autoimmune disease pathogenesis. |
| miR-148b | Chr12 | ↑ | CaMKIIα, Gadd45α, PTEN, Bim | miR-148b suppresses TLR-induced cytokine and IFN-I production, impairing DC-medicated innate responses. It also promotes DNA hypomethylation and survival of autoreactive B cells, contributing systemic autoimmunity. |
| miR-155 | Chr21 | ↑ |
MAPK, INS, Wnt, NF-κB, BIC, Pu.1, c-Maf, c-Fos, IFNγRα, c-Rel, c-Fos, Peli1, p27kip1, KPC1, SOCS1 |
miR-155 regulates immune cell homeostasis, Th1 differentiation, tolerance, and development. It supports B cell maturation, isotype switching, germinal center formation, high-affinity IgG1 production, DC activation, apoptosis, and IL-12 production, promoting autoimmune susceptibility. |
|
miR-183 |
Chr7 | ↑ | FoxP3, RHOA, FCGR1, IL-2, CD138, CEACAM1 | miR-183 modulates immune and inflammatory responses, contributing to susceptibility to SLE and influencing its onset, clinical heterogeneity, and progression. |
|
miR-379 |
Chr14 | ↑ | FoxP3, RHOA, FCGR1, IL-2, CD138, CEACAM1 | miR-379 modulates immune and inflammatory responses, influencing SLE susceptibility, onset, heterogeneity, and progression. |
| let-7e | Chr19 | ↓ | SOCS1,TLR6, TLR9 | let-7e targets SOCS1 and TLR pathway components, regulating cytokine signaling and innate immunity. Estrogens suppress let-7e in B cells enhancing immune activation. |
| let-7f | Chr9/ ChrX |
↓ |
IL-23R, NLRP3, IL-6, A20 (TNFAIP3) |
let-7f-5p targets NLRP3, mitigating inflammation in bone marrow-derived mesenchymal stem cells, and also regulates IL-6 and A20, key modulators of inflammatory and NF-κB signaling pathways. Estradiol and progesterone suppress let-7f and upregulate IL-23R, enhancing IL-17A production in Th17 cells, thereby promoting Th17 differentiation and inflammation. |
*↑, over-expressed; ↓, under-expressed.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



