Submitted:
03 June 2025
Posted:
04 June 2025
You are already at the latest version
Abstract
This study explores the self-assembly and optical properties of close-packed quantum dots (QDs) within Langmuir-Blodgett (LB) films, focusing on strain-driven interactions and potential inter-atom diffusion. Monolayers of yellow-emitting QD450 and red-emitting QD645 were subjected to continuous laser illumination, revealing significant inter-dot coupling effects and time-dependent photoluminescence (PL) dynamics. The exponential shifts in PL peak positions and broadening of emission spectra suggest strain-induced modulation of optical properties. Differences in Se/S mole ratios and anisotropic strain at the core/shell interfaces of the QDs are proposed to drive inter-atom diffusion, leading to localized structural and electronic changes. Additionally, nonlinear variations in band-gap energy under varying laser intensities highlight a complex interplay between strain, optical excitation, and material properties. These findings provide insights into strain-driven atomic rearrangements and their impact on the photophysical behavior of QD systems.
Keywords:
Quantum dots (QDs)
; Langmuir-Blodgett films
; PL
; Inter-dot coupling effects
1. Introduction
Since their initial use in Light-Emitting Diodes (LEDs) by Colvin et al. [1], semiconductor quantum dots (QDs) have become essential in optoelectronics due to their size-tunable, narrow, and bright emission. They enable high-performance displays—such as QD color filters and ultra-high-display screens[2]—and efficient QD-LEDs, with external quantum efficiency (EQEs) exceeding 18% through optimized architectures[3]. In photovoltaics, PbS and CdSe QDs offer tunable bandgaps and multiple exciton generation, achieving over 15% efficiency in third-generation solar cells[4,5]. Their stable fluorescence and high surface area make QDs ideal for chemical sensing via FRET, PET, and charge transfer, with CdTe QDs detecting Cu²⁺ at trace levels[6]. In biomedicine, QDs pioneered by Bruchez et al. and Chan & Nie[7,8] are widely used for labeling, imaging, and diagnostics, including sensitive detection of biomarkers like miRNA-21 linked to cancer and psychiatric disorders[9], positioning QDs as versatile platforms across advanced technologies[10,11,12,13].
However, the conventional binary QDs have size-dependent emission properties, which restricts independent control over optical wavelength and particle size. Alloyed QDs, such as CdSeS and CdSeTe, provide a solution to this limitation by offering bandgap tunability via compositional adjustment at a fixed size. This decoupling is particularly valuable in applications requiring monodisperse particles size with precise optical outputs. Chen et al. showed that CdSeS alloyed QDs could be systematically tuned from green to red emission by adjusting the Se/S ratio while preserving nanocrystal size and stability[14]. These alloyed systems also exhibit reduced lattice mismatch and improved photostability when protected with suitable shells like ZnS.
Beyond intrinsic tunability, the optical properties of alloyed QDs are highly sensitive to external environmental conditions. Factors such as substrate interaction, film morphology, interlayer coupling, and ligand integrity can substantially influence emission characteristics. Layer-by-layer (LbL) assembled films where monolayers of QDs are deposited sequentially onto substrates present opportunities for engineered light–matter interactions but also introduce complexity through interdot coupling, strain accumulation, and photoinduced modifications. Previous studies have shown that photoluminescence (PL) from QDs can shift in wavelength, broaden in linewidth, or fluctuate in intensity depending on excitation power, ambient conditions, or substrate chemistry[7,14,15,16,17,18,19]. We have demonstrated in the past that strain-driven atomic interdiffusion of alloyed quantum dots at interfaces during film formation significantly alters their emission properties, highlighting the importance of post-synthetic stability studies and their implications for long-term optoelectronic and biomedical imaging applications.[17] Yet, detailed investigations into photophysical behaviors and bandgap modulation effects in layered alloyed QDs remain scarce, especially for CdSeS/ZnS core/shell systems under continuous optical excitation.
In this work, we investigate the temporal and spectral evolution of photoluminescence in layered CdSeS/ZnS quantum dot films assembled via Langmuir–Blodgett (LB) deposition on oxide-coated silicon substrates. The layered structure consists of a bottom layer of blue-emitting QD450 (∼9 nm) and a top layer of red-emitting QD645 (∼12 nm), with oleic acid ligands serving as optically transparent spacer layer. Under continuous 532 nm laser excitation focused on the top layer, we observe an unexpected combination of PL intensity oscillation, progressive blue-shift in peak wavelength, and broadening of the emission linewidth (FWHM). These effects are absent or minimal in control architectures lacking interlayer heterojunctions or compositional asymmetry. Through this, we aim to uncover new insights into the environmental effect on the exitonic behavior of QD heterostructures, and inform the design of future photonic, biosensing, and optoelectronic applications using engineered nanocrystal films.
2. Materials and Methods
2.1. Materials
A10 mg/mL suspension in chloroform of CdSSe/ZnS or CdSe/ZnS core/shell quantum dots (QDs) consisting of a monolayer of oleic acid coating were obtained from Ocean NanoTech LLC, San Diego, CA, USA and used as received. The QDs have an emission wavelength peak ranging from 450 nm to 645 nm and with quantum yield >50%.
2.2. Preparation of QD Film by Langmuir-Blodgett (LB) Techniques
Quantum dot thin films were deposited on oxide-coated silicon wafers and cover glass substrates. These substrates were cleaned by sonicating in acetone, isopropanol, and ultrapure water for 5 minutes each, followed by a 5-minute ultraviolet ozone treatment (Novascan Technologies, Inc.). Then the substrate was immersed vertically in ultrapure DI water subphase in Langmuir-Blodgett (LB) trough to a pre-programmed height and let the water surface clean and stable. 10 -20 microliters of a 10 mg/mL solution of quantum dots in a chloroform was gently applied to the water surface using Hamilton microliter syringe and was allowed to stabilize for 20 to 30 minutes. The film was then compressed at a typical barrier speed of 10 mm/min until a target surface pressure of 20−40 mN/m was attained and form close-packed monolayer film of QDs at the air/water interface. The film is allowed to be stabilized for about 5 min. Then the film was transferred to a substrate as it moved automatically vertically out of the subphase at a speed of 5 mm/min under a constant surface pressure.
2.3. Optical Characterization
A collimated, linearly polarized light of lasers ( 405 nm and 532 nm diode lasers) were attenuated and directed to a microscope (Olympus GX51) and focused on the sample using 20x objective. The collected PL is passed through the same objective, and the signal is sent to a spectrometer (IsoPlane Spectrograph of Princeton Instruments) with a cooled CCD camera, while a long-pass filter removes any remaining source of light. This setup allows PL collection from a fixed area by switching excitation sources without moving the components or the sample.
2.4. Structural Characterization
Atomic force microscope (AFM).
Atomic force microscope (AFM) images were recorded in air using Neaspec-GmbH instrument operated in a tapping mode with tapping amplitude ≈50 nm near the resonance oscillation frequency of the cantilever. The cantilevers used were NCHR series of standard and supersharp silicon (Nanoworld, NanoAndmore, USA), with a resonance frequency of 250-390 kHz and an average tip radius of 5 nm and 12 nm respectively, for super sharp and standard AFM tip.
3. Results and Discussion
The structural organization, thickness, and optical properties of the self-assembled QD layers are depicted in Figure 1a-c. The data in the Figure serves as a foundational characterization of the LB QD film, demonstrating its structural integrity, precise thickness control, and distinct photoluminescence properties. These attributes set the stage for subsequent investigations into optical coupling as well as the photophysical and photochemical dynamics.
The AFM image in Figure 1a reveals the morphology of the QD monolayers deposited onto an oxide-coated silicon wafer. The image distinctly shows the monolayer of QD645 (orange layer) on top of QD450 (red layer), with the underlying silicon substrate appearing in black. The LB technique enables the formation of a well-defined, closely packed monolayer structure, ensuring controlled stacking of the QDs. The AFM image confirms the uniformity of the film and the successful deposition of multiple QD layers with minimal aggregation, which is crucial for achieving consistent optical properties. Figure 1b provides a cross-sectional thickness profile of the LB film, measured along a white dashed line in the AFM image. The thickness of the individual QD monolayers is determined by the AFM scan, which captures the height variations across the sample and the height is consistent with the size of individual quantum dots (Figure S1a-b supporting information). The thickness profile suggests that the spacing between the QDs is maintained by an oleic acid monolayer, which enforces a 1.1 nm separation between adjacent dots. This precise control over layer thickness is essential for modulating inter-dot interactions and optimizing the optical properties of the assembled structure.
Figure 1c shows the photoluminescence spectra, highlighting the emission characteristics of the two types of QDs in the LB film. QD450 exhibits a peak emission at approximately 450 nm (red region), while QD645 emits at around 645 nm (orange region). Spectral analysis indicates that both quantum dots maintain their unique optical characteristics within the monolayer assembly. The absence of significant spectral overlap indicates minimal self-quenching or undesirable energy transfer between the layers under initial conditions. However, further experimental analysis suggests that prolonged laser illumination induces photophysical changes, the detail of which is described in the next section.
3.1. Oscillatory Temporal Dynamics in PL Emission
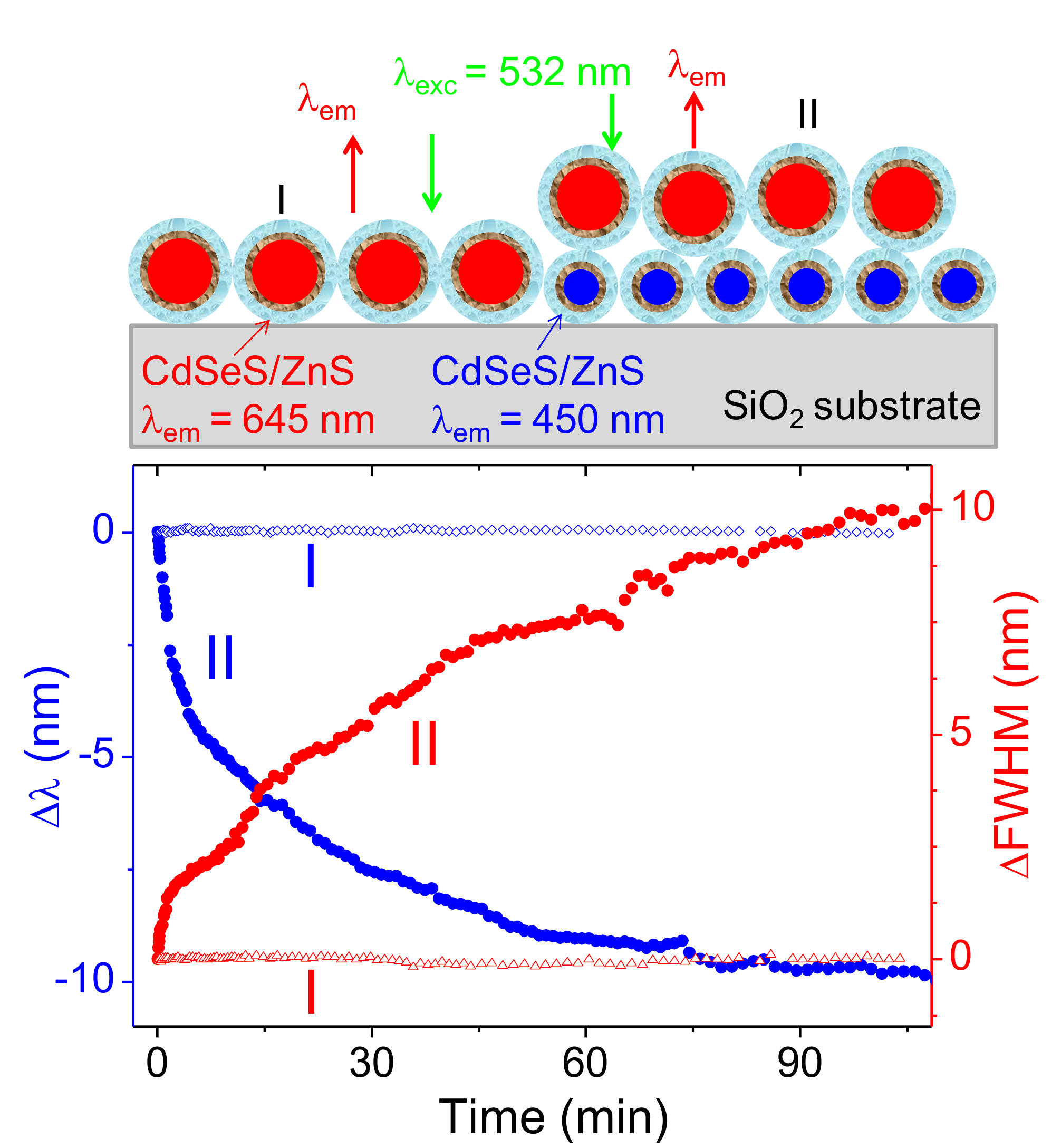
Figure 2 displays the time evolution of photoluminescence (PL) characteristics from the top layer ternary CdSeS/ZnS quantum dot (QD) film with a nominal diameter of 12 nm under continuous 532 nm laser excitation. The upper panel in the figure (Figure 2a) simultaneously tracks the peak wavelength shift (blue trace) and full width at half maximum (FWHM, green trace), while the lower panel (Figure 2b) shows the PL intensity decay (red trace). All measurements are plotted against time in minutes.
Over the course of the 120-minute measurement, the PL emission characteristics exhibit a clear oscillatory behavior superimposed on their general trends. The emission peak wavelength shows a net blueshift, progressing up to approximately 10 nm, suggesting an increasing average recombination energy over time. Simultaneously, the FWHM undergoes gradual broadening, rising from near zero (change at initial time) to about 6 nm, indicating that the spectral profile becomes increasingly disordered or diverse. These spectral features mirror the underlying carrier dynamics and structural reorganization of the QD ensemble. However, excitation of the QDs monolayers on the blue absorption edge results in drastic red-shift in emission peak positions with time(data not shown).
Most notably, both the wavelength shift and the FWHM trace display recurrent modulations with subtle amplitude. These non-random fluctuations reflect slow, damped oscillations around the mean trend line, suggesting a dynamic feedback loop in which the local band structure, strain field, or charge distribution periodically adjusts under continuous excitation. The oscillatory modulation in FWHM hints at transient narrowing and broadening cycles, which likely correspond to alternating phases of localized and delocalized recombination across spatially varying emission sites.
In the lower panel, the PL intensity likewise decreases over time but with visible quasi-periodic dips and partial recoveries, indicating that recombination efficiency is not monotonically declining. These dips appear to be phase-shifted relative to the spectral changes—periods of minimum intensity often align with blueshifted emission and broader spectral width. This antiphase relationship supports a model in which recombination becomes less efficient as the system shifts toward higher-energy, more disordered states. Together, these oscillatory trends point to a coupled photophysical system in which trap carriers, stress relaxation, and band-edge modulation co-evolve under prolonged photoexcitation.
3.2. Exponential Fitting of Temporal PL Dynamics
Figure 3 presents a detailed analysis of the underlying temporal decay trends extracted from the same dataset shown in Figure 2. Each panel displays experimental data (colored dots) overlaid with fitted exponential models (black or dashed lines) to isolate and quantify the relaxation dynamics governing photoluminescence (PL) intensity, emission peak shift, and spectral broadening over time.
In the top panel (Figure 3a), the PL intensity (red) is modeled using a bi-exponential decay function, reflecting the presence of two distinct recombination pathways. The initial rapid decay (τ₁ ≈ 953 s) likely corresponds to direct excitonic recombination, where photoexcited electrons and holes recombine efficiently in well-passivated QDs. The slower component (τ₂ ≈ 8821 s) is attributed to trap-assisted relaxation or long-lived carrier retention, where charge carriers are transiently localized in sub-bandgap states or migrate across energetically disordered regions before recombination. This long tail is consistent with the persistent low-level emission observed even after extended illumination, and it sets the timescale for structural and environmental changes within the QD layer.
The middle panel (Figure 3b) illustrates the peak wavelength shift (blue), which also fits well to a bi-exponential decay. The overall blueshift trajectory suggests that recombination pathways shift from lower-energy, possibly trapped states toward higher-energy states over time. The fast component (τ₁ ≈ 523 s) may reflect early-time carrier redistribution and filling of shallower traps, while the slower component (τ₂ ≈ 5541 s) is associated with gradual changes in the QD energy landscape, possibly due to photoinduced structural relaxation or band-edge modulation arising from accumulated excitation stress.
In contrast, the bottom panel (Figure 3c) shows that FWHM broadening (green) is best described by a single-exponential growth model with a decay constant of approximately τ ≈ 2231 s. The monotonic increase in spectral width suggests that the emission arises from an increasingly diverse set of recombination sites, either due to progressive delocalization of carriers or the activation of QDs with slightly different bandgap energies. Unlike the PL intensity and wavelength shift, no significant secondary component was needed to capture the broadening trend, indicating that a single, slow process—likely involving the gradual introduction of energetic disorder—is sufficient to explain the FWHM evolution.
Collectively, the exponential fitting in Figure 3 disentangles the decay and broadening behavior of the PL signal into physically meaningful timescales. These fits confirm that multiple relaxation pathways govern the optical behavior of the ternary QD layer, and that structural and energetic reorganization unfolds over tens of minutes to hours, underpinning the damped oscillatory modulations observed in Figure 2. These findings reinforce the role of internal feedback mechanisms—such as carrier migration, trap cycling, and stress modulation—as key drivers of temporal and spectral variability in this system.
The emergence of damp oscillations and a sustained blueshift in the PL response of layered ternary quantum dot (QD) films presents a deviation from canonical photoluminescence behavior under continuous excitation. These findings suggest the presence of underlying feedback mechanisms or slow dynamic processes that modulate the optical properties over time. Several hypotheses may account for these observations, though each remains incomplete.
While several hypotheses including potential gradients, strain relaxation, internal electric fields, and trap-state cycling could plausibly account for the observed PL oscillations, their mechanisms remain speculative and experimentally unverified. To isolate the role of compositional asymmetry and interfacial coupling, we conducted control experiments using symmetric and binary QD bilayer systems under identical conditions as shown in Section 3.4.
3.3. Control Experiment
To substantiate the hypothesis proposed in Figure 2 and Figure 3 namely, that the temporal oscillatory behavior in PL emission arises from interfacial processes between QD layers of differing composition, we performed control experiments using alternate quantum dot configurations. The results are summarized in Figure 4a,b, and Supporting Information Figure S2a-c.
Figure 4a displays the time-dependent shift in PL peak wavelength under continuous 532 nm excitation for four different QD film systems. The heterostructured QD645/QD450 bilayer (blue traces), which combines a ternary CdSeS/ZnS (QD645) top layer with a CdSeS/ZnS (QD450) bottom layer, exhibits a clear oscillatory blue-shift—consistent with compositional evolution and bandgap modulation over time. In contrast, all three control systems exhibit negligible wavelength shifts throughout the same duration. The QD645/Glass (black squares, Figure 4a) is an LB monolayer CdSeS/ZnS QDs fabricated directly on a substrate. The QD645/QD645 (green triangles) is symmetric LB bilayer, and QD630/QD450 (orange diamonds) is a binary CdSe/ZnS QD LB film on a molayer LB film of CdSeS/ZnS (QD450). This indicates that neither prolonged illumination nor substrate effects alone can account for the spectral behavior observed in the asymmetric bilayer, underscoring the necessity of compositional asymmetry and inter-QD contact for triggering dynamic interfacial processes.
Figure 4b reinforces this conclusion by examining the evolution of the full width at half maximum (FWHM) of the PL emission spectrum, plotted as the change relative to its initial value. As clearly observed in Figure 2 the QD645/QD450 film shows a gradual and oscillatory increase in ΔFWHM, consistent with an increasing heterogeneity in recombination sites or evolving electronic disorder across the film. Meanwhile, the control systems maintain stable spectral widths, indicating that the observed broadening in the test system arises from interfacial structural reorganization, not general photobleaching or optical instability.
Figure S3 (Supporting Information) shows normalized PL intensity trends under continuous 532 nm excitation. The asymmetric QD645/QD450 bilayer exhibits a characteristic oscillatory decay, mirroring the spectral dynamics seen in Figure 4a and Figure 4b. In contrast, all control systems show an increase in PL intensity over time, albeit to varying degrees. This rise is minimal in QD645/QD645 and QD630/QD450, but pronounced in the QD645/Glass sample, suggesting a strong photobrightening effect.
Interestingly, while the QD645/QD645 bilayer appears stable in terms of peak wavelength (Figure 4a), its FWHM (Figure 4b) exhibits a slight narrowing trend over time. This subtle but reproducible behavior may reflect interlayer energy transfer or self-organization processes that enhance spectral uniformity, even in the absence of compositional asymmetry. Such narrowing is absent in other controls, indicating that interlayer communication in symmetric films, though weaker, may still lead to measurable spectral refinement over time.
Together, these control experiments validate the central claim that the PL oscillations and spectral changes reported in Figures 2 and Figure 3 are not universal effects of illumination or film fabrication but are instead the result of strain-induced, compositionally driven interfacial diffusion and dynamic bandgap restructuring in the asymmetric bilayer system.
3.4. Laser Power Dependence
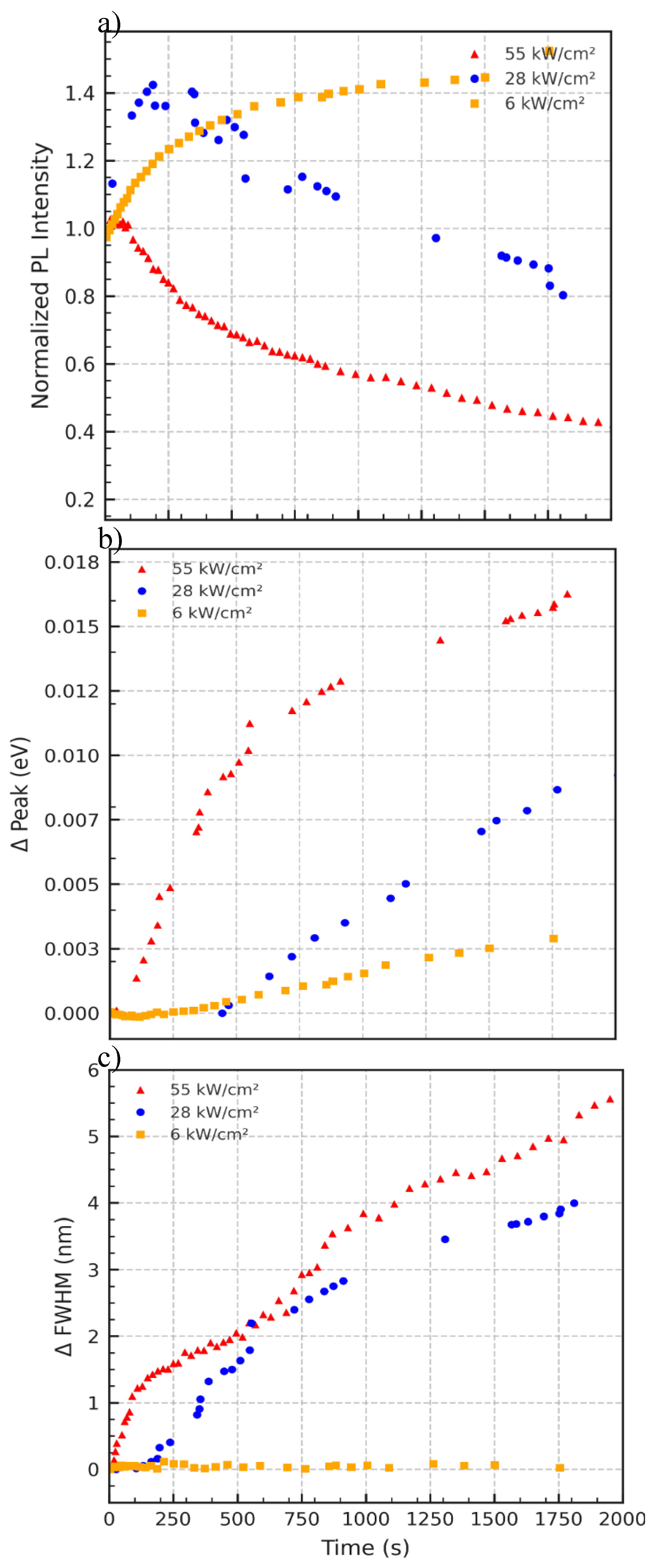
The Data in Figure 5a exhibits power-dependent PL intensity behavior. At low power (6 kW/cm²), intensity slightly increases over time, suggesting mild reorganization or trap passivation. At high power (55 kW/cm²), intensity decays more rapidly, consistent with photobleaching and enhanced nonradiative recombination pathways. Figure 5b depicts progressive blueshift in the emission peak, particularly at higher excitation powers. Figure 5c shows that Spectral broadening is most pronounced at high excitation power. The ΔFWHM increases steadily with time, indicating increased energetic disorder, due to structural relaxation and interdot interaction. At lower powers, the FWHM remains more stable. However, the data in this trend supports a mechanism involving compositional redistribution (e.g., Se/S exchange) and bandgap widening, driven by strain and atomic diffusion between coupled quantum dots.
4. Discussion
In our Langmuir–Blodgett (LB) assembled bilayers of ternary CdSeS/ZnS quantum dots (QDs), we observe striking oscillatory photoluminescence (PL) behavior under continuous excitation (Figure 2 and Figure 3). Rather than a simple photobleaching or monotonic shift, the PL intensity and peak wavelength fluctuate periodically over time. For example, the PL intensity rises and falls in an oscillatory fashion, and the emission peak of the bilayer alternately red-shifts and blue-shifts in successive cycles. Such oscillations are unprecedented in static QD films and point to active photophysical and photochemical processes occurring within the LB film during illumination.
This unusual behavior can be understood by considering the unique interlayer interactions enabled by the LB assembly. The LB technique produces extremely close-packed QD monolayers, and when two different QD monolayers (QD645 and QD450) are sequentially deposited to form a bilayer, the QDs from opposing layers come into intimate contact at the interface. Crucially, the LB process likely compromises the native surface ligands, bringing QD surfaces closer than in solution or thick films. It is known that exposing QDs to polar environments or short-chain solvents can strip away long-chain ligands, dramatically reducing inter-dot distances and quenching PL[20]. In our LB films, a similar effect is expected: the compression and transfer steps can displace or disorder the oleate ligands, allowing direct QD–QD contacts across the bilayer interface. Within a single monolayer, by contrast, neighboring QDs remain separated by whatever ligand shell remains, limiting any atomic exchange or charge transfer to relatively weak dipolar couplings. Thus, intralayer interactions are minimal, whereas interlayer interactions are pronounced in the bilayer. The experimental fact that oscillatory PL emerges only in the mixed bilayer (Figure 2 and Figure 3) and not in single-component films underscores that this effect originates from an interface-specific phenomenon introduced by the LB assembly.
At the interface between the two QD monolayers, we propose that a unique atom interchange process takes place, driven by the compositional asymmetry between the contacting QD645 and QD450 nanocrystals. Although both QD types are chemically similar alloyed CdSeS/ZnS heterostructures, their different emission wavelengths indicate different internal compositions and size (e.g., QD645 likely has a larger, more Se-rich core, whereas QD450 has a smaller, more S-rich core). When these dissimilar QDs are pressed into contact (with minimal ligand barriers), a thermodynamic driving force exists for atomic interdiffusion at the interface. In essence, the two QDs strive to reach a common composition at their juncture—a process akin to an alloying or cation/anion exchange localized at the contact region. We hypothesize that chalcogen exchange (Se↔S) or cation mixing (Cd↔Zn) occurs across the interface, altering the core compositions of both QDs in the contact pair. This photoinduced interdiffusion is plausible, given the energy input from the laser and the close proximity of the crystals. Analogous diffusion processes are well documented in core/shell QDs under thermal or photonic stimulus. For instance, under elevated temperatures, Zn[2]+ has been observed to diffuse from a ZnS shell into a CdSe-based core, leading to a continuous blue-shift of the emission peak.[21] Such interdiffusion is often accompanied by lattice strain effects; as the shell material mixes into the core, the lattice mismatch is reduced, which can further shift the bandgap.[21] Indeed, in situ heating of a CdSe–CdS–ZnS core–multishell system causes Zn and Cd intermixing between layers, transforming the structure into an alloyed CdZnSe/CdZnS and changing its optical output.[22] By parallel, in our LB bilayer, the initial contact between a CdSeS-rich QD645 and a CdSeS-lean QD450 induces a “change–equilibrium–change” sequence: a rapid atomic rearrangement (change) at the interface as the two QDs partially exchange constituents, a temporary equilibrium when their interface compositions balance, and subsequent changes if new driving forces arise (e.g., further illumination or heating). This sequence will manifest as an oscillatory PL if each stage alters the emission in opposite directions. Notably, an initial intermixing could red-shift the blue QD’s emission (by adding Se or increasing its size) while blue-shifting the red QD’s emission (by adding S or reducing quantum confinement), bringing their peak wavelengths closer. If the process overshoots or reverses (for example, due to stress relaxation or reversible ligand reattachment), the peaks might then diverge again, producing a cyclical shift. The net result is a photoinduced oscillation in the optical properties, as observed.
The oscillatory PL intensity can be rationalized in a similar framework. Initially, the close packing and partial ligand stripping in the bilayer likely introduce nonradiative surface defect sites, causing an abrupt PL drop (first “valley” in the oscillation)—consistent with reports that removing native X-type ligands sharply reduces QY.[20] However, as interfacial atoms exchange and a more favorable bonding configuration sets in, some defects may be annealed out or new radiative recombination pathways might form, leading to PL recovery (“peak” in intensity). This mirrors behaviors seen in other QD systems where PL can darken then brighten under sustained illumination depending on surface chemistry.[23] For example, partial oxidation or alloying can initially trap excitons (diminishing PL) but subsequently passivate deep traps (recovering PL).[24] In our case, the first atomic rearrangement at the QD–QD interface might alleviate strain or neutralize dangling bonds, temporarily restoring PL intensity. Subsequent cycles of intensity decrease, and increase could arise from repeating structural/chemical oscillations—for instance, recurrent formation and healing of interfacial trap states. It is conceivable that local strain plays a role in this feedback: the lattice mismatch between QD645 and QD450 imposes stress at the interface, which could drive further atom exchange or defect creation to relieve that strain. In epitaxially fused QD superlattices[25,26], it is known that the “necks” connecting QDs introduce new electronic states and regions of different bandgap.[27] Those neck regions, under strain, have distinctly larger bandgaps than the dot interiors, creating potential barriers that facilitate strong coupling between dots.[27] By analogy, the interfacial region in our bilayer behaves as a “neck” between two different QDs—any lattice strain or compositional gradient there could modulate carrier localization and recombination rates. Initially, the strain (and associated defects) might quench the PL.[22] But as atoms interchange to form a more compositionally graded interface (similar to inserting a graded CdS layer to mitigate CdSe/ZnS strain), the strain is partly relieved and the trap density drops, boosting the PL. This interplay of strain-driven defect formation and strain relief through interdiffusion could naturally lead to oscillatory PL output as the system finds a dynamic balance.[28]
Control experiments (Figure 4) reinforce that the observed oscillatory photoluminescence dynamics are unique to the compositional and structural asymmetry of the QD645/QD450 LB bilayer. Neither symmetric LB bilayers of QD645/QD645 nor mixed bilayers of binary CdSe QD630 with QD450 exhibit such behavior, indicating that both alloyed composition and interlayer heterogeneity are critical. These findings suggest that strain-coupled interfacial atom exchange—enabled by compromised ligand coverage during LB assembly—drives the reversible bandgap modulation underlying the observed photophysical oscillations.
The experimental data in Figure 5 reveal that laser power strongly influences the dynamic optical response of the layered CdSeS/ZnS quantum dot (QD) films. At low excitation intensity (6 kW/cm²), minimal changes are observed in both emission peak position and FWHM, and photoluminescence (PL) intensity remains relatively stable. As excitation power increases to 28 kW/cm² and 55 kW/cm², the system exhibits enhanced blue-shifting of the PL peak and significant spectral broadening—both of which are classic hallmarks of thermally activated processes.
The PL intensity trends (Figure 5a) reinforce this interpretation: at low laser power, a modest increase in PL output is observed, likely due to trap passivation or photoactivation of emissive states. In contrast, under high-power excitation, the PL decay dominates, suggesting thermal quenching, irreversible trap formation, or structural reorganization of the QD layer. These observations are consistent with models where ligand disorder, interdot coupling, and thermal gradients co-modulate charge recombination dynamics.[29]
In our LB-assembled QD heterostructures, where ligand shells are partially compromised and interlayer distances minimized, this thermal effect is compounded by enhanced interdot atomic communication. The power-dependent emission blue-shift (Figure 5b) is indicative of dynamic compositional exchange at the interface—most plausibly Se migration out of QD645 and S enrichment from QD450—driven by photoinduced strain and asymmetric chemical potentials. Such atom exchange has previously been reported in post-synthetic alloying or ripening of QDs under laser excitation[30], and is known to induce shifts in bandgap energies by modifying local stoichiometry.
In particular, the monotonic increase in FWHM under high-power excitation (Figure 5c) suggests thermal broadening of the electronic states within the QDs, a phenomenon attributed to elevated local temperatures in the film. This heating softens the energetic landscape of the nanocrystal ensemble, increasing electron–phonon interactions and enabling carriers to access a broader range of recombination pathways. This behavior is consistent with recent reports on quantum dot superlattices where thermally induced broadening and carrier delocalization were observed under moderate excitation power, particularly in epitaxially fused PbSe QD arrays[31].
Together, the power-dependent modulation of optical properties in layered QDs underscores a complex interplay of photoinduced strain relaxation, thermal disorder, and compositional diffusion—distinct from behaviors seen in monolayer or solution-phase QD systems. These insights not only enhance our understanding of QD films under optical stress but also highlight pathways for engineering dynamic optoelectronic responses in layered nanostructures.
5. Mechanistic Considerations and Future Outlook
The photophysical and photochemical behavior revealed by these LB-assembled QD bilayers is rich and complex. We propose that the LB film formation—by achieving close packing and compromised ligand passivation—enables a form of interlayer “communication” between QDs that is absent in well-passivated, isolated QDs. Through this interlayer coupling, one QD can influence its neighbor’s structure and emission via direct atomic exchange or strain transfer, a phenomenon reminiscent of coupled quantum dot superlattices where lattice distortions and electronic states propagate across particles.[32,33] In our system, the compositional asymmetry between the two QD species provides the driving force for an interface reaction: the QD pair essentially acts to reduce the free energy by evening out composition gradients (an entropic and enthalpic gain) at their juncture. Lattice contraction and enhanced interdot electronic coupling[34], as a result of ligand removal is in agreement with our observations of emission dynamics in the LB films.
We acknowledge that we do not yet have a comprehensive model that quantifies this oscillatory PL behavior. The interplay of ligand dynamics, atomic diffusion, trap state formation/annealing, and thermal feedback is inherently complex. The observations, however, open up several avenues for future investigation. For instance, time-resolved spectroscopic studies (e.g., pump–probe or single-particle PL tracking) could shed light on whether the two QD components in the bilayer undergo synchronous emissivity changes or if one population leads the other. In situ structural characterization, such as TEM or X-ray scattering on a laser-illuminated bilayer, could directly reveal any interfacial alloy formation or periodic lattice expansion/contraction corresponding to the optical oscillations. Recent advances in operando X-ray diffraction have successfully captured light-induced structural dynamics in QD solids[35], offering a potential roadmap for such experiments. Our findings suggest that photo-annealing (via laser heating) in a hetero-QD bilayer can accomplish a similar feat in a highly localized, cyclic manner. There are also intriguing connections to strain engineering in QDs: imposing asymmetric strain on core/shell QDs has been shown to modify their electronic structure and eliminate certain nonradiative pathways.[36,37] In a colloidal bilayer, the strain generated at a heterointerface might shift energy levels or activate new recombination routes, contributing to the oscillatory output.
6. Conclusions
In summary, the oscillatory PL behavior of the CdSeS/ZnS QD bilayers is a photoinduced phenomenon enabled by the unique structural milieu of the LB film. The LB assembly provides the tight interlayer contact and partial ligand removal needed for direct QD–QD interactions, while the compositional difference between the two QD types drives a dynamic change–equilibrium–change process at the interface. The result is a self-modulating optical response: the bilayer’s emission toggles through different states (different intensities and peak positions) as it absorbs energy. We attribute this to a cycle of atomistic and defect-state transformations—likely involving interdiffusion (atom exchange) to relieve strain and align compositions. The experimental evidence (oscillations only in mixed bilayers, sensitivity to laser power, and the role of LB-induced ligand manipulation) all support this interpretation. Nevertheless, the exact mechanistic pathway remains to be fully elucidated. Our work motivates further mechanistic studies, for example, to determine the role of lattice strain vs. chemical reaction, to quantify the kinetics of the interchange, and to explore if the oscillation can be controlled (or quenched) by environmental parameters like temperature or atmosphere. Unraveling this mechanism will not only explain the present observations but also deepen our understanding of photochemical reactions in nanocrystal solids—an emerging frontier where nanoparticles communicate and transform under illumination. Ultimately, the ability to induce and control such interparticle interactions could lead to new approaches in optoelectronic devices and smart materials, where an external stimulus (light, heat) actively tunes material properties in real time. Our findings, though currently without a complete model, lay the groundwork for these exciting future directions.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
References
- Alivisatos, A. P.; Schlamp, M. C.; Colvin, V. L., Light-emitting diodes made from cadmium selenide nanocrystals and a semiconducting polymer. Nature 1994, 370, 354-357. [CrossRef]
- Lee, D. C.; Lee, S. Y.; Kim, L., Color conversion materials for QD-based displays: Current status and perspectives. Advanced Optical Materials 2022, 10 (1), 2101180.
- Klimov, V. I.; Pietryga, J. M.; Lee, C.; Robel, I.; McDaniel, H.; Padilha, L. A.; Lee, D.; Lim, J.; Park, Y. S.; Bae, W. K., Controlling the influence of Auger recombination on the performance of quantum-dot light-emitting diodes. Nature Communications 2013, 4, 2661. [CrossRef]
- Klimov, V. I.; Schaller, R. D., High Efficiency Carrier Multiplication in PbSe Nanocrystals: Implications for Solar Energy Conversion. Physical Review Letters 2004, 92 (18), 186601. [CrossRef]
- Sargent, E. H.; Talapin, D. V.; Garcia de Arquer, F. P., Colloidal quantum dot photovoltaics. Nature Reviews Materials 2021, 6, 413-430.
- Wang, X.; Ding, L.; Huang, Y.; Wang, C.; Zhang, Y.; Li, H., Sensitive fluorescence probe based on CdTe QDs for detecting copper ion. Sensors and Actuators B: Chemical 2017, 241, 61-67.
- Alivisatos, A. P.; Weiss, S.; Gin, P.; Moronne, M.; Bruchez, M., Semiconductor Nanocrystals as Fluorescent Biological Labels. Science 1998, 281, 2013-2016.
- Nie, S.; Chan, W. C. W., Quantum Dot Bioconjugates for Ultrasensitive Nonisotopic Detection. Science 1998, 281, 2016-2018. [CrossRef]
- Wang, H.; Wang, L.; Tang, Z.; Yu, J.; Tang, M.; Wang, Y., Quantum dot-based lateral flow test strip for detection of miRNA-21 in gastric cancer. Biosensors and Bioelectronics 2016, 85, 641-646.
- Verma, A. K.; Noumani, A.; Yadav, A. K.; Solanki, P. R. FRET Based Biosensor: Principle Applications Recent Advances and Challenges Diagnostics [Online], 2023.
- Zhang, W.; Li, B.; Chang, C.; Chen, F.; Zhang, Q.; Lin, Q.; Wang, L.; Yan, J.; Wang, F.; Chong, Y.; Du, Z.; Fan, F.; Shen, H., Stable and efficient pure blue quantum-dot LEDs enabled by inserting an anti-oxidation layer. Nature Communications 2024, 15 (1), 783.
- Quesada-González, D.; Merkoçi, A., Quantum dots for biosensing: Classification and applications. Biosensors and Bioelectronics 2025, 273, 117180. [CrossRef]
- Sharma, S. N.; Semalti, P.; Bhawna; Rao, A. S., Pioneering advancements in quantum dot solar cells: Innovations in synthesis and cutting-edge applications. Current Opinion in Colloid & Interface Science 2025, 77, 101905. [CrossRef]
- Sun, Y.; Zhou, S.; Li, X.; Gao, Y.; Zhao, X.; Chen, B., Bandgap engineering and enhanced stability of composition-tunable CdSeS alloyed nanocrystals. Nanoscale 2016, 8, 13288-13296.
- Chung, D. S.; Lee, J.; Choi, S. H.; Lee, H.; Kim, J., Bandgap modulation of quantum dot films by strain and substrate interaction. ACS Nano 2012, 6, 11273-11280.
- Bawendi, M. G.; Supran, G. J.; Shirasaki, Y., Emergence of colloidal quantum-dot light-emitting technologies. Nature Photonics 2013, 7, 13-23.
- Kafle, B.; Tesema, T. E.; Kazemi, A.; Habteyes, T. G., Stripping and Transforming Alloyed Semiconductor Quantum Dots via Atomic Interdiffusion. The Journal of Physical Chemistry C 2016, 120 (23), 12850-12859. [CrossRef]
- Park, E. Y.; Adegoke, O., Virus detection using nanoparticles and surface enhanced Raman spectroscopy. Biosensors and Bioelectronics 2016, 77, 799-806.
- Wang, Y.; Wu, L.; Liu, L.; Zhang, D.; Zhang, Z.; Sun, Q., Modulation of photoluminescence in quantum dots by temperature and surrounding matrix. Journal of Materials Chemistry C 2022, 10, 14301-14310.
- Hassinen, A.; Moreels, I.; De Nolf, K.; Smet, P. F.; Martins, J. C.; Hens, Z., Short-Chain Alcohols Strip X-Type Ligands and Quench the Luminescence of PbSe and CdSe Quantum Dots, Acetonitrile Does Not. Journal of the American Chemical Society 2012, 134 (51), 20705-20712. [CrossRef]
- Zhang, A.; Dong, C.; Li, L.; Yin, J.; Liu, H.; Huang, X.; Ren, J., Non-blinking (Zn)CuInS/ZnS Quantum Dots Prepared by In Situ Interfacial Alloying Approach. Scientific Reports 2015, 5 (1), 15227. [CrossRef]
- Yalcin, A. O.; Goris, B.; van Dijk-Moes, R. J. A.; Fan, Z.; Erdamar, A. K.; Tichelaar, F. D.; Vlugt, T. J. H.; Van Tendeloo, G.; Bals, S.; Vanmaekelbergh, D.; Zandbergen, H. W.; van Huis, M. A., Heat-induced transformation of CdSe–CdS–ZnS core–multishell quantum dots by Zn diffusion into inner layers. Chemical Communications 2015, 51 (16), 3320-3323.
- Asami, H.; Abe, Y.; Ohtsu, T.; Kamiya, I.; Hara, M., Surface State Analysis of Photobrightening in CdSe Nanocrystal Thin Films. The Journal of Physical Chemistry B 2003, 107, 12566.
- Emara, M.; Van Patten, G., Effect of oxygen and polymer matrix on photo-induced changes in CdSe quantum dots. Materials Chemistry and Physics 2020, 256, 123652.
- Abelson, A.; Qian, C.; Salk, T.; Luan, Z.; Fu, K.; Zheng, J.-G.; Wardini, J. L.; Law, M., Collective topo-epitaxy in the self-assembly of a 3D quantum dot superlattice. Nature Materials 2020, 19 (1), 49-55. [CrossRef]
- Chu, X.; Heidari, H.; Abelson, A.; Unruh, D.; Hansen, C.; Qian, C.; Zimanyi, G.; Law, M.; Moulé, A. J., Structural characterization of a polycrystalline epitaxially-fused colloidal quantum dot superlattice by electron tomography. Journal of Materials Chemistry A 2020, 8 (35), 18254-18265. [CrossRef]
- Kavrik, M. S.; Hachtel, J. A.; Ko, W.; Qian, C.; Abelson, A.; Unlu, E. B.; Kashyap, H.; Li, A.-P.; Idrobo, J. C.; Law, M., Emergence of distinct electronic states in epitaxially-fused PbSe quantum dot superlattices. Nature Communications 2022, 13 (1), 6802.
- Boles, M. A.; Engel, M.; Talapin, D. V., Self-Assembly of Colloidal Nanocrystals: From Intricate Structures to Functional Materials. Chemical Reviews 2016, 116 (18), 11220-11289. [CrossRef]
- Talapin, D. V.; Lee, J.-S.; Kovalenko, M. V.; Shevchenko, E. V., Prospects of Colloidal Nanocrystals for Electronic and Optoelectronic Applications. Chemical Reviews 2010, 110 (1), 389-458. [CrossRef]
- Choudhury, T. H.; Zhang, X.; Al Balushi, Z. Y.; Chubarov, M.; Redwing, J. M., Epitaxial Growth of Two-Dimensional Layered Transition Metal Dichalcogenides. Annual Review of Materials Research 2020, 50 (Volume 50, 2020), 155-177.
- Koh, W.-k.; Koposov, A. Y.; Stewart, J. T.; Pal, B. N.; Robel, I.; Pietryga, J. M.; Klimov, V. I., Heavily doped n-type PbSe and PbS nanocrystals using ground-state charge transfer from cobaltocene. Scientific Reports 2013, 3 (1), 2004. [CrossRef]
- Whitham, K.; Yang, J.; Savitzky, B. H.; Kourkoutis, L. F.; Wise, F.; Hanrath, T., Charge transport and localization in atomically coherent quantum dot solids. Nat Mater 2016, 15 (5), 557-63.
- Puthirath Balan, A.; Radhakrishnan, S.; Woellner, C. F.; Sinha, S. K.; Deng, L.; Reyes, C. d. l.; Rao, B. M.; Paulose, M.; Neupane, R.; Apte, A.; Kochat, V.; Vajtai, R.; Harutyunyan, A. R.; Chu, C.-W.; Costin, G.; Galvao, D. S.; Martí, A. A.; van Aken, P. A.; Varghese, O. K.; Tiwary, C. S.; Malie Madom Ramaswamy Iyer, A.; Ajayan, P. M., Exfoliation of a non-van der Waals material from iron ore hematite. Nature Nanotechnology 2018, 13 (7), 602-609. [CrossRef]
- Mameli, A.; Karasulu, B.; Verheijen, M. A.; Barcones, B.; Macco, B.; Mackus, A. J. M.; Kessels, W. M. M. E.; Roozeboom, F., Area-Selective Atomic Layer Deposition of ZnO by Area Activation Using Electron Beam-Induced Deposition. Chemistry of Materials 2019, 31 (4), 1250-1257. [CrossRef]
- Marino, E.; Rosen, D. J.; Yang, S.; Tsai, E. H. R.; Murray, C. B., Temperature-Controlled Reversible Formation and Phase Transformation of 3D Nanocrystal Superlattices Through In Situ Small-Angle X-ray Scattering. Nano Letters 2023, 23 (10), 4250-4257.
- Pile, D. F. P., One-way chiral light. Nature Photonics 2019, 13 (3), 139-139.
- Moloney, E. G.; Yeddu, V.; Saidaminov, M. I., Strain Engineering in Halide Perovskites. ACS Materials Letters 2020, 2 (11), 1495-1508. [CrossRef]
Figure 1.
Schematic illustration of the experimental configuration used for layer-by-layer assembly and optical probing of quantum dot (QD) bilayers. Panel (a) is an atomic force microscopy (AFM) image of a close-packed Langmuir–Blodgett monolayers of CdSeS/ZnS QD450 (red layer) and CdSeS/ZnS QD645 (orange layer) deposited on oxide-coated silicon substrate (black layer). Panel (b) is an AFM topographic profiling of the layer measured along a white dashed line in panel (a). The Photoluminescence (PL) spectrum of QD450 (blue peak) and QD645(red peak) measured by exciting the sample using 405 nm laser.
Figure 1.
Schematic illustration of the experimental configuration used for layer-by-layer assembly and optical probing of quantum dot (QD) bilayers. Panel (a) is an atomic force microscopy (AFM) image of a close-packed Langmuir–Blodgett monolayers of CdSeS/ZnS QD450 (red layer) and CdSeS/ZnS QD645 (orange layer) deposited on oxide-coated silicon substrate (black layer). Panel (b) is an AFM topographic profiling of the layer measured along a white dashed line in panel (a). The Photoluminescence (PL) spectrum of QD450 (blue peak) and QD645(red peak) measured by exciting the sample using 405 nm laser.
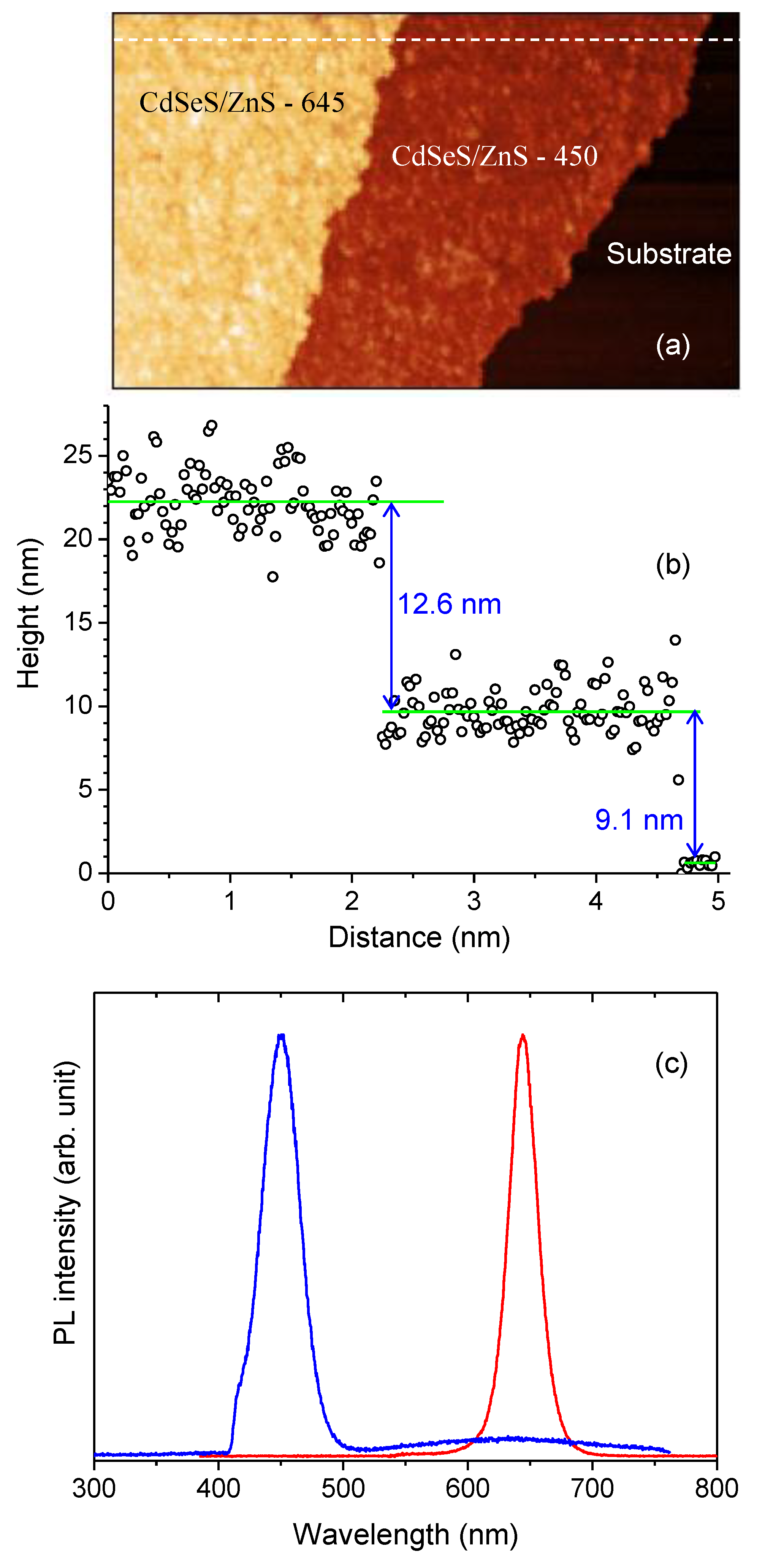
Figure 2.
Time-resolved photoluminescence (PL) characteristics of a QD645/QD450 bilayer excited at 532 nm. (a) The peak wavelength shift is shown as blue dots, while full width at half maximum (FWHM) is plotted as green dots. (b) Normalized PL intensity is plotted using red dots. Time is in minutes; spectral observables are tracked continuously during 120 minutes of excitation.
Figure 2.
Time-resolved photoluminescence (PL) characteristics of a QD645/QD450 bilayer excited at 532 nm. (a) The peak wavelength shift is shown as blue dots, while full width at half maximum (FWHM) is plotted as green dots. (b) Normalized PL intensity is plotted using red dots. Time is in minutes; spectral observables are tracked continuously during 120 minutes of excitation.
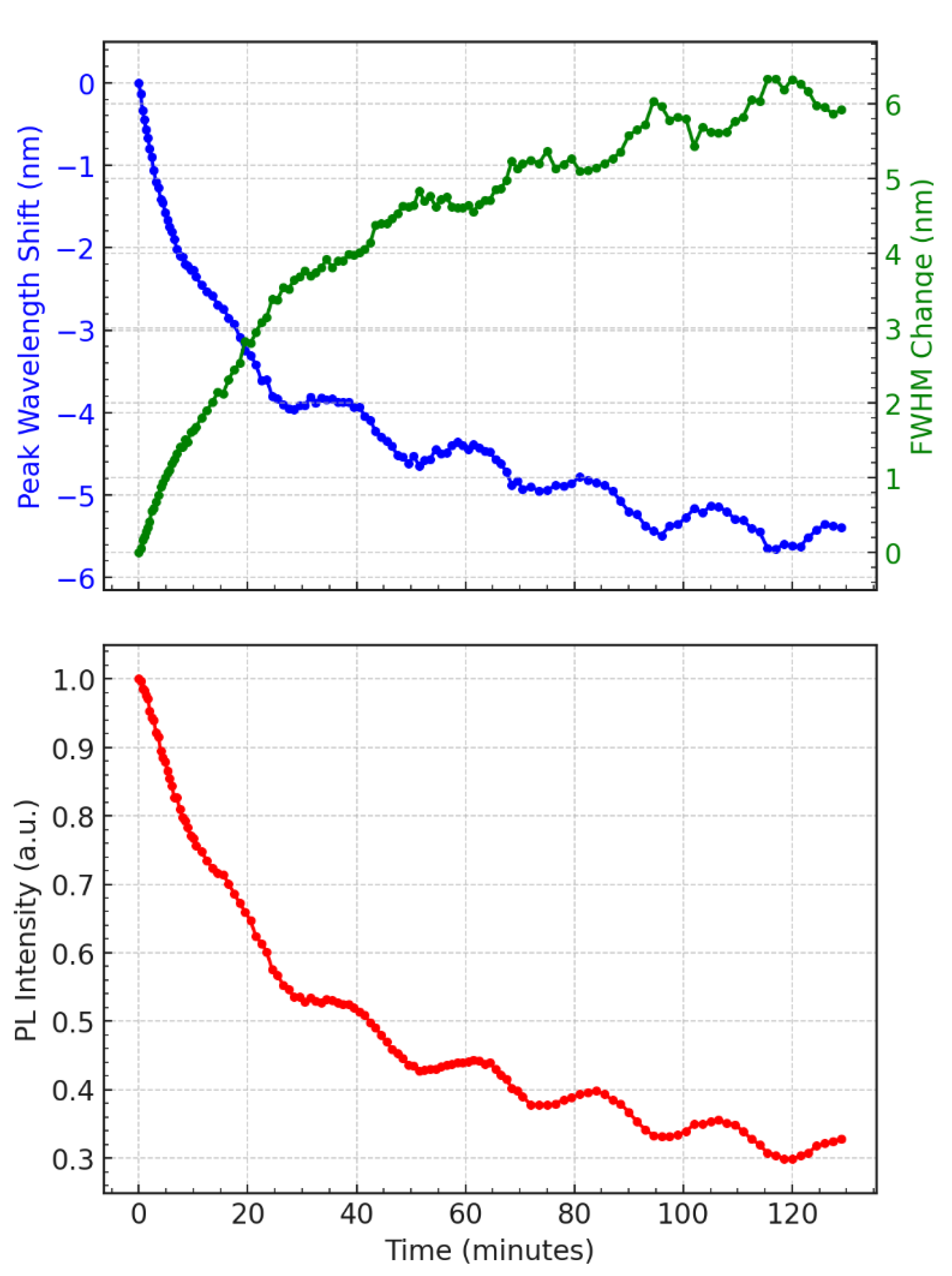
Figure 3.
Exponential model fitting of PL data presented in Figure 2. (a) Red circles represent experimental PL intensity; the solid black line is a biexponential fit. (b) Blue dots show the experimental peak shift, with a solid black biexponential fit. (c) Green dots indicate FWHM broadening over time, with a single-exponential model overlaid in a dashed black line. All measurements correspond to 532 nm excitation.
Figure 3.
Exponential model fitting of PL data presented in Figure 2. (a) Red circles represent experimental PL intensity; the solid black line is a biexponential fit. (b) Blue dots show the experimental peak shift, with a solid black biexponential fit. (c) Green dots indicate FWHM broadening over time, with a single-exponential model overlaid in a dashed black line. All measurements correspond to 532 nm excitation.
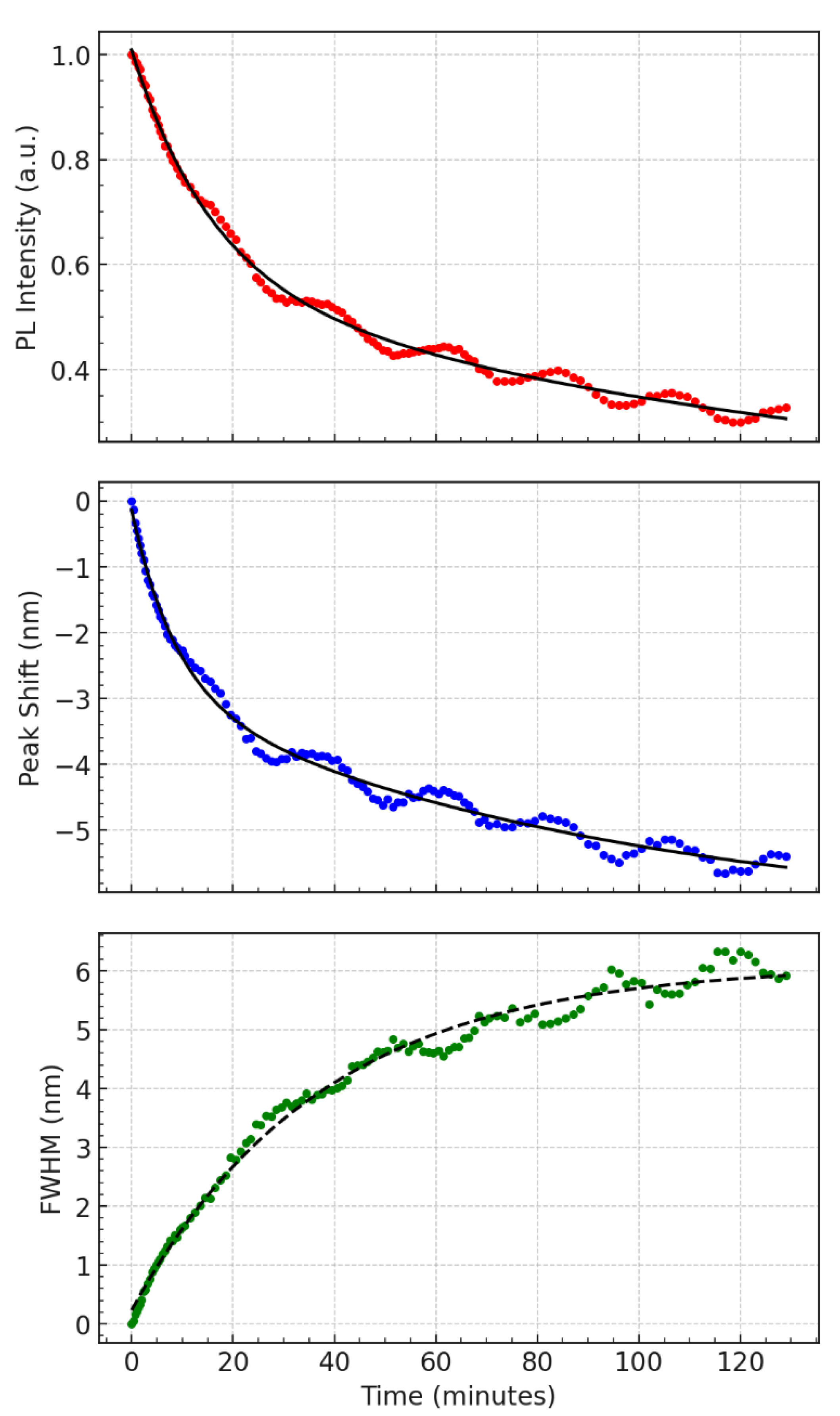
Figure 4.
Control experiments for QD bilayers under 532 nm excitation. (a) Blue dots: QD645/QD450; black squares: QD645/glass; green triangles: QD645/QD645; orange diamonds: QD630/QD450. Panel (a) shows peak wavelength shifts over time; (b) displays the corresponding change in FWHM.
Figure 4.
Control experiments for QD bilayers under 532 nm excitation. (a) Blue dots: QD645/QD450; black squares: QD645/glass; green triangles: QD645/QD645; orange diamonds: QD630/QD450. Panel (a) shows peak wavelength shifts over time; (b) displays the corresponding change in FWHM.
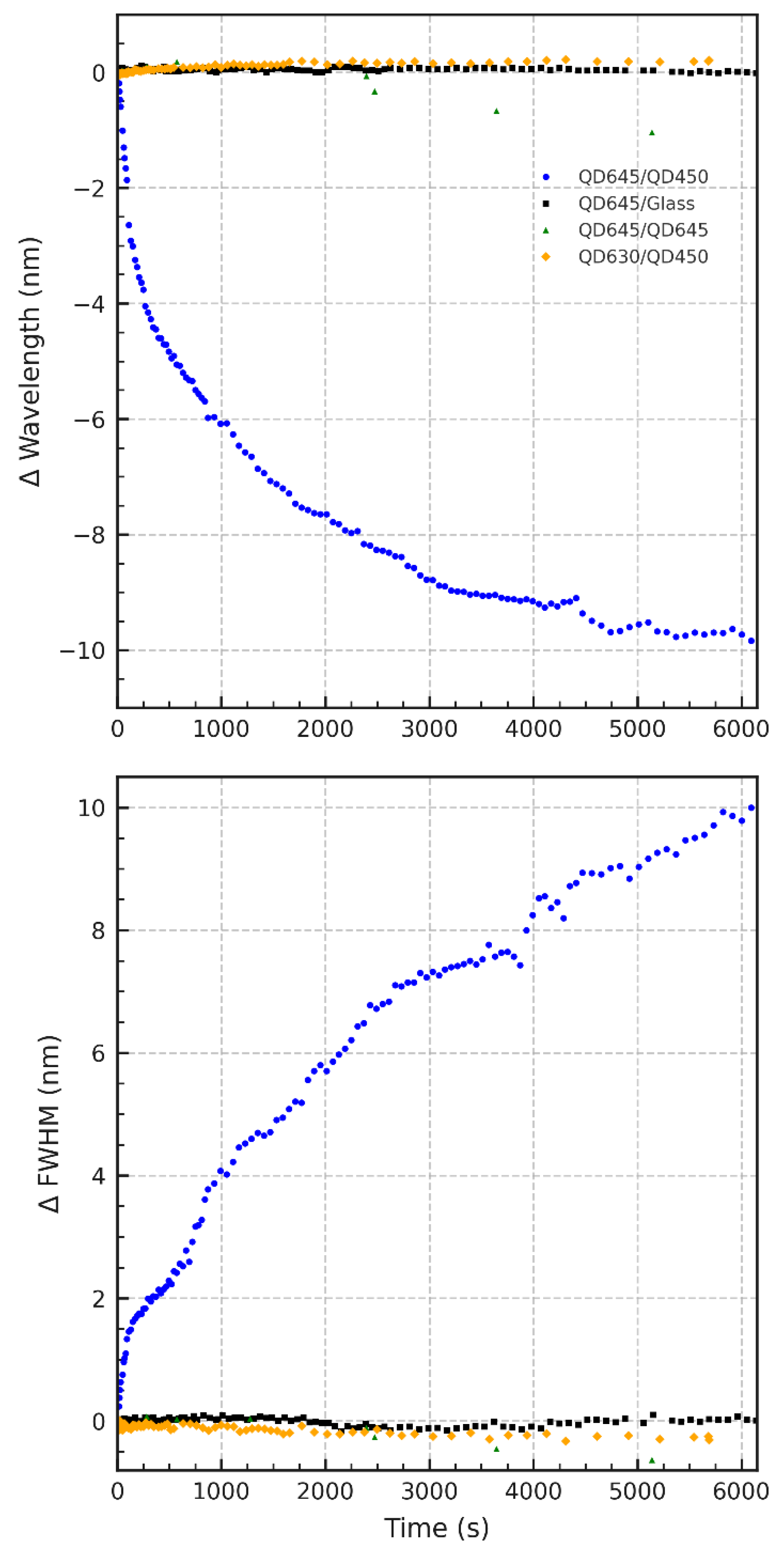
Figure 5.
Excitation power-dependent evolution of PL characteristics from QD645/QD450 bilayers excited at 532 nm. (a) Red triangles, blue circles, and orange squares represent PL intensity for 55, 28, and 6 kW/cm², respectively. (b) Peak emission energy shift (in eV) plotted with the same color/symbol coding. (c) Change in FWHM for the same power levels, using identical symbols.
Figure 5.
Excitation power-dependent evolution of PL characteristics from QD645/QD450 bilayers excited at 532 nm. (a) Red triangles, blue circles, and orange squares represent PL intensity for 55, 28, and 6 kW/cm², respectively. (b) Peak emission energy shift (in eV) plotted with the same color/symbol coding. (c) Change in FWHM for the same power levels, using identical symbols.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



