Submitted:
30 May 2025
Posted:
04 June 2025
You are already at the latest version
Abstract
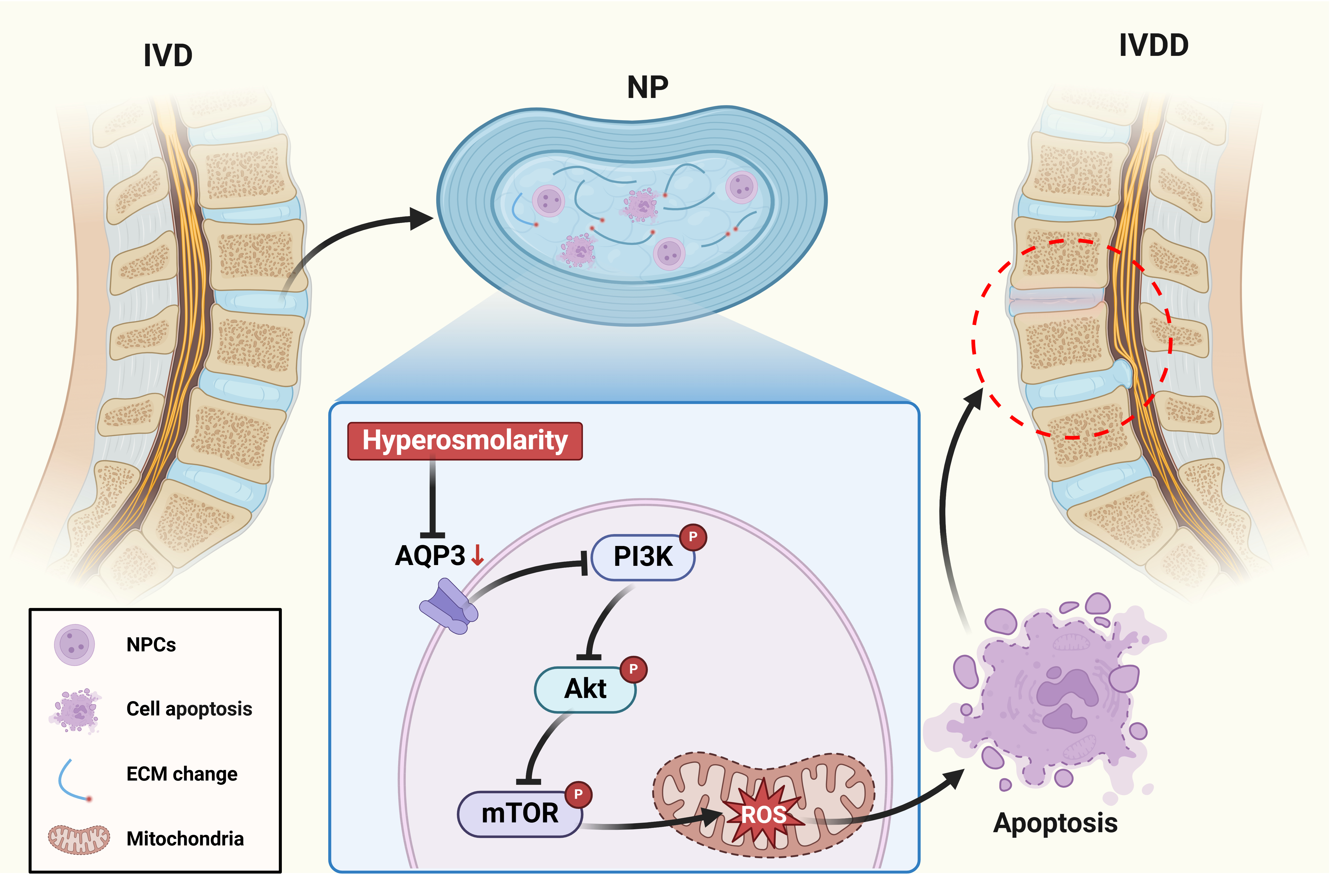
Hyperosmolarity has emerged as a pathogenic driver of nucleus pulposus cells (NPCs) apoptosis during intervertebral disc degeneration (IVDD) pathogenesis. Aquaporin 3 (AQP3), an integral membrane channel within the aquaporin superfamily, orchestrates cellular osmotic balance through selective transport of water and osmolytes. Although AQP3 depletion correlates with disc degeneration, the mechanistic interplay between AQP3-mediated osmoregulation and apoptotic signaling in NPCs under hyperosmotic microenvironment remains undefined. Through comprehensive investigation combining molecular interrogation and functional validation, we delineated the cytoprotective mechanism of AQP3 in counteracting hyperosmolarity-induced disc degeneration. Experimental evidence revealed that hyperosmolarity triggers NPCs apoptosis via concurrent AQP3 depletion and PI3K/AKT/mTOR pathway suppression, culminating in mitochondrial impairment and reactive oxygen species (ROS) accumulation. Intriguingly, lentiviral-mediated AQP3 restoration attenuated oxidative damage and apoptotic execution through PI3K/AKT/mTOR pathway reactivation, effectively preserving disc integrity in IVDD rat models. Conversely, pharmacological AQP3 blockade exacerbated extracellular matrix (ECM) catabolism and accelerated nucleus pulposus (NP) tissue destruction. Mechanistic dissection established that hyperosmolarity-induced AQP3 deficiency initiates the PI3K/AKT/mTOR signaling cascade, creating a pathological feedback loop that amplifies NPCs dysfunction. This work not only elucidates the osmoprotective role of AQP3 in maintaining NP homeostasis but also identifies its dual regulatory function in mitochondrial quality control. Our findings provide groundbreaking insights into IVDD pathomechanisms while proposing AQP3-mediated osmoregulation as a promising therapeutic paradigm for disc degenerative disorders.
Keywords:
IVDD
; NPCs
; hyperosmolarity
; AQP3
; PI3K/Akt/mTOR pathway
; ROS
1. Introduction
Lower back pain (LBP), ranked as the leading global cause of disability, originates predominantly from intervertebral disc degeneration (IVDD) - a progressive musculoskeletal disorder affecting 40% of adults aged over 40. Beyond its debilitating impact on quality-adjusted life years, IVDD imposes substantial socioeconomic burdens [1]. The multifactorial pathogenesis of IVDD involves complex interplay including cellular senescence, biomechanical stressors, inflammatory cascades and genetic predisposition [2,3,4,5]. Despite these mechanistic insights, the precise spatiotemporal regulation of disc degeneration remains elusive.Current therapeutic paradigms remain palliative, offering transient symptom relief through physical rehabilitation protocols, pharmacologic interventions and invasive surgical procedures [6,7,8,9].This critical gap in disease-modifying therapies underscores the urgent need for targeted molecular interventions addressing the underlying pathobiology of IVDD progression.
The intervertebral disc (IVD) represents a sophisticated tri-lamellar structure comprising the proteoglycan-rich nucleus pulposus (NP), the collagenous annulus fibrosus (AF) and the cartilaginous endplates.This unique architectural organization enables the IVD to fulfill its biomechanical functions as a viscoelastic shock absorber, distributing compressive forces while preserving spinal flexibility and range of motion [10]. The pathognomonic features of IVDD encompass progressive cellular depletion and extracellular matrix (ECM) degradation, particularly within the NP compartment [11]. NPCs persist within one of the most physiologically challenging microenvironments in mammalian systems, characterized by hyperosmolarity, chronic hypoxia, acidic pH and excessive mechanical stress [12]. Within the intervertebral disc microenvironment, the remarkably elevated osmotic pressure primarily arises from proteoglycans accumulated in the ECM. Notably, the physiological osmotic pressure in NP tissue maintains dynamic fluctuations between 450-550 mOsm/kg, which is much higher than that of conventional extracellular fluids in mammalian systems. Mounting evidence indicates that this pathological hyperosmolarity exerts profound regulatory effects on NPCs homeostasis, including metabolic activity, apoptosis resistance, and phenotypic maintenance. However, the precise molecular mechanisms underlying these osmotic adaptations remain elusive, particularly in the context of IVDD [13,14,15]. Elucidating hyperosmolarity governing NPCs responses may therefore provide critical insights into IVDD pathophysiology and identify novel therapeutic targets.
The IVD, when subjected to dynamic fluctuations in local osmotic pressure, engages a complex interplay of proteins and molecular mechanisms to facilitate osmotic adaptation in resident cells. Among these regulatory components, the aquaporins (AQPs) family has been identified as a pivotal contributor to cellular osmoregulation within this microenvironment [16]. As transmembrane channel proteins specialized in facilitating water molecule transport across lipid bilayers, AQPs serve as critical regulators of cellular osmotic homeostasis [17,18,19]. Emerging evidence from IVDD pathogenesis research reveals that AQP expression in embryonic notochordal cells exhibits microenvironmental plasticity, with transcriptional dynamics modulated by local osmotic gradients to orchestrate lineage commitment and apoptotic regulation in murine intervertebral disc models [20]. Of particular pathophysiological relevance, aquaporin 3 (AQP3), a multifunctional aquaglyceroporin, demonstrates tissue-specific expression patterns in human IVD compartments [20,21]. Intriguingly, proteomic profiling of degenerative disc specimens reveals a marked downregulation of AQP3 concomitant with disrupted transmembrane glycerol/water homeostasis. This phenotypic shift suggests a mechano-osmotic coupling mechanism whereby AQP3-mediated transmembrane flux may transduce biophysical signals (mechanobiology, osmolarity and matrix hydration) into cytoarchitectural remodeling during disc degeneration cascades [20,22]. To address this knowledge gap, the present study aims to elucidate the pathophysiological role of AQP3 in governing NPCs fate determination under osmotically challenged conditions, with particular emphasis on molecular mechanisms underlying osmoadaptive reprogramming.
2. Materials and Methods
2.1. Extraction of Rat NPCs
Thirty Sprague-Dawley rats (female, 200-220 g, 12-week-old) were purchased from Guangdong Sijia Jingda Biotechnology Co., Ltd. (SCXK (Xiang) 2021-0002) and acclimatized for 7 days in specific-pathogen-free (SPF) conditions with ad libitum access to autoclaved feed and water. All procedures were performed at Guangzhou Forevergen Biosciences Medical Laboratory Animal Center following protocol approval by the Institutional Animal Care and Use Committee (SYXK (Yue) 2023-0186).
Euthanasia was performed using gradual CO2 displacement. Lumbar intervertebral discs (Co6/7-Co8/9) were aseptically excised within 5 min post-mortem under laminar flow. NP tissue was microdissected from the innermost annular region under a dissecting microscope. Sequential enzymatic digestion was performed in 0.25% trypsin-EDTA (T4049, Sigma-Aldrich, USA) at 37°C for 5 min with 150 rpm orbital shaking and 0.25% collagenase type II (C2-BIOC, Sigma-Aldrich, USA) for 10 min under identical conditions. Digestion was terminated with 10% fetal bovine serum (FBS) (A3160802, Gibco, USA)-containing DMEM/F12 (11320033, Gibco, USA). Cell suspensions were filtered through 70 μm nylon mesh (352350, Corning, USA), centrifuged at 300×g for 5 min, and resuspended in complete medium: DMEM/F12 supplemented with 10% FBS, 1% penicillin-streptomycin (15140122, Gibco, USA), and 25 μg/mL ascorbic acid (A4544, Sigma-Aldrich, USA). Primary NPCs were maintained at 37°C in 5% CO2 with medium changes every 48 h until 80% confluency.
2.2. Culture of Rat NPCs Under Different Osmotic Pressure Environments
NPCs were resuspended in DMEM/F12 medium containing 10% FBS and 1% penicillin-streptomycin, then maintained in a humidified incubator with standard atmospheric conditions (37°C, 21% O2, and 5% CO2). To establish different osmotic pressure conditions, sodium chloride (S805280, Macklin, China) was dissolved in the culture medium to achieve osmolality levels of 330 mOsm/kg (physiological level) and 550 mOsm/kg (hyperosmolarity level). The final osmolality was quantitatively confirmed through triplicate measurements using a freezing-point osmometer (FM-8P, Shanghai Medical College Instrument Co., Ltd., China).
2.3. Detection of Apoptosis in NPCs by Flow Cytometry
NPCs were subjected to osmotic stress induction through incubation in the osmolarity of 330 mOsm/kg and 550 mOsm/kg. Following treatment, cells were enzymatically dissociated using 0.25% trypsin, washed twice with PBS, and collected by centrifugation at 300×g for 5 min at 4°C. Apoptosis analysis was performed using the Annexin V-FITC/PE Apoptosis Detection Kit (KGA1102, KeyGEN BioTECH, China) according to the manufacturer's protocol. Briefly, 1×105 cells/mL were resuspended in 100 μL binding buffer and dual-stained with 5 μL Annexin V-FITC and 10 μL PE for 15 min at 25°C under light-protected conditions. Quantitative flow cytometric analysis was conducted using a FACSCalibur system (BD Biosciences, CA). Fluorescence signals were detected through 530/30 nm (FITC) and 585/42 nm (PE) bandpass filters, with data acquisition performed using CellQuest Pro software.
2.4. Detection of Caspase-3 Activity by Flow Cytometry
NPCs were plated at a density of 1×105 cells per well and maintained in a humidified incubator (37°C, 5% CO2). Upon reaching 80-90% confluence, cell monolayers were washed twice with buffer. Caspase-3 proteolytic activity was measured using the Caspase-3 Activity Assay Kit (C1168S, Beyotime Biotechnology, China), strictly adhering to the manufacturer's protocol.
2.5. Detection of Apoptosis in NPCs by Immunofluorescence Double Staining
Cultured NPCs were enzymatically dissociated using 0.25% EDTA-free trypsin for 3 min at 37°C. After centrifugation at 300×g for 5 min, cellular pellets were resuspended in cold Annexin V binding buffer. Aliquots containing 1×105 cells were dual-stained with 5 μL Annexin V-FITC and 2 μL PI working solutions. The stained cell suspensions were incubated in light-protected conditions for 10 min at 25°C. Fluorescence imaging was performed using a Leica DMi8 inverted microscope (Leica Microsystems, Germany). Digital images were captured using LAS X software with identical exposure settings. Apoptotic cells were identified by characteristic membrane staining patterns: green fluorescence (Annexin V-FITC) indicating phosphatidylserine externalization, and red nuclear staining (PI) denoting membrane integrity loss.
2.6. Western Blot Assay
Protein expression levels were quantified using Western blot analysis with β-actin serving as the endogenous loading control. Cellular proteins were extracted from NPCs using ice-cold radioimmunoprecipitation assay (RIPA) lysis buffer (P0013B, Beyotime Biotechnology, China). Protein concentrations were subsequently measured using a bicinchoninic acid (BCA) assay kit (23225, Thermo Fisher Scientific, USA) according to the manufacturer's protocol. Equal amounts of protein lysates were resolved through 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; P0012A, Beyotime Biotechnology, China) and subsequently transferred onto polyvinylidene difluoride (PVDF) membranes (IPVH00010, Merck Millipore, USA) using a semi-dry transfer system. Primary antibodies include anti-Bax (1:1000; ab32503, Abcam, UK); anti-Bcl-2 (1:1000; ab59348, Abcam, UK); anti-type II collagen (1:5000; 600-403-104, Thermo Fisher Scientific, USA); anti-MMP3 (1:1000; ab52915, Abcam, UK); anti-AQP3 (1:1000; ab307969, Abcam, UK); anti-mTOR (1:1000; 2983, Cell Signaling Technology, USA); anti-p-mTOR (1:1000; 5536, Cell Signaling Technology, USA); anti-PI3K (1:1000; 4249, Cell Signaling Technology, USA); anti-p-PI3K (1:1000; 13857, Cell Signaling Technology, USA); anti-Akt (1:2000; 2920, Cell Signaling Technology, USA); anti-p-Akt (1:2000; 4060, Cell Signaling Technology, USA); anti-β-actin (1:5000; 93473, Cell Signaling Technology, USA). Following primary antibody incubation, membranes were washed three times with Tris-buffered saline containing 0.1% Tween-20 (TBST; ST671, Beyotime Biotechnology, China) under gentle agitation. Subsequently, membranes were probed with species-matched horseradish peroxidase (HRP)-conjugated secondary antibodies (Goat anti-mouse IgG (1:2000; 7076, Cell Signaling Technology, USA) and Goat anti-rabbit IgG (1:2000; 7074, Cell Signaling Technology, USA)) at room temperature for 2 h with continuous shaking. After three additional TBST washes, immunoreactive bands were visualized using an enhanced chemiluminescence (ECL) kit (A38556, Thermo Fisher Scientific, USA). Protein band intensities were quantified through densitometric analysis with ImageJ software (National Institutes of Health, USA), normalized to β-actin expression levels.
2.7. CCK-8 Assay
The proliferation of NPCs was assessed using the Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Japan). Briefly, NPCs were seeded at a density of 5×103 cells per well in 96-well flat-bottom plates and allowed to adhere overnight in a humidified incubator (37°C, 5% CO2). Following experimental treatments, 20 μL of CCK-8 reagent was added to each well, and plates were incubated for 2 h under standard culture conditions. Absorbance was measured at 450 nm using a SpectraMax M5 microplate reader with a reference wavelength of 650 nm to correct for background interference. Cell viability was calculated using the following formula: Cell viability (%) = experimental OD value/control OD value×100%.
2.8. Detection of ROS Levels in NPCs by Flow Cytometry
Intracellular reactive oxygen species (ROS) levels were quantified using the 2',7'-dichlorodihydrofluorescein diacetate (DCFDA) Cellular ROS Detection Assay Kit (ab113851, Abcam, UK), with protocol optimization. Briefly, NPCs were incubated with 10 μM DCFDA in serum-free RPMI-1640 medium and incubated at 37°C under 5% CO2 workstation for precise environmental control. Following 30 min dark incubation, cells were washed twice with thermostated PBS and resuspended in phenol red-free DMEM/F12 to minimize autofluorescence. Data were acquired using BD FACSDiva software with threshold set at FSC-H 200. The 2',7'-dichlorofluorescein (DCF) mean fluorescence intensity (MFI) was quantified after spectral overlap compensation using single-stained controls. ROS levels were expressed as fold change relative to vehicle-treated controls.
2.9. Detection of ROS Levels in NPCs by Immunofluorescence
Mitochondrial ROS were quantified using a dual-fluorescence colocalization assay. Cells were costained with MitoSOX Red mitochondrial superoxide indicator (M36008, Thermo Fisher Scientific, MA) and MitoTracker Green FM (C1048, Beyotime Biotechnology, China) according to the manufacturers' protocols with optimization. Briefly, cells were incubated with the dye mixture in Hank's Balanced Salt Solution at 37°C under 5% CO2 for 30 min, followed by three washes with pre-warmed HBSS. Live-cell imaging was performed using a Leica DMi8 inverted fluorescence microscope. Image acquisition was conducted using LAS X software with identical exposure settings. Colocalization analysis was performed using ImageJ .
2.10. Determination of GPx and MDA Levels
Malondialdehyde (MDA) concentrations and glutathione peroxidase (GPx) activity were quantified using standardized colorimetric and enzymatic assays, respectively. The MDA levels were determined via the thiobarbituric acid reactive substances (TBARS) method using a commercial kit (RK09070, ABclonal, China), while GPx activity was measured through NADPH oxidation kinetics with a kit (EEA010, Thermo Fisher Scientific, MA). Briefly, cell lysates were centrifuged at 12,000×g for 15 min at 4°C to obtain supernatant fractions. Absorbance readings were acquired using a Synergy H1 microplate reader. The lower detection limits were validated as 0.1 μM for MDA and 5 U/mL for GPx using serial dilutions of reference standards.
2.11. MMP Assessment
Mitochondrial depolarization was evaluated using the JC-1 assay kit (C2006, Beyotime Biotechnology, China) following established protocols. Treated NPCs were incubated with JC-1 working solution at 37°C under dark conditions for 30 min. Following triple washing with PBS, cells were resuspended in fresh complete medium and immediately analyzed using an inverted fluorescence microscope (DMi8, Leica Microsystems, Germany). Mitochondrial integrity was determined by quantifying the red/green fluorescence intensity ratio.
Quantitative MMP measurement was performed using JC-1 staining coupled with flow cytometry. Apoptosis-induced NPCs were harvested and washed with ice-cold PBS, then resuspended in 1× assay buffer containing JC-1 . After 20 min incubation at 37°C in the dark, cells were centrifuged and washed twice with warm PBS. Cellular fluorescence was immediately analyzed using a FACSCalibur flow cytometer (BD Biosciences, USA).
2.12. RNA Sequencing
RNA extraction from NPCs was performed using TRIzol (15596026, Thermo Fisher Scientific, USA), followed by determination of RNA concentration and purity by Nanodrop2000 microspectrophotometer. The integrity of RNA samples was examined by the Labchip GX touch microfluidic capillary system. The raw RNA sequencing data were subjected to quality control (QC) to assess the suitability of the sequencing data for subsequent analysis. RNA sequencing was performed on the Illumina HiSeq platform using next-generation sequencing (NGS) technology. To evaluate the functionality of candidate targets in IVDD, GO and KEGG pathway enrichment analysis of co-expressed genes was performed using the clusterProfiler software package in R software. GO and KEGG pathways with P < 0.05 were considered to be significantly enriched.
2.13. Transfection of Expression Vectors into NPCs AAV Vector Construction and Transfection
The pAAV-AQP3 expression plasmid was engineered by subcloning full-length human AQP3 (GenBank accession NM_004925) cDNA into the multiple cloning site of the pAAV-MCS vector (Agilent Technologies, USA) under control of the CMV immediate-early promoter. The insert orientation and sequence fidelity were verified through bidirectional Sanger sequencing (GENEWIZ, China) using vector-specific primers (forward: 5'-CGCAAATGGGCGGTAGGCGTG-3'; reverse: 5'-CTCAGTTGGCGAGCTCGGATC-3'). NPCs were plated at a density of 2.0×105 cells/well on poly-L-lysine-coated glass coverslips (0111520, Thermo Fisher Scientific, USA) in 6-well plates (140675, Nunc, Denmark) and cultured overnight in DMEM/F-12 medium supplemented with 10% FBS. Transfection complexes were prepared by mixing 2.5 μg pAAV-AQP3 plasmid with 7.5 μL Lipofectamine 2000 (11668019, Thermo Fisher Scientific, USA) in Opti-MEM reduced serum medium (31985062, Gibco, USA), following a 3:1 lipid:DNA ratio. After 20 min incubation at room temperature, the complexes were added to cells maintained in antibiotic-free medium. Following 6 h incubation at 37°C with 5% CO2, the transfection medium was replaced with complete growth medium for 48 h prior to subsequent analyses.
2.14. AAV-Mediated Expression of AQP3
Recombinant adeno-associated virus (rAAV) vectors were generated through triple-plasmid co-transfection in HEK293T cells (CRL-3216, ATCC, USA). The payload plasmid pAAV-TBG-AQP3-GFP contained human AQP3 cDNA fused with enhanced GFP reporter, hepatocyte-specific thyroxine-binding globulin (TBG) promoter and AAV2 inverted terminal repeats (ITRs). The packaging system consisted of pAAV-RC2/8 providing AAV2 replication (Rep) and AAV8 capsid (Cap) proteins and pHelper supplying adenoviral E2A, E4, and VA RNA genes.
72 h post-transfection, cells were lysed in 0.5% sodium deoxycholate (D6750, Sigma-Aldrich, USA) and subjected to cesium chloride gradient ultracentrifugation (Optima XE-90, Beckman Coulter, USA) at 210,000×g for 48 h at 4°C. Viral fractions with refractive indices between 1.365-1.371 were collected and dialyzed against PBS containing 5% sorbitol (S3889, Sigma-Aldrich, USA). Genome titers (1.2×1013 vg/mL) were determined by absolute quantification using ITR-targeted qPCR (Forward: 5'-GGAACCCCTAGTGATGGAGTT-3'; Reverse: 5'-CGGCCTCAGTGAGCGA-3') with linearized plasmid standards.
2.15. Animal Experiment
Forty adult female Sprague-Dawley rats were randomly allocated into four experimental groups using stratified randomization based on body weight: Control, Annulus fibrosus puncture (AFP)+DMSO, AFP+AQP3 inhibitor DFP00173 (20 mg/kg; HY-126073, MedChemExpress, China), AFP+AAV-AQP3 (1×109 TU/mL lentiviral vectors).
Animals were anesthetized via intraperitoneal injection of sodium pentobarbital (40 mg/kg; P3761, Sigma-Aldrich, USA) and positioned prone on a warming pad. Under fluoroscopic guidance (C-arm Ziehm Vision RFD 3D, Germany), the caudal intervertebral disc (Co6/7 level) was exposed through a posterolateral approach. A 26-gauge spinal needle (405210, B. Braun, Germany) was inserted perpendicular to the annulus fibrosus at 5 mm depth, followed by 360° rotation with 30-second dwell time to induce controlled annular injury.
2 μL of therapeutic agents were intradiscally injected using a microsyringe. Postoperative analgesia was maintained with meloxicam (1 mg/kg SC; M3937, Sigma-Aldrich, USA) for 72 h. Weekly booster injections were administered under isoflurane anesthesia. Body weights were recorded biweekly using calibrated scales (XB220A, Precisa, Switzerland).
2.16. Micro-CT and MRI
Following 5-week postoperative recovery, animals underwent in vivo imaging under isoflurane anesthesia (3% induction, 1.5% maintenance). High-resolution Micro-computed tomography (micro-CT) scans were acquired using a SkyScan 1276 system (Bruker, Germany).
T2-weighted imaging was conducted using a 3T clinical scanner (MAGNETOM Prisma; Siemens Healthineers, Germany) equipped with a 16-channel phased-array spine coil. Sequence parameters were optimized for intervertebral disc assessment (repletion time 2000 ms; echo time 76 ms; field of view 260×320 mm; slice thickness 0.8 mm).
2.17. Histopathologic Staining
IVD specimens were fixed in 4% paraformaldehyde (PFA; P6148, Sigma-Aldrich, USA) for 48 h at 4°C, followed by decalcification in 10% ethylenediaminetetraacetic acid (EDTA; E4884, Sigma-Aldrich, USA) solution for 28 days. Tissues were dehydrated through a graded ethanol series (70%-100%), cleared in xylene (534056, Sigma-Aldrich, USA), and embedded in paraffin (76242, Leica, Germany) using a HistoStar embedding station (Thermo Fisher Scientific, MA).
Serial 5 μm sagittal sections were cut using a RM2255 microtome (Leica) and mounted on poly-L-lysine-coated slides (S8902, Solarbio, China). >Sections stained with Mayer's hematoxylin (HXG732, Baso, China) for 8 min and eosin Y (HT110232, Sigma-Aldrich, USA) for 1 min. Sections stained with 0.1% Safranin O (S8884, Sigma-Aldrich, USA) for 5 min followed by 0.02% Fast Green FCF (FCF-1, Sigma-Aldrich, USA) for 3 min. Blinded histological analysis was performed by two independent pathologists using a histological grading scale system [23].
2.18. Statistical Analysis
All graphical representations were generated using GraphPad Prism v8.0.2 (GraphPad Software, USA). Continuous variables with normal distribution (assessed by Shapiro-Wilk test) are presented as mean±standard deviation (SD), while non-normally distributed data are expressed as median with interquartile range. Student’s-test, one-way ANOVA or two-way ANOVA analysis of variance were used to compare data from the in vitro experimental group. Non-parametric analyses were applied to ordinal histological scores and heteroscedastic datasets. Type I error rate was controlled at α=0.05 with two-tailed testing. Multiplicity-adjusted p-values are denoted as * P < 0.05 (95% CI excludes null), ** P < 0.01, ** * P < 0.001). All experiments included ≥3 independent biological replicates with technical triplicates. Randomization and blinding protocols were implemented during data collection and analysis phases.
3. Results
3.1. Hyperosmolarity Inhibited Proliferation and Induced Apoptosis in NPCs
The pathogenesis of IVDD has been associated with altered osmotic homeostasis. To investigate hyperosmolar effects on NPCs, primary rat NPC cultures were established and subjected to graded osmotic stress (330, 550, and 600 mOsm/kg) for 48 hr. Quantitative apoptosis assessment through flow cytometry revealed substantial cell death potentiation under hyperosmolarity conditions (550 and 600 mOsm/kg), with apoptosis rates increased respectively compared to physiological osmolality controls (330 mOsm/kg) (Figure 1A,B). This apoptotic activation was corroborated by significant elevation of cleaved caspase-3 expression, as quantified through both flow cytometric analysis (Figure 1C,D). The immunofluorescence results further demonstrated that hyperosmotic stress promotes apoptosis in NPCs (Figure 1E,F). Mechanistic investigation via western blotting demonstrated marked upregulation of pro-apoptotic Bax protein coupled with concomitant downregulation of anti-apoptotic Bcl-2 in the 550 mOsm/kg group (Figure 1G,H). Furthermore, ECM degradation was evidenced by significant reductions in type II collagen and MMP3 expression levels. These findings collectively establish that sustained hypertonic stress induces apoptotic cascades in NPCs while disrupting matrix homeostasis.
3.2. Hyperosmolality Promotes Mitochondrial Oxidative Damage and ROS Accumulation in NPCs
To elucidate the mechanisms underlying hyperosmolarity-induced cellular damage, we investigated the role of reactive oxygen species (ROS) in NPCs, given their established involvement in apoptotic pathways [24]. Flow cytometric analysis revealed distinct alterations in oxidative stress markers across different osmotic conditions. Specifically, NPCs exposed to 550 mOsm/kg demonstrated a marked elevation in ROS levels (Figure 2A–C), concomitant with a significant reduction in GPx activity (Figure 2D) and increased MDA content (Figure 2E) when compared to the 330 mOsm/kg control group.
Considering the mitochondrial origin of ROS generation, we employed dual immunofluorescence staining using Mito-SOX (red) and Mito-Tracker (green) to specifically assess mitochondrial ROS production. Quantitative analysis demonstrated a substantial enhancement in Mito-SOX fluorescence intensity in NPCs subjected to 550 mOsm/kg relative to the control group (Figure 2F,G), indicating heightened mitochondrial oxidative stress. To further evaluate mitochondrial integrity, we examined the mitochondrial membrane potential (MMP), a critical parameter of mitochondrial function, using JC-1 staining. Immunofluorescence quantification revealed the MMP reduced in NPCs under an osmotic pressure of 550 mOsm/kg compared to the physiological osmotic pressure of 330 mOsm/kg (Figure 2H). These collective findings strongly suggest that hyperosmolarity induces mitochondrial dysfunction in NPCs, leading to excessive ROS accumulation and subsequent cellular damage.
3.3. Overexpression of AQP3 Mitigated Mitochondrial Oxidative Stress and Attenuated ROS Accumulation in NPCs Under a High Osmotic Pressure Condition
AQP3, an aquaporin family member responsible for transporting water and small solutes, is notably reduced in aging IVD [20,22]. To investigate its role under hyperosmolarity, we assessed AQP3 expression in NPCs. Quantitative analysis revealed a marked downregulation of AQP3 expression in NPCs exposed to 550 mOsm/kg compared to those under 330 mOsm/kg (Figure 3A). Consistent with this, Western blot demonstrated significantly reduced AQP3 protein levels under hyperosmotic conditions (550 mOsm/kg; Figure 3B,C), indicating osmotic stress suppresses AQP3 expression.
To evaluate the functional impact of AQP3 on NPCs survival, lentiviral AQP3 overexpression (AAV-AQP3) was performed. Flow cytometry analysis showed that AAV-AQP3-transfected NPCs exhibited a lower apoptotic rate under 550 mOsm/kg compared to controls (Figure 3D). Concurrently, JC-1 staining revealed elevated MMP (Figure 3E,F), while ROS accumulation was significantly attenuated in the AAV-AQP3 group (Figure 3G,H). Immunofluorescence further confirmed reduced Mito-SOX fluorescence intensity in AQP3-overexpressing NPCs (Figure 3I,J). Collectively, these findings demonstrate that AQP3 overexpression alleviates mitochondrial dysfunction, suppresses ROS production, and enhances NPCs viability under hyperosmolarity.
3.4. Downregulation of AQP3 Exacerbates NPCs Apoptosis via PI3K/Akt/mTOR Pathway Inhibition Under Hyperosmolarity
To elucidate the mechanism linking AQP3 downregulation to NPCs apoptosis, RNA sequencing and pathway enrichment analyses were conducted. KEGG and GSEA revealed suppression of PI3K/Akt and mTOR signaling pathways in NPCs under 550 mOsm/kg compared to 330 mOsm/kg (Figure 4A,B). Western blot confirmed reduced PI3K, Akt, and mTOR phosphorylation levels under hyperosmotic conditions. Notably, AQP3 overexpression restored their expression (Figure 4C,D), suggesting AQP3 modulates these pathways. These results indicate that hyperosmolarity-induced AQP3 depletion promotes NPC apoptosis by impairing PI3K/Akt/mTOR pathway.
3.5. Overexpression of AQP3 Ameliorated IVDD Progression in Rat Model
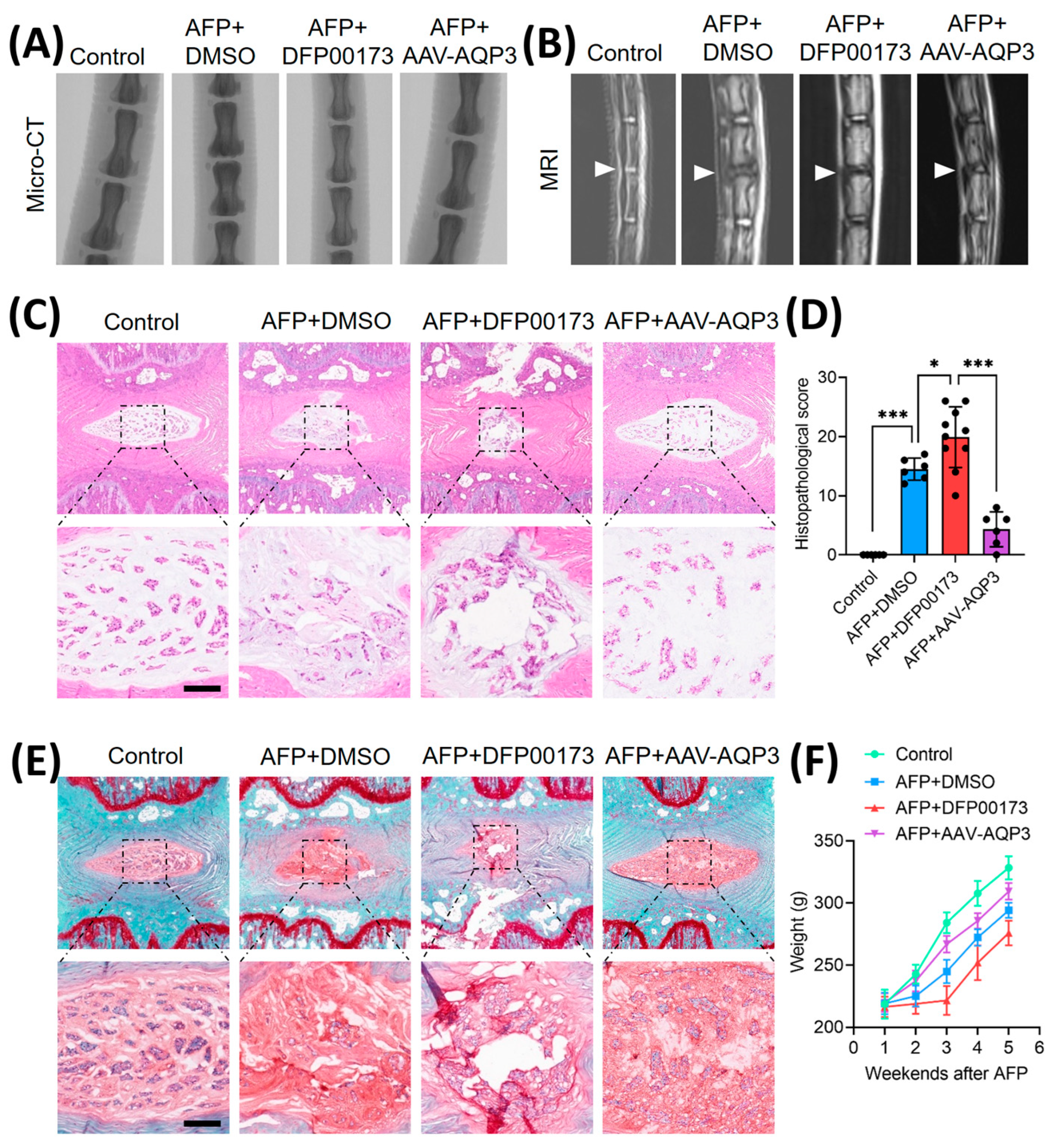
To validate the therapeutic potential of AQP3 in vivo, an IVDD model was established via annulus fibrosus puncture (AFP). Rats received weekly injections of AQP3 inhibitor (DFP00173) or AAV-AQP3. After 5 weeks, micro-CT imaging showed narrowed intervertebral disc spaces in IVDD rats, which were partially restored by AQP3 overexpression (Figure 5A). MRI analysis revealed uneven disc intensity and diminished T2-weighted signals in IVDD rats, whereas AAV-AQP3 treatment elevated T2 signals (Figure 5B), indicating improved disc hydration.
Histopathological assessment demonstrated severe structural degeneration in AFP-induced IVDD rats, including collapsed disc height, NPCs loss, and fibrosis. In contrast, AAV-AQP3 administration ameliorated tissue architecture, reduced fibrosis, and lowered histological scores (Figure 5C–E). Furthermore, AQP3-overexpressing rats exhibited improved weight gain compared to the IVDD group (Figure 5F). These results confirm that AQP3 overexpression mitigates IVDD progression in vivo.
4. Discussion
LBP secondary to IVDD represents a predominant contributor to global disability burdens. The degenerative cascade is initiated through multifactorial interactions involving risk factors such as advancing age, genetic predisposition, inflammatory damage, immune dysfunction, hyperosmolarity, and nutritional deficits. These pathogenic stimuli progressively trigger NPCs apoptosis and AF cellular disruption, ultimately leading to intervertebral space narrowing, herniated nucleus pulposus, and subsequent nerve root or spinal cord compression - pathological hallmarks directly associated with chronic pain manifestation [25,26]. The IVDD pathogenesis is characterized by dysregulation of ECM biosynthesis, enhanced catabolism with concomitant cellular senescence, and progressive depletion of functional NPCs populations through apoptotic pathways [27,28,29]. Notably, microenvironmental hyperosmolarity has been identified as a critical modulator of NPCs. Experimental evidence demonstrates that sustained exposure to the osmolarity of 550 mOsm/kg induces characteristic apoptotic morphology in NPCs, including nuclear fragmentation, chromatin condensation, and cytoplasmic organelle disintegration. In contrast, physiological osmolarity level approximating 450 mOsm/kg - comparable to native nucleus pulposus tissue conditions - exhibit minimal apoptotic effects on cellular viability [14].
Emerging evidence indicates osmotic stress-mediated AQP3 dysregulation in NPCs, with previous studies empirically demonstrating a 40% reduction in AQP3 expression at 550 mOsm/kg compared to physiological 330 mOsm/kg conditions [30]. While this osmotic sensitivity was documented, the precise mechanistic link between AQP3 downregulation and NPC apoptosis remained unresolved. Our investigation systematically evaluates both apoptotic dynamics and AQP3 expression patterns across osmotic gradients, mechanistically linking this phenomenon to ROS-mediated mitochondrial dysfunction through PI3K/Akt/mTOR pathway inhibition. Three principal findings emerge from this study: First, hyperosmolarity (550 mOsm/kg) induces significant AQP3 suppression, correlating with elevated intracellular ROS levels and increased apoptosis rate. Second, pharmacological interventions confirm the causal relationship - AQP3 overexpression restores redox homeostasis by enhancing mitochondrial membrane potential stability and reduces caspase-3 activation. Third, pathway analysis reveals hyperosmolarity disrupts PI3K/Akt/mTOR pathway, with phosphorylated Akt levels decreasing proportionally to osmotic intensity.
The pathophysiological relevance of these findings is underscored by clinical data showing 60% lower AQP3 expression in IVDD patient specimens versus healthy controls [31]. As a transmembrane glycerol-aquaporin dual transporter, AQP3's osmotic regulatory capacity extends beyond water homeostasis to encompass antioxidant defense mechanisms [32]. This dual functionality explains its protective effects against H2O2-induced oxidative damage in rodent models, where AQP3 overexpression reduced lipid peroxidation and improved cell viability [33]. Our data further position AQP3 as a critical molecular nexus integrating osmotic adaptation, redox regulation, and apoptotic signaling in IVDD pathogenesis.
Our experimental data demonstrate a distinct osmotic threshold response in NPCs, with osmotic pressure of 550 mOsm/kg inducing increased apoptosis rates and reduced proliferative capacity compared to physiological 330 mOsm/kg conditions, corroborating previous findings by Li's group regarding osmotic sensitivity in disc cells (30). This pathological progression appears mediated through redox imbalance mechanisms, where hyperosmolarity-induced mitochondrial oxidative damage establishes a self-perpetuating cycle of ROS accumulation and cellular dysfunction. The oxidative stress cascade in IVDD manifests as a three-phase molecular derangement, including inflammatory mediators and neovascularization-derived inflammatory cells generate primary ROS bursts [34], vascular endothelial growth factor (VEGF)-mediated neoangiogenesis disrupts the avascular NP microenvironment, introducing erythrocyte-derived iron ions that catalyze Fenton reactions [35], and mitochondrial permeability transition pore (mPTP) opening precipitates cytochrome c release, activating caspase-3 apoptotic pathways. Notably, our findings extend Chen's observation of N-acetylcysteine (NAC)-mediated ROS reduction by mechanistically linking osmotic stress [36].
Magnesium boride-alginate (MB-ALG) hydrogels have been demonstrated to promote the proliferation of senescent cells within rat IVD through activation of the PI3K/Akt/mTOR signaling cascade. Concurrently, molecular hydrogen demonstrated remarkable antioxidative efficacy by effectively neutralizing ROS, thereby protecting NPCs from oxidative stress-induced damage [37]. Building upon previous investigations by Sun et al., which demonstrated the PI3K/Akt/mTOR pathway suppression under hyperosmolar conditions and subsequent recovery following osteogenic protein-1 (OP-1) intervention, resulting in reduced NPCs apoptosis [38], we further explored the molecular mechanisms associated with AQP3 downregulation and its involvement in NPCs apoptosis under a high osmotic pressure condition. Transcriptomic analysis through RNA sequencing revealed substantial inhibition of the PI3K/Akt/mTOR signaling axis in rat NPCs subjected to hyperosmolar conditions. Importantly, adenoviral vector-mediated AQP3 overexpression successfully reversed this pathway suppression, leading to a pronounced decrease in NPCs apoptosis. These molecular interventions translated into significant improvements in the IVDD rat model, highlighting the therapeutic promise of AQP3 modulation and PI3K/Akt/mTOR pathway targeting for IVDD management.
Previous investigations by Palacio-Mancheno et al. revealed elevated AQP3 expression in notochordal cell lineages following 14-day hyperosmotic conditioning of murine intervertebral disc cell cultures [19], presenting divergent outcomes from our experimental findings. This observed disparity may stem from inherent differences in osmotic stress responsiveness between notochordal cells and NPCs, though rigorous comparative analyses remain imperative to substantiate this mechanistic interpretation. Notably, the inherent cellular heterogeneity of rodent nucleus pulposus tissue - comprising both NPCs and persistent notochordal remnants - introduces critical methodological considerations. Current technical constraints in distinguishing these populations through definitive molecular signatures complicate the exclusion of notochordal cell contamination during conventional NPCs isolation protocols. Establishing standardized protocols employing lineage-specific biomarkers for NPCs identification represents an essential advancement for future investigations, ensuring cellular purity and minimizing confounding variables associated with interspecies cellular admixture.
5. Conclusions
This study elucidates the mechanism by which downregulation of AQP3 mediates NPCs apoptosis under a high osmotic pressure condition. Our experimental findings demonstrate that hyperosmolarity significantly enhances rat NPC apoptosis while suppressing AQP3 expression. Notably, AQP3 upregulation exhibits a rescuing effect on hyperosmolarity-induced NPC apoptosis through activation of the PI3K/Akt/mTOR signaling pathway. Mechanistically, these results clarify the pathophysiological role of AQP3 in IVDD and establish a molecular foundation for targeting AQP3 in clinical therapeutic strategies.
Author Contributions
Data curation, Y.S., HQ.Z. and JJ.W.; Investigation, Y.S., HQ.Z. JJ.W., C.L. and JB.Z.; Methodology, Y.S., and C.L.; Writing—review and editing, Y.S., WB.X., KH.L., YC.X. and G.H.; Validation, HQ.Z., C.L. and JB.Z.; Formal analysis, JJ.W.; Conceptualisation, WB.X., H.Y., KH.L., P.L., DP.W. and YC.X.; Resources, WB.X.; Supervision, H.Y.; Writing—original draft, KH.L., YC.X. and G.H.; Funding acquisition, P.L. and G.H..
Funding
This work was supported by the Basic and Applied Basic Research Foundation of Guangdong Province [Grant Number 2019A1515010209 and 2023A1515011572].
Institutional Review Board Statement
Animal handling and experimental procedures were performed at Guangzhou Forevergen Biosciences Medical Laboratory Animal Center following protocol approval by the Institutional Animal Care and Use Committee (IACUC Approval No. SYXK(Yue)2023-0186).
Data Availability Statement
The datasets generated and analyzed during the current study are either included in this published article or available from the corresponding author upon reasonable request. All authors have verified the availability and integrity of the data supporting this study's findings.
Acknowledgments
We gratefully acknowledge the Third Affiliated Hospital of Sun Yat-sen University for providing access to their state-of-the-art research facilities.
Conflicts of Interest
The authors declare no conflicts of interest associated with the manuscript.
Abbreviations
| LBP | Lower back pain |
| IVDD | Intervertebral disc degeneration |
| IVD | Intervertebral disc |
| NP | Nucleus pulposus |
| AF | Annulus fibrosus |
| ECM | Extracellular matrix |
| NPCs | Nucleus pulposus cells |
| AQPs | Aquaporins |
| AQP3 | Aquaporin-3 |
| SPF | Specific-pathogen-free |
| FBS | Fetal bovine serum |
| RIPA | Radioimmunoprecipitation assay |
| BCA | Bicinchoninic acid |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| PVDF | Polyvinylidene difluoride |
| HRP | Horseradish peroxidase |
| ECL | Enhanced chemiluminescence |
| CCK-8 | Cell Counting Kit-8 |
| ROS | Reactive oxygen species |
| DCFDA | 2',7'-dichlorodihydrofluorescein diacetate |
| DCF | 2',7'-dichlorofluorescein |
| MFI | Mean fluorescence intensity |
| MDA | Malondialdehyde |
| GPx | Glutathione peroxidase |
| TBARS | Thiobarbituric acid reactive substances |
| QC | Quality control |
| NGS | Next-generation sequencing |
| rAAV | Recombinant adeno-associated virus |
| TBG | Thyroxine-binding globulin |
| ITRs | Inverted terminal repeats |
| AFP | Annulus fibrosus puncture |
| micro-CT | micro-computed tomography |
| MRI | Magnetic resonance imaging |
| H&E | Hematoxylin and eosin |
| SD | Standard deviation |
| MMP | Mitochondrial membrane potential |
| VEGF | Vascular endothelial growth factor |
| mPTP | mitochondrial permeability transition pore |
| NAC | N-acetylcysteine |
| MB-ALG | Magnesium boride-alginate |
| OP-1 | Osteogenic protein-1 |
References
- Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [CrossRef]
- Wang, F.; Cai, F.; Shi, R.; Wang, X.H.; Wu, X.T. Aging and age related stresses: A senescence mechanism of intervertebral disc degeneration. Osteoarthritis and cartilage. 2016, 24, 398–408. [Google Scholar] [CrossRef]
- Xiang, Z.; Zhang, P.; Jia, C.; Xu, R.; Cao, D.; Xu, Z.; et al. Piezo1 channel exaggerates ferroptosis of nucleus pulposus cells by mediating mechanical stress-induced iron influx. Bone research. 2024, 12, 20. [Google Scholar] [CrossRef]
- Zheng, D.; Chen, W.; Chen, T.; Chen, X.; Liang, J.; Chen, H.; et al. Hydrogen Ion Capturing Hydrogel Microspheres for Reversing Inflammaging. Advanced materials 2024, 36, e2306105. [Google Scholar] [CrossRef]
- Feng, Y.; Egan, B.; Wang, J. Genetic Factors in Intervertebral Disc Degeneration. Genes & diseases. 2016, 3, 178–185. [Google Scholar] [CrossRef]
- Song, C.; Hu, P.; Peng, R.; Li, F.; Fang, Z.; Xu, Y. Bioenergetic dysfunction in the pathogenesis of intervertebral disc degeneration. Pharmacological research. 2024, 202, 107119. [Google Scholar] [CrossRef]
- Mroz, T.E.; Lubelski, D.; Williams, S.K.; O'Rourke, C.; Obuchowski, N.A.; Wang, J.C.; et al. Differences in the surgical treatment of recurrent lumbar disc herniation among spine surgeons in the United States. The spine journal : Official journal of the North American Spine Society. 2014, 14, 2334–2343. [Google Scholar] [CrossRef]
- Bhadouria, N.; Holguin, N. Osteoporosis treatments for intervertebral disc degeneration and back pain: A perspective. JBMR plus. 2024, 8, ziae048. [Google Scholar] [CrossRef]
- Chou, R.; Huffman, L.H. Medications for acute and chronic low back pain: A review of the evidence for an American Pain Society/American College of Physicians clinical practice guideline. Annals of internal medicine. 2007, 147, 505–514. [Google Scholar] [CrossRef]
- Roberts, S.; Evans, H.; Trivedi, J.; Menage, J. Histology and pathology of the human intervertebral disc. The Journal of bone and joint surgery American volume. 2006, 88 (Suppl. 2), 10–14. [Google Scholar] [CrossRef]
- Vadalà, G.; Ambrosio, L.; Russo, F.; Papalia, R.; Denaro, V. Interaction between Mesenchymal Stem Cells and Intervertebral Disc Microenvironment: From Cell Therapy to Tissue Engineering. Stem cells international. 2019, 2019, 2376172. [Google Scholar] [CrossRef] [PubMed]
- Urban, J.P.; Roberts, S. Degeneration of the intervertebral disc. Arthritis research & therapy. 2003, 5, 120–130. [Google Scholar] [CrossRef]
- Li, P.; Gan, Y.; Xu, Y.; Li, S.; Song, L.; Li, S.; et al. Osmolarity affects matrix synthesis in the nucleus pulposus associated with the involvement of MAPK pathways: A study of ex vivo disc organ culture system. Journal of orthopaedic research : Official publication of the Orthopaedic Research Society. 2016, 34, 1092–1100. [Google Scholar] [CrossRef]
- Li, P.; Gan, Y.; Wang, H.; Xu, Y.; Li, S.; Song, L.; et al. Role of the ERK1/2 pathway in osmolarity effects on nucleus pulposus cell apoptosis in a disc perfusion culture. Journal of orthopaedic research : Official publication of the Orthopaedic Research Society. 2017, 35, 86–92. [Google Scholar] [CrossRef]
- Ebi, H.; Costa, C.; Faber, A.C.; Nishtala, M.; Kotani, H.; Juric, D.; et al. PI3K regulates MEK/ERK signaling in breast cancer via the Rac-GEF, P-Rex1. Proceedings of the National Academy of Sciences of the United States of America. 2013, 110, 21124–21129. [Google Scholar] [CrossRef]
- Sadowska, A.; Kameda, T.; Krupkova, O.; Wuertz-Kozak, K. Osmosensing, osmosignalling and inflammation: How intervertebral disc cells respond to altered osmolarity. European cells & materials. 2018, 36, 231–250. [Google Scholar] [CrossRef]
- Kozono, D.; Yasui, M.; King, L.S.; Agre, P. Aquaporin water channels: Atomic structure molecular dynamics meet clinical medicine. The Journal of clinical investigation. 2002, 109, 1395–1399. [Google Scholar] [CrossRef]
- Gao, H.; Gui, J.; Wang, L.; Xu, Y.; Jiang, Y.; Xiong, M.; et al. Aquaporin 1 contributes to chondrocyte apoptosis in a rat model of osteoarthritis. International journal of molecular medicine. 2016, 38, 1752–1758. [Google Scholar] [CrossRef]
- Palacio-Mancheno, P.E.; Evashwick-Rogler, T.W.; Laudier, D.M.; Purmessur, D.; Iatridis, J.C. Hyperosmolarity induces notochordal cell differentiation with aquaporin3 upregulation and reduced N-cadherin expression. Journal of orthopaedic research : Official publication of the Orthopaedic Research Society. 2018, 36, 788–798. [Google Scholar] [CrossRef]
- Richardson, S.M.; Knowles, R.; Marples, D.; Hoyland, J.A.; Mobasheri, A. Aquaporin expression in the human intervertebral disc. Journal of molecular histology. 2008, 39, 303–309. [Google Scholar] [CrossRef]
- Mobasheri, A.; Trujillo, E.; Bell, S.; Carter, S.D.; Clegg, P.D.; Martín-Vasallo, P.; et al. Aquaporin water channels AQP1 and AQP3, are expressed in equine articular chondrocytes. Veterinary journal (London, England: 1997). 2004, 168, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Jing, Y.; Xia, J.; Wang, X.; You, C.; Yan, J. Aquaporin 3 protects against lumbar intervertebral disc degeneration via the Wnt/β-catenin pathway. International journal of molecular medicine. 2016, 37, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Masuda, K.; Aota, Y.; Muehleman, C.; Imai, Y.; Okuma, M.; Thonar, E.J.; et al. A novel rabbit model of mild, reproducible disc degeneration by an anulus needle puncture: Correlation between the degree of disc injury and radiological and histological appearances of disc degeneration. Spine. 2005, 30, 5–14. [Google Scholar] [CrossRef]
- Cheng, Z.; Gan, W.; Xiang, Q.; Zhao, K.; Gao, H.; Chen, Y.; et al. Impaired degradation of PLCG1 by chaperone-mediated autophagy promotes cellular senescence and intervertebral disc degeneration. Autophagy. 2025, 21, 352–373. [Google Scholar] [CrossRef]
- Knezevic, N.N.; Candido, K.D.; Vlaeyen, J.W.S.; Van Zundert, J.; Cohen, S.P. Low back pain. Lancet 2021, 398, 78–92. [Google Scholar] [CrossRef]
- Risbud, M.V.; Shapiro, I.M. Role of cytokines in intervertebral disc degeneration: Pain and disc content. Nature reviews Rheumatology. 2014, 10, 44–56. [Google Scholar] [CrossRef]
- Silagi, E.S.; Shapiro, I.M.; Risbud, M.V. Glycosaminoglycan synthesis in the nucleus pulposus: Dysregulation and the pathogenesis of disc degeneration. Matrix biology : Journal of the International Society for Matrix Biology. 2018, 71–72, 368–379. [Google Scholar] [CrossRef]
- Choi, H.; Tessier, S.; Silagi, E.S.; Kyada, R.; Yousefi, F.; Pleshko, N.; et al. A novel mouse model of intervertebral disc degeneration shows altered cell fate and matrix homeostasis. Matrix biology : Journal of the International Society for Matrix Biology. 2018, 70, 102–122. [Google Scholar] [CrossRef]
- Tessier, S.; Tran, V.A.; Ottone, O.K.; Novais, E.J.; Doolittle, A.; DiMuzio, M.J.; et al. TonEBP-deficiency accelerates intervertebral disc degeneration underscored by matrix remodeling, cytoskeletal rearrangements, and changes in proinflammatory gene expression. Matrix biology : Journal of the International Society for Matrix Biology. 2020, 87, 94–111. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhao, C.; Zhang, R.; Wang, Y.; Hu, Y.; Zhou, Q.; et al. Overexpression of Aquaporin-3 Alleviates Hyperosmolarity-Induced Nucleus Pulposus Cell Apoptosis via Regulating the ERK1/2 Pathway. Pain research & management. 2022, 2022, 1639560. [Google Scholar] [CrossRef]
- Li, S.B.; Yang, K.S.; Zhang, Y.T. Expression of aquaporins 1 and 3 in degenerative tissue of the lumbar intervertebral disc. Genetics and molecular research: GMR. 2014, 13, 8225–8233. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Peng, H.; Lei, L.; Zhang, Y.; Kuang, H.; Cao, Y.; et al. Aquaporin3 is a sperm water channel essential for postcopulatory sperm osmoadaptation and migration. Cell research. 2011, 21, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Hara-Chikuma, M.; Satooka, H.; Watanabe, S.; Honda, T.; Miyachi, Y.; Watanabe, T.; et al. Aquaporin-3-mediated hydrogen peroxide transport is required for NF-κB signalling in keratinocytes and development of psoriasis. Nature communications. 2015, 6, 7454. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Fujita, N.; Hosogane, N.; Watanabe, K.; Ishii, K.; Toyama, Y.; et al. Excessive reactive oxygen species are therapeutic targets for intervertebral disc degeneration. Arthritis research & therapy. 2015, 17, 316. [Google Scholar]
- Ali, R.; Le Maitre, C.L.; Richardson, S.M.; Hoyland, J.A.; Freemont, A.J. Connective tissue growth factor expression in human intervertebral disc: Implications for angiogenesis in intervertebral disc degeneration. Biotechnic & histochemistry : Official publication of the Biological Stain Commission. 2008, 83, 239–245. [Google Scholar] [CrossRef]
- Xu, J.; Li, H.; Yang, K.; Guo, S.; Wang, J.; Feng, C.; et al. Hyper-osmolarity environment-induced oxidative stress injury promotes nucleus pulposus cell senescence in vitro. Bioscience reports. 2019, 39. [Google Scholar] [CrossRef]
- Han, Y.; Zheng, D.; Ji, Y.; Feng, Y.; Chen, Z.; Chen, L.; et al. Active Magnesium Boride/Alginate Hydrogels Rejuvenate Senescent Cells. ACS nano. 2024, 18, 23566–23578. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, X.; Liu, Z.; Xiao, X.; Hu, W.; Sun, Z. Osteogenic protein-1 attenuates nucleus pulposus cell apoptosis through activating the PI3K/Akt/mTOR pathway in a hyperosmotic culture. Bioscience reports. 2018, 38. [Google Scholar] [CrossRef]
Figure 1.
Hyperosmolarity inhibited proliferation and induced apoptosis in rat NPC. (A, B) Quantitative analysis of apoptosis rates in NPCs under different osmotic pressures (330, 550, and 600 mOsm/kg) using flow cytometry. (C, D) Flow cytometric detection of cleaved caspase-3 activation in apoptotic NPCs through intracellular staining with specific antibodies. (E, F) Dual parameter assessment of phosphatidylserine externalization using Annexin V-FITC/PI double staining. (G, H) Western blot analysis of apoptosis-related proteins (Bax, Bcl-2) and extracellular matrix components (type II collagen, MMP3) under osmotic pressures of 330 mOsm/kg and 550 mOsm/kg. Representative immunoblots and densitometric quantification normalized to β-actin (42 kDa) show protein expression profiles. Molecular weight markers indicated in kDa. Data are expressed as mean ± SD. Significant differences between different groups are indicated as *P<0.05, **P<0.01.
Figure 1.
Hyperosmolarity inhibited proliferation and induced apoptosis in rat NPC. (A, B) Quantitative analysis of apoptosis rates in NPCs under different osmotic pressures (330, 550, and 600 mOsm/kg) using flow cytometry. (C, D) Flow cytometric detection of cleaved caspase-3 activation in apoptotic NPCs through intracellular staining with specific antibodies. (E, F) Dual parameter assessment of phosphatidylserine externalization using Annexin V-FITC/PI double staining. (G, H) Western blot analysis of apoptosis-related proteins (Bax, Bcl-2) and extracellular matrix components (type II collagen, MMP3) under osmotic pressures of 330 mOsm/kg and 550 mOsm/kg. Representative immunoblots and densitometric quantification normalized to β-actin (42 kDa) show protein expression profiles. Molecular weight markers indicated in kDa. Data are expressed as mean ± SD. Significant differences between different groups are indicated as *P<0.05, **P<0.01.
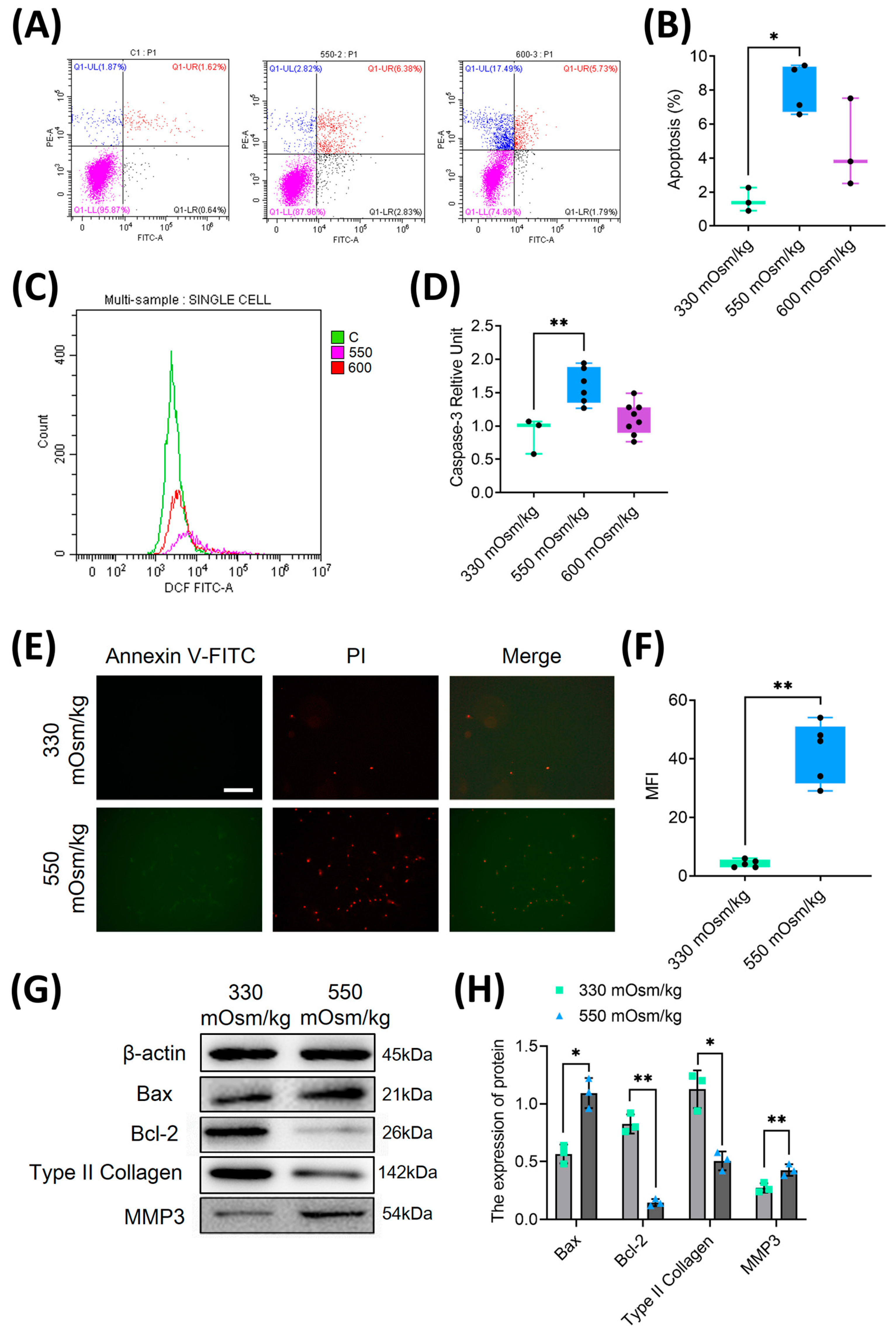
Figure 2.
Hyperosmolality induces mitochondrial oxidative damage and ROS accumulation in NPCs. (A) Dose-dependent suppression of cellular viability in NPCs exposed to different osmotic pressures assessed by CCK-8 assay. (B, C) Flow cytometric quantification of intracellular ROS levels under osmotic pressures of 330 mOsm/kg, 550 mOsm/kg and 600 mOsm/kg. (D) The activity of GPx measured by colorimetric assay showing enzymatic antioxidant capacity attenuation under different osmotic pressures (330 mOsm/kg and 550 mOsm/kg). (E) MDA content quantification via thiobarbituric acid reaction, reflecting lipid peroxidation levels under different osmotic pressures (330 mOsm/kg and 550 mOsm/kg). (F, G) Confocal imaging of mitochondrial superoxide (MitoSOX, Red) and mitochondrial morphology (MitoTracker, Green) co-localization. Scar bar: 200 μm. (H) MMP assessment using JC-1 fluorescence shift (red/green ratio). Scar bar: 100 μm. Data represent mean ± SD from three biological replicates. Significant differences between different groups are indicated as *P<0.05, **P<0.01, ***P<0.001.
Figure 2.
Hyperosmolality induces mitochondrial oxidative damage and ROS accumulation in NPCs. (A) Dose-dependent suppression of cellular viability in NPCs exposed to different osmotic pressures assessed by CCK-8 assay. (B, C) Flow cytometric quantification of intracellular ROS levels under osmotic pressures of 330 mOsm/kg, 550 mOsm/kg and 600 mOsm/kg. (D) The activity of GPx measured by colorimetric assay showing enzymatic antioxidant capacity attenuation under different osmotic pressures (330 mOsm/kg and 550 mOsm/kg). (E) MDA content quantification via thiobarbituric acid reaction, reflecting lipid peroxidation levels under different osmotic pressures (330 mOsm/kg and 550 mOsm/kg). (F, G) Confocal imaging of mitochondrial superoxide (MitoSOX, Red) and mitochondrial morphology (MitoTracker, Green) co-localization. Scar bar: 200 μm. (H) MMP assessment using JC-1 fluorescence shift (red/green ratio). Scar bar: 100 μm. Data represent mean ± SD from three biological replicates. Significant differences between different groups are indicated as *P<0.05, **P<0.01, ***P<0.001.
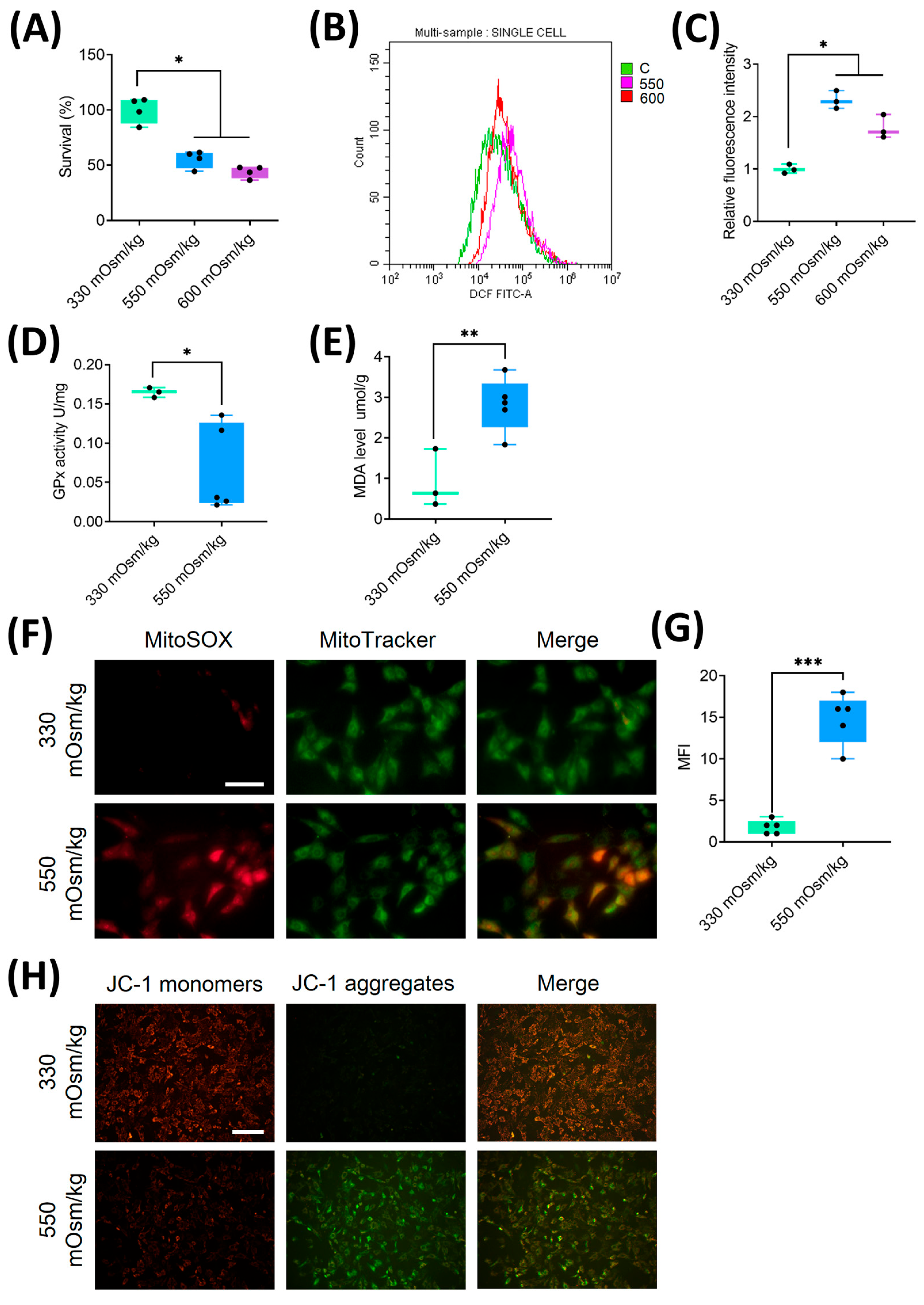
Figure 3.
Overexpression of AQP3 mitigated mitochondrial oxidative stress and attenuated ROS accumulation in NPCs under a high osmotic pressure condition. (A) Transcriptomic profiling of AQP3 expression by qPCR in NPCs under different osmotic pressures (330 mOsm/kg and 550 mOsm/kg) (n=3 biological replicates). (B, C) Western blot analysis of AQP3 under different osmotic pressures (330 mOsm/kg and 550 mOsm/kg). Representative blots and densitometric quantification normalized to β-actin (42 kDa) are shown. (D) The quantitative statistics of apoptotic NPCs. (E, F) MMP preservation assessed by flow cytometry in NPCs of each group. (G, H) Intracellular ROS levels measured using flow cytometry. (I, J) Confocal microscopy visualization of mitochondrial superoxide (MitoSOX, Red) and mitochondrial morphology (MitoTracker, Green) co-localization. Scar bar: 200 μm. Data represent mean ± SD from three biological replicates. Significant differences between different groups are indicated as *P<0.05, **P<0.01, ***P<0.001.
Figure 3.
Overexpression of AQP3 mitigated mitochondrial oxidative stress and attenuated ROS accumulation in NPCs under a high osmotic pressure condition. (A) Transcriptomic profiling of AQP3 expression by qPCR in NPCs under different osmotic pressures (330 mOsm/kg and 550 mOsm/kg) (n=3 biological replicates). (B, C) Western blot analysis of AQP3 under different osmotic pressures (330 mOsm/kg and 550 mOsm/kg). Representative blots and densitometric quantification normalized to β-actin (42 kDa) are shown. (D) The quantitative statistics of apoptotic NPCs. (E, F) MMP preservation assessed by flow cytometry in NPCs of each group. (G, H) Intracellular ROS levels measured using flow cytometry. (I, J) Confocal microscopy visualization of mitochondrial superoxide (MitoSOX, Red) and mitochondrial morphology (MitoTracker, Green) co-localization. Scar bar: 200 μm. Data represent mean ± SD from three biological replicates. Significant differences between different groups are indicated as *P<0.05, **P<0.01, ***P<0.001.
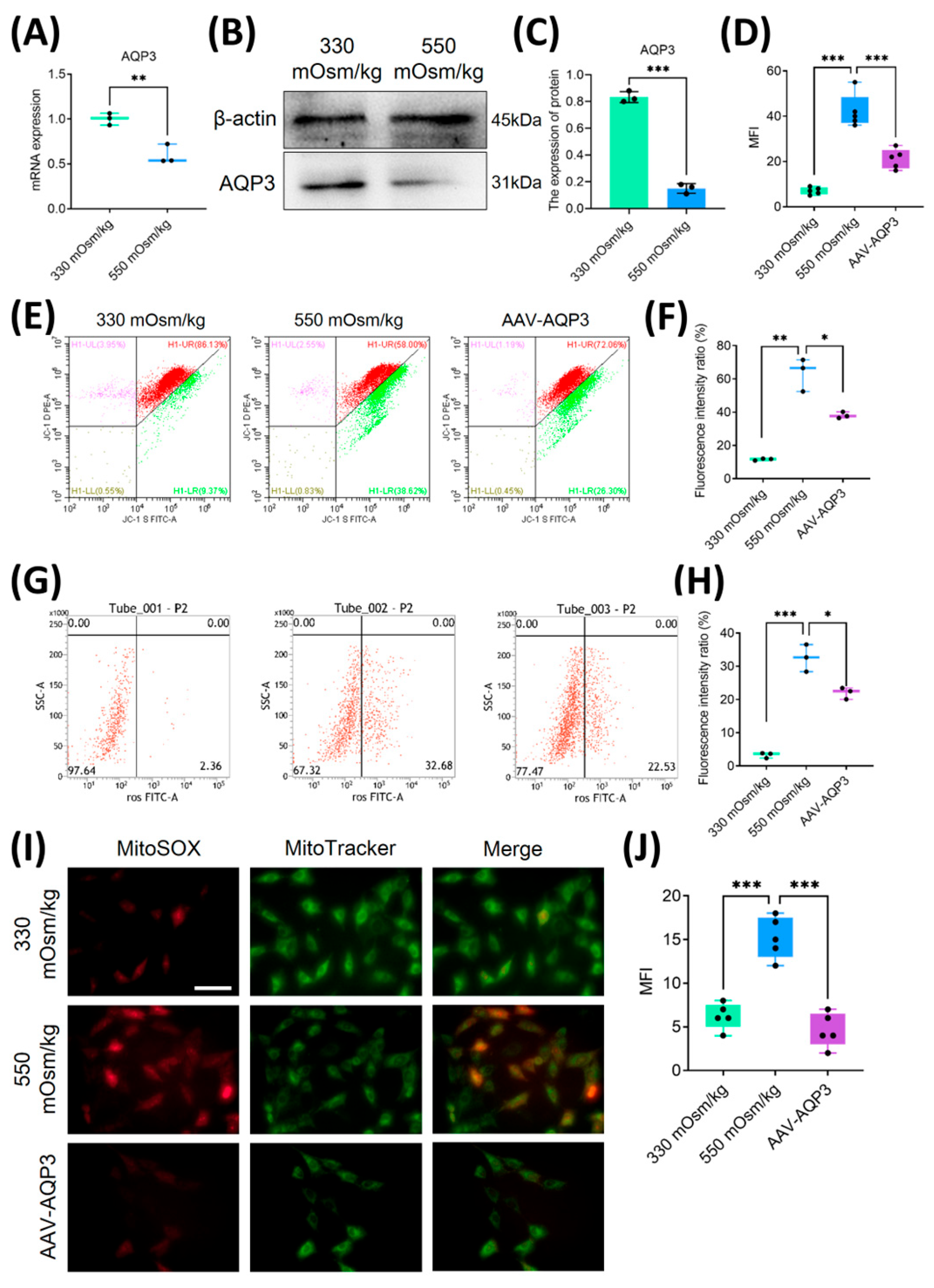
Figure 4.
Downregulation of AQP3 exacerbates NPCs apoptosis via PI3K/Akt/mTOR pathway inhibition under hyperosmolarity. (A) Top 20 enriched KEGG pathways of downregulated differentially expressed genes (DEGs) identified by RNA sequencing. Spot diameter corresponds to gene count, while color gradient reflects statistical significance (-log10 (p-value)). (B) Gene Set Enrichment Analysis (GSEA) demonstrating coordinated suppression of PI3K-Akt-mTOR signaling components. The y-axis represents enrichment score, and the x-axis denotes genes represented in gene sets. (C, D) Western blot analysis of p-mTOR, p-PI3K and p-Akt level in rat NPCs of each group. Data represent mean±SD from three biological replicates. Significant differences between different groups are indicated as *P<0.05, **P<0.01, ***P<0.001.
Figure 4.
Downregulation of AQP3 exacerbates NPCs apoptosis via PI3K/Akt/mTOR pathway inhibition under hyperosmolarity. (A) Top 20 enriched KEGG pathways of downregulated differentially expressed genes (DEGs) identified by RNA sequencing. Spot diameter corresponds to gene count, while color gradient reflects statistical significance (-log10 (p-value)). (B) Gene Set Enrichment Analysis (GSEA) demonstrating coordinated suppression of PI3K-Akt-mTOR signaling components. The y-axis represents enrichment score, and the x-axis denotes genes represented in gene sets. (C, D) Western blot analysis of p-mTOR, p-PI3K and p-Akt level in rat NPCs of each group. Data represent mean±SD from three biological replicates. Significant differences between different groups are indicated as *P<0.05, **P<0.01, ***P<0.001.
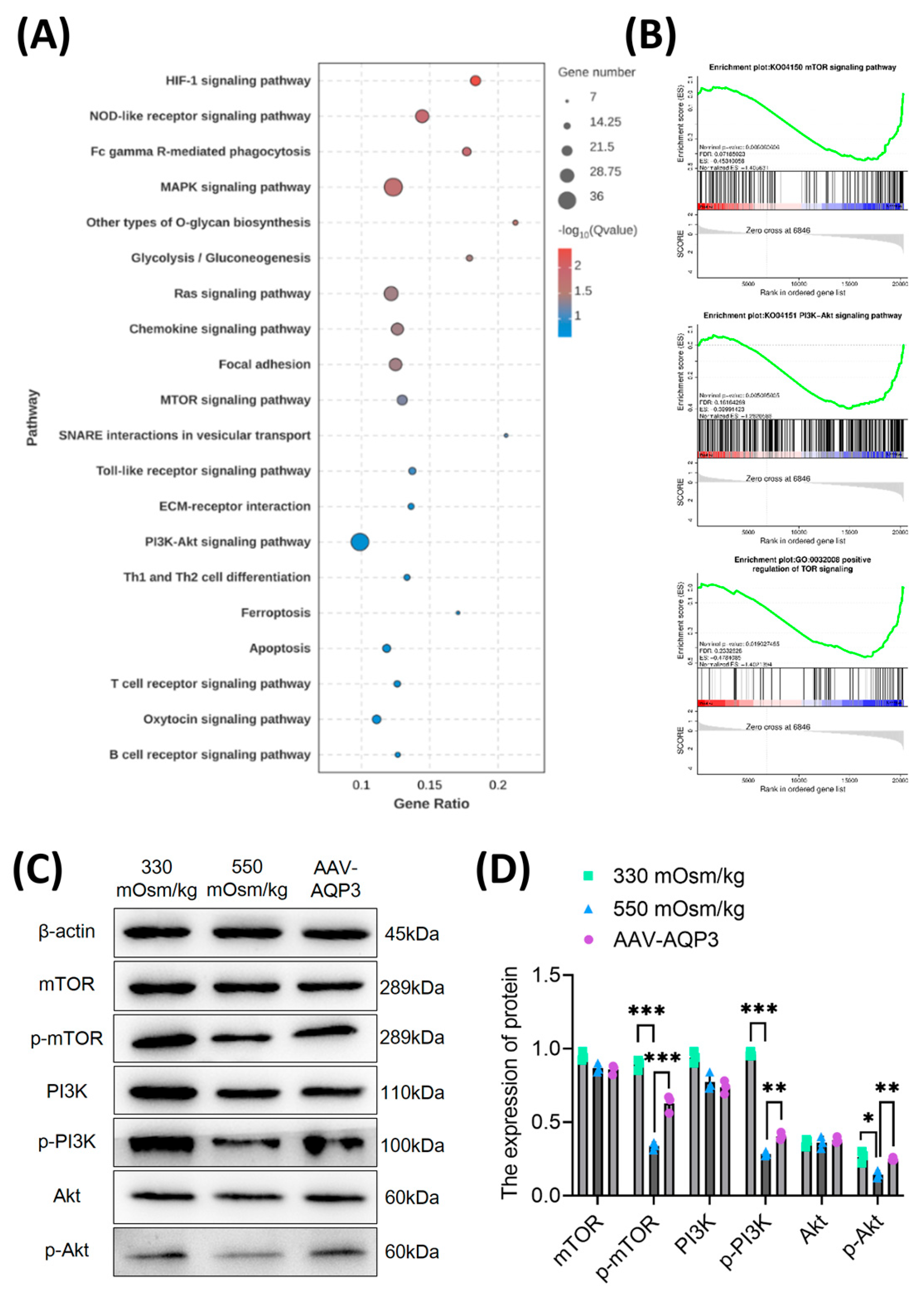
Figure 5.
Overexpression of AQP3 ameliorated IVDD progression in rat model. (A) Schematic representation of experimental design: Control (n=6), AFP+DMSO (n=6), AFP+DFP00173 (AQP3 inhibitor, 20 mg/kg, n=10), and AFP+AAV-AQP3 (n=6) groups. Interventions were administered via intradiscal injection weekly for 5 weeks post-puncture. Micro-CT sagittal reconstructions demonstrating intervertebral space height preservation. (B) T2-weighted MRI axial sections. Operated levels (Co6/7) are demarcated by white triangles. (C-E) Histopathological evaluation of NP tissues integrity. Representative H&E staining and Safranin-O/fast green staining with semi-quantitative histomorphometric analysis. Scale bar: 500 μm. (F) Average growth weight curves of rats in each group. Data represent mean±SD. Significant differences between different groups are indicated as *P<0.05, ***P<0.001.
Figure 5.
Overexpression of AQP3 ameliorated IVDD progression in rat model. (A) Schematic representation of experimental design: Control (n=6), AFP+DMSO (n=6), AFP+DFP00173 (AQP3 inhibitor, 20 mg/kg, n=10), and AFP+AAV-AQP3 (n=6) groups. Interventions were administered via intradiscal injection weekly for 5 weeks post-puncture. Micro-CT sagittal reconstructions demonstrating intervertebral space height preservation. (B) T2-weighted MRI axial sections. Operated levels (Co6/7) are demarcated by white triangles. (C-E) Histopathological evaluation of NP tissues integrity. Representative H&E staining and Safranin-O/fast green staining with semi-quantitative histomorphometric analysis. Scale bar: 500 μm. (F) Average growth weight curves of rats in each group. Data represent mean±SD. Significant differences between different groups are indicated as *P<0.05, ***P<0.001.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



