
Submitted:
03 June 2025
Posted:
03 June 2025
You are already at the latest version
Abstract
Graphical Abstract:
Abstract: Sudden Unexpected Death in Epilepsy (SUDEP) is a leading cause of mortality among individuals with epilepsy, particularly those with drug-resistant forms. This re-view explores the complex multisystem mechanisms underpinning SUDEP, integrating recent findings on brain, cardiac, and pulmonary dysfunctions. Background/Objectives: The main objective of this review is to elucidate how seizures disrupt critical physiological systems, especially the brainstem, heart, and lungs, contributing to SUDEP, with empha-sis on respiratory control failure and autonomic instability. Methods: We systematically examined literature from experimental models, clinical observations, neuroimaging stud-ies, and genetic analyses. Sources were drawn from recent peer-reviewed publications cataloged in a supplementary reference matrix. Results: SUDEP is frequently preceded by generalized tonic-clonic seizures, which trigger central and obstructive apnea, hypoventi-lation, and cardiac arrhythmias. Brainstem dysfunction, particularly in areas such as the pre-Bötzinger complex and nucleus tractus solitarius, plays a central role. Genetic muta-tions affecting ion channels (e.g., SCN1A, KCNQ1) and neurotransmitter imbalances (no-tably serotonin and GABA) exacerbate autonomic dysregulation. Risk is compounded by prone sleeping position, reduced arousal capacity, and impaired ventilatory responses. Conclusions: SUDEP arises from a cascade of interrelated failures in respiratory and car-diac regulation initiated by seizure activity. Recognition of modifiable risk factors, imple-mentation of monitoring technologies, and targeted therapies such as serotonergic agents may reduce mortality. Multidisciplinary approaches integrating neurology, cardiology, and respiratory medicine are essential for effective prevention strategies.
Keywords:
SUDEP
; Epilepsy
; Autonomic nervous system
; Central apnea
; Hypoventilation
; Neuro-degeneration
1. Introduction
Sudden Unexpected Death in Epilepsy (SUDEP) refers to the sudden death of a person with epilepsy, not caused by trauma, drowning, or a prolonged seizure. Often, there is evidence of a seizure close to the time of death, but the exact cause remains unclear. SUDEP is one of the main causes of death in people with epilepsy [1], with the risk being up to 20 times higher than in the general population. In one study of childhood-onset epilepsy, 55% of deaths were epilepsy-related, and 30% were due to sudden, unexplained causes—resulting in a 7% risk by age 40 [2]. What makes SUDEP even more challenging is that autopsies often reveal no clear cause [2].
SUDEP can happen at any age but is most common in adults between 20 and 45 years old [3,4]. Higher risks have been observed in patients with uncontrolled epilepsy, those treated at epilepsy centers, people in care facilities, and those who had brain surgery [5]. Being male [5] and having childhood-onset epilepsy may also raise the risk. Socioeconomic factors like poor access to healthcare and medications also contribute [3]. Interestingly, people who only experience absence or myoclonic seizures do not seem to have an increased risk [6]. However, having generalized tonic-clonic seizures (GTCS) significantly increases the risk—by as much as tenfold—especially in people who live alone. Substance or alcohol abuse can also double the risk [7].
Research has identified several specific risk factors. Having more than three GTCS per year can increase SUDEP risk by 10 to 15 times [8]. Other risks include epilepsy that begins early in life, seizures that occur during sleep, and a long history of epilepsy. In children, developmental delay and early-onset epilepsy are additional concerns [9]. Some risks cannot be changed—such as having severe epilepsy or genetic mutations affecting heart-related ion channels like KCNQ1, SCN1A, or SCN5A [10].
A major study known as MORTEMUS found that SUDEP often follows a seizure that leads to brain shutdown (EEG suppression), breathing stops (apnea), and then heart failure [11]. This highlights the critical role of breathing problems in SUDEP, alongside heart and brain function shutdown. The brainstem, which controls breathing, may be suppressed after seizures, leading to dangerous conditions like severe acid buildup, airway spasms, and lack of oxygen [12]. Sleeping in a facedown (prone) position, often seen in SUDEP cases, can worsen these breathing problems [11].
In addition to affecting the brain, epilepsy also puts a strain on the heart. People with generalized or drug-resistant epilepsy are at higher risk for heart issues like irregular heartbeats (arrhythmias), problems with the body's automatic control system (autonomic imbalance), and structural heart changes [228,229]. During a seizure, the nervous system can become unstable, triggering dangerous changes in heart rhythm [230]. Cardiac problems are a major reason why SUDEP occurs and contribute to early death in people with epilepsy [231].
Key mechanisms in SUDEP include stopped breathing (central apnea), slow heart rate (bradycardia), complete heart stoppage (asystole), and irregular rhythms, which may happen during or after a seizure. One critical event is postictal generalized EEG suppression (PGES), where the brain temporarily shuts down after a seizure. This affects the medulla oblongata, the brain region that controls breathing and heart rate. When this area fails to work, fatal heart problems can follow [232,233].
Another key factor is autonomic imbalance, where the body's stress system (sympathetic) becomes overactive, and the calming system (parasympathetic) is suppressed. This weakens the heart over time and increases the risk of deadly arrhythmias [234]. Disruption in key brain areas like the insular cortex and brainstem also make these irregular heart rhythms more likely, further raising the risk of sudden cardiac arrest [235].
2. Materials and Methods
This review was conducted through a comprehensive and integrative analysis of current literature related to Sudden Unexpected Death in Epilepsy (SUDEP). A systematic search was performed using databases including PubMed, Scopus, and Web of Science to identify peer-reviewed studies published up to May 2025. Keywords were used such as SUDEP, epilepsy, brainstem dysfunction, cardiac arrhythmia, respiratory failure, central apnea, hypoventilation, autonomic nervous system, and neurotransmitter imbalance. Primary research articles, reviews, meta-analyses, animal studies, and clinical trials were considered.
Selection criteria emphasized high-impact studies that provided mechanistic insights into SUDEP, especially those highlighting interactions among the brain, lungs, and heart. Data from experimental epilepsy models (e.g., pilocarpine, kainic acid, Scn1a−/−, Kcna1−/− mice), human neuroimaging studies, post-mortem pathology reports, and electrophysiological recordings were synthesized to map the pathophysiological cascade of SUDEP. The references cited were cross validated against an external bibliography matrix provided in supplementary data (Excel file), ensuring accuracy and coverage.
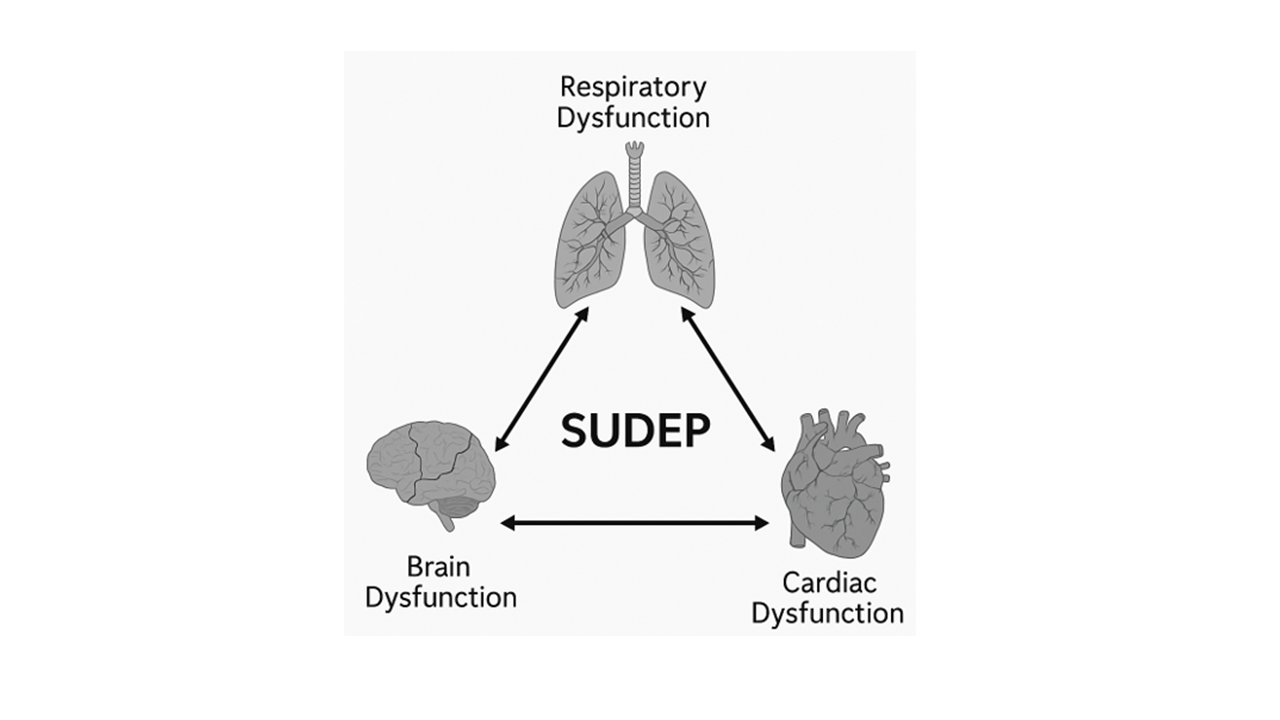
The extracted findings were organized thematically into respiratory, cardiac, and neurological dysfunctions, with additional focus on genetic, molecular, and structural contributors. Emphasis was placed on the translational relevance of preclinical findings to human pathology. Finally, conceptual integration was guided by the triangular framework (brain–lung–heart axis shown in graphical abstract figure), which was used to illustrate the dynamic interplay among the three organ systems implicated in epilepsy linked SUDEP.
3. Results
Based on the extensive literature reviewed and organized in the reference matrix, the following sections explore the key physiological systems implicated in SUDEP: neurological, respiratory, and cardiac. These categories were assigned based on each study’s primary focus, determined through a detailed screening of article titles and abstract content.
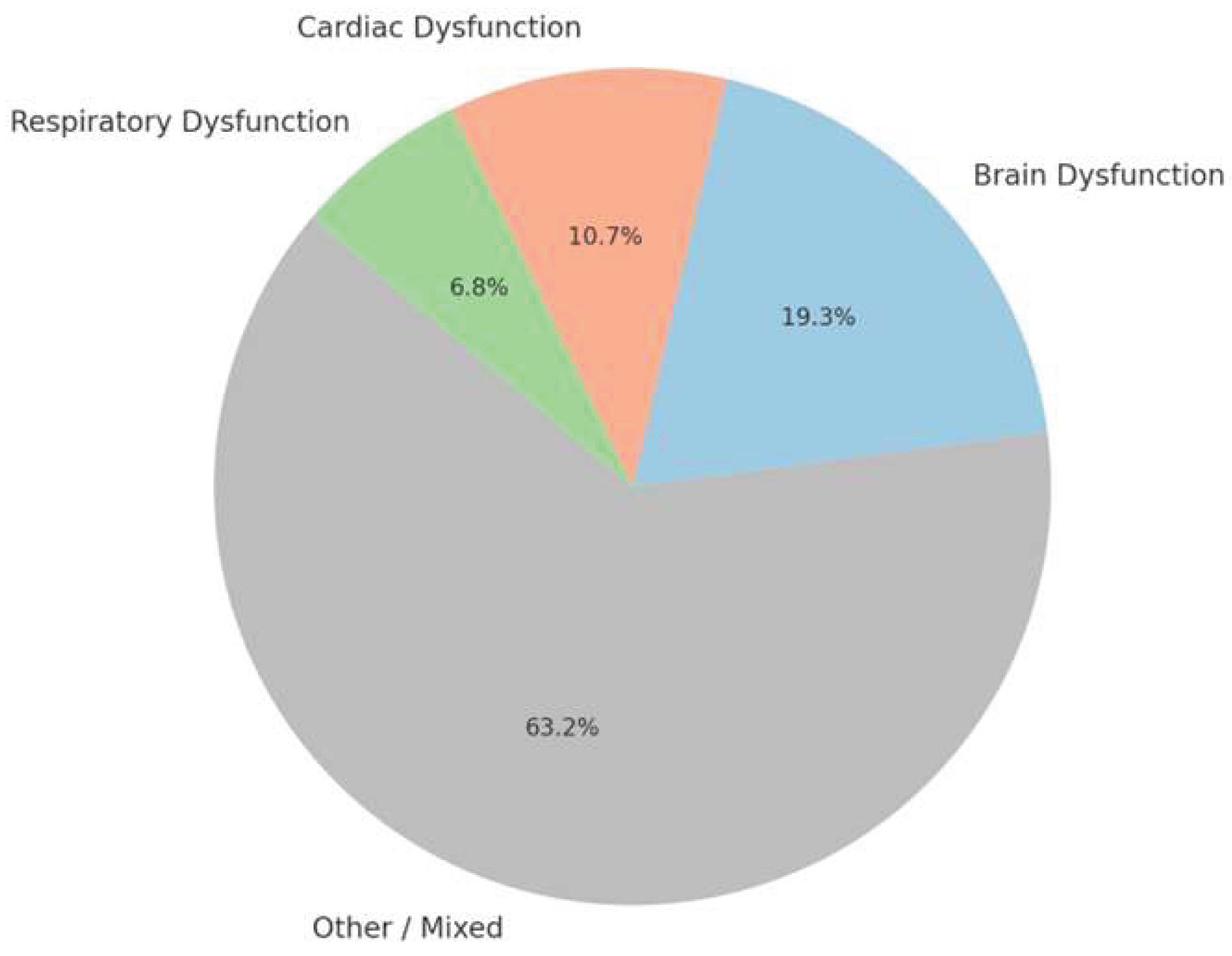
To visually represent the scope and focus of the literature included in this review, Pi chart shown below illustrates the distribution of references across these three domains. As shown, most studies span multiple systems or fall into general or overlapping themes (Other/Mixed). However, a significant portion of the evidence specifically targets brain-related mechanisms (~19%), followed by cardiac (~11%) and respiratory (~7%) dysfunction.
This classification demonstrates that although SUDEP is widely recognized as a multisystem failure, distinct patterns and mechanistic clusters can be extracted from the literature guiding the structure of the following subsections.
3.1. Respiratory Dysfunction in Epilepsy-Related Sudden Deaths
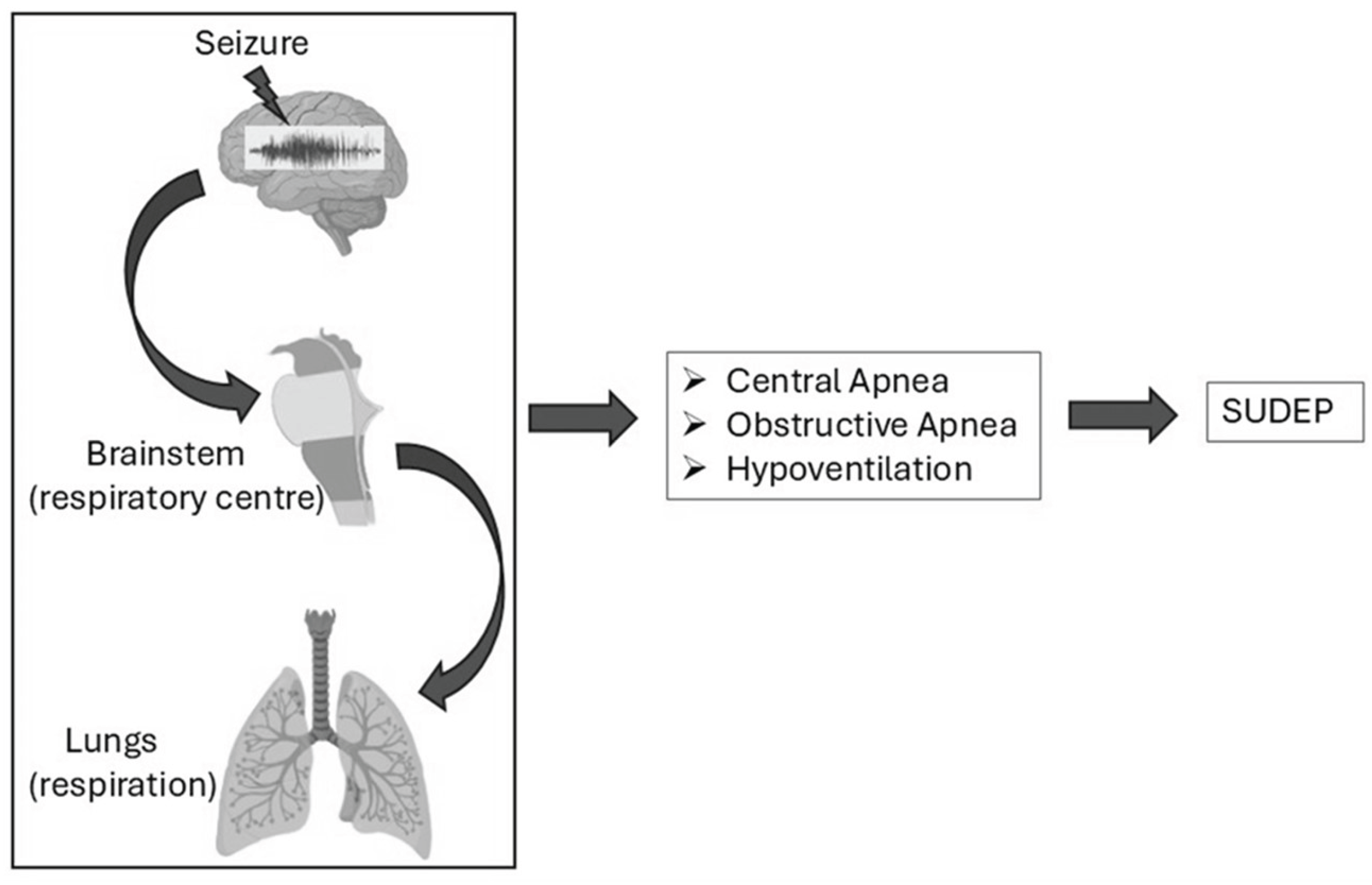
Figure 1.
Figure: Seizure-induced respiratory dysfunction contributing to SUDEP. Seizures can impair brainstem respiratory centers, affecting lung function and leading to central apnea, obstructive apnea, or hypoventilation—key factors associated with SUDEP risk.
Figure 1.
Figure: Seizure-induced respiratory dysfunction contributing to SUDEP. Seizures can impair brainstem respiratory centers, affecting lung function and leading to central apnea, obstructive apnea, or hypoventilation—key factors associated with SUDEP risk.
3.1.1. Central Apnea
Central apnea, defined as the cessation of breathing due to a lack of respiratory effort, is a critical factor in seizure-related respiratory dysfunction and a significant contributor to sudden unexpected death in Epilepsy (SUDEP). [1,2] .While historically SUDEP was primarily attributed to cardiovascular failure, data from epilepsy monitoring units (EMUs) have revealed that central apnea frequently precedes asystole, implicating a primary respiratory mechanism [11] This phenomenon is surprisingly common, affecting nearly half of focal epilepsy patients monitored in EMUs [12,13] Severe and prolonged episodes of central apnea, occurring during (ictal) or immediately after (postictal) seizures, can have fatal consequences, as has been observed in both SUDEP and near-SUDEP cases within these monitoring units [13] Both ictal and postictal apnea are well-documented phenomena, with reports of oxygen desaturation and respiratory arrests during seizures [14] These apneic episodes lead to hypoxemia, a state of low oxygen levels, which further exacerbates neurological damage and significantly elevates the risk of SUDEP.
Seizures disrupt normal respiratory function by impacting key brain regions that regulate respiration, including the brainstem and central autonomic networks. This disruption is thought to occur through several interconnected mechanisms. One key mechanism involves the direct disruption of brainstem respiratory centers. During seizures, the intense electrical activity in the forebrain propagates down to the brainstem, targeting crucial areas such as the pre-Bötzinger complex, which is responsible for generating the rhythmic pattern of breathing, and the parabrachial nucleus, involved in modulating respiratory signals [15] Dysfunction in these critical brainstem regions can directly lead to central apnea. Another proposed mechanism centers on the disruption of central respiratory control, which involves the complex coordination between forebrain structures and brainstem centers, particularly the pons and medulla, which are responsible for autonomic and respiratory regulation [16] In temporal lobe epilepsy (TLE), specific regions within the limbic system and temporal lobe have been strongly associated with both desaturation events and ictal central apnea [13,14] The study by Isabel D. Derera et al. focuses on a specific brainstem region crucial for cardiorespiratory control: the nucleus tractus solitarii (NTS). The NTS is the primary brainstem center for integrating visceral autonomic information, including respiratory and cardiovascular control [17, 18} Prior work has demonstrated that GABAergic neurons within the NTS become hyper excitable in a mouse model of acquired TLE, potentially increasing SUDEP risk [19] This hyper excitability may be a key link between seizure activity and the respiratory and cardiac dysfunction observed in SUDEP.
Specifically, the study examined the role of A-type potassium (K+) currents, mediated by the Kv4 family of K+ channels, in NTS neurons [20,21] These currents are critical for regulating neuronal excitability [22] Alterations in the Kv4.2 subunit have been previously linked to TLE and increased seizure susceptibility [23] Using a pilocarpine-induced status epilepticus (pilo-SE) mouse model of TLE, the researchers found a reduction in A-type K+ current and Kv4 channel function in GABAergic NTS neurons [19,24] This reduction was associated with increased action potential frequency and half-width, indicating enhanced neuronal excitability. This increased excitability, coupled with increased glutamate release onto these neurons [19] likely contributes to the observed hyperexcitability of GABAergic NTS neurons in TLE. This finding is highly relevant to SUDEP because the NTS plays a critical role in cardiorespiratory control. Increased excitability of GABAergic NTS neurons can disrupt these control mechanisms, potentially leading to respiratory dysfunction, including hypoventilation or apnea, and cardiac arrhythmias, both of which are implicated in SUDEP. Moreover, the increased neuronal excitability in the NTS may lower the threshold for spreading depolarization, a wave of neuronal excitation followed by suppression, which, when occurring in the brainstem, can trigger cardiorespiratory collapse and sudden death [25] The study distinguishes itself from previous research on genetic epilepsy models with channelopathies in the NTS [26] by demonstrating that these changes in K+ channel function develop concurrently with epileptogenesis in an acquired epilepsy model, suggesting a direct link between the development of epilepsy and the observed autonomic dysfunction. The observed changes are unlikely due to the direct pharmacological effects of pilocarpine [27] pointing instead to long-term cellular changes related to SE and/or spontaneous recurrent seizures. Several brainstem regions play critical roles in respiratory control and are implicated in SUDEP and ictal central apnea. Studies using quantitative MRI and stereological methods have revealed volume loss in specific medullary regions in SUDEP cases, particularly in the ventrolateral medulla (VLM), which houses key respiratory control centers, including the pre-Bötzinger complex (pre-BötC) [28]. This volume reduction, especially in the rostral medulla, aligns with the location of the pre-BötC, the primary generator of respiratory rhythm. Further investigations have identified specific neuronal changes within the VLM. A significant reduction in somatostatin-expressing neurons, particularly those with synaptic terminal labeling, was observed in the rostral VLM of SUDEP cases [29] Additionally, a reduction in neurokinin 1 receptor (NK1R) expression, another pre-BötC marker, and a decrease in total VLM neuron number were found in the caudal medulla of SUDEP cases These findings, along with increased reticular zone volumes in the caudal medulla of SUDEP cases suggest broader pathology within the ventral respiratory groups, which control respiratory motor output [29,30] Also altered serotonergic signaling has been strongly implicated in SUDEP [31,32] Post-mortem studies have shown reduced 5-HT synthesizing capacity (TPH2 expression) in the rostral VLM and decreased 5-HT reuptake (SERT expression) in the raphe nucleus of SUDEP cases [29] potentially reducing 5-HT availability in the pre-BötC during the vulnerable postictal period. While catecholaminergic neurons (TH-expressing) in the VLM (including C1 adrenergic neurons) influence respiratory drive and arousal [33] and are affected in other conditions associated with sudden death [34] no significant differences in TH neuron number were found in SUDEP cases [29] However, altered neuronal activation, as indicated by c-fos expression, has been observed in medullary autonomic regions, including TH and TPH cells, in SUDEP [24] Finally, changes in galaninergic signaling, which modulates noradrenergic and serotonergic networks and the pre-BötC have also been implicated, with decreased galanin immunolabeling observed in the VLM of SUDEP cases [29,35] Changes in medullary glial cell density, which also contribute to respiratory homeostasis [36,37] have also been observed in SUDEP, specifically in the VLM, medullary surface, and raphe [38,39] These collective findings highlight the complex interplay of neuronal and glial changes within multiple brainstem regions, contributing to respiratory dysfunction and increased SUDEP risk.
Within these regions, the amygdala appears to play a particularly crucial role. Studies involving electrical stimulation of the amygdala have consistently shown that it can trigger central apnea, and there have been documented cases where seizure spread to the amygdala resulted in the onset of apnea [40,41,42] The amygdala's extensive connections to brainstem respiratory centers, including the pre-Bötzinger complex and the pontine respiratory group, underscore its critical role in mediating respiratory rhythms and directly linking seizures to respiratory dysfunction. Supporting this idea, research by Nobis et al. has shown that seizures without central apnea typically involve minimal or no amygdala activity. Further research in rodent models has solidified the amygdala's influence on respiratory control, demonstrating its ability to modulate respiratory efforts under both normal conditions and during seizures [43,44] Research by Lacuey et al. has also shown that stimulation of the amygdala and hippocampus preferentially disrupts inspiration, the active phase of breathing requiring neural coordination, while expiration, a more passive process, is comparatively less affected. This immediate apnea induced by amygdalohippocampal stimulation is believed to result from the inhibition or disruption of brainstem inspiratory neurons, particularly within the pre-Bötzinger complex [45].
The insula, known to regulate both respiratory and cardiac activity, is hypothesized to play a significant role in SUDEP, where respiratory depression and cardiac arrhythmias are considered major direct causes [46]. Insular lobe epilepsy, due to its complex presentation and spread through epileptogenic networks, is often misdiagnosed as temporal, frontal, or parietal lobe epilepsy [47]. This diagnostic challenge is highlighted by studies showing that surgical outcomes for temporal lobe epilepsy are significantly improved when the insula is also addressed [48]. In some cases, the true epileptogenic focus may reside in the insula rather than the temporal lobe, as evidenced by persistent seizure activity after temporal lobectomy and continued propagation of temporal seizure discharges to the insula [48]. Structural damage to the insula has also been observed in many patients with insular lobe epilepsy [49], further supporting its central role in seizures. Direct stimulation studies have identified characteristic features of insular lobe seizures, including preserved consciousness, preictal premonitory symptoms (e.g., feelings of electrical current or burning around the mouth), paresthesias, retrosternal pain, abdominal symptoms, breathing difficulties, and pharyngeal motor and sensory symptoms often accompanied by contralateral hand scratching [48,49,50]. These symptoms reflect the role of insula in autonomic nervous system function, and disruption of cardiovascular and respiratory coordination due to insular epilepsy has been linked to fatal outcomes [51]. Therefore, ictal discharges originating in the cortex can involve the insula, primarily or secondarily leading to cardiac and respiratory dysfunction, central respiratory inhibition and apnea, arrhythmias, and potentially SUDEP.
A seminal study by Ryvlin et al. detailed a characteristic progression to SUDEP in humans, often beginning with a terminal generalized tonic-clonic (GTC) seizure, followed by a brief period of tachypnea, or rapid breathing, which then transitions into bouts of apnea, ultimately culminating in complete respiratory arrest [52] This specific sequence closely mirrors the respiratory patterns observed in established animal models of SUDEP, such as Kcna1−/− mice, where tachypnea can similarly progress to apnea. Tachypnea was particularly prominent in Kcna1−/− mice, occasionally escalating to hyperventilation, characterized by increases in both respiration rate and depth. This was evidenced by elevated tidal volume and peak inspiratory and expiratory flow [53] While under normal physiological conditions, tachypnea or hyperventilation would trigger arousal as a protective mechanism to restore normal breathing, in cases of chemo responsive instability, where the body's response to changes in blood carbon dioxide and oxygen levels is impaired [54,55], this protective mechanism fails, instead leading to apnea. Asphyxia caused by sustained oxygen deprivation can rapidly induce cardiac arrest, with life-threatening consequences occurring within approximately five minutes [56] These combined findings underscore the critical role of central apnea in seizure-related respiratory dysfunction and ultimately in the occurrence of SUDEP. The amygdala appears to be a key target for future therapeutic interventions aimed at mitigating the risk of respiratory failure and SUDEP. Further investigation into the precise mechanisms underlying chemo responsive instability is crucial for developing effective preventative strategies, and a deeper understanding of the neural pathways connecting the amygdala, hippocampus, and brainstem is essential for identifying potential therapeutic targets.
3.1.2. Obstructive Apnea
Airway obstruction, distinct from central apnea, can significantly exacerbate respiratory dysfunction during and after seizures. Phenomena such as laryngospasm, often triggered by recurrent laryngeal nerve hyperactivity during seizures, and postictal immobility, particularly in prone patients hindering head repositioning, can contribute to airway obstruction and subsequent respiratory distress [57,58]. These events can precipitate a deleterious cascade of hypoxia, hypercapnia, and blood gas abnormalities, potentially culminating in fatal cardiac dysfunction and death [59]. The intricate interplay between the amygdala and brainstem in central apnea is of particular interest. Seizures involving the amygdala have been frequently associated with central apnea, as evidenced by both experimental stimulation studies and clinical observations [40,41]. Notably, during these apneic episodes, patients often remain unaware of their respiratory compromise yet retain the capacity for voluntary respiration when prompted, suggesting a disruption of involuntary ventilatory drive rather than a fundamental motor output dysfunction [40]. This hypothesis is further corroborated by studies demonstrating seizure-induced suppression of medullary serotonergic neuronal activity, which is crucial for respiratory rhythmogenesis, leading to transient apnea [60].
Studies utilizing DBA/2J mice, employing survival tracheostomy as a mechanism to circumvent death, have illuminated the pivotal role of obstructive apnea. These investigations identified obstructive events, such as seizure-induced laryngospasm, as a primary instigator of respiratory failure. However, it was observed that even some mice with tracheal tubes succumbed to seizures due to severe spastic contraction of thoracic skeletal muscles, imposing further constraints on respiratory movements. This dual mechanism—laryngospasm inducing obstructive apnea and thoracic spasm inhibiting respiratory effort—accentuates the complex interplay between peripheral respiratory failure and potential central nervous system contributions. Critically, low doses of ketamine/xylazine were found to mitigate thoracic spasm but were neither necessary nor independently sufficient to ensure survival, underscoring that the definitive protective intervention was the elimination of obstructive apnea via tracheostomy [61]. Laryngospasm has thus emerged as a potentially significant factor in the mechanisms underlying sudden unexpected death in epilepsy (SUDEP), particularly within the context of obstructive apnea. Recent evidence gleaned from both animal models and clinical observations underscores the critical role of laryngospasm in SUDEP pathophysiology. For instance, Stewart et al. characterized laryngospasm as a compelling SUDEP biomarker in a rat model, defined by high frequency electromyographic (EMG) artifacts indicative of persistent respiratory effort during obstructive events. Analogous artifacts, observed in EEG and EKG channels, reflect progressive escalations in inspiratory effort, consistent with the physiological manifestations of laryngospasm during seizures. This phenomenon has also been observed in human patients with epilepsy, with reports indicating a diminished awareness or response to breathing distress during seizures [62,63]. This respiratory agnosia emphasizes the imperative need for video-EEG monitoring and polygraphy in patients with refractory epilepsy to detect potentially life-threatening events such as laryngospasm, which may be under-recognized in SUDEP cases.
The synergistic interplay between obstructive apnea resulting from laryngospasm and cardiac manifestations, such as bradycardia, has been postulated as a particularly detrimental combination in the context of SUDEP. Nashef et al. and Stewart et al. have proposed that these two factors may act synergistically to precipitate fatal outcomes. The MORTEMUS study, which meticulously analyzed SUDEP cases, provided evidence suggesting that laryngospasm might have played a crucial role in some deaths, further reinforcing its significance in SUDEP pathogenesis [64]. The study by Stewart et al employed a sophisticated combination of invasive and non-invasive methodologies to investigate the physiological cascade driving respiratory and cardiac dysfunction during seizure activity. Their findings elucidated that seizure-induced increases in recurrent laryngeal nerve (RLN) activity lead to laryngospasm, which can partially or completely occlude the airway, resulting in obstructive apnea. While seizure activity was shown to alter various respiratory parameters, including frequency, amplitude, and variability, and in some instances induce central apnea, the study definitively established that only obstructive apnea caused by laryngospasm is uniquely associated with rapid hypoxemia and the fatal sequence culminating in SUDEP [64]. This finding is of paramount importance as it identifies obstructive apnea as a more critical determinant than central apnea in the context of seizure-related sudden death. The mechanism underlying obstructive apneas lethal potential resides in the profound autonomic response triggered when the respiratory system attempts to overcome a mechanically obstructed airway. This response amplifies the already heightened autonomic tone induced by seizure activity, initiating a cascade of events that ultimately leads to cardiopulmonary collapse. Experimental models further substantiate this understanding. Manual airway occlusion in non-seizing rats replicated a sequence of oxygen desaturation, bradycardia, respiratory arrest, and eventual cardiac failure, closely mirroring the pathophysiology observed during seizure-induced obstructive apnea. These findings underscore that it is the physical airway occlusion, rather than the seizure activity per se, that precipitates the fatal outcome. The stark contrast in outcomes between central and obstructive apnea further accentuates the critical role of autonomic activation. Central apneas are generally considered relatively benign due to their association with a lack of respiratory drive and minimal systemic autonomic activation. Conversely, obstructive apneas are catastrophic due to the pronounced autonomic response elicited by the concerted effort to breathe against a closed airway, compounding the stress induced by the seizure. Unlike voluntary breath-holding, which is characterized by reduced respiratory drive and minimal autonomic involvement, the forced inspiratory effort during obstructive apnea triggers significant autonomic co-activation, leading to a potentially lethal. The physiological basis of this lethal process can be traced to seizure activity propagating along specific neural pathways. The dissemination of seizures from the subiculum to the paraventricular nucleus (PVN) of the hypothalamus and subsequently to medullary regions likely impacts respiratory centers, autonomic nuclei, and laryngeal motor neurons [65]. This intricate neural involvement underscores the centrality of seizure-induced obstructive apnea in SUDEP pathogenesis. In the MORTEMUS study, it is noteworthy that 9 of 10 patients exhibited the most substantial heart rate drop at the termination of seizure activity and prior to terminal apnea onset. This temporal alignment corresponds with the late obstruction phase observed in the rat experiment conducted by Stewart et al, suggesting that obstructive apnea may have played a previously underappreciated role in SUDEP cases and validating the utility of the rat model employed in their study for investigating obstructive apnea. Importantly, the experiments conducted in DBA/2J mice demonstrated that seizure activity in these animals likely does not induce central apnea as the primary mechanism of respiratory arrest. Had central apnea been the predominant cause, tracheostomy would have conferred no survival advantage, even in animals with patent airways. These findings suggest those alterations in serotonergic neuronal activity [66,67,68,69] and/or spreading depolarization act as downstream contributors to respiratory arrest following an obstructive event rather than initiating independent triggers. This intricate interplay between obstructive apnea, central mechanisms, and cardiac consequences underscores the complex pathophysiology of SUDEP. The findings presented here strongly implicate respiratory dysfunction, particularly obstructive apnea driven by laryngospasm, as a critical component in the lethal cascade leading to SUDEP, highlighting the crucial interplay between the lungs, brain, and heart in this devastating outcome.
3.1.3. Hyperventilation
Hyperventilation, a process of increased breathing rate and depth beyond what is physiologically necessary, has a long-established association with seizures, even predating the use of EEG. It was historically the first activation method used in EEG to aid epilepsy diagnosis, activating epileptiform spiking activity, and less frequently, clinical seizures, in susceptible individuals [70,71], While hyperventilation can trigger seizures in up to 50% of patients with generalized epilepsy, particularly children with absence seizures, it is less effective in focal epilepsy [72]. The mechanisms by which hyperventilation triggers seizures are primarily attributed to hypocapnia, the reduction of carbon dioxide (CO2) levels in the blood [73,74]. This leads to respiratory alkalosis, decreased cerebral blood flow, and increased neuronal excitability, making neurons more prone to spontaneous discharges [75,76]. The change in pCO2, rather than the absolute level, appears to be the critical factor, with patient-specific sensitivities to hypocapnia. The autonomic nervous system, particularly the sympathetic division, may also play a role. Studies have shown increased sympathetic responses to hyperventilation in patients with mesial temporal lobe epilepsy, suggesting that sympathetic overactivation may contribute to seizure triggering in some focal epilepsies [77,78,79]. Changes in brain diffusion have also been observed during hyperventilation in patients with temporal lobe epilepsy and hippocampal sclerosis, but not in those without sclerosis or in controls [80]. Hyperventilation is more effective in activating epileptiform activity in generalized epilepsies, particularly in untreated children with absence epilepsies [81], while less common in focal epilepsies [36], hyperventilation can still be useful in some cases, particularly in medically intractable focal epilepsies [9], and may reflect the pathophysiology of the epileptogenic area [82]. The position of patient during hyperventilation (supine vs. sitting) may also influence the occurrence of absence seizures [83] Further research is needed to fully elucidate the complex interplay between hyperventilation, neuronal excitability, autonomic function, and seizure generation in various epilepsy syndromes.
3.1.4. Hypoventilation
Hypoventilation during seizures, characterized by a reduced rate and depth of breathing, results in insufficient carbon dioxide (CO2) expulsion from the body. This physiological derangement can arise from seizure activity impacting brain regions governing respiration, encompassing the brainstem and the autonomic nervous system. The ensuing hypoxemia (low blood oxygen) and hypercapnia (elevated blood CO2) exacerbate the neurological sequelae of seizures, contributing to adverse outcomes and constituting a recognized risk factor for SUDEP [11,14]. Notably, seizure-induced hypoventilation during sleep, compounded by prone positioning and the potential for pulmonary edema, is a frequent observation in monitored SUDEP cases. While congenital central hypoventilation syndrome, a condition marked by impaired ventilatory response and linked to PHOX2B mutations, has been hypothesized as a potential contributor to SUDEP, investigations of SUDEP cohorts have not identified significant PHOX2B variants or mutations in related genes, suggesting the involvement of alternative pathophysiological mechanisms [84,85].
Beyond PHOX2B, other developmental genes influencing respiratory control warrant investigation. HOXA4, crucial for hindbrain development and the formation of the retrotrapezoid nucleus (RTN), a key respiratory chemosensor, represents one such candidate [86] Studies in mouse models with disrupted MeCP2 function, a protein involved in neuronal regulation, have shown complex effects on breathing. While MeCP2 in the caudal, HOXA4-derived hindbrain is essential for survival, it's not solely responsible for normal breathing rate, apnea prevention, or regular breathing patterns. Disrupting MeCP2 function in various parts of the breathing control circuitry, including the PRG and rVRG, can paradoxically lead to hyperventilation, possibly due to imbalances in neurotransmitter signaling [87,88]. Age-dependent changes in breathing rate and other parameters were observed, suggesting progressive deficits in multiple respiratory components and dynamic interactions within the network [89]. MeCP2 in the HoxB1 domain, including the BötC and preBötC, is critical for preventing apneas and irregularity, with partial rescue observed in medullary slices containing the preBötC [90] , the hypoxic ventilatory response (HVR) is also affected by MeCP2, with its function in the HoxA4 domain being necessary for normal HVR, and its deficiency leading to failed transition into proper HVD [86,91]. Although severe breathing irregularity doesn't directly correlate with early lethality, cardiac dysfunction is a major candidate [92] These findings underscore the functionally diverse respiratory network and suggest that Rett syndrome breathing disorders result from interactions within the entire network, not just a single component's malfunction.
Respiratory impairments are frequently documented in epilepsy models, implicating their role as either a causative or contributory factor in SUDEP. Among these impairments, hypoventilation emerges as a prominent candidate, often reported during both ictal and postictal periods. However, the precise underlying mechanisms remain incompletely understood, with prevailing hypotheses implicating seizure-induced disruption of respiratory control centers or alterations in basal breathing patterns associated with epilepsy [14,93,94]. Experimental models provide crucial insights into these complex processes. For instance, a study by Campos et al. evaluated interictal respiratory function in rats with chronic epilepsy induced by the pilocarpine model. Their findings revealed that epileptic rats exhibited impaired compensation for arterial CO2 fluctuations, particularly during hypoventilation, suggesting potential central chemoreceptor dysfunction [95] This finding is corroborated by research on chemoreceptor dysfunction in epilepsy and its potential link to SUDEP [96] This chemoreceptor dysfunction may involve alterations in pH sensitivity or changes in the expression of chemo sensitive ion channels.
In a SUDEP model involving sheep undergoing status epilepticus (SE) induced by bicuculline, Johnson et al. observed hypoventilation in animals that succumbed shortly after SE onset. While the initial study assessed respiratory function primarily through blood gas levels, leaving the precise nature of hypoventilation (obstructive or central) ambiguous [97], a subsequent investigation using tracheotomized sheep revealed increased tidal volume, hypercapnia, and hypoxia, strongly suggesting compromised ventilation or perfusion and further underscoring the role of hypoventilation as a critical factor in SUDEP [98]. This is consistent with more recent research emphasizing the importance of both ventilation and perfusion matching in maintaining adequate gas exchange. Further studies have consistently documented hypoventilation during seizures, associating it with an augmented mortality risk. Hypoventilation, often occurring alongside central or obstructive apnea, represents a hallmark of ictal and postictal respiratory dysfunctions, potentially triggered by seizure-activation of cardiorespiratory control centers [99,100,101,102,103]. These impairments are hypothesized to originate from disturbances in adenosinergic and serotonergic pathways, brainstem spreading depolarization, or autonomic dysregulation [104] These disruptions can affect neuronal excitability and synaptic transmission within respiratory control networks.
Animal models have been instrumental in understanding how seizures impact respiration and contribute to SUDEP. The DBA/1 and DBA/2 mouse audiogenic seizure (AGS) models recapitulate seizure-induced respiratory arrest (S-IRA), which is influenced by serotonin (5-HT) and norepinephrine (NE) signaling [69,100,105,106]
Genetic mouse models, such as those with Dravet syndrome-related SCN1A mutations or KCNA1 deletions, demonstrate peri-ictal and postictal respiratory dysfunction, including hypoventilation, apnea, and abnormal responses to CO2 [53,107]. Large animal models, like sheep and baboons, further support the role of respiratory dysfunction and pulmonary complications in SUDEP [98,108].
A study of Wistar audiogenic rats (WARs), a model of epileptic seizures, revealed elevated resting blood pressure and heart rate, along with increased sympathetic cardiovascular modulation, independent of seizures [109,110]. These rats also showed neuroendocrine abnormalities, including adrenal hyperplasia, altered ACTH circadian cycle, and higher corticosterone levels, potentially contributing to the increased sympathetic tone [111,112]. While baroreflex function was preserved in these young WARs, possibly as a compensatory mechanism, these animals exhibit increased susceptibility to status epilepticus and mortality following pilocarpine-induced seizures [113], highlighting a heightened cardiovascular risk. This sustained sympathetic overactivity, linked to life-threatening cardiovascular events, aligns with the prominent role of autonomic discharges in SUDEP [114,115].
A study using the tetanic toxin (TeTX) model of chronic temporal lobe epilepsy (TLE) in rats investigated the relationship between seizure occurrence and sleep-wake states (SOVs) [116]. Surprisingly, a disproportionate number of seizures (47%) occurred during REM sleep, despite rats spending only 6.1% of their time in this state. Conversely, few seizures initiated during NREM sleep (6.8% of seizures during 39% of time [117]. A significant proportion (34%) also occurred during wakefulness with high hippocampal theta activity (Wakeθ), suggesting a link between theta rhythms and seizure susceptibility. This contrasts with the prevailing view that NREM favors seizures and REM/active wake prevents them [117,118,119,120]. While the mechanisms underlying this increased seizure susceptibility during theta oscillations, especially in REM, remain to be fully elucidated, potential factors include compromised dentate gyrus function, increased hippocampal excitability, and altered interneuron activity leading to potassium buildup and runaway excitation [121,122].
A study of Scn1aR1407X/+ mice, a Dravet syndrome (DS) model, revealed high premature mortality associated with increased spontaneous generalized tonic-clonic seizures (GTCSs) prior to death [123,124,125]. Both seizures and SUDEP in these mice occurred more frequently at night, though during their active period, unlike human SUDEP which is often associated with sleep [53,126]. While sleep state could be a contributing factor, other variables like circadian influences, motor activity, light, or body temperature may also play a role [117]. A surge in seizure frequency was observed in the 24 hours preceding SUDEP, though the pattern varied between individuals. Surprisingly, a ketogenic diet (KD) significantly reduced mortality without affecting seizure frequency or severity [52,127]. This suggests that the KD's protective effect is likely not due to seizure reduction but potentially through preventing seizure propagation to brainstem cardiorespiratory control centers [128]. or by mitigating epilepsy-related cytological changes [129]. These findings challenge the notion that uncontrolled GTCSs are the primary SUDEP risk factor
A retrospective study of naturally occurring deaths in a captive baboon colony identified key features relevant to SUDEP. Epileptic baboons (SZ) died younger than controls (CTL) and predominantly in adolescence/young adulthood, while CTL baboons died in adulthood [130]. While infection was a common cause of death in both groups, a subgroup of epileptic baboons with unknown cause of death (SZ-UKN) exhibited distinct pathological findings, including pulmonary edema, serosanguinous bronchial secretions, and chronic myocardial fibrosis, like human SUDEP [131]. These findings were less common in other groups. The SZ-UKN group also had more frequent seizures and longer epilepsy duration, though not statistically significant, likely due to their longer lifespan compared to the SZ group with known causes of death (SZ-KN). The study suggests a possible natural SUDEP model in primates, given the similarities in age, sex distribution, seizure history, and pathological findings to human SUDEP [132,133,134]. Further electrophysiological monitoring is needed to investigate potential ictal or interictal arrhythmias in these baboons.
Beyond these mechanisms, the pilocarpine-induced SE model in mice offers insights into SUDEP risk in temporal lobe epilepsy (TLE), the most common form of epilepsy. While some mice die acutely after pilocarpine-induced SE, likely due to the initial insult, those surviving the first week develop spontaneous seizures and exhibit a high rate of sudden unexpected death, mirroring SUDEP in humans [135]. This model, distinct from genetic epilepsy models with inherent channelopathies [25,136]. highlights the role of reactive neuroplasticity in seizure generation and SUDEP. Specifically, pilocarpine-induced seizures trigger ion channel and synaptic reorganization in cortical structures [137] and, importantly, in brainstem neurons, particularly within the NTS (nucleus of the solitary tract). Increased glutamate release and neuronal excitability in GABAergic NTS neurons are observed after SE, potentially disrupting autonomic control of thoracic and abdominal viscera [138,139]. This increased activity, eliminated by glutamate receptor blockade, suggests long-term synaptic function changes associated with epileptogenesis. These changes might involve altered presynaptic release properties or new synapse formation [140]. Such increased synaptic excitation of GABAergic NTS neurons could inhibit parasympathetic motor output and disinhibit sympathetic output, impacting respiratory centers [141] Furthermore, this heightened excitability may predispose NTS neurons to depolarization block and spreading depression, potentially leading to cardiorespiratory collapse and SUDEP, especially during seizures that spread to the brainstem or during strong reflex activation [25,142,143].
Adding to the complexity of seizure-related respiratory compromise, studies using the kainic acid model have revealed a critical role for seizure-induced laryngospasm and obstructive apnea in sudden death. The kainic acid (KA) model is valuable for studying acute temporal lobe epilepsy-like seizure [57,144]. However, it does not perfectly model SUDEP because it induces seizures and status epilepticus in otherwise healthy brains. Variability in death mechanisms following KA administration, even at the same dose, makes it difficult to determine which outcomes truly mimic SUDEP. While the ability of the KA model to replicate acute seizure-related deaths is unique, these findings require validation in chronic epilepsy models [145].
The interplay between hypoventilation and other SUDEP risk factors has also been explored. For example, studies have investigated the interaction between hypoventilation and cardiac arrhythmias [146]. suggesting that the combination of these two factors can significantly increase the risk of sudden death Furthermore, the impact of genetic factors on hypoventilation susceptibility in epilepsy is an area of ongoing research [147]. Thus, hypoventilation stands as a critical factor linking respiratory dysfunction and SUDEP, warranting further investigation through both clinical and experimental epilepsy models. A deeper understanding of the mechanisms underlying seizure-induced hypoventilation, its interaction with other physiological systems, and its contribution to the lethal cascade in SUDEP is crucial for developing effective preventative strategies aimed at mitigating this devastating outcome.
3.2. Brain Dysfunction in Epilepsy-Related Sudden Deaths
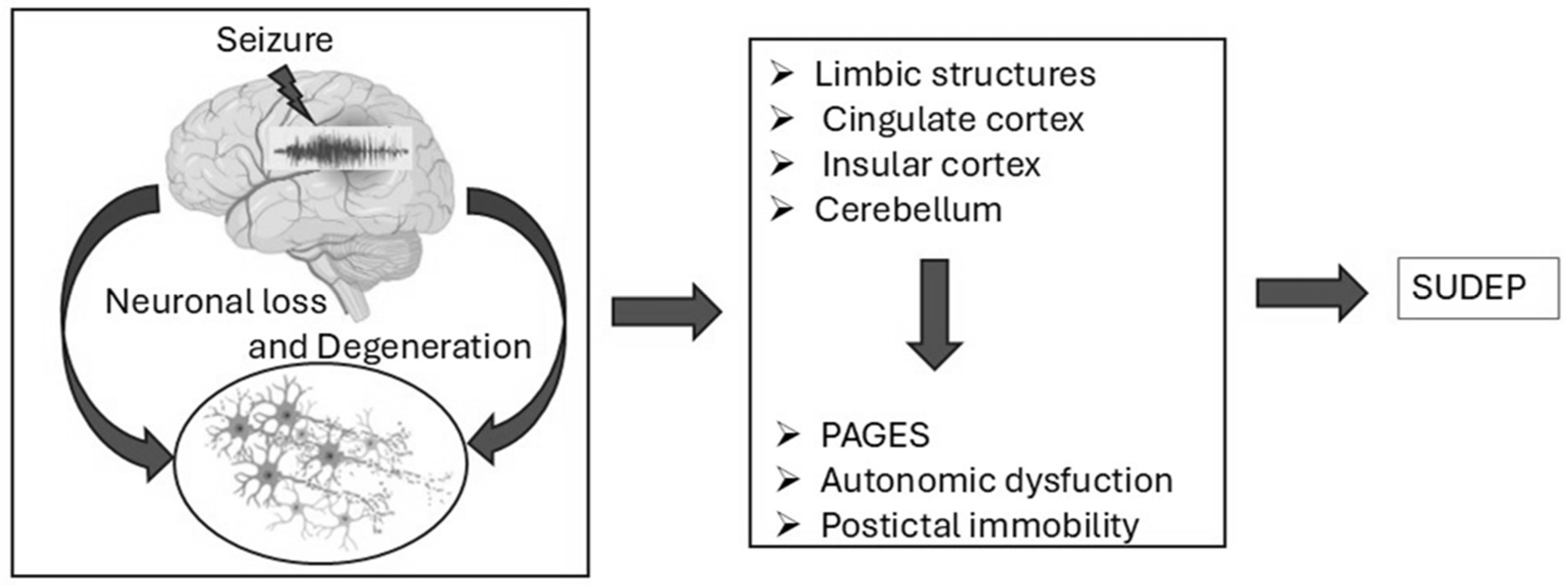
Figure 2.
Schematic representation of potential mechanisms leading to SUDEP. Seizures contribute to neuronal loss and degeneration, particularly in regions such as the limbic system, cingulate cortex, insular cortex, and cerebellum. This leads to pathological outcomes including PAGES, autonomic dysfunction, and postictal immobility, ultimately increasing SUDEP risk.
Figure 2.
Schematic representation of potential mechanisms leading to SUDEP. Seizures contribute to neuronal loss and degeneration, particularly in regions such as the limbic system, cingulate cortex, insular cortex, and cerebellum. This leads to pathological outcomes including PAGES, autonomic dysfunction, and postictal immobility, ultimately increasing SUDEP risk.
Brain dysfunction plays a significant role in epilepsy-related sudden deaths and SUDEP. Prolonged and recurrent seizures can lead to neurodegeneration and brain damage over time, impairing cognitive functions, memory, and overall brain health, ultimately influencing an individual's susceptibility to SUDEP and other complications. This neurodegeneration affects key brain regions involved in autonomic control, directly impacting the interplay between the brain, lungs, and heart—the central theme of SUDEP pathophysiology [148,149].
3.2.1. Neurotransmitter Imbalances
Neurotransmitters, the chemical messengers of brain, are fundamental to the regulation of neuronal function. Imbalances in these signaling molecules are critically implicated in the pathophysiology of epilepsy. Specifically, a reduction in gamma-aminobutyric acid (GABA), the principal inhibitory neurotransmitter, coupled with an elevation in glutamate, the primary excitatory neurotransmitter, contributes significantly to seizure activity. These disruptions not only initiate and propagate seizures but also exert profound effects on overall brain health and function. Numerous genetic mutations associated with altered GABA metabolism have been linked to developmental and epileptic encephalopathies (DEEs), with majority of these mutations observed in genes encoding GABA receptors, such as GABRA, GABRB, and GABRG (encoding GABAA receptors), and GABBR (encoding GABAB receptors). Mutations affecting any stage of GABA metabolism can disrupt the delicate balance between neuronal excitation and inhibition, predisposing individuals to epilepsy. Dysregulation of GABAergic pathways can result in neuronal hyperexcitability, thereby increasing susceptibility to seizures and associated autonomic dysfunctions that may contribute to SUDEP. Therapeutic interventions targeting GABAergic signaling aim to restore this crucial balance and mitigate seizure activity. For instance, valproic acid (VPA) exerts its effects by inhibiting GABA degradation and enhancing its synthesis, effectively increasing synaptic GABA levels. Similarly, vigabatrin (VGB), an irreversible inhibitor of GABA transaminase (GABA-T), prevents GABA breakdown, while tiagabine (TGB) blocks GABA transporter 1 (GAT1), reducing GABA reuptake into neurons and glial cells. These mechanisms collectively enhance synaptic GABA concentrations, providing a stabilizing influence on neuronal networks. In epilepsy syndromes linked to GABAA receptor mutations, such as DEEs, enhancing receptor function has demonstrated therapeutic potential. Identifying these specific genetic mutations facilitates the implementation of precision medicine approaches, offering opportunities for targeted treatments [150]. These findings underscore the critical importance of addressing GABAergic dysregulation not only for seizure control but also for its potential influence on autonomic and respiratory regulation, both of which are crucial factors in SUDEP pathophysiology. Addressing neurotransmitter imbalances, particularly impaired GABAergic signaling, constitutes a vital component of SUDEP research, emphasizing the interconnectedness of brain excitability, respiratory control, and cardiovascular stability. As research continues to elucidate these complex mechanisms, therapeutic strategies tailored to individual neurochemical profiles of epilepsy patients may emerge, potentially reducing SUDEP risk and improving overall disease management.
Glutamate, the predominant excitatory neurotransmitter, and GABA maintain the critical equilibrium between excitation and inhibition in the central nervous system. Disruption of this balance represents a hallmark of epileptic activity and may contribute to SUDEP pathophysiology. Elevations in extracellular glutamate levels have been consistently reported in epileptogenic regions during ictal, peri-ictal, and interictal phases. Microdialysis studies in focal epilepsies have revealed significantly higher glutamate concentrations in epileptogenic regions compared to non-epileptogenic regions or baseline measures, suggesting region-specific dysregulation of excitatory signalling [151,152,153,154,155]. This excessive glutamate is closely linked to dysfunction of the glutamate-glutamine cycle. Under normal physiological conditions, glutamate released into the synaptic cleft is efficiently taken up by astrocytes, converted to glutamine-by-glutamine synthetase, and subsequently recycled back to neurons. However, studies have demonstrated reduced glutamine synthetase expression and activity in epileptogenic hippocampi, resulting in insufficient glutamate clearance and heightened excitotoxicity [153,156]. This dysfunction can exacerbate neuronal hyperexcitability and promote seizure activity, ultimately increasing the risk of seizure-induced mortality, including SUDEP. While glutamate promotes excitation, GABA counteracts this effect through its inhibitory actions. This imbalance between delayed GABA-mediated inhibition and preictal glutamate surges may create a hyperexcitable neuronal milieu, increasing the propensity for prolonged seizures and seizure-related cardiorespiratory dysfunction, both potential precursors to SUDEP. The interplay between glutamate and GABA is further complicated by the presence of mitochondrial dysfunction in epilepsy. Several studies have noted opposing relationships between GABA levels and mitochondrial function in mesial temporal lobe epilepsy and neocortical epilepsy, suggesting differential metabolic regulation between epileptic subtypes [154]. The dysregulation of glutamate and GABA can directly impact neuronal excitability and indirectly influence autonomic and cardiorespiratory control, mechanisms critically implicated in SUDEP. Elevated glutamate levels may precipitate seizures that disrupt brainstem regulatory centers, thus impairing respiration and cardiac function. Similarly, inadequate GABAergic inhibition during seizures could fail to counteract excessive excitation, exacerbating the likelihood of fatal outcomes. Furthermore, glutamate-mediated excitotoxicity and oxidative stress could contribute to neurodegenerative changes that compromise vital autonomic functions over time [153,157]. Recent studies have also implicated alterations in glutamate transporter expression and function in SUDEP, further highlighting the importance of glutamate homeostasis in this condition.
Abnormalities in serotonin (5-HT) neurotransmission have been increasingly implicated in SUDEP pathogenesis. This association arises from serotonin's crucial role in modulating respiratory and arousal functions, both of which are integral to brainstem activity and are demonstrably disrupted in SUDEP. Evidence supporting the involvement of 5-HT in SUDEP, while largely circumstantial, is nonetheless compelling. 5-HT neurotransmission participates in seizure suppression, with studies demonstrating that 5-HT decreases seizure frequency and severity in both animal models and human epilepsy patients [158]. Many commonly prescribed antiepileptic drugs are known to elevate extracellular 5-HT concentrations, which may partially explain their therapeutic efficacy. Reduced 5-HT levels observed post-seizure in animal models further suggest that serotonin dysfunction may contribute to postictal respiratory suppression [159]. Animal studies provide further evidence for the protective role of serotonin against seizure-induced respiratory arrest. Pretreatment with selective serotonin reuptake inhibitors (SSRIs) has been shown to prevent seizure-related respiratory arrest in DBA mice, a well-established model of SUDEP [67,160]. Similarly, mice deficient in 5-HT2C receptors exhibit a heightened incidence of audiogenic seizures and are prone to respiratory arrest following seizures [161]. These findings underscore the importance of 5-HT signaling in maintaining respiratory stability during and after seizures. In human studies, the administration of SSRIs has been associated with reduced oxygen desaturation during seizures, a recognized biomarker for SUDEP risk. This observation highlights the clinical relevance of enhancing 5-HT activity to mitigate SUDEP risk. The parallels drawn between SUDEP and sudden infant death syndrome (SIDS), which is also linked to brainstem 5-HT system abnormalities, further strengthens the hypothesis of serotonin involvement in SUDEP pathogenesis [46,162]. Although direct evidence definitively linking 5-HT dysfunction to human SUDEP remains limited, the convergence of animal and clinical data strongly suggests that serotonin plays a critical role in regulating the complex interplay between seizures, respiratory function, and arousal. This positions the 5-HT system as a crucial focus for both understanding and mitigating SUDEP. Serotonin (5-HT) dysfunction has been implicated not only in SUDEP but also in SIDS, highlighting overlapping mechanisms related to arousal and respiratory failure. Infants who succumb to SIDS often exhibit defects in the 5-HT system, including reduced binding of 5-HT1A receptor ligands in the raphe nuclei, a brainstem region vital for serotonergic regulation of respiration and arousal [163]. Furthermore, an increased number of immature 5-HT neurons and decreased 5-HT levels in the medulla have been observed in SIDS victims, suggesting developmental delays in serotonergic maturation [162,164]. A key similarity between SIDS and SUDEP lies in the context of the fatal events: both conditions involve impaired arousal and respiratory responses during states of central nervous system depression—sleep in SIDS and the postictal state in SUDEP. In SIDS, these deficits are attributed to delayed maturation of 5-HT neurons and diminished neuronal firing during sleep. In SUDEP, transient dysfunction of 5-HT neurons is hypothesized to occur due to seizure activity propagating into the brainstem, thereby disrupting serotonergic modulation of respiratory and arousal mechanisms. Recent research has investigated the role of specific 5-HT receptor subtypes, such as 5-HT1A and 5-HT2A receptors, in SUDEP, suggesting that these receptors may be promising therapeutic targets. Importantly, preventive measures in SIDS, such as the “Back to Sleep” campaign advocating supine sleeping positions, and have significantly reduced its incidence, further implicating positional factors in the role of 5-HT dysfunction. Similarly, in SUDEP, targeting serotonergic dysfunction through interventions such as SSRIs has shown promise in mitigating postictal respiratory depression and seizure-induced death in animal models [67,160]. These parallels underscore the crucial role of 5-HT in regulating responses to external stressors during vulnerable states, thus reinforcing the need to explore serotonergic pathways for therapeutic intervention in SUDEP. In summary, dysregulation of these key neurotransmitter systems disrupts the intricate communication pathways within the brain, directly impacting respiratory and cardiac control and contributing to the lethal cascade observed in SUDEP. This emphasizes the crucial interplay between the brain, lungs, and heart in this complex condition.
3.2.2. Neurodegeneration and Brain Damage
Structural and functional alterations, particularly in the brainstem, hippocampus, amygdala, and insular cortex, have been linked to increased SUDEP risk. Brainstem atrophy, especially when extending into the midbrain, impairs autonomic control, a critical factor in SUDEP [148,165]. Seizure spread to the amygdala may contribute to respiratory depression via its functional connection with the medullary respiratory network [40], directly impacting lung function. Structural changes, such as increased gray matter volume in the right anterior hippocampus/amygdala and parahippocampus, and decreased volume in the posterior thalamus, further suggest compromised oxygen regulation in individuals at higher SUDEP risk, again highlighting the brain-lung connection. Intrinsic or acquired insular damage in refractory epilepsy patients is also a potential risk factor, as insular dysfunction correlates with autonomic nervous system abnormalities and peri-ictal respiratory or cardiac impairments, contributing to SUDEP [166,167]. Neuroimaging studies in temporal lobe epilepsy (TLE) patients have further identified altered functional connectivity in brain regions involved in autonomic, respiratory, and cardiac regulation, potentially serving as biomarkers for SUDEP risk [148,168].
Specifically, patients at high risk for SUDEP exhibit reduced connectivity in a subnetwork encompassing the thalamus, brainstem, anterior cingulate cortex, putamen, and amygdala, and increased connectivity in regions like the medial/orbital frontal cortex, insula, limbic areas, subcallosal cortex, brainstem, thalamus, caudate, and putamen [168]. The posterior thalamus, crucial for oxygen sensing and breathing (the lung-brain connection), demonstrates disrupted links with the brainstem in high-risk patients, potentially contributing to respiratory failure [169]. Reduced thalamic–cingulate connectivity further implicates disruptions in cardiorespiratory and blood pressure regulation (linking brain, heart, and lungs), which could lead to prolonged hypotension, a possible SUDEP mechanism [170]. The putamen, integral to autonomic and motor regulation, shows reduced connectivity with the cingulate cortex, impairing critical ANS communication [171] Similarly, diminished connectivity between the amygdala and brainstem in high-risk patients may result in sustained apnea or failure to recover from seizure-induced hypoventilation 148, 172].
Several other factors contribute to this complex interplay. The prone position exacerbates respiratory compromise, as impaired arousal and compromised brainstem autoresuscitation mechanisms prevent patients from clearing airway obstructions caused by bedding [173]. Serotonin dysfunction has been implicated in SUDEP, with defective serotonin-producing neurons in epilepsy patients reducing ventilatory responses to rising CO₂ levels and compromising arousal mechanisms, leading to fatal outcomes during airway obstruction [44,60,174]. Endogenous adenosine, which increases during seizures, provides anticonvulsant effects but also inhibits cardiac and respiratory functions. Prolonged seizures combined with impaired adenosine clearance may trigger excessive adenosine release, contributing to SUDEP [101,175,176,177,178]. Similarly, seizures may activate endogenous opioids, producing central hypoventilation and postictal apnea, which heighten SUDEP risk [179].
Seizure spread to the amygdala has been specifically linked to respiratory arrest due to disruption of the medullary respiratory network, leading to loss of spontaneous breathing and awareness of dyspnea, further emphasizing the brain's pivotal role in SUDEP pathophysiology. Ion channel gene mutations, including those in sodium (e.g., SCN1A, SCN8A) and potassium channels (e.g., KCNA1, KCNQ2), disrupt autonomic control and postictal recovery, increasing SUDEP risk [180,181]. Central hypoventilation and apnea may also result from the seizure-induced release of endogenous opioids, with polymorphisms in the ARRB2 gene amplifying the desensitization of brainstem opioid receptors, leading to severe postictal apnea and higher SUDEP susceptibility. Genetic variants affecting glutamatergic and GABAergic neurotransmission can also disrupt the excitatory-inhibitory balance, influencing seizure severity and centrally mediated autonomic dysfunction, thereby increasing the risk of SUDEP [179]. These findings highlight the genetic underpinnings of autonomic dysfunction and their pivotal role in seizure-related respiratory compromise. Structural neuroimaging studies have revealed reduced posterior thalamic gray matter and increased right hippocampal and amygdala volumes in high-risk SUDEP patients [182]. Significant volume loss in the dorsal mesencephalon and damage to central autonomic control regions, such as the limbic system, disrupt critical autonomic and respiratory regulation [183,184] Seizure-related autonomic dysfunction, including impaired cardiac and respiratory control, is consistently linked to SUDEP risk [185].
Functional connectivity (FC) studies in TLE patients at high risk of SUDEP reveal significant disruptions in brain networks involved in autonomic and respiratory regulation. Reduced FC has been identified in key regions, including the thalamus, brainstem, ACC, bilateral putamen, and left amygdala, all of which play critical roles in regulating breathing, cardiac function, and blood pressure [168,169]. The posterior thalamus, essential for oxygen sensing and relaying respiratory signals, exhibits disrupted connections with the brainstem, aligning with volumetric studies showing reduced gray matter in the thalamus of high-risk SUDEP patients [186]. The putamen shows diminished connectivity with the ACC, potentially impairing communication between motor and autonomic pathways [187] Similarly, reduced connectivity between the amygdala and brainstem is concerning, given the amygdala's influence on respiratory nuclei and its association with terminal apnea in SUDEP cases [41,172]. Conversely, high-risk SUDEP patients also show enhanced FC, primarily involving connections between the medial/orbital frontal cortex, insula, and limbic regions (amygdala and hippocampus). These changes could represent compensatory or maladaptive adaptations in autonomic regulation pathways [168].
Temporal lobe epilepsy (TLE), with seizure foci in temporal lobe structures such as the hippocampus, amygdala, entorhinal cortex, and subiculum [188,189], is characterized by significant neurodegeneration and brain damage. Hippocampal neurodegeneration, a hallmark of TLE, leads to structural and functional impairments [190]. Neurodegenerative changes in TLE include aberrant mossy fiber sprouting [191] granule cell dispersion and astrogliosis [192], all of which contribute to altered neural circuitry and seizure propagation. The neurodegenerative processes in epilepsy are intricately linked to SUDEP. Recurrent seizures and associated excitotoxicity drive neuronal loss, metabolic dysfunction, and progressive structural damage, particularly in the hippocampus. This damage is often exacerbated by ictal and interictal activity, contributing to oxidative stress and inflammatory responses [193]. In TLE, higher seizure frequency and prolonged seizure duration correlate with severe hippocampal atrophy, which impairs the brain's ability to regulate critical autonomic functions, including respiration and cardiac activity [194]. TLE patients are particularly vulnerable to cognitive deficits, with the nature and severity of these deficits depending on the location and extent of underlying neuropathology [195,196]. In TLE, hippocampal sclerosis (HS) is a common neuropathological feature associated with widespread cognitive impairments, including verbal and visual memory deficits, language difficulties, and post-ictal psychosis, particularly when both hemispheres are affected [197,198]. Ictal and interictal activities further exacerbate these cognitive disturbances by contributing to excitotoxic damage, inflammatory responses, and disruptions in neural network integrity.
This brain damage plays a pivotal role in the pathophysiology of SUDEP, as the hippocampus and other affected regions are involved in the central regulation of breathing and cardiovascular functions. Dysregulation in these systems due to neurodegeneration can result in impaired responses to seizure-induced apnea or cardiac arrhythmias, which are critical events in SUDEP. Thus, understanding the mechanisms of neurodegeneration in epilepsy provides essential insights into the vulnerability of brain to fatal outcomes such as SUDEP.
3.2.3. Genetic Mutations and Aberrant Neurogenesis
Mutations in the Kv1.1 (Kcna1) subunit of voltage-gated potassium channels cause significant brain damage and neurodegeneration in epilepsy, with direct implications for SUDEP. In humans, these mutations are associated with temporal lobe epilepsy (TLE) and episodic ataxia type I [195]. Mouse models recapitulate these phenotypes, exhibiting aberrant postnatal neurogenesis in the dentate gyrus, contributing to hippocampal enlargement—an early hallmark of epileptogenesis [199]. This aberrant neurogenesis stems from intrinsic defects in progenitor cell depolarization and extrinsic excitatory inputs, such as NMDA receptor activation and dysregulated GABA signaling, accelerating cellular proliferation [200]. This hyperactive neurogenesis integrates immature neurons into hippocampal circuits, promoting aberrant synchronization and recurrent seizures, destabilizing network excitability, and potentially driving further neurodegeneration and elevating SUDEP risk [197].
System x-c (Sxc), a sodium-independent cystine-glutamate antiporter, a heterodimeric complex composed of xCT and 4F2 chains, primarily located on astrocytes, and possibly other glial cells and neurons [201] is implicated in this process. Upregulated during epileptogenesis, Sxc modulates extracellular glutamate, linking excitotoxicity, inflammation, and oxidative stress to neurodegeneration [196]. In a Kcna1 knockout mouse model, genetic deletion of Sxc decreased aberrant neurogenesis in the dentate gyrus, preventing hippocampal enlargement despite persistent severe epilepsy [202]. Increased xCT protein expression has been observed in resected hippocampi from patients with mesial temporal lobe epilepsy [203] and xCT-deficient mice have shown increased chemoconvulsant seizure threshold. This role is further supported by studies investigating the effects of Sxc manipulation on epileptogenesis. For instance, xCT deletion prolongs the latency to the first spontaneous seizure and reduces the number of spontaneous seizures after self-sustained status epilepticus (SSSE). Similarly, in the corneal kindling model, xCT deletion decreases the number of focal to bilateral tonic-clonic seizures (FBTCS) and lowers mean seizure scores, although these effects are modest. In the pentylenetetrazol (PTZ) kindling model, xCT deletion reduced the percentage of mice that became fully kindled [204]. Furthermore, pharmacological inhibition of Sxc with sulfasalazine (SAS) reduced seizures in mice that had undergone pilocarpine-induced SE but not in control mice, suggesting that Sxc plays a role in modulating the seizure circuit resulting from epileptogenesis [205] These findings suggest that Sxc contributes to the long-term changes associated with epilepsy development and progression, rather than simply influencing acute seizure activity. While xCT deletion reduced microglial and astrocytic activation after SSSE [205].
3.2.4. Role of Adenosine and Purinergic Signaling
Mesial temporal lobe epilepsy (MTLE) is characterized by significant brain damage, including irreversible biochemical and structural changes in the hippocampus and neocortical regions, contributing to epilepsy pathophysiology and increased SUDEP risk. Dysregulation of extracellular ATP and adenosine is a critical factor in epilepsy progression and neurodegeneration. High-frequency neuronal firing during seizures increases ATP and adenosine levels, which act on adenosine receptors to modulate neuronal excitability. The adenosine A2A receptor (A2AR), expressed in the hippocampus and other brain regions, plays a crucial role in controlling synaptic transmission and plasticity [206]. A2AR activation increases glutamate release and impairs glutamate uptake, leading to an imbalance in excitatory synaptic transmission that may contribute to the hyperexcitability seen in epilepsy. A2AR activation in astrocytes and microglia is also important for modulating neuronal network activity during intense or prolonged seizures. Astrocytes, through A2AR signaling, can influence synaptic glutamate levels and are involved in memory formation [168]. In epilepsy models, including kainate-induced seizures, A2AR levels are upregulated in astrocytes, suggesting a role in the maladaptive neuroplasticity and neurodegeneration associated with epilepsy [207]. These findings indicate that A2AR, by modulating glutamate dynamics, contributes to the progressive neuronal damage and dysfunction seen in MTLE, which may also increase SUDEP susceptibility [208]. Neurodegeneration in epilepsy, particularly MTLE, is thus driven by a combination of altered purine signaling, astrocyte and microglial activation, and glutamate dysregulation. The upregulation of A2AR in both neurons and glial cells contributes to network hyperexcitability and sustained neuronal damage, central to the development of drug-resistant epilepsy and the heightened risk of SUDEP.
In sclerotic TLE, characterized by tonic-clonic convulsions causing progressive brain damage and exacerbating seizures [209] the adenosine modulation system plays a significant role [210]. Increased neuronal activity, especially during seizures, elevates extracellular adenosine levels. While the A1 adenosine receptor (A1R) system is generally considered neuroprotective, reducing seizures and protecting against neuronal damage [211] repeated seizures reduce A1R density and function, limiting its effectiveness [212,213]. This loss of A1R function contributes to progressive brain damage in epilepsy [214]. Conversely, A2AR is upregulated in sclerotic brain regions in epilepsy models and TLE patients [215]. A2AR activation increases glutamate release, enhances NMDA receptor activity, and promotes neuroinflammation [207,216,217]. These effects contribute to pathological changes in the hippocampus during epileptic episodes. While A2AR antagonism has shown neuroprotective effects in various brain conditions [206,218], however the exact role in seizure-induced neurodegeneration remains uncertain [219,220]. The dysregulation of adenosine receptors, particularly A1R and A2AR, contributes to chronic brain damage and increased SUDEP risk.
3.2.5. Mossy Fiber Sprouting, Dynorphin, and CDKL5 Deficiency
Aberrant mossy fiber sprouting, where mossy fibers project into the inner molecular layer, is observed in TLE. This sprouting, originating from the dentate gyrus and associated with hilar mossy cell loss, is thought to exacerbate epileptic activity [190], contributing to seizure perpetuation and neuronal damage. Perturbation of glutamatergic signaling, particularly through AMPA receptors and mossy fiber sprouting, plays a critical role in excitotoxic damage and neurodegeneration in epilepsy and may contribute to SUDEP.
Dynorphin A (1–17), an opioid peptide normally found in the hippocampal mossy fiber system, exhibits altered distribution in TLE. In TLE specimens, Dyn-IR structures appear in the molecular and granule cell layers, showing distinct distribution patterns. The extent of these aberrant Dyn-IR structures correlates with the degree of cell loss in the polymorph and CA3 regions, as well as granule cell dispersion. This sprouting of mossy fibers and their axon collaterals suggests the formation of new, potentially excitatory circuits. These reorganized fibers, containing dynorphin, could contribute to recurrent excitatory circuits that facilitate synchronous firing and epileptiform activity. This aligns with observations in experimental epilepsy models, where mossy fiber sprouting is implicated in seizure propagation and brain damage, potentially contributing to SUDEP [221]
Studies using Emx1- and CamK2α-derived Cdkl5 conditional knockout (cKO) hemizygous male mice have revealed recurrent spontaneous seizures and aberrant mossy fiber sprouting in the hippocampus of Emx1-derived Cdkl5 cKO mice [221,222]. Increased frequencies of spontaneous and miniature excitatory postsynaptic currents in dentate granule cells of the Emx1-cKO mice further support the epileptic phenotype, suggesting that hyperexcitability in glutamatergic neurons plays a role in the seizures observed in CDKL5 deficiency disorder (CDD) [223,227]. Thus, CDKL5 disruption in glutamatergic neurons not only leads to spontaneous seizures but also enhances excitatory signaling, potentially fostering neurodegeneration and exacerbating SUDEP risk.
These diverse mechanisms—genetic mutations, aberrant neurogenesis, altered purinergic signaling, mossy fiber sprouting, and CDKL5 deficiency—converge on a common pathway: neurodegeneration and disruption of neuronal networks crucial for autonomic control. This disruption directly impacts the delicate balance between the brain, lungs, and heart, making individuals with epilepsy, particularly those with TLE and related conditions, more vulnerable to SUDEP. The interplay of these factors underscores the complexity of SUDEP pathophysiology and highlights the need for further research to develop targeted therapies.
3.3. Cardiac Dysfunction in Epilepsy-Related Sudden Deaths
Seizures, particularly generalized tonic-clonic seizures, can significantly disrupt autonomic regulation, leading to cardiac arrhythmias such as tachycardia, bradycardia, or asystole [236,232]. The insular cortex and brainstem, critical regions for autonomic control, are frequently impacted during seizures. The insular cortex manages signals related to heart rate and blood pressure, while the brainstem coordinates vital autonomic reflexes. When seizures affect these areas, they cause imbalances between sympathetic and parasympathetic activity. Overactivation of the sympathetic system results in rapid heart rates (tachycardia), whereas parasympathetic overstimulation can lead to dangerously slow heart rates (bradycardia) or temporary cardiac arrest (asystole), all of which substantially increase the risk of cardiac arrest during or following seizures [35]. As depicted in Figure 1, seizures disrupt autonomic regulation, provoking arrhythmias such as asystole, atrial fibrillation, and QT interval abnormalities, which are key contributors to SUDEP.
Figure 3.
Seizure-induced cardiac arrhythmias and their role in SUDEP. Seizures can trigger cardiac dysfunction, including asystole, atrial fibrillation, tachycardia, and QT interval abnormalities, all of which may contribute to the risk of sudden unexpected death in epilepsy.
Figure 3.
Seizure-induced cardiac arrhythmias and their role in SUDEP. Seizures can trigger cardiac dysfunction, including asystole, atrial fibrillation, tachycardia, and QT interval abnormalities, all of which may contribute to the risk of sudden unexpected death in epilepsy.
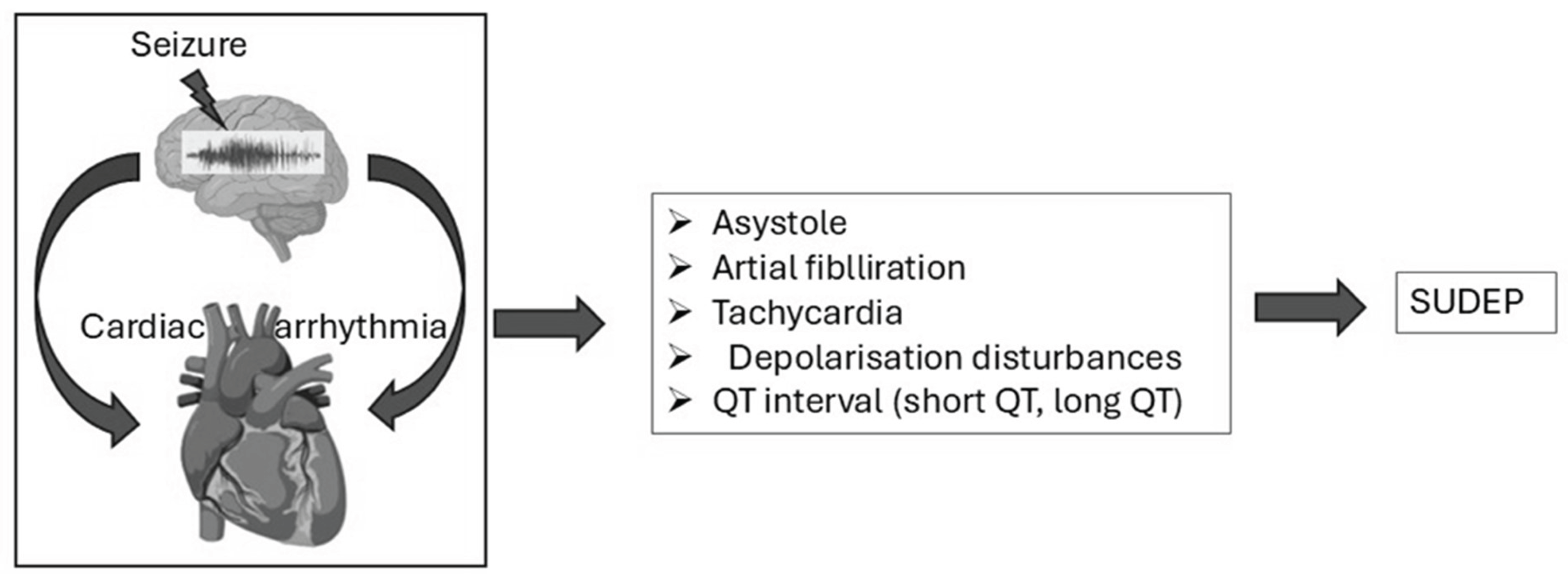
Seizures also cause increase in catecholamines including adrenaline and noradrenaline, which can temporarily impair heart function in a condition termed "neurogenic stunned myocardium." This transient myocardial dysfunction arises due to the excessive catecholamine release during the seizures. The resulting calcium overload in cardiac myocytes reduces contractility and predisposes the heart to the ventricular arrhythmias. Patients experiencing frequent or drug-resistant seizures are particularly susceptible to this phenomenon and increase their SUDEP risk [237]. These effects are further aggravated by the genetic predispositions such as mutations in ion channel genes (e.g., SCN5A, KCNQ1), which compromise the electrical stability of cardiac cells, thereby increasing the risk of arrhythmias [228,231]. Genetic factors also play a critical role in associating epilepsy to the cardiac risk. Mutations in genes like SCN1A is commonly associated with Dravet syndrome, which disrupt neuronal and cardiac excitability and increase the likelihood of the prolonged seizures and autonomic instability. This instability may lead to bradycardia and the other life-threatening arrhythmias [238,239]. Similarly, KCNA1 mutations, which impair potassium ion flow, can provoke early after depolarizations and arrhythmias that affect both epilepsy and non-epilepsy populations. These insights emphasize the need for routine cardiac monitoring in epilepsy patients, particularly those at the elevated risk. Tools like continuous electrocardiogram (ECG) monitoring, wearable seizure detection devices and heart rate variability (HRV) analysis provide valuable information about autonomic dysfunction. Early interventions such as beta-blockers to manage sympathetic overactivity or antiepileptic drugs to reduce seizure frequency, can reduce arrhythmia risks and improve the overall outcomes [240,241].
3.3.1. Autonomic Nervous System Dysregulation and SUDEP Risk
The ANS plays an important role in the connection between the seizures and SUDEP. Seizures that affect critical areas such as the insular cortex and brainstem disrupt the delicate balance between the sympathetic and parasympathetic branches of the ANS. This imbalance results in irregular heart rhythms including tachycardia and bradycardia, which substantially elevate the risk of SUDEP [230,233]. A key factor contributing to this autonomic instability during the seizures is impaired baroreceptor reflex sensitivity. This dysfunction amplifies sympathetic overactivity while reducing parasympathetic recovery, which makes individuals more prone to arrhythmias. Baroreflex gain analysis during interictal, and postictal phases holds promise as a tool for identifying the patients at higher risk of autonomic instability and SUDEP [242]. Excessive sympathetic activation during seizures can lead to elevated heart rates, vasoconstriction and increased blood pressure, which place significant strain on the cardiovascular system. While such responses are typically adaptive under normal conditions, their increased activation during seizures results in chaotic heart rhythms that increase the risk of arrhythmias and cardiac arrest [231,240]. Conversely, parasympathetic over activation during the seizure, particularly those which originate in the temporal lobe can cause a dramatic slowing of the heart, occasionally leading to asystole. These abrupt and extreme shifts in the autonomic output increase the chances of fatal outcomes [235,242]
3.3.2. Genetic Contributions to Autonomic Dysregulation
Genetic factors significantly intensify the relationship between epilepsy, ANS dysregulation and cardiac risks. Mutations in the ion channel genes such as SCN1A and KCNA1 can interfere with the excitability of both neurons and the cardiac cells. SCN1A mutations, often associated to Dravet syndrome, disrupt sodium channel function and lead to prolonged seizures and the autonomic instability that increase the risk of bradycardia and other arrhythmias [238]. Similarly, KCNA1 mutations effect potassium channels can compromise the heart's electrical stability and raise the likelihood of early after-depolarizations and arrhythmias. The release of the stress hormones such as epinephrine and norepinephrine during the seizures amplifies these autonomic imbalances. The increase in these hormones increase the risk of the arrhythmias particularly in individuals with drug resistant epilepsy and frequent seizures [234,241,243] The combination of the hormonal fluctuations and the genetic predisposition create a state of increased susceptibility to SUDEP.
3.3.3. Monitoring and Biomarkers of ANS Dysregulation
Understanding role of the ANS in SUDEP has highlighted the importance of monitoring the autonomic function. Tools-like heart HRV analysis provide insights into the ANS stability. A decrease in HRV during and after the seizures signals impaired autonomic regulation is associated with an elevated risk of the SUDEP. Low HRV reflects increased sympathetic activity or inadequate parasympathetic recovery, both of which are critical contributors to cardiac dysfunction in epilepsy patients [244,245]. For high-risk patients, monitoring autonomic biomarkers such as HRV and catecholamine levels can serve as early indicators of developing cardiac dysfunction. This can facilitate timely interventions including the beta blocker therapy to control the sympathetic overactivity and the targeted antiepileptic treatments to reduce the seizure frequency. Such personalized strategies may help lower the risk of SUDEP and improve overall outcomes for the epilepsy patients [240].
3.3.4. Postictal Cardiac Dysfunction and the Role of Continuous Monitoring
3.3.4.1. Postictal Phase and Cardiac Health
The postictal phase, the period immediately following a seizure, is an important time for assessing cardiac health. During this phase, autonomic function is often severely compromised, which leads to the cardiac complications such as prolonged QT intervals, bradyarrhythmias and other arrhythmogenic conditions that contribute to elevate the risk of SUDEP [246,247]. These issues arise from persistent sympathetic overactivation or parasympathetic withdrawal, both of which destabilize cardiac rhythms and predispose patients to fatal arrhythmias.
Postictal generalized EEG suppression (PGES), characterized by reduced brain activity after seizures, is closely associated to brainstem dysfunction. Figure 4 illustrates the mechanistic relationship between PGES, brainstem dysfunction, and SUDEP. PGES leads to impaired autonomic control via brainstem dysfunction, which then contributes to both respiratory failure and cardiac instability, ultimately increasing the risk of SUDEP.
PGES-induced brainstem dysfunction affects both respiratory and cardiac functions, increasing SUDEP risk. Monitoring postictal cardiac and respiratory markers may aid in identifying high-risk individuals. The medulla oblongata, which controls important cardiorespiratory functions and becomes impaired during PGES leads to bradycardia, asystole, or central apnea. These mechanisms play a significant role in SUDEP by disrupting heart rhythm during the vulnerable postictal phase [232,233]. Additionally, patients experiencing severe PGES mostly exhibit electrocardiographic markers of cardiac instability, such as QT dispersion and T-wave alternans. These markers provide valuable insight into cardiac vulnerability. Monitoring such parameters during the postictal phase may enable early identification of high-risk individuals [248]
3.3.4.2. Importance of Postictal Monitoring
Real-time monitoring of cardiac function during the postictal phase gives important insights into seizure-related cardiac abnormalities. Ambulatory electrocardiogram (ECG) devices enable the detection of arrhythmic events such as heart rate variability disturbances, bradyarrhythmias or QT prolongation, which might not be apparent during routine clinical evaluations [249,250]. Early detection of these disruptions lowers the risk of SUDEP and enables timely treatments. It provides details about heart dysfunction by combining postictal ECG data with advanced autonomic evaluations like HRV analysis. The balance between sympathetic and parasympathetic activity is examined by HRV analysis, and it is linked to the risk of SUDEP and the severity of seizures [251].
3.3.4.3. Emerging Role of Wearable Monitoring Devices
The management of cardiac risks associated with epilepsy is being transformed by wearable technologies, such as adhesive chest sensors and wrist-worn ECG monitors. Continuous cardiac monitoring in real-time environments is made possible by these technologies, allowing for the early diagnosis of autonomic changes or seizure-induced arrhythmias [252,253]. Data collected by these devices can be transmitted in real-time to healthcare providers, facilitating prompt intervention when abnormalities are identified [254,255]. Recent advancements have enabled the development of multi-parameter systems that integrate cardiac, respiratory, and neurological data to provide a comprehensive view of patient health [256]. These systems are particularly beneficial for identifying subtle postictal changes that may otherwise go unnoticed [257]. The integration of wearable devices into routine epilepsy management has the potential to significantly enhance long-term outcomes for patients and reduce the risk of SUDEP [258]. The incorporation of machine learning algorithms into wearable technology has further improved their capacity to detect cardiac risks associated with seizures [259]. These systems can analyze multi-parameter data, such as heart rate variability and motion patterns, to predict seizure onset and autonomic instability [260]. Devices such as Empatica’s Embrace2 have shown effectiveness in clinical assessments, offering real-time notifications to carers and healthcare professionals, thereby facilitating a prompt response [254,261].
3.3.5. Mechanisms Linking Respiratory and Cardiac Dysfunction in SUDEP
3.3.5.1. Overlap Between Respiratory and Cardiac Dysfunction
Respiratory failure is recognized as an important factor in SUDEP, although its interplay with cardiac dysfunction causes a dangerous overlap that markedly increases risk during and after seizures [261]. Respiratory abnormalities resulting from seizures, including central apnea, hypoventilation, and obstructive airway events, lead to hypoxia and hypercapnia, imposing significant strain on the cardiovascular system [262,263,264]. These alterations aggravate arrhythmogenic conditions by increasing sympathetic activity and suppressing parasympathetic recovery, hence elevating the chance of fatal cardiac consequences [265]. Central apnea, frequently triggered by brainstem impairment during convulsions, significantly affects cardiorespiratory stability [230]. The medulla oblongata, responsible for regulating respiration and heart rate, becomes compromised, resulting in concurrent respiratory arrest and bradyarrhythmias [232]. This dual dysfunction is a characteristic mechanism in SUDEP, as hypoxia resulting from apnea might increase the likelihood of fatal arrhythmias [266,267]. Obstructive and central apneas during seizures lead to severe hypoxemia, which increases sympathetic overactivation and raises arrhythmogenic risk [268]. Myocardial stress from hypoxemia diminishes oxygen supply to heart tissue, elevating the risk of fatal ventricular arrhythmias [235,269]. These findings emphasize the significance of continuous monitoring of respiratory and cardiac functions to enhance understanding of SUDEP pathogenesis. Ongoing monitoring of these characteristics may facilitate the identification of high-risk episodes and suggest preventive measures [270].
3.3.5.2. Neurotransmitter Release and Autonomic Imbalance
The excessive release of neurotransmitters like serotonin, glutamate, and catecholamines during seizures significantly disrupts the balance between the respiratory and cardiac systems [271]. Serotonin, for instance, influences respiratory drive and autonomic tone, and its dysregulation during seizures may lead to both apnea and arrhythmias [272]. Glutamate-induced excitotoxicity disrupts brainstem function, impairing central respiratory control and triggering sympathetic overdrive [273,274]. Catecholamine surges, which frequently occur during generalized seizures, increase myocardial stress by increasing heart rate and blood pressure. This increased sympathetic activity raises the likelihood of arrhythmias and simultaneously increases oxygen demand, resulting in a detrimental cycle of cardiorespiratory instability [258,266].
3.3.5.3. Importance of Multimodal Monitoring
The intricate relationship between respiratory and cardiac dysfunction emphasises the need for integrated monitoring strategies in epilepsy management. Multimodal systems that evaluate respiratory parameters, including oxygen saturation and end-tidal CO₂ levels, along with ECG and HRV, offer a complete picture of the patient's cardiorespiratory health [275]. These devices allow clinicians to identify apnea-induced arrhythmias and intervene effectively to avoid negative consequences [276]. The integration of wearable and portable monitoring devices into clinical practice provides an effective way of obtaining real-time data. Devices that monitor respiratory and cardiac parameters are essential for identifying at-risk individuals in healthcare settings. These tools enhance the accuracy of SUDEP risk classification and provide personalized management strategies to improve patient safety through continuous observation [230].
4. Discussion
Sudden Unexpected Death in Epilepsy (SUDEP) is a devastating outcome for individuals with epilepsy, especially those with uncontrolled generalized tonic-clonic seizures (GTCS). The phenomenon is now widely acknowledged to result from a complex interaction between the brain, heart, and lungs, with failures in one or more of these systems contributing to sudden fatality. Emerging evidence strongly suggests that respiratory compromise, autonomic imbalance, and seizure propagation into critical brainstem centers form a lethal cascade culminating in SUDEP.
Among the most critical events observed in SUDEP is central apnea, characterized by a cessation of respiratory effort due to neural suppression. Monitoring data from epilepsy units have shown that central apnea often precedes cardiac arrest, suggesting it may be an initiating event rather than a secondary consequence of seizure activity [11,13]. Brainstem structures such as the pre-Bötzinger complex and the nucleus tractus solitarius (NTS) play pivotal roles in maintaining respiratory rhythm. Seizure propagation into these areas can suppress their function, thereby triggering central apnea [14,15]. Specifically, hyperexcitability of GABAergic neurons in the NTS has been demonstrated in pilocarpine-induced epilepsy models, which is directly linked to impaired cardiorespiratory regulation and increased SUDEP vulnerability [19,24]. This hyperexcitability also reduces the threshold for spreading depolarization, a pathological neuronal event that can lead to acute respiratory and cardiac shutdown [25].
In addition to central mechanisms, obstructive apnea, particularly resulting from laryngospasm, is increasingly recognized as a major factor in SUDEP. Seizure-induced laryngospasm leads to intense inspiratory efforts against a closed airway, resulting in progressive hypoxemia and autonomic overload. Animal models have provided compelling evidence of this mechanism: Stewart et al. identified high-frequency EMG signatures of laryngospasm in both EEG and EKG recordings during seizures, closely correlating with SUDEP-like outcomes [61]. Further supporting this, human cases also show signs of obstructive apnea with absent arousal responses during the postictal period [62,63]. Importantly, in models where tracheostomy bypasses the upper airway, survival increases significantly, confirming that mechanical airway occlusion is often the immediate cause of death [64].
Cardiac dysfunction also plays a crucial role. Seizures induce profound shifts in autonomic balance, with sympathetic overactivation or vagal dominance capable of inducing bradycardia, asystole, or ventricular arrhythmias [59,60]. Genetic mutations, particularly in SCN1A and KCNA1, compromise ion channel function in both neurons and cardiomyocytes. These mutations are seen in high-risk epilepsy syndromes such as Dravet syndrome and lead to reduced cardiac excitability, increasing the likelihood of arrhythmias during or after seizures [52,53].
A unique focus has been placed on the amygdala, a limbic structure that interfaces directly with brainstem respiratory centers. Electrical stimulation of the amygdala in both animal and human studies has been shown to induce central apnea, with patients sometimes unaware of the breathing arrest, a condition referred to as respiratory agnosia [40,41,42]. These findings support the hypothesis that seizure spread to the amygdala can silence involuntary respiratory drive without affecting voluntary motor output, greatly complicating postictal recovery.
Another area of growing interest is the serotonergic system, particularly the role of 5-HT in maintaining postictal respiration. In mouse models, pretreatment with SSRIs significantly reduces the incidence of seizure-induced respiratory arrest [66,67]. Human studies show reduced oxygen desaturation during seizures in patients taking SSRIs, suggesting a protective effect [160]. This parallels observations in sudden infant death syndrome (SIDS), which is also associated with impaired serotonergic function in the medulla, a region crucial for arousal and respiratory control [162,163,164].
Moreover, the integration of these dysfunctions creates a vulnerable scenario, especially in high-risk contexts like sleep, the prone position, or when patients are unattended. The transition from a seizure to a fatal event often involves a combination of impaired arousal, autonomic instability, and compromised respiratory control, all occurring within a critical timeframe where intervention is difficult.
5. Conclusions
The overall understanding is that SUDEP is not the result of a single system failure but rather a cascading interplay between central apnea, obstructive airway events, and cardiac dysregulation, each potentially exacerbated by seizure-induced neural suppression or genetic susceptibility. Interventions aimed at reducing seizure spread to brainstem structures, enhancing serotonergic tone, and mitigating airway obstruction hold promise in reducing SUDEP risk. Continued research into neural cardiorespiratory circuits, genetic biomarkers, and pharmacological modulation is essential to develop effective, personalized preventive strategies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, M.Y.M., writing—original draft preparation, M.Y.M.; B.A.S., S.Z, writing—review and editing, M.Y.M., A.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| SUDEP. | Sudden Unexpected Death in Epilepsy |
| EEG | Electroencephalography |
| GTCS | Generalized Tonic-Clonic Seizures |
| ANS | Autonomic Nervous System |
| PGES | Postictal Generalized EEG Suppression |
| RLN | Recurrent Laryngeal Nerve |
| HVR | Hypoxic Ventilatory Response |
| TLE | Temporal Lobe Epilepsy |
| DEE | Developmental and Epileptic Encephalopathy |
| GABA | Gamma-Aminobutyric Acid |
| VPA | Valproic Acid |
| VGB | Vigabatrin |
| TGB | Tiagabine |
| SSRI | Selective Serotonin Reuptake Inhibitor |
| SIDS | Sudden Infant Death Syndrome |
| 5-HT | Serotonin (5-Hydroxytryptamine) |
| Sxc | System x-c (Cystine-Glutamate Antiporter) |
| SAS | Sulfasalazine |
| A1R | Adenosine A1 Receptor |
| A2AR | Adenosine A2A Receptor |
| MTLE | Mesial Temporal Lobe Epilepsy |
| SE | Status Epilepticus |
| FBCTS | Focal to Bilateral Tonic-Clonic Seizures |
| CDKL5 | Cyclin-Dependent Kinase-Like 5 |
| KD | Ketogenic Diet |
| HRV | Heart Rate Variability |
References
- Devinsky, O.; Bundock, E.; Hesdorffer, D.; Donner, E.; Moseley, B.; Cihan, E.; Hussain, F.; Friedman, D. Resolving Ambiguities in SUDEP Classification. Epilepsia 2018, 59, 1220–1233. [Google Scholar] [CrossRef] [PubMed]
- Sillanpää, M.; Shinnar, S. Long-Term Mortality in Childhood-Onset Epilepsy. N. Engl. J. Med. 2010, 363, 2522–2529. [Google Scholar] [CrossRef]
- Sveinsson, O.; Andersson, T.; Carlsson, S.; Tomson, T. The Incidence of SUDEP: A Nationwide Population-Based Cohort Study. Neurology 2017, 89, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Nashef, L.; Walker, F.; Allen, P.; Sander, J.W.; Shorvon, S.D.; Fish, D.R. Apnoea and Bradycardia During Epileptic Seizures: Relation to Sudden Death in Epilepsy. J. Neurol. Neurosurg. Psychiatry 1996, 60, 297–300. [Google Scholar] [CrossRef]
- Hesdorffer, D.C.; Tomson, T.; Benn, E.; Sander, J.W.; Nilsson, L.; Langan, Y.; Walczak, T.S.; Beghi, E.; Brodie, M.J.; Hauser, A.; For the ILAE Commission on Epidemiology; Subcommission on Mortality. Combined Analysis of Risk Factors for SUDEP: Combined SUDEP Analysis. Epilepsia 2011, 52, 1150–1159. [Google Scholar] [CrossRef]
- Appleton, R.E. Mortality in Paediatric Epilepsy. Arch. Dis. Child. 2003, 88, 1091–1094. [Google Scholar] [CrossRef] [PubMed]
- Sveinsson, O.; Andersson, T.; Mattsson, P.; Carlsson, S.; Tomson, T. Clinical Risk Factors in SUDEP: A Nationwide Population-Based Case-Control Study. Neurology 2020, 94, e419–e429. [Google Scholar] [CrossRef]
- Harden, C.; Tomson, T.; Gloss, D.; Buchhalter, J.; Cross, J.H.; Donner, E.; French, J.A.; Gil-Nagel, A.; Hesdorffer, D.C.; Smithson, W.H.; Spitz, M.C.; Walczak, T.S.; Sander, J.W.; Ryvlin, P. Practice Guideline Summary: Sudden Unexpected Death in Epilepsy Incidence Rates and Risk Factors: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2017, 88, 1674–1680. [Google Scholar] [CrossRef]
- Saxena, A.; Jones, L.; Shankar, R.; McLean, B.; Newman, C.G.; Hamandi, K. Sudden Unexpected Death in Epilepsy in Children: A Focused Review of Incidence and Risk Factors. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1064–1070. [Google Scholar] [CrossRef]
- Chahal, C.A.A.; Salloum, M.N.; Alahdab, F.; Gottwald, J.A.; Tester, D.J.; Anwer, L.A.; So, E.L.; Murad, M.H.; St Louis, E.K.; Ackerman, M.J.; Somers, V.K. Systematic Review of the Genetics of Sudden Unexpected Death in Epilepsy: Potential Overlap With Sudden Cardiac Death and Arrhythmia-Related Genes. J. Am. Heart Assoc. 2020, 9, e012264. [Google Scholar] [CrossRef]
- Ryvlin, P.; Nashef, L.; Lhatoo, S.D.; Bateman, L.M.; Bird, J.; Bleasel, A.; Boon, P.; Crespel, A.; Dworetzky, B.A.; Høgenhaven, H. Incidence and Mechanisms of Cardiorespiratory Arrests in Epilepsy Monitoring Units (MORTEMUS): A Retrospective Study. Lancet Neurol. 2013, 12, 966–977. [Google Scholar] [CrossRef]
- Michalak, Z.; Obari, D.; Ellis, M.; Thom, M.; Sisodiya, S.M. Neuropathology of SUDEP: Role of Inflammation, Blood-Brain Barrier Impairment, and Hypoxia. Neurology 2017, 88, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Lacuey, N.; Zonjy, B.; Hampson, J.P.; Rani, M.R.S.; Zaremba, A.; Sainju, R.K.; Gehlbach, B.K.; Schuele, S.; Friedman, D.; Devinsky, O.; Nei, M.; Harper, R.M.; Allen, L.; Diehl, B.; Millichap, J.J.; Bateman, L.; Granner, M.A.; Dragon, D.N.; Richerson, G.B.; Lhatoo, S.D. The Incidence and Significance of Periictal Apnea in Epileptic Seizures. Epilepsia 2018, 59, 573–582. [Google Scholar] [CrossRef]
- Bateman, L.M.; Li, C.-S.; Seyal, M. Ictal Hypoxemia in Localization-Related Epilepsy: Analysis of Incidence, Severity and Risk Factors. Brain 2008, 131, 3239–3245. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.C.; Ellenberger, H.H.; Ballanyi, K.; Richter, D.W.; Feldman, J.L. Pre-Bötzinger Complex: A Brainstem Region That May Generate Respiratory Rhythm in Mammals. Science 1991, 254, 726–729. [Google Scholar] [CrossRef]
- Richter, D.W.; Smith, J.C. Respiratory Rhythm Generation In Vivo. Physiology 2014, 29, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Affleck, V.S.; Coote, J.H.; Pyner, S. The Projection and Synaptic Organisation of NTS Afferent Connections with Presympathetic Neurons, GABA and nNOS Neurons in the Paraventricular Nucleus of the Hypothalamus. Neuroscience 2012, 219, 48–61. [Google Scholar] [CrossRef]
- Browning, K.N.; Travagli, R.A. Plasticity of Vagal Brainstem Circuits in the Control of Gastric Function: Receptor Trafficking in the DVC. Neurogastroenterol. Motil. 2010, 22, 1154–1163. [Google Scholar] [CrossRef]
- Derera, I.D.; Delisle, B.P.; Smith, B.N. Functional Neuroplasticity in the Nucleus Tractus Solitarius and Increased Risk of Sudden Death in Mice with Acquired Temporal Lobe Epilepsy. eNeuro 2017, 4, ENEURO.0319–17.2017. [Google Scholar] [CrossRef]
- Bailey, T.W.; Hermes, S.M.; Whittier, K.L.; Aicher, S.A.; Andresen, M.C. A-Type Potassium Channels Differentially Tune Afferent Pathways from Rat Solitary Tract Nucleus to Caudal Ventrolateral Medulla or Paraventricular Hypothalamus. J. Physiol. 2007, 582, 613–628. [Google Scholar] [CrossRef]
- Strube, C.; Saliba, L.; Moubarak, E.; Penalba, V.; Martin-Eauclaire, M.-F.; Tell, F.; Clerc, N. Kv4 Channels Underlie A-Currents with Highly Variable Inactivation Time Courses but Homogeneous Other Gating Properties in the Nucleus Tractus Solitarii. Pflugers Arch. 2015, 467, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Villa, C.; Combi, R. Potassium Channels and Human Epileptic Phenotypes: An Updated Overview. Front. Cell. Neurosci. 2016, 10, 81. [Google Scholar] [CrossRef]
- Lee, H.; Lin, M.A.; Kornblum, H.I.; Papazian, D.M.; Nelson, S.F. Exome Sequencing Identifies de Novo Gain of Function Missense Mutation in KCND2 in Identical Twins with Autism and Seizures That Slows Potassium Channel Inactivation. Hum. Mol. Genet. 2014, 23, 3481–3489. [Google Scholar] [CrossRef]
- Su, T.; Cong, W.D.; Long, Y.S.; Luo, A.H.; Sun, W.W.; Deng, W.Y.; Liao, W.P. Altered Expression of Voltage-Gated Potassium Channel 4.2 and Voltage-Gated Potassium Channel 4-Interacting Protein, and Changes in Intracellular Calcium Levels Following Lithium-Pilocarpine-Induced Status Epilepticus. Neuroscience 2008, 157, 566–576. [Google Scholar] [CrossRef]
- Aiba, I.; Noebels, J.L. Spreading Depolarization in the Brainstem Mediates Sudden Cardiorespiratory Arrest in Mouse SUDEP Models. Sci. Transl. Med. 2015, 7, 282ra46. [Google Scholar] [CrossRef]
- Vanhoof-Villalba, S.L.; Gautier, N.M.; Mishra, V.; Glasscock, E. Pharmacogenetics of KCNQ Channel Activation in 2 Potassium Channelopathy Mouse Models of Epilepsy. Epilepsia 2018, 59, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Römermann, K.; Bankstahl, J.P.; Löscher, W.; Bankstahl, M. Pilocarpine-Induced Convulsive Activity Is Limited by Multidrug Transporters at the Rodent Blood-Brain Barrier. J. Pharmacol. Exp. Ther. 2015, 353, 351–359. [Google Scholar] [CrossRef]
- Patodia, S.; Tachrount, M.; Somani, A.; Scheffer, I.; Yousry, T.; Golay, X.; Sisodiya, S.M.; Thom, M. MRI and Pathology Correlations in the Medulla in Sudden Unexpected Death in Epilepsy (SUDEP): A Postmortem Study. Neuropathol. Appl. Neurobiol. 2021, 47, 157–170. [Google Scholar] [CrossRef]
- Patodia, S.; Somani, A.; O’Hare, M.; Venkateswaran, R.; Liu, J.; Michalak, Z.; Ellis, M.; Scheffer, I.E.; Diehl, B.; Sisodiya, S.M. The Ventrolateral Medulla and Medullary Raphe in Sudden Unexpected Death in Epilepsy. Brain 2018, 141, 1719–1733. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Zhao, Y.; Wong-Riley, M.T.T.; Ju, G.; Liu, Y. Synaptic Relationship between Somatostatin- and Neurokinin-1 Receptor-Immunoreactive Neurons in the Pre-Bötzinger Complex of Rats. J. Neurochem. 2012, 122, 923–933. [Google Scholar] [CrossRef]
- Murugesan, A.; Rani, M.R.S.; Hampson, J.; Zonjy, B.; Lacuey, N.; Faingold, C.L.; Friedman, D.; Devinsky, O.; Sainju, R.K.; Schuele, S.; Diehl, B.; Nei, M.; Harper, R.M.; Bateman, L.M.; Richerson, G.; Lhatoo, S.D. Serum Serotonin Levels in Patients with Epileptic Seizures. Epilepsia 2018, 59, 1152–1161. [Google Scholar] [CrossRef]
- Petrucci, A.N.; Joyal, K.G.; Chou, J.W.; Li, R.; Vencer, K.M.; Buchanan, G.F. Post-Ictal Generalized EEG Suppression Is Reduced by Enhancing Dorsal Raphe Serotonergic Neurotransmission. Neuroscience 2021, 453, 206–221. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-J.; Liang, W.-H.; Lam, C.-S.; Huang, X.-F.; Yang, S.-J.; Wong-Riley, M.T.T.; Fung, M.-L.; Liu, Y.-Y. Catecholaminergic Neurons in Synaptic Connections with Pre-Bötzinger Complex Neurons in the Rostral Ventrolateral Medulla in Normoxic and Daily Acute Intermittent Hypoxic Rats. Exp. Neurol. 2017, 287, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Kakita, A.; Toyoshima, Y.; Onodera, O.; Ozawa, T.; Morita, T.; Nishizawa, M.; Takahashi, H. Depletion of Medullary Serotonergic Neurons in Patients with Multiple System Atrophy Who Succumbed to Sudden Death. Brain 2009, 132, 1810–1819. [Google Scholar] [CrossRef]
- Spirovski, D.; Li, Q.; Pilowsky, P.M. Brainstem Galanin-Synthesizing Neurons Are Differentially Activated by Chemoreceptor Stimuli and Represent a Subpopulation of Respiratory Neurons. J. Comp. Neurol. 2012, 520, 154–173. [Google Scholar] [CrossRef]
- Czeisler, C.M.; Silva, T.M.; Fair, S.R.; Liu, J.; Tupal, S.; Kaya, B.; Cowgill, A.; Mahajan, S.; Silva, P.E.; Wang, Y.; Blissett, A.R.; Göksel, M.; Borniger, J.C.; Zhang, N.; Fernandes-Junior, S.A.; Catacutan, F.; Alves, M.J.; Nelson, R.J.; Sundaresan, V.; Rekling, J.; Takakura, A.C.; Moreira, T.S.; Otero, J.J. The Role of PHOX2B-Derived Astrocytes in Chemosensory Control of Breathing and Sleep Homeostasis. J. Physiol. 2019, 597, 2225–2251. [Google Scholar] [CrossRef]
- Falquetto, B.; Oliveira, L.M.; Takakura, A.C.; Mulkey, D.K.; Moreira, T.S. Inhibition of the Hypercapnic Ventilatory Response by Adenosine in the Retrotrapezoid Nucleus in Awake Rats. Neuropharmacology 2018, 138, 47–56. [Google Scholar] [CrossRef]
- Patodia, S.; Paradiso, B.; Ellis, M.; Somani, A.; Sisodiya, S.M.; Devinsky, O.; Thom, M. Characterisation of Medullary Astrocytic Populations in Respiratory Nuclei and Alterations in Sudden Unexpected Death in Epilepsy. Epilepsy Res. 2019, 157, 106213. [Google Scholar] [CrossRef]
- SheikhBahaei, S.; Morris, B.; Collina, J.; Anjum, S.; Znati, S.; Gamarra, J.; Zhang, R.; Gourine, A.V.; Smith, J.C. Morphometric Analysis of Astrocytes in Brainstem Respiratory Regions. J. Comp. Neurol. 2018, 526, 2032–2047. [Google Scholar] [CrossRef]
- Dlouhy, B.J.; Gehlbach, B.K.; Kreple, C.J.; Kawasaki, H.; Oya, H.; Buzza, C.; Granner, M.A.; Welsh, M.J.; Howard, M.A.; Wemmie, J.A. Breathing Inhibited When Seizures Spread to the Amygdala and upon Amygdala Stimulation. J. Neurosci. 2015, 35, 10281–10289. [Google Scholar] [CrossRef]
- Lacuey, N.; Zonjy, B.; Londono, L.; Lhatoo, S.D. Amygdala and Hippocampus Are Symptomatogenic Zones for Central Apneic Seizures. Neurology 2017, 88, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Nobis, W.P.; Schuele, S.; Templer, J.W.; Zhou, G.; Lane, G.; Rosenow, J.M.; Zelano, C. Amygdala-Stimulation-Induced Apnea Is Attention and Nasal-Breathing Dependent. Ann. Neurol. 2018, 83, 460–471. [Google Scholar] [CrossRef]
- Applegate, C.D.; Kapp, B.S.; Underwood, M.D.; McNall, C.L. Autonomic and Somatomotor Effects of Amygdala Central N. Stimulation in Awake Rabbits. Physiol. Behav. 1983, 31, 353–360. [Google Scholar] [CrossRef]
- Seyal, M.; Bateman, L.M. Ictal Apnea Linked to Contralateral Spread of Temporal Lobe Seizures: Intracranial EEG Recordings in Refractory Temporal Lobe Epilepsy. Epilepsia 2009, 50, 2557–2562. [Google Scholar] [CrossRef]
- Feldman, J.L.; Mitchell, G.S.; Nattie, E.E. Breathing: Rhythmicity, Plasticity, Chemosensitivity. Annu. Rev. Neurosci. 2003, 26, 239–266. [Google Scholar] [CrossRef]
- Sowers, L.P.; Massey, C.A.; Gehlbach, B.K.; Granner, M.A.; Richerson, G.B. Sudden Unexpected Death in Epilepsy: Fatal Post-Ictal Respiratory and Arousal Mechanisms. Respir. Physiol. Neurobiol. 2013, 189, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Kriegel, M.F.; Roberts, D.W.; Jobst, B.C. Orbitofrontal and Insular Epilepsy. J. Clin. Neurophysiol. 2012, 29, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Isnard, J.; Guénot, M.; Ostrowsky, K.; Sindou, M.; Mauguière, F. The Role of the Insular Cortex in Temporal Lobe Epilepsy. Ann. Neurol. 2000, 48, 614–623. [Google Scholar] [CrossRef]
- Ryvlin, P. Avoid Falling into the Depths of the Insular Trap. Epileptic Disord. 2006, 8, S33–S36. [Google Scholar] [CrossRef]
- Sun, T.; Wang, F.; Cui, J. Insular Epilepsy; People’s Medical Publishing House: Beijing, China, 2013. [Google Scholar]
- Oppenheimer, S. Forebrain Lateralization and the Cardiovascular Correlates of Epilepsy. Brain 2001, 124, 2345–2346. [Google Scholar] [CrossRef]
- Smart, S.L.; Lopantsev, V.; Zhang, C.L.; Robbins, C.A.; Wang, H.; Chiu, S.Y.; Schwartzkroin, P.A.; Messing, A.; Tempel, B.L. Deletion of the Kv1.1 Potassium Channel Causes Epilepsy in Mice. Neuron 1998, 20, 809–819. [Google Scholar] [PubMed]
- Kim, Y.; Bravo, E.; Thirnbeck, C.K.; Smith-Mellecker, L.A.; Kim, S.H.; Gehlbach, B.K.; Laux, L.C.; Zhou, X.; Nordli, D.R.; Richerson, G.B. Severe Peri-Ictal Respiratory Dysfunction Is Common in Dravet Syndrome. J. Clin. Invest. 2018, 128, 1141–1153. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Wong, B.; Phillipson, E.A.; Slutsky, A.S.; Bradley, T.D. Interaction of Hyperventilation and Arousal in the Pathogenesis of Idiopathic Central Sleep Apnea. Am. J. Respir. Crit. Care Med. 1994, 150, 489–495. [Google Scholar] [CrossRef]
- Xie, A.; Rankin, F.; Rutherford, R.; Bradley, T.D. Effects of Inhaled CO₂ and Added Dead Space on Idiopathic Central Sleep Apnea. J. Appl. Physiol. 1997, 82, 918–926. [Google Scholar] [CrossRef]
- Li, D.; Mabrouk, O.S.; Liu, T.; Tian, F.; Xu, G.; Rengifo, S.; Choi, S.J.; Mathur, A.; Crooks, C.P.; Kennedy, R.T.; Wang, M.M.; Ghanbari, H.; Borjigin, J. Asphyxia-Activated Corticocardiac Signaling Accelerates Onset of Cardiac Arrest. Proc. Natl. Acad. Sci. USA 2015, 112, 4995–5000. [Google Scholar] [CrossRef] [PubMed]
- Nakase, K.; Kollmar, R.; Lazar, J.; Arjomandi, H.; Sundaram, K.; Silverman, J.; Orman, R.; Weedon, J.; Stefanov, D.; Savoca, E. Laryngospasm, Central and Obstructive Apnea during Seizures: Defining Pathophysiology for Sudden Death in a Rat Model. Epilepsy Res. 2016, 128, 126–139. [Google Scholar] [CrossRef]
- Peng, W.; Danison, J.L.; Seyal, M. Postictal Generalized EEG Suppression and Respiratory Dysfunction Following Generalized Tonic–Clonic Seizures in Sleep and Wakefulness. Epilepsia 2017, 58, 1409–1414. [Google Scholar] [CrossRef]
- Massey, C.A.; Sowers, L.P.; Dlouhy, B.J.; Richerson, G.B. Mechanisms of Sudden Unexpected Death in Epilepsy: The Pathway to Prevention. Nat. Rev. Neurol. 2014, 10, 271–282. [Google Scholar] [CrossRef]
- Zhan, Q.; Buchanan, G.F.; Motelow, J.E.; Andrews, J.; Vitkovskiy, P.; Chen, W.C.; Serout, F.; Gummadavelli, A.; Kundishora, A.; Furman, M.; Li, W.; Bo, X.; Richerson, G.B.; Blumenfeld, H. Impaired Serotonergic Brainstem Function during and after Seizures. J. Neurosci. 2016, 36, 2711–2722. [Google Scholar] [CrossRef]
- Irizarry, R.; Sukato, D.; Kollmar, R.; Schild, S.; Silverman, J.; Sundaram, K.; Stephenson, S.; Stewart, M. Seizures Induce Obstructive Apnea in DBA/2J Audiogenic Seizure-Prone Mice: Lifesaving Impact of Tracheal Implants. Epilepsia 2020, 61, 365–375. [Google Scholar] [CrossRef]
- Ravindran, M. Temporal Lobe Seizure Presenting as “Laryngospasm”. Clin. Electroencephalogr. 1981, 12, 139–140. [Google Scholar] [CrossRef]
- Amir, J.; Ashkenazi, S.; Schonfeld, T.; Weitz, R.; Nitzan, M. Laryngospasm as a Single Manifestation of Epilepsy. Arch. Dis. Child. 1983, 58, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.; Kollmar, R.; Nakase, K.; Silverman, J.; Sundaram, K.; Orman, R.; Lazar, J. Obstructive Apnea Due to Laryngospasm Links Ictal to Postictal Events in SUDEP Cases and Offers Practical Biomarkers for Review of Past Cases and Prevention of New Ones. Epilepsia 2017, 58, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Geerling, J.C.; Shin, J.; Chimenti, P.C.; Loewy, A.D. Paraventricular Hypothalamic Nucleus: Axonal Projections to the Brainstem. J. Comp. Neurol. 2010, 518, 1460–1499. [Google Scholar] [CrossRef]
- Richerson, G.B.; Buchanan, G.F. The Serotonin Axis: Shared Mechanisms in Seizures, Depression, and SUDEP. Epilepsia 2011, 52, 28–38. [Google Scholar] [CrossRef]
- Tupal, S.; Faingold, C.L. Evidence Supporting a Role of Serotonin in Modulation of Sudden Death Induced by Seizures in DBA/2 Mice. Epilepsia 2006, 47, 21–26. [Google Scholar] [CrossRef]
- Uteshev, V.V.; Tupal, S.; Mhaskar, Y.; Faingold, C.L. Abnormal Serotonin Receptor Expression in DBA/2 Mice Associated with Susceptibility to Sudden Death Due to Respiratory Arrest. Epilepsy Res. 2010, 88, 183–188. [Google Scholar] [CrossRef]
- Buchanan, G.F.; Murray, N.M.; Hajek, M.A.; Richerson, G.B. Serotonin Neurones Have Anti-Convulsant Effects and Reduce Seizure-Induced Mortality. J. Physiol. 2014, 592, 4395–4410. [Google Scholar] [CrossRef]
- Foerster, O. Hyperventilationsepilepsie. Dtsch. Z. Nervenheilkd. 1925, 83, 347–356. [Google Scholar] [CrossRef]
- Lennox, W.G.; Gibbs, F.A.; Gibbs, E.L. Effect on the Electro-Encephalogram of Drugs and Conditions Which Influence Seizures. Arch. Neurol. Psychiatry 1936, 36, 1236–1250. [Google Scholar] [CrossRef]
- Wirrell, E.C.; Camfield, P.R.; Gordon, K.E.; Camfield, C.S.; Dooley, J.M.; Hanna, B.D. Will a Critical Level of Hyperventilation-Induced Hypocapnia Always Induce an Absence Seizure? Epilepsia 1996, 37, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Guaranha, M.S.B.; Garzon, E.; Buchpiguel, C.A.; Tazima, S.; Yacubian, E.M.T.; Sakamoto, A.C. Hyperventilation Revisited: Physiological Effects and Efficacy on Focal Seizure Activation in the Era of Video-EEG Monitoring. Epilepsia 2005, 46, 69–75. [Google Scholar] [CrossRef]
- Rockstroh, B. Hyperventilation-Induced EEG Changes in Humans and Their Modulation by an Anticonvulsant Drug. Epilepsy Res. 1990, 7, 146–154. [Google Scholar] [CrossRef]
- Lee, J.; Taira, T.; Pihlaja, P.; Ransom, B.R.; Kaila, K. Effects of CO₂ on Excitatory Transmission Apparently Caused by Changes in Intracellular pH in the Rat Hippocampal Slice. Brain Res. 1996, 706, 210–216. [Google Scholar] [CrossRef]
- Weinand, M.E.; Carter, L.P.; Oommen, K.J.; Hutzler, R.; Labiner, D.M.; Talwar, D.; El-Saadany, W.; Ahern, G.L. Response of Human Epileptic Temporal Lobe Cortical Blood Flow to Hyperventilation. Epilepsy Res. 1995, 21, 221–226. [Google Scholar] [CrossRef]
- Labuz-Roszak, B.; Pierzchała, K. Assessment of Autonomic Nervous System in Patients with Epilepsy in the Interictal State. A Pilot Study. Neurol. Neurochir. Pol. 2009, 43, 330–336. [Google Scholar]
- Jansen, K.; Vandeput, S.; Milosevic, M.; Ceulemans, B.; Van Huffel, S.; Brown, L.; Penders, J.; Lagae, L. Autonomic Effects of Refractory Epilepsy on Heart Rate Variability in Children: Influence of Intermittent Vagus Nerve Stimulation. Dev. Med. Child Neurol. 2011, 53, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Assenza, G.; Mecarelli, O.; Tombini, M.; Pulitano, P.; Pellegrino, G.; Benvenga, A.; Assenza, F.; Campana, C.; Di Pino, G.; Di Lazzaro, V. Hyperventilation Induces Sympathetic Overactivation in Mesial Temporal Epilepsy. Epilepsy Res. 2015, 110, 221–227. [Google Scholar] [CrossRef]
- Leonhardt, G.; de Greiff, A.; Marks, S.; Ludwig, T.; Doerfler, A.; Forsting, M.; Konermann, S.; Hufnagel, A. Brain Diffusion during Hyperventilation: Diffusion-Weighted MR-Monitoring in Patients with Temporal Lobe Epilepsy and in Healthy Volunteers. Epilepsy Res. 2002, 51, 269–278. [Google Scholar] [CrossRef]
- Seneviratne, U.; Cook, M.; D’Souza, W. Consistent Topography and Amplitude Symmetry Are More Typical than Morphology of Epileptiform Discharges in Genetic Generalized Epilepsy. Clin. Neurophysiol. 2016, 127, 1138–1146. [Google Scholar] [CrossRef]
- Kjaer, T.W.; Madsen, F.F.; Moltke, F.B.; Uldall, P.; Hogenhaven, H. Intraoperative Hyperventilation vs Remifentanil during Electrocorticography for Epilepsy Surgery - A Case Report: Finding Ictal Onset Zones during Surgery. Acta Neurol. Scand. 2010, 121, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Marrosu, F.; Puligheddu, M.; Giagheddu, M.; Cossu, G.; Piga, M. Correlation between Cerebral Perfusion and Hyperventilation Enhanced Focal Spiking Activity. Epilepsy Res. 2000, 40, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Bagnall, R.D.; Crompton, D.E.; Petrovski, S.; Lam, L.; Cutmore, C.; Garry, S.I.; Sadleir, L.G.; Dibbens, L.M.; Cairns, A.; Kivity, S.; Afawi, Z.; Regan, B.M.; Duflou, J.; Berkovic, S.F.; Scheffer, I.E.; Semsarian, C. Exome-Based Analysis of Cardiac Arrhythmia, Respiratory Control, and Epilepsy Genes in Sudden Unexpected Death in Epilepsy. Ann. Neurol. 2016, 79, 522–534. [Google Scholar] [CrossRef]
- Bagnall, R.D.; Crompton, D.E.; Cutmore, C.; Regan, B.M.; Berkovic, S.F.; Scheffer, I.E.; Semsarian, C. Genetic Analysis of PHOX2B in Sudden Unexpected Death in Epilepsy Cases. Neurology 2014, 83, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.S.; Arvide, E.M.; Huang, T.-W.; Yoo, J.; Noebels, J.L.; Neul, J.L. MeCP2 Is Critical within HoxB1-Derived Tissues of Mice for Normal Lifespan. J. Neurosci. 2011, 31, 10359–10370. [Google Scholar] [CrossRef]
- El-Khoury, R.; Panayotis, N.; Matagne, V.; Ghata, A.; Villard, L.; Roux, J.-C. GABA and Glutamate Pathways Are Spatially and Developmentally Affected in the Brain of Mecp2-Deficient Mice. PLoS ONE 2014, 9, e92169. [Google Scholar] [CrossRef]
- Viemari, J.-C.; Roux, J.-C.; Tryba, A.K.; Saywell, V.; Burnet, H.; Peña, F.; Zanella, S.; Bévengut, M.; Barthelemy-Requin, M.; Herzing, L.B.K.; Moncla, A.; Mancini, J.; Ramirez, J.-M.; Villard, L.; Hilaire, G. Mecp2 Deficiency Disrupts Norepinephrine and Respiratory Systems in Mice. J. Neurosci. 2005, 25, 11521–11530. [Google Scholar] [CrossRef]
- Ren, J.; Ding, X.; Funk, G.D.; Greer, J.J. Anxiety-Related Mechanisms of Respiratory Dysfunction in a Mouse Model of Rett Syndrome. J. Neurosci. 2012, 32, 17230–17240. [Google Scholar] [CrossRef]
- Smith, J.C.; Abdala, A.P.; Borgmann, A.; Rybak, I.A.; Paton, J.F. Brainstem Respiratory Networks: Building Blocks and Microcircuits. Trends Neurosci. 2013, 36, 152–162. [Google Scholar] [CrossRef]
- Ramirez, J.-M.; Ward, C.S.; Neul, J.L. Breathing Challenges in Rett Syndrome: Lessons Learned from Humans and Animal Models. Respir. Physiol. Neurobiol. 2013, 189, 280–287. [Google Scholar] [CrossRef]
- McCauley, M.D.; Wang, T.; Mike, E.; Herrera, J.; Beavers, D.L.; Huang, T.-W.; Ward, C.S.; Skinner, S.; Percy, A.K.; Glaze, D.G.; Wehrens, X.H.T.; Neul, J.L. Pathogenesis of Lethal Cardiac Arrhythmias in Mecp2 Mutant Mice: Implication for Therapy in Rett Syndrome. Sci. Transl. Med. 2011, 3, 113ra125. [Google Scholar] [CrossRef] [PubMed]
- Seyal, M.; Bateman, L.M.; Li, C. Impact of Periictal Interventions on Respiratory Dysfunction, Postictal EEG Suppression, and Postictal Immobility. Epilepsia 2013, 54, 377–382. [Google Scholar] [CrossRef]
- Moore, B.M.; Jou, C.J.; Tatalovic, M.; Kaufman, E.S.; Kline, D.D.; Kunze, D.L. The Kv1.1 Null Mouse, a Model of Sudden Unexpected Death in Epilepsy (SUDEP). Epilepsia 2014, 55, 1808–1816. [Google Scholar] [CrossRef] [PubMed]
- Campos, R.R.; Tolentino-Silva, F.R.P.; Mello, L.E.A.M. Respiratory Pattern in a Rat Model of Epilepsy. Epilepsia 2003, 44, 712–717. [Google Scholar] [CrossRef]
- Richerson, G.B. Serotonergic Neurons as Carbon Dioxide Sensors That Maintain PH Homeostasis. Nat. Rev. Neurosci. 2004, 5, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.C.; Horn, J.K.; Valente, J.; Simon, R.P. The Role of Hypoventilation in a Sheep Model of Epileptic Sudden Death. Ann. Neurol. 1995, 37, 531–537. [Google Scholar] [CrossRef]
- Johnston, S.C.; Siedenberg, R.; Min, J.K.; Jerome, E.H.; Laxer, K.D. Central Apnea and Acute Cardiac Ischemia in a Sheep Model of Epileptic Sudden Death. Ann. Neurol. 1997, 42, 588–594. [Google Scholar] [CrossRef]
- Faingold, C.L.; Randall, M.; Tupal, S. DBA/1 Mice Exhibit Chronic Susceptibility to Audiogenic Seizures Followed by Sudden Death Associated with Respiratory Arrest. Epilepsy Behav. 2010, 17, 436–440. [Google Scholar] [CrossRef]
- Feng, H.-J.; Faingold, C.L. Abnormalities of Serotonergic Neurotransmission in Animal Models of SUDEP. Epilepsy Behav. 2017, 71, 174–180. [Google Scholar] [CrossRef]
- During, M.J.; Spencer, D.D. Adenosine: A Potential Mediator of Seizure Arrest and Postictal Refractoriness. Ann. Neurol. 1992, 32, 618–624. [Google Scholar] [CrossRef]
- Paydarfar, D.; Eldridge, F.L.; Scott, S.C.; Dowell, R.T.; Wagner, P.G. Respiratory Responses to Focal and Generalized Seizures in Cats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1991, 260, R934–R940. [Google Scholar] [CrossRef] [PubMed]
- Paydarfar, D.; Eldridge, F.L.; Wagner, P.G.; Dowell, R.T. Neural Respiratory Responses to Cortically Induced Seizures in Cats. Respir. Physiol. 1992, 89, 225–237. [Google Scholar] [CrossRef]
- Lutas, A.; Yellen, G. The Ketogenic Diet: Metabolic Influences on Brain Excitability and Epilepsy. Trends Neurosci. 2013, 36, 32–40. [Google Scholar] [CrossRef]
- Faingold, C.L.; Randall, M.; Mhaskar, Y.; Uteshev, V.V. Differences in Serotonin Receptor Expression in the Brainstem May Explain the Differential Ability of a Serotonin Agonist to Block Seizure-Induced Sudden Death in DBA/2 vs. DBA/1 Mice. Brain Res. 2011, 1418, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, H.; Feng, H.-J. Atomoxetine, a Norepinephrine Reuptake Inhibitor, Reduces Seizure-Induced Respiratory Arrest. Epilepsy Behav. 2017, 73, 6–9. [Google Scholar] [CrossRef]
- Kuo, F.-S.; Cleary, C.M.; LoTurco, J.J.; Chen, X.; Mulkey, D.K. Disordered Breathing in a Mouse Model of Dravet Syndrome. eLife 2019, 8, e43387. [Google Scholar] [CrossRef] [PubMed]
- Szabó, C.Á.; Knape, K.D.; Leland, M.M.; Feldman, J.; McCoy, K.J.M.; Hubbard, G.B.; Williams, J.T. Mortality in Captive Baboons with Seizures: A New Model for SUDEP? Epilepsia 2009, 50, 1995–1998. [Google Scholar] [CrossRef]
- Kalume, F.; Westenbroek, R.E.; Cheah, C.S.; Yu, F.H.; Oakley, J.C.; Scheuer, T.; Catterall, W.A. Sudden Unexpected Death in a Mouse Model of Dravet Syndrome. J. Clin. Investig. 2013, 123, 1798–1808. [Google Scholar] [CrossRef]
- Auerbach, D.S.; Jones, J.; Clawson, B.C.; Offord, J.; Lenk, G.M.; Ogiwara, I.; Yamakawa, K.; Meisler, M.H.; Parent, J.M.; Isom, L.L. Altered Cardiac Electrophysiology and SUDEP in a Model of Dravet Syndrome. PLoS ONE 2013, 8, e77843. [Google Scholar] [CrossRef]
- Iyer, S.H.; Matthews, S.A.; Simeone, T.A.; Maganti, R.; Simeone, K.A. Accumulation of rest deficiency precedes sudden death of epileptic Kv1.1 knockout mice, a model of sudden unexpected death in epilepsy. Epilepsia 2018, 59, 92–105. [Google Scholar] [CrossRef]
- Ren, Y.; Chang, J.; Li, C.; Jia, C.; Li, P.; Wang, Y.; Chu, X.-P. The effects of ketogenic diet treatment in Kcna1-null mouse, a model of sudden unexpected death in epilepsy. Front. Neurol. 2019, 10, 744. [Google Scholar] [CrossRef] [PubMed]
- Foley, J.; Burnham, V.; Tedoldi, M.; Danial, N.N.; Yellen, G. BAD knockout provides metabolic seizure resistance in a genetic model of epilepsy with sudden unexplained death in epilepsy. Epilepsia 2018, 59, e1–e4. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.; Karumuri, B.K.; Gautier, N.M.; Liu, R.; Hutson, T.N.; Vanhoof-Villalba, S.L.; Vlachos, I.; Iasemidis, L.; Glasscock, E. Scn2a deletion improves survival and brain–heart dynamics in the Kcna1-null mouse model of sudden unexpected death in epilepsy (SUDEP). Hum. Mol. Genet. 2017, 26, 2091–2103. [Google Scholar] [CrossRef]
- Dhaibar, H.; Gautier, N.M.; Chernyshev, O.Y.; Dominic, P.; Glasscock, E. Cardiorespiratory profiling reveals primary breathing dysfunction in Kcna1-null mice: Implications for sudden unexpected death in epilepsy. Neurobiol. Dis. 2019, 127, 502–511. [Google Scholar] [CrossRef]
- Cook, M.J.; O’Brien, T.J.; Berkovic, S.F.; Murphy, M.; Morokoff, A.; Fabinyi, G.; D’Souza, W.; Yerra, R.; Archer, J.; Litewka, L. Prediction of seizure likelihood with a long-term, implanted seizure advisory system in patients with drug-resistant epilepsy: A first-in-man study. Lancet Neurol. 2013, 12, 563–571. [Google Scholar] [CrossRef]
- Ng, M.; Pavlova, M. Why are seizures rare in rapid eye movement sleep? Review of the frequency of seizures in different sleep stages. Epilepsy Res. Treat. 2013, 2013, 932790. [Google Scholar] [CrossRef]
- Kitchigina, V.F.; Butuzova, M.V. Theta activity of septal neurons during different epileptic phases: The same frequency but different significance? Exp. Neurol. 2009, 216, 449–458. [Google Scholar] [CrossRef]
- Roliz, A.H.; Kothare, S. The interaction between sleep and epilepsy. Curr. Neurol. Neurosci. Rep. 2022, 22, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Derry, C.P.; Duncan, S. Sleep and epilepsy. Epilepsy Behav. 2013, 26, 394–404. [Google Scholar] [CrossRef]
- Žiburkus, J.; Cressman, J.R.; Schiff, S.J. Seizures as imbalanced up states: Excitatory and inhibitory conductances during seizure-like events. J. Neurophysiol. 2013, 109, 1296–1306.CrossRef. [Google Scholar] [CrossRef]
- Finnerty, G.T.; Whittington, M.A.; Jefferys, J.G.R. Altered dentate filtering during the transition to seizure in the rat tetanus toxin model of epilepsy. J. Neurophysiol. 2001, 86, 2748–2753.CrossRef. [Google Scholar] [CrossRef] [PubMed]
- Lamberts, R.J.; Thijs, R.D.; Laffan, A.; Langan, Y.; Sander, J.W. Sudden unexpected death in epilepsy: People with nocturnal seizures may be at highest risk. Epilepsia 2012, 53, 253–257.CrossRef. [Google Scholar] [CrossRef] [PubMed]
- Hajek, M.A.; Buchanan, G.F. Influence of vigilance state on physiological consequences of seizures and seizure-induced death in mice. J. Neurophysiol. 2016, 115, 2286–2293.CrossRef. [Google Scholar] [CrossRef]
- Nobili, L.; Proserpio, P.; Rubboli, G.; Montano, N.; Didato, G.; Tassinari, C.A. Sudden unexpected death in epilepsy (SUDEP) and sleep. Sleep Med. Rev. 2011, 15, 237–246.CrossRef. [Google Scholar] [CrossRef]
- Purnell, B.S.; Hajek, M.A.; Buchanan, G.F. Time-of-day influences on respiratory sequelae following maximal electroshock-induced seizures in mice. J. Neurophysiol. 2017, 118, 2592–2600.CrossRef. [Google Scholar] [CrossRef]
- Dutton, S.B.B.; Sawyer, N.T.; Kalume, F.; Jumbo-Lucioni, P.; Borges, K.; Catterall, W.A.; Escayg, A. Protective effect of the ketogenic diet in Scn1a mutant mice. Epilepsia 2011, 52, 2050–2056.CrossRef. [Google Scholar] [CrossRef]
- Richerson, G.B.; Boison, D.; Faingold, C.L.; Ryvlin, P. From unwitnessed fatality to witnessed rescue: Pharmacologic intervention in sudden unexpected death in epilepsy. Epilepsia.
- Dallérac, G.; Moulard, J.; Benoist, J.-F.; Rouach, S.; Auvin, S.; Guilbot, A.; Lenoir, L.; Rouach, N. Non-ketogenic combination of nutritional strategies provides robust protection against seizures. Sci. Rep. 2017, 7, 5496. [Google Scholar] [CrossRef]
- Szabó, C.Á.; Knape, K.D.; Leland, M.M.; Cwikla, D.J.; Williams-Blangero, S.; Williams, J.T. Epidemiology and characterization of seizures in a pedigreed baboon colony. Comp. Med. 2012, 62, 535–538. [Google Scholar] [PubMed]
- Kloster, R.; Engelskjøn, T. Sudden unexpected death in epilepsy (SUDEP): A clinical perspective and a search for risk factors. J. Neurol. Neurosurg. Psychiatry 1999, 67, 439–444. [Google Scholar] [CrossRef]
- Leung, H.; Kwan, P.; Elger, C.E. Finding the missing link between ictal bradyarrhythmia, ictal asystole, and sudden unexpected death in epilepsy. Epilepsy Behav. 2006, 9, 19–30. [Google Scholar] [CrossRef]
- Earnest, M.P.; Thomas, G.E.; Eden, R.A.; Hossack, K.F. The sudden unexplained death syndrome in epilepsy: Demographic, clinical, and postmortem features. Epilepsia 1992, 33, 310–316. [Google Scholar] [CrossRef]
- Langan, Y.; Nashef, L.; Sander, J.W. Case-control study of SUDEP. Neurology 2005, 64, 1131–1133. [Google Scholar] [CrossRef] [PubMed]
- Bhaskaran, M.D.; Smith, B.N. Effects of TRPV1 activation on synaptic excitation in the dentate gyrus of a mouse model of temporal lobe epilepsy. Exp. Neurol. 2010, 223, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Goldman, A.M.; Glasscock, E.; Yoo, J.; Chen, T.T.; Klassen, T.L.; Noebels, J.L. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci. Transl. Med. 2009, 1, 2ra6. [Google Scholar] [CrossRef]
- Shibley, H.; Smith, B.N. Pilocarpine-induced status epilepticus results in mossy fiber sprouting and spontaneous seizures in C57BL/6 and CD-1 mice. Epilepsy Res. 2002, 49, 109–120. [Google Scholar] [CrossRef]
- Bach, E.C.; Halmos, K.C.; Smith, B.N. Enhanced NMDA receptor-mediated modulation of excitatory neurotransmission in the dorsal vagal complex of streptozotocin-treated, chronically hyperglycemic mice. PLoS ONE 2015, 10, e0121022. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Zhang, J.; Mifflin, S. Hypertension alters GABA receptor-mediated inhibition of neurons in the nucleus of the solitary tract. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R1276–R1286. [Google Scholar] [CrossRef]
- Glatzer, N.R.; Derbenev, A.V.; Banfield, B.W.; Smith, B.N. Endomorphin-1 modulates intrinsic inhibition in the dorsal vagal complex. J. Neurophysiol. 2007, 98, 1591–1599. [Google Scholar] [CrossRef]
- Card, J.P.; Sved, J.C.; Craig, B.; Raizada, M.; Vazquez, J.; Sved, A.F. Efferent projections of rat rostroventrolateral medulla C1 catecholamine neurons: Implications for the central control of cardiovascular regulation. J. Comp. Neurol. 2006, 499, 840–859. [Google Scholar] [CrossRef]
- Sawant-Pokam, P.M.; Suryavanshi, P.; Mendez, J.M.; Dudek, F.E.; Brennan, K.C. Mechanisms of neuronal silencing after cortical spreading depression. Cereb. Cortex 2017, 27, 1311–1325. [Google Scholar] [CrossRef]
- Fong, A.Y.; Stornetta, R.L.; Foley, C.M.; Potts, J.T. Immunohistochemical localization of GAD67-expressing neurons and processes in the rat brainstem: Subregional distribution in the nucleus tractus solitarius. J. Comp. Neurol. 2005, 493, 274–290. [Google Scholar] [CrossRef] [PubMed]
- Naggar, I.; Stewart, M. A rat model for exploring the contributions of ventricular arrhythmias to sudden death in epilepsy. In Sudden Unexpected Death in Epilepsy: Mechanisms and New Methods for Analyzing Risks; Lathers, C.M., Schraeder, P.L., Leestma, J.E., Wannamaker, B.B., Verrier, R.L., Schachter, S.C., Eds.; Taylor & Francis: Boca Raton, FL, USA, 2015; pp. 241–250. [Google Scholar]
- Budde, R.B.; Arafat, M.A.; Pederson, D.J.; Lovick, T.A.; Jefferys, J.G.R.; Irazoqui, P.P. Acid reflux induced laryngospasm as a potential mechanism of sudden death in epilepsy. Epilepsy Res. 2018, 148, 23–31. [Google Scholar] [CrossRef]
- Tomson, T.; Walczak, T.; Sillanpää, M.; Sander, J.W.A.S. Sudden unexpected death in epilepsy: A review of incidence and risk factors. Epilepsia 2005, 46, 54–61. [Google Scholar] [CrossRef]
- Helbig, I.; Scheffer, I.E.; Mulley, J.C.; Berkovic, S.F. Navigating the channels and beyond: Unravelling the genetics of the epilepsies. Lancet Neurol. 2008, 7, 231–245. [Google Scholar] [CrossRef]
- Allen, L.A.; Harper, R.M.; Kumar, R.; Guye, M.; Ogren, J.A.; Lhatoo, S.D.; Lemieux, L.; Scott, C.A.; Vos, S.B.; Rani, S.; Diehl, B. Dysfunctional brain networking among autonomic regulatory structures in temporal lobe epilepsy patients at high risk of sudden unexpected death in epilepsy. Front. Neurol. 2017, 8, 544. [Google Scholar] [CrossRef]
- Patodia, S.; Somani, A.; Thom, M. Neuropathology findings in autonomic brain regions in SUDEP and future research directions. Auton. Neurosci. 2021, 235, 102862. [Google Scholar] [CrossRef]
- Feng, Y.; Wei, Z.-H.; Liu, C.; Li, G.-Y.; Qiao, X.-Z.; Gan, Y.-J.; Zhang, C.-C.; Deng, Y.-C. Genetic variations in GABA metabolism and epilepsy. Seizure 2022, 101, 22–29. [Google Scholar] [CrossRef] [PubMed]
- During, M.J. In vivo neurochemistry of the conscious human brain: Intrahippocampal microdialysis in epilepsy. In Techniques in the Behavioral and Neural Sciences; Elsevier, 1991; Vol. 7, pp. 425–442. [Google Scholar]
- During, M.J.; Spencer, D.D. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 1993, 341, 1607–1610. [Google Scholar] [CrossRef] [PubMed]
- Cavus, I.; Kasoff, W.S.; Cassaday, M.P.; Jacob, R.; Gueorguieva, R.; Sherwin, R.S.; Krystal, J.H.; Spencer, D.D.; Abi-Saab, W.M. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann. Neurol. 2005, 57, 226–235. [Google Scholar] [CrossRef]
- Pan, J.W.; Cavus, I.; Kim, J.; Hetherington, H.P.; Spencer, D.D. Hippocampal extracellular GABA correlates with metabolism in human epilepsy. Metab. Brain Dis. 2008, 23, 457–468. [Google Scholar] [CrossRef]
- Çavuş, I.; Romanyshyn, J.C.; Kennard, J.T.; Farooque, P.; Williamson, A.; Eid, T.; Spencer, S.S.; Duckrow, R.; Dziura, J.; Spencer, D.D. Elevated basal glutamate and unchanged glutamine and GABA in refractory epilepsy: Microdialysis study of 79 patients at the Yale Epilepsy Surgery Program. Ann. Neurol. 2016, 80, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.R.; Felix, L.; Zeug, A.; Dietrich, D.; Reiner, A.; Henneberger, C. Astroglial glutamate signaling and uptake in the hippocampus. Front. Mol. Neurosci. 2018, 10, 451. [Google Scholar] [CrossRef]
- Eid, T.; Thomas, M.J.; Spencer, D.D.; Runden-Pran, E.; Lai, J.C.K.; Malthankar, G.V.; Kim, J.H.; Danbolt, N.C.; Ottersen, O.P.; De Lanerolle, N.C. Loss of glutamine synthetase in the human epileptogenic hippocampus: Possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 2004, 363, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Bagdy, G.; Kecskemeti, V.; Riba, P.; Jakus, R. Serotonin and epilepsy. J. Neurochem. 2007, 100, 857–873. [Google Scholar] [CrossRef]
- Ferraz, A.C.; Anselmo-Franci, J.A.; Perosa, S.R.; de Castro-Neto, E.F.; Bellissimo, M.I.; de Oliveira, B.H.; Cavalheiro, E.A.; da Graça Naffah-Mazzacoratti, M.; Da Cunha, C. Amino acid and monoamine alterations in the cerebral cortex and hippocampus of mice submitted to ricinine-induced seizures. Pharmacol. Biochem. Behav. 2002, 72, 779–786. [Google Scholar] [CrossRef]
- Faingold, C.L.; Tupal, S.; Randall, M. Prevention of seizure-induced sudden death in a chronic SUDEP model by semichronic administration of a selective serotonin reuptake inhibitor. Epilepsy Behav. 2011, 22, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Brennan, T.J.; Seeley, W.W.; Kilgard, M.; Schreiner, C.E.; Tecott, L.H. Sound-Induced Seizures in Serotonin 5-HT2C Receptor Mutant Mice. Nat. Genet. 1997, 16, 387–390. [Google Scholar] [CrossRef]
- Kinney, H.C.; Richerson, G.B.; Dymecki, S.M.; Darnall, R.A.; Nattie, E.E. The Brainstem and Serotonin in the Sudden Infant Death Syndrome. Annu. Rev. Pathol. 2009, 4, 517–550. [Google Scholar] [CrossRef]
- Paterson, D.S.; Trachtenberg, F.L.; Thompson, E.G.; Belliveau, R.A.; Beggs, A.H.; Darnall, R.; Chadwick, A.E.; Krous, H.F.; Kinney, H.C. Multiple Serotonergic Brainstem Abnormalities in Sudden Infant Death Syndrome. JAMA 2006, 296, 2124–2132. [Google Scholar] [CrossRef]
- Duncan, J.R.; Paterson, D.S.; Hoffman, J.M.; Mokler, D.J.; Borenstein, N.S.; Belliveau, R.A.; Krous, H.F.; Haas, E.A.; Stanley, C.; Nattie, E.E.; et al. Brainstem Serotonergic Deficiency in Sudden Infant Death Syndrome. JAMA 2010, 303, 430–437. [Google Scholar] [CrossRef]
- Mueller, S.G.; Nei, M.; Bateman, L.M.; Knowlton, R.; Laxer, K.D.; Friedman, D.; Devinsky, O.; Goldman, A.M. Brainstem Network Disruption: A Pathway to Sudden Unexplained Death in Epilepsy? Hum. Brain Mapp. 2018, 39, 4820–4830. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ming, Q.; Lin, W. The Insula Lobe and Sudden Unexpected Death in Epilepsy: A Hypothesis. Epileptic Disord. 2017, 19, 10–14. [Google Scholar] [CrossRef]
- Lacuey, N.; Zonjy, B.; Theerannaew, W.; Loparo, K.A.; Tatsuoka, C.; Sahadevan, J.; Lhatoo, S.D. Left-Insular Damage, Autonomic Instability, and Sudden Unexpected Death in Epilepsy. Epilepsy Behav. 2016, 55, 170–173. [Google Scholar] [CrossRef]
- Tang, Y.; Chen, Q.; Yu, X.; Xia, W.; Luo, C.; Huang, X.; Tang, H.; Gong, Q.; Zhou, D. A Resting-State Functional Connectivity Study in Patients at High Risk for Sudden Unexpected Death in Epilepsy. Epilepsy Behav. 2014, 41, 33–38. [Google Scholar] [CrossRef]
- Harper, R.M.; Kumar, R.; Macey, P.M.; Harper, R.K.; Ogren, J.A. Impaired Neural Structure and Function Contributing to Autonomic Symptoms in Congenital Central Hypoventilation Syndrome. Front. Neurosci. 2015, 9, 415. [Google Scholar] [CrossRef] [PubMed]
- Bozorgi, A.; Chung, S.; Kaffashi, F.; Loparo, K.A.; Sahoo, S.; Zhang, G.Q.; Kaiboriboon, K.; Lhatoo, S.D. Significant Postictal Hypotension: Expanding the Spectrum of Seizure-Induced Autonomic Dysregulation. Epilepsia 2013, 54, e127–e130. [Google Scholar] [CrossRef]
- Kimmerly, D.S.; O’Leary, D.D.; Menon, R.S.; Gati, J.S.; Shoemaker, J.K. Cortical Regions Associated with Autonomic Cardiovascular Regulation during Lower Body Negative Pressure in Humans. J. Physiol. 2005, 569, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Harper, R.M.; Gozal, D.; Bandler, R.; Spriggs, D.; Lee, J.; Alger, J. Regional Brain Activation in Humans during Respiratory and Blood Pressure Challenges. Clin. Exp. Pharmacol. Physiol. 1998, 25, 483–486. [Google Scholar] [CrossRef]
- Liebenthal, J.A.; Wu, S.; Rose, S.; Ebersole, J.S.; Tao, J.X. Association of Prone Position with Sudden Unexpected Death in Epilepsy. Neurology 2015, 84, 703–709. [Google Scholar] [CrossRef]
- Richerson, G.B. Serotonin: The Anti-SuddenDeathAmine?: Serotonin & SUDEP. Epilepsy Curr. 2013, 13, 241–244. [Google Scholar]
- Benarroch, E.E. Adenosine and Its Receptors: Multiple Modulatory Functions and Potential Therapeutic Targets for Neurologic Disease. Neurology 2008, 70, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Zuchora, B.; Wielosz, M.; Urbańska, E.M. Adenosine A1 Receptors and the Anticonvulsant Potential of Drugs Effective in the Model of 3-Nitropropionic Acid-Induced Seizures in Mice. Eur. Neuropsychopharmacol. 2005, 15, 85–93. [Google Scholar] [CrossRef]
- Weltha, L.; Reemmer, J.; Boison, D. The Role of Adenosine in Epilepsy. Brain Res. Bull. 2019, 151, 46–54. [Google Scholar] [CrossRef]
- Shen, H.; Li, T.; Boison, D. A Novel Mouse Model for Sudden Unexpected Death in Epilepsy (SUDEP): Role of Impaired Adenosine Clearance. Epilepsia 2010, 51, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.; Kannan, K.; Faustin, A.; Shroff, S.; Thomas, C.; Heguy, A.; Serrano, J.; Snuderl, M.; Devinsky, O. Cardiac Arrhythmia and Neuroexcitability Gene Variants in Resected Brain Tissue from Patients with Sudden Unexpected Death in Epilepsy (SUDEP). NPJ Genom. Med. 2018, 3, 9. [Google Scholar] [CrossRef]
- Goldman, A.M.; Behr, E.R.; Semsarian, C.; Bagnall, R.D.; Sisodiya, S.; Cooper, P.N. Sudden Unexpected Death in Epilepsy Genetics: Molecular Diagnostics and Prevention. Epilepsia 2016, 57, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Gano, L.B.; Grabenstatter, H.L. Modulation of Abnormal Sodium Channel Currents in Heart and Brain: Hope for SUDEP Prevention and Seizure Reduction. Epilepsy Curr. 2017, 17, 306–310. [Google Scholar] [CrossRef]
- Wandschneider, B.; Koepp, M.; Scott, C.; Micallef, C.; Balestrini, S.; Sisodiya, S.M.; Thom, M.; Harper, R.M.; Sander, J.W.; Vos, S.B. Structural Imaging Biomarkers of Sudden Unexpected Death in Epilepsy. Brain 2015, 138, 2907–2919. [Google Scholar] [CrossRef]
- Wilson, C.L.; Isokawa, M.; Babb, T.L.; Crandall, P.H. Functional Connections in the Human Temporal Lobe: I. Analysis of Limbic System Pathways Using Neuronal Responses Evoked by Electrical Stimulation. Exp. Brain Res. 1990, 82, 279–292. [Google Scholar] [CrossRef]
- Rugg-Gunn, F.J.; Holdright, D. Epilepsy and the Heart. Br. J. Cardiol. 2010, 17, 223–229. [Google Scholar]
- Devinsky, O. Effects of Seizures on Autonomic and Cardiovascular Function. Epilepsy Curr. 2004, 4, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Koos, B.J.; Chau, A.; Matsuura, M.; Punla, O.; Kruger, L. Thalamic Locus Mediates Hypoxic Inhibition of Breathing in Fetal Sheep. J. Neurophysiol. 1998, 79, 2383–2393. [Google Scholar] [CrossRef]
- Saper, C.B. Convergence of Autonomic and Limbic Connections in the Insular Cortex of the Rat. J. Comp. Neurol. 1982, 210, 163–173. [Google Scholar] [CrossRef]
- Engel, J., Jr. Introduction to Temporal Lobe Epilepsy. Epilepsy Res. 1996, 26, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Bertram, E.H. Temporal Lobe Epilepsy: Where Do the Seizures Really Begin? Epilepsy Behav. 2009, 14, 32–37. [Google Scholar] [CrossRef]
- Sutula, T.; Cascino, G.; Cavazos, J.; Parada, I.; Ramirez, L. Mossy Fiber Synaptic Reorganization in the Epileptic Human Temporal Lobe. Ann. Neurol. 1989, 26, 321–330. [Google Scholar] [CrossRef]
- Wozny, C.; Gabriel, S.; Jandova, K.; Schulze, K.; Heinemann, U.; Behr, J. Entorhinal Cortex Entrains Epileptiform Activity in CA1 in Pilocarpine-Treated Rats. Neurobiol. Dis. 2005, 19, 451–460. [Google Scholar] [CrossRef]
- Steinhäuser, C.; Seifert, G.; Bedner, P. Astrocyte Dysfunction in Temporal Lobe Epilepsy: K⁺ Channels and Gap Junction Coupling. Glia 2012, 60, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Theodore, W.H.; Bhatia, S.; Hatta, J.; Fazilat, S.; DeCarli, C.; Bookheimer, S.Y.; Gaillard, W.D. Hippocampal Atrophy, Epilepsy Duration, and Febrile Seizures in Patients with Partial Seizures. Neurology 1999, 52, 132–132. [Google Scholar] [CrossRef]
- O’Brien, T.J.; So, E.L.; Meyer, F.B.; Parisi, J.E.; Jack, C.R. Progressive Hippocampal Atrophy in Chronic Intractable Temporal Lobe Epilepsy. Ann. Neurol. 1999, 45, 526–529. [Google Scholar] [CrossRef]
- Hudson, J.M.; Flowers, K.A.; Walster, K.L. Attentional Control in Patients with Temporal Lobe Epilepsy. J. Neuropsychol. 2014, 8, 140–146. [Google Scholar] [CrossRef]
- Meador, K.J. Cognitive Outcomes and Predictive Factors in Epilepsy. Neurology 2002, 58, S21–S26. [Google Scholar] [CrossRef] [PubMed]
- Bell, B.; Lin, J.J.; Seidenberg, M.; Hermann, B. The Neurobiology of Cognitive Disorders in Temporal Lobe Epilepsy. Nat. Rev. Neurol. 2011, 7, 154–164. [Google Scholar] [CrossRef] [PubMed]
- York, M.K.; Rettig, G.M.; Grossman, R.G.; Hamilton, W.J.; Armstrong, D.D.; Levin, H.S.; Mizrahi, E.M. Seizure Control and Cognitive Outcome after Temporal Lobectomy: A Comparison of Classic Ammon’s Horn Sclerosis, Atypical Mesial Temporal Sclerosis, and Tumoral Pathologies. Epilepsia 2003, 44, 387–398. [Google Scholar] [CrossRef]
- Pulliainen, V.; Kuikka, P.; Jokelainen, M. Motor and Cognitive Functions in Newly Diagnosed Adult Seizure Patients before Antiepileptic Medication: Cognitive Functions in Newly Diagnosed Seizure Patients. Acta Neurol. Scand. 2000, 101, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, S.E.; Park, C.; Yoo, J.H.; Lee, H.W. Gray and White Matter Volumes and Cognitive Dysfunction in Drug-Naïve Newly Diagnosed Pediatric Epilepsy. Biomed. Res. Int. 2015, 2015, 1–10. [Google Scholar] [CrossRef]
- Soria, F.N.; Pérez-Samartín, A.; Martin, A.; Gona, K.B.; Llop, J.; Szczupak, B.; Chara, J.C.; Matute, C.; Domercq, M. Extrasynaptic Glutamate Release through Cystine/Glutamate Antiporter Contributes to Ischemic Damage. J. Clin. Invest. 2014, 124, 3645–3655. [Google Scholar] [CrossRef]
- Kim, E.-H.; Ko, T.-S. Cognitive Impairment in Childhood Onset Epilepsy: Up-to-Date Information about Its Causes. Korean J. Pediatr. 2016, 59, 155–164. [Google Scholar] [CrossRef]
- Lewerenz, J.; Baxter, P.; Kassubek, R.; Albrecht, P.; Van Liefferinge, J.; Westhoff, M.-A.; Halatsch, M.-E.; Karpel-Massler, G.; Meakin, P.J.; Hayes, J.D.; Aronica, E.; Smolders, I.; Ludolph, A.C.; Methner, A.; Conrad, M.; Massie, A.; Hardingham, G.E.; Maher, P. Phosphoinositide 3-Kinases Upregulate System xc⁻ via Eukaryotic Initiation Factor 2α and Activating Transcription Factor 4 – A Pathway Active in Glioblastomas and Epilepsy. Antioxid. Redox Signal. 2014, 20, 2907–2922. [Google Scholar] [CrossRef]
- Sears, S.M.S.; Hewett, J.A.; Hewett, S.J. Decreased Epileptogenesis in Mice Lacking the System xc⁻ Transporter Occurs in Association with a Reduction in AMPA Receptor Subunit GluA1. Epilepsia Open 2019, 4, 133–143. [Google Scholar] [CrossRef]
- Leclercq, K.; Van Liefferinge, J.; Albertini, G.; Neveux, M.; Dardenne, S.; Mairet-Coello, G.; Vandenplas, C.; Deprez, T.; Chong, S.; Foerch, P.; Bentea, E.; Sato, H.; Maher, P.; Massie, A.; Smolders, I.; Kaminski, R.M. Anticonvulsant and Antiepileptogenic Effects of System Xc⁻ Inactivation in Chronic Epilepsy Models. Epilepsia 2019, 60, 1412–1423. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-F.; Sonsalla, P.K.; Pedata, F.; Melani, A.; Domenici, M.R.; Popoli, P.; Geiger, J.; Lopes, L.V.; De Mendonça, A. Adenosine A2A Receptors and Brain Injury: Broad Spectrum of Neuroprotection, Multifaceted Actions and “Fine Tuning” Modulation. Prog. Neurobiol. 2007, 83, 310–331. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.V.; Cunha, R.A.; Kull, B.; Fredholm, B.B.; Ribeiro, J.A. Adenosine A2A Receptor Facilitation of Hippocampal Synaptic Transmission Is Dependent on Tonic A1 Receptor Inhibition. Neuroscience 2002, 112, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.-Y.; Baer, S.B.; Gesese, R.; Cook, J.M.; Weltha, L.; Coffman, S.Q.; Wu, J.; Chen, J.-F.; Gao, M.; Ji, T. Adenosine-A2A Receptor Signaling Plays a Crucial Role in Sudden Unexpected Death in Epilepsy. Front. Pharmacol. 2022, 13, 910535. [Google Scholar] [CrossRef] [PubMed]
- Pitkänen, A.; Sutula, T.P. Is Epilepsy a Progressive Disorder? Prospects for New Therapeutic Approaches in Temporal-Lobe Epilepsy. Lancet Neurol. 2002, 1, 173–181. [Google Scholar] [CrossRef]
- Boison, D. Adenosinergic Signaling in Epilepsy. Neuropharmacology 2016, 104, 131–139. [Google Scholar] [CrossRef]
- Dragunow, M. Purinergic Mechanisms in Epilepsy. Prog. Neurobiol. 1988, 31, 85–108. [Google Scholar] [CrossRef]
- Ochiishi, T.; Takita, M.; Ikemoto, M.; Nakata, H.; Suzuki, S.S. Immunohistochemical Analysis on the Role of Adenosine A1 Receptors in Epilepsy. Neuroreport 1999, 10, 3535–3541. [Google Scholar] [CrossRef]
- Young, D.; Dragunow, M. Status Epilepticus May Be Caused by Loss of Adenosine Anticonvulsant Mechanisms. Neuroscience 1994, 58, 245–261. [Google Scholar] [CrossRef]
- Von Lubitz, D.K. Adenosine in the Treatment of Stroke: Yes, Maybe, or Absolutely Not? Expert Opin. Investig. Drugs 2001, 10, 619–632. [Google Scholar] [CrossRef]
- Barros-Barbosa, A.R.; Ferreirinha, F.; Oliveira, Â.; Mendes, M.; Lobo, M.G.; Santos, A.; Rangel, R.; Pelletier, J.; Sévigny, J.; Cordeiro, J.M.; Correia-de-Sá, P. Adenosine A2A Receptor and Ecto-5′-Nucleotidase/CD73 Are Upregulated in Hippocampal Astrocytes of Human Patients with Mesial Temporal Lobe Epilepsy (MTLE). Purinergic Signal. 2016, 12, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Rebola, N.; Lujan, R.; Cunha, R.A.; Mulle, C. Adenosine A2A Receptors Are Essential for Long-Term Potentiation of NMDA-EPSCs at Hippocampal Mossy Fiber Synapses. Neuron 2008, 57, 121–134. [Google Scholar] [CrossRef]
- Marchi, M.; Raiteri, L.; Risso, F.; Vallarino, A.; Bonfanti, A.; Monopoli, A.; Ongini, E.; Raiteri, M. Effects of Adenosine A1 and A2A Receptor Activation on the Evoked Release of Glutamate from Rat Cerebrocortical Synaptosomes. Br. J. Pharmacol. 2002, 136, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.A. How Does Adenosine Control Neuronal Dysfunction and Neurodegeneration? J. Neurochem. 2016, 139, 1019–1055. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-K.; Choi, S.-S.; Han, K.-J.; Han, E.-J.; Suh, H.-W. Roles of Adenosine Receptors in the Regulation of Kainic Acid-Induced Neurotoxic Responses in Mice. Mol. Brain Res. 2004, 125, 76–85. [Google Scholar] [CrossRef]
- Rosim, F.E.; Persike, D.S.; Nehlig, A.; Amorim, R.P.; de Oliveira, D.M.; da Silva Fernandes, M.J. Differential Neuroprotection by A1 Receptor Activation and A2A Receptor Inhibition Following Pilocarpine-Induced Status Epilepticus. Epilepsy Behav. 2011, 22, 207–213. [Google Scholar] [CrossRef]
- Houser, C.R.; Miyashiro, J.E.; Swartz, B.E.; Walsh, G.O.; Rich, J.R.; Delgado-Escueta, A.V. Altered Patterns of Dynorphin Immunoreactivity Suggest Mossy Fiber Reorganization in Human Hippocampal Epilepsy. J. Neurosci. 1990, 10, 267–282. [Google Scholar] [CrossRef]
- Lynd-Balta, E.; Pilcher, W.H.; Joseph, S.A. AMPA Receptor Alterations Precede Mossy Fiber Sprouting in Young Children with Temporal Lobe Epilepsy. Neuroscience 2004, 126, 105–114. [Google Scholar] [CrossRef]
- Wang, I.-T.J.; Allen, M.; Goffin, D.; Zhu, X.; Fairless, A.H.; Brodkin, E.S.; Siegel, S.J.; Marsh, E.D.; Blendy, J.A.; Zhou, Z. Loss of CDKL5 Disrupts Kinome Profile and Event-Related Potentials Leading to Autistic-like Phenotypes in Mice. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 21516–21521. [Google Scholar] [CrossRef]
- Amendola, E.; Zhan, Y.; Mattucci, C.; Castroflorio, E.; Calcagno, E.; Fuchs, C.; Lonetti, G.; Silingardi, D.; Vyssotski, A.L.; Farley, D.; Ciani, E.; Pizzorusso, T.; Gross, C.T.; Giustetto, M. Mapping Pathological Phenotypes in a Mouse Model of CDKL5 Disorder. PLOS ONE 2014, 9, e91613. [Google Scholar] [CrossRef]
- Okuda, K.; Kobayashi, S.; Fukaya, M.; Watanabe, A.; Murakami, T.; Hagiwara, M.; Sato, T.; Ueno, H.; Ogonuki, N.; Komano-Inoue, S. CDKL5 Controls Postsynaptic Localization of GluN2B-Containing NMDA Receptors in the Hippocampus and Regulates Seizure Susceptibility. Neurobiol. Dis. 2017, 106, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Yennawar, M.; White, R.S.; Jensen, F.E. AMPA Receptor Dysregulation and Therapeutic Interventions in a Mouse Model of CDKL5 Deficiency Disorder. J. Neurosci. 2019, 39, 4814–4828. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Terzic, B.; Wang, I.-T.J.; Sarmiento, N.; Sizov, K.; Cui, Y.; Takano, H.; Marsh, E.D.; Zhou, Z.; Coulter, D.A. Altered NMDAR Signaling Underlies Autistic-like Features in Mouse Models of CDKL5 Deficiency Disorder. Nat. Commun. 2019, 10, 2655. [Google Scholar] [CrossRef] [PubMed]
- Koubeissi, M.Z.; Abou-Khalil, B. Autonomic and Cardiac Risk Factors for SUDEP. Clin. Neurophysiol. 2020, 131, 1351–1360. [Google Scholar]
- Glasscock, E. Cardiac Dysfunction in Epilepsy: From Cellular Mechanisms to Clinical Risk. Epilepsia 2021, 62, 234–248. [Google Scholar]
- Ryvlin, P.; Nashef, L.; Lhatoo, S.D.; Bateman, L.M.; Bird, J.; Bleasel, A.; et al. Incidence and Mechanisms of Cardiorespiratory Arrests in Epilepsy Monitoring Units (MORTEMUS): A Retrospective Study. Lancet Neurol. 2013, 12, 966–977. [Google Scholar] [CrossRef]
- Bagnall, R.D.; Crompton, D.E.; Petrovski, S.; et al. Exome-Based Analysis of Cardiac Arrhythmia, Respiratory Control, and Epilepsy Genes in SUDEP. Ann. Neurol. 2016, 79, 522–534. [Google Scholar] [CrossRef]
- Massey, C.A.; Sowers, L.P.; Dlouhy, B.J.; Richerson, G.B. Mechanisms of SUDEP: The Pathway to Prevention. Nat. Rev. Neurol. 2014, 10, 271–282. [Google Scholar] [CrossRef]
- Bozorgi, A.; Jafari, A.; Massey, C.A. Ictal and Postictal Arrhythmias in Epilepsy and SUDEP Risk. Epilepsy Res. 2021, 172, 106577. [Google Scholar]
- Surges, R.; Sander, J.W.; Thijs, R.D.; Tan, H.L.; Sander, J.W. Sudden Unexpected Death in Epilepsy: Risk Factors and Potential Pathomechanisms. Nat. Rev. Neurol. 2009, 5, 492–504. [Google Scholar] [CrossRef]
- Devinsky, O.; Hesdorffer, D.C.; Thurman, D.J.; Lhatoo, S.; Richerson, G. Sudden Unexpected Death in Epilepsy: Epidemiology, Mechanisms, and Prevention. Lancet Neurol. 2016, 15, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Verrier, R.L.; Pang, T.D.; Nearing, B.D.; Schachter, S.C. The Epileptic Heart: Concept and Clinical Evidence. Epilepsy Behav. 2020, 105, 106946. [Google Scholar] [CrossRef]
- Stöllberger, C.; Finsterer, J. Cardiorespiratory Findings in Sudden Unexplained/Unexpected Death in Epilepsy (SUDEP). Epilepsy Res. 2004, 59, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Delogu, A.B.; Spinelli, A.; Battaglia, D.; Dravet, C.; De Nisco, A.; Saracino, A.; Romagnoli, C.; Lanza, G.A.; Crea, F. Electrical and Autonomic Cardiac Function in Patients with Dravet Syndrome. Epilepsia 2011, 52 (Suppl. 2), 55–58. [Google Scholar] [CrossRef] [PubMed]
- Trosclair, K.; Dhaibar, H.A.; Gautier, N.M.; Mishra, V.; Glasscock, E. Neuron-Specific Kv1.1 Deficiency Is Sufficient to Cause Epilepsy, Premature Death, and Cardiorespiratory Dysregulation. Neurobiol. Dis. 2020, 137, 104759. [Google Scholar] [CrossRef]
- Thijs, R.D.; Surges, R.; O'Brien, T.J.; Sander, J.W. Epilepsy in Adults. Lancet 2019, 393, 689–701. [Google Scholar] [CrossRef]
- Rossi, M.; Janse van Rensburg, M.; Santangelo, G.; Ricciardi, D.; Vico, C.; Russo, M.; et al. Wearable Devices in Epilepsy: From Seizure Detection to SUDEP Prevention. Epilepsy Behav. 2021, 104, 106935. [Google Scholar]
- Esmaeili, B.; Kaffashi, F.; Theeranaew, W.; Dabir, A.; Lhatoo, S.D.; Loparo, K.A. Post-Ictal Modulation of Baroreflex Sensitivity in Patients with Intractable Epilepsy. Front. Neurol. 2018, 9, 793. [Google Scholar] [CrossRef]
- Glasscock, E. Kv1.1 Potassium Channel Deficiency Reveals Brain-Driven Cardiac Dysfunction as a Candidate Mechanism for Sudden Unexplained Death in Epilepsy. J. Neurosci. 2010, 30, 5167–5175. [Google Scholar] [CrossRef]
- Lotufo, P.A.; Valiengo, L.; Benseñor, I.M.; Brunoni, A.R. A Systematic Review and Meta-Analysis of Heart Rate Variability in Epilepsy and Antiepileptic Drugs. Epilepsia 2012, 53, 272–282. [Google Scholar] [CrossRef]
- Zaccara, G.; Franciotta, D.; Lattanzi, S. The Seizure-Heart Connection: Perspectives and Potential Impact of Heart Rate Variability (HRV) in Epilepsy. Epilepsia 2018, 59, 1969–1981. [Google Scholar]
- Moseley, B.D.; Nickels, K.C.; Britton, J.W.; et al. Sudden Unexpected Death in Epilepsy: An Analysis of 24 Years of Autopsies from a Single Center. Epilepsia 2013, 54, 119–125. [Google Scholar]
- Hirsch, L.J.; LaRoche, S.M.; Gaspard, N.; et al. Risk Factors for Sudden Unexpected Death in Epilepsy: Current Understanding and Future Directions. Epilepsy Curr. 2021, 21, 13–23. [Google Scholar]
- Aurlien, D.; Leren, T.P.; Taubøll, E.; Gjerstad, L. New Directions in the Prevention of Sudden Unexpected Death in Epilepsy. Seizure 2016, 38, 30–36. [Google Scholar]
- Nashef, L.; So, E.L.; Ryvlin, P.; et al. Unifying the Definitions of Sudden Unexpected Death in Epilepsy. Epilepsia 2012, 53, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Dewar, S.; Moura, F.; Thompson, R.; et al. Postictal Arrhythmias: Implications for Monitoring and Prevention of SUDEP. Epileptic Disord. 2022, 24, 701–709. [Google Scholar]
- Nei, M.; Ho, R.T.; Abou-Khalil, B.W.; Drislane, F.W.; Liporace, J.D.; Romeo, A.; Schmitt, S.E.; Singh, G.; Spencer, D.C.; Sperling, M.R.; et al. Postictal Autonomic Dysfunction and Cardiac Arrhythmias in Epilepsy. Neurology 2023, 100, e1804–e1815. [Google Scholar]
- Cui, W.; Wang, Y.; Hu, Y.; Gao, Y.; Zhang, Y.; Li, Y.; Wang, J. Wearable Electrocardiogram Monitoring Devices for Epilepsy Patients: Advancing Real-Time Seizure Management. J. Biomed. Inform. 2021, 121, 103882. [Google Scholar]
- van Westrhenen, A.; Geertsema, E.E.; van Rootselaar, A.F.; Stolker, R.J.; Hendrikse, J. Cardiac Effects of Epilepsy and Antiepileptic Drugs: A Systematic Review. CNS Drugs 2020, 34, 233–256. [Google Scholar]
- Beniczky, S.; Ryvlin, P. Standards for Testing and Clinical Validation of Seizure Detection Devices. Epilepsia 2018, 59 (Suppl 1), 9–13. [Google Scholar] [CrossRef]
- Myers, K.A.; Bello-Espinosa, L.; Ladbon-Bernard, D.; Farrell, K.; Gofton, T.; Mackenzie, A.; McLachlan, R.S.; Wirrell, E.C. Cardiac Monitoring in Epilepsy: Are We Utilizing It Effectively? Seizure 2018, 58, 47–54. [Google Scholar]
- Magruder, K.P.; Grant, L.; Spencer, D.C. Wearable Devices for Epilepsy: The Use of Heart Rate and Machine Learning in Monitoring Seizures. Epilepsy Res. 2019, 156, 106181. [Google Scholar]
- Rugg-Gunn, F.J. Adapting to New Technologies in Epilepsy Monitoring: The Future of Seizure Detection. J. Neurol. Neurosurg. Psychiatry 2021, 92, 301–310. [Google Scholar]
- Surges, R.; Strzelczyk, A.; Scott, C.A.; Walker, M.C. Postictal Generalized EEG Suppression and SUDEP: Mechanisms and Implications. Epilepsy Behav. 2018, 86, 123–127. [Google Scholar]
- Cogan, D.; Birjandtalab, J.; Nourani, M. Machine Learning Applications in Seizure Prediction and Epilepsy Management. IEEE Rev. Biomed. Eng. 2020, 13, 30–46. [Google Scholar]
- Milosevic, M.; Van de Vel, A.; Bonroy, B.; Ceulemans, B.; Lagae, L.; Van Huffel, S.; Vanrumste, B. Automated Detection of Tonic-Clonic Seizures Using a Wearable Accelerometer Device. Epilepsia 2021, 62, 1187–1196. [Google Scholar]
- Conradsen, I.; Beniczky, S.; Wolf, P.; Kjaer, T.W.; Sams, T.; Sørensen, H.B.D. Automatic Multi-Modal Intelligent Seizure Acquisition (MISA) System for Detection of Motor Seizures from Electromyographic Data and Motion Data. Comput. Methods Programs Biomed. 2012, 107, 97–110. [Google Scholar] [CrossRef]
- Vilella, L.; Lacuey, N.; Hampson, J.P.; Rani, M.S.; Vyas, K.; Hupp, N.J.; et al. Incidence, Recurrence, and Risk Factors for Peri-Ictal Central Apnea and Sudden Unexpected Death in Epilepsy. Front. Neurol. 2019, 10, 166. [Google Scholar] [CrossRef]
- Devinsky, O.; Friedman, D.; Lai, Y.C.; Macher, K.; Tran, T.T.; Philbrook, B. Combined Cardiac and Respiratory Monitoring to Predict Sudden Unexpected Death in Epilepsy. Epilepsia 2018, 59, 1904–1914. [Google Scholar]
- Shorvon, S.; Tomson, T. Sudden Unexpected Death in Epilepsy. Lancet 2011, 378, 2028–2038. [Google Scholar] [CrossRef]
- Hesdorffer, D.C.; Tomson, T.; Benn, E.; Sander, J.W.; Nilsson, L.; Langan, Y. Combined Analysis of Risk Factors for SUDEP. Epilepsia 2020, 61, e27–e32. [Google Scholar] [CrossRef] [PubMed]
- and Prevention. Curr. Opin. Neurol. 2012, 25, 201–207.
- Lhatoo, S.D.; Faulkner, H.J.; Dembny, K.; Trippick, K.; Johnson, C.; Bird, J.M. An Electroclinical Case-Control Study of Sudden Unexpected Death in Epilepsy. Ann. Neurol. 2010, 68, 787–796. [Google Scholar] [CrossRef]
- Purnell, B.S.; Thijs, R.D.; Buchanan, G.F. Deadly Rhythms: Neurological Modulation of Heart Rate Precedes Sudden Unexpected Death in Epilepsy. Front. Neurol. 2021, 12, 654576. [Google Scholar]
- Ali, A.; Ali, I.; Keil, A.; Diaz, G.; Kabbani, H.; Detyniecki, K. Peri-Ictal Breathing Patterns and SUDEP Risk. Epilepsy Behav. 2017, 77, 96–100. [Google Scholar]
- Lucey, B.P.; Nelson, S.E.; Lopez, J.E.; Howlett, J.A.; Green, A.L.; Koehler, P.J. Cardiorespiratory Changes Associated with Seizures in Adults. J. Clin. Neurophysiol. 2016, 33, 282–289. [Google Scholar]
- Nobili, L.; Proserpio, P.; Cicolin, A.; Sforza, E.; Braghiroli, A.; Nobili, F. Cardiorespiratory and Autonomic Alterations in Epilepsy: Implications for SUDEP Prevention. Clin. Neurophysiol. Pract. 2020, 5, 129–138. [Google Scholar]
- Richerson, G.B.; Buchanan, G.F. The Serotonin Axis: Shared Mechanisms in Seizures, Depression, and SUDEP. Epilepsia 2011, 52 (Suppl 1), 28–38. [Google Scholar] [CrossRef]
- Tupal, S.; Faingold, C.L. Serotonin and Sudden Death: Differential Effects of Serotonin Receptor Subtypes in the Brainstem. Neurosci. Lett. 2018, 703, 147–153. [Google Scholar]
- Glasscock, E. The Role of Respiratory and Autonomic Dysfunction in Sudden Unexpected Death in Epilepsy (SUDEP). J. Physiol. 2018, 596, 2247–2260. [Google Scholar]
- Kinney, H.C.; Poduri, A.H.; Cryan, J.B.; Haynes, R.L.; Teot, L.; Sleeper, L.A.; Holm, I.A.; Berry, G.T.; Prabhu, S.P.; Warfield, S.K.; Brownstein, C. Hippocampal Formation Maldevelopment and Sudden Unexpected Death Across the Pediatric Age Spectrum. J. Neuropathol. Exp. Neurol. 2016, 75, 981–997. [Google Scholar] [CrossRef]
- Dempsey, J.A.; Smith, C.A.; Blain, G.M.; Xie, A. Role of Chemoreception in Sudden Unexpected Death in Epilepsy (SUDEP). J. Physiol. 2020, 598, 2335–2347. [Google Scholar]
- Dewar, S.; Moura, F.; Thompson, R.; et al. Postictal Arrhythmias: Implications for Monitoring and Prevention of SUDEP. Epileptic Disord. 2022, 24, 701–709. [Google Scholar]
- Cooper, M.S.; McIntosh, A.; Crompton, D.E.; Campbell, C.A.; Brodie, M.J.; Berkovic, S.F.; et al. Mortality in Dravet Syndrome. Epilepsy Res. 2021, 169, 106525. [Google Scholar] [CrossRef] [PubMed]
- Goldman, A.M.; Glasscock, E.; Yoo, J.; Chen, T.T.; Klassen, T.L.; Noebels, J.L. Arrhythmia in Heart and Brain: KCNQ1 Mutations Link Epilepsy and Sudden Unexplained Death. Sci. Transl. Med. 2009, 1, 2ra6. [Google Scholar] [CrossRef]
- Hanna, P.; Blackwell, K.; Taylor, J.C.; Hall, A.; Kiss, S.; Florez-Caravas, J.A.; et al. Genetic Risk Factors for Sudden Unexpected Death in Epilepsy: A Case-Control Sequencing Study. Lancet Neurol. 2022, 21, 650–659. [Google Scholar]
Figure 4.
Pathway from PGES to SUDEP via brainstem dysfunction, respiratory failure, and cardiac instability.
Figure 4.
Pathway from PGES to SUDEP via brainstem dysfunction, respiratory failure, and cardiac instability.
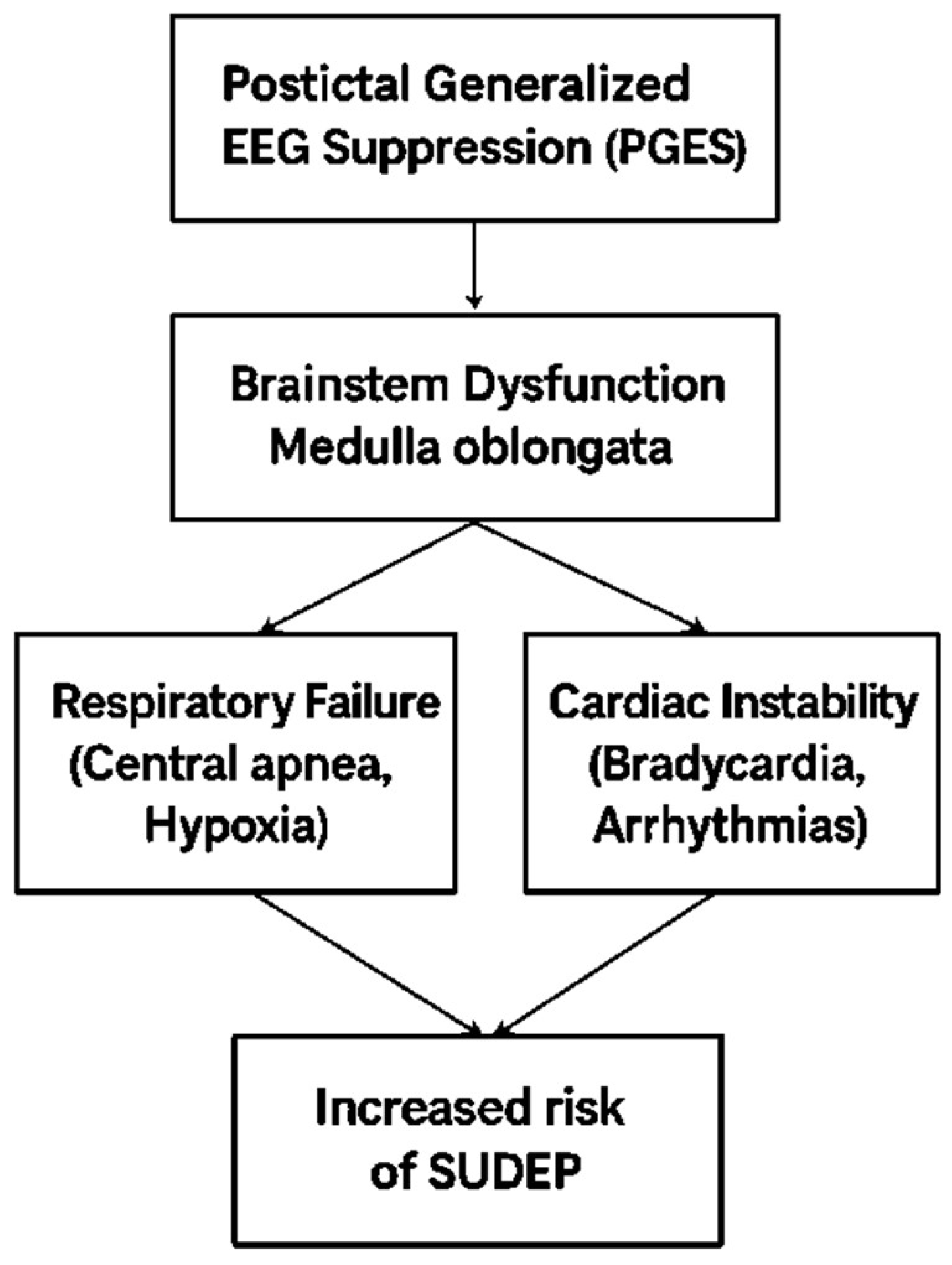
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



