
Submitted:
14 May 2025
Posted:
15 May 2025
You are already at the latest version
Abstract
In chronic respiratory diseases (CRDs), oxidative stress and inflammation exhibit a strong pathophysiological interplay, playing essential roles in the onset and progression of these conditions and their associated comorbidities. Oxidative stress can activate inflammatory pathways, leading to sustained airway inflammation. In turn, chronic inflammation can increase the production of reactive oxygen species (ROS), thereby exacerbating oxidative stress. This vicious cycle contributes to airway remodeling, reduced lung function, and disease progression. This review emphasizes the central roles of inflammation and oxidative stress in two of the most prevalent hypoxemic conditions, chronic obstructive pulmonary disease (COPD) and obstructive sleep apnea (OSA), as well as their involvement in the development of major comorbidities. A comprehensive understanding of these processes is crucial to developing therapeutic strategies aimed at reducing oxidative stress and inflammation for the effective management and treatment of CRDs and their comorbidities.
It also underscores the crucial role of hypoxia and its transcriptional effects, mediated by hypoxia-inducible factors (HIF), as a primary driver of disease progression through the promotion of oxidative stress and inflammation. Another key element is the rapid advancement of Artificial Intelligence (AI), which presents promising strategies to mitigate the global burden of CRDs. AI holds potential for reducing healthcare costs while improving diagnostic specificity, enabling earlier detection, and optimizing disease monitoring and management. This review explores the current landscape and future prospects of AI in transforming the diagnosis, clinical management, and prognosis of these respira-tory disorders.
Keywords:
Chronic obstructive respiratory disease
; obstructive sleep apnea
; hypoxia
; HIF
; oxidative stress
; inflammation
; comorbidities
; cancer
; pulmonary hypertension
; artificial intelligence
1. Introduction
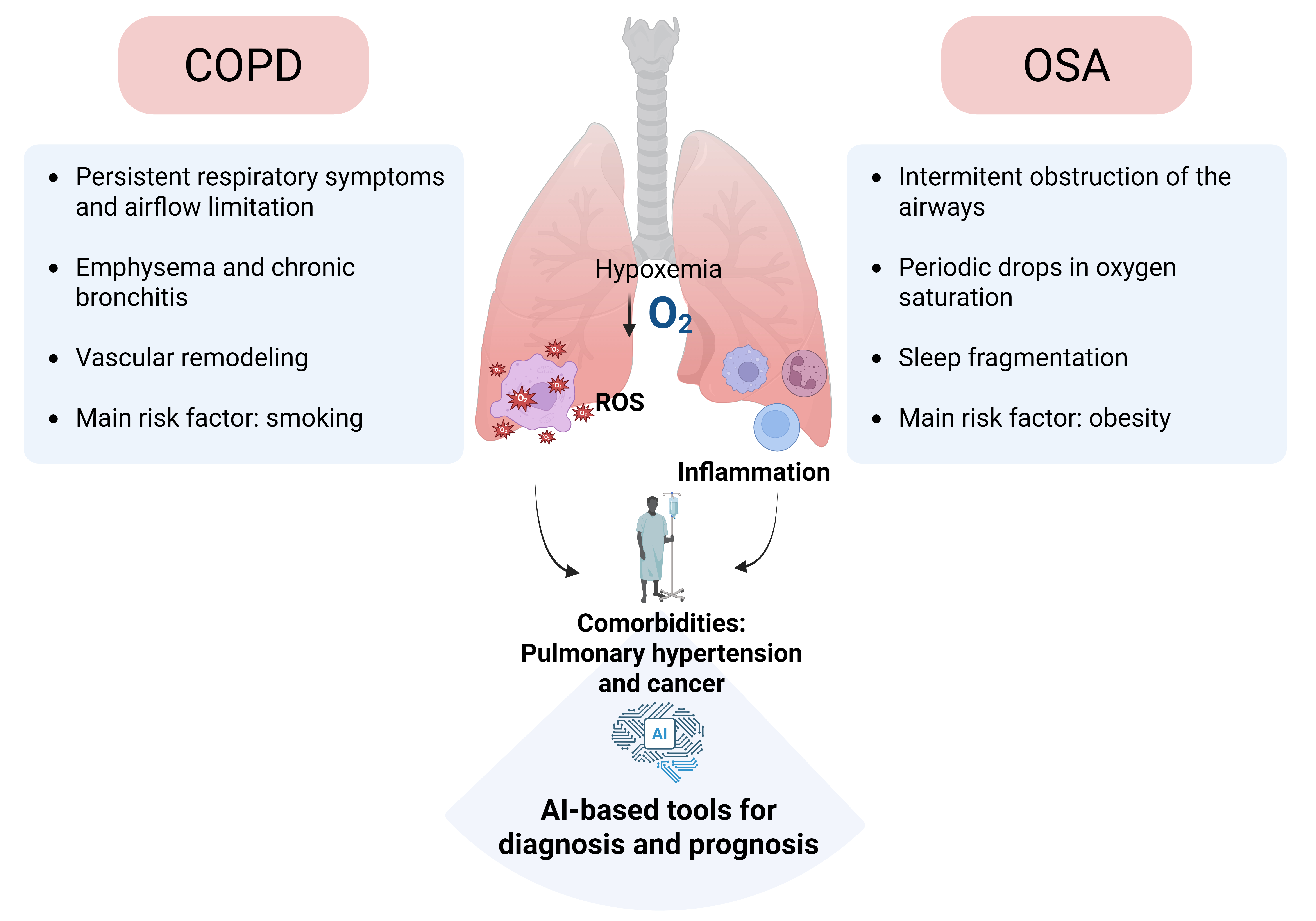
Overview of chronic respiratory diseases and comorbidities related to COPD and OSA. Globally, chronic respiratory diseases (CRDs) are estimated to cause one death every 10 seconds (equivalent to 3 million annually) and are projected to become the third leading cause of mortality by 2030, according to the World Health Organization (WHO). Data from the Global Burden of Diseases, Injuries, and Risk Factors Study [1] indicates that total deaths and the prevalence of CRDs have increased by 28.5% and 39.8%, respectively [1]. Chronic obstructive pulmonary disease (COPD) and obstructive sleep apnea (OSA) are highly prevalent chronic respiratory conditions associated with significant morbidity. The global prevalence of COPD among adults is estimated at approximately 12% [2]; [3], whereas the prevalence of OSA may reach up to 38% [4]. Both conditions are characterized by reduced arterial oxygen tension (hypoxemia), increased production of ROS, and an exacerbated inflammatory response, all of which shape the clinical manifestations observed in these patients. Notably, the clinical course of these individuals is highly variable and largely influenced by the development of comorbidities, including cardiovascular, metabolic, and neoplastic diseases, that are often the primary cause of mortality. Despite this, the underlying mechanisms that govern the onset and progression of these comorbidities remain poorly defined, and robust biomarkers to predict comorbidome risk have yet to be identified.
COPD is defined by the presence of respiratory symptoms and persistent airflow limitation resulting from structural abnormalities in the airways and/or alveoli [5]. Although traditionally associated with older adults, COPD can also arise from early-life developmental alterations [6,7]. While smoking remains the primary etiological factor and is strongly associated with adverse clinical outcomes and increased mortality in genetically susceptible individuals worldwide, only approximately 15% of smokers develop an exaggerated local inflammatory response to ROS in cigarette smoke. This leads to irreversible pulmonary damage [8,9] and the emergence of a systemic pro-inflammatory state [10]. This pathological condition contributes to the progression and prognosis of COPD, promoting the development of comorbidities linked to systemic dysfunction, reduced quality of life, increased hospitalizations, suboptimal therapeutic responses, and elevated mortality rates [11].
Frequent comorbidities observed in COPD patients include cardiovascular diseases, metabolic disorders, pulmonary fibrosis, and lung cancer [12,13,14]. It is estimated that 78.6% of individuals with COPD present with at least one clinically relevant comorbidity, 68.8% with two or more, and nearly 47.9% with three or more. Although patients with advanced airflow limitation often succumb to respiratory failure, the majority of deaths among COPD patients are due to non-respiratory causes, chiefly cardiovascular disease and cancer [13]. Despite these clinical correlations, the pathophysiological mechanisms underlying the association between COPD and its comorbidities remain poorly understood. Beyond aging, sedentary lifestyle, and tobacco use, both hypoxia and systemic inflammation, hallmarks of COPD, are believed to significantly contribute to the high burden of comorbid conditions in this population [15,16].
OSA is another prevalent respiratory disorder characterized by repeated collapse of the upper airways during sleep, leading to intermittent hypoxia (periodic drops in oxygen saturation) and sleep fragmentation (micro-awakenings). These disturbances result in non-restorative sleep, causing fatigue and daytime sleepiness, and contribute to the development of respiratory, neuropsychiatric, metabolic, cardiovascular, and oncological disorders [17]. OSA is also associated with a high mortality rate, primarily linked to daytime sleepiness and the onset of comorbidities, particularly cardiovascular and metabolic conditions [18,19,20,21]. Severe OSA has been specifically associated to an increased risk of dyslipidemia, non-alcoholic fatty liver disease, and, most notably, diabetes [22]. Additionally, it is also associated with a higher risk of arrhythmias, hypertension, atherosclerosis, ischemic heart disease, and stroke [23,24]. Although the high prevalence of hypertension in OSA patients, exceeding 50% in some cohorts, has been proposed as the main driver of its cardiovascular complications [25], other risk factors, particularly those promoting atherosclerosis, also play a critical role [26,27]. Among these, dyslipidemia and inflammation, both highly prevalent in OSA, stand out as key pro-atherosclerotic contributors [28,29].
Moreover, several epidemiological studies, patient cohort analyses, and animal models have shown that OSA is associated with an increased incidence, aggressiveness, and mortality of various types of cancer [30,31]. Intermittent hypoxia has been proposed as a potential mechanism, promoting tumor vascularization, facilitating tumor growth, and modulating immune responses by disrupting immune surveillance, thus allowing the emergence of more aggressive tumors [32,33,34]. In addition, the repetitive cycles of hypoxia and reoxygenation alter redox homeostasis in OSA, promoting ROS production and reducing endogenous antioxidant defenses. This oxidative imbalance correlates with disease severity [35].
The co-occurrence of COPD and OSA, referred to as overlap syndrome, is relatively common and presents a unique pattern of nocturnal hypoxemia, leading to worse clinical outcomes than either condition alone. Recent studies have demonstrated that patients with overlap syndrome exhibit elevated levels of systemic inflammatory biomarkers compared to individuals with isolated COPD or OSA, potentially contributing to the increased cardiovascular risk observed in this subgroup [36]. However, inflammation alone may not fully account for the heightened cardiovascular risk, highlighting the need for further investigation into alternative pathogenic mechanisms.
In the context of CRDs, persistent hypoxic conditions trigger a key adaptive response mediated by transcriptional programs governed primarily by hypoxia-inducible factors (HIFs). HIF-1α and HIF-2α are central mediators of cellular adaptation to low oxygen levels, regulating a wide array of genes involved in essential adaptive processes [37,38]. However, in the context of CRDs, this regulation is complex and significantly contributes to disease pathogenesis and progression. While initially protective, chronic activation of HIF pathways can become maladaptive, initiating a cascade of detrimental effects. In this review, we highlight several HIF-mediated pathophysiological responses, with a particular focus on pulmonary vascular adaptation to hypoxia.
Artificial Intelligence (AI), and specifically Machine Learning (ML), has emerged as a transformative tool across multiple disciplines, including medicine. Its applications range from disease diagnosis and risk prediction to treatment optimization. Recent advances in Deep Learning (DL) and Generative AI, coupled with the availability of large-scale multimodal datasets and increased computational power, have opened new frontiers in medical research and practice. These technologies are becoming especially relevant in respiratory medicine, significantly improving diagnosis and prognosis in diseases such as COPD [39] and OSA [40].
ML models, which learn functional relationships between input data (e.g., clinical features, physiological signals, or imaging) and clinical outcomes, have evolved from traditional algorithms, such as random forests, support vector machines, and logistic regression, to more complex DL architectures capable of processing high-dimensional data, including medical images and biosignals [39,40,41,42]. Traditional ML approaches have demonstrated high efficacy in analyzing temporal physiological signals from portable sensors, imaging data (notably radiomics features from chest CT scans), and structured clinical information, achieving robust diagnostic and prognostic performance for COPD and OSA. These models have been particularly effective in predicting exacerbations, disease severity, and mortality by integrating diverse data sources, sometimes outperforming deep learning methods in low-dimensional, standardized clinical datasets. DL models [43], including convolutional neural networks (CNNs), recurrent neural networks (RNNs), and transformer-based architectures [44], have further enhanced the analysis of complex data such as medical images, audio, and sequential signals, often achieving or exceeding expert-level performance and receiving regulatory approval for autonomous diagnostic use. In COPD and OSA, DL has improved the classification of disease phenotypes, early risk prediction, and accurate event detection from polysomnography and ECG signals, while also enabling advanced image-based diagnostics and prognosis. Recently, foundational models, large, self-supervised neural networks such as transformers, have emerged, capable of integrating multimodal data (text, imaging, biosignals) and adapting to diverse clinical tasks via transfer learning. The recent advances illustrate the potential of these models to unify and advance diagnostic and prognostic frameworks in respiratory medicine, although their full clinical impact is still under investigation. Overall, the integration of AI methodologies is transforming respiratory medicine, enabling more precise, data-driven clinical decision-making and fostering the development of generalist models for comprehensive patient care.
2. Role of Oxidative Stress and Inflammation in CRDS
As mentioned above, oxidative stress is a key mechanism in CRDs development, making the identification of oxidative stress-related biomarkers beneficial for improving its diagnosis and treatment. Repeated cycles of hypoxia and reoxygenation, along with exposure to indoor and outdoor air pollutants, and other toxic chemicals, are key factors that stimulate the endogenous production of ROS in respiratory diseases such as OSA and COPD [9,45,46]. At low concentrations, ROS act as secondary messengers, playing a crucial role in redox signaling and regulating a variety of essential physiological processes, including bacterial killing during phagocytosis, vasodilation, tissue repair, and regeneration [47,48]. However, when present at elevated levels, ROS can cause irreversible damage to cellular biomolecules, such as lipids, carbohydrates, and nucleic acids, impairing their function, altering cellular metabolism, and ultimately leading to cell and tissue injury [45]. If not properly regulated, oxidative stress can drive the onset and progression of inflammatory conditions, particularly those affecting the respiratory system [49].
Recent findings from multivariate logistic regression models have revealed a negative correlation between oxidative balance scores and the likelihood of COPD prevalence [50]. This imbalance is partly attributable to free radicals in cigarette smoke, which enhance endogenous ROS production and trigger inflammatory responses at the cellular level. Consequently, the respiratory systems of smokers exhibit age-related characteristics, partially due to increased oxidative stress, accumulation of damaged proteins, and elevated inflammation. On this matter, expression of the ROS generating enzyme NADPH oxidase (NOX)-4 is enhanced in airway smooth muscle from patients with COPD, which is also correlated with disease severity and lung function decline [51]. Indeed, in end-stage COPD, not only NOX4 but also other isoforms such as NOX1, NOX2, and NOX5 remain active. Among them, NOX1 and NOX4 are notably involved in oxidative stress and inflammation following acute exposure to cigarette smoke [52]. Thus, NOX enzymes are considered potential therapeutic targets for COPD. Supporting this, apocynin, a selective NOX inhibitor partially improved cigarette smoke-induced lung neutrophilia and reversed systemic inflammation and oxidative stress [53] in mice, although clinical trials have not been reported yet in COPD.
COPD patients exhibit a distinctive inflammatory profile characterized by elevated levels of macrophages, neutrophils, and both T and B lymphocytes in airway secretions [54,55]. This inflammatory response involves both the innate and adaptive immune systems, interconnected through dendritic cell activation. Compared to smokers without airway obstruction, COPD patients show a heightened inflammatory response that persists even after smoking cessation [56]. Although smoking is the major environmental risk factor, only a subset of smokers develops COPD, likely due to genetic predispositions, epigenetic modifications, and oxidative stress, all of which may amplify inflammation [55]. Persistent oxidative stress and inflammation accelerate epithelial cell senescence, weaken barrier integrity, and drive chronic airway inflammation and tissue remodeling [57]. During acute exacerbations, systemic inflammation often worsens [58,59], and population studies link elevated systemic inflammatory markers to higher risks of diabetes, cardiovascular disease, lung cancer, and mortality [56,60].
OSA is another chronic respiratory condition wherein oxidative stress and inflammation play significant roles. The cyclical pattern of oxygen deprivation and reoxygenation intrinsic to OSA leads to excessive ROS production, overwhelming cellular antioxidant defenses and triggering oxidative stress. Such stress contributes to OSA-related complications, including cardiovascular disorders [35,61]. Despite evidence of elevated ROS in OSA, no consensus exists on reliable oxidative stress biomarkers or effective antioxidant therapies, highlighting the need for further research.
Unlike continuous hypoxia, OSA-associated intermittent hypoxia includes a post-hypoxic reoxygenation phase (ROX), marked by frequent, severe oxygen desaturation and fluctuations [62]. This pattern amplifies ROS production, enhancing oxidative stress and inflammation. Oxidative stress also activates NF-κB, promoting the release of pro-inflammatory cytokines such as IL-1α, IL-1β, IL-6, and TNF-α [63,64]. Recent studies demonstrate that intermittent hypoxia in OSA patients induces cardiomyocyte apoptosis, through the calcium permeable cation channel TRPC5, contributing to increase ROS production, disrupting mitochondrial function, and disturbing calcium balance. These findings shed light on the mechanisms of TRPC5-mediated cardiac damage and highlight its potential as both a diagnostic biomarker and therapeutic target for OSA-related cardiovascular complications [65].
Continuous positive airway pressure (CPAP) therapy, the gold standard for OSA treatment, effectively minimizes the frequency and severity of hypoxic episodes and reduces apneas and hypopneas while lowering oxidative stress [66,67]. However, the damage caused by oxidative stress in OSA patients is not limited to the airway; it extends to vital organs, including the cardiovascular system. Chronic oxidative damage is a key factor linking OSA to conditions such as hypertension, atherosclerosis, and other cardiovascular diseases. Despite the beneficial effects of CPAP therapy on reducing hypoxic episodes, it does not directly address the underlying oxidative stress or reverse the oxidative damage already incurred. Indeed, some individuals may continue to exhibit residual oxidative stress despite consistent and effective use of CPAP. In line with this, the combination of polyphenol-rich antioxidants sourced from Aronia melanocarpa berries with CPAP therapy has shown synergistic benefits in the treatment of OSA by lowering the levels of pro-inflammatory cytokines, such as TNF-α and IL-6, which are commonly elevated in OSA patients [68]. This anti-inflammatory effect may contribute to improved cardiovascular health and a lower risk of long-term complications. Moreover, the polyphenols in Aronia enhance endothelial function by increasing nitric oxide (NO) availability. This supports vascular tone regulation and helps reduce blood pressure, thereby potentially lowering the risk of atherosclerosis and other cardiovascular issues, supporting improved overall health outcomes in OSA patients [69]. Similar studies show that Tempol effectively reduces inflammation and oxidative stress, significantly alleviating intermittent hypoxia-induced lung injury by modulating the miR-145-5p/Nrf2 signaling pathway [70]. Additionally, antioxidant vitamins have shown potential in lowering oxidative and carbonyl stress, key contributors to cardiovascular complications in OSA [67]. Thus, the combination of CPAP with antioxidant therapies might be a potential therapeutic approach to reduce the risk of health complications by further reducing oxidative stress in OSA.
3. Oxidative Stress and Inflammation in Comorbidities of Hypoxemic Respiratory Diseases (COPD and OSA)
3.1. Role of Inflammation in Cancer Associated with Respiratory Diseases
A growing body of evidence highlights the pivotal role of inflammation in cancer development, particularly by facilitating key processes involved in tumor progression [71]. Inflammation contributes to tumor initiation and promotes the accumulation of oncogenic mutations. Furthermore, the inflammatory conditions elevate oxidative stress, resulting in enhanced DNA damage and increased mutation rates [72]. Inflammatory mediators are also implicated in processes such as epithelial-to-mesenchymal transition and epigenetic alterations that favor oncogenesis. In addition, chronic inflammation leads to immune exhaustion, fostering the development of an immunosuppressive tumor microenvironment and facilitating immune evasion [73]. Patients with CRDs, including COPD and OSA, typically present a persistent low-grade inflammatory state, partly driven by hypoxemia [36,56,74].
In the context of COPD, the combination of chronic hypoxia, oxidative stress, and inflammation promote a tumor-promoting environment in the airways, especially increasing the risk of lung cancer [75]. Although lung cancer can occur regardless of airflow limitation severity, numerous studies have underscored chronic inflammation as a central factor in promoting cellular transformation and disrupting immune regulation, thereby facilitating tumor initiation and progression [56,76]. In COPD patients, inflammatory processes are thought to support carcinogenesis via several mechanisms, including oxidative damage, immune dysregulation, genomic instability, and tissue remodeling [77,78].
Similarly, several studies indicate that OSA is associated with increased cancer incidence, aggressiveness, and mortality [31]. Intermittent hypoxia experienced during sleep in OSA patients induces oxidative stress and promotes tumorigenic processes, including angiogenesis and epithelial-to-mesenchymal transition [34,79,80,81]. More importantly, there is solid evidence elucidating how the immune surveillance in OSA patients is disrupted due to chronic inflammation, which modulates immune cell subsets and their immune functionality. For instance, a switch to tumor-associated macrophages and a defect in NK cell maturation have been reported as well as suppression of T cells function mediated by overexpression of immune checkpoints axes [34,82,83,84,85].
3.2. Role of Inflammation in Cardiovascular Complications Associated to CRDs
Chronic inflammation is a central pathological mechanism implicated in a wide range of metabolic and cardiovascular disorders. Defined as a prolonged and persistent immune activation, chronic inflammation results in tissue injury, oxidative stress, mitochondrial dysfunction, and disruption of physiological homeostasis [86,87,88]. Several inflammatory pathways link respiratory diseases with cardiometabolic comorbidities, with particular emphasis on NF-κB signaling, NLRP3 inflammasome activation, and oxidative stress [88,89]. Although both hypoxemia and ROS are known to stimulate NF-κB and NLRP3, current evidence points to NLRP3 as a major driver of systemic inflammation [90,91]. Sterile stimulation activates the inflammasome complex, enhancing the production of inflammatory cytokines such as IL-18 and IL-1β [92]. These cytokines further exacerbate oxidative stress and hypoxemia. Altogether, chronic inflammation contributes to every phase of cardiovascular disease development, from early onset to overt clinical complications [93,94]. Furthermore, in CRDs, inflammation acts as a persistent noxious stimulus that promotes tissue remodeling, exacerbates inflammatory responses, and induces immune dysregulation.
Cardiovascular disease and COPD share various risk factors, including smoking, and overlapping mechanisms such as accelerated aging, making cardiovascular mortality more common than respiratory failure in COPD patients [95,96]. COPD is identified in 7–28% of individuals experiencing acute myocardial infarction [97], and cardiovascular disease accounts for approximately one-third of COPD-related deaths, primarily due to ischemic heart disease. Moreover, once COPD is established, the risk of cardiovascular disease further increases due to a confluence of systemic inflammation, hypoxemia, oxidative stress, physical inactivity, and other contributing factors. These combined effects contribute to impaired cardiac function, endothelial dysfunction, and autonomic imbalance, making individuals with COPD more susceptible to cardiovascular events such as myocardial infarction, heart failure, stroke, and arrhythmias [14].
Indeed, the incidence of major adverse cardiovascular events is 25% higher in patients with COPD compared to those without the condition [98]. Although shared risk factors such as age, smoking, and socioeconomic status contribute to this association, they do not fully explain it. Pathophysiological mechanisms such as hypoxemia, oxidative stress, and systemic inflammation are likely key contributors. In line with this, systematic review by Chen et al. reported that individuals with COPD are at significantly higher risk of developing cardiovascular disease than the general population (OR 2.46, 95% CI 2.02–3.00) [98,99,100].
OSA patients also exhibit a chronic inflammatory phenotype characterized by oxidative stress and elevated levels of pro-inflammatory cytokines and adhesion molecules [101]. This inflammatory state is known to induce or exacerbate cardiovascular and metabolic comorbidities such as type 2 diabetes mellitus, dyslipidemia, atherosclerosis, and ischemic heart disease [17,102,103,104]. This “sterile” inflammation occurs in response to DAMPs (damage-associated molecular patterns), such as high mobility group box 1 protein (HMGB1) and oxidized low-density lipoprotein (oxLDL) [105], often as a consequence of intermittent hypoxia and oxidative stress [106,107]. These stimuli activate inflammatory pathways, including NF-κB and the NLRP3 inflammasome [64,108]. The inflammasome activation results in elevated levels of IL-1β, IL-18, and tissue factor, enhancing systemic inflammation and coagulability [109,110]. Additionally, oxLDL potentiates NF-κB signaling and promotes foam cell formation, contributing to the progression of atherosclerosis [111]. Altogether, this network of intermittent hypoxia, oxidative stress, and systemic inflammation fosters endothelial dysfunction and promotes cardiovascular complications in OSA patients.
3.3. Role of Oxidative Stress and Inflammation in Pulmonary Hypertension Associated to CRDs
Pulmonary hypertension (PH) is one of the most significant cardiovascular complications associated with CRDs. This condition is defined by an elevation in mean pulmonary arterial pressure exceeding 20 mmHg [112]. Group 3 PH, as classified by the World Symposium on Pulmonary Hypertension, encompasses PH related to CRDs and represents one of the most prevalent subtypes. It includes conditions with diverse etiologies such as COPD, interstitial lung diseases (e.g., pulmonary fibrosis), non-parenchymal restrictive disorders (e.g., OSA), hypoxia without underlying lung disease (e.g., high-altitude exposure), and developmental lung conditions (e.g., bronchopulmonary dysplasia) [113].
The development of PH in the context of CRDs is associated with increased morbidity, reduced quality of life, and poorer prognosis [114]. The underlying pathophysiology is complex and multifactorial, involving disease-specific mechanisms [115]. Hypoxia represents a major common factor in these conditions and much of the insight concerning the molecular mechanisms responsible for pulmonary vasculopathy in CRDs comes from experimental animal models with exposure to hypoxia [116]. Acute hypoxia induces a compensatory response known as hypoxic pulmonary vasoconstriction (HPV), aimed at optimizing ventilation-perfusion matching. However, sustained hypoxia promotes persistent HPV and vascular remodeling, resulting in elevated pulmonary vascular resistance and PH development [116,117]. The involvement of ROS in HPV has been widely documented, although some controversy remains regarding whether ROS levels increase or decrease under hypoxic conditions. Current evidence predominantly supports an increase in ROS, which activates multiple downstream effectors that elevate intracellular calcium and induce contraction of pulmonary artery smooth muscle cells [118,119,120,121]. Mitochondria-derived ROS are considered pivotal in HPV [120,121,122], although other sources, such as NADPH oxidase, may amplify the response [118,119].In chronic hypoxia, persistently elevated ROS levels are well-established contributors to pulmonary arterial remodeling and enhanced pulmonary vasoconstriction, as observed in chronic hypoxia-induced PH [123,124,125]. Similar mechanisms are also implicated in chronic intermittent hypoxia, a widely used model for OSA-induced PH [126,127].
Mitochondrial respiratory chain [128] and members of the NAPDH oxidase family [129] are considered key sources of this hypoxic ROS generation. Intriguingly, there is also evidence of decreased ROS in pulmonary artery smooth muscle cells after chronic hypoxia [121]. Excessive oxidative stress has also been postulated as a key driver of lung fibrosis [130,131]. Indeed, TGF-β, a key mediator of pulmonary fibrosis and associated pulmonary vasculopathy, enhances the production of ROS through mitochondrial pathways and NADPH oxidase activation [130]. Conversely, ROS can further stimulate the release of TGF-β from alveolar cells, creating a self-amplifying cycle [132]. Moreover, the protective effects of PDE5A inhibition in bleomycin-induced pulmonary fibrosis and PH were associated with inhibition of ROS formation [133]. In the perinatal period, ROS-mediated injury to the developing lung is also recognized as a critical factor in the pathogenesis of neonatal pulmonary vascular diseases, including persistent pulmonary hypertension of the newborn and bronchopulmonary dysplasia [119,134,135,136].
As already mentioned, inflammation is increasingly recognized as a key contributor to pulmonary vascular disease and plays a significant role in the pathogenesis of various forms of PH, including those associated with CRD [115,117,137]. Many immune cells, and multiple cytokines, adhesion molecules, and differentiation factors have been suggested to participate in the complex inflammatory responses promoting the pulmonary vascular remodeling characteristic of PH [115,117,138]. Thus, elevated levels of inflammatory cytokines and chemokines, including TNFα, IL-1β, IL-6, Il-8, IL-17, IL-21, CCl2 and CX3CL1, among others, originating from a variety of inflammatory cell types are considered hallmarks of CRD-associated PH [126,139,140,141,142,143]. Extensive evidence highlights monocytes and macrophages as central mediators of lung inflammation and vascular remodeling in hypoxia-induced PH [142,144,145,146]. Upon hypoxic exposure, circulatory monocytes are recruited into the lungs where they differentiate into interstitial macrophages. Both, non-classical [142,144] and classical monocytes [143,147] have been proposed as precursors of recruited interstitial macrophages. The expression of thrombospondin-1 (TSP-1) in these recruited interstitial macrophages, was shown to activate TGF-β, resulting in enhanced pulmonary vasoconstriction and remodeling [143,145,147]. There is also involvement of lymphoid cells in hypoxia-induced PH. Thus, CD4+ T cells, specifically Th17 cells have been demonstrated to contribute to the development of hypoxia-induced PH in murine models [141,148] and their inhibition lowers right ventricle systolic pressure in established PH [148]. Similarly, Th17 cells have been suggested to play a detrimental role in PH secondary to lung fibrosis [126].
In addition to hypoxia, other pathogenic environmental factors (e.g cigarette smoke, hyperoxia, etc…) involved in the development of respiratory diseases have been shown to produce pulmonary vascular injury through increased ROS or inflammation. Thus, cigarette smoke, a major risk factor in COPD, induces pulmonary vascular dysfunction through the secretion of inflammatory molecules and increased ROS production [149]. In particular, cigarette smoke promotes the production of mitochondria-derived ROS, affecting the redox state of the soluble guanylyl cyclase, which severely impairs the NO- soluble guanylyl cyclase signaling pathway in the pulmonary vasculature [9]. Using a murine model of nicotine/hypoxia co-exposure to better illustrate the pathological conditions of COPD-PH, Truong et al. have recently identified a major role of Rieske iron-sulfur protein (RISP)-mediated mitochondrial ROS and NF-κB-dependent inflammation in PH development [150]. Other sources of ROS such as xanthine oxidoreductase have also been shown to contribute to the harmful effects of cigarette smoke in pulmonary vascular endothelial cells [151]. In addition, cigarette smoke has been shown to promote dysregulated inflammatory signaling due to increased recruitment of inflammatory cells or ROS-induced inflammation in pulmonary arteries [149,150,152]. An excess of ROS production is also recognized as a critical factor for hyperoxia-induced pulmonary vascular damage in several conditions such as respiratory distress syndrome [153] or pulmonary brochodysplasia [154].
Many efforts have been made to target oxidative stress and the inflammatory component underlying the pulmonary vascular injury linked to CRDs. Thus, targeting ROS with unspecific antioxidants such as N-acetylcysteine [155] or with strategies to specifically reduce mitochondrial ROS [123,128] show protective effects in chronic hypoxia-PH. Likewise, numerous intervention strategies for inflammation have been proven as effective in attenuating hypoxic-PH. These include among others: glucocorticoids [143], blocking IL-6 [139,140,141] CX3CR1 [142], the SDF-1/CXCR4 axis [156], the CCR2-CCL2 axis [143], inhibiting macrophages [141], switching macrophage polarity toward an anti-inflammatory phenotype [146] or adoptive transfer of M2 regulatory macrophages [157].
Despite substantial evidence supporting the pathogenic role of oxidative stress and inflammation in pulmonary vascular damage associated CRDs, and the demonstrated benefits of therapies targeting these pathways in preclinical studies, these strategies have yet to be successfully translated into clinical practice.
4. Transcriptional Response to Sustained and Intermittent Hypoxia in CRDS
4.1. HIF Signaling and Gene Expression Dynamics in Sustained and Intermittent Hypoxia
As previously mentioned, hypoxemia is believed to be a major contributor to CRDs progression, driving metabolic reprogramming, tissue remodeling, and inflammation. To maintain oxygen homeostasis, the body employs various mechanisms, broadly categorized into immediate physiological adjustments [158] and longer-term transcriptional responses [159]. Acute responses are triggered within seconds of oxygen deprivation, such as the activation of chemoreceptors like the carotid body to modulate respiration, and vascular changes that enhance oxygen delivery. However, these mechanisms are insufficient for adaptation to prolonged hypoxia. Instead, transcriptional responses become central over hours or days, adjusting metabolism and oxygen supply and playing a critical role in adapting to CRDs. Additionally, hypoxia-induced transcriptional reprogramming may help precondition tissues to better tolerate future hypoxic episodes [160,161], providing an essential adaptive mechanism in chronic respiratory conditions where tissues are repeatedly subjected to low oxygen levels.
A universal property of nucleated cells is their ability to autonomously sense oxygen levels and regulate members of the HIF family of transcription factors, which orchestrate a broad gene expression response to hypoxia. HIFs, consisting of an oxygen-regulated alpha subunit and a constitutively expressed beta subunit (ARNT), drive a metabolic shift from oxidative phosphorylation to glycolysis, reducing both oxygen consumption and the generation of ROS. They also promote the transcription of genes involved in angiogenesis and erythropoiesis, facilitating oxygen delivery to hypoxic tissues [159]. While these adaptations enhance survival, as mentioned above chronic HIF activation is implicated in pathological processes including tissue remodeling, inflammation, and abnormal vascularization, hallmarks of chronic respiratory and cardiovascular diseases [38].
The oxygen-dependent regulation of HIF and its role in hypoxia adaptation have been extensively characterized, a contribution recognized by the 2019 Nobel Prize in Physiology or Medicine. The HIF pathway depends on a set of oxygen-sensitive enzymes that act as cellular oxygen sensors [162]. Under normoxic conditions, EGLN family prolyl hydroxylases hydroxylate specific proline residues on HIF-α, marking it for recognition by a ubiquitin ligase complex containing the von Hippel-Lindau (VHL) tumor suppressor protein and targeting it for proteasomal degradation [162]. These EGLN enzymes require iron (Fe²⁺), 2-oxoglutarate (2-OG), and molecular oxygen for their activity, making them highly sensitive to oxygen levels. Even mild hypoxia inhibits EGLN activity, leading to HIF-α stabilization and activation of its downstream transcriptional targets. HIF transcriptional activity is also regulated by another oxygen-dependent enzyme, factor inhibiting HIF (FIH), which hydroxylates an asparagine residue on HIF-α [162]. The central role of HIFs in hypoxia is further reinforced by early studies showing that most hypoxia-induced genes are dependent on HIF activity [163]
Nevertheless, while HIFs are central players, other transcription factors also contribute to the hypoxic gene expression program by integrating diverse cellular signals. NF-κB is a key example, especially relevant for inflammatory responses to hypoxia. Its activation is closely linked to the inhibition of EGLN enzymes, paralleling HIF regulation [164]. Importantly, a recent report suggests a large part of the hypoxia-regulated genes depend, at least in part, of NF-kB [165]. This interplay between NF-κB and HIF is particularly significant in CRDs, where inflammation and hypoxia coexist, with NF-κB acting as a key transcription factor in inflammatory responses and HIF playing a central role in the cellular response to hypoxia. FOXO3 is another transcription factor regulated under hypoxia through prolyl hydroxylation [166], modulating responses such as oxidative stress resistance and metabolic adaptation [167]. Notch signaling also intersects with hypoxic pathways via direct interactions between HIF1α and the Notch intracellular domain (NICD), resulting in the hypoxic induction of Notch target genes [168];direct hydroxylation of NICD in an oxygen-dependent manner has also been reported. AP-1, in contrast, becomes more prominent following prolonged hypoxic exposure [169], suggesting a role in later stages of adaptation rather than in the immediate response [170,171]. Similarly, the Hippo pathway, which regulates TEAD transcription factors, is modulated by oxygen levels, potentially through the hypoxia-inducible ubiquitin ligase Siah2 [172]. This interaction may contribute to the development of pulmonary hypertension [173,174] and other cardiovascular diseases [175], which are common comorbidities associated with CRDs. Other transcription factors, such as TP53 and Nrf2, may respond to hypoxia in more context-dependent ways. TP53 is generally induced in severe hypoxia or anoxia, which may explain conflicting findings regarding its regulation by oxygen [176]. Nrf2, a master regulator of oxidative stress, is also likely to be indirectly involved in hypoxia adaptation [177], particularly during reoxygenation and in conditions that promote ROS production [178]. Given the critical role of ROS in the progression of CRDs, the involvement of Nrf2 in hypoxia-related responses, albeit indirect, may be especially important in this context.
Thus, while HIFs are central, a diverse network of transcription factors contributes to the hypoxic response, integrating cues from inflammation, metabolism, and cellular stress. Beyond transcription factor regulation, oxygen availability also influences gene expression via epigenetic mechanisms, adding another layer of dynamic control during hypoxic adaptation.
Epigenetic modifications are crucial for modulating hypoxia-induced gene expression by altering chromatin accessibility. Seminal studies have demonstrated that hypoxia inhibits histone demethylases such as KDM5A and KDM6A, leading to increased levels of histone marks like H3K4me3 and H3K27me, respectively, modifications that contribute to hypoxic responses independently of HIF [179,180]. These and other findings indicate that DNA and histone methylation states shift dynamically in response to oxygen levels. Many demethylases are 2-oxoglutarate-dependent dioxygenases that require oxygen as a co-substrate, positioning them as oxygen sensors analogous to HIF prolyl hydroxylases [181,182]. Members of the KDM4, KDM5, and KDM6 families, which have a high affinity for oxygen, are particularly susceptible to inhibition under physiological hypoxia. As a result, hypoxia can profoundly alter chromatin landscapes, modulating gene expression in ways that extend beyond the HIF pathway [181,182]. These insights highlight the importance of oxygen-dependent epigenetic regulation in shaping hypoxic responses and potentially influencing long-term disease progression.
The convergence of these transcriptional and epigenetic regulators underscores the complexity of the cellular response to hypoxia and the importance of considering multiple regulatory pathways. However, caution is warranted when extrapolating findings to CRDs. A significant limitation is that much of the current knowledge comes from in vitro cellular models, often derived from tumor cell lines that are not representative of lung tissue. For example, a query of the NCBI Gene Expression Omnibus (GEO) for hypoxia-related transcriptional profiles revealed that 83% (504 out of 609) of datasets originate from in vitro experiments, and only 5% (30 out of 609) pertain to lung tissue (unpublished data). This raises uncertainty regarding the contribution of these transcriptional mechanisms to hypoxia adaptation in pulmonary cell types in vivo.
Moreover, most studies focus on sustained hypoxia, even though oxygen levels fluctuate in CRDs. OSA represents an extreme case, with severe patients experiencing more than 30 apnea episodes per hour, and some exceeding 80 [183]. These cycles of hypoxia and reoxygenation create a physiological context distinct from continuous hypoxia. Whether intermittent hypoxia elicits the same transcriptional program as sustained hypoxia remains unclear, especially in cases involving high-frequency, short-duration hypoxic episodes. Evidence suggests that intermittent hypoxia preferentially activates NF-κB over HIFs, particularly in OSA models, where inflammation rather than canonical HIF-driven adaptation appears to dominate [184]. Supporting this, intermittent hypoxia has been associated with NF-κB-mediated inflammatory gene expression in adipose tissue, a key contributor to the cardiovascular complications of OSA [185]. These observations highlight the need for further studies to systematically compare the transcriptional effects of sustained versus intermittent hypoxia, particularly in CRDs, where oxygenation patterns have critical implications for disease progression and treatment strategies.
4.2. Role of HIFs in Vascular Dysfunction in COPD and OSA.
The bronchial and alveolar epithelium constitute the primary cellular interface with inhaled oxygen. However, the role of HIFs in lung epithelial cell biology remains largely unexplored. In contrast, the pulmonary response to hypoxia has been more extensively studied in the vascular endothelium, where hypoxic exposure leads to increased coverage of pulmonary vessels by smooth muscle cells (PASMCs). This vascular remodeling is responsible for the increase in pulmonary arterial pressure, which may ultimately result in right ventricular hypertrophy and heart failure.
Previous studies have demonstrated that hypoxia-induced pulmonary vascular remodeling is mediated by HIFs. Specifically, HIF-induced vascular remodeling involves the emergence of PASMCs in alveolar wall vessels (which are normally devoid of smooth muscle under physiological conditions). It also involves the thickening of the tunica media and adventitia in vessels already covered by smooth muscle cells under normoxic conditions. In this context, mice with heterozygous germline deletion of HIF1α or HIF2α exhibit attenuated development of pulmonary hypertension upon chronic hypoxic exposure [186,187,188]. Further investigations have confirmed the pivotal role of endothelial HIF2α in hypoxia-driven pulmonary hypertension in which endothelial cell-specific deletion of HIF2α protects against the development of hypoxia-induced pulmonary hypertension [162,189,190,191,192,193]. Conversely, sustained activation of HIF2α in pulmonary artery endothelial cells in mice leads to pulmonary hypertension and right ventricular hypertrophy [189,192,194].
In line with these findings, both human studies and mouse models carrying gain-of-function mutations in HIF2α have demonstrated severe pulmonary hypertension phenotypes [195,196]. Additionally, HIF2α regulates the expression of key endothelial genes that enhance vasoconstriction and impair vasodilation, including endothelin-1, apelin, and arginase 1 and 2. It also upregulates molecules such as CXCL12, which promote the proliferation and migration of pulmonary vascular smooth muscle cells [192,193,197]
Apart from the role of HIF2α overactivation in pulmonary hypertension, decreased HIF2α activity in lung endothelial cells also plays an important role in other pathological conditions, these including COPD. Notably, these patients exhibit decreased expression of HIF2α in the lungs [198]. Consistent with these findings, an independent study further demonstrated that lung tissue from COPD patients contains a reduced number of CD31⁺ endothelial cells, along with lower HIF2α expression per CD31⁺ cell, an effect that is particularly pronounced in advanced stages of the disease [199]. Similarly, diminished endothelial HIF2α expression has been observed in murine models of COPD following chronic exposure to cigarette smoke [198,199].
In agreement with these results, genetic inactivation of HIF2α specifically in pulmonary endothelial cells results in an emphysematous phenotype characterized by enlarged alveolar spaces and increased apoptosis of alveolar structural cells [199]. These findings suggest that reduced HIF2α expression in pulmonary endothelial cells may act as a pathogenic driver in the progression of COPD. Moreover, its deficiency in lung endothelial cells leads to a concomitant reduction in both endothelial cells and pericytes, accompanied by increased expression of pro-inflammatory cytokines such as TNFα and IL-1β. Importantly, this emphysematous phenotype is not observed following endothelial-specific deletion of HIF1α, underscoring the pivotal role of HIF2α in preserving lung vascular homeostasis [199]. Collectively, these findings support the idea that endothelial HIF2α plays a critical protective role in maintaining alveolar architecture and limiting pathological inflammation in the lung. Nevertheless, excessive HIF2α activation in endothelial cells, such as that observed in pulmonary hypertension, can be detrimental, which highlights the importance of well-balanced HIF2α levels in the endothelium [198].
In addition to the role of the HIF pathway in pulmonary hypertension, HIF isoforms have been also involved in systemic hypertension in OSA [200]. Indeed, IH-dependent HIF1α activity has been proposed to hyperactivate the central and peripheral nervous system leading to hypertension. At molecular level HIF1α induces the prooxidant protein NOX2, which provokes oxidative stress and subsequent inhibition of heme-oxygenase 2 (HO-2) and generation of hydrogen sulfide (H2S) [200,201]. In contrast, HIF2α counteracts this pathological action of HIF1α by inducing the antioxidant enzyme SOD2, which can counteract HIF1α-dependent oxidative stress. In this line, IH evoked a degradation of HIF2α therefore facilitating oxidative stress-dependent hypertension [202]. In addition to the central and peripheral nervous system, IH induces HIF1α in heart and aortic tissue, which leads to oxidative stress, vascular smooth muscle cell migration, vascular remodeling and systemic hypertension [203]. This HIF1α-dependent vascular remodelling has been associated to the activation of the Hippo-YAP pathway [203].
4.3. Role of HIF Pathway in Airway Inflammation
HIF signaling is also activated in other pulmonary cell types, including epithelial Club cells. where HIF2α promotes epithelial cell proliferation [204]. Notably, the progression of COPD is associated with a chronic inflammatory state in the airways [205], a process in which HIF activity may play a significant role. Supporting this notion, activation of HIF2α in Club cells triggers a type 2 helper T cell (Th2)-skewed inflammatory response in the lung, characterized by the activation of group 2 innate lymphoid cells (ILC2s) [206]. At the molecular level, this effect is mediated through HIF2α-induced expression of adrenomedullin (ADM), a gene which contains a hypoxia response element (HRE) in its proximal promoter [206]. ADM enhances ILC2 activation by increasing the expression of surface markers such as Sca-1 and KLRG1, and by amplifying their capacity to secrete interleukin-5 (IL-5) [206].
These findings establish a novel link between HIF2α activity, ILC2 biology, and Th2-driven airway inflammation. Additionally, recent clinical evidence demonstrates that patients with severe COPD present elevated numbers of ILC2s in sputum samples [207]. In parallel, circulating ADM levels have been identified as a potential biomarker for COPD exacerbation [208], while pro-adrenomedullin (proADM) has been proposed as a predictor of survival in COPD patients [209]. Collectively, these data suggest that aberrant HIF2α activity in the bronchial epithelium may exacerbate COPD by promoting airway inflammation through the ADM–ILC2 axis.
In addition to epithelial cells, HIF transcription factors also modulate immune responses in alveolar macrophages, which are essential for maintaining immune homeostasis within the alveolar space. HIF1α expression in myeloid cells has been shown to be critical for sustaining inflammatory responses [210], with HIF1α generally associated with M1 macrophage polarization, whereas HIF2α has been linked to M2 polarization [211]. However, this paradigm appears more complex in the context of alveolar macrophages. Constitutive activation of HIF1α signaling, through deletion of tumor suppressor gene VHL, leads to impaired Th2 responses, evidenced by reduced eosinophil recruitment and lower IL-5 and IL-13 production [212]. Furthermore, VHL inactivation in alveolar macrophages induces an immature phenotype and impairs their capacity for surfactant clearance [213].
These findings indicate that, contrary to its role in promoting pro-inflammatory M1 phenotypes in other contexts, HIF1α may act suppressing specific immune functions of alveolar macrophages. Importantly, alveolar macrophage function is known to be compromised in COPD [214]; however, the potential contribution of HIF signaling to this dysfunction remains unexplored and requires further investigation.
5. Relevance of AI in Respiratory Medicine
Machine Learning (ML) is a subfield of artificial intelligence that enables computer systems to learn patterns from data and make predictions or decisions without being explicitly programmed. At the core of any ML system is a model, typically represented by a function h, which maps an input x (e.g., patient data, medical images, or sensor readings) to an output y (e.g., a diagnosis, risk score, or recommended treatment). This function h is not predefined; rather, it is learned from a dataset during a training phase, where the model adjusts its internal parameters to approximate the underlying relationship between x and y. The different approaches vary depending on these variables (x, y, h).
In medicine, the application of ML has grown exponentially, evolving from traditional methods to advanced Deep Learning (DL) models. Traditional ML models learn patterns from clinical features such as symptoms, spirometry values, or imaging metrics, demonstrating effectiveness in both diagnostic and prognostic predictions for conditions such as COPD and OSA, and more modern Deep Learning models can learn from images, audio or complex tabular data [39,40,41,42].
5.1. Applications of Traditional Machine Learning in Diagnosis and Prognosis
In respiratory diseases such as COPD and OSA, traditional ML methods effectively leverage temporal physiological signals, imaging data, and structured clinical information, either individually or combined, to enhance diagnostic accuracy and prognostic prediction.
Temporal physiological signals from portable sensors have shown effectiveness in diagnosing and prognosticating respiratory diseases. In COPD, models such as XGBoost, Random Forest (RF), and Support Vector Machines (SVM) have demonstrated high performance using data from capnography, personal air quality monitors, remote spirometry, and electronic stethoscopes, achieving remarkable performance figures for detection and exacerbation prediction [215,216,217]. In OSA, typical inputs come from polysomnography (e.g. ECG, airflow, muscle activity), as well as alternative sources such as voice and tracheal sounds [40,42], which have been successfully analysed using SVM, RF, and logistic regression (LR) models [218,219,220].
Imaging data, particularly radiomics features extracted from chest Computed Tomography (CT) scans, have shown strong potential in diagnosing and prognosticating COPD and OSA when combined with traditional ML algorithms such as LR, SVM, and tree ensembles such as RF. These approaches quantify airway texture, density, and morphology from chest CT scans, achieving high diagnostic accuracy and severity classification, often outperforming demographic-based models or basic emphysema quantification [221,222,223]. Radiomics has also successfully differentiated COPD from asthma and predicted disease progression, particularly when combined with clinical data [224,225]. Notably, ML models that rely on carefully selected radiomic and clinical features have occasionally outperformed deep learning-based CNN approaches [226]
Tabular clinical data is one of the most extensively explored input modalities in combination with traditional ML models for both diagnosis and prognosis in COPD and OSA. In the case of COPD, models such as XGBoost, RF and LightGBM have demonstrated high predictive performance in estimating critical outcomes, including acute respiratory failure, mechanical ventilation requirements, mortality and severe exacerbations. These predictions are based on structured clinical variables such as demographics, comorbidities, functional tests, and laboratory results, which are used either alone or in combination with imaging and sensor data [227,228,229]. These models have also been used to identify early risk factors for airflow obstruction, which underscores the preventive potential of ML in respiratory medicine Muro S et al., 2021. In OSA, anthropometric and clinical features have been effectively used by neural networks, SVMs, RF and LR models to predict the apnea–hypopnea index (AHI), detect the presence of apnea and phenotype individuals based on anatomical characteristics of the upper airway [40,42,230].
5.2. Deep Learning Approaches in Diagnosis and Prognosis
Recent advances in Deep Learning (DL) have allowed models to achieve or exceed human expert performance in clinical tasks, with some systems receiving regulatory approval as autonomous diagnostic tools [231]Abramoff M et al., 2018. DL architectures such as Convolutional Neural Networks (CNNs) are effective for medical imaging and 1D biomedical signals [232]; Recurrent Neural Networks (RNNs), particularly Long Short-Term Memory Networks (LSTM), excel at analyzing temporal data like respiratory or polysomnography signals [40]; and transformer-based models, trained via self-supervised learning on large datasets, have emerged as powerful generalist approaches [233,234]. These models demonstrate their potential in medical applications when leveraging large-scale data [235,236].
In the specific context of COPD and OSA, temporal physiological signals have proven valuable when combined with DL architectures such as CNNs, RNNs and hybrid CNN-LSTM models, showing high effectiveness in classifying respiratory conditions based on lung and cough sounds [237,238], as CRDs typically alter acoustic patterns and key functional variables like airflow and lung capacity. DL has improved spirometry-based analysis, both in phenotype classification [239] and early COPD risk prediction [240], outperforming traditional approaches. More recently, transformer-based architectures have demonstrated additional improvements, confirming their strength in sequential signal processing [241]. In the case of OSA, DL models have effectively leveraged PSG and ECG data for apnea detection. CNN and LSTM architectures have demonstrated high performance in accurately identifying apnea events using portable PSG signals, recurrence plot images, and time–frequency representations such as scalograms and spectrograms derived from ECG signals portable sleep apnea; [242,243,244,245,246,247]. Transformer-based architectures have also emerged as promising alternatives for apnea detection [248].
Image data, particularly chest CT and X-rays, has been used for the diagnosis and prognosis of COPD and OSA. CNN-based models, including deep residual networks, as well as Graph Convolutional Networks (GCNs), have been applied to COPD detection from low-dose CT scans, leveraging spatial relationships within pulmonary structures to enhance early diagnosis. These models have outperformed both traditional ML approaches and other DL baselines [222,232,249]. In addition, weakly supervised approaches—such as multiple instance learning [250,251]—and self-supervised learning strategies, [252] have been explored to enable training with limited annotation, improving both robustness and generalizability. CNNs have also been applied to COPD severity staging and prognosis, leveraging CT and Chest X-Ray (CXR) data to classify GOLD stages (proposed by the Global Initiative for Chronic Obstructive Lung Disease, GOLD), predict acute respiratory events, and estimate mortality risk based on airway features or biological age models [253,254,255,256]. For OSA detection, CNN-based methods have demonstrated high effectiveness, both independently and in combination with techniques such as SVM or Multivariate Gaussian Process Regression (MVGPR) [257]. These approaches leverage a wide range of data sources, including lateral cephalometric radiographs, airway morphology features extracted from medical images, and image-based representations of ECG and polysomnography signals (e.g., scalograms, spectrograms, and recurrence plots), achieving accurate apnea detection [242,243,245,246,247,257,258].
Tabular clinical data has also been explored using DL for COPD and OSA prognosis, often through multilayer perceptrons (MLPs), RNNs, and, more recently, attention-based models applied to Electronic Health Record (EHR) sequences—though their benefits over traditional machine learning remain limited. The HiTANet system, for instance, combines local transformer-based attention with global key-query mechanisms and has outperformed classical and baseline DL models in predicting the onset of chronic diseases such as COPD [259]. Likewise, MLPs trained on hospital data have accurately estimated readmission risk in patients with COPD and asthma exacerbations [260]. However, traditional ML approaches remain highly competitive. As noted in recent reviews and meta-analyses, DL has not yet demonstrated substantial improvements over classical methods when applied to structured tabular data for COPD or OSA prognosis—likely due to the low dimensionality and standardisation of clinical features [40,41,42].
5.3. Foundational Models for Diagnosis and Prognosis
Foundational models are large neural networks—typically Transformer-based architectures with attention mechanisms— trained on massive, diverse datasets using self-supervised learning, that are typically available with diverse levels of openness, to be adapted to downstream tasks with transfer learning techniques such as so-called fine-tuning [233]. It is important to distinguish between specialized models—designed for narrow, well-defined tasks and highly optimized for specific objectives (the majority of the studies referenced in earlier sections)—and generalist models, which aim to handle a wide range of inputs and tasks within a single system. This distinction reflects a broader shift towards more general AI systems in medicine. Examples include large language models (LLMs) such as ChatGPT, which has demonstrated remarkable capabilities in clinical contexts—passing medical licensing exams and generating human-level clinical text the Lancet Digital Health 2023. Other foundation models include multimodal architectures like CLIP [261]and domain-specific transformers such as BioBERT, which enhances biomedical text mining [262]. In the context of COPD and OSA, foundational models represent a promising pathway for integrating multimodal data—including text, imaging, and biosignals—into unified diagnostic and prognostic frameworks. Although still under development, these models are beginning to show potential in areas such as medical imaging, clinical text interpretation, and multisensor data analysis.
For instance, in the domain of temporal signals such as audio, a notable example is the OPERA framework [263], which introduced three pretrained models using self-supervised learning on over 136,000 unlabeled audio samples (~400 hours) from 5 public datasets, including breathing, coughing, and lung auscultation sounds. Fine-tuning on downstream respiratory datasets further boosted performance across most tasks. The models are: i) OPERA-CT a contrastive transformer model (31M parameters); ii) OPERA-CE a lightweight CNN-based model (4M parameters), suitable for low-power devices; and iii) OPERA-GT a generative model using a Vision Treansformer (33M parameters). All models take images that represent audio as input. They were benchmarked across 19 respiratory health tasks, including 12 health-state classification tasks (e.g., COPD detection and severity grading) and 7 pulmonary function estimation tasks. OPERA models present reasonable performance in general, even when compared to other domain-specific and general-purpose baseline models such as OpenSMILE [264], VGGish [265], AudioMAE [266], and CLAP [267].
Foundational models have also been developed for medical imaging analysis. A notable example is M3FM [268], a general-purpose foundation model designed to perform multiple chest-related clinical tasks using low-dose 3D CT scans and multimodal clinical data. M3FM employed a scalable, question-answering framework for multimodal reasoning. Although not COPD-specific, M3FM covered several relevant tasks such as the detection of emphysema, fibrosis, reticulonodular opacities, atelectasis, and consolidation—key features for differential diagnosis, longitudinal monitoring, and phenotyping in COPD patients. Its architecture comprises 4 main components: i) a Vision Transformer for multiscale 3D CT volumes (CTViT), ii) a clinical text and question encoder, iii) a task encoder that fuses inputs from previous modules and enables handling multiple tasks in a question–answering format, and iv) a set of predictors trained for 17 clinical tasks. The model was pretrained using self-supervised learning on the OpenM3Chest dataset (128,693 CT scans from multiple sources) and later fine-tuned on task-specific annotated data. M3FM consistently outperformed prior models—including both task-specific and generalist approaches such as Sybil [269], Tri2D-Net [270], and GPT-4o—achieving up to 20% improvements in tasks like lung cancer prediction, cardiovascular disease diagnosis, and emphysema detection.
In addition, models developed for natural language processing have been successfully adapted to clinical domains. A notable example is BEHRT [271], a transformer-based model built upon BERT and pre-trained using masked language modeling on longitudinal EHRs. BEHRT was trained and evaluated on ~1.6 million patients from the UK Clinical Practice Research Datalink (CPRD) [272], one of the world’s largest and most respected databases of anonymized primary care health records. The model predicted the future onset of 301 conditions—including COPD—formulated as a multi-label classification task with multiple time horizons (next visit, 6 months, 12 months). BEHRT outperformed previous EHR-based models such as Deepr (CNN-based) [273] and RETAIN (RNN with reverse-time attention) [274], achieving improvements in performance. The gains were particularly notable in predicting the first incidence of diseases—a challenging task in clinical prognosis. Moreover, its self-attention mechanism enabled personalized interpretation and model transparency.
Taken together, these foundational models show promise in unifying multimodal diagnostic and prognostic tools for chronic respiratory care. Notably, they also show adaptability: the same core model can be fine-tuned or prompted to address COPD or OSA among various conditions, supporting the development of more generalist AI systems for pulmonary care. Overall, the findings across imaging, text, audio, and multimodal data suggest that integrating diverse inputs and large training corpora can enhance deep learning model performance and versatility.
5.4. Clinical Databases for COPD and OSA Research
ML applications in COPD and OSA research have been made possible by recent publications of extensive clinical datasets. For COPD, some of the most prominent cohorts include COPDGene [275], ECLIPSE [276], SPIROMICS [277], and CanCOLD [278], which offer clinical data, genetic profiles, spirometry results, biomarkers, and chest Computed Tomography (CT) imaging from patients with and without COPD, covering diverse stages of disease severity. Additionally, intensive care databases like MIMIC-III [279], MIMIC-IV [280], eICU [281], and HiRID [282] provide detailed respiratory data specific to patients with critical respiratory conditions, including those with acute exacerbations of COPD [283]. In sleep apnea research, prominent databases including Apnea-ECG [284], UCD Sleep Apnea Database [285], and SHHS [286] offer detailed polysomnographic recordings, ECG, EEG, EOG signals, apnea event annotations, and extensive longitudinal clinical data, enabling a wide range of analytical approaches.
5.5. Risk Assessment of AI for Health. Can AI Be Used as a New Tool in Therapeutic Decision-Making?
AI, particularly generative AI, is becoming an increasingly powerful tool in medicine, with the potential to function as a comprehensive assistant or even as an autonomous system in clinical environments. These models can handle multiple tasks with minimal adjustment, suggesting a future in which medical AI may play an integral role in therapeutic decision-making. However, despite this promise, AI also presents considerable risks and challenges, especially when used in high-stakes contexts such as healthcare [233,287].
One of the most pressing concerns regarding modern large generative models involves AI hallucinations — instances where generative models produce outputs that are plausible in form but factually incorrect or fabricated [288,289]. This phenomenon is well-documented in LLMs, which may fabricate clinical details or provide incorrect recommendations with high confidence. In healthcare, such hallucinations pose serious dangers that can directly affect patient safety.
A closely related issue is the lack of explainability. Many AI models — particularly DL networks — operate as “black boxes,” offering little insight into how decisions are made [290]. This opacity undermines clinician and patient trust and impedes the adoption of AI in medical decisions. Regulators and practitioners increasingly demand interpretability or, at minimum, reliable confidence metrics before integrating AI into clinical workflows.
Bias is another critical concern. AI models learn from data, which may underrepresent specific populations, leading to less accurate predictions for women, ethnic minorities, or other underserved groups [289]. This could perpetuate or even exacerbate existing disparities in healthcare delivery and outcomes.
Beyond these, there are growing concerns about privacy, security, and accountability. AI systems trained on patient data must comply with strict data protection laws such as HIPAA and GDPR. They must also be resilient against adversarial attacks and ensure robust mechanisms for ethical and legal responsibility when outcomes are incorrect or harmful.
5.6. Regulatory Landscape
To mitigate these risks, regulatory frameworks are evolving. The European Union has classified these systems as high-risk technologies, subject to strict compliance requirements under the EU Artificial Intelligence Act. [291]. This Act, the world’s first comprehensive AI legislation, adopts a risk-based approach and mandates the following for medical AI applications (e.g., diagnostic tools for COPD or OSA): 1) Adequate risk assessment and mitigation plans. 2) High-quality training datasets to minimize bias. 3) Logging capabilities to ensure traceability. 4) Comprehensive documentation for regulatory review. 5) Clear deployment instructions and information. 6) Human oversight mechanisms. 7) High levels of robustness, accuracy, and cybersecurity.
In contrast, the United States currently lacks a comprehensive AI law. However, the FDA leads the regulation of AI- and ML-based medical devices. The FDA requires clinical evidence of safety and efficacy, typically through validation studies, before approving or authorizing such systems. Notably, the FDA has approved autonomous AI systems [231], marking an important precedent.
Currently (2025), regulatory approvals for AI in respiratory medicine remain focused on assistive systems, rather than fully autonomous diagnostic tools. Approval for autonomous diagnosis remains subject to regulatory caution.
5.7. Integration of the AI in Practitioner’s Decisions
Decision-making is a fundamental process in medical practice, particularly in clinical settings, and can be significantly enhanced through the integration of artificial intelligence (AI). This integration must be approached with the understanding that the healthcare professional is (and must remain) ultimately responsible for decisions, while AI should be seen as a tool to support, rather than replace or overshadow, them. In this regard, decision theory [292] offers a formal framework for selecting optimal actions under uncertainty, which is essential in healthcare, where decisions can profoundly affect patient outcomes, resource allocation, and societal costs. In high-stakes environments such as oncology, emergency medicine, or intensive care, decisions must be informed not only by clinical expertise but also by models that systematically address uncertainty and the trade-offs between possible outcomes. This also applies to other disciplines where diagnosis and treatment can have a significant impact on patient well-being, such as hypoxemic respiratory diseases, including COPD and OSA.
To effectively integrate the knowledge produced by AI models into the practitioners’ decision-making process, interpreting model outputs is essential, as the knowledge extracted and distilled by these models can only be effectively utilized by practitioners if they clearly understand the meaning of those outcomes. Probabilistic machine learning models, which generate well-calibrated probability distributions over their predictions, are fundamental to enabling this interpretability. These models allow for the rigorous estimation of expected utilities or losses, thereby supporting personalized and cost-effective treatment planning, while also mitigating the cognitive and systematic biases inherent in human judgment [293]. However, the utility of probabilistic models in healthcare critically depends on their calibration—i.e., the alignment between predicted probabilities and empirical outcomes [294,295]. Poorly calibrated models can lead to overconfident or underconfident decisions, resulting in either overtreatment or missed interventions, both of which carry significant ethical and economic costs. Probabilistic calibration techniques such as Platt scaling, isotonic regression, and Bayesian post-processing are thus essential to ensure the reliability of predictive probabilities [296], especially in imbalanced or distributionally-shifted clinical data. Integrating calibrated probabilistic predictions into decision-theoretic frameworks not only enhances interpretability and trustworthiness of AI systems in healthcare but also supports actionable decision-making that aligns with patient-specific goals and institutional priorities under uncertainty. Moreover, the ability to calibrate any AI model—regardless of its underlying complexity—makes calibration a ubiquitous and interoperable component in any context where AI supports clinical practitioners in making decisions within a formal and rigorous framework.
6. Conclusions, Future Research and Directions
COPD and OSA are both hypoxemic lung diseases that share overlapping pathophysiological mechanisms, including hypoxia-induced oxidative stress and systemic inflammation, which exacerbate disease progression and comorbidities. In OSA, recurrent apneic episodes during sleep result in intermittent hypoxia, which in turn promotes excessive ROS production and systemic inflammation. Similarly, in COPD, chronic hypoxia and exposure to environmental insults such as cigarette smoke promote oxidative stress and a persistent inflammatory response. These common pathways contribute to endothelial dysfunction, vascular remodeling, and elevated cardiovascular risk. While antioxidant therapies offer promising strategies for mitigating oxidative stress-related lung damage, a thorough understanding of the evolving role of ROS in pulmonary inflammation is essential for the development of more effective treatments. In addition, the high prevalence of pulmonary hypertension in both conditions underscores the need to elucidate shared mechanisms and identify new therapeutic strategies.
In this respect, understanding the role of HIFs in CRDs provides opportunities for targeted therapies. While initially adaptive, sustained HIF activation may exacerbate inflammation and tissue remodeling, suggesting a dual role that must be carefully navigated in therapeutic design. Despite the promise of antioxidant and anti-inflammatory treatments, their clinical translation remains limited due to the complexity of redox and inflammatory signaling networks, highlighting the need for integrative and multidisciplinary approaches.
In this context, artificial intelligence (AI) has emerged as a powerful tool to manage the clinical and biological complexity of OSA and COPD. Machine learning and deep learning models enable accurate phenotyping, risk stratification, and treatment personalization by integrating multimodal data, including clinical, imaging, and physiological signals. AI-based models have improved the classification of disease subtypes, predicted treatment responses, and enhanced diagnostic consistency, particularly in pulmonary function testing and imaging analysis. Foundational models like OPERA and M3FM exemplify AI potential to unify heterogeneous data sources and bridge the gap between molecular insights and clinical practice.
In conclusion, the convergence of OSA and COPD pathophysiology reflects shared systemic consequences of hypoxia, oxidative stress, and inflammation. Advances in molecular understanding, particularly of HIF signaling, and the integration of AI into clinical workflows offer promising avenues for precision medicine. Interdisciplinary strategies combining biomedical research and computational innovation are essential to improve outcomes in these prevalent and complex respiratory diseases.
Author Contributions
Abstract: J.R., R.A., M.C.; Introduction: J.R., R.A., R.D., A.L., A.M., D.R., M.C.; Role of oxidative stress and inflammation in CRDS: J.R., R.A., E.D., C.C., L.P., F.G., M.C.; Oxidative stress and inflammation in comorbidities of hypoxemic respiratory diseases (COPD and OSA): A.H., E.D., C.C., L.P., A.C., F.G., M.C.; Transcriptional response to sustained and intermittent hypoxia in CRDS: L.F., E.D., C.C., J.A., L.P., F.G.; Relevance of AI in respiratory medicine: R.D., A.L., A.M., D.R.,; Conclusions, future research and directions: M.C..
Acknowledgments
Supporting grants: Redes en Biomedicina from Comunidad Autonoma of Madrid; INSPIRACM (P2022/BMD7224).
Conflicts of Interest
The authors have no interest related to the content of the manuscript to declare.
References
- GBD 2019 Chronic Respiratory Diseases Collaborators. Global burden of chronic respiratory diseases and risk factors, 1990-2019: an update from the Global Burden of Disease Study 2019. EClinicalMedicine. 2023;59:101936.
- Soriano JB, Alfageme I, Miravitlles M, de Lucas P, Soler-Cataluña JJ, García-Río F, et al. Prevalence and Determinants of COPD in Spain: EPISCAN II. Arch Bronconeumol. 2021;57:61–9.
- Safiri S, Carson-Chahhoud K, Noori M, Nejadghaderi SA, Sullman MJM, Ahmadian Heris J, et al. Burden of chronic obstructive pulmonary disease and its attributable risk factors in 204 countries and territories, 1990-2019: results from the Global Burden of Disease Study 2019. BMJ. 2022;378:e069679.
- Benjafield A V, Ayas NT, Eastwood PR, Heinzer R, Ip MSM, Morrell MJ, et al. Estimation of the global prevalence and burden of obstructive sleep apnoea: a literature-based analysis. Lancet Respir Med. 2019;7:687–98.
- Bagdonas E, Raudoniute J, Bruzauskaite I, Aldonyte R. Novel aspects of pathogenesis and regeneration mechanisms in COPD. Int J Chron Obstruct Pulmon Dis. 2015;10:995–1013.
- Duijts L, Reiss IK, Brusselle G, de Jongste JC. Early origins of chronic obstructive lung diseases across the life course. Eur J Epidemiol. 2014;29:871–85.
- Fonseca W, Lukacs NW, Elesela S, Malinczak C-A. Role of ILC2 in Viral-Induced Lung Pathogenesis. Front Immunol. 2021;12:675169.
- MacNee W. Pulmonary and systemic oxidant/antioxidant imbalance in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005;2:50–60.
- Sevilla-Montero J, Munar-Rubert O, Pino-Fadón J, Aguilar-Latorre C, Villegas-Esguevillas M, Climent B, et al. Cigarette smoke induces pulmonary arterial dysfunction through an imbalance in the redox status of the soluble guanylyl cyclase. Free Radic Biol Med. 2022;193:9–22.
- Garcia-Rio F, Miravitlles M, Soriano JB, Muñoz L, Duran-Tauleria E, Sánchez G, et al. Systemic inflammation in chronic obstructive pulmonary disease: a population-based study. Respir Res. 2010;11:63.
- Divo MJ, Casanova C, Marin JM, Pinto-Plata VM, de-Torres JP, Zulueta JJ, et al. COPD comorbidities network. Eur Respir J. 2015;46:640–50.
- Smith MC, Wrobel JP. Epidemiology and clinical impact of major comorbidities in patients with COPD. Int J Chron Obstruct Pulmon Dis. 2014;9:871–88.
- Santiago Díaz C, Medrano FJ, Muñoz-Rivas N, Castilla Guerra L, Alonso Ortiz MB, en representación de los grupos de trabajo de EPOC y de Riesgo Vascular de la Sociedad Española de Medicina Interna. COPD and cardiovascular risk. Clin Investig Arterioscler. 2025;500757.
- Polman R, Hurst JR, Uysal OF, Mandal S, Linz D, Simons S. Cardiovascular disease and risk in COPD: a state of the art review. Expert Rev Cardiovasc Ther. 2024;22:177–91.
- Camiciottoli G, Bigazzi F, Magni C, Bonti V, Diciotti S, Bartolucci M, et al. Prevalence of comorbidities according to predominant phenotype and severity of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2016;11:2229–36.
- Safiri S, Carson-Chahhoud K, Noori M, Nejadghaderi SA, Sullman MJM, Ahmadian Heris J, et al. Burden of chronic obstructive pulmonary disease and its attributable risk factors in 204 countries and territories, 1990-2019: results from the Global Burden of Disease Study 2019. BMJ. 2022;378:e069679.
- Gottlieb DJ, Punjabi NM. Diagnosis and Management of Obstructive Sleep Apnea: A Review. JAMA. 2020;323:1389–400.
- Parra O, Arboix A, Montserrat JM, Quintó L, Bechich S, García-Eroles L. Sleep-related breathing disorders: impact on mortality of cerebrovascular disease. Eur Respir J. 2004;24:267–72.
- Martínez-García MA, Soler-Cataluña JJ, Ejarque-Martínez L, Soriano Y, Román-Sánchez P, Illa FB, et al. Continuous positive airway pressure treatment reduces mortality in patients with ischemic stroke and obstructive sleep apnea: a 5-year follow-up study. Am J Respir Crit Care Med. 2009;180:36–41.
- Marin JM, Carrizo SJ, Vicente E, Agusti AGN. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet. 2005;365:1046–53.
- Labarca G, Vena D, Hu W-H, Esmaeili N, Gell L, Yang HC, et al. Sleep Apnea Physiological Burdens and Cardiovascular Morbidity and Mortality. Am J Respir Crit Care Med. 2023;208:802–13.
- Martínez Cerón E, Casitas Mateos R, García-Río F. Sleep apnea-hypopnea syndrome and type 2 diabetes. A reciprocal relationship? Arch Bronconeumol. 2015;51:128–39.
- Nieto FJ, Young TB, Lind BK, Shahar E, Samet JM, Redline S, et al. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. JAMA. 2000;283:1829–36.
- Loke YK, Brown JWL, Kwok CS, Niruban A, Myint PK. Association of obstructive sleep apnea with risk of serious cardiovascular events: a systematic review and meta-analysis. Circ Cardiovasc Qual Outcomes. 2012;5:720–8.
- Cai A, Wang L, Zhou Y. Hypertension and obstructive sleep apnea. Hypertens Res. 2016;39:391–5.
- Nakanishi R, Baskaran L, Gransar H, Budoff MJ, Achenbach S, Al-Mallah M, et al. Relationship of Hypertension to Coronary Atherosclerosis and Cardiac Events in Patients With Coronary Computed Tomographic Angiography. Hypertension. 2017;70:293–9.
- Al-Mashhadi RH, Al-Mashhadi AL, Nasr ZP, Mortensen MB, Lewis EA, Camafeita E, et al. Local Pressure Drives Low-Density Lipoprotein Accumulation and Coronary Atherosclerosis in Hypertensive Minipigs. J Am Coll Cardiol. 2021;77:575–89.
- Baguet J-P, Hammer L, Lévy P, Pierre H, Launois S, Mallion J-M, et al. The severity of oxygen desaturation is predictive of carotid wall thickening and plaque occurrence. Chest. 2005;128:3407–12.
- Savransky V, Nanayakkara A, Li J, Bevans S, Smith PL, Rodriguez A, et al. Chronic intermittent hypoxia induces atherosclerosis. Am J Respir Crit Care Med. 2007;175:1290–7.
- Campos-Rodriguez F, Martinez-Garcia MA, Martinez M, Duran-Cantolla J, Peña M de la, Masdeu MJ, et al. Association between obstructive sleep apnea and cancer incidence in a large multicenter Spanish cohort. Am J Respir Crit Care Med. 2013;187:99–105.
- Martinez-Garcia MA, Campos-Rodriguez F, Almendros I, Garcia-Rio F, Sanchez-de-la-Torre M, Farre R, et al. Cancer and Sleep Apnea: Cutaneous Melanoma as a Case Study. Am J Respir Crit Care Med. 2019;200:1345–53.
- Cubillos-Zapata C, Martínez-García MÁ, Campos-Rodríguez F, Sánchez de la Torre M, Nagore E, Martorell-Calatayud A, et al. Soluble PD-L1 is a potential biomarker of cutaneous melanoma aggressiveness and metastasis in obstructive sleep apnoea patients. Eur Respir J. 2019;53.
- Cubillos-Zapata C, Martínez-García MÁ, Díaz-García E, Jaureguizar A, Campos-Rodríguez F, Sánchez-de-la-Torre M, et al. Obesity attenuates the effect of sleep apnea on active TGF-ß1 levels and tumor aggressiveness in patients with melanoma. Sci Rep. 2020;10:15528.
- Cubillos-Zapata C, Martínez-García MÁ, Díaz-García E, Toledano V, Campos-Rodríguez F, Sánchez-de-la-Torre M, et al. Proangiogenic factor midkine is increased in melanoma patients with sleep apnea and induces tumor cell proliferation. The FASEB Journal. 2020;34:16179–90.
- Olszewska E, Rogalska J, Brzóska MM. The Association of Oxidative Stress in the Uvular Mucosa with Obstructive Sleep Apnea Syndrome: A Clinical Study. J Clin Med. 2021;10:1132.
- Sanchez-Azofra A, Gu W, Masso-Silva JA, Sanz-Rubio D, Marin-Oto M, Cubero P, et al. Inflammation biomarkers in OSA, chronic obstructive pulmonary disease, and chronic obstructive pulmonary disease/OSA overlap syndrome. J Clin Sleep Med. 2023;19:1447–56.
- Ratcliffe PJ. Oxygen sensing and hypoxia signalling pathways in animals: the implications of physiology for cancer. J Physiol. 2013;591:2027–42.
- Luo Z, Tian M, Yang G, Tan Q, Chen Y, Li G, et al. Hypoxia signaling in human health and diseases: implications and prospects for therapeutics. Signal Transduct Target Ther. 2022;7:218.
- Chen Z, Hao J, Sun H, Li M, Zhang Y, Qian Q. Applications of digital health technologies and artificial intelligence algorithms in COPD: systematic review. BMC Med Inform Decis Mak. 2025;25:77.
- Bazoukis G, Bollepalli SC, Chung CT, Li X, Tse G, Bartley BL, et al. Application of artificial intelligence in the diagnosis of sleep apnea. Journal of Clinical Sleep Medicine. 2023;19:1337–63.
- Smith LA, Oakden-Rayner L, Bird A, Zeng M, To M-S, Mukherjee S, et al. Machine learning and deep learning predictive models for long-term prognosis in patients with chronic obstructive pulmonary disease: a systematic review and meta-analysis. Lancet Digit Health. 2023;5:e872–81.
- Vigil L, Zapata T, Grau A, Bonet M, Montaña M, Piñar M. Innovación en sueño. Open Respiratory Archives. 2024;6:100402.
- LeCun Y, Bengio Y, Hinton G. Deep learning. Nature. 2015;521:436–44.
- Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez AN, et al. Attention Is All You Need. 2017;
- Meliante PG, Zoccali F, Cascone F, Di Stefano V, Greco A, de Vincentiis M, et al. Molecular Pathology, Oxidative Stress, and Biomarkers in Obstructive Sleep Apnea. Int J Mol Sci. 2023;24.
- Lavalle S, Masiello E, Iannella G, Magliulo G, Pace A, Lechien JR, et al. Unraveling the Complexities of Oxidative Stress and Inflammation Biomarkers in Obstructive Sleep Apnea Syndrome: A Comprehensive Review. Life (Basel). 2024;14.
- Antosova M, Mokra D, Pepucha L, Plevkova J, Buday T, Sterusky M, et al. Physiology of nitric oxide in the respiratory system. Physiol Res. 2017;66:S159–72.
- Bernard K, Hecker L, Luckhardt TR, Cheng G, Thannickal VJ. NADPH oxidases in lung health and disease. Antioxid Redox Signal. 2014;20:2838–53.
- Al Ghouleh I, Khoo NKH, Knaus UG, Griendling KK, Touyz RM, Thannickal VJ, et al. Oxidases and peroxidases in cardiovascular and lung disease: new concepts in reactive oxygen species signaling. Free Radic Biol Med. 2011;51:1271–88.
- Chen W, Zhang W. Association between oxidative balance score and chronic obstructive pulmonary disease: A cross-sectional study. Medicine. 2024;103:e39883.
- Liu X, Hao B, Ma A, He J, Liu X, Chen J. The Expression of NOX4 in Smooth Muscles of Small Airway Correlates with the Disease Severity of COPD. Biomed Res Int. 2016;2016:1–17.
- Wang X, Murugesan P, Zhang P, Xu S, Peng L, Wang C, et al. NADPH Oxidase Isoforms in COPD Patients and Acute Cigarette Smoke-Exposed Mice: Induction of Oxidative Stress and Lung Inflammation. Antioxidants. 2022;11:1539.
- Alateeq R, Akhtar A, De Luca SN, Chan SMH, Vlahos R. Apocynin Prevents Cigarette Smoke-Induced Anxiety-Like Behavior and Preserves Microglial Profiles in Male Mice. Antioxidants. 2024;13:855.
- Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The Nature of Small-Airway Obstruction in Chronic Obstructive Pulmonary Disease. New England Journal of Medicine. 2004;350:2645–53.
- Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. The Lancet. 2011;378:1015–26.
- Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. Journal of Allergy and Clinical Immunology. 2016;138:16–27.
- Zuo L, He F, Sergakis GG, Koozehchian MS, Stimpfl JN, Rong Y, et al. Interrelated role of cigarette smoking, oxidative stress, and immune response in COPD and corresponding treatments. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2014;307:L205–18.
- Gan WQ. Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax. 2004;59:574–80.
- Agustí A, Edwards LD, Rennard SI, MacNee W, Tal-Singer R, Miller BE, et al. Persistent Systemic Inflammation is Associated with Poor Clinical Outcomes in COPD: A Novel Phenotype. PLoS One. 2012;7:e37483.
- Thomsen M, Dahl M, Lange P, Vestbo J, Nordestgaard BG. Inflammatory Biomarkers and Comorbidities in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2012;186:982–8.
- Mastino P, Rosati D, de Soccio G, Romeo M, Pentangelo D, Venarubea S, et al. Oxidative Stress in Obstructive Sleep Apnea Syndrome: Putative Pathways to Hearing System Impairment. Antioxidants (Basel). 2023;12.
- Abourjeili J, Salameh E, Noureddine M, Bou Khalil P, Eid AA. Obstructive sleep apnea: Beyond the dogma of obesity! Respir Med. 2024;222:107512.
- Alharbi KS, Fuloria NK, Fuloria S, Rahman SB, Al-Malki WH, Javed Shaikh MA, et al. Nuclear factor-kappa B and its role in inflammatory lung disease. Chem Biol Interact. 2021;345:109568.
- Israel LP, Benharoch D, Gopas J, Goldbart AD. A Pro-Inflammatory Role for Nuclear Factor Kappa B in Childhood Obstructive Sleep Apnea Syndrome. Sleep. 2013;36:1947–55.
- Qiu X, Yao Y, Chen Y, Li Y, Sun X, Zhu X. TRPC5 Promotes Intermittent Hypoxia-Induced Cardiomyocyte Injury Through Oxidative Stress. Nat Sci Sleep. 2024;16:2125–41.
- Karamanlı H, Özol D, Ugur KS, Yıldırım Z, Armutçu F, Bozkurt B, et al. Influence of CPAP treatment on airway and systemic inflammation in OSAS patients. Sleep and Breathing. 2014;18:251–6.
- Celec P, Jurkovičová I, Buchta R, Bartík I, Gardlík R, Pálffy R, et al. Antioxidant vitamins prevent oxidative and carbonyl stress in an animal model of obstructive sleep apnea. Sleep and Breathing. 2013;17:867–71.
- Jang B-K, Lee J-W, Choi H, Yim S-V. Aronia melanocarpa Fruit Bioactive Fraction Attenuates LPS-Induced Inflammatory Response in Human Bronchial Epithelial Cells. Antioxidants. 2020;9:816.
- Jelska A, Polecka A, Zahorodnii A, Olszewska E. The Role of Oxidative Stress and the Potential Therapeutic Benefits of Aronia melanocarpa Supplementation in Obstructive Sleep Apnea Syndrome: A Comprehensive Literature Review. Antioxidants (Basel). 2024;13.
- Ai L, Li Y, Liu Z, Li R, Wang X, Hai B. Tempol attenuates chronic intermittent hypoxia-induced lung injury through the miR-145-5p/Nrf2 signaling pathway. Cell Mol Biol (Noisy-le-grand). 2023;69:225–34.
- Greten FR, Grivennikov SI. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity. 2019;51:27–41.
- Kay J, Thadhani E, Samson L, Engelward B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair (Amst). 2019;83:102673.
- Fang L, Liu K, Liu C, Wang X, Ma W, Xu W, et al. Tumor accomplice: T cell exhaustion induced by chronic inflammation. Front Immunol. 2022;13.
- Vicente E, Marin JM, Carrizo SJ, Osuna CS, González R, Marin-Oto M, et al. Upper airway and systemic inflammation in obstructive sleep apnoea. European Respiratory Journal. 2016;48:1108–17.
- Houghton AM. Mechanistic links between COPD and lung cancer. Nat Rev Cancer. 2013;13:233–45.
- Dubinett SM, Spira AE. Impact of Chronic Obstructive Pulmonary Disease on Immune-based Treatment for Lung Cancer. Moving toward Disease Interception. Am J Respir Crit Care Med. 2018;197:278–80.
- Zaynagetdinov R, Sherrill TP, Gleaves LA, Hunt P, Han W, McLoed AG, et al. Chronic NF-κB activation links COPD and lung cancer through generation of an immunosuppressive microenvironment in the lungs. Oncotarget. 2016;7:5470–82.
- Forder A, Zhuang R, Souza VGP, Brockley LJ, Pewarchuk ME, Telkar N, et al. Mechanisms Contributing to the Comorbidity of COPD and Lung Cancer. Int J Mol Sci. 2023;24:2859.
- Díaz-García E, García-Tovar S, Casitas R, Jaureguizar A, Zamarrón E, Sánchez-Sánchez B, et al. Intermittent Hypoxia Mediates Paraspeckle Protein-1 Upregulation in Sleep Apnea. Cancers (Basel). 2021;13:3888.
- Cubillos-Zapata C, Martínez-García MÁ, Díaz-García E, García-Tovar S, Campos-Rodríguez F, Sánchez-de-la-Torre M, et al. Obstructive sleep apnoea is related to melanoma aggressiveness through paraspeckle protein-1 upregulation. European Respiratory Journal. 2023;61:2200707.
- Gozal D, Lipton AJ, Jones KL. Circulating Vascular Endothelial Growth Factor Levels in Patients with Obstructive Sleep Apnea. Sleep. 2002;25:59–65.
- Díaz-García E, García-Sánchez A, Alfaro E, López-Fernández C, Mañas E, Cano-Pumarega I, et al. PSGL-1: a novel immune checkpoint driving T-cell dysfunction in obstructive sleep apnea. Front Immunol. 2023;14.
- Hernández-Jiménez E, Cubillos-Zapata C, Toledano V, Pérez de Diego R, Fernández-Navarro I, Casitas R, et al. Monocytes inhibit NK activity via TGF-β in patients with obstructive sleep apnoea. European Respiratory Journal. 2017;49:1602456.
- Verduzco D, Lloyd M, Xu L, Ibrahim-Hashim A, Balagurunathan Y, Gatenby RA, et al. Intermittent Hypoxia Selects for Genotypes and Phenotypes That Increase Survival, Invasion, and Therapy Resistance. PLoS One. 2015;10:e0120958.
- Almendros I, Wang Y, Becker L, Lennon FE, Zheng J, Coats BR, et al. Intermittent Hypoxia-induced Changes in Tumor-associated Macrophages and Tumor Malignancy in a Mouse Model of Sleep Apnea. Am J Respir Crit Care Med. 2014;189:593–601.
- Pahwa R, Goyal A, Jialal I. Chronic Inflammation. 2025.
- Abderrazak A, Syrovets T, Couchie D, El Hadri K, Friguet B, Simmet T, et al. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015;4:296–307.
- Kim J, Kim H-S, Chung JH. Molecular mechanisms of mitochondrial DNA release and activation of the cGAS-STING pathway. Exp Mol Med. 2023;55:510–9.
- Sharma BR, Kanneganti T-D. NLRP3 inflammasome in cancer and metabolic diseases. Nat Immunol. 2021;22:550–9.
- Lamkanfi M, Dixit VM. Mechanisms and Functions of Inflammasomes. Cell. 2014;157:1013–22.
- Gupta N, Sahu A, Prabhakar A, Chatterjee T, Tyagi T, Kumari B, et al. Activation of NLRP3 inflammasome complex potentiates venous thrombosis in response to hypoxia. Proceedings of the National Academy of Sciences. 2017;114:4763–8.
- Li Y, Huang H, Liu B, Zhang Y, Pan X, Yu X-Y, et al. Inflammasomes as therapeutic targets in human diseases. Signal Transduct Target Ther. 2021;6:247.
- Youm Y-H, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A, et al. Canonical Nlrp3 Inflammasome Links Systemic Low-Grade Inflammation to Functional Decline in Aging. Cell Metab. 2013;18:519–32.
- Sutterwala FS, Ogura Y, Flavell RA. The inflammasome in pathogen recognition and inflammation. J Leukoc Biol. 2007;82:259–64.
- Campo G, Pavasini R, Malagù M, Mascetti S, Biscaglia S, Ceconi C, et al. Chronic Obstructive Pulmonary Disease and Ischemic Heart Disease Comorbidity: Overview of Mechanisms and Clinical Management. Cardiovasc Drugs Ther. 2015;29:147–57.
- Divo M, Cote C, de Torres JP, Casanova C, Marin JM, Pinto-Plata V, et al. Comorbidities and Risk of Mortality in Patients with Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2012;186:155–61.
- Goedemans L, Bax JJ, Delgado V. COPD and acute myocardial infarction. European Respiratory Review. 2020;29:190139.
- Onishi K. Total management of chronic obstructive pulmonary disease (COPD) as an independent risk factor for cardiovascular disease. J Cardiol. 2017;70:128–34.
- Morgan AD, Zakeri R, Quint JK. Defining the relationship between COPD and CVD: what are the implications for clinical practice? Ther Adv Respir Dis. 2018;12.
- Chen W, Thomas J, Sadatsafavi M, FitzGerald JM. Risk of cardiovascular comorbidity in patients with chronic obstructive pulmonary disease: a systematic review and meta-analysis. Lancet Respir Med. 2015;3:631–9.
- Kheirandish-Gozal L, Gozal D. Obstructive Sleep Apnea and Inflammation: Proof of Concept Based on Two Illustrative Cytokines. Int J Mol Sci. 2019;20:459.
- Yeghiazarians Y, Jneid H, Tietjens JR, Redline S, Brown DL, El-Sherif N, et al. Obstructive Sleep Apnea and Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation. 2021;144.
- Tietjens JR, Claman D, Kezirian EJ, De Marco T, Mirzayan A, Sadroonri B, et al. Obstructive Sleep Apnea in Cardiovascular Disease: A Review of the Literature and Proposed Multidisciplinary Clinical Management Strategy. J Am Heart Assoc. 2019;8.
- Yang L, Zhang H, Cai M, Zou Y, Jiang X, Song L, et al. Effect of spironolactone on patients with resistant hypertension and obstructive sleep apnea. Clin Exp Hypertens. 2016;38:464–8.
- Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. 2020;20:95–112.
- Lavie L, Lavie P. Molecular mechanisms of cardiovascular disease in OSAHS: the oxidative stress link. European Respiratory Journal. 2009;33:1467–84.
- Feres MC, Fonseca FAH, Cintra FD, Mello-Fujita L, de Souza AL, De Martino MC, et al. An assessment of oxidized LDL in the lipid profiles of patients with obstructive sleep apnea and its association with both hypertension and dyslipidemia, and the impact of treatment with CPAP. Atherosclerosis. 2015;241:342–9.
- Imamura T, Poulsen O, Haddad GG. Intermittent hypoxia induces murine macrophage foam cell formation by IKK-β-dependent NF-κB pathway activation. J Appl Physiol. 2016;121:670–7.
- Díaz-García E, García-Tovar S, Alfaro E, Jaureguizar A, Casitas R, Sánchez-Sánchez B, et al. Inflammasome Activation: A Keystone of Proinflammatory Response in Obstructive Sleep Apnea. Am J Respir Crit Care Med. 2022;205:1337–48.
- Díaz-García E, Sanz-Rubio D, García-Tovar S, Alfaro E, Cubero P, Gil A V., et al. Inflammasome activation mediated by oxidised low-density lipoprotein in patients with sleep apnoea and early subclinical atherosclerosis. European Respiratory Journal. 2023;61:2201401.
- Wolf D, Ley K. Immunity and Inflammation in Atherosclerosis. Circ Res. 2019;124:315–27.
- Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53.
- Kovacs G, Bartolome S, Denton CP, Gatzoulis MA, Gu S, Khanna D, et al. Definition, classification and diagnosis of pulmonary hypertension. Eur Respir J. 2024;64.
- Krompa A, Marino P. Diagnosis and management of pulmonary hypertension related to chronic respiratory disease. Breathe (Sheff). 2022;18:220205.
- McNicholas WT. Chronic obstructive pulmonary disease and obstructive sleep apnea: overlaps in pathophysiology, systemic inflammation, and cardiovascular disease. Am J Respir Crit Care Med. 2009;180:692–700.
- Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1013-32.
- Pugliese SC, Poth JM, Fini MA, Olschewski A, El Kasmi KC, Stenmark KR. The role of inflammation in hypoxic pulmonary hypertension: from cellular mechanisms to clinical phenotypes. Am J Physiol Lung Cell Mol Physiol. 2015;308:L229-52.
- Frazziano G, Moreno L, Moral-Sanz J, Menendez C, Escolano L, Gonzalez C, et al. Neutral sphingomyelinase, NADPH oxidase and reactive oxygen species. Role in acute hypoxic pulmonary vasoconstriction. J Cell Physiol. 2011;226:2633–40.
- Villamor E, Moreno L, Mohammed R, Pérez-Vizcaíno F, Cogolludo A. Reactive oxygen species as mediators of oxygen signaling during fetal-to-neonatal circulatory transition. Free Radic Biol Med. 2019;142:82–96.
- Hernansanz-Agustín P, Choya-Foces C, Carregal-Romero S, Ramos E, Oliva T, Villa-Piña T, et al. Na+ controls hypoxic signalling by the mitochondrial respiratory chain. Nature. 2020;586:287–91.
- Pak O, Scheibe S, Esfandiary A, Gierhardt M, Sydykov A, Logan A, et al. Impact of the mitochondria-targeted antioxidant MitoQ on hypoxia-induced pulmonary hypertension. Eur Respir J. 2018;51.
- Smith KA, Schumacker PT. Sensors and signals: the role of reactive oxygen species in hypoxic pulmonary vasoconstriction. J Physiol. 2019;597:1033–43.
- Adesina SE, Kang B-Y, Bijli KM, Ma J, Cheng J, Murphy TC, et al. Targeting mitochondrial reactive oxygen species to modulate hypoxia-induced pulmonary hypertension. Free Radic Biol Med. 2015;87:36–47.
- Yan S, Sheak JR, Walker BR, Jernigan NL, Resta TC. Contribution of Mitochondrial Reactive Oxygen Species to Chronic Hypoxia-Induced Pulmonary Hypertension. Antioxidants (Basel). 2023;12.
- Liu JQ, Erbynn EM, Folz RJ. Chronic hypoxia-enhanced murine pulmonary vasoconstriction: role of superoxide and gp91phox. Chest. 2005;128:594S-596S.
- Li X, Zhang X, Hou X, Bing X, Zhu F, Wu X, et al. Obstructive sleep apnea-increased DEC1 regulates systemic inflammation and oxidative stress that promotes development of pulmonary arterial hypertension. Apoptosis. 2023;28:432–46.
- Nisbet RE, Graves AS, Kleinhenz DJ, Rupnow HL, Reed AL, Fan T-HM, et al. The role of NADPH oxidase in chronic intermittent hypoxia-induced pulmonary hypertension in mice. Am J Respir Cell Mol Biol. 2009;40:601–9.
- Mei L, Zheng Y-M, Song T, Yadav VR, Joseph LC, Truong L, et al. Rieske iron-sulfur protein induces FKBP12.6/RyR2 complex remodeling and subsequent pulmonary hypertension through NF-κB/cyclin D1 pathway. Nat Commun. 2020;11:3527.
- Nagaraj C, Tabeling C, Nagy BM, Jain PP, Marsh LM, Papp R, et al. Hypoxic vascular response and ventilation/perfusion matching in end-stage COPD may depend on p22phox. Eur Respir J. 2017;50.
- Gonzalez-Gonzalez FJ, Chandel NS, Jain M, Budinger GRS. Reactive oxygen species as signaling molecules in the development of lung fibrosis. Transl Res. 2017;190:61–8.
- Kinnula VL, Fattman CL, Tan RJ, Oury TD. Oxidative stress in pulmonary fibrosis: a possible role for redox modulatory therapy. Am J Respir Crit Care Med. 2005;172:417–22.
- Bellocq A, Azoulay E, Marullo S, Flahault A, Fouqueray B, Philippe C, et al. Reactive oxygen and nitrogen intermediates increase transforming growth factor-beta1 release from human epithelial alveolar cells through two different mechanisms. Am J Respir Cell Mol Biol. 1999;21:128–36.
- Hemnes AR, Zaiman A, Champion HC. PDE5A inhibition attenuates bleomycin-induced pulmonary fibrosis and pulmonary hypertension through inhibition of ROS generation and RhoA/Rho kinase activation. Am J Physiol Lung Cell Mol Physiol. 2008;294:L24-33.
- Behnke J, Dippel CM, Choi Y, Rekers L, Schmidt A, Lauer T, et al. Oxygen Toxicity to the Immature Lung-Part II: The Unmet Clinical Need for Causal Therapy. Int J Mol Sci. 2021;22.
- Wedgwood S, Steinhorn RH. Role of reactive oxygen species in neonatal pulmonary vascular disease. Antioxid Redox Signal. 2014;21:1926–42.
- Huizing MJ, Cavallaro G, Moonen RM, González-Luis GE, Mosca F, Vento M, et al. Is the C242T Polymorphism of the CYBA Gene Linked with Oxidative Stress-Associated Complications of Prematurity? Antioxid Redox Signal. 2017;27:1432–8.
- Wang R-R, Yuan T-Y, Wang J-M, Chen Y-C, Zhao J-L, Li M-T, et al. Immunity and inflammation in pulmonary arterial hypertension: From pathophysiology mechanisms to treatment perspective. Pharmacol Res. 2022;180:106238.
- Ye Y, Xu Q, Wuren T. Inflammation and immunity in the pathogenesis of hypoxic pulmonary hypertension. Front Immunol. 2023;14:1162556.
- Lin D, Hu L, Wei D, Li Y, Yu Y, Wang Q, et al. Peli1 Deficiency in Macrophages Attenuates Pulmonary Hypertension by Enhancing Foxp1-Mediated Transcriptional Inhibition of IL-6. Hypertension. 2025;82:445–59.
- Savale L, Tu L, Rideau D, Izziki M, Maitre B, Adnot S, et al. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir Res. 2009;10:6.
- Hashimoto-Kataoka T, Hosen N, Sonobe T, Arita Y, Yasui T, Masaki T, et al. Interleukin-6/interleukin-21 signaling axis is critical in the pathogenesis of pulmonary arterial hypertension. Proc Natl Acad Sci U S A. 2015;112:E2677-86.
- Amsellem V, Abid S, Poupel L, Parpaleix A, Rodero M, Gary-Bobo G, et al. Roles for the CX3CL1/CX3CR1 and CCL2/CCR2 Chemokine Systems in Hypoxic Pulmonary Hypertension. Am J Respir Cell Mol Biol. 2017;56:597–608.
- Kumar R, Nolan K, Kassa B, Chanana N, Palmo T, Sharma K, et al. Monocytes and interstitial macrophages contribute to hypoxic pulmonary hypertension. J Clin Invest. 2025;135.
- Yu Y-RA, Malakhau Y, Yu C-HA, Phelan S-LJ, Cumming RI, Kan MJ, et al. Nonclassical Monocytes Sense Hypoxia, Regulate Pulmonary Vascular Remodeling, and Promote Pulmonary Hypertension. J Immunol. 2020;204:1474–85.
- Kumar R, Mickael C, Kassa B, Gebreab L, Robinson JC, Koyanagi DE, et al. TGF-β activation by bone marrow-derived thrombospondin-1 causes Schistosoma- and hypoxia-induced pulmonary hypertension. Nat Commun. 2017;8:15494.
- Vergadi E, Chang MS, Lee C, Liang OD, Liu X, Fernandez-Gonzalez A, et al. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation. 2011;123:1986–95.
- Kumar R, Mickael C, Kassa B, Sanders L, Hernandez-Saavedra D, Koyanagi DE, et al. Interstitial macrophage-derived thrombospondin-1 contributes to hypoxia-induced pulmonary hypertension. Cardiovasc Res. 2020;116:2021–30.
- Maston LD, Jones DT, Giermakowska W, Howard TA, Cannon JL, Wang W, et al. Central role of T helper 17 cells in chronic hypoxia-induced pulmonary hypertension. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2017;312:L609–24.
- Sevilla-Montero J, Labrousse-Arias D, Fernández-Pérez C, Fernández-Blanco L, Barreira B, Mondéjar-Parreño G, et al. Cigarette Smoke Directly Promotes Pulmonary Arterial Remodeling and Kv7.4 Channel Dysfunction. Am J Respir Crit Care Med. 2021;203:1290–305.
- Truong LN, Wilson Santos E, Zheng Y-M, Wang Y-X. Rieske Iron-Sulfur Protein Mediates Pulmonary Hypertension Following Nicotine/Hypoxia Coexposure. Am J Respir Cell Mol Biol. 2024;70:193–202.
- Kim BS, Serebreni L, Hamdan O, Wang L, Parniani A, Sussan T, et al. Xanthine oxidoreductase is a critical mediator of cigarette smoke-induced endothelial cell DNA damage and apoptosis. Free Radic Biol Med. 2013;60:336–46.
- Peinado VI, Barberá JA, Abate P, Ramírez J, Roca J, Santos S, et al. Inflammatory reaction in pulmonary muscular arteries of patients with mild chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;159:1605–11.
- Lim EY, Lee S-Y, Shin HS, Kim G-D. Reactive Oxygen Species and Strategies for Antioxidant Intervention in Acute Respiratory Distress Syndrome. Antioxidants (Basel). 2023;12.
- Wang J, Dong W. Oxidative stress and bronchopulmonary dysplasia. Gene. 2018;678:177–83.
- Lachmanová V, Hnilicková O, Povýsilová V, Hampl V, Herget J. N-acetylcysteine inhibits hypoxic pulmonary hypertension most effectively in the initial phase of chronic hypoxia. Life Sci. 2005;77:175–82.
- Young KC, Torres E, Hatzistergos KE, Hehre D, Suguihara C, Hare JM. Inhibition of the SDF-1/CXCR4 axis attenuates neonatal hypoxia-induced pulmonary hypertension. Circ Res. 2009;104:1293–301.
- Fernandez-Gonzalez A, Mukhia A, Nadkarni J, Willis GR, Reis M, Zhumka K, et al. Immunoregulatory Macrophages Modify Local Pulmonary Immunity and Ameliorate Hypoxic Pulmonary Hypertension. Arterioscler Thromb Vasc Biol. 2024;44:e288–303.
- López-Barneo J, González-Rodríguez P, Gao L, Fernández-Agüera MC, Pardal R, Ortega-Sáenz P. Oxygen sensing by the carotid body: mechanisms and role in adaptation to hypoxia. Am J Physiol Cell Physiol. 2016;310:C629-42.
- Ratcliffe PJ. Oxygen sensing and hypoxia signalling pathways in animals: the implications of physiology for cancer. J Physiol. 2013;591:2027–42.
- Pescador N, Villar D, Cifuentes D, Garcia-Rocha M, Ortiz-Barahona A, Vazquez S, et al. Hypoxia promotes glycogen accumulation through hypoxia inducible factor (HIF)-mediated induction of glycogen synthase 1. PLoS One. 2010;5:e9644.
- Arias CF, Acosta FJ, Bertocchini F, Fernández-Arias C. Redefining the role of hypoxia-inducible factors (HIFs) in oxygen homeostasis. Commun Biol. 2025;8:446.
- Bishop T, Ratcliffe PJ. Signaling hypoxia by hypoxia-inducible factor protein hydroxylases: a historical overview and future perspectives. Hypoxia (Auckl). 2014;2:197–213.
- Elvidge GP, Glenny L, Appelhoff RJ, Ratcliffe PJ, Ragoussis J, Gleadle JM. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: the role of HIF-1alpha, HIF-2alpha, and other pathways. J Biol Chem. 2006;281:15215–26.
- Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–11.
- Shakir D, Batie M, Kwok C-S, Kenneth NS, Rocha S. NF-κB is a Central Regulator of Hypoxia-Induced Gene Expression. 2025.
- Li L, Kang H, Zhang Q, D’Agati VD, Al-Awqati Q, Lin F. FoxO3 activation in hypoxic tubules prevents chronic kidney disease. J Clin Invest. 2019;129:2374–89.
- Jensen KS, Binderup T, Jensen KT, Therkelsen I, Borup R, Nilsson E, et al. FoxO3A promotes metabolic adaptation to hypoxia by antagonizing Myc function. EMBO J. 2011;30:4554–70.
- Gustafsson M V, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005;9:617–28.
- Laderoute KR, Calaoagan JM, Gustafson-Brown C, Knapp AM, Li G-C, Mendonca HL, et al. The response of c-jun/AP-1 to chronic hypoxia is hypoxia-inducible factor 1 alpha dependent. Mol Cell Biol. 2002;22:2515–23.
- Michiels C, Minet E, Michel G, Mottet D, Piret JP, Raes M. HIF-1 and AP-1 cooperate to increase gene expression in hypoxia: role of MAP kinases. IUBMB Life. 2001;52:49–53.
- Villar D, Ortiz-Barahona A, Gómez-Maldonado L, Pescador N, Sánchez-Cabo F, Hackl H, et al. Cooperativity of stress-responsive transcription factors in core hypoxia-inducible factor binding regions. PLoS One. 2012;7:e45708.
- Ma B, Chen Y, Chen L, Cheng H, Mu C, Li J, et al. Hypoxia regulates Hippo signalling through the SIAH2 ubiquitin E3 ligase. Nat Cell Biol. 2015;17:95–103.
- Wu Z, Zhu L, Nie X, Wei L, Qi Y. USP15 promotes pulmonary vascular remodeling in pulmonary hypertension in a YAP1/TAZ-dependent manner. Exp Mol Med. 2023;55:183–95.
- Chen J, Lockett A, Zhao S, Huang LS, Wang Y, Wu W, et al. Sphingosine Kinase 1 Deficiency in Smooth Muscle Cells Protects against Hypoxia-Mediated Pulmonary Hypertension via YAP1 Signaling. Int J Mol Sci. 2022;23.
- Islam R, Hong Z. YAP/TAZ as mechanobiological signaling pathway in cardiovascular physiological regulation and pathogenesis. Mechanobiology in medicine. 2024;2.
- Sermeus A, Michiels C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2011;2:e164.
- Potteti HR, Noone PM, Tamatam CR, Ankireddy A, Noel S, Rabb H, et al. Nrf2 mediates hypoxia-inducible HIF1α activation in kidney tubular epithelial cells. Am J Physiol Renal Physiol. 2021;320:F464–74.
- Bae T, Hallis SP, Kwak M-K. Hypoxia, oxidative stress, and the interplay of HIFs and NRF2 signaling in cancer. Exp Mol Med. 2024;56:501–14.
- Batie M, Frost J, Frost M, Wilson JW, Schofield P, Rocha S. Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science. 2019;363:1222–6.
- Chakraborty AA, Laukka T, Myllykoski M, Ringel AE, Booker MA, Tolstorukov MY, et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science. 2019;363:1217–22.
- Kindrick JD, Mole DR. Hypoxic Regulation of Gene Transcription and Chromatin: Cause and Effect. Int J Mol Sci. 2020;21.
- Batie M, Rocha S. Gene transcription and chromatin regulation in hypoxia. Biochem Soc Trans. 2020;48:1121–8.
- Romero-Peralta S, García-Rio F, Resano Barrio P, Viejo-Ayuso E, Izquierdo JL, Sabroso R, et al. Defining the Heterogeneity of Sleep Apnea Syndrome: A Cluster Analysis With Implications for Patient Management. Arch Bronconeumol. 2022;58:125–34.
- Ryan S, Taylor CT, McNicholas WT. Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation. 2005;112:2660–7.
- Taylor CT, Kent BD, Crinion SJ, McNicholas WT, Ryan S. Human adipocytes are highly sensitive to intermittent hypoxia induced NF-kappaB activity and subsequent inflammatory gene expression. Biochem Biophys Res Commun. 2014;447:660–5.
- Yu AY, Shimoda LA, Iyer N V, Huso DL, Sun X, McWilliams R, et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J Clin Invest. 1999;103:691–6.
- Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D, et al. Heterozygous deficiency of hypoxia-inducible factor-2alpha protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest. 2003;111:1519–27.
- Sheikh AQ, Saddouk FZ, Ntokou A, Mazurek R, Greif DM. Cell Autonomous and Non-cell Autonomous Regulation of SMC Progenitors in Pulmonary Hypertension. Cell Rep. 2018;23:1152–65.
- Kapitsinou PP, Rajendran G, Astleford L, Michael M, Schonfeld MP, Fields T, et al. The Endothelial Prolyl-4-Hydroxylase Domain 2/Hypoxia-Inducible Factor 2 Axis Regulates Pulmonary Artery Pressure in Mice. Mol Cell Biol. 2016;36:1584–94.
- Pullamsetti SS, Mamazhakypov A, Weissmann N, Seeger W, Savai R. Hypoxia-inducible factor signaling in pulmonary hypertension. J Clin Invest. 2020;130:5638–51.
- Hu C-J, Poth JM, Zhang H, Flockton A, Laux A, Kumar S, et al. Suppression of HIF2 signalling attenuates the initiation of hypoxia-induced pulmonary hypertension. Eur Respir J. 2019;54.
- Dai Z, Li M, Wharton J, Zhu MM, Zhao Y-Y. Prolyl-4 Hydroxylase 2 (PHD2) Deficiency in Endothelial Cells and Hematopoietic Cells Induces Obliterative Vascular Remodeling and Severe Pulmonary Arterial Hypertension in Mice and Humans Through Hypoxia-Inducible Factor-2α. Circulation. 2016;133:2447–58.
- Cowburn AS, Crosby A, Macias D, Branco C, Colaço RDDR, Southwood M, et al. HIF2α-arginase axis is essential for the development of pulmonary hypertension. Proc Natl Acad Sci U S A. 2016;113:8801–6.
- Wang S, Zeng H, Xie X-J, Tao Y-K, He X, Roman RJ, et al. Loss of prolyl hydroxylase domain protein 2 in vascular endothelium increases pericyte coverage and promotes pulmonary arterial remodeling. Oncotarget. 2016;7:58848–61.
- Gale DP, Harten SK, Reid CDL, Tuddenham EGD, Maxwell PH. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 alpha mutation. Blood. 2008;112:919–21.
- Tan Q, Kerestes H, Percy MJ, Pietrofesa R, Chen L, Khurana TS, et al. Erythrocytosis and pulmonary hypertension in a mouse model of human HIF2A gain of function mutation. J Biol Chem. 2013;288:17134–44.
- Urrutia AA, Aragonés J. HIF Oxygen Sensing Pathways in Lung Biology. Biomedicines. 2018;6.
- Pasupneti S, Tian W, Tu AB, Dahms P, Granucci E, Gandjeva A, et al. Endothelial HIF-2α as a Key Endogenous Mediator Preventing Emphysema. Am J Respir Crit Care Med. 2020;202:983–95.
- Yoo S, Takikawa S, Geraghty P, Argmann C, Campbell J, Lin L, et al. Integrative analysis of DNA methylation and gene expression data identifies EPAS1 as a key regulator of COPD. PLoS Genet. 2015;11:e1004898.
- Prabhakar NR, Peng Y-J, Nanduri J. Hypoxia-inducible factors and obstructive sleep apnea. J Clin Invest. 2020;130:5042–51.
- Yuan G, Khan SA, Luo W, Nanduri J, Semenza GL, Prabhakar NR. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J Cell Physiol. 2011;226:2925–33.
- Nanduri J, Wang N, Yuan G, Khan SA, Souvannakitti D, Peng Y-J, et al. Intermittent hypoxia degrades HIF-2alpha via calpains resulting in oxidative stress: implications for recurrent apnea-induced morbidities. Proc Natl Acad Sci U S A. 2009;106:1199–204.
- Zhang S, Zhao Y, Dong Z, Jin M, Lu Y, Xu M, et al. HIF-1α mediates hypertension and vascular remodeling in sleep apnea via hippo-YAP pathway activation. Mol Med. 2024;30:281.
- Torres-Capelli M, Marsboom G, Li QOY, Tello D, Rodriguez FM, Alonso T, et al. Role Of Hif2α Oxygen Sensing Pathway In Bronchial Epithelial Club Cell Proliferation. Sci Rep. 2016;6:25357.
- Woodruff PG, Agusti A, Roche N, Singh D, Martinez FJ. Current concepts in targeting chronic obstructive pulmonary disease pharmacotherapy: making progress towards personalised management. Lancet. 2015;385:1789–98.
- Han J, Wan Q, Seo G-Y, Kim K, El Baghdady S, Lee JH, et al. Hypoxia induces adrenomedullin from lung epithelia, stimulating ILC2 inflammation and immunity. J Exp Med. 2022;219.
- Flayer CH, Linderholm AL, Ge MQ, Juarez M, Franzi L, Tham T, et al. COPD with elevated sputum group 2 innate lymphoid cells is characterized by severe disease. medRxiv. 2024;
- Meng D-Q, Li X-J, Song X-Y, Xin J-B, Yang W-B. Diagnostic and prognostic value of plasma adrenomedullin in COPD exacerbation. Respir Care. 2014;59:1542–9.
- Stolz D, Christ-Crain M, Morgenthaler NG, Miedinger D, Leuppi J, Müller C, et al. Plasma pro-adrenomedullin but not plasma pro-endothelin predicts survival in exacerbations of COPD. Chest. 2008;134:263–72.
- Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–57.
- Takeda N, O’Dea EL, Doedens A, Kim J, Weidemann A, Stockmann C, et al. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010;24:491–501.
- Zhang W, Li Q, Li D, Li J, Aki D, Liu Y-C. The E3 ligase VHL controls alveolar macrophage function via metabolic-epigenetic regulation. J Exp Med. 2018;215:3180–93.
- Izquierdo HM, Brandi P, Gómez M-J, Conde-Garrosa R, Priego E, Enamorado M, et al. Von Hippel-Lindau Protein Is Required for Optimal Alveolar Macrophage Terminal Differentiation, Self-Renewal, and Function. Cell Rep. 2018;24:1738–46.
- Wu J, Zhao X, Xiao C, Xiong G, Ye X, Li L, et al. The role of lung macrophages in chronic obstructive pulmonary disease. Respir Med. 2022;205:107035.
- Talker L, Dogan C, Neville D, Lim RH, Broomfield H, Lambert G, et al. Diagnosis and Severity Assessment of COPD Using a Novel Fast-Response Capnometer and Interpretable Machine Learning. COPD: Journal of Chronic Obstructive Pulmonary Disease. 2024;21.
- Atzeni M, Cappon G, Quint JK, Kelly F, Barratt B, Vettoretti M. A machine learning framework for short-term prediction of chronic obstructive pulmonary disease exacerbations using personal air quality monitors and lifestyle data. Sci Rep. 2025;15:2385.
- Yin H, Wang K, Yang R, Tan Y, Li Q, Zhu W, et al. A machine learning model for predicting acute exacerbation of in-home chronic obstructive pulmonary disease patients. Comput Methods Programs Biomed. 2024;246:108005.
- Almazaydeh L, Elleithy K, Faezipour M. Obstructive sleep apnea detection using SVM-based classification of ECG signal features. 2012 Annual International Conference of the IEEE Engineering in Medicine and Biology Society. IEEE; 2012. p. 4938–41.
- Espinoza-Cuadros F, Fernández-Pozo R, Toledano DT, Alcázar-Ramírez JD, López-Gonzalo E, Hernández-Gómez LA. Reviewing the connection between speech and obstructive sleep apnea. Biomed Eng Online. 2016;15:20.
- Hajipour F, Jozani MJ, Moussavi Z. A comparison of regularized logistic regression and random forest machine learning models for daytime diagnosis of obstructive sleep apnea. Med Biol Eng Comput. 2020;58:2517–29.
- Cheplygina V, Pena IP, Pedersen JH, Lynch DA, Sorensen L, de Bruijne M. Transfer Learning for Multicenter Classification of Chronic Obstructive Pulmonary Disease. IEEE J Biomed Health Inform. 2018;22:1486–96.
- Li Z, Liu L, Zhang Z, Yang X, Li X, Gao Y, et al. A Novel CT-Based Radiomics Features Analysis for Identification and Severity Staging of COPD. Acad Radiol. 2022;29:663–73.
- Amudala Puchakayala PR, Sthanam VL, Nakhmani A, Chaudhary MFA, Kizhakke Puliyakote A, Reinhardt JM, et al. Radiomics for Improved Detection of Chronic Obstructive Pulmonary Disease in Low-Dose and Standard-Dose Chest CT Scans. Radiology. 2023;307.
- Moslemi A, Kontogianni K, Brock J, Wood S, Herth F, Kirby M. Differentiating COPD and asthma using quantitative CT imaging and machine learning. European Respiratory Journal. 2022;60:2103078.
- Makimoto K, Hogg JC, Bourbeau J, Tan WC, Kirby M. CT Imaging With Machine Learning for Predicting Progression to COPD in Individuals at Risk. Chest. 2023;164:1139–49.
- Guan Y, Zhang D, Zhou X, Xia Y, Lu Y, Zheng X, et al. Comparison of deep-learning and radiomics-based machine-learning methods for the identification of chronic obstructive pulmonary disease on low-dose computed tomography images. Quant Imaging Med Surg. 2024;14:2485–98.
- Liao K-M, Liu C-F, Chen C-J, Shen Y-T. Machine Learning Approaches for Predicting Acute Respiratory Failure, Ventilator Dependence, and Mortality in Chronic Obstructive Pulmonary Disease. Diagnostics. 2021;11:2396.
- Moll M, Qiao D, Regan EA, Hunninghake GM, Make BJ, Tal-Singer R, et al. Machine Learning and Prediction of All-Cause Mortality in COPD. Chest. 2020;158:952–64.
- Zeng S, Arjomandi M, Tong Y, Liao ZC, Luo G. Developing a Machine Learning Model to Predict Severe Chronic Obstructive Pulmonary Disease Exacerbations: Retrospective Cohort Study. J Med Internet Res. 2022;24:e28953.
- Chen H, Emami E, Kauffmann C, Rompré P, Almeida F, Schmittbuhl M, et al. Airway Phenotypes and Nocturnal Wearing of Dentures in Elders with Sleep Apnea. J Dent Res. 2023;102:263–9.
- Abràmoff MD, Lavin PT, Birch M, Shah N, Folk JC. Pivotal trial of an autonomous AI-based diagnostic system for detection of diabetic retinopathy in primary care offices. NPJ Digit Med. 2018;1:39.
- Wu Y, Xia S, Liang Z, Chen R, Qi S. Artificial intelligence in COPD CT images: identification, staging, and quantitation. Respir Res. 2024;25:319.
- Quer G, Topol EJ. The potential for large language models to transform cardiovascular medicine. Lancet Digit Health. 2024;6:e767–71.
- Moor M, Banerjee O, Abad ZSH, Krumholz HM, Leskovec J, Topol EJ, et al. Foundation models for generalist medical artificial intelligence. Nature. 2023;616:259–65.
- Rasmy L, Xiang Y, Xie Z, Tao C, Zhi D. Med-BERT: pretrained contextualized embeddings on large-scale structured electronic health records for disease prediction. NPJ Digit Med. 2021;4:86.
- Yang S, Yang X, Lyu T, Huang JL, Chen A, He X, et al. Extracting Pulmonary Nodules and Nodule Characteristics from Radiology Reports of Lung Cancer Screening Patients Using Transformer Models. J Healthc Inform Res. 2024;8:463–77.
- Roy A, Gyanchandani B, Oza A, Singh A. TriSpectraKAN: a novel approach for COPD detection via lung sound analysis. Sci Rep. 2025;15:6296.
- Vodnala N, Yarlagadda PS, Bhuvana K S, Ch M, Sailaja K. Novel Deep Learning Approaches to Differentiate Asthma and COPD Based on Cough Sounds. 2024 Parul International Conference on Engineering and Technology (PICET). IEEE; 2024. p. 1–4.
- Bodduluri S, Nakhmani A, Reinhardt JM, Wilson CG, McDonald M-L, Rudraraju R, et al. Deep neural network analyses of spirometry for structural phenotyping of chronic obstructive pulmonary disease. JCI Insight. 2020;5.
- Mei S, Li X, Zhou Y, Xu J, Zhang Y, Wan Y, et al. Deep learning for detecting and early predicting chronic obstructive pulmonary disease from spirogram time series. NPJ Syst Biol Appl. 2025;11:18.
- Gadgil S, Galanter J, Negahdar M. Transformer-based Time-Series Biomarker Discovery for COPD Diagnosis. 2024;
- Taghizadegan Y, Jafarnia Dabanloo N, Maghooli K, Sheikhani A. Prediction of obstructive sleep apnea using ensemble of recurrence plot convolutional neural networks (RPCNNs) from polysomnography signals. Med Hypotheses. 2021;154:110659.
- Niroshana SMI, Zhu X, Nakamura K, Chen W. A fused-image-based approach to detect obstructive sleep apnea using a single-lead ECG and a 2D convolutional neural network. PLoS One. 2021;16:e0250618.
- Mukherjee D, Dhar K, Schwenker F, Sarkar R. Ensemble of Deep Learning Models for Sleep Apnea Detection: An Experimental Study. Sensors. 2021;21:5425.
- Mashrur FR, Islam MdS, Saha DK, Islam SMR, Moni MA. SCNN: Scalogram-based convolutional neural network to detect obstructive sleep apnea using single-lead electrocardiogram signals. Comput Biol Med. 2021;134:104532.
- Wang T, Lu C, Shen G, Hong F. Sleep apnea detection from a single-lead ECG signal with automatic feature-extraction through a modified LeNet-5 convolutional neural network. PeerJ. 2019;7:e7731.
- Chang H-Y, Yeh C-Y, Lee C-T, Lin C-C. A Sleep Apnea Detection System Based on a One-Dimensional Deep Convolution Neural Network Model Using Single-Lead Electrocardiogram. Sensors. 2020;20:4157.
- Zhang Y, Zhou L, Zhu S, Zhou Y, Wang Z, Ma L, et al. Deep Learning for Obstructive Sleep Apnea Detection and Severity Assessment: A Multimodal Signals Fusion Multiscale Transformer Model. Nat Sci Sleep. 2025;Volume 17:1–15.
- Tang LYW, Coxson HO, Lam S, Leipsic J, Tam RC, Sin DD. Towards large-scale case-finding: training and validation of residual networks for detection of chronic obstructive pulmonary disease using low-dose CT. Lancet Digit Health. 2020;2:e259–67.
- Wu Y, Qi S, Feng J, Chang R, Pang H, Hou J, et al. Attention-guided multiple instance learning for COPD identification: To combine the intensity and morphology. Biocybern Biomed Eng. 2023;43:568–85.
- Xu C, Qi S, Feng J, Xia S, Kang Y, Yao Y, et al. DCT-MIL: Deep CNN transferred multiple instance learning for COPD identification using CT images. Phys Med Biol. 2020;65:145011.
- Yu K, Sun L, Chen J, Reynolds M, Chaudhary T, Batmanghelich K. DrasCLR: A self-supervised framework of learning disease-related and anatomy-specific representation for 3D lung CT images. Med Image Anal. 2024;92:103062.
- González G, Ash SY, Vegas-Sánchez-Ferrero G, Onieva Onieva J, Rahaghi FN, Ross JC, et al. Disease Staging and Prognosis in Smokers Using Deep Learning in Chest Computed Tomography. Am J Respir Crit Care Med. 2018;197:193–203.
- Zou X, Ren Y, Yang H, Zou M, Meng P, Zhang L, et al. Screening and staging of chronic obstructive pulmonary disease with deep learning based on chest X-ray images and clinical parameters. BMC Pulm Med. 2024;24:153.
- Schroeder JD, Bigolin Lanfredi R, Li T, Chan J, Vachet C, Paine R, et al. Prediction of Obstructive Lung Disease from Chest Radiographs via Deep Learning Trained on Pulmonary Function Data. Int J Chron Obstruct Pulmon Dis. 2021;Volume 15:3455–66.
- Weiss J, Raghu VK, Bontempi D, Christiani DC, Mak RH, Lu MT, et al. Deep learning to estimate lung disease mortality from chest radiographs. Nat Commun. 2023;14:2797.
- Ryu S, Kim JH, Yu H, Jung H-D, Chang SW, Park JJ, et al. Diagnosis of obstructive sleep apnea with prediction of flow characteristics according to airway morphology automatically extracted from medical images: Computational fluid dynamics and artificial intelligence approach. Comput Methods Programs Biomed. 2021;208:106243.
- Tsuiki S, Nagaoka T, Fukuda T, Sakamoto Y, Almeida FR, Nakayama H, et al. Machine learning for image-based detection of patients with obstructive sleep apnea: an exploratory study. Sleep and Breathing. 2021;25:2297–305.
- Luo J, Ye M, Xiao C, Ma F. HiTANet: Hierarchical Time-Aware Attention Networks for Risk Prediction on Electronic Health Records. Proceedings of the 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining. New York, NY, USA: ACM; 2020. p. 647–56.
- Lopez K, Li H, Lipkin-Moore Z, Kay S, Rajeevan H, Davis JL, et al. Deep learning prediction of hospital readmissions for asthma and COPD. Respir Res. 2023;24:311.
- Radford A, Kim JW, Hallacy C, Ramesh A, Goh G, Agarwal S, et al. Learning Transferable Visual Models From Natural Language Supervision. 2021;
- Lee J, Yoon W, Kim S, Kim D, Kim S, So CH, et al. BioBERT: a pre-trained biomedical language representation model for biomedical text mining. Bioinformatics. 2020;36:1234–40.
- Zhang Y, Xia T, Han J, Wu Y, Rizos G, Liu Y, et al. Towards Open Respiratory Acoustic Foundation Models: Pretraining and Benchmarking. 2024;
- Eyben F, Wöllmer M, Schuller B. Opensmile. Proceedings of the 18th ACM international conference on Multimedia. New York, NY, USA: ACM; 2010. p. 1459–62.
- Hershey S, Chaudhuri S, Ellis DPW, Gemmeke JF, Jansen A, Moore RC, et al. CNN architectures for large-scale audio classification. 2017 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP). IEEE; 2017. p. 131–5.
- Po-Yao Huang, Hu Xu, Juncheng Li, Alexei Baevski, Michael Auli, Wojciech Galuba, et al. Masked Autoencoders that Listen. NeurIPS Proceedings. 2022;
- Elizalde B, Deshmukh S, Ismail M Al, Wang H. CLAP Learning Audio Concepts from Natural Language Supervision. ICASSP 2023 - 2023 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP). IEEE; 2023. p. 1–5.
- Niu C, Lyu Q, Carothers CD, Kaviani P, Tan J, Yan P, et al. Medical multimodal multitask foundation model for lung cancer screening. Nat Commun. 2025;16:1523.
- Mikhael PG, Wohlwend J, Yala A, Karstens L, Xiang J, Takigami AK, et al. Sybil: A Validated Deep Learning Model to Predict Future Lung Cancer Risk From a Single Low-Dose Chest Computed Tomography. Journal of Clinical Oncology. 2023;41:2191–200.
- Chao H, Shan H, Homayounieh F, Singh R, Khera RD, Guo H, et al. Deep learning predicts cardiovascular disease risks from lung cancer screening low dose computed tomography. Nat Commun. 2021;12:2963.
- Li Y, Rao S, Solares JRA, Hassaine A, Ramakrishnan R, Canoy D, et al. BEHRT: Transformer for Electronic Health Records. Sci Rep. 2020;10:7155.
- Herrett E, Gallagher AM, Bhaskaran K, Forbes H, Mathur R, van Staa T, et al. Data Resource Profile: Clinical Practice Research Datalink (CPRD). Int J Epidemiol. 2015;44:827–36.
- Miotto R, Li L, Kidd BA, Dudley JT. Deep Patient: An Unsupervised Representation to Predict the Future of Patients from the Electronic Health Records. Sci Rep. 2016;6:26094.
- Choi E, Bahadori MT, Kulas JA, Schuetz A, Stewart WF, Sun J. RETAIN: An Interpretable Predictive Model for Healthcare using Reverse Time Attention Mechanism. 2016;
- Regan EA, Hokanson JE, Murphy JR, Make B, Lynch DA, Beaty TH, et al. Genetic Epidemiology of COPD (COPDGene) Study Design. COPD: Journal of Chronic Obstructive Pulmonary Disease. 2011;7:32–43.
- Vestbo J, Anderson W, Coxson HO, Crim C, Dawber F, Edwards L, et al. Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points (ECLIPSE). European Respiratory Journal. 2008;31:869–73.
- Couper D, LaVange LM, Han M, Barr RG, Bleecker E, Hoffman EA, et al. Design of the Subpopulations and Intermediate Outcomes in COPD Study (SPIROMICS): Table 1. Thorax. 2014;69:492–5.
- Bourbeau J, Tan WC, Benedetti A, Aaron SD, Chapman KR, Coxson HO, et al. Canadian Cohort Obstructive Lung Disease (CanCOLD): Fulfilling the Need for Longitudinal Observational Studies in COPD. COPD: Journal of Chronic Obstructive Pulmonary Disease. 2014;11:125–32.
- Johnson AEW, Pollard TJ, Shen L, Lehman LH, Feng M, Ghassemi M, et al. MIMIC-III, a freely accessible critical care database. Sci Data. 2016;3:160035.
- Johnson AEW, Bulgarelli L, Shen L, Gayles A, Shammout A, Horng S, et al. Author Correction: MIMIC-IV, a freely accessible electronic health record dataset. Sci Data. 2023;10:219.
- Pollard TJ, Johnson AEW, Raffa JD, Celi LA, Mark RG, Badawi O. The eICU Collaborative Research Database, a freely available multi-center database for critical care research. Sci Data. 2018;5:180178.
- Faltys M, Zimmermann M, Lyu X, Hüser M, Hyland S, Rätsch G, et al. HiRID, a high time-resolution ICU dataset. PhysioNet. 2021;
- Wu Z, Jiang Y, Li S, Li A. Enhanced machine learning predictive modeling for delirium in elderly ICU patients with COPD and respiratory failure: A retrospective study based on MIMIC-IV. PLoS One. 2025;20:e0319297.
- Penzel T, Moody GB, Mark RG, Goldberger AL, Peter JH. The apnea-ECG database. Computers in Cardiology 2000 Vol27 (Cat 00CH37163). IEEE; 2002. p. 255–8.
- Goldberger A, Amaral L, Glass L, Hausdorff J, Ivanov PC, Mark R, et al. St. Vincent’s University Hospital / University College Dublin Sleep Apnea Database. PhysioNet. 2007.
- Quan SF, Howard B V, Iber C, Kiley JP, Nieto FJ, O’Connor GT, et al. The Sleep Heart Health Study: design, rationale, and methods. Sleep. 1997;20:1077–85.
- Topol EJ. High-performance medicine: the convergence of human and artificial intelligence. Nat Med. 2019;25:44–56.
- Hatem R, Simmons B, Thornton JE. A Call to Address AI “Hallucinations” and How Healthcare Professionals Can Mitigate Their Risks. Cureus. 2023;
- Science & Tech Spotlight: Generative AI in Health Care. U.S. Government Accountability Office. 2024.
- AI Act. European Commision. 2025.
- Regulation (EU) 2024/1689 of the European Parliament and of the Council of 13 June 2024 laying down harmonised rules on artificial intelligence and amending Regulations (EC) No 300/2008, (EU) No 167/2013, (EU) No 168/2013, (EU) 2018/858, (EU) 2018/1139 and (EU) 2019/2144 and Directives 2014/90/EU, (EU) 2016/797 and (EU) 2020/1828 (Artificial Intelligence Act) (Text with EEA relevance). European Union; 2024.
- DeGroot MH. Optimal Statistical Decisions. Wiley; 2004.
- Kevin P. Murphy. Probabilistic Machine Learning: An introduction. MIT Press; 2022.
- Dawid AP. The Well-Calibrated Bayesian. J Am Stat Assoc. 1982;77:605–10.
- Luciana Ferrer, Daniel Ramos. Evaluating Posterior Probabilities: Decision Theory, Proper Scoring Rules, and Calibration. Transactions on Machine Learning Research. 2025;
- Guo C, Pleiss G, Sun Y, Weinberger KQ. On calibration of modern neural networks. Proceedings of the 34th International Conference on Machine Learning. JMLR.org; 2017. p. 1321–30.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



