
Submitted:
13 May 2025
Posted:
15 May 2025
You are already at the latest version
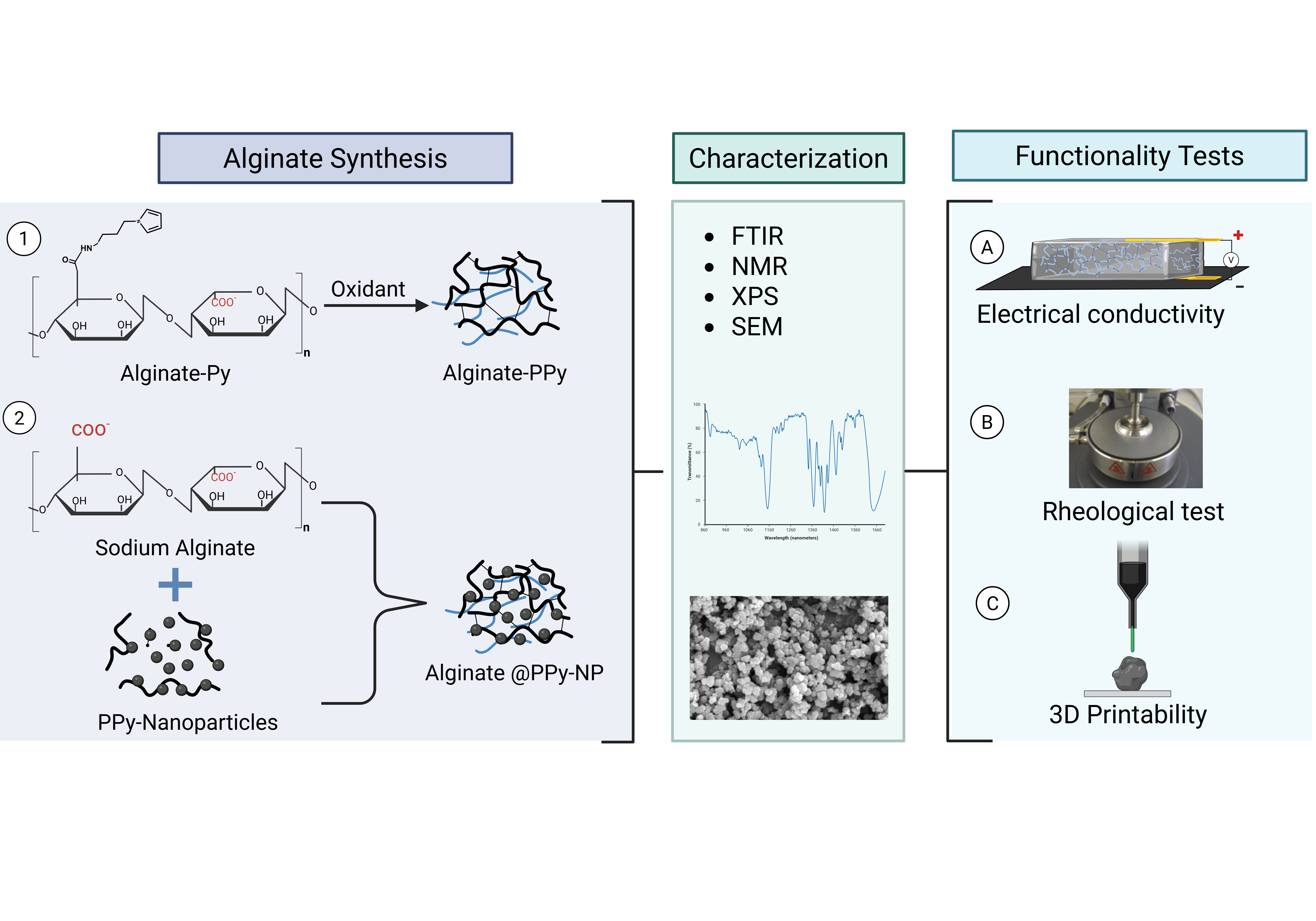
Abstract
Electrically conductive hydrogels are gaining attention for their applications in biosensing, cellular interfaces, and tissue engineering. However, conventional hydrogels often lack adequate electrical conductivity. Here, we present two novel conductive alginate-based hydrogels designed for extrusion-based 3D bioprinting: (i) covalently synthesized alginate-polypyrrole (alginate-PPy) via EDC/NHS-mediated conjugation with 3-aminopropyl pyrrole and (ii) nanoparticle-reinforced alginate blended with polypyrrole nanoparticles (alginate@PPy-NP). Both systems exhibit shear-thinning behavior, tunable viscoelasticity, and excellent printability. Alginate@PPy-NP demonstrated superior compressive strength and shape fidelity, whereas alginate-PPy showed enhanced elastic moduli (G′/G″), reflecting a more uniform gel network. The electrical conductivity increased with increasing pyrrole content in both formulations. Optimization of the composition and printing conditions enabled the fabrication of fibroblast-laden constructs with high structural integrity. This work highlights the potential of alginate-polypyrrole hydrogels as customizable, conductive bioinks for 3D bioprinting in regenerative medicine.
Keywords:
Alginate-pyrrole
; 3D bioprinting
; polypyrrole
; conductive hydrogel
; bioink
; electrical conductivity
; rheological properties
1. Introduction
Three-dimensional (3D) (bio)printing is a cutting-edge technology for the precise deposition of (bio)materials onto specified locations with a high resolution. This process involves fabricating objects layer-by-layer using a printer head, nozzle, or similar technology [1]. The convergence of additive manufacturing with biomaterials has introduced new paradigms in the field of biotechnology. The emergence of new printing materials and various 3D printers that integrate sensing layers with complex scaffold geometries has opened new avenues for biosensor development [2,3]. 3D bioprinting now extends to critical applications, including embedding active biomolecular recognition elements into 3D printed objects for (bio)sensing [2,3,4,5], applications in tissue engineering [6], wound healing [6], and the production of various medical implants and models [7]. The major challenges commonly encountered during printing processes when working with bio-based hydrogels are structural collapse and loss of shape fidelity, depending on the viscoelastic property of the printing material [8]. Rapidly solidifying ‘inks’ are desirable for printing to prevent the deformation of the printed structure. An ideal hydrogel ink should flow through a nozzle during printing and retain its shape after printing and curing; this requires custom materials with tailored properties to be formulated for each application. Therefore, many hydrogels with varying viscosities have been adapted for 3D printing via microextrusion [9].
Hydrogels, particularly those derived from natural polymers, such as alginate, gelatin, and chitosan, are widely utilized in bioprinting because of their biocompatibility and ability in an aqueous environment, which is essential for cell viability and growth [10,11]. Hydrogels are hydrophilic 3D polymeric networks with a high degree of flexibility and ability to absorb and retain large amounts of aqueous liquids while retaining mechanical stability [1,12,13]. Physical hydrogels have several advantages, such as easy preparation and sol-gel transition [13,14]. However, such hydrogels often exhibit mechanical weaknesses in terms of gelation and printability, which limits their application in additive manufacturing processes. Therefore, significant efforts have been directed toward enhancing the mechanical properties of hydrogels, thereby improving their printability and functionality in tissue engineering. For instance, using a composite approach, alginate-gelatin composite bioinks, together with the incorporation of nanoparticles, have significantly improved mechanical strength, enhancing printability and cellular support in tissue engineering applications [15].
There are numerous challenges associated with working with conventional hydrogels, such as dynamic rheological behavior under different printing conditions, which leads to a loss of printing fidelity and structural integrity post-printing [16,17]. To address such challenges, efforts such as the use of composite polymers and blending of inorganic nanoparticles with biopolymers are being made for structural and functional enhancement. In addition, the use of sacrificial copolymers and printing into sacrificial support baths, whereby the sacrificial copolymer and support bath are removed after printing, is being actively investigated[18,19]. Furthermore, tissue engineering applications that cultivate electrically excitable cells (such as cardiomyocytes, neurons, and myocytes) would benefit from improved cell adhesion, cell-cell communication, proliferation, and differentiation when the hydrogel is made conductive [20,21]. However, a critical research question remains to be answered: how can we engineer a hydrogel that retains the biocompatibility, printability, and rheological properties of alginate while enhancing its electrical conductivity properties?
Research on conductive hydrogels is advancing because of their potential applications in various fields including bioelectronics, drug delivery systems, tissue engineering, and biosensors [3]. Despite these advances, current hydrogel-based bioinks fail to fully support the cultivation and electrical stimulation of electrically excitable cells, such as neurons and cardiomyocytes, owing to insufficient electrical conductivity. This need limits the potential of 3D bioprinted tissues to mimic the native electroactive tissues. Therefore, continuous research efforts are directed towards incorporating conductive materials into hydrogel bioinks by doping the hydrogel with intrinsically conductive materials (ICMs), such as polypyrrole, poly(3,4-ethylenedioxythiophene): poly(styrene sulfonate) (PEDOT: PSS) [22,23], graphene oxide [24], polythiophene, and nanoparticles. While the incorporation of ICMs into hydrogels has been explored and shown to enhance the electrical properties while maintaining the unique characteristics of the hydrogels, such as flexibility and biocompatibility [24], conductive hydrogels prepared via doping ICMs are presented with a significant setback: uncontrolled leaching of the conductive unit from the hydrogel due to the non-covalent nature of the incorporation [25]. Thus, we hypothesized that covalently binding the intrinsically conductive unit to the alginate polymer could lead to a stable 3D printable electrically conductive hydrogel.
Sodium alginate is an ideal biopolymer candidate because of its excellent biocompatibility, non-toxicity, and ability to rapidly form 3D hydrogels upon interaction with divalent cations [26]. Furthermore, its thixotropic properties, including its shear-thinning behavior and viscosity recovery [27], make it particularly suitable for 3D printing applications, either as a standalone material or in combination with other polymers.
Despite their documented advantages, alginate hydrogels, like other biopolymer-based hydrogels, lack sufficient electrical conductivity, limiting their widespread application in bioelectronics, biosensors, and electroactive tissue scaffolds. Thus, we propose the integration of polypyrrole into alginate hydrogels to confer electrical properties to alginate while maintaining its rheological features, making it amenable to 3D bioprinting. This study introduces a new approach for creating an electrically conductive alginate-based composite hydrogel by covalently binding sodium alginate to a pyrrole monomer, from which conductive polypyrrole is chemically synthesized either in situ or ex situ. Specifically, an N-substituted pyrrole derivative, aminopropyl pyrrole, was synthesized and conjugated with alginate using carbodiimide chemistry. The resulting alginate-pyrrole conjugate was characterized by its rheological properties, electrical conductivity, and 3D printability.
Wright et al. showed the printability of alginate mixed with pyrrole monomer, which was subsequently polymerized into polypyrrole, and reported uncontrolled leaching of polypyrrole and the brittleness of the resulting hydrogel [25]. Therefore, two strategies were explored in this study to incorporate polypyrrole into the alginate matrix: (1) oxidative polymerization of pyrrole directly within the 3D printed alginate-pyrrole network (in situ polymerization), referred to as alginate-PPy, and (2) pre-synthesis of polypyrrole nanoparticles (PPy-NP), which were subsequently blended with alginate (as alginate@PPy-NP) prior to 3D printing. This is the first time that this approach has been employed to create an alginate-polypyrrole-based conductive hydrogel. The effects of these strategies on the printability, mechanical integrity, and electrical properties of hydrogels were examined. Notably, the electrical conductivity increased with pyrrole content in both cases, and both types of alginate-polypyrrole were 3D printable, while alginate mixed with already-made polypyrrole nanoparticles displayed appreciably higher compressibility, without leaching out of the ICMs, highlighting the potential of this covalently functionalized alginate as an electrically conductive bio-ink.
2. Materials and Methods
2.1. Materials
1-(2-cyanoethyl)-pyrrole, ether, LiAlH4, low-viscosity sodium alginate (A2158-250G), 1-ethyl-[3-(dimethyl amino)propyl]-3-ethyl carbodiimide HCl (EDAC; E-1769), 2-[N-morpholino] ethane sulfonic acid (MES) buffer (M-8250), ammonium persulfate, phosphate-buffered saline (PBS) (P-3813), calcium chloride (C-5426), and N-hydroxysulfo-succinimide (NHSS; 24510) were of analytical grade.
2.2. Synthesis of Aminopropyl Pyrrole
A solution of 1-(2-cyanoethyl) pyrrole (0.02 mol) was added dropwise to anhydrous ether (15 mL) in a suspension of LiAlH4 (0.05 mol), prepared in anhydrous ether (150 mL), refluxed for 10 h, and cooled. The excess hydride was quenched by successive addition of water (1.7 mL), a solution of 15% (w/v) NaOH (1.7 mL), and water (5.1 mL), and then heated at 40 °C for two h. The resulting solution was filtered through celite and the filtrate was evaporated to dryness. The synthesis of N-(3-aminopropyl)-pyrrole was confirmed by ¹H NMR (400 MHz, CDCl₃) δ: 6.70 (s, 2H, H–α), 6.24 (s, 2H, H–β), 3.25 (t, J = 6.8 Hz, 2H, –CH₂–NH₂), 2.65 (t, J = 7.2 Hz, 2H, –CH₂–CH₂–NH₂), 1.85 (m, 2H, –CH₂–CH₂–CH₂–) (supplementary section Figure S1), FTIR (Figure 2), and mass (supplementary section, Figure S2) spectroscopies.
Figure 1.
Covalent conjugation of pyrrole monomers to sodium alginate
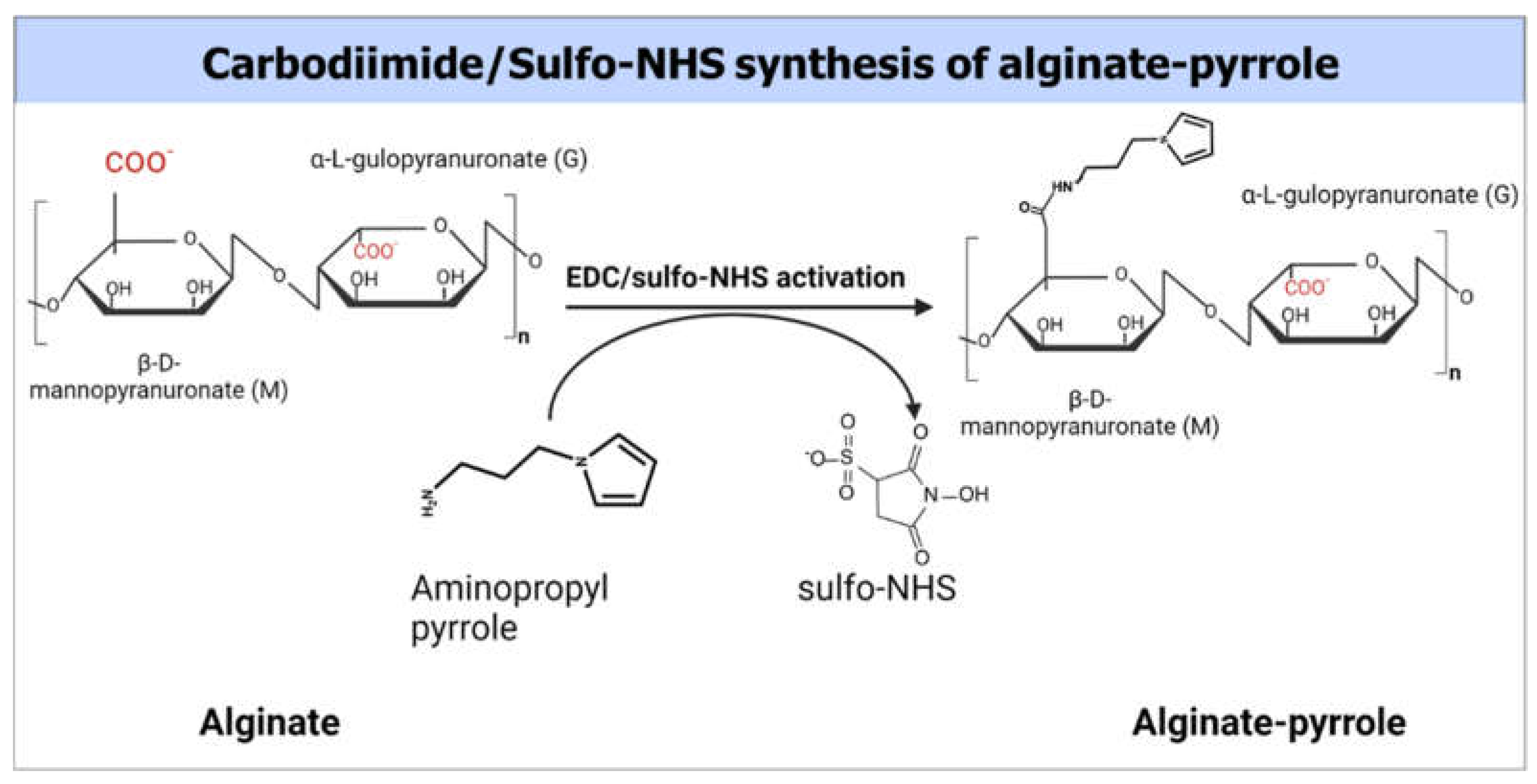
2.3. Synthesis of Alginate-Pyrrole
An alginate solution (2.5% w/v) was prepared by dissolving 500 mg of alginate in 20 ml of double-distilled water (DDW) with a resistivity of 18.2 MΩ and sterile filtered through a 0.22 µm filter (Stericup and Steritop EMD Millipore Corp., Billerica, USA). To the filtered alginate solution, 852 mg MES buffer and 400 mg NaCl were added and stirred well, and the pH was adjusted to 6.5. Next, the carboxyl groups of the alginate were activated using 1 mmol of EDAC and 0.5 mmol NHSS as a co-reactant (the calculation was for the ratios of reagents that would produce a theoretical 50% molar modification of the number of carboxylic groups of alginates) and stirred for 3 h at room temperature. mmol) of N-(3-aminopropyl) pyrrole (480 mg, 3 mmol) were added to a solution of activated alginate and stirred overnight at room temperature (Figure 1). The resulting polymer composite was dialyzed against doubly distilled water using a 3.5 kDa MWCO membrane (Spectrum Lab Membrane Filtration Products, Inc., TX, U.S.A.), with the water changed twice a day for 3 days. The dialysate was then lyophilized. The extent of pyrrole incorporation was determined by UV-vis spectroscopy using a pedestal function of NanoDrop ONEC desktop spectrophotometer (Thermo Scientific, USA). Briefly, the extent of alginate modification by N-(3-aminopropyl)-pyrrole was determined by dissolving the pyrrole-alginate samples to produce a 0.01% (w/v) pyrrole-alginate solution and measuring the absorbance. The extent of alginate modification was determined from the calibration curve by measuring the absorbance of different amounts of N-(3-aminopropyl)-pyrrole in 0.01% (w/v) alginate solution. A standard solution of alginate at a concentration of 0.01% (w/v) was used as blank (Supplementary Figure S5).
2.4. Attenuated Total Reflection Fourier-Transform Infrared Spectroscopy (ATR-FTIR).
Infrared spectra of the synthesized aminopropyl pyrrole and alginate-pyrrole were recorded using a Smart iTR™ Diamond ATR-FTIR sampling accessory and Nicolet 6700 spectrometer to characterize the functional groups of the synthetic products. All measurements were performed using a built-in diamond-attenuated total-reflection (ATR) crystal. The FTIR spectra were acquired over the range of 4000–650 cm−1 at a resolution of 4 cm−1 and represented an average of 36 scans. The spectrum of the clean, dry diamond ATR crystal in ambient atmosphere (air) was used as the background for the infrared measurements.
2.5. X-Ray Photoelectron Spectroscopy (XPS) Analysis
X-ray photoelectron spectroscopy (XPS) analysis was conducted on lyophilized samples of alginate-pyrrole and alginate using an “ESCALAB Xi+” instrument (Thermo Fisher Scientific). The ionization energies of oxygen, nitrogen, and carbon present in the lyophilized samples were recorded as previously described for alginate [28]. The analysis was performed at an ambient pressure < 1 × 10 -10 mbar. An aluminum Kα radiation source (1486.68 eV) was used for photoelectron emission at room temperature, and spectra were collected at an angle of 90° from the X-ray source. A low-energy electron flood gun was employed to minimize surface charging and measurements were performed with a spot diameter of 650 µm. Spectral analysis was performed using the Avantage software version 6.6.0, provided by Thermo Fisher Scientific). High-resolution spectra of the C1s, O1s, and N1s peaks and a survey spectrum at a pass energy of 20 eV were obtained.
2.6. Chemical Synthesis of Polypyrrole Nanoparticles (PPy-NP)
Polypyrrole (PPy) was synthesized via chemical oxidative polymerization using ammonium persulfate (APS) as the oxidant with a monomer-to-oxidant molar ratio of 1:1.5. The final APS concentration was 212 mM, and the reaction was conducted at 4 °C for 120 min. Briefly, 1.0 mL of pyrrole monomer (14.4 mmol) was added to 50 mL of ice-cold deionized water (DDW) under continuous stirring. Separately, 4.83 g APS (21.6 mmol) was dissolved in 10 mL DDW and added dropwise to the pyrrole solution. The total reaction volume was adjusted to 100 mL using DDW and the mixture was magnetically stirred. Aliquots of 5 mL were withdrawn at defined time intervals to monitor the polymerization progress via UV-Vis spectroscopy at 460 nm (Supplementary Figure S4). Following polymerization, the black precipitate of PPy was collected by vacuum filtration, thoroughly washed with DDW and ethanol to remove residual monomers and oxidants, and dried under vacuum at 50 °C overnight. The surface morphology of the synthesized PPy was examined using scanning electron microscopy (SEM). Samples were sputter-coated with gold and imaged at various magnifications to assess surface features and particle aggregation (Figure 12).
2.7. Preparation of Alginate-Pyrrole as Bioink
2.7.1. Alginate-Pyrrole/Pyrrole (Alginate-Py) as Bioink
To create an optimal ink formulation, different stoichiometric ratios of pyrrole in alginate were prepared (Table 1) using double-distilled water (DDW) with a resistivity of 18.2 MΩ. Lyophilized alginate-pyrrole and sodium alginate were separately dissolved in double-distilled water to a final concentration of 2.5% (w/v) under stirring for 2 h. Primary crosslinking of the alginate was achieved by mixing 1.2 ml of alginate-pyrrole, 1.2 ml of alginate, 0.3 ml of CaCl2 (0.1 M), and 0.021 ml of pyrrole monomer (14.125 M), using a homogenizer to equally distribute the calcium ions throughout the solution (Table 1). The mixture was then stirred at RT until further use. The partially crosslinked alginate-pyrrole/pyrrole was transferred to a sterile syringe.
For the cell-laden bioink, 0.28 ml of Dulbecco's Modified Eagle Medium (DMEM) containing 2.6 × 106 cells/ml of isolated GFP-expressing fibroblasts mixed with DMEM was loaded into an additional sterile syringe, which was immediately connected to the alginate-pyrrole/pyrrole-loaded syringe through a Luer-to-Luer connector. The solutions were mixed by gently pushing the pistons back and forth for 1 min until they were homogeneously mixed. The final bioink solution consisted of 2% alginate-pyrrole (w/v), 0.01 M CaCl2 (or 0.36% w/v calcium gluconate), and 2.6 × 106 cells/mL.
2.7.2. Alginate@polypyrrole Nanoparticles (alginate@PPy-NP) as Bioink
The possibility of blending PPy-NP into an alginate hydrogel as a bioink (alginate@PPy-NP) for 3D printing was explored. Different solutions of 3 ml (2% w/v) alginate containing PPy-NP at final concentrations of 1, 0.33, and 0.17% w/v, corresponding to solutions with PPy-NP/alginate ratios of 8.3, 16.7, and 50% w/w, respectively, were prepared. The primary crosslinking was performed by dropwise addition of 0.05 M CaCl2 to reach a final concentration of 0.01 M and was stirred overnight and subsequently used for 3D printing or further processed for rheological testing and electrical conductivity testing.
2.8. 3D Printing of Alginate-Pyrrole Conjugate and Alginate@polypyrrole Composites
Basic 3D models of the scaffold construct were designed and spliced using Perfectory RP and software supplied by the Envision TEC 3D bio-plotter. Using the VisualMachine BP interface software (V2.2; EnvisionTEC GmbH), the designs were printed using a 3D Bioplotter Manufacturer Series system (EnvisionTEC GmbH). Scaffolds were 11 mm × 11 × 2.5 mm with an inner strand distance of 2.25 mm and inner strand angles of 0° and 90°. The strands were dispensed layer by layer using pneumatic pressure in a piston-based system through 25-gauge blunt dispense needles (EFD Nordson, Switzerland). The printing parameters were optimized in terms of the printing speed and pressure in the range of 10-22 millimeter per second and 0.5-1.0 bar respectively.
Prior to bioprinting, as previously described [19], the bioink was deposited into a sterilized 30 mL printer cylindrical cartridge sealed with a fitted plunger utilizing a Luer-to-Luer connector. The printer cartridge was subsequently sealed and centrifuged for 1 min at 150 g to eliminate any remaining air bubbles. A sterile 25-gauge needle tip was attached to the cartridge and a cap connecting the print head to the barrel was affixed. The barrel was placed in the low-temperature head of the bioprinter (EnvisionTEC 3D-bioplotter Developer Series, Germany), set to 25°C. The bioink solution was allowed to equilibrate to the printing head temperature for 30 min prior to bioprinting. The 3D printing was conducted by extrusion over the range of 0.5-0.8 bar and printing speed of 10-22 mm/s, followed by secondary crosslinking with 0.1 M CaCl2. In addition, the possibility of printing using the freeform reversible embedding of suspended hydrogels (FRESH), as described by Hinton et al. [18], was evaluated.
To eliminate the gelatin support bath, the bioprinted constructs were incubated for 1 h at 37°C and 5% CO2. The constructs were then washed and incubated for 10 min at 37°C in DMEM supplemented with 0.1 M CaCl2 to further crosslink the 3D-bioprinted constructs, after which the constructs were incubated in DMEM supplemented with 20% fetal bovine serum (FBS) in a new 12-well plate.
2.8.1. Semi-Quantification of Printability
The printability of the alginate-polypyrrole inks to achieve square-shaped pores was quantified from light microscopy images using the ImageJ software. The printability of the bioink was determined according to the method described by Ouyang et al. [29], based on the understanding of the circularity (C) of an enclosed area. The circularity of an enclosed area is defined as
Where L is the perimeter and A is the area.
The circles exhibit the highest circularity (C = 1). The closer the value of C is to one, the closer the shape is to a circle. For a square shape, the circularity is π/4 [29]. Because the model designed in this study was square, the bioink printability (Pr) based on the square shape was defined according to the following equation [29]
In this study, the printability factor (Pf) is defined as
In this context, three statuses of Pf are defined as follows: Pf < 1 corresponds to under-gelation and usually rounded pore corners because of the fusion of layers and swelling; Pf = 1 corresponds to proper gelation with ideal square-shaped pores; and Pf > 1 corresponds to over-gelation, causing shrinkage of the construct [30].
2.9. Rheological Characterization
The Viscoelastic behavior of the alginate and alginate-pyrrole solutions and hydrogels was assessed using a stress-controlled advanced rheometer (AR 2000; TA Instruments). A 40 mm diameter 3°58 steel cone geometry with a truncation gap height of 104 μm was used. The storage modulus (G′), loss modulus (G″), and complex viscosity (|η∗|) were recorded as a function of time through oscillatory measurements in a frequency range of 0.1–10 Hz, and the strain was determined to be in the linear viscoelastic region. In addition, the apparent viscosities (Pa · s) of the hydrogel solutions were assessed at shear rates between 0.1 − 150 s−1. Compressibility was measured using a rheometer (TA Instruments, model AR 2000) operated in flat-plate mode with a 40 mm diameter in an increasing normal force.
2.10. Electrical Conductivity Characterization
Electrical measurements were performed using a sandwich configuration. A partially crosslinked alginate, alginate-Py (later polymerized to alginate-PPy after crosslinking), and alginate@PPy-NP hydrogel were individually cast into a custom-made mold to fully crosslink overnight by adding a crosslinker (150 mM calcium ions). A hydrogel with a volume of 1 cm³ was placed on a stainless-steel plate (1 × 1 cm²), and a second stainless-steel plate was placed on top of the hydrogel. Prior to device assembly, the stainless steel plates underwent a thorough cleaning process involving sequential sonication in acetone, methanol, isopropyl alcohol (IPA), and deionized water (DDW) for 15 min each, followed by nitrogen drying.
The device was placed in a probe station (JANIS ST-500, USA) and tightly secured by applying pressure to the top probe. Current-voltage (I-V) measurements were conducted using a Keithley 2635 Source-Meter Unit, with data acquisition controlled by a custom MATLAB program. The measurements spanned a voltage range from -1 V to 1 V in 0.05 V increments. At least three devices were tested for each sample type, to ensure reproducibility.
2.11. Statistical Analysis
A two-way or one-way ANOVA, followed by a Tukey multiple comparisons test, was conducted using GraphPad Prism version 8.0.2 for Windows (GraphPad Software, San Diego, CA, USA, www.graphpad.com, accessed on 26 November 2024)
3. Results
3.1. ATR-FTIR and NMR Spectra of Aminopropyl Pyrrole
Aminopropyl pyrrole is composed of a primary amine (NH2), repeated CH2 units, and a pyrrole ring. Primary amines undergo stretching and bending (scissoring and wagging, respectively). As shown in Figure 2, the peaks at 3325, 2929, and 1278 are respectively assigned to symmetric, asymmetric NH2 stretching, and C-N stretching, respectively. NH2 scissoring and wagging, which are detectable within 750-600 and 1650-1630 were found at 729 and 1627 respectively, while the in-plane and out-of-plane N-H bending appeared at 1567 and 720 cm-1, respectively [31].
The FT-IR spectra of sodium alginate and alginate-pyrrole were compared to confirm the structural transformation imposed on alginate by the incorporation of covalent pyrrole into alginate (Supplementary Figure S3). The broad band at approximately 3600-3060 cm−1 was assigned to hydrogen-bonded O–H stretching vibrations, and the two peaks at 2914 and 1591 were attributed to C-H stretching and C=O asymmetric stretching, respectively. The peak at 1404 cm-1 was assigned to both C-OH deformation and C=O asymmetric vibrations of the carboxylate groups [32]. In addition, additional peaks characteristic of alginate were observed at 1025 and 1053 cm-1, attributed to C-O stretching vibrations and C-O + C-C stretching vibrations of the pyranose rings, respectively. Additionally, the bands at 812 and 880 cm -1 were respectively assigned to mannopyranuronic acid and α-L-gulopyranuronic acid asymmetric ring vibrations, respectively, and were attributed to the α1-4 bond [32,33]. The pyrrole monomer has certain bonds whose vibrational state changes during IR spectroscopy and occurs in identical regions in the electromagnetic spectrum as alginate and pyrrole ring deformation at 730 cm-1.
Figure 2.
ATR-FTIR Spectra of aminopropyl pyrrole.
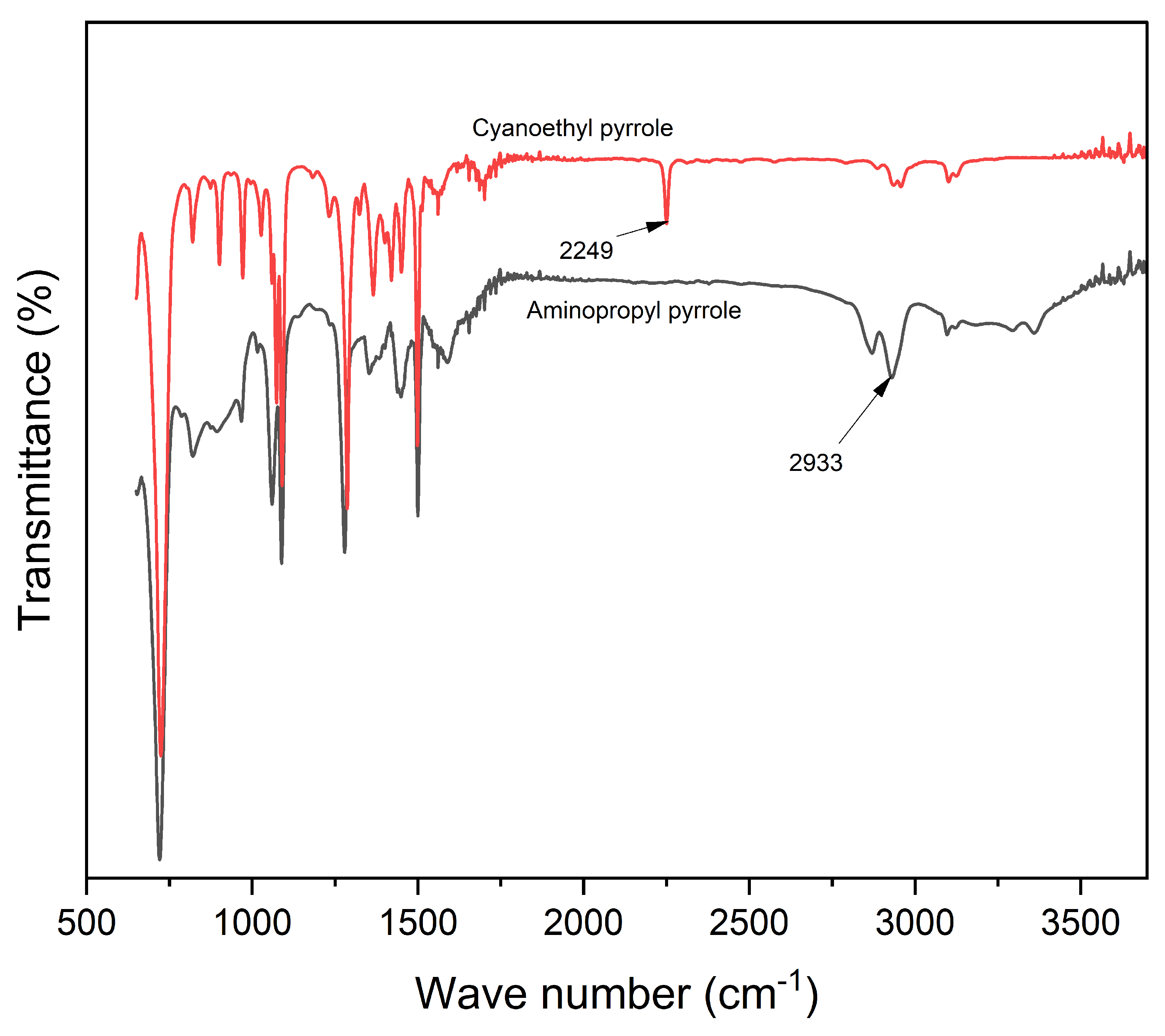
For instance, the N-H in-plane stretching, C-C in-ring stretching modes, and ring vibrations of pyrrole have been reported to occur at 3396, 1571, and 1467 cm-1, respectively, which overlap with some regions previously assigned to alginate [34,35]. In addition, the peaks attributable to amide bonds formed during alginate-pyrrole conjugation are reported to be located at approximately 1650 cm-1, which overlaps well with the C-H stretching of the carboxylate group of alginates. Therefore, it is desirable to distinguish between pyrrole and alginate. Such a feature is the peak that is detected at 729 cm-1 in the alginate-pyrrole and aminopropyl pyrrole spectra, but absent in the alginate spectrum. This peak is attributed to the C-H vibration of the 2,5 hydrogen atoms of the pyrrole ring [34].
The 1H-NMR spectrum revealed different hydrogens in the synthesized aminopropyl pyrrole (Figure 2 b) Hydrogens attached to an amine appear ~ at 0.5-5.0 ppm, whereas those on carbons directly bonded to an amine appear at ~2.3-3.0 ppm. The latter hydrogens are de-shielded by the electron-withdrawing effects of nitrogen and appear downfield in the NMR spectrum compared to the alkane hydrogens. This finding was further corroborated by the 13C NMR spectrum of the synthesized aminopropyl pyrrole (Supplementary Figure S1).
3.2. X-Ray Photoelectric Spectroscopy (XPS) of Alginate-Pyrrole
XPS was used to quantitatively determine the elemental and chemical compositions of alginate and synthesized alginate-pyrrole. High-resolution spectra were recorded for carbon, oxygen, and nitrogen, as shown in Figure 3. The compositions were calculated from the XPS spectra recorded in survey mode and expressed as relative atomic percentages.
Figure 3.
Comparison of deconvoluted XPS spectrum peaks (A-C) and survey analysis (D) of lyophilized alginate-pyrrole (alginate-Py). The deconvoluted peaks were assigned to the chemical groups based on the binding energies of the peaks (N1s, O1s, and C1s).
Figure 3.
Comparison of deconvoluted XPS spectrum peaks (A-C) and survey analysis (D) of lyophilized alginate-pyrrole (alginate-Py). The deconvoluted peaks were assigned to the chemical groups based on the binding energies of the peaks (N1s, O1s, and C1s).
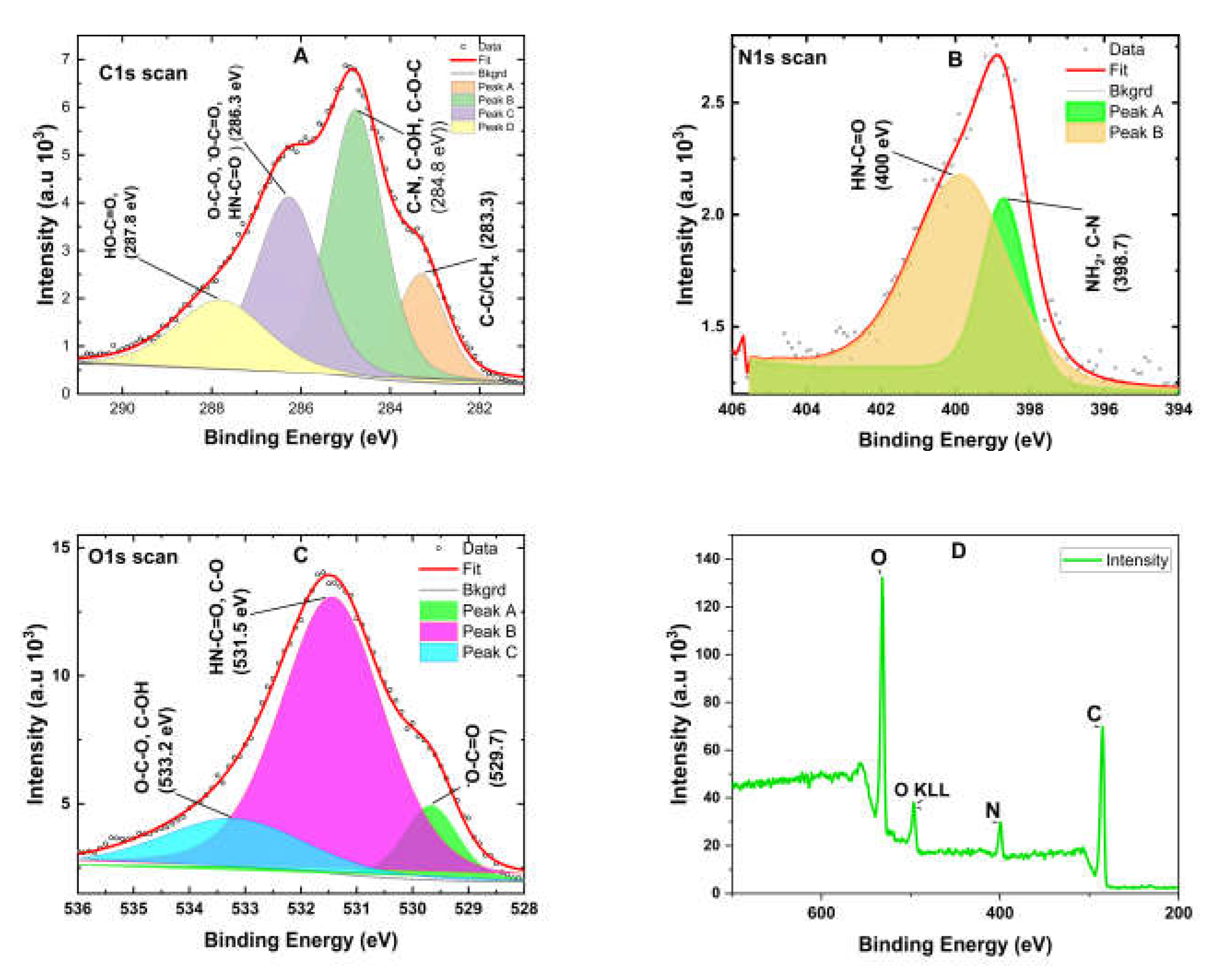
Elemental composition analysis of unmodified alginate and pyrrole-modified alginate (alginate-pyrrole) revealed that alginate-pyrrole consisted of 68.46% carbon, 23.44% oxygen, and 8.1% nitrogen, whereas unmodified alginate contained 53.58% carbon and 46.6% oxygen. A comparative evaluation of the carbon-to-oxygen (C/O) ratio showed a significant increase from 1.15 in unmodified alginate to 2.70 in alginate-pyrrole, representing a 42.59% increase when the C/O ratio of alginate was divided by that of alginate-pyrrole. This increase is attributed to the additional carbon introduced by aminopropyl-pyrrole conjugation and the reduction in oxygen content, likely due to the loss of water (H₂O) during amide bond formation.
3.3. Rheological Behavior of Calcium Alginate-Pyrrole Hydrogels
A key requirement for the successful three-dimensional (3D) bioprinting of hydrogels is the capability of the bio-ink to undergo a liquid-to-gel phase transition. The bioink should be injectable and gel rapidly upon extrusion from its surface. Alginate hydrogels crosslink through calcium ion-mediated electrostatic interactions, which determine the sol-gel phase-transition conditions. Additionally, components that modify crosslinking, such as partial covalent modification of the carboxyl group, affect the mechanical properties of the hydrogel, necessitating characterization of the rheological properties of chemically functionalized physical calcium alginate hydrogels.
3.3.1. Viscosity and Stored (G′) and Loss (G″) Moduli
To evaluate the rheological behavior of the alginate and pyrrole-modified alginate (alginate-PPy and alginate@PPy-NP) hydrogels, they were subjected to primary crosslinking at a final Ca2+ concentration of 10 mM (because primary crosslinking was performed prior to 3D printing). The mechanical properties of hydrogels, particularly their stored modulus (G'), indicate their ability to withstand deformation without permanent damage. For instance, hydrogels with a G' are significantly more significant than their loss modulus (G"), demonstrating a stable cross-linked structure, which is essential for maintaining shape and function under stress [36]. The time-dependent viscoelastic behavior of the alginate-PPy and alginate@PPy-NP formulations was evaluated through oscillatory rheology by monitoring the progression of the storage modulus (G′), loss modulus (G″), and complex viscosity (|η*|) as functions of time. As shown in Figure 5, both G′ and G″ gradually increased over time, and a sharp increase was observed after 60 s. It is noteworthy to mention the increase in G' with the amount of pyrrole (Figure 5 a-d) and ex-situ-generated polypyrrole nanoparticles added to alginate (alginate@PPy-NP).
Figure 4.
Oscillatory time sweep test for CA-alginate-pyrrole conjugates (alginate-Py) and Ca-alginate (a-d).
Figure 4.
Oscillatory time sweep test for CA-alginate-pyrrole conjugates (alginate-Py) and Ca-alginate (a-d).
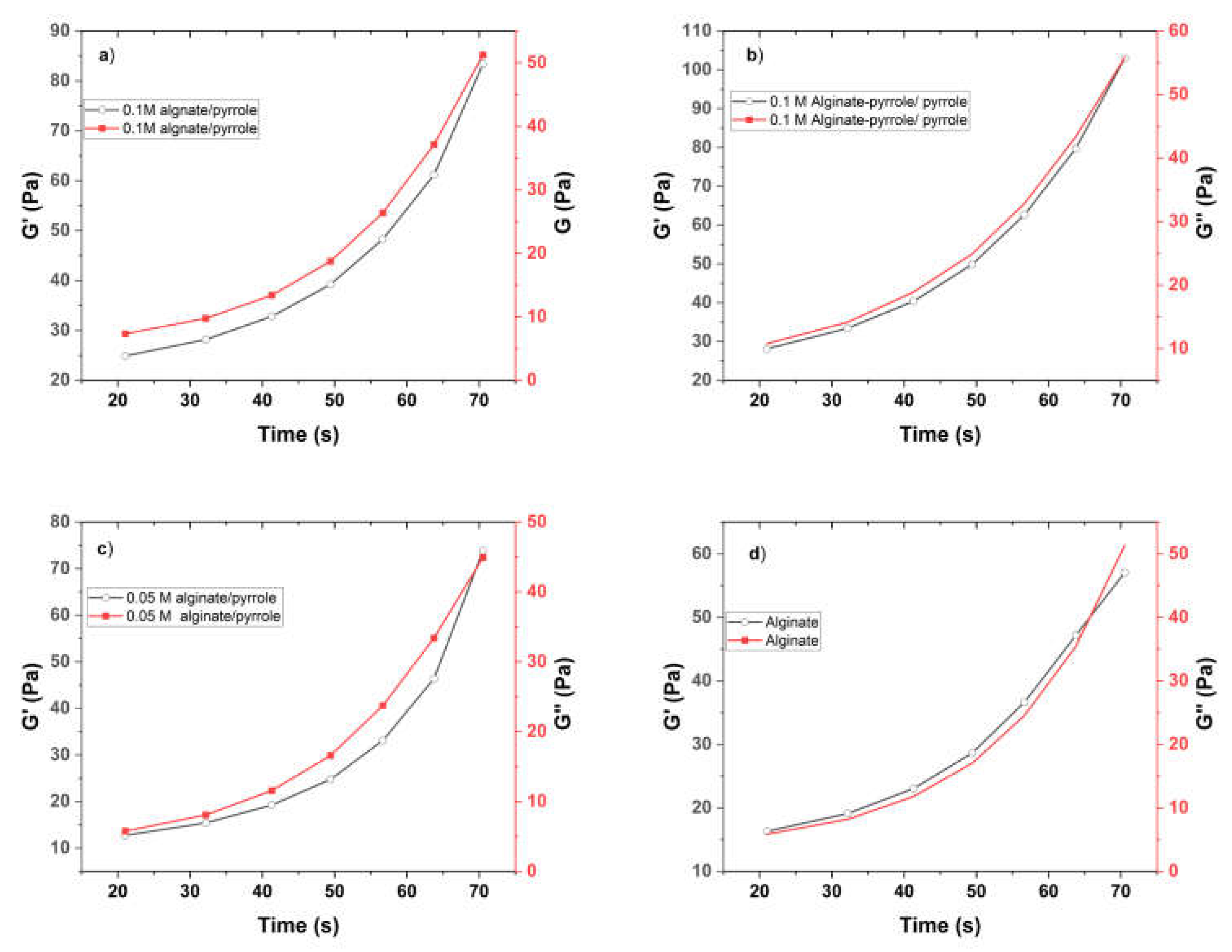
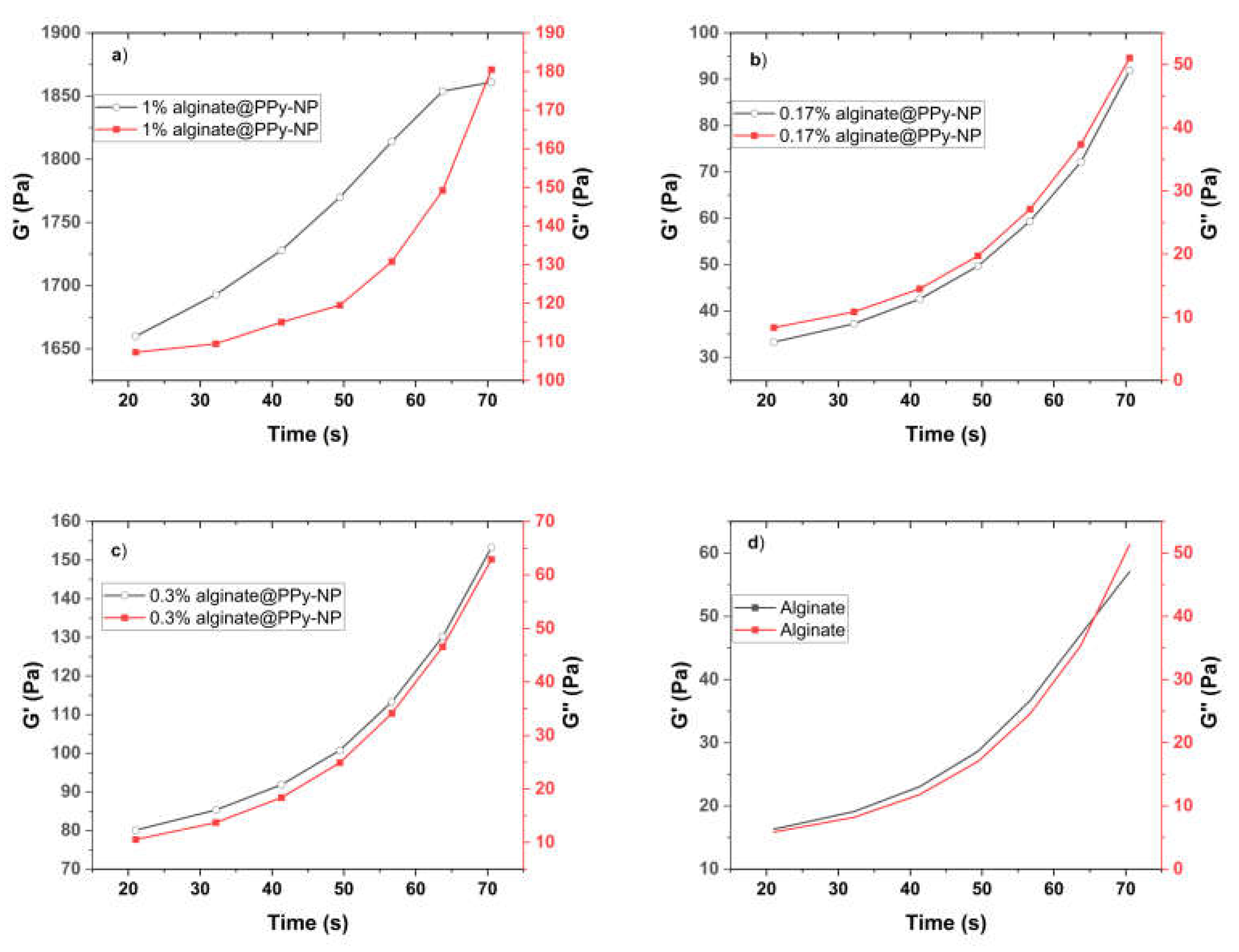
For the alginate-PPy system, the incorporation of pyrrole enhanced the gelation behavior compared to plain alginate. The alginate-PPy composite exhibited a marked increase in G′ from 28.11 Pa to 103.00 Pa, while |η*| decreased steadily from 47.24 Pa·s to 1.86 Pa·s over time, indicating the formation of a stable, elastic network. Alginate/pyrrole samples (0.05 M and 0.1 M) followed a similar but less pronounced trend, with final G′ values of 45.25 Pa and 83.38 Pa, respectively. In contrast, plain alginate showed limited structural evolution, with G′ reaching only 57.05 Pa and G″ approaching parity. Conversely, the alginate@PPy-NP system, which incorporated pyrrole nanoparticles at concentrations of 0.17%, 0.3%, and 1% (w/v), exhibited concentration-dependent enhancements in viscoelastic properties. The formulation containing 1% PPy-NP demonstrated the most significant elastic response, with the storage modulus (G′) increasing from 1660 to 1861 Pa, while the complex viscosity (|η*|) markedly decreased from 2648 to 29.76 Pa·s. Lower concentrations, specifically 0.3% and 0.17%, displayed similar, but progressively diminished trends. In contrast, alginate control exhibited a minimal elastic structure.
Taken together, these results reveal that pyrrole-based modifications of alginate—both molecular (alginate-PPy) and particulate (alginate@PPy-NP)–not only preserve but also enhance the time-dependent gelation and elastic behavior of the conductive pyrrole-modified alginate, with potential implications for improved structural fidelity in 3D bioprinting applications.
Figure 5.
Oscillatory time sweep test for Ca-alginate/polypyrrole (alginate@PPy-NP) and Ca-alginate (a-d).
Figure 5.
Oscillatory time sweep test for Ca-alginate/polypyrrole (alginate@PPy-NP) and Ca-alginate (a-d).
The storage modulus (G′) and loss modulus (G″) of the alginate and its pyrrole composite hydrogels as a function of frequency (Hz) at 25°C are shown in Supplementary Figure S6 (a-d) for the alginate-pyrrole (alginate-PPy) conjugate and Figure S7 (a-d) for alginate mixed with polypyrrole nanoparticles (alginate@PPy-NP). The G′ and G″ values of all the composite hydrogels increased with increasing frequency over the entire frequency range tested. Notably, the storage modulus G′ was greater than the corresponding loss modulus G″ over the entire frequency range, indicating the possible formation of a crosslinked structure by hydrogels with good stability.
Figure 6.
Frequency sweep test (a-d) and viscosities (e-h) of Ca-alginate, Ca-alginate-pyrrole conjugates, and Ca-alginate/pyrrole.
Figure 6.
Frequency sweep test (a-d) and viscosities (e-h) of Ca-alginate, Ca-alginate-pyrrole conjugates, and Ca-alginate/pyrrole.
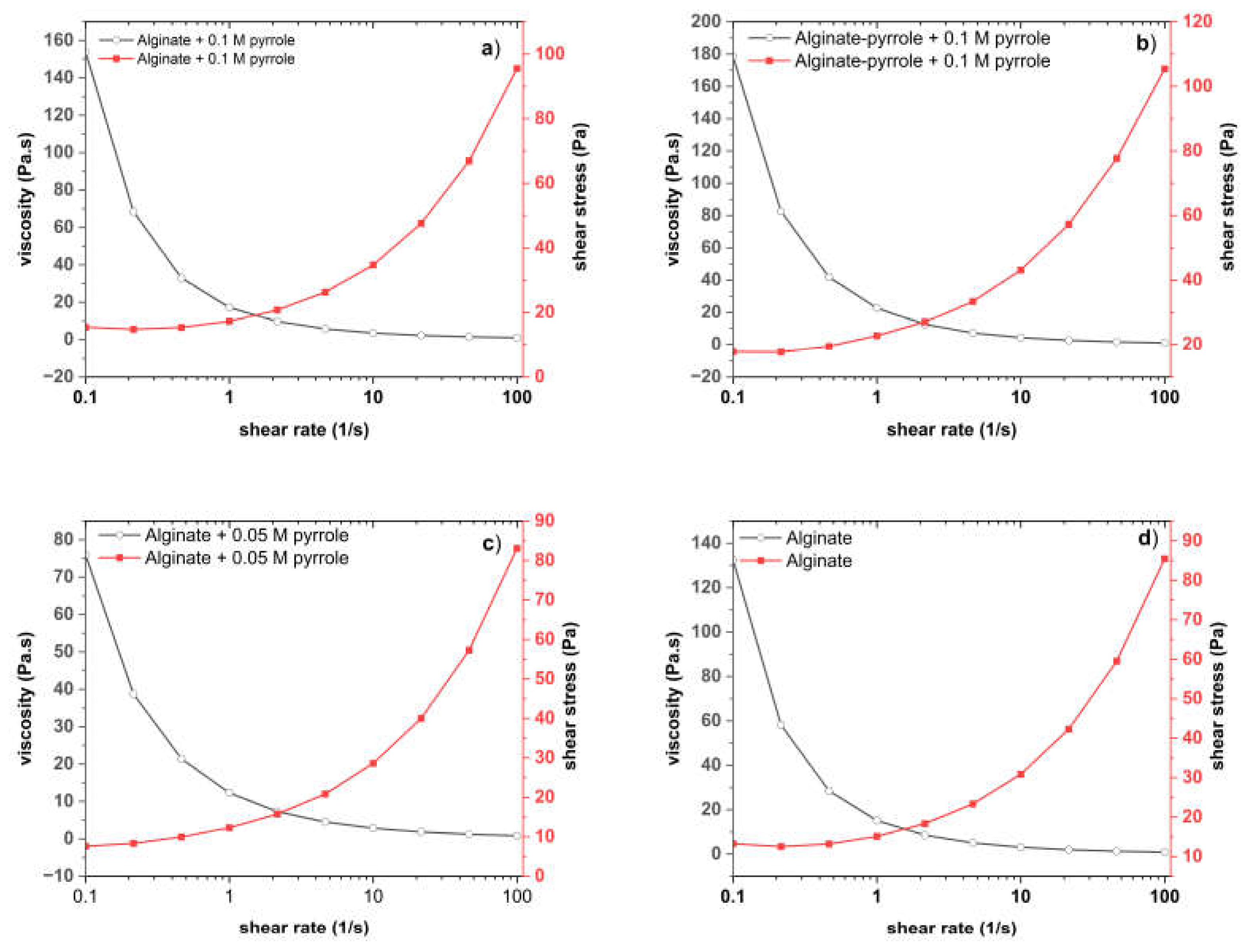
The plot of viscosity against shear rate shows that all samples exhibit shear-thinning (non-Newtonian) behavior, as the viscosity decreases with increasing shear rate. This is typical of polymeric and gel-like systems. Shear-thinning behavior was observed for all the samples, and higher pyrrole concentrations led to increased viscosity and shear stress. As shown in Figure 6 (a) and (b), the covalently modified alginate-pyrrole (Figure 6b) showed a higher viscosity than its counterpart (Figure 6a).
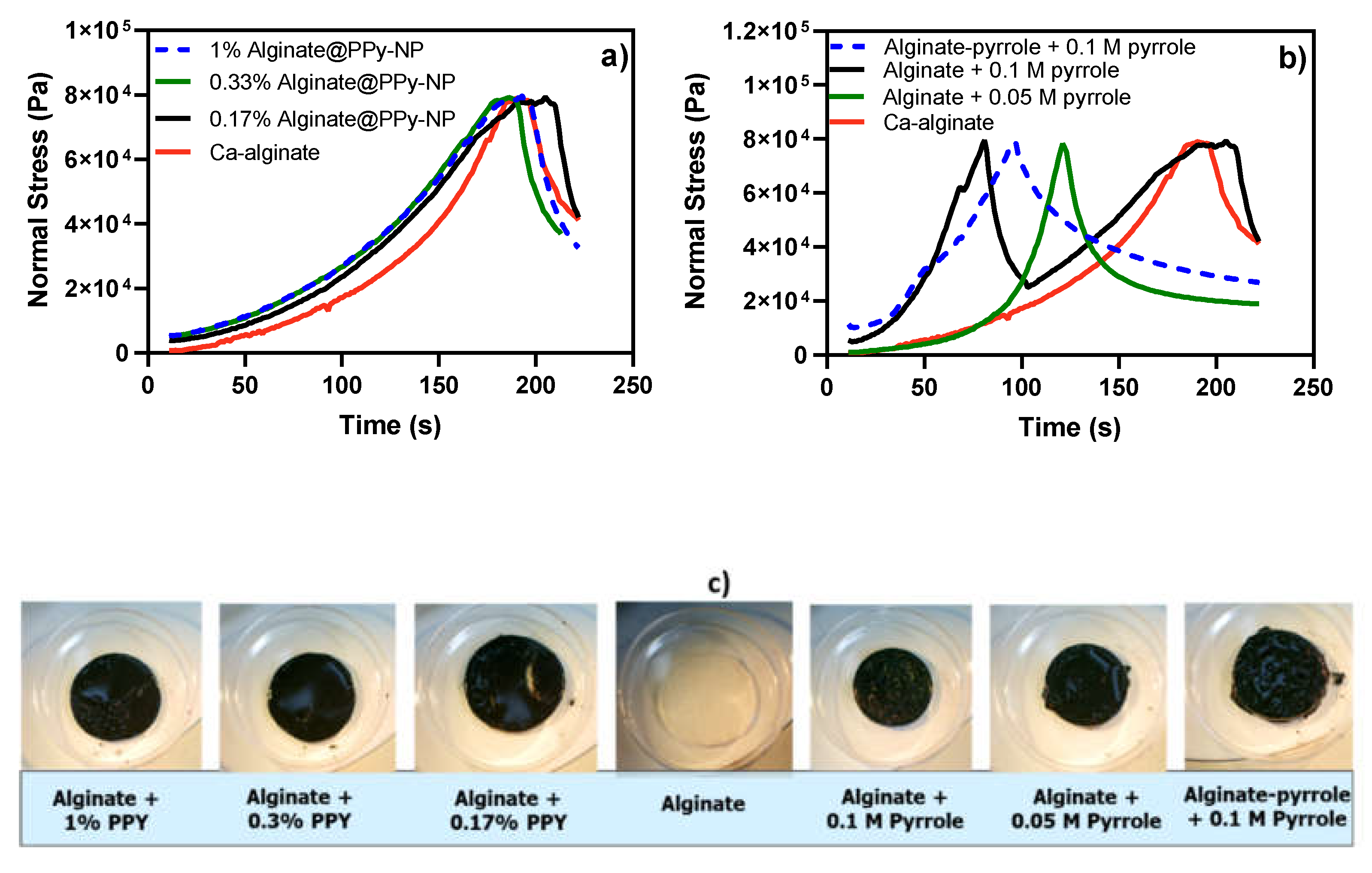
3.3.2. Compressibility Test
The mechanical properties of the composite hydrogels are shown in Figure 7 (a-c). The compressibility test was performed at increasing normal force (Pa) over time (s). Alginate and alginate with ex situ-generated polypyrrole displayed a characteristic compressive stress of 80 kPa and a gradual permanent deformation when the time exceeded 180 s (Figure 7a). In addition, alginate with an in-situ generated polypyrrole had a characteristic compressive stress of 80 kPa and a sharp permanent deformation when the time exceeded 90 s, which is half the time required for a permanent deformation to set in in the counterpart composite (Figure 7b). The lower the pyrrole content, the longer it takes to achieve permanent deformation.
Figure 7.
Compressibility test for (a) ex situ-generated alginate/polypyrrole composite, (b) in situ-generated alginate-polypyrrole composite, and (c) images of the composite hydrogels after the compressive stress was removed.
Figure 7.
Compressibility test for (a) ex situ-generated alginate/polypyrrole composite, (b) in situ-generated alginate-polypyrrole composite, and (c) images of the composite hydrogels after the compressive stress was removed.
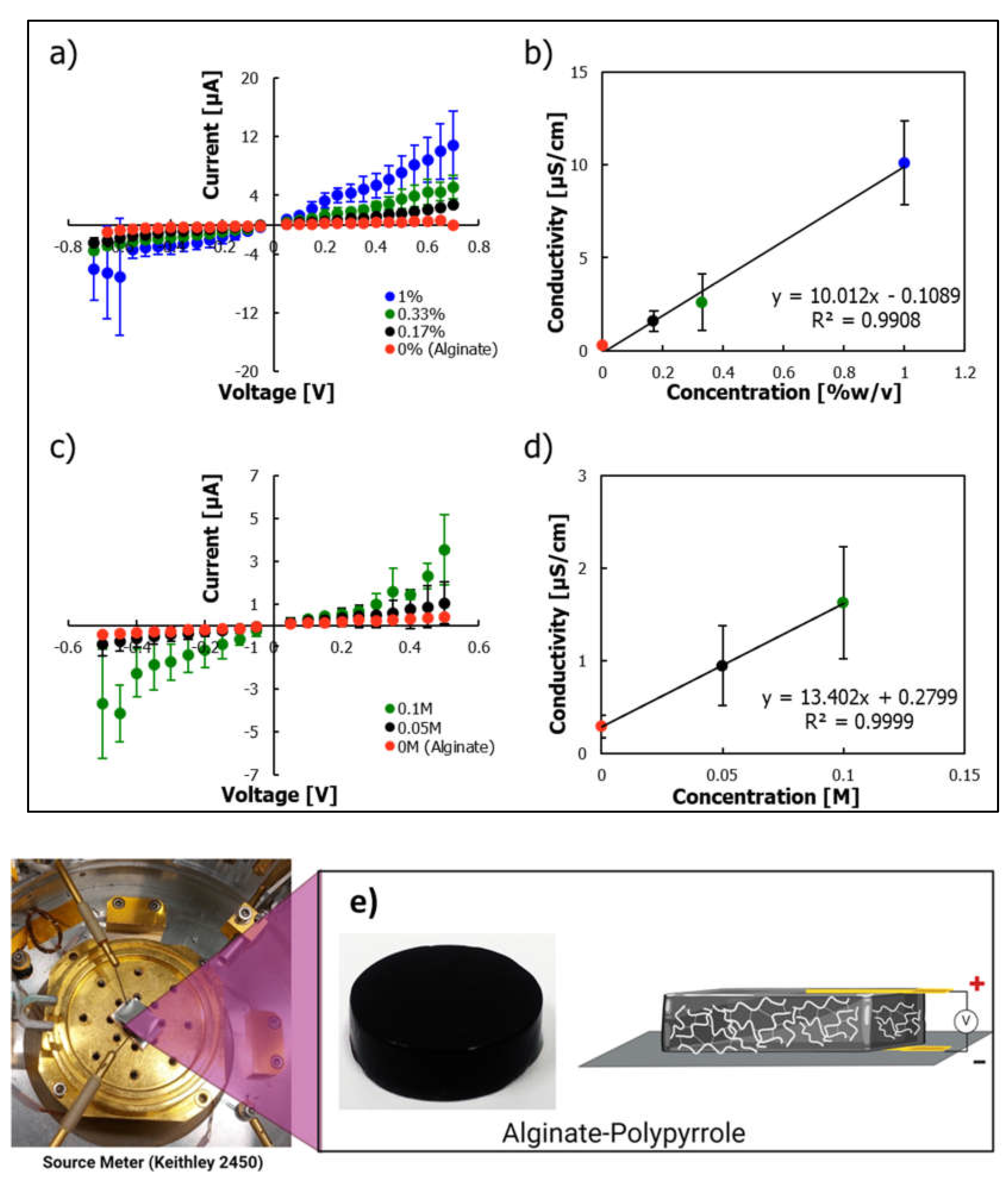
3.4. Electrical Conductivity
To investigate the electrical conductivity of the alginate-pyrrole samples, current-voltage curves were extracted for devices in which the hydrogel was sandwiched between stainless steel disks as contacts (Figure 8e). A linear increase in conductivity with an increase in pyrrole loading led to a proportional increase in the conductivity (Figure 7b), which was extracted as the inverse of the slope of the current-voltage curve, and was observed for both ex situ, alginate@PPy-NP (Figure 8 a and b), and in situ alginate-PPy (Figure 8 c & d) samples. This increase in conductivity can be attributed to electron transport within the pyrrole network. A conductivity of 10 µS/cm was achieved at a polypyrrole loading of 1% w/v. Polymerizing pyrrole within the pyrrole-alginate conjugate led to a larger increase in conductivity compared with similar pyrrole loading levels (Supplementary Figure S10). However, the conductivity reached a maximum of 2 µS/cm at a pyrrole-loading level of 0.1 M. It should be noted that adding pyrrole to the pyrrole conjugate can also lead to a decrease in conductivity at a higher concentration [25]. The reduction in the conductivity when a higher loading of pyrrole was used suggests a decrease in the ion conductivity within the hydrogel [25]. Notably, the polymerization of pyrrole within the gel led to a higher level of water hydrolysis when higher voltages were applied to the sample (not shown).
Figure 8.
Electrical conductivity test. (a) Current-voltage plot of alginate@PPy-NP expressed as percentage (PPY %) b) plot of concentration-dependent conductivity of alginate@PPy-NP in percentage (PPY %) c) Current-voltage plot of alginate-PPy mixed with pyrrole monomer (expressed in molar, M) polypyrrole generated in situ d) plot of concentration-dependent conductivity of alginate mixed with pyrrole monomer (expressed in molar, M) polypyrrole generated in situ.
Figure 8.
Electrical conductivity test. (a) Current-voltage plot of alginate@PPy-NP expressed as percentage (PPY %) b) plot of concentration-dependent conductivity of alginate@PPy-NP in percentage (PPY %) c) Current-voltage plot of alginate-PPy mixed with pyrrole monomer (expressed in molar, M) polypyrrole generated in situ d) plot of concentration-dependent conductivity of alginate mixed with pyrrole monomer (expressed in molar, M) polypyrrole generated in situ.
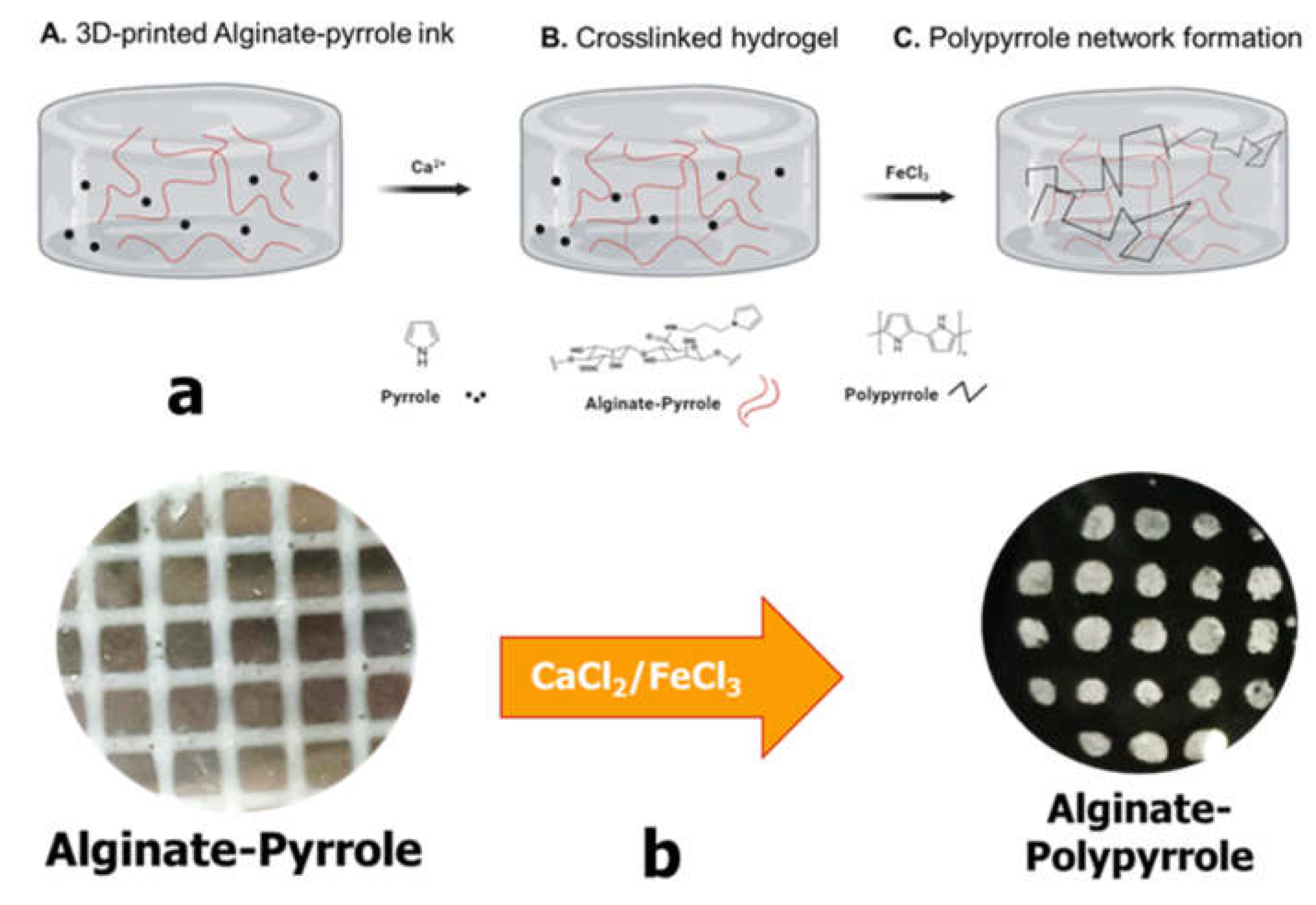
3.5. 3D Printability Characterization
Bioink printability was assessed by evaluating the integrity of printed multilayer constructs. Three gelation states have been described for printed bioinks: under-gelation, proper-gelation, and over-gelation [29,30]. When the bioink is under ideal gelation conditions, the extruded structure should maintain constant and uniform filaments, resulting in a standard grid construct with distinguishable layers and dimensions similar to the designed layout. As shown in Figure 9, extrusion was followed by cross-linking with calcium ions and polymerization of the pyrrole anchored within the hydrogel.
Figure 9.
Scheme of post-printing steps (a) and image of printed grids followed by secondary crosslinking and in situ polypyrrole synthesis (b).
Figure 9.
Scheme of post-printing steps (a) and image of printed grids followed by secondary crosslinking and in situ polypyrrole synthesis (b).
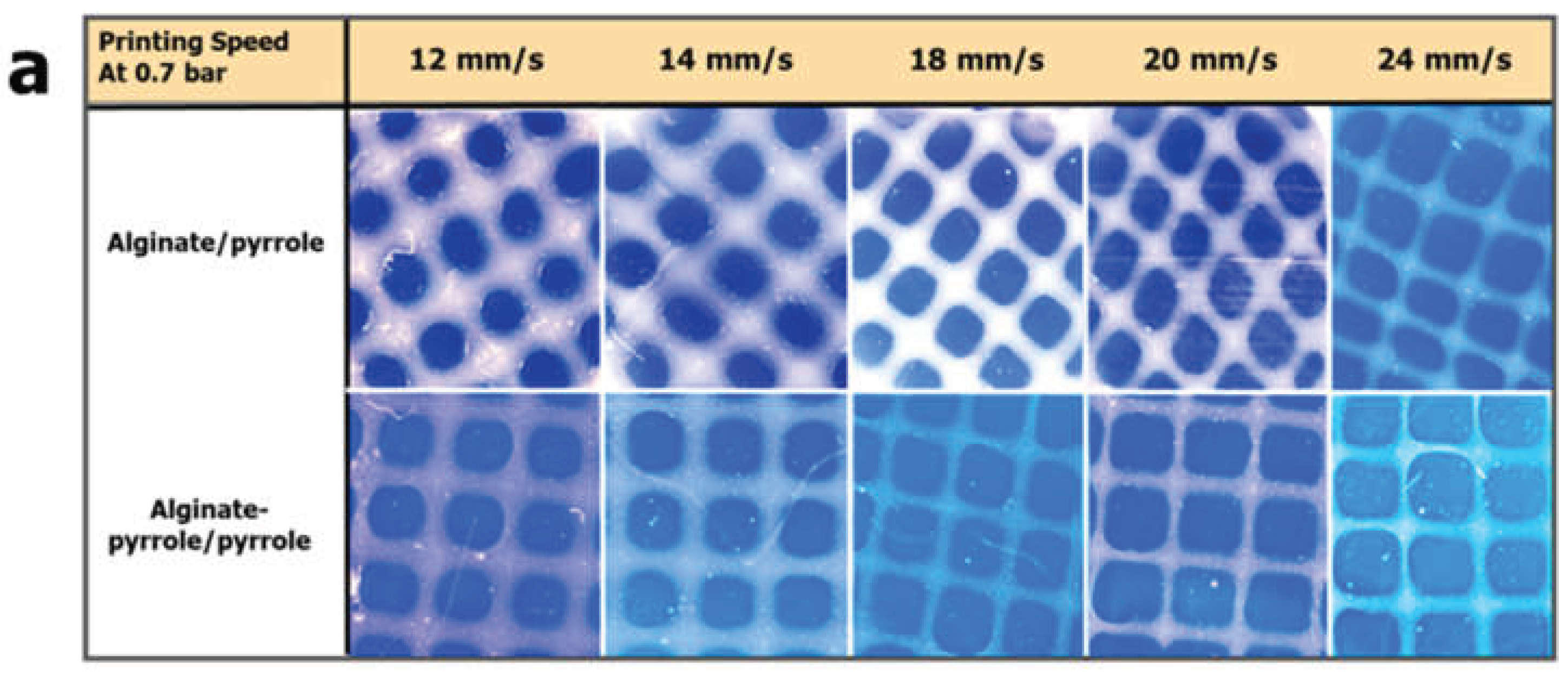
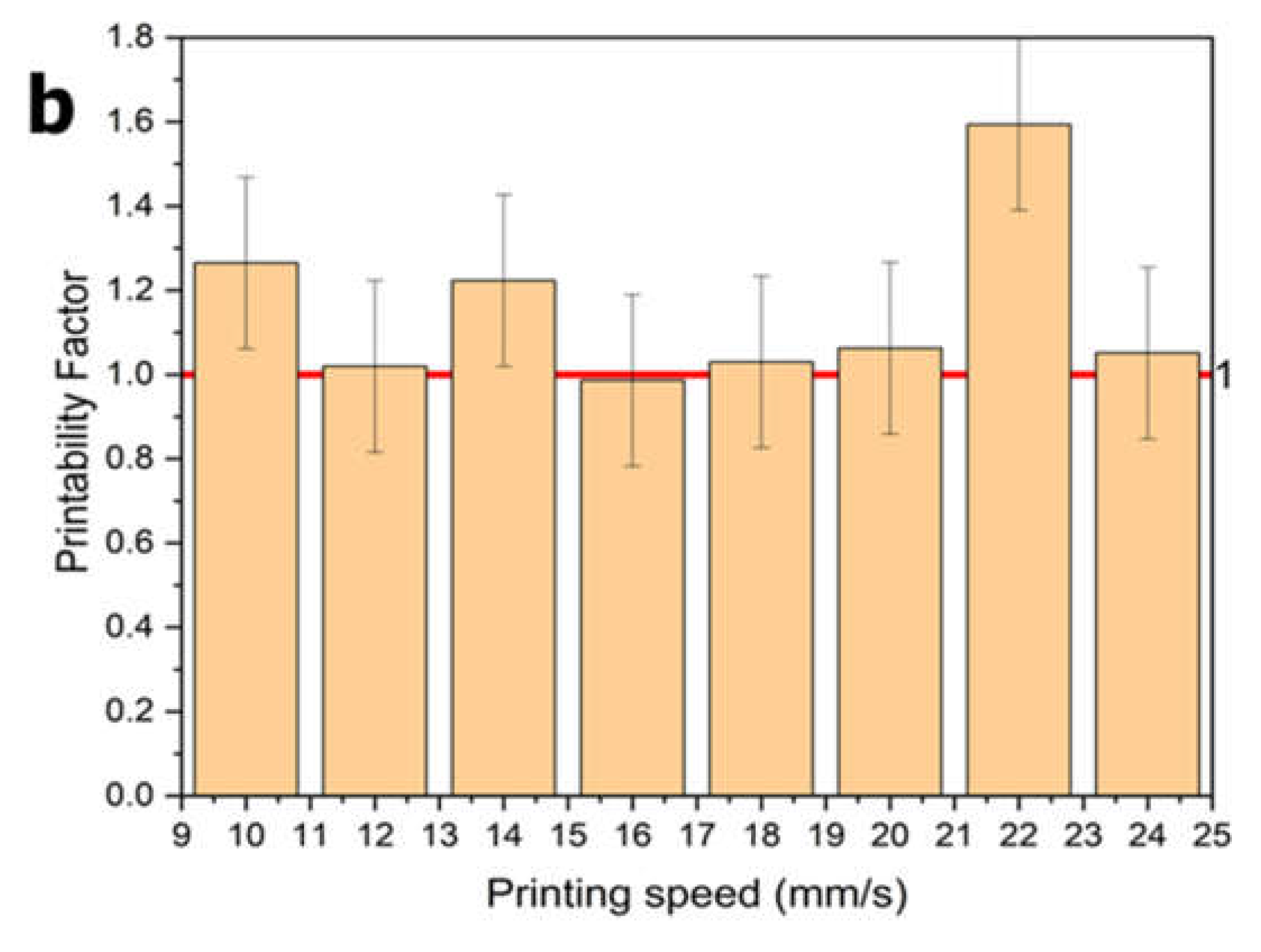
The printing pressure and speed were evaluated during the printing. Printability was calculated using Equation (2): the printability factor (Pf) was within 0.9-1.1 was considered acceptable, demonstrating structural and morphological stability. Figure 10(a) shows images of the various constructs of alginate/pyrrole and alginate-pyrrole/pyrrole extruded at a constant printing pressure (0.7 bar) and printing speed ranging from 12 mm/s to 24 mm/s. A plot of the printability factor with respect to printing speed is shown in Figure 10(b). Printing speeds of 16, 18, and 20 mm/s consistently demonstrated the printability of different alginate-pyrrole bioink formulations.
Figure 10.
Printability studies as a function of printing speed (a) images of the construct od alginate-Py and alginate-Py/pyrrole, and (b) the printability of alginate-pyrrole/polypyrrole
Figure 10.
Printability studies as a function of printing speed (a) images of the construct od alginate-Py and alginate-Py/pyrrole, and (b) the printability of alginate-pyrrole/polypyrrole
The possibility of extruding the bioink directly into the support bath was evaluated using calcium-ion-supplemented free-form reversible embedding of suspended hydrogels (FRESH), as described by Hinton et al. [18]. As shown in Figure 11(a), the construct was extruded into a support bath at a printing speed of 18 mm/s. After the sacrificial FRESH support was removed, images were captured by incubation at 37 °C for 60 min.
Figure 11.
(Bio)Printing in a support bath (FRESH). (a) Construct printed with support, (b) construct printed without support, and (c) construct of bioink loaded with GFP-expressing human fibroblasts after removing FRESH.
Figure 11.
(Bio)Printing in a support bath (FRESH). (a) Construct printed with support, (b) construct printed without support, and (c) construct of bioink loaded with GFP-expressing human fibroblasts after removing FRESH.
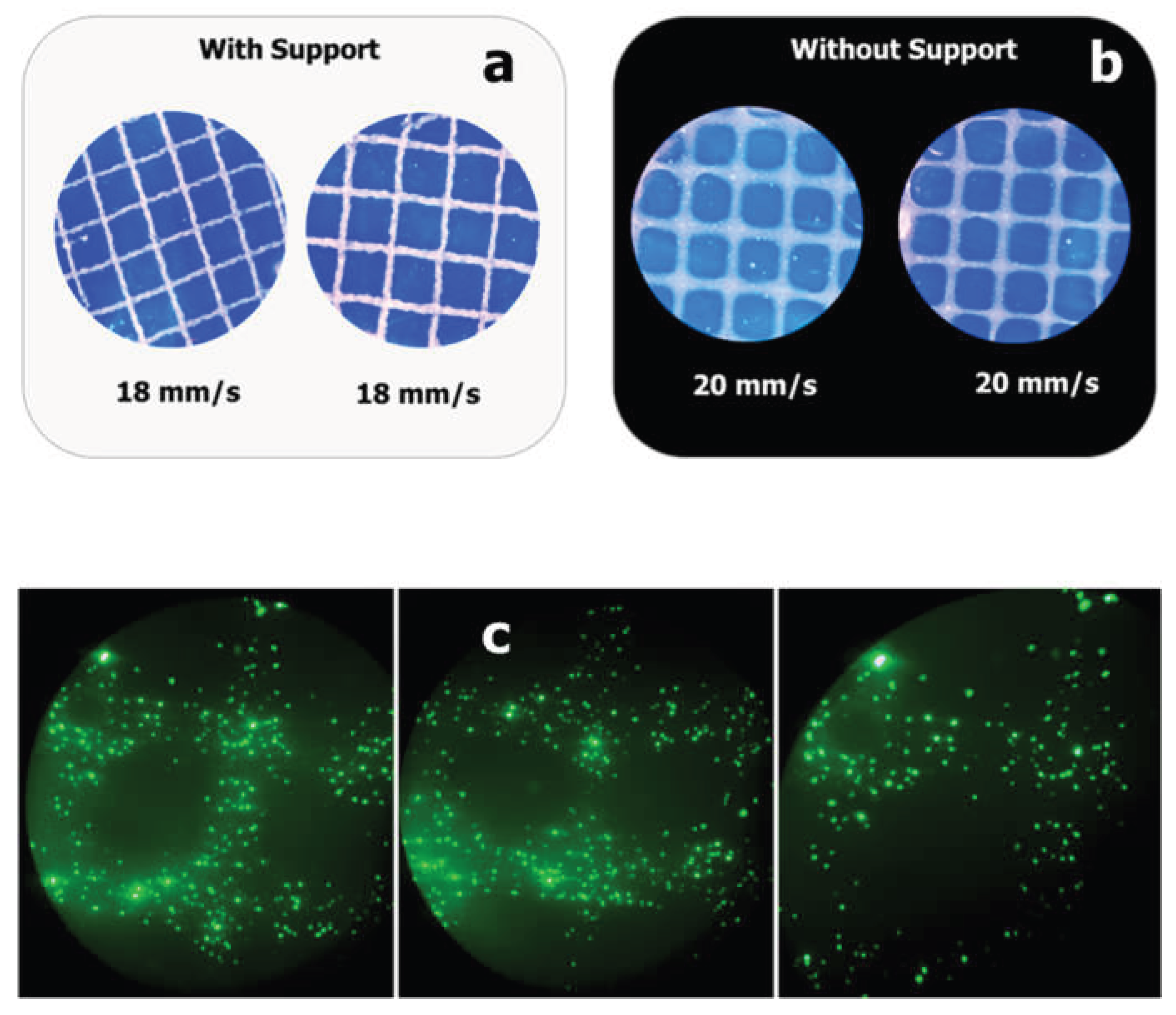
As shown in Figure 11(b), the construct was extruded at a printing speed of 20 mm/s without a support bath followed by Ca2+ ion-mediated crosslinking. Figure 11(a) and (b) show the practicality of printing alginate-pyrrole with or without support, depending on the application needs. Finally, alginate pyrrole was loaded into GFP-expressing human fibroblasts and printed onto a calcium-containing FRESH support. After printing, the support was removed, and the cell-laden construct was incubated in fetal bovine serum (FBS) -supplemented DMEM in an incubator maintained at 37 °C and 5% CO2 (Figure 11c).
3.5.1. Morphology of PPy-NP and Printability of Alginate@PPy-NP Constructs
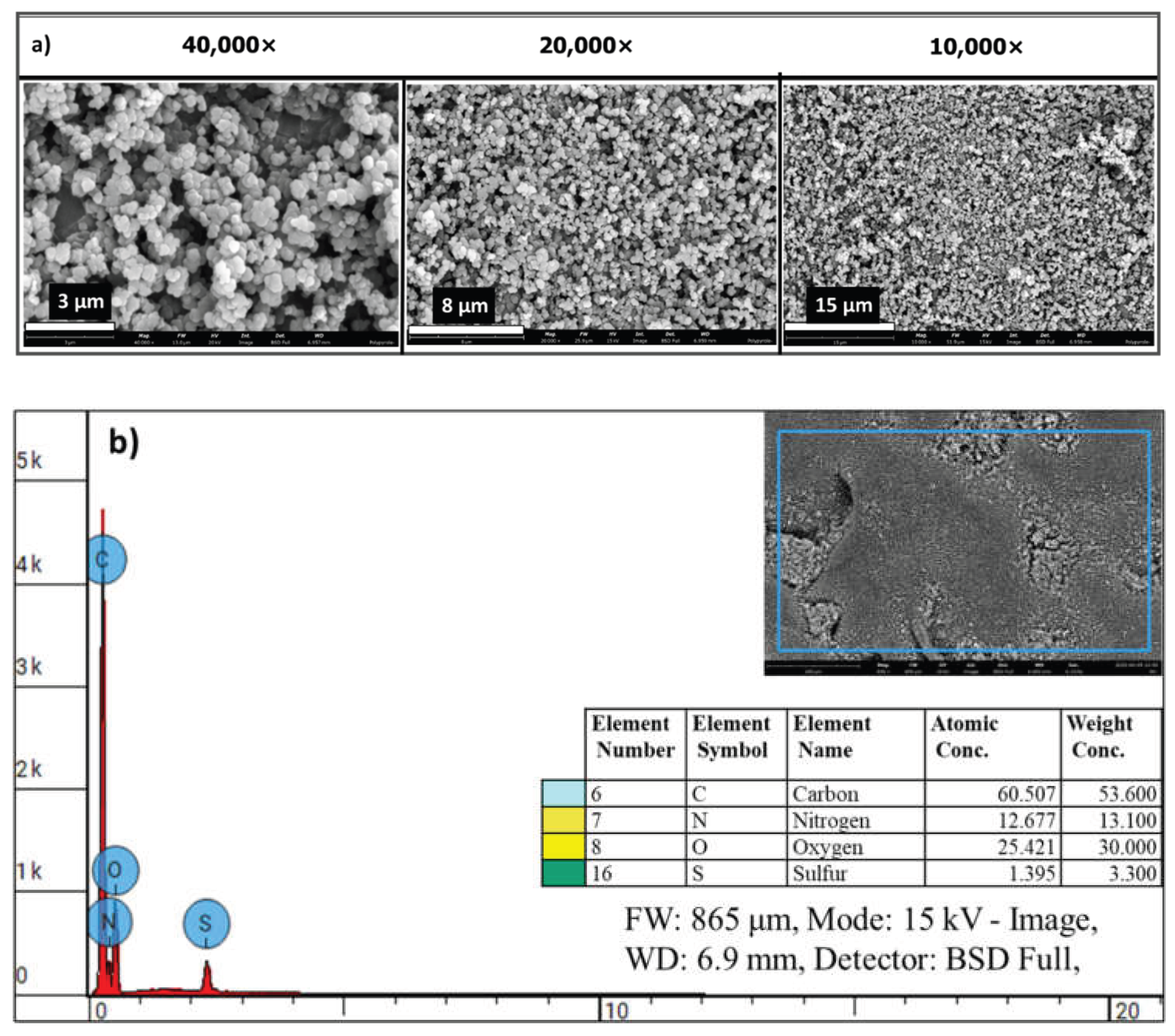
Scanning electron microscopy (SEM) revealed the successful synthesis of polypyrrole nanoparticles (PPy-NPs) with uniform spherical morphology and submicron size distribution (Figure 12). At increasing magnifications (10,000× to 40,000×), the particles exhibited dense packing with slight surface roughness, supporting their suitability as reinforcing fillers within the hydrogel matrices.
Figure 12.
SEM micrographs of chemically prepared polypyrrole nanoparticles (PPy-NP). (a) The micrographs at different magnifications b) the EDS elemental analyses of the synthesized PPy-NP
Figure 12.
SEM micrographs of chemically prepared polypyrrole nanoparticles (PPy-NP). (a) The micrographs at different magnifications b) the EDS elemental analyses of the synthesized PPy-NP
Extrusion-based 3D printing of alginate@PPy-NP was performed at different printing pressures and speeds. The resulting constructs showed good printability across all the PPy-NP concentrations tested (Figure 13). At 1% (w/v) PPy-NP, the printed lines displayed appreciable structural fidelity, whereas lower concentrations (0.33% and 0.17%) resulted in reduced printing resolution and minor spreading at intersections, suggesting a concentration-dependent improvement in shape fidelity and filament stability. These observations are in agreement with the enhanced rheological and mechanical properties observed at higher PPy-NP loadings.
Figure 13.
Extrusion-based 3D construct of alginate@PPy-NP at a Printing Speed of 18 mm/s and a pressure of 0.7 bar. Extrusion printing of the composite was carried out at different printing speeds and pressures.
Figure 13.
Extrusion-based 3D construct of alginate@PPy-NP at a Printing Speed of 18 mm/s and a pressure of 0.7 bar. Extrusion printing of the composite was carried out at different printing speeds and pressures.

4. Discussion
In the category of conducting polymers, polypyrrole (PPy) is widely used because of its advantages such as low-cost preparation, excellent conductivity, and large surface area. PPy, a conductive polymer, facilitates the immobilization of various metallic nanoparticles on the surface of electrodes through π−π stacking, electrostatic interactions, or entrapment procedures [37]. In this study, we synthesized an N-substituted pyrrole (aminopropyl pyrrole) and its covalent conjugation to alginate, which can be polymerized in situ into an alginate-polypyrrole (alginate-PPy) conjugate. In addition, polypyrrole nanoparticles (PPy-NPs) were chemically synthesized and blended with alginate to produce a stable conductive alginate polypyrrole composite (alginate@PPy-NP), which was used independently together with alginate-PPy as inks for extrusion-based 3D printing.
Spectroscopic analysis confirmed the successful incorporation of PPy into the alginate matrix via both in situ polymerization and nanoparticle embedding. FTIR, mass spectrometry (Supplementary Figure S2), and NMR confirmed the success of aminopropyl pyrrole synthesis, while X-ray photoelectron spectroscopy (XPS) revealed characteristic binding energies for C–C, C–O, and N–H species, confirming the presence of polypyrrole and its interaction with the alginate backbone. Rheological measurements demonstrated that both the alginate-PPy and alginate@PPy-NP systems exhibited time-dependent increases in the storage modulus (G′) and reductions in the complex viscosity (|η*|), indicating progressive gelation (Figure 5). The in situ polymerized alginate-PPy system showed moderate improvements in viscoelasticity, whereas the alginate@PPy-NP system exhibited more substantial enhancements, particularly at 1% (w/v) PPy-NP. A higher G' indicates that the hydrogel exhibits greater elasticity and structural integrity, which is crucial for mechanical stability and resilience applications [38,39]. This characteristic is particularly beneficial in applications, such as 3D printing and tissue scaffolding, where mechanical stability and resilience are necessary to support printing fidelity, cellular activity, and tissue regeneration. [40]. The ability of hydrogels to maintain their structural integrity under load is further emphasized by studies showing that higher G' correlates with increased gel strength and reduced water absorbency under pressure [40]. This concentration-dependent behavior suggests that the nanoparticles acted as physical crosslinkers, promoting network densification and elastic recovery. Shear-thinning behavior was observed across all formulations, which was favorable for extrusion-based bioprinting.
Mechanical analysis using compressibility tests further supported these rheological findings (Figure 7). The alginate@PPy-NP hydrogels (Figure 7a) showed a clear concentration-dependent increase in compressive strength and deformation resistance, with the 1% NP variant achieving the highest mechanical robustness. In contrast, alginate-PPy hydrogels displayed symmetric stress–strain profiles, indicative of homogenous, yet brittle networks. Notably, the deformation pattern of alginate@PPy-NP extended beyond 180 s, whereas alginate-PPy (Figure 7b) exhibited earlier yielding, reflecting differences in the network architecture. A higher cross-linking density generally leads to an increased compressive modulus, which enhances the ability of the hydrogel to resist deformation under the applied loads. As shown in Figure 7, the compressive stress for both types of alginate-PPy (or alginate@PPy-NP) was 80 kPa, with a permanent deformation setting at different times. Alginate@PPy-NP resulted in a more elastic composite hydrogel than the composite alginate-polypyrrole. These results suggest potential nanoparticle-mediated reinforcement of the hydrogels, consistent with prior reports of enhanced compressive performance in nanocomposite hydrogels [38,41]. Conversely, the alginate-PPy system (Figure 7b) shows moderate improvements in mechanical integrity, with a single symmetric stress–strain peak that shifts slightly with increasing pyrrole content. Alginate-PPy exhibited a sharp deformation pattern over a relatively short time (less than 100 s), whereas alginate@PPy-NP exhibited a curve-like deformation behavior beyond 180 s. This is similar to the report by Wright et al. [25], who reported that in situ generated polypyrrole resulted in a brittle hydrogel film at certain concentrations.
Conductive hydrogels can be engineered by incorporating conductive materials such as conductive polymers or nanoparticles into the hydrogel matrix. In this study, we demonstrated the possibility of improving the electrical conductivity of hydrogels through covalent addition of a conductive polypyrrole polymer. Electrical characterization showed that the integration of PPy imparted conductivity to the hydrogels, with a clear trend of increasing conductivity with increasing PPy content (Figure 8). Different oxidants, such as hydrogen peroxide, ferric chloride, and ammonium persulfate (APS), have been used to synthesize polypyrrole from its monomer precursor. Ferric chloride and APS gave a fast reaction rate (Figure S4), and because ferric ions, as multivalent cations, can interfere with alginate crosslinking, APS was selected as the preferred oxidant to prepare polypyrrole [42] due of its compatibility with alginate crosslinking, avoiding interference from Fe³⁺ ions. Polypyrrole-incorporated alginate displayed electrical conductivity that increased with increasing polypyrrole content. The observed conductivity shown in Figure 7b falls within the range of the conductivity observed in polysaccharide-based hydrogels. Guo et al. [43] noted that the conductivity of pure chitosan was approximately 3.13 x 10-8 S/cm, which increased to 2.97 x 10-5 S/cm with the addition of 10% aniline as an intrinsically conducting unit [43]. Crosslinked alginate gels conduct electricity via movement of mobile ions within the gel, which is enhanced by the introduction of a polypyrrole backbone with conjugated π-electrons. These electroconductive features render the material suitable for electrically active tissue-engineering applications.
Scanning electron microscopy (SEM) confirmed the nanoscale morphology of the chemically synthesized conductive PPy-NPs, revealing uniform spherical particles (Figure 12a) with rough surfaces, stable integration within the alginate matrix, and elemental distribution (Figure 12b). The influence of these nanoparticles is evident in extrusion-based 3D printing and enhanced mechanical properties. The alginate@PPy-NP constructs, particularly at 1% loading, exhibited high shape fidelity with well-defined pores and minimal filament spreading (Figure 13). Lower concentrations resulted in a poorer resolution and structural collapse. Printability was further optimized by primary ionic crosslinking with 10 mM Ca²⁺, which improved ink viscosity and extrusion stability. Additionally, printing within a support bath preserved the grid geometry, even at higher speeds, similar to unsupported printing. This demonstrates the robustness of printability amenable to different 3D printing applications in tissue engineering.
Putting all together, the integration of polypyrrole into alginate via covalent polymerization or nanoparticle dispersion significantly enhanced the physicochemical and functional properties of the resulting electrically conductive hydrogels. These modifications improve the rheological behavior, mechanical integrity, electrical conductivity, and print fidelity, all of which are critical parameters for bioprinting applications. The ability to tailor these properties through pyrrole concentration and formulation method makes these bioinks promising candidates for extrusion-based tissue engineering, particularly in scenarios requiring both mechanical robustness and electrical activity, such as in neural or bone tissue regeneration.
5. Conclusions
Alginate-polypyrrole hydrogels were developed via EDC/NHS-mediated conjugation with 3-aminopropyl pyrrole, and their physicochemical, rheological, and electrical properties were characterized. Pyrrole incorporation enhanced viscoelasticity, printability, and conductivity, enabling the optimization of 3D bioprinting. Alginate-PPy formed soft, uniform gels ideal for cell encapsulation, whereas alginate@PPy-NP provided mechanically robust scaffolds suitable for load-bearing applications. These findings demonstrate the potential of alginate-polypyrrole composites as tunable, conductive bioinks for electrically responsive tissue regeneration.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, A.A.P. and R.S.M.; methodology, A.A.P., O.K, H.M, AKB.; investigation, A.A.P, H.M., A.K.B., O.K.; resources, R.S.M., N.A., S.C..; writing—original draft preparation, A.A.P.; writing—review and editing, A.A.P., H.M, O.K., A.K.B., R.S.M., N.A., S.C.; visualization, A.A.P., H.M.; supervision, R.S.M., N.A., S.C..; project administration, R.S.M., N.A., S.C.; funding acquisition, R.S.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original results presented in this study are included in this article and the supplementary materials, and further inquiries in terms of data can be found in our records in footprints (https://footprints-b291f.web.app/); authorization for access may be granted by the corresponding author (rsmarks@bgu.ac.il).
Acknowledgments
The authors acknowledge financial support from the Avram and Stella Goldstein-Goren funds. The authors thank Mark Karpasa for helping with the rheological analysis and Lee Shelly for his help with the XPS analysis. The authors equally thank Kathelina Kristollari for her help with BioRender illustrations, and Victor Multanen for his help during microscopy. During the preparation of this manuscript, the author(s) used Grammarly V1.2.158.1662 and Paperpal 3.4.1 for the purpose of improving the grammatical accuracy and readability of the manuscript. The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| DDW | Double-distilled water |
| DMEM | Dulbecco's Modified Eagle Medium |
| FRESH | Freeform Reversible Embedding of Suspended Hydrogels |
| NMR | Nuclear magnetic resonance |
| PPy-NP | Polypyrrole Nanoparticles |
| SEM | Scanning Electron Microscope |
| XPS | X-ray photoelectron spectroscopy |
References
- Leppiniemi, J.; Lahtinen, P.; Paajanen, A.; Mahlberg, R.; Metsä-Kortelainen, S.; Pinomaa, T.; Pajari, H.; Vikholm-Lundin, I.; Pursula, P.; Hytönen, V.P.J.A.a.m.; et al. 3D-printable bioactivated nanocellulose–alginate hydrogels. 2017, 9, 21959–21970. [Google Scholar] [CrossRef]
- Trampe, E.; Koren, K.; Akkineni, A.R.; Senwitz, C.; Krujatz, F.; Lode, A.; Gelinsky, M.; Kühl, M.J.A.F.M. Functionalized bioink with optical sensor nanoparticles for O2 imaging in 3D-bioprinted constructs. 2018, 28, 1804411. [Google Scholar] [CrossRef]
- Paul, A.A.; Aladese, A.D.; Marks, R.S.J.B. Additive manufacturing applications in biosensors technologies. 2024, 14, 60. [Google Scholar] [CrossRef]
- Mandon, C.A.; Blum, L.J.; Marquette, C.A.J.A.c. Adding biomolecular recognition capability to 3D printed objects. 2016, 88, 10767–10772. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Feng, J. Skin-inspired multifunctional luminescent hydrogel containing layered rare-earth hydroxide with 3D printability for human motion sensing. 2020, 12, 6797–6805. [Google Scholar] [CrossRef] [PubMed]
- Rees, A.; Powell, L.C.; Chinga-Carrasco, G.; Gethin, D.T.; Syverud, K.; Hill, K.E.; Thomas, D.W.J.B.r.i. 3D bioprinting of carboxymethylated-periodate oxidized nanocellulose constructs for wound dressing applications. 2015, 2015. [Google Scholar] [CrossRef]
- Murphy, S.V.; Atala, A.J.N.b. 3D bioprinting of tissues and organs. 2014, 32, 773–785. [Google Scholar] [CrossRef]
- Markstedt, K.; Mantas, A.; Tournier, I.; Martínez Ávila, H.; Hagg, D.; Gatenholm, P.J.B. 3D bioprinting human chondrocytes with nanocellulose–alginate bioink for cartilage tissue engineering applications. 2015, 16, 1489–1496. [Google Scholar] [CrossRef]
- Gungor-Ozkerim, P.S.; Inci, I.; Zhang, Y.S.; Khademhosseini, A.; Dokmeci, M.R.J.B.s. Bioinks for 3D bioprinting: an overview. 2018, 6, 915–946. [Google Scholar] [CrossRef]
- Xu, M.; Zhang, L.; Zhou, Q.; Luo, L. Automated Quantitative Assessment of Three-Dimensional Bioprinted Hydrogel Scaffolds Using Optical Coherence Tomography. Biomedical Optics Express 2016, 7, 894. [Google Scholar] [CrossRef]
- Li, H.; Tan, Y.; Leong, K.F.; Li, L. 3D Bioprinting of Highly Thixotropic Alginate/Methylcellulose Hydrogel With Strong Interface Bonding. Acs Applied Materials & Interfaces 2017, 9, 20086–20097. [Google Scholar] [CrossRef]
- Hoffman, A.S.J.A.d.d.r. Hydrogels for biomedical applications. 2012, 64, 18–23. [Google Scholar] [CrossRef]
- Rajabi, M.; McConnell, M.; Cabral, J.; Ali, M.A.J.C.P. Chitosan hydrogels in 3D printing for biomedical applications. 2021, 260, 117768. [Google Scholar] [CrossRef]
- Sacco, P.; Furlani, F.; De Marzo, G.; Marsich, E.; Paoletti, S.; Donati, I.J.G. Concepts for developing physical gels of chitosan and of chitosan derivatives. 2018, 4, 67. [Google Scholar] [CrossRef]
- Wei, L.; Li, Z.; Li, J.; Zhang, Y.; Yao, B.; Liu, Y.; Song, W.; Fu, X.; Wu, X.; Huang, S. An Approach for Mechanical Property Optimization of Cell-Laden Alginate–gelatin Composite Bioink With Bioactive Glass Nanoparticles. Journal of Materials Science Materials in Medicine 2020, 31. [Google Scholar] [CrossRef]
- Bercea, M.J.M. Rheology as a tool for fine-tuning the properties of printable bioinspired gels. 2023, 28, 2766. [Google Scholar] [CrossRef] [PubMed]
- Barrulas, R.V.; Corvo, M.C.J.G. Rheology in product development: an insight into 3D printing of hydrogels and aerogels. 2023, 9, 986. [Google Scholar] [CrossRef]
- Hinton, T.J.; Jallerat, Q.; Palchesko, R.N.; Park, J.H.; Grodzicki, M.S.; Shue, H.-J.; Ramadan, M.H.; Hudson, A.R.; Feinberg, A.W.J.S.a. Three-dimensional printing of complex biological structures by freeform reversible embedding of suspended hydrogels. 2015, 1, e1500758. [Google Scholar] [CrossRef] [PubMed]
- Bar, A.; Kryukov, O.; Etzion, S.; Cohen, S.J.I.J.o.B. Engineered extracellular vesicle-mediated delivery of miR-199a-3p increases the viability of 3D-printed cardiac patches. 2023, 9. [Google Scholar] [CrossRef]
- Amiryaghoubi, N.; Fathi, M.J.B.B. Bioscaffolds of graphene based-polymeric hybrid materials for myocardial tissue engineering. 2023, 14, 27684. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, J.; Gao, Y.; Xu, Z.; Dai, C.; Li, G.; Sun, C.; Yang, Y.; Zhang, K.J.J.o.M.C.B. Conductive biocomposite hydrogels with multiple biophysical cues regulate schwann cell behaviors. 2022, 10, 1582–1590. [Google Scholar] [CrossRef]
- Rastin, H.; Zhang, B.; Bi, J.; Hassan, K.; Tung, T.T.; Losic, D.J.J.o.M.C.B. 3D printing of cell-laden electroconductive bioinks for tissue engineering applications. 2020, 8, 5862–5876. [Google Scholar] [CrossRef] [PubMed]
- Kasimu, A.; Zhu, H.; Meng, Z.; Qiu, Z.; Wang, Y.; Li, D.; He, J.J.A.B. Development of Electro-Conductive Composite Bioinks for Electrohydrodynamic Bioprintin g with Microscale Resolution. 2023, 7, 2300056. [Google Scholar]
- Kang, M.S.; Kang, J.I.; Le Thi, P.; Park, K.M.; Hong, S.W.; Choi, Y.S.; Han, D.-W.; Park, K.D.J.A.M.L. Three-dimensional printable gelatin hydrogels incorporating graphene oxide to enable spontaneous myogenic differentiation. 2021, 10, 426–432. [Google Scholar] [CrossRef]
- Wright, C.J.; Molino, B.Z.; Chung, J.H.; Pannell, J.T.; Kuester, M.; Molino, P.J.; Hanks, T.W.J.G. Synthesis and 3D printing of conducting alginate–polypyrrole ionomers. 2020, 6, 13. [Google Scholar] [CrossRef]
- Drury, J.L.; Mooney, D.J.J.B. Hydrogels for tissue engineering: scaffold design variables and applications. 2003, 24, 4337–4351. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Lin, Y.; Chen, X.; Zhao, B.; Zhang, J.J.F.H. Flow behavior, thixotropy and dynamical viscoelasticity of sodium alginate aqueous solutions. 2014, 38, 119–128. [Google Scholar] [CrossRef]
- Paul, A.A.; Kadosh, Y.S.; Kushmaro, A.; Marks, R.S.J.B. Microbead-Encapsulated Luminescent Bioreporter Screening of P. aeruginosa via Its Secreted Quorum-Sensing Molecules. 2024, 14, 383. [Google Scholar] [CrossRef]
- Ouyang, L.; Yao, R.; Zhao, Y.; Sun, W.J.B. Effect of bioink properties on printability and cell viability for 3D bioplotting of embryonic stem cells. 2016, 8, 035020. [Google Scholar] [CrossRef]
- Distler, T.; Polley, C.; Shi, F.; Schneidereit, D.; Ashton, M.D.; Friedrich, O.; Kolb, J.F.; Hardy, J.G.; Detsch, R.; Seitz, H.J.A.H.M. Electrically conductive and 3D-printable oxidized alginate-gelatin polypyrrole: PSS hydrogels for tissue engineering. 2021, 10, 2001876. [Google Scholar]
- Smith, B.C. Organic nitrogen compounds, VII: Amides—the rest of the story. 2020. [Google Scholar]
- Chandıa, N.; Matsuhiro, B.; Vásquez, A.J.C.P. Alginic acids in Lessonia trabeculata: characterization by formic acid hydrolysis and FT-IR spectroscopy. 2001, 46, 81–87. [Google Scholar] [CrossRef]
- Fenoradosoa, T.A.; Ali, G.; Delattre, C.; Laroche, C.; Petit, E.; Wadouachi, A.; Michaud, P.J.J.o.a.p. Extraction and characterization of an alginate from the brown seaweed Sargassum turbinarioides Grunow. 2010, 22, 131–137. [Google Scholar] [CrossRef]
- Cui, Z.; Coletta, C.; Dazzi, A.; Lefrancois, P.; Gervais, M.; Néron, S.; Remita, S.J.L. Radiolytic method as a novel approach for the synthesis of nanostructured conducting polypyrrole. 2014, 30, 14086–14094. [Google Scholar] [CrossRef]
- Scienza, L.C.; Thompson, G.E.J.P. Preparation and surface analysis of PPY/SDBS films on aluminum substrates. 2001, 11, 142–148. [Google Scholar] [CrossRef]
- Dou, Y.; Wang, X.; Liu, Z.; Kong, F.; Wang, S.J.J.o.A.P.S. Effect of different modified lignins on the properties of xylan composite hydrogels. 2024, 141, e54910. [Google Scholar] [CrossRef]
- Tian, W.; Mao, X.; Brown, P.; Rutledge, G.C.; Hatton, T.A.J.A.F.M. Electrochemically nanostructured polyvinylferrocene/polypyrrole hybrids with synergy for energy storage. 2015, 25, 4803–4813. [Google Scholar] [CrossRef]
- Sharma, S.; Bhende, M.; Verma, H.R.; Mulwani, P.J.P.E. ; Science. Impact of multi-walled carbon nanotubes on the thermal, mechanical, and electrical properties of guar gum hydrogel. 2024, 64, 3414–3423. [Google Scholar]
- Chen, T.; Hou, K.; Ren, Q.; Chen, G.; Wei, P.; Zhu, M.J.M.R.C. Nanoparticle–polymer synergies in nanocomposite hydrogels: from design to application. 2018, 39, 1800337. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhao, J.; Sun, X.; Li, S.; Hou, X.; Yuan, X.; Yuan, X.J.B. Rapid gelling chitosan/polylysine hydrogel with enhanced bulk cohesive and interfacial adhesive force: mimicking features of epineurial matrix for peripheral nerve anastomosis. 2016, 17, 622–630. [Google Scholar] [CrossRef]
- Xie, L.; Jiang, M.; Dong, X.; Bai, X.; Tong, J.; Zhou, J.J.J.o.A.P.S. Controlled mechanical and swelling properties of poly (vinyl alcohol)/sodium alginate blend hydrogels prepared by freeze–thaw followed by Ca2+ crosslinking. 2012, 124, 823–831. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, Q.; Gao, X.; Piao, G.J.I.J.o.P.S. A nanocellulose polypyrrole composite based on tunicate cellulose. 2013, 2013, 175609. [Google Scholar] [CrossRef]
- Guo, B.; Finne-Wistrand, A.; Albertsson, A.-C.J.B. Facile synthesis of degradable and electrically conductive polysaccharide hydrogels. 2011, 12, 2601–2609. [Google Scholar] [CrossRef] [PubMed]

Table 1.
Bioink formulation.
| Sample | Materials | [Stock] | [Final] |
| A | Alginate | 2.5% | 2% |
| Ca2+ | 0.1 M | 0.01 M | |
| Pyrrole | 14.125 M | 0.1 M | |
| DDW | |||
| B | Alginate-pyrrole | 2.5% | 2.0% |
| Alginate | 2.5% | 2% | |
| Ca2+ | 0.1 M | 0.01 M | |
| Pyrrole | 14.125 M | 0.1 M | |
| DDW |
DDW is double-distilled water.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



