Submitted:
06 May 2025
Posted:
07 May 2025
You are already at the latest version
Abstract
Hydrogen-bonded main-chain Azobenzene (azo) photoactive polymers have broad application prospects in flexible actuators, optical actuators and other fields. Hydro-gen bonds, as dynamic reversible physical crosslinking points and crystal networks, limit the macro flow of polymer chains in rigid micro regions, and can improve the stability and mechanical strength of materials. In this paper, three kinds of hydrogen bonded main chain azobenzene polyamide ester (PEAs)semi-crystalline polymers with different flexible chain lengths were synthesized by Michael addition polymerization. The mechanical properties and light response time of the flexible chain length on the main chain polymer were explored. Azobenzene oriented films with a strain of 235% were obtained by a simple solution casting/mechanical stretching method, and uniaxially oriented polymer films with high orientation (48.85%) and fast response (5 s under ultraviolet light, 55 s under visible light) were obtained. These films exhibit light induced deformation not only in air, but also in polar solvents such as water, methanol, ethanol, ordinary saline solutions (such as normal saline, 0.9 wt% NaCl) and simulated seawater (3.5 wt% NaCl) The typical application range of photodeformation has been successfully extended from air to polar solvents. After soaking in water/brine for one month, polarized optical microscopy (POM) showed that the brightness and orientation of the texture of the film remained stable (Δ brightness< 5%), the light response time was stable (6 s under UV light, 65 s under visible light), and the retention rate of photoinduced stress was 95%.
Keywords:
azobenzene
; main-chain semi-crystalline polymers
; physically cross-linked
; photodeformation
; solvent resistance
1. Introduction
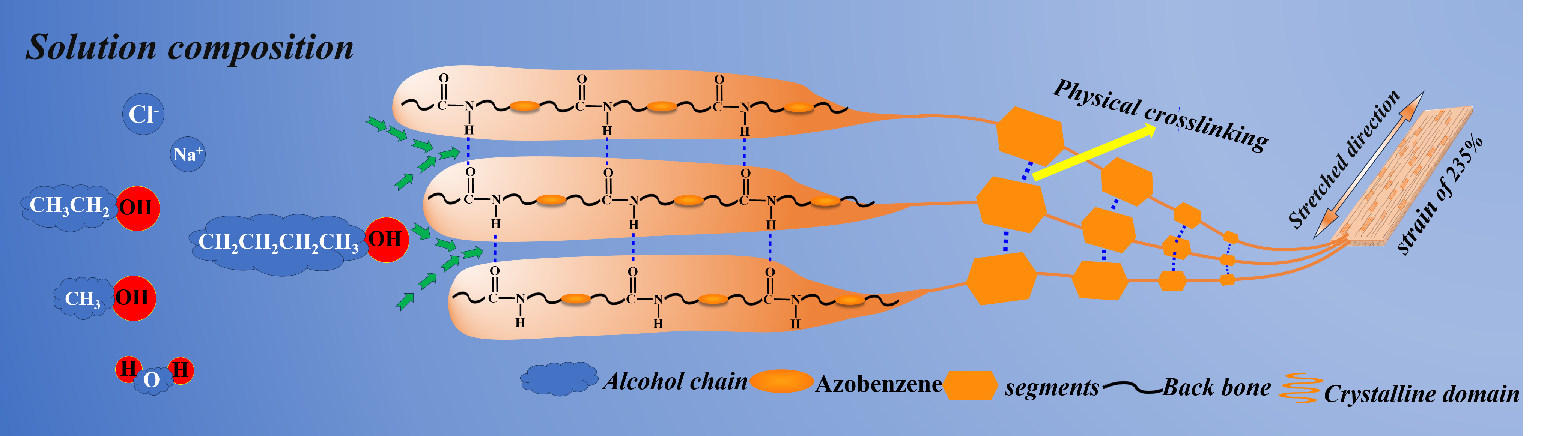
Photodeformable azo polymers, as a core system in the field of intelligent responsive materials, have demonstrated irreplaceable application value in optical driven microdevices, biomimetic actuators, and adaptive optics systems [1,2]. The core of these materials lies in the photoisomerization behavior of the azobenzene group: under specific wavelength illumination, the azobenzene group can undergo reversible transitions between cis and trans configurations, triggering geometric changes at the molecular scale and rearrangement of dipole moments [3,4]. However, the light responsiveness of the azobenzene group alone is insufficient to achieve macroscopic deformation of the material; the key is how to effectively transmit microscopic molecular motion to the macroscopic scale through rational polymer structural design [5,6]. In this context, the introduction of cross-linked structures has become a core strategy for regulating light deformation performance, as it not only imparts excellent mechanical stability to the material but also achieves efficient conversion of microscopic isomerization behavior to macroscopic deformation by fixing the orientation of the azobenzene groups and accumulating photo-induced stress [7].
At present, researchers have developed two main strategies for constructing photo induced deformation polymer networks: covalent crosslinking and dynamic non covalent crosslinking. Although traditional covalent crosslinking systems (such as epoxy resin crosslinking [8]and free radical polymerization crosslinking[9]) construct three-dimensional networks through high-strength covalent bonds (bond energies of 200-400 kJ/mol), endowing materials with excellent mechanical properties and thermal stability, permanent chemical bonds form rigid networks. However, irreversible crosslinking results in the inability of materials to undergo thermoforming or solvent reprocessing after curing, severely restricting device reconstruction and resource cycling. Although dynamic covalent crosslinking[10,11,12,13,14] (such as Diels Alder reversible bonds[15,16] and disulfide bond exchange [8,17]) endows materials with a certain degree of reparability, it requires complex molecular design and precise synthesis control. In contrast, systems based on dynamic non covalent crosslinking such as hydrogen bonding, electrostatic interactions[18,19,20,21,22] , π - π interactions[23,24], and self-assembly induced physical crosslinking can form stable networks through reversible interactions while retaining the material's thermoplastic processing properties, providing new ideas for developing reproducible photo deformable materials. The semi-crystalline polymer crosslinked by hydrogen bonding constructs a dynamic crosslinked structure through non covalent interactions such as hydrogen bonding and crystalline networks, which not only preserves the material's pho-to-responsiveness but also enhances the polymer's processability and cycling stability. Zhou [25] used Michael addition polymerization to prepare a semi crystalline polymer crosslinked by hydrogen bonds in the main chain. The hydrogen bonds induced by amide units and the crystalline domains in the prepared films and fibers endow them with dynamic but stable crosslinking points, making them easily programmable to transform into various three-dimensional (3D) shapes at room temperature. Woojin Lee [26] prepared a dual dynamic supramolecular network by using semi-crystalline star-shaped poly(ε-caprolactone) s (USPs) functionalized with urea pyrimidinone (UPy) end effectors. The appropriate arm length balance of USP can provide a mechanically rigid semi crystalline supra-molecular network. Its reversible dual dynamic properties, stemming from the reassociation of UPy quadruple hydrogen bonds and the recovery of crystal physical bonds during healing, endow it with efficient healing properties. In addition, by doping polydopamine (PDA) nanoparticles into the main chain azobenzene semi-crystalline poly (ester amide) (PEAs), a photo actuator with UV-NIR dual photoresponsiveness, room temperature 3D shape reprogramming and reprocessing, and photothermal healing properties is manufactured through hydrogen bonding between PEA and the polymer[27].In recent years, non-covalently crosslinked photo deformable azo polymers have shown two major development directions: side-chain[28] and main-chain[29]. Side chain systems (such as polyacrylate grafted azobenzene[30,31,32,33,34]) can achieve millisecond level response speed by connecting photoactive units through flexible spacers (such as -(CH2)6-)[35], but their applications are still limited[36].The main chain design directly embeds azo monomers into the polymer main chain (such as polyamide azo-benzene alternating copolymer[37]) to form continuous stress transfer channels. Combined with π - π stacking between the main chains [23] and hydrogen bond arrays, a hierarchical network is constructed, which improves the optical strain efficiency by 2-3 times[38] and the cycling stability exceeds 100 times without attenuation[39]. This structural advantage has attracted much attention in fields such as optically driven pumps that require continuous mechanical output.
In addition, the strength of hydrogen bonds between the main chains can be flexibly regulated through the design of functional groups, such as polymers containing both ester and secondary amine groups (i.e., poly (ester secondary amine) or PesAs)[40], polymers containing both ester and amide groups in the skeleton (such as poly (ester amide) or PEAs)[25,37,41], and polymers containing both ester and urea groups[42] in the skeleton and external environment. The regulation of these microstructures pro-vides possibilities for the multifunctionality of materials. Therefore, this structural advantage makes the main chain type material significantly superior to traditional side chain systems in terms of cycling stability and deformation amplitude. At present, research on hydrogen bonded crosslinked azobenzene materials mainly focuses on air, with only a small portion being tested in water [27,43] and non-polar reagents [44].
Although progress has been made in crosslinking azo polymers with hydrogen bonds in the main chain, their long-term stability in solvent environments remains a key but not yet fully explored challenge. The existing research mainly focuses on the photo deformation performance in air or short-term solvent exposure (<24 hours), and there is little systematic study on solvent resistance, especially the ability to maintain photo actuation efficiency after prolonged immersion (several days to weeks). This gap severely limits their application in biomedical devices that require physiological saline compatibility or industrial actuators that operate in organic media. To address this issue, we synthesized a dynamic and robust network system through Michael addition polymerization between azobenzene monomer with acrylic ester on one end and acrylamide on the other end, and dithiol with different chain lengths (n=2, 4, 6), as shown in Scheme 1 below.
The aims of this work include the following three aspects: (i) The mechanical properties and light response time as a function of flexible chain length in the main-chain polymer were explored, and a newly designed three-dimensional programmable and recyclable driver at room temperature was developed. (ii) The regularity of light deformation time and bending angle in uniaxially oriented azo polymer films was systematically evaluated under immersion in water (25, 30, 40, and 50 ℃), NaCl aqueous solutions (0.9 wt% and 3.5 wt% at 25 and 50 ℃), and common alcohol solutions (methanol, ethanol, and n-butanol). (iii) The persistent solvent resistance of photoinduced deformation in main-chain azo polymers across different polar solvents was investigated. Firstly, solvent-polymer interactions were analyzed via Fourier transform infrared spectroscopy to characterize the absorption changes of functional groups in polymer films after immersion. Additionally, time-dependent solvent resistance tests (soaking for up to 30 days) were conducted to characterize the maximum light deformation angle, variations in photoinduced stress, orientation changes, and quantitative POM brightness alterations of the films under different soaking durations.
2. Results and Discussion
2.1. Synthesis and Characterization of the Main-Chain Azo Semi-Crystalline Poly(ester-amide)S (PEAs)
We prepared acrylate-amido azo monomer and the Michael addition polymerization of M-EA with three dithiols (including 1,2-ethanedithiol, 1,4-butane-dithiol and 1,6-hexane-dithiol) was carried out in THF at 60 ℃ (i.e., M-EA, Scheme 1). The polymerization system was homogeneous during the reaction. Orange PEAs with different flexible spacers (PEA-nT, n = 2, 4, 6) were obtained in high yield (90–92%) within 3 hours of polymerization (Table 1). TLC verification confirmed no monomer residue in the polymer products.
The azo monomer M-EA and azo polymers PEA-nT (Scheme S1) were characterized by 1H NMR spectroscopy (Figure 1). As shown in Figure 1, the proton signal of the acrylate-amide group in M-EA is barely detectable in the 1H NMR spectrum of PEA-6. The number-average molecular weight (Mn, NMR) was calculated using the formula: Mn, NMR = x × M repeat unit = [(Sg/Su)/2] × M repeat unit
In the formula, ‘x’ denotes the number of repeating units; M repeat unit is the molecular weight of the repeating units; Su and Sg correspond to the peak integrals at 5.86-5.92 ppm (proton ‘u’ in the terminal acrylate group) and the peak value of 7.10 ppm (proton ‘g’ in the benzene ring), respectively. The number average molecular weight (Mn) is obtained.
The thermal stability and phase transition behaviors of PEA-nT (n = 2, 4, 6) were then characterized by TGA, DSC, POM, and XRD. To directly assess the thermal stability, the TGA was performed. The thermal decomposition temperature (Td) of the polymer was determined by monitoring mass changes with temperature during heating, which reflects the material’s high-temperature stability. TGA analyses revealed that PEAs exhibit high thermal stability, with 5% weight loss temperatures of 288, 292, and 294 °C for n = 2, 4, and 6, respectively (Table 1). DSC was used to quantitatively analyze phase transition behavior and thermodynamic properties, including glass transition temperature (Tg) and melting temperature (Tm), which were determined by measuring endothermic or exothermic peaks during heating/cooling cycles. The DSC thermogram of PEAs showed a relatively weak glass transition step (only during the second heating scan) and a phase transition peak (during the first cooling and second heating scans) (Figure 2a). Their glass transition temperatures (Tg) were 16.4, 12.6, and 8.3 °C for n = 2, 4, and 6, respectively, all lower than room temperature (25 °C). The DSC results indicated that in the PEA-nT polymers, both the first cooling and second heating curves exhibited a glass transition temperature. Tg is the temperature at which the transition from a glassy to a highly elastic state occurs. Glass transition is an inherent property of amorphous polymers and a macroscopic manifestation of changes in polymer molecular motion. It directly affects the material's performance and processability. POM (polarizing optical microscopy) utilizes birefringence to visualize the morphology, size, and texture of crystalline regions in polymers. POM images of PEA-nT are shown in Figure S1. XRD was used to quantitatively calculate crystallinity and correlate phase transition behavior with crystal structure. The XRD spectra of PEA-nT exhibited sharp diffraction peaks in the wide-angle region (2θ ≈ 21.5°), suggesting a highly ordered semi-crystalline phase (Figure 2a). Assuming that a purely crystalline sample has a typical Δ Hm ≈ 150−300 J/g, the crystalline degree of PEAs was derived to be around 10-20%[45] (Figure 2b).
The photoresponsivity of a representative azo polymer (PEA-6T) thin film was investigated. The photoresponse of PEA-6T thin films is shown in Figure 3a,b. UV-vis spectral changes were observed under ultraviolet (λ = 365 nm, 40 mW cm-2) and visible light (λ > 510 nm, 30 mWcm-2) irradiation. There is a strong absorption peak at 325 nm and a weak absorption peak near 450 nm, corresponding to the π-π* electron transition (trans isomer) and n - π* electron transition (cis isomer) of the nitrogen-nitrogen double bond (-N=N-) in the azobenzene group, respectively.
As shown in Figure 3a, under ultraviolet light irradiation, the intensity of the π-π*electron transition absorption peak near 325 nm decreases significantly, while the intensity of the n-π* electron transition peak near 450 nm increases slightly. After 65 seconds of ultraviolet irradiation, the PEA-6T film reaches a photostable state. The UV-vis spectrum shows two equal absorption points, indicating that the film undergoes only cis-trans isomerization of the azobenzene group under UV irradiation, without side reactions [46]. Subsequently, the aforementioned photostable film was exposed to visible light (λ > 510 nm, 30 mW cm⁻²) to investigate its recovery behavior. Figure 3b shows that under visible light irradiation, the absorption peak intensity at 325 nm increases, while that near 450 nm decreases slightly, indicating the gradual recovery of azobenzene groups from cis to trans isomers. After 110 seconds of visible light irradiation, the PEA-6T film reached a photostable state. According to the absorbance values, approximately 89% of the cis isomer converts to the trans isomer, but the film cannot fully recover its initial state before UV irradiation, as previously reported [47,48]. The maximum absorption peak of the azobenzene polymer in the solid film state exhibits a blue shift, from 365 nm in solution to 325 nm in the film. This is attributed to restricted azobenzene group mobility in the solid film state and the formation of H-aggregates [49]. In addition, as shown in Figure S2, ten UV-visible light irradiation cycles were performed on the thin film. Similarly, azo polymer PEA-2T and PEA-4T thin films exhibited the same photoresponsivity, as shown in Figure S3 and S4.
2.2 Fabrication of Uniaxially Oriented Films from Main-Chain Azo PEAs and Study on Mechan-Ical and Photomechanical Properties in Air
The main chain azobenzene polymer films were first prepared by the solution casting method, and the mechanical properties of the unstretched films were studied (Figure 4 and Table 2). The lengths and widths of the uniaxially oriented azobenzene polymer films were 10 mm and 2 mm, respectively, and the thicknesses of PEA-2T, PEA-4T and PEA-6T were 63, 63 and 64 μm, respectively. And the photoinduced stress was measured by the stretched PEA-nT films with a strain of 235%. An external force of 500 kPa was applied to the films. The dimensions (length × width × thickness) of the azobenzene polymer stretched films are: PEA-2T (10 mm×1 mm×37 μm), PEA-4T (10 mm×1 mm×38 μm), and PEA-6T (10 mm×1 mm×37 μm).
The uniaxially oriented PEA-nT films showed homogeneous azo mesogen alignment along the film axes, as revealed by a sharp contrast inversion every 45° upon rotating the samples with respect to the analyzer under crossed POM observation, as shown in Figure S5.
The photoinduced stress curves of uniaxially oriented PEA-nT (n = 2, 4, 6) were recorded. To ensure test accuracy, the two ends of each film were clamped to secure the film’s position, and an initial stress of 500 kPa was applied to keep its length constant. Subsequently, time-dependent photo-stress values of the films under ultraviolet light (λ=365 nm, 40 mW cm⁻²) were recorded. For PEA-2T, PEA-4T, and PEA-6T, the values were 582.7 ± 43.1, 722.5 ± 48.6, and 835.9 ± 48.2 kPa, respectively, at ambient temperature. The photoinduced stress of the films increased with the elongation of the flexible spacer length, and the degree of orientation increased accordingly.
By measuring small-angle X-ray scattering (SAXS) (Figures S6 and S7), the degree of orientation in PEA-nT for n = 2, 4, and 6 was determined to be 46.41%, 47.16%, and 48.85%, respectively.
For air-induced deformation testing, we obtained a PEA-6T polymer film with a thickness of ~63 μm (measured by metallographic microscope) and a strain of 235%. The dimensions were 12 mm × 1 mm × 37 μm, and a 2-mm segment of the film was clamped with a flat clip. The uniaxially oriented PEA-6T film bent to the left toward the UV light source (λ=365 nm, 40 mW cm⁻²), reaching maximum bending within 5 seconds. The bent film returned to its initial straightness after irradiation, and was irradiated with visible light (λ>510 nm, 30 mW cm⁻²) for 55 seconds, as shown in Figure 5. Thus, the maximum response time for UV/visible light, bending/non-bending duration, and bending amplitude/angle were obtained. Results consistent with the photoinduced stress measurements were obtained through testing.
In addition, the uniaxially oriented PEA-6T film exhibits excellent fatigue resistance through its reversible deformation over 100 cycles, with nearly constant bending/extension times and bending amplitudes, as shown in Table 3. Similar to the uniaxially oriented PEA-6T film, PEA-2T (Figure S8) and PEA-4T (Figure S9) stretched films (with a strain of 235%) also exhibit significant and reversible photo-induced bending and stretching. Due to its excellent reversibility and fast photoresponsiveness, PEA-6T was selected as the experimental material to study the adaptability of azo main-chain polymer films in different polar solvents and their ability to resist solvents over a prolonged period.
2.3. Room Temperature Three-Dimensional Shape Programmability and Recyclability of Uniaxial Oriented PEA-6T Films
In addition, we investigated the three-dimensional shape programmability of uniaxial oriented PEA-6T films. Similar to the main-chain azo polymers reported earlier [50], stable 3D photo-drivers were easily obtained by reprogramming all oriented films prepared from main-chain azo LCPs strained at room temperature, as shown in Figure S8.
Next, the high recyclability and reprocessability of uniaxially oriented films were demonstrated. For PEA-6T films, the original film was dissolved in hexafluoro isopropanol, the solvent was evaporated to dryness, and then the residue was vacuum-dried (all steps were carried out at room temperature). This simple room-temperature recovery capability of PEA-6T-based photo-actuators offers a clear advantage over photo-actuators that can only be recovered by heating or reversible chemical reactions, primarily because the polymer remains intact during this gentle recovery process. In addition, the recovered PEA-6T can be further reprocessed into uniaxially oriented films by solution pouring/mechanical drawing and can display reversible photoinduced bending/non-bending with high fatigue resistance (over 100 cycles), exactly like the original film, as shown in Figure S10.
The recycling method for PEA-6T thin films involved dissolving the films in DCM, followed by evaporation of the solvent to dryness and drying under vacuum until a constant weight was achieved (all procedures were conducted at room temperature), as shown in Figure S11. The recovered product is then characterized using 1H NMR, as shown in Figure S12.
2.4. Photomechanical Properties and Solvent Resistance of Uniaxially Oriented Films of Main-Chain Azo PEAs Under Common Hydrogen Bonding Solvents
The presence of hydrogen bonding and crystalline domains in polymer chains endows main-chain physically cross-linked polymers with photodeformation, providing an intrinsic driving force for its micro to macro photo induced bending. Most researchers focus on studying different light intensities and temperatures [8,50]; however, few investigate the adaptability of photodeformation in air or explore long-term solvent resistance in polar environments [52]. Therefore, it is extremely important to systematically evaluate adaptability in different solvents (such as water, methanol, ethanol, butanol, and saline solutions), including initial photodeformation time and bending angle.
Firstly, a uniaxially oriented PEA-6T film (235% strain) with dimensions of 12 mm × 1 mm × 36 μm (length × width × thickness) was obtained via solution casting/mechanical stretching, with a 2-mm segment clamped by tweezers. We tested the UV-visible light cycling of the polymers in water (25 °C, 30 °C, 40 °C, 50 °C), saline solutions of different concentrations (0.9 wt%, 3.5 wt%) at 25 °C and 50 °C, methanol, ethanol, and butanol. The maximum bending distance (L) and the time required for light deformation to reach maximum bending (t) were recorded using a mobile phone, as shown in Table 3.
In order to study the relationship between solvent polarity and photodeformation time, we first analyzed various parameters of the following solvents. Uniaxially oriented PEA-6T film (235% strain) was tested for contact angle in different solvents, as shown in Figure S13 and Table S1. The water contact angle was relatively large—indicating weak wettability—which suggested partial compatibility between the solvent and the polymer surface but incomplete penetration. However, the bending and non-bending rates of the uniaxially oriented PEA-6T film in water were slightly slower than in air, likely due to the greater viscous resistance of water to the film. Therefore, measurements of the infrared spectra of uniaxially oriented PEA-6T film before and after light deformation in water showed that the intensity ratio of the sur-rounding bands was basically the same, ranging from 3306 cm-1 to 3377 cm-1 (which originated from hydrogen bonds and "free" N-H extending from amide units, and the band near 1639 cm-1 was approximately 1670 cm-1 (corresponding to hydrogen bonds and "free" C=O extending from amide units, respectively), revealing the hydrogen bonds between amide units in the PEA-6T film caused by water, as shown in Figure S14.
The moderate wettability of methanol and ethanol, as indicated by contact angles, allows solvent molecules to partially swell the material surface, reducing the energy barrier for chain segment movement while maintaining the overall stability of the hydrogen-bond cross-linked network. Therefore, the film could be cycled up to 100 times in methanol/ethanol, with a lower degree of change compared to water. Dynamic UV light overlay (every 0.5 seconds) and visible light overlay (every 5 seconds) were recorded, as shown in Figure S15. Although the contact angle of butanol is close to those of methanol and ethanol, the polymer’s photodeformation experienced strong viscous resistance—twice that of water—and its Hansen parameter was the smallest, reducing its photodeformation speed by ~33.3% compared to ethanol.
Mechanism analysis revealed faster photodeformation in physiological saline (0.9% NaCl) than in pure water, while high-concentration saline (3.5% NaCl) showed slower speeds (Table 3, Entries 8 and 9). The "ion shielding effect" of 0.9% NaCl refers to partial shielding of the polarity of amide bonds (–CONH–) in polymers by low-concentration salt ions (Na⁺/Cl⁻), weakening intramolecular/intermolecular hydrogen-bond cross-linking and reducing segment motion resistance. Dynamic enhancement of hydrogen-bond networks facilitates segment rearrangement driven by azobenzene photoisomerization, accelerating photodeformation; while strong hydration of high-concentration salt ions reduces free water, weakening solvent-amide hydrogen bond competition. Meanwhile, excessive salt ions cause polymer chains to aggregate through electrostatic attraction (similar to the "salting out effect"), leading to network rigidity and suppressing photoresponsive deformation.
We tested the photoresponse of pure water under different temperature gradients. The primary driver of accelerated photodeformation is the activation of chain segment motion. Increasing temperature (25–40 °C) provides thermal energy, reduces the potential barrier for polymer chain segment motion, and promotes the translation of local configuration changes—induced by photo-induced cis-trans isomerization (trans-cis) of azobenzene groups—into macroscopic deformation. Dynamic enhancement of hydrogen bonds involves thermal weakening of amide-amide hydrogen bonding, which induces "temporary softening" of the crosslinked network, enabling segment rearrangement and accelerating deformation response. Enhanced solvent permeability arises from temperature-induced viscosity reduction, promoting water/saltwater infiltration into the material surface, which enhances swelling, free volume, and deformation capacity.
While increasing temperature accelerates light response, it also disrupts hydrogen bonds—since hydrogen bond energy is typically 25–40 kJ/mol—leading to loss of weak intermolecular interactions at excessively high temperatures. Notably, the above-mentioned uniaxially oriented PEA-6T film exhibits a larger bending angle than previously reported (Table S4). It was not only oriented and stretched at room temperature but also maintained good photoinduced deformation and reversible cycling in various solvents. This is also the significance of developing and exploring the photoresponsivity of such polymer films in different solvents
Photoresponsive drivers in air exhibit long-lasting and stable responses. The solvent resistance of the prepared main-chain uniaxially oriented PEA-6T polymer in photodeformation tests was evaluated as follows.
To investigate the persistent solvent resistance of physically crosslinked photodeformable main-chain azo polymers in different polar solvents, we conducted long-term soaking tests. Firstly, the films were placed in corresponding test solvents for 7, 14, and 30 days, and their maximum bending angles were measured after each soaking duration. (Note: Soaking time for 7 hours means directly placing the uniaxially oriented polymer films into a solvent to test the results of 100 cycles, as shown in Table 3)
As shown in Figure S16, the maximum bending angle varies with the immersion time of the films in solvents. Statistical results, including those in Figure 6a and Table S3, are presented below. Notably, consistent with previous tests, the maximum bending angle in water (64.5 ± 0.8°) was greater than in other solvents and remained stable over time. However, the maximum bending angle fluctuated more significantly in water than in saline solutions over time. The maximum bending angle in saltwater solutions of different concentrations remained stable, likely due to dynamic hydrogen bond equilibrium between ions and the main-chain azobenzene polymer films. It is worth noting that it can be clearly seen that the maximum bending angle of the polymer film in the alcohol solvents decreases to varying degrees with the prolongation of soaking time. After soaking in methanol for 30 days, the polymer film exhibited significant changes. The polymer film showed significant changes after soaking in ethanol and butanol for 14 days and 7 hours, respectively. To investigate the reasons, we tested the changes in orientation and photo induced stress of the polymer film soaked for different times, as shown in Figure 6b, 6c and Table S3. To ensure test accuracy, the two ends of the film were clamped to secure its position, and an initial stress of 500 kPa was applied to maintain constant length. Subsequently, the time-dependent photo-stress of the film under ultraviolet light (λ=365 nm, 40 mW cm-²) was recorded and shown in Figure 6c. Photoinduced stress results showed distinct magnitudes and trends for polymer films soaked in different solvents over different times. In water, the larger contact angle resisted infiltration, thus minimizing photoinduced stress changes. In NaCl aqueous solutions, slightly smaller contact angles caused moderate film damage. Methanol, ethanol, and butanol had similar small contact angles; however, their different solubility parameters caused distinct degrees of film damage.
The orientation degree of saltwater is lower than that of water, which may be attributed to the dynamic equilibrium of hydrogen bonds between ions and the main chain azo-benzene polymer film, and its maximum bending angle remains unchanged; It is worth mentioning that from Figure 6a, the maximum bending angle in saltwater is more stable. Alongside Figure 6a results, analysis of orientation and photoinduced stress in methanol-soaked films revealed minimal damage to hydrogen bonds and film crystallinity. That is, even though orientation changed slightly, the maximum bending angle of uniaxially oriented films remained nearly unchanged (only a 3.3% reduction). However, the solvent resistance of the polymer films in ethanol/butanol was poor. From day 14 onward, orientation in ethanol-soaked films decreased significantly, accompanied by a 16.2% reduction in maximum bending angle. The change in the maximum bending angle of the polymer film after soaking in butanol was significant, indicating severe disruption of hydrogen bonds and crystallinity in the polymer films.
Figure 7 shows the Polarizing Optical Microscopy (POM) of polymer films soaked in different solvents for different durations. These tests were conducted at room temperature using polymer films with a strain of 235%.Specifically, the polymer films were observed after immersion in water (a0-a4), 0.9% NaCl aqueous solution (b0-b4), 3.5% NaCl aqueous solution (c0-c4), methanol (d0-d4), ethanol (e0-e4), and butanol (f0-f4).The angles θ between these membranes and the analyzer are 0 ° (a0, b0, c0, d0, e0, f0) and 45 ° (a1-a4, b1-b4, c1-c4, d1-d4, e1-e4, f1-f4), respectively. Among them, a1–f1 correspond to 7 hours, a2–f2 to 7 days, a3–f3 to 14 days, and a4–f4 to 30 days of soaking time, respectively. Microscopic images allowed clear observation of the films' microstructure and orientation. The scale bar in the images is 500 μm. As soaking time increased, POM images of methanol-soaked (30 days), ethanol-soaked (14 days), and butanol-soaked films showed significant changes consistent with the aforementioned photoinduced stress and orientation results. The results clearly indicate that the solvent affects the overall orientation of the films by influencing their crystalline network. In addition, POM images show that the films expanded to varying degrees in methanol, ethanol, and butanol, consistent with Hansen parameters (Figure S17). Brightness levels in POM images under different soaking times were analyzed using Image J, as shown in Figure 6d and Table S2.
FTIR spectra were recorded (Figure S18) in the range of 3306–3377 cm−1, corresponding to hydrogen-bonded and "free" N-H stretching vibrations of amide units. A band near 1639 cm−1 shifted to ~1670 cm−1, indicating hydrogen-bonded and "free" C=O stretching vibrations, respectively. These changes revealed alcohol-induced hydrogen bonding between amide units in PEA-6T films. Notably, a strong absorption peak at 1011 cm−1 originated from "free" C-O stretching in alcohols. This indicates that solvents can induce local changes in the films' network by affecting hydrogen bonds, leading to POM darkening and a decrease in orientation and photoinduced stress.
3. Materials and Methods
3.1. Materials and Reagents
Tetrahydrofuran (THF, Tianjin Chemical Reagent Co., Ltd., Tianjin, China, analytical grade (AR)) was refluxed over sodium and then distilled. Methanol (Tianjin Jiangtian Chemical Technology Co., Ltd., AR) and dichloromethane (CH2Cl2, Tianjin Chemical Reagent Co., Ltd., AR) were refluxed over calcium hydride and then distilled. 4-((4-(ω- hydroxyhexyloxy)) phenylazo) benzoic acid (H6AzoC), was synthesized according to previously reported procedure [25]. 1,2-Ethanedithiol (Macklin, 97%), 1,4-butanedithiol (Aladdin, 97%), 1,6-hexanedithiol (Aladdin, 98%), 1,8- diazabicyclo [5.4.0] undec-7-ene (DBU, Tianjin Heowns Biochem Technologies, LLC, 98%), 1-Ethyl-3- (3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI, Heowns Biochem Technologies, LLC, Tianjin, 98%), 4-(dimethylamino)pyridine (DMAP, Merck, AR), diethyl ether (AR), 4-amino-1-butanol (Heowns Biochem Technologies, LLC, Tianjin, 97%), acryloyl chloride (Heowns Biochem Technologies, LLC, Tianjin, 98%) and all the other chemicals were commercially available and used directly without further purification.
3.2. Synthesis of N- (4-hydroxybutyl) Acrylamide
4-Amino-1-butanol (3.565 g, 40 mmol) was dissolved in 75 mL of dry chloroform solution, and the solution was cooled to 0 ℃ by an ice water bath. Dissolve acryloyl chloride (1.625 mL, 20 mmol) in 20 mL of dry chloroform solution. The ice water bath was added with a constant pressure drip funnel, and then continued to stir for 4 h under this condition. After the reaction, 5 times the amount of silica gel was added directly to remove the solvent, and EA: MeOH = 10:1 was used as the eluant. The white solid product was purified by silica gel column chromatography, and the yield was 71%. 1H NMR (400 MHz, DMSO-d6): δ(PPM) = 8.07 (s, 1 H - NH -), 6.28 6.24 (dd, 1 H, CH = CH -), 6.12 6.06 (d, 1 H, CH2 = CH -), 5.64 5.61 (d, 1 H, CH2 = CH -), 4.54 4.32 (t, 1 H, - OH), 3.27 2.97 (s, 2 H - NH - CH2 -), 1.53 1.43 (s, 4 H - (CH2) 2 -).
3.3. Synthesis of M-EA
N- (4-hydroxybutyl) acrylamide (1.43 g, 10.00 mmol), A6AzoC (1.98 g, 5.00 mmol) and DMAP (61.08 mg, 0.50 mmol) were dissolved in 55 mL of dehydrated dichloromethane, and the solution was cooled to 0 ℃ by an ice water bath. EDCI (1.15 g, 6.00 mmol) was dissolved in 25 mL of dehydrated methylene chloride solution and slowly added to the reaction system under the condition of ice water bath stirring. After the drip is finished, continue to stir under this condition for 1 h, and then stir at room temperature 25 ℃ for 24 h. After the reaction, it was washed three times with 10 mL 0.01M hydrochloric acid, and then washed three times with saturated salt to remove excess EDCI and reaction catalyst DMAP. Concentrate the filtrate and add silica gel (200-300) 10 g. The liquid mixture EA: PE (v: v) = 1:1 was used as eluent by dry method and purified by silica gel column chromatography. The orange yellow solid product was obtained with yield of 62%. UV-Vis (DMAc): λmax/nm (ε/L mol-1 cm-1) = 362 (26670). 1H NMR (400 MHz, DMSO-d6): δ (PPM) = 8.20 8.07 (m, 3 H, Ar - and H - NH - CO -), 7.99 7.86 (dd, 4 H, Ar - H), 7.21 7.10 (d, 2 H, Ar - H), (6.38 6.02 m, 4 H, CH = CH - COO - and CH = CH - CON -) 5.98 5.88 (dd, 1 H, CH = C - COO), 5.63 5.54 (dd, 1 h, CH = C - CON -), 4.40 4.26 (t, 2 h, Ar - the COO - CH2 -), 4.18 4.04 (m, 4 H, CH2 = CH - COOCH2 - and - CH2 - O - Ar), 3.27 3.15 (q, 2 H, CH2 - NH -), (1.85 1.33 m, 12 H, -(CH2)4- and -(CH2)2-).
3.4. Synthesis of PEA-nT (n=2, 4, 6)
M-EA (72.38 mg, 0.14 mmol) was added with dried tetrahydrofuran (THF, 2.25 mL) and 1, 8-diazecyclic [5,4,0] undecene-7 (DBU, 25 uL, 0.16 mmol) in a 10 ml single-necked round-bottomed flask, stirring well until dissolved. Under the conditions of an ice water bath, argon gas is injected to remove oxygen for 5 minutes. Next, an equal molar amount of dimercaptan (0.14 mmol) was added and argon was continued for 10 minutes. Subsequently, the reaction bottle was sealed and placed in an oil bath at 60 ℃, and the reaction was carried out under dark conditions for 3 hours. After the reaction is completed, the reaction liquid is slowly added to the ice methanol, and the precipitate is obtained by centrifugation. After the solid was collected and dissolved in tetrahydrofuran, it was settled in ice methanol again and repeated operation, and there was no monomer residue on the silicone plate. The centrifuged solids were collected and dried in a vacuum drying oven at 40 ℃ until their constant weight was obtained. Finally, the orange polymer PET-nT (n=2,4,6,) was obtained.
3.5. Preparation of the Thin PEA-nT Film for the Photoresponsivity Study
A thin PEA-6T film was cast from a PEA-6T solution in hexafluoroisopropanol (2.0 mg mL- 1, 100 μL) on a clean quartz glass plate (12 × 45 mm). After the solvent was evaporated slowly at ambient temperature for 12 h, a transparent light-yellow film was formed on the quartz glass plate. The thickness (l) of the polymer film is estimated to be 740 nm by using the equation l= VC/ρS, where V is the volume of the polymer solution cast on the quartz glass plate, C is the concentration of the polymer solution, ρ is the density of the solid polymer film (ρ is assumed to be 1 g mL-1), and S is the surface area of the quartz glass plate. The photoresponsivity of the resulting thin PEA-6T film was studied by first irradiating it with UV light (365 nm, 40 mW cm-2) until its photostationary state was reached. The photostationary PEA-6T film was then irradiated with visible light (λ > 510 nm, 30 mW cm-2). The UV-vis spectra of the sample were recorded during the above studies. The used UV light and visible light were obtained from a high-pressure mercury lamp (USHIO SP-7, glass filters were used to obtain the desired light). And thin PEA-2T film and thin PEA-4T film were prepared using the same method as described above.
3.6. Preparation of the XRD Characterization for the Azo Polymer Powders
The crystallinity of the main chain azobenzene polymer was characterized by a Rigaku Smart Lab 9 kW (copper target) X-ray diffractometer from Nippon Science Corporation. Test conditions: Tube voltage 40 kV, tube current 150 mA, 2θ scanning range 1.5-40 °, scanning rate 5 °/min.
The sample preparation procedure for measuring XRD spectra of polymer powders includes first heating the polymer to a temperature higher than its anisotropy temperature (+10 ℃ min-1), then cooling it to a temperature 2 ℃ lower than the exothermic peak temperature in DSC spectra (-10 ℃ min-1) during the first cooling process, and quenching it with liquid nitrogen to maintain its phase structure (all heating and cooling are carried out under a nitrogen atmosphere).
3.7. Preparation of Uniaxially Oriented Main-Chain Azo Polymer Films
Uniaxially oriented PEA-6T films were prepared by solution pouring/mechanical stretching method using the method [53]. Take 100 mg polymer and fill 1 mL with hexafluoro isopropanol. Take 180 uL and place the bottom of the polytetrafluoroethylene mold with length * width * height (15 mm*10 mm*3 mm). The top was covered with a glass petri dish, volatilized at room temperature 25 ℃ for 24 h, and then the polymer film was cut into a width * thickness 2 mm*12 mm*~63 um. The polymer film with width * thickness of 2mm * 12mm * 63um was mechanically stretched at room temperature and cut into a width * thickness 1 mm*12 mm*~36 um (a strain of 235%) film. Used to measure photoinduced stress, photodeformation and degree of orientation.
3.8. Determination for Degree of Orientation in Uniaxially Oriented Main-Chain Azo Polymer Films
Degree of orientation of the uniaxially oriented PEA-nT films were tested by using Xeuss 3.0 UHR from Xenocs S.A.S in full power state with a power supply of 50 kV and 0.6 mA[54,55]. And the preparation method of uniaxially oriented PEA nT films were as described above [56]. Apply carefully the non-oriented/uniaxially oriented polymer film horizontally onto the sample stage using polyimide tape (a strain of 235%), which has circular holes with a diameter of 2 mm on the sample stage.
4. Conclusions
Low Tg (<room temperature) and high-temperature (Td > 288 ℃) resistant polymers were effectively synthesized via Michael addition polymerization of semi-crystalline main-chain azo polyester amides (PEAs). Uniaxially oriented polymer films physically cross-linked by solution casting/mechanical stretching exhibit good mechanical strength. Within a certain range, the length of flexible polymer chains significantly influences both mechanical strength and photomechanical properties. Elastic modulus ranges from 179.2 to 210.3 MPa, rupture strength from 12.6 to 18.0 MPa, elongation at break from 472.9% to 632.9%, and photoinduced stress from 582.7 to 835.9 kPa.
Photodeformation tests of uniaxially oriented PEA-6T polymer films in different solvents yielded the following conclusions: First, contact angle and viscosity index influence the response speed and stability of optical deformation. When compared to methanol, the photo response speed of butanol, with a higher viscosity index, is reduced by 33%. Second, the polarity constant of the solution directly affects the strength and kinetics of hydrogen bonds: high-polarity solvents (such as water) do not significantly weaken hydrogen bonds, whereas low-polarity solvents (such as ethanol and butanol) may destabilize the cross-linked network, thereby influencing light response rates and deformation reversibility. This correlates with Hansen's solubility parameters. At room temperature, changes in POM brightness, photoinduced stress, photoresponsiveness, and orientation of uniaxially oriented polymer films soaked in water or brine for 30 days remain below 5%. In contrast, films soaked in methanol for 30 days, ethanol for 14 days, and butanol even after short-term immersion exhibited significant decreases in POM brightness, maximum bending angle, film orientation, and photoinduced stress. Additionally, polymer films swelled to varying degrees in non-polar solvents. Solvent resistance tests indicated that retarded responses primarily stem from the disruption of hydrogen bonds and crystalline domains.
The study of photodeformation in polar reagents is expected to simulate material performance in saline plasma environments, such as biological fluids or industrial corrosion scenarios, thereby laying the foundation for marine equipment coatings or biomedical actuators. Additionally, solvent adaptability research can uncover general design principles for hydrogen-bond cross-linked networks, facilitating cross-medium applications in flexible electronics, environmental sensing, and other fields. Thus, investigating the photodeformation behavior and tolerance of hydrogen-bonded crosslinked azobenzene polymers in various solvents not only bridges the research gap in non-aqueous systems but also provides a theoretical foundation and technical support for developing smart materials with high environmental adaptability.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Scheme S1: Chemical structure and synthetic procedure of M-EA; Figure S1: POM images of PEA-nT (n =2, 4, 6): (a) PEA-2T upon cooling to 133 ℃ and annealing for 30 min (during the first cooling process); (b) PEA-4T upon cooling to 112 ℃ and annealing for 30 min (during the first cooling process); (c) PEA-6T upon cooling to 97 ℃ and then annealed for 30 min (during the first cooling process). The scale bar is 20 μm; Figure S2: UV and visible light-induced photoisomerization cycles of the PEA-6T thin film at 25 ℃. (in each cycle, the ultraviolet irradiation time is 65 s and the visible light irradiation time is 110s); Figure S3. Photoresponsivity of the thin PEA-2T film in the UV (a) and visible light (b) at25 ℃; Figure S4. Photoresponsivity of the thin PEA-4T film in the UV (a) and visible light (b) at 25 ℃; Figure S5: Photographs of azo polymer films in POM, Unstretched (a1, b1, c1) and stretched (a2, b2, c2) at room temperature; Photos of PEA-2T polymer films (a1, a2), PEA-4T polymer films (b1, b2), PEA-6T polymer films (c1, c2), at angles θ of 0 ° (left figure) and 45 ° (right figure) to the analyzer under orthogonal POM, with a scale size of 500 μm; Figure S6. SAXS of the Unstretched PEA-2T-2 film (a1), PEA-4T film (b1) and PEA-6T film (c1) and the uniaxially oriented PEA-2T film (a2), PEA-4T film (b2) and PEA-6T film (c2) (a strain of 235%); Figure S7. SAXS of integration on the uniaxially oriented PEA-2T film, PEA-4T film and PEA-6T film (a strain of 235%); Figure S8. Photo of photo induced bending/non bending of azobenzene polymer PEA-2T stretch film under ultraviolet light (365 nm, 40 mWcm-2) and visible light (>510 nm, 30 mWcm-2) irradiation at room temperature; Figure S9. Photo of photo induced bending/non bending of azobenzene polymer PEA-4T stretch film under ultraviolet light (365 nm, 40 mWcm-2) and visible light (>510 nm, 30 mWcm-2) irradiation at room temperature; Figure S10. 3D shape reprogramming of uniaxially oriented PEA-6T thin films and the optical deformation behavior of the resulting reshaped optical actuators. (a) Photo of polymer film obtained by solution casting method, length x width x thickness: 11 mm ×4 mm × 64 μm; (b) the uniaxially oriented PEA-6T film (235% strain, 37 mm × 2 mm × 37 μm; (c)Reshape film with one end containing approximately 1 mm of unstretched film) into a helical band at 26 ℃; (d) place it at 26 or 80 ℃ for 20 days (or at 100 ℃ for 2 days); (e-i) The optical deformation behavior of helical bands at room temperature under irradiation of ultraviolet light (365 nm, 40 mW cm-2) (e-f) and visible light (λ>510 nm, 30 mWcm-2) (h-i). The scale in the figure is 10 mm; Figure S11. The recycling process of light deformed PEA-6T film. Firstly, dissolve the chopped PEA-6T film in hexafluoro isopropanol, pour the solution into a PTFE mold, evaporate the solvent, cut the peeled film into strips, and then stretch it to 235 % strain (all processes are carried out at room temperature). The photo deformation properties of reprocessed PEA-6T film strips (10 mm × 1 mm × 37 μm) under ultraviolet light (365 nm, 40 mWcm-2) and visible light (λ>510 nm, 30 mW cm-2). The scale in the picture is 10 mm; Figure S12. 1H NMR spectra of PEA-6T and recovered polymer; Figure S13. Contact angles under different solutions at 25 ℃; Table S1. Contact angles under different solutions; Figure S14. FTIR of the uniaxially orientated PEA- 6T films in air and water at 25 ℃; Table S2. Brightness Level of POM in different solvents in different soaking time; Figure S15. (a) For UV dynamic overlay every 0.5 seconds, (b) Visible light dynamic overlay every 5 seconds. This includes air (a1, b1), water (a2, b2), methanol (a3, b3), ethanol (a4, b4), 0.9 wt% (a5, b5) and 3.5 wt% (a6, b6) sodium chloride aqueous solutions under ultraviolet light (365 nm, 40 mWcm-2) and visible light (λ>510 nm, 30 mWcm-2); Table S3. The data of Uniaxially oriented PEA-6T film (a strain of 235%); Figure S16. Photos of uniaxially orientated films in light deformation with different solvent in different soaking time. (a) water (b) 0.9 wt% NaCl (c) 3.5 wt% NaCl (d) Methanol (e) Ethanol (f) N-butanol at 25℃Figure S17.Comparison diagram of polymer film swelling to different degrees after soaking in solvent for 30 days. The red dotted line is the length of the uniaxially oriented film soaked in water for 30 days, and the red arrow points to the swelling direction. POM in different solvent in different soaking time. (a) water (b) 0.9 wt% NaCl (c) 3.5 wt% NaCl (d) Methanol (e) Ethanol (f) N-butanol at 25 ℃ The swelling rate is determined by the film width after the film (except water) is soaked for 30 days and the film width after the film is soaked in water for 30 days. Figure S18. FTIR of the uniaxially orientated PEA- 6T films in Methanol, Ethanol and Butanol at 25℃. Table S4. Comparison of the Photodeformation Behaviors of the Azo films
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, J.G.; methodology, Z.Z; software, Z.Z.; validation, Z.Z and S.M; formal analysis, Z.Z.; investigation, Z.Z.; resources, J.G.; data curation, J.G.; writing—original draft preparation, Z.Z.; writing—review and editing, Z.Z.; visualization, Z.Z.; supervision, J.G.; project administration, J.G.; funding acquisition, J.G. All authors have read and agreed to the published version of the manuscript.”. Authorship must be limited to those who have contributed substantially to the work reported.
Funding
This research received no external funding
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not applicable.
References
- Yu, H.; Ikeda, T., Photocontrollable liquid-crystalline actuators. Adv. Mater. 2011, 23, (19), 2149-2180. [CrossRef]
- White, T. J.; Broer, D. J., Programmable and adaptive mechanics with liquid crystal polymer networks and elastomers. Nat. Mater. 2015, 14, (11), 1087-1098. [CrossRef]
- Natansohn, A.; Rochon, P., Photoinduced motions in azo-containing polymers. Chem. Rev. 2002, 102(11), 4139-4176. [CrossRef]
- Mysliwiec, J.; Czajkowski, M.; Miniewicz, A.; Bartkiewicz, S.; Kochalska, A.; Polakova, L.; Sedlakova, Z.; Nespurek, S., Dynamics of photoinduced motions in azobenzene grafted polybutadienes. Opt. Mater. 2011, 33, (9), 1398-1404. [CrossRef]
- Iamsaard, S.; Anger, E.; Asshoff, S. J.; Depauw, A.; Fletcher, S. P.; Katsonis, N., Fluorinated azobenzenes for shape-persistent liquid crystal polymer networks. Angew. Chem. Int. Ed. 2016, 55, (34), 9908-12.
- Tkachenko, I. M.; Kurioz, Y. I.; Kravchuk, R. M.; Kobzar, Y. L.; Litoshenko, D. V.; Glushchenko, A. V.; Shevchenko, V. V.; Nazarenko, V. G., Photoinduced birefringence and liquid crystal orientation on polymers with different azobenzene content in the main chain. ACS Appl. Mater. Interfaces 2024, 16, (39), 52945-52957. [CrossRef]
- Wang, D. H.; Lee, K. M.; Yu, Z.; Koerner, H.; Vaia, R. A.; White, T. J.; Tan, L.-S., Photomechanical response of glassy azobenzene polyimide networks. Macromolecules 2011, 44, (10), 3840-3846. [CrossRef]
- Dong, H.; Liu, G.; Zhang, H., Preparation of photodeformable azobenzene polymer fibers by post-crosslinking strategy: Understanding the structure-property relationship. Eur. Polym. J. 2020, 135: 109863. [CrossRef]
- Zhu, Y.; Xu, Z.; Wu, F.; Wang, M.; Chen, L., Liquid-crystal elastomers based on covalent adaptable networks: From molecular design to applications. Science China Materials 2023, 66, (8), 3004-3021. [CrossRef]
- Bin Rusayyis, M. A.; Fenimore, L. M.; Purwanto, N. S.; Torkelson, J. M., Reprocessable, creep-resistant covalent adaptable networks synthesized using conventional free-radical polymerization conditions with piperidine-based and non-piperidine-based dynamic dialkylamino disulfide chemistry. Polym. Chem. 2023, 14, (30), 3519-3534. [CrossRef]
- Zhang, Z. P.; Rong, M. Z.; Zhang, M. Q., Polymer engineering based on reversible covalent chemistry: A promising innovative pathway towards new materials and new functionalities. Prog. Polym. Sci. 2018, 80, 39-93. [CrossRef]
- Van Zee, N. J.; Nicolaÿ, R., Vitrimers: Permanently crosslinked polymers with dynamic network topology. Prog. Polym. Sci. 2020, 104: 101233.
- Chakma, P.; Konkolewicz, D., Dynamic Covalent Bonds in Polymeric Materials. Angew. Chem. Int. Ed. 2019, 58(29), 9682-9695. [CrossRef]
- Podgórski, M.; Fairbanks, B. D.; Kirkpatrick, B. E.; McBride, M.; Martinez, A.; Dobson, A.; Bongiardina, N. J.; Bowman, C. N., Toward stimuli-responsive dynamic thermosets through continuous development and improvements in covalent adaptable networks (CANs). Adv. Mater. 2020, 32(20): 1906876. [CrossRef]
- Jiang, Z. C.; Xiao, Y. Y.; Yin, L.; Han, L.; Zhao, Y., "Self-Lockable" liquid crystalline diels-alder dynamic network actuators with room temperature programmability and solution reprocessability. Angew. Chem. Int. Ed. 2020, 59, (12), 4925-4931.
- Huang, X.; Qin, L.; Wang, J.; Zhang, X.; Peng, B.; Yu, Y., Multiple Shape Manipulation of Liquid Crystal Polymers Containing Diels-Alder Network. Adv. Funct. Mater. 2022, 32(51): 2208312. [CrossRef]
- Han, G.; Nie, J.; Zhang, H., Facile preparation of recyclable photodeformable azobenzene polymer fibers with chemically crosslinked networks. Polym. Chem. 2016, 7, (32), 5088-5092. [CrossRef]
- Qin, C.; Feng, Y.; Luo, W.; Cao, C.; Hu, W.; Feng, W., A supramolecular assembly of cross-linked azobenzene/polymers for a high-performance light-driven actuator. J. Mater. Chem. A 2015, 3, (32), 16453-16460. [CrossRef]
- Qin, C.; Feng, Y.; An, H.; Han, J.; Cao, C.; Feng, W., Tetracarboxylated azobenzene/polymer supramolecular assemblies as high-performance multiresponsive actuators. ACS Appl. Mater. Interfaces 2017, 9, (4), 4066-4073. [CrossRef]
- Vapaavuori, J.; Bazuin, C. G.; Priimagi, A., Supramolecular design principles for efficient photoresponsive polymer–azobenzene complexes. J. Mater. Chem. C 2018, 6, (9), 2168-2188. [CrossRef]
- Yu, H.-T.; Tang, J.-W.; Feng, Y.-Y.; Feng, W., Structural design and application of azo-based supramolecular polymer systems. Chin. J. Polym. Sci. 2019, 37, (12), 1183-1199. [CrossRef]
- Houben, S. J. A.; Lugger, S. J. D.; van Raak, R. J. H.; Schenning, A. P. H. J., A ph-responsive liquid crystal hydrogel actuator with calcium-induced reprogrammable shape fixing. ACS Appl. Polym. Mater. 2022, 4, (2), 1298-1304. [CrossRef]
- Zhong, H.-Y.; Chen, L.; Yang, R.; Meng, Z.-Y.; Ding, X.-M.; Liu, X.-F.; Wang, Y.-Z., Azobenzene-containing liquid crystalline polyester with π–π interactions: diverse thermo- and photo-responsive behaviours. J. Mater. Chem. C 2017, 5, (13), 3306-3314. [CrossRef]
- Zhong, H.-Y.; Chen, L.; Liu, X.-F.; Yang, R.; Wang, Y.-Z., Novel liquid crystalline copolyester containing amphi-mesogenic units toward multiple stimuli-response behaviors. J. Mater. Chem. C 2017, 5, (37), 9702-9711. [CrossRef]
- Zhou, Y.; Wang, L.; Ma, S.; Zhang, H. Fully room-temperature reprogrammable, reprocessable, and photomobile soft actuators from a high-molecular-weight main-chain azobenzene crystalline poly(ester-amide). ACS Appl. Mater. Interfaces 2022, 14, (2), 3264-3273. [CrossRef]
- Lee, W.; Kwak, S.-Y.; Chung, J. W., Arm-length-dependent phase transformation and dual dynamic healing behavior of supramolecular networks consisting of ureidopyrimidinone-end-functionalized semi-crystalline star polymers. Eur. Polym. J. 2020, 138: 109976. [CrossRef]
- Ma, S.; Zhou, Y.; Wang, L.; Zhang, H., Multifunctional UV-NIR dual light-responsive soft actuators from a main-chain azobenzene semi-crystalline poly(ester-amide) doped with polydopamine nanoparticles. Chem. Eur. J. 2024, 30(8): e202303306.
- Li, G.; Xu, M.; Zhang, S.; Yang, G.; Li, W., Reversible controlling the supramolecular chirality of side chain azobenzene polymers: chiral induction and modulation. Macromol. Rapid Commun. 2022, 43(6): 2100904. [CrossRef]
- Zhang, H. Reprocessable photodeformable azobenzene polymers. Molecules 2021, 26(15): 4455. [CrossRef]
- Fang, L.; Han, G.; Zhang, J.; Zhang, H.; Zhang, H., Synthesis of well-defined easily crosslinkable azobenzene side-chain liquid crystalline polymers via reversible addition–fragmentation chain transfer polymerization and photomechanical properties of their post-crosslinked fibers. Eur. Polym. J. 2015, 69, 592-604. [CrossRef]
- Han, G.; Zhang, H.; Chen, J.; Sun, Q.; Zhang, Y.; Zhang, H., Easily crosslinkable side-chain azobenzene polymers for fast and persistent fixation of surface relief gratings. New J. Chem. 2015, 39, (2), 1410-1420. [CrossRef]
- Li, X.; Fang, L.; Hou, L.; Zhu, L.; Zhang, Y.; Zhang, B.; Zhang, H., Photoresponsive side-chain liquid crystalline polymers with amide group-substituted azobenzene mesogens: effects of hydrogen bonding, flexible spacers, and terminal tails. Soft Matter 2012, 8(20), 5532-5542. [CrossRef]
- Li, X.; Wen, R.; Zhang, Y.; Zhu, L.; Zhang, B.; Zhang, H., Photoresponsive side-chain liquid crystalline polymers with an easily cross-linkable azobenzene mesogen. J. Mater. Chem. 2009, 19, (2), 236-245. [CrossRef]
- Li, Z.; Zhang, Y.; Zhu, L.; Shen, T.; Zhang, H., Efficient synthesis of photoresponsive azobenzene-containing side-chain liquid crystalline polymers with high molecular weights by click chemistry. Polym. Chem. 2010, 1(9), 1501-1511. [CrossRef]
- Wani, O. M.; Zeng, H.; Priimagi, A., A light-driven artificial flytrap. Nat. Commun. 2017, 8(1): 15546. [CrossRef]
- Guo, C.; Gao, J.; Ma, S.; Zhang, H., Efficient preparation of chemically crosslinked recyclable photodeformable azobenzene polymer fibers with high processability and reconstruction ability via a facile post-crosslinking method. Eur. Polym. J. 2020, 139: 109998. [CrossRef]
- Nie, J.; Liu, X.; Yan, Y.; Zhang, H. Supramolecular hydrogen-bonded photodriven actuators based on an azobenzene-containing main-chain liquid crystalline poly(ester-amide). J. Mater. Chem. C 2017, 5, (39), 10391-10398. [CrossRef]
- Tan, S.; Sha, Y.; Zhu, T.; Rahman, M. A.; Tang, C., Photoresponsive supramolecular polymers based on quadruple hydrogen-bonding and a photochromic azobenzene motif. Polym. Chem. 2018, 9, (44), 5395-5401. [CrossRef]
- Ube, T.; Nakayama, R.; Ikeda, T., Photoinduced motions of thermoplastic polyurethanes containing azobenzene moieties in main chains. Macromolecules 2022, 55, (2), 413-420. [CrossRef]
- Ma, S.; Wang, L.; Zhou, Y.; Zhang, H., Fully room temperature reprogrammable, recyclable, and photomobile soft actuators from physically cross-linked main-chain azobenzene liquid crystalline polymers. Molecules 2023, 28, (10): 4174. [CrossRef]
- Zhou, Y.; Wang, L.; Zhang, H. Enhancing the performances of physically cross-linked photodeformable main-chain azobenzene poly(ester-amide)s via chemical structure engineering. Polym. Chem. 2022, 13, (24), 3713-3725. [CrossRef]
- Wang, L.; Zhou, Y.; Ma, S.; Zhang, H. Reprocessable and healable room temperature photoactuators based on a main-chain. [CrossRef]
- azobenzene liquid crystalline poly(ester-urea). J. Mater. Chem. C 2021, 9, (38), 13255-13265.
- Pilz da Cunha, M.; van Thoor, E. A. J.; Debije, M. G.; Broer, D. J.; Schenning, A. P. H. J., Unravelling the photothermal and photomechanical contributions to actuation of azobenzene-doped liquid crystal polymers in air and water. J. Mater. Chem. C 2019, 7, (43), 13502-13509. [CrossRef]
- Zhang, P.; Lan, Z.; Wei, J.; Yu, Y., Photodeformable azobenzene-containing polyimide with flexible linkers and molecular alignment. ACS Macro Lett. 2021, 10, (4), 469-475. [CrossRef]
- Kuenstler, A. S.; Clark, K. D.; de Alaniz, J. R.; Hayward, R. C., Reversible actuation via photoisomerization-induced melting of a semicrystal line poly(azobenzene). ACS Macro Lett. 2020, 9, (6), 902-909.
- Niemann, M.; Ritter, H., Comb-like methacrylamide polymers containing condensates of amino acids and azobenzene moieties in the side chains. Makromol. Chem. 1993, 194, (4), 1169-1181.
- Wang, G.-J.; Tong, X.; Zhao, Y. J. M., Preparation of azobenzene-containing amphiphilic diblock copolymers for light-responsive micellar aggregates. Macromolecules 2004, 37, 8911-8917. [CrossRef]
- Akiyama, H.; Tamaoki, N. Synthesis and photoinduced phase transitions of poly(N-isopropylacrylamide) derivative functionalized with terminal azobenzene units. Macromolecules 2007, 40, 5129–5132. [CrossRef]
- Wei, R.; He, Y.; Wang, X., Diblock copolymers composed of a liquid crystalline azo block and a poly(dimethylsiloxane) block: synthesis, morphology and photoresponsive properties. RSC Adv. 2014, 4, (102), 58386-58396. [CrossRef]
- Wie, J. J.; Wang, D. H.; Lee, K. M.; White, T. J.; Tan, L. S., The contribution of hydrogen bonding to the photomechanical response of azobenzene-functionalized polyamides. J. Mater. Chem. C, 2018, 6(22), 5964-5974. [CrossRef]
- Wang, Z.; Huang, H.; Hsu, C.; Wang, X., Azo molecular glass patterning from chiral submicron pillar array to self-organized topographic transition via irradiation with circularly polarized light. Adv. Opt. Mater. 2021, 9, (21): 2100922. [CrossRef]
- Corrado, F.; Bruno, U.; Prato, M.; Carella, A.; Criscuolo, V.; Massaro, A.; Pavone, M.; Muñoz-García, A. B.; Forti, S.; Coletti, C.; Bettucci, O.; Santoro, F., Azobenzene-based optoelectronic transistors for neurohybrid building blocks. Nat. Commun. 2023, 14, (1): 6760.
- Kizhakidathazhath, R.; Geng, Y.; Jampani, V. S. R.; Charni, C.; Sharma, A.; Lagerwall, J. P. F., Facile anisotropic deswelling method for realizing large-area cholesteric liquid crystal elastomers with uniform structural color and broad-range mechanochromic response. Adv. Funct. Mater. 2019, 30(7): 1909537. [CrossRef]
- Kaniyoor, A.; Gspann, T. S.; Mizen, J. E.; Elliott, J. A., Quantifying alignment in carbon nanotube yarns and similar two-dimensional anisotropic systems. J. Appl. Polym. Sci. 2021, 138, (37): 50939. [CrossRef]
- Patil, N.; Balzano, L.; Portale, G.; Rastogi, S., Influence of nanoparticles on the rheological behaviour and initial stages of crystal growth in linear polyethylene. Macromol. Chem. Phys. 2009, 210, (24), 2174-2187. [CrossRef]
- Cinader, D. K.; Burghardt, W. R., X-ray scattering studies of orientation in channel flows of a lyotropic liquid crystalline polymer. Polymer 1999, 40, (15), 4169-4180.
Scheme 1.
Synthetic routes and chemical structures of the main-chain azo semi-crystalline PEA-nT (prepared via the Michael addition polymerization of M-EA with different chain lengths of dithiol (i.e., HS(CH2) n SH (n = 2, 4, 6)).
Scheme 1.
Synthetic routes and chemical structures of the main-chain azo semi-crystalline PEA-nT (prepared via the Michael addition polymerization of M-EA with different chain lengths of dithiol (i.e., HS(CH2) n SH (n = 2, 4, 6)).
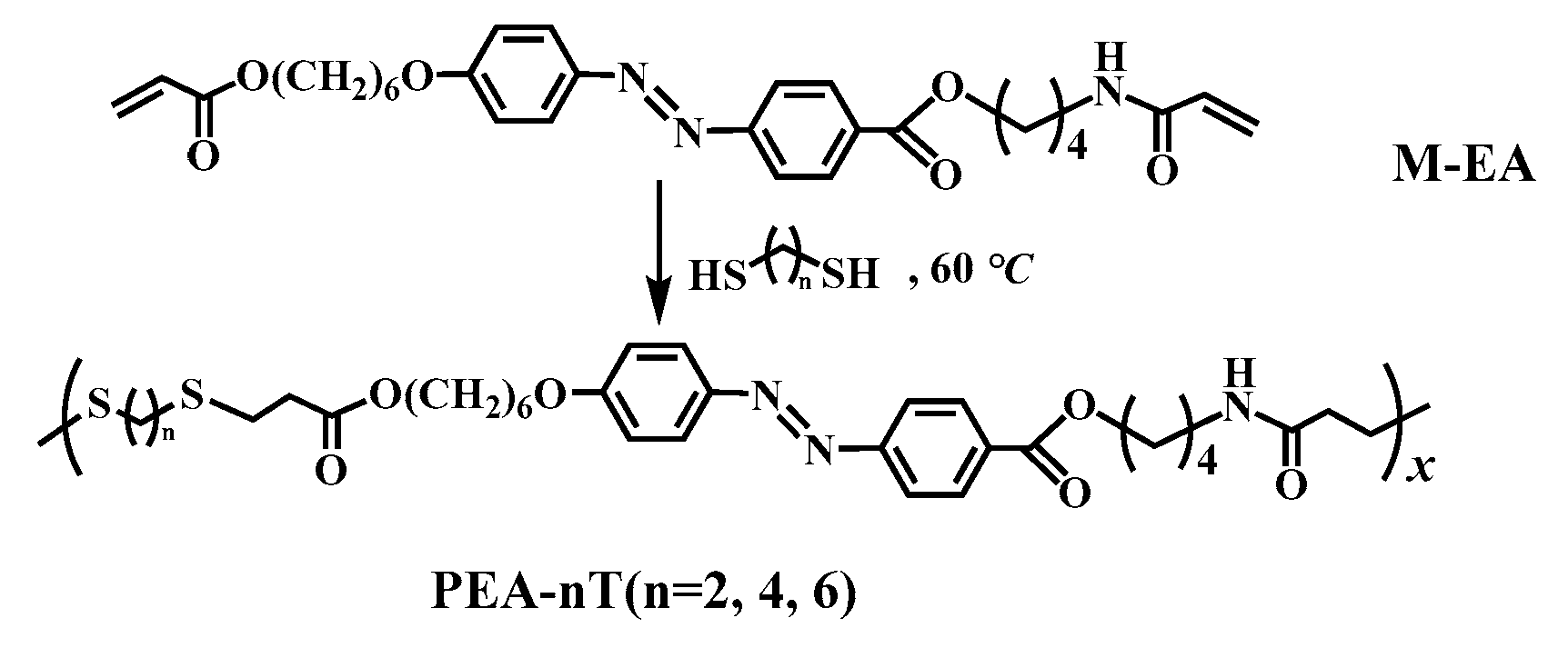
Figure 1.
1H NMR spectra of M-EA (A), PEA-2T (B), PEA-4T (C), PEA-6T (D)and in DMSO under 80 ℃.
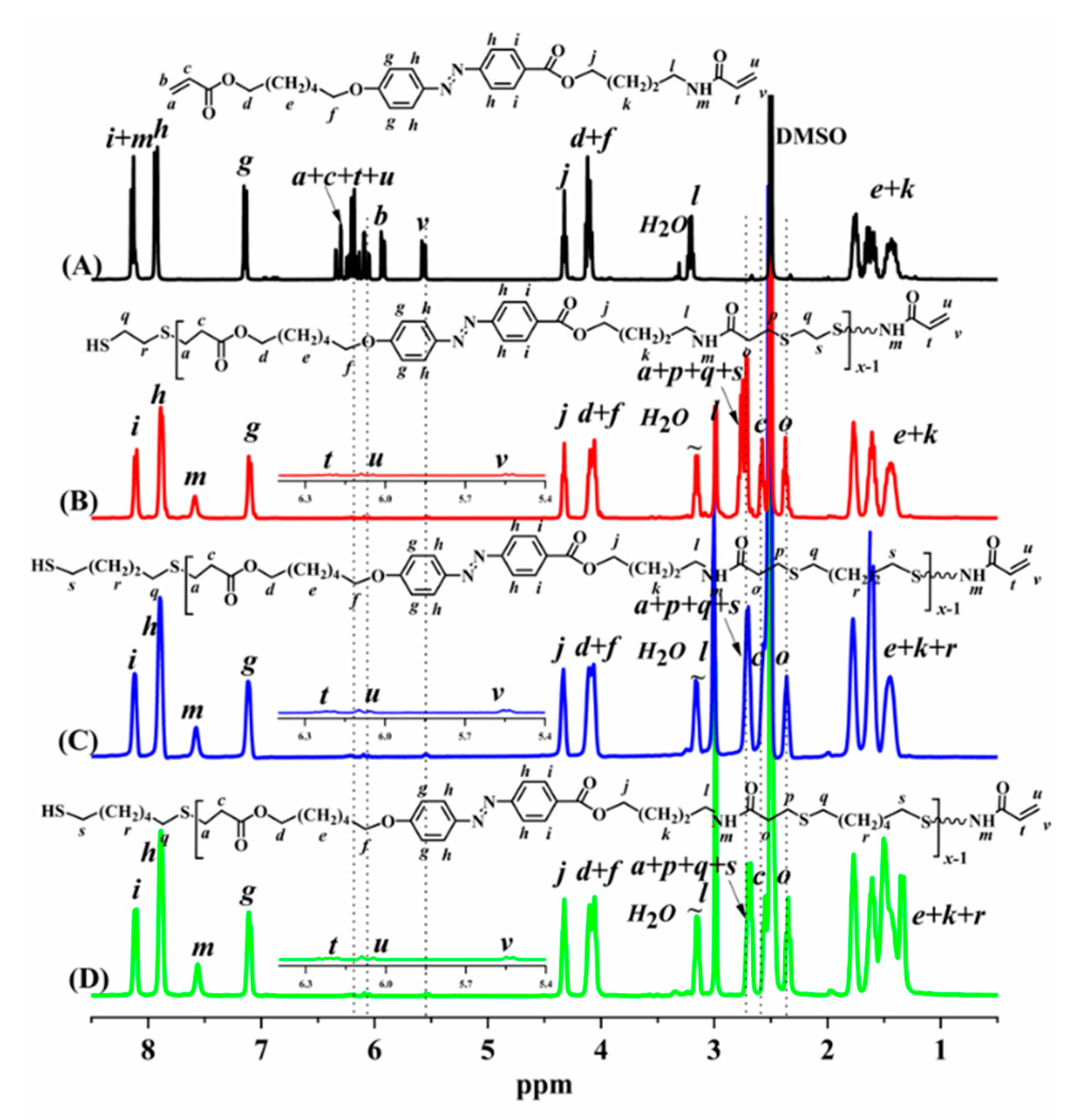
Figure 2.
(a) The DSC thermogram of PEA-nT (n=2 (a1, a4), n=4 (a2, a5) and n=6 (a3, a6)) from the second heating scan(a1-a3) and first cooling scan (a4–a6) (±10 ◦C min−1) While second heating scans is a1, a2 and a3. (b) The XRD spectra of PEA-nT (n=2 (b1), n=4 (b2) and n=6 (b3)).
Figure 2.
(a) The DSC thermogram of PEA-nT (n=2 (a1, a4), n=4 (a2, a5) and n=6 (a3, a6)) from the second heating scan(a1-a3) and first cooling scan (a4–a6) (±10 ◦C min−1) While second heating scans is a1, a2 and a3. (b) The XRD spectra of PEA-nT (n=2 (b1), n=4 (b2) and n=6 (b3)).
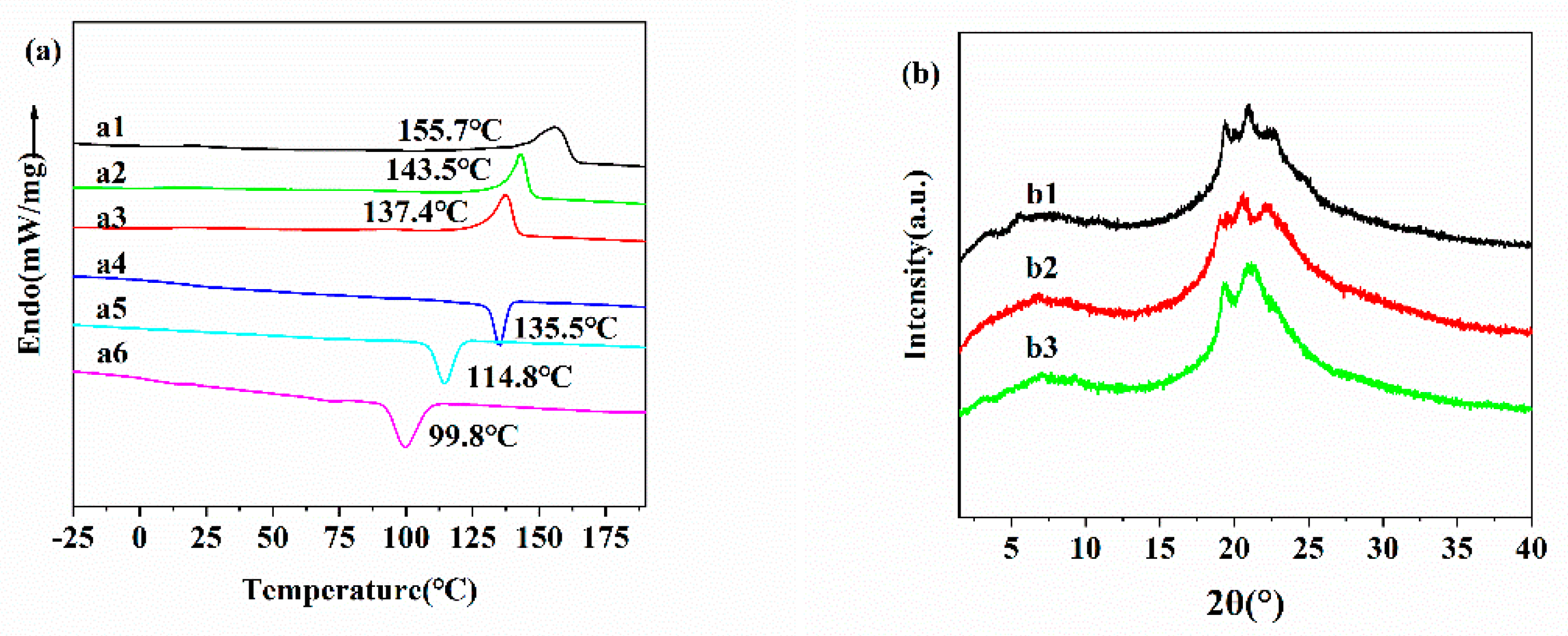
Figure 3.
Photoresponsivity of the PEA-6T thin film in the UV (a) and visible light (b) UV-vis spectral changes in dependence of time for the thin PEA-6T film (cast on the quartz glass plate, film thickness: 740 nm) upon its exposure to 365 nm of UV light (40 mW cm−2) and upon irradiating the polymer film at the photostationary state with visible light (λ > 510 nm, 30 mW cm−2) at 25 ◦C.
Figure 3.
Photoresponsivity of the PEA-6T thin film in the UV (a) and visible light (b) UV-vis spectral changes in dependence of time for the thin PEA-6T film (cast on the quartz glass plate, film thickness: 740 nm) upon its exposure to 365 nm of UV light (40 mW cm−2) and upon irradiating the polymer film at the photostationary state with visible light (λ > 510 nm, 30 mW cm−2) at 25 ◦C.
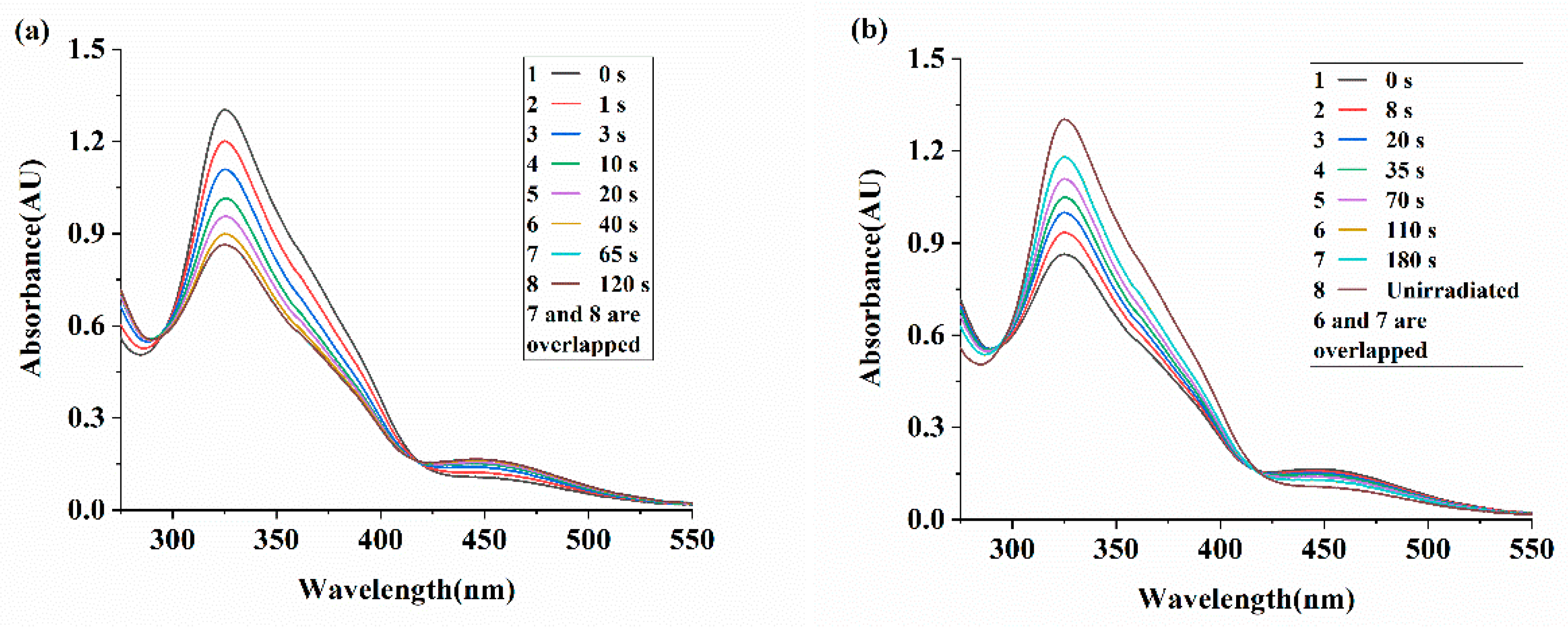
Figure 4.
(a) Stress-strain curves of unoriented main-chain azobenzene polymer PEA-nT films and photoinduced stress of uniaxially oriented azobenzene polymer PEA-nT films (a strain of 235%) were measured at 25 ℃ at a stretching rate of 10 mm min-1. (b) The photoinduced stress of azobenzene PEA-nT (n= 2, 4, 6) stretched films with time under ultraviolet irradiation (λ = 365 nm, 40 mW cm-2) at 25 ℃ was investigated.
Figure 4.
(a) Stress-strain curves of unoriented main-chain azobenzene polymer PEA-nT films and photoinduced stress of uniaxially oriented azobenzene polymer PEA-nT films (a strain of 235%) were measured at 25 ℃ at a stretching rate of 10 mm min-1. (b) The photoinduced stress of azobenzene PEA-nT (n= 2, 4, 6) stretched films with time under ultraviolet irradiation (λ = 365 nm, 40 mW cm-2) at 25 ℃ was investigated.
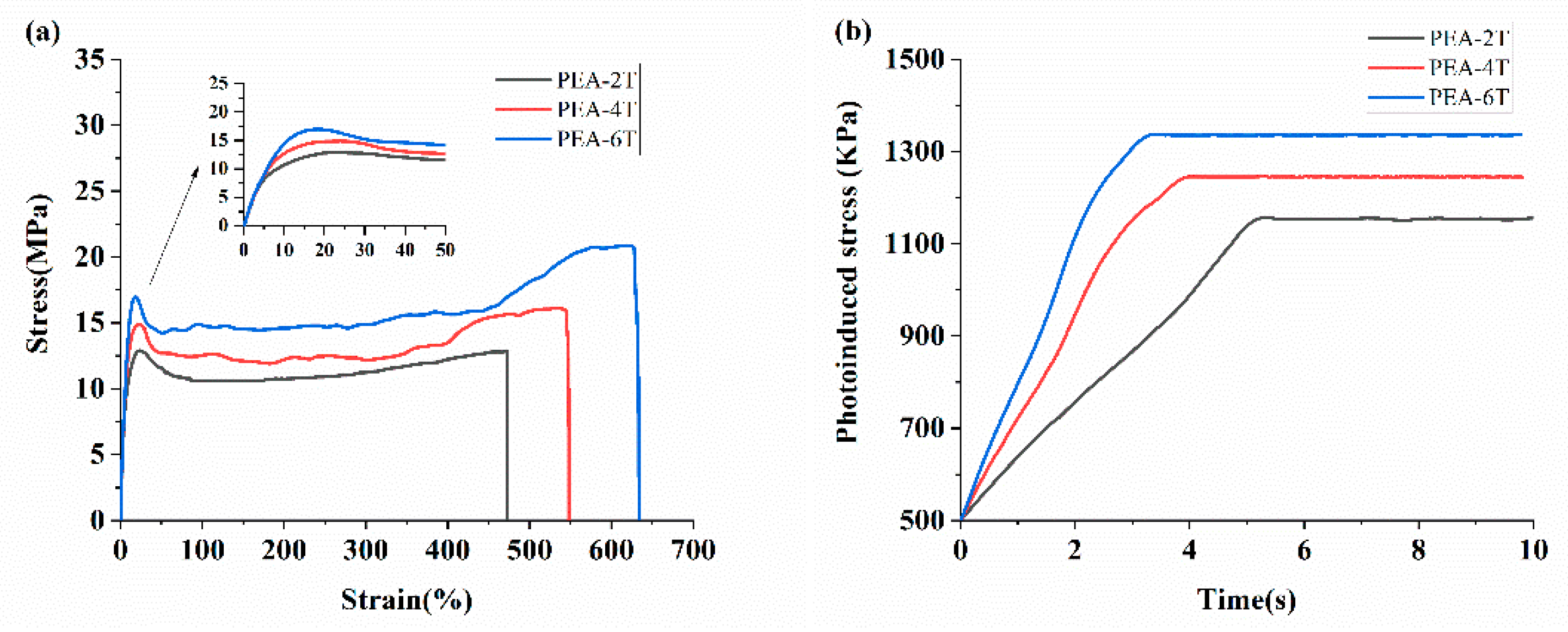
Figure 5.
(a)Photo of photo induced bending/non bending of azobenzene polymer PEA-6T stretch film under 365 nm ultraviolet light (40 mW cm-2) and visible light (>510 nm, 30 mWcm-2) irradiation at room temperature. (b)The reversible deformation of the PEA-6T film characterized by tracing the bent distance from its straight state at room temperature.
Figure 5.
(a)Photo of photo induced bending/non bending of azobenzene polymer PEA-6T stretch film under 365 nm ultraviolet light (40 mW cm-2) and visible light (>510 nm, 30 mWcm-2) irradiation at room temperature. (b)The reversible deformation of the PEA-6T film characterized by tracing the bent distance from its straight state at room temperature.
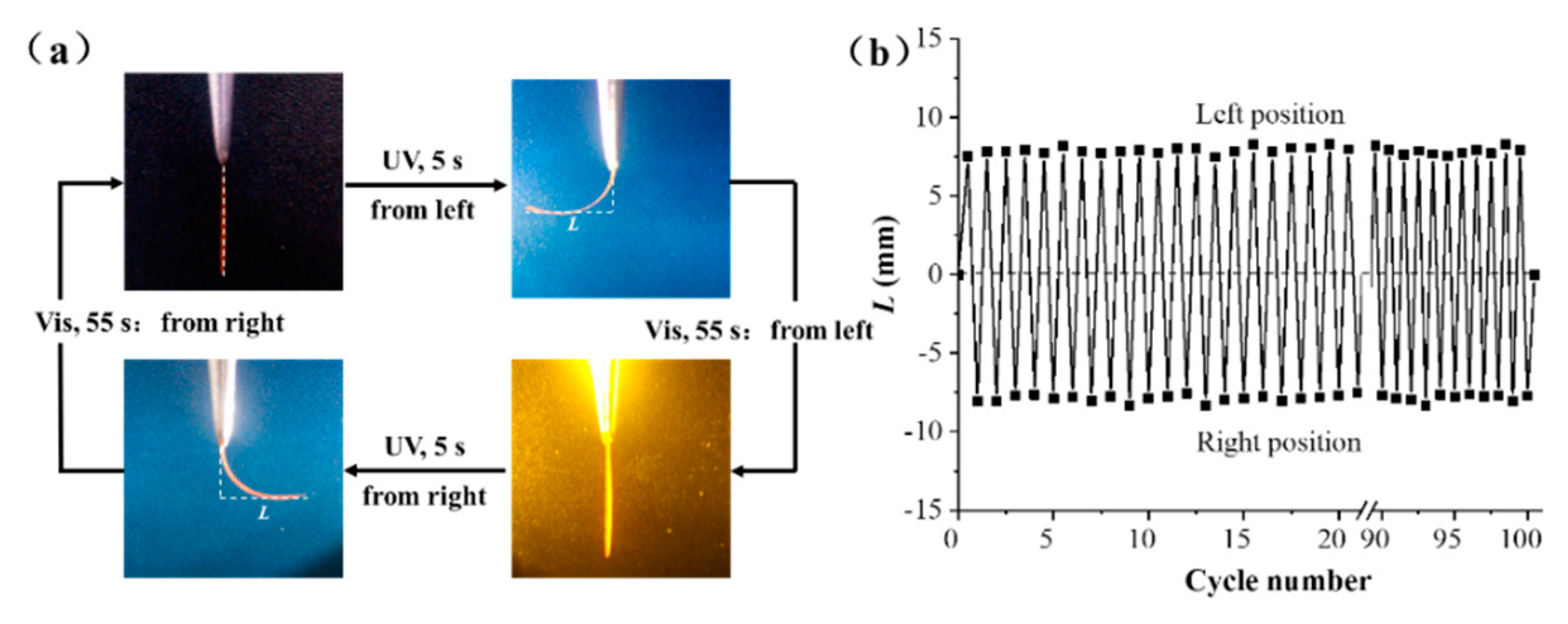
Figure 6.
Figure 6. (a) Maximum bending angle in different solvent in different soaking time. (b) orientation of polymer films in different solvent in different soaking time. (c) photoinduced stress in different solvent in different soaking time. (d) Brightness level of POM in different solvent in different soaking time. (Soaking time for 7 hours means directly placing the uniaxially oriented polymer films into a solvent to test the results of 100 cycles, as shown in Table 3).
Figure 6.
Figure 6. (a) Maximum bending angle in different solvent in different soaking time. (b) orientation of polymer films in different solvent in different soaking time. (c) photoinduced stress in different solvent in different soaking time. (d) Brightness level of POM in different solvent in different soaking time. (Soaking time for 7 hours means directly placing the uniaxially oriented polymer films into a solvent to test the results of 100 cycles, as shown in Table 3).
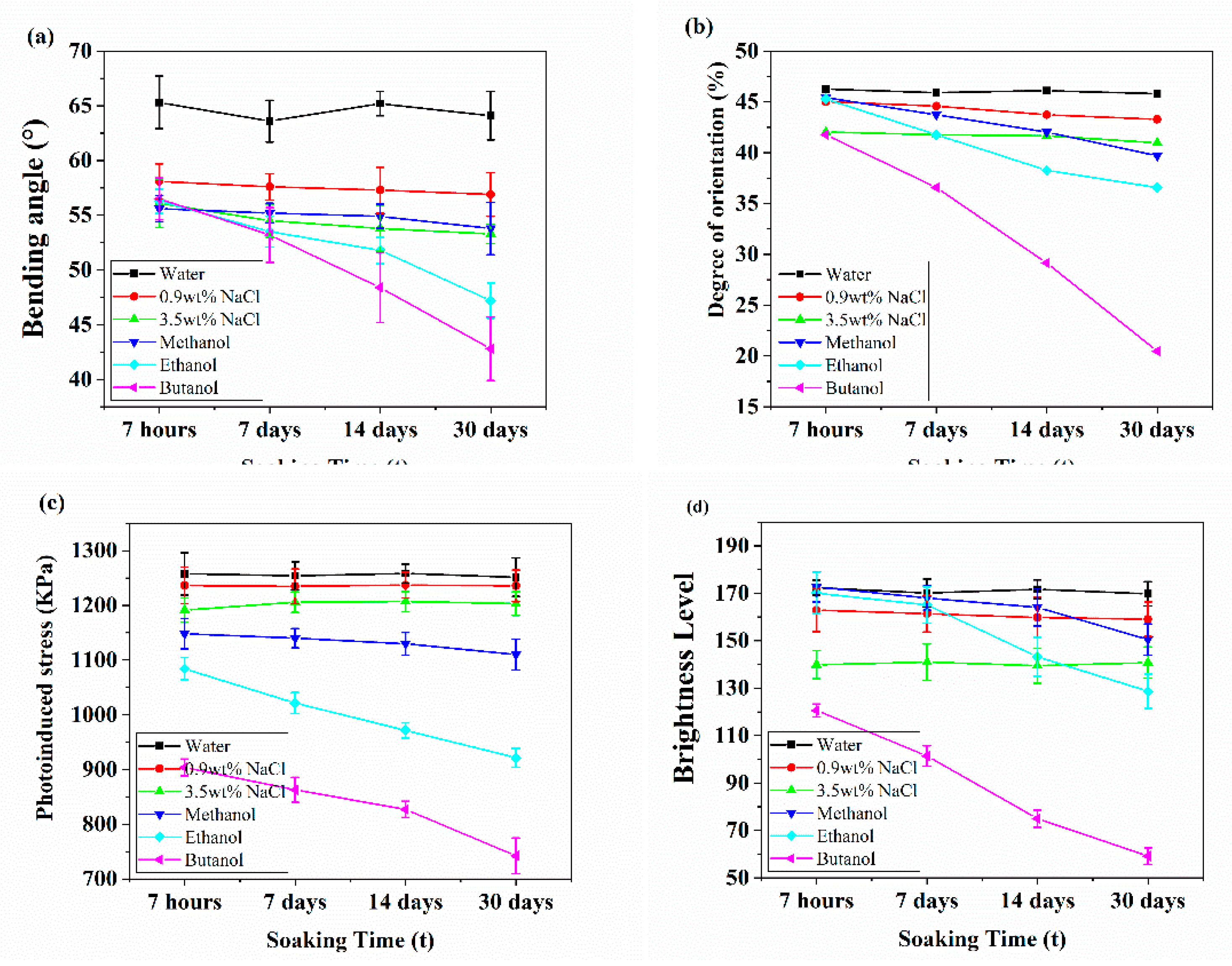
Figure 7.
POM of uniaxially oriented azobenzene polymer PEA-6T in different solvent in different soaking time.
Figure 7.
POM of uniaxially oriented azobenzene polymer PEA-6T in different solvent in different soaking time.


Table 1.
Synthetic and characterization data of the main-chain azo semi-crystalline PEAs.
| Entry | Samplea | Yield (%) |
Degree of polymerization (DP)b |
Mn, NMR (g·mol−1)c |
Thermal transition T (℃)d |
Δ Hsi (J·g−1)g |
Td (℃)h |
| 1 | PEA-2T | 90 | 47 | 28400 | G 16.3 Cr 153.7 I e I 134.0 Cr 10.5 G f |
31.3 -26.4 |
288 |
| 2 | PEA-4T | 91 | 48 | 30900 | G 12.6 Cr 143.5 I e I 114.8 Cr 4.5 G f |
29.4 -24.4 |
292 |
| 3 | PEA-6T | 90 | 49 | 32900 | G 8.3 Cr 133.4 I e I 99.8 Cr 1.4 G f |
28.6 -25.5 |
294 |
a All the main-chain azo polymers were obtained via Michael addition polymerization of the azo monomer and different chain lengths of dithiol with DBU as the catalyst at 60 ℃ in THF. b Degree of polymerization calculated by 1H NMR. c The number-average molecular weight determined by 1H NMR. d G = glassy, Cr = crystalline, and I = isotropic. e DSC second heating scan under nitrogen (10 ℃ min−1). f DSC first cooling scan under nitrogen (−10 ℃ min−1). g Enthalpy of the phase transitions. h The temperature at 5% weight loss of the polymer under nitrogen determined by TGA heating experiment (10 ℃min−1).
Table 2.
Mechanical properties of main-chain azobenzene polymer films and photoinduced stress of uniaxially oriented films.
Table 2.
Mechanical properties of main-chain azobenzene polymer films and photoinduced stress of uniaxially oriented films.
|
Entry |
Sample |
Elastic modulus (MPa) |
Yield strength (MPa) | Rupture strength (MPa) |
Elongation at break (%) | Toughness (MJ/m3) | Photoinduced stress (kPa) |
| 1 | 1,2-film | 179.2 ± 4.2 | 12.6 ± 1.1 | 12.6 ± 0.7 | 472.9 ± 28.7 | 53.7 ± 8.4 | 582.7 ± 43.1 |
| 2 | 1,4-film | 197.5 ± 3.2 | 14.8 ± 0.9 | 16.1 ± 1.1 | 547.6 ± 22.7 | 72.7± 5.2 | 722.5 ± 48.6 |
| 3 | 1,6-film | 210.3 ± 4.0 | 16.9 ± 1.1 | 22.1 ± 2.1 | 632.9 ± 15.4 | 108.1 ± 2.3 | 835.9 ± 48.2 |
Table 3.
Light induced bending/non bending of uniaxially oriented azobenzene polymer PEA-nT films under different environments and temperatures.
Table 3.
Light induced bending/non bending of uniaxially oriented azobenzene polymer PEA-nT films under different environments and temperatures.
| Entry | Sample | Environment | Temperature (℃) | Induced bending time (s) a | Bending amplitude L(mm) b |
Cycling number (times) c |
|
| UV | Vis | ||||||
| 1 | PEA-2T | Air | 25 | 12 | 110 | 6.8 ± 0.2 | >100 |
| 2 | PEA-4T | Air | 25 | 10 | 90 | 7.0 ± 0.3 | >100 |
| 3 | PEA-6T | Air | 25 | 5 | 55 | 7.9 ± 0.4 | >100 |
| 4 | Water | 25 | 6 | 60 | 7.9 ± 0.3 | >100 | |
| 30 | 6 | 55 | 7.6 ± 0.2 | >100 | |||
| 40 | 5.5 | 51 | 7.6 ± 0.3 | >100 | |||
| 50 | 4 | 46 | 7.5 ± 0.5 | ~40 | |||
| 5 | Methanol | 25 | 6 | 65 | 7.8 ± 0.2 | >100 | |
| 6 | Ethanol | 25 | 6.5 | 73 | 7.8 ± 0.2 | >100 | |
| 7 | N-butanol | 25 | 8 | 90 | 7.6 ± 0.2 | ~40 | |
| 8 | (0.9 wt.%) NaCl | 25 | 5.5 | 55 | 7.6 ± 0.2 | >100 | |
| 50 | 3.5 | 42 | 7.1 ± 0.6 | ~55 | |||
| 9 | (3.5 wt.%) NaCl | 25 | 6 | 65 | 7.7 ± 0.2 | >100 | |
| 50 | 4 | 33 | 6.8 ± 0.7 | ~60 | |||
a Induced bending time of PEA-nT films were tested under ultraviolet (λ = 365 nm, 40 mW cm-2) and visible light irradiation (λ > 510 nm, 30 mW cm-2). b Bending amplitude was calculated according to its uniform cycle times, which are 40 times for 50 ℃ water, 40 times for N-butanol, 40 times for 0.9 wt% NaCl, and 60 times for 3.5 wt% NaCl. c Cycling number was calculated according to its uniform cycle times. If the cycle exceeds 100 times, the results of 100 times will be used for calculation > 100.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



