
Submitted:
30 April 2025
Posted:
02 May 2025
You are already at the latest version
Abstract
Cyclic di-GMP (bis-(3′ → 5′) cyclic dimeric guanosine monophosphate) is a ubiquitous bacterial second messenger that regulates a wide range of cellular processes, including biofilm formation, motility, virulence, and environmental adaptation. Its intracellular levels are dynamically controlled by diguanylate cyclases (DGCs), which synthesize c-di-GMP from GTP, and phosphodiesterases (PDEs), which degrade it into linear pGpG or GMP. The functional effects of cytoplasmic c-di-GMP are mediated through diverse effector proteins, including PilZ domain-containing receptors, transcription factors, and riboswitches. In Leptospira interrogans, a major pathogenic species responsible for leptospirosis, the regulatory roles of c-di-GMP remain poorly understood. Here, we performed a comprehensive bioinformatics and structural analysis of all predicted c-di-GMP related proteins in L. interrogans serovar Copenhageni strain Fiocruz L1-130. Our analysis identified 17 proteins containing GGDEF domain, five proteins containing both GGDEF and EAL domains, four proteins containing EAL domain, five proteins containing HD-GYP domain, 12 proteins containing PilZ domain, and one protein containing an MshEN domain. Comparative analysis with well-characterized bacterial homologs suggests that L. interrogans possess a complex c-di-GMP network, likely involved in modulating biofilm formation, host-pathogen interactions, and environmental survival. These findings provide new insights into the c-di-GMP regulatory network and on signal transduction in Leptospira and lay the foundation for future functional studies aimed at understanding its role in physiology, virulence and persistence.
Keywords:
GGDEF
; PilZ
; EAL
; HD-GYP
; c-di-GMP receptors
1. Introduction
Leptospirosis is a globally re-emerging zoonotic disease caused by pathogenic bacteria of the genus Leptospira, which infect mainly mammals, including humans [1]. Leptospira interrogans is the predominant pathogenic species in the genus, responsible for most human leptospirosis cases and the most severe clinical manifestations of the disease [2,3,4,5]. Leptospirosis symptoms are nonspecific and often mimic other tropical diseases such as dengue, influenza, malaria, rickettsiosis, acute Chagas disease, toxoplasmosis, COVID-19, and typhoid fever [6]. The clinical presentation varies widely, ranging from asymptomatic or mild cases—characterized by fever, myalgia (particularly calf’s pain), headache, nausea/vomiting, diarrhea, arthralgia, conjunctival redness, photophobia, and occasionally rash—to severe manifestations like Weil’s syndrome, which involves jaundice, acute kidney injury, hepatic dysfunction, meningitis, pulmonary hemorrhage, Acute Respiratory Distress Syndrome (ARDS), hypotension, and potentially fatal multi-organ failure [6,7,8].
Leptospirosis is more prevalent in tropical regions, where warm climates and heavy rainfall create favorable conditions for its transmission. However, leptospirosis is also re-emerging in Europe and other countries in the Northern Hemisphere [9,10]. These spirochetes are maintained in nature through chronic renal colonization of asymptomatic reservoir hosts, particularly rodents, which continuously excrete viable bacteria into the environment via urine. Upon release, Leptospira can persist in moist soil and aquatic environments for several weeks and, under certain conditions, may exhibit limited replication. Environmental transmission plays a central role in the epidemiology of leptospirosis, with outbreaks frequently associated with heavy rainfall, flooding, and other extreme weather events that increase human exposure to contaminated water [1]. Climate change has intensified these conditions, with rising temperatures and increased rainfall frequency and intensity—especially in tropical areas—further facilitating the spread of the bacteria. An example of this trend is the situation in Rio Grande do Sul (Brazil) in 2024, where extreme flooding affected approximately 96% of the state’s municipalities and displaced 600,000 people. That year, the state recorded 1,313 cases of leptospirosis, resulting in 53 deaths—a 2.7-fold increase in infections and double the number of fatalities compared to 2023. These deaths accounted for approximately 14% of all fatalities linked to the 2024 flooding and disease outbreak in Rio Grande do Sul [11,12,13,14].
The adaptation mechanisms used by Leptospira while exposed to the environment remain poorly understood. Recent efforts have focused on elucidating the biological and environmental factors that contribute to the survival, dissemination, and infectivity of these pathogens outside the host [15,16,17,18,19,20]. Davignon and co-authors showed a global transcriptomic response of L. interrogans, gene expression levels of a planktonic phase and biofilm mature. This study revealed an important modulation of the expression of c-di-GMP related genes in two growth conditions, clarifying the importance of c-di-GMP in L. interrogans biofilm [1]. In addition, the increase in c-di-GMP triggers biofilm formation in L. interrogans and promotes increased survival to environmental stress [16]. Xiao and collaborators showed that shift from lower to higher temperature resulted in low c-di-GMP concentration in L. interrogans and most of the c-di-GMP metabolic genes exhibited differential temperature regulation. This was the first work to demonstrate the relevance of c-di-GMP networks in the environmental adaptation for this bacterium. Furthermore, infection of murine J774A.1 macrophage-like cells led to a reduction in intracellular c-di-GMP levels, despite the absence of significant transcriptional changes in genes involved in c-di-GMP metabolism during the course of infection [18]. Such results suggest changes in c-di-GMP levels probably allows these bacteria to better adapt to different complex microenvironments, such as in the environment and infecting mammalian hosts [15,16,18]. However, despite these intriguing observations, the complete repertoire of c-di-GMP-related proteins in L. interrogans remains uncharacterized, leaving significant gaps in our understanding of how this pathogen regulates its virulence and environmental persistence.
In bacteria, c-di-GMP is a ubiquitous second messenger that regulates diverse cellular processes [21,22]. In general, elevate c-di-GMP intracellular concentrations promote biofilm formation through extracellular matrix production, while low levels enhance motility via flagellar activation [23,24]. Such precise control of bacterial behavior makes c-di-GMP signaling crucial for environmental adaptation and pathogenicity [19,22]. The c-di-GMP regulatory network operates through three main components: diguanylate cyclases (DGCs) containing GGDEF domains that synthesize c-di-GMP from two molecules of GTP, while phosphodiesterases (PDEs) with EAL or HD-GYP domains degrade c-di-GMP to pGpG or GMP, respectively [19,22,25,26] (Figure 1). These enzymes work with diverse effectors that translate c-di-GMP signals into cellular responses, such as riboswitches, PilZ proteins and other c-di-GMP binding macromolecules present in transcription factors, enzymes, and other targets [20,22,26,27]. The PilZ domain was the first protein domain identified as a c-di-GMP receptor and remains the most well-characterized to date [19,20,22]. It can play a key role in coordinating the transition to biofilm formation and the expression of virulence factor. In pathogens like Pseudomonas aeruginosa, the production of c-di-GMP controls virulence factor expression and antibiotic resistance mechanisms [28], highlighting its clinical relevance.
Herein, we present a comprehensive bioinformatic and structural characterization of the c-di-GMP signaling network in L. interrogans serovar Copenhageni strain Fiocruz L1-130. Through systematic analysis of protein domains and comparative genomics, we identified: 17 GGDEF-containing proteins (putative DGCs), four EAL and five HD-GYP domain proteins (putative PDEs), five GGDEF-EAL hybrids, and 13 potential receptors (12 proteins containing PilZ domains and one protein containing an MshEN domain). This study provides a systematic and comprehensive characterization of the c-di-GMP signaling repertoire in L. interrogans, establishing a molecular framework for future mechanistic studies of c-di-GMP-mediated regulatory networks in this neglected zoonotic human pathogen.
2. Materials and Methods
2.1. Potential Proteins Involved in c-di-GMP Signaling in L. interrogans: Bioinformatic Analysis and Structural Prediction Models
To identify proteins associated with c-di-GMP signaling in the Leptospira genus, a systematic search was conducted between February and March 2024. Searches were performed using the UniProt database 2 [29], employing relevant keywords (Table S1) associated with c-di-GMP signaling domains, including GGDEF, EAL, HD-GYP, PilZ, and MshEN, in combination with the term “Leptospira”. This yielded 5,359 protein sequences that potentially contain domains associated with c-di-GMP signaling. Additionally, complementary searches were conducted in the NCBI c-di-GMP database [22,30,31,32] to identify genes related to c-di-GMP signaling that have already been characterized or annotated within the Leptospira genus. Following the construction of the database, a BLASTp [33] was performed using the genome of L. interrogans serovar Copenhageni strain Fiocruz L1-130 (Lic) (accessible at NCBI Genome ID 179) [34] as the query. The search was conducted against the identified proteins, with an e-value threshold less or equal than 10⁻⁵ and sequence identity equal to or higher than 40%. Based on the database of potential proteins identified in Lic containing PilZ, MshEN, GGDEF, EAL, and/or HD-GYP domains, three-dimensional structure predictions were performed for all proteins using AlphaFold (version 3) [35]. Subsequently, the predicted structures were evaluated for their association with c-di-GMP signaling proteins by identifying homologs through searches conducted in FoldSeek, DALI, or HHpred server databases [36,37,38]. The analysis of domains was performed by Conserved Domain Database (CDD) [39]. Additionally, alternative domain analysis methods and structural similarity assessments were employed to identify other domains present in these proteins.
2.2. Functional and Structural Characterization of Identified Proteins: An Analytical Approach
2.3. Multiple Amino acid Sequence Alignment using Three-dimensional Structure Predictions
Multiple sequence alignments (MSAs) were performed using the predicted tertiary structures of each domain generated by the AlphaFold (version 3) [35] as an input for the DALI server [37] to obtain structure-based alignments. MSAs were visualized and curated using the Jalview program (version 2.11.2.7) [45]. Each domain was compared to well-characterized reference proteins to infer their potential functional characteristics. The GGDEF domain was compared to PleD (locus_tag CCNA_02546, PDB 1W25 [46]) from Caulobacter vibrioides (synonym C. crescentus). The EAL domain was compared to the RocR (locus_tag PA3947, PDB 3SY8 [47]) from P. aeruginosa, and HD-GYP domain was referenced to PmGH (locus_tag PERMA_0986, PDB 4MDZ [48]) from Persephonella maritima. For the PilZ domain, the comparison was performed with MapZ (locus_tag PA4608, PDB 5XLY [49]) from P. aeruginosa [50], and the MshEN domain was compared with VcMshEN (locus_tag VC_0405) from Vibrio cholerae [32].
2.4. Graphical, Structural, and Imaging Software tools
The graphs presented in this study were generated using the R software (version 4.4.1) [51] employing the packages BiocManager [52], ComplexHeatmap [53], and ape 5.0 [54]. The images were constructed using the Inkscape program (version 1.3.2) [55] and protein visualization and editing were performed using the Pymol software (version 3.0, Schrödinger, LLC) [56] or Chimera X (1.18) [57].
3. Results
3.1. Identification of c-di-GMP-Related Genes in L. interrogans
To elucidate the c-di-GMP signaling network in L. interrogans serovar Copenhageni strain Fiocruz L1-130 (Lic), we conducted a systematic search of the UniProt database using domain-specific keywords (GGDEF, EAL, HD-GYP, PilZ, and MshEN) alongside the term “Leptospira” in order to identify candidate proteins potentially involved in c-di-GMP signaling (Supplementary file S1). This search yielded 5,359 protein sequences. Complementary searches in the NCBI c-di-GMP database did not yield additional annotated proteins within the Leptospira genus, suggesting our initial dataset was comprehensive. We then performed a BLASTp analysis of these candidates against the Lic genome, applying stringent thresholds (e-value ≤ 10⁻⁵, identity ≥ 40%), which narrowed the pool to 4,913 potential homologs. Domain validation was achieved through a combination of AlphaFold-predicted structures and FoldSeek-based structural analysis, followed by manual inspection. Through our comprehensive bioinformatics approach (Figure 2), we identified and confirmed the presence of at least 44 proteins in the Lic genome that contain the target domains associated with c-di-GMP signaling (Figure 3). Notably, seven previously uncharacterized proteins (LIC_10321, LIC_10122, LIC_11167, LIC_11247, LIC_11447, LIC_11706, and LIC_20136) and two misannotated domains (LIC_20106 and LIC_20198) were identified. One putative PilZ domain protein (LIC_11628) was excluded due to lack of structural homology with the PilZ domain based on analysis performed using FoldSeek and DALI server.
The proteins were classified by domain composition: 17 contained GGDEF domains, four possessed EAL domains, five contained both GGDEF and EAL domains, five harbored HD-GYP domains, 12 featured PilZ domains, and one carried an MshEN domain. Structural analysis using AlphaFold, FoldSeek, and the DALI server revealed conserved folds and potential functional diversification. Further characterization via UniProt, CDD, DeepTMHMM, and SignalP-5.0 provided insights into domain organization, transmembrane topology, and secretion signals. These results significantly expand the catalog of c-di-GMP-related proteins in L. interrogans, highlighting previously unrecognized components. The integration of structural prediction and multi-database annotation offers a robust framework for future experimental studies targeting specific domains or proteins.
3.3. Diversity of Sensor Domains Present in c-di-GMP-Related Proteins
Proteins involved in c-di-GMP signaling in general respond to diverse environmental and intracellular stimuli, modulating their activity to promote survival through behavioral or physiological adaptations [26]. To ensure rapid and efficient responses, sensor or signaling domains are frequently associated with c-di-GMP-related proteins, regulating enzymatic or receptor functions via signal transduction pathways [58]. While various signal transduction domains are associated with c-di-GMP signaling proteins, the occurrence of c-di-GMP-related genes without sensor domains is rare; nonetheless, these genes may remain functional [59]. To define the sensor domains associated with c-di-GMP-related proteins in Lic, we generated structural models of these domains separately using AlphaFold. The resulting structures were analyzed using FoldSeek, DALI server, or HHpred in order to characterize the domain or infer its function based on three-dimensional structural homology. The identified sensor and signaling domains present in Lic are summarized in Table S3.
3.4. Diguanylate Cyclases
The GGDEF domain is a highly conserved bacterial enzyme that functions as a diguanylate cyclase enzyme (DGC), catalyzing the synthesis of the secondary messenger cyclic di-GMP (c-di-GMP) from two guanosine-5’-triphosphate (GTP) molecules, via a bi-ter reaction yielding one c-di-GMP and two pyrophosphates. Catalysis requires dimerization to form an active site where the GG(D/E)EF motif (comprising Gly-Gly(Asp/Glu)-Glu-Phe) creates an active site that enables nucleophilic attack on the α-phosphate of each GTP substrate. Structurally, the domain adopts an α/β fold (α0-β1-α1-α2-β2-β3-α3-β4-β5-β6-α4-β7 topology) with five central β-strands flanked by five α-helices [17,19,22,26]. The GG(D/E)EF motif is located in the loop between β2 and β3 of the domain. The glutamic acid residue in this motif plays a key role in catalysis by binding the α-phosphate group of the GTP molecule and coordinating one of the cations in the binding site. In the case of the PleD GGDEF domain of C. crescentus, the catalytic site accommodates two magnesium cations, which are coordinated by E370, D327 (both form the GG(D/E)EF motif), and the backbone of I328. These metal ions are essential for stabilizing the negative charges on the phosphate groups during the catalysis process. The N335 and D344 residues are involved in binding the guanine base of the GTP substrate, contributing to substrate specificity. Meanwhile, the side chains of L294, L347, E370, K442, R446, and the backbone residues of F330, F331, and K332 interact with the phosphate groups of the GTP molecule, further stabilizing the transition state to form the c-di-GMP product [26,60,61].
GGDEF domain activity is regulated through two primary mechanisms: (1) upstream sensor domains that often mediate dimerization, and (2) allosteric inhibition by c-di-GMP binding. The dimeric enzyme contains two symmetrical allosteric sites (I and I’), each composed of four critical residues: the RxxD motif (R359/D362) and R390 from one monomer. The fourth residue comes from the adjacent GGDEF monomer (e.g., R313 in PleD) [26,60]. When c-di-GMP binds these sites, it crosslinks and immobilizes the GGDEF dimer in an inactive conformation. This noncompetitive product inhibition prevents GTP hydrolysis, thereby controlling cellular c-di-GMP levels. A subclass of GGDEF enzymes, referred to as Hybrid promiscuous (Hypr) GGDEF enzymes, exhibits broader substrate specificity. These enzymes predominantly produce cGAMP (cyclic GMP-AMP), but they can also generate c-di-AMP and c-di-GMP depending on active site modifications. The ability to synthesize multiple cyclic nucleotides is attributed to a specific residue substitution: in the case of the PleD protein, the aspartate residue at position D344 (located within the α2 helix) is replaced by a serine, which affects the enzyme’s substrate preference by altering how the guanine base of GTP is recognized and bound. Some GGDEF domains have become catalytically degenerate while acquiring novel functions: a protein from C. crescentus (locus_tag CC_3396) acts as a GTP sensor that allosterically activates an adjacent EAL domain’s phosphodiesterase activity, while B. subtilis YybT has evolved ATPase activity, completely diverging from c-di-GMP synthesis. Moreover, the YybT that has a PAS-GGDEF-DHH-DHHA1 domain architecture also clives c-di-AMP and c-di-GMP by the DHH-DHHA1 domains [62]. These examples demonstrate the remarkable functional plasticity of GGDEF domains in bacterial signaling networks [22,62].
Our structural and functional analysis of the 22 putative diguanylate cyclases identified in Lic revealed 17 proteins containing only the GGDEF domain and five proteins with both GGDEF and EAL domains (Figure 3). Using AlphaFold, we predicted the protein structures and isolated the GGDEF domains with PyMOL for further analysis. From the protein sequences containing the GGDEF domain (as described in Materials and Methods), we predicted the protein structures using AlphaFold. Subsequently, we isolated the GGDEF domain by removing other domains using the PyMol program. Structural alignment against the reference GGDEF domain from C. crescentus PleD, via DALI server [46], demonstrated conservation of key catalytic residues, including the GG(D/E)EF motif (positions 368-372), metal-coordinating residues (D327, E370), substrate-binding residues (N335, D344), and transition-state stabilizers (L294, L347, K442, R446). The allosteric regulation sites (RxxD motif, R390, R313) were also preserved in most proteins, though some showed variations suggesting functional divergence. Figure 4 highlights these structural alignments, emphasizing both the conserved features and unique adaptations among Lic GGDEF domains compared to PleD. This comprehensive analysis provides insights into the potential catalytic activity and regulatory mechanisms of these proteins in L. interrogans. The residue numbers are from PleD protein from C. crescentus. Figure 4 illustrates the GGDEF domain alignment, highlighting conserved residues essential for catalytic activity, relative to the reference protein PleD.
Based on this analysis, 13 out of the 22 proteins containing the GGDEF domain are possibly functional, as they retain the key residues essential for enzymatic catalytic activity. This includes the enzymes within the PAS-GGDEF cluster (LIC_11125, LIC_11126, LIC_11127, LIC_11128, LIC_11129, LIC_11130, and LIC_11131), as well as LIC_11300, LIC_11444, LIC_12273, LIC_13137Lcd1, LIC_20181, and LIC_20182 (Figure 4). Absence of the catalytic residues would render these proteins to be considered degenerated. Still, it is notable that the remaining proteins, including LIC_10321, LIC_11167, LIC_11247, LIC_11706, and all proteins containing both GGDEF and EAL domains, LIC_12505, LIC_13120, LIC_20106, LIC_20180, and LIC_20196, lack the residues necessary for catalytic activity and are therefore probably unable to synthesize c-di-GMP.
Analysis of the allosteric site revealed that the consensus sequence RxxD and the corresponding R390 of PelD are present in most of the canonical GGDEF proteins from Lic, with exception of LIC_12273 that has a degenerated allosteric site. Nevertheless, Lic proteins that have the two symmetrical allosteric sites (I and I’ sites) of are in LIC_11300, LIC_11444, LIC_11706, and LIC_13137Lcd1. Based on this analysis, probably 12 out of 13 potential active DGCs have an allosteric regulation. Among the proteins containing the GGDEF domain and predicted to be inactive, we analyzed the presence of the allosteric site to assess their potential as c-di-GMP receptors rather than diguanylate cyclases (DGCs). Notably, LIC_11706 retained a conserved allosteric site, suggesting a possible role as a c-di-GMP receptor (Figure 4). Interestingly, the structural similarity matrix obtained from the structural models of the 22 GGDEF domains (Figure S1A) revealed that the GGDEF domain structures of the paralogs LIC_11125–LIC_11131 are closely related, as are LIC_20181–LIC_20182 and LIC_12505–LIC_20180. In contrast, two groups of paralogs—LIC_13120 with LIC_20198, and LIC_11167 with LIC_12273 and LIC_13137Lcd1—did not cluster together, suggesting that these paralogs have already diverged.
3.5. Proteins Containing EAL Domains
The Lic genome encodes two types of phosphodiesterases (PDEs) capable of degrading cyclic di-GMP (c-di-GMP): (1) those with EAL domains and others with HD-GYP domains [63]. EAL domain-containing proteins are the most extensively studied and best-characterized PDEs, compared to proteins containing HD-GYP domain [25,61,64,65]. EAL domain-containing proteins primarily function as phosphodiesterases, catalyzing the degradation of c-di-GMP into linear 5’-pGpG (PDE-A activity). While some EAL domains may exhibit weak PDE-B activity (converting pGpG to GMP), this secondary function occurs at rates considered physiologically insignificant in most bacteria [61,66,67]. Complete c-di-GMP degradation therefore typically requires two enzymatic steps: initial cleavage by PDE-As followed by pGpG hydrolysis by dedicated PDE-Bs enzymes like oligoribonuclease Orn (e.g., VC0341 in V. cholerae containing the RNase_T domain) [68]. Interestingly, accumulated pGpG can inhibit PDE-A activity, making PDE-B enzymes crucial for maintaining c-di-GMP homeostasis. Notably, our analysis revealed no RNase_T domain-containing proteins in the L. interrogans genome, suggesting alternative pGpG degradation mechanisms may operate in this organism. It is possible that proteins containing HD-GYP domains may hydrolyze pGpG in two GMP to complete the second step in Leptospira genus. Another possibility is that other proteins not yet described have the PDE-B activity in this group of bacteria.
A comparison with the RocR protein from P. aeruginosa, a model for the EAL domain [69], reveals that this domain is characterized by the ExL motif, residues for binding divalent cations (Mg2+ or Mn2+), catalytic residues necessary for c-di-GMP cleavage, including glutamic acid (Glu352), and loop 6 19,22,61, [69,70,71]. Loop 6, containing the DFG(T/A)GYSS motif, functional active site loop (loop 6) not only mediates the EAL dimerization but it is also important for regulation of substrate and cofactor binding, and it is therefore essential for enzymatic activity 69,72. The three-dimensional structure of EAL domain-containing proteins adopts a modified TIM-barrel (β/α)₈ fold [61,72]. The enzymatic activity of these phosphodiesterases is in general inhibited by ions of Ca²⁺ and Zn²⁺ [22,61,64,71]. EAL domain proteins exhibit activity as monomers, dimers, and oligomers in vitro assays [22,64,69,71,72,73]. However, homodimer formation is likely the predominant state in vivo, as it enhances protein stability and it is crucial for PDE activity in response to environmental stimuli [61,64,69,74].
Through searches in the NCBI database looking for proteins containing EAL domains in the genome of Lic as well as in genomes of other Leptospira species, we found nine proteins containing the EAL domain. Of these, five are proteins containing both GGDEF and EAL domains (Figure 3). Using the RocR protein from P. aeruginosa as a reference, only LIC_20198 has the ExL motif degenerated. Nevertheless, LIC_10996 does not have the serine located in the Loop 6 (S302 in RocR), a residue important for enzyme activity (Figure 5). Based on RocR, mutations at S302 for an alanine abolished the enzyme activity, as this residue interacts with glutamic acid at position 268, stabilizing loop 6 and enabling catalysis [61,69,70]. All identified enzymes appear capable of binding c-di-GMP, except LIC_20198, which does not have most of the important residues for c-di-GMP binding and catalysis (Figure 5). These alterations may disrupt substrate binding and, consequently, the enzyme’s catalytic function [69,70]. Therefore, eight out of nine proteins containing the EAL domain have the most residues involved in PDE-A activity and they are probably functional, while LIC_20198 is a degenerated EAL domain. LIC_12505 and LIC_20180 have YQG instead of YQP motif (YQP in RocR, important to substrate interaction). LIC_12505 and LIC_20180 have alanine and serine instead of glutamic acid (E355 in RocR, important to magnesium interaction), respectively. These modifications may not affect the PDE-A activity since the other residues are conserved (Figure 5).
Interestingly, the structural similarity matrix derived from the models of the nine EAL domains (Figure S1B) revealed that, unlike GGDEF domains, EAL domain structures do not cluster with proteins that share the same domain architecture. Notably, the EAL domain of LIC_20198 shares greater structural similarity with LIC_10996, whereas LIC_13120 is more structurally similar to LIC_11203. The Leptospira proteins containing EAL domains that share the same domain architecture are: (i) LIC_10641 and LIC_11921, which possess an EAL-YkuI_C domain architecture; (ii) LIC_20198 and LIC_13120, which feature a GGDEF-EAL domain architecture; and (iii) LIC_12505 and LIC_20180, which exhibit a REC-GGDEF-EAL domain architecture (Figure 3).
3.6. The Proteins Containing HD-GYP Domain Characterization
The genome of Lic encodes four annotated HD-GYP domain-containing proteins: LIC_10138, LIC_10139, LIC_11189, and LIC_11563. A fifth protein was identified through a BLASTp search in the NCBI database using the Leptospira biflexa LEPBI_I1560 HD-GYP protein as a query, revealing the protein LIC_10122. To facilitate sequence comparison, structural alignments of the HD-GYP domains were performed using Foldseek, with AlphaFold-predicted models as input. The HD-GYP domain from the P. marina PmGH protein (PDB 4MDZ [48]) served as the reference structure for a canonical HD-GYP domain.
The catalytic HD-GYP domain belongs to the HD domain superfamily and features a characteristic five α-helix core (α6–α10), which provides the structural framework for coordinating a bi or trimetal center. In PmGH protein this coordination is mediated by eight conserved side-chain residues: E185, H189, H221, D222, H250, H276, H277, and D305, with K225 also contributing to the stabilization of the trinuclear iron center. The HD motif (H221 and D222), a defining feature of this domain, is located on helix α7. Additionally, the HD-GYP domain includes two extra C-terminal helices (α11 and α12), which allows it to pack against helices α6 and α10. The loop region connecting helices α9 and α10 contains the conserved GYP motif, with Y285 positioned towards the metal-binding center within this loop. A sequence-based conserved motif in HD-GYP proteins identified as HHExxDGxGYPxxxxxxxI, which includes a conserved isoleucine residue (I294 in PmGH) [48]. The c-di-GMP substrate interacts with the middle iron site (Me-site) via one of its non-bridging phosphate oxygens and the hydroxyl group of Y285, the signature tyrosine of the GYP motif. Additional interactions occur with the side chains of K235, R314, and K317, as well as residues G284, P286, and I294. Mutational analysis revealed that substitutions at key metal-coordinating residues (H189A, H221A, D222A, H250A, H276A, and H277A) significantly reduced phosphodiesterase activity, while mutations at E185A and D305A reduce the c-di-GMP hydrolysis. Interestingly, mutations in the GYP motif and conserved residues involved in c-di-GMP recognition (G284A, Y285A, P286A, I294A, R314A, and K317A) did not substantially affect catalytic activity. However, substitutions in conserved residues near the metal center (D183A, D308A, and K225A) significantly disrupted PDE function.
Among the five proteins, only LIC_11189 retains the conserved residues characteristic of a canonical HD-GYP domain. LIC_11563 has most of the key residues suggesting a potential PDE (Table S4). In contrast, LIC_10138, LIC_10139, and LIC_10122 exhibit significant substitutions in catalytic residues, potentially impairing metal-binding capacity and phosphodiesterase activity. Nevertheless, it has been described that HD-GYP domains may accept more diversifications in the key residues without losing its enzymatic activity (Table S4). Notably, none of the HD-GYP proteins, including LIC_11189, possess the essential glutamate required for coordinating the third metal ion in a trinuclear catalytic center, suggesting that LIC_11189 may be restricted to a bimetal catalytic mechanism [48,75]. Furthermore, except for LIC_11189, the other HD-GYP proteins lack key residues necessary for substrate interaction, indicating that they are not only catalytically inactive but also unlikely to interact with c-di-GMP molecules (Figure 6A and Table S4). The five HD-GYP proteins share a three-domain architecture with distinct variations. LIC_10122, LIC_10138, LIC_10139, and LIC_11563 harbor the HD-GYP domain as the central domain, flanked by N- and C-terminal domains. In contrast, LIC_11189 localizes its HD-GYP domain to the C-terminal region, preceded by two sensory domains: REC and GAF (Figures 3 and S2). LIC_10138 and LIC_10139 are highly similar in sequence and structure (Figure 6B and Figure S1C), consistent with a recent gene duplication event. In contrast, LIC_10122 is the most divergent, displaying the lowest sequence similarity to the other four proteins (Figure S1C). LIC_10138 and LIC_10139 each contain two additional helices immediately following the mutated GYP motif. LIC_10122, however, harbors a poorly conserved GYP motif within a long, flexible C-terminal loop, with minimal structural similarity to PmGH (Figure S2). Interestingly, LIC_11189 features a unique insertion of a subdomain comprising two long helices, two short helices, and one hairpin in the middle of the domain, further distinguishing it from the other HD-GYP proteins (Figure 6B and Figure S3).
3.7. The Insertion in the HD-GYP of LIC_11189 Is Widely Distributed in Response-Regulator HD-GYP Proteins
LIC_11189, the unique canonical HD-GYP protein in L. interrogans, is characterized by a REC-GAF-HD-GYP domain architecture. Foldseek and DALI searches using the HD-GYP insertion (LIC_11189 residues K416 to D536) as a query failed to identify homologous structures in the PDB, yielding only low-confidence matches aligning primarily with the two long helices. For example, DALI identified succinate dehydrogenase 2 from Mycobacterium smegmatis (PDB 6LUM [76]; z-score of 7.0 and RMSD equal to 2.5 Å), while FoldSeek PDB100 matched the RNAP-SutA complex from P. aeruginosa (PDB: 7XL3 [77]; probability and e-value of 0.25 and ~9.1). In contrast, Foldseek AFDB50 revealed highly similar structures across diverse bacterial species, with the insertion consistently embedded within HD-GYP domains. Strikingly, despite using only the insertion sequence, most retrieved proteins exhibited a domain architecture closely resembling LIC_11189, spanning bacteria beyond the Spirochaetia phylum. In phylogenetically distant species, including Vibrio mediterranei, Vibrio variabilis, Paraburkholderia caballeronis, and Thalassomonas viridans, the insertion contained an extended flexible loop between the two short β-strains that forms a hairpin (Figures S3 and S4). The HD-GYP insertion domain was found closely associated with a preceding GAF domain, which was sometimes incomplete, and N-terminal domains that were frequently highly or moderately structurally similar to REC domains. Interestingly, this insertion appears strongly linked to HD-GYP domains fused to response regulator domains, suggesting a potential ancient role in two-component system signaling.
Sequence conservation analysis revealed conserved hydrophobic residues likely mediating apolar interactions that stabilize the connection between the two long helices with the short secondary structure elements. In addition, highly conserved polar residues are positioned near the phosphodiesterase active site, adjacent to the substrate-binding pocket. Notably, such structure has not been described in previously characterized HD-GYP proteins. This unique architecture may represent a novel mechanism for interaction with c-di-GMP or confer increased permissiveness for binding other cyclic dinucleotides (Figure S5). We named this domain the HD-GYP insertion domain (HdiD).
3.8. Potential Distant Members of DUF3391 Family
The limited information available on the DUF3391 (PF11871) domain poses challenges in elucidating its relationship with the N-terminal domain of HD-GYP-containing proteins from L. interrogans such as LIC_10122, LIC_10138, LIC_10139, and LIC_11563. According to InterPro, DUF3391 is an uncharacterized N-terminal domain frequently associated with HD-GYP proteins across diverse bacterial taxa. Among the HD-GYP proteins in L. interrogans, only LIC_11563 has its N-terminal domain annotated as DUF3391 in the CDD database. CDD and InterPro analyses failed to identify domains in the N-terminal regions of LIC_10122, LIC_10138, and LIC_10139. Notably, the DUF3391 domain, including that of LIC_11563, features a characteristic long C-terminal helix linking it to the HD-GYP domain. Excluding the HD-GYP domain, the N-terminal portions of the four proteins exhibit comparable lengths, ranging from 119 to 132 residues (Figure S6A). BLASTp searches using these N-terminal regions identified homologs exclusively within the Spirochaetota phylum, predominantly in the Leptospira genus. Matches corresponded to N-terminal regions of HD-GYP-containing proteins, but only LIC_11563 returned hits with the N-terminal domain explicitly annotated as DUF3391 in the NCBI database.
For LIC_11563 (residues M1–V131), the search identified proteins annotated with DUF3391, with high scores (probability 1.0; e-values from 1.3x10-18 to 5.7x10-3). When the full-length LIC_11563 was used as a query, the PA4108 HD-GYP protein from Pseudomonas aeruginosa PAO1, which also contains a DUF3391 domain, returned with a probability of 1.0 and an e-value of 4.3x10-20. For LIC_10122, LIC_10138, and LIC_10139, matches similarly corresponded to N-terminal regions of HD-GYP-containing proteins, although most lacked explicit annotations. Among the matches for LIC_10122, COS84_03205 encoded a standalone DUF3391 protein (not fused to an HD-GYP domain) with moderate similarity (probability 0.97; e-value 9.0x10-2). A subsequent FoldSeek AFDB50 search identified G3N55_00320 — another standalone DUF3391 protein — with a higher similarity score (probability of 1.0 and e-value of 8.6x10-8). LIC_10138 matched to E4G96_08570 from a Chrysiogenales bacterium (metagenomic data), containing a DUF3391 domain with lower similarity scores (probability=0.69 and e-value = 1.7x10-1). Using this sequence as a query, FE240_05590 from Aeromonas simiae, containing a DUF3391 domain, returned with high confidence (probability of 1.0). Due to the strong sequence and structural similarity between LIC_10138 and LIC_10139, FoldSeek results for LIC_10139 were nearly identical to those for LIC_10138, including matches to some DUF3391 domains. Topology representations of LIC_10122, LIC_10138, LIC_10139, and LIC_11563, alongside PA4108, Dvul_2450, FE240_05590, F0M16_21925, and G3N55_00320 as DUF3391 representatives, were generated using PDBsum. Owing to the low confidence of AlphaFold-predicted models, topology analysis of the L. interrogans domains was limited. Canonical DUF3391 domains exhibited 4–5 antiparallel β-strands and 1–2 α-helices within the globular domain, followed by a C-terminal α-helix. In some cases, DUF3391 domains appeared independently, without an HD-GYP domain; however, genomic analyses often revealed downstream HD domains separated by frameshift mutations, as observed in G3N55_00320, which contains a premature stop codon disrupting the HDOD domain (Figure S6B). The globular domains of L. interrogans proteins were predicted to contain 4–6 antiparallel β-strands with a C-terminal α-helix. Notably, LIC_10122 and LIC_11563 contained an additional α-helix preceding the last β-strand, while LIC_10138 and LIC_10139 exhibited topologies similar to canonical DUF3391 domains but with an α-helix preceding β4, followed by two additional β-strands before the C-terminal α-helix (Figure S6A).
A multiple sequence alignment, as described by Galperin and co-authors revealed high conservation of key hydrophobic residues among the four L. interrogans N-terminal domains and canonical DUF3391 domains (Figure S7A) [63]. Residue substitutions observed in these positions preserved their apolar characteristics, suggesting functional significance. Interestingly, despite its DUF3391 annotation, LIC_11563 clustered phylogenetically with the other L. interrogans proteins rather than with canonical DUF3391 domains (Figure S7B). The DUF3391 domain remains poorly characterized, with substantial variability across members. Structural predictions and sequence alignments suggest that many unannotated N-terminal regions fused to HD-GYP domains may belong to this family. Additionally, inspection of the domain architectures of hits InterPro for the DUF3391 Pfam model, revealed that DUF3391 always appear as the N-terminal region in proteins with fusions to other domain families, such as T2SSB, a PilP superfamily member, isolated instances of some enzymes, such as the glucose/sorbosone dehydrogenases and the thioredoxin-like alkyl hydroperoxide reductase (AhpC/TSA). Interestingly, in addition to the many fusions to HD homologs, some examples of fusions to other signal transduction effector domains are also observed, such as fusions to C-terminal SpoIIE and methyl-accepting chemotaxis proteins (MCPsignal). These patterns of C-terminal fusions suggest that DUF3391 could have a regulatory role, acting as a sensor that regulates the activity of the enzymatic/effectors domains. Still, additional, experimental and computational studies will be necessary to clarify the biological role of these domains and refine their classification. Accordingly, we propose that the N-terminal domains of L. interrogans HD-GYP-containing proteins represent a divergent subgroup within the DUF3391 family.
3.9. A novel C-Terminal Domain in HD-GYP-Containing Proteins
In addition to the N-terminal domain DUF3391 present in LIC_10122, LIC_10138, LIC_10139, and LIC_11563, all four proteins contain an unannotated C-terminal domain from 68 to 120 amino acids in length. While CDD and InterPro analyses failed to identify this region as a known domain, AlphaFold predictions yielded models with low confidence for LIC_10122E325-K433 and LIC_11563L326-A394 but high confidence for LIC_10138S371-A489 and LIC_10139L383-A50. PDBsum topology representations revealed conserved secondary structures in LIC_11563L326-A394, LIC_10138S371-A489, and LIC_10139L383-A503, characterized by 5–6 antiparallel β-strands followed by a C-terminal α-helix, forming a barrel-like structure. In contrast, LIC_10122E325-K433 is predicted to begin with a long helix preceding two pairs of β-strands (Figure S8). Dali PDB90 and Foldseek PDB100 searches using LIC_10122E325-K433 identified small structural similarities with the human cohesin-NIPBL-DNA complex (PDB 6WG3 [78], z = 3.2, RMSD = 4.0 Å) and the human inner kinetochore CCAN complex (PDB 7XHO [79], probability = 0.1, e-value = 4.3x10-1). Foldseek AFDB50 analysis identified 25 proteins, 12 with significant scores (probability = 1.0, e-value between 1.7x10-18 and 3.9x10-4), including hypothetical proteins from Leptospira species and one HD-GYP protein from Turneriella parva DSM 21527 (probability = 1.0, e-value = 2.8x10-6). Consistent with Foldseek, BLASTp analysis revealed homologs only in closely related Leptospira species, including L. interrogans, L. kirschneri, L. noguchii, L. weilii, and L. santarosai (100% query coverage, 95.4–100% identity). Residue conservation analysis of representative sequences revealed low sequence variability, limiting insights into the functional importance of specific residues (Figure S9). Based on the monomer prediction of the three-dimensional structure using AlphaFold suggested that the long N-terminal α-helix prevents direct interaction between the small globular domain and the degenerated HD-GYP domain (Figure S2).
Homology searches for LIC_11563L326-A394, LIC_10138S371-A489, and LIC_10139L383-A503 identified matches exclusively with the C-terminal regions of HD-GYP proteins from diverse bacterial species beyond the Spirochaetota phylum. Dali PDB90 analysis of LIC_11563L326-A394 returned structural matches with Pyrococcus furiosus transcription elongation factor Spt4/5 (PDB 3P8B [80]), human TDRD1 extended Tudor domain (PDB 5M9N, unpublished article ), and Saccharomyces cerevisiae Ski238 complex (PDB 8Q9T [81]) with z-score ranging from 5.8 to 5.6 and RMSDCα values ranging from 2.2 to 2.4 Å. Foldseek PDB50 analysis identified matches with the human nucleolar pre-60S ribosomal subunit (PDB 8FKR [82]) and Nanoarchaeum equitans ATP synthase core complex (PDB 5BN5 [83]) (both with probability of 0.6 and e-value of 7.3x10-1 and 3.7x10-1, respectively). For LIC_10138S371-A489 and LIC_10139L383-A503, DALI analysis returned human SGF29 in complex with R2AK4me3 (PDB 3MEV [84]) and a hypothetical protein from an uncultured marine organism (PDB 3BY7 [85]) as top results (z-score = 5.3, RMSD = 3.1 Å). LIC_10139L383-A503 yielded identical DALI matches to LIC_10138S371-A489, reflecting structural similarity. Foldseek PDB50 analysis for LIC_10139L383-A503 identified the mitochondrial ribosome from Polytomella magna (PDB 8APO [86]) as the best hit (probability = 0.84, e-value = 6.8x10-2), a match also identified for LIC_10138S371-A489, although with lower confidence (probability = 0.41, e-value of 1.4x10-1) (Figures S9 and S10).
Despite structural similarities, sequence alignments with these homologs were poor, likely due to the generic barrel-like architecture of the domain, which may contribute to false-positive homolog identification and moderate confidence scores. BLASTp analysis of LIC_11563L326-A394 revealed numerous HD-GYP proteins containing this C-terminal domain, including sequences outside the Spirochaetota phylum. Divergent sequences were manually curated, aligned using Clustal, and analyzed for residue conservation with WebLogo. Highly conserved residues were identified, with prolines (P328, P351, P354) likely disrupting secondary structures and forming flexible loops, while inward-facing apolar residues (I333, L335, V343, L375, I383) stabilized β-sheet folding through hydrophobic interactions. Conserved surface-exposed residues (R353, R357, K360, K377) formed stabilizing intradomain hydrogen bonds (R353, K360) or remained solvent-accessible, suggesting roles in protein–protein interactions or ligand binding (Figure S11).
BLASTp analysis of LIC_10138S371-A489 and LIC_10139L383-A503 identified homologs exclusively within the Leptospira genus. Forty representative hits were selected, aligned, and filtered for redundancy (95% threshold). Residue conservation analysis revealed no strong correlation with LIC_11563L326-A394, aside from conserved R-P-X-X-R and R-P-X-X-X-R motifs in LIC_11563L326-A394 and LIC_10138/LIC_10139, respectively (Figures S11–S13). All three proteins contained five β-strands with inward-facing apolar side chains, inducing curvature and forming a hydrophobic pocket within the incomplete barrel-like structure (Figure S14). Unlike LIC_10122E325-K433, which features a long N-terminal helix separating it from the HD-GYP domain, LIC_11563L326-A394, LIC_10138S371-A489, and LIC_10139L383-A503 are in close proximity to their HD-GYP domains. LIC_10138S371-A489 and LIC_10139L383-A503 also possess longer β4 and β5 strands near the degenerated c-di-GMP binding pocket (Figure S2). Absolutely conserved residues, such as K430 in LIC_10138 and K443 in LIC_10139, mediate interactions with the HD-GYP domain via hydrogen bonding with E325 and E338, respectively. These glutamic acid residues are oriented toward the c-di-GMP binding pocket, suggesting a potential role in nucleotide interaction, analogous to that proposed for LIC_11189. Additionally, a hydrophobic pocket along the extended β-sheets mediates apolar interactions with the HD-GYP domain, with absolute conservation of interacting residues suggesting their importance for structural integrity (Figure S15). Based on these analyses, the C-terminal domains of the proteins LIC_10122, LIC_10138, and LIC_11563 show divergent sequences. The models of their three-dimensional structures do not clearly display structural similarities that would allow us to infer that these domains are homologous, and the amino acid conservation patterns of each protein are not similar among them. These data do not provide robust information to discuss whether these domains are homologous. Therefore, further studies are needed to better understand the nature of these domains found in the C-terminal region of these proteins containing the HD-GYP domain of L. interrogans.
3.10. Proteins Containing PilZ Domain
The PilZ domain was one of the first specific binding domains identified for the c-di-GMP and remains among the most extensively studied c-di-GMP effector molecules [87]. PilZ domains are characterized by a closed β-barrel containing six antiparallel β-strands followed by an α-helix [21]. This unique structural architecture, encoded in most bacterial genomes, underscores the importance of the PilZ domain in regulating cellular processes mediated by c-di-GMP. Canonical PilZ domains contain two conserved motifs essential for c-di-GMP binding. The first motif, a arginine-rich sequence (RxxxR) located in a loop that precede the N-terminal regions of the domain, while the second motif, (D/N)xSxxG, lies at the end of the second β-strand [88]. Binding of c-di-GMP often induces conformational changes in the PilZ domain, facilitating interactions with other proteins or modulating cellular activities. Hydrophobic residues within the domain core are critical for maintaining structural integrity and regulating the activity of associated domains [87]. Genome analysis of Lic, using NCBI database searches, local alignments, and orthology investigations, identified 12 proteins containing PilZ domains (Figure 3). These proteins either possess isolated PilZ domains or PilZ domains coupled with additional domains. Among them, LIC_10128 emerged as a functional PilZ, containing the conserved RxxxR and (D/N)xSxxG motifs necessary for c-di-GMP interaction. In contrast, five proteins (LIC_20173, LIC_20136, LIC_12491, LIC_11993, and LIC_11447) were classified as non-functional PilZ variants due to the absence of the conserved arginines required for c-di-GMP binding (Figure 7).
Six out of twelve identified proteins are annotated in UniProt as containing the DUF1577 domain, which, through in-depth bioinformatic analyses, was revealed to be a fusion of three distinct domains: the PilZN domain (formerly YcgR_N), a GAF-like domain (known as GAZ domain), and a C-terminal PilZ domain. The PilZ domain has the presence of the GAZ domain, which has lost its dimerization capability due to the absence of the α1 secondary structure element [20]. Despite the domain insertion, the c-di-GMP binding regions remain conserved, preserving functionality. Notably, the PilZ domains in LIC_11920 [20] retain the ability to bind c-di-GMP [20]. Additionally, two novel domain architectures were identified in Lic PilZ proteins. Based on this analysis, 7 out of twelve are predicted to be c-di-GMP effectors while the other 5 are degenerated PilZ domains (Figure 7). The group of PilZ proteins with the same domain architecture are: YcgRGAZ proteins (LIC_10049, LIC_11920, LIC_12546, LIC_12723, LIC_12994, LIC_14002), and four PilZ proteins (LIC_10128, LIC_11447, LIC_11993, and LIC_12491) (Figure 3 and 7). The analysis of structure similarities showed that YcgRGAZ proteins clustered together but the PilZ proteins containing only the PilZ domain are structurally divergent (Figure S1D). LIC_20136 contains a D1 domain in the N-terminal region, a non-functional PilZ domain, and a D2 domain in the C-terminal region. In the case of LIC_20173, this protein contains an N-terminal PrsW-protease domain and a non-functional C-terminal PilZ domain. These previously unreported architectures suggest potential diversification of PilZ-related signaling mechanisms within Leptospira.
3.11. The LIC_20136 and LIC_20173 Represent Novel PilZ-like Families with Unique Domains Architecture
DeepTMHMM predictions and the top-ranked AlphaFold model for LIC_20136 indicate that this protein contains a transmembrane domain (residues 16–38), followed by a globular pentahelical domain (residues 39–131, designated as D1), which is connected to a non-canonical PilZ domain (residues 132–241) and a long C-terminal domain (residues 243–707, designated D2) (Figure S16A). Structural similarity searches of D1, conducted using the DALI, yielded borderline significant alignments (z-score less than 5.6). The closest structural match was to the C-terminal domain described as ribosome-associated complex head domain (RAC_head, PF_16717) of the Zuotin protein (PDB 7X34 [89]) with a Cα RMSD of 3.3 Å and 10% identity of sequence to this domain. The RAC_head has also been described as the C-terminal four-helix bundle (4HB) domain [90,91]. Notably, the final α-helix of D1 is absent in the 4HB domain, indicating a degree of structural divergence. The first helix of the 4HB domain mediates interactions with ES12 (helix 44 of 18S rRNA) through six lysine residues, which are not present in LIC_20136 D1 (Figure S16B). Nevertheless, the presence of positive residues in this helix, along with the multiple sequence alignment representation by WebLogo diagram shows conserved positively charged residues in this region (Figure S16D, residues in bright blue), suggests a potential RNA-binding function. YcgR homologs containing NpzN domains (“N-terminal to PilZN”), referred to as YcgRNpzN, share a conserved domain architecture. This consists of a transmembrane helix, followed by the NpzN domain—characterized by four α-helices arranged in perpendicular pairs—and the classical PilZN and PilZ domains typical of the YcgR family. Although the D1 domain resembles the NpzN domain, we do not observe the same domain architecture [20]. This discrepancy may be due to limitations in the structural prediction of the D1 domain.
In the case of the D2 domain, residues 239 to 707, searched by domain characterized using the interPro and CD-search, did not identify any domain. Searches for homology detection and structure prediction by HMM-HMM comparison (HHpred) also did not identify any domain. Structural similarity analysis using the DALI server and the structure predicted by AlphaFold observed structural similarities of residues 413 to 650 of the D2 domain with: protein Zmp1 from Clostridioides difficile (strain 630) that has the Pro-Pro endopeptidase domain described by the PFAM as ATLF domain (Anthrax toxin lethal factor, N- and C-terminal domain, PF07737, IPRO14781) with Z-score of 6.4 (PDB 6R4Z [92]); the ATLF domain of the anthrax lethal factor protein (residues 63 to 279) with Z-score of 5.8 (PDB 1JKY [93]); the ATLF domain of the certhrax toxin from Bacillus cereus, residues 2 to 226, with Z-score of 5.7 (PDB 4FXQ [94]); the Peptidase_M4 domain (Thermolysin metallopeptidase, catalytic domain) of the zinc metalloprotease ProA of Legionella pneumophila, residues from 222 to 385, with Z-score of 5.5 (PDB 6YA1 [95]); the ATLF-like domain of the Edema factor exotoxin of the Anthrax bacteria, residues 60 to 273, with Z-score of 5.5 (PDB 1XFU [96]) (Figure S16C). The active site of Zmp1 is built by the H142E143xxH146 motif, along with residues E140, W103, and Y178 (Figure S16C). However, only Y178, corresponding to Y608, is present in the D2 domain. This analysis suggests that the D2 domain is unlikely to bind zinc or function as a protease in the same manner as the Zmp1 protein. The ATLF domain of the anthrax lethal factor (LF) protein binds to the membrane-translocating component of anthrax toxin, the protective antigen (PA), which is crucial for host cell binding and facilitates the entry of LF. Notably, the ATLF domain lacks the HExxH motif required for zinc binding and protease activity. The same thing happens with the ATLF domain of the certhrax toxin from B. cereus. In the case of the toxin ProA of L. pneumophila the Peptidase_M4 domain is responsible for cleaving a broad spectrum of substrates such as casein or gelatin and promotes infection of human lung tissue. The protease activity is mediated by the same motifs as the ATLF domain, the HExxH motif and other accessory residues, most of which are absent in the D2 domain. Based on this analysis the D2 domain is homologous to the ATLF domain but lacks the residues important to zinc coordination and catalysis and may instead work, as proposed for the ATLF domain of the anthrax lethal factor (LF) protein and the certhrax toxin from B. cereus that are involved in protein-protein interactions. LIC_20136 is widely distributed within the genus Leptospira, but we did not identify it in other genera (Figure S16E).
Domain characterization using InterPro and CD-Search revealed that LIC_20173 contains a PrsW domain in its N-terminal region (residues 1–210) but no additional domains were detected. To further investigate its domain architecture, we analyzed the AlphaFold-predicted structural model of LIC_20173 using the DALI and FoldSeek server, which identified a PilZ domain in the C-terminal region (Figure S17A, with domain PrsW of LIC_20173 in pink and PilZ shows in magenta). Additionally, DeepTMHMM predictions indicated that the protein possesses nine transmembrane helices: seven within the PrsW domain and two additional transmembrane helices, spanning residues P268–P337 (Figure S17A, with the additional helices showed in blue). The only functionally characterized PrsW domain is the protein YpdC of Bacillus subtilis, which has four motifs, correspond to E75E76xxK79, followed by F110xxxE114, a conserved histidine (H138) as the third motif, and the fourth motif H175xxxD/N179 [97] (Figure S17A e B, with domain PrsW of YpdC in green). The motifs E75E76xxK79 and H175xxxD/N179 are located in a large extracytoplasmic loop. The PrsW domain of YpdC and LIC_20173 have an N-terminal domain started in periplasm, with these proteins set, probably, by an insertase protein YidC. The YidC insertase follows the positive inside rule to set the protein in the inner membrane [25].
Site-directed mutagenesis in YpdC indicates that either double point mutation of the two conserved glutamates in the first motif (E75A/E76A), or a single mutation of the conserved histidine in the fourth motif (H175A), are of functional importance. Using the YpdC of B. subtilis as a model of a PrsW domain’s active site, we see in the alignment of these two amino acid sequences (Figure S17C), that LIC_20173 has the two glutamates in the first motif (E75A/E76A), and a histidine in the fourth motif (H175A), suggesting that the PrsW domain of LIC_20173 is active. The PrsW domain of YpdC and probably in the case of LIC_20173 have an N-terminal domain starting in the periplasm, with these proteins set, probably, by an insertase protein YidC. The YidC insertase follows the positive inside rule to set the protein position in the inner membrane [25]. Probably the protein LIC_20136 assembles in the inner membrane in the same way as LIC_20173, with the N-terminal region located in the periplasm and the C-terminal domains located in the cytoplasm.
3.12. Proteins Containing the MshEN Domain in L. interrogans Serovar Copenhageni Strain Fiocruz L1-130
Through searches in the NCBI database, we searched for orthologs in the genomes of other strains, as well as species within the genus Leptospira. These analyses led to the identification of a protein containing the MshEN domain in the genome of Lic. The only protein found to contain a MshEN domain in its N-terminal region was LIC_11571. In the MshEN domain, c-di-GMP is accommodated by two motifs connected by a 5-residue linker [32] (Figure 8). Wang and collaborators studied the MshEN domain from V. cholerae (locus tag VC0405), a protein associated with the formation of the mannose-sensitive hemagglutinin type IV pilus (T4P) [32,98]. A VC0405 homolog from P. aeruginosa, PA14_29490, also contains a T2SSE ATPase domain and is involved in the type II secretion system (T2SS) [88]. Both proteins interact with c-di-GMP through their MshEN receptor domains. MshE proteins exhibit high-affinity binding to c-di-GMP, with the interaction occurring in their N-terminal T2SSE_N domains (hereafter referred to as MshEN). In contrast, the ATPase domain, which binds ATP, does not interact with c-di-GMP. Notably, residues Arg9 and Gln32 of MshEN from V. cholerae play pivotal roles in c-di-GMP binding. Indeed, crystallographic studies have revealed that MshEN contains two 24-residue motifs connected by five non-conserved residues, cooperatively forming a 53-residue domain that interacts with c-di-GMP [32]. This c-di-GMP-binding domain is found across various bacteria, often fused with ATPase, glycosyltransferase, or other domains. These proteins exhibit c-di-GMP binding affinities with dissociation constants (KD) ranging from 14 nM to 0.5 μM, highlighting MshEN as a highly sensitive c-di-GMP receptor capable of participating in diverse c-di-GMP-mediated bacterial processes [32]. The crystal structure of the MshEN-c-di-GMP complex from V. cholerae reveals two subdomains: an N-terminal MshEN_N four-helix bundle (α1-α4) and a C-terminal MshEN_C subdomain featuring antiparallel β-strands (β1-β3) flanked by three helices. MshEN_N subdomain binds c-di-GMP primarily and the D108 in MshEN_C may also contact the guanine base of c-di-GMP.
The electrostatic surface of the MshEN_N subdomain of V. cholerae is predominantly positive, composed of residues K5, R7, K8, R9, and R38, which stabilize c-di-GMP interactions via electrostatic forces while the guanine bases of c-di-GMP are stabilized by hydrophobic interactions with L25, L29, and L39. Mutation analysis showed that R9A/D12A, R88A/R89A, and D108A/D111A variants reduced c-di-GMP interactions, while R146A/R147A and E191A/D192A did not affect c-di-GMP binding. Figure 8 illustrates the conserved residues of this protein, highlighting the critical roles of R9, L14, and Q32 in c-di-GMP binding, with hydrophobic interactions and hydrogen bonds further stabilizing the complex. The LIC_11571 does not have any arginines at the motif RLG in the motif 1 and 2, suggesting that it is unlikely to function as a c-di-GMP receptor.
4. Discussion
The production of cyclic di-GMP (c-di-GMP) in bacteria orchestrates diverse physiological processes, including biofilm formation, enhanced resistance to environmental stressors, modulation of antibiotic susceptibility, and regulation of virulence [16,22,28]. The precise regulation of c-di-GMP synthesis is crucial for bacterial fitness, as it sustains a coordinated signaling network by maintaining well-defined intracellular concentrations of this second messenger, thereby preserving cellular homeostasis [100,101]. To elucidate how c-di-GMP influences the behavior of L. interrogans, it is essential to identify the proteins involved in its signaling pathways. In L. interrogans, c-di-GMP production is associated with biofilm formation, contributing to infection persistence within hosts and reservoir animals, as well as survival under adverse physical and chemical conditions, including high UV radiation, fluctuating salinity, pH variations, and antimicrobial exposure [15,16]. In this study, 44 proteins associated with c-di-GMP signaling were identified in L. interrogans, including 7 previously undescribed in the literature (Figure 3). The bacterium exhibits a high density of proteins involved in c-di-GMP metabolism, encompassing 17 proteins with GGDEF domains, four with EAL domains, five with both GGDEF and EAL domains, five with HD-GYP domains, 12 with PilZ domain, and one with MshEN domain. This corresponds to a c-di-GMP intelligence quotient (IQ) — the density of turnover domains per megabase pair (Mbp) of genome — of 6.6 (31/4.69) per Mbp, surpassing the average of many bacteria (4.69 Mbp) [61,102,103,104,105]. This high c-di-GMP IQ, suggesting that L. interrogans has a highly complex and likely finely tuned regulatory network for c-di-GMP, consistent with its need to adapt between environmental and host-associated lifestyles. The present study focused on identifying GGDEF, EAL, HD-GYP, PilZ, and MshEN domains through structural homology, leveraging their established catalytic or c-di-GMP binding residues as curated in the NCBI c-di-GMP database [22,30,31,32]. All analyses were conducted in silico, including assessments of degenerate domains. However, predicting novel receptors remains a considerable challenge using bioinformatics alone, owing to the structural and functional diversity of these receptors. Many c-di-GMP receptors lack well-defined conserved motifs, and binding can occur at multiple sites depending on the c-di-GMP receptor domain via distinct molecular interactions, complicating identification of new c-di-GMP receptors based solely on known sequences or structural templates [22,28,106].
In L. interrogans, 22 diguanylate cyclases (DGCs) have been identified, 13 are predicted to be functional based on the conservation of residues essential for enzymatic activity (Figure 4). Even degenerate GGDEF domains can act as GTP or c-di-GMP sensors or regulate associated EAL domains, despite losing catalytic activity [19,22]. Experimental validation is needed to confirm these findings since bacterial species exhibit diversity in motif functionality. Moreover, some proteins with GGDEF domain containing the canonical catalytic residues can be inactive such as GdpS from Staphylococcus aureus and S. epidemitis, c21220 and B54690 from Sinorhizobium fredii, HmsT from Yersinia pestis [22,107,108,109]. In L. interrogans, many DGCs can not bind GTP due to mutations in guanosine-binding residues but they may still function as c-di-GMP receptors or participate in protein-protein interactions. For instance, the FimX protein of Xanthomonas citri pv. citri contains EAL and GGDEF degenerate domains yet modulates pilus-mediated twitching motility by EAL c-di-GMP interaction [110]. The synthesis of c-di-GMP is often modulated by negative feedback through two inhibitory allosteric sites: the I and I’ sites. Binding of c-di-GMP to these sites immobilizes DGCs in a non-catalytic state [22,111]. Most L. interrogans DGCs appear to have functional inhibitory sites, with the exception of LIC_12273. Overall, the functionality of L. interrogans GGDEF domain proteins is shaped by a complex interplay of catalytic site conservation, motif variation, and regulatory feedback mechanisms. Experimental studies will be essential to unravel the physiological relevance of these DGCs and their contributions to bacterial signaling and adaptation.
The EAL domain is widely recognized for its role in proteins that regulate cyclic diguanylate monophosphate (c-di-GMP) metabolism. This domain is associated with phosphodiesterase activity, catalyzing the degradation of c-di-GMP into 5’-phosphoguanylyl-(3’–5’)-guanosine, which is subsequently hydrolyzed into two molecules of GMP (Figure 1). Bacteria often encode multiple proteins containing EAL domains (EXLXR motif), which may exist either as standalone proteins or fused with other signaling or output domains. This structural diversity enables EAL-containing proteins to participate in complex regulatory networks, where different domain combinations confer distinct functional properties, including sensitivity to various stimuli or interactions with other signaling molecules. Thus, EAL domains, whether isolated or combined with other domains, play diverse roles in regulating cellular processes mediated by c-di-GMP.
Genome analysis of L. interrogans identified nine EAL family members containing the conserved EXLXR and DFG(T/A)GYSS motifs, which are critical for coordinating metal ions like magnesium (Mg²⁺) or manganese (Mn²⁺). The glutamate in loop 6 and arginine residues within the EXLXR motif directly participate in this coordination, stabilizing the cofactors essential for phosphodiesterase activity [69,70]. These metal ions play a key role for catalyzing c-di-GMP hydrolysis, ensuring proper EAL domain function, and regulating intracellular signaling pathways. Using the RocR protein from P. aeruginosa as a reference, several Lic proteins appear capable of binding c-di-GMP, except LIC_20198 and LIC_10996, which does not have most of the important residues for c-di-GMP binding and catalysis (Figure 5). Interestingly, LIC_20198 carries an S302Q substitution, along with substitutions of RocR tyrosine 160 for isoleucine and arginine 179 for glycine, which may disrupt substrate binding and enzymatic activity [61,69,70]. However, transposon insertion studies by Thibeaux and co-authors revealed that disrupting the EAL domains of LMANV2_v2_270021 (LIC_20180) and LMANV2_v2_90001 (LIC_20198) increases biofilm production, suggesting these enzymes likely degrade c-di-GMP in vivo [16]. Additional functional experiments are necessary to clarify the activity and regulatory roles of these enzymes. Negative feedback regulation of c-di-GMP degradation is vital for bacterial homeostasis, maintaining appropriate intracellular concentrations of this secondary messenger. EAL domain-containing phosphodiesterases play an important role in this process by hydrolyzing c-di-GMP to pGpG, while responding to intracellular levels of pGpG, thus balancing synthesis and degradation, and preserving cellular equilibrium.
Among c-di-GMP turnover domains, HD-GYP is notably less common and less studied than EAL domains, and therefore, on occasion, described as the “neglected small sibling” [63,112]. The HD-GYP catalytic mechanism depends on di-metal or tri-metal centers coordinated by specific amino acid residues. The HxxxHDxxxHxxxHxxxD motif is essential for phosphodiesterase activity, as it organizes the dinuclear structure [75,112]. A third ion may be coordinated by an additional glutamate residue, such as E185 in PmGH [48]. In L. interrogans, none of the HD-GYP proteins contain this extra glutamate necessary for tri-metal coordination. Among them, LIC_11189 is the only protein retaining canonical residues required for di-metal center formation, whereas LIC_10122, LIC_10138, and LIC_10139 contain mutations in critical residues, such as the histidine within the HD motif. LIC_11563 has some key residues for PDE activity being therefore a possible PDE enzyme. Still, previous studies demonstrated that site-directed mutagenesis of any residue responsible for metal ion coordination abolishes phosphodiesterase activity [48]. This strongly suggests that LIC_10122, LIC_10138, and LIC_10139 are catalytically inactive due to their inability to coordinate the di-metal center. Furthermore, only LIC_11189 and LIC_11563 conserve the residues required for c-di-GMP binding, whereas LIC_10122, LIC_10138, and LIC_10139 lack these residues, rendering them incapable of binding either metal ions or c-di-GMP [63]. Notably, LIC_10122 appears to be fully degenerated as an HD-GYP protein (Figure 6 and Table S4). Interestingly, the HD-GYP proteins of L. interrogans exhibit unique features not yet described in the literature. LIC_11189, the only protein predicted to be catalytically active, contains an insertion within the HD-GYP domain that does not disrupt its folding. Highly conserved polar residues near the c-di-GMP binding pocket suggest a potentially novel interaction mechanism with this cyclic dinucleotide. Similarly, the extended β-sheets in the C-terminal domains of LIC_10138 and LIC_10139 may create a comparable effect despite the absence of canonical c-di-GMP binding residues. The presence of a highly conserved lysine residue in both proteins could implicate interactions through an new unconventional mechanism, potentially compensating for the missing binding residues. Although CDD and InterPro did not annotate LIC_11563 beyond its N-terminal domain, structural similarities suggest that all four proteins may represent a divergent group within the DUF3391 family. DUF3391, an uncharacterized domain commonly found in the N-terminal region of HD-GYP proteins, is structurally solved in Bdellovibrio bacteriovorus (PDB 3TM8 [113]). This domain features a secondary structure resembling that of the Leptospira proteins, comprising a C-terminal S-helix and a globular domain formed by four antiparallel β-strands and an α-helix preceding the final strand. Previous studies revealed that DUF3391 deletion in some HD-GYP proteins did not affect enzymatic activity and the domain does not participate in c-di-GMP binding, leaving its biological role unclear [63,114,115]. Additionally, this work identifies similar previously undescribed domains in the C-terminal regions of LIC_10122, LIC_10138, LIC_10139, and LIC_11563. Structural predictions suggest these regions fold into compact globular domains. Among them, LIC_10122 is the most divergent, exhibiting a predicted structure distinct from the 5–6 antiparallel β-strands and single α-helix observed in the other three proteins. This analysis highlights the structural and functional diversity of HD-GYP proteins in L. interrogans, expanding our understanding of this enigmatic protein family and suggesting potential novel mechanisms of c-di-GMP interaction beyond canonical binding motifs.
Through bioinformatics and structural biology analyses, we identified twelve proteins containing a PilZ domain in L. interrogans (Figure 3 and 7). Among these, LIC_10128 lacks accessory domains but retains critical residues, including arginines at positions 19 and 23 in the N-terminal region, as well as the (D/N)xSxxG motif in the C-terminal region. These features suggest that this PilZ domain is functional, potentially binding c-di-GMP. Studies in other bacteria support this notion. In P. aeruginosa, the Alg44 protein interacts with c-di-GMP, activating the alginate secretion complex [116,117]. In C. crescentus, the DgrA protein, a PilZ homolog, binds c-di-GMP to regulate the transition from a motile, flagellated cell to an adherent, sessile form, a crucial step in the bacterium’s life cycle [118]. Similarly, in E. coli, the YcgR protein, upon binding c-di-GMP (YcgR–c-di-GMP), interacts with MotA and FliG at their interface [119]. This interaction increases resistance and reduces or extinguishes energy transfer between the stator and rotor of the flagellar motor, resulting in decreased or arrested swimming motility [119]. Notably, this motility inhibition occurs only when YcgR interacts with both MotA and FliG; mutations disrupting either interaction site lead to a reduced counterclockwise bias and restoration of swimming speed [120]. In Xanthomonas campestris, the PilZ domain directly binds c-di-GMP, inducing a conformational change that disrupts interactions with proteins controlling pilus formation, thereby adjusting the bacterium’s motility in response to environmental cues [121]. Interestingly, X. campestris contains an atypical, tetrameric PilZ family (tPilZ), also found in Campylobacter jejuni. This tPilZ family lacks the canonical c-di-GMP binding motifs but appears to interact with the flagellar machinery similarly to YcgR [122]. Remarkably, the Campylobacterota phylum lacks GGDEF domains and does not produce c-di-GMP [122]. Nevertheless, interaction predictions and Cryo-ET experiments indicate that even non-canonical PilZ domains can bind MotA and potentially interact with FliG, with conserved interaction residues despite the absence of the (D/N)xSxxG motif [122]. It is possible that these non-canonical PilZ domains, stabilized by two additional α-helices between the first and second beta-strands, evolved to function independently of c-di-GMP signaling [122]. Despite the diversity of PilZ domain configurations, we found no evidence for the presence of the tetrameric PilZ family in L. interrogans. Neither sequence homology searches nor structural fold analyses of orthologs revealed this family in the L. interrogans genome, suggesting that the PilZ domains in this organism are more likely to function through classical c-di-GMP binding rather than through an alternative, c-di-GMP-independent mechanism.
PilZ domains play fundamental roles in controlling gene expression, exemplified by the interaction of c-di-GMP with the MrkH protein in Klebsiella pneumoniae [123]. In this regard, MrkH functions as a transcriptional activator, facilitating the association of RNA polymerase with suboptimal promoters that regulate the expression of type III fimbriae [123]. Structurally, MrkH is characterized by a C-terminal PilZ domain, specialized in binding c-di-GMP, and an N-terminal β-barrel domain, similar to other PilZ and YcgRN domains [124]. Notably, the PilZC domain of MrkH contains a positively charged helix that promotes interaction with DNA, indicating that the C-terminal motif plays a crucial role in binding additional partners beyond c-di-GMP [21]. These examples illustrate the structural and functional diversity of non-canonical PilZ domains, enabling bacteria to respond more broadly to c-di-GMP-mediated signals. The LIC_20173 protein exemplifies functional diversity in L. interrogans. It is a membrane protein composed of a non-canonical PilZ domain associated with a PrsW domain, a metalloprotease similar to that found in Bacillus subtilis (Figure S17). However, while PrsW in Bacillus functions independently, in Leptospira, it is fused to the PilZ domain. Its primary function is to cleave the anti-sigma factor RsiW, activating genes involved in stress response and antimicrobial defense [125,126,127].
Extracytoplasmic function (ECF) sigma factors constitute a specialized signaling system that regulates the response to environmental stresses. Under normal conditions, their activity is inhibited by anti-sigma factors, which prevent sigma factor interaction with RNA polymerase. The detection of an external signal triggers intramembrane proteolysis, releasing the sigma factor and promoting the expression of genes associated with cellular adaptation [128,129,130]. LIC_20173 shares similarities with PrsW from B. subtilis, whose function is to cleave RsiW, activating genes involved in stress response, including resistance to antimicrobial peptides and cell wall integrity [127]. In Clostridium difficile, PrsW plays a similar role [131]. Indeed, mutation studies have shown that deletion of this gene reduces ECF sigma factor expression, increasing sensitivity to antimicrobial peptides and impairing colonization ability, highlighting the importance of this regulatory pathway [131]. The PilZ domain of LIC_20173 is classified as non-canonical, as it lacks the arginine residues (RxxxR) and the [D/N]xSxxG motifs characteristic of its conventional function. Despite this divergence, it may play additional roles in the cell. Studies indicate that some proteins containing this domain, even without interaction with c-di-GMP, still regulate their targets through protein-protein interactions [20,88,132]. The PrsW domain of LIC_20173 is predicted to be an active protease based on our analysis (Figure S17).
Exploring further into the evolution of PilZ domains, there are also truncated PilZ domains, characterized by the presence of a GAF-like domain within their core structure, known as GAZ domain. This insertion results in a unique architecture that differs from canonical PilZ domains. The insertion of the GAZ domain may influence the function of the PilZ domain, altering its interactions and its response to c-di-GMP. Additionally, these proteins feature a third domain in the N-terminal region, PilZN (also known as YcgR_N), which may play an additional role in the functional regulation of these proteins [115]. The L. interrogans proteins with this structural configuration (YcgRGAZ) include LIC_10049, LIC_11920, LIC_12546, LIC_12723, LIC_12994, and LIC_14002. Due to their distinct architecture, these proteins have been annotated in the database as belonging to the DUF1577 domain. This classification reflects the structural and functional uniqueness of the truncated PilZ domains that contain an inserted GAF-like domain. Although the exact function of these proteins is not yet fully elucidated, their structure suggests a potential role in cellular signaling and adaptation to different environmental conditions. However, upon analyzing the sequences of proteins annotated as DUF1577, the presence of the conserved motifs RxxxR and (D/N)xSxxG is evident. This conservation suggests that, despite the insertion of the GAF-like domain, these proteins still have the potential to interact with c-di-GMP. Our recent study showed that LIC_11920, one of the proteins annotated as DUF1577, is capable of interacting with the c-di-GMP molecule [20]. This interaction occurs through the arginines located at positions 112 and 116 of the first β-strand of the PilZ domain. When these arginines were substituted with alanines, the protein completely loses its ability to bind c-di-GMP, emphasizing the critical role of these residues in forming the binding interface and highlighting their significance in c-di-GMP-mediated signaling. Another important finding in this study is that the GAF-like domain of LIC_11920 completely loses its dimerization capability [20]. This limitation is attributed to the absence of an α-helix in the GAF-like domain of LIC_11920. The absence of this essential structure prevents dimer formation. This observation underscores the evolutionary implications of structural modifications in GAF-like domains and their consequences for the biology of proteins associated with c-di-GMP [20]. The abundance of proteins containing PilZ domains in L. interrogans highlights the complexity of the c-di-GMP signaling network in this bacterium and the importance to investigate the biological function of each c-di-GMP circuit. Figure 9 summarizes the enzymes described in this work and the prediction of their activity. Interestingly, L. interrogans grown in EMJH medium expresses a diverse set of c-di-GMP signaling components, including six proteins with GGDEF domains associated with diguanylate cyclase activity, seven phosphodiesterases (comprising five proteins with EAL domains and two with HD-GYP domains), and five predicted c-di-GMP receptors containing PilZ domains. This repertoire highlights the complexity and potential regulatory significance of c-di-GMP signaling in L. interrogans physiology and pathogenicity.
6. Conclusions
This study provides a comprehensive analysis of the c-di-GMP signaling network in L. interrogans, revealing an intricate regulatory system composed of 44 proteins involved in cyclic di-GMP metabolism, including diguanylate cyclases (DGCs), phosphodiesterases (PDEs), and c-di-GMP-binding effectors. Our findings highlight the remarkable c-di-GMP signaling capacity of L. interrogans, with a high turnover domain density (IQ of 6.7 per Mbp), exceeding the bacterial average. This suggests that c-di-GMP plays a pivotal role in bacterial adaptation, biofilm formation, and survival under environmental stressors. Through structural homology and sequence conservation analysis, we identified 22 putative DGCs, 13 of which are predicted to be enzymatically active. Many degenerate GGDEF domains, while lacking catalytic activity, may still function as c-di-GMP receptors. Similarly, among the nine identified EAL domain proteins, several retain functional residues critical for phosphodiesterase activity, whereas others appear inactive but may regulate c-di-GMP signaling via protein-protein interactions. The HD-GYP proteins exhibit a striking degree of structural diversity, with one catalytically active candidate (LIC_11189) and unique structural features that could contribute to non-canonical c-di-GMP interactions. The identification of 12 PilZ domain-containing proteins suggests multiple regulatory roles for c-di-GMP in L. interrogans, likely influencing motility, biofilm formation, and gene expression. Interestingly, LIC_20173 represents a non-canonical PilZ-containing protease with a putative role in stress response regulation, emphasizing the functional versatility of c-di-GMP effectors in bacterial adaptation. Altogether, this study expands our understanding of the c-di-GMP regulatory network in L. interrogans, providing insights into the functional complexity of this second messenger system. However, experimental validation is essential to confirm the enzymatic activities, regulatory interactions, and physiological relevance of these proteins. Future studies integrating biochemical, genetic, and structural approaches will be crucial to elucidate the full scope of c-di-GMP-mediated regulation in L. interrogans and its implications for pathogenesis and environmental persistence.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. The supporting information has the Figures S1 to S17. Tables S1 to S4.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors acknowledge the National Council for Scientific and Technological Development (CNPq), the Coordination for the Improvement of Higher Education Personnel (CAPES, grants 88887.374931/2019-00, 88887.842544/2023-00 Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Finance Code 01), and the State of São Paulo Research Foundation (FAPESP, grants 2019/00195-2, 2020/04680-0, 2016/09047-8, 2022/08730-7, 2017/17303-7, 2021/10577-0, 2021/05262-0, 2017/17303-7, 2017/06394-1, 2024/10763-6, 2023/10432-7, 2022/04601-8, 2023/13894-1, 2023/18211-0, 2021/10577-0), for financial support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Davignon, G.; et al. Leptospira interrogans biofilm transcriptome highlights adaption to starvation and general stress while maintaining virulence. Npj Biofilms Microbiomes 2024, 10, 95. [Google Scholar] [CrossRef] [PubMed]
- Bonhomme, D.; Werts, C. Host and Species-Specificities of Pattern Recognition Receptors Upon Infection With Leptospira interrogans. Front. Cell. Infect. Microbiol. 2022, 12, 932137. [Google Scholar] [CrossRef]
- Philip, N.; et al. Leptospira interrogans and Leptospira kirschneri are the dominant Leptospira species causing human leptospirosis in Central Malaysia. PLoS Negl. Trop. Dis. 2020, 14, e0008197. [Google Scholar] [CrossRef]
- Pětrošová, H.; et al. Lipid A structural diversity among members of the genus Leptospira. Front. Microbiol. 2023, 14, 1181034. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.T.; et al. Revisiting the taxonomy and evolution of pathogenicity of the genus Leptospira through the prism of genomics. PLoS Negl. Trop. Dis. 2019, 13, e0007270. [Google Scholar] [CrossRef] [PubMed]
- Brazil. Ministry of Health. Leptospirosis. Available online: https://www.gov.br/saude/pt-br/assuntos/saude-de-a-a-z/l/leptospirose#:~:text=A%20leptospirose%20%C3%A9%20uma%20doen%C3%A7a,contaminada%20ou%20por%20meio%20de (accessed on 28 April 2025).
- Rajapakse, S. Leptospirosis: Clinical aspects. Clin. Med. 2022, 22, 14–17. [Google Scholar] [CrossRef]
- Bharti, A.R.; et al. Leptospirosis: A zoonotic disease of global importance. Lancet Infect. Dis. 2003, 3, 757–771. [Google Scholar] [CrossRef]
- Dupouey, J.; et al. Human leptospirosis: An emerging risk in Europe? Comp. Immunol. Microbiol. Infect. Dis. 2014, 37, 77–83. [Google Scholar] [CrossRef]
- Smith, A.M.; Stull, J.W.; Moore, G.E. Potential Drivers for the Re-Emergence of Canine Leptospirosis in the United States and Canada. Trop. Med. Infect. Dis. 2022, 7, 377. [Google Scholar] [CrossRef]
- OCHA; United Nations. Brazil: Floods in Rio Grande do Sul - United Nations Situation Report, as of 20 September 2024. Available online: https://www.unocha.org/publications/report/brazil/brazil-floods-rio-grande-do-sul-united-nations-situation-report-20-september-2024 (accessed on 28 April 2025).
- PAHO, World Health Organization (WHO); United Nations. PAHO supports emergency response following flooding in Rio Grande do Sul, Brazil. Available online: https://www.paho.org/en/news/3-7-2024-paho-supports-emergency-response-following-flooding-rio-grande-do-sul-brazil (accessed on 28 April 2025).
- Martins-Filho, P.R.; Croda, J.; Araújo, A.A.D.S.; Correia, D.; Quintans-Júnior, L.J. Catastrophic Floods in Rio Grande do Sul, Brazil: The Need for Public Health Responses to Potential Infectious Disease Outbreaks. Rev. Soc. Bras. Med. Trop. 2024, 57, e00603-2024. [Google Scholar] [CrossRef]
- Ziliotto, M.; Chies, J.A.B.; Ellwanger, J.H. Extreme Weather Events and Pathogen Pollution Fuel Infectious Diseases: The 2024 Flood-Related Leptospirosis Outbreak in Southern Brazil and Other Red Lights. Pollutants 2024, 4, 424–433. [Google Scholar] [CrossRef]
- Davignon, G.; et al. Leptospira interrogans biofilm transcriptome highlights adaption to starvation and general stress while maintaining virulence. Npj Biofilms Microbiomes 2024, 10, 95. [Google Scholar] [CrossRef]
- Thibeaux, R.; et al. The zoonotic pathogen Leptospira interrogans mitigates environmental stress through cyclic-di-GMP-controlled biofilm production. Npj Biofilms Microbiomes 2020, 6, 24. [Google Scholar] [CrossRef]
- Da Costa Vasconcelos, F.N.; et al. Structural and Enzymatic Characterization of a cAMP-Dependent Diguanylate Cyclase from Pathogenic Leptospira Species. J. Mol. Biol. 2017, 429, 2337–2352. [Google Scholar] [CrossRef]
- Xiao, G.; et al. Identification and Characterization of c-di-GMP Metabolic Enzymes of Leptospira interrogans and c-di-GMP Fluctuations After Thermal Shift and Infection. Front. Microbiol. 2018, 9, 764. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, L.; et al. Genomic insights into the c-di-GMP signaling and biofilm development in the saprophytic spirochete Leptospira biflexa. Arch. Microbiol. 2023, 205, 180. [Google Scholar] [CrossRef]
- Visnardi, A.B.; et al. Insertion of a Divergent GAF-like Domain Defines a Novel Family of YcgR Homologues That Bind c-di-GMP in Leptospirales. ACS Omega 2025, 10, 3988–4006. [Google Scholar] [CrossRef]
- Cheang, Q.W.; Xin, L.; Chea, R.Y.F.; Liang, Z.-X. Emerging paradigms for PilZ domain-mediated C-di-GMP signaling. Biochem. Soc. Trans. 2019, 47, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Römling, U.; Galperin, M.Y.; Gomelsky, M. Cyclic di-GMP: The First 25 Years of a Universal Bacterial Second Messenger. Microbiol. Mol. Biol. Rev. 2013, 77, 1–52. [Google Scholar] [CrossRef]
- Park, S.; Sauer, K. Controlling Biofilm Development Through Cyclic di-GMP Signaling. In Pseudomonas aeruginosa; Filloux, A., Ramos, J.-L., Eds.; Springer International Publishing: Cham, Switzerland, 2022; Volume 1386; pp. 69–94. [Google Scholar]
- Blanco-Romero, E.; et al. Role of extracellular matrix components in biofilm formation and adaptation of Pseudomonas ogarae F113 to the rhizosphere environment. Front. Microbiol. 2024, 15, 1341728. [Google Scholar] [CrossRef]
- Schirmer, T.; Jenal, U. Structural and mechanistic determinants of c-di-GMP signalling. Nat. Rev. Microbiol. 2009, 7, 724–735. [Google Scholar] [CrossRef]
- Purificação, A.D.D.; Azevedo, N.M.D.; Araujo, G.G.D.; Souza, R.F.D.; Guzzo, C.R. The World of Cyclic Dinucleotides in Bacterial Behavior. Molecules 2020, 25, 2462. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; et al. The Multiple Regulatory Relationship Between RNA-Chaperone Hfq and the Second Messenger c-di-GMP. Front. Microbiol. 2021, 12, 689619. [Google Scholar] [CrossRef] [PubMed]
- Valentini, M.; Filloux, A. Biofilms and Cyclic di-GMP (c-di-GMP) Signaling: Lessons from Pseudomonas aeruginosa and Other Bacteria. J. Biol. Chem. 2016, 291, 12547–12555. [Google Scholar] [CrossRef]
- The UniProt, Consortium; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.-H.; Galperin, M.Y. Diversity of Cyclic Di-GMP-Binding Proteins and Mechanisms. J. Bacteriol. 2016, 198, 32–46. [Google Scholar] [CrossRef]
- Roelofs, K.G.; et al. Systematic Identification of Cyclic-di-GMP Binding Proteins in Vibrio cholerae Reveals a Novel Class of Cyclic-di-GMP-Binding ATPases Associated with Type II Secretion Systems. PLOS Pathog. 2015, 11, e1005232. [Google Scholar] [CrossRef]
- Wang, Y.-C.; et al. Nucleotide binding by the widespread high-affinity cyclic di-GMP receptor MshEN domain. Nat. Commun. 2016, 7, 12481. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Nascimento, A.L.T.O.; et al. Genome features of Leptospira interrogans serovar Copenhageni. Braz. J. Med. Biol. Res. 2004, 37, 459–477. [Google Scholar] [CrossRef]
- Abramson, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Van Kempen, M.; et al. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 2024, 42, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Holm, L. Dali server: Structural unification of protein families. Nucleic Acids Res. 2022, 50, W210–W215. [Google Scholar] [CrossRef]
- Hildebrand, A.; Remmert, M.; Biegert, A.; Söding, J. Fast and accurate automatic structure prediction with HHpred. Proteins Struct. Funct. Bioinforma. 2009, 77, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; et al. The conserved domain database in 2023. Nucleic Acids Res. 2023, 51, D384–D388. [Google Scholar] [CrossRef]
- Yu, C.; Chen, Y.; Lu, C.; Hwang, J. Prediction of protein subcellular localization. Proteins Struct. Funct. Bioinforma. 2006, 64, 643–651. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-X.; Pan, X.; Shen, H.-B. Signal-3L 3.0: Improving Signal Peptide Prediction through Combining Attention Deep Learning with Window-Based Scoring. J. Chem. Inf. Model. 2020, 60, 3679–3686. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes11Edited by F. Cohen. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Hallgren, J.; et al. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. bioRxiv 2022. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.; et al. Structural basis of activity and allosteric control of diguanylate cyclase. Proc. Natl. Acad. Sci. USA 2004, 101, 17084–17089. [Google Scholar] [CrossRef]
- Chen, M.W.; et al. Structural Insights into the Regulatory Mechanism of the Response Regulator RocR from Pseudomonas aeruginosa in Cyclic Di-GMP Signaling. J. Bacteriol. 2012, 194, 4837–4846. [Google Scholar] [CrossRef] [PubMed]
- Bellini, D.; et al. Crystal structure of an HD-GYP domain cyclic-di- GMP phosphodiesterase reveals an enzyme with a novel trinuclear catalytic iron centre. Mol. Microbiol. 2014, 91, 26–38. [Google Scholar] [CrossRef]
- Zhu, Y.; Yuan, Z.; Gu, L. Structural basis for the regulation of chemotaxis by MapZ in the presence of c-di-GMP. Acta Crystallogr. Sect. Struct. Biol. 2017, 73, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Sheng, S.; et al. The MapZ-Mediated Methylation of Chemoreceptors Contributes to Pathogenicity of Pseudomonas aeruginosa. Front. Microbiol. 2019, 10, 67. [Google Scholar] [CrossRef] [PubMed]
- Shim, S.R.; Kim, S.-J.; Lee, J.; Rücker, G. Network meta-analysis: Application and practice using R software. Epidemiol. Health 2019, 41, e2019013. [Google Scholar] [CrossRef]
- Morgan, M.; Ramos, M. BiocManager: Access the Bioconductor Project Package Repository. 1.30.25. 2018. [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Inkscape Project. Inkscape. 2020. Available online: https://inkscape.org.
- Schrödinger, L.; DeLano, W. PyMOL. 2020. Available online: http://www.pymol.org/pymol.
- Meng, E.C.; et al. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef]
- Krasteva, P.V.; Giglio, K.M.; Sondermann, H. Sensing the messenger: The diverse ways that bacteria signal through c-di-GMP. Protein Sci. 2012, 21, 929–948. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; et al. Phenotypic–genotypic analysis of GGDEF/EAL/HD-GYP domain-encoding genes in Pseudomonas putida. Environ. Microbiol. Rep. 2020, 12, 38–48. [Google Scholar] [CrossRef]
- Yang, C.-Y.; et al. The structure and inhibition of a GGDEF diguanylate cyclase complexed with (c-di-GMP)2 at the active site. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Römling, U.; Liang, Z.-X.; Dow, J.M. Progress in Understanding the Molecular Basis Underlying Functional Diversification of Cyclic Dinucleotide Turnover Proteins. J. Bacteriol. 2017, 199. [Google Scholar] [CrossRef]
- Rao, F.; et al. YybT Is a Signaling Protein That Contains a Cyclic Dinucleotide Phosphodiesterase Domain and a GGDEF Domain with ATPase Activity. J. Biol. Chem. 2010, 285, 473–482. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Chou, S.-H. Sequence Conservation, Domain Architectures, and Phylogenetic Distribution of the HD-GYP Type c-di-GMP Phosphodiesterases. J. Bacteriol. 2022, 204, e00561-21. [Google Scholar] [CrossRef]
- Schmidt, A.J.; Ryjenkov, D.A.; Gomelsky, M. The Ubiquitous Protein Domain EAL Is a Cyclic Diguanylate-Specific Phosphodiesterase: Enzymatically Active and Inactive EAL Domains. J. Bacteriol. 2005, 187, 4774–4781. [Google Scholar] [CrossRef] [PubMed]
- Tischler, A.D.; Camilli, A. Cyclic diguanylate (c-di-GMP) regulates Vibrio cholerae biofilm formation. Mol. Microbiol. 2004, 53, 857–869. [Google Scholar] [CrossRef]
- Römling, U.; Gomelsky, M.; Galperin, M.Y. C-di-GMP: The dawning of a novel bacterial signalling system. Mol. Microbiol. 2005, 57, 629–639. [Google Scholar] [CrossRef]
- Tamayo, R.; Tischler, A.D.; Camilli, A. The EAL Domain Protein VieA Is a Cyclic Diguanylate Phosphodiesterase. J. Biol. Chem. 2005, 280, 33324–33330. [Google Scholar] [CrossRef]
- Heo, K.; et al. A pGpG-specific phosphodiesterase regulates cyclic di-GMP signaling in Vibrio cholerae. J. Biol. Chem. 2022, 298, 101626. [Google Scholar] [CrossRef]
- Rao, F.; et al. The Functional Role of a Conserved Loop in EAL Domain-Based Cyclic di-GMP-Specific Phosphodiesterase. J. Bacteriol. 2009, 191, 4722–4731. [Google Scholar] [CrossRef] [PubMed]
- Römling, U. Rationalizing the Evolution of EAL Domain-Based Cyclic di-GMP-Specific Phosphodiesterases. J. Bacteriol. 2009, 191, 4697–4700. [Google Scholar] [CrossRef]
- Tchigvintsev, A.; et al. Structural Insight into the Mechanism of c-di-GMP Hydrolysis by EAL Domain Phosphodiesterases. J. Mol. Biol. 2010, 402, 524–538. [Google Scholar] [CrossRef] [PubMed]
- Barends, T.R.M.; et al. Structure and mechanism of a bacterial light-regulated cyclic nucleotide phosphodiesterase. Nature 2009, 459, 1015–1018. [Google Scholar] [CrossRef]
- Tarutina, M.; Ryjenkov, D.A.; Gomelsky, M. An Unorthodox Bacteriophytochrome from Rhodobacter sphaeroides Involved in Turnover of the Second Messenger c-di-GMP. J. Biol. Chem. 2006, 281, 34751–34758. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.G.; Murphy, C.N.; Sippy, J.; Johnson, T.J.; Clegg, S. Type 3 Fimbriae and Biofilm Formation Are Regulated by the Transcriptional Regulators MrkHI in Klebsiella pneumoniae. J. Bacteriol. 2011, 193, 3453–3460. [Google Scholar] [CrossRef]
- Sun, S.; Pandelia, M.-E. HD-[HD-GYP] Phosphodiesterases: Activities and Evolutionary Diversification within the HD-GYP Family. Biochemistry 2020, 59, 2340–2350. [Google Scholar] [CrossRef]
- Gong, H.; et al. Cryo-EM structure of trimeric Mycobacterium smegmatis succinate dehydrogenase with a membrane-anchor SdhF. Nat. Commun. 2020, 11, 4245. [Google Scholar] [CrossRef]
- He, D.; et al. Pseudomonas aeruginosa SutA wedges RNAP lobe domain open to facilitate promoter DNA unwinding. Nat. Commun. 2022, 13, 4204. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Gao, H.; Bai, X.; Yu, H. Cryo-EM structure of the human cohesin-NIPBL-DNA complex. Science 2020, 368, 1454–1459. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; et al. Structural insights into human CCAN complex assembled onto DNA. Cell Discov. 2022, 8, 90. [Google Scholar] [CrossRef]
- Klein, B.J.; et al. RNA polymerase and transcription elongation factor Spt4/5 complex structure. Proc. Natl. Acad. Sci. 2011, 108, 546–550. [Google Scholar] [CrossRef]
- Keidel, A.; et al. Concerted structural rearrangements enable RNA channeling into the cytoplasmic Ski238-Ski7-exosome assembly. Mol. Cell 2023, 83, 4093–4105.e7. [Google Scholar] [CrossRef] [PubMed]
- Vanden Broeck, A.; Klinge, S. Principles of human pre-60 S biogenesis. Science 2023, 381, eadh3892. [Google Scholar] [CrossRef]
- Mohanty, S.; et al. Structural Basis for a Unique ATP Synthase Core Complex from Nanoarcheaum equitans. J. Biol. Chem. 2015, 290, 27280–27296. [Google Scholar] [CrossRef]
- Bian, C.; et al. Sgf29 binds histone H3K4me2/3 and is required for SAGA complex recruitment and histone H3 acetylation: Sgf29 functions as an H3K4me2/3 binder in SAGA. EMBO J. 2011, 30, 2829–2842. [Google Scholar] [CrossRef]
- Das, D.; et al. Crystal structure of a novel Sm-like protein of putative cyanophage origin at 2.60 Å resolution. Proteins Struct. Funct. Bioinforma. 2009, 75, 296–307. [Google Scholar] [CrossRef]
- Tobiasson, V.; Berzina, I.; Amunts, A. Structure of a mitochondrial ribosome with fragmented rRNA in complex with membrane-targeting elements. Nat. Commun. 2022, 13, 6132. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Chou, S.-H. Structural Conservation and Diversity of PilZ-Related Domains. J. Bacteriol. 2020, 202. [Google Scholar] [CrossRef] [PubMed]
- Amikam, D.; Galperin, M.Y. PilZ domain is part of the bacterial c-di-GMP binding protein. Bioinformatics 2006, 22, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tsai, B.; Li, N.; Gao, N. Structural remodeling of ribosome associated Hsp40-Hsp70 chaperones during co-translational folding. Nat. Commun. 2022, 13, 3410. [Google Scholar] [CrossRef]
- Koplin, A.; et al. A dual function for chaperones SSB–RAC and the NAC nascent polypeptide–associated complex on ribosomes. J. Cell Biol. 2010, 189, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Peisker, K.; et al. Ribosome-associated Complex Binds to Ribosomes in Close Proximity of Rpl31 at the Exit of the Polypeptide Tunnel in Yeast. Mol. Biol. Cell 2008, 19, 5279–5288. [Google Scholar] [CrossRef]
- Pichlo, C.; et al. Molecular determinants of the mechanism and substrate specificity of Clostridium difficile proline-proline endopeptidase-1. J. Biol. Chem. 2019, 294, 11525–11535. [Google Scholar] [CrossRef]
- Pannifer, A.D.; et al. Crystal structure of the anthrax lethal factor. Nature 2001, 414, 229–233. [Google Scholar] [CrossRef]
- Visschedyk, D.; et al. Certhrax Toxin, an Anthrax-related ADP-ribosyltransferase from Bacillus cereus. J. Biol. Chem. 2012, 287, 41089–41102. [Google Scholar] [CrossRef]
- Scheithauer, L.; et al. Zinc metalloprotease ProA of Legionella pneumophila increases alveolar septal thickness in human lung tissue explants by collagen IV degradation. Cell. Microbiol. 2021, 23. [Google Scholar] [CrossRef]
- Shen, Y.; Zhukovskaya, N.L.; Guo, Q.; Florián, J.; Tang, W.-J. Calcium-independent calmodulin binding and two-metal–ion catalytic mechanism of anthrax edema factor. EMBO J. 2005, 24, 929–941. [Google Scholar] [CrossRef]
- Pei, J.; Mitchell, D.A.; Dixon, J.E.; Grishin, N.V. Expansion of Type II CAAX Proteases Reveals Evolutionary Origin of γ-Secretase Subunit APH-1. J. Mol. Biol. 2011, 410, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.J.; et al. C-di-GMP Regulates Motile to Sessile Transition by Modulating MshA Pili Biogenesis and Near-Surface Motility Behavior in Vibrio cholerae. PLOS Pathog. 2015, 11, e1005068. [Google Scholar] [CrossRef]
- Chen, Y.; et al. Structure and Function of the XpsE N-Terminal Domain, an Essential Component of the Xanthomonas campestris Type II Secretion System. J. Biol. Chem. 2005, 280, 42356–42363. [Google Scholar] [CrossRef] [PubMed]
- Christen, M.; Christen, B.; Folcher, M.; Schauerte, A.; Jenal, U. Identification and Characterization of a Cyclic di-GMP-specific Phosphodiesterase and Its Allosteric Control by GTP. J. Biol. Chem. 2005, 280, 30829–30837. [Google Scholar] [CrossRef]
- Jenal, U.; Reinders, A.; Lori, C. Cyclic di-GMP: Second messenger extraordinaire. Nat. Rev. Microbiol. 2017, 15, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Martín-Rodríguez, A.J.; et al. Comparative Genomics of Cyclic di-GMP Metabolism and Chemosensory Pathways in Shewanella algae Strains: Novel Bacterial Sensory Domains and Functional Insights into Lifestyle Regulation. mSystems 2022, 7, e01518-21. [Google Scholar] [CrossRef]
- Galperin, M.Y. A census of membrane-bound and intracellular signal transduction proteins in bacteria: Bacterial IQ, extroverts and introverts. BMC Microbiol. 2005, 5, 35. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Higdon, R.; Kolker, E. Interplay of heritage and habitat in the distribution of bacterial signal transduction systems. Mol. Biosyst. 2010, 6, 721. [Google Scholar] [CrossRef]
- Römling, U.; Cao, L.-Y.; Bai, F.-W. Evolution of cyclic di-GMP signalling on a short and long term time scale. Microbiology 2023, 169. [Google Scholar] [CrossRef]
- Khan, F.; Jeong, G.-J.; Tabassum, N.; Kim, Y.-M. Functional diversity of c-di-GMP receptors in prokaryotic and eukaryotic systems. Cell Commun. Signal. 2023, 21, 259. [Google Scholar] [CrossRef]
- Holland, L.M.; et al. A Staphylococcal GGDEF Domain Protein Regulates Biofilm Formation Independently of Cyclic Dimeric GMP. J. Bacteriol. 2008, 190, 5178–5189. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-L.; et al. Global Transcriptional Repression of Diguanylate Cyclases by MucR1 Is Essential for Sinorhizobium -Soybean Symbiosis. mBio 2021, 12, e01192-21. [Google Scholar] [CrossRef]
- Bobrov, A.G.; et al. Systematic analysis of cyclic di-GMP signalling enzymes and their role in biofilm formation and virulence in Yersinia pestis. Mol. Microbiol. 2011, 79, 533–551. [Google Scholar] [CrossRef]
- Guzzo, C.R.; Salinas, R.K.; Andrade, M.O.; Farah, C.S. PILZ Protein Structure and Interactions with PILB and the FIMX EAL Domain: Implications for Control of Type IV Pilus Biogenesis. J. Mol. Biol. 2009, 393, 848–866. [Google Scholar] [CrossRef]
- Bordeleau, E.; Fortier, L.-C.; Malouin, F.; Burrus, V. c-di-GMP Turn-Over in Clostridium difficile Is Controlled by a Plethora of Diguanylate Cyclases and Phosphodiesterases. PLoS Genet. 2011, 7, e1002039. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Koonin, E.V. The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem. Sci. 1998, 23, 469–472. [Google Scholar] [CrossRef]
- Lovering, A.L.; Capeness, M.J.; Lambert, C.; Hobley, L.; Sockett, R.E. The Structure of an Unconventional HD-GYP Protein from Bdellovibrio Reveals the Roles of Conserved Residues in this Class of Cyclic-di-GMP Phosphodiesterases. mBio 2011, 2, e00163-11. [Google Scholar] [CrossRef] [PubMed]
- McKee, R.W.; Kariisa, A.; Mudrak, B.; Whitaker, C.; Tamayo, R. A systematic analysis of the in vitro and in vivo functions of the HD-GYP domain proteins of Vibrio cholerae. BMC Microbiol. 2014, 14, 272. [Google Scholar] [CrossRef]
- Stelitano, V.; et al. C-di-GMP Hydrolysis by Pseudomonas aeruginosa HD-GYP Phosphodiesterases: Analysis of the Reaction Mechanism and Novel Roles for pGpG. PLoS ONE 2013, 8, e74920. [Google Scholar] [CrossRef]
- Remminghorst, U.; Rehm, B.H.A. Alg44, a unique protein required for alginate biosynthesis in Pseudomonas aeruginosa. FEBS Lett. 2006, 580, 3883–3888. [Google Scholar] [CrossRef]
- Moradali, M.F.; Donati, I.; Sims, I.M.; Ghods, S.; Rehm, B.H.A. Alginate Polymerization and Modification Are Linked in Pseudomonas aeruginosa. mBio 2015, 6, e00453-15. [Google Scholar] [CrossRef] [PubMed]
- Christen, M.; et al. DgrA is a member of a new family of cyclic diguanosine monophosphate receptors and controls flagellar motor function in Caulobacter crescentus. Proc. Natl. Acad. Sci. USA 2007, 104, 4112–4117. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.-J.; et al. Structural insights into the mechanism of c-di-GMP–bound YcgR regulating flagellar motility in Escherichia coli. J. Biol. Chem. 2020, 295, 808–821. [Google Scholar] [CrossRef]
- Han, Q.; et al. Flagellar brake protein YcgR interacts with motor proteins MotA and FliG to regulate the flagellar rotation speed and direction. Front. Microbiol. 2023, 14, 1159974. [Google Scholar] [CrossRef]
- Randall, T.E.; et al. Sensory Perception in Bacterial Cyclic Diguanylate Signal Transduction. J. Bacteriol. 2022, 204, e00433-21. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; et al. Tetrameric PilZ protein stabilizes stator ring in complex flagellar motor and is required for motility in Campylobacter jejuni. Proc. Natl. Acad. Sci. USA 2025, 122, e2412594121. [Google Scholar] [CrossRef]
- Wilksch, J.J.; et al. MrkH, a Novel c-di-GMP-Dependent Transcriptional Activator, Controls Klebsiella pneumoniae Biofilm Formation by Regulating Type 3 Fimbriae Expression. PLoS Pathog. 2011, 7, e1002204. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.A.; Zeng, W. Structures of the activator of K. pneumonia biofilm formation, MrkH, indicates PilZ domains involved in c-di-GMP and DNA binding. Proc. Natl. Acad. Sci. 2016, 113, 10067–10072. [Google Scholar] [CrossRef]
- Devkota, S.R.; Kwon, E.; Ha, S.C.; Chang, H.W.; Kim, D.Y. Structural insights into the regulation of Bacillus subtilis SigW activity by anti-sigma RsiW. PLoS ONE 2017, 12, e0174284. [Google Scholar] [CrossRef]
- Ellermeier, C.D.; Losick, R. Evidence for a novel protease governing regulated intramembrane proteolysis and resistance to antimicrobial peptides in Bacillus subtilis. Genes Dev. 2006, 20, 1911–1922. [Google Scholar] [CrossRef]
- Heinrich, J.; Hein, K.; Wiegert, T. Two proteolytic modules are involved in regulated intramembrane proteolysis of Bacillus subtilis RsiW. Mol. Microbiol. 2009, 74, 1412–1426. [Google Scholar] [CrossRef] [PubMed]
- Pannullo, A.G.; Ellermeier, C.D. Activation of the Extracytoplasmic Function σ Factor σV in Clostridioides difficile Requires Regulated Intramembrane Proteolysis of the Anti-σ Factor RsiV. mSphere 2022, 7, e00092–22. [Google Scholar] [CrossRef] [PubMed]
- Helmann, J.D. Bacillus subtilis extracytoplasmic function (ECF) sigma factors and defense of the cell envelope. Curr. Opin. Microbiol. 2016, 30, 122–132. [Google Scholar] [CrossRef] [PubMed]
- De Bruijn, F.J. Stress and Environmental Regulation of Gene Expression and Adaptation in Bacteria; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016. [Google Scholar]
- Ho, T.D.; Ellermeier, C.D. PrsW Is Required for Colonization, Resistance to Antimicrobial Peptides, and Expression of Extracytoplasmic Function σ Factors in Clostridium difficile. Infect. Immun. 2011, 79, 3229–3238. [Google Scholar] [CrossRef]
- Merighi, M.; Lee, V.T.; Hyodo, M.; Hayakawa, Y.; Lory, S. The second messenger bis-(3′-5′)-cyclic-GMP and its PilZ domain-containing receptor Alg44 are required for alginate biosynthesis in Pseudomonas aeruginosa. Mol. Microbiol. 2007, 65, 876–895. [Google Scholar] [CrossRef]
- Malmström, J.; et al. Proteome-wide cellular protein concentrations of the human pathogen Leptospira interrogans. Nature 2009, 460, 762–765. [Google Scholar] [CrossRef]
Figure 1.
Synthesis and degradation of c-di-GMP. The synthesis of c-di-GMP is catalyzed by diguanylate cyclases (DGCs) through the cooperative action of their catalytic GGDEF domains (green), promoting the conversion of two GTP molecules into one c-di-GMP molecule and two pyrophosphate molecules. The degradation of c-di-GMP is carried out by specific phosphodiesterases (PDEs), which contain EAL or HD-GYP domains (red). These enzymes hydrolyze c-di-GMP into phospho-guanylyl-(3′-5′)-guanosine (pGpG) or into two guanosine monophosphate (GMP) molecules, respectively. The regulation of cellular processes mediated by c-di-GMP occurs through the interaction of this molecule with various effectors, including proteins containing PilZ domains, transcription factors, riboswitches, and degenerate GGDEF and EAL domains. In this regard, c-di-GMP influences essential processes such as motility, adhesion, biofilm formation, virulence, and other bacterial behaviors.
Figure 1.
Synthesis and degradation of c-di-GMP. The synthesis of c-di-GMP is catalyzed by diguanylate cyclases (DGCs) through the cooperative action of their catalytic GGDEF domains (green), promoting the conversion of two GTP molecules into one c-di-GMP molecule and two pyrophosphate molecules. The degradation of c-di-GMP is carried out by specific phosphodiesterases (PDEs), which contain EAL or HD-GYP domains (red). These enzymes hydrolyze c-di-GMP into phospho-guanylyl-(3′-5′)-guanosine (pGpG) or into two guanosine monophosphate (GMP) molecules, respectively. The regulation of cellular processes mediated by c-di-GMP occurs through the interaction of this molecule with various effectors, including proteins containing PilZ domains, transcription factors, riboswitches, and degenerate GGDEF and EAL domains. In this regard, c-di-GMP influences essential processes such as motility, adhesion, biofilm formation, virulence, and other bacterial behaviors.
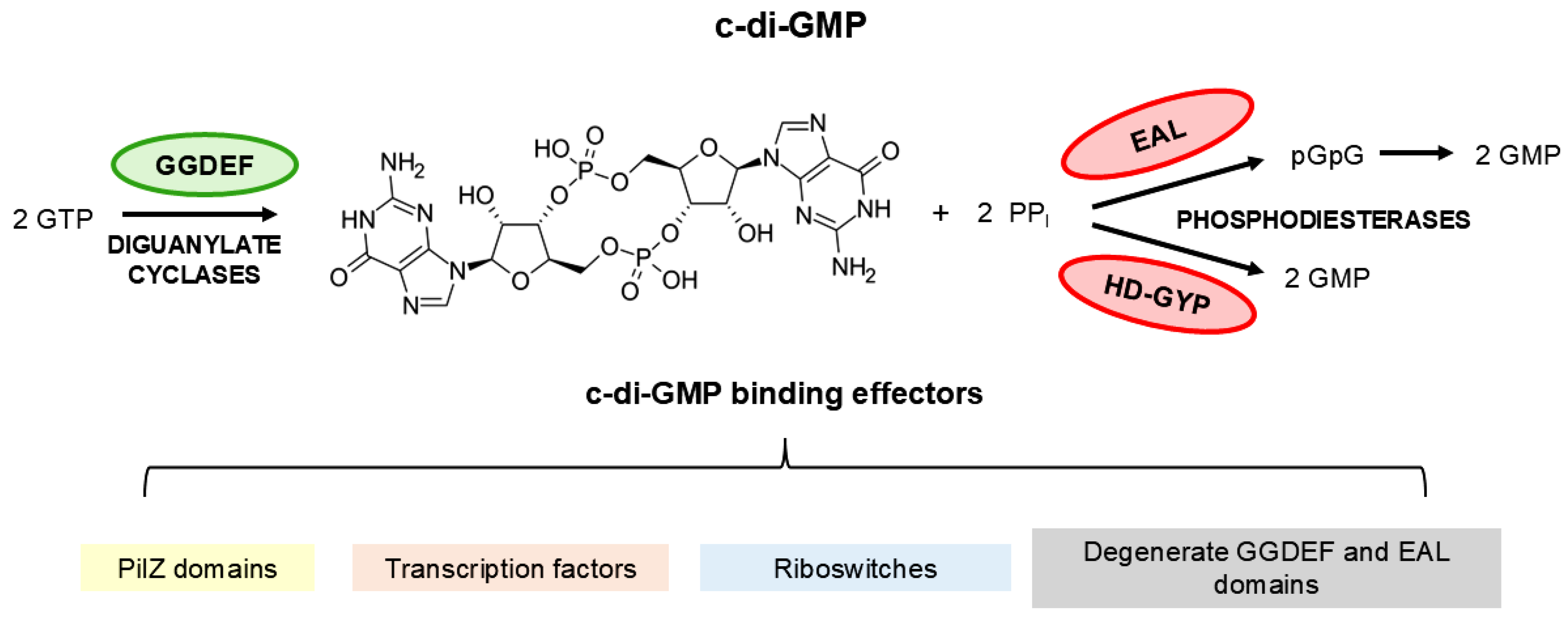
Figure 2.
Workflow of the bioinformatics analysis of c-di-GMP-related genes. The database was built by searching the NCBI c-di-GMP database, Uniprot and the literature. The identified proteins were submitted to BLASTP analysis, yielding 75 candidate proteins potentially associated with c-di-GMP signaling. These proteins were subsequently analyzed using AlphaFold for structural prediction and FoldSeek for structural homology assessment. The domains present in the proteins were analyzed and annotated, in addition to analyzing the presence of signal peptides, transmembrane regions, and predicting the cellular sub-localization of the protein. Overall, resulting in 44 proteins.
Figure 2.
Workflow of the bioinformatics analysis of c-di-GMP-related genes. The database was built by searching the NCBI c-di-GMP database, Uniprot and the literature. The identified proteins were submitted to BLASTP analysis, yielding 75 candidate proteins potentially associated with c-di-GMP signaling. These proteins were subsequently analyzed using AlphaFold for structural prediction and FoldSeek for structural homology assessment. The domains present in the proteins were analyzed and annotated, in addition to analyzing the presence of signal peptides, transmembrane regions, and predicting the cellular sub-localization of the protein. Overall, resulting in 44 proteins.
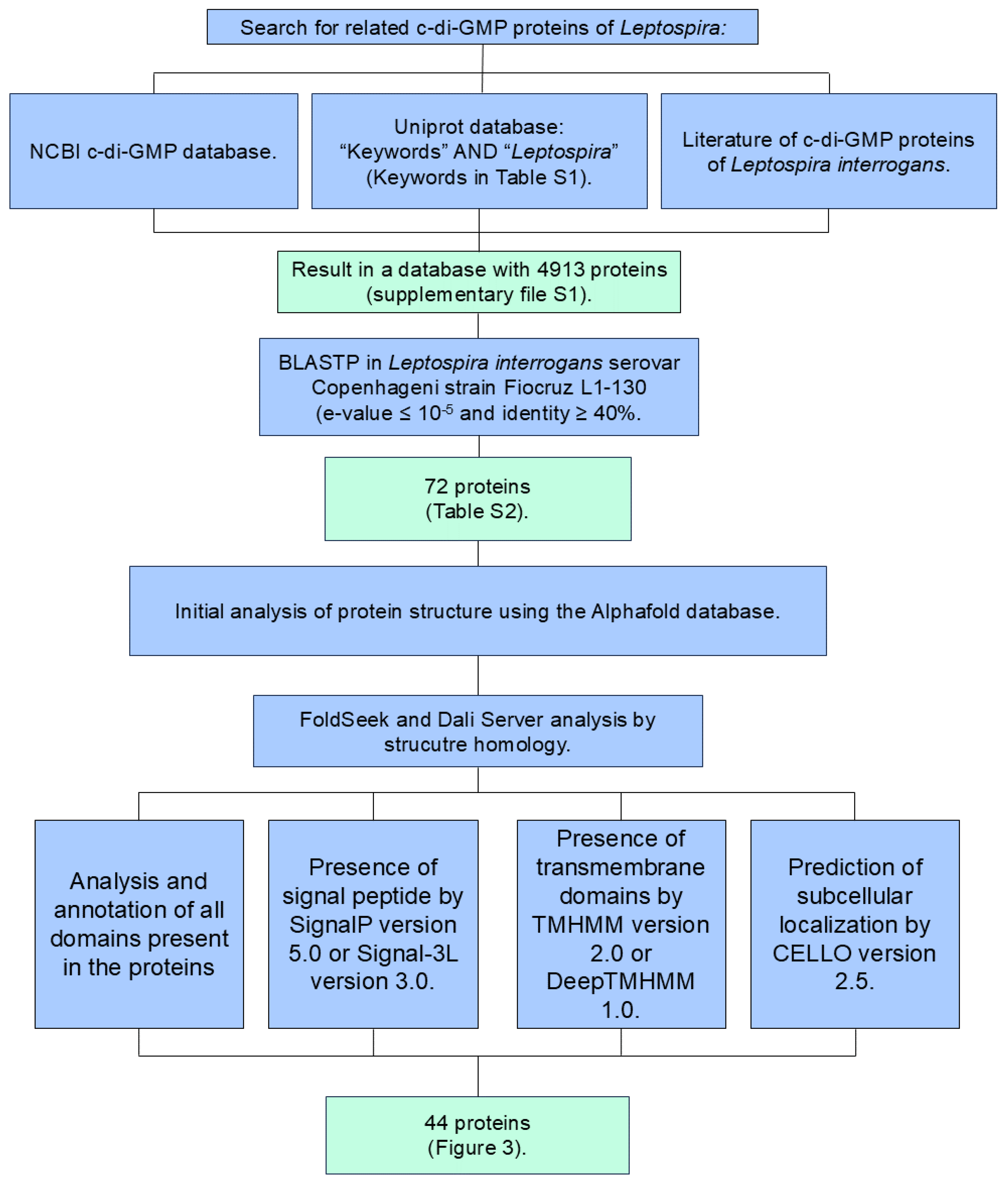
Figure 3.
Proteins identified in the genome of L. interrogans serovar Copenhageni strain Fiocruz L1-130 containing GGDEF, EAL, PilZ, HD-GYP, and MshEN domains. Domain architecture of c-di-GMP-related proteins in Lic is shown. Protein names are listed on the left, with the corresponding number of amino acids on the right. The protein sequence analysis, domain identification, transmembrane domain predictions, signal peptide detection were conducted using UniProt, CDD, DeepTMHMM 1.0, and SignalIP-5.0. The genes LIC_10049, LIC_11920, LIC_12546, LIC_12723, LIC_12994, LIC_14002 (not annotated in L. interrogans serovar Copenhageni strain Fiocruz L1-130 genome; the locus_tag corresponds to RefSeqGene LIC14002 or LIC_RS11585, and this gene is orthologous to LA_1489) were identified. The gene with locus_tag LIC_13137 was experimentally characterized and named Lcd1 protein [17]. The paralog group, including LIC_10049, LIC_11920, LIC_12546, LIC_12723, LIC_12994, and LIC_14002, was recognized as members of the YcgRGAZ family [20]. Additionally, LIC_11571 is annotated as GspE, associated with the type II secretion system (T2SS). *Possible DUF3391.
Figure 3.
Proteins identified in the genome of L. interrogans serovar Copenhageni strain Fiocruz L1-130 containing GGDEF, EAL, PilZ, HD-GYP, and MshEN domains. Domain architecture of c-di-GMP-related proteins in Lic is shown. Protein names are listed on the left, with the corresponding number of amino acids on the right. The protein sequence analysis, domain identification, transmembrane domain predictions, signal peptide detection were conducted using UniProt, CDD, DeepTMHMM 1.0, and SignalIP-5.0. The genes LIC_10049, LIC_11920, LIC_12546, LIC_12723, LIC_12994, LIC_14002 (not annotated in L. interrogans serovar Copenhageni strain Fiocruz L1-130 genome; the locus_tag corresponds to RefSeqGene LIC14002 or LIC_RS11585, and this gene is orthologous to LA_1489) were identified. The gene with locus_tag LIC_13137 was experimentally characterized and named Lcd1 protein [17]. The paralog group, including LIC_10049, LIC_11920, LIC_12546, LIC_12723, LIC_12994, and LIC_14002, was recognized as members of the YcgRGAZ family [20]. Additionally, LIC_11571 is annotated as GspE, associated with the type II secretion system (T2SS). *Possible DUF3391.

Figure 4.
Characterization of the GGDEF domain of different proteins from L. interrogans serovar Copenhageni strain Fiocruz L1-130. Structural alignments of GGDEF domains from L. interrogans serovar Copenhageni strain Fiocruz L1-130 via the DALI server, using the protein PleD (locus_tag CCNA_02546, PDB 1W25 [46]) of C. crescentus as a reference.
Figure 4.
Characterization of the GGDEF domain of different proteins from L. interrogans serovar Copenhageni strain Fiocruz L1-130. Structural alignments of GGDEF domains from L. interrogans serovar Copenhageni strain Fiocruz L1-130 via the DALI server, using the protein PleD (locus_tag CCNA_02546, PDB 1W25 [46]) of C. crescentus as a reference.

Figure 5.
Characterization of the EAL domain of proteins from L. interrogans serovar Copenhageni strain Fiocruz L1-130. Structural alignments of EAL domains from L. interrogans serovar Copenhageni strain Fiocruz L1-130 via the DALI server, using the RocR (locus_tag PA3947, PDB 3SY8 [47]) from P. aeruginosa as a reference.
Figure 5.
Characterization of the EAL domain of proteins from L. interrogans serovar Copenhageni strain Fiocruz L1-130. Structural alignments of EAL domains from L. interrogans serovar Copenhageni strain Fiocruz L1-130 via the DALI server, using the RocR (locus_tag PA3947, PDB 3SY8 [47]) from P. aeruginosa as a reference.
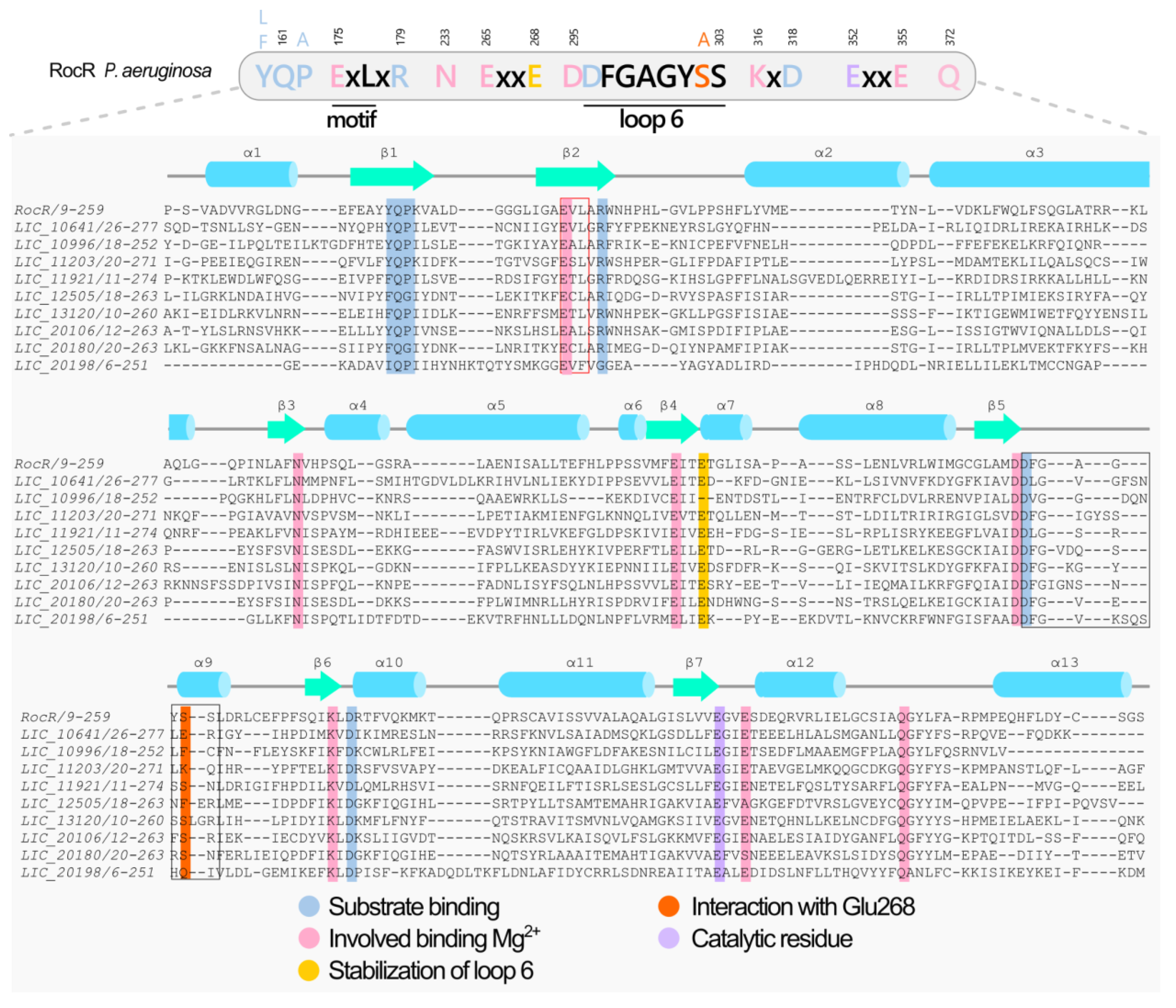
Figure 6.
Characterization of the HD-GYP-containing proteins from L. interrogans serovar Copenhageni strain Fiocruz L1-130. (A) Sequence alignment of HD-GYPs from L interrogans serovar Copenhageni strain Fiocruz L1-130 based on structure performed on DALI using the PmGH (locus_tag PERMA_0986, PDB 4MDZ [48]) as reference. (B) Cartoon representation of the HD-GYP domains from L. interrogans predicted by AlphaFold (green). Extensions/insertions (in comparison to PmGH) are colored in red.
Figure 6.
Characterization of the HD-GYP-containing proteins from L. interrogans serovar Copenhageni strain Fiocruz L1-130. (A) Sequence alignment of HD-GYPs from L interrogans serovar Copenhageni strain Fiocruz L1-130 based on structure performed on DALI using the PmGH (locus_tag PERMA_0986, PDB 4MDZ [48]) as reference. (B) Cartoon representation of the HD-GYP domains from L. interrogans predicted by AlphaFold (green). Extensions/insertions (in comparison to PmGH) are colored in red.
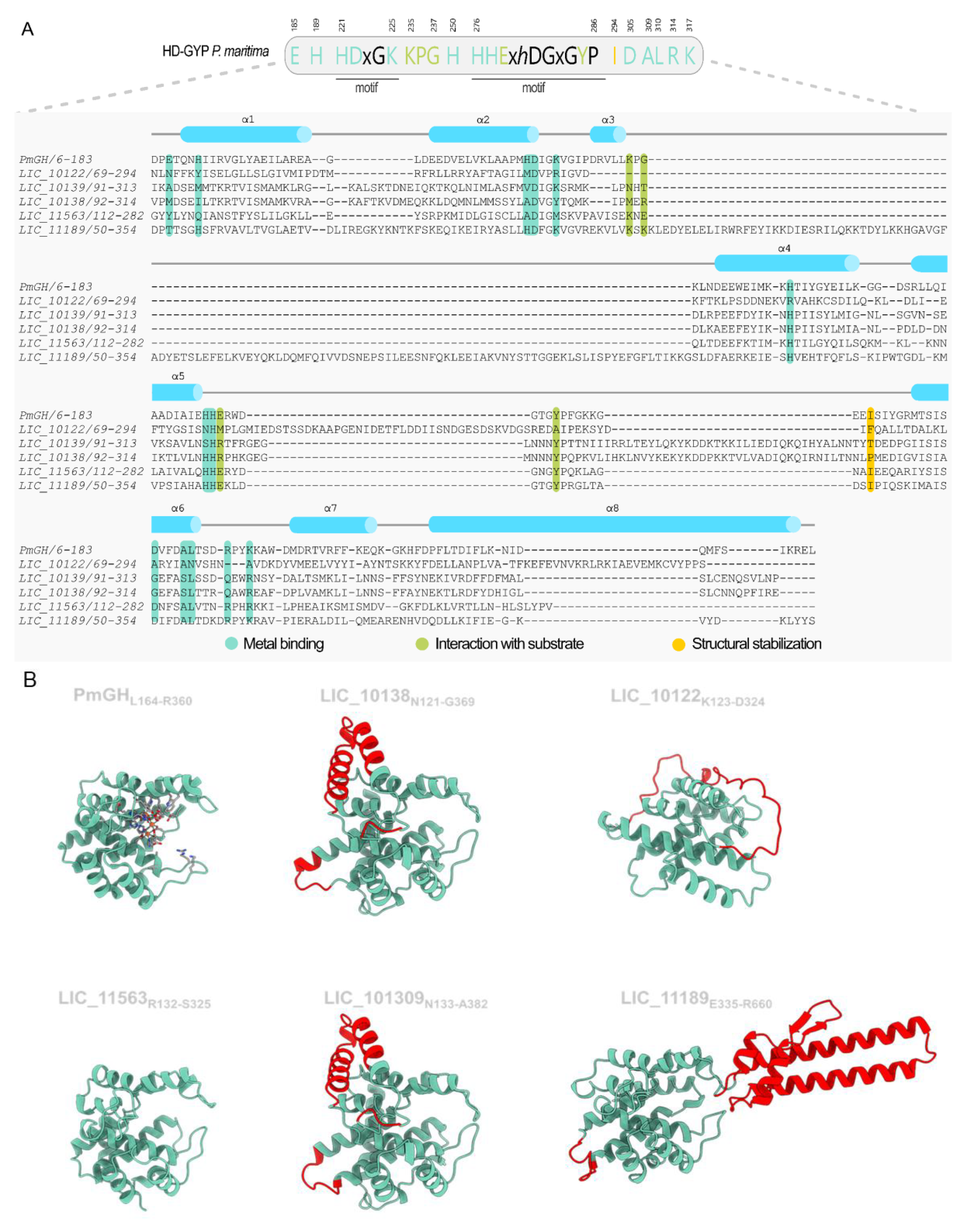
Figure 7.
Characterization of the PilZ domain of proteins from L. interrogans serovar Copenhageni strain Fiocruz L1-130. Structural alignments of PilZ domains from L. interrogans serovar Copenhageni strain Fiocruz L1-130 via the DALI server, using the protein MapZ (locus_tag PA4608, PDB 5HTL [50]) from P. aeruginosa as a reference.
Figure 7.
Characterization of the PilZ domain of proteins from L. interrogans serovar Copenhageni strain Fiocruz L1-130. Structural alignments of PilZ domains from L. interrogans serovar Copenhageni strain Fiocruz L1-130 via the DALI server, using the protein MapZ (locus_tag PA4608, PDB 5HTL [50]) from P. aeruginosa as a reference.
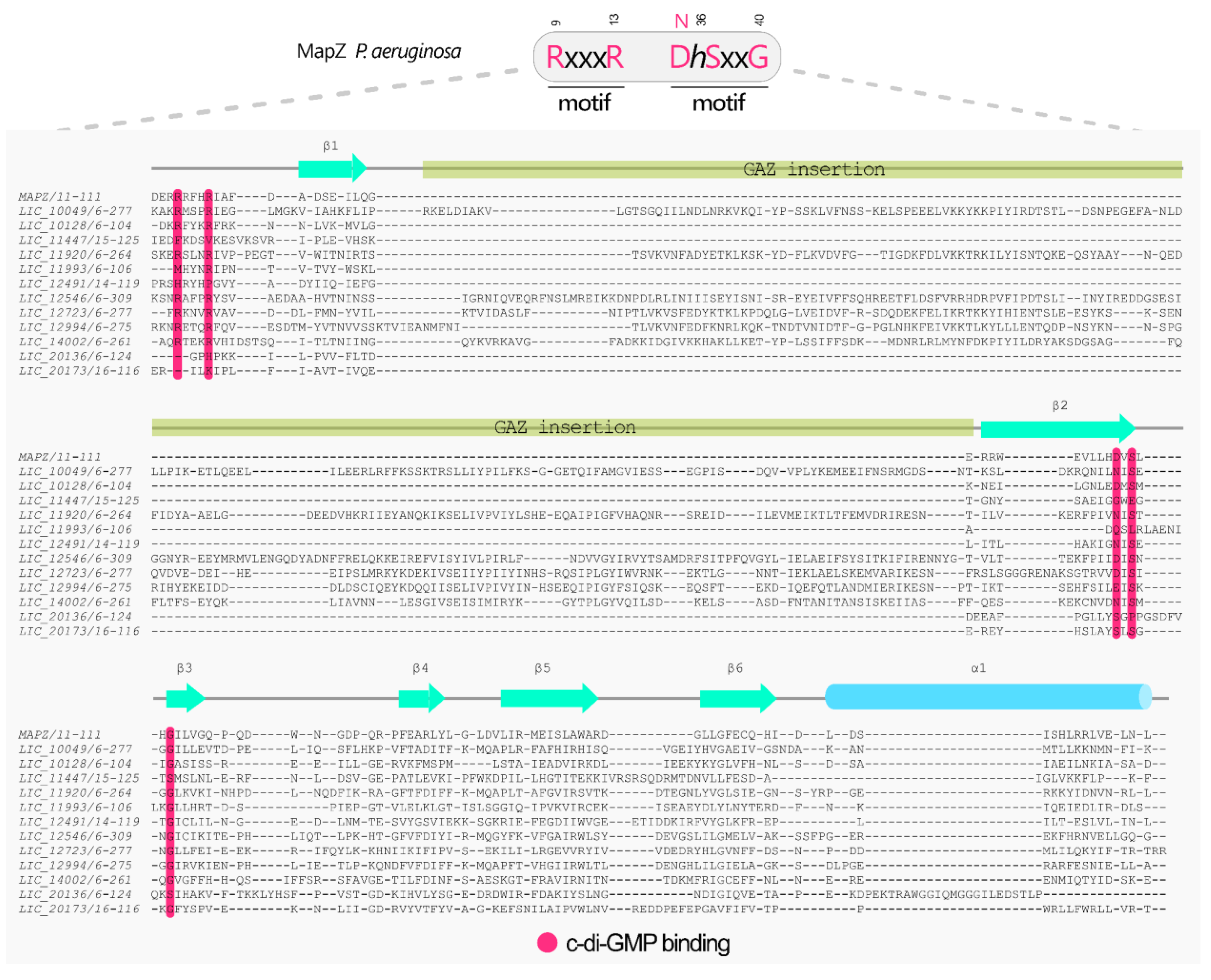
Figure 8.
Characterization of the MshEN domain of proteins from L. interrogans serovar Copenhageni strain Fiocruz L1-130. In the MshEN domain, c-di-GMP is accommodated by two 24-residue motifs [RLGxxL(L/V/I)xxG(L/V/I/F)(L/V/I)xxxxLxxxLxxQ], connected by a 5-residue linker, forming a complete 53-residue-long domain. The MshEN domain uses has a model is MshEN_N (locus_tag VC_0405, PDB: 5HTL [32]) of V. cholerae, PA3740 (probably functional) and PA14_29490 (experimentally demonstrated to be functional) of P. aeruginosa [31,32,98], XspE of X. campestris strain 17 (PDB: 2D27), which has been shown that it is a degenerate MshEN domain [99].
Figure 8.
Characterization of the MshEN domain of proteins from L. interrogans serovar Copenhageni strain Fiocruz L1-130. In the MshEN domain, c-di-GMP is accommodated by two 24-residue motifs [RLGxxL(L/V/I)xxG(L/V/I/F)(L/V/I)xxxxLxxxLxxQ], connected by a 5-residue linker, forming a complete 53-residue-long domain. The MshEN domain uses has a model is MshEN_N (locus_tag VC_0405, PDB: 5HTL [32]) of V. cholerae, PA3740 (probably functional) and PA14_29490 (experimentally demonstrated to be functional) of P. aeruginosa [31,32,98], XspE of X. campestris strain 17 (PDB: 2D27), which has been shown that it is a degenerate MshEN domain [99].
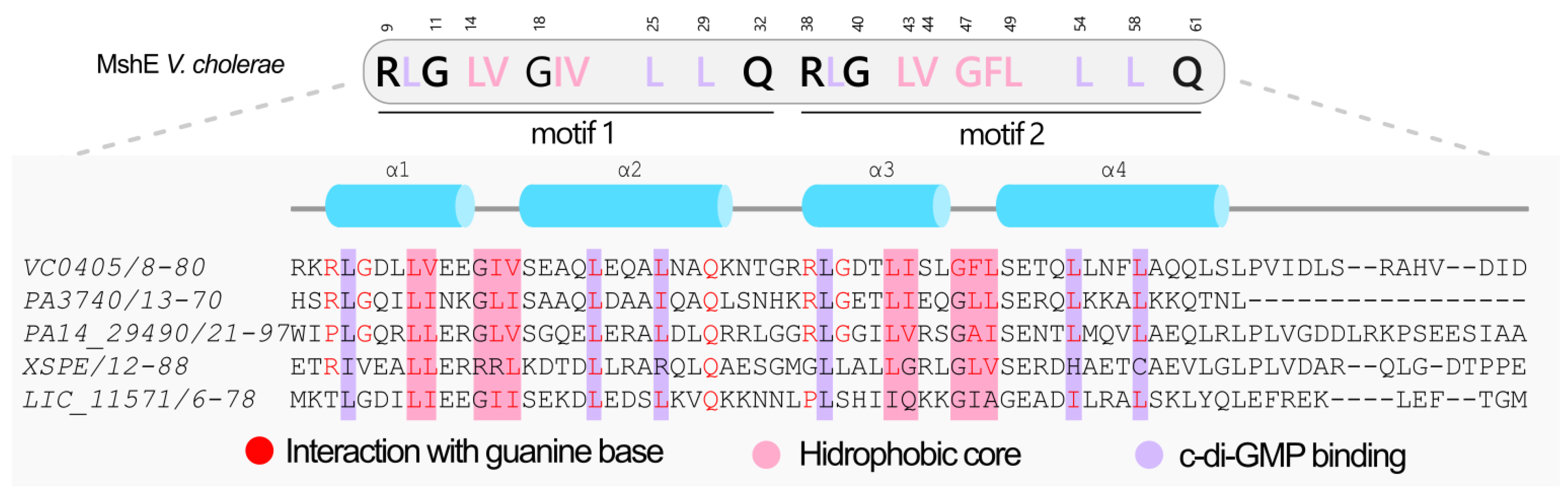
Figure 9.
Number of proteins containing GGDEF, EAL or PilZ domains in L. interrogans serovar Copenhageni Fiocruz L1-130. Experimental evidence was obtained by several authors, in different ways: from recombinant proteins and heterologous expression in E. coli [17,18,20] or by producing knockouts in L. interrogans [16]. Data of copies per cell was from a proteome of the cell sample subjected to extensive mapping via LC-MS/MS experiments [133]. (-) means that this functional isn’t applied to this protein; (?) we could not identify the protein in the list table of the article. (ND) no data was found for this protein. (NI) not identified.
Figure 9.
Number of proteins containing GGDEF, EAL or PilZ domains in L. interrogans serovar Copenhageni Fiocruz L1-130. Experimental evidence was obtained by several authors, in different ways: from recombinant proteins and heterologous expression in E. coli [17,18,20] or by producing knockouts in L. interrogans [16]. Data of copies per cell was from a proteome of the cell sample subjected to extensive mapping via LC-MS/MS experiments [133]. (-) means that this functional isn’t applied to this protein; (?) we could not identify the protein in the list table of the article. (ND) no data was found for this protein. (NI) not identified.
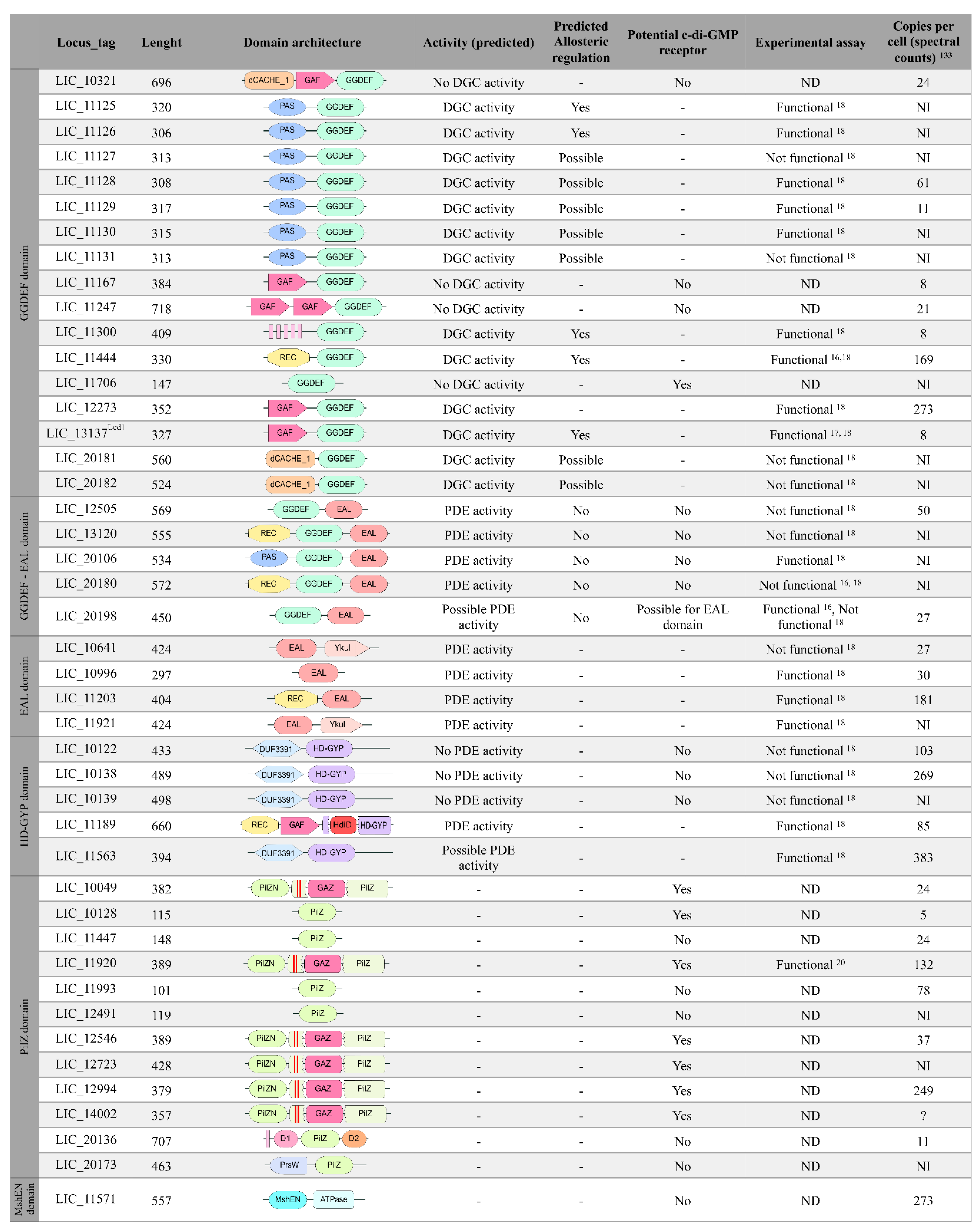
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



