
Submitted:
30 April 2025
Posted:
02 May 2025
You are already at the latest version
Abstract
Endosomal dysfunction is one of the earliest cellular signs in Alzheimer’s disease. Tumor susceptibility gene 101 protein (TSG101) is a component of the Endosomal Sorting Complex Required for Transport (ESCRT)-I, which plays a key role in sorting ubiquitinated cell-surface proteins and lipids onto intraluminal vesicles of multivesicular bodies for trafficking to lysosomes or autophagosomes for degradation, or to the plasma membrane for exosomal secretion. TSG101-dependent trafficking has been implicated in the propagation and spread of misfolded proteins associated with neurodegenerative diseases. We used transgenesis in mice to study the in vivo consequences of disrupting TSG101-dependent trafficking in adult neurons. Mice lacking Tsg101 in forebrain neurons (Tsg101ck2-null) showed rapid loss of hippocampal neurons and progressive forebrain atrophy. Astrogliosis was apparent in the dentate gyrus within 1 week of deleting Tsg101, followed by apoptosis of hippocampal CA3 neurons and accumulation of the autophagy adapter P62/SQSTM1 and ubiquitinated proteins. Failure to detect lipidated LC3 indicated autophagy was impaired rather than upregulated. Endosomal markers (RAB5 and RAB7) and amyloid protein also accumulated in hippocampal neurons of Tsg101ck2-null mice. Our data establish a critical role for TSG101 in neuronal survival and demonstrate the importance of in vivo assessment of gene/protein function.
Keywords:
tumor susceptibility gene 101 (Tsg101/TSG101)
; endosomal trafficking
; neurodegeneration
; amyloid beta
1. Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disorder, with an estimated 55 million people worldwide currently living with AD. It is associated with the accumulation of extracellular amyloid β (Aβ) plaques and intracellular neurofibrillary tangles of hyperphosphorylated tau protein (p-tau), but one of the earliest cellular features is endosomal dysfunction in neurons [1,2,3,4]. Studies have detected abnormally enlarged early endosomes and elevated expression of the small guanosine triphosphatase (GTPase), ras-related proteins RAB5 (early endosome marker) and RAB7 (late endosome marker) in hippocampal neurons in individuals with mild cognitive impairment and AD [4,5,6]. In multiple cell types from AD patients or mouse models, endocytic activation associated with increased RAB5 activity led to late endosomal dysfunction [7,8], which suggests that increased RAB5 activity could in turn lead to elevated RAB7 expression. RAB5 is aberrantly activated by APP-βCTF (secretase-cleaved C-terminal fragment (CTF) of amyloid precursor protein, APP), and transgenic expression of activated RAB5 in mouse neurons led to enlargement and mistrafficking of early endosomes as well as synaptic plasticity deficits, tau hyperphosphorylation, and cognitive deficits [7,9,10]. Thus, endosomal defects may play a direct role in AD pathogenesis.
Tumor susceptibility gene 101 (TSG101) is a housekeeping gene that encodes a multidomain protein with many functions in different cellular compartments. TSG101 has been proposed to play roles in transcriptional regulation, cell proliferation, cell division, retroviral particle assembly, and viral budding, but it is best characterized as a component of the Endosomal Sorting Complex Required for Transport (ESCRT)-I [11,12,13,14,15,16,17,18,19,20,21]. The ESCRT machinery sorts ubiquitinated proteins onto intraluminal vesicles (ILVs) of multivesicular bodies (MVBs) for trafficking either to the lysosome, for degradation, or to the plasma membrane, whereupon ILVs are secreted as a subclass of nanosized extracellular vesicles (EVs) called exosomes [22,23,24]. The cellular mechanisms that determine whether MVBs traffic their ubiquitinated cargoes to the lysosome or the plasma membrane remain poorly understood. APP is ubiquitinated and sorted through the endosomal system in an ESCRT-dependent manner [25,26,27,28], and Tsg101 depletion in cultured cells inhibited the sorting of APP onto ILVs and to the lysosome, resulting in intracellular accumulation of Aβ and a concomitant decrease in Aβ secretion [26].
There are many additional links between the endo-lysosomal pathway and AD. APP cleavage to form amyloid-β (Aβ) and CTFs is believed to occur, at least in part, in early endosomes [29,30,31,32], and the acidic environment of late endosomes/lysosomes would promote the self-assembly of Aβ peptides into fibrils that seed plaque formation when released from the cell, perhaps via exosomes or other EVs [33,34,35]. Neuronal cells in the brains of human AD patients and mouse and rat AD models, as well as in cell culture, have been shown to accumulate aggregation-prone and oligomerized Aβ peptides in MVBs, and to secrete exosomes containing full-length APP as well as CTFs and Aβ [36,37,38,39,40,41]. Neurally-derived exosomes isolated from the blood of AD patients were reported to contain significantly higher levels of lysosomal proteins (cathepsin D and LAMP-1) and ubiquitinated proteins, indicative of endo-lysosomal dysfunction [42]. ESCRT components were associated with amyloid plaques in AD transgenic mice [35]. Knockdown of ESCRT proteins, including TSG101, promoted tau aggregation [43], and endosomally-derived exosomes contain and may propagate the spread of p-tau [39,44,45,46,47].
Most studies on the molecular, biochemical, and intracellular functions of TSG101 have emphasized the relevance of their specific findings to diseases like cancer and neurodegeneration, but the proposed mechanisms are rarely validated in genetically defined, in vivo disease models or primary human tissue samples. Consequently, the biological significance of the multifaceted functions of TSG101 in normal development, differentiation, and tissue homeostasis, as well as the potential roles of TSG101-mediated pathways in disease pathogenesis, are poorly defined [22]. In the present study, we used tamoxifen-induced conditional mutagenesis to delete Tsg101 from forebrain neurons in adult mice. Neuronal deletion of Tsg101 resulted in reactive astrogliosis in the dentate gyrus starting within 1-week of Tsg101 deletion, followed by apoptosis of hippocampal CA3 neurons and forebrain atrophy. No upregulation of autophagy was detected. Impaired endosomal trafficking was indicated by the accumulation of ubiquitinated proteins, RAB5, and RAB7, and there was an increase in intracellular APP and/or its proteolytic derivatives in hippocampal neurons. This study supports the hypothesis that disrupted ESCRT function affects the trafficking of APP and its proteolytic derivatives, such as Aβ, and demonstrates that impairing TSG101-dependent processes leads to rapid onset neurodegeneration in brain areas impacted in AD.
2. Materials and Methods
2.1. Mice
Animals were housed under standard conditions in the Animal Resource Center at the McLaughlin Research Institute. Tsg101tm1-Kuw (Tsg101fl) conditional knockout mice, which were created and maintained on a 129/SvJ genetic background, have been described previously [19]. As this allele has loxP sites 3kb upstream and 230 bp downstream of the first coding exon of Tsg101, Cre recombinase excises the proximal promoter region and first exon of Tsg101, resulting in a null allele. Tsg101fl/fl mice were crossed to the tamoxifen-inducible Cre transgenic line, B6.FVB-Tg(Camk2a-cre/ERT2)2Gsc/Ieg (referred to as Camk2a-cre/ERT2), which expresses CreERT2 in forebrain neurons [48]. Camk2a-cre/ERT2 mice were obtained as frozen embryos from the European Mutant Mouse Archive, Munich, Germany (EMMA ID EM:02125) and rederived by the McLaughlin Research Institute Transgenic Facility (RRID:SCR_023108). Cre-positive F1’s were backcrossed to Tsg101fl/fl animals to produce Tsg101fl/fl and Tsg101fl/+ mice segregating for the Cre transgene. Tamoxifen treated Tsg101fl/+; Camk2a-cre/ERT2(+) or Tsg101fl/fl without the Camk2a-cre/ERT2 transgene, or vehicle-treated Tsg101fl/fl; Camk2a-cre/ERT2(+) were used as controls, and no significant differences in brain pathology were observed between these groups. Despite the mixed genetic background, no differences were observed in brain pathology between animals within the same genotype/treatment group. All animal procedures adhered to Association for Assessment and Accreditation of Laboratory Animal Care guidelines and were approved by the Institutional Animal Care and Use Committee of the McLaughlin Research Institute.
2.2. Genotyping
Animals were genotyped using PCR GoTaq Green 2X Master Mix (Promega). Tsg101 genotyping primers were: Tsg101wt GTTCGCTGAAGTAGAGCAGCCAG and CATTTCTGGAGTCCGATGCGCAG; Tsg101fl AGAGGCTATTCGGCTATGACTG and TTCGTCCAGATCATCCTGATC; Tsg101null GATGGTCATACCTGGTTAGAAAGC and CATTTCTGGAGTCCGATGCGCAG. The Camk2a-cre/ERT2 transgene was detected using forward primer GGTTCTCCGTTTGCACTCAGGA and reverse primer GCTTGCAGGTACAGGAGGTAGT.
2.3. Activation and Verification of Cre Recombinase Activity
Tsg101 deletion was induced at 12 weeks of age by twice-daily intraperitoneal injection of 0.025 mg tamoxifen (Sigma) per kg of body weight for three consecutive days, except for the oldest cohort (brains collected 140 days post-injection), which received twice-daily injections of 0.025 mg/kg for five consecutive days. Tamoxifen treated Tsg101fl/fl; Camk2a-cre/ERT2(+) mice are referred to hereafter as Tsg101ck2-null. Tamoxifen was dissolved to 5 μg/ml in a 1:9 mixture of absolute ethanol and pharmacological grade sunflower oil (Sigma). Control animals received injections of the 1:9 ethanol/oil vehicle mixture without tamoxifen. As Tsg101 deletion caused weight loss in Tsg101fl/fl; Camk2a-cre/ERT2(+) mice, excessive weight loss (≥20% of initial body weight) was used as a humane end-point criterion. A liquid dietary supplement (Ensure Plus or Esbilac) was provided to treated animals as needed to support the maintenance of a healthy body weight.
Cre recombinase-mediated deletion of the floxed Tsg101 sequence was verified by PCR using the genotyping primers described above and a template of 10 ng genomic DNA isolated from dissected brain regions of 3 vehicle- and 3 tamoxifen-injected animals (2 weeks post-treatment), as well as from brains of 3 Camk2a-cre/ERT2 negative animals, as a further negative control. Reduction of TSG101 protein levels was confirmed by immunoblotting of brain lysates isolated two weeks after tamoxifen treatment (n=3 per genotype/treatment), using rabbit anti-TSG101 (Proteintech Cat# 14497-1-AP, RRID:AB_2208090). Brain lysates were prepared by homogenizing brain tissue in solubilization buffer (50 mM Tris-HCl pH 8.0, 1 mM EDTA, 1% Igepal CA-630) containing cOmplete Protease Inhibitor Cocktail (Roche). Blots were imaged using ECL Plus chemiluminescent substrate (Pierce).
2.4. Histology
Brains were fixed in formalin, processed for histology by standard methods and embedded in paraffin. Experimental and control brains were processed together in each batch, with care taken to avoid long exposure to 70% ethanol as this can cause artifactual vacuolation in rodent nervous tissue [49]. Paraffin-embedded brains were sectioned at 3 or 5 mm and stained with hematoxylin and eosin (H&E). For the 1- and 2-weeks-post-injection cohort, brains of at least 4 animals were examined for each genotype/treatment combination. Brains from at least 3 animals of each genotype/treatment combination were examined for later time points except 20 weeks, as only two Tsg101ck2-null mice survived that long. Immunohistochemistry (IHC) was performed on paraffin sections, which were pretreated for antigen retrieval using 10mM sodium citrate (pH 6.0, 100 C, 10 minutes). Antibody information is provided in section 2.6. Immunolabelling was detected using fluorescent secondary antibodies (Vector Labs, CA), horseradish peroxidase (HRP)-conjugated secondary antibodies (anti-rabbit IgG-peroxidase, Sigma-Aldrich Cat# A0545, RRID:AB_257896), or HRP-conjugated avidin and biotinylated secondary antibodies of the appropriate isotype (Vector Labs, CA) and the peroxidase substrates diaminobenzidine (DAB) (Trevigen) and/or NovaRed (Vector Labs). Sections were counterstained with hematoxylin. For all studies, experimental and control samples were processed together to eliminate inter-experimental variability and at least 3 independent samples were examined for each genotype. Stained sections were imaged on a Zeiss AxioImagerM1 microscope using a PixieLink A623C color camera or on an Olympus Fluoview 1000 confocal microscope.
2.5. Immunoblotting
For western blotting, brain tissue was homogenized in RIPA lysis buffer (50 mM Tris, 150 mM NaCl, 1% NP40, 0.1% sodium deoxycholate) supplemented with cOmplete Protease Inhibitor Cocktail (Roche). Cellular debris was pelleted by centrifugation and the supernatant diluted in 2X SDS loading buffer. Proteins were electrophoresed through 8% or 12% SDS-polyacrylamide gels and transferred to Immobilon P membrane (Millipore). Immunoblotting was performed following standard protocols. Briefly, membranes were blocked with 5% nonfat dry milk in tris-buffered saline with Tween 20 (TTBS). Antibodies were diluted in TTBS and HRP-conjugated secondary antibodies (goat anti-rabbit IgG-peroxidase, Sigma-Aldrich Cat# A0545, RRID:AB_257896 or goat anti-mouse IgG, BD Biosciences Cat# 554002, RRID:AB_395198) were imaged using Clarity ECL Western Blotting Substrate (BioRad) and either a Bio-Rad ChemiDoc imaging system or the rapid capture setting on an Azure 300 Imager (Azure Biosystems). For LC3 western, the Azure software to verify the signal was not saturated, then bands were quantified using AzureSpot Pro (Azure Biosystems). Antibody information is provided in section 2.6.
2.6. Antibodies
Primary antibodies used were: rabbit anti-cleaved caspase-3 (Trevigen Cat# 2305-PC-100, RRID:AB_2665453), mouse anti-NeuN (Millipore Cat# MAB377, RRID:AB_2298772), rabbit anti-GFAP (Proteintech Cat# 16825-1-AP, RRID:AB_2109646), mouse anti-mono and polyubiuquitinated conjugates (Enzo Life Sciences Cat# BML-PW8810, RRID:AB_10541840), rabbit anti-RAB5 (LifeSpan Biosciences LS-C138527, now sold by Abcam, Cat# ab109534, RRID:AB_10865740), guinea pig anti-p62 (ARP American Research Products Cat# 03-GP62-C, RRID:AB_1542690), rabbit anti-LC3A (Novus Cat# NB100-2331, RRID:AB_10001955), rabbit anti-LC3 (RRID:AB_2716621, a gift from Dr. Masahiro Shibata), mouse anti-RAB7 (Abcam Cat# ab58029, RRID:AB_945127), and mouse monoclonal 4G8 (BioLegend Cat#800701, RRID:AB_2564633) which is reactive to amino acids 17-24 of Aβ and to APP. For immunoblotting, mouse anti-beta-tubulin-III (3F3-G2) (Santa Cruz Biotechnology Cat# sc-53140, RRID:AB_793543) was used as a loading control.
2.6. Statistics
Brain weight, body weight, food consumption, and protein levels were assessed for statistically significant differences using Student’s t-test. Minimum sample sizes were selected to provide at least 90% power of detecting a significant difference (p = 0.05) if at least 90% of animals within a genotype/treatment group gave a consistent phenotype or result. As some of the Tsg101ck2-null mice were lost within 14 days of Tsg101 deletion, an excess number of Tsg101fl/fl; Camk2a-cre/ERT2(+) mice were treated with tamoxifen to collect the desired number of mice at later time-points.
3. Results
Tsg101 was ablated in forebrain neurons of adult Tsg101 conditional knockout mice carrying the Camk2a-cre/ERT2 transgene by treating them with tamoxifen at 12 weeks of age (Figure 1A). This cre transgene has previously been shown to have low activity in the hippocampus in the absence of tamoxifen and robust activity, especially in neurons of the cortex, dentate gyrus, and hippocampus upon tamoxifen treatment [48]. Initially, Tsg101fl/fl; Camk2a-cre/ERT2+ and control (Tsg101fl/fl; Camk2a-cre/ERT2- and Tsg101fl/-; Camk2a-cre/ERT2+) mice were given twice daily IP injections of tamoxifen or vehicle control (1:9 ethanol/sunflower oil) for 5 days. Deletion of Tsg101 in tamoxifen-treated mice was verified by PCR on DNA isolated from dissected regions of the CNS and the sciatic nerve using primers that distinguish the endogenous wild-type allele, the floxed allele, and the recombined (deleted) allele (Figure 1B). Immunoblotting confirmed a reduction in TSG101 protein levels in the brains of tamoxifen-treated Tsg101fl/fl; Camk2a-cre/ERT2+ mice, referred to hereafter as Tsg101ck2-null (Figure 1C). In initial studies, these mice showed significant, progressive weight loss and reduced food intake relative to controls, starting 9 days after the initiation of tamoxifen treatment (Figure 2A,B). By 13-14 days post-tamoxifen, Tsg101ck2-null mice lost ≥ 20% of their body weight and were humanely euthanized. Home cage observation indicated that reduced food consumption coincided with onset of a head “shaking” or bobbing behavior, which may have made it difficult for the mice to feed from the overhead hopper or on pieces of food on the cage floor, but they also appeared to generally lose interest in even trying to eat. The tamoxifen treatment regime was reduced to 2 injections per day for 3 consecutive days and Tsg101ck2-null mice were offered a pipette of liquid Ensure meal replacement or Esbilac supplement every 3-4 hours for ~2 weeks, starting 8 days after the initiation of tamoxifen injections. Under these conditions, the anorexia and weight loss experienced by Tsg101ck2-null mice appeared to be transient as, after liquid oral supplementation, most of the mice resumed appropriate feeding behavior and weight gain, although some still appeared frail and had reduced lifespan. As brain histology did not discern any differences between Tsg101ck2-null mice generated using either tamoxifen treatment regime, samples were used interchangeably.
Twenty weeks after tamoxifen treatment was initiated, Tsg101ck2-null mice showed a substantial reduction in forebrain size, apparent even upon gross inspection (Figure 3A,B). Hematoxylin and eosin (H&E)-stained coronal sections revealed marked reductions in the volume of the hippocampal formation (Figure 3C–H) and cortex layer 1 (Figure 3I–K). The granule cell layers of the dentate gyrus and hippocampus were much thinner in Tsg101ck2-null brains (Figure 3C–H), consistent with loss of neurons. To investigate the time course of neurodegeneration, Tsg101ck2-null and control brain sections were examined 1, 2, 4, and 6 weeks following tamoxifen treatment, using H&E staining to assess overall morphology and IHC for glial fibrillary acidic protein (GFAP) to detect reactive astrogliosis. One week after tamoxifen treatment, Tsg101ck2-null brains showed a small number of neurons with pyknotic nuclei in the CA3 subfield of the hippocampus and elevated GFAP immunoreactivity surrounding the granule cell layer of the dentate gyrus, indicative of reactive astrogliosis. The latter suggests neuronal homeostasis was disrupted in the dentate gyrus, despite no overt neuron loss (Figure 4A–D). Within 2 weeks of tamoxifen treatment, Tsg101ck2-null brains showed extensive loss of NeuN-positive neurons in the CA3 subfield of the hippocampus, associated with the presence of pyknotic nuclei (Figure 4E–J). Astrogliosis was still concentrated in the dentate gyrus, with weaker GFAP staining present more broadly throughout the hippocampal formation (Figure 4K,L). By 4 weeks after tamoxifen treatment, the pyramidal layers of all regions of the hippocampus as well as the dentate gyrus were thin and showed the presence of pyknotic nuclei (Figure 4M,N), and GFAP staining remained strongest in the dentate gyrus (Figure 4O,P). By the 6-week time point, only a thin layer of neurons remained in the granule cell layers of the hippocampus and dentate gyrus (Figure 4Q,R). At this time-point, astrogliosis was observed throughout the hippocampal formation and the dentate gyrus continued to show stronger GFAP staining than other regions (Figure 4S,T). Although cortex layer 1 was significantly thinner in the brains of Tsg101ck2-null mice 20 weeks after tamoxifen treatment, no difference was observed up to the 6-week time point (Figure 4U,V). Consistent with a significant loss of neurons, brains of Tsg101ck2-null mice weighed significantly less than those of control mice by the 2-week time point (Figure 4W) and atrophy was progressive, as indicated by the significant reduction in the brain weight of Tsg101ck2-null mice between the 2 and 6 week time points (Figure 4W). By 6-weeks post-tamoxifen, there was a ~20% reduction in total brain mass in Tsg101ck2-null mice relative to controls.
Degenerating pyramidal cells in the CA3 region of the hippocampus of Tsg101ck2-null mice showed morphological and immunohistochemical evidence of apoptosis, such as an abundance of pyknotic nuclei (Figure 4H,J) and expression of the cleaved (active) form of the apoptosis effector caspase-3 (CC3) (Figure 5A–C). CA3 pyramidal cells also showed increased staining for mono- and poly-ubiquitinated proteins (Figure 5D–F), consistent with impaired sorting of ubiquitinated proteins into MVBs, a process that includes substrate deubiquitination. As loss of TSG101 function has been reported to up-regulate autophagy and dysregulated autophagy contributes to caspase-dependent neuronal apoptosis in cultured cells [51,52], we examined expression of the autophagy adapter protein, p62/SQSTM1 (Sequestosome-1), as well as LC3 (microtubule-associated protein 1A/1B-light chain 3), which undergoes conversion from LC3-I to LC3-II via the addition of a phosphatidylethanolamine group to the C terminus prior to being recruited to autophagosomes. In Tsg101ck2-null brains collected 1 week following the initiation of tamoxifen treatment, SQSTM1-positive punctae were observed in some pyramidal cells in the dentate gyrus, and more frequently in the hippocampal CA3 region (Figure 5G–K). As SQSTM1 is degraded through autophagy, elevated levels could indicate impairment of this pathway. IHC for LC3B did not reveal punctae of staining in Tsg101ck2-null brains (data not shown). Immunoblotting indicated reduced expression of LC3-I in Tsg101ck2-null hippocampi but a band consistent in size with LC3-II was not detected, even on long exposures or on blots of subcellular fractions expected to contain autophagosomes, prepared as described by Lai et al. [53] (Figure 5L,M and data not shown). This result was replicated using two different antibodies against LC3. The reduced expression of LC3-I was unlikely to be due to a loss of neurons as expression was normalized against a neuron-specific marker, TUJ1. Taken together, these data suggest that either autophagy was not upregulated in Tsg101ck2-null neurons, or that it was a very early and transient effect since no LC3-II could be detected within one week of induction of Tsg101 deletion.
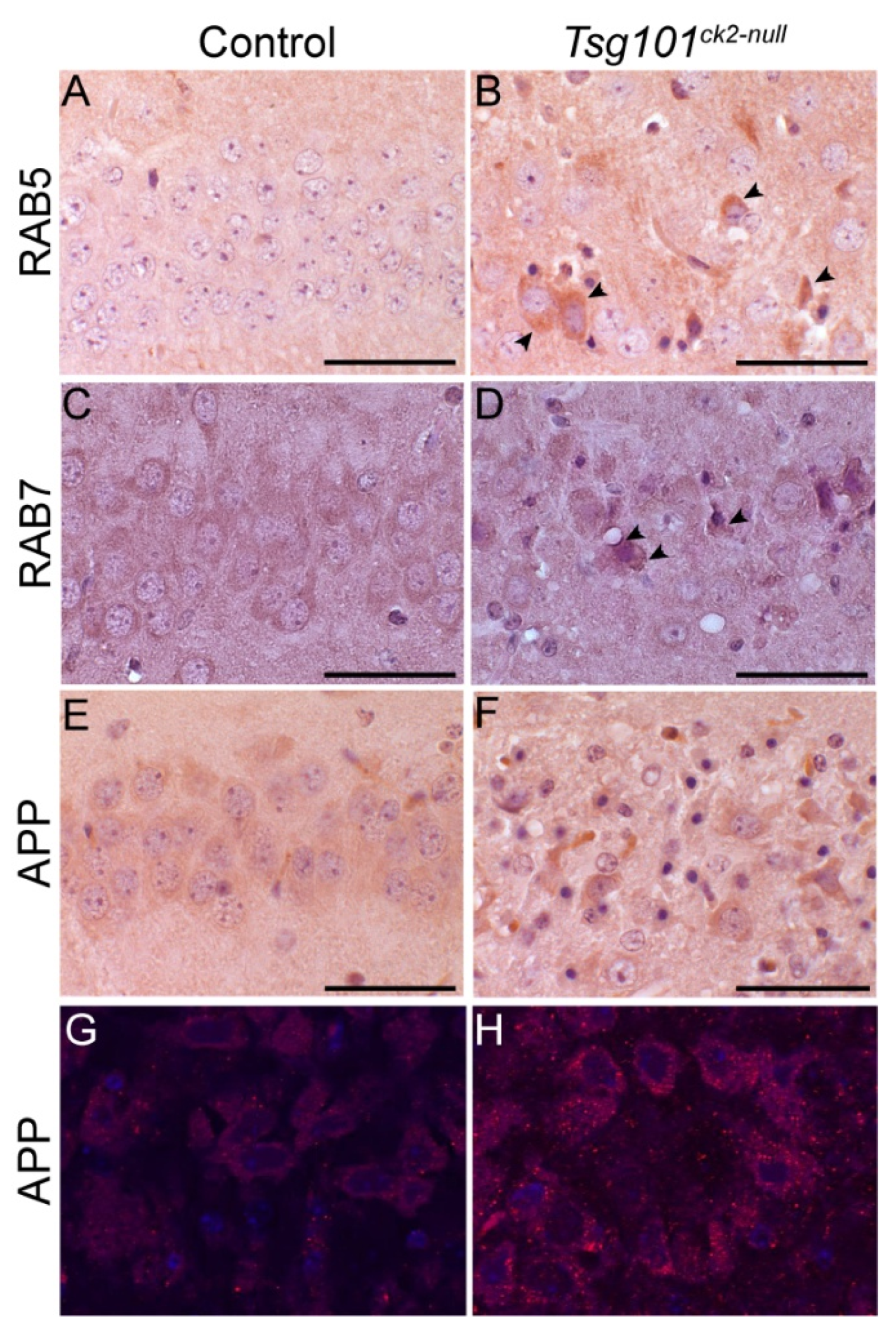
Our mouse model provided the opportunity to examine, in vivo, whether neuronal depletion of Tsg101 leads to endosomal defects similar to those observed in AD brains. IHC on coronal sections from Tsg101ck2-null and control brains collected 2-weeks post-tamoxifen revealed accumulation of RAB5 and RAB7 in hippocampal CA3 neurons (Figure 6A–D). Staining using an antibody reactive to APP amino acids 17-24 (clone 4G8), thus recognizing APP and Aβ, revealed more abundant intraneuronal staining in Tsg101ck2-null brains relative to controls (Figure 6E–H), consistent with impaired trafficking of APP/Aβ.
4. Discussion
The results of this study demonstrate that deleting Tsg101 from Camk2a-expressing neurons led to severe, progressive neurodegeneration. Within 1 week of Tsg101 deletion, significant GFAP staining was observed around the granule cell layer of the dentate gyrus and remained most pronounced in this region through all stages examined. Astrogliosis in the dentate gyrus would disrupt its ability to filter information passing through to the CA3 region of the hippocampus from the entorhinal cortex, which can lead to increased neuronal activity in the CA3 due to reduced recycling of glutamate and potassium from the synaptic cleft by activated astrocytes [54,55,56,57,58]. Hippocampal CA3 neurons showed caspase-dependent apoptosis within two-weeks of deleting Tsg101, consistent with astrogliosis in the dentate gyrus leading to excitotoxicity and apoptosis in the CA3. Although embryonic fibroblasts showed a transient upregulation of autophagy prior to apoptosis following the conditional deletion of Tsg101 [59], we did not detect lipidated LC3 (LC3-II) in Tsg101ck2-null brains 1 week after tamoxifen treatment was initiated. This suggests that, in vivo, either autophagy is not upregulated in neurons as a result of loss of Tsg101, or it occurs early and is very brief. The presence of reactive astrocytes in the dentate gyrus may explain the difference in cell death pathways triggered in neurons in culture and neurons in vivo. Reactive astrogliosis in the dentate gyrus could reflect either local effects of loss of Tsg101 from neurons in this region or disruption of neuronal signaling in the entorhinal cortex, which projects to and can induce astrocyte activation in the dentate gyrus. The entorhinal cortex is believed to play a role in the early stages of AD and often shows the earliest histopathological alterations, including the presence of neurofibrillary tangles and cell death [60]. We hypothesize that CA neurons may be the earliest to die in Tsg101ck2-null mice because the dentate gyrus provides excitatory inputs to the CA3, and impaired glutamate uptake by activated astrocytes in the dentate gyrus would be predicted to lead to excitotoxicity and apoptosis in CA3 neurons.
Endosomal abnormalities are one of the earliest pathological features of AD, characterized by enlarged RAB5-positive early endosomes and RAB7-positive late endosomes and progressive accumulation of MVBs, lysosomes and autophagosomes [4,8,61]. APP is known to traffic through the endosomal system, and the accumulation of APP and/or its N-terminal proteolytic derivatives in Tsg101ck2-null hippocampal neurons supports a role for the ESCRT pathway in normal APP trafficking. Our in vivo results are consistent with in vitro studies that demonstrated inhibited sorting of APP onto ILVs and to the lysosome, with intracellular accumulation of Aβ and a concomitant decrease in Aβ secretion when Tsg101 was depleted [26]. It is not clear whether accumulation of APP in Tsg101ck2-null neurons was due to reduced lysosomal degradation or impaired exosomal release, as both can be MVB-dependent. Exosomes isolated from mouse brain are enriched with toxic APP fragments [36], suggesting exosomal secretion may clear them from cells, but also deposit them into the extracellular space where they may contribute to Aβ plaques. Mice homozygous for the human apolipoprotein E4 (ApoE4) variant, which is the greatest genetic risk factor for AD in people, showed an age-dependent decrease in exosomes associated with reduced levels of TSG101 and RAB35 [62], both of which are involved in exosome biosynthesis and release [24,63]. The role of exosomes and other EVs in AD pathogenesis remains to be fully elucidated.
Generally, ESCRT dysfunction is associated with neurodegeneration. Mutations in the gene encoding the ESCRT-III component Charged Multivesicular Body Protein 2B (CHMP2B) are associated with frontotemporal dementia (FTD), FTD with motor neuron disease, and Amyotrophic Lateral Sclerosis (ALS) [64,65,66]. Studies using transgenic mice suggest Chmp2b mutations act through a gain-of-function mechanism [67,68]. Deleting the gene encoding the ESCRT-0 protein, HGF-regulated tyrosine kinase substrate, HGS (formerly referred to as HRS), from Synapsin-1-expressing neurons in mice resulted in apoptotic loss of hippocampal CA3 neurons and reduced locomotor activity and learning ability [69], but their phenotype was less severe than that observed in Tsg101ck2-null mice. Deleting Hgs from mouse forebrain neurons using a constitutive Camk2a-cre resulted in a phenotype more similar to what we report here for Tsg101ck2-null mice, including a marked accumulation of ubiquitinated proteins by 5 weeks of age and apoptotic and necrotic loss of hippocampal neurons by 7 weeks of age, with the CA3 region being most severely affected [70]. We have also demonstrated that deleting Tsg101 from Schwann cells in the mouse peripheral nervous system resulted in severe, rapid onset peripheral neuropathy associated with “onion bulb” formations and de/dysmyelination [71]. Mice lacking Hgs in Schwann cells displayed a much milder phenotype, displaying mild motor and sensory defects, fewer myelinated axons, and thinner and aberrantly folded myelin sheaths in the sciatic nerve [72]. Our group also previously demonstrated that conditional ablation of Tsg101 in oligodendroglia of the central nervous system (CNS) resulted in severe and rapid onset spongiform neurodegeneration and de/dys-myelination [73]. In the same study, we showed that oligodendroglial deletion of the gene encoding RAB7 did not produce any histopathological abnormalities, suggesting that the severe phenotypes associated with TSG101 deficiency were not due to defects in late endosomal, lysosomal, or autophagosomal functions, which are RAB7-dependent [74,75,76]. Taken together, these studies demonstrate critical roles for TSG101 in a variety of nervous system cell types.
Although HGS interacts with TSG101 to recruit ESCRT-I to endosomes [77,78,79], in multiple mammalian cell lines, knockdown of TSG101 or HGS had significantly different effects on epidermal growth factor receptor (EGFR) trafficking and degradation and endosomal and MVB morphology [59,77,78,80,81]. Depleting TSG101 resulted in the formation of multicisternal early endosomes and defects in protein sorting, as well as endoplasmic reticulum (ER) stress, ER membrane remodeling, and upregulation of autophagy [59,80,81,82,83]. Down-regulation of TSG101 has also been reported to result in reduced secretion of exosomes [24,84]. Depleting HGS did not induce early endosome tubulation and led to the production of enlarged MVBs containing fewer ILVs [17,80,82]. Thus, TSG101 appears to be required for the formation of stable vacuolar domains within the early endosome that subsequently develop into MVBs, while HGS is more important in the formation and/or accumulation of ILVs within MVBs. TSG101 deletion also disrupts the recycling of membrane proteins by decoupling early endosomes from recycling endosomes [22]. Given their roles in MVB production, it is not surprising that HGS and TSG101 are both involved in exosome production, with HGS depletion resulting in smaller exosomes and TSG101 depletion leading to fewer exosomes and a concomitant increase in other EV subtypes [24,80].
5. Conclusions
The phenotype we observed in Tsg101ck2-null mice validated some findings from cell culture studies, such as the intracellular accumulation of APP and/or its proteolytic derivatives in Tsg101-depleted neurons. Other findings in the whole brain contrasted with observations in cell culture models. For example, we did not observe lipidated LC3 in vivo within 1 week of Tsg101 deletion, which was prior to detecting autophagy in hippocampal CA3 neurons, suggesting that if autophagy was upregulated in vivo, it was not sustained. Assessing neuronal depletion of Tsg101 in the context of the whole brain also provided important insight into the influence of neuronal connectivity in disease pathogenesis and progression. Although all Camk2a-expressing forebrains neurons lost Tsg101, hippocampal CA3 pyramidal neurons were the first to undergo apoptosis and showed a more robust accumulation of ubiquitinated proteins and p62/SQTM1 by the 2-week timepoint, whereas the dentate gyrus showed the earliest and consistently most pronounced reactive astrocytosis. We hypothesize that depleting Tsg101 from neurons in the dentate gyrus disrupted neurotrophic signaling, triggering reactive astrogliosis to support neuronal survival and repair. The ability of activated astrocytes to execute normal functions, such as uptake of excess glutamate from the synaptic cleft, would be impaired and we propose that this leads to excitotoxicity and apoptosis in CA3 neurons. Our results stress the importance of in vivo studies to fully assess gene/protein function, especially for cells that contribute to organs like the brain, where communication between cells and different cell types is crucial.
These studies provide important proof-of-concept data toward the elucidation of complex neurodegenerative pathways in diseases such as AD. Endolysosomal abnormalities have been noted in AD-affected brain tissue and are hypothesized to contribute to the pathology of AD. Our results lend support to this hypothesis by demonstrating that impairment of the endo-lysosomal sorting machinery at TSG101 is sufficient to induce neurodegeneration in the hippocampal neurons that are most susceptible in AD, as well as to recapitulate a subset of features of AD pathology, including the accumulation of endosomal markers and Aβ. Finally, these studies demonstrate that TSG 101-dependent trafficking in neurons is critical for maintaining neuronal homeostasis.
Author Contributions
Conceptualization, W.P.W. and T.M.G.; methodology, W.P.W., T.M.G., K-U.W.; validation, W.P.W., M.L.R-M., T.M.G.; formal analysis, W.P.W. and T.M.G.; investigation, W.P.W., M.L.R-M., TMG; resources, K-U.W.; writing—original draft preparation, W.P.W. and T.M.G.; writing—review and editing, W.P.W., M.L.R-M., K-U.W., T.M.G.; visualization, W.P.W. and T.M.G.; supervision, T.M.G.; project administration, W.P.W. and T.M.G.; funding acquisition, T.M.G. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by The McLaughlin Research Institute and its generous supporters. W.P.W. was supported by the Oakland family and the Montana Department of Commerce. K-U.W. is the Lloyd and Marilyn Smith Endowed Professor for Breast Cancer Research at the Karmanos Comprehensive Cancer Center.
Acknowledgments
Thanks to Sarah Anderson and Derek Silvius for technical assistance, and Anita Pecukonis and Rachel Marden for exceptional animal care.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| TSG101 | Tumor susceptibility gene 101 |
| ESCRT | Endosomal sorting complex required for transport |
| p62/SQTM1 | Sequestosome-1 |
| RAB5 | Ras-related protein 5 |
| RAB7 | Ras-related protein 7 |
| LC3 | Microtubule-associated protein 1 light chain 3 beta (MAP1LC3B, ATG8) |
| CA3 | Cornu Ammonis subfield 3 of the hippocampus |
| AD | Alzheimer’s Disease |
| Aβ | A-beta or amyloid beta (proteolytic product of APP) |
| p-tau | Phosphorylated microtubule-associated protein tau (MAPT) |
| GTPase | guanosine triphosphatase |
| APP | Amyloid precursor protein |
| APP-βCTF | APP β-cleavage C-terminal fragment (precursor to Aβ) |
| MVB | Multivesicular body |
| ILV | Intraluminal vesicle |
| EV | Extracellular vesicle |
| PCR | Polymerase chain reaction |
| Camk2a | Calcium/calmodulin-dependent protein kinase type II subunit alpha |
| Cre/ERT2 | Tamoxifen inducible cre recombinase |
| IHC | immunohistochemistry |
| HRP | horseradish peroxidase |
| DAB | 3,3′-Diaminobenzidine |
| RIPA | Radioimmunoprecipitation assay buffer |
| TTBS | Tween Tris-buffered saline |
| CC3 | Cleaved caspase 3 |
| GFAP | Glial fibrillary acidic protein |
| NeuN | Neuronal nuclear antigen |
| 4G8 | Antibody reactive to amino acids 17-24 of APP and Aβ |
| CNS | Central nervous system |
| DNA | Deoxyribonucleic acid |
| H&E | Hematoxylin and eosin |
| TUJ1 | tubulin, beta 3 class III (TUBB3) |
References
- Ginsberg, S.D.; Alldred, M.J.; Counts, S.E.; Cataldo, A.M.; Neve, R.L.; Jiang, Y.; Wuu, J.; Chao, M.V.; Mufson, E.J.; Nixon, R.A.; et al. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol. Psychiatry 2010, 68, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.; Rebeck, G.W.; Ghetri, B.; Hulette, C.; Lippa, C.; Van Broeckhoven, C.; van Duijn, C.; Cras, P.; Bogdanovic, N.; Bird, T.; et al. Endocytic disturbances distinguish among subtypes of Alzheimer’s disease and related disorders. Ann. Neurol. 2001, 50, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Hamilton, D.J.; Barnett, J.L.; Paskevich, P.A.; Nixon, R.A. Properties of the endosomal-lysosomal system in the human central nervous system: disturbances mark most neurons in populations at risk to degenerate in Alzheimer’s disease. J. Neurosci. 1996, 16, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Peterhoff, C.M.; Troncoso, J.C.; Gomez-Isla, T.; Hyman, B.T.; Nixon, R.A. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 2000, 157, 277–286. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Mufson, E.J.; Alldred, M.J.; Counts, S.E.; Wuu, J.; Nixon, R.A.; Che, S. Upregulation of select rab GTPases in cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. J. Chem. Neuroanat. 2011, 42, 102–110. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Petanceska, S.; Peterhoff, C.M.; Terio, N.B.; Epstein, C.J.; Villar, A.; Carlson, E.J.; Staufenbiel, M.; Nixon, R.A. App gene dosage modulates endosomal abnormalities of Alzheimer’s disease in a segmental trisomy 16 mouse model of down syndrome. J. Neurosci. 2003, 23, 6788–6792. [Google Scholar] [CrossRef]
- Xu, W.; Weissmiller, A.M.; White, J.A., 2nd; Fang, F.; Wang, X.; Wu, Y.; Pearn, M.L.; Zhao, X.; Sawa, M.; Chen, S.; et al. Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J. Clin. Invest. 2016, 126, 1815–1833. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Mathews, P.M.; Boiteau, A.B.; Hassinger, L.C.; Peterhoff, C.M.; Jiang, Y.; Mullaney, K.; Neve, R.L.; Gruenberg, J.; Nixon, R.A. Down syndrome fibroblast model of Alzheimer-related endosome pathology: accelerated endocytosis promotes late endocytic defects. Am. J. Pathol. 2008, 173, 370–384. [Google Scholar] [CrossRef]
- Pensalfini, A.; Kim, S.; Subbanna, S.; Bleiwas, C.; Goulbourne, C.N.; Stavrides, P.H.; Jiang, Y.; Lee, J.H.; Darji, S.; Pawlik, M.; et al. Endosomal Dysfunction Induced by Directly Overactivating Rab5 Recapitulates Prodromal and Neurodegenerative Features of Alzheimer’s Disease. Cell reports 2020, 33, 108420. [Google Scholar] [CrossRef]
- Grbovic, O.M.; Mathews, P.M.; Jiang, Y.; Schmidt, S.D.; Dinakar, R.; Summers-Terio, N.B.; Ceresa, B.P.; Nixon, R.A.; Cataldo, A.M. Rab5-stimulated up-regulation of the endocytic pathway increases intracellular beta-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Abeta production. J. Biol. Chem. 2003, 278, 31261–31268. [Google Scholar] [CrossRef]
- Xie, W.; Li, L.; Cohen, S.N. Cell cycle-dependent subcellular localization of the TSG101 protein and mitotic and nuclear abnormalities associated with TSG101 deficiency. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 1595–1600. [Google Scholar] [CrossRef] [PubMed]
- Babst, M.; Odorizzi, G.; Estepa, E.J.; Emr, S.D. Mammalian tumor susceptibility gene 101 (TSG101) and the yeast homologue, Vps23p, both function in late endosomal trafficking. Traffic 2000, 1, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Bishop, N.; Woodman, P. TSG101/mammalian VPS23 and mammalian VPS28 interact directly and are recruited to VPS4-induced endosomes. J. Biol. Chem. 2001, 276, 11735–11742. [Google Scholar] [CrossRef] [PubMed]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Cote, M.; Rich, R.L.; et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef]
- Martin-Serrano, J.; Zang, T.; Bieniasz, P.D. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 2001, 7, 1313–1319. [Google Scholar] [CrossRef]
- Ruland, J.; Sirard, C.; Elia, A.; MacPherson, D.; Wakeham, A.; Li, L.; de la Pompa, J.L.; Cohen, S.N.; Mak, T.W. p53 accumulation, defective cell proliferation, and early embryonic lethality in mice lacking tsg101. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 1859–1864. [Google Scholar] [CrossRef] [PubMed]
- Bishop, N.; Horman, A.; Woodman, P. Mammalian class E vps proteins recognize ubiquitin and act in the removal of endosomal protein-ubiquitin conjugates. J. Cell Biol. 2002, 157, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Licata, J.M.; Simpson-Holley, M.; Wright, N.T.; Han, Z.; Paragas, J.; Harty, R.N. Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: involvement of host proteins TSG101 and VPS-4. J. Virol. 2003, 77, 1812–1819. [Google Scholar] [CrossRef]
- Wagner, K.U.; Krempler, A.; Qi, Y.; Park, K.; Henry, M.D.; Triplett, A.A.; Riedlinger, G.; Rucker, I.E.; Hennighausen, L. Tsg101 is essential for cell growth, proliferation, and cell survival of embryonic and adult tissues. Mol. Cell. Biol. 2003, 23, 150–162. [Google Scholar] [CrossRef]
- Oh, K.B.; Stanton, M.J.; West, W.W.; Todd, G.L.; Wagner, K.U. Tsg101 is upregulated in a subset of invasive human breast cancers and its targeted overexpression in transgenic mice reveals weak oncogenic properties for mammary cancer initiation. Oncogene 2007, 26, 5950–5959. [Google Scholar] [CrossRef]
- Dennaoui, R.; Radler, P.D.; Wicker, M.N.; Vistisen, K.; Ferraiuolo, R.; Triplett, A.A.; Shrestha, H.; Liner, T.A.; Manthey, K.C.; Rui, H.; et al. Overexpression of TSG101 causes the development of adenosquamousmammary carcinoma. Breast Cancer Research 2025. [Google Scholar]
- Ferraiuolo, R.M.; Manthey, K.C.; Stanton, M.J.; Triplett, A.A.; Wagner, K.U. The Multifaceted Roles of the Tumor Susceptibility Gene 101 (TSG101) in Normal Development and Disease. Cancers 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Théry, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef] [PubMed]
- Morel, E.; Chamoun, Z.; Lasiecka, Z.M.; Chan, R.B.; Williamson, R.L.; Vetanovetz, C.; Dall’Armi, C.; Simoes, S.; Point Du Jour, K.S.; McCabe, B.D.; et al. Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat Commun 2013, 4, 2250. [Google Scholar] [CrossRef]
- Edgar, J.R.; Willen, K.; Gouras, G.K.; Futter, C.E. ESCRTs regulate amyloid precursor protein sorting in multivesicular bodies and intracellular amyloid-beta accumulation. J. Cell Sci. 2015, 128, 2520–2528. [Google Scholar] [CrossRef]
- Haass, C.; Koo, E.H.; Mellon, A.; Hung, A.Y.; Selkoe, D.J. Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature 1992, 357, 500–503. [Google Scholar] [CrossRef]
- Toh, W.H.; Tan, J.Z.; Zulkefli, K.L.; Houghton, F.J.; Gleeson, P.A. Amyloid precursor protein traffics from the Golgi directly to early endosomes in an Arl5b- and AP4-dependent pathway. Traffic 2017, 18, 159–175. [Google Scholar] [CrossRef]
- Li, J.; Kanekiyo, T.; Shinohara, M.; Zhang, Y.; LaDu, M.J.; Xu, H.; Bu, G. Differential regulation of amyloid-beta endocytic trafficking and lysosomal degradation by apolipoprotein E isoforms. J. Biol. Chem. 2012, 287, 44593–44601. [Google Scholar] [CrossRef]
- Yamazaki, T.; Koo, E.H.; Selkoe, D.J. Trafficking of cell-surface amyloid beta-protein precursor. II. Endocytosis, recycling and lysosomal targeting detected by immunolocalization. J. Cell Sci. 1996, 109 Pt 5, 999–1008. [Google Scholar] [CrossRef]
- Yu, Y.; Gao, Y.; Winblad, B.; Tjernberg, L.O.; Schedin-Weiss, S. A Super-Resolved View of the Alzheimer’s Disease-Related Amyloidogenic Pathway in Hippocampal Neurons. J Alzheimers Dis 2021, 83, 833–852. [Google Scholar] [CrossRef] [PubMed]
- Koo, E.H.; Squazzo, S.L. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 1994, 269, 17386–17389. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Crick, S.L.; Bu, G.; Frieden, C.; Pappu, R.V.; Lee, J.M. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 20324–20329. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, R.P.; Tepper, K.; Rönicke, R.; Soom, M.; Westermann, M.; Reymann, K.; Kaether, C.; Fändrich, M. Mechanism of amyloid plaque formation suggests an intracellular basis of Abeta pathogenicity. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 1942–1947. [Google Scholar] [CrossRef]
- Willén, K.; Edgar, J.R.; Hasegawa, T.; Tanaka, N.; Futter, C.E.; Gouras, G.K. Aβ accumulation causes MVB enlargement and is modelled by dominant negative VPS4A. Molecular neurodegeneration 2017, 12, 61. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gonzalez, R.; Gauthier, S.A.; Kumar, A.; Levy, E. The exosome secretory pathway transports amyloid precursor protein carboxyl-terminal fragments from the cell into the brain extracellular space. J. Biol. Chem. 2012, 287, 43108–43115. [Google Scholar] [CrossRef]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 11172–11177. [Google Scholar] [CrossRef]
- Sardar Sinha, M.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol 2018, 136, 41–56. [Google Scholar] [CrossRef]
- Winston, C.N.; Goetzl, E.J.; Akers, J.C.; Carter, B.S.; Rockenstein, E.M.; Galasko, D.; Masliah, E.; Rissman, R.A. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimer’s & dementia (Amsterdam, Netherlands) 2016, 3, 63–72. [Google Scholar] [CrossRef]
- Laulagnier, K.; Javalet, C.; Hemming, F.J.; Chivet, M.; Lachenal, G.; Blot, B.; Chatellard, C.; Sadoul, R. Amyloid precursor protein products concentrate in a subset of exosomes specifically endocytosed by neurons. Cell. Mol. Life Sci. 2018, 75, 757–773. [Google Scholar] [CrossRef]
- Takahashi, R.H.; Milner, T.A.; Li, F.; Nam, E.E.; Edgar, M.A.; Yamaguchi, H.; Beal, M.F.; Xu, H.; Greengard, P.; Gouras, G.K. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am. J. Pathol. 2002, 161, 1869–1879. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Kapogiannis, D. Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 2015, 85, 40–47. [Google Scholar] [CrossRef]
- Chen, J.J.; Nathaniel, D.L.; Raghavan, P.; Nelson, M.; Tian, R.; Tse, E.; Hong, J.Y.; See, S.K.; Mok, S.A.; Hein, M.Y.; et al. Compromised function of the ESCRT pathway promotes endolysosomal escape of tau seeds and propagation of tau aggregation. J. Biol. Chem. 2019, 294, 18952–18966. [Google Scholar] [CrossRef]
- Polanco, J.C.; Scicluna, B.J.; Hill, A.F.; Götz, J. Extracellular Vesicles Isolated from the Brains of rTg4510 Mice Seed Tau Protein Aggregation in a Threshold-dependent Manner. J. Biol. Chem. 2016, 291, 12445–12466. [Google Scholar] [CrossRef]
- Baker, S.; Polanco, J.C.; Götz, J. Extracellular Vesicles Containing P301L Mutant Tau Accelerate Pathological Tau Phosphorylation and Oligomer Formation but Do Not Seed Mature Neurofibrillary Tangles in ALZ17 Mice. J Alzheimers Dis 2016, 54, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Jackson, N.A.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L. The prion-like transmission of tau oligomers via exosomes. Frontiers in aging neuroscience 2022, 14, 974414. [Google Scholar] [CrossRef]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of Tau via exosomes. Molecular neurodegeneration 2017, 12, 5. [Google Scholar] [CrossRef]
- Erdmann, G.; Schutz, G.; Berger, S. Inducible gene inactivation in neurons of the adult mouse forebrain. BMC Neurosci 2007, 8, 63. [Google Scholar] [CrossRef]
- Wells, G.A.; Wells, M. Neuropil vacuolation in brain: a reproducible histological processing artefact. J. Comp. Pathol. 1989, 101, 355–362. [Google Scholar] [CrossRef]
- Allen Reference Atlas - Mouse Brain [brain atlas]. Available from atlas.brain-map.org. 2004.
- Cheng, J.T.; Liu, P.F.; Yang, H.C.; Huang, S.J.; Griffith, M.; Morgan, P.; Shu, C.W. Tumor Susceptibility Gene 101 facilitates rapamycin-induced autophagic flux in neuron cells. Biomed. Pharmacother. 2021, 134, 111106. [Google Scholar] [CrossRef]
- Chung, Y.; Lee, J.; Jung, S.; Lee, Y.; Cho, J.W.; Oh, Y.J. Dysregulated autophagy contributes to caspase-dependent neuronal apoptosis. Cell Death Dis 2018, 9, 1189. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Hickey, R.W.; Chen, Y.; Bayir, H.; Sullivan, M.L.; Chu, C.T.; Kochanek, P.M.; Dixon, C.E.; Jenkins, L.W.; Graham, S.H.; et al. Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J. Cereb. Blood Flow Metab. 2008, 28, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Amaral, D.G.; Scharfman, H.E.; Lavenex, P. The dentate gyrus: fundamental neuroanatomical organization (dentate gyrus for dummies). Prog. Brain Res. 2007, 163, 3–22. [Google Scholar] [CrossRef]
- Hsu, D. The dentate gyrus as a filter or gate: a look back and a look ahead. Prog. Brain Res. 2007, 163, 601–613. [Google Scholar] [CrossRef]
- Price, B.R.; Johnson, L.A.; Norris, C.M. Reactive astrocytes: The nexus of pathological and clinical hallmarks of Alzheimer’s disease. Ageing research reviews 2021, 68, 101335. [Google Scholar] [CrossRef]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8. [Google Scholar] [CrossRef]
- Mira, R.G.; Cerpa, W. Building a Bridge Between NMDAR-Mediated Excitotoxicity and Mitochondrial Dysfunction in Chronic and Acute Diseases. Cell. Mol. Neurobiol. 2021, 41, 1413–1430. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.R.; Stanton, M.J.; Manthey, K.C.; Oh, K.B.; Wagner, K.U. A Knockout of the Tsg101 Gene Leads to Decreased Expression of ErbB Receptor Tyrosine Kinases and Induction of Autophagy Prior to Cell Death. PLoS One 2012, 7, e34308. [Google Scholar] [CrossRef]
- Igarashi, K.M. Entorhinal cortex dysfunction in Alzheimer’s disease. Trends Neurosci. 2023, 46, 124–136. [Google Scholar] [CrossRef]
- Peric, A.; Annaert, W. Early etiology of Alzheimer’s disease: tipping the balance toward autophagy or endosomal dysfunction? Acta Neuropathol 2015, 129, 363–381. [Google Scholar] [CrossRef]
- Peng, K.Y.; Pérez-González, R.; Alldred, M.J.; Goulbourne, C.N.; Morales-Corraliza, J.; Saito, M.; Saito, M.; Ginsberg, S.D.; Mathews, P.M.; Levy, E. Apolipoprotein E4 genotype compromises brain exosome production. Brain 2019, 142, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.; Morohashi, Y.; Yoshimura, S.; Manrique-Hoyos, N.; Jung, S.; Lauterbach, M.A.; Bakhti, M.; Grønborg, M.; Möbius, W.; Rhee, J.; et al. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A-C. J. Cell Biol. 2010, 189, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, G.; Parkinson, N.J.; Brown, J.M.; Chakrabarti, L.; Lloyd, S.L.; Hummerich, H.; Nielsen, J.E.; Hodges, J.R.; Spillantini, M.G.; Thusgaard, T.; et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 2005, 37, 806–808. [Google Scholar] [CrossRef]
- Cox, L.E.; Ferraiuolo, L.; Goodall, E.F.; Heath, P.R.; Higginbottom, A.; Mortiboys, H.; Hollinger, H.C.; Hartley, J.A.; Brockington, A.; Burness, C.E.; et al. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS One 2010, 5, e9872. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, A.M.; Johannsen, P.; Holm, I.; Nielsen, J.E. Frontotemporal dementia caused by CHMP2B mutations. Curr Alzheimer Res 2011, 8, 246–251. [Google Scholar] [CrossRef]
- Ghazi-Noori, S.; Froud, K.E.; Mizielinska, S.; Powell, C.; Smidak, M.; Fernandez de Marco, M.; O’Malley, C.; Farmer, M.; Parkinson, N.; Fisher, E.M.; et al. Progressive neuronal inclusion formation and axonal degeneration in CHMP2B mutant transgenic mice. Brain 2012, 135, 819–832. [Google Scholar] [CrossRef]
- Vernay, A.; Therreau, L.; Blot, B.; Risson, V.; Dirrig-Grosch, S.; Waegaert, R.; Lequeu, T.; Sellal, F.; Schaeffer, L.; Sadoul, R.; et al. A transgenic mouse expressing CHMP2Bintron5 mutant in neurons develops histological and behavioural features of amyotrophic lateral sclerosis and frontotemporal dementia. Hum. Mol. Genet. 2016, 25, 3341–3360. [Google Scholar] [CrossRef]
- Tamai, K.; Toyoshima, M.; Tanaka, N.; Yamamoto, N.; Owada, Y.; Kiyonari, H.; Murata, K.; Ueno, Y.; Ono, M.; Shimosegawa, T.; et al. Loss of hrs in the central nervous system causes accumulation of ubiquitinated proteins and neurodegeneration. Am. J. Pathol. 2008, 173, 1806–1817. [Google Scholar] [CrossRef]
- Oshima, R.; Hasegawa, T.; Tamai, K.; Sugeno, N.; Yoshida, S.; Kobayashi, J.; Kikuchi, A.; Baba, T.; Futatsugi, A.; Sato, I.; et al. ESCRT-0 dysfunction compromises autophagic degradation of protein aggregates and facilitates ER stress-mediated neurodegeneration via apoptotic and necroptotic pathways. Sci Rep 2016, 6, 24997. [Google Scholar] [CrossRef]
- Silvius, D.; Hurley, E.; Poitelon, Y.; Wagner, K.U.; Feltri, M.L.; Gunn, T.M. Schwann cell deletion of Tumor Susceptibility Gene 101 ( Tsg101 ) in mice results in severe peripheral neuropathy. microPublication biology 2025, 2025. [Google Scholar] [CrossRef]
- McLean, J.W.; Wilson, J.A.; Tian, T.; Watson, J.A.; VanHart, M.; Bean, A.J.; Scherer, S.S.; Crossman, D.K.; Ubogu, E.; Wilson, S.M. Disruption of Endosomal Sorting in Schwann Cells Leads to Defective Myelination and Endosomal Abnormalities Observed in Charcot-Marie-Tooth Disease. J. Neurosci. 2022, 42, 5085–5101. [Google Scholar] [CrossRef] [PubMed]
- Walker, W.P.; Oehler, A.; Edinger, A.L.; Wagner, K.U.; Gunn, T.M. Oligodendroglial deletion of ESCRT-I component TSG101 causes spongiform encephalopathy. Biol. Cell. 2016, 108, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Bucci, C.; Thomsen, P.; Nicoziani, P.; McCarthy, J.; van Deurs, B. Rab7: a key to lysosome biogenesis. Mol. Biol. Cell 2000, 11, 467–480. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Munafó, D.B.; Berón, W.; Colombo, M.I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004, 117, 2687–2697. [Google Scholar] [CrossRef]
- Hyttinen, J.M.; Niittykoski, M.; Salminen, A.; Kaarniranta, K. Maturation of autophagosomes and endosomes: a key role for Rab7. Biochim. Biophys. Acta 2013, 1833, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Bache, K.G.; Brech, A.; Mehlum, A.; Stenmark, H. Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J. Cell Biol. 2003, 162, 435–442. [Google Scholar] [CrossRef]
- Lu, Q.; Hope, L.W.; Brasch, M.; Reinhard, C.; Cohen, S.N. TSG101 interaction with HRS mediates endosomal trafficking and receptor down-regulation. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 7626–7631. [Google Scholar] [CrossRef]
- Pornillos, O.; Higginson, D.S.; Stray, K.M.; Fisher, R.D.; Garrus, J.E.; Payne, M.; He, G.P.; Wang, H.E.; Morham, S.G.; Sundquist, W.I. HIV Gag mimics the Tsg101-recruiting activity of the human Hrs protein. J. Cell Biol. 2003, 162, 425–434. [Google Scholar] [CrossRef]
- Razi, M.; Futter, C.E. Distinct roles for Tsg101 and Hrs in multivesicular body formation and inward vesiculation. Mol Biol Cell 2006, 17, 3469–3483. [Google Scholar] [CrossRef]
- Raiborg, C.; Malerod, L.; Pedersen, N.M.; Stenmark, H. Differential functions of Hrs and ESCRT proteins in endocytic membrane trafficking. Exp. Cell Res. 2008, 314, 801–813. [Google Scholar] [CrossRef]
- Doyotte, A.; Russell, M.R.; Hopkins, C.R.; Woodman, P.G. Depletion of TSG101 forms a mammalian “Class E” compartment: a multicisternal early endosome with multiple sorting defects. J. Cell Sci. 2005, 118, 3003–3017. [Google Scholar] [CrossRef] [PubMed]
- Kaul, Z.; Mookherjee, D.; Das, S.; Chatterjee, D.; Chakrabarti, S.; Chakrabarti, O. Loss of tumor susceptibility gene 101 (TSG101) perturbs endoplasmic reticulum structure and function. Biochimica et biophysica acta. Molecular cell research 2020, 1867, 118741. [Google Scholar] [CrossRef] [PubMed]
- Kilinc, S.; Paisner, R.; Camarda, R.; Gupta, S.; Momcilovic, O.; Kohnz, R.A.; Avsaroglu, B.; L’Etoile, N.D.; Perera, R.M.; Nomura, D.K.; et al. Oncogene-regulated release of extracellular vesicles. Dev Cell 2021, 56, 1989–2006. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Generation and validation of the Tsg10ck2-null allele. (A) Schematic representation of the 5′ region of the Tsg101fl conditional allele, showing loxP sites flanking exon 1 (top, in blue) and deletion of exon 1 in mice expressing the Camk2a-cre/ERT2 transgene following tamoxifen treatment (bottom, in red). (B) Allele-specific PCR results from amplification of the Tsg101fl (top) or Tsg101ck2-null allele (bottom) from DNA isolated from indicated tissues dissected from Camk2a-cre/ERT2 transgenic and non-transgenic Tsg101fl/+ animals two weeks after administration of tamoxifen or oil. Results shown are representative of 4 replicates. (C) Western blots of forebrain lysates from 3 Tsg101ck-null mice and 3 controls (Tsg101fl/fl without the Camk2a-cre/ERT2 transgene), showing a reduction in TSG101 protein levels in Cre(+) brain tissues 2 weeks after the initiation of tamoxifen treatment.
Figure 1.
Generation and validation of the Tsg10ck2-null allele. (A) Schematic representation of the 5′ region of the Tsg101fl conditional allele, showing loxP sites flanking exon 1 (top, in blue) and deletion of exon 1 in mice expressing the Camk2a-cre/ERT2 transgene following tamoxifen treatment (bottom, in red). (B) Allele-specific PCR results from amplification of the Tsg101fl (top) or Tsg101ck2-null allele (bottom) from DNA isolated from indicated tissues dissected from Camk2a-cre/ERT2 transgenic and non-transgenic Tsg101fl/+ animals two weeks after administration of tamoxifen or oil. Results shown are representative of 4 replicates. (C) Western blots of forebrain lysates from 3 Tsg101ck-null mice and 3 controls (Tsg101fl/fl without the Camk2a-cre/ERT2 transgene), showing a reduction in TSG101 protein levels in Cre(+) brain tissues 2 weeks after the initiation of tamoxifen treatment.
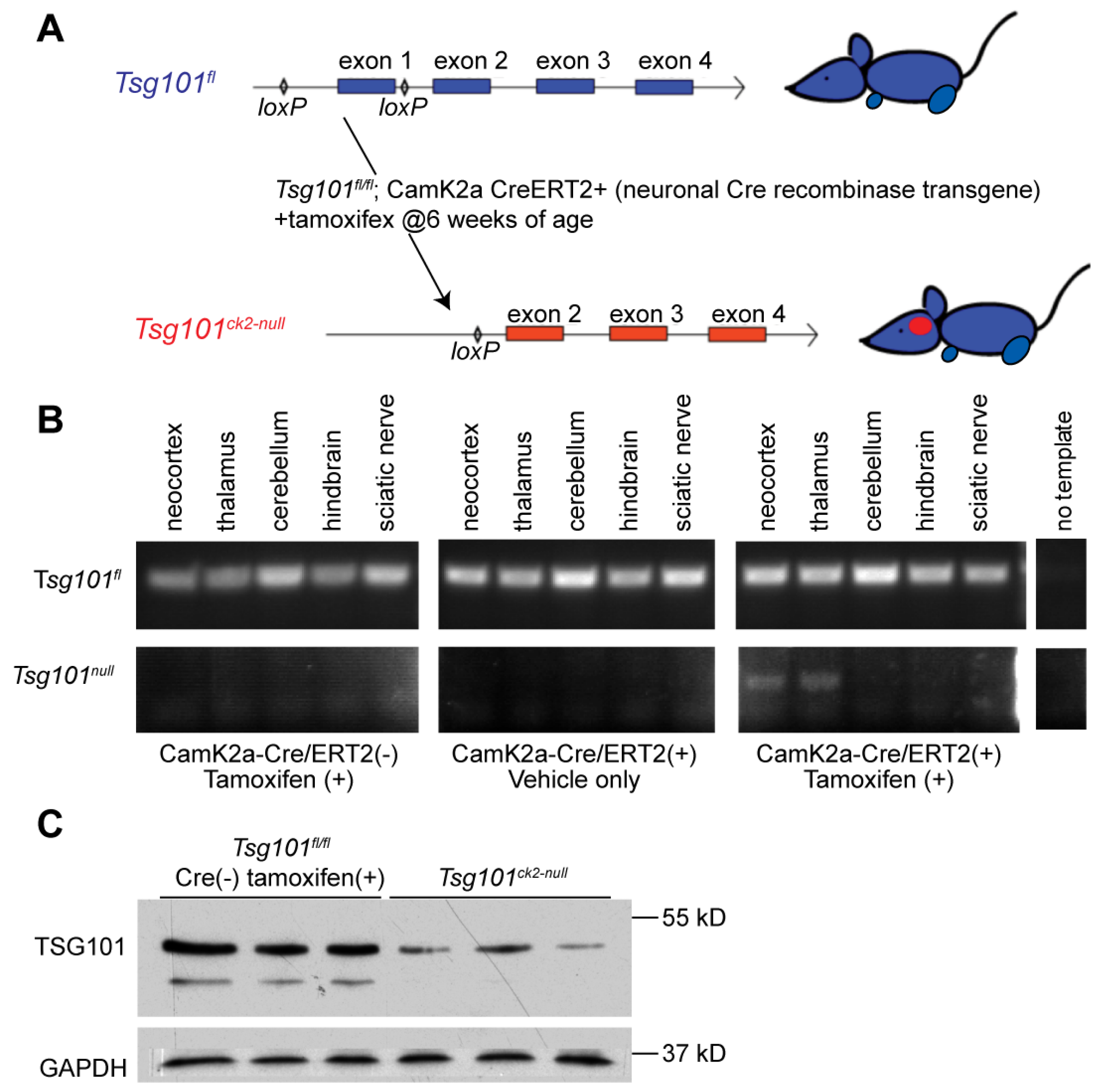
Figure 2.
Effects of neuronal ablation of Tsg101 on body weight and food intake. (A) Tsg101ck2-null mice showed a significant decline in body weight starting 9 days after tamoxifen treatment. (B) Tsg101ck2-null mice also showed a significant decline in food consumption starting 9 days after tamoxifen treatment.
Figure 2.
Effects of neuronal ablation of Tsg101 on body weight and food intake. (A) Tsg101ck2-null mice showed a significant decline in body weight starting 9 days after tamoxifen treatment. (B) Tsg101ck2-null mice also showed a significant decline in food consumption starting 9 days after tamoxifen treatment.
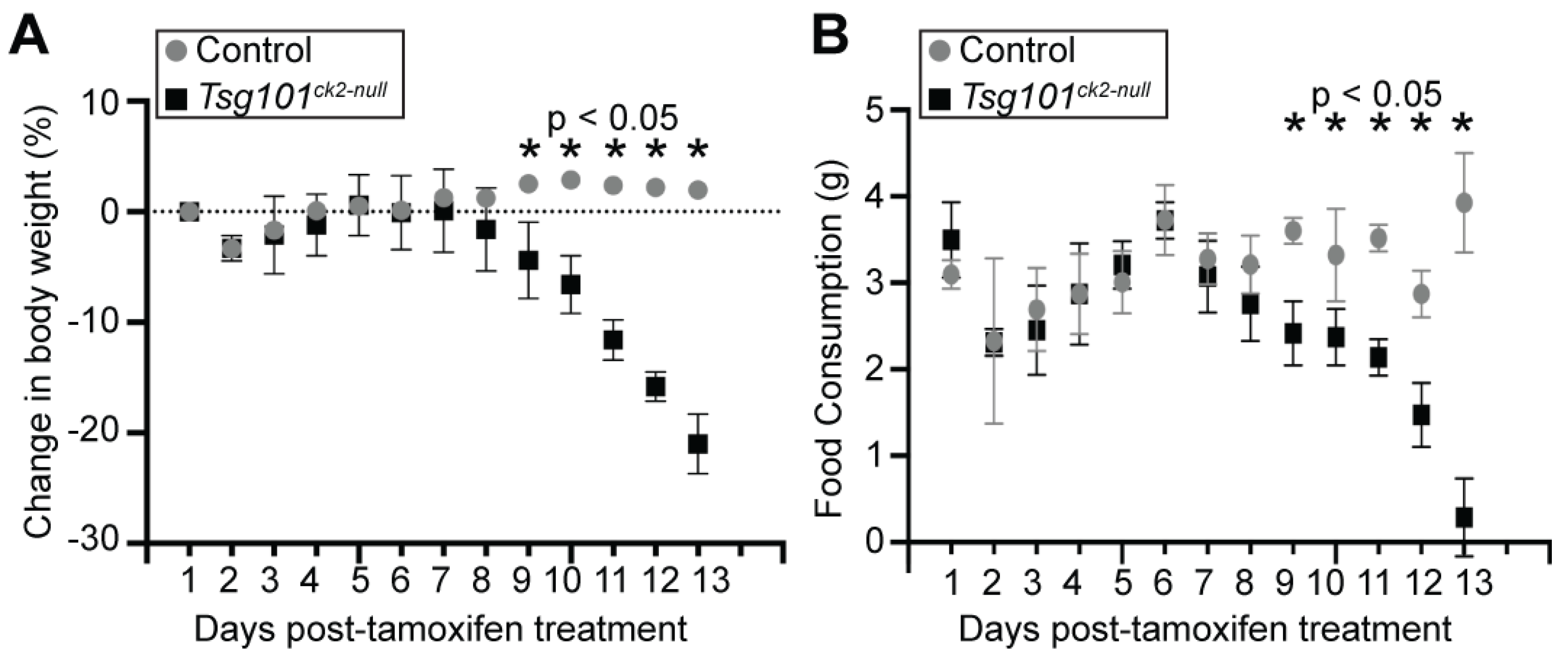
Figure 3.
Significant neurodegeneration occurs within 20 weeks of deleting Tsg101 from forebrain neurons. (A-B) Gross analysis of brains from tamoxifen-treated control (A) and Tsg101ck2-null (B) mice demonstrates severe neocortical atrophy. (C-H) Histological assessment of the hippocampus shows significant loss of pyramidal neurons in the hippocampus and dentate gyrus (E), shrinkage of the hippocampus (distance indicated by arrows in G and H), and reduction in the thickness of cortical layer 1 (indicated by * in J and K) in Tsg101ck2-null brains relative to controls (D, G, J, respectively). Panels C, F and I are from the Allen Mouse Brain Atlas [50] and the boxed areas indicate the regions shown at higher magnification in D-E, G-H, and J-K, respectively.
Figure 3.
Significant neurodegeneration occurs within 20 weeks of deleting Tsg101 from forebrain neurons. (A-B) Gross analysis of brains from tamoxifen-treated control (A) and Tsg101ck2-null (B) mice demonstrates severe neocortical atrophy. (C-H) Histological assessment of the hippocampus shows significant loss of pyramidal neurons in the hippocampus and dentate gyrus (E), shrinkage of the hippocampus (distance indicated by arrows in G and H), and reduction in the thickness of cortical layer 1 (indicated by * in J and K) in Tsg101ck2-null brains relative to controls (D, G, J, respectively). Panels C, F and I are from the Allen Mouse Brain Atlas [50] and the boxed areas indicate the regions shown at higher magnification in D-E, G-H, and J-K, respectively.
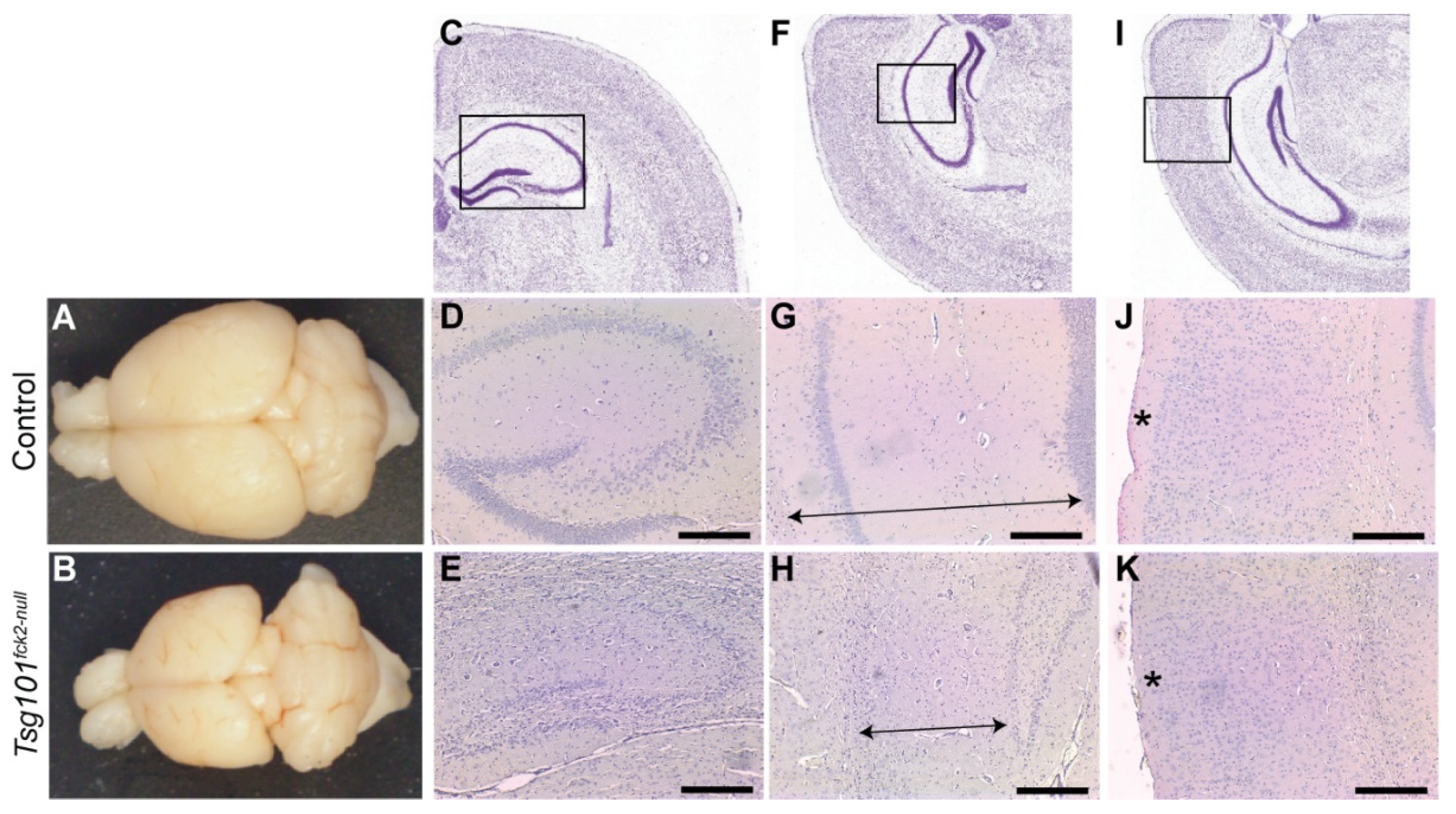
Figure 4.
Tsg101ck2-null brains show progressive loss of hippocampal neurons associated with reactive astrogliosis in the dentate gyrus. (A-T) Hippocampal sections from control and Tsg101ck2-null brains (as specified above each column) were H&E stained (A-B,E-H,M-N,Q-R) or subjected to IHC staining for GFAP (C,D,K,L,O,P,S,T) or NeuN (I-J) 1, 2, 4, and 6 weeks after mice were given tamoxifen (as indicated to the left of each row). (G-H) Pyknotic nuclei (arrowheads in H) were present in the CA3 region 2-weeks after Tsg101 deletion was induced by tamoxifen (I-J) Loss of pyramidal neurons and presence of pyknotic nuclei (arrowheads in J) were apparent in the CA3 region of Tsg101ck2-null mice at the 2-week timepoint. (U-V) H&E staining of the cortex indicated no difference in cortical layer 1 thickness by the 6-week timepoint. (W) Whole brain weight of Tsg101ck2-null and control mice 2- and 6- weeks after tamoxifen (+) or vehicle (−) injection. Statistically significant (p < 0.05) comparisons are indicated by asterisks. Scale bars: 500 µm in A-F and K-V, 50 µm in G-J.
Figure 4.
Tsg101ck2-null brains show progressive loss of hippocampal neurons associated with reactive astrogliosis in the dentate gyrus. (A-T) Hippocampal sections from control and Tsg101ck2-null brains (as specified above each column) were H&E stained (A-B,E-H,M-N,Q-R) or subjected to IHC staining for GFAP (C,D,K,L,O,P,S,T) or NeuN (I-J) 1, 2, 4, and 6 weeks after mice were given tamoxifen (as indicated to the left of each row). (G-H) Pyknotic nuclei (arrowheads in H) were present in the CA3 region 2-weeks after Tsg101 deletion was induced by tamoxifen (I-J) Loss of pyramidal neurons and presence of pyknotic nuclei (arrowheads in J) were apparent in the CA3 region of Tsg101ck2-null mice at the 2-week timepoint. (U-V) H&E staining of the cortex indicated no difference in cortical layer 1 thickness by the 6-week timepoint. (W) Whole brain weight of Tsg101ck2-null and control mice 2- and 6- weeks after tamoxifen (+) or vehicle (−) injection. Statistically significant (p < 0.05) comparisons are indicated by asterisks. Scale bars: 500 µm in A-F and K-V, 50 µm in G-J.
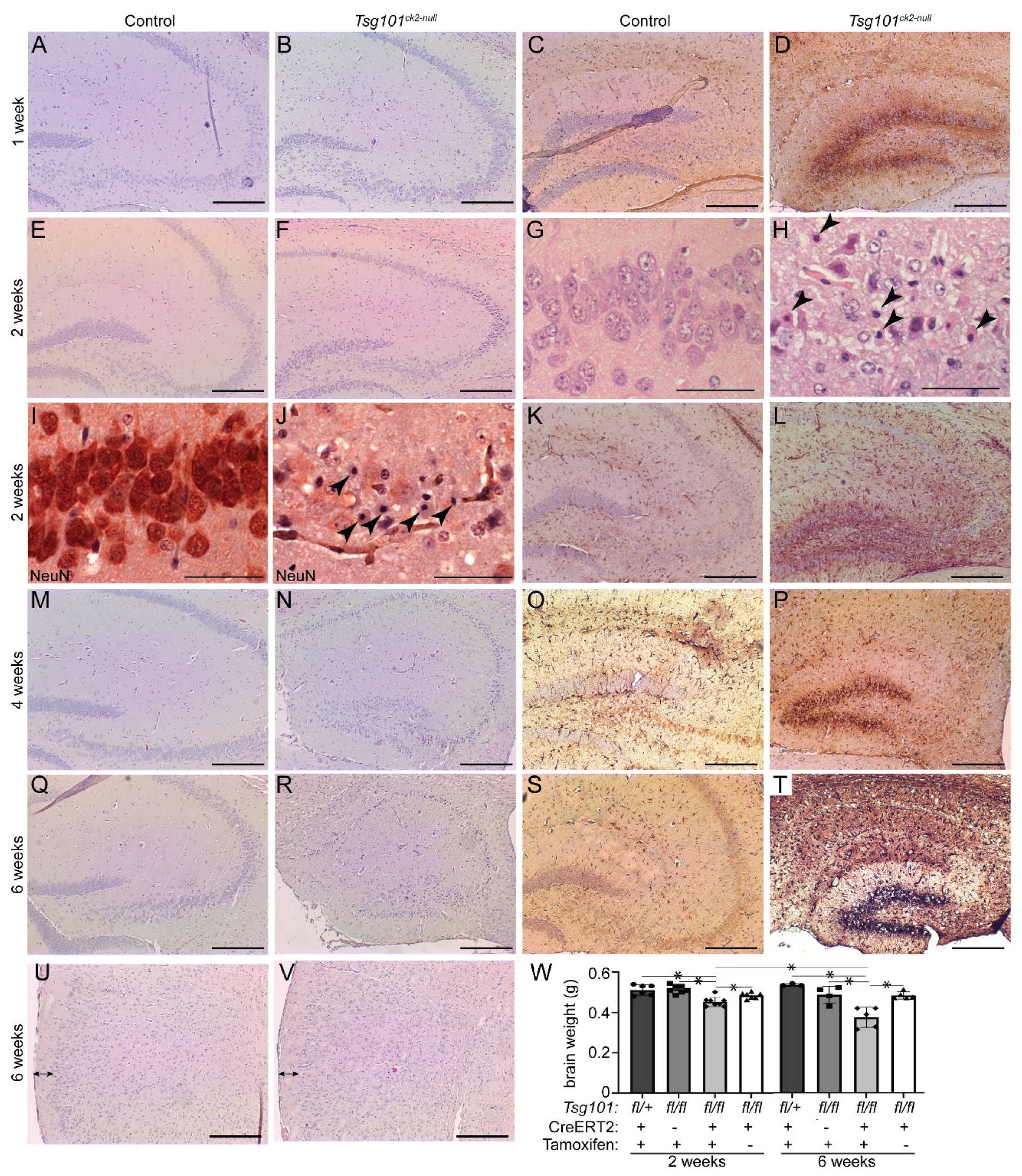
Figure 5.
Characterization of ubiquitination and cell death pathways in Tsg101ck2-null hippocampi 2-weeks after tamoxifen treatment. (A-C) IHC for activated cleaved caspase 3 (CC3) identified accumulation CC3-positive punctae (arrowheads) in Tsg101ck2-null hippocampal neurons. Boxed area in B is shown at higher magnification in C. (D-F) Ubiquitinated proteins were detected by IHC using the FK2 antibody. Tsg101ck2-null hippocampal neurons displayed elevated staining relative to controls, consistent with accumulation of ubiquitinated inclusions. Area boxed in E is shown at higher magnification in F. (G-K) IHC for p62/SQSTM1 revealed accumulation of p62 in CA3 neurons of Tsg101ck2-null mice. Boxed area in G is shown at higher magnification in J. Boxed areas in H are shown at higher magnification in I and K. (L-M) Hippocampi dissected from Tsg101ck2-null and control mice 1 week after tamoxifen treatment was initiated were subjected to western blotting for LC3-II (LC3B) and TUJ1. A band consistent in size with LC3B-I but not LC3B-II was detected, and when normalized against TUJ1, was significantly reduced in Tsg101ck2-null hippocampi, p = 0.0002. Scale bars represent 500 µm.
Figure 5.
Characterization of ubiquitination and cell death pathways in Tsg101ck2-null hippocampi 2-weeks after tamoxifen treatment. (A-C) IHC for activated cleaved caspase 3 (CC3) identified accumulation CC3-positive punctae (arrowheads) in Tsg101ck2-null hippocampal neurons. Boxed area in B is shown at higher magnification in C. (D-F) Ubiquitinated proteins were detected by IHC using the FK2 antibody. Tsg101ck2-null hippocampal neurons displayed elevated staining relative to controls, consistent with accumulation of ubiquitinated inclusions. Area boxed in E is shown at higher magnification in F. (G-K) IHC for p62/SQSTM1 revealed accumulation of p62 in CA3 neurons of Tsg101ck2-null mice. Boxed area in G is shown at higher magnification in J. Boxed areas in H are shown at higher magnification in I and K. (L-M) Hippocampi dissected from Tsg101ck2-null and control mice 1 week after tamoxifen treatment was initiated were subjected to western blotting for LC3-II (LC3B) and TUJ1. A band consistent in size with LC3B-I but not LC3B-II was detected, and when normalized against TUJ1, was significantly reduced in Tsg101ck2-null hippocampi, p = 0.0002. Scale bars represent 500 µm.
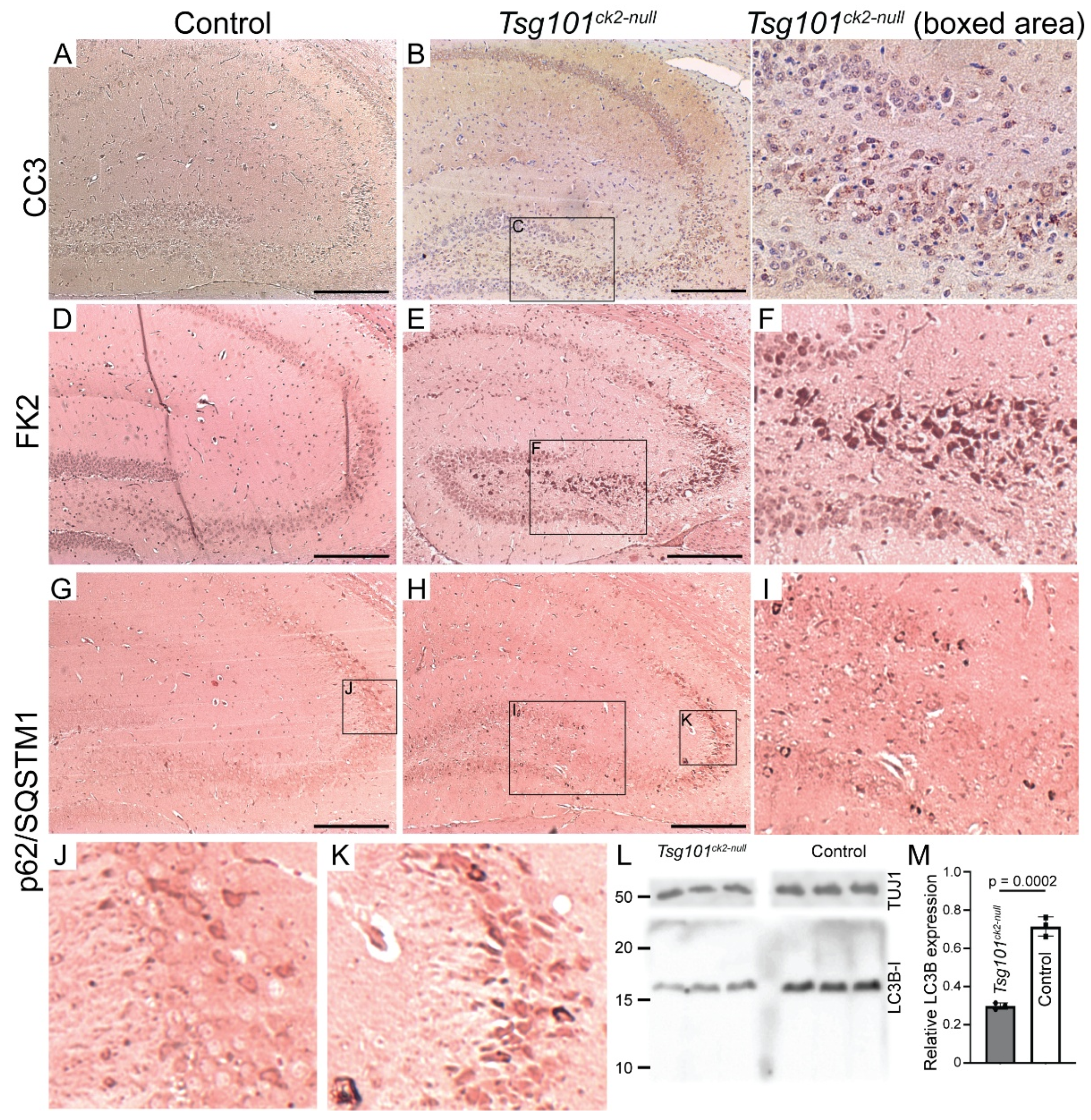
Figure 6.
Endosomal markers and intracellular APP accumulated in hippocampal neurons within 2 weeks of deleting Tsg101 from forebrain neurons. (A-D) Tsg101ck2-null hippocampal CA3 neurons accumulated early (RAB5, A-B) and late (RAB7, C-D) endosomal markers. Arrowheads in B and D indicate representative cells with elevated RAB5 or RAB7 expression, respectively. (E-H) IHC with the 4G8 antibody against APP (which also detects Aβ) showed more intense staining in Tsg101ck2-null hippocampal CA3 neurons relative to controls (E-F). Immunofluorescence staining revealed more 4G8-positive punctae in Tsg101ck2-null CA3 neurons relative to controls (G-H). Images shown in G and H were captured from slides stained at the same time, using the 40X objective and identical PMT voltage settings on an Olympus Fluoview 1000 confocal microscope, and are representative of 4 independent samples for each genotype. Scale bars: 50 µm.
Figure 6.
Endosomal markers and intracellular APP accumulated in hippocampal neurons within 2 weeks of deleting Tsg101 from forebrain neurons. (A-D) Tsg101ck2-null hippocampal CA3 neurons accumulated early (RAB5, A-B) and late (RAB7, C-D) endosomal markers. Arrowheads in B and D indicate representative cells with elevated RAB5 or RAB7 expression, respectively. (E-H) IHC with the 4G8 antibody against APP (which also detects Aβ) showed more intense staining in Tsg101ck2-null hippocampal CA3 neurons relative to controls (E-F). Immunofluorescence staining revealed more 4G8-positive punctae in Tsg101ck2-null CA3 neurons relative to controls (G-H). Images shown in G and H were captured from slides stained at the same time, using the 40X objective and identical PMT voltage settings on an Olympus Fluoview 1000 confocal microscope, and are representative of 4 independent samples for each genotype. Scale bars: 50 µm.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



