
Submitted:
17 April 2025
Posted:
21 April 2025
You are already at the latest version
Abstract
Sepsis and cancer, though distinct in their clinical manifestations, share profound pathophysiological overlaps that underscore their interconnectedness in disease progression and outcomes. Here we discuss the intricate biological mechanisms linking these two conditions, focusing on the roles of inflammation, immune dysregulation, and metabolic alterations. In sepsis, an uncontrolled immune response to infection leads to a cytokine storm, tissue damage, and immune paralysis, while cancer exploits chronic inflammation and immunosuppressive pathways to promote tumor growth and metastasis. Both conditions exhibit metabolic reprogramming, such as the Warburg effect in cancer and glycolysis-driven immune cell activation in sepsis, which fuels disease progression and complicates treatment. Sepsis can exacerbate cancer progression by inducing genomic instability, epigenetic modifications, and a pro-tumorigenic microenvironment, while cancer increases susceptibility to sepsis through immunosuppression and treatment-related complications. The shared pathways between sepsis and cancer present unique opportunities for therapeutic intervention, including anti-inflammatory agents, immune checkpoint inhibitors, and metabolic modulators. Anti-inflammatory therapies, such as IL-6 and TNF-α inhibitors, show promise in mitigating inflammation, while immune checkpoint inhibitors like anti-PD-1 and anti-CTLA-4 antibodies are being explored to restore immune function in sepsis and enhance anti-tumor immunity in cancer. Metabolic modulators, including glycolysis and glutaminolysis inhibitors, target the metabolic reprogramming common to both conditions, though their dual roles in normal and pathological processes necessitate careful consideration. Additionally, antimicrobial peptides (AMPs) represent a versatile therapeutic option with their dual antimicrobial and anti-tumor properties. In this review ,we also highlight the critical need for integrated approaches to understanding and managing the complex interactions between sepsis and cancer. By bridging the gap between sepsis and cancer research, this work aims to inspire interdisciplinary collaboration and advance the development of targeted therapies that address the shared mechanisms driving these devastating diseases. Ultimately, these insights may pave the way for novel diagnostic tools and therapeutic strategies to improve outcomes for patients affected by both conditions.
Keywords:
sepsis
; cancer
; inflammation
; immunosuppression
; tumor microenvironment
; cytokines
; immune response
; therapeutic targets
1. Introduction
Sepsis and cancer represent two major global health burdens with complex interconnections. Sepsis affects nearly 50 million people annually, causing over 11 million deaths worldwide, while cancer accounts for 9.7 million annual deaths, with projections suggesting a 77% increase in cases by 2050 [1]. Sepsis, a life-threatening condition caused by dysregulated host responses to infection, affects millions annually and is a leading cause of death in intensive care units [2]. Despite advances in antimicrobial therapies and critical care, sepsis remains a formidable clinical challenge due to its complex pathophysiology and high risk of multi-organ failure. On the other hand, cancer, characterized by uncontrolled cell proliferation and metastasis, is a leading cause of death globally, with an estimated 10 million deaths in 2020 alone [3]. The burden of cancer continues to rise, driven by aging populations, lifestyle factors, and environmental exposures.While sepsis and cancer are traditionally viewed as distinct entities, emerging evidence suggests a profound and bidirectional relationship between the two conditions. Sepsis can act as a trigger for cancer progression by inducing chronic inflammation, immune suppression, and genomic instability [4,5]. Conversely, cancer patients are at heightened risk of developing sepsis due to immunosuppression caused by the malignancy itself or its treatments, such as chemotherapy, radiotherapy, and surgery [6]. This interplay between sepsis and cancer has significant implications for patient outcomes, complicating diagnosis, treatment, and long-term prognosis [7].The growing recognition of this bidirectional relationship has sparked interest in understanding the shared mechanisms underlying both conditions. Chronic inflammation, immune dysregulation, and metabolic alterations are common features of sepsis and cancer, suggesting potential overlapping therapeutic targets [8]. Moreover, the clinical management of patients with both sepsis and cancer presents unique challenges, necessitating a nuanced approach to balance immune activation and suppression.By synthesizing current evidence and identifying gaps in knowledge, we aim to provide a thorough understanding of the sepsis-cancer connection. Ultimately, this knowledge may pave the way for novel diagnostic tools and therapeutic strategies to improve outcomes for patients affected by these intertwined conditions.
2. Pathophysiological Overlap Between Sepsis and Cancer and Therapeutic Implications
Both Sepsis and cancer share complex biological processes involving inflammation, immune system dysfunction, and metabolic alterations. Understanding these overlapping mechanisms not only elucidates their interconnectedness but also opens new possibilities for treating both conditions. Pathologically, Extracellular Toll-like receptors (TLRs), including TLR1, TLR2, and TLR4, are located on the cell surface and recognize bacterial pathogens, initiating signaling pathways that lead to dysregulated production of cytokines, chemokines, and interferons (IFNs). This excessive immune activation can result in a cytokine storm, which is a central driver of sepsis.Endosomal TLRs—such as TLR3, TLR7, TLR8, and TLR9—are situated within endosomes and detect bacterial and viral genetic material, further amplifying immune responses. Endogenous negative regulators of TLR signaling act to suppress overactivation of these pathways. However, when this regulatory control fails, unchecked TLR signaling leads to systemic inflammation, sepsis, and ultimately, death (Figure 1).
2.1. Cytokine Storm in Sepsis
In sepsis, the immune system's response to infection can become dysregulated, leading to a phenomenon known as the "cytokine storm" [9]. Chronic inflammation in sepsis is also associated with the development of long-term complications, such as post-sepsis syndrome, which includes cognitive impairment, physical disability, and an increased risk of late mortality [10]. The hyperinflammatory phase in sepsis is characterized by the excessive and uncontrolled release of pro-inflammatory cytokines, such as interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α). These cytokines, which are essential for coordinating the immune response, can spiral out of control, causing widespread systemic inflammation, tissue damage, and multi-organ failure [11,12]. For example, IL-6 plays a critical role in the acute phase response, but its overproduction during sepsis can lead to endothelial dysfunction, vascular leakage, and hypotension, contributing to septic shock [13]. Similarly, TNF-α, a potent pro-inflammatory cytokine, can induce apoptosis in endothelial cells and disrupt the vascular barrier, exacerbating organ damage [14]. Studies have shown that elevated levels of IL-6 and TNF-α are strongly associated with poor outcomes in sepsis patients, highlighting their central role in the pathophysiology of the disease [10,15].
2.2. Cytokines in Cancer
In cancer, the same cytokines that drive inflammation in sepsis are often co-opted by tumors to promote their growth and survival. For instance, cancer cells can produce cytokines and chemokines that recruit immune cells, such as macrophages and neutrophils, to the tumor site. These immune cells, in turn, release additional cytokines and growth factors that promote tumor cell proliferation, angiogenesis, and invasion [16]. The inflammatory microenvironment also plays a key role in immune evasion, a hallmark of cancer. Tumors can manipulate the immune system to suppress anti-tumor responses and create an immunosuppressive milieu. It has been observed that tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) are often recruited to the tumor site, where they release immunosuppressive cytokines, such as IL-10 and transforming growth factor-beta (TGF-β), that inhibit the activity of cytotoxic T cells and natural killer (NK) cells [17]. Other cytokines such as IL-6 in cancer could be a potent growth factor for many types of cancer cells [18]. They activate signaling pathways such as JAK/STAT, which promote cell proliferation, inhibit apoptosis, and enhance angiogenesis. In ovarian cancer, elevated IL-6 levels have been linked to tumor progression and poor prognosis [19]. TNF-α, while initially identified for its ability to induce tumor cell death, has a more complex role in cancer biology. In some contexts, TNF-α promotes chronic inflammation, which creates a tumor-friendly microenvironment. This chronic inflammation may lead to DNA damage, genomic instability, and the activation of oncogenic pathways, all of which contribute to cancer initiation and progression [20]. Specifically, in colorectal cancer, TNF-α has been shown to enhance the invasive and metastatic potential of cancer cells by promoting epithelial-mesenchymal transition (EMT) [21]. Thus, both cancer and sepsis can lead to chronic inflammation which is a persistent, low-grade inflammatory state that is a common feature of both sepsis and cancer (Figure 2).
This central role of cytokines like IL-6 and TNF-α in both sepsis and cancer has led to the development of targeted therapies aimed at modulating their activity. For example, monoclonal antibodies against IL-6 or its receptor, such as tocilizumab, have shown efficacy in dampening the cytokine storm in sepsis and are also being explored as treatments for certain cancers [22]. In sepsis, tocilizumab has been investigated in clinical trials for its potential to reduce inflammation and improve outcomes [23]. Similarly, TNF-α inhibitors, which are widely used in autoimmune diseases, are being investigated for their potential to reduce inflammation-driven tumor progression in cancer [24].
However, the dual roles of these cytokines in both protective and pathological processes pose significant challenges. For instance, while inhibiting IL-6 or TNF-α may reduce inflammation and tumor growth, it could also compromise the body's ability to fight infections or control tumor progression [25]. This highlights the need for a nuanced approach to therapy, one that balances the need to control inflammation without compromising essential immune functions (Table 1).
3. Immunosuppression in Sepsis and Cancer and Therapeutic Implications
3.1. Immune Paralysis in Sepsis
Sepsis typically induces a biphasic immune response: an initial hyperinflammatory phase followed by a prolonged state of immune suppression, known as "immune paralysis." During this phase, the immune system becomes less effective at fighting infections, and patients are highly susceptible to secondary infections [26]. Immune paralysis is characterized by the dysfunction of key immune cells, such as T-cells, B-cells, and dendritic cells, which are essential for mounting an effective immune response [27].
T-cells, in particular, play a critical role in the adaptive immune response, but in sepsis, they often become anergic or exhausted, losing their ability to proliferate and produce cytokines [28]. This T cell dysfunction is partly mediated by the upregulation of immune checkpoint molecules, such as programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), which act as brakes on the immune system. For example, PD-1 is upregulated on T cells during sepsis, and its interaction with its ligand PD-L1 inhibits T cell activation and promotes T cell exhaustion [29]. Similarly, CTLA-4, which is expressed on regulatory T cells (Tregs) and activated T cells, can suppress T cell responses by competing with the co-stimulatory molecule CD28 for binding to B7 molecules on antigen-presenting cells [30].
In addition to T-cell dysfunction, sepsis can lead to the apoptosis of immune cells, further depleting the body's ability to respond to infections. This immune paralysis is a major contributor to the high mortality rate observed in sepsis patients, particularly in the later stages of the disease.
3.2. Immunosuppression in Cancer
Similar to sepsis, cancer exploits immunosuppression as a critical mechanism to evade immune detection and destruction, enabling tumor survival and progression. Tumors create an immunosuppressive microenvironment by recruiting regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), which inhibit the activity of cytotoxic T cells and natural killer (NK) cells [31].
Tregs, which normally function to maintain immune tolerance and prevent autoimmunity, are co-opted by tumors to suppress anti-tumor immune responses. Tregs inhibit the activity of effector T cells through the release of immunosuppressive cytokines, such as interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β), and through direct cell-to-cell contact. Similarly, MDSCs, which are immature myeloid cells, release immunosuppressive cytokines and metabolites, such as arginase and reactive oxygen species (ROS), that inhibit T-cell function and promote tumor growth [32].
Tumors also exploit immune checkpoint pathways to evade immune surveillance. For example, the interaction between PD-1 on T cells and its ligand PD-L1 on tumor cells can inhibit T cell activation and promote T cell exhaustion. Similarly, CTLA-4, which is expressed on Tregs and activated T cells, can suppress T cell responses by competing with the co-stimulatory molecule CD28 for binding to B7 molecules on antigen-presenting cells.
The shared mechanisms of immune suppression in sepsis and cancer suggest that therapies targeting these pathways could be beneficial in both conditions. Immune checkpoint inhibitors, such as anti-PD-1 and anti-CTLA-4 antibodies, have shown remarkable efficacy in enhancing anti-tumor immune responses and improving patient outcomes in cancer. For example, pembrolizumab (anti-PD-1) and ipilimumab (anti-CTLA-4) have been approved for the treatment of various cancers, including melanoma and non-small cell lung cancer [33,34].
In sepsis, immune checkpoint inhibitors are being explored as potential treatments to reverse immune paralysis and restore immune function. Preclinical studies have shown that blocking PD-1 or CTLA-4 can improve survival in animal models of sepsis by enhancing T-cell responses and reducing susceptibility to secondary infections [35]. However, the complex interplay between inflammation and immune suppression in sepsis poses significant challenges, and further research is needed to fully understand the potential benefits and risks of these therapies.
4. Metabolic Alterations
4.1. The Warburg Effect on Cancer
One of the most well-known metabolic alterations in cancer is the "Warburg effect," named after Otto Warburg, who first observed that cancer cells preferentially rely on glycolysis for energy production, even in the presence of sufficient oxygen (aerobic glycolysis) [36]. This phenomenon is counterintuitive because glycolysis is far less efficient than oxidative phosphorylation in terms of ATP production. However, the Warburg effect provides cancer cells with several advantages. First, glycolysis generates ATP rapidly, which supports the high energy demands of rapidly proliferating cancer cells. Second, the intermediates of glycolysis can be diverted into biosynthetic pathways, providing the building blocks (e.g., nucleotides, amino acids, and lipids) needed for cell growth and division. Third, the production of lactate, a byproduct of glycolysis, creates an acidic tumor microenvironment that promotes invasion, metastasis, and immune evasion [37].
The Warburg effect is driven by oncogenic signaling pathways, such as PI3K/AKT/mTOR and MYC, which upregulate glycolytic enzymes and glucose transporters (e.g., GLUT1) [38]. Additionally, mutations in tumor suppressor genes, such as TP53, can further enhance glycolytic flux by disrupting mitochondrial function and promoting the shift toward glycolysis [39]. This metabolic reprogramming not only supports tumor growth but also contributes to the resistance of cancer cells to apoptosis and chemotherapy.
4.2. Metabolic Shifts in Sepsis
In sepsis, immune cells, particularly macrophages and neutrophils, also undergo a metabolic shift toward glycolysis [40,41]. This shift is essential for meeting the high energy demands of these cells as they respond to infection. Glycolysis allows immune cells to rapidly generate ATP and produce biosynthetic precursors needed for the production of cytokines, chemokines, and reactive oxygen species (ROS), which are critical for pathogen clearance.
However, this metabolic reprogramming can become dysregulated, leading to detrimental effects. In the early hyperinflammatory phase of sepsis, excessive immune cell activation drives a significant upregulation of glycolysis, contributing to tissue damage and organ dysfunction. The overproduction of lactate, a byproduct of glycolysis, contributes to lactic acidosis, a common complication of sepsis that is associated with poor outcomes [42]. Additionally, the persistent activation of glycolysis can deplete cellular energy reserves and impair mitochondrial function, leading to a state of cellular exhaustion and contributing to the later phase of immune paralysis.
4.3. Shared Metabolic Pathways and Therapeutic Implications
The metabolic alterations in sepsis and cancer extend beyond glycolysis and involve disruptions in the metabolism of amino acids, lipids, and other nutrients. These shared pathways highlight the interconnectedness of metabolic and immune dysregulation in both conditions.
4.4. Glutamine Metabolism
Glutamine, the most abundant amino acid in the blood, plays a critical role in both cancer and sepsis. In cancer, glutamine is a key nutrient that fuels the growth and survival of tumor cells [43,44]. Glutamine is converted to glutamate by the enzyme glutaminase, and glutamate is further metabolized to α-ketoglutarate, which enters the tricarboxylic acid (TCA) cycle to support ATP production and biosynthetic pathways. This process, known as glutaminolysis, is particularly important in cancer cells with defective mitochondria that rely on alternative pathways for energy production [45].
Glutamine enters cancer cells through specific transporters such as SLC1A5 (ASCT2) and SLC7A5 [45]. Once inside the cell, glutamine is metabolized through various pathways, including the glutaminase I and II pathways, to support cellular energetics and biosynthesis [46]. The glutaminase II pathway, which has been recently identified in some cancer types, provides an alternative route for glutamine metabolism and may be particularly important in hypoxic tumor region [47].
In sepsis, immune cells also depend on glutamine to support their function. Glutamine is used as a fuel for proliferation and as a precursor for the synthesis of nucleotides, proteins, and antioxidants. However, the high demand for glutamine during sepsis can lead to its depletion, impairing immune cell function and contributing to immune paralysis. Additionally, the metabolism of glutamine generates ammonia, which can be toxic to cells and contribute to organ dysfunction.
4.5. Lipid Metabolism
Lipid metabolism is another shared pathway that is dysregulated in both sepsis and cancer. In cancer, alterations in lipid metabolism support tumor growth, survival, and metastasis. Cancer cells often exhibit increased lipogenesis (the synthesis of fatty acids) and lipid uptake, which provide the lipids needed for membrane synthesis, signaling molecules, and energy storage [48]. Additionally, lipid metabolism plays a role in immune evasion, as lipid droplets in cancer cells can sequester pro-inflammatory signaling molecules and inhibit anti-tumor immune responses [49].
In sepsis, lipid metabolism is also disrupted, contributing to inflammation and immune suppression. During sepsis, there is an increase in lipolysis (the breakdown of fats), which releases free fatty acids (FFAs) into the bloodstream [50]. While FFAs can serve as an energy source, their excessive accumulation can lead to lipotoxicity, mitochondrial dysfunction, and the production of pro-inflammatory lipid mediators, such as prostaglandins and leukotrienes. These lipid mediators can exacerbate inflammation and contribute to organ damage.The shared metabolic pathways in sepsis and cancer present opportunities for targeted therapies (Table 2). For example, inhibiting glycolysis or glutaminolysis could potentially slow tumor growth in cancer and reduce the hyperinflammatory response in sepsis. Drugs that target glycolytic enzymes, such as hexokinase 2 (HK2) or lactate dehydrogenase A (LDHA), are being explored as cancer therapies and could also be tested in sepsis [51]. Similarly, inhibitors of glutaminase, such as CB-839, are being investigated in clinical trials for cancer and may have potential applications in sepsis [52].Targeting lipid metabolism is another promising strategy. Inhibitors of fatty acid synthesis, such as FASN inhibitors, or drugs that modulate lipid signaling pathways, such as cyclooxygenase-2 (COX-2) inhibitors, could be used to disrupt the metabolic adaptations that support tumor growth and inflammation [53]. However, the complexity of metabolic networks and their dual roles in both normal and pathological processes pose significant challenges. For example, inhibiting glycolysis or glutaminolysis could impair the function of immune cells, which also rely on these pathways to fight infections.
6. Clinical Implications of the Sepsis-Cancer Connection
6.1. Impact of Sepsis on Tumor Microenvironment
The tumor microenvironment (TME) is a dynamic network of cancer cells, immune cells, stromal cells, blood vessels, and extracellular matrix (ECM) that influences tumor behavior [54]. Sepsis often leads to systemic hypoxia, a condition characterized by insufficient oxygen supply to tissues, due to microcirculatory dysfunction, impaired oxygen delivery, and mitochondrial dysfunction. Within the tumor microenvironment (TME), hypoxia activates hypoxia-inducible factor 1-alpha (HIF-1α), a master transcriptional regulator of cellular responses to low oxygen levels. HIF-1α upregulates a wide array of genes involved in angiogenesis, metabolism, and survival, enabling cancer cells to adapt to and thrive in low-oxygen conditions [55]. This adaptive response not only supports tumor survival but also contributes to its aggressiveness and resistance to therapy.
A central role of HIF-1α is the promotion of vascular endothelial growth factor (VEGF), a critical mediator of angiogenesis—the process of forming new blood vessels. VEGF stimulates the proliferation and migration of endothelial cells, leading to the formation of new blood vessels that supply tumors with oxygen and nutrients [56]. However, these newly formed vessels are often structurally abnormal, leaky, and dysfunctional, which further exacerbates hypoxia and creates a pro-tumorigenic environment. For example, in breast cancer models, sepsis-induced hypoxia has been shown to significantly increase VEGF expression, leading to enhanced tumor vascularization and metastasis [57]. This abnormal vasculature not only supports tumor growth but also facilitates the spread of cancer cells to distant organs.
Hypoxia does more than just support tumor growth; it also promotes metastasis by selecting for more aggressive cancer cell populations. Hypoxic conditions induce the expression of genes involved in invasion and migration, such as LOX (lysyl oxidase) and CXCR4 (C-X-C chemokine receptor type 4). LOX, for instance, modifies the extracellular matrix to facilitate cancer cell invasion, while CXCR4 promotes the migration of cancer cells to distant sites. In colorectal cancer, hypoxia-driven VEGF expression has been linked to increased liver metastasis, underscoring the role of sepsis-induced hypoxia in promoting tumor spread [23]. These findings highlight how hypoxia, driven by sepsis, creates a vicious cycle that fuels tumor progression and metastasis.
Moreover, the interplay between hypoxia and angiogenesis is further complicated by the immunosuppressive effects of the hypoxic TME. Hypoxia recruits immunosuppressive cells, such as myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs), which inhibit anti-tumor immune responses [58]. This immunosuppressive milieu not only allows cancer cells to evade immune detection but also creates a favorable environment for tumor growth and metastasis.
Thus, sepsis-induced hypoxia activates HIF-1α, which drives VEGF-mediated angiogenesis and promotes the expression of genes involved in invasion and metastasis [59]. While these adaptations enable cancer cells to survive in low-oxygen conditions, they also contribute to the structural and functional abnormalities of tumor vasculature, exacerbating hypoxia and creating a pro-tumorigenic environment. The resulting increase in tumor aggressiveness and metastatic potential underscores the critical role of hypoxia and angiogenesis in the sepsis-cancer connection. Understanding these mechanisms provides valuable insights into potential therapeutic targets, such as HIF-1α and VEGF inhibitors, which could disrupt this cycle and improve outcomes for patients with sepsis and cancer.
6.2. Inflammatory Cytokines and Reactive Oxygen Species (ROS)
Sepsis also triggers a massive release of pro-inflammatory cytokines, such as interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), and interleukin-1 beta (IL-1β), as well as reactive oxygen species (ROS). These molecules play a dual role in the tumor microenvironment (TME), simultaneously promoting tumor progression and suppressing anti-tumor immunity, thereby creating a favorable environment for cancer growth and metastasis. ROS, including superoxide anions and hydrogen peroxide, are highly reactive molecules that can directly damage cellular DNA, leading to double-strand breaks, base modifications, and chromosomal aberrations. In cancer cells, such genomic alterations can result in the activation of oncogenes or the inactivation of tumor suppressor genes, driving tumor aggressiveness and resistance to therapy. For example, in colorectal cancer, sepsis-induced ROS has been shown to promote mutations in the APC gene, a key regulator of the Wnt signaling pathway, enhancing tumor progression by disrupting normal cellular processes and promoting uncontrolled cell proliferation [60]. The accumulation of DNA damage and genomic instability not only accelerates tumor growth but also increases the likelihood of metastasis and therapy resistance, making it a critical factor in the sepsis-cancer connection.
The inflammatory milieu created by sepsis also profoundly suppresses anti-tumor immune responses, further facilitating cancer progression. Sepsis recruits immunosuppressive cells, such as myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs), to the TME. These cells play a pivotal role in creating an immune-tolerant environment by inhibiting the activity of cytotoxic T cells and natural killer (NK) cells, which are essential for detecting and destroying cancer cells. For instance, sepsis-induced IL-6 has been shown to promote the expansion of MDSCs, a heterogeneous population of immature myeloid cells that suppress T cell function and promote tumor growth. In melanoma, the expansion of MDSCs driven by sepsis-induced IL-6 has been linked to accelerated tumor growth and reduced patient survival [61]. Similarly, Tregs, which normally function to maintain immune tolerance, are co-opted by tumors to suppress anti-tumor immune responses. The recruitment and activation of these immunosuppressive cells create a microenvironment that allows cancer cells to evade immune surveillance and proliferate unchecked.
Inflammatory cytokines and reactive oxygen species (ROS) play a complex, dual role in the tumor microenvironment (TME), simultaneously promoting cancer progression and suppressing anti-tumor immunity. These mediators fuel tumor growth by inducing DNA damage, genomic instability, and activating oncogenic pathways such as NF-κB and STAT3. Concurrently, they impair host defenses by recruiting immunosuppressive cells (e.g., Tregs, MDSCs) and inhibiting effector immune cell function through mechanisms like PD-L1 upregulation. This interplay creates a self-reinforcing cycle that accelerates tumor progression and metastasis, making it a critical target for therapeutic intervention. Emerging strategies that address sepsis-associated inflammatory mediators offer promising avenues for disrupting this cycle. For instance, IL-6 signaling inhibitors like tocilizumab may reduce STAT3-driven immunosuppression, while targeted antioxidants could mitigate oxidative DNA damage without compromising essential immune signaling. These approaches underscore the potential for translating insights from sepsis biology into novel cancer immunotherapies, highlighting the interconnected nature of these seemingly distinct pathologies. [62]. Additionally, therapies aimed at depleting MDSCs or blocking their immunosuppressive activity could restore anti-tumor immunity and improve outcomes for cancer patients with a history of sepsis.
7. Long-Term Consequences of Sepsis in Cancer Patients
Sepsis has profound and lasting effects on cancer patients, creating a conducive environment for cancer progression and recurrence through mechanisms such as chronic inflammation, immune suppression, genomic instability, and epigenetic modifications. Persistent inflammation post-sepsis, characterized by the sustained release of pro-tumorigenic cytokines like IL-6 and TNF-α, fuels tumor growth and immune evasion, increasing the risk of recurrence. For example, in ovarian cancer patients, post-sepsis chronic inflammation has been correlated with higher rates of tumor recurrence, underscoring the role of inflammation in driving cancer progression [63]. Sepsis-induced immune paralysis further exacerbates this issue by compromising anti-tumor immunity. The dysfunction of key immune cells, such as T cells, natural killer (NK) cells, and dendritic cells, impairs the body’s ability to detect and eliminate cancer cells. In melanoma patients, sepsis-induced immune suppression has been associated with accelerated tumor progression and reduced survival, highlighting the detrimental impact of immune dysfunction on cancer outcomes [29]. Additionally, sepsis can directly damage cellular DNA through the overproduction of reactive oxygen and nitrogen species (ROS/RNS), leading to double-strand breaks, base modifications, and other forms of DNA damage. In cancer cells, these alterations result in genomic instability and the accumulation of oncogenic mutations, such as those in the APC gene in colorectal cancer, which drive tumor aggressiveness and therapy resistance [60]. Sepsis also induces epigenetic changes, including DNA methylation and histone acetylation, which alter gene expression patterns without modifying the DNA sequence itself. For instance, sepsis-induced hypermethylation of the PTEN gene, a tumor suppressor, has been linked to tumor growth and metastasis in prostate cancer [64].Together, these long-term consequences of sepsis—chronic inflammation, immune suppression, genomic instability, and epigenetic modifications—create a pro-tumorigenic environment that not only promotes cancer progression but also increases the risk of recurrence, emphasizing the need for targeted therapeutic strategies to mitigate these effects and improve outcomes for cancer patients with a history of sepsis.
8. Therapeutic Opportunities
The shared mechanisms between sepsis and cancer present unique opportunities for therapeutic intervention, with several promising strategies emerging from the overlapping pathways of inflammation, immune dysregulation, and metabolic reprogramming. Anti-inflammatory therapies, such as targeting cytokines like IL-6 and TNF-α, have shown potential in mitigating inflammation in both conditions. For instance, monoclonal antibodies like tocilizumab (anti-IL-6) have been effective in dampening the cytokine storm in sepsis and are being explored in cancer, while TNF-α inhibitors like infliximab are used in autoimmune diseases and are being investigated for their ability to reduce inflammation-driven tumor progression. However, the dual roles of these cytokines in both protective and pathological processes pose challenges, as their inhibition may compromise the body’s ability to fight infections or control tumor growth. Immune checkpoint inhibitors, such as anti-PD-1 (e.g., pembrolizumab) and anti-CTLA-4 (e.g., ipilimumab) antibodies, have revolutionized cancer treatment by reactivating anti-tumor immunity and are now being explored in sepsis to reverse immune paralysis and restore immune function. Preclinical studies have demonstrated that blocking PD-1 or CTLA-4 can improve survival in sepsis models by enhancing T-cell responses, though overactivation of the immune system remains a concern. Metabolic modulators, including inhibitors of glycolysis (e.g., 2-deoxy-D-glucose) and glutaminolysis (e.g., CB-839), offer another therapeutic avenue. These drugs target the metabolic reprogramming seen in both cancer cells, which rely on aerobic glycolysis (the Warburg effect), and immune cells in sepsis, which undergo a glycolytic shift to meet energy demands during infection. However, the dual roles of these pathways in normal and pathological processes necessitate careful consideration, as their inhibition could impair immune cell function. In Table 3, we list the drugs that have been studied in both sepsis and cancer models for research. Antimicrobial peptides (AMPs), such as cathelicidins and defensins, represent a versatile therapeutic option due to their antimicrobial and anti-tumor properties [65,66,67]. In sepsis, AMPs such as LL-37 can effectively neutralize pathogens by disrupting microbial membranes and modulating the immune response to reduce inflammation [68]. In cancer, AMPs exhibit selective cytotoxicity toward cancer cells, inhibiting key processes like angiogenesis and tumor growth. Their ability to distinguish between malignant and normal cells makes them valuable candidates for targeted cancer therapy. Several peptides derived from the naturally occurring proteins, including Myeloid Differentiation Factor 2 (MD2) [69,70], MyD88 [71], MIEN1 [72,73], and Annexin A2 [74,75], have been identified to possess either anticancer or anti-inflammatory properties — or both. These peptides can be leveraged to develop dual therapeutics that target the shared inflammatory and immune pathways involved in both sepsis and cancer. For example, MD2 and MyD88 play key roles in the Toll-like receptor (TLR) signaling pathway, which regulates immune responses to pathogens and tumors. MIEN1 and Annexin A2 have been implicated in cancer progression and metastasis, but their modulation can also dampen inflammatory responses, suggesting potential for integrated sepsis and cancer therapies [76,77].Structural motifs within proteins, such as phenylalanine zippers and conserved motifs like GXXXXG, offer valuable tools for designing synthetic peptides with enhanced specificity and stability [78,79]. Phenylalanine zippers can stabilize peptide structures, increasing their ability to penetrate cell membranes and improve therapeutic efficacy [80]. Similarly, the GXXXXG motif has been shown to enhance the antimicrobial and anticancer activity of certain peptides while reducing cytotoxicity toward normal cells. These structural elements can be engineered as molecular "switches" to create dual-function peptides that simultaneously exhibit anti-endotoxin and anti-inflammatory properties [81].Moreover, AMPs enhance anti-tumor immunity by recruiting and activating immune cells such as macrophages, natural killer (NK) cells, and T cells. This immunomodulatory effect increases the body’s ability to recognize and eliminate malignant cells, further enhancing the therapeutic potential of AMPs in cancer treatment. However, despite their promise, challenges remain in optimizing the stability, specificity, and delivery of AMPs for clinical applications. Issues such as peptide degradation in physiological conditions, off-target effects, and limited bioavailability need to be carefully addressed to translate preclinical successes into clinical outcomes.
Together, these strategies underscore the potential for integrated approaches to target the shared pathophysiology of sepsis and cancer. By leveraging the dual roles of AMPs and naturally occurring proteins, it may be possible to develop novel, multi-functional therapies that not only treat infections and inflammation but also inhibit tumor progression. However, a careful balance of these mechanisms is essential to maximize therapeutic benefits while minimizing potential risks.
9. Conclusion
The emerging evidences clearly reveal that Sepsis and cancer, though distinct in their clinical presentations, are deeply interconnected through shared biological mechanisms, including inflammation, immune dysregulation, and metabolic reprogramming. These overlapping pathways not only drive disease progression but also present unique opportunities for therapeutic intervention. The shared mechanisms between these conditions offer promising avenues for targeted therapies. Anti-inflammatory agents, such as IL-6 and TNF-α inhibitors, have shown potential in mitigating inflammation in both sepsis and cancer, though their dual roles in protective and pathological processes require careful consideration. Immune checkpoint inhibitors, including anti-PD-1 and anti-CTLA-4 antibodies, have revolutionized cancer treatment and are now being explored in sepsis to reverse immune paralysis and restore immune function. Metabolic modulators, such as glycolysis and glutaminolysis inhibitors, target the metabolic reprogramming common to both conditions, though their impact on normal cellular functions must be carefully balanced. Antimicrobial peptides (AMPs), with their dual antimicrobial and anti-tumor properties, represent a versatile therapeutic option, though challenges in stability and delivery remain. Understanding the intricate interplay between sepsis and cancer is critical for developing integrated therapeutic strategies that address their shared pathophysiology. By targeting overlapping mechanisms, such as inflammation, immune suppression, and metabolic alterations, we can improve outcomes for patients affected by these devastating diseases. However, the dual roles of these pathways in both protective and pathological processes highlight the need for a nuanced approach to therapy, one that balances the need to control disease progression without compromising essential immune and metabolic functions. Future research should focus on elucidating the context-specific roles of these shared mechanisms and developing personalized therapies that account for the complex interplay between sepsis and cancer. By bridging the gap between sepsis and cancer research, we can inspire interdisciplinary collaboration and advance the development of innovative diagnostic tools and therapeutic strategies. Ultimately, this integrated approach has the potential to transform the management of sepsis and cancer, improving survival and quality of life for patients worldwide.
Author Contributions
Conceptualization; original draft preparation; editing by A.K.T. Review, and figures by Y.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- La Via, L., et al., The Global Burden of Sepsis and Septic Shock. Epidemiologia (Basel), 2024. 5(3): p. 456-478.
- Schlapbach, L.J., et al., World Sepsis Day: a global agenda to target a leading cause of morbidity and mortality. Am J Physiol Lung Cell Mol Physiol, 2020. 319(3): p. L518-L522. [CrossRef]
- Sung, H., et al., Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin, 2021. 71(3): p. 209-249. [CrossRef]
- Staudinger, T. and F. Pene, Current insights into severe sepsis in cancer patients. Rev Bras Ter Intensiva, 2014. 26(4): p. 335-8. [CrossRef]
- Gudiol, C., et al., Understanding and Managing Sepsis in Patients With Cancer in the Era of Antimicrobial Resistance. Front Med (Lausanne), 2021. 8: p. 636547. [CrossRef]
- Said, S.A., et al., Impact of Sepsis on the Oncologic Outcomes of Advanced Epithelial Ovarian Cancer Patients: A Multicenter Observational Study. Cancers (Basel), 2023. 15(18). [CrossRef]
- Hensley, M.K., et al., Epidemiology and Outcomes of Cancer-Related Versus Non-Cancer-Related Sepsis Hospitalizations. Crit Care Med, 2019. 47(10): p. 1310-1316. [CrossRef]
- Chesney, J.A., R.A. Mitchell, and K. Yaddanapudi, Myeloid-derived suppressor cells-a new therapeutic target to overcome resistance to cancer immunotherapy. J Leukoc Biol, 2017. 102(3): p. 727-740. [CrossRef]
- Chaudhry, H., et al., Role of cytokines as a double-edged sword in sepsis. In Vivo, 2013. 27(6): p. 669-84.
- van der Poll, T., et al., The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol, 2017. 17(7): p. 407-420.
- Schulte, W., J. Bernhagen, and R. Bucala, Cytokines in sepsis: potent immunoregulators and potential therapeutic targets--an updated view. Mediators Inflamm, 2013. 2013: p. 165974. [CrossRef]
- Damas, P., et al., Cytokine serum level during severe sepsis in human IL-6 as a marker of severity. Ann Surg, 1992. 215(4): p. 356-62.
- Kang, S., et al., IL-6 trans-signaling induces plasminogen activator inhibitor-1 from vascular endothelial cells in cytokine release syndrome. Proc Natl Acad Sci U S A, 2020. 117(36): p. 22351-22356. [CrossRef]
- Liang, Y., et al., Elevated levels of plasma TNF-alpha are associated with microvascular endothelial dysfunction in patients with sepsis through activating the NF-kappaB and p38 mitogen-activated protein kinase in endothelial cells. Shock, 2014. 41(4): p. 275-81. [CrossRef]
- Riedemann, N.C., et al., Protective effects of IL-6 blockade in sepsis are linked to reduced C5a receptor expression. J Immunol, 2003. 170(1): p. 503-7. [CrossRef]
- Zhao, H., et al., Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther, 2021. 6(1): p. 263. [CrossRef]
- Mantovani, A., The growing diversity and spectrum of action of myeloid-derived suppressor cells. Eur J Immunol, 2010. 40(12): p. 3317-20. [CrossRef]
- Raskova, M., et al., The Role of IL-6 in Cancer Cell Invasiveness and Metastasis-Overview and Therapeutic Opportunities. Cells, 2022. 11(22). [CrossRef]
- Coward, J., et al., Interleukin-6 as a therapeutic target in human ovarian cancer. Clin Cancer Res, 2011. 17(18): p. 6083-96.
- Yan, B., et al., Tumor necrosis factor-alpha is a potent endogenous mutagen that promotes cellular transformation. Cancer Res, 2006. 66(24): p. 11565-70.
- Balkwill, F., Tumour necrosis factor and cancer. Nat Rev Cancer, 2009. 9(5): p. 361-71.
- Du, Y., et al., Tocilizumab for Advanced Non-Small-Cell Lung Cancer With Concomitant Cachexia: An Observational Study. J Cachexia Sarcopenia Muscle, 2024. 15(6): p. 2815-2825. [CrossRef]
- Tavaci, T., et al., The impact of tocilizumab treatment on the severity of inflammation and survival rates in sepsis is significantly influence by the timing of administration. Inflammopharmacology, 2025.
- Jang, D.I., et al., The Role of Tumor Necrosis Factor Alpha (TNF-alpha) in Autoimmune Disease and Current TNF-alpha Inhibitors in Therapeutics. Int J Mol Sci, 2021. 22(5). [CrossRef]
- Bilal, J., et al., Risk of Infections and Cancer in Patients With Rheumatologic Diseases Receiving Interleukin Inhibitors: A Systematic Review and Meta-analysis. JAMA Netw Open, 2019. 2(10): p. e1913102. [CrossRef]
- Nakamori, Y., E.J. Park, and M. Shimaoka, Immune Deregulation in Sepsis and Septic Shock: Reversing Immune Paralysis by Targeting PD-1/PD-L1 Pathway. Front Immunol, 2020. 11: p. 624279.
- Boomer, J.S., J.M. Green, and R.S. Hotchkiss, The changing immune system in sepsis: is individualized immuno-modulatory therapy the answer? Virulence, 2014. 5(1): p. 45-56.
- Jensen, I.J., et al., Sepsis-Induced T Cell Immunoparalysis: The Ins and Outs of Impaired T Cell Immunity. J Immunol, 2018. 200(5): p. 1543-1553. [CrossRef]
- Hotchkiss, R.S., G. Monneret, and D. Payen, Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol, 2013. 13(12): p. 862-74. [CrossRef]
- Jain, N., et al., Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc Natl Acad Sci U S A, 2010. 107(4): p. 1524-8. [CrossRef]
- Togashi, Y., K. Shitara, and H. Nishikawa, Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol, 2019. 16(6): p. 356-371. [CrossRef]
- Jimenez-Morales, S., et al., Mechanisms of Immunosuppressive Tumor Evasion: Focus on Acute Lymphoblastic Leukemia. Front Immunol, 2021. 12: p. 737340. [CrossRef]
- Topalian, S.L., C.G. Drake, and D.M. Pardoll, Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell, 2015. 27(4): p. 450-61. [CrossRef]
- Kang, S.P., et al., Pembrolizumab KEYNOTE-001: an adaptive study leading to accelerated approval for two indications and a companion diagnostic. Ann Oncol, 2017. 28(6): p. 1388-1398. [CrossRef]
- Chang, K.C., et al., Blockade of the negative co-stimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Crit Care, 2013. 17(3): p. R85. [CrossRef]
- Koppenol, W.H., P.L. Bounds, and C.V. Dang, Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer, 2011. 11(5): p. 325-37.
- Liberti, M.V. and J.W. Locasale, The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci, 2016. 41(3): p. 211-218.
- Cairns, R.A., I.S. Harris, and T.W. Mak, Regulation of cancer cell metabolism. Nat Rev Cancer, 2011. 11(2): p. 85-95.
- Harami-Papp, H., et al., TP53 mutation hits energy metabolism and increases glycolysis in breast cancer. Oncotarget, 2016. 7(41): p. 67183-67195. [CrossRef]
- Ferreira, B.L., et al., Glucose metabolism is upregulated in the mononuclear cell proteome during sepsis and supports endotoxin-tolerant cell function. Front Immunol, 2022. 13: p. 1051514. [CrossRef]
- Liu, J., et al., Metabolic reprogramming consequences of sepsis: adaptations and contradictions. Cell Mol Life Sci, 2022. 79(8): p. 456. [CrossRef]
- Suetrong, B. and K.R. Walley, Lactic Acidosis in Sepsis: It's Not All Anaerobic: Implications for Diagnosis and Management. Chest, 2016. 149(1): p. 252-61.
- Fan, Y., et al., Exploiting the Achilles' heel of cancer: disrupting glutamine metabolism for effective cancer treatment. Front Pharmacol, 2024. 15: p. 1345522. [CrossRef]
- Jin, J., et al., Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp Mol Med, 2023. 55(4): p. 706-715. [CrossRef]
- Jin, L., G.N. Alesi, and S. Kang, Glutaminolysis as a target for cancer therapy. Oncogene, 2016. 35(28): p. 3619-25.
- Ni, R., et al., Rethinking glutamine metabolism and the regulation of glutamine addiction by oncogenes in cancer. Front Oncol, 2023. 13: p. 1143798. [CrossRef]
- Cooper, A.J.L., et al., Metabolic Heterogeneity, Plasticity, and Adaptation to "Glutamine Addiction" in Cancer Cells: The Role of Glutaminase and the GTomegaA [Glutamine Transaminase-omega-Amidase (Glutaminase II)] Pathway. Biology (Basel), 2023. 12(8).
- Munir, R., et al., Lipid metabolism in cancer cells under metabolic stress. Br J Cancer, 2019. 120(12): p. 1090-1098. [CrossRef]
- Soula, M., et al., Glycosphingolipid synthesis mediates immune evasion in KRAS-driven cancer. Nature, 2024. 633(8029): p. 451-458. [CrossRef]
- Van Wyngene, L., et al., Hepatic PPARalpha function and lipid metabolic pathways are dysregulated in polymicrobial sepsis. EMBO Mol Med, 2020. 12(2): p. e11319.
- Ganapathy-Kanniappan, S. and J.F. Geschwind, Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer, 2013. 12: p. 152. [CrossRef]
- Wicker, C.A., et al., Glutaminase inhibition with telaglenastat (CB-839) improves treatment response in combination with ionizing radiation in head and neck squamous cell carcinoma models. Cancer Lett, 2021. 502: p. 180-188. [CrossRef]
- Jin, Z., Y.D. Chai, and S. Hu, Fatty Acid Metabolism and Cancer. Adv Exp Med Biol, 2021. 1280: p. 231-241.
- Xiao, Y. and D. Yu, Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther, 2021. 221: p. 107753. [CrossRef]
- Chen, G., et al., Role of hypoxia in the tumor microenvironment and targeted therapy. Front Oncol, 2022. 12: p. 961637. [CrossRef]
- Dong, G., et al., Intermittent hypoxia alleviates increased VEGF and pro-angiogenic potential in liver cancer cells. Oncol Lett, 2019. 18(2): p. 1831-1839. [CrossRef]
- Semenza, G.L., Hypoxia-inducible factors in physiology and medicine. Cell, 2012. 148(3): p. 399-408. [CrossRef]
- Fu, Z., et al., Tumour Hypoxia-Mediated Immunosuppression: Mechanisms and Therapeutic Approaches to Improve Cancer Immunotherapy. Cells, 2021. 10(5). [CrossRef]
- Jin, F., et al., Impairment of hypoxia-induced angiogenesis by LDL involves a HIF-centered signaling network linking inflammatory TNFalpha and angiogenic VEGF. Aging (Albany NY), 2019. 11(2): p. 328-349. [CrossRef]
- Grivennikov, S.I., F.R. Greten, and M. Karin, Immunity, inflammation, and cancer. Cell, 2010. 140(6): p. 883-99.
- Gabrilovich, D.I. and S. Nagaraj, Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol, 2009. 9(3): p. 162-74. [CrossRef]
- Soler, M.F., et al., New perspectives in cancer immunotherapy: targeting IL-6 cytokine family. J Immunother Cancer, 2023. 11(11). [CrossRef]
- Mantovani, A., et al., Cancer-related inflammation. Nature, 2008. 454(7203): p. 436-44. [CrossRef]
- Baylin, S.B. and P.A. Jones, A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer, 2011. 11(10): p. 726-34. [CrossRef]
- Verma, D.P., A.K. Tripathi, and A.K. Thakur, Innovative Strategies and Methodologies in Antimicrobial Peptide Design. J Funct Biomater, 2024. 15(11). [CrossRef]
- Tripathi, A.K., et al., Shaping the Future of Antimicrobial Therapy: Harnessing the Power of Antimicrobial Peptides in Biomedical Applications. J Funct Biomater, 2023. 14(11). [CrossRef]
- Tripathi, A.K. and J.K. Vishwanatha, Role of Anti-Cancer Peptides as Immunomodulatory Agents: Potential and Design Strategy. Pharmaceutics, 2022. 14(12). [CrossRef]
- Kumar, A., et al., Single Amino Acid Substitutions at Specific Positions of the Heptad Repeat Sequence of Piscidin-1 Yielded Novel Analogs That Show Low Cytotoxicity and In Vitro and In Vivo Antiendotoxin Activity. Antimicrob Agents Chemother, 2016. 60(6): p. 3687-99. [CrossRef]
- Tandon, A., et al., Characterization of a Myeloid Differentiation Factor 2-Derived Peptide that Facilitates THP-1 Macrophage-Mediated Phagocytosis of Gram-Negative Bacteria. ACS Infect Dis, 2024. 10(3): p. 845-857. [CrossRef]
- Tandon, A., et al., An MD2-derived peptide promotes LPS aggregation, facilitates its internalization in THP-1 cells, and inhibits LPS-induced pro-inflammatory responses. Cell Mol Life Sci, 2018. 75(13): p. 2431-2446.
- Kumari, T., et al., 10-Residue MyD88-Peptide Adopts beta-Sheet Structure, Self-Assembles, Binds to Lipopolysaccharides, and Rescues Mice from Endotoxin-Mediated Lung-Infection and Death. ACS Chem Biol, 2022. 17(12): p. 3420-3434. [CrossRef]
- Tripathi, A.K., et al., Short peptides based on the conserved regions of MIEN1 protein exhibit anticancer activity by targeting the MIEN1 signaling pathway. J Biol Chem, 2024. 300(3): p. 105680. [CrossRef]
- Vishwanatha, J.K.F.W., TX, US), Tripathi, Amit Kumar (Fort Worth, TX, US), DESIGN AND CHARACTERIZATION OF INHIBITORY PEPTIDES (IPEPS) DERIVED FROM OF MIEN 1 PROTEIN SEQUENCE. 2024, The University of North Texas Health Sciences Cente (Fort Worth, TX, US): United States.
- Desai, P.P., et al., Combination of Small Extracellular Vesicle-Derived Annexin A2 Protein and mRNA as a Potential Predictive Biomarker for Chemotherapy Responsiveness in Aggressive Triple-Negative Breast Cancer. Cancers (Basel), 2022. 15(1). [CrossRef]
- Guo, C., et al., Higher Expression of Annexin A2 in Metastatic Bladder Urothelial Carcinoma Promotes Migration and Invasion. Cancers (Basel), 2022. 14(22). [CrossRef]
- Tripathi, A.K. and J.K. Vishwanatha, Abstract 2184: Short Peptides derived from MIEN1 and their analogs exhibit anti-cancer activity in breast and prostate cancer cells. Cancer Research, 2023. 83(7_Supplement): p. 2184-2184. [CrossRef]
- Tripathi, A.K. and J.K. Vishwanatha, Abstract LB034: First-in-class compounds targeting the MIEN1 signaling pathway: A breakthrough approach for breast and prostate cancer therapy. Cancer Research, 2024. 84(7_Supplement): p. LB034-LB034. [CrossRef]
- Tripathi, A.K., et al., Identification of GXXXXG motif in Chrysophsin-1 and its implication in the design of analogs with cell-selective antimicrobial and anti-endotoxin activities. Sci Rep, 2017. 7(1): p. 3384. [CrossRef]
- Saxena, R., et al., HIV-1 Nef CAWLEAQ motif: a regulator of monocytes invasion through ENO1 modulation. Mol Cell Biochem, 2018. 447(1-2): p. 151-164. [CrossRef]
- Tripathi, A.K., et al., Selective phenylalanine to proline substitution for improved antimicrobial and anticancer activities of peptides designed on phenylalanine heptad repeat. Acta Biomater, 2017. 57: p. 170-186. [CrossRef]
- Srivastava, S., et al., Modulation of anti-endotoxin property of Temporin L by minor amino acid substitution in identified phenylalanine zipper sequence. Biochem J, 2016. 473(21): p. 4045-4062. [CrossRef]
- Rumienczyk, I., et al., Oncology Drug Repurposing for Sepsis Treatment. Biomedicines, 2022. 10(4). [CrossRef]
Figure 1.
TLR-Mediated Sepsis Pathogenesis. Extracellular TLRs (TLR1, TLR2, and TLR4) recognize bacterial pathogens, leading to dysregulated cytokine, chemokine, and IFN production and subsequent cytokine storm. Endosomal TLRs (TLR3, TLR7, TLR8, and TLR9) detect bacterial/viral genetic material, further contributing to inflammation. Endogenous negative regulators of TLR signaling aim to control this process, but failure leads to sepsis and death.
Figure 1.
TLR-Mediated Sepsis Pathogenesis. Extracellular TLRs (TLR1, TLR2, and TLR4) recognize bacterial pathogens, leading to dysregulated cytokine, chemokine, and IFN production and subsequent cytokine storm. Endosomal TLRs (TLR3, TLR7, TLR8, and TLR9) detect bacterial/viral genetic material, further contributing to inflammation. Endogenous negative regulators of TLR signaling aim to control this process, but failure leads to sepsis and death.
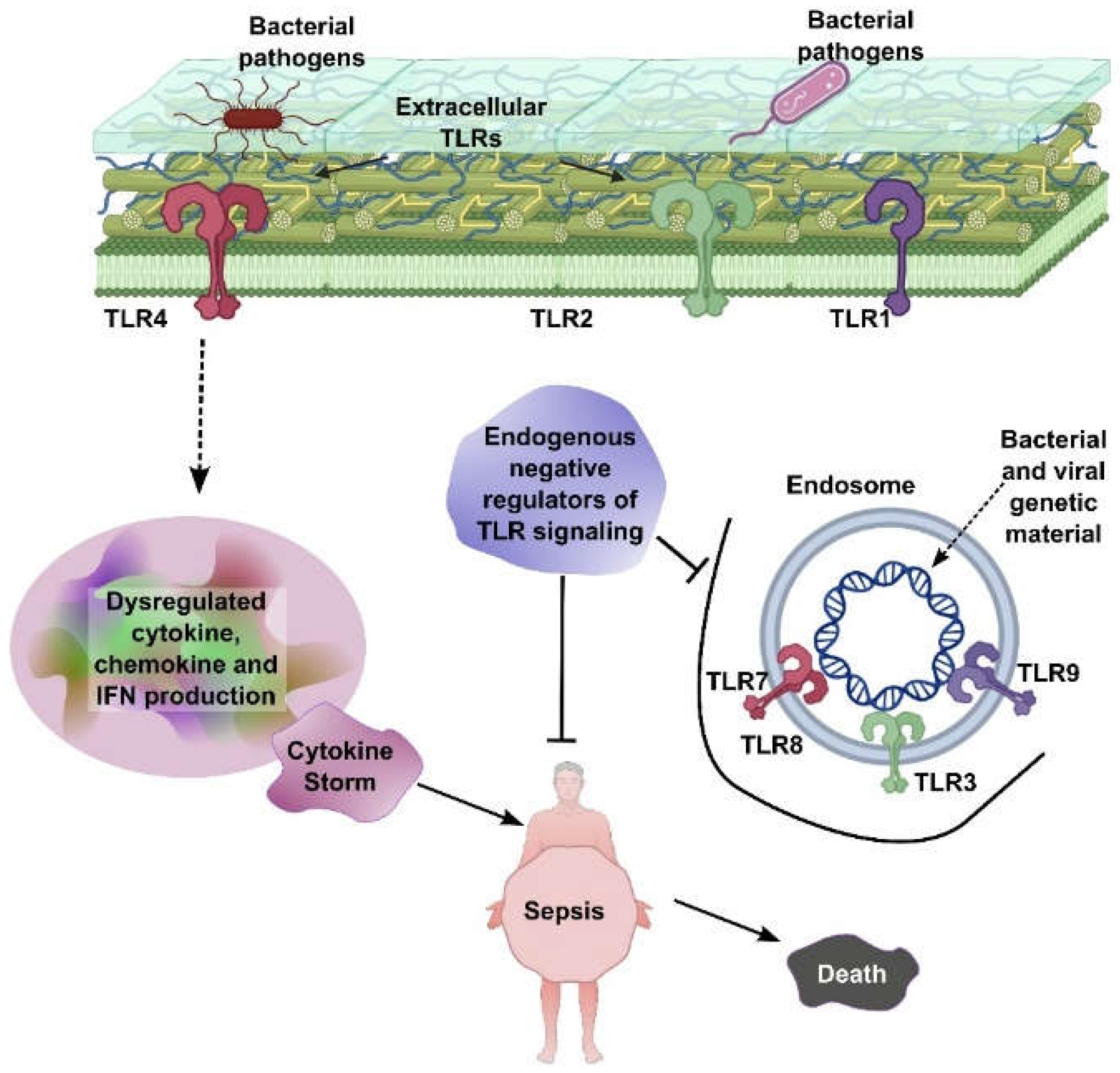
Figure 2.
Cytokine storm progression in cancer and sepsis. The six shared stages of cytokine storm in cancer and sepsis: (1) Initial Trigger by pathogens or tumor signals, (2) Early Pro-Inflammatory Phase with cytokine release, (3) Systemic Inflammation causing vascular leakage and microthrombosis, (4) Cytokine Storm with excessive cytokines and tissue damage, (5) Immunosuppressive Phase marked by anti-inflammatory responses and immune exhaustion, and (6) Multi-Organ Dysfunction Syndrome (MODS) leading to organ failure and death. Despite different triggers, both conditions follow a similar inflammatory trajectory.
Figure 2.
Cytokine storm progression in cancer and sepsis. The six shared stages of cytokine storm in cancer and sepsis: (1) Initial Trigger by pathogens or tumor signals, (2) Early Pro-Inflammatory Phase with cytokine release, (3) Systemic Inflammation causing vascular leakage and microthrombosis, (4) Cytokine Storm with excessive cytokines and tissue damage, (5) Immunosuppressive Phase marked by anti-inflammatory responses and immune exhaustion, and (6) Multi-Organ Dysfunction Syndrome (MODS) leading to organ failure and death. Despite different triggers, both conditions follow a similar inflammatory trajectory.
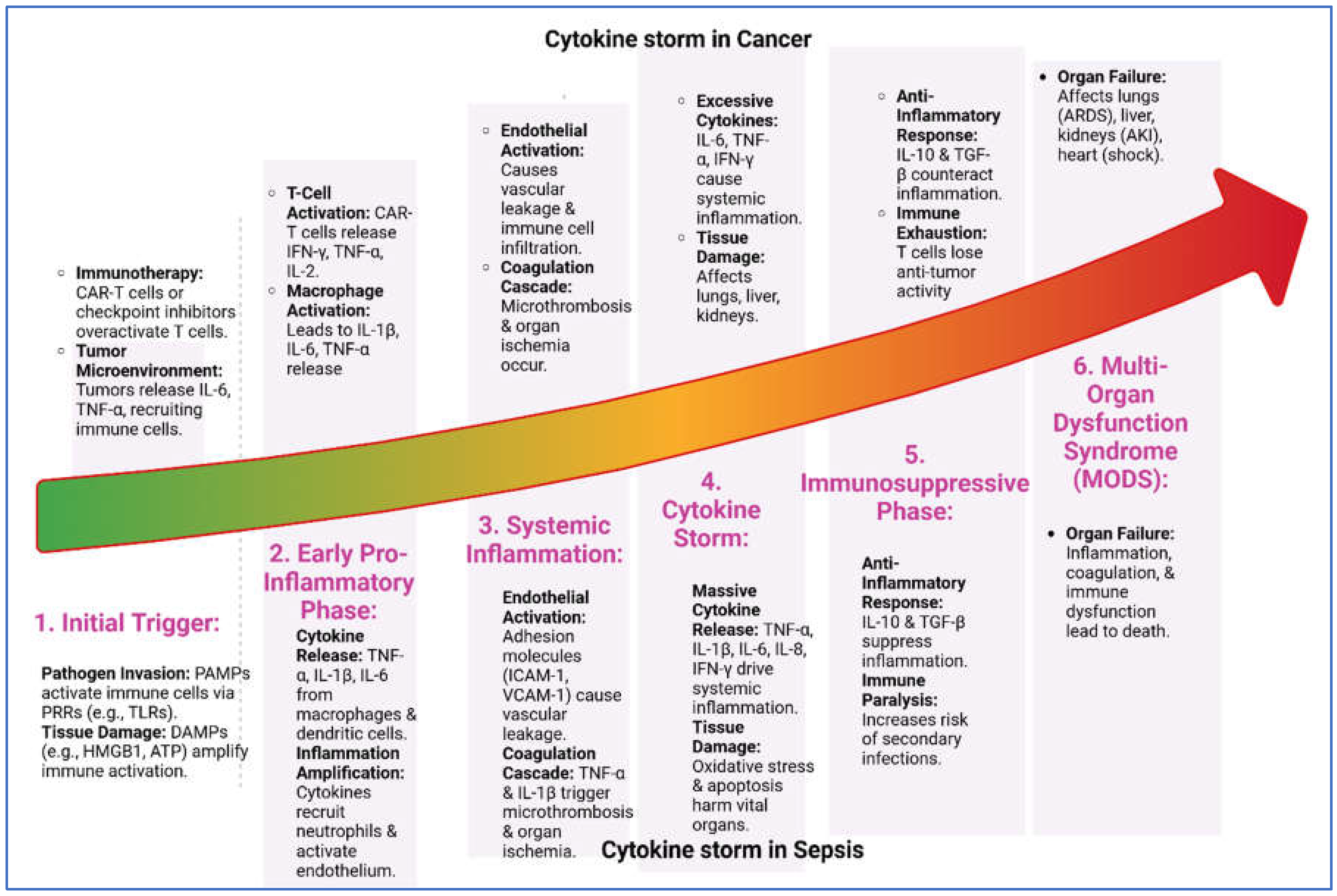

Table 1.
Cytokines Linking Sepsis and Cancer: Common cytokines involved in both conditions, with their roles in inflammation, immune regulation, and disease progression.
Table 1.
Cytokines Linking Sepsis and Cancer: Common cytokines involved in both conditions, with their roles in inflammation, immune regulation, and disease progression.
| Cytokine | Role in Sepsis | Role in Cancer |
| TNF-α | - Early pro-inflammatory mediator. - Promotes inflammation, endothelial activation, and organ dysfunction. |
- Promotes tumor progression, angiogenesis, and metastasis. - Can induce tumor cell death in high concentrations. |
| IL-1β | - Induces fever, vasodilation, and immune cell recruitment. - Contributes to tissue damage. |
- Promotes tumor growth, angiogenesis, and metastasis. - Enhances immunosuppressive microenvironment. |
| IL-6 | - Key mediator of acute-phase response. - Correlates with severity and mortality. |
- Promotes tumor growth, survival, and metastasis. - Drives chronic inflammation and immune evasion. |
| IL-8 (CXCL8) | - Chemoattractant for neutrophils. - Contributes to tissue injury. |
- Promotes angiogenesis, tumor growth, and metastasis. - Attracts immunosuppressive cells to the tumor microenvironment. |
| IL-10 | - Anti-inflammatory cytokine. - Suppresses pro-inflammatory responses. - Can lead to immunosuppression. |
- Promotes immune evasion by suppressing anti-tumor immunity. - Enhances tumor progression. |
| IL-17 | - Produced by Th17 cells. - Promotes neutrophil recruitment and inflammation. |
- Promotes tumor growth, angiogenesis, and metastasis. - Contributes to chronic inflammation. |
| IL-23 | - Promotes Th17 cell differentiation and IL-17 production. - Amplifies inflammation. |
- Promotes tumor growth and immune evasion. - Enhances chronic inflammation. |
| IFN-γ | - Activates macrophages and enhances pro-inflammatory responses. - Contributes to tissue damage. |
- Can have anti-tumor effects by activating immune cells. - May promote tumor immune evasion in chronic settings. |
| TGF-β | - Anti-inflammatory cytokine. - Promotes tissue repair and immunosuppression. |
- Promotes tumor progression, immune evasion, and metastasis. - Induces epithelial-mesenchymal transition (EMT). |
| VEGF | - Promotes vascular permeability and endothelial dysfunction. | - Drives angiogenesis, supporting tumor growth and metastasis. |
| HMGB1 | - Late-phase mediator of sepsis. - Sustains inflammation and organ damage. |
- Promotes tumor growth, metastasis, and immune evasion. - Acts as a damage-associated molecular pattern (DAMP). |
| PD-1/PD-L1 | - Contributes to T-cell exhaustion and immunosuppression in sepsis. | - Key immune checkpoint in cancer. - Promotes immune evasion and tumor progression. |
| G-CSF | - Stimulates neutrophil production and mobilization. | - Promotes tumor growth and metastasis. - Enhances myeloid-derived suppressor cells (MDSCs). |
| MCP-1 (CCL2) | - Recruits monocytes and macrophages to sites of inflammation. | - Recruits tumor-associated macrophages (TAMs), promoting tumor progression and immune evasion. |
Table 2.
Metabolic Similarities in Sepsis and Cancer. Comparison of key metabolic changes in sepsis and cancer, emphasizing shared pathways and clinical implications for potential therapeutic strategies.
Table 2.
Metabolic Similarities in Sepsis and Cancer. Comparison of key metabolic changes in sepsis and cancer, emphasizing shared pathways and clinical implications for potential therapeutic strategies.
| Aspect | Sepsis | Cancer | Clinical Implications |
| Glucose Metabolism | - Hyperglycemia: Insulin resistance and increased gluconeogenesis. - Warburg Effect: Increased glycolysis. |
- Warburg Effect: Aerobic glycolysis for rapid ATP production. - Increased Glucose Uptake: Enhanced by GLUT transporters. |
- Targeting Glycolysis: Inhibitors like 2-DG may help in both conditions. - Glucose Control: Tight glucose management improves outcomes in sepsis. |
| Lactate Production | - Lactic Acidosis: Excessive glycolysis leads to lactate accumulation. | - High Lactate Levels: Lactate contributes to tumor microenvironment acidosis. | - Lactate as a Biomarker: High lactate levels correlate with poor prognosis in both conditions. |
| Lipid Metabolism | - Lipolysis: Increased breakdown of fats for energy. - Hyperlipidemia: Elevated free fatty acids. |
- Lipid Synthesis: Increased de novo lipogenesis. - Fatty Acid Oxidation: Some cancers rely on fatty acids for energy. |
- Lipid-Targeting Therapies: Inhibitors of lipogenesis (e.g., FASN inhibitors) are explored in cancer. |
| Protein Metabolism | - Protein Catabolism: Muscle breakdown for gluconeogenesis. - Negative Nitrogen Balance. |
- Increased Protein Synthesis: Supports cell proliferation. - Amino Acid Dependency: Reliance on glutamine. |
- Nutritional Support: Glutamine supplementation may benefit both conditions. |
| Glutamine Metabolism | - Glutamine Utilization: Supports immune cell function and energy production. | - Glutamine Addiction: Used for anaplerosis and nucleotide synthesis. | - Glutaminase Inhibitors: CB-839 is being tested in cancer and may have potential in sepsis. |
| Mitochondrial Dysfunction | - Impaired Oxidative Phosphorylation: Reduced ATP production. - ROS Production: Contributes to tissue damage. |
- Altered Mitochondrial Function: Dysfunction or upregulation depending on cancer type. - ROS Signaling: Promotes tumor growth. |
- Antioxidant Therapies: May help mitigate ROS-induced damage in both conditions. |
| Ketone Body Metabolism | - Increased Ketogenesis: In response to energy demands. | - Ketone Utilization: Some cancers use ketone bodies as an energy source. | - Ketogenic Diets: May benefit cancer patients and potentially sepsis patients. |
| Immune Cell Metabolism | - Metabolic Reprogramming: Immune cells shift to glycolysis. - Immunosuppression: M2 macrophages rely on oxidative metabolism. |
- Tumor-Associated Immune Cells: TAMs and Tregs exhibit metabolic changes supporting tumor growth. | - Immunometabolism Targeting: Modulating immune cell metabolism may improve outcomes. |
| Hypoxia Response | - HIF-1α Activation: Promotes glycolysis and angiogenesis. | - HIF-1α Activation: Drives angiogenesis and tumor progression. | - HIF-1α Inhibitors: Potential therapeutic target in both conditions. |
| Insulin Resistance | - Peripheral Insulin Resistance: Reduces glucose uptake in muscle and adipose tissue. | - Altered Insulin Signaling: Some cancers exhibit insulin resistance or upregulate insulin/IGF-1 signaling. | - Insulin Sensitizers: May improve outcomes in sepsis and certain cancers. |
| Acidosis | - Metabolic Acidosis: Due to lactate accumulation and impaired renal function. | - Tumor Microenvironment Acidosis: Results from high lactate production. | - pH Modulation: Alkalinizing agents may help mitigate acidosis in both conditions. |
| Energy Demand | - Increased Energy Demand: To support hypermetabolic state and immune responses. | - Increased Energy Demand: To support rapid cell proliferation and tumor growth. | - Nutritional Support: High-calorie diets may benefit patients in both conditions. |
Table 3.
Drugs Used in Both Sepsis and Cancer Models: Listed drugs, that have shown therapeutic potential in both sepsis and cancer across various experimental and clinical models.
Table 3.
Drugs Used in Both Sepsis and Cancer Models: Listed drugs, that have shown therapeutic potential in both sepsis and cancer across various experimental and clinical models.
| Drug Name | Cancer Indication | Sepsis Application |
| Irinotecan | Ovarian, small cell lung, cervical cancer | Bacterial-induced sepsis models [82] |
| Topotecan | Ovarian, small cell lung, cervical cancer | LPS and S. aureus infection models [82] |
| Olaparib | Ovarian, breast, prostate, pancreatic cancer | CLP and LPS sepsis models [82] |
| Trametinib | Melanoma | CLP and LPS sepsis models [82] |
| SCH772984 | Melanoma, colon cancer | CLP and LPS sepsis models [82] |
| Ceritinib | Non-small-cell lung cancer | CLP and LPS sepsis models [82] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



