Submitted:
16 April 2025
Posted:
17 April 2025
You are already at the latest version
Abstract
We investigate the thermal conductivity of graphene Moiré superlattices formed by twisting bilayer graphene (TBG) at small angles, employing approach-to-equilibrium molecular dynamics and lattice dynamics calculations based on the Boltzmann Transport Equation. Our simulations reveal a non-monotonic dependence of the thermal conductivity on the twisting angle, with a local minimum near the first magic angle (θ∼1.1∘). This behavior is attributed to the evolution of local stacking configurations—AA, AB, and saddle-point (SP)—across the Moiré superlattice, which strongly affect phonon transport. A detailed analysis of phonon mean free paths and mode-resolved thermal conductivity shows that AA stacking suppresses thermal transport while, AB and SP stackings exhibit enhanced conductivity owing to more efficient low-frequency phonon transport. Furthermore, we establish a direct correlation between the thermal conductivity of twisted structures and the relative abundance of stacking domains within the Moiré supercell. Our results demonstrate that even very small changes in twisting angle (<2∘) can lead to thermal conductivity variations of over 30%, emphasizing the high tunability of thermal transport in TBG.
Keywords:
graphene
; Moiré
; molecular dynamics
; lattice dynamics
1. Introduction
Thermal management is a fundamental challenge in the electronics industry, especially in the design of next-generation integrated circuits. Efficient heat dissipation is crucial for the performance and reliability of ultra-large-scale integrated circuits, where excess heat can degrade functionality and lifespan [1]. Graphene, with its extraordinarily high intrinsic thermal conductivity, offers a promising solution for thermal management applications, ranging from heat spreaders to thermoelectric energy conversion and advanced cooling systems [2,3].
Despite its high thermal conductivity, several factors influence heat transport in graphene, including defects, grain boundaries, isotopic composition, strain, and substrate interactions [1,2,3,4,5,6,7,8,9]. In particular, nanostructuration techniques, such as nanopatterning and chemical functionalization (e.g., hydrogenation, fluorination), have been explored to modulate graphene phonon transport properties [10,11,12,13,14]. These methods introduce phonon scattering sites that can either suppress or enhance heat conduction depending on the specific engineering approach. A recent and highly effective method for tuning graphene thermal conductivity is the creation of Moiré superlattices: these are formed when two graphene layers are stacked with a twisting angle, creating periodic interference patterns that significantly alter electronic and phononic properties [15]. In 2011, theoretical studies identified that specific twisting angles, known as "magic angles," result in flat electronic bands in twisted bilayer graphene (TBG), with the first magic angle commonly reported around [16]. This discovery led to the observation of exotic phenomena such as correlated insulating states, unconventional superconductivity, and magnetic ordering in TBG and other 2D material heterostructures [17,18].
Beyond electronic properties, recent studies have shown that Moiré superlattices also impact phonon transport and thermal conductivity in TBG. Similarly to other nanostructures, where thermal conductivity can be tweaked by imposing a superperiodicity [19,20], the realization of Moiré pattern can enhance or suppress thermal transport properties. Molecular dynamics simulations indicate that the thermal conductivity of TBG varies non-monotonically with twisting angle [21]. Specifically, the conductivity decreases as the twisting angle increases, reaching a local minimum at approximately , followed by an increase up to , after which it stabilizes. This non-trivial dependence is attributed to the emergence of Moiré phonons—phonon modes induced by interlayer twisting—that modify phonon dispersion and introduce additional scattering mechanisms [22]. Theoretical studies on phonons in twisted bilayer graphene reveal that the stacking order plays a critical role in phonon transport. Phonon dispersion in AA-stacked, AB-stacked, and twisted bilayer graphene with various rotation angles shows that the stacking configuration predominantly affects the out-of-plane acoustic phonon modes [23]. The introduction of Moiré superlattices leads to new phonon branches—entangled phonons—that originate from the mixing of phonon modes from different high-symmetry directions in the Brillouin zone. The frequencies of these entangled phonons depend strongly on the twisting angle and can serve as a mean for non-contact identification of twisting angles in graphene samples. Furthermore, the emergence of these Moiré phonons significantly influences the thermal conductivity of twisted bilayer graphene, modifying phonon scattering mechanisms and transport properties [23]. Additionally, twisted multilayer SnSe2 Moiré superlattices have been experimentally synthesized and demonstrate a substantial reduction in thermal conductivity compared to their untwisted counterparts [15]. This is primarily due to enhanced phonon scattering, increased lattice mismatch, and the emergence of localized phonon modes. These findings highlight the potential of superperiodic structures and Moiré engineering for precise control over heat transport in 2D materials.
Despite these advancements, research on the thermal properties of Moiré superlattices remains in its early stages, with a limited number of studies focusing on small twisting angles and their phonon interactions. In this work, we explore the twisting angle dependence of thermal transport in TBG, with an emphasis on the first magic angle, . Furthermore, we aim to establish a connection between the variations in thermal conductivity and the underlying phonon characteristics of Moiré superlattices. By investigating the role of Moiré phonons and their influence on heat transport, this study provides new insights into the fundamental mechanisms governing thermal conductivity in twisted 2D materials.
2. Methods
In this study, we employed classical molecular dynamics (MD) simulations to calculate the thermal conductivity of TBG at various twisting angles using the approach-to-equilibrium molecular dynamics (AEMD) method [24,25,26]. TBG samples were prepared by first initializing the atomic coordinates of monolayer graphene sheets with an interatomic spacing of nm [27,28]. The sheets were then duplicated and separated along the z-axis by nm [29] to form bilayers. We then rotated one layer by a precise twisting angle around a common center, forming a Moiré superlattice. The lattice parameters of the resulting structure were determined using the relation [30]:
where a is the lattice constant of graphene ( nm). The number of atoms per Moiré unit cell was calculated as:
We considered nine twisting angles : °, °, °, °, °, °, °, and °, corresponding to Moiré unit cells with longitudinal dimensions L ranging from 15.9 to 26.9 nm and lateral dimensions d from 9.2 to 15.5 nm. The number of atoms in these unit cells varied between 11096 and 30248 (see Table 1). To construct the simulation supercells, the Moiré unit cells were replicated along their longest edge, forming samples with lengths L and widths d, containing between one and seven Moiré unit cells. To avoid artificial strains or stresses, an energy minimization step (box relaxation) was performed before each simulation.
To implement AEMD approach we defined separated regions within our simulation cell, as illustrated in Figure 1b. A step-like temperature profile is initially realized by thermalizing the top and bottom halves at temperatures K and K, respectively. The system is then evolved in the NVE ensemble while monitoring the temperature difference between the average temperatures of the two halves of the supercell. The temperature difference follows an exponential decay, given by [24,26]
where was used as fitting parameter. The thermal conductivity is finally calculated according to
where N is the number of atoms in the system, and J K−1 is the Boltzmann constant. The simulation cell section S is estimated as the width d of the sample multiplied by the interlayer distance and the number of layers. The length, width, and interlayer distance, are obtained from a fully relaxed box.
In order to assess the convergence of our results, we adopted the approach proposed by in Ref. [24]:
- the converged thermal conductivity is estimated for an infinitely long NVE simulation, according to
- the thermal conductivity for an infinitely long sample is obtained by a finite-size extrapolation, a commonly used approach in non-equilibrium simulations [31]
MD simulations were performed using LAMMPS [31], employing the AIREBO potential [32,33] with a time step of 1 fs. The cutoff radius for the Lennard-Jones term was set to 1.02 nm1. NVE runs lasted between 25 to 35 ps, depending of the simulation cell size. To showcase the adopted protocol in detail, we consider here the smallest sample of AA-stacked bilayer graphene as a case study. We remark that the same procedure was applied to all the samples investigated in this work. First, was fitted using Eq. (3) for different fitting times, , ranging from 1 ps (the first 1000 time-steps of the NVE run) to 25 ps, effectively covering the entire NVE run.
The obtained values of along with their corresponding uncertainties, , are used to obtain and an associated error using Eq. (4), as shown in Figure 2a. We found our simulation results to be converged using a number of terms in the sum of Eq. (3), although even a lower yielded nearly identical: for example, the results for and differ by less than 0.1%, within the uncertainty margin. This is in line with the findings of Melis et al. [34], who also observed rapid convergence with increasing . The calculated for each sample were determined by performing a weighted average on 4 statistically independent run with different initial velocities. The averaged values were reported as a function of L on a vs. plot: Figure 2b shows the case of the AA-stacked graphene sample. A final extrapolation via Eq. (6) up to the second order of the Taylor expansion, was then used to produce the final value of for that specific configuration. In what follows, we will refer to the extrapolated thermal conductivity and its uncertainty simply as and .
3. Results and Discussion
Using this procedure, we initially estimated the thermal conductivities of the graphene monolayer and bilayers in both the AA and AB stacking configurations, as reported in Table 2:
As stated in the introduction, the thermal conductivity of monolayer graphene remains an area of active research, with reported values ranging from 600 to 5000 W·K−1 [28,35,36,37,38]. However, our prediction of W/mK can be limited by size effects, as phonon mean free paths (MFP) in monolayer graphene can extend up to 10 m [39]. However, the study of these size effects is beyond the scope of the present work. Our result is in good agreement with Han and Ruan [40], in which the thermal conductivity at room temperatures converges to 1300 W/mK for macroscopic sizes. Interestingly, the stacking of the layers appears to significantly influence the thermal conductivity of bilayer graphene: in Figure Table 2 we compare our data with those in Han and Ruan [40]. Although there is no general consensus about thermal conductivity in AA and AB stack [41,42], we remark that the value reported here are influenced by the adopted interatomic potential. According to the AIREBO potential, the AA stacking configuration results in an energy of eV per atom, while for the AB stacking, eV per atom, confirming the AB-stacked bilayer as the most stable configuration, as also reported by other studies [43]. However, the energy difference is less than 0.4 meV per atom—well below the thermal energy at ambient temperature (approximately 25 meV at 300 K). Given this small difference, we exclude the large variation in to depend exclusively on the relative stability of the two configurations (equivalent calculations performed on ab initio system using PBE exchange-correlation functional confirmed AB as the most stable structure with an energy difference of meV). Furthermore, the interlayer distances between the two configuration differ by only Å (Å and Å). Therefore, we cannot attribute the observed differences in thermal conductivity solely to variations in interlayer distance. We conclude that the significant difference in thermal conductivity is primarily driven by the stacking of the layers, rather than indirect factors such as equilibrium energy, interlayer distance, or size. The different stacking configurations likely modulate phonon transport through the bilayer, potentially suppressing or promoting it.
We report the thermal conductivity values for the TBG as a function of the twisting angle in Figure 3b. While the AB stacking configuration remains the most conductive, the conductivity values show a considerable variability with respect to the twisting angle. When comparing our results with those of [44] for the same temperature of 300 K, we observe a significant discrepancy, with their values being approximately twice as large as ours. As already pointed out, differences can be attributed by the choice of different force fields, which can have a substantial impact on the computed thermal transport properties of graphene [45,46,47]. While a non-monotonic trend with respect to can be identified, we also observe that a minimum in occurs at . Our results suggest a more irregular trend in the thermal conductivity of TBG for small twisting angles than initially anticipated, with very small angle variation resulting in large fluctuation in thermal conductivity. We cannot rule out the possibility that a more detailed study might reveal additional maxima and minima in this trend. Thermal conductivity of Moiré superlattices of graphene appears to be a highly-tunable property: in particular, variations of % from the AB stack are observed within less than . We attempt a qualitative explanation from our estimations for the thermal conductivity of the AA and AB stacks. In fact, Moiré superlattices in graphene are characterized by the presence of both the stacking types: the great difference in thermal conductivity manifested by AA and AB stacks can then have a major role in such tunability. The observed variation in thermal conductivity in TBG can be understood by considering the local stacking configurations that emerge within the Moiré superstructure. Due to the periodicity of the Moiré pattern, different stacking regions coexist, each exhibiting distinct phonon transport properties. We quantified the contribution of different configurations, by estimating the thermal conductivity for three notable stacking configurations in bilayer graphene—AA, AB, and SP (saddle-point) stacking (shown in Figure 4a,b,c)—by solving the Boltzmann Transport Equation (BTE) using the ALDo code [48,49]. ALDo computes phonon transport properties using the finite displacement method, which estimates interatomic force constants (IFCs) by considering harmonic and anharmonic lattice vibrations, to diagonalize the dynamical matric and solve the BTE.
The analysis of phonon dispersion curves reported in Figure 4d, e, and g for the AA, AB, and SP configurations respectively, does not reveal any significant variation in the phonon frequencies across the different stackings. However, more subtle features exhibit marked differences that critically impact the thermal transport. In particular, the analysis of the MFP distribution (Fig. Figure 4g) and the mode-resolved conductivity for the three stacking configurations (Fig. Figure 4h) results in some dependence especially in the low frequency region. The MFP spectral distribution reveals that the AA stacking has lower values than those of AB and SP, mainly occurring at low frequencies corresponding to acoustic phonon modes. While the phonon dispersion curves show negligible changes in phonon dispersion and group velocities, the observed differences in the acoustic regime are reflected in the mode-resolved conductivity: in agreement with the MFP analysis, AB and SP distribution is highly peaked around 3-4 THz, where low-frequency modes (known as quasi-acoustic, low-frequency modes often fundamental for the propagation of sound-like waves at long wavelengths [50]) contribute with higher values of thermal conductivity compared to AA. This enhancement is due to their larger MFP and more efficient phonon transport in the acoustic range. The cumulative conductivity as a function of the MFP, shown in the right panel of Figure 4h, provides an additional point of view of the phenomenon: while phonons in the whole MFP range transport heat more effectively than the SP configuration, AA saturates at lower values for phonons with a reduced extension.
The calculated values for total thermal conductivity from the BTE calculations are:
- AA: 1027.406 W·K−1
- AB: 1274.969 W·m−1·K−1
- SP: 1217.723 W·m−1·K−1
The results indicate that the SP configuration closely resembles the AB stacking in terms of thermal transport properties, even when considering the offset between the two atomic planes. These results confirm the findings obtained through the AEMD simulations, both in terms of absolute thermal conductivity values and the overall trend. From this comparison, it emerges that the SP configuration is closer to the AB than to the AA stacking. AA stacking has the highest symmetry among the three, which could lead to fewer restrictions on phonon-phonon scattering processes (like Umklapp scattering). The lower symmetry of AB and SP configurations imposes stricter selection rules on which phonon interactions are allowed and this can suppress certain scattering channels that limit thermal conductivity, effectively increasing the phonon lifetime and thus compared to AA [51]. This reflection symmetry is usually altered by interlayer interactions, which allows more scattering processes. These data suggest that the overall thermal conductivity for intermediate twisting angles is strongly influenced by the percentage of atoms that are in an intermediate configuration between the two stacking limits. In fact, although these stacking modes where previously considered as scattering sites that reduce thermal conductivity in the non-uniform structure [21], we infer from our observations that the thermal conductivity of TBG samples is intimately related to the composition in intermediate stacking configurations. In fact, since AB and SP represent structures where the perfect atom-on-atom stacking of AA is broken by a shift, the thermal conductivity of all the possible stacking configurations occurring between the AA and AB should be lower than in the extreme cases. The primary hypothesis to explain this behavior is based on an analogy with silicon/germanium alloys, where thermal conductivity varies according to the stoichiometry of the mixture [52]. The direct calculation via BTE cannot be performed, since these intermediate configurations are found to be metastable with respect to AA, AB, and SP (which we remark to be almost degenerate with respect to AB, as also found in [53]). Hence, to verify our hypothesis, we quantified for each twisting angle configuration the percentage of atoms that are exactly in one of the three notable stacking phases. This estimate was obtained by evaluating the x and y distances of corresponding atoms in the two layers and adopting a tolerance of 0.05Å. The result of this analysis (Fig. Figure 5) shows a trend similar to that of thermal conductivity, with a minimum that closely reproduces the minimum observed in the angular dependence of .
It is worth noting that, although there is a clear correlation between the percentage of atoms in defined stacking configurations and thermal conductivity, the two curves do not perfectly overlap. This discrepancy can be attributed to several factors: limited statistics, as the error bars in the simulations remain relatively large and may require more samples to improve precision; non-linear phonon effects, since the phononic states in intermediate configurations may not follow a simple interpolating trend between the two extreme stackings; complex atomic dynamics, as the assumption of stacking configuration stability, particularly for the AA phase at finite temperature, may not be entirely valid. Furthermore, as already mentioned, a treatment based on the BTE would be impractical for the intermediate stacking configurations and even less feasible for the TBG, due to the very large number of IFCs required to solve the equations.
4. Conclusions
In this work, we investigated the thermal conductivity of twisted bilayer graphene (TBG) as a function of the twisting angle using both classical molecular dynamics simulations and lattice dynamics calculations via the Boltzmann Transport Equation. Our results confirm that the thermal transport properties of TBG are highly sensitive to the twisting angle, with variations up to 30% observed over a range of less than 2°. The thermal conductivity reaches a minimum near the first magic angle and displays a non-monotonic dependence that correlates with the evolution of local stacking configurations.
By analyzing ideal AA, AB, and SP stacking arrangements, we showed that the reduced conductivity of AA stacking is primarily due to reduced mean free paths for low-frequency acoustic modes. In contrast, both AB and SP stackings are characterized by extended vibrational modes, particularly in the acoustic regime, which enhances their ability to conduct heat. These observations were corroborated by mode-resolved and cumulative thermal conductivity analyses. In addition, we showed that the thermal conductivity of TBG can be qualitatively explained by the different distribution of stacking configurations within the Moiré supercell. In particular, we found that the local proportion of AA, AB, and SP stacking regions plays a critical role in determining the effective thermal transport. Regions with higher AA, AB and SP content contribute more efficiently to heat conduction, while areas characterized by intermediate stacking arrangement can act as bottlenecks due to suppressed acoustic phonon transport. The correlation between the relative abundance of these stacking configurations and the total thermal conductivity underscores the highly tunable nature of heat transport in TBG, strongly affected by the microscopic structural composition of the Moiré pattern.
Data Availability Statement
The simulation cells for all the structures investigated in this paper and the input files are available at https://github.com/Manunz15/moire-superlattice
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Balandin, A.A. Thermal properties of graphene and nanostructured carbon materials. Nat. Mater. 2011, 10, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Calizo, I.; Teweldebrhan, D.; Pokatilov, E.; Nika, D.; Balandin, A. Extremely high thermal conductivity of graphene: Prospects for thermal management applications in nanoelectronic circuits. Appl. Phys. Lett. 2008, 92, 151911. [Google Scholar] [CrossRef]
- Seol, J.; Jo, I.; Moore, A.; Lindsay, L.; Aitken, Z.; Pettes, M.; Li, X.; Yao, Z.; Huang, R.; Broido, D. Two-dimensional phonon transport in supported graphene. Science 2010, 328, 213–216. [Google Scholar] [CrossRef]
- Nika, D.L.; Balandin, A.A. Two-dimensional phonon transport in graphene. J. Physics: Condens. Matter 2012, 24, 233203. [Google Scholar] [CrossRef]
- Xu, X.; Pereira, L.F.C.; Wang, Y.; Wu, J.; Zhang, K.; Zhao, X.; Bae, S.; Tinh Bui, C.; Xie, R.; Thong, J.T.L.; et al. Length-dependent thermal conductivity in suspended single-layer graphene. Nat. Commun. 2014, 5, 3689. [Google Scholar] [CrossRef]
- Chen, S.; Moore, A.; Cai, W.; Suk, J.; An, J.; Mishra, C.; Amos, C.; Magnuson, C.; Kang, J.; Shi, L.; et al. Raman measurements of thermal transport in suspended monolayer graphene of variable sizes in vacuum and gaseous environments. ACS Nano 2011, 5, 321–328. [Google Scholar] [CrossRef]
- Nika, D.L.; Pokatilov, E.P.; Askerov, A.S.; Balandin, A.A. Phonon thermal conduction in graphene: Role of umklapp and edge roughness scattering. Phys. Rev. B 2009, 79, 155413. [Google Scholar] [CrossRef]
- Yan, Z.; Liu, G.; Khan, J.; Balandin, A. Thermal properties of graphene and multilayer graphene: Applications in thermal interface materials. MRS Bull. 2012, 37, 1245–1251. [Google Scholar] [CrossRef]
- Kong, B.; Paul, S.; Nardelli, M.; Kim, K. First-principles analysis of lattice thermal conductivity in monolayer and bilayer graphene. Phys. Rev. B 2009, 80, 033406. [Google Scholar] [CrossRef]
- Barbarino, G.; Melis, C.; Colombo, L. Effect of hydrogenation on graphene thermal transport. Carbon 2014, 80, 167–173. [Google Scholar] [CrossRef]
- Hahn, K.; Melis, C.; Colombo, L. Thermal transport in nanocrystalline graphene investigated by approach-to-equilibrium molecular dynamics simulations. Carbon 2016, 96, 429–438. [Google Scholar] [CrossRef]
- Li, C.; Wu, R.; Zhao, X.; Feng, K.; Huang, W. Interfacial Thermal Transport in Functionalized Graphene Layers: The Role of Fluorination. J. Phys. Chem. C 2023, 127, 72353–72361. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, Z.; Wang, J.; Guo, Y.; Wei, J. Seamless Graphene/Fluorinated Graphene Lateral Heterostructures for Enhanced Heat Dissipation in Transistor Devices. Adv. Sci. 2024, 11, 2401586. [Google Scholar] [CrossRef]
- Yang, W.; Zhang, Y.; Li, T.; Wang, H.; Chen, Y. Lattice thermal conductivity and phonon transport properties of monolayer fluorographene: A first-principles study. J. Appl. Phys. 2023, 136, 134305. [Google Scholar] [CrossRef]
- Ran, Y.; Meng, C.; Ma, Y.; Li, Q.; Zhu, H. Reduced Thermal Conductivity in SnSe2 Moiré Superlattices. ACS Nano 2025. [Google Scholar] [CrossRef]
- Bistritzer, R.; MacDonald, A.H. Moiré bands in twisted double-layer graphene. Proc. Natl. Acad. Sci. 2011, 108, 12233–12237. [Google Scholar] [CrossRef]
- Cao, Y.; Fatemi, V.; Fang, S.; Watanabe, K.; Taniguchi, T.; Kaxiras, E.; Jarillo-Herrero, P. Unconventional superconductivity in magic-angle graphene superlattices. Nature 2018, 556, 43–50. [Google Scholar] [CrossRef]
- Cao, Y.; Fatemi, V.; Demir, A.; Fang, S.; Tomarken, S.L.; Luo, J.Y.; Sanchez-Yamagishi, J.D.; Watanabe, K.; Taniguchi, T.; Kaxiras, E.; et al. Correlated insulator behaviour at half-filling in magic-angle graphene superlattices. Nature 2018, 556, 80–84. [Google Scholar] [CrossRef]
- Dettori, R.; Melis, C.; Colombo, L. SixGe1-x alloy as efficient phonon barrier in Ge/Si superlattices for thermoelectric applications. Eur. Phys. J. B 2015, 88. [Google Scholar] [CrossRef]
- Dettori, R.; Siddi, F.; Colombo, L.; Melis, C. Superlattice Thermal Modulation in MoS2MoS2 by Defect Engineering. Adv. Theory Simulations 2024, 8. [Google Scholar] [CrossRef]
- Cheng, Y.; Fan, Z.; Zhang, T.; Nomura, M.; Volz, S.; Zhu, G.; Li, B.; Xiong, S. Magic angle in thermal conductivity of twisted bilayer graphene. Mater. Today Phys. 2023, 35, 101093. [Google Scholar] [CrossRef]
- Han, W.; Li, Y.; Chen, H.; Zhou, J.; Zhang, G. Twist-Angle Dependent Phonon Transport in Twisted Bilayer Graphene. Adv. Mater. 2023, 35, e2209876. [Google Scholar] [CrossRef]
- Cocemasov, A.I.; Nika, D.L.; Balandin, A.A. Phonons in Twisted Bilayer Graphene. Phys. Rev. B 2013, 88, 035428. [Google Scholar] [CrossRef]
- Melis, C.; Dettori, R.; Vandermeulen, S.; Colombo, L. Calculating thermal conductivity in a transient conduction regime: theory and implementation. Eur. Phys. J. B 2014, 87, 1–9. [Google Scholar] [CrossRef]
- Lampin, E.; Palla, P.L.; Francioso, P.A.; Cleri, F. Thermal conductivity from approach-to-equilibrium molecular dynamics. J. Appl. Phys. 2013, 114. [Google Scholar] [CrossRef]
- Cappai, A.; Melis, C.; Colombo, L.; Dettori, R. Molecular Dynamics Simulations of Thermal Transport in Solid State Systems. In Comprehensive Computational Chemistry; Elsevier, 2024; pp. 804–820. [Google Scholar] [CrossRef]
- Yang, G.; Li, L.; Lee, W.B.; Ng, M.C. Structure of graphene and its disorders: A review. Sci. Technol. Adv. Mater. 2018, 19, 613–648. [Google Scholar] [CrossRef]
- Walimbe, P.; Chaudhari, M. State-of-the-art advancements in studies and applications of graphene: A comprehensive review. Mater. Today Sustain. 2019, 6, 100026. [Google Scholar] [CrossRef]
- Chung, D.D.L. Review graphite. J. Mater. Sci. 2002, 37, 1475–1489. [Google Scholar] [CrossRef]
- Feuerbacher, M. Moiré, Euler, and self-similarity—the lattice parameters of twisted hexagonal crystals. arXiv preprint arXiv:2007.03542, arXiv:2007.03542 2020.
- LAMMPS. LAMMPS Molecular Dynamics Simulator, Thu 22th August 2024.
- Stuart, S.J.; Tutein, A.B.; Harrison, J.A. A reactive potential for hydrocarbons with intermolecular interactions. J. Chem. Phys. 2000, 112, 6472–6486. [Google Scholar] [CrossRef]
- LAMMPS. pair_style airebo command, Sat 3rd August 2024.
- Melis, C.; Dettori, R.; Vandermeulen, S.; Colombo, L. Calculating thermal conductivity in a transient conduction regime: theory and implementation. Eur. Phys. J. B 2014, 87, 1–9. [Google Scholar] [CrossRef]
- Chen, S.; Moore, A.L.; Cai, W.; Suk, J.W.; An, J.; Mishra, C.; Amos, C.; Magnuson, C.W.; Kang, J.; Shi, L.; et al. Raman measurements of thermal transport in suspended monolayer graphene of variable sizes in vacuum and gaseous environments. ACS Nano 2011, 5, 321–328. [Google Scholar] [CrossRef]
- Cai, W.; Moore, A.L.; Zhu, Y.; Li, X.; Chen, S.; Shi, L.; Ruoff, R.S. Thermal transport in suspended and supported monolayer graphene grown by chemical vapor deposition. Nano Lett. 2010, 10, 1645–1651. [Google Scholar] [CrossRef]
- Chen, S.; Wu, Q.; Mishra, C.; Kang, J.; Zhang, H.; Cho, K.; Cai, W.; Balandin, A.A.; Ruoff, R.S. Thermal conductivity of isotopically modified graphene. Nat. Mater. 2012, 11, 203–207. [Google Scholar] [CrossRef]
- Balandin, A.A.; Ghosh, S.; Bao, W.; Calizo, I.; Teweldebrhan, D.; Miao, F.; Lau, C.N. Superior thermal conductivity of single-layer graphene. Nano Lett. 2008, 8, 902–907. [Google Scholar] [CrossRef]
- Barbarino, G.; Melis, C.; Colombo, L. Intrinsic thermal conductivity in monolayer graphene is ultimately upper limited: A direct estimation by atomistic simulations. Phys. Rev. B 2015, 91. [Google Scholar] [CrossRef]
- Han, Z.; Ruan, X. Thermal conductivity of monolayer graphene: Convergent and lower than diamond. Phys. Rev. B 2023, 108, L121412. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, H.; Liao, N.; Yang, P. Investigation on thermal conductivity of bilayer graphene nanoribbons. RSC Adv. 2014, 4, 54474–54479. [Google Scholar] [CrossRef]
- Rezania, H.; Yarmohammadi, M. Dynamical thermal conductivity of bilayer graphene in the presence of bias voltage. Phys. E: Low-Dimens. Syst. Nanostructures 2016, 75, 125–135. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Sboychakov, A.O.; Rakhmanov, A.L.; Nori, F. Electronic properties of graphene-based bilayer systems. Phys. Rep. 2016, 648, 1–104. [Google Scholar] [CrossRef]
- Cheng, Y.; Fan, Z.; Zhang, T.; Nomura, M.; Volz, S.; Zhu, G.; Li, B.; Xiong, S. Magic angle in thermal conductivity of twisted bilayer graphene. Mater. Today Phys. 2023, 35, 101093. [Google Scholar] [CrossRef]
- Si, C.; Wang, X.D.; Fan, Z.; Feng, Z.H.; Cao, B.Y. Impacts of potential models on calculating the thermal conductivity of graphene using non-equilibrium molecular dynamics simulations. Int. J. Heat Mass Transf. 2017, 107, 450–460. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, Z.; Chen, H.; Xu, L. Comparative studies of thermal conductivity for bilayer graphene with different potential functions in molecular dynamic simulations. Results Phys. 2021, 22, 103894. [Google Scholar] [CrossRef]
- Diao, C.; Dong, Y.; Lin, J. Reactive force field simulation on thermal conductivities of carbon nanotubes and graphene. Int. J. Heat Mass Transf. 2017, 112, 903–912. [Google Scholar] [CrossRef]
- Barbalinardo, G.; Chen, Z.; Lundgren, N.W.; Donadio, D. Efficient anharmonic lattice dynamics calculations of thermal transport in crystalline and disordered solids. J. Appl. Phys. 2020, 128, 135104–12. [Google Scholar] [CrossRef]
- Barbalinardo, G.; Chen, Z.; Lundgren, N.W.; Donadio, D. kALDo: Advanced Thermal Property Predictions via Lattice Dynamics and Machine Learning for Nanoscale Materials. https://github.com/nanotheorygroup/kaldo. Accessed: 2025-03-11.
- Siddi, F.; Cappai, A.; Colombo, L.; Melis, C. An Ab Initio Investigation of Ultra-Low Thermal Conductivity in Organically Functionalized TaS2TaS2. Adv. Theory Simulations 2024, 7. [Google Scholar] [CrossRef]
- Lindsay, L.; Broido, D.A.; Mingo, N. Flexural phonons and thermal transport in graphene. Phys. Rev. B 2010, 82. [Google Scholar] [CrossRef]
- Melis, C.; Colombo, L. Lattice Thermal Conductivity of Si1-xGex Nanocomposites. Phys. Rev. Lett. 2014, 112. [Google Scholar] [CrossRef]
- Lee, J.K.; Kim, J.G.; Hembram, K.P.S.S.; Kim, Y.I.; Min, B.K.; Park, Y.; Lee, J.K.; Moon, D.J.; Lee, W.; Lee, S.G.; et al. The Nature of Metastable AA’ Graphite: Low Dimensional Nano- and Single-Crystalline Forms. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
| 1 | The cutoff radius in LAMMPS AIREBO pair style [33] is defined as a fixed sigma value nm multiplied by a scale factor set to 3. |
Figure 1.
(a) Example of structure of conventional Moiré unit cells, the twisting angle is . (b) A schematic represenation of the subregion used in the simulations. The top half, red, is initially set at the temperature K, while the bottom half, blue, at K. During all the simulation, atoms in the two non-highlighted edges are constrained to avoid relative translations or rotations between the layers; their width is equal to 0.2 nm.
Figure 1.
(a) Example of structure of conventional Moiré unit cells, the twisting angle is . (b) A schematic represenation of the subregion used in the simulations. The top half, red, is initially set at the temperature K, while the bottom half, blue, at K. During all the simulation, atoms in the two non-highlighted edges are constrained to avoid relative translations or rotations between the layers; their width is equal to 0.2 nm.
Figure 2.
(a) Estimation of as a function of the fitting time for the smallest sample of AA-stacked bilayer graphene. Here, (see text). Convergence was studied by fitting the points with Eq. (5).(b) The inverse of the thermal conductivity as a function of the inverse of the length L for AA-stacked bilayer graphene. The fit was performed using the second-order approximation of the Taylor series of Eq. (6)
Figure 2.
(a) Estimation of as a function of the fitting time for the smallest sample of AA-stacked bilayer graphene. Here, (see text). Convergence was studied by fitting the points with Eq. (5).(b) The inverse of the thermal conductivity as a function of the inverse of the length L for AA-stacked bilayer graphene. The fit was performed using the second-order approximation of the Taylor series of Eq. (6)
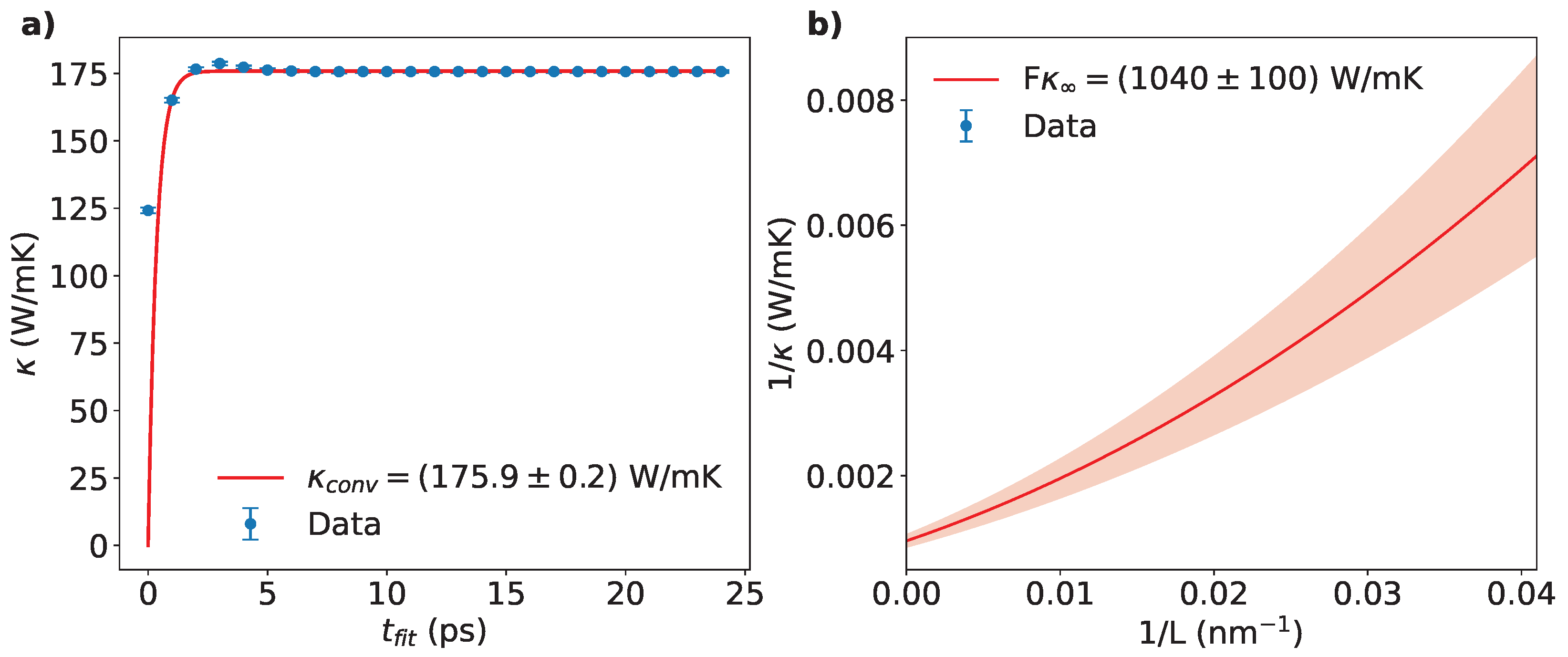
Figure 3.
(a) Thermal conductivity for monolayer, AA-stacked and AB-stacked bilayer graphene compared to the value reported by Han and Ruan [40] for the monolayer. Lines are just a guide for the eye. (b) Thermal conductivity as a function of the twisting angle in TBG simulated with the AIREBO potential. Lines are just a guide for the eye.
Figure 3.
(a) Thermal conductivity for monolayer, AA-stacked and AB-stacked bilayer graphene compared to the value reported by Han and Ruan [40] for the monolayer. Lines are just a guide for the eye. (b) Thermal conductivity as a function of the twisting angle in TBG simulated with the AIREBO potential. Lines are just a guide for the eye.
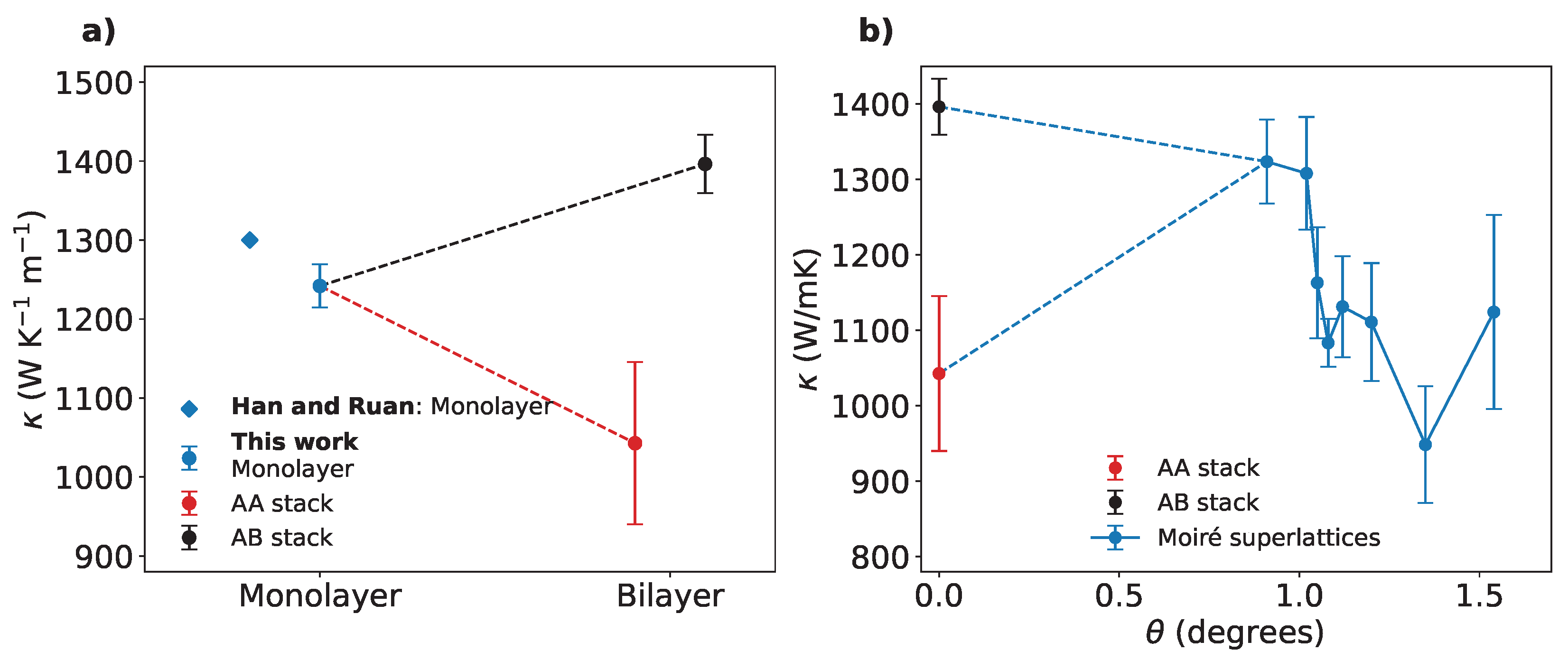
Figure 4.
(a, b, c) Schematic represetation of the AA, AB and SP stacking configuration, respectively. (d, e, f) Corresponding phonon dispersion relations. (g) MFP spectral distribution, (h) mode-resolved conductivity, and (i) cumulative conductivity (right) for different stacking configurations.
Figure 4.
(a, b, c) Schematic represetation of the AA, AB and SP stacking configuration, respectively. (d, e, f) Corresponding phonon dispersion relations. (g) MFP spectral distribution, (h) mode-resolved conductivity, and (i) cumulative conductivity (right) for different stacking configurations.
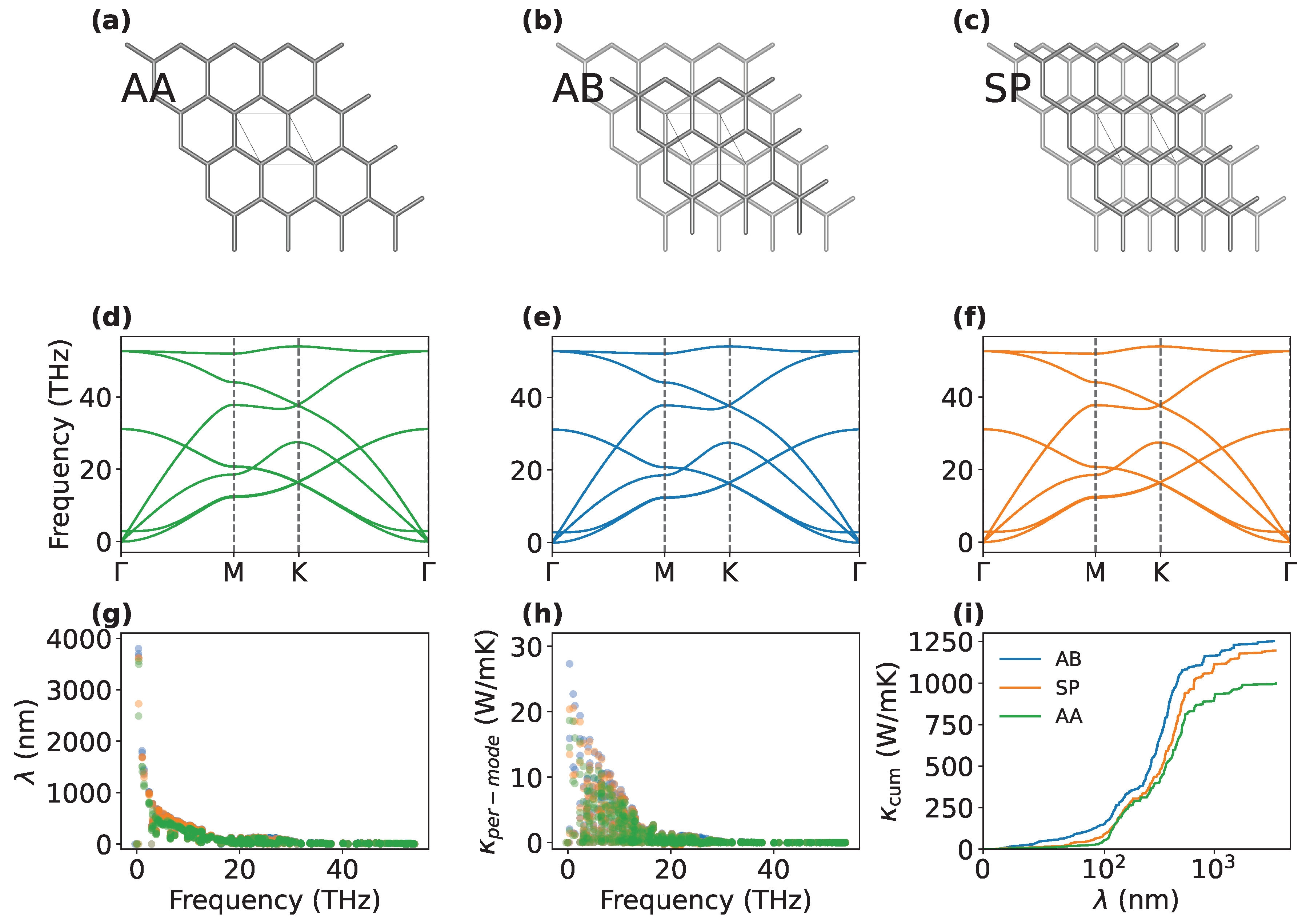
Figure 5.
Direct comparison between the thermal conductivity of TBG as a function of the twisting angle (right y-axis, blue dots) and the relative number of perfect AA, AB, and SP stacking configuration in each TBG sample (left y-axis, red dots) calculated as , where N is the total number of atoms in each sample.
Figure 5.
Direct comparison between the thermal conductivity of TBG as a function of the twisting angle (right y-axis, blue dots) and the relative number of perfect AA, AB, and SP stacking configuration in each TBG sample (left y-axis, red dots) calculated as , where N is the total number of atoms in each sample.
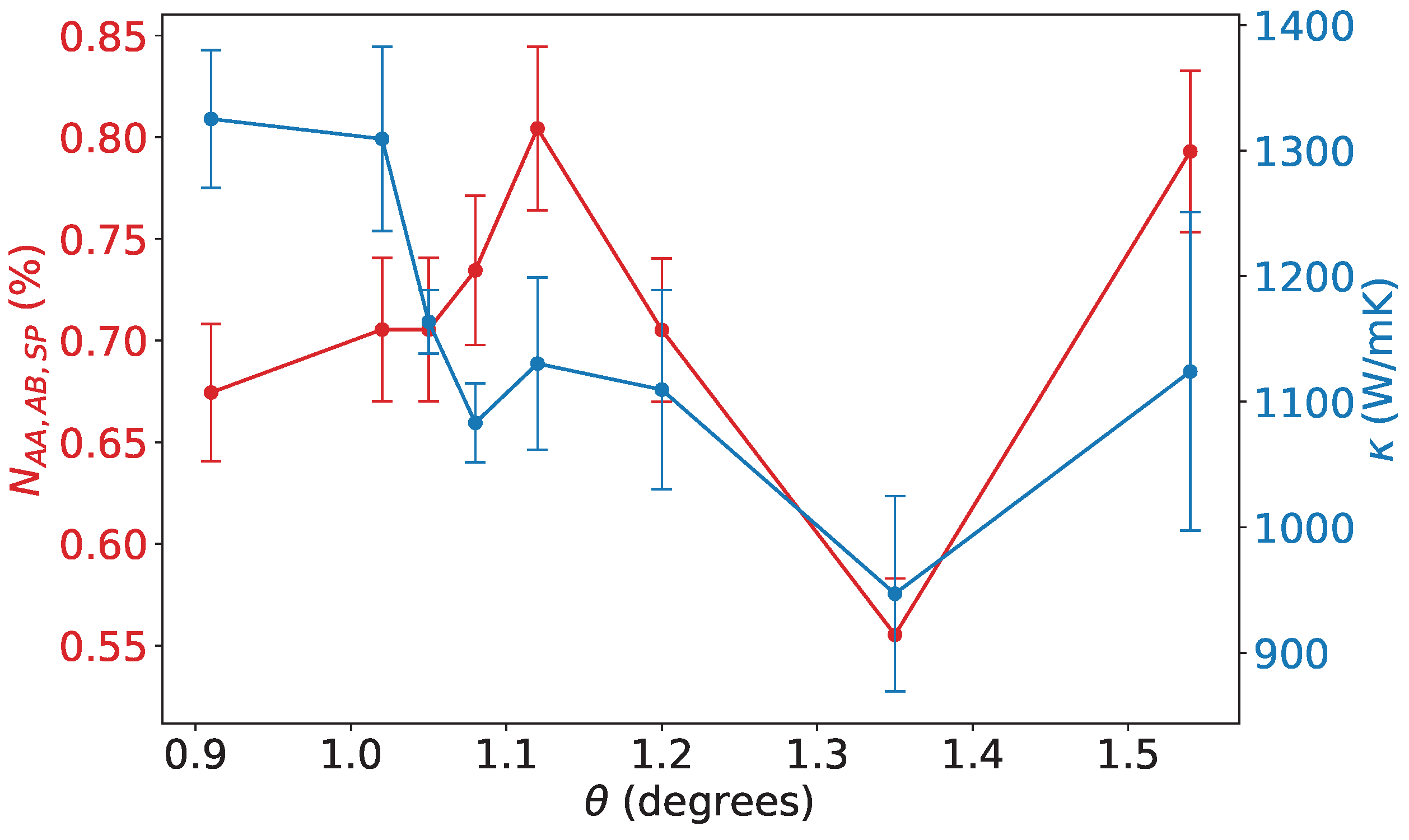

Table 1.
Moiré cells used in simulations, including twisting angle, number of atoms, length L, and width d.
Table 1.
Moiré cells used in simulations, including twisting angle, number of atoms, length L, and width d.
| 0° | ° | ° | ° | ° | ° | ° | ° | ° | |
|---|---|---|---|---|---|---|---|---|---|
| N | 39688 | 30248 | 25352 | 23816 | 22328 | 20888 | 18152 | 14408 | 11096 |
| L (nm) | 29.7 | 26.9 | 24.0 | 23.2 | 22.5 | 21.8 | 20.3 | 18.1 | 15.9 |
| d (nm) | 17.5 | 15.5 | 17.2 | 13.4 | 13.0 | 12.6 | 11.7 | 10.4 | 9.2 |
Table 2.
Thermal conductivity expressed in W/mK for monolayer, AA-stacked and AB-stacked bilayers of graphene simulated with the AIREBO potential.
Table 2.
Thermal conductivity expressed in W/mK for monolayer, AA-stacked and AB-stacked bilayers of graphene simulated with the AIREBO potential.
| Monolayer | AA stack | AB stack |
|---|---|---|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



