
Submitted:
07 April 2025
Posted:
08 April 2025
You are already at the latest version
Abstract
Iron dysregulation has emerged as a pivotal factor in neurodegenerative pathologies, especially through its capacity to promote ferroptosis, a unique form of regulated cell death driven by iron-catalyzed lipid peroxidation. This review synthesizes current evidence on the molecular underpinnings of ferroptosis, focusing on how disruptions in iron homeostasis interact with key antioxidant defenses, such as the system Xc^−–glutathione–GPX4 axis, to tip neurons toward lethal oxidative damage. Building on these mechanistic foundations, we explore how ferroptosis intersects with hallmark pathologies in Alzheimer’s disease (AD) and Parkinson’s disease (PD) and examine how iron accumulation in vulnerable brain regions may fuel disease-specific protein aggregation and neurodegeneration. We further surveyed the distinct components of ferroptosis, highlighting the role of lipid peroxidation enzymes, mitochondrial dysfunction, and recently discovered parallel pathways that either exacerbate or mitigate neuronal death. Finally, we discuss how these insights open new avenues for neuroprotective strategies, including iron chelation and lipid peroxidation inhibitors. By highlighting open questions, this review seeks to clarify the current state of knowledge and proposes directions to harness ferroptosis modulation for disease intervention.
Keywords:
Ferroptosis
; Iron homeostasis
; Cell death
; Neurodegeneration
; Alzheimer’s disease
; Parkinson’s disease
; Amyotropic lateral sclerosis
; Huntington's disease
; Multiple sclerosis
1. Introduction
Cell death is a fundamental biological process with both physiological and pathological implications. While accidental cell death (ACD) occurs under extreme conditions such as severe mechanical stress or toxic chemical exposure, regulated cell death (RCD) follows orchestrated molecular pathways [1]. Among the various forms of RCD, apoptosis is the most extensively characterized [2]. Apoptosis plays a central role in embryonic development, tissue remodeling, immune regulation, and aging. It is a programmed mechanism designed for the controlled removal of unwanted, damaged, or potentially harmful cells. Apoptosis is characterized by distinctive morphological features, such as cell shrinkage, membrane blebbing, nuclear condensation, and the formation of apoptotic bodies, and relies on the activation of caspases, a family of proteolytic enzymes that ensure that apoptosis proceeds in an orderly, often noninflammatory manner, facilitating tissue maintenance and immune tolerance [1,2].
In recent decades, several additional RCD pathways, including ferroptosis, pyroptosis, parthanatos, and autophagic cell death, have been identified. Each of these pathways involves unique molecular mechanisms and cellular outcomes and employs mechanisms distinct from those of apoptosis [1,3,4,5]. Among RCD mechanisms, ferroptosis, an iron-dependent process characterized by lethal lipid peroxidation that was first described by Stockwell and colleagues in 2012 [3], has emerged as particularly relevant to neurodegenerative disorders [6,7,8].
Ferroptosis differs fundamentally from other types of cell death in that it operates primarily through metabolic disruption rather than dedicated protein cascades. This process culminates in the oxidative destruction of cellular membranes through the convergence of three key mechanisms: disrupted antioxidant systems, dysregulated iron metabolism, and accelerated lipid peroxidation [9].
2. Significance and Scope
The brain presents a unique environment for iron homeostasis and ferroptosis regulation. Owing to its high oxygen consumption, abundant polyunsaturated fatty acids, and robust iron utilization for neurotransmitter synthesis and myelin formation, the central nervous system is especially vulnerable to ferroptotic damage. This vulnerability is further complicated by the blood–brain barrier (BBB), which tightly regulates iron influx and efflux, creating distinct compartmentalization of iron metabolism [10].
Neurodegenerative disorders represent a group of disorders characterized by the progressive loss of neuronal structure and function, manifesting as cognitive decline, motor dysfunction, and eventually neuronal death [11]. Major conditions include Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), multiple sclerosis (MS), and amyotrophic lateral sclerosis (ALS) [11]. These disorders share dysregulated iron homeostasis as a common feature.
Iron, which has long been recognized as a double-edged sword in biology, plays an indispensable role in central nervous system (CNS) functions and participates in multiple essential processes, such as oxygen transport, DNA synthesis, mitochondrial respiration, and neurotransmitter synthesis [12,13]. However, increased free iron can become neurotoxic priming neurons for ferroptotic death [12,13].
Owing to their high metabolic demands, oxygen consumption, and lipid-rich membranes, neurons are especially vulnerable to oxidative damage. This vulnerability is exacerbated by the fine balance between the intracellular iron levels required for maintaining essential cellular functions and the conditions that promote iron overload [12,13]. When this balance is disrupted, the accumulation of free iron catalyzes harmful reactions that culminate in lipid peroxidation and ultimately results in ferroptotic cell death.
This vulnerability is further complicated by the blood–brain barrier (BBB), which tightly regulates iron influx and efflux, creating distinct compartmentalization of iron metabolism. Disruptions in BBB integrity, a common feature in many neurodegenerative conditions, can lead to aberrant iron accumulation within the brain parenchyma [10,14,15,16].
The importance of ferroptosis in neurodegenerative diseases extends beyond being merely a marker of pathology; it functions as an active mediator of disease progression. Under these conditions, characteristic patterns of iron accumulation precede or accompany neuronal degeneration, suggesting that iron-induced lipid peroxidation may be a critical upstream event driving pathological cascades.
Consequently, targeting ferroptosis regulatory pathways, whether by modulating iron homeostasis, enhancing antioxidant defenses, or inhibiting lipid peroxidation, represents a promising therapeutic strategy to mitigate the progressive neuronal loss characteristic of these diseases. However, despite considerable advances, significant knowledge gaps remain in our understanding of ferroptosis within the context of neurodegeneration. Our current understanding of this process is largely derived from studies in nonneuronal systems or simplified in vitro models, which fail to fully capture the complexity of neuronal iron metabolism or the unique microenvironment of the brain.
This review synthesizes current knowledge on the molecular mechanisms underlying ferroptosis and examines their roles in neurodegenerative disease pathology, with a particular focus on AD and PD, where ferroptosis has been best characterized. We further explored potential therapeutic strategies for modulating this cell death pathway. By addressing the molecular mechanisms of iron homeostasis, lipid peroxidation, and BBB function in the brain, we aim to provide insights that will guide future research and therapeutic development in the field of neurodegeneration.
3. Ferroptosis: Core Mechanisms and Iron Regulation
3.1. The System Xc-—GSH—GPX4 Regulatory Axis
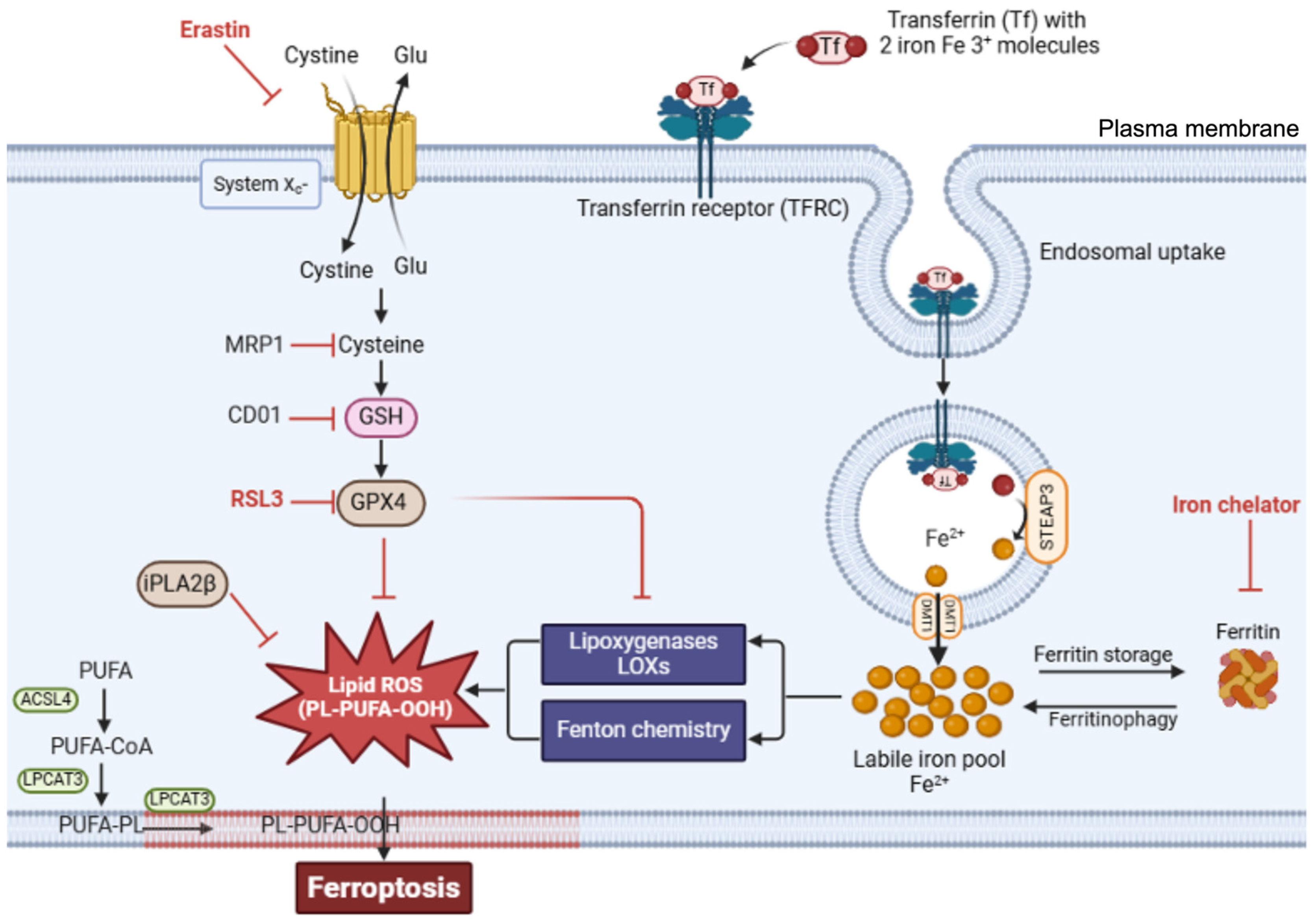
Ferroptosis occurs through the convergence of three fundamental mechanisms: disrupted antioxidant systems, dysregulated iron metabolism, and accelerated lipid peroxidation [9]. The primary antioxidant defense against ferroptosis involves glutathione peroxidase 4 (GPX4). GPX4 acts as part of the system Xc--GSH--GPX4 axis, which comprises the cystine/glutamate antiporter system Xc⁻, glutathione (GSH), and glutathione peroxidase 4 (GPX4), and is crucial for preventing ferroptosis. GPX4, a unique member of the oxidoreductase family, is a selenoenzyme that plays an essential role in protection against ferroptosis by converting toxic lipid hydroperoxides (L-OOH) into nontoxic lipid alcohols (L-OH) [9,17,18]. It utilizes glutathione (GSH) as a substrate for the reaction. GPX4 stands out among the glutathione peroxidase family as the sole enzyme capable of reducing complex lipid hydroperoxides within biological membranes. This unique capability makes GPX4 the primary defense against ferroptotic cell death, managing oxidative stress and maintaining membrane integrity by converting potentially lethal lipid hydroperoxides into nontoxic lipid alcohols (Figure 1) [19].
In addition to its direct antioxidant function, GPX4 also regulates the activity of lipoxygenases (LOXs), which are enzymes that promote the peroxidation of polyunsaturated fatty acids (PUFAs), underscoring the pivotal role of GPX4 as a key regulator of ferroptosis [20].
System Xc-, formed by the SLC7A11 and SLC3A2 subunits, functions as a cystine/glutamate antiporter that imports extracellular cystine in exchange for intracellular glutamate [21]. Once inside the cell, cystine is reduced to cysteine, an essential precursor for GSH synthesis. This GSH pool is the reducing substrate for GPX4, allowing it to detoxify lipid peroxides and inhibit ferroptosis. As a result, GSH levels, which are directly influenced by system Xc- activity, are central to ferroptosis regulation [19].
When the protective action of the system Xc--GSH--GPX4 protective axis fails, cells become vulnerable to ferroptotic death. For example, the inhibition of system Xc- depletes cellular GSH reserves, whereas direct GPX4 inhibition permits the uncontrolled accumulation of lipid peroxides. Accordingly, treatment with erastin, a well-characterized ferroptosis inducer, blocks cystine uptake by inhibiting system Xc-, resulting in GSH depletion and ferroptosis (Figure 1) [3,22]. Furthermore, inducing GSH depletion through alternative routes can also be a catalyst for ferroptosis. Accordingly, the overexpression of multidrug resistance-associated protein 1 (MRP1), which exports endogenous GSH, can sensitize cells to ferroptosis [23]. Similarly, the upregulation of cysteine dioxygenase 1 (CDO1) was shown to increase vulnerability to ferroptosis by inducing cysteine catabolism and, by extension, GSH depletion [24]. Moreover, direct inhibition of GPX4, whether via the use of the inhibitory molecule RSL3 (RAS-selective lethal) or RNA interference (RNAi), also induces ferroptosis by allowing lipid peroxides to accumulate and damage cell membranes [25]. This GPX4 inhibition leads to an increase in reactive oxygen species (ROS) and promotes the conditions necessary for ferroptotic cell death [22].
The vulnerability of the system Xc--GSH--GPX4 protective axis has significant implications for both disease mechanisms and therapeutic strategies, particularly in contexts where cells face high oxidative stress or increased metabolic demands [26,27,28,29]. On the one hand, therapeutic inhibition of the system Xc--GSH--GPX4 axis is promising for inducing ferroptosis in cancer cells that have developed resistance to conventional apoptosis-inducing treatments. On the other hand, enhancing the system’s function could be beneficial in degenerative diseases where ferroptosis contributes to pathology, such as neurodegenerative diseases, which are mostly associated with oxidative stress and cell death.
While GPX4 was long thought to be the only enzyme that converts potentially lethal lipid hydroperoxides into nonlethal forms, genetic and chemical screens recently identified a complementary protective pathway. Ferroptosis suppressor protein 1 (FSP1, also known as AIFM2) defines a separate, parallel defense line that does not rely on GPX4 or glutathione [30,31,32]. Instead, FSP1 functions by reducing coenzyme Q10 (ubiquinone) to its antioxidant form, ubiquinol, which uses NADPH as an electron donor [30,31,32]. This regeneration of ubiquinol in the lipid bilayer prevents the propagation of lipid peroxidation independent of the GPX4/GSH axis. Thus, even when GPX4 is inhibited or GSH is depleted, FSP1 can maintain the redox integrity of membranes by limiting lipid peroxidation through the ubiquinone–ubiquinol cycle.
3.2. Lipid Peroxidation Drives Ferroptosis
Lipid peroxidation in ferroptosis follows a specific sequence of events beginning with the activation of polyunsaturated fatty acids (PUFAs). The enzyme ACSL4 first converts free PUFAs to acyl-CoA derivatives, which LPCAT3 then incorporates into membrane phospholipids. These membrane-integrated PUFAs become primary targets for iron-catalyzed peroxidation.
The execution of ferroptosis requires the peroxidation of membrane-localized polyunsaturated fatty acids (FAs), a process that is enabled through the inhibition of GPX4 [6,33]. Nonetheless, compromised GPX4 function alone is insufficient for lipid peroxidation and subsequent ferroptosis; PUFAs must first be activated and incorporated into cellular membranes. In the initial phase, synthesized free PUFAs, including substances such as arachidonic acid, are transformed into acyl-coenzyme A thioesters (PUFA-CoAs) by the enzyme acyl-CoA synthetase long-chain family member 4 (ACSL4), a reaction necessary for PUFA utilization in membrane lipid synthesis (Figure 2) [34,35]. Subsequently, PUFA-CoAs undergo remodeling mediated by the enzyme lysophosphatidylcholine acyltransferase 3 (LPCAT3), thereby forming PUFA-containing phospholipids (PUFA-PLs), which are then incorporated into cell membranes [34,36]. Then, membrane-localized PUFA-PLs can be oxidized to generate toxic lipid peroxides (Figure 2).
Genetic screening of cells that are resistant to GPX4-inhibiting drugs suggested that ACSL4 and LPCAT3 are essential for ferroptosis triggered by the suppression of GPX4 activity [37]. In line with these findings, the inhibition of LPCAT3 by small-molecule inhibitors confers partial resistance to ferroptosis facilitated by GPX4 inhibition (Figure 2) [38]. Similarly, ACSL4-deficient cells exhibit resistance to ferroptosis upon knockout or inhibition of GPX4 [39]. On the other hand, the upregulation of ACSL4 has been linked to increased sensitivity to ferroptosis in different disease models [40,41,42,43,44,45].
The balance of lipid peroxidation is further modulated by calcium-independent phospholipase A2β (iPLA2β), which hydrolyzes peroxidized PUFA-PLs, thereby curtailing ferroptotic cell death [46]. This protective action highlights the importance of membrane-incorporated lipid peroxides as executors of ferroptosis; notably, supplementation with PUFA-PL peroxides, but not free PUFA peroxides, restores ferroptosis in ACSL4-deficient cells, demonstrating the critical role of membrane-localized lipid peroxides in ferroptosis [34,46,47].
In summary, the processes of PUFA remodeling and incorporation into phospholipids generate PUFA-PLs, the primary substrates of peroxidation in ferroptosis. The resulting membrane-localized lipid peroxides are the drivers of ferroptosis.
3.3. Reactive Oxygen Species (ROS)
The peroxidation process is accelerated through a self-propagating chain reaction. Iron, particularly in its ferrous (Fe²⁺) form, catalyzes the formation of ROS through the Fenton reaction [9], where ferrous iron (Fe2+) reacts with hydrogen peroxide to produce ferric iron (Fe3+) and hydroxyl radicals. These highly reactive hydroxyl radicals initiate the lipid peroxidation of polyunsaturated fatty acids (PUFAs) in cell membranes [48,49]. Lipid peroxides are unstable molecules that can decompose into various reactive aldehydes, further amplifying damage to cellular components and enhancing ROS production. The main sources of ROS are enzymes such as lipoxygenases (LOXs) and cytochrome P450 enzymes, which are key in the enzymatic production of ROS during ferroptosis by contributing to the formation of lipid hydroperoxides from PUFAs [50].
The ROS generated in ferroptosis propagate a chain reaction of lipid peroxidation, which leads to further ROS production and cellular damage. This propagation can be self-amplifying unless interrupted by lipophilic ROS-trapping antioxidants such as vitamin E or inhibitors of lipid peroxidation, such as ferrostatins or liproxstatins [51,52,53]. Furthermore, the inactivation of GPX, which catalyzes the reduction of lipid hydroperoxides to nontoxic alcohols, enhances susceptibility to ferroptosis [54].
Ultimately, the reactive species attack membrane-bound PUFAs, generating lipid radicals that perpetuate the peroxidation cascade [7,8,55]. This process specifically targets membrane phospholipids containing arachidonic and adrenic acids, explaining why cells enriched with these fatty acids show increased susceptibility to ferroptosis.
The final stages of ferroptosis involve catastrophic membrane failure leading to cell death. Unlike the controlled membrane blebbing observed in apoptosis, ferroptotic cell death results from widespread membrane peroxidation and subsequent rupture. This process can trigger inflammatory responses in surrounding tissue, as cellular contents leak into the extracellular space.
3.4. Mitochondrial Contribution to Ferroptosis
In addition to their function in energy production, mitochondria play crucial roles in ferroptosis [56,57]. During normal respiratory function, electrons occasionally leak from complexes I and III, leading to the partial reduction of oxygen and the formation of superoxide radicals. NADPH oxidases (NOX), membrane-bound enzymes that are capable of transferring electrons from NADPH to oxygen, can provide an additional source of ROS, particularly under stress conditions [57]. Furthermore, ferroptotic stress induces a significant shift in energy production with increased oxidative phosphorylation and ATP generation, whereas glucose metabolism through glycolysis decreases. This causes a surge in ROS formation and oxidative stress, leading to permanent mitochondrial damage [57].
As mitochondrial membranes suffer oxidative damage, the ETC becomes increasingly dysfunctional, leading to further electron leakage and ROS production [56,57]. This vicious cycle is exacerbated by the presence of free iron, which catalyzes the Fenton reaction, generating highly reactive hydroxyl radicals. The mitochondrial iron pool, which is normally tightly regulated for heme synthesis and iron–sulfur cluster assembly, becomes labile during ferroptosis, driving ROS production and contributing to energy failure [57].
Mounting evidence suggests that the mitochondrial control of ferroptosis through iron homeostasis, ROS production, and metabolic regulation is particularly important for neurodegenerative processes.
The impact of ferroptosis on mitochondria extends beyond ROS generation to the balance of mitochondrial fusion and division and the removal of damaged mitochondria through mitophagy, which makes neurons particularly vulnerable to ferroptosis.
4. Iron Homeostasis and Ferroptosis Regulation
Iron homeostasis represents a critical control point in ferroptotic cell death and involves tightly coordinated regulation of iron import, storage, utilization, and export pathways. Each of these processes is controlled by specific proteins and regulatory factors that collectively maintain appropriate iron levels while preventing excessive accumulation.
The intracellular metabolism of iron can be roughly categorized into the uptake, storage, utilization, and export of iron—each orchestrated by regulatory proteins to maintain the cellular redox balance and avoid oxidative damage.
4.1. Regulation of Iron Uptake
Iron import primarily occurs via transferrin receptor 1 (TfR), which binds to transferrin-bound iron on the cell surface [58]. Iron in the blood is bound by transferrin (Tf), a natural chelating plasma protein. When iron-laden transferrin binds to TfR on the cell surface, the resulting complex (TFRC) undergoes clathrin-mediated internalization into endosomes (Figure 3). Within the endosome, the lower pH of the environment prompts iron (Fe3+) to dissociate from transferrin and undergo reduction from Fe³⁺ to Fe²⁺, which is catalyzed by the ferrireductase STEAP3. The reduced iron is then transported across the endosomal membrane into the cytosol by divalent metal transporter 1 (DMT1) [58,59], while the transferrin-TfR complex is recycled back to the cell surface for additional rounds of iron uptake.
TfR1 recycling to the cell surface is mediated by the endosomal sorting machinery, particularly the retromer complex, in association with the adaptor sorting nexin 3 (SNX3) [60,61]. This complex recognizes specific motifs on TfR1 and facilitates its return to the plasma membrane. Disruption of this recycling pathway can alter cellular iron uptake and has been implicated in altered ferroptosis sensitivity. Accordingly, SNX3 overexpression confers increased ferroptosis sensitivity, whereas SNX3 loss confers ferroptosis resistance [62]. Consequently, the accumulation of TfRs on the plasma membrane has been established as an accurate marker of ferroptosis [63].
Another way in which cells regulate the intracellular concentration of iron is by modulating the abundance of TfRs by altering the expression of TfRs. For example, during iron deficiency, iron regulatory proteins (IRPs, see below) can promote increased TfR expression by stabilizing TfR mRNA [64].
4.2. Regulation of Iron Storage and Mobilization
Once in the cytosol, iron exists either as part of the labile iron pool or in storage complexes. The labile iron pool, which consists of redox-active Fe²⁺, requires strict regulation because of its potential to generate harmful ROS through Fenton chemistry. To prevent excessive accumulation of free iron, cells utilize the iron storage protein ferritin [65,66], which can sequester thousands of iron atoms in a relatively inert form.
The ferritin complex, which is composed of 24 subunits of heavy (FTH) and light (FTL) chains, plays a central role in iron storage [66]. FTH possesses ferroxidase activity that converts Fe²⁺ to Fe³⁺, whereas FTL provides nucleation sites for mineralized iron core formation [66]. The ratio of FTH to FTL varies among tissues and can be modulated in response to oxidative stress and inflammation, allowing cells to fine-tune their iron storage capacity and antioxidant defenses.
The regulation of iron storage critically impacts ferroptosis: ferroptosis sensitivity can be significantly influenced by agents that reduce iron availability, such as the iron chelator deferoxamine, as well as by changes in iron-regulating proteins that modulate the cellular labile iron pool [3]. For example, ferroptosis sensitivity can be increased by overexpression of TfR, which enhances iron uptake, and by decreased expression of FTH1 and FTL, which lowers ferritin levels and thereby reduces iron storage [25].
Ferritinophagy, the selective autophagic degradation of ferritin, represents an additional regulatory mechanism in cellular iron homeostasis and ferroptosis sensitivity [67]. This process involves nuclear receptor coactivator 4 (NCOA4), which functions as a selective cargo receptor that recognizes and delivers ferritin to form autophagosomes [67].
The interaction between NCOA4 and ferritin is regulated by cellular iron levels, with high iron concentrations promoting NCOA4 degradation through an iron-dependent ubiquitination mechanism mediated by the E3 ubiquitin ligase HERC2 [67,68,69]. Under conditions of iron deficiency, increased NCOA4-mediated ferritinophagy helps maintain adequate levels of bioavailable iron by releasing it from ferritin stores [69,70]. Conversely, oxidative stress can modulate ferritinophagy through effects on the autophagy machinery and NCOA4 stability [70].
The role of ferritinophagy in ferroptosis is significant, as increased ferritinophagy can promote ferroptotic cell death by expanding the labile iron pool. Inhibition of NCOA4 can protect cells from ferroptosis by preventing the release of iron from ferritin stores, whereas conditions that increase ferritinophagy can sensitize cells to ferroptotic death [71,72].
4.3. Iron Regulatory Network
Iron homeostasis is further regulated through an iron regulatory network that adjusts iron-dependent gene expression through iron regulatory proteins (IRPs) [64]. IRPs respond to the cellular iron status by controlling the expression of iron transport, storage, and export proteins through interactions with iron-responsive elements (IREs) in their mRNAs [64]. IRP1 contains an iron–sulfur cluster that, when assembled under iron-replete conditions, converts the protein into cytosolic aconitate hydratase, preventing IRE binding [64]. In contrast, IRP2 is regulated through iron-dependent proteasomal degradation mediated by the E3 ubiquitin ligase FBXL5, which acts as a direct iron sensor through its hemerythrin domain [64].
When cellular iron levels are low, IRPs bind to IREs in the untranslated regions (UTRs) of target mRNAs. The position of the IRE determines the regulatory outcome: IRP binding to 5′ UTR IREs blocks translation initiation, whereas binding to 3′ UTR IREs stabilizes the mRNA [64]. This dual mechanism allows the coordinated regulation of iron metabolism proteins. For example, under low-iron conditions, IRPs simultaneously repress the translation of ferritin and ferroportin (5′ IRE) while stabilizing transferrin receptor (3′ IRE) mRNA, thereby promoting iron acquisition while preventing iron storage and export.
4.4. Regulation of Iron Export
Iron export represents another important regulatory point and is mediated by ferroportin (SLC40A1), the sole known cellular iron exporter in vertebrates. This transmembrane protein, encoded by FPN1, facilitates the translocation of ferrous iron (Fe²⁺) across the plasma membrane, working in concert with ferroxidases that convert the exported Fe²⁺ to Fe³⁺ for safe transport in the circulation [73].
Ferroportin is regulated at the systemic level by the peptide hormone hepcidin, which binds directly to ferroportin and its ubiquitination, internalization, and subsequent lysosomal degradation, effectively blocking cellular iron export [74]. At the cellular level, ferroportin expression is controlled by the IRP/IRE system, with IRPs repressing ferroportin translation under low-iron conditions [65,75].
Hepcidin is produced primarily by the liver in response to high iron levels, inflammation, or other physiological signals and provides rapid control over cellular iron export, allowing a quick response to changing physiological conditions or environmental challenges [76,77]. Blockage of hepcidin-mediated regulation of ferroportin, therefore, can lead to disorders caused by iron overload [78].
Alterations in ferroportin expression or function can significantly impact cellular iron levels and subsequent susceptibility to ferroptotic cell death. For example, inflammatory signals can suppress ferroportin transcription during infection or inflammation, sensitizing cells to ferroptosis through intracellular iron accumulation [65]. Conversely, increased ferroportin expression or activity may protect against ferroptosis by reducing cellular iron levels, although this protection must be balanced against the requirements for essential iron-dependent cellular processes [65].
Figure 4.
Summary of the key mechanism underlying ferroptotic cell death. Ferroptosis is primarily governed by three interrelated pathways: the GSH/GPX4 antioxidant system, iron metabolism, and lipid peroxidation. The initiation of ferroptosis is driven by two critical events: suppression of the SLC7A11/GSH/GPX4 axis and the accumulation of intracellular free iron. Iron is imported into cells via transferrin-mediated endocytosis after binding to the transferrin receptor (TFR) forming the TFR complex (TFRC). Once inside the cell, ferric iron () is converted to ferrous iron (Fe²⁺) and transported into the cytoplasm via the action of STEAP3 and DMT1, respectively. Fe²⁺, once imported into the cytoplasm, either enters the labile iron pool, where it participates in redox reactions such as the Fenton reaction, or is sequestered and stored in a redox-inactive form by ferritin to prevent oxidative damage. The accumulation of redox-active iron promotes the formation of lipid peroxides and thus contributes to ferroptotic cell death. The system Xc⁻ antiporter imports extracellular cystine in exchange for intracellular glutamate at a 1:1 ratio. Intracellularly, cystine is reduced to cysteine, which serves as a precursor for glutathione (GSH) synthesis. This process is catalyzed by glutathione synthase (GSS). Glutathione peroxidase 4 (GPX4) plays a central role by reducing lipid hydroperoxides (PL-OOH) to non-toxic lipid alcohols (PL-OH) using GSH as a reducing agent. The lipid composition of cellular membranes also determines susceptibility to ferroptosis. Long-chain fatty acyl-CoA synthetase 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) promote the incorporation of polyunsaturated fatty acids (PUFAs) into membrane phospholipids, generating PUFA-containing phospholipids (PUFA-PLs), which are are highly susceptible to oxidation by reactive oxygen species (ROS), thereby promoting lipid peroxidation and ferroptosis. Several chemical and genetic modulators influence ferroptotic sensitivity. Iron chelators such as deferoxamine inhibit ferroptosis by reducing the labile iron pool and preventing iron-catalyzed oxidative damage. Erastin promotes ferroptosis by inhibiting system Xc⁻, leading to cystine and glutathione depletion and impaired GPX4 function. Similarly, RSL3 directly inhibits GPX4, resulting in lipid peroxide accumulation. Multidrug resistance-associated protein 1 (MRP1) exacerbates ferroptosis by exporting intracellular glutathione, diminishing antioxidant capacity. Cysteine dioxygenase 1 (CDO1) shifts cysteine metabolism away from glutathione synthesis toward taurine production, thereby reducing cellular defense against oxidative stress. In contrast, calcium-independent phospholipase A2 beta (iPLA2β) acts as a suppressor of ferroptosis by hydrolyzing oxidized phospholipids and attenuating lipid peroxidation. Created via BioRender.com (accessed 1 December, 2024).
Figure 4.
Summary of the key mechanism underlying ferroptotic cell death. Ferroptosis is primarily governed by three interrelated pathways: the GSH/GPX4 antioxidant system, iron metabolism, and lipid peroxidation. The initiation of ferroptosis is driven by two critical events: suppression of the SLC7A11/GSH/GPX4 axis and the accumulation of intracellular free iron. Iron is imported into cells via transferrin-mediated endocytosis after binding to the transferrin receptor (TFR) forming the TFR complex (TFRC). Once inside the cell, ferric iron () is converted to ferrous iron (Fe²⁺) and transported into the cytoplasm via the action of STEAP3 and DMT1, respectively. Fe²⁺, once imported into the cytoplasm, either enters the labile iron pool, where it participates in redox reactions such as the Fenton reaction, or is sequestered and stored in a redox-inactive form by ferritin to prevent oxidative damage. The accumulation of redox-active iron promotes the formation of lipid peroxides and thus contributes to ferroptotic cell death. The system Xc⁻ antiporter imports extracellular cystine in exchange for intracellular glutamate at a 1:1 ratio. Intracellularly, cystine is reduced to cysteine, which serves as a precursor for glutathione (GSH) synthesis. This process is catalyzed by glutathione synthase (GSS). Glutathione peroxidase 4 (GPX4) plays a central role by reducing lipid hydroperoxides (PL-OOH) to non-toxic lipid alcohols (PL-OH) using GSH as a reducing agent. The lipid composition of cellular membranes also determines susceptibility to ferroptosis. Long-chain fatty acyl-CoA synthetase 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) promote the incorporation of polyunsaturated fatty acids (PUFAs) into membrane phospholipids, generating PUFA-containing phospholipids (PUFA-PLs), which are are highly susceptible to oxidation by reactive oxygen species (ROS), thereby promoting lipid peroxidation and ferroptosis. Several chemical and genetic modulators influence ferroptotic sensitivity. Iron chelators such as deferoxamine inhibit ferroptosis by reducing the labile iron pool and preventing iron-catalyzed oxidative damage. Erastin promotes ferroptosis by inhibiting system Xc⁻, leading to cystine and glutathione depletion and impaired GPX4 function. Similarly, RSL3 directly inhibits GPX4, resulting in lipid peroxide accumulation. Multidrug resistance-associated protein 1 (MRP1) exacerbates ferroptosis by exporting intracellular glutathione, diminishing antioxidant capacity. Cysteine dioxygenase 1 (CDO1) shifts cysteine metabolism away from glutathione synthesis toward taurine production, thereby reducing cellular defense against oxidative stress. In contrast, calcium-independent phospholipase A2 beta (iPLA2β) acts as a suppressor of ferroptosis by hydrolyzing oxidized phospholipids and attenuating lipid peroxidation. Created via BioRender.com (accessed 1 December, 2024).
5. Brain-Specific Iron Homeostasis: The Critical Role of the Blood–Brain Barrier
5.1. Iron Functions in the Central Nervous System
Iron plays crucial roles in the CNS by sustaining neuronal energy metabolism and facilitating neural communication and plasticity [12]. It serves as an essential cofactor for enzymes involved in neurotransmitter synthesis, including tyrosine hydroxylase for dopamine production and tryptophan hydroxylase for serotonin synthesis [12]. Iron is also vital for myelination, as oligodendrocytes require iron for their development and for the production of myelin components [12].
Despite these essential functions, the high oxygen consumption and lipid-rich composition of the CNS render it particularly vulnerable to iron-induced oxidative damage [12]. Consequently, the brain has evolved specialized mechanisms to regulate iron uptake, distribution, and storage, with the blood–brain barrier (BBB) serving as the primary interface for controlled iron transport between systemic circulation and brain tissue.
5.2. Structure and Function of the Blood–Brain Barrier in Iron Transport
The BBB is a highly specialized, selectively permeable barrier composed primarily of brain microvascular endothelial cells joined by tight junctions alongside pericytes, astrocytic end-feet, and a supporting basement membrane (Figure 5) [10]. This complex structure tightly regulates the movement of substances, including iron, between the bloodstream and the CNS [10].
Systemic iron originates primarily from dietary sources and is absorbed in the duodenum [79]. Once in circulation, it binds to transferrin, which prevents excessive redox activity in the bloodstream. Iron uptake across the BBB primarily occurs through TfR-mediated endocytosis. Brain capillary endothelial cells express high levels of TfR1 on their luminal side, which mediates the uptake of transferrin-bound iron from the circulation through endocytosis [80,81,82]. Within acidic endosomes, iron is released from transferrin and reduced from Fe³⁺ to Fe²⁺, which is often facilitated by STEAP proteins [80,81,82]. The reduced iron is then exported into the cytosol via DMT1 and subsequently transported into the brain parenchyma through ferroportin (Figure 5) [10,14,80,83]. Ferrous iron (Fe²⁺) is then converted to ferric iron (Fe³⁺) through the action of ferroxidases such as hephaestin (HEPH) or ceruloplasmin (Figure 5) [14,84,85,86].
Astrocytes and pericytes contribute to BBB integrity and function by regulating tight junction formation and transporter expression [10]. Astrocytes, in particular, can influence iron transport across the BBB through the secretion of regulatory factors that modulate endothelial cell functions [10].
To meet increased iron demands or respond to fluctuations, particularly under conditions of stress, the CNS can also engage nontransferrin-bound iron uptake pathways [14]. These alternative mechanisms include the involvement of lactoferrin receptors and other transporters capable of binding nontransferrin-bound iron.
5.3. Iron Distribution and Regulation in the Brain Parenchyma
Once in the brain parenchyma, iron is distributed among neurons, astrocytes, microglia, and oligodendrocytes, each with distinct iron requirements and regulatory mechanisms [12,14]. Neurons express TfR, STEAP, and DMT1 for iron uptake, and some neuronal populations also express ferroportin for iron export [87].
Astrocytes play dual roles in brain iron homeostasis, both by storing iron in ferritin and supplying iron to neurons as needed [88]. They express both iron importers and exporters, allowing them to respond dynamically to changes in local iron demand [12,14,88]. Oligodendrocytes, which are responsible for myelin production, contain high levels of iron and express ferritin for iron storage [12,14,88]. The high iron requirement of these cells makes them particularly vulnerable to iron-mediated oxidative stress and ferroptosis.
Microglia, as resident immune cells of the CNS, can phagocytose damaged cells and degrade iron-containing proteins, thereby contributing to iron recycling within the brain [12,14]. During neuroinflammation, activated microglia can release stored iron, potentially contributing to oxidative stress in the surrounding tissue [12,14].
The regulation of iron within the brain parenchyma involves cell-specific expression patterns of iron transporters, storage proteins, and regulatory factors, allowing fine-tuned control of local iron concentrations. Iron efflux from the brain may occur via ferroportin-mediated export back across the BBB [89,90] or through the glymphatic system [91], which contributes to the clearance of metabolic waste from the brain.
Taken together, key mediators determining iron levels in the brain include TfR, which can increase neuronal iron uptake if overexpressed; ferroportin, whose reduced expression can impede iron export; and ferritin, which serves as the major iron storage protein but may be excessively degraded through ferritinophagy. Dysregulation of any of these control points can promote iron overload in the brain.
6. Disruption of Iron Homeostasis in Neurodegenerative Diseases
6.1. Common Pathways of Iron Dysregulation in Neurodegeneration
Neurodegenerative diseases share common pathways of iron dysregulation despite their distinct clinical and pathological features [12,92]. These include BBB dysfunction, altered expression of iron transporters and storage proteins, impaired iron export mechanisms, and disrupted cellular iron handling.
BBB dysfunction, a feature of many neurodegenerative conditions, or altered expression of iron transporters (e.g., increased TfR1 or DMT1 levels) can lead to unregulated iron influx into the brain parenchyma [10].
Within the brain parenchyma, dysregulated expression of iron regulatory proteins is commonly observed [12,92]. Increased expression of iron import proteins, coupled with decreased expression of storage proteins or export mechanisms, can lead to iron accumulation in vulnerable regions and, potentially, promote ferroptosis.
Additionally, neuroinflammation, a hallmark of neurodegenerative diseases, and age-related changes in iron metabolism can exacerbate iron overload and trigger ferroptosis [93,94].
The role of iron overload and ferroptosis in disease pathogenesis has been most extensively studied in AD and PD, revealing both shared and unique characteristics that suggest that ferroptosis is a potential driver of cell death. Below, we examine the evidence linking iron dyshomeostasis (through BBB disruption and altered iron handling in the brain) and iron-mediated neuronal cell death (influenced by mitochondrial ROS, neuroinflammation, and aging) to the disease pathology of AD and PD.
6.2. Blood-Brain Barrier Dysfunction in Alzheimer’s and Parkinson’s Disease
Dysfunction of the BBB is an important contributing factor in neurodegenerative diseases. BBB breakdown through the disruption of tight junctions between endothelial cells potentially allows unregulated paracellular iron influx, bypassing normal regulatory mechanisms [95].
In AD, the breakdown of tight junction complexes has been identified as a key feature of BBB dysfunction [96,97,98]. Studies using human postmortem tissues confirmed selective reductions in tight junction protein levels in cortical regions of AD brains, which correlated with synaptic degeneration and amyloid-beta (Aβ) accumulation [97,99]. Importantly, BBB disruption was shown to be an early marker of cognitive impairment in AD, regardless of Aβ or tau pathology or other signs of vascular disease [100].
Changes in BBB permeability in AD are also linked to dysregulated transport mechanisms, which facilitate Aβ influx into the brain and impair Aβ clearance [101,102]. Indeed, Aβ may further modulate iron transport across the BBB. Accordingly, Aβ-exposed astrocytes stimulate an increase in endothelial TfR1 and DMT1 expression in patients’ brain regions vulnerable to AD pathology, such as the hippocampus and cortex, thereby potentially increasing transferrin-mediated iron uptake by endothelial cells [103].
Additionally, genetic factors, particularly the apolipoprotein E (APOE) ε4 allele, have been strongly implicated in BBB breakdown in AD [98,104]. APOEε4 carriers exhibit increased BBB permeability in vitro and in vivo, as demonstrated through diverse approaches, including MRI-based imaging, biomarker analyses, and an induced pluripotent stem cell (iPSC) three-dimensional model, whereas studies on postmortem brain tissue have revealed pericyte loss or vascular degeneration linked to APOEε4-driven neuroinflammation [104,105].
BBB dysfunction in PD is less characterized. While tight junction disruption and transporter remodeling are well defined in AD, parallel investigations into PD remain underdeveloped, with fewer human studies examining tight junction protein loss or transporter dysfunction in relation to α-synuclein pathology [95,106]. iPSC-based models suggest that familial PD mutations may compromise BBB integrity [107]; however, direct human data remain sparse. Furthermore, inflammation-driven BBB disruption appears to be a shared feature between AD and PD, with elevated cytokine and oxidative stress responses contributing to vascular damage in both diseases, although the specific molecular pathways involved remain better characterized in AD [96,98,108].
In summary, aberrantly regulated iron uptake mechanisms at the BBB play important roles in the pathogenesis of AD and, potentially, PD. The dysregulation of TfR1, DMT1, and other iron transporters, in conjunction with BBB compromise and neuroinflammatory signals, may potentiate oxidative stress and thereby render neurons particularly susceptible to ferroptotic cell death.
6.3. Iron Homeostasis and Ferroptosis Susceptibility in Alzheimer’s and Parkinson’s Disease
Iron dyshomeostasis is increasingly recognized as a significant contributor to AD pathogenesis, with multiple lines of evidence showing disrupted iron regulation in affected brain regions, particularly those crucial for memory and cognition [109].
Amyloid precursor protein (APP) has been implicated in the modulation of iron export through interactions with ferroportin [110,111]. Studies have shown that APP facilitates iron export by interacting with ferroportin, particularly in cells lacking ceruloplasmin, such as cortical neurons [111]. The absence of APP leads to iron retention and oxidative stress, highlighting its role in iron metabolism. Notably, APP possesses ferroxidase activity, similar to ceruloplasmin, which is essential for oxidizing Fe²⁺ to Fe³⁺ before it can be exported via ferroportin [111]. This activity is important for loading iron onto transferrin and preventing oxidative stress. The alteration in APP processing observed in AD may disrupt this function, contributing to iron accumulation. Furthermore, as described in more detail below, Aβ plaques and tau pathology intersect with iron homeostasis [112,113,114], as these protein aggregates can bind iron, creating local concentrations of redox-active iron that promote oxidative stress.
Genetic factors associated with AD risk are also mechanistically connected to iron homeostasis. The APOE ε4 allele, the strongest genetic risk factor for late-onset AD, has been associated with increased brain iron accumulation in human patients and significantly elevated ferritin levels in their cerebrospinal fluid, which is associated with accelerated AD pathology [115,116,117]. Variants in other genes involved in iron transport and storage have been identified as potential modifiers of AD risk and progression [118]. Furthermore, in mice, APOE is required for iron homeostasis in the brain because it modulates TfR1, IRPs, Fpn1, aconitase and hepcidin in the hippocampus and basal ganglia [119].
Like AD, iron overload is a well-established feature of PD. However, iron overload in PD is predominantly associated with α-synuclein aggregation, neuroinflammation, and mitochondrial dysfunction [120,121]. The SNpc, the brain area most vulnerable to neuron loss in PD, is naturally rich in iron [122,123]. This selective vulnerability reflects the unique characteristics of dopaminergic neurons, including their high iron requirements for dopamine synthesis, the presence of neuromelanin that can bind iron, their high polyunsaturated fatty acid content, and their elevated metabolic rate [124,125,126,127,128]. These characteristics create an environment where even small disruptions in iron homeostasis can have significant consequences [129,130,131].
Furthermore, dopamine metabolism itself contributes to the vulnerability of SNpc neurons to iron-mediated damage. Dopamine autooxidation generates ROS and quinones, which can interact with iron to increase oxidative stress [121]. Additionally, dopamine metabolism through monoamine oxidase produces hydrogen peroxide as a byproduct [121], which can participate in mitochondrial iron-catalyzed Fenton reactions [132].
PD brains, like AD brains, also show altered expression of iron regulatory proteins. Increased expression of TfR1 and DMT1 has been observed in the SNpc of PD animal models and PD patients [133], potentially enhancing iron uptake. Studies also report reduced expression of ferroportin, which impairs iron export from neurons [134]. Additionally, changes in ferritin expression and increases in iron-responsive element binding proteins have been documented [135], reflecting disrupted iron storage and regulation.
Furthermore, genetic factors associated with PD are significantly associated with iron homeostasis and increased susceptibility to ferroptosis. Mutations in several PD-associated genes, including LRRK2, PINK1, Parkin, and DJ-1, affect mitochondrial function and iron metabolism [136,137,138]. For example, PINK1 and Parkin mutations can disrupt mitochondrial iron handling and increase oxidative stress, potentially increasing susceptibility to ferroptotic cell death [139,140,141,142,143,144,145,146,147,148]. DJ-1 mutations may compromise cellular antioxidant defenses, increasing the vulnerability of neurons to iron-dependent oxidative damage [136,149,150,151].
Taken together, the combination of elevated baseline iron levels and disease-related dysregulation of iron metabolism may create a toxic environment that underlies the selective vulnerability of dopaminergic neurons in PD. Indeed, increased mitochondrial oxidant stress in human SNc neurons has been shown to initiate a dopamine- and iron-dependent toxic cascade, leading to lysosomal dysfunction and α-synuclein accumulation in the presence of a PD-associated DJ-1 mutation—thus establishing a causal link among key pathological features of PD [152,153].
In summary, both AD and PD patients exhibit altered expression of iron transport and storage proteins in the brain. Specifically, increased DMT1 and TfR1 expression in vulnerable regions may enhance iron uptake, while decreased ferroportin expression can impair iron export, further promoting iron accumulation. Additionally, changes in ferritin levels and composition may reflect disrupted iron storage capacity. Finally, disease-specific factors and genetic mutations may contribute to multiple pathological mechanisms across these disorders.
7. Linking Iron Dysregulation, Ferroptosis, and Neurodegeneration
7.1. Regional Selectivity of Iron Accumulation and Neuronal Vulnerability
A striking feature of neurodegenerative diseases is their regional selectivity, with specific brain areas showing greater vulnerability to pathology. This selectivity coincides with patterns of iron accumulation, suggesting a relationship between regional iron handling and disease progression.
Neuroimaging studies using techniques such as quantitative susceptibility mapping and magnetic resonance imaging have consistently revealed elevated iron levels in critical regions [154,155,156]. In AD, iron accumulation is particularly pronounced in the hippocampus, associated cortices, and certain subcortical nuclei [156], regions critical for memory and cognition. This pattern aligns with the distribution of tau pathology and neurodegeneration. Importantly, these changes can be detected even in the prodromal or mild cognitive impairment (MCI) stages, supporting the notion that iron dysregulation is likely an early event in AD pathogenesis [157,158].
PD shows a distinct pattern of iron accumulation primarily centered on the SNpc [126,159], which coincides with the primary site of dopaminergic neuron loss [129,130,131]. Similar to AD, even in the early stages of PD, iron metabolism is disturbed, and iron begins to redistribute in the brain, particularly in regions such as the SNpc [160,161,162].
The distinct regional accumulation of iron in AD and PD patients may reflect differences in baseline iron content, variations in the expression of iron regulatory proteins, or region-specific responses to aging and inflammation. Localized iron leakage across a compromised BBB may generate ROS in affected regions, which further damages BBB integrity, creating a detrimental feedback loop. Notably, BBB dysfunction does not occur uniformly across the brain: the hippocampus and certain cortical regions exhibit an early and more severe BBB breakdown in patients with AD [100]. As described earlier, this breakdown is associated with cognitive impairment and can occur independently of Aβ pathology [100]. Similarly, the midbrain, including the substantia nigra, also exhibits a localized increase in BBB permeability in patients with PD [163].
7.2. Ferroptosis Signatures in Alzheimer’s and Parkinson’s Disease
Disruption of iron metabolism and increased susceptibility to ferroptotic pathways are common features of AD and PD. Neurons in the brains of AD and PD patients present multiple hallmarks of ferroptotic stress in affected regions, including increased lipid peroxidation products and disrupted expression of proteins that regulate iron homeostasis [93,94,164,165,166]. These changes are also observed in animal models of AD. For example, ferroportin was downregulated in the brains of AD patients and in APPswe/PS1dE9 mice [110,167]. Moreover, loss of ferroportin was associated with memory impairment in a second murine AD model [167].
While the molecular signatures are broadly similar in both diseases, the cellular context differs. In AD, iron dysregulation predominates in cortical and hippocampal regions, impacting both neurons and glia (astrocytes and microglia). In contrast, PD pathology centers on the SNpc, where dopaminergic neurons and surrounding glial cells are particularly susceptible. Regional differences in the expression of ferroptosis regulators, such as GPX4, system Xc⁻, or FSP1, and their heterogeneous expression in different cell types likely underlie these distinct vulnerabilities.
Recent advances in single-cell and spatial omics have enabled high-resolution dissection of ferroptotic mechanisms in neurodegeneration. In AD, Dang et al. (2022) [168] used single-cell RNA sequencing to dissect ferroptosis in astrocytes and identified FTH1 and SAT1 as key drivers of iron-induced lipid peroxidation, thereby linking ferroptosis to Aβ toxicity. Subsequent transcriptomic analyses revealed additional ferroptosis-related genes associated with hippocampal degeneration in AD [169,170].
In PD, single-nuclei RNA sequencing of a human stem cell–derived tri-culture system (microglia, neurons, and astrocytes) revealed that SEC24B is a regulator of iron-driven ferroptosis in microglia [171]. Zhang et al. (2024) [172] further linked α-synuclein aggregation to ferroptotic neuronal loss. Spatial transcriptomics was used to map transcriptional alterations in midbrain dopamine neurons, identifying molecular subtypes uniquely vulnerable to PD degeneration [173]. These spatial profiles confirmed the colocalization of α-synuclein aggregates with astrocyte clusters expressing low SLC7A11 and high SAT1 and ACSL4, implicating localized iron dyshomeostasis in disease progression [168].
Despite these insights, many studies rely on bulk transcriptomics and bioinformatics analyses, limiting cell type-specific and spatial resolution. Furthermore, comparative analyses between AD and PD and across disease stages remain scarce. Emerging single-cell studies are beginning to fill these gaps. Shwab et al. (2024) [174] and Bhattarchaya et al. (2025) [175] investigated transcriptional changes in AD and PD brains at single-cell resolution. These studies replicated the roles of important AD or PD genes and pathways and identified shared alterations in cellular energy metabolism and stress response, mitochondrial ROS, inflammation, lipid signaling, protein folding, and protein degradation [174,175]. Notably, oligodendrocytes, the primary myelin-producing cells, emerged as uniquely involved in PD pathology, whereas AD pathology was more strongly associated with microglia and astrocytes [174,175,176].
Significantly, dysfunction of oligodendrocytes may be an early hallmark of neurodegeneration [174,175,177]. Oligodendrocytes are particularly vulnerable to neurodegenerative conditions because of their high metabolic demands and sensitivity to oxidative stress [178,179]. As oligodendrocytes are the primary iron-containing cells in the brain and play a crucial role in maintaining iron homeostasis for myelin synthesis through the expression of transferrin and ferritin [180], their degeneration can release excess iron into the local environment, potentially exacerbating oxidative damage and ferroptosis.
Overall, single-cell and spatial omics approaches have revealed that while AD and PD share a common mechanism—excess iron–driven oxidative damage and ferroptosis—their cellular and regional contexts differ markedly, emphasizing the roles of microglia, astrocytes, and oligodendrocytes in ferroptosis. Future studies should therefore prioritize comparative, spatially resolved analyses across neurodegenerative diseases and disease stages to clarify temporal dynamics and pinpoint therapeutic targets.
7.3. Iron, Protein Aggregation, and Liquid–Liquid–Phase Separation (LLPS)
AD is characterized by the accumulation of Aβ plaques and neurofibrillary tangles composed of hyperphosphorylated tau protein [181]. Protein aggregates in AD may act as centers for iron accumulation and ROS production, potentially creating local environments conducive to ferroptotic cell death.
Studies have shown that Aβ plaques serve as sites of iron accumulation, with metal ions becoming entrapped within the fibrillar structure of these protein aggregates [182]. Metals, in turn, may further promote Aβ aggregation by inducing conformational changes that enhance oligomerization and fibril formation [182,183,184]. Similar bidirectional relationships exist between iron and tau pathology, with iron promoting tau hyperphosphorylation and aggregation [114,185], whereas hyperphosphorylated tau disrupts normal axonal transport mechanisms, potentially including those responsible for iron trafficking [114].
In PD, the relationship between iron and disease pathology is bidirectional [186]. Iron dysregulation in PD is closely linked to the aggregation of α-synuclein, the primary component of Lewy bodies [130]. Iron directly influences α-synuclein aggregation by promoting protein oligomerization and fibril formation REF. Additionally, the SNCA mRNA has an IRE in its 5′ untranslated region that controls its translation, suggesting that iron also regulates α-synuclein levels [187]. Conversely, α-synuclein aggregates can bind iron and act as a local source of oxidative stress through iron-catalyzed reactions [188]. As in AD, this bidirectional relationship may create a self-reinforcing cycle in which iron promotes α-synuclein aggregation, and the resulting aggregates further disrupt iron homeostasis.
Recent research has highlighted the role of LLPS, a mechanism that controls biological and biochemical processes [189], in protein aggregation in neurodegenerative diseases. Many disease-associated proteins, including tau [190,191,192,193], Aβ [194,195,196], and α-synuclein [130,197,198,199,200,201], can undergo LLPS to form liquid droplets, which may represent early stages in the pathway leading to pathological aggregation [130,190,191,192,193,197,198,199,200,201]. The liquid condensates can mature over time into more solid-like structures, eventually leading to the formation of toxic aggregates and fibrils [202].
Iron appears to modulate this process, with iron binding potentially accelerating the transition from liquid droplets to more stable fibrillar aggregates [190,203,204]. Early reports connected LLPS-driven protein aggregation with ferroptotic stress, implying that iron not only promotes lipid peroxidation but also modulates how vulnerable proteins transition from liquid-like droplets to insoluble deposits [205].
The interplay between iron, protein aggregation, and LLPS creates a complex pathological landscape in which these processes reinforce each other, potentially accelerating disease progression. Targeting this interplay represents a promising therapeutic approach for interrupting the neurodegenerative cascade.
7.4. Mitochondrial ROS
The brain may also be uniquely vulnerable to ferroptosis in part because of its mitochondria-rich neurons and high energy demands [56]. The mitochondrial iron pool, which is normally tightly regulated for heme synthesis and iron–sulfur cluster assembly, becomes dysregulated under conditions of iron overload [206,207,208], increasing the formation of hydroxyl radicals via the Fenton reaction and leading to oxidative damage to mitochondrial DNA, proteins, and lipids [56,209].
A self-perpetuating cycle of ROS production and oxidative stress is particularly evident in AD and PD, where mitochondrial dysfunction, characterized by deficits in respiratory chain complexes, altered mitochondrial morphology, and impaired mitophagy, is well documented [56,210]. These early changes in disease progression heighten neuronal susceptibility to ferroptosis by increasing ROS production and compromising energy metabolism. In turn, iron overload and lipid peroxidation exacerbate mitochondrial damage, while dysfunctional mitochondria release additional ROS and labile iron, collectively fueling ferroptotic processes.
7.5. Neuroinflammation and Ferroptosis
Neuroinflammation is another well-established feature of both AD and PD, and accumulating evidence suggests that it contributes to disease progression partly through effects on iron homeostasis and ferroptosis sensitivity [93,211]. Neuroinflammation is triggered by the activation of microglia and astrocytes, which release proinflammatory factors that exacerbate neuronal damage [212]. Proinflammatory cytokines such as IL-1β, TNF-α, and IL-6 can alter the expression of iron transporters and storage proteins, leading to increased cellular iron uptake and decreased iron export [212] through the upregulation of TfR1 and DMT1 expression and the downregulation of ferroportin expression [212,213]. Iron overload, in turn, contributes to the labile iron pool, which, in the presence of ROS, catalyzes the Fenton reaction and exacerbates lipid peroxidation.
Microglia are professional phagocytes of the central nervous system and are essential for removing a range of debris, including apoptotic neurons, myelin fragments, oxidized lipids, and degenerating synapses. Significantly, their phagocytic capacity is modulated by AD risk genes and protective variants, including APOE4 and APOE3 [214,215,216,217,218,219]. RNA sequencing analysis further revealed a possible link between lipid metabolism, ferroptosis, and phagocytosis in microglia [171]. Activated microglia can also release iron from their stores, increasing extracellular iron levels in the microenvironment surrounding neurons [220]. This iron can be taken up by neurons, potentially overwhelming their iron regulatory mechanisms and promoting ferroptotic cell death.
Inflammatory cascades typically involve reciprocal signaling among glial cells. The release of damage-associated molecular patterns from ferroptotic neurons [221] can further activate microglia and astrocytes, creating a feedforward cycle of inflammation and cell death. Oligodendrocytes can also modulate astrocyte and microglial activity via secreted factors; conversely, astrocytes and microglia produce signals that influence oligodendrocyte maturation and survival [171,211,222]. This intercellular cross-talk can perpetuate neuroinflammation.
7.6. Aging and Ferroptosis Susceptibility
As individuals age, different iron complexes tend to accumulate in brain regions associated with motor and cognitive decline [94,223]. The disruption of normal iron homeostasis may contribute to the altered cellular iron distribution and subsequent regional accumulation observed in neurodegenerative disorders.
The vulnerability of neurons to ferroptosis can also be exacerbated by age-related decreases in antioxidant defenses. For example, aging-related changes in iron handling and antioxidant defenses may contribute to the age-dependent increase in AD risk. AD and PD neurons exhibit decreased activity of the GPX4 system, which normally protects against ferroptosis by converting lipid hydroperoxides into nontoxic compounds [164,165].
This phenomenon may be particularly relevant in PD, as the high content of polyunsaturated fatty acids in neuronal membranes, combined with the oxidative environment created by dopamine metabolism and iron accumulation, makes these cells especially susceptible to ferroptotic damage [224,225,226,227]. In addition, age-related disruption of BBB function may lead to increased iron accumulation in the brain, further enhancing neuronal vulnerability to ferroptosis [228,229].
8. Iron Dyshomeostasis and Ferroptosis: Implications for Other Neurodegenerative Conditions
While AD and PD are the two primary examples of neurodegenerative diseases linked to iron overload and ferroptosis, converging evidence increasingly establishes ferroptosis and iron dyshomeostasis as critical pathogenic mechanisms underlying other neurodegenerative conditions, including ALS, HD, and MS. Each disease exhibits unique features - demyelination, motor neuron loss, or striatal atrophy - but may share this common mechanism of iron-induced oxidative cell death, warranting further investigation.
8.1. Amyotrophic Lateral Sclerosis (ALS) – Ferroptotic Motor Neuron Loss
ALS is characterized by the selective degeneration of motor neurons in the cortex, brainstem, and spinal cord [230]. Neuroimaging and histological studies have provided evidence of significant iron accumulation in affected regions in both patients and animal models, suggesting a role for ferroptosis in the distinctive pattern of motor neuron loss in ALS. For example, MRI has revealed abnormal hypointense signals in the primary motor cortex of ALS patients, which pathological analysis has attributed to iron-loaded microglia in the deep cortical layers [231]. This iron deposition in the motor cortex correlated with upper motor neuron impairment and was apparent even in early (premanifest) stages of disease [231]. Similarly, postmortem analyses and susceptibility-weighted imaging have revealed excessive iron in the spinal cord and various subcortical nuclei of ALS patients [232]. In transgenic ALS mouse models (e.g., SOD1 mutations), ventral spinal motor neurons have shown progressive iron accumulation correlating with disease progression [232]. These patterns indicate that both neurons and glia in ALS patients show enhanced iron loading in regions undergoing neurodegeneration.
At the molecular level, recent findings strongly link ferroptosis to ALS pathogenesis. In spinal cord tissue from both sporadic and familial ALS patients, researchers have reported a marked depletion of GPX4 [233], indicating an impaired ability to detoxify lipid peroxides. Notably, the same signature is recapitulated in multiple ALS mouse models, where GPX4 loss in the spinal cord occurs early and universally during disease development [233]. The loss of this ferroptosis suppressor, together with the dysregulation of other antioxidant pathways, such as NRF2 signaling and glutathione synthesis, creates high susceptibility to iron-mediated lipid peroxidation in ALS neurons [233]. Indeed, lipid peroxidation damage, which is consistent with ferroptotic injury, is a prominent feature of degenerating motor neurons in individuals with ALS [234]. Conversely, genetic or pharmacological enhancement of GPX4 activity has been shown to delay the onset of paralysis, preserve motor neurons, and prolong survival in ALS models (SOD1G93A) [235]. This neuroprotection was accompanied by an attenuation of lipid peroxidation in the spinal cord.
In addition to neurons, glial cells in ALS also contribute to ferroptosis-related pathology. Iron import and storage proteins (DMT1, transferrin receptor, ferritin) are abnormally upregulated in ALS models, indicating that microglia and astrocytes may sequester excess iron and propagate oxidative stress [232]. Therefore, iron accumulation may amplify neuroinflammation, further contributing to motor neuron vulnerability via ferroptotic mechanisms. Indeed, a ferroptosis signature identified in a recent patient study has been shown to act as a prognositic biomarker panel for ALS [236]. Together, these data support a model in which iron overload and impaired antioxidant defenses trigger ferroptosis of motor neurons in ALS. The characteristic motor neuron loss in ALS patients, both in the cortex and the spinal cord, may therefore stem from iron-induced lipid peroxidation.
8.2. Huntington’s Disease (HD) – Iron, Mitochondria, and Striatal Neurodegeneration
HD is an inherited neurodegenerative disorder marked by progressive loss of medium spiny neurons in the striatum (caudate and putamen), leading to motor dysfunction (chorea) and cognitive decline [237]. Emerging evidence implicates iron dyshomeostasis and ferroptosis in the distinctive striatal pathology of HD. Compared with healthy controls, brain imaging studies have consistently revealed elevated iron content in the basal ganglia, particularly in the caudate, putamen, and globus pallidus, of both premanifest and symptomatic HD individuals [238]. This accumulation of iron in the striatum increases with disease severity and duration, suggesting that it is both an early event and a continuing process throughout HD progression [234]. Postmortem analyses have confirmed increased ferric iron in the striatal neurons of HD patients [238].
Mechanistically, the mutant huntingtin protein appears to disrupt cellular iron handling by increasing the expression of iron importers and promoting iron accumulation in neuronal mitochondria [239]. This mitochondrial iron overload contributes to lipid ROS generation, potentially linking iron directly to the energy deficits and oxidative damage characteristic of HD neurons [239,240]. Both HD mouse models and patient tissues exhibit increased lipid peroxidation and oxidative damage in striatal cells, which is correlated with the degree of neurodegeneration and severity of motor symptoms [240,241,242,243]. The accumulation of toxic lipid peroxidation byproducts in the corpus striatum has been [244,245] associated with worsened motor performance, suggesting that ferroptosis is a plausible mechanism for the selective vulnerability of the striatum in HD.
Transcriptomic studies further support this connection, revealing the dysregulation of multiple ferroptosis regulators in HD patient brains. These include altered expression of antioxidant enzymes and iron transport proteins and activation of lipid-peroxide-producing enzymes such as 5-lipoxygenase (ALOX5) in both neurons and microglia of the striatum [240,246]. These changes suggest that neurons and glial cells in HD may engage in ferroptotic pathways that contribute to the overall neurodegenerative process. In summary, HD pathology features regional iron accumulation that triggers mitochondrial dysfunction and oxidative stress, ultimately leading to ferroptosis of vulnerable striatal neurons.
8.3. Multiple Sclerosis (MS) – Iron, Ferroptosis, and Demyelination
MS is an inflammatory demyelinating disease [247] and converging evidence suggests that iron dysregulation contributes significantly to its pathology. Abnormal iron accumulation is observed in so-called “iron rim” lesions, where iron-laden microglia and oligodendrocytes contribute to chronic tissue damage[248,249]. Postmortem and magnetic resonance imaging (MRI) studies of progressive MS have revealed chronic active plaques with iron-laden microglia at the edges that are more destructive, expand over time, and are characterized by a ferroptosis signature [250,251,252]. This regional iron overload may accelerate oxidative injury and impair remyelination processes.
The cellular mechanisms connecting iron and MS pathology are becoming increasingly clear. Oligodendrocytes, which are essential for myelin production, are naturally enriched in iron for myelin synthesis, which may render them particularly susceptible to ferroptosis under conditions of oxidative stress [253]. Excess iron in these cells promotes oxidative stress and lipid peroxidation, creating ideal conditions for ferroptotic cell death. Single-cell RNA sequencing and animal studies have confirmed that oligodendrocyte death in MS is accompanied by molecular hallmarks of ferroptosis [251,254]. At the molecular level, experimental autoimmune encephalomyelitis (EAE) mouse models have demonstrated increased expression of iron import proteins (e.g., TfR1) and markers of ferritinophagy (NCOA4), which are correlated with oligodendrocyte death. Pathological iron accumulation appears to result from altered expression and function of iron transport proteins at the blood–brain barrier, exacerbated by inflammatory cytokines and oxidative stress that modulate key iron transporters such as transferrin receptor, divalent metal transporter 1, and ferroportin. These changes lead to increased iron influx or impaired efflux, promoting tissue iron accumulation.
Importantly, therapeutic approaches targeting ferroptosis have shown promise in MS models. Treatment of EAE mice with small-molecule ferroptosis inhibitors (liproxstatin-1 or ferrostatin-1) significantly alleviates clinical severity and demyelination [251,255]. Consistent with ferroptotic mechanisms, neurons in EAE patients exhibit reduced levels of the anti-ferroptotic enzyme glutathione peroxidase 4 (GPX4) and elevated lipid peroxidation, with GPX4 depletion correlated with neuronal death [251,255]. An elegant study revealed that a histone methyltransferase (G9a) can drive MS progression by repressing neuronal GPX4 and other anti-ferroptosis genes, underscoring the disease relevance of ferroptotic pathways [251,256].
The ferroptosis–inflammation connection appears to be bidirectional in MS. As neurons undergo ferroptosis, they can secrete factors that further activate infiltrating T cells (Th1/Th17), exacerbating autoimmune inflammation [251,257]. This creates a vicious cycle linking iron overload directly to the demyelination and neuroinflammation that characterize MS pathology. These findings collectively suggest that iron-mediated ferroptosis in oligodendrocytes and neurons may be a fundamental mechanism underlying MS progression.
8.4. Converging Mechanisms: Iron-Mediated Ferroptosis as a Common Pathway in Neurodegeneration
The evidence presented for ALS, HD, and MS reveals striking parallels with the iron dysregulation previously discussed in AD and PD, suggesting that iron-mediated ferroptosis represents a unifying mechanism in neurodegeneration. Despite their distinct clinical presentations and affected neural populations, these conditions share several key features: region-specific iron accumulation, impaired antioxidant defenses, and vulnerability to lipid peroxidation. In each disease, iron dysregulation occurs in precisely the most affected regions, motor neurons in ALS patients, striatal neurons in HD patients, and oligodendrocytes in MS patients, suggesting that iron-catalyzed oxidative damage may determine regional vulnerability patterns. Moreover, the molecular signatures of ferroptosis, particularly GPX4 depletion and lipid peroxidation, appear consistently across these disorders, often preceding overt neurodegeneration.
9. Clinical Translation: Iron Chelation and Overcoming the Blood–Brain Barrier
Given the growing evidence implicating ferroptosis in neurodegeneration, targeting this cell-death pathway therapeutically offers a promising approach for treating different neurodegenerative diseases. Translating knowledge of iron-driven pathology into patient therapies has focused largely on iron chelators, which bind and sequester excess iron before it can participate in damaging Fenton reactions [258,259].
Among these, deferiprone has attracted particular attention for its potential to slow or modify the course of PD. Deferiprone is already approved for treating systemic iron overload in β-thalassemia patients [260] and has shown promising results in preliminary clinical trials aimed at reducing iron levels in the substantia nigra of PD patients. While deferoxamine has shown positive results in preclinical models, its clinical utility in treating neurodegeneration has been limited by poor brain penetrance and systemic side effects [259,261]. Moreover, while early studies suggested modest neuroprotective effects and a slowdown in motor symptom progression, the trials were relatively small, and improvements were not universally observed. Few clinical trials have investigated iron chelation therapy for AD thus far, partly because AD pathology is more diffuse, and the direct correlation between iron deposition and clinical symptoms is less straightforward.
In addition to chelation, enhancing neuronal antioxidant defenses, such as GPX4 activity [262,263,264,265,266,267,268], or alternative pathways, such as FSP1-mediated ubiquinol regeneration, could suppress ferroptosis. Since reactive astrocytes, microglia, oligodendrocytes appear to be early hallmarks of neurodegeneration and contribute to iron dysregulation and the inflammatory microenvironment, therapies aimed at normalizing their function could also be beneficial. Combination therapies that address both iron imbalance and oxidative stress (for example, the use of ferrostatin or mitochondrion-targeted antioxidants) may be especially promising [269,270,271]. Early intervention is also likely key, as iron accumulation and ferroptotic stress can precede overt clinical symptoms.
Targeting ferroptosis through iron chelators, antioxidants, or inhibitors of lipid peroxidation has also shown promise in preclinical models of MS, ALS, and HD, suggesting that modulating iron-driven cell death pathways could be a viable therapeutic strategy to slow or mitigate these otherwise intractable neurodegenerative diseases. Anti-ferroptotic interventions have shown promise in animal models of each condition, suggesting that targeting iron homeostasis and lipid peroxidation could offer neuroprotective strategies applicable across multiple neurodegenerative diseases. The cell-specific vulnerabilities of motor neurons in ALS, medium spiny neurons in HD, and oligodendrocytes in MS may reflect different cellular capacities for managing iron and combating lipid peroxidation. Understanding these differences within the common framework of iron-mediated ferroptosis could guide the development of both broad-spectrum and disease-specific therapeutic approaches. As research continues to elucidate the precise mechanisms linking iron dysregulation to cell death in these disorders, ferroptosis inhibition has emerged as a promising strategy that could transform our approach to treating the underlying pathology rather than merely addressing symptoms of neurodegeneration.
A key hurdle in any therapeutic approach targeting ferroptosis in the brain is ensuring that the drug actually reaches its neuronal targets. The BBB restricts the entry of most small molecules, including many iron chelators. Agents such as deferiprone do exhibit some capacity to cross the BBB, but others with higher molecular weights or different chemical structures are often excluded. This limitation has driven the development of novel iron chelators designed specifically for neurological applications with optimized molecular properties to increase BBB permeability [259,261,272].
New drug delivery strategies, such as encapsulating chelators or ferroptosis inhibitors in nanoscale carriers coated with BBB-penetrating ligands, are being pursued [273,274]. Nanocarriers constructed from lipids, polymers, or biocompatible metals can release their cargo in a controlled manner within the brain, potentially increasing both the efficacy and safety profiles. Another emerging approach is to modify small-molecule inhibitors, either chemically or by linking them to shuttle peptides so that they exploit endogenous transport systems across the BBB [275].
The partial successes observed with deferiprone in PD and the more sporadic attempts in AD underscore both the promise and the complexity of ferroptosis-focused therapies. As research advances, the refinement of BBB-penetrating platforms and the identification of biomarkers that identify patients who stand to benefit most may well tip the balance toward successful clinical translation.
10. Challenges and Future Directions
Despite these advances, many open questions and knowledge gaps remain. While an increasing number of studies support an association between iron overload, ferroptosis, and neurodegeneration, this association is not causal. The precise temporal sequence linking iron accumulation to protein aggregation is still under investigation, and reliable biomarkers of ferroptosis in humans are still lacking. Future research integrating single-cell omics, longitudinal neuroimaging, and clinical trials will be crucial for refining our understanding of a causative link and the underlying molecular mechanisms. Such studies will also be necessary for devising therapeutic strategies and for identifying the patients most likely to benefit from ferroptosis-targeting interventions. Moreover, an emerging area of interest is the link between the gut microbiome, ferroptosis, and neurodegenerative diseases [276,277,278].
Many challenges also remain in the development of more effective ferroptosis-targeted therapeutics [263]. The complexity of neurodegenerative diseases, involving multiple cell types and pathological processes, necessitates careful consideration of timing and regional targeting in therapeutic approaches.
11. Conclusions
Ferroptosis represents a compelling molecular pathway that unites iron dyshomeostasis and oxidative stress with neuronal loss in AD and PD. The central nervous system’s high metabolic demands, lipid-rich membranes, and reliance on tight iron regulation set the stage for iron-driven damage when homeostasis falters. Mounting data suggest that pathologic iron accumulation not only correlates with hallmark protein aggregates but also actively drives disease progression via ferroptotic death.
Continued efforts to elucidate ferroptosis mechanisms in neurons, refine iron-targeting therapies, and improve drug delivery across the BBB hold promise for more effective disease-modifying treatments. By focusing on iron homeostasis in the brain and linking it directly to ferroptosis, we gain a more cohesive and translational perspective on the pathophysiology of AD and PD—and on potential strategies to halt or slow these devastating disorders.
Author Contributions
Conceptualization, N.A., and J.K.; writing—original draft preparation, N.A., K.K., and J.K.; visualization, N.A., K.K. and J.K.; funding acquisition, J.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a Collaborative Research Program Grant, grant number 021220CRP1722, awarded by Nazarbayev University to J.K.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther 2021, 6, 128. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Hirata, Y.; Cai, R.; Volchuk, A.; Steinberg, B.E.; Saito, Y.; Matsuzawa, A.; Grinstein, S.; Freeman, S.A. Lipid peroxidation increases membrane tension, Piezo1 gating, and cation permeability to execute ferroptosis. Curr Biol 2023, 33, 1282–1294 e1285. [Google Scholar] [CrossRef]
- Balakrishnan, M.; Kenworthy, A.K. Lipid Peroxidation Drives Liquid-Liquid Phase Separation and Disrupts Raft Protein Partitioning in Biological Membranes. J Am Chem Soc 2024, 146, 1374–1387. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem Biophys Res Commun 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol Rev 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Wilson, D.M., 3rd; Cookson, M.R.; Van Den Bosch, L.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of neurodegenerative diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef]
- Gao, G.; You, L.; Zhang, J.; Chang, Y.Z.; Yu, P. Brain Iron Metabolism, Redox Balance and Neurological Diseases. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Masaldan, S.; Bush, A.I.; Devos, D.; Rolland, A.S.; Moreau, C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic Biol Med 2019, 133, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Moos, T.; Rosengren Nielsen, T.; Skjorringe, T.; Morgan, E.H. Iron trafficking inside the brain. J Neurochem 2007, 103, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jung, U.J.; Kim, S.R. The Crucial Role of the Blood-Brain Barrier in Neurodegenerative Diseases: Mechanisms of Disruption and Therapeutic Implications. J Clin Med 2025, 14. [Google Scholar] [CrossRef] [PubMed]
- Pedder, J.H.; Sonabend, A.M.; Cearns, M.D.; Michael, B.D.; Zakaria, R.; Heimberger, A.B.; Jenkinson, M.D.; Dickens, D. Crossing the blood-brain barrier: emerging therapeutic strategies for neurological disease. Lancet Neurol 2025, 24, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Friedmann Angeli, J.P. Glutathione peroxidase 4 (Gpx4) and ferroptosis: what’s so special about it? Mol Cell Oncol 2015, 2, e995047. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Brigelius-Flohe, R.; Maiorino, M. Glutathione peroxidases. Biochim Biophys Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem 1999, 274, 11455–11458. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.Y.; Poddar, A.; Magtanong, L.; Lumb, J.H.; Mileur, T.R.; Reid, M.A.; Dovey, C.M.; Wang, J.; Locasale, J.W.; Stone, E.; et al. A Genome-wide Haploid Genetic Screen Identifies Regulators of Glutathione Abundance and Ferroptosis Sensitivity. Cell Rep 2019, 26, 1544–1556 e1548. [Google Scholar] [CrossRef]
- Hao, S.; Yu, J.; He, W.; Huang, Q.; Zhao, Y.; Liang, B.; Zhang, S.; Wen, Z.; Dong, S.; Rao, J.; et al. Cysteine Dioxygenase 1 Mediates Erastin-Induced Ferroptosis in Human Gastric Cancer Cells. Neoplasia 2017, 19, 1022–1032. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 2008, 15, 234–245. [Google Scholar] [CrossRef]
- Banjac, A.; Perisic, T.; Sato, H.; Seiler, A.; Bannai, S.; Weiss, N.; Kolle, P.; Tschoep, K.; Issels, R.D.; Daniel, P.T.; et al. The cystine/cysteine cycle: a redox cycle regulating susceptibility versus resistance to cell death. Oncogene 2008, 27, 1618–1628. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Dar, N.J.; Na, R.; McLane, K.D.; Yoo, K.; Han, X.; Ran, Q. Enhanced defense against ferroptosis ameliorates cognitive impairment and reduces neurodegeneration in 5xFAD mice. Free Radic Biol Med 2022, 180, 1–12. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Krysko, D.V.; Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Nakamura, T.; Mishima, E.; Yamada, N.; Mourao, A.S.D.; Trumbach, D.; Doll, S.; Wanninger, J.; Lytton, E.; Sennhenn, P.; Nishida Xavier da Silva, T.; et al. Integrated chemical and genetic screens unveil FSP1 mechanisms of ferroptosis regulation. Nat Struct Mol Biol 2023, 30, 1806–1815. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Hadian, K.; Stockwell, B.R. The therapeutic potential of targeting regulated non-apoptotic cell death. Nat Rev Drug Discov 2023, 22, 723–742. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, H.; Hara, S. Role of acyl-CoA synthetase ACSL4 in arachidonic acid metabolism. Prostaglandins Other Lipid Mediat 2019, 144, 106363. [Google Scholar] [CrossRef]
- Hashidate-Yoshida, T.; Harayama, T.; Hishikawa, D.; Morimoto, R.; Hamano, F.; Tokuoka, S.M.; Eto, M.; Tamura-Nakano, M.; Yanobu-Takanashi, R.; Mukumoto, Y.; et al. Fatty acid remodeling by LPCAT3 enriches arachidonate in phospholipid membranes and regulates triglyceride transport. Elife 2015, 4. [Google Scholar] [CrossRef]
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem Biol 2015, 10, 1604–1609. [Google Scholar] [CrossRef]
- Reed, A.; Ichu, T.A.; Milosevich, N.; Melillo, B.; Schafroth, M.A.; Otsuka, Y.; Scampavia, L.; Spicer, T.P.; Cravatt, B.F. LPCAT3 Inhibitors Remodel the Polyunsaturated Phospholipid Content of Human Cells and Protect from Ferroptosis. ACS Chem Biol 2022, 17, 1607–1618. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Cheng, J.; Fan, Y.Q.; Liu, B.H.; Zhou, H.; Wang, J.M.; Chen, Q.X. ACSL4 suppresses glioma cells proliferation via activating ferroptosis. Oncol Rep 2020, 43, 147–158. [Google Scholar] [CrossRef]
- Liu, L.; Kang, X.X. ACSL4 is overexpressed in psoriasis and enhances inflammatory responses by activating ferroptosis. Biochem Biophys Res Commun 2022, 623, 1–8. [Google Scholar] [CrossRef]
- Miao, Z.; Tian, W.; Ye, Y.; Gu, W.; Bao, Z.; Xu, L.; Sun, G.; Li, C.; Tu, Y.; Chao, H.; et al. Hsp90 induces Acsl4-dependent glioma ferroptosis via dephosphorylating Ser637 at Drp1. Cell Death Dis 2022, 13, 548. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.F.; Liang, T.Y.; Wu, D.G.; Lai, N.S.; Deng, R.M.; Ma, C.; Li, X.; Li, H.Y.; Liu, Y.Z.; Shen, H.T.; et al. Acyl-CoA synthetase long chain family member 4 plays detrimental role in early brain injury after subarachnoid hemorrhage in rats by inducing ferroptosis. CNS Neurosci Ther 2021, 27, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Hu, B.X.; Li, Z.L.; Du, T.; Shan, J.L.; Ye, Z.P.; Peng, X.D.; Li, X.; Huang, Y.; Zhu, X.Y.; et al. PKCbetaII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell Biol 2022, 24, 88–98. [Google Scholar] [CrossRef]
- Wu, X.; Deng, F.; Li, Y.; Daniels, G.; Du, X.; Ren, Q.; Wang, J.; Wang, L.H.; Yang, Y.; Zhang, V.; et al. ACSL4 promotes prostate cancer growth, invasion and hormonal resistance. Oncotarget 2015, 6, 44849–44863. [Google Scholar] [CrossRef]
- Chen, D.; Chu, B.; Yang, X.; Liu, Z.; Jin, Y.; Kon, N.; Rabadan, R.; Jiang, X.; Stockwell, B.R.; Gu, W. iPLA2beta-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat Commun 2021, 12, 3644. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.Y.; Tyurin, V.A.; Mikulska-Ruminska, K.; Shrivastava, I.H.; Anthonymuthu, T.S.; Zhai, Y.J.; Pan, M.H.; Gong, H.B.; Lu, D.H.; Sun, J.; et al. Phospholipase iPLA(2)beta averts ferroptosis by eliminating a redox lipid death signal. Nat Chem Biol 2021, 17, 465–476. [Google Scholar] [CrossRef]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations With Regulated Cell Death: A Review. Front Physiol 2019, 10, 139. [Google Scholar] [CrossRef]
- Toyokuni, S.; Ito, F.; Yamashita, K.; Okazaki, Y.; Akatsuka, S. Iron and thiol redox signaling in cancer: An exquisite balance to escape ferroptosis. Free Radic Biol Med 2017, 108, 610–626. [Google Scholar] [CrossRef]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent Sci 2018, 4, 387–396. [Google Scholar] [CrossRef]
- Angeli, J.P.F.; Shah, R.; Pratt, D.A.; Conrad, M. Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol Sci 2017, 38, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Davies, K.J.; Ursini, F. How do nutritional antioxidants really work: nucleophilic tone and para-hormesis versus free radical scavenging in vivo. Free Radic Biol Med 2014, 66, 24–35. [Google Scholar] [CrossRef]
- Shah, R.; Margison, K.; Pratt, D.A. The Potency of Diarylamine Radical-Trapping Antioxidants as Inhibitors of Ferroptosis Underscores the Role of Autoxidation in the Mechanism of Cell Death. ACS Chem Biol 2017, 12, 2538–2545. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Van Kessel, A.T.M.; Cosa, G. Lipid-derived electrophiles inhibit the function of membrane channels during ferroptosis. Proc Natl Acad Sci U S A 2024, 121, e2317616121. [Google Scholar] [CrossRef]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol Cell 2019, 73, 354–363 e353. [Google Scholar] [CrossRef]
- Fu, C.; Cao, N.; Zeng, S.; Zhu, W.; Fu, X.; Liu, W.; Fan, S. Role of mitochondria in the regulation of ferroptosis and disease. Front Med (Lausanne) 2023, 10, 1301822. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P. Transferrin receptor 1. Int J Biochem Cell Biol 2004, 36, 2137–2143. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet 2005, 37, 1264–1269. [Google Scholar] [CrossRef]
- Cullen, P.J.; Steinberg, F. To degrade or not to degrade: mechanisms and significance of endocytic recycling. Nat Rev Mol Cell Biol 2018, 19, 679–696. [Google Scholar] [CrossRef]
- Chen, C.; Garcia-Santos, D.; Ishikawa, Y.; Seguin, A.; Li, L.; Fegan, K.H.; Hildick-Smith, G.J.; Shah, D.I.; Cooney, J.D.; Chen, W.; et al. Snx3 regulates recycling of the transferrin receptor and iron assimilation. Cell Metab 2013, 17, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Hu, Y.; Liu, Z.; Guo, K.; Ma, D.; Peng, M.; Wang, Y.; Zhang, J.; Zhang, X.; Wang, P.; et al. Sorting nexin 3 exacerbates doxorubicin-induced cardiomyopathy via regulation of TFRC-dependent ferroptosis. Acta Pharm Sin B 2023, 13, 4875–4892. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, C.E.; Hoffstrom, B.G.; Decker, A.M.; Rajbhandari, P.; Stokes, M.E.; Bender, H.G.; Csuka, J.M.; et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep 2020, 30, 3411–3423 e3417. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.P.; Shen, M.; Eisenstein, R.S.; Leibold, E.A. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim Biophys Acta 2012, 1823, 1468–1483. [Google Scholar] [CrossRef]
- Drakesmith, H.; Nemeth, E.; Ganz, T. Ironing out Ferroportin. Cell Metab 2015, 22, 777–787. [Google Scholar] [CrossRef]
- Arosio, P.; Ingrassia, R.; Cavadini, P. Ferritins: a family of molecules for iron storage, antioxidation and more. Biochim Biophys Acta 2009, 1790, 589–599. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Zhao, H.; Lu, Y.; Zhang, J.; Sun, Z.; Cheng, C.; Liu, Y.; Wu, L.; Zhang, M.; He, W.; Hao, S.; et al. NCOA4 requires a [3Fe-4S] to sense and maintain the iron homeostasis. J Biol Chem 2024, 300, 105612. [Google Scholar] [CrossRef]
- Mancias, J.D.; Pontano Vaites, L.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife 2015, 4. [Google Scholar] [CrossRef]
- Galy, B.; Conrad, M.; Muckenthaler, M. Mechanisms controlling cellular and systemic iron homeostasis. Nat Rev Mol Cell Biol 2024, 25, 133–155. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res 2016, 26, 1021–1032. [Google Scholar] [CrossRef]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 2005, 1, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.L.; Hughes, R.M.; Ollivierre-Wilson, H.; Ghosh, M.C.; Rouault, T.A. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metab 2009, 9, 461–473. [Google Scholar] [CrossRef]
- Fleming, R.E. Hepcidin activation during inflammation: make it STAT. Gastroenterology 2007, 132, 447–449. [Google Scholar] [CrossRef]
- Ganz, T. Hepcidin--a regulator of intestinal iron absorption and iron recycling by macrophages. Best Pract Res Clin Haematol 2005, 18, 171–182. [Google Scholar] [CrossRef]
- Altamura, S.; Kessler, R.; Grone, H.J.; Gretz, N.; Hentze, M.W.; Galy, B.; Muckenthaler, M.U. Resistance of ferroportin to hepcidin binding causes exocrine pancreatic failure and fatal iron overload. Cell Metab 2014, 20, 359–367. [Google Scholar] [CrossRef]
- Ganz, T. Systemic iron homeostasis. Physiol Rev 2013, 93, 1721–1741. [Google Scholar] [CrossRef]
- Bien-Ly, N.; Yu, Y.J.; Bumbaca, D.; Elstrott, J.; Boswell, C.A.; Zhang, Y.; Luk, W.; Lu, Y.; Dennis, M.S.; Weimer, R.M.; et al. Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J Exp Med 2014, 211, 233–244. [Google Scholar] [CrossRef]
- Bradbury, M.W. Transport of iron in the blood-brain-cerebrospinal fluid system. J Neurochem 1997, 69, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Qian, Z.M. Brain iron metabolism: neurobiology and neurochemistry. Prog Neurobiol 2007, 83, 149–173. [Google Scholar] [CrossRef]
- Skjorringe, T.; Burkhart, A.; Johnsen, K.B.; Moos, T. Divalent metal transporter 1 (DMT1) in the brain: implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front Mol Neurosci 2015, 8, 19. [Google Scholar] [CrossRef]
- Vulpe, C.D.; Kuo, Y.M.; Murphy, T.L.; Cowley, L.; Askwith, C.; Libina, N.; Gitschier, J.; Anderson, G.J. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 1999, 21, 195–199. [Google Scholar] [CrossRef]
- Rouault, T.A. Iron metabolism in the CNS: implications for neurodegenerative diseases. Nat Rev Neurosci 2013, 14, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Attieh, Z.K.; Mukhopadhyay, C.K.; Seshadri, V.; Tripoulas, N.A.; Fox, P.L. Ceruloplasmin ferroxidase activity stimulates cellular iron uptake by a trivalent cation-specific transport mechanism. J Biol Chem 1999, 274, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Kosman, D.J. Molecular mechanisms of non-transferrin-bound and transferring-bound iron uptake in primary hippocampal neurons. J Neurochem 2015, 133, 668–683. [Google Scholar] [CrossRef]
- Cheli, V.T.; Correale, J.; Paez, P.M.; Pasquini, J.M. Iron Metabolism in Oligodendrocytes and Astrocytes, Implications for Myelination and Remyelination. ASN Neuro 2020, 12, 1759091420962681. [Google Scholar] [CrossRef]
- Baumann, B.H.; Shu, W.; Song, Y.; Simpson, E.M.; Lakhal-Littleton, S.; Dunaief, J.L. Ferroportin-mediated iron export from vascular endothelial cells in retina and brain. Exp Eye Res 2019, 187, 107728. [Google Scholar] [CrossRef]
- McCarthy, R.C.; Kosman, D.J. Ferroportin and exocytoplasmic ferroxidase activity are required for brain microvascular endothelial cell iron efflux. J Biol Chem 2013, 288, 17932–17940. [Google Scholar] [CrossRef]
- Zhou, W.; Shen, B.; Shen, W.Q.; Chen, H.; Zheng, Y.F.; Fei, J.J. Dysfunction of the Glymphatic System Might Be Related to Iron Deposition in the Normal Aging Brain. Front Aging Neurosci 2020, 12, 559603. [Google Scholar] [CrossRef] [PubMed]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics. J Neurochem 2016, 139 Suppl 1, 179–197. [Google Scholar] [CrossRef]
- Ward, R.J.; Dexter, D.T.; Crichton, R.R. Iron, Neuroinflammation and Neurodegeneration. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol 2014, 13, 1045–1060. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Xiao, Z.J.; Yang, B.; Lan, Z.; Fang, F. Blood-Brain Barrier: More Contributor to Disruption of Central Nervous System Homeostasis Than Victim in Neurological Disorders. Front Neurosci 2020, 14, 764. [Google Scholar] [CrossRef]
- Keaney, J.; Walsh, D.M.; O’Malley, T.; Hudson, N.; Crosbie, D.E.; Loftus, T.; Sheehan, F.; McDaid, J.; Humphries, M.M.; Callanan, J.J.; et al. Autoregulated paracellular clearance of amyloid-beta across the blood-brain barrier. Sci Adv 2015, 1, e1500472. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Shinohara, M.; Shinohara, M.; Yamazaki, A.; Murray, M.E.; Liesinger, A.M.; Heckman, M.G.; Lesser, E.R.; Parisi, J.E.; Petersen, R.C.; et al. Selective loss of cortical endothelial tight junction proteins during Alzheimer’s disease progression. Brain 2019, 142, 1077–1092. [Google Scholar] [CrossRef]
- Alkhalifa, A.E.; Al-Ghraiybah, N.F.; Odum, J.; Shunnarah, J.G.; Austin, N.; Kaddoumi, A. Blood-Brain Barrier Breakdown in Alzheimer’s Disease: Mechanisms and Targeted Strategies. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Kurz, C.; Walker, L.; Rauchmann, B.S.; Perneczky, R. Dysfunction of the blood-brain barrier in Alzheimer’s disease: Evidence from human studies. Neuropathol Appl Neurobiol 2022, 48, e12782. [Google Scholar] [CrossRef]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Provias, J.; Jeynes, B. The role of the blood-brain barrier in the pathogenesis of senile plaques in Alzheimer’s disease. Int J Alzheimers Dis 2014, 2014, 191863. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Qiao, P.F.; Wan, C.Q.; Cai, M.; Zhou, N.K.; Li, Q. Role of Blood-Brain Barrier in Alzheimer’s Disease. J Alzheimers Dis 2018, 63, 1223–1234. [Google Scholar] [CrossRef]
- Baringer, S.L.; Lukacher, A.S.; Palsa, K.; Kim, H.; Lippmann, E.S.; Spiegelman, V.S.; Simpson, I.A.; Connor, J.R. Amyloid-beta exposed astrocytes induce iron transport from endothelial cells at the blood-brain barrier by altering the ratio of apo- and holo-transferrin. J Neurochem 2023, 167, 248–261. [Google Scholar] [CrossRef]
- Alruwais, N.M.; Rusted, J.M.; Tabet, N.; Dowell, N.G. Evidence of emerging BBB changes in mid-age apolipoprotein E epsilon-4 carriers. Brain Behav 2022, 12, e2806. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, J.W.; Bula, M.; Davila-Velderrain, J.; Akay, L.A.; Zhu, L.; Frank, A.; Victor, M.B.; Bonner, J.M.; Mathys, H.; Lin, Y.T.; et al. Reconstruction of the human blood-brain barrier in vitro reveals a pathogenic mechanism of APOE4 in pericytes. Nat Med 2020, 26, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Khor, S.L.Q.; Ng, K.Y.; Koh, R.Y.; Chye, S.M. Blood-brain Barrier and Neurovascular Unit Dysfunction in Parkinson’s Disease: From Clinical Insights to Pathogenic Mechanisms and Novel Therapeutic Approaches. CNS Neurol Disord Drug Targets 2024, 23, 315–330. [Google Scholar] [CrossRef]
- Katt, M.E.; Mayo, L.N.; Ellis, S.E.; Mahairaki, V.; Rothstein, J.D.; Cheng, L.; Searson, P.C. The role of mutations associated with familial neurodegenerative disorders on blood-brain barrier function in an iPSC model. Fluids Barriers CNS 2019, 16, 20. [Google Scholar] [CrossRef]
- Carrano, A.; Hoozemans, J.J.; van der Vies, S.M.; van Horssen, J.; de Vries, H.E.; Rozemuller, A.J. Neuroinflammation and blood-brain barrier changes in capillary amyloid angiopathy. Neurodegener Dis 2012, 10, 329–331. [Google Scholar] [CrossRef]
- Connor, J.R.; Snyder, B.S.; Beard, J.L.; Fine, R.E.; Mufson, E.J. Regional distribution of iron and iron-regulatory proteins in the brain in aging and Alzheimer’s disease. J Neurosci Res 1992, 31, 327–335. [Google Scholar] [CrossRef]
- Raha, A.A.; Vaishnav, R.A.; Friedland, R.P.; Bomford, A.; Raha-Chowdhury, R. The systemic iron-regulatory proteins hepcidin and ferroportin are reduced in the brain in Alzheimer’s disease. Acta Neuropathol Commun 2013, 1, 55. [Google Scholar] [CrossRef]
- Duce, J.A.; Tsatsanis, A.; Cater, M.A.; James, S.A.; Robb, E.; Wikhe, K.; Leong, S.L.; Perez, K.; Johanssen, T.; Greenough, M.A.; et al. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 2010, 142, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Spotorno, N.; Acosta-Cabronero, J.; Stomrud, E.; Lampinen, B.; Strandberg, O.T.; van Westen, D.; Hansson, O. Relationship between cortical iron and tau aggregation in Alzheimer’s disease. Brain 2020, 143, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- An, X.; He, J.; Xie, P.; Li, C.; Xia, M.; Guo, D.; Bi, B.; Wu, G.; Xu, J.; Yu, W.; et al. The effect of tau K677 lactylation on ferritinophagy and ferroptosis in Alzheimer’s disease. Free Radic Biol Med 2024, 224, 685–706. [Google Scholar] [CrossRef]
- Yamamoto, A.; Shin, R.W.; Hasegawa, K.; Naiki, H.; Sato, H.; Yoshimasu, F.; Kitamoto, T. Iron (III) induces aggregation of hyperphosphorylated tau and its reduction to iron (II) reverses the aggregation: implications in the formation of neurofibrillary tangles of Alzheimer’s disease. J Neurochem 2002, 82, 1137–1147. [Google Scholar] [CrossRef]
- Ayton, S.; Diouf, I.; Bush, A.I.; Alzheimer’s disease Neuroimaging, I. Evidence that iron accelerates Alzheimer’s pathology: a CSF biomarker study. J Neurol Neurosurg Psychiatry 2018, 89, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Faux, N.G.; Bush, A.I. Association of Cerebrospinal Fluid Ferritin Level With Preclinical Cognitive Decline in APOE-epsilon4 Carriers. JAMA Neurol 2017, 74, 122–125. [Google Scholar] [CrossRef]
- Ayton, S.; Faux, N.G.; Bush, A.I.; Alzheimer’s Disease Neuroimaging, I. Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nat Commun 2015, 6, 6760. [Google Scholar] [CrossRef]
- Bartzokis, G.; Lu, P.H.; Tishler, T.A.; Peters, D.G.; Kosenko, A.; Barrall, K.A.; Finn, J.P.; Villablanca, P.; Laub, G.; Altshuler, L.L.; et al. Prevalent iron metabolism gene variants associated with increased brain ferritin iron in healthy older men. J Alzheimers Dis 2010, 20, 333–341. [Google Scholar] [CrossRef]
- Ma, J.; Guo, Q.; Shen, M.Q.; Li, W.; Zhong, Q.X.; Qian, Z.M. Apolipoprotein E is required for brain iron homeostasis in mice. Redox Biol 2023, 64, 102779. [Google Scholar] [CrossRef]
- Ryan, B.J.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci 2015, 40, 200–210. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front Neuroanat 2015, 9, 91. [Google Scholar] [CrossRef]
- Berman, S.B.; Zigmond, M.J.; Hastings, T.G. Modification of dopamine transporter function: effect of reactive oxygen species and dopamine. J Neurochem 1996, 67, 593–600. [Google Scholar] [CrossRef]
- Berman, S.B.; Hastings, T.G. Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson’s disease. J Neurochem 1999, 73, 1127–1137. [Google Scholar] [CrossRef]
- Bartels, M.; Weckbecker, D.; Kuhn, P.H.; Ryazanov, S.; Leonov, A.; Griesinger, C.; Lichtenthaler, S.F.; Botzel, K.; Giese, A. Iron-mediated aggregation and toxicity in a novel neuronal cell culture model with inducible alpha-synuclein expression. Sci Rep 2019, 9, 9100. [Google Scholar] [CrossRef] [PubMed]
- He, N.; Langley, J.; Huddleston, D.E.; Chen, S.; Huang, P.; Ling, H.; Yan, F.; Hu, X. Increased iron-deposition in lateral-ventral substantia nigra pars compacta: A promising neuroimaging marker for Parkinson’s disease. Neuroimage Clin 2020, 28, 102391. [Google Scholar] [CrossRef]
- Griffiths, P.D.; Dobson, B.R.; Jones, G.R.; Clarke, D.T. Iron in the basal ganglia in Parkinson’s disease. An in vitro study using extended X-ray absorption fine structure and cryo-electron microscopy. Brain 1999, 122 Pt 4, 667–673. [Google Scholar] [CrossRef]
- Oakley, A.E.; Collingwood, J.F.; Dobson, J.; Love, G.; Perrott, H.R.; Edwardson, J.A.; Elstner, M.; Morris, C.M. Individual dopaminergic neurons show raised iron levels in Parkinson disease. Neurology 2007, 68, 1820–1825. [Google Scholar] [CrossRef] [PubMed]
- Fedorow, H.; Tribl, F.; Halliday, G.; Gerlach, M.; Riederer, P.; Double, K.L. Neuromelanin in human dopamine neurons: comparison with peripheral melanins and relevance to Parkinson’s disease. Prog Neurobiol 2005, 75, 109–124. [Google Scholar] [CrossRef]
- Arawaka, S.; Saito, Y.; Murayama, S.; Mori, H. Lewy body in neurodegeneration with brain iron accumulation type 1 is immunoreactive for alpha-synuclein. Neurology 1998, 51, 887–889. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.J.; Siedlak, S.L.; Perry, G.; Smith, M.A. Sequestration of iron by Lewy bodies in Parkinson’s disease. Acta Neuropathol 2000, 100, 111–114. [Google Scholar] [CrossRef]
- Wang, J.Y.; Zhuang, Q.Q.; Zhu, L.B.; Zhu, H.; Li, T.; Li, R.; Chen, S.F.; Huang, C.P.; Zhang, X.; Zhu, J.H. Meta-analysis of brain iron levels of Parkinson’s disease patients determined by postmortem and MRI measurements. Sci Rep 2016, 6, 36669. [Google Scholar] [CrossRef]
- Graves, S.M.; Xie, Z.; Stout, K.A.; Zampese, E.; Burbulla, L.F.; Shih, J.C.; Kondapalli, J.; Patriarchi, T.; Tian, L.; Brichta, L.; et al. Dopamine metabolism by a monoamine oxidase mitochondrial shuttle activates the electron transport chain. Nat Neurosci 2020, 23, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.; Mena, N.; Hunot, S.; Prigent, A.; Alvarez-Fischer, D.; Arredondo, M.; Duyckaerts, C.; Sazdovitch, V.; Zhao, L.; Garrick, L.M.; et al. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson’s disease. Proc Natl Acad Sci U S A 2008, 105, 18578–18583. [Google Scholar] [CrossRef]
- Raha, A.A.; Biswas, A.; Henderson, J.; Chakraborty, S.; Holland, A.; Friedland, R.P.; Mukaetova-Ladinska, E.; Zaman, S.; Raha-Chowdhury, R. Interplay of Ferritin Accumulation and Ferroportin Loss in Ageing Brain: Implication for Protein Aggregation in Down Syndrome Dementia, Alzheimer’s, and Parkinson’s Diseases. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Matak, P.; Matak, A.; Moustafa, S.; Aryal, D.K.; Benner, E.J.; Wetsel, W.; Andrews, N.C. Disrupted iron homeostasis causes dopaminergic neurodegeneration in mice. Proc Natl Acad Sci U S A 2016, 113, 3428–3435. [Google Scholar] [CrossRef]
- Lin, R.R.; Tao, Q.Q.; Wu, Z.Y. Early-Onset Parkinson’s Disease and Brain Iron Accumulation Caused by a Novel Homozygous DJ-1 Mutation. J Parkinsons Dis 2022, 12, 813–819. [Google Scholar] [CrossRef]
- Zheng, Z.; Zhang, S.; Liu, X.; Wang, X.; Xue, C.; Wu, X.; Zhang, X.; Xu, X.; Liu, Z.; Yao, L.; et al. LRRK2 regulates ferroptosis through the system Xc-GSH-GPX4 pathway in the neuroinflammatory mechanism of Parkinson’s disease. J Cell Physiol 2024, 239, e31250. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Arcos, M.; Goodla, L.; Kim, H.; Desai, S.P.; Liu, R.; Yin, K.; Liu, Z.; Martin, D.R.; Xue, X. PINK1-deficiency facilitates mitochondrial iron accumulation and colon tumorigenesis. Autophagy 2024, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Bitar, S.; Baumann, T.; Weber, C.; Abusaada, M.; Rojas-Charry, L.; Ziegler, P.; Schettgen, T.; Randerath, I.E.; Venkataramani, V.; Michalke, B.; et al. Iron-sulfur cluster loss in mitochondrial CISD1 mediates PINK1 loss-of-function phenotypes. Elife 2024, 13. [Google Scholar] [CrossRef]
- Chang, B.; Su, Y.; Li, T.; Zheng, Y.; Yang, R.; Lu, H.; Wang, H.; Ding, Y. Mito-TEMPO Ameliorates Sodium Palmitate Induced Ferroptosis in MIN6 Cells through PINK1/Parkin-Mediated Mitophagy. Biomed Environ Sci 2024, 37, 1128–1141. [Google Scholar] [CrossRef]
- Kang, R.; Xie, Y.; Zeh, H.J.; Klionsky, D.J.; Tang, D. Mitochondrial quality control mediated by PINK1 and PRKN: links to iron metabolism and tumor immunity. Autophagy 2019, 15, 172–173. [Google Scholar] [CrossRef] [PubMed]
- Key, J.; Sen, N.E.; Arsovic, A.; Kramer, S.; Hulse, R.; Khan, N.N.; Meierhofer, D.; Gispert, S.; Koepf, G.; Auburger, G. Systematic Surveys of Iron Homeostasis Mechanisms Reveal Ferritin Superfamily and Nucleotide Surveillance Regulation to be Modified by PINK1 Absence. Cells 2020, 9. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Refai, F.S.; Xie, S.P.; Ng, S.H.; Chan, C.H.; Ho, P.G.; Zhang, X.D.; Lim, T.M.; Tan, E.K. Mutant PINK1 upregulates tyrosine hydroxylase and dopamine levels, leading to vulnerability of dopaminergic neurons. Free Radic Biol Med 2014, 68, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Li, X.; Du, X.; Bi, M.; Ma, F.; Xie, J.; Jiang, H. The S-nitrosylation of parkin attenuated the ubiquitination of divalent metal transporter 1 in MPP(+)-treated SH-SY5Y cells. Sci Rep 2020, 10, 15542. [Google Scholar] [CrossRef]
- Ganguly, U.; Banerjee, A.; Chakrabarti, S.S.; Kaur, U.; Sen, O.; Cappai, R.; Chakrabarti, S. Interaction of alpha-synuclein and Parkin in iron toxicity on SH-SY5Y cells: implications in the pathogenesis of Parkinson’s disease. Biochem J 2020, 477, 1109–1122. [Google Scholar] [CrossRef]
- Pyatigorskaya, N.; Sharman, M.; Corvol, J.C.; Valabregue, R.; Yahia-Cherif, L.; Poupon, F.; Cormier-Dequaire, F.; Siebner, H.; Klebe, S.; Vidailhet, M.; et al. High nigral iron deposition in LRRK2 and Parkin mutation carriers using R2* relaxometry. Mov Disord 2015, 30, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.A.; Singleton, S.; Feng, J.; Garrick, M.; Paradkar, P.N. Parkin regulates metal transport via proteasomal degradation of the 1B isoforms of divalent metal transporter 1. J Neurochem 2010, 113, 454–464. [Google Scholar] [CrossRef]
- Li, Y.; Chen, T.; Xue, Y.; Wang, Y.; Peng, L.; Wang, C.; Yu, S. DJ-1 inhibits ferroptosis in cerebral ischemia-reperfusion via ATF4/HSPA5 pathway. Neurochem Int 2023, 171, 105628. [Google Scholar] [CrossRef]
- Liao, T.; Xu, X.; Ye, X.; Yan, J. DJ-1 upregulates the Nrf2/GPX4 signal pathway to inhibit trophoblast ferroptosis in the pathogenesis of preeclampsia. Sci Rep 2022, 12, 2934. [Google Scholar] [CrossRef]
- Cao, J.; Chen, X.; Jiang, L.; Lu, B.; Yuan, M.; Zhu, D.; Zhu, H.; He, Q.; Yang, B.; Ying, M. DJ-1 suppresses ferroptosis through preserving the activity of S-adenosyl homocysteine hydrolase. Nat Commun 2020, 11, 1251. [Google Scholar] [CrossRef]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef]
- Song, N.; Xie, J. Iron, Dopamine, and alpha-Synuclein Interactions in at-Risk Dopaminergic Neurons in Parkinson’s Disease. Neurosci Bull 2018, 34, 382–384. [Google Scholar] [CrossRef]
- Ghaderi, S.; Mohammadi, S.; Nezhad, N.J.; Karami, S.; Sayehmiri, F. Iron quantification in basal ganglia: quantitative susceptibility mapping as a potential biomarker for Alzheimer’s disease - a systematic review and meta-analysis. Front Neurosci 2024, 18, 1338891. [Google Scholar] [CrossRef]
- Mohammadi, S.; Ghaderi, S. Parkinson’s disease and Parkinsonism syndromes: Evaluating iron deposition in the putamen using magnetic susceptibility MRI techniques - A systematic review and literature analysis. Heliyon 2024, 10, e27950. [Google Scholar] [CrossRef]
- Yang, A.; Luan, J.; Xu, M.; Du, L.; Lv, K.; Hu, P.; Shu, N.; Yuan, Z.; Shmuel, A.; Ma, G. Regional brain iron correlates with transcriptional and cellular signatures in Alzheimer’s disease. Alzheimers Dement 2025, 21, e14459. [Google Scholar] [CrossRef]
- van Bergen, J.M.; Li, X.; Hua, J.; Schreiner, S.J.; Steininger, S.C.; Quevenco, F.C.; Wyss, M.; Gietl, A.F.; Treyer, V.; Leh, S.E.; et al. Colocalization of cerebral iron with Amyloid beta in Mild Cognitive Impairment. Sci Rep 2016, 6, 35514. [Google Scholar] [CrossRef]
- Zhi, Y.; Huang, T.; Liu, S.; Li, M.; Hu, H.; Liang, X.; Jiang, Z.; Zhu, J.; Liu, R. Correlation between iron deposition and cognitive function in mild to moderate Alzheimer’s disease based on quantitative susceptibility mapping. Front Aging Neurosci 2024, 16, 1485530. [Google Scholar] [CrossRef]
- Atasoy, H.T.; Nuyan, O.; Tunc, T.; Yorubulut, M.; Unal, A.E.; Inan, L.E. T2-weighted MRI in Parkinson’s disease; substantia nigra pars compacta hypointensity correlates with the clinical scores. Neurol India 2004, 52, 332–337. [Google Scholar]
- Liu, Z.; Shen, H.C.; Lian, T.H.; Mao, L.; Tang, S.X.; Sun, L.; Huang, X.Y.; Guo, P.; Cao, C.J.; Yu, S.Y.; et al. Iron deposition in substantia nigra: abnormal iron metabolism, neuroinflammatory mechanism and clinical relevance. Sci Rep 2017, 7, 14973. [Google Scholar] [CrossRef]
- Fu, X.; Deng, W.; Cui, X.; Zhou, X.; Song, W.; Pan, M.; Chi, X.; Xu, J.; Jiang, Y.; Wang, Q.; et al. Time-Specific Pattern of Iron Deposition in Different Regions in Parkinson’s Disease Measured by Quantitative Susceptibility Mapping. Front Neurol 2021, 12, 631210. [Google Scholar] [CrossRef]
- Thomas, G.E.C.; Leyland, L.A.; Schrag, A.E.; Lees, A.J.; Acosta-Cabronero, J.; Weil, R.S. Brain iron deposition is linked with cognitive severity in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2020, 91, 418–425. [Google Scholar] [CrossRef]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J Cereb Blood Flow Metab 2015, 35, 747–750. [Google Scholar] [CrossRef]
- da Rocha, T.J.; Silva Alves, M.; Guisso, C.C.; de Andrade, F.M.; Camozzato, A.; de Oliveira, A.A.; Fiegenbaum, M. Association of GPX1 and GPX4 polymorphisms with episodic memory and Alzheimer’s disease. Neurosci Lett 2018, 666, 32–37. [Google Scholar] [CrossRef]
- Baruah, P.; Moorthy, H.; Ramesh, M.; Padhi, D.; Govindaraju, T. A natural polyphenol activates and enhances GPX4 to mitigate amyloid-beta induced ferroptosis in Alzheimer’s disease. Chem Sci 2023, 14, 9427–9438. [Google Scholar] [CrossRef]
- Dar, N.J.; John, U.; Bano, N.; Khan, S.; Bhat, S.A. Oxytosis/Ferroptosis in Neurodegeneration: the Underlying Role of Master Regulator Glutathione Peroxidase 4 (GPX4). Mol Neurobiol 2024, 61, 1507–1526. [Google Scholar] [CrossRef]
- Bao, W.D.; Pang, P.; Zhou, X.T.; Hu, F.; Xiong, W.; Chen, K.; Wang, J.; Wang, F.; Xie, D.; Hu, Y.Z.; et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ 2021, 28, 1548–1562. [Google Scholar] [CrossRef]
- Dang, Y.; He, Q.; Yang, S.; Sun, H.; Liu, Y.; Li, W.; Tang, Y.; Zheng, Y.; Wu, T. FTH1- and SAT1-Induced Astrocytic Ferroptosis Is Involved in Alzheimer’s Disease: Evidence from Single-Cell Transcriptomic Analysis. Pharmaceuticals (Basel) 2022, 15. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, J.; Li, Z.; Wang, S.; Yu, G.; Wang, L. Identification ferroptosis-related hub genes and diagnostic model in Alzheimer’s disease. Front Mol Neurosci 2023, 16, 1280639. [Google Scholar] [CrossRef]
- Zhang, L.; Fang, J.; Tang, Z.; Luo, Y. A Bioinformatics Perspective on the Dysregulation of Ferroptosis and Ferroptosis-related Immune Cell Infiltration in Alzheimer’s Disease. Int J Med Sci 2022, 19, 1888–1902. [Google Scholar] [CrossRef]
- Ryan, S.K.; Zelic, M.; Han, Y.; Teeple, E.; Chen, L.; Sadeghi, M.; Shankara, S.; Guo, L.; Li, C.; Pontarelli, F.; et al. Microglia ferroptosis is regulated by SEC24B and contributes to neurodegeneration. Nat Neurosci 2023, 26, 12–26. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, K.; Dai, Y.; Luo, X.; Xiong, Z.; Zhang, W.; Huang, X.; So, K.F.; Zhang, L. Human alpha-synuclein aggregation activates ferroptosis leading to parvalbumin interneuron degeneration and motor learning impairment. Commun Biol 2024, 7, 1227. [Google Scholar] [CrossRef]
- Gaertner, Z.; Oram, C.; Schneeweis, A.; Schonfeld, E.; Bolduc, C.; Chen, C.; Dombeck, D.; Parisiadou, L.; Poulin, J.F.; Awatramani, R. Molecular and spatial transcriptomic classification of midbrain dopamine neurons and their alterations in a LRRK2(G2019S) model of Parkinson’s disease. bioRxiv 2024. [Google Scholar] [CrossRef]
- Shwab, E.K.; Man, Z.; Gingerich, D.C.; Gamache, J.; Garrett, M.E.; Serrano, G.E.; Beach, T.G.; Crawford, G.E.; Ashley-Koch, A.E.; Chiba-Falek, O. Comparative mapping of single-cell transcriptomic landscapes in neurodegenerative diseases. bioRxiv 2024. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Fon, E.A.; Dagher, A.; Iturria-Medina, Y.; Stratton, J.A.; Savignac, C.; Stanley, J.; Hodgson, L.; Hammou, B.A.; Bennett, D.A.; et al. Cell type transcriptomics reveal shared genetic mechanisms in Alzheimer’s and Parkinson’s disease. bioRxiv 2025. [Google Scholar] [CrossRef]
- Agarwal, D.; Sandor, C.; Volpato, V.; Caffrey, T.M.; Monzon-Sandoval, J.; Bowden, R.; Alegre-Abarrategui, J.; Wade-Martins, R.; Webber, C. A single-cell atlas of the human substantia nigra reveals cell-specific pathways associated with neurological disorders. Nat Commun 2020, 11, 4183. [Google Scholar] [CrossRef]
- Han, S.; Gim, Y.; Jang, E.H.; Hur, E.M. Functions and dysfunctions of oligodendrocytes in neurodegenerative diseases. Front Cell Neurosci 2022, 16, 1083159. [Google Scholar] [CrossRef]
- Philips, T.; Rothstein, J.D. Oligodendroglia: metabolic supporters of neurons. J Clin Invest 2017, 127, 3271–3280. [Google Scholar] [CrossRef]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat Rev Immunol 2022, 22, 657–673. [Google Scholar] [CrossRef]
- Todorich, B.; Pasquini, J.M.; Garcia, C.I.; Paez, P.M.; Connor, J.R. Oligodendrocytes and myelination: the role of iron. Glia 2009, 57, 467–478. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat Rev Dis Primers 2015, 1, 15056. [Google Scholar] [CrossRef]
- Liu, Y.; Nguyen, M.; Robert, A.; Meunier, B. Metal Ions in Alzheimer’s Disease: A Key Role or Not? Acc Chem Res 2019, 52, 2026–2035. [Google Scholar] [CrossRef]
- Abelein, A. Metal Binding of Alzheimer’s Amyloid-beta and Its Effect on Peptide Self-Assembly. Acc Chem Res 2023, 56, 2653–2663. [Google Scholar] [CrossRef]
- Boopathi, S.; Kolandaivel, P. Fe(2+) binding on amyloid beta-peptide promotes aggregation. Proteins 2016, 84, 1257–1274. [Google Scholar] [CrossRef]
- Wan, W.; Cao, L.; Kalionis, B.; Murthi, P.; Xia, S.; Guan, Y. Iron Deposition Leads to Hyperphosphorylation of Tau and Disruption of Insulin Signaling. Front Neurol 2019, 10, 607. [Google Scholar] [CrossRef]
- Kenkhuis, B.; Bush, A.I.; Ayton, S. How iron can drive neurodegeneration. Trends Neurosci 2023, 46, 333–335. [Google Scholar] [CrossRef]
- Friedlich, A.L.; Tanzi, R.E.; Rogers, J.T. The 5′-untranslated region of Parkinson’s disease alpha-synuclein messengerRNA contains a predicted iron responsive element. Mol Psychiatry 2007, 12, 222–223. [Google Scholar] [CrossRef]
- Angelova, P.R.; Choi, M.L.; Berezhnov, A.V.; Horrocks, M.H.; Hughes, C.D.; De, S.; Rodrigues, M.; Yapom, R.; Little, D.; Dolt, K.S.; et al. Alpha synuclein aggregation drives ferroptosis: an interplay of iron, calcium and lipid peroxidation. Cell Death Differ 2020, 27, 2781–2796. [Google Scholar] [CrossRef]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef]
- Boyko, S.; Surewicz, W.K. Tau liquid-liquid phase separation in neurodegenerative diseases. Trends Cell Biol 2022, 32, 611–623. [Google Scholar] [CrossRef]
- Hernandez-Vega, A.; Braun, M.; Scharrel, L.; Jahnel, M.; Wegmann, S.; Hyman, B.T.; Alberti, S.; Diez, S.; Hyman, A.A. Local Nucleation of Microtubule Bundles through Tubulin Concentration into a Condensed Tau Phase. Cell Rep 2017, 20, 2304–2312. [Google Scholar] [CrossRef]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J 2018, 37. [Google Scholar] [CrossRef]
- Savastano, A.; Flores, D.; Kadavath, H.; Biernat, J.; Mandelkow, E.; Zweckstetter, M. Disease-Associated Tau Phosphorylation Hinders Tubulin Assembly within Tau Condensates. Angew Chem Int Ed Engl 2021, 60, 726–730. [Google Scholar] [CrossRef]
- Gui, X.; Feng, S.; Li, Z.; Li, Y.; Reif, B.; Shi, B.; Niu, Z. Liquid-liquid phase separation of amyloid-beta oligomers modulates amyloid fibrils formation. J Biol Chem 2023, 299, 102926. [Google Scholar] [CrossRef]
- Sudhakar, S.; Manohar, A.; Mani, E. Liquid-Liquid Phase Separation (LLPS)-Driven Fibrilization of Amyloid-beta Protein. ACS Chem Neurosci 2023, 14, 3655–3664. [Google Scholar] [CrossRef]
- Schutzmann, M.P.; Hasecke, F.; Bachmann, S.; Zielinski, M.; Hansch, S.; Schroder, G.F.; Zempel, H.; Hoyer, W. Endo-lysosomal Abeta concentration and pH trigger formation of Abeta oligomers that potently induce Tau missorting. Nat Commun 2021, 12, 4634. [Google Scholar] [CrossRef]
- Galvagnion, C.; Topgaard, D.; Makasewicz, K.; Buell, A.K.; Linse, S.; Sparr, E.; Dobson, C.M. Lipid Dynamics and Phase Transition within alpha-Synuclein Amyloid Fibrils. J Phys Chem Lett 2019, 10, 7872–7877. [Google Scholar] [CrossRef]
- Li, X.; Yu, L.; Liu, X.; Shi, T.; Zhang, Y.; Xiao, Y.; Wang, C.; Song, L.; Li, N.; Liu, X.; et al. beta-synuclein regulates the phase transitions and amyloid conversion of alpha-synuclein. Nat Commun 2024, 15, 8748. [Google Scholar] [CrossRef]
- Ziaunys, M.; Sulskis, D.; Veiveris, D.; Kopustas, A.; Snieckute, R.; Mikalauskaite, K.; Sakalauskas, A.; Tutkus, M.; Smirnovas, V. Liquid-liquid phase separation of alpha-synuclein increases the structural variability of fibrils formed during amyloid aggregation. FEBS J 2024, 291, 4522–4538. [Google Scholar] [CrossRef]
- Xu, B.; Fan, F.; Liu, Y.; Liu, Y.; Zhou, L.; Yu, H. Distinct Effects of Familial Parkinson’s Disease-Associated Mutations on alpha-Synuclein Phase Separation and Amyloid Aggregation. Biomolecules 2023, 13. [Google Scholar] [CrossRef]
- Ray, S.; Singh, N.; Kumar, R.; Patel, K.; Pandey, S.; Datta, D.; Mahato, J.; Panigrahi, R.; Navalkar, A.; Mehra, S.; et al. alpha-Synuclein aggregation nucleates through liquid-liquid phase separation. Nat Chem 2020, 12, 705–716. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Hamel, C.; Grabinski, T.; Combs, B. Liquid-liquid phase separation induces pathogenic tau conformations in vitro. Nat Commun 2020, 11, 2809. [Google Scholar] [CrossRef]
- Zbinden, A.; Perez-Berlanga, M.; De Rossi, P.; Polymenidou, M. Phase Separation and Neurodegenerative Diseases: A Disturbance in the Force. Dev Cell 2020, 55, 45–68. [Google Scholar] [CrossRef]
- Nedelsky, N.B.; Taylor, J.P. Bridging biophysics and neurology: aberrant phase transitions in neurodegenerative disease. Nat Rev Neurol 2019, 15, 272–286. [Google Scholar] [CrossRef]
- Nakamura, T.; Hipp, C.; Santos Dias Mourao, A.; Borggrafe, J.; Aldrovandi, M.; Henkelmann, B.; Wanninger, J.; Mishima, E.; Lytton, E.; Emler, D.; et al. Phase separation of FSP1 promotes ferroptosis. Nature 2023, 619, 371–377. [Google Scholar] [CrossRef]
- Liu, G.; Sil, D.; Maio, N.; Tong, W.H.; Bollinger, J.M., Jr.; Krebs, C.; Rouault, T.A. Heme biosynthesis depends on previously unrecognized acquisition of iron-sulfur cofactors in human amino-levulinic acid dehydratase. Nat Commun 2020, 11, 6310. [Google Scholar] [CrossRef]
- Wolff, N.A.; Garrick, M.D.; Zhao, L.; Garrick, L.M.; Ghio, A.J.; Thevenod, F. A role for divalent metal transporter (DMT1) in mitochondrial uptake of iron and manganese. Sci Rep 2018, 8, 211. [Google Scholar] [CrossRef]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol Cell Biol 2009, 29, 1007–1016. [Google Scholar] [CrossRef]
- Arosio, P.; Levi, S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim Biophys Acta 2010, 1800, 783–792. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, X.; Zeng, Y.; Mo, X.; Hong, S.; He, H.; Li, J.; Fatima, S.; Liu, Q. Oxidative stress induces mitochondrial iron overload and ferroptotic cell death. Sci Rep 2023, 13, 15515. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants (Basel) 2020, 9. [Google Scholar] [CrossRef]
- Guo, X.; Wei, R.; Yin, X.; Yang, G. Crosstalk between neuroinflammation and ferroptosis: Implications for Parkinson’s disease progression. Frontiers in Pharmacology 2025, 16. [Google Scholar] [CrossRef]
- Liu, S.; Gao, X.; Zhou, S. New Target for Prevention and Treatment of Neuroinflammation: Microglia Iron Accumulation and Ferroptosis. ASN Neuro 2022, 14, 17590914221133236. [Google Scholar] [CrossRef]
- Kaji, S.; Berghoff, S.A.; Spieth, L.; Schlaphoff, L.; Sasmita, A.O.; Vitale, S.; Buschgens, L.; Kedia, S.; Zirngibl, M.; Nazarenko, T.; et al. Apolipoprotein E aggregation in microglia initiates Alzheimer’s disease pathology by seeding beta-amyloidosis. Immunity 2024, 57, 2651–2668 e2612. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.S.; Palovics, R.; Munson, C.N.; Long, C.; Johansson, P.K.; Yip, O.; Dong, W.; Rawat, E.; West, E.; Schlachetzki, J.C.M.; et al. APOE4/4 is linked to damaging lipid droplets in Alzheimer’s disease microglia. Nature 2024, 628, 154–161. [Google Scholar] [CrossRef]
- Wogram, E.; Prinz, M. APOE set the microglia free. Nat Immunol 2023, 24, 1790–1791. [Google Scholar] [CrossRef] [PubMed]
- Kiani, L. ApoE attracts microglia to amyloid-beta plaques. Nat Rev Neurol 2023, 19, 639. [Google Scholar] [CrossRef]
- Podlesny-Drabiniok, A.; Marcora, E.; Goate, A.M. Microglial Phagocytosis: A Disease-Associated Process Emerging from Alzheimer’s Disease Genetics. Trends Neurosci 2020, 43, 965–979. [Google Scholar] [CrossRef]
- Chen, Y.; Song, S.; Parhizkar, S.; Lord, J.; Zhu, Y.; Strickland, M.R.; Wang, C.; Park, J.; Tabor, G.T.; Jiang, H.; et al. APOE3ch alters microglial response and suppresses Abeta-induced tau seeding and spread. Cell 2024, 187, 428–445 e420. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Y.; Song, N.; Wang, J.; Jiang, H.; Xie, J. New Progress on the Role of Glia in Iron Metabolism and Iron-Induced Degeneration of Dopamine Neurons in Parkinson’s Disease. Front Mol Neurosci 2017, 10, 455. [Google Scholar] [CrossRef]
- Cheng, Y.; Song, Y.; Chen, H.; Li, Q.; Gao, Y.; Lu, G.; Luo, C. Ferroptosis Mediated by Lipid Reactive Oxygen Species: A Possible Causal Link of Neuroinflammation to Neurological Disorders. Oxid Med Cell Longev 2021, 2021, 5005136. [Google Scholar] [CrossRef]
- Gutierre, R.C.; Rocha, P.R.; Graciani, A.L.; Coppi, A.A.; Arida, R.M. Tau, amyloid, iron, oligodendrocytes ferroptosis, and inflammaging in the hippocampal formation of aged rats submitted to an aerobic exercise program. Brain Res 2025, 1850, 149419. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Cabronero, J.; Betts, M.J.; Cardenas-Blanco, A.; Yang, S.; Nestor, P.J. In Vivo MRI Mapping of Brain Iron Deposition across the Adult Lifespan. J Neurosci 2016, 36, 364–374. [Google Scholar] [CrossRef]
- Naoi, M.; Maruyama, W. Cell death of dopamine neurons in aging and Parkinson’s disease. Mech Ageing Dev 1999, 111, 175–188. [Google Scholar] [CrossRef]
- Adamczyk, A.; Kazmierczak, A.; Strosznajder, J.B. Alpha-synuclein and its neurotoxic fragment inhibit dopamine uptake into rat striatal synaptosomes. Relationship to nitric oxide. Neurochem Int 2006, 49, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Morales, I.; Rodriguez-Sabate, C.; Sanchez, A.; Castro, R.; Brito, J.M.; Sabate, M. The degeneration and replacement of dopamine cells in Parkinson’s disease: the role of aging. Front Neuroanat 2014, 8, 80. [Google Scholar] [CrossRef] [PubMed]
- Coleman, C.R.; Pallos, J.; Arreola-Bustos, A.; Wang, L.; Raftery, D.; Promislow, D.E.L.; Martin, I. Natural Variation in Age-Related Dopamine Neuron Degeneration is Glutathione-Dependent and Linked to Life Span. bioRxiv 2024. [Google Scholar] [CrossRef]
- Banks, W.A.; Reed, M.J.; Logsdon, A.F.; Rhea, E.M.; Erickson, M.A. Healthy aging and the blood-brain barrier. Nat Aging 2021, 1, 243–254. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef]
- Kwan, J.Y.; Jeong, S.Y.; Van Gelderen, P.; Deng, H.X.; Quezado, M.M.; Danielian, L.E.; Butman, J.A.; Chen, L.; Bayat, E.; Russell, J.; et al. Iron accumulation in deep cortical layers accounts for MRI signal abnormalities in ALS: correlating 7 tesla MRI and pathology. PLoS One 2012, 7, e35241. [Google Scholar] [CrossRef]
- Petillon, C.; Hergesheimer, R.; Puy, H.; Corcia, P.; Vourc’h, P.; Andres, C.; Karim, Z.; Blasco, H. The Relevancy of Data Regarding the Metabolism of Iron to Our Understanding of Deregulated Mechanisms in ALS; Hypotheses and Pitfalls. Front Neurosci 2018, 12, 1031. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Tomas, D.; Perera, N.D.; Cuic, B.; Luikinga, S.; Viden, A.; Barton, S.K.; McLean, C.A.; Samson, A.L.; Southon, A.; et al. Ferroptosis mediates selective motor neuron death in amyotrophic lateral sclerosis. Cell Death Differ 2022, 29, 1187–1198. [Google Scholar] [CrossRef]
- Wang, D.; Liang, W.; Huo, D.; Wang, H.; Wang, Y.; Cong, C.; Zhang, C.; Yan, S.; Gao, M.; Su, X.; et al. SPY1 inhibits neuronal ferroptosis in amyotrophic lateral sclerosis by reducing lipid peroxidation through regulation of GCH1 and TFR1. Cell Death Differ 2023, 30, 369–382. [Google Scholar] [CrossRef]
- Chen, L.; Na, R.; Danae McLane, K.; Thompson, C.S.; Gao, J.; Wang, X.; Ran, Q. Overexpression of ferroptosis defense enzyme Gpx4 retards motor neuron disease of SOD1G93A mice. Sci Rep 2021, 11, 12890. [Google Scholar] [CrossRef]
- Devos, D.; Moreau, C.; Kyheng, M.; Garcon, G.; Rolland, A.S.; Blasco, H.; Gele, P.; Timothee Lenglet, T.; Veyrat-Durebex, C.; Corcia, P.; et al. A ferroptosis-based panel of prognostic biomarkers for Amyotrophic Lateral Sclerosis. Sci Rep 2019, 9, 2918. [Google Scholar] [CrossRef]
- Tong, H.; Yang, T.; Xu, S.; Li, X.; Liu, L.; Zhou, G.; Yang, S.; Yin, S.; Li, X.J.; Li, S. Huntington’s Disease: Complex Pathogenesis and Therapeutic Strategies. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, J.F.; Ng, A.C.; Poudel, G.; Stout, J.C.; Churchyard, A.; Chua, P.; Egan, G.F.; Georgiou-Karistianis, N. Iron accumulation in the basal ganglia in Huntington’s disease: cross-sectional data from the IMAGE-HD study. J Neurol Neurosurg Psychiatry 2016, 87, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Fox, J.; Thyagarajan, B.; Fox, J.H. Brain mitochondrial iron accumulates in Huntington’s disease, mediates mitochondrial dysfunction, and can be removed pharmacologically. Free Radic Biol Med 2018, 120, 317–329. [Google Scholar] [CrossRef]
- Liu, M.; Zhao, J.; Xue, C.; Yang, J.; Ying, L. Uncovering the ferroptosis related mechanism of laduviglusib in the cell-type-specific targets of the striatum in Huntington’s disease. BMC Genomics 2024, 25, 633. [Google Scholar] [CrossRef]
- Stack, E.C.; Matson, W.R.; Ferrante, R.J. Evidence of oxidant damage in Huntington’s disease: translational strategies using antioxidants. Ann N Y Acad Sci 2008, 1147, 79–92. [Google Scholar] [CrossRef]
- Perez-Severiano, F.; Rios, C.; Segovia, J. Striatal oxidative damage parallels the expression of a neurological phenotype in mice transgenic for the mutation of Huntington’s disease. Brain Res 2000, 862, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kosaras, B.; Del Signore, S.J.; Cormier, K.; McKee, A.; Ratan, R.R.; Kowall, N.W.; Ryu, H. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice. Acta Neuropathol 2011, 121, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Snyder, S.H. Impaired Redox Signaling in Huntington’s Disease: Therapeutic Implications. Front Mol Neurosci 2019, 12, 68. [Google Scholar] [CrossRef]
- Kumar, A.; Ratan, R.R. Oxidative Stress and Huntington’s Disease: The Good, The Bad, and The Ugly. J Huntingtons Dis 2016, 5, 217–237. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Su, Z.; Kon, N.; Chu, B.; Li, H.; Jiang, X.; Luo, J.; Stockwell, B.R.; Gu, W. ALOX5-mediated ferroptosis acts as a distinct cell death pathway upon oxidative stress in Huntington’s disease. Genes Dev 2023, 37, 204–217. [Google Scholar] [CrossRef]
- Garton, T.; Gadani, S.P.; Gill, A.J.; Calabresi, P.A. Neurodegeneration and demyelination in multiple sclerosis. Neuron 2024, 112, 3231–3251. [Google Scholar] [CrossRef]
- Dal-Bianco, A.; Grabner, G.; Kronnerwetter, C.; Weber, M.; Kornek, B.; Kasprian, G.; Berger, T.; Leutmezer, F.; Rommer, P.S.; Trattnig, S.; et al. Long-term evolution of multiple sclerosis iron rim lesions in 7 T MRI. Brain 2021, 144, 833–847. [Google Scholar] [CrossRef]
- Kim, S.; Lee, E.K.; Song, C.J.; Sohn, E. Iron Rim Lesions as a Specific and Prognostic Biomarker of Multiple Sclerosis: 3T-Based Susceptibility-Weighted Imaging. Diagnostics (Basel) 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Jhelum, P.; Zandee, S.; Ryan, F.; Zarruk, J.G.; Michalke, B.; Venkataramani, V.; Curran, L.; Klement, W.; Prat, A.; David, S. Ferroptosis induces detrimental effects in chronic EAE and its implications for progressive MS. Acta Neuropathol Commun 2023, 11, 121. [Google Scholar] [CrossRef]
- Duarte-Silva, E.; Meuth, S.G.; Peixoto, C.A. The role of iron metabolism in the pathogenesis and treatment of multiple sclerosis. Front Immunol 2023, 14, 1137635. [Google Scholar] [CrossRef]
- Wu, T.; Ning, S.; Zhang, H.; Cao, Y.; Li, X.; Hao, J.; Wang, L. Role of ferroptosis in neuroimmunity and neurodegeneration in multiple sclerosis revealed by multi-omics data. J Cell Mol Med 2024, 28, e18396. [Google Scholar] [CrossRef] [PubMed]
- Jhelum, P.; Santos-Nogueira, E.; Teo, W.; Haumont, A.; Lenoel, I.; Stys, P.K.; David, S. Ferroptosis Mediates Cuprizone-Induced Loss of Oligodendrocytes and Demyelination. J Neurosci 2020, 40, 9327–9341. [Google Scholar] [CrossRef]
- Stojkovic, L.; Jovanovic, I.; Dincic, E.; Djordjevic, A.; Kuveljic, J.; Djuric, T.; Stankovic, A.; Vojinovic, S.; Zivkovic, M. Targeted RNAseq Revealed the Gene Expression Signature of Ferroptosis-Related Processes Associated with Disease Severity in Patients with Multiple Sclerosis. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef]
- Van San, E.; Debruyne, A.C.; Veeckmans, G.; Tyurina, Y.Y.; Tyurin, V.A.; Zheng, H.; Choi, S.M.; Augustyns, K.; van Loo, G.; Michalke, B.; et al. Ferroptosis contributes to multiple sclerosis and its pharmacological targeting suppresses experimental disease progression. Cell Death Differ 2023, 30, 2092–2103. [Google Scholar] [CrossRef]
- Rothammer, N.; Woo, M.S.; Bauer, S.; Binkle-Ladisch, L.; Di Liberto, G.; Egervari, K.; Wagner, I.; Haferkamp, U.; Pless, O.; Merkler, D.; et al. G9a dictates neuronal vulnerability to inflammatory stress via transcriptional control of ferroptosis. Sci Adv 2022, 8, eabm5500. [Google Scholar] [CrossRef] [PubMed]
- Luoqian, J.; Yang, W.; Ding, X.; Tuo, Q.Z.; Xiang, Z.; Zheng, Z.; Guo, Y.J.; Li, L.; Guan, P.; Ayton, S.; et al. Ferroptosis promotes T-cell activation-induced neurodegeneration in multiple sclerosis. Cell Mol Immunol 2022, 19, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Marupudi, N.; Xiong, M.P. Genetic Targets and Applications of Iron Chelators for Neurodegeneration with Brain Iron Accumulation. ACS Bio Med Chem Au 2024, 4, 119–130. [Google Scholar] [CrossRef]
- Crichton, R.R.; Ward, R.J.; Hider, R.C. The Efficacy of Iron Chelators for Removing Iron from Specific Brain Regions and the Pituitary-Ironing out the Brain. Pharmaceuticals (Basel) 2019, 12. [Google Scholar] [CrossRef]
- Barman Balfour, J.A.; Foster, R.H. Deferiprone: a review of its clinical potential in iron overload in beta-thalassaemia major and other transfusion-dependent diseases. Drugs 1999, 58, 553–578. [Google Scholar] [CrossRef]
- Shachar, D.B.; Kahana, N.; Kampel, V.; Warshawsky, A.; Youdim, M.B. Neuroprotection by a novel brain permeable iron chelator, VK-28, against 6-hydroxydopamine lession in rats. Neuropharmacology 2004, 46, 254–263. [Google Scholar] [CrossRef]
- Liu, J.P.; Cen, S.Y.; Xue, Z.; Wang, T.X.; Gao, Y.; Zheng, J.; Zhang, C.; Hu, J.; Nie, S.; Xiong, Y.; et al. A Class of Disulfide Compounds Suppresses Ferroptosis by Stabilizing GPX4. ACS Chem Biol 2022, 17, 3389–3406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Luo, Y.L.; Xiang, Y.; Bai, X.Y.; Qiang, R.R.; Zhang, X.; Yang, Y.L.; Liu, X.L. Ferroptosis inhibitors: past, present and future. Front Pharmacol 2024, 15, 1407335. [Google Scholar] [CrossRef]
- Li, B.; Cheng, K.; Wang, T.; Peng, X.; Xu, P.; Liu, G.; Xue, D.; Jiao, N.; Wang, C. Research progress on GPX4 targeted compounds. Eur J Med Chem 2024, 274, 116548. [Google Scholar] [CrossRef]
- Alkandari, A.F.; Madhyastha, S.; Rao, M.S. N-Acetylcysteine Amide against Abeta-Induced Alzheimer’s-like Pathology in Rats. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Tardiolo, G.; Bramanti, P.; Mazzon, E. Overview on the Effects of N-Acetylcysteine in Neurodegenerative Diseases. Molecules 2018, 23. [Google Scholar] [CrossRef]
- Hammerschmidt, T.G.; Guerreiro, G.B.; Donida, B.; Raabe, M.; Kessler, R.G.; Ferro, M.B.; Moura, D.J.; Giugliani, R.; Vargas, C.R. Beneficial in vitro effect of N-acetylcysteine and coenzyme Q10 on DNA damage in neurodegenerative Niemann-Pick type C 1 disease: preliminary results. Naunyn Schmiedebergs Arch Pharmacol 2023, 396, 1563–1569. [Google Scholar] [CrossRef]
- Morgan, A.M.; Hassanen, E.I.; Ogaly, H.A.; Al Dulmani, S.A.; Al-Zahrani, F.A.M.; Galal, M.K.; Kamel, S.; Rashad, M.M.; Ibrahim, M.A.; Hussien, A.M. The ameliorative effect of N-acetylcysteine against penconazole induced neurodegenerative and neuroinflammatory disorders in rats. J Biochem Mol Toxicol 2021, 35, e22884. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Shen, J.; Jiang, J.; Wang, F.; Min, J. Targeting ferroptosis opens new avenues for the development of novel therapeutics. Signal Transduct Target Ther 2023, 8, 372. [Google Scholar] [CrossRef]
- Mukem, S.; Thongbuakaew, T.; Khornchatri, K. Mito-Tempo suppresses autophagic flux via the PI3K/Akt/mTOR signaling pathway in neuroblastoma SH-SY5Y cells. Heliyon 2021, 7, e07310. [Google Scholar] [CrossRef]
- Zhelev, Z.; Bakalova, R.; Aoki, I.; Lazarova, D.; Saga, T. Imaging of superoxide generation in the dopaminergic area of the brain in Parkinson’s disease, using mito-TEMPO. ACS Chem Neurosci 2013, 4, 1439–1445. [Google Scholar] [CrossRef]
- Mandal, P.K.; Maroon, J.C.; Samkaria, A.; Arora, Y.; Sharma, S.; Pandey, A. Iron Chelators and Alzheimer’s Disease Clinical Trials. J Alzheimers Dis 2024, 100, S243–S249. [Google Scholar] [CrossRef] [PubMed]
- Zha, S.; Liu, H.; Li, H.; Li, H.; Wong, K.L.; All, A.H. Functionalized Nanomaterials Capable of Crossing the Blood-Brain Barrier. ACS Nano 2024, 18, 1820–1845. [Google Scholar] [CrossRef] [PubMed]
- Maher, R.; Moreno-Borrallo, A.; Jindal, D.; Mai, B.T.; Ruiz-Hernandez, E.; Harkin, A. Intranasal Polymeric and Lipid-Based Nanocarriers for CNS Drug Delivery. Pharmaceutics 2023, 15. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab 2012, 32, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Zha, X.; Liu, X.; Wei, M.; Huang, H.; Cao, J.; Liu, S.; Bian, X.; Zhang, Y.; Xiao, F.; Xie, Y.; et al. Microbiota-derived lysophosphatidylcholine alleviates Alzheimer’s disease pathology via suppressing ferroptosis. Cell Metab 2025, 37, 169–186 e169. [Google Scholar] [CrossRef]
- Forster, P.M.; Jakob, M.O.; Yusuf, D.; Bubeck, M.; Limberger, H.; Luo, Y.; Thieme, P.; Polici, A.; Sterczyk, N.; Boulekou, S.; et al. A transcriptional atlas of gut-innervating neurons reveals activation of interferon signaling and ferroptosis during intestinal inflammation. Neuron 2025. [Google Scholar] [CrossRef]
- Jian, J.; Wei, J. Ferroptosis: A New Pathway in the Interaction between Gut Microbiota and Multiple Sclerosis. Front Biosci (Landmark Ed) 2025, 30, 26265. [Google Scholar] [CrossRef]
Figure 1.
The system Xc--GSH--GPX4 axis negatively regulates ferroptosis by suppressing excessive peroxidation of phospholipids. Lipid peroxidation is maintained by ferroptosis defense mechanisms mediated by GSH and GPX4, with GPX4 utilizing GSH to neutralize lipid peroxides, and cystine uptake mediated by system Xc– provides the key precursor cysteine for GSH synthesis. The upregulation of either MRP1 or CDO1 counteracts this activity and decreases GSH synthesis. Suppression of system Xc--GSH--GPX4 activity by the ferroptosis inducers erastin and RSL3 results in excessive lipid peroxidation, leading to membrane rupture and cell death. Abbreviations: CDO1, cysteine dioxygenase 1; GSH, reduced glutathione; GPX4, glutathione peroxidase 4; LOX, lipoxygenase; MRP1, multidrug resistance-associated protein 1; RSL3, RAS-selective lethal. Created at https://BioRender.com (accessed on 10 March, 2025).
Figure 1.
The system Xc--GSH--GPX4 axis negatively regulates ferroptosis by suppressing excessive peroxidation of phospholipids. Lipid peroxidation is maintained by ferroptosis defense mechanisms mediated by GSH and GPX4, with GPX4 utilizing GSH to neutralize lipid peroxides, and cystine uptake mediated by system Xc– provides the key precursor cysteine for GSH synthesis. The upregulation of either MRP1 or CDO1 counteracts this activity and decreases GSH synthesis. Suppression of system Xc--GSH--GPX4 activity by the ferroptosis inducers erastin and RSL3 results in excessive lipid peroxidation, leading to membrane rupture and cell death. Abbreviations: CDO1, cysteine dioxygenase 1; GSH, reduced glutathione; GPX4, glutathione peroxidase 4; LOX, lipoxygenase; MRP1, multidrug resistance-associated protein 1; RSL3, RAS-selective lethal. Created at https://BioRender.com (accessed on 10 March, 2025).
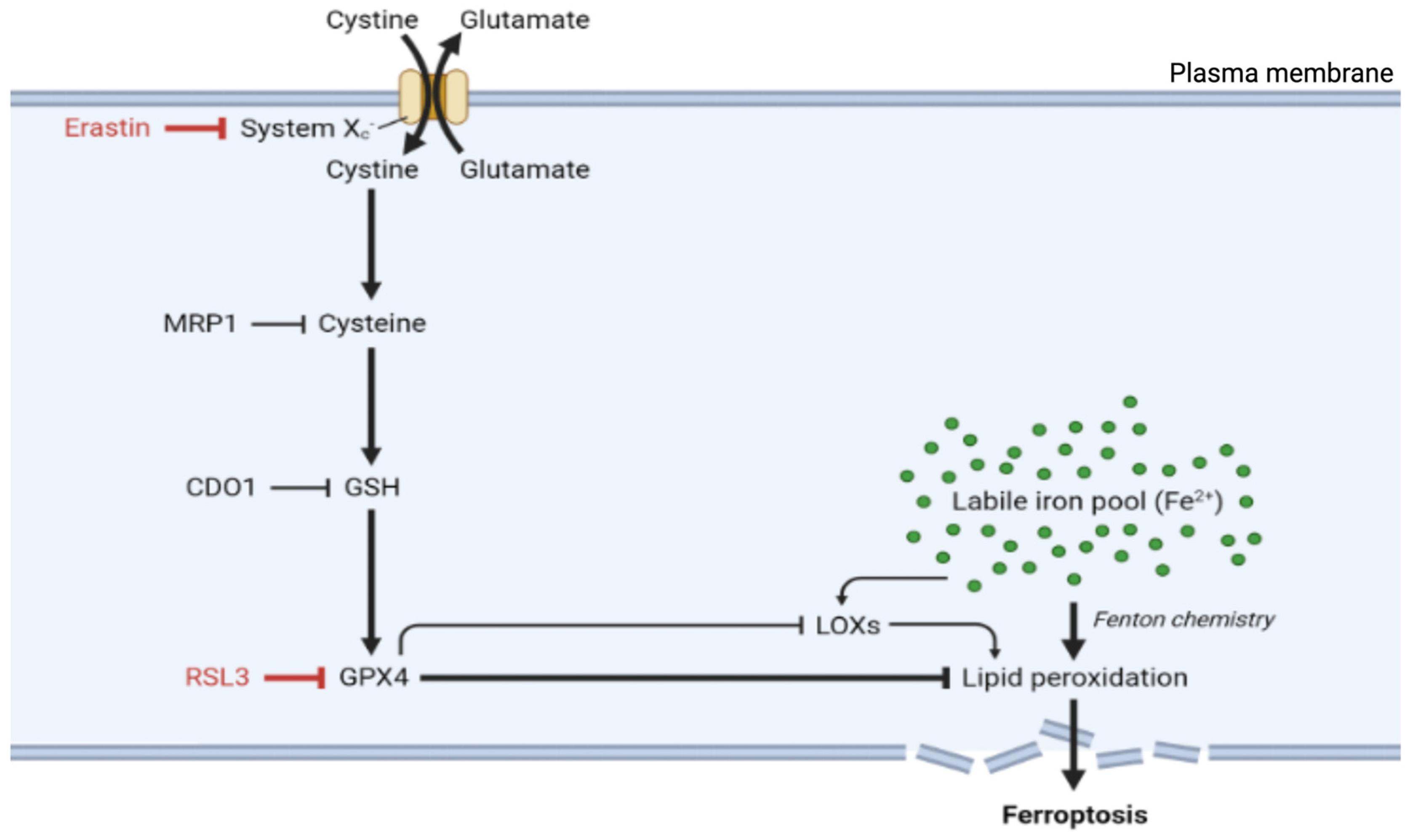
Figure 2.
Lipid metabolism generates substrates for lipid peroxidation during ferroptosis. PUFA remodeling and incorporation into phospholipids through the actions of ACSL4 and LPCAT3 generate PUFA-PLs, the primary substrates of peroxidation in ferroptosis. The accumulation of lipid peroxides leads to profound cellular consequences, beginning with alterations in membrane properties. These changes include decreased membrane fluidity, disrupted membrane protein function, altered membrane organization, and ultimately, membrane rupture and ferroptosis. Abbreviations: ACSL4, acyl-coenzyme A synthetase long-chain family member 4; GPX4, glutathione peroxidase 4; iPLA2β, calcium-independent phospholipase A2β; LPCAT3, lysophosphatidylcholine acyltransferase 3; LOX, lipoxygenase; PUFA, polyunsaturated fatty acid; PL, phospholipid. Created at https://BioRender.com (accessed on 10 March, 2025).
Figure 2.
Lipid metabolism generates substrates for lipid peroxidation during ferroptosis. PUFA remodeling and incorporation into phospholipids through the actions of ACSL4 and LPCAT3 generate PUFA-PLs, the primary substrates of peroxidation in ferroptosis. The accumulation of lipid peroxides leads to profound cellular consequences, beginning with alterations in membrane properties. These changes include decreased membrane fluidity, disrupted membrane protein function, altered membrane organization, and ultimately, membrane rupture and ferroptosis. Abbreviations: ACSL4, acyl-coenzyme A synthetase long-chain family member 4; GPX4, glutathione peroxidase 4; iPLA2β, calcium-independent phospholipase A2β; LPCAT3, lysophosphatidylcholine acyltransferase 3; LOX, lipoxygenase; PUFA, polyunsaturated fatty acid; PL, phospholipid. Created at https://BioRender.com (accessed on 10 March, 2025).
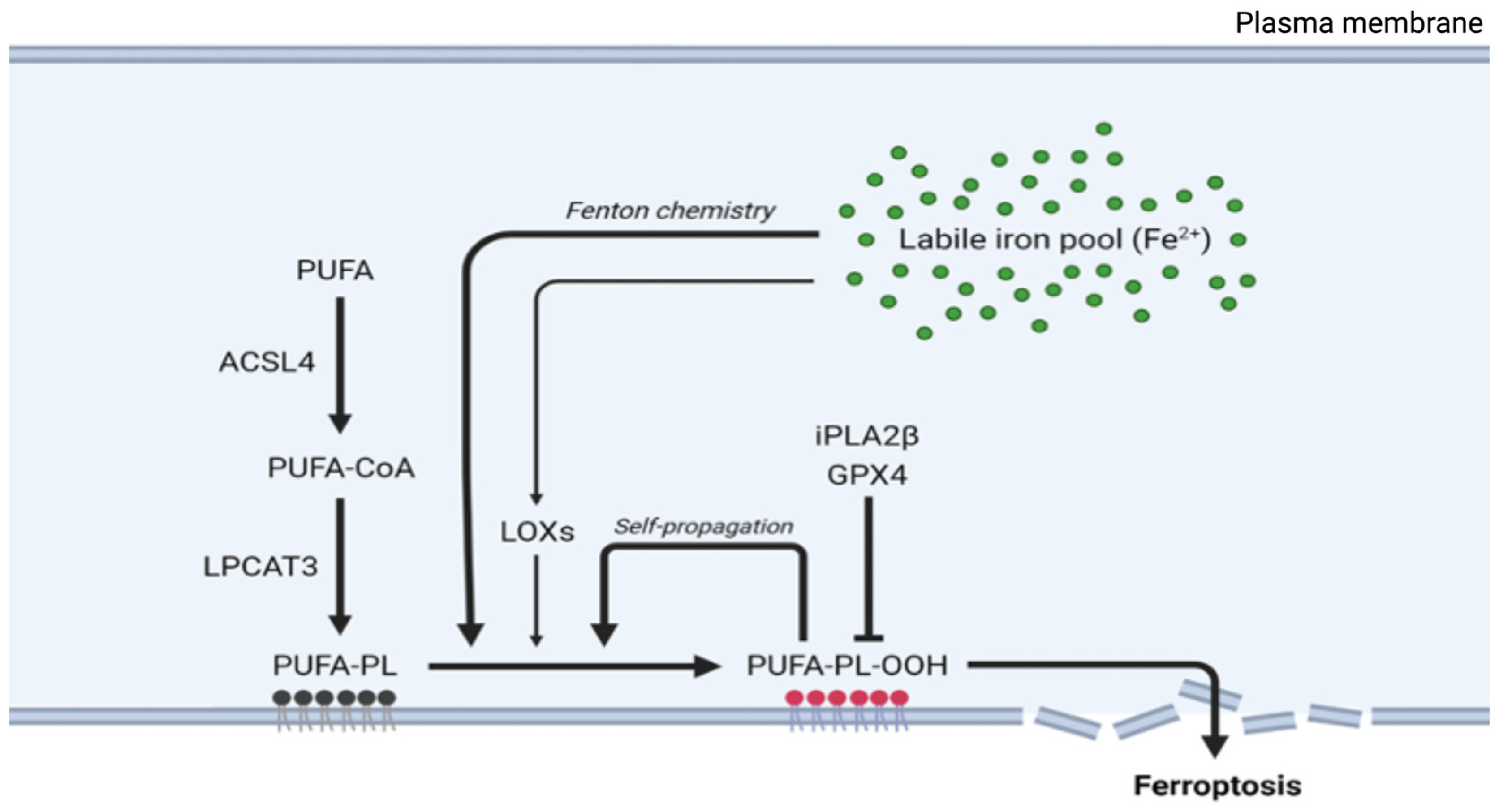
Figure 3.
Iron metabolism facilitates lipid peroxidation in ferroptosis. Extracellular Fe3+ attached to transferrin (Tf) binds to the transferrin receptor, leading to the internalization of the transferrin receptor complex (TFRC). Once inside the endosome, STEAP3, a metalloreductase, converts Fe3+ into Fe2+. DMT1 then shuttles Fe2+ from the endosome to a cytoplasmic labile iron pool, where Fe2+ generates ROS through the Fenton reaction. Fe2+ can also be exported via the FPN. Abbreviations: DMT1, divalent metal transporter 1; FPN, ferroportin; LOX, lipoxygenase; ROS, reactive oxygen species; STEAP3, six-transmembrane epithelial antigen of the prostate 3; Tf, transferrin; TFRC, transferrin receptor complex. Created at https://BioRender.com (accessed on 10 March, 2025).
Figure 3.
Iron metabolism facilitates lipid peroxidation in ferroptosis. Extracellular Fe3+ attached to transferrin (Tf) binds to the transferrin receptor, leading to the internalization of the transferrin receptor complex (TFRC). Once inside the endosome, STEAP3, a metalloreductase, converts Fe3+ into Fe2+. DMT1 then shuttles Fe2+ from the endosome to a cytoplasmic labile iron pool, where Fe2+ generates ROS through the Fenton reaction. Fe2+ can also be exported via the FPN. Abbreviations: DMT1, divalent metal transporter 1; FPN, ferroportin; LOX, lipoxygenase; ROS, reactive oxygen species; STEAP3, six-transmembrane epithelial antigen of the prostate 3; Tf, transferrin; TFRC, transferrin receptor complex. Created at https://BioRender.com (accessed on 10 March, 2025).
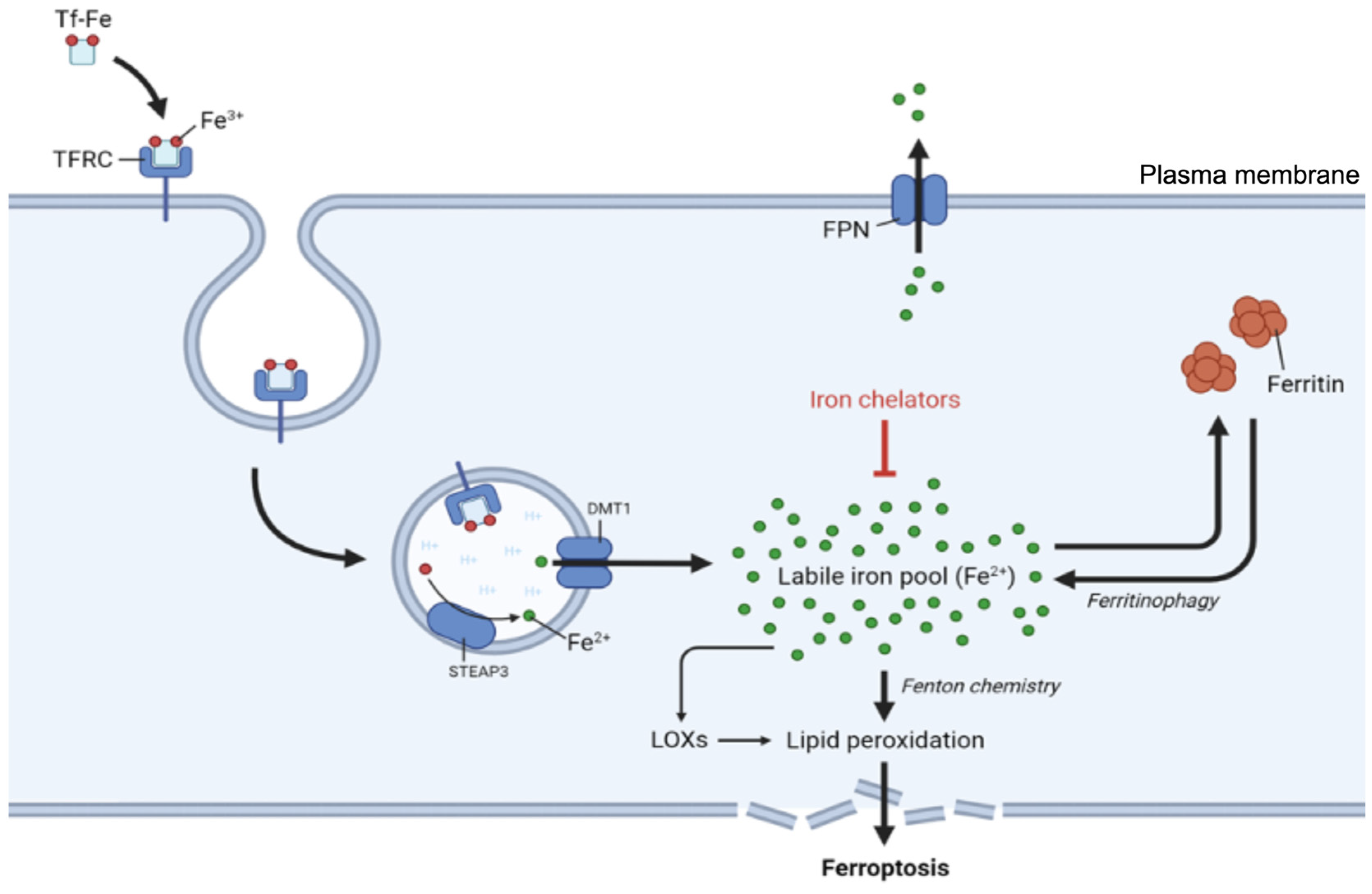
Figure 5.
Model depicting the neovascular unit of the brain with a cross-section delineating the arrangement of cell types forming the blood–brain barrier (BBB). A magnified image of a section of the BBB is shown below, depicting two capillary endothelial cells connected by tight junctions along with a section of an astrocyte end-foot. The main routes of iron uptake by capillary endothelial cells involve three main processes: uptake by endocytosis, transcytosis, and FPN-mediated export. Initially, transferrin (Tf, red), which is iron bound, binds to the transferrin receptor (TfR) on the luminal membrane of endothelial cells, and the resulting transferrin receptor complex (TFRC) is subsequently endocytosed. Following internalization, the complex can proceed via two distinct routes. In the first route, termed transcytosis (1), the TFRC is directly shuttled to the abluminal membrane, where Tf is then released into the extracellular fluid of the brain. Alternatively, the TFRC is dissociated within the acidic environment of the endosome. In this case, ferric (Fe³⁺) iron is released from Tf and reduced to ferrous (Fe²⁺) iron via the action of STEAP proteins, yielding apo-transferrin (apo-Tf, blue). The apo-Tf is then recycled back to the luminal membrane along with the TfR. Ferrous (Fe²⁺) iron is subsequently released into the cytoplasm via DMT1, where it can be exported via the ferroportin (FPN) export pathway (2). The ferroxidase hephaestin (HEPH) oxidizes Fe²⁺ to ferric (Fe³⁺) iron, enabling apo-Tf to bind iron for utilization by other cells. Created at https://BioRender.com (accessed on 12 March, 2025).
Figure 5.
Model depicting the neovascular unit of the brain with a cross-section delineating the arrangement of cell types forming the blood–brain barrier (BBB). A magnified image of a section of the BBB is shown below, depicting two capillary endothelial cells connected by tight junctions along with a section of an astrocyte end-foot. The main routes of iron uptake by capillary endothelial cells involve three main processes: uptake by endocytosis, transcytosis, and FPN-mediated export. Initially, transferrin (Tf, red), which is iron bound, binds to the transferrin receptor (TfR) on the luminal membrane of endothelial cells, and the resulting transferrin receptor complex (TFRC) is subsequently endocytosed. Following internalization, the complex can proceed via two distinct routes. In the first route, termed transcytosis (1), the TFRC is directly shuttled to the abluminal membrane, where Tf is then released into the extracellular fluid of the brain. Alternatively, the TFRC is dissociated within the acidic environment of the endosome. In this case, ferric (Fe³⁺) iron is released from Tf and reduced to ferrous (Fe²⁺) iron via the action of STEAP proteins, yielding apo-transferrin (apo-Tf, blue). The apo-Tf is then recycled back to the luminal membrane along with the TfR. Ferrous (Fe²⁺) iron is subsequently released into the cytoplasm via DMT1, where it can be exported via the ferroportin (FPN) export pathway (2). The ferroxidase hephaestin (HEPH) oxidizes Fe²⁺ to ferric (Fe³⁺) iron, enabling apo-Tf to bind iron for utilization by other cells. Created at https://BioRender.com (accessed on 12 March, 2025).
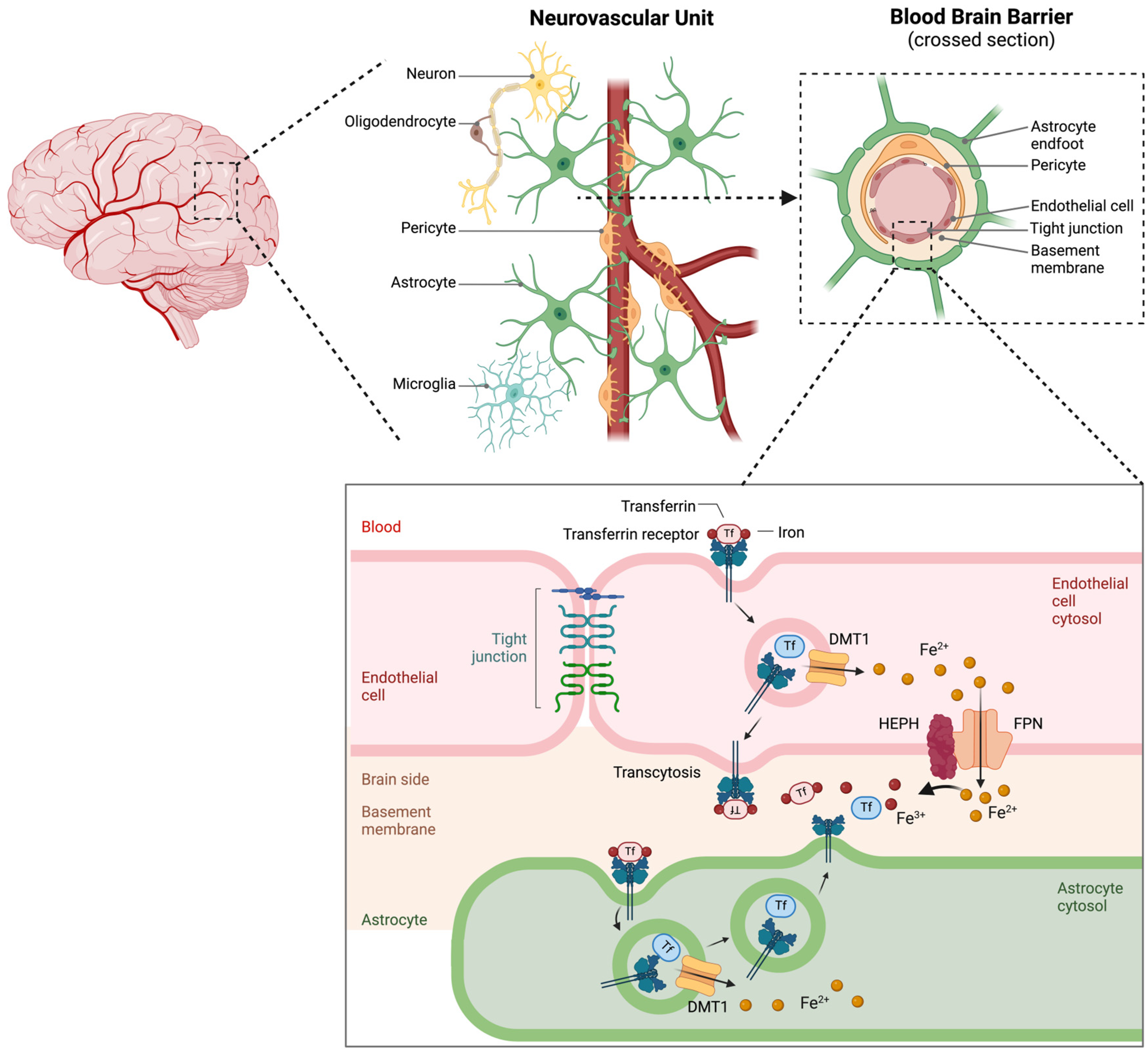
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



