
Submitted:
24 March 2025
Posted:
25 March 2025
You are already at the latest version
Abstract
Fusarium oxysporum f. sp. cubense (Foc) causes Fusarium wilt, a devastating epidemic disease that has caused widespread damage to banana crops worldwide. We report the draft genomes of Foc race 1 (16117) and Foc tropical race 4 (Fusarium odoratissimum) (CNSD1) isolates from China, assembled using PacBio HiFi sequencing reads, with functional annotation performed. The strains group in distinct lineages within the Fusarium oxysporum species complex. This genetic resource will contribute towards understanding the pathogenicity and evolutionary dynamics of Foc populations in banana-growing regions around the world.
Keywords:
Fusarium wilt of banana
; Pacbio HiFi
; genome assembly
; fungal effectors
; pathogenicity
; Fusarium oxysporum species complex
; whole-genome phylogenetics
Introduction
Fusarium wilt of banana, caused by Fusarium oxysporum f. sp. cubense (Foc) is a devastating disease affecting banana production worldwide [1,2]. This disease, also known as Panama disease, occurs when Foc infects the vascular system of the banana plant, blocking water and nutrient flow leading to plant wilting and eventual death [3]. The diversity within Foc populations can be differentiated using vegetative compatibility grouping (VCG), a classification system that shows association with virulence, host range and geographic distribution of Foc populations [4]. Currently, at least 24 VCGs have been identified for Foc, underscoring the complexity and adaptability of this pathogen [5,6]. Alternatively, Foc can be classified into a race structure based on its virulence against specific banana cultivars it infects. Foc race 1 caused the pandemic that led to the demise of the ‘Gros Michel’ banana in the mid-20th Century and also affects several other cultivars including 'Lady Finger' and 'Silk' bananas [3]. The virulent pathogen race, Foc tropical race 4 (TR4) has threatened the global banana production due to the widespread reliance on the Cavendish cultivars. The spread of Foc TR4 to Southeast Asia, the Middle East, Africa, and Latin America has caused serious concerns about its impact on banana production globally [7,8].
Foc TR4 and Foc race 1 both cause Fusarium wilt in banana but differ significantly in their host range, virulence, and impact on the banana industry. Understanding these differences is essential for developing diagnostic tools to aid the management strategies in the control and deterrence of Foc TR4. To this end, two Foc isolates, confirmed as Foc Race 1 and Foc TR4, were sequenced using the Pac-Bio long-read HiFi technology. These genome resources will contribute towards understanding differences in host specificity and virulence between these strains, thereby allowing improved strategies to manage Foc TR4 in the future.
Materials and Methods
Sample collecting and Fungal Isolates
Banana pseudostem samples showing symptoms of Fusarium wilt were collected from fields in Baini, Foshan, Guangdong (23°2′48′′N, 112°52′50′′E) on dwarf banana plants and from fields in Wuming, Nanning, Guangxi, China (23°19′98′′N, 108°16′68′′E) on Cavendish banana. The pseudostem sections were surface-sterilized and cultured on potato dextrose agar (PDA) at 28°C for 3 days to isolate the fungus. Single spore isolation was then performed to generate monoconidial cultures for each of these isolates. Prior to this study, vegetative compatibility group (VCG) testing confirmed the identities of both isolate 16117 (Baini) and CNSD1 (Wuming) as Foc Race 1 (VCGs 0120/15, 01218) and Foc TR4 (VCG 01213/16), respectively. The Foc TR4 isolate CNSD1 was used in a previous study to assess transcriptome reprogramming in response to Foc TR4 in resistant and susceptible banana cultivars [9].
DNA extraction and genome sequencing
Monoconidial cultures of isolates 16117 and CNSD1 were incubated on PDA medium for 7 days and mycelia were scrapped off the plates and used for DNA extraction. Genomic DNA of the two isolates were then extracted using the MagAttract HMW DNA kit (Qiagen, Hilden, Germany) according to manufacturer’s instructions, and the purified DNA quantified with Qubit (4.0) fluorimeter (Life Technologies, Carlsbad, CA, USA). The isolates were sequenced on a PacBio SMRT flow cell. Libraries were prepared by Personalbio Technology Co. Ltd. (Shanghai, China) using a standard PacBio gDNA library preparation kit. Briefly, the high molecular weight genomic DNA was sheared by g-TUBE to an average size between 10-15 Kb, ligated with known adapters and digested by enzyme reaction. The BluePippin (Sage Science, Beverley, MA, USA) was used to select the DNA fragment with target size of 10-15 Kb to generate SMRTbell structure libraries. The purified libraries were analyzed by Agilent 2100 Bioanalyzer System (Agilent technologies, Santa Clara, CA, USA) before sequencing on the PacBio Sequel II platform.
Genome assembly and gene prediction
The resulting long reads were subject to de novo assembly using hifiasm v0.18.5 under the Galaxy Australia compute environment (Version 0.24.0), with default settings including -k 51 and -l0 [10]. The assemblies were initially evaluated using QUAST [11]. The genome assembly completeness were further assessed using BUSCO v5.4.5 with AUGUSTUS species training mode set to Fusarium graminearum against the Ascomycota lineage [12].
The assemblies were then annotated for elements including repeat sequences and protein-coding genes. Repeats in the genomes were identified using RepeatModeler (v2.0.4) and then masked using RepeatMasker (v4.1.4) software [13]. Protein-coding gene prediction was performed by GlimmerHMM (v3.0.4) [14], AUGUSTUS (version 2.5.5) [15] and GeneMark-ES (v4.71) software [16]. To refine gene annotations, additional gene structure evidence was obtained by performing homology-dependent alignments to closely related species using exonerate (v2.2.0) [17]. The predicted gene models were then integrated into a weighted consensus gene set using EvidenceModeler v 2.0.0 [18] to generate a final high-confidence gene annotation set.
Chromosome alignments
Telomere repeats of 5'-TAACCC-'3 were first detected using the search function of tidk (version 0.2.63) and then visualised using its plot function [21].
The assembly scaffolds were aligned chromosome-level reference genomes using the nucmer function of the MUMmer software (version 4.0.1), with default settings [22]. An interactive web plot viewer Dot (https://github.com/marianattestad/dot, accessed on 12 February 2025) was then used to visualise an ordered set of reference and query alignments, passing in the nucmer outputs using the script DotPrep.py, and applying filtering to display aligned regions of greater than 2 kb.
Functional annotation
Genes were functionally annotated by performing searches against multiple databases. Specifically, genes were searched in BLAST against the non-redundant protein database to identify homologous proteins [23]. GO annotation was performed using InterPro (version 66.0, release 2017.11.23) [24]. The results were then processed in InterPro2GO to obtain GO terms, which were then mapped to a list of selected terms (GO slims) using map2slim (https://github.com/elhumble/map2slim, accessed on 18 February 2025). Database searches using eggNOG (http://eggnogdb.embl.de/) and an E-value threshold of 1×10-6 to infer orthologous groups and functional annotations, the KEGG database (https://www.kegg.jp/) to associate genes with metabolic and signalling pathways, and the CAZy database (http://www.cazy.org/) to classify carbohydrate-active enzymes, were performed using Diamond (v2.0.14) [25].
To further characterise protein function, localisation signals including signal peptides were predicted from the draft genomes of strains 16117 and CNSD1 using SignalP (v5.0) [26] and TargetP (v2.0) [27]. Membrane protein topology was predicted using TMHMM version 2.0 [28]. Additionally, potential secreted effectors, proteins that may play a role in host-pathogen interactions, were predicted using EffectorP (v3.0) [29].
Phylogenetic analysis
The phylogenetic analysis was performed using the public available genomes of 152 F. oxysporum strains, including special forms on banana [30,31,32] and other plant hosts, as well as a F. verticillioides (isolate 7600) strain, used to anchor the whole phylogeny. The retrieval of the assemblies, AUGUSTUS annotation and the subsequent phylogenetic analysis was performed using a workflow described in another study [33].
Briefly, all genomes were loaded into the Galaxy web platform, via the public server at usegalaxy.org.au to analyse the data [20]. De novo gene annotation was performed using AUGUSTUS (version 3.4.0) [15,34,35], with Fusarium graminearum splice models. Only genes without internal stop codons were retained. BUSCO was then performed on these coding sequences to retain only complete, single-copy conserved genes [12], using a custom bash script. Single-copy genes across all 152 genomes were identified using seqkit grep (version 2.9.0) [36], aligned using MAFFT (version 7.520) [37] with default settings, with poorly aligned regions removed using trimAI (version v1.5.rev0) [38]. The final edited alignments were concatenated using the seqkit concat command.
Phylogenetic reconstruction was conducted using RAxML GUI (version 2.0) [39]. The best-fit model, GTR+I+G4, was selected and applied for maximum likelihood tree inference, with 100 bootstrap replicates performed to assess branch support. The F. verticillioides isolate 7600 was designated as the outgroup. The resulting phylogenetic tree was then imported into the Interactive Tree of Life (iTOL) [40](Letunic and Bork 2024).
Genome-wide Profiling of SIX gene effectors
The Fusarium oxysporum Effector Clustering (FoEC2) pipeline was run using the Foc genomes retrieved from the public databases and the ones obtained in this study [41]. Fourteen SIX gene nucleotide sequences previously obtained from a Fol strain was also used as a query [42]. Clustering in the pipeline was performed using default settings which included binary distance matrix and average distance calculation. TBLASTN search of Fol-SIX protein sequences against all Foc genomes was performed using the command line version of NCBI-BLAST+ (version v2.12.0) and an e-value cut-off score of 1×10-10.
Results and Discussion
Figure 1.
Local banana production regions in Guangxi, China. (A) A local farm holder selling freshly produced bananas from the ‘AAA’ cultivar group, including Musa acuminata ‘Red Dacca’ and ‘Williams’ Cavendish in Wuming, Nanning, Guangxi, China. (B) Symptomatic banana plants infected with Fusarium wilt in Wuming, Nanning, Guangxi, China, where Fusarium oxysporum f. sp. cubense isolate CNSD1 was collected. Fusarium oxysporum f. sp. cubense monoconidial isolates (C) 16117 and (D) CNSD1 grown on potato dextrose agar.
Figure 1.
Local banana production regions in Guangxi, China. (A) A local farm holder selling freshly produced bananas from the ‘AAA’ cultivar group, including Musa acuminata ‘Red Dacca’ and ‘Williams’ Cavendish in Wuming, Nanning, Guangxi, China. (B) Symptomatic banana plants infected with Fusarium wilt in Wuming, Nanning, Guangxi, China, where Fusarium oxysporum f. sp. cubense isolate CNSD1 was collected. Fusarium oxysporum f. sp. cubense monoconidial isolates (C) 16117 and (D) CNSD1 grown on potato dextrose agar.

Figure 2.
Whole-genome contig alignment of Fusarium oxysporum f. sp. cubense TR4 isolate CNSD1 (A), and Race 1 isolate 16117 (B), to each of the 15 chromosomes of the F. oxysporum f. sp. lycopersici strain 4287 (Fol4287) genome. (C) Contig alignment of CNSD1 to Foc TR4 II5. (D) Contig alignment of 16117 to Foc R1 GD02. Unique alignments are shown in blue (forward) and green (reverse complement). Repetitive alignments are shown in orange. Only alignments greater than 2 Kb are shown. Red arrows indicate the presence of telomere repeats detected at the terminal ends of the contigs.
Figure 2.
Whole-genome contig alignment of Fusarium oxysporum f. sp. cubense TR4 isolate CNSD1 (A), and Race 1 isolate 16117 (B), to each of the 15 chromosomes of the F. oxysporum f. sp. lycopersici strain 4287 (Fol4287) genome. (C) Contig alignment of CNSD1 to Foc TR4 II5. (D) Contig alignment of 16117 to Foc R1 GD02. Unique alignments are shown in blue (forward) and green (reverse complement). Repetitive alignments are shown in orange. Only alignments greater than 2 Kb are shown. Red arrows indicate the presence of telomere repeats detected at the terminal ends of the contigs.

Figure 3.
Whole genome-based phylogenetic reconstruction based on a BUSCO set of 894 conserved single-copy orthologs shared among 154 representatives of the Fusarium oxysporum species complex. Most Foc genomes and VCG information were retrieved from previous studies [30,31]. R1, R2, STR4, and TR4 annotates Race 1, Race 2, Subtropical Race 4, and Tropical Race 4, respectively. Red highlight indicates the strains sequenced in this study. The host of origin is indicated, along with the race and VCG designations for banana isolates (marked with orange boxes) where this information could be found in NCBI databases or publications. Branch support based on bootstrap analysis (percentage) is indicated by circles scaled relative to a 0-100% bootstrap value.
Figure 3.
Whole genome-based phylogenetic reconstruction based on a BUSCO set of 894 conserved single-copy orthologs shared among 154 representatives of the Fusarium oxysporum species complex. Most Foc genomes and VCG information were retrieved from previous studies [30,31]. R1, R2, STR4, and TR4 annotates Race 1, Race 2, Subtropical Race 4, and Tropical Race 4, respectively. Red highlight indicates the strains sequenced in this study. The host of origin is indicated, along with the race and VCG designations for banana isolates (marked with orange boxes) where this information could be found in NCBI databases or publications. Branch support based on bootstrap analysis (percentage) is indicated by circles scaled relative to a 0-100% bootstrap value.

Pathogen isolation
Fusarium wilt caused by Foc TR4 has placed a heavy burden on local farm holders who rely on fresh locally grown bananas as a primary source of income in Wuming, Guangxi China (Figure 1A). Foc TR4 isolate CNSD1 was isolated from plants in a local banana plantation in Wuming, which exhibited severe Fusarium wilt symptoms including leaf yellowing, necrosis and vascular wilt of entire plants (Figure 1B). The Foc race 1 isolate 16117 was isolated from a dwarf banana plant in Baini, Foshan, Guangdong. After single spore isolation, both isolates showed a light pink colour, with aerial hyphae observed, when grown on PDA (Figure 1C-D).
Mitochondrial genomes
The mitochondrial genomes of 16117 and CNSD1 were 45,628 bp and 49,694 bp respectively (Figure S1, Additional files 1-2). Analysis of Fusarium oxysporum mitochondrial genomes suggests the presence of a large variable region, which comes in the form of three distinct haplotypes or variants [43]. The mitochondrial genomes of 16117 and CNSD1 are similar to that of Fusarium oxysporum f. sp cubense race 4 strain B2 (LT571433), carrying the haplotype as large variable region 1 [43].
Nuclear genomes
Nuclear genomes of both isolates were assembled into primary contigs with N50s of 4.5 Mbp (16117) and 4.2 Mbp (CNSD1), resembling the size of entire chromosomes in Fusarium oxysporum species (Table 1). Genome completeness analysis using BUSCO showed that both genomes encoded near-complete sets of conserved genes (Table 1). A total of 19 and 11 telomere regions containing the 5'-TAACCC-3' repeats were identified on the contigs corresponding to the 11 core chromosomes of CNSD1 and 16117, respectively (Figure 2, Figure S2-S3).
Both isolates appeared to have the equivalents for 11 of the 15 chromosomes in F. oxysporum f. sp. lycopersici (Fol) strain 4287, while lacking the equivalents of Fol chromosomes 3, 6, 14 and 15 (Figure 2A-B). The accessory sequences of these genomes did not align with any chromosomes in Fol strain 4287. When aligned to a near-complete genome of Foc TR4 isolate II-5 and Foc Race 1 isolate GD02 derived from a previous study [31], all 11 core chromosomes of Foc strains 16117 and CNSD1 aligned well to their counterparts (Figure 3C-D). The accessory sequence from CNSD1 also aligned well to its counterpart in II-5 (Figure 3C). In the race 1 comparison, these sequences appeared fragmented and variable in size (Figure 3D). This accessory sequence has been examined in terms of its structural variation and gene content in several studies [30,31].
Chromosome rearrangements
When aligned to the Foc TR4 II-5 genome, CNSD1 showed an inversion in contig ptg000011l that is otherwise absent in its alignment to Fol4287 (Figure 2A, C). In the Foc race 1 assembly 16117, a putative reciprocal translocation event was detected in contigs ptg000002l and ptg000004l, when it is aligned to the genomes of Fol4287 and Foc race 1 GD02 (Figure 2B, D). Telomeric repeats were also identified on the ends of all three contigs involved in these rearrangements. The assemblies will have to be validated by other means to confirm whether these rearrangements are genuine events or assembly errors.
Functional Annotations
A total of 15,943 and 15,247 protein-coding genes were annotated in 16117 and CNSD1, respectively (Table 1). GO classification for these two genomes includes an abundance of terms associated with carbohydrate, lipid, nitrogen metabolism, cell wall biogenesis, and enzymes and transport activities (Figure S4-5, additional files 3-4). This is also evident in the representative groups (carbohydrate, lipid and amino acid) within metabolism and cellular processes identified in the KEGG pathway classifications for each isolate (Figure S6-7, additional files 5-6). For both isolates, the three most abundant KOG categories are carbohydrate transport and metabolism, secondary metabolites biosynthesis, transport and catabolism, and amino acid transport and metabolism (Table S1-2, additional files 7-8).
CAZymes (Carbohydrate-active enzymes) are a diverse group of enzymes that play key roles in the breakdown, modification, and synthesis of carbohydrates. Some of these enzymes can be considered components of pathogen-secreted proteins involved in infection processes. Classification into the six CAZyme subfamilies [44] revealed that both isolates contained approximately 900 carbohydrate-active enzymes, with glycoside hydrolases being the most abundant, numbering over 300 (Table S3-4, additional files 9-10).
Phylogenetic analysis
A phylogenetic tree was constructed using a total of 154 genomes, including the two newly sequenced in this study. The analysis encompassed 1.49×106 nucleotide sites, with nearly 10% of the positions exhibiting variation across the alignment, highlighting substantial genetic diversity within the dataset. The F. oxysporum f. sp. cubense isolate CNSD1 clustered together with other Foc TR4 isolates in a TR4-specific phylogroup (Figure 3). This group exhibited little genetic variations among its members (Figure 3). In contrast, F. oxysporum f. sp. cubense isolate 16117 grouped with three other Foc isolates, two of which are classified as race 1. The positioning of this phylogroup was distinct from other clusters containing Foc race 1 strains, suggesting an independent evolutionary origin. This finding underscores the genomic diversity of Foc race 1 isolates, consistent with previous reports [45]. Overall, the phylogenetic analysis supports the polyphyletic nature of banana-infecting F. oxysporum strains. The presence of multiple, independently evolved lineages within F. oxysporum species complex suggests that these pathogens have evolved independently, potentially driven by horizontal gene transfer [46]. These findings reinforce the complex evolutionary dynamics underlying the genetic diversity of F. oxysporum strains infecting banana.
Effector annotation
Fungal effectors are small, secreted proteins that aid fungal pathogens to infect their hosts by suppressing host immune responses. These effectors are typically characterised by their small size, and a high cysteine content, which contributes to stability.
To define the secretomes of F. oxysporum f. sp. cubense isolates 16117 and CNSD1, proteins carrying signal peptides were independently identified using the prediction programs SignalP and TargetP. A total of 1,357 and 1,270 genes encoding secreted proteins were identified for isolates 16117 and CNSD1, respectively, after excluding those containing transmembrane domains (Supplemental Table 5) (Additional files 9-10).
Effector profiling with EffectorP predicted a total of 420 and 417 apoplastic effectors within the genomes of isolates 16117 and CNSD1, respectively (Additional files 11-12). Of these, 265 effectors in isolate 16117 and 256 in isolate CNSD1 were found to be secreted proteins containing signal peptides, as determined by comparison against each isolate's respective secretome. These numbers are consistent with effector predictions previously reported for other Fusarium oxysporum isolates and Fusarium species [29,47]. The identification of these effectors will provide valuable insights into the molecular mechanisms driving their pathogenicity in banana.
SIX gene profiles of Fusarium oxysporum f.sp. cubense genomes
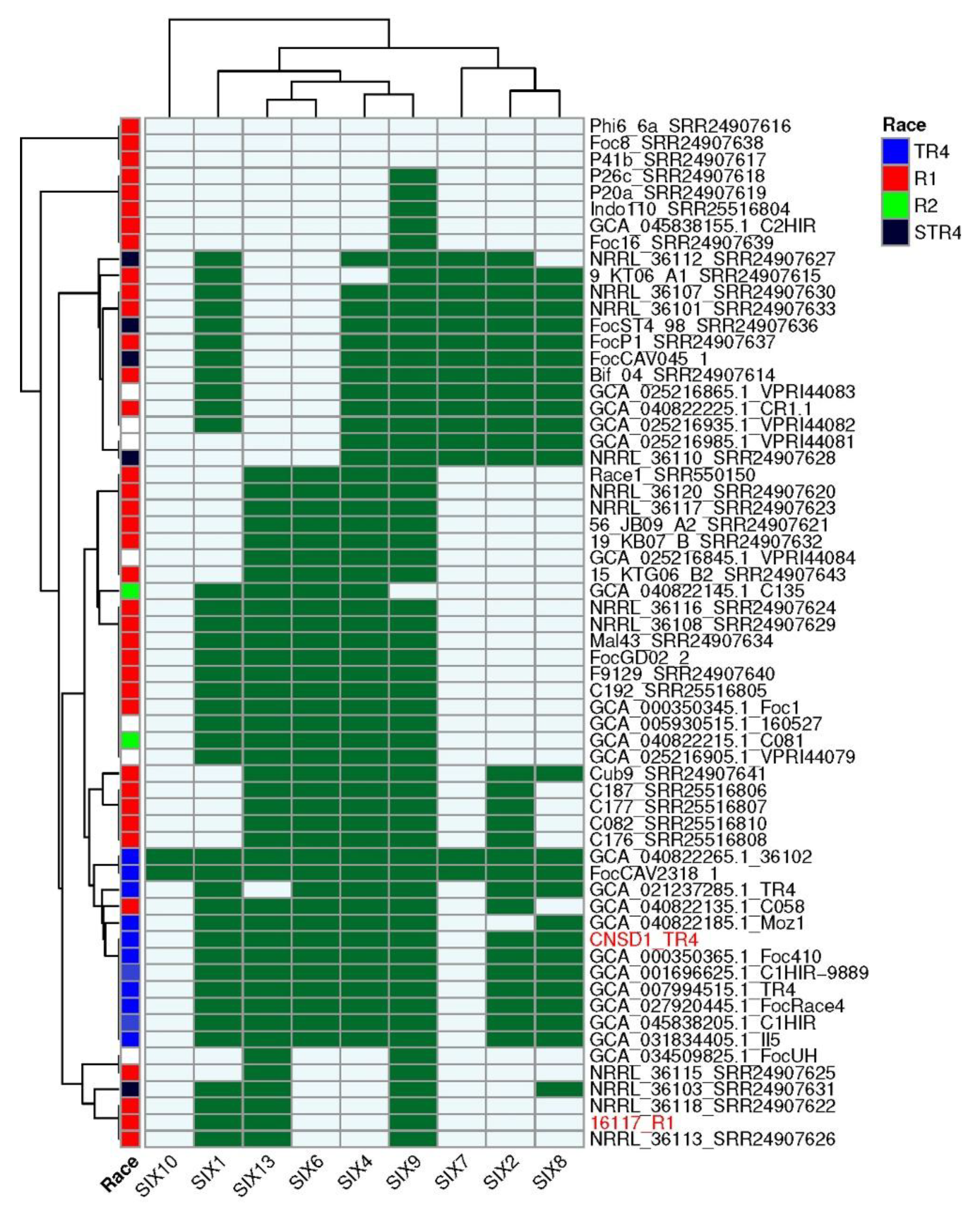
The genome of CNSD1 encoded homologs for SIX1, SIX2, SIX4, SIX6, SIX8, SIX9 and SIX13, whereas the 16117 genome appeared to carry homologs for only SIX1, SIX9 and SIX13 (Table S6). In CNSD1, multiple copies of SIX genes, including three copies of SIX1, two copies of SIX8, three copies of SIX9 and two copies of SIX13 were detected. Out of the 13 SIX gene homologs detected in CNSD1, ten are located on the contig ptg000003l. Isolate 16117, on the other hand, carried two copies of SIX1, three copies of SIX9 and two copies of SIX13 (Table S6).
Analysis of the presence and absence of SIX genes across all 62 Foc genomes revealed that CNSD1 clustered with the other known Foc TR4 genomes, consistent with both the number of SIX genes typically present in TR4 genomes and the position of this phylogroup in the phylogenetic tree derived from conserved genes (Figure 4). TR4 strains FocCAV2318_1 and 36102 have additional homologs corresponding to SIX7 and SIX10 genes that are otherwise absent in all other TR4 strains. Isolate C058 clustered within the TR4 phylogroup, based on its SIX gene profile, despite having been designated as a race 1 isolate.
The presence and absence of SIX genes in the race 1 strains confirmed their polyphyletic nature, with six phylogroups being apparent (Figure 4). Phylogroups that lacked any SIX genes (Phi6_6a, Foc8, and P41b), those possessing a single SIX gene (P26a, P20a, Indo110, C2HIR and Foc16), and those grouping with 16117, possessing two to four SIX genes (NRRL_36115, NRRL_36103, NRRL_36118, NRRL_36113) are incongruent with their positions in the phylogenetic tree derived from conserved genes (Figure 3 and Figure 4). However, the larger race 1 phylogroups that contained four to six SIX genes, and interspersed with either race 2 or STR4 strains, are largely congruent with the conserved gene phylogeny.
Figure 4.
SIX gene profiles of 62 Fusarium oxysporum f. sp. cubense genomes determined using the FoEC2 pipeline. Foc race designation for each isolate is provided where available [30,31]. Dark green and light blue boxes respectively indicate the presence and absence of a SIX gene homolog. SIX homologs that are absent in this collection (SIX3, SIX5, SIX11, SIX12 and SIX14) are not shown. Red highlight indicates the strains sequenced in this study.
Figure 4.
SIX gene profiles of 62 Fusarium oxysporum f. sp. cubense genomes determined using the FoEC2 pipeline. Foc race designation for each isolate is provided where available [30,31]. Dark green and light blue boxes respectively indicate the presence and absence of a SIX gene homolog. SIX homologs that are absent in this collection (SIX3, SIX5, SIX11, SIX12 and SIX14) are not shown. Red highlight indicates the strains sequenced in this study.
Conclusion
The fungal genome assemblies presented in this study are essential for managing Fusarium wilt in bananas. They provide valuable insights into the pathogenic mechanisms of Foc TR4 and race 1, their evolutionary origins, and banana-Fusarium interactions, all of which will aid in the development of resistant banana cultivars. Additionally, these resources support the improvement of disease diagnostics and the formulation of sustainable strategies to combat Fusarium wilt in banana.
Supplementary Materials
The following supporting information can be downloaded at Preprints.org.
Author Contributions
Jiaman Sun: Conceptualization, Data curation, Formal analysis, Methodology, Writing – original draft , Writing – review & editing. Jinzhong Zhang: Data curation, Formal analysis, Funding acquisition, Investigation, Writing – original draft , Writing – review & editing. Donald M. Gardiner: Data curation, Supervision, Formal analysis, Methodology, Investigation, Writing – review & editing. Peter van Dam: Data curation, Supervision, Formal analysis, Methodology, Investigation, Writing – review & editing. Gang Fu, Resources, Project administration, Writing – review & editing. Brett J. Ferguson: Supervision, Writing – review & editing. Elizabeth Aitken, Project administration, Supervision, Writing – review & editing. Andrew Chen, Conceptualization, Formal analysis, Methodology, Supervision, Writing – original draft , Writing – review & editing.
Funding
The research was supported by Guangxi Natural Science Foundation (2021GXNSFAA196014) and Funding of Jiaying University (2022RC97). The bioinformatic analyses were supported by Australian Research Council Research Hub for Sustainable Crop Protection (IH190100022) funded by the Australian Government. A.C. and E.A.B.A. were also supported by The Bill and Melinda Gates Foundation (Project Grant ID: OPP1093845) through its grant to the International Institute of Tropical Agriculture (IITA) under the project Accelerated Breeding of Better Bananas, grant number IITA 20600.15/0008-8—Phase II as well as Hort Innovation Australia through grant ‘BA21000’, using the banana research and development levy and contributions from the Australian Government. Hort Innovation is the grower-owned, not-for-profit research and development corporation for Australian horticulture.
Data Availability Statement
The original raw data presented in the study are openly available in NCBI Sequence Read Archive (SRA) under accession numbers SRR31177457 (16117) and SRR31177522 (CNSD1). The genome assemblies described in this study are available in GenBank under Bioproject accession numbers PRJNA1178358 for strain 16117 and PRJNA1174872 for CNSD1. The data analysis outputs presented in this study are included in the supplementary material/additional files.
Acknowledgments
We are grateful for Altus Viljoen of Stellenbosch University in the VCG identification of the Foc isolate CNSD1.
Declaration of Interest Statement
The authors declare no competing interests.
References
- Dita, M.; Barquero, M.; Heck, D.; Mizubuti, E.S.; Staver, C.P. Fusarium wilt of banana: current knowledge on epidemiology and research needs toward sustainable disease management. Front. Plant Sci. 2018, 9, 1468. [CrossRef]
- Ploetz, R.C. Fusarium wilt of banana. Phytopathology 2015, 105, 1512-1521. [CrossRef]
- Pegg, K.G.; Coates, L.M.; O’Neill, W.T.; Turner, D.W. The epidemiology of Fusarium wilt of banana. Front. Plant Sci. 2019, 10, 1395. [CrossRef]
- Islands, C.; Province, M. Vegetative compatibility among races of Fusarium oxysporum f. sp. cubense. Plant Dis. 1988, 72, 325-328.
- Fourie, G.; Steenkamp, E.T.; Ploetz, R.C.; Gordon, T.; Viljoen, A. Current status of the taxonomic position of Fusarium oxysporum formae specialis cubense within the Fusarium oxysporum complex. Infect. Genet. Evol. 2011, 11, 533-542. [CrossRef]
- Mostert, D.; Molina, A.B.; Daniells, J.; Fourie, G.; Hermanto, C.; Chao, C.-P.; Fabregar, E.; Sinohin, V.G.; Masdek, N.; Thangavelu, R. The distribution and host range of the banana Fusarium wilt fungus, Fusarium oxysporum f. sp. cubense, in Asia. PLoS One 2017, 12, e0181630. [CrossRef]
- Munhoz, T.; Vargas, J.; Teixeira, L.; Staver, C.; Dita, M. Fusarium Tropical Race 4 in Latin America and the Caribbean: status and global research advances towards disease management. Front. Plant Sci. 2024, 15. [CrossRef]
- van Westerhoven, A.C.; Meijer, H.J.; Seidl, M.F.; Kema, G.H. Uncontained spread of Fusarium wilt of banana threatens African food security. PLoS Pathog. 2022, 18, e1010769. [CrossRef]
- Sun, J.; Zhang, J.; Fang, H.; Peng, L.; Wei, S.; Li, C.; Zheng, S.; Lu, J. Comparative transcriptome analysis reveals resistance-related genes and pathways in Musa acuminata banana 'Guijiao 9' in response to Fusarium wilt. Plant Physiol.Biochem. 2019, 141, 83-94. [CrossRef]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 2021, 18, 170-175. [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072-1075. [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Zdobnov, E.M. BUSCO: Assessing genomic data quality and beyond. Curr. Protoc. 2021, 1, e323. [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Nat. Acad. Sci. 2020, 117, 9451-9457. [CrossRef]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878-2879. [CrossRef]
- Stanke, M.; Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465-W467. DOI:org/10.1093/nar/gki458.
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008, 18, 1979-1990. [CrossRef]
- Slater, G.S.C.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 2005, 6, 31. [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, 1-22. [CrossRef]
- Uliano-Silva, M.; Ferreira, J.G.R.N.; Krasheninnikova, K.; Blaxter, M.; Mieszkowska, N.; Hall, N.; Holland, P.; Durbin, R.; Richards, T.; Kersey, P.; et al. MitoHiFi: a python pipeline for mitochondrial genome assembly from PacBio high fidelity reads. BMC Bioinformatics 2023, 24, 288. [CrossRef]
- Community, T.G. The Galaxy platform for accessible, reproducible, and collaborative data analyses: 2024 update. Nucleic Acids Res. 2024, 52, W83-W94. [CrossRef]
- Brown, M.R.; Manuel Gonzalez de La Rosa, P.; Blaxter, M. tidk: a toolkit to rapidly identify telomeric repeats from genomic datasets. Bioinformatics 2025, 41. [CrossRef]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [CrossRef]
- Blake, J.D.; Cohen, F.E. Pairwise sequence alignment below the twilight zone. J. Mol. Biol. 2001, 307, 721-735. [CrossRef]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.-Y.; Dosztányi, Z.; El-Gebali, S.; Fraser, M.; et al. InterPro in 2017—beyond protein family and domain annotations. Nucleic Acids Res. 2016, 45, D190-D199. [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59-60. [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420-423. [CrossRef]
- Emanuelsson, O.; Brunak, S.; von Heijne, G.; Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007, 2, 953-971. [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001, 305, 567-580. [CrossRef]
- Sperschneider, J.; Dodds, P.N. EffectorP 3.0: prediction of apoplastic and cytoplasmic effectors in fungi and oomycetes. Mol. Plant Microbe Interact. 2022, 35, 146-156. [CrossRef]
- van Westerhoven, A.C.; Aguilera-Galvez, C.; Nakasato-Tagami, G.; Shi-Kunne, X.; Martinez de la Parte, E.; Chavarro-Carrero, E.; Meijer, H.J.G.; Feurtey, A.; Maryani, N.; Ordóñez, N.; et al. Segmental duplications drive the evolution of accessory regions in a major crop pathogen. New Phytol. 2024, 242, 610-625. [CrossRef]
- Zhang, Y.; Liu, S.; Mostert, D.; Yu, H.; Zhuo, M.; Li, G.; Zuo, C.; Haridas, S.; Webster, K.; Li, M.; et al. Virulence of banana wilt-causing fungal pathogen Fusarium oxysporum tropical race 4 is mediated by nitric oxide biosynthesis and accessory genes. Nat. Microbiol. 2024, 9, 2232-2243. [CrossRef]
- Kaliapan, K.; Mazlin, S.N.A.; Chua, K.O.; Rejab, N.A.; Mohd-Yusuf, Y. Secreted in Xylem (SIX) genes in Fusarium oxysporum f. sp. cubense (Foc) unravels the potential biomarkers for early detection of Fusarium wilt disease. Arch. Microbiol. 2024, 206, 271. [CrossRef]
- Gardiner, D.P.; Aitken, E.A.B.; Le, D.P.; Smith, L.J.; Chen, A. De novo long-read assembly and annotation for genomes of two cotton-associated Fusarium oxysporum isolates (Submitted). 2025.
- Keller, O.; Kollmar, M.; Stanke, M.; Waack, S. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 2011, 27, 757-763. [CrossRef]
- Stanke, M.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003, 19, 215-225.
- Shen, W.; Sipos, B.; Zhao, L. SeqKit2: A Swiss army knife for sequence and alignment processing. iMeta 2024, e191. [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.i.; Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059-3066. [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972-1973. [CrossRef]
- Edler, D.; Klein, J.; Antonelli, A.; Silvestro, D. raxmlGUI 2.0: a graphical interface and toolkit for phylogenetic analyses using RAxML. Methods Ecol. Evol. 2021, 12, 373-377. [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 2024. [CrossRef]
- Brenes Guallar, M.A.; Fokkens, L.; Rep, M.; Berke, L.; van Dam, P. Fusarium oxysporum effector clustering version 2: An updated pipeline to infer host range. Front. Plant Sci. 2022, 13. [CrossRef]
- van Dam, P.; Fokkens, L.; Schmidt, S.M.; Linmans, J.H.; Kistler, H.C.; Ma, L.J.; Rep, M. Effector profiles distinguish formae speciales of Fusarium oxysporum. Environ. Microbiol. 2016, 18, 4087-4102. [CrossRef]
- Brankovics, B.; van Dam, P.; Rep, M.; de Hoog, G.S.; J. van der Lee, T.A.; Waalwijk, C.; van Diepeningen, A.D. Mitochondrial genomes reveal recombination in the presumed asexual Fusarium oxysporum species complex. BMC Genomics 2017, 18, 735. [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233-238. [CrossRef]
- Ordonez, N.; Seidl, M.F.; Waalwijk, C.; Drenth, A.; Kilian, A.; Thomma, B.P.H.J.; Ploetz, R.C.; Kema, G.H.J. Worse comes to worst: bananas and Panama disease—when plant and pathogen clones meet. PLoS Pathog. 2015, 11, e1005197. [CrossRef]
- Czislowski, E.; Fraser-Smith, S.; Zander, M.; O'Neill, W.T.; Meldrum, R.A.; Tran-Nguyen, L.T.; Batley, J.; Aitken, E.A. Investigation of the diversity of effector genes in the banana pathogen, Fusarium oxysporum f. sp. cubense, reveals evidence of horizontal gene transfer. Mol. Plant pathol. 2018, 19, 1155-1171. [CrossRef]
- Gardiner, D.M.; McDonald, M.C.; Covarelli, L.; Solomon, P.S.; Rusu, A.G.; Marshall, M.; Kazan, K.; Chakraborty, S.; McDonald, B.A.; Manners, J.M. Comparative Pathogenomics reveals horizontally acquired novel virulence genes in fungi infecting cereal hosts. PLoS Pathog. 2012, 8, e1002952. [CrossRef]

Table 1.
Assembly statistics for Fusarium oxysporum f. sp. cubense isolate 16117 and CNSD1.
| Statistics | 16117 (Race 1) | CNSD1 (TR4) |
|---|---|---|
| Assembly | ||
| Total sequence data (Gbp) | 8.46 | 10.5 |
| Coverage (fold) | 170 | 223 |
| Assembly size (bp) | 51,695,064 | 49,684,144 |
| No. of contigs | 92 | 77 |
| Largest contig (bp) | 5,598,307 | 6,666,412 |
| N50 contig length (bp) | 4,227,447 | 4,512,489 |
| Contig L50 | 6 | 5 |
| Contig L90 | 14 | 11 |
| GC content (%) | 47.87 | 47.72 |
| BUSCO coverage (%) | 98.4 | 98.4 |
| Total no. of BUSCOs | 1706 | 1706 |
| No. of duplicated BUSCOs | 9 | 6 |
| No. of fragmented BUSCOs | 8 | 8 |
| No. of missing BUSCOs | 19 | 20 |
| Gene models | ||
| Total no. of genes | 15,943 | 15,247 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



