Submitted:
13 March 2025
Posted:
14 March 2025
You are already at the latest version
Abstract
The ubiquitin-proteasome system (UPS) is a critical cellular mechanism involved in protein turnover, influencing processes such as cell cycle regulation, gene expression, and stress responses. Dysregulation of the UPS contributes to the pathogenesis of chronic diseases, including neurodegeneration, cardiovascular disorders, and cancer. This review explores the potential of circulating phenolic metabolites — bioactive metabolites derived from dietary (poly)phenols — as modulators of UPS activity. Using comprehensive analysis, we summarize evidence linking phenolic metabolites, such as valerolactones, benzoic acids, urolithins, and hydroxycinnamic acids, to proteasome modulation through mechanisms like autophagy activation, inflammatory mediator reduction, and oxidative stress prevention. Despite the growing interest, significant gaps remain, including limited data on physiological concentrations, metabolite availability, and exposure conditions in cellular and in vivo models. Advancing the understanding of phenolic metabolites in UPS regulation could unlock new therapeutic strategies for managing chronic diseases, highlighting the need for further research integrating clinical and mechanistic insights.
Keywords:
cancer
; cardiovascular diseases
; neurodegenerative diseases
; polyphenols
; proteasome
; small molecules
1. Introduction
The emergence of chronic diseases as the predominant challenge to global health is undisputed. As reported by the World Health Organization (WHO), two-thirds of deaths worldwide are due to chronic diseases, which include cancer, cardiovascular disease, diabetes, and mental health disorders [1]. Chronic diseases are defined by their prolonged duration, often lasting at least one year, and their potential to progressively worsen over time [2]. These conditions arise from genetic alterations, environmental factors, behavioral patterns, infections, and other risk factors [3]. Lifestyle habits such as smoking, alcohol consumption, diet, and physical activity play a significant role in their development. Additionally, increased life expectancy has led to a larger aging population with multiple chronic conditions [4]. Consequently, individuals with chronic diseases place a substantial burden on healthcare systems, both financially and socially, making their management a critical global priority.
At the molecular level, the accumulation of aberrantly folded proteins, which results in increased proteotoxic stress, is considered a key pathological factor in chronic disorders [5]. Cellular health relies on the proper function of thousands of proteins involved in diverse biological processes. A typical mammalian cell expresses from 10,000 to 20,000 proteins, which are prone to become structurally destabilized or misfolded due to mutations, or a part of them can even lack ordered structure and generate toxic aggregates [6]. Maintaining proteome homeostasis — proteostasis — is therefore essential for cellular and organismal health [7]. Proteostasis is orchestrated by an interconnected network known as the proteostasis network (PN), which includes cellular machinery responsible for protein synthesis, folding, and degradation [8]. However, the capacity of the PN declines with age, increasing susceptibility to chronic diseases [9]. To counteract proteotoxic stress, two primary degradation systems, the ubiquitin–proteasome system (UPS) and the autophagy–lysosome system (ALS), have evolved [10].
Diet has garnered considerable attention as a preventive strategy for age-related cognitive decline and other chronic conditions. Numerous nutrients and food components have demonstrated the potential to enhance cognitive function and support brain health [11]. Meta-analyses and epidemiological studies suggest that diets rich in plant-based products reduce the risk of developing type 2 diabetes and cardiovascular diseases [12,13]. Additionally, dietary choices significantly influence carcinogenesis. According to the World Cancer Research Fund (WCRF), 30–50% of cancer cases are preventable through healthy diet and lifestyle practices [14]. The Mediterranean diet, characterized by high consumption of plant-based foods, cereals, legumes, and olive oil, alongside low intake of meat, fish, and dairy products, exemplifies this protective effect. This dietary pattern promotes increased intake of monounsaturated fatty acids (MUFAs), polyunsaturated fatty acids (PUFAs), and (poly)phenols [15]. In this regard, epidemiological studies have described the inverse association between a high intake of (poly)phenols and the incidence of various chronic diseases [16,17,18]. The biological activity of (poly)phenols extends beyond their antioxidant properties, encompassing pleiotropic effects that influence diverse cellular mechanisms and signaling pathways [19,20].
Given the pivotal role of the UPS in maintaining cellular homeostasis, proteasome dysfunction is implicated in a wide range of age-related diseases. As a result, the UPS has emerged as an attractive target for therapeutic intervention. Although many studies have highlighted the impact of (poly)phenols on proteasomal protein degradation pathways, this review aims to explore the role of the UPS in chronic conditions such as neurodegeneration, cardiovascular diseases and cancer — leading causes of mortality and morbidity worldwide — while focusing on phenolic metabolites detected in circulation. These circulating phenolic metabolites, derived from dietary sources could be promising in modulating UPS-related mechanisms, thus offering potential new avenues for these diseases.
2. Role of Ubiquitin-Proteasome System (UPS) in Chronic Diseases
The UPS is a cornerstone of cellular homeostasis, orchestrating the degradation of misfolded and damaged proteins. Discovered in the late 1970s to early 1980s, the UPS has since been recognized as a central component of the cellular protein-degradation machinery [21]. In eukaryotes, it is a multisubunit complex of ∼2.5 MDa (Figure 1), known as 26S proteasome, composed of a 20S core particle (CP) attached to one or both ends by a 19S regulatory particle (RP). The CP consists of two outer α-subunits (1-7) and two inner β-subunits (1-7) arranged in four stacked heteroheptameric (Tsakiri and Trougakos, 2015). Proteolytic activity is carried out by β1, β2, and β5 subunits, which possess the caspase-like, trypsin-like, and chymotrypsin-like activities, respectively. The RP is composed of the lid (Rpn3, Rpn5, Rpn6, Rpn7, Rpn8, Rpn9, Rpn11, Rpn12, Rpn15) and the base (ATPase subunits Rpt1-6, and Rpn1, Rpn2, Rpn10, Rpn13). The association of the RP to the CP is a tightly controlled process since RP controls essential steps in substrate recognition and ubiquitination [22]. Polyubiquitination, however, is not always a prerequisite for degradation by the ubiquitin-proteasome system. While the 26S proteasome primarily recognizes proteins tagged with polyubiquitin chains, certain monoubiquitinated or even non-ubiquitinated proteins can also be targeted for degradation through specific mechanisms. Examples include monoubiquitinated proteins such as p21 and HIF-1α, as well as non-ubiquitinated proteins like ornithine decarboxylase (ODC) and tau, particularly those with intrinsically disordered regions under certain conditions. Substrates are subsequently deubiquitinated by deubiquitinating enzymes (DUBs), which recycle ubiquitin to maintain cellular ubiquitin pools [22].
Figure 1.
– Protein degradation by the Ubiquitin-Proteasome Pathway. The ubiquitin-proteasome system is responsible for selective protein degradation in eukaryotic cells. In most cases, proteins targeted for degradation are first conjugated to multiple ubiquitin (Ub) molecules through a multi-enzymatic cascade involving the Ub-activating enzyme (E1), Ub-conjugating enzyme (E2), and Ub-ligase enzyme (E3). The polyubiquitinated protein is then recognized by the 26S proteasome, an ATP-dependent proteolytic complex consisting of a 20S catalytic core and two 19S regulatory caps. The 19S subunit facilitates substrate recognition, deubiquitination, unfolding, and translocation into the 20S core. Deubiquitinases (DUBs) cleave ubiquitin chains, allowing ubiquitin recycling. Inside 20S core, protein degradation is mediated by the three catalytically active β-subunits: β1 (caspase-like), β2 (trypsin-like), and β5 (chymotrypsin-like). These subunits cleave proteins into oligopeptides composed of 7–9 residues, and are the major targets for small-molecule proteasome inhibitors. The ubiquitin-proteasome system ensures efficient cellular proteostasis by tightly regulating protein turnover.
Figure 1.
– Protein degradation by the Ubiquitin-Proteasome Pathway. The ubiquitin-proteasome system is responsible for selective protein degradation in eukaryotic cells. In most cases, proteins targeted for degradation are first conjugated to multiple ubiquitin (Ub) molecules through a multi-enzymatic cascade involving the Ub-activating enzyme (E1), Ub-conjugating enzyme (E2), and Ub-ligase enzyme (E3). The polyubiquitinated protein is then recognized by the 26S proteasome, an ATP-dependent proteolytic complex consisting of a 20S catalytic core and two 19S regulatory caps. The 19S subunit facilitates substrate recognition, deubiquitination, unfolding, and translocation into the 20S core. Deubiquitinases (DUBs) cleave ubiquitin chains, allowing ubiquitin recycling. Inside 20S core, protein degradation is mediated by the three catalytically active β-subunits: β1 (caspase-like), β2 (trypsin-like), and β5 (chymotrypsin-like). These subunits cleave proteins into oligopeptides composed of 7–9 residues, and are the major targets for small-molecule proteasome inhibitors. The ubiquitin-proteasome system ensures efficient cellular proteostasis by tightly regulating protein turnover.

Proteasome dysfunction – caused by decreased subunit expression, mutations or modifications in proteasome subunits, or inactivation by protein aggregates – has been linked to a wide spectrum of diseases, including neurodegeneration, cardiovascular diseases and cancer [23]. Phenolic compounds may influence proteostasis by interacting with specific UPS components, thereby mitigating dysfunctions associated with chronic diseases. Altogether, this underscores the importance of integrating dietary strategies to target UPS-related pathways in disease prevention and management.
2.1. Involvement of UPS in Neurodegenerative Diseases (NDs)
The UPS is a key regulator of neuronal plasticity and long-term survival during physiological aging. However, its competence declines with age, contributing to the accumulation of misfolded proteins, neurotoxic oligomers, and their diffusion in the central nervous system (CNS) [24]. This dysfunction is linked to neurodegenerative diseases (NDs), including Parkinson’s disease (PD), Huntington’s disease (HD) and Alzheimer’s disease (AD). For instance, protein misfolding involving mutant α-synuclein in PD, mutant huntingtin in HD, and Aβ and hyperphosphorylated Tau in AD, leads to UPS dysregulation, proteotoxic stress, and neuronal death [25,26]. Neuronal death and dysfunction are associated with oxidative stress in NDs. The disturbed equilibrium between pro-oxidant and antioxidant molecules can generate an accumulation of reactive oxygen species (ROS) and free radicals, which are detrimental to neurons. It should be pointed out that UPS is closely associated with the endoplasmic reticulum (ER) and autophagy [27]. For instance, sustained ER stress contributes to the loss of ER integrity, hampering cellular proteasomal activity, and ultimately leading to the induction of neuronal apoptosis [28]. In this regard, the interaction between oxidative stress, ER stress, and UPS dysfunction seems to be crucial in embellishing the development of potential therapeutics against NDs. UPS has been described as one of the main pathways to eliminate these neurotoxic proteins in different brain cells [29].
The potential relationship between UPS dysfunction and cell apoptosis during 1-methyl-4-phenylpyridinium (MPP+) insult has been described in a PD in vitro model, confirming that the degradation of protein by UPS plays a critical role in the MPP+-induced apoptosis [30]. Furthermore, in a variety of experimental animal models, UPS dysfunction has been associated with PD. For instance, mutations in the E3 ubiquitin ligase parkin are associated with familial forms of the disease, illustrating the critical role of UPS in maintaining neuronal health. The loss of parkin function impairs the clearance of damaged mitochondria via mitophagy, leading to neuronal dysfunction and dopaminergic cell death [31]. Several genes, such as Parkin, UCH-L1, and FBXO7, which are involved in the UPS degradation pathway, are associated with the onset of NDs [32,33,34]. Additionally, proteasomal degradation of α-synuclein aggregates is impaired in sporadic PD, promoting the formation of toxic oligomers and Lewy bodies. A study showed that MG132 treatment reduced α-synuclein clearance in PD models, highlighting the importance of UPS in aggregate removal [35].
In HD, expanded polyglutamine (polyQ) tracts in the huntingtin protein overwhelm the UPS, leading to proteasomal inhibition and neuronal death. Studies in transgenic mouse models of HD demonstrated that enhancing UPS function improved motor performance and reduced huntingtin aggregates [36]. However, chronic proteasome activation may disrupt the degradation of essential neuronal proteins, emphasizing the need for precise therapeutic modulation.
Regarding AD, it has been demonstrated that proteasome inhibition exacerbates β-amyloid aggregation in cultured neurons while enhancing proteasome activity reduces its accumulation [37]. Extensive evidence supports a close association between AD and type 2 diabetes mellitus (T2DM). Aβ and Islet Amyloid Polypeptide (IAPP) – a highly amyloidogenic hormone secreted by pancreatic β-cells in response to glucose – were shown to co-deposit in the brain, forming toxic heterocomplexes that contribute to AD onset and progression. Molecular interactions between Tau and IAPP were also documented. Importantly, ER stress and impairment of proteasome function have been associated with human IAPP-induced toxicity. In vitro binding and activity assays showed an intrinsic and strong ability of IAPP to interact with the 20S core complex thereby modulating its proteolytic activity. IAPP-mediated proteasomal activity decline was described to reduce clearance of misfolded proteins, accumulation of ubiquitinated proteins and exacerbation of proteotoxic stress. This impairment has been linked to reduced expression of key proteasomal regulators, including cytosolic chaperone Hsp90 and the deubiquitination enzyme UCH-L1. Conversely, the proteasome appears to play a role in IAPP degradation, as its inhibition increases IAPP accumulation and toxicity, while its activation mitigates these effects. Such findings highlight a vicious loop, where IAPP aggregation promotes proteasomal dysfunction, which in turn exacerbates the accumulation of toxic IAPP species and other misfolded proteins, contributing to cellular damage and degeneration. This cycle may be particularly detrimental in β-cells and neurons, potentially worsening conditions like type 2 diabetes and neurodegenerative diseases [38].
Therapeutic strategies targeting the UPS in NDs focus on enhancing its proteolytic capacity or preventing the degradation of neuroprotective proteins. For example, small molecules like IU1, which inhibit specific deubiquitinases, have been shown to increase proteasomal degradation of toxic aggregates while sparing functional proteins [39]. These findings highlight the potential of UPS modulation to restore proteostasis in neurodegenerative disorders.
2.2. Involvement of UPS in Cardiovascular Diseases (CVDs)
A general picture of the UPS as an isolated system no longer adequately describes its wide array of biological functions in the cardiovascular system. During the development of the vascular system, UPS is known to modify many transcription factors, receptors, and structural proteins involved in common embryogenesis-related signaling pathways, such as Notch, VEGF, and HIF-1α [40,41]. In the heart, the UPS is described to degrade stress-sensitive transcriptional regulators, including NF-κB and inducible cAMP early repressor [42,43]. Furthermore, its role in the turnover of contractile proteins and sarcomeric components has been extensively documented [44,45].
The UPS also regulates the functionality of voltage-gated ion channels, including hERG1, a channel associated with familial long QT syndromes [46]. Highlighting its significance in blood pressure regulation, clinical studies have identified potential cardiotoxic effects, such as hypertension, in individuals undergoing proteasome inhibitor therapy for multiple myeloma [47]. Additionally, the UPS attenuates MAPK activation and directly influences apoptosis through the degradation of caspases and tumor suppressor proteins [42,48]. For instance, low-density lipoprotein (LDL) aggregates induce ubiquitin-mediated degradation of p53, thereby suppressing macrophage apoptosis and promoting foam cell formation, a hallmark of atherosclerosis. Proteasome inhibition reverses this antiapoptotic effect by increasing p53 stability [49]. Contrarily, in vascular cells, proteasome inhibition exacerbates oxidized LDL (oxLDL)-induced toxicity, suggesting a pro-apoptotic role for the UPS under these conditions [50]. Similarly, oxLDL has been shown to downregulate the insulin-like growth factor-1 receptor (IGF-1R) in smooth muscle cells, leading to apoptosis through Nedd4-mediated ubiquitination, albeit independently of the proteasome pathway [51,52]. Such conflicting findings imply that the role of UPS in cellular apoptosis may vary depending on the apoptosis mechanism and cell type involved.
Beyond apoptosis, the UPS modulates inflammatory and vascular cell functions. Macrophages, critical players in innate immunity and inflammation, rely on proteasome activity for their functionality. Proteasome inhibitors reduce endotoxin-induced gene expression, including Toll-like receptor 2, and protect mice from LPS-induced shock [53]. The involvement of UPS in the regulation of soluble inflammatory mediators has also been described [54]. In carotid artery plaques, protein ubiquitination and 20S proteasome activity were associated with the presence of the inflammatory cytokine TNF-α [55]. In endothelial and smooth muscle cells, TNF-α stimulated the expression of the de-ubiquitinating enzyme cylindromatosis (CYLD). Overexpression of CYLD inhibited cell viability and neointima formation in a rat model of carotid artery injury [56].
A hallmark of endothelial dysfunction is the imbalance between vasoconstrictors (e.g., endothelin-1, ET-1) and vasodilators (e.g., nitric oxide, NO). The UPS has been implicated in endothelial dysfunction by regulating the endothelin system [57]. In hypertensive rats, proteasome inhibitors reduced aortic ET-1 levels and lowered systolic blood pressure [58]. Similarly, low doses of MG132 decreased ET-1 expression in human umbilical vein endothelial cells (HUVECs) [59]. Proteasome inhibition also enhanced eNOS expression and NO production, leading to improved endothelial-dependent vasorelaxation in rat aortic rings. However, the effects were dose-dependent, with high inhibitor concentrations downregulating eNOS, while low doses upregulated its activity [59,60]. This dose-dependency, along with other variables such as treatment duration, species, and cell type, underscores the complexity of UPS regulation in endothelial function.
The UPS also [59] a pivotal role in oxidative stress in vascular cells. In HUVECs, non-toxic MG132 concentrations suppressed ROS generation by downregulating NADPH oxidase subunits and enhancing antioxidant defenses, such as SOD-1 [59] On the other hand, in a study using female pigs under a high-cholesterol diet, chronic proteasome inhibition treatment induced increased expression of eNOS and was associated with higher levels of oxidative stress parameters [61,62].
These findings suggest that prolonged proteasome inhibition in conditions of elevated ROS may impair compensatory mechanisms, aggravating oxidative stress and endothelial dysfunction.
While the UPS has protective roles under certain conditions, its inhibition can exacerbate pathological processes, particularly during sustained oxidative stress. These dual effects emphasize the need for precise therapeutic modulation of the UPS to address cardiovascular diseases effectively.
The cardiovascular system is intimately linked to diabetes, with systemic effects that extend beyond glycemic control. Recent evidence has highlighted the pivotal role of IAPP aggregation in this interplay, suggesting its contribution to both cardiac and metabolic dysfunction [63]. Upon its production in pancreas, IAPP enters the bloodstream and may provokes diabetes-related complications in extra-pancreatic tissues (e.g., brain, kidney and heart). Elevated levels of circulating IAPP have been linked to amyloid deposition in the heart of diabetic and obese patients [64]. At cardiac myocytes, IAPP aggregation triggers inflammation and Ca2+ dysregulation. In contrast, mitigating myocardial IAPP deposition improves heart function in diabetic human IAPP transgenic rats [65]. In human pancreatic islets, treatment with human IAPP induced ER stress and reduced proteasome activity with accumulation of ubiquitinated proteins. While proteasome-mediated degradation is essential for clearing misfolded proteins, the specific role of proteasomal activity towards IAPP aggregation in the heart remains poorly explored. Therefore, further research into the proteasomal activity in cardiac tissues is critical to uncovering potential therapeutic targets for addressing IAPP-related cardiovascular dysfunction in diabetes.
2.3. Involvement of UPS in Cancer
The heightened activity of the UPS in cancer cells is closely linked to their rapid proliferation and metabolic demands. This system supports tumor progression by maintaining a delicate balance between the synthesis and degradation of proteins essential for cellular growth and survival. Specifically, the UPS regulates the degradation of tumor suppressor proteins, such as p53 and members of the pro-apoptotic Bcl-2 family, while stabilizing oncogenic proteins [66]. Dysregulation of this process contributes to hallmark features of cancer, including evasion of apoptosis, sustained angiogenesis, and unchecked proliferation.
One critical pathway influenced by the UPS involves the transcription factor NF-κB, a key mediator of inflammation and tumor progression. Under normal conditions, NF-κB is sequestered in the cytoplasm by its inhibitor, IκB. Proteasomal degradation of IκB allows NF-κB to translocate into the nucleus, where it activates genes promoting angiogenesis, cell migration, and anti-apoptotic signaling [67,68]. Since E3s and DUBs are involved in the regulation of fundamental cellular outcomes, the deregulation of the signaling pathways that underpin these functions plays a role in conferring a survival advantage to cancer cells, thus linking E3s and DUBs to tumorigenesis. On the other hand, interfering with the activation of ubiquitin ligases, such as the E3 ligase Anaphase Promoting Complex/Cyclosome [69] can induce mitotic catastrophe in cancer cells [70,71].
Proteasome inhibitors, such as MG132 and bortezomib, block this degradation, effectively reducing NF-κB activity. For instance, in clinical trials bortezomib has demonstrated efficacy in reducing cytokine production, alleviating inflammation, and restoring p53 stability by preventing its ubiquitination and degradation, inducing apoptosis in multiple myeloma cells [72]. Carfilzomib, another inhibitor of the 26S Proteasome and Ixazomib, a reversible inhibitor of the 20S Proteasome (β5) have been approved by the FDA for the treatment of lymphomas and multiple myeloma, respectively [73].
In the tumor microenvironment, macrophages rely on UPS activity to maintain cellular homeostasis during inflammatory responses. Dysregulated UPS activity in macrophages has been associated with excessive inflammation, leading to chronic conditions such as rheumatoid arthritis and contributing to the tumor-promoting inflammatory milieu [74]. Proteasome inhibition in macrophages has been shown to impair the clearance of oxidized proteins, resulting in intracellular aggregation and functional deficits [75].
The UPS also regulates the stability of hypoxia-inducible factors (HIFs), particularly HIF-1α, which plays a pivotal role in tumor adaptation to hypoxic conditions. By modulating HIF-1α degradation, the UPS indirectly promotes vascular endothelial growth factor (VEGF) expression and tumor angiogenesis. Dysregulation of UPS activity enhances HIF-1α stability, driving angiogenesis and supporting tumor survival in oxygen-deprived environments [76].
Proteasome inhibitors have emerged as a therapeutic strategy in cancer, with notable success in hematological malignancies. However, the therapeutic efficacy of proteasome inhibitors in solid tumors is less consistent and influenced by factors such as tumor vascularization and proteasome subunit composition. For instance, carfilzomib has shown limited efficacy in hypovascularized solid tumors compared to bortezomib, highlighting the need for tumor-specific approaches [77].
Cancer often manifests as an aging-related disease, involving molecular alterations at cellular, tissue, and systemic levels. UPS dysregulation contributes to age-related oncogenesis by affecting key processes such as oxidative stress, chronic inflammation, and post-translational modifications (e.g., ubiquitination, phosphorylation, acetylation, and SUMOylation) [78,79]. These modifications influence cellular signaling pathways, DNA repair mechanisms, and protein quality control, collectively driving cancer progression.
Despite its therapeutic promise, the clinical application of proteasome inhibitors remains challenging. Emerging evidence suggests that their efficacy depends on multiple variables, including tumor type, microenvironment, and treatment regimen. Understanding these context-dependent effects is critical to optimizing UPS-targeted therapies and overcoming resistance mechanisms in cancer.
3. (Poly)phenols as a Potential Strategy for Chronic Diseases Through UPS Modulation
The UPS is a critical regulator of protein homeostasis and is involved in the degradation of approximately 80% of intracellular proteins, including misfolded, damaged, or short-lived regulatory proteins. Given its central role in maintaining cellular function, dysregulation of the UPS has been implicated in numerous chronic diseases, including NDs, CVDs, and cancer. Dietary (poly)phenols, widely recognized for their pleiotropic health benefits, have emerged as promising candidates for modulating the UPS and addressing related pathologies.
3.1. UPS Modulators
The UPS governs essential cellular processes such as cell cycle regulation, gene expression, stress responses, metabolism, protein quality control, and inflammation [5,62]. UPS-modulating drugs can either increase or inhibit proteasome activity. Enhanced proteasome activity can promote the degradation of pathological proteins and, therefore, can be an effective therapeutic agent for NDs and CVDs. On the other hand, compounds that decrease proteasome activity have also a significant therapeutic value and are a promising anticancer strategy given that cancer cells often require high levels of proteasome activity [80].
The dual-edged nature of UPS modulation requires careful balancing to avoid disrupting protein homeostasis, which could lead to unintended cytotoxicity. For example, bortezomib, the first FDA-approved proteasome inhibitor (PI) for multiple myeloma, exemplifies the potential and limitations of UPS-targeting drugs. While effective in hematological malignancies, resistance to bortezomib and its adverse effects, such as peripheral neuropathy and cardiac toxicity, highlight the need for alternative approaches [81,82].
Emerging technologies, such as PROteolysis TArgeting Chimeras (PROTACs), represent a novel UPS-modulating strategy. PROTACs utilize bifunctional molecules to recruit specific proteins to E3 ubiquitin ligases for targeted degradation. Although promising in treating cancer and immune disorders, challenges remain, including optimizing pharmacological properties and expanding the repertoire of E3 ligases [83,84].
Despite the remarkable proteasome-targeting therapies to date, the potential for improvement and for the development of new and optimal UPS-modulating drugs remains challenging. Dietary (poly)phenols offer an attractive and safer alternative for UPS modulation. These plant-derived compounds exhibit the potential to directly or indirectly influence UPS activity, with additional benefits such as reducing oxidative stress and modulating inflammation [85,86].
3.2. The Role of (Poly)phenols in Modulating the UPS
Proteasome inhibitors (PIs) have significantly advanced cancer treatment, especially for blood cancers. However, issues such as treatment resistance, off-target effects, and limited effectiveness in solid tumors persist. Therefore, alternative strategies are needed to modulate the UPS safely.
The capability of many (poly)phenols to inhibit the UPS has been extensively reviewed [85]. Resveratrol, curcumin, and flavonoids are the most studied compounds in different preclinical models.
In cancer, for example, UPS activity is often upregulated to support the high turnover of proteins required for tumor growth and survival. This includes the degradation of tumor suppressor proteins, such as p53, and the stabilization of oncoproteins. Certain (poly)phenols, such as quercetin, apigenin, and myricetin, have been shown to inhibit proteasome activity in cancer cells, leading to the accumulation of pro-apoptotic proteins and suppression of tumor growth. For instance, quercetin has demonstrated pro-apoptotic effects by impairing the proteasomal degradation of Bax, a pro-apoptotic member of the Bcl-2 family, thus sensitizing cancer cells to programmed cell death [87]. (Poly)phenols from green tea such as epigallocatechin gallate (EGCG) have shown a interaction with UPS, decreasing the expression of 19S and 20S proteasomal proteins, as well as ubiquitin ligases MuRF1 and MAFbx, reducing proteasome chymotrypsin-like activity in animals [88].
(Poly)phenols have also been reported to exert multiple protective effects against CVDs, partly through their interaction with the UPS. Resveratrol has been shown to attenuate cardiac hypertrophy and dysfunction in mice, an effect attributed to its ability to decrease proteasome caspase-like, trypsin-like and chymotrypsin-like activities [89]. Similarly, quercetin was shown to inhibit 26S proteasome function in a cardiomyocytes culture [90], while EGCG modulated the proteasome chymotrypsin-like activity in both in vitro and in vivo models [91]. Hydroxytyrosol, a (poly)phenol present in olive oil, has shown effects on oxidative stress and wound repair of vascular endothelial cells, increasing the Nrf2 protein stability by preventing its degradation via UPS [92]. Beyond cardiovascular protection, (poly)phenols have also been linked to metabolic regulation (Gu et al., 2024). Epidemiological evidence showed an inverse correlation between the high intake of (poly)phenols and diabetes progression in elderly individuals at high risk of CVD [93]. By targeting different components of the UPS, polyphenolic compounds have been described to mitigate dysregulated metabolic states. For instance, EGCG and curcumin were recently described to modulate E3 ligase and 26S proteasome, thus regulating lipid metabolism, insulin resistance and exerting antioxidative and anti-inflammatory effects. Resveratrol, in turn, increased the ubiquitin-proteasome-dependent degradation of PPARy protein in adipocytes, leading to reduced lipid synthesis and improved insulin sensitivity. Additionally, compounds such as resveratrol and the ellagitannin pentagalloyl-glucose (PGG) have also been described to prevent the aggregation of IAPP, and to reduce its proteotoxic effects in in vitro models.
Another mechanism by which (poly)phenols influence the UPS is through their impact on oxidative stress, a key regulator of proteasome activity. Oxidative damage to proteins can impair UPS function, creating a vicious cycle of proteotoxic stress. (Poly)phenols, with their potent antioxidant properties, can mitigate oxidative damage and restore proteasome functionality. For example, EGCG has been reported to reduce oxidative stress markers and upregulate proteasome activity in cellular models of neurodegeneration [94]. In NDs, proteasome activity is often compromised, leading to the accumulation of toxic protein aggregates that exacerbate disease progression.
Beyond direct proteasome interactions, (poly)phenols also modulate UPS-related pathways. For instance, resveratrol has been shown to activate sirtuins and AMP-activated protein kinase (AMPK), both of which regulate cellular energy homeostasis and stress responses. These pathways intersect with the UPS, influencing protein degradation and cellular adaptation to stress. Recently, the anticancer activity of certain dietary (poly)phenols (e.g., apigenin, curcumin, EGCG, kaempferol, quercetin, and resveratrol) has been reviewed [85]. These compounds regulate key signaling pathways involved in cancer development, progression, and metastasis, including Akt, MAPK, mTOR, PI3K, RAS, and those influencing p53 tumor suppressor gene expression [78]. Certain (poly)phenols have been implicated in the alteration of protein turnover such as oligonol, a (poly)phenol derived from lychee (Litchi chinensis Sonn.) fruits [95]. This (poly)phenol seems to stimulate the expression of genes encoding protein components of the UPS as well as the muscle ring-finger protein-1 and Forkhead box class O family member proteins (FoxOs) of transcription factors [95].
Altogether, it makes (poly)phenols attractive candidates for multifaceted therapeutic strategies targeting both the UPS and other cellular systems. The therapeutic potential of (poly)phenols in UPS modulation is supported by preclinical and clinical studies. For example, clinical trials have demonstrated that dietary (poly)phenols, such as curcumin and genistein, can improve markers of oxidative stress and inflammation in individuals with chronic diseases, conditions where UPS dysregulation plays a critical role [96,97]. However, the precise mechanisms through which these compounds influence the UPS remain an active area of research.
However, it is important to note that while most studies focus on the parent (poly)phenols, their bioavailability and bioactivity in in vivo settings largely depend on their metabolic transformations. In in vivo models or human interventions, the observed effects may not be directly attributable to the native compounds but rather to their circulating metabolites. In contrast, in vitro studies primarily assess the direct effects of the parent compounds, requiring controlled delivery to achieve biologically relevant concentrations.
In conclusion, dietary (poly)phenols represent a promising, multifaceted strategy for regulating UPS activity in chronic diseases. Nonetheless, the precise mechanisms underlying their UPS modulation remain an active area of research, particularly concerning their bioavailability and metabolic transformation in vivo. Importantly, distinguishing between the effects of parent (poly)phenols and their metabolites is essential for accurately assessing their therapeutic potential. Future studies should also explore the synergy between (poly)phenols and pharmacological proteasome inhibitors to enhance therapeutic efficacy while mitigating adverse effects associated with high-dose treatments.
3.3. Metabolism and Bioavailability of Dietary (Poly)phenols
After ingestion, only a small fraction of (poly)phenols is absorbed in the small intestine, where they undergo rapid phase II metabolism to form glucuronidated, sulfated, or methylated conjugates. The majority (90-95%) of dietary (poly)phenols, however, reach the colon largely unabsorbed, where they are further metabolized by the gut microbiota into smaller phenolic compounds [98,99]. These smaller compounds or phenolic metabolites, such as urolithins derived from ellagitannins or dihydroxyphelacetic acids from flavonoids, are more easily absorbed and exhibit distinct biological activities compared to their parent compounds [98,99]. Factors such as the food matrix, interactions with other dietary components, and interindividual variability in the gut microbiota composition significantly influence the absorption and metabolism of (poly)phenols [100,101,102].
The biotransformation of dietary (poly)phenols into active metabolites is particularly relevant in the context of UPS modulation. While the parent compounds may have limited systemic availability, their circulating metabolites could change the interaction with cellular pathways, including those regulated by the UPS.
Focusing on phenolic metabolites rather than parent compounds is crucial for developing targeted therapies, as these are the bioactive forms that reach tissues at physiologically relevant concentrations. Understanding their interaction with the UPS could pave the way for novel strategies to prevent or treat chronic diseases linked to UPS dysfunction.
3.4. Phenolic Metabolites as Potential UPS Modulators in Chronic Diseases
Growing evidence identifies phenolic metabolites as key contributors to the health benefits of (poly)phenol consumption [103]. These metabolites originate from diverse pathways, including direct absorption from dietary sources, microbial catabolism, and endogenous metabolism [104].
We carried out a literature survey applying the following criteria to identify the (poly)phenol metabolites with potential activity in modulating the UPS: i) phenolic metabolites among 108 identified compounds in human circulation [99], including phase II conjugates derived from acyl-quinic acids, ellagitannins, and major dietary flavonoids such as anthocyanins, flavones, flavonols, flavanones, and flavan-3-ols; and ii) studies explicitly linking these metabolites to the proteasome pathway. Articles focusing on inflammation without a direct connection to the proteasome were excluded. This review offers an unprecedented synthesis of studies investigating phenolic metabolites detected in humans within the framework of UPS modulation, with a summary of the current evidence provided in Table 1.
An elegant study by Cecarini et al. evaluated the effects in the proteasome of flavan-3-ols-derived metabolites valerolactones in silico, as well as their activity in a cell-free system and their activity in neuroblastoma cells [105]. This study revealed that 5-(4ʹ-hydroxyphenyl)-γ-valerolactone, 5-(3ʹ,4ʹ-dihydroxyphenyl)-γ-valerolactone and 5-(3ʹ-dihydroxyphenyl)-γ-valerolactone-4ʹ-sulfate have moderate binding affinity for proteasome catalytic subunits. At 10 µM, these compounds inhibited the β2 subunit (5-(4′-hydroxyphenyl)-γ-valerolactone and 5-(3′-dihydroxyphenyl)-γ-valerolactone-4′-sulfate) and β5 subunit (5-(3′,4′-dihydroxyphenyl)-γ-valerolactone) of the proteasome (Table 1). These metabolites also promoted autophagy activation and reduced amyloid formation, demonstrating their neuroprotective effects (Figure 2A).
Flavonoid-derived metabolites, such as 4-hydroxy-3,5-dimethoxycinnamic acid (sinapic acid), 4-hydroxy-3-methoxycinnamic acid (ferulic acid), and 4-hydroxy-3-methoxybenzoic acid (vanillic acid), exhibited diverse effects on proteasome activity, although these effects were observed at high doses, starting the assays from 10 µM. Sinapic and ferulic acid showed no direct effect on chymotrypsin-like (ChT-L) activity of the proteasome but reduced the production of nitric oxide (NO), an important regulator of inflammatory events [106]. Vanillic acid inhibited proteasome activity through ChT-L suppression in macrophages (Figure 2B), demonstrating a more direct interaction with UPS components [106]. Additionally, 2,4,5-trimethoxybenzoic acid (asaronic acid) enhanced UPS-mediated degradation of misfolded proteins in murine macrophages, contributing to ER stress alleviation [107].
Hippuric acid, a metabolite derived both endogenously and from dietary (poly)phenols, enhanced E3 ubiquitin ligase activity, NRF2 ubiquitination, and 26S proteasome degradation in HK-2 renal cells (Table 1) [108], disrupting redox homeostasis (Figure 2C). Vanillin, one of the most extensively studied phenolic metabolites, demonstrated anticancer effects through Akt ubiquitination and proteasomal degradation (Figure 2D) in lung cancer cells [109]. Additionally, in colorectal cancer models (animals and in vitro model), vanillin suppressed proteasome activity and downregulated proteasome-related gene expression, contributing to tumor suppression [110]. Protocatechuic acid (3,4-dihydroxybenzoic acid) exhibited anticancer activity by inhibiting 26S proteasome activity in vivo (Figure 2D), affecting key events in the initiation and promotion stages of carcinogenesis [111]. However, it is important to note that this effect was observed following topical application, suggesting a pharmacological action rather than a nutritional approach.
Other metabolites, such as syringic acid, demonstrated strong binding affinity to the 20S proteasome in yeast models [112]. Caffeic acid displayed neuroprotective effects via proteasome inhibition in primary neurons, while ferulic acid offered neuroprotection independently of proteasome modulation [113].
Among benzoic acid derivatives, 4-hydroxybenzoic acid and 4-methoxybenzoic acid exhibited strong binding affinity to procathepsin B and L, enhancing cathepsin activity and promoting UPS function in human fibroblasts [114]. DOPAC (3,4-dihydroxyphenylacetic acid) inhibited proteasome activity in a cell-free model [115], though at a supraphysiological concentration of 0.19 mM (Table 1), whereas the maximum concentration detected in the bloodstream is 0.0005 mM [99]. Gallic acid (3,4,5-trihydroxybenzoic acid) induced the accumulation of ubiquitinated proteins and reduced chymotrypsin-like proteasome activity in endothelial cells (Figure 2E), highlighting its preventive potential [116]. However, it is important to note that the doses used by these authors are supraphysiological.
Urolithins, metabolites of ellagitannins, have been studied in muscle cells. Urolithin A, at 15 µM, prevented UPS activation in TNF-α-stimulated cells [117]. Additionally, a clinical trial demonstrated its ability to improve muscle performance on overweight individuals by increasing the levels of ubiquitin-conjugating enzymes and proteasomal components necessary for mitochondrial quality control, following an oral daily dose for 4 months [118]. This phenolic metabolite has been detected in skeletal muscle following oral consumption in humans, suggesting its potential biodistribution in this tissue [119] Urolithin B, meanwhile, repressed UPS activity in murine cells, promoting myotube growth and differentiation and reducing protein degradation (Figure 2F) [117].
The findings summarized in Table 1 underscore the potential of phenolic metabolites as versatile UPS modulators. While much of the evidence stems from in vitro and in silico studies, these metabolites present promising therapeutic avenues for chronic diseases linked to proteasome dysfunction. Expanding research to include comprehensive in vivo models and human clinical trials will be pivotal in advancing our understanding of their mechanisms and therapeutic applications.

Table 1.
Circulating phenolic metabolites and their potential impact on the UPS system.
| Model |
Dose/ Duration |
Mechanism of action | Main outcomes | Reference |
| Valerolactone derivatives | ||||
| 5-(4ʹ-hydroxyphenyl)-γ-valerolactone | ||||
| In silico | - | Binding with β1, β2, and β5 subunits of human constitutive 20S proteasome (pdb ID: 6rgq). Binding with β1i, β2i, and β5i subunits of human immunoproteasome (pdb ID: 6e5b). |
Moderate binding affinity for proteasome catalytic subunits. | [105] |
| Cell-free | 0 ̶ 10 µM for 90 min | Inhibits catalytic subunits of proteasome at 10 µM: - Chymotrypsin-like (ChT-L) and branched chain amino acids preferring (BrAAP) (associated with the β5 subunit). - Trypsin-like (T-L) (β2 subunit). - Peptidyl glutamyl-peptide hydrolyzing (PGPH) (β1 subunit). |
Proteasome inhibition promoting autophagy activation. | [105] |
| Human neuroblastoma SH-SY5Y cells | 1 ̶ 5µM for 24h | Strongly affects the functionality of enzymatic complex, mainly catalytic subunit ChT-L of both 20S and 26S proteasome. Increases the levels of Ub-conjugates and p27. |
Affect the functionality of proteosomal enzymatic complex. Decrease in amyloid formation. |
[105] |
| 5-(3ʹ,4ʹ-dihydroxyphenyl)-γ-valerolactone | ||||
| In silico | - | Binding with β1, β2, and β5 subunits of human constitutive 20S proteasome (pdb ID: 6rgq) Binding with β1i, β2i, and β5i subunits of human immunoproteasome (pdb ID: 6e5b). |
Moderate binding affinity for proteasome catalytic subunits. | [105] |
| Cell-free | 0 ̶ 10 µM for 90 min | Inhibits catalytic subunits of proteasome at 10 µM: - ChT-L and BrAAP (associated with the β5 subunit). - T-L (β2 subunit). - PGPH (β1 subunit). |
Proteasome inhibition promotes autophagy activation. |
[105] |
| Human neuroblastoma SH-SY5Y cells | 1 ̶ 5µM for 24h | Low affectation in the enzymatic complex (ChT-L of both 20S and 26S proteasome). | Proteasome inhibition promotes autophagy activation. Decrease in amyloid formation. | [105] |
| 5-(3ʹ-dihydroxyphenyl)-γ-valerolactone-4ʹ-sulfate | ||||
| In silico | - | Binding with β1, β2, and β5 subunits of human constitutive 20S proteasome (pdb ID: 6rgq). Binding with β1i, β2i, and β5i subunits of human immunoproteasome (pdb ID: 6e5b). |
Moderate binding affinity for proteasome catalytic subunits. | [105] |
| Cell-free | 0 ̶ 10 µM for 90 min | Inhibits catalytic subunits of proteasome at 10 µM: - ChT-L and BrAAP (associated with the β5 subunit). - T-L (β2 subunit). - PGPH (β1 subunit). |
Proteasome inhibition. Promotes autophagy activation. |
[105] |
| Human neuroblastoma SH-SY5Y cells | 1 ̶ 5µM for 24h | Strongly affects the functionality of enzymatic complex, mainly ChT-L of both 20S and 26S proteasome. | Affectation the functionality of proteosomal enzymatic complex. Decrease in amyloid formation. |
[105] |
| Benzoic acid derivatives | ||||
| 4-hydroxybenzoic acid | ||||
| In silico | - | Binding with pro-cathepsin B (PDBid: 2PBH) and pro-cathepsin L (PDBid: 1CS8). | Binders of both procathepsin B and L, and thus suggest a likely direct effect on cathepsins activity, which enhances the activity of UPS system. | [114] |
| Human foreskin fibroblast cells | 5 μM for 24h | Proteasome proteolytic activities on ChT-L (β5 subunit) and caspase-like (C-L) (β1 subunit). | ↑Activity of the two main cell protein degradation systems, namely ALP and UPS and especially the activity of cathepsins B and L. | [114] |
| 3,4-dihydroxybenzoic acid (Protocatechuic acid) | ||||
| Balb/c mice with tumors induced by 12-O-tetradecanoylphorbol-13-acetate | 16 μM in 0.2 mL of acetone topical application | Reduction of 20S proteasome trypsin-like (T-L) activity. | Suppression of proteasome 20S activities in mouse epidermis. Affect several key events of initiation and the promotion stage of carcinogenesis |
[111] |
| 3,4,5-trihydroxybenzoic acid (Gallic acid) | ||||
| EA.hy926 human cardiovascular endothelial cell and HBEC-5i human cerebrovascular endothelial cells | Pretreatment (before cell death induced by homocysteine, adenosine and TNFα) 1 ̶ 100 μM for 4h | Accumulation of ubiquitinated protein aggregates and ↓ in ChT-L (β5 subunit) proteasome activities. | ↓Cytotoxicity . Reversed DNA methyltransferase 1 (DNMT1) depletions at the protein level. Anti-apoptotic effects. ↓Microparticle formation and proteasome activity inhibition. |
[116] |
| 4-methoxybenzoic acid | ||||
| In silico | - | Binding with pro-cathepsin B (PDBid: 2PBH) and pro-cathepsin L (PDBid: 1CS8). | Binders of both procathepsin B and L, and thus suggest a likely direct effect on cathepsins activity, which enhances the activity of UPS system. | [114] |
| Human foreskin fibroblast cells | 5 μM for 24h | Proteasome proteolytic activities on ChT-L (β5 subunit) and caspase-like (C-L) (β1 subunit). | ↑Activity of the two main cell protein degradation systems, namely ALP and UPS and especially the activity of cathepsins B and L. | [114] |
| 2,4,5-Trimethoxybenzoic acid (Asaronic acid) | ||||
| J774A.1 murine macrophage cells | 1 ̶ 20 μM up to 24h | ↑UPS degradation of non-native proteins dislocated to the cytosol. | ↓Oxysterol-induced expression of EDEM1, OS9, Sel1L-Hrd1 and p97/VCP1 Activation of the ER stress sensors of ATF6, IRE1 and PERK stimulated by 7β-hydroxycholesterol. |
[107] |
| 4-hydroxy-3-methoxybenzoic acid (vanillic acid) | ||||
| Raw 264.7 macrophage cells | 10 ̶ 100 μM for 60 min | Blocking the proteasome through inhibition of ChT-L activity. | Inhibition of proteasome activity. | [106] |
| 3,5-dimethoxy-4-hydroxybenzoic acid (syringic acid) | ||||
| In silico | - | Eukaryotic yeast 20S proteasome crystal structure (PDB code: 2 F16). | Good affinity of syringic acid and different proposed derivatives. | [112] |
| Benzaldehyde derivatives | ||||
| 3-methoxy-4-hydroxybenzaldehyde (vanillin) | ||||
| Human non-small cell lung cancer NCI-H460 | 0 ̶ 50 μM for 1 or 3 days | Akt degradation through the ubiquitin-proteasomal pathway. | Downregulation of different cancer stem cells markers (CD133, ALDH1A1) and transcription factors (Oct4 and Nanog). Akt-proteasomal degradation. |
[109] |
| Six-week-old BALB/c mice with colitis-associated colon cancer | 10, 50, and 100 mg/kg in distilled water for 13 weeks | ↓ Proteasome expression in colon tissues (Proteasome β5 subunit) and ↓ Psma1, Psma4, Psmb2, Psmb5, Psmb9, Psmb10, Psmc4, Psmd3, Psmd8 gene expression. | ↓ Tumor number and growth. Affects gene expression profiles of six biological pathways involved in protein folding and degradation (proteasome and ER-associated degradation), transcription (spliceosome), immune system (Fcγ-mediated phagocytosis), cell motility (regulation of actin cytoskeleton), and glycan metabolism (N-glycan biosynthesis) |
[110] |
| Human colorectal cancer HCT-116 cells | 0.01 ̶ 10000 µM for 2h | ↓ Proteasome β5 activity. | Inhibition of proteasome activity. | [110] |
| 3′,4′-dihydroxycinnamic acid (caffeic acid) | ||||
| Cerebellar granule neurons isolated from Sprague-Dawley rats | 50 µM for 24h | Blocking of the proteasome inhibitor PS-341, which causes cell death | ↓Oxidative and nitrosative stress and excitotoxicity. Protection against intrinsic apoptosis and proteasome inhibition. |
[113] |
| 4-hydroxy-3-methoxycinnamic acid (ferulic acid) | ||||
| Raw 264.7 macrophage cells | 10 ̶ 100 μM for 60 min | No affectation of proteasome activity via ChT-L. | ↓ NO production. No inhibition of proteasome activity. |
[106] |
| Cerebellar granule neurons isolated from Sprague-Dawley rats | 50 µM for 24h | No effects on blocking the proteasome inhibitor PS-341, which causes cell death. | Significantly protected neurons from excitotoxicity and glutamate-induced cell death, independent of proteasome inhibition. | [113] |
| 4-Hydroxy-3,5-dimethoxycinnamic acid (sinapic acid) | ||||
| Raw 264.7 macrophage cells | 10 ̶ 100 μM/ 60 minutes | No affectation of proteasome activity via ChT-L. | ↓ NO production. No inhibition of proteasome activity. |
[106] |
| Phenylacetic acid derivatives | ||||
| 3,4-dihydroxyphenylacetic acid (DOPAC) | ||||
| Untreated rabbit reticulocyte lysate | 0.19 mM for 30, 60 and 180 min | Protective role for NQO1 in protecting against dopamine-induced proteasomal inhibition. | Inhibition of proteasome activity. | [115] |
| Hippuric acid derivatives | ||||
| Hippuric acid | ||||
| Human renal proximal tubule HK-2 cells | 0 ̶ 1000 µM for 24h | ↑E3 ubiquitin activity ligase by strengthening the NRF2–KEAP1–CUL3 interactions. ↑NRF2 ubiquitination and degradation by 26S proteosome. |
Disruption of redox homeostasis by NRF2 antioxidant activity. | [108] |
| Urolithin derivatives | ||||
| Urolithin A | ||||
| Vastus lateralis skeletal muscle from overweight adults (n=88) | 500 mg oral daily dose for 4 months | Increases the levels of ubiquitin-conjugating enzymes and proteasomal components, which are required for Parkin-mediated degradation of dysfunctional mitochondria and damaged proteins. | Improvement of muscle performance. | [118] |
| C2C12 murine skeletal muscle myoblasts | 15 μM for 24h |
Prevents the activation of NF-kB signalling and ubiquitin proteasome pathway. | No affectation on differentiation of C2C12 myotubes. | [117] |
| Urolithin B | ||||
| C2C12 murine skeletal muscle myoblasts | 15 μM for 24h |
Represses UPS through downregulation of transcription factors (FoxO1 and FoxO3) and ubiquitin ligases (MAFbx and MuRF1). | Enhances the growth and differentiation of C2C12 myotubes. Potential for treatment of muscle mass loss. Decreases protein degradation rate. |
[117] |
| Twelve-week-old C57/Bl6 J | 10 μg/day (subcutaneous) for 28 days | ↑p-mTOR. | ↑Muscle hypertrophy and ↓muscle atrophy after the sciatic nerve section. | [117] |
4. Conclusions and Existing Gaps to Address in the Future
To the best of our knowledge, this is the first review compiling the effects of different physiological phenolics as potential modulators of the UPS. Despite the large number of studies conducted with dietary polyphenols or enriched extracts, there are only a few studies exploring relevant phenolics (those found in human circulation). The limited availability of these metabolites, the suitability of cell models, and uncertainties regarding the metabolic forms of (poly)phenols, their physiological concentrations, and appropriate exposure times all contribute to existing gaps in understanding their roles in proteasome activity.
Moreover, the suitability of cell models used in these studies remains a major concern. Many experiments rely on immortalized or cancer-derived cell lines that lack key metabolic features, failing to accurately represent normal human physiology. These models often do not account for phase I and II metabolism, efflux mechanisms, or cellular uptake processes that influence polyphenol bioactivity. Consequently, the effects observed in these systems may not be directly translatable to human physiology. Future studies should prioritize models that better mimic human metabolism, such as primary cells, organoids, or co-culture systems that include relevant metabolic enzymes.
Another critical limitation is the use of supraphysiological concentrations, which can lead to misleading conclusions about the real impact of these compounds. Many studies use phenolic metabolite concentrations in the micromolar range (often above 10 µM), which exceed those typically found in plasma after dietary intake. While these high doses may provide insights into mechanistic interactions with the UPS, they do not necessarily reflect the actual physiological effects. More studies should focus on metabolite concentrations that are realistically achievable in humans to ensure the clinical relevance of the findings.
A promising approach to bridge the gap between preclinical and human studies is the direct administration of physiologically relevant metabolites rather than parent compounds. This would allow researchers to assess their actual biological activity without interference from metabolic conversion, providing more accurate data for translational applications. Additionally, in vivo models should incorporate physiological routes of administration, considering the complexity of polyphenol metabolism, tissue distribution, and long-term bioavailability.
To fully understand the role of phenolic metabolites in chronic diseases and their potential as UPS modulators, new approaches are required. This includes i) developing reliable methods for producing and stabilizing relevant metabolites for experimental use; ii) refining cell and animal models to better mimic human metabolism and physiological conditions; iii) conducting long-term studies to assess chronic exposure effects and metabolic adaptations, and iv) exploring synergistic effects between phenolic metabolites.
Addressing these gaps will pave the way for a deeper understanding of how phenolic metabolites interact with the UPS and their potential therapeutic applications in chronic diseases.
.
Author Contributions
Sofia Ferreira: Writing – review & editing, Data curation. Regina Menezes: Writing – original draft, Writing – review & editing, Supervision. Ioannis Trougakos: Writing – review & editing; Sentiljana Gumeni: Writing – review & editing. Victor Bolanos-García: Writing – review & editing. Cláudia Nunes dos Santos: Writing – review & editing. María Ángeles Ávila-Gálvez: Writing – review & editing, Writing – original draft, Data curation, Conceptualization and Supervision.
Acknowledgments
This article/publication is based upon work from COST Action ProteoCure, CA20113, supported by COST (European Cooperation in Science and Technology).
References
- Bauer UE, Briss PA, Goodman RA, Bowman BA. Prevention of chronic disease in the 21st century: elimination of the leading preventable causes of premature death and disability in the USA. The Lancet 2014;384:45–52. [CrossRef]
- Helgeson VS, Zajdel M. Adjusting to Chronic Health Conditions. Annu Rev Psychol 2017;68:545–71. [CrossRef]
- Shelton RC, Philbin MM, Ramanadhan S. Qualitative Research Methods in Chronic Disease: Introduction and Opportunities to Promote Health Equity. Annu Rev Public Health 2022;43:37–57. [CrossRef]
- Kassis A, Fichot M-C, Horcajada M-N, Horstman AMH, Duncan P, Bergonzelli G, et al. Nutritional and lifestyle management of the aging journey: A narrative review. Front Nutr 2023;9. [CrossRef]
- Schmidt M, Finley D. Regulation of proteasome activity in health and disease. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2014;1843:13–25. [CrossRef]
- Dunker AK, Silman I, Uversky VN, Sussman JL. Function and structure of inherently disordered proteins. Curr Opin Struct Biol 2008;18:756–64. [CrossRef]
- Balchin D, Hayer-Hartl M, Hartl FU. In vivo aspects of protein folding and quality control. Science (1979) 2016;353. [CrossRef]
- Jayaraj GG, Hipp MS, Hartl FU. Functional Modules of the Proteostasis Network. Cold Spring Harb Perspect Biol 2020;12:a033951. [CrossRef]
- Höhn A, Tramutola A, Cascella R. Proteostasis Failure in Neurodegenerative Diseases: Focus on Oxidative Stress. Oxid Med Cell Longev 2020;2020:1–21. [CrossRef]
- Dikic I. Proteasomal and Autophagic Degradation Systems. Annu Rev Biochem 2017;86:193–224. [CrossRef]
- Scarmeas N, Anastasiou CA, Yannakoulia M. Nutrition and prevention of cognitive impairment. Lancet Neurol 2018;17:1006–15. [CrossRef]
- Chiavaroli L, Nishi SK, Khan TA, Braunstein CR, Glenn AJ, Mejia SB, et al. Portfolio Dietary Pattern and Cardiovascular Disease: A Systematic Review and Meta-analysis of Controlled Trials. Prog Cardiovasc Dis 2018;61:43–53. [CrossRef]
- Poulsen NB, Lambert MNT, Jeppesen PB. The Effect of Plant Derived Bioactive Compounds on Inflammation: A Systematic Review and Meta-Analysis. Mol Nutr Food Res 2020;64. [CrossRef]
- Clinton SK, Giovannucci EL, Hursting SD. The World Cancer Research Fund/American Institute for Cancer Research Third Expert Report on Diet, Nutrition, Physical Activity, and Cancer: Impact and Future Directions. J Nutr 2020;150:663–71. [CrossRef]
- Amiot MJ, Riva C, Vinet A. Effects of dietary polyphenols on metabolic syndrome features in humans: a systematic review. Obesity Reviews 2016;17:573–86. [CrossRef]
- Tresserra-Rimbau A, Rimm EB, Medina-Remón A, Martínez-González MA, de la Torre R, Corella D, et al. Inverse association between habitual polyphenol intake and incidence of cardiovascular events in the PREDIMED study. Nutrition, Metabolism and Cardiovascular Diseases 2014;24:639–47. [CrossRef]
- Zamora-Ros R, Knaze V, Rothwell JA, Hémon B, Moskal A, Overvad K, et al. Dietary polyphenol intake in Europe: the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Eur J Nutr 2016;55:1359–75. [CrossRef]
- Rienks J, Barbaresko J, Nöthlings U. Association of polyphenol biomarkers with cardiovascular disease and mortality risk: A systematic review and meta-analysis of observational studies. Nutrients 2017;9:415.
- Kishimoto Y, Tani M, Kondo K. Pleiotropic preventive effects of dietary polyphenols in cardiovascular diseases. Eur J Clin Nutr 2013;67:532–5. [CrossRef]
- Yan L, Guo MS, Zhang Y, Yu L, Wu JM, Tang Y, et al. Dietary Plant Polyphenols as the Potential Drugs in Neurodegenerative Diseases: Current Evidence, Advances, and Opportunities. Oxid Med Cell Longev 2022;2022. [CrossRef]
- Manasanch EE, Orlowski RZ. Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol 2017;14:417–33. [CrossRef]
- Collins GA, Goldberg AL. The Logic of the 26S Proteasome. Cell 2017;169:792–806. [CrossRef]
- Andersson V, Hanzén S, Liu B, Molin M, Nyström T. Enhancing protein disaggregation restores proteasome activity in aged cells. Aging 2013;5:802–12. [CrossRef]
- Tsakiri EN, Trougakos IP. Chapter Five - The Amazing Ubiquitin-Proteasome System: Structural Components and Implication in Aging. In: Jeon KW, editor. Int Rev Cell Mol Biol, vol. 314, Academic Press; 2015, p. 171–237. [CrossRef]
- Narasimhan S, Guo JL, Changolkar L, Stieber A, McBride JD, Silva L V., et al. Pathological Tau Strains from Human Brains Recapitulate the Diversity of Tauopathies in Nontransgenic Mouse Brain. The Journal of Neuroscience 2017;37:11406–23. [CrossRef]
- Heilbronner G, Eisele YS, Langer F, Kaeser SA, Novotny R, Nagarathinam A, et al. Seeded strain-like transmission of β-amyloid morphotypes in APP transgenic mice. EMBO Rep 2013;14:1017–22. [CrossRef]
- Smith DM. Could a Common Mechanism of Protein Degradation Impairment Underlie Many Neurodegenerative Diseases? J Exp Neurosci 2018;12. [CrossRef]
- Kumar P. Role of Oxidative Stress, ER Stress and Ubiquitin Proteasome System in Neurodegeneration. MOJ Cell Science & Report 2014;1. [CrossRef]
- Nedelsky NB, Todd PK, Taylor JP. Autophagy and the ubiquitin-proteasome system: Collaborators in neuroprotection. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2008;1782:691–9. [CrossRef]
- Cheng B, Martinez AA, Morado J, Scofield V, Roberts JL, Maffi SK. Retinoic acid protects against proteasome inhibition associated cell death in SH-SY5Y cells via the AKT pathway. Neurochem Int 2013;62:31–42. [CrossRef]
- Pickrell AM, Youle RJ. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015;85:257–73. [CrossRef]
- Itier J-M, Ibanez P, Mena MA, Abbas N, Cohen-Salmon C, Bohme GA, et al. Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet 2003;12:2277–91.
- Setsuie R, Wang Y-L, Mochizuki H, Osaka H, Hayakawa H, Ichihara N, et al. Dopaminergic neuronal loss in transgenic mice expressing the Parkinson’s disease-associated UCH-L1 I93M mutant. Neurochem Int 2007;50:119–29. [CrossRef]
- Marshall RS, Vierstra RD. Dynamic Regulation of the 26S Proteasome: From Synthesis to Degradation. Front Mol Biosci 2019;6. [CrossRef]
- LAN D, WANG W, ZHUANG J, ZHAO Z. Proteasome inhibitor-induced autophagy in PC12 cells overexpressing A53T mutant α-synuclein. Mol Med Rep 2015;11:1655–60. [CrossRef]
- Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol 2014;112:24–49. [CrossRef]
- Dantuma NP, Bott LC. The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Front Mol Neurosci 2014;7. [CrossRef]
- Press M, Jung T, König J, Grune T, Höhn A. Protein aggregates and proteostasis in aging: Amylin and β-cell function. Mech Ageing Dev 2019;177:46–54. [CrossRef]
- Lee B-H, Lee MJ, Park S, Oh D-C, Elsasser S, Chen P-C, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010;467:179–84. [CrossRef]
- Willis MS, Townley-Tilson WHD, Kang EY, Homeister JW, Patterson C. Sent to Destroy. Circ Res 2010;106:463–78. [CrossRef]
- Iso T, Hamamori Y, Kedes L. Notch Signaling in Vascular Development. Arterioscler Thromb Vasc Biol 2003;23:543–53. [CrossRef]
- de Moissac D, Mustapha S, Greenberg AH, Kirshenbaum LA. Bcl-2 Activates the Transcription Factor NFκB through the Degradation of the Cytoplasmic Inhibitor IκBα. Journal of Biological Chemistry 1998;273:23946–51. [CrossRef]
- FOLCO JE, KOREN G. Degradation of the inducible cAMP early repressor (ICER) by the ubiquitin–proteasome pathway. Biochemical Journal 1997;328:37–43. [CrossRef]
- Eble DM, Spragia ML, Ferguson AG, Samarel AM. Sarcomeric myosin heavy chain is degraded by the proteasome. Cell Tissue Res 1999;296:541. [CrossRef]
- Taylor RG, Tassy C, Briand M, Robert N, Briand Y, Ouali A. Proteolytic activity of proteasome on myofibrillar structures. Mol Biol Rep 1995;21:71–3. [CrossRef]
- Gong Q, Keeney DR, Molinari M, Zhou Z. Degradation of Trafficking-defective Long QT Syndrome Type II Mutant Channels by the Ubiquitin-Proteasome Pathway. Journal of Biological Chemistry 2005;280:19419–25. [CrossRef]
- Waxman AJ, Clasen S, Hwang W-T, Garfall A, Vogl DT, Carver J, et al. Carfilzomib-Associated Cardiovascular Adverse Events. JAMA Oncol 2018;4:e174519. [CrossRef]
- Laine A, Ronai Z. Ubiquitin Chains in the Ladder of MAPK Signaling. Science’s STKE 2005;2005. [CrossRef]
- Kikuchi J, Furukawa Y, Kubo N, Tokura A, Hayashi N, Nakamura M, et al. Induction of Ubiquitin-Conjugating Enzyme by Aggregated Low Density Lipoprotein in Human Macrophages and Its Implications for Atherosclerosis. Arterioscler Thromb Vasc Biol 2000;20:128–34. [CrossRef]
- VIEIRA O, ESCARGUEIL-BLANC I, JÜRGENS G, BORNER C, ALMEIDA L, SALVAYRE R, et al. Oxidized LDLs alter the activity of the ubiquitin-proteasome pathway: potential role in oxidized LDL-induced apoptosis. The FASEB Journal 2000;14:532–42. [CrossRef]
- Scheidegger KJ. Differential effects of low density lipoproteins on IGF-1 and IGF-1R expression in vascular smooth muscle cells. Journal of Biological Chemistry 2000. [CrossRef]
- Higashi Y, Sukhanov S, Parthasarathy S, Delafontaine P. The ubiquitin ligase Nedd4 mediates oxidized low-density lipoprotein-induced downregulation of insulin-like growth factor-1 receptor. American Journal of Physiology-Heart and Circulatory Physiology 2008;295:H1684–9. [CrossRef]
- Qureshi N, Vogel SN, Van Way C, Papasian CJ, Qureshi AA, Morrison DC. The Proteasome: A Central Regulator of Inflammation and Macrophage Function. Immunol Res 2005;31:243–60. [CrossRef]
- Marfella R, D’Amico M, Di Filippo C, Baldi A, Siniscalchi M, Sasso FC, et al. Increased Activity of the Ubiquitin-Proteasome System in Patients With Symptomatic Carotid Disease Is Associated With Enhanced Inflammation and May Destabilize the Atherosclerotic Plaque. J Am Coll Cardiol 2006;47:2444–55. [CrossRef]
- Marfella R, Di Filippo C, Portoghese M, Ferraraccio F, Crescenzi B, Siniscalchi M, et al. Proteasome Activity as a Target of Hormone Replacement Therapy–Dependent Plaque Stabilization in Postmenopausal Women. Hypertension 2008;51:1135–41. [CrossRef]
- Takami Y, Nakagami H, Morishita R, Katsuya T, Hayashi H, Mori M, et al. Potential Role of CYLD (Cylindromatosis) as a Deubiquitinating Enzyme in Vascular Cells. Am J Pathol 2008;172:818–29. [CrossRef]
- Stangl K, Stangl V. The ubiquitin-proteasome pathway and endothelial (dys)function. Cardiovasc Res 2010;85:281–90. [CrossRef]
- Okamoto H, Takaoka M, Ohkita M, Itoh M, Nishioka M, Matsumura Y. A proteasome inhibitor lessens the increased aortic endothelin-1 content in deoxycorticosterone acetate-salt hypertensive rats. Eur J Pharmacol 1998;350:R11–2. [CrossRef]
- Meiners S, Ludwig A, Lorenz M, Dreger H, Baumann G, Stangl V, et al. Nontoxic proteasome inhibition activates a protective antioxidant defense response in endothelial cells. Free Radic Biol Med 2006;40:2232–41. [CrossRef]
- Stangl V, Lorenz M, Meiners S, Ludwig A, Bartsch C, Moobed M, et al. Long-term up-regulation of eNOS and improvement of endothelial function by inhibition of the ubiquitin–proteasome pathway. The FASEB Journal 2004;18:272–9. [CrossRef]
- Chade AR, Herrmann J, Zhu X, Krier JD, Lerman A, Lerman LO. Effects of Proteasome Inhibition on the Kidney in Experimental Hypercholesterolemia. Journal of the American Society of Nephrology 2005;16:1005–12. [CrossRef]
- Herrmann J, Saguner AM, Versari D, Peterson TE, Chade A, Olson M, et al. Chronic Proteasome Inhibition Contributes to Coronary Atherosclerosis. Circ Res 2007;101:865–74. [CrossRef]
- Reza MI, Syed AA, Kumariya S, Singh P, Husain A, Gayen JR. Pancreastatin induces islet amyloid peptide aggregation in the pancreas, liver, and skeletal muscle: An implication for type 2 diabetes. Int J Biol Macromol 2021;182:760–71. [CrossRef]
- Despa S, Margulies KB, Chen L, Knowlton AA, Havel PJ, Taegtmeyer H, et al. Hyperamylinemia Contributes to Cardiac Dysfunction in Obesity and Diabetes. Circ Res 2012;110:598–608. [CrossRef]
- Marmentini C, Branco RCS, Boschero AC, Kurauti MA. Islet amyloid toxicity: From genesis to counteracting mechanisms. J Cell Physiol 2022;237:1119–42. [CrossRef]
- Diehl JA, Fuchs SY, Haines DS. Ubiquitin and Cancer: New Discussions for a New Journal. Genes Cancer 2010;1:679–80. [CrossRef]
- Németh ZH, Wong HR, Odoms K, Deitch EA, Szabó C, Vizi ES, et al. Proteasome Inhibitors Induce Inhibitory κB (IκB) Kinase Activation, IκBα Degradation, and Nuclear Factor κB Activation in HT-29 Cells. Mol Pharmacol 2004;65:342–9. [CrossRef]
- Rastogi N, Mishra DP. Therapeutic targeting of cancer cell cycle using proteasome inhibitors. Cell Div 2012;7:26. [CrossRef]
- Curtis NL, Bolanos-Garcia VM. The Anaphase Promoting Complex/Cyclosome (APC/C): A Versatile E3 Ubiquitin Ligase, 2019, p. 539–623. [CrossRef]
- Mayah A, Arenas RB, Bastida A, Bolanos-Garcia VM. The Use of APC/C Antagonists to Promote Mitotic Catastrophe in Cancer Cells, 2025, p. 207–13. [CrossRef]
- Kapanidou M, Curtis NL, Diaz-Minguez SS, Agudo-Alvarez S, Rus Sanchez A, Mayah A, et al. Targeting APC/C Ubiquitin E3-Ligase Activation with Pyrimidinethylcarbamate Apcin Analogues for the Treatment of Breast Cancer. Biomolecules 2024;14:1439. [CrossRef]
- Zhang S, Kulkarni AA, Xu B, Chu H, Kourelis T, Go RS, et al. Bortezomib-based consolidation or maintenance therapy for multiple myeloma: a meta-analysis. Blood Cancer J 2020;10:33. [CrossRef]
- Spano D, Catara G. Targeting the Ubiquitin–Proteasome System and Recent Advances in Cancer Therapy. Cells 2023;13:29. [CrossRef]
- Goldring MB, Marcu KB. Epigenomic and microRNA-mediated regulation in cartilage development, homeostasis, and osteoarthritis. Trends Mol Med 2012;18:109–18. [CrossRef]
- Hu J, Lin SL, Schachner M. A fragment of cell adhesion molecule L1 reduces amyloid-β plaques in a mouse model of Alzheimer’s disease. Cell Death Dis 2022;13:48. [CrossRef]
- Zhang J, Yao M, Xia S, Zeng F, Liu Q. Systematic and comprehensive insights into HIF-1 stabilization under normoxic conditions: implications for cellular adaptation and therapeutic strategies in cancer. Cell Mol Biol Lett 2025;30:2. [CrossRef]
- Li R, Liu M, Yang Z, Li J, Gao Y, Tan R. Proteolysis-Targeting Chimeras (PROTACs) in Cancer Therapy: Present and Future. Molecules 2022;27:8828. [CrossRef]
- Cháirez-Ramírez MH, de la Cruz-López KG, García-Carrancá A. Polyphenols as Antitumor Agents Targeting Key Players in Cancer-Driving Signaling Pathways. Front Pharmacol 2021;12. [CrossRef]
- Floris A, Mazarei M, Yang X, Robinson A, Zhou J, Barberis A, et al. SUMOylation Protects FASN Against Proteasomal Degradation in Breast Cancer Cells Treated with Grape Leaf Extract. Biomolecules 2020;10:529. [CrossRef]
- Rousseau A, Bertolotti A. Regulation of proteasome assembly and activity in health and disease. Nat Rev Mol Cell Biol 2018;19:697–712. [CrossRef]
- Dimopoulos MA, Moreau P, Palumbo A, Joshua D, Pour L, Hájek R, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol 2016;17:27–38. [CrossRef]
- Papanagnou E, Gumeni S, Sklirou AD, Rafeletou A, Terpos E, Keklikoglou K, et al. Autophagy activation can partially rescue proteasome dysfunction-mediated cardiac toxicity. Aging Cell 2022;21. [CrossRef]
- An S, Fu L. Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. EBioMedicine 2018;36:553–62. [CrossRef]
- Liu Y, Yang J, Wang T, Luo M, Chen Y, Chen C, et al. Expanding PROTACtable genome universe of E3 ligases. Nat Commun 2023;14:6509. [CrossRef]
- Golonko A, Pienkowski T, Swislocka R, Lazny R, Roszko M, Lewandowski W. Another look at phenolic compounds in cancer therapy the effect of polyphenols on ubiquitin-proteasome system. Eur J Med Chem 2019;167:291–311. [CrossRef]
- Gu W, Wu G, Chen G, Meng X, Xie Z, Cai S. Polyphenols alleviate metabolic disorders: the role of ubiquitin-proteasome system. Front Nutr 2024;11. [CrossRef]
- Chen D, Daniel KG, Chen MS, Kuhn DJ, Landis-Piwowar KR, Dou QP. Dietary flavonoids as proteasome inhibitors and apoptosis inducers in human leukemia cells. Biochem Pharmacol 2005;69:1421–32. [CrossRef]
- MEADOR BM, MIRZA KA, TIAN M, SKELDING MB, REAVES LA, EDENS NK, et al. THE GREEN TEA POLYPHENOL EPIGALLOCATECHIN-3-GALLATE (EGCG) ATTENUATES SKELETAL MUSCLE ATROPHY IN A RAT MODEL OF SARCOPENIA. Journal of Frailty & Aging 2015:1–7. [CrossRef]
- Chen C, Zou L-X, Lin Q-Y, Yan X, Bi H-L, Xie X, et al. Resveratrol as a new inhibitor of immunoproteasome prevents PTEN degradation and attenuates cardiac hypertrophy after pressure overload. Redox Biol 2019;20:390–401. [CrossRef]
- Dosenko VE, Nagibin VS, Tumanovskaya L V., Zagorii VYu, Moibenko AA. Effect of quercetin on the activity of purified 20S and 26S proteasomes and proteasomal activity in isolated cardiomyocytes. Biochem Mosc Suppl B Biomed Chem 2007;1:40–4. [CrossRef]
- Bonfili L, Cecarini V, Amici M, Cuccioloni M, Angeletti M, Keller JN, et al. Natural polyphenols as proteasome modulators and their role as anti-cancer compounds. FEBS J 2008;275:5512–26. [CrossRef]
- Zrelli H, Kusunoki M, Miyazaki H. Role of Hydroxytyrosol-dependent Regulation of HO-1 Expression in Promoting Wound Healing of Vascular Endothelial Cells via Nrf2 De Novo Synthesis and Stabilization. Phytotherapy Research 2015;29:1011–8. [CrossRef]
- Raimundo AF, Félix F, Andrade R, García-Conesa M-T, González-Sarrías A, Gilsa-Lopes J, et al. Combined effect of interventions with pure or enriched mixtures of (poly)phenols and anti-diabetic medication in type 2 diabetes management: a meta-analysis of randomized controlled human trials. Eur J Nutr 2020;59:1329–43. [CrossRef]
- Zhou ZD, Xie SP, Saw WT, Ho PGH, Wang HY, Zhou L, et al. The Therapeutic Implications of Tea Polyphenols against Dopamine (DA) Neuron Degeneration in Parkinson’s Disease (PD). Cells 2019;8:911. [CrossRef]
- Kim Y-M, Abas F, Park Y-S, Park Y-K, Ham K-S, Kang S-G, et al. Bioactivities of Phenolic Compounds from Kiwifruit and Persimmon. Molecules 2021;26:4405. [CrossRef]
- Pierzynowska K, Gaffke L, Jankowska E, Rintz E, Witkowska J, Wieczerzak E, et al. Proteasome Composition and Activity Changes in Cultured Fibroblasts Derived From Mucopolysaccharidoses Patients and Their Modulation by Genistein. Front Cell Dev Biol 2020;8. [CrossRef]
- Cardaci TD, Machek SB, Wilburn DT, Hwang PS, Willoughby DS. Ubiquitin Proteasome System Activity is Suppressed by Curcumin following Exercise-Induced Muscle Damage in Human Skeletal Muscle. J Am Coll Nutr 2021;40:401–11. [CrossRef]
- González-Sarrías A, Espín JC, Tomás-Barberán FA. Non-extractable polyphenols produce gut microbiota metabolites that persist in circulation and show anti-inflammatory and free radical-scavenging effects. Trends Food Sci Technol 2017;69:281–8. [CrossRef]
- Carregosa D, Pinto C, Ávila-Gálvez MÁ, Bastos P, Berry D, Santos CN. A look beyond dietary (poly)phenols: The low molecular weight phenolic metabolites and their concentrations in human circulation. Compr Rev Food Sci Food Saf 2022;21:3931–62. [CrossRef]
- Tomás-Barberán FA, Espín JC. Effect of food structure and processing on (Poly) phenol–gut microbiota interactions and the effects on human health. Annu Rev Food Sci Technol 2019;10:221–38.
- Espín JC, González-Sarrías A, Tomás-Barberán FA. The gut microbiota: A key factor in the therapeutic effects of (poly)phenols. Biochem Pharmacol 2017;139:82–93. [CrossRef]
- Di Lorenzo C, Colombo F, Biella S, Stockley C, Restani P. Polyphenols and Human Health: The Role of Bioavailability. Nutrients 2021;13:273. [CrossRef]
- Cortés-Martín A, Selma MV, Tomás-Barberán FA, González-Sarrías A, Espín JC. Where to Look into the Puzzle of Polyphenols and Health? The Postbiotics and Gut Microbiota Associated with Human Metabotypes. Mol Nutr Food Res 2020;64. [CrossRef]
- Koppel N, Maini Rekdal V, Balskus EP. Chemical transformation of xenobiotics by the human gut microbiota. Science (1979) 2017;356. [CrossRef]
- Cecarini V, Cuccioloni M, Zheng Y, Bonfili L, Gong C, Angeletti M, et al. Flavan-3-ol Microbial Metabolites Modulate Proteolysis in Neuronal Cells Reducing Amyloid-beta (1-42) Levels. Mol Nutr Food Res 2021;65. [CrossRef]
- Johansson E, Lange S, Oshalim M, Lönnroth I. Anti-Inflammatory Substances in Wheat Malt Inducing Antisecretory Factor. Plant Foods for Human Nutrition 2019;74:489–94. [CrossRef]
- Oh H, Kang M-K, Park S-H, Kim DY, Kim S-I, Oh SY, et al. Asaronic acid inhibits ER stress sensors and boosts functionality of ubiquitin-proteasomal degradation in 7β-hydroxycholesterol-loaded macrophages. Phytomedicine 2021;92:153763. [CrossRef]
- Sun B, Wang X, Liu X, Wang L, Ren F, Wang X, et al. Hippuric Acid Promotes Renal Fibrosis by Disrupting Redox Homeostasis via Facilitation of NRF2–KEAP1–CUL3 Interactions in Chronic Kidney Disease. Antioxidants 2020;9:783. [CrossRef]
- Srinual S, Chanvorachote P, Pongrakhananon V. Suppression of cancer stem-like phenotypes in NCI-H460 lung cancer cells by vanillin through an Akt-dependent pathway. Int J Oncol 2017;50:1341–51. [CrossRef]
- Li J-M, Lee Y-C, Li C-C, Lo H-Y, Chen F-Y, Chen Y-S, et al. Vanillin-Ameliorated Development of Azoxymethane/Dextran Sodium Sulfate-Induced Murine Colorectal Cancer: The Involvement of Proteasome/Nuclear Factor-κB/Mitogen-Activated Protein Kinase Pathways. J Agric Food Chem 2018;66:5563–73. [CrossRef]
- Cichocki M, Blumczyńska J, Baer-Dubowska W. Naturally occurring phenolic acids inhibit 12-O-tetradecanoylphorbol-13-acetate induced NF-κB, iNOS and COX-2 activation in mouse epidermis. Toxicology 2010;268:118–24. [CrossRef]
- Orabi KY, Abaza MS, Sayed KA El, Elnagar AY, Al-Attiyah R, Guleri RP. Selective growth inhibition of human malignant melanoma cells by syringic acid-derived proteasome inhibitors. Cancer Cell Int 2013;13:82. [CrossRef]
- Taram F, Winter AN, Linseman DA. Neuroprotection comparison of chlorogenic acid and its metabolites against mechanistically distinct cell death-inducing agents in cultured cerebellar granule neurons. Brain Res 2016;1648:69–80. [CrossRef]
- Georgousaki K, Tsafantakis N, Gumeni S, Lambrinidis G, González-Menéndez V, Tormo JR, et al. Biological Evaluation and In Silico Study of Benzoic Acid Derivatives from Bjerkandera adusta Targeting Proteostasis Network Modules. Molecules 2020;25:666. [CrossRef]
- Zafar KS, Siegel D, Ross D. A Potential Role for Cyclized Quinones Derived from Dopamine, DOPA, and 3,4-Dihydroxyphenylacetic Acid in Proteasomal Inhibition. Mol Pharmacol 2006;70:1079–86. [CrossRef]
- Kam A, Li KM, Razmovski-Naumovski V, Nammi S, Chan K, Li GQ. Gallic acid protects against endothelial injury by restoring the depletion of DNA methyltransferase 1 and inhibiting proteasome activities. Int J Cardiol 2014;171:231–42. [CrossRef]
- Rodriguez J, Caille O, Ferreira D, Francaux M. Pomegranate extract prevents skeletal muscle of mice against wasting induced by acute TNF-α injection. Mol Nutr Food Res 2017;61. [CrossRef]
- Singh A, D’Amico D, Andreux PA, Fouassier AM, Blanco-Bose W, Evans M, et al. Urolithin A improves muscle strength, exercise performance, and biomarkers of mitochondrial health in a randomized trial in middle-aged adults. Cell Rep Med 2022;3:100633. [CrossRef]
- Andreux PA, Blanco-Bose W, Ryu D, Burdet F, Ibberson M, Aebischer P, et al. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat Metab 2019;1:595–603. [CrossRef]
Figure 2.
– Modulation of UPS activity by circulating phenolic metabolites in different cell models. (A) In neuroblastoma cells expressing mutant APP, UPS inhibition by 5-(4ʹ-hydroxyphenyl)-γ-valerolactone, 5-(3ʹ,4ʹ-dihydroxyphenyl)-γ-valerolactone and 5-(3ʹ-dihydroxyphenyl)-γ-valerolactone-4ʹ-sulfate increases the levels of p27 and ubiquitin conjugates, and promotes autophagy, leading to reduced amyloid formation. (B) In macrophage cells, the inhibition of UPS by sinapic, ferulic and vanillic acids metabolites reduces the secretion of inflammatory mediators. (C) In renal cells, enhancement of E3 ubiquitin activity by hippuric acid action leads to NRF2 ubiquitination and consequent proteasomal degradation, thus preventing oxidative stress. (D) In colorectal adenocarcinoma cells, protocatechuic acid, reduces proteasome activity in Balb/c mice with tumors. Such effect impacts the initiation state of carcinogenesis. In non-small lung cancer cells, the presence of vanillin increases Akt ubiquitination and proteasomal degradation, leading to the suppression of cancer stemness phenotype. In colon tissues of Balb/c mice, the same metabolite shows an anticancer effect that is associated with down-regulation of proteasome genes expression, nuclear factor NF-kB, and reduced phosphorylation of p65. (E) In endothelial cells, gallic acid reduces proteasome chymotrypsin-like activity. This culminates in an anti-apoptotic effect that is important for wound repair of vascular endothelial cells. (F) In muscle cells, urolithin A augments the levels of ubiquitin conjugating enzymes, which are required for parkin-mediated degradation of damaged mitochondria. Removal of dysfunctional mitochondria has an important role in the improvement of muscle performance. Additionally, urolithin B represses UPS, and decreases protein degradation rate, thus ameliorating the growth and differentiation of myotubes.
Figure 2.
– Modulation of UPS activity by circulating phenolic metabolites in different cell models. (A) In neuroblastoma cells expressing mutant APP, UPS inhibition by 5-(4ʹ-hydroxyphenyl)-γ-valerolactone, 5-(3ʹ,4ʹ-dihydroxyphenyl)-γ-valerolactone and 5-(3ʹ-dihydroxyphenyl)-γ-valerolactone-4ʹ-sulfate increases the levels of p27 and ubiquitin conjugates, and promotes autophagy, leading to reduced amyloid formation. (B) In macrophage cells, the inhibition of UPS by sinapic, ferulic and vanillic acids metabolites reduces the secretion of inflammatory mediators. (C) In renal cells, enhancement of E3 ubiquitin activity by hippuric acid action leads to NRF2 ubiquitination and consequent proteasomal degradation, thus preventing oxidative stress. (D) In colorectal adenocarcinoma cells, protocatechuic acid, reduces proteasome activity in Balb/c mice with tumors. Such effect impacts the initiation state of carcinogenesis. In non-small lung cancer cells, the presence of vanillin increases Akt ubiquitination and proteasomal degradation, leading to the suppression of cancer stemness phenotype. In colon tissues of Balb/c mice, the same metabolite shows an anticancer effect that is associated with down-regulation of proteasome genes expression, nuclear factor NF-kB, and reduced phosphorylation of p65. (E) In endothelial cells, gallic acid reduces proteasome chymotrypsin-like activity. This culminates in an anti-apoptotic effect that is important for wound repair of vascular endothelial cells. (F) In muscle cells, urolithin A augments the levels of ubiquitin conjugating enzymes, which are required for parkin-mediated degradation of damaged mitochondria. Removal of dysfunctional mitochondria has an important role in the improvement of muscle performance. Additionally, urolithin B represses UPS, and decreases protein degradation rate, thus ameliorating the growth and differentiation of myotubes.
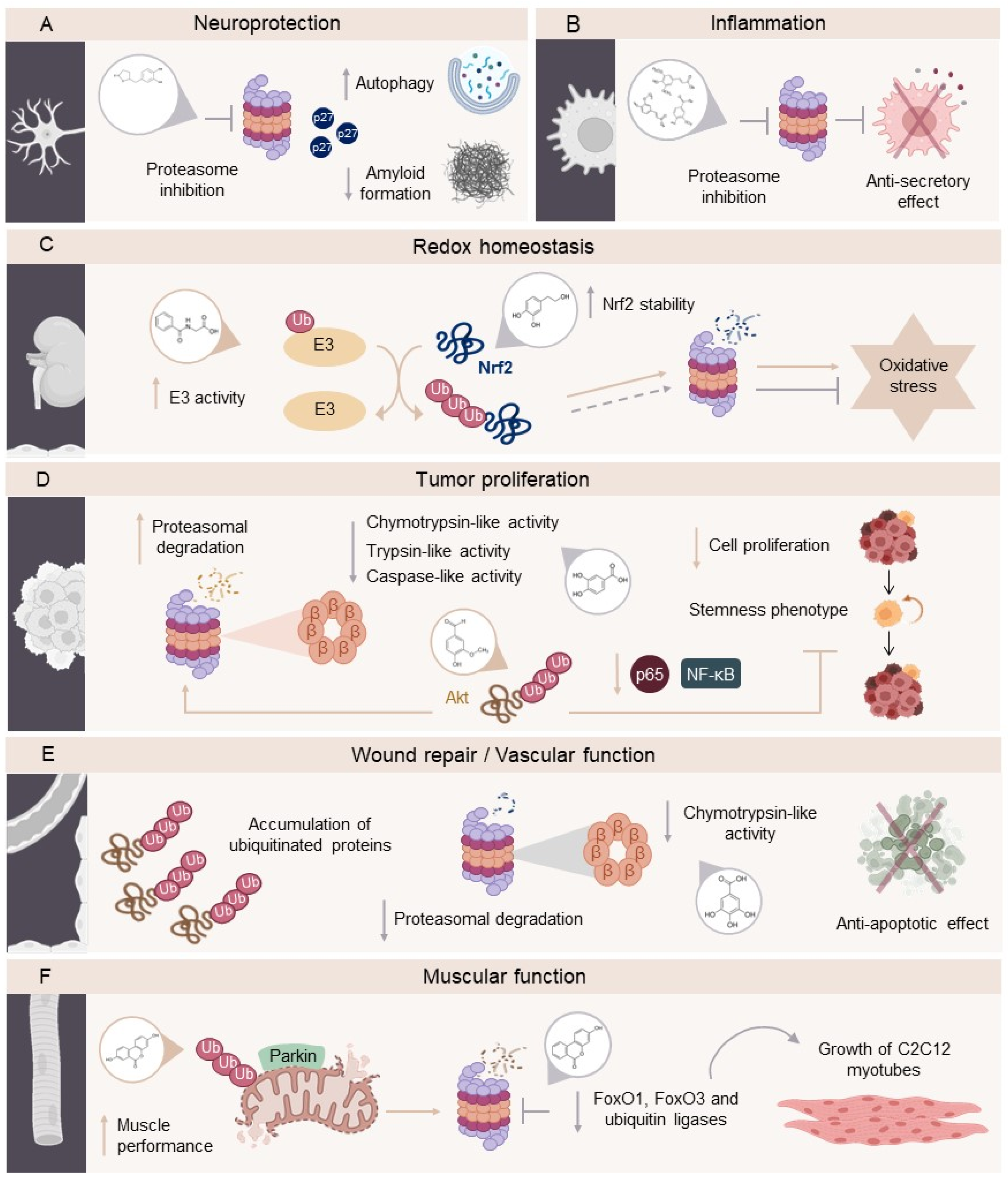
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



