
Submitted:
19 February 2025
Posted:
20 February 2025
You are already at the latest version
Abstract
Our previous research has demonstrated that the spinal cord undergoes epigenetic and molecular alterations following non-severe burn injury. However, the primary somatosensory cortex (S1), crucial for pain perception, remains unexplored in this context. Here, we investigated transcriptomic alterations in the S1 cortex of mice subjected to burn injury or formalin treatment, utilizing RNA sequencing (RNA-seq) one hour after injury. RNA-seq identified 1116 differentially expressed genes (DEGs) in burn injury and 136 DEGs in inflammatory pain. Notably, 82.4% of DEGs in burn injury and 32.4% in inflammatory pain were downregulated. A limited number of overlapping DEGs were found, suggesting significant differences in cortical processing of pain with different origins. Gene Ontology Enrichment Analysis revealed that upregulated genes in burn injury were associated with mitochondrial functions and gene expression, whereas downregulated genes were related to axon guidance, synaptic plasticity, and neurotransmission. KEGG pathway analysis highlighted the role of retrograde endocannabinoid signaling in the response to burn injury. Formalin treatment mainly impacted metabolic processes, particularly protein digestion and purine metabolism. These findings demonstrate that transcriptomic remodeling in the S1 cortex is dependent on the sensory modality and suggest that the retrograde endocannabinoid network is activated during the acute pain response following burn injury.
Keywords:
burn injury
; primary somatosensory cortex
; RNA sequencing
; inflammatory pain
; retrograde endocannabinoid signaling
1. Introduction
A burn injury can significantly reduce the quality of life, leading to lasting changes not only at the site of the injury site but also resulting in pain and neurological alterations due to chronic neuroplasticity [1,2,3,4]. Various brain regions, including the anterior cingulate cortex (ACC), insular cortex, medial prefrontal cortex, and primary and secondary somatosensory cortices (S1 and S2), are involved in the experience of pain [5,6,7], even though they are temporally and spatially distant from the initial injury. Epidemiological data show that burn patients, whether they have severe burns (≥20% total body surface area; TBSA) or non-severe burns (<20% TBSA), require extended hospital stays due to neurological dysfunction [2,4]. Despite the functional and structural changes observed in the central nervous system (CNS) of burn patients [1,2,3,4], our understanding of the molecular basis underlying these burn-induced functional consequences remains limited.
The sensory cortex, particularly the primary somatosensory cortex (S1), plays a crucial role in encoding the sensory-discriminative aspects of noxious input, such as identifying the location of the pain stimulus and the perceptual discrimination of pain intensity [8,9,10]. Currently, a PubMed search for RNA sequencing, somatosensory cortex, and pain reveals only four publications [11,12,13,14]. These studies investigate various models of neuropathic pain, including tibial nerve injury and infraorbital nerve transection [12,13]. In addition, other studies have demonstrated a significant association between specific chronic pain models (e.g., spared nerve injury, chronic constriction injury (CCI), postherpetic neuralgia, tibial nerve damage) and transcriptional changes in various brain regions, including the anterior cingulate cortex (ACC), prefrontal cortex, and primary somatosensory cortex [5,6,15,16,17,18]. Although the neurobiological aspects of nociception are well established [19], the molecular mechanisms underlying pain processing in the somatosensory cortex, particularly in relation to burn-induced pain, remain poorly understood.
Our recent findings have revealed significant differences in the central processing of pain models of various origins at the spinal cord level [20]. This suggests that the transcriptomic patterns in the primary somatosensory cortex may vary between the burn injury model and the inflammatory pain model induced by formalin treatment. To test this hypothesis, we utilized next-generation RNA sequencing (RNA-seq) to examine alterations in gene expression in the S1 cortex of adult male mice one hour after a non-severe burn injury or formalin application to the hindpaw. We compared the gene expression patterns from burn injury to those induced by formalin-induced inflammatory pain models in adult male mice. It is important to note that, due to ethical considerations concerning animal welfare, the survival time was limited to one-hour post-injury. This comparative analysis allowed us to identify differentially expressed genes (DEGs) and to delineate key signaling pathways and functional gene clusters through Gene Ontology enrichment analysis and KEGG Pathway analysis during the acute phase of noxious stimuli. Additionally, immunofluorescent studies targeting the CB1 receptor, and Ggamma14 were conducted to validate the gene expression results.
This study enhances our understanding of the molecular mechanisms associated with burn injury. It demonstrates that different painful stimuli activate distinct transcriptional programs in the S1 cortex depending on the sensory modality, with minimal overlap between pain models. The identified genes and molecular pathways are pivotal in determining the functional outcomes of various pain conditions and possess significant clinical relevance. They also offer potential targets for further experimental hypotheses and therapeutic interventions.
2. Results
2.1. Burn Injury Significantly Alters the Activity of Different Cortical Neuron Populations in the S1 Cortex, as Assessed by c-Fos Immunohistochemistry
Neuronal activation was investigated in the S1 cortex following burn injury using c-Fos immunohistochemistry. c-Fos is an immediate early gene and a well-known marker for neuronal activation [21,22]. Its expression along the pain pathway is often utilized to study immediate pain responses [21,22]. Therefore, comparing c-Fos expression and intensity between the control and burn injury-treated samples was a logical first step (Figure 1a).
c-Fos expression in the spinal cord typically increases rapidly after a painful stimulus, peaking at 1 to 2 hours and returning to baseline within 4 to 6 hours. This expression is elevated in specific regions of the central nervous system, including the primary somatosensory cortex, in response to neuropathic or inflammatory pain [21,22,23]. In murine models, the medial aspect of the primary somatosensory cortex is associated with the representation of the hindpaw region [24]. A schematic representation of the coronal section of the mouse brain, illustrating the S1 cortical area, is depicted in Figure 1b. Figure 1c displays representative confocal images of c-Fos immunoreactivity (IR) from a control mouse and those with burn injury. In the area of the contralateral S1 cortex that represents the lower limb, 6.8 ± 1.6% of DAPI-labeled cells exhibited c-Fos-IR following a burn injury, with an average count of 494 out of 7253 cells (5-6 slices from 2 animals per group). Conversely, this proportion was approximately 3% on the ipsilateral side of the brain following the burn injury as well as in control animals, with average counts of 225 out of 7178 and 280 out of 8987 cells, respectively (5-6 slices from 2 animals per group). The percentage of c-Fos-IR nuclei was 144 ± 17% in the contralateral side of the S1 cortex after the burn injury compared to the control (p = 0.031; Figure 1d). In contrast, the percentage on the ipsilateral side was 84.7 ± 39.3% (Figure 1d). Additionally, the fluorescence intensity values, also normalized to the control values, were 155.4 ± 25.7% and 114.4 ± 13.1% for the contralateral and for the ipsilateral-sided S1 cortex, respectively (p = 0.029; Figure 1e).
While parvalbumin (Pvalb)-expressing neurons are distributed throughout the entire length of the S1 cortex, both calbindin (Calb)- and calretinin (CR)-positive interneurons are primarily located in the superficial layers, particularly in layers 2/3. Consequently, colocalization analysis between c-Fos and the various subgroups of interneurons was conducted in layers 2/3. In the contralateral S1 cortex, 11.8% of c-Fos-IR cells were found to be positive for parvalbumin, while 3.22% were positive for calretinin, which was not significantly different from the control group (12.9% for Pvalb and 5.5% for CR). For a representative confocal image of a burn-injured mouse, please refer to Figure 1f. However, the co-localization of c-Fos and calbindin significantly increased from 12.09 ± 1.2% to 38.7 ± 2.3% after burn injury (p = 0.021; Figure 1g).
These findings indicate that burn injury significantly influences the activities of various cortical neuron subpopulations, with a pronounced effect on calbindin-positive interneurons.
2.2. Burn Injury Is Accompanied by Moderate Transcriptomic Changes in the Primary Somatosensory (S1) Cortex as Early as One-Hour Post-Burn Injury
RNA sequencing (RNA-seq) was performed to evaluate the impact of burn injury (BI) on pain-related gene expression profiles. We also investigated changes in gene expression in an inflammatory pain model induced by formalin application (FA) to assess whether there are differences in gene expression patterns between the two types of painful stimuli (Figure 2a). Samples were collected from the primary somatosensory cortex of wild-type mice that had been exposed to painful stimuli. Our RNA-seq analysis revealed significant differences at the transcriptomic level between the treated and untreated control groups. The corresponding findings are summarized in Figure 2 and the online Supplementary Tables S1-S3.
Using next-generation RNA-seq and applying the criteria (-1 < logFC < 1), we identified a total of 1116 differentially expressed genes (DEGs) in the S1 cortex of BI mice, while 136 DEGs were detected in FA mice compared to the untreated control sample. Furthermore, we noted that only a small number of genes overlapped between the experimental groups. This comprehensive analysis, using a fold change (FC) of ≥ 2, revealed that 196 genes were upregulated in the mouse S1 cortex following burn injury and 92 genes after inflammatory pain (Figure 2b). Among these, 42 genes were elevated in both pain models, accounting for 21.4% of the upregulated genes in BI and 45.6% in inflammatory pain (Figure 2b). According to the criteria (-1 > logFC), 920 genes were downregulated in the BI samples, while 44 were downregulated in the FA samples (Figure 2b). Only 17 genes were negatively affected by both nociceptive stimuli, accounting for 1.8% of the BI group and 38.6% of the FA group (Figure 1b).
Next, the pain-related DEGs in the primary somatosensory cortex of BI and FA mice were analyzed using cluster analysis to create heat maps (Figure 2c-d). Heat maps illustrate the 15 most upregulated and downregulated genes in response to burn injury and formalin application. RNA-seq showed moderate changes in gene expression in response to painful stimuli compared to the control group. After burn injury, the log2 fold changes in gene expression ranged from 2.69 to -3.42, while formalin treatment resulted in log2 fold changes ranging from 1.84 to -1.53 (Supplementary Tables S1–S3). Among the transcripts that were exclusively affected by burn injury, MT-ATP6 demonstrated the most substantial increase in expression, whereas DCC exhibited the most pronounced decrease (as detailed in Supplementary Table S1; Figure 2c-d). In contrast, PCP2 was the most significantly upregulated gene due to formalin treatment, and peripherin was the most downregulated gene affected exclusively by formalin injection (refer to Supplementary Table S2; Figure 2c-d). Among the genes with well-known functions that are regulated similarly by noxious stimuli, MT-ATP8 showed the highest level of upregulation, as illustrated in Figure 2c and Supplementary Table S3. In contrast, PRSS56, which is a serine protease, displayed the greatest level of downregulation, regardless of the treatment applied (Figure 2d; Supplementary Table S3).
In conclusion, burn injury elicits moderate alterations in the transcriptomic profile of the S1 cortex as early as one-hour post-injury. These changes differ significantly from those observed in the inflammatory pain model established by formalin treatment.
2.3. Differential Cortical Mechanisms in Acute Pain Processing Within the Primary Somatosensory Cortex Following Burn Injury and Inflammatory Pain
Heatmaps of gene expression revealed that both formalin application (FA) and burn injury (BI) significantly altered the gene expression profile in the S1 cortex (Figure 2). In the subsequent in silico experiments, we conducted Gene Ontology enrichment analysis (GOEA) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis to elucidate nervous system-specific GO terms associated with differentially expressed genes (DEGs). We utilized the criteria of -0.5 < logFC < 0.5 to compare wild-type mice subjected to either treatment (BI or FA) with untreated controls.
The results of the GOEA are shown in Figure 3, which includes the Gene Ontology biological process (GO_BP), cellular compartment (GO_CC), molecular function (GO_MF), and KEGG pathways. In the context of burn injury (Figure 3a), the GO_BP analysis identified two enriched terms associated with upregulated DEGs (ATP metabolic process and gene expression) and eighteen enriched terms for downregulated DEGs. The GO_CC analysis revealed four enriched terms for upregulated DEGs and twenty-eight enriched terms for downregulated DEGs. Regarding the GO_MF analysis, only one enriched term was identified for upregulated DEGs, while nine enriched terms were identified for downregulated DEGs. In relation to burn injury, two terms were enriched among upregulated DEGs (i.e. retrograde endocannabinoid signaling and ribosome), and twenty-four terms were enriched among downregulated DEGs according to KEGG pathways analysis.
In contrast to burn injury, the formalin-induced inflammatory pain model (FA; Figure 3b) predominantly exhibited upregulated terms. The GO_BP analysis identified nine enriched terms for upregulated DEGs, while the GO_CC analysis found 14 enriched terms for upregulated DEGs and three for downregulated DEGs. The GO_MF analysis revealed four enriched terms for upregulated DEGs and two for downregulated DEGs. In the KEGG pathway analysis, ten enriched terms were upregulated, such as purine metabolism and protein digestion and absorption, while seven terms were downregulated (Figure 3b).
In conclusion, the Gene Ontology (GO) enrichment analysis, along with the KEGG pathway analysis, showed a significant difference in the expression of genes involved in various nervous system-specific GO terms between the two pain models. In the burn injury model, the majority of GO terms were downregulated, with only a limited number of terms being upregulated. Conversely, upregulation dominated over downregulation following formalin treatment. This observation suggests that the molecular mechanisms underlying the pain response in these two models are fundamentally distinct.
2.4. Differential Pain-Related Signaling in the S1 Cortex: Insights from KEGG Pathway Analysis Across Sensory Modalities
KEGG enrichment analysis was performed on the Cytoscape platform using the criteria -0.5 < logFC < 0.5 (Figure 4). Figure 4a provides a comprehensive list of burn injury-affected KEGG pathways linked to synaptic functions and neuronal morphology, including axon guidance, focal adhesion, and extracellular matrix (ECM)-receptor interaction. Notably, retrograde endocannabinoid signaling is significantly upregulated, as evidenced by its relatively high enrichment. Interestingly, DEGs associated with axon guidance were downregulated following burn injury (Figure 4a). Despite the involvement of fewer genes in the serotonergic synapse, burn injury had the most significant impact on serotonergic synaptic processes, as indicated by its high -log10(FDR) value (Figure 4a). Figure 4b presents a comprehensive list of KEGG pathways associated with signaling processes in response to burn injury. Among these pathways, retrograde endocannabinoid signaling is the most upregulated, as indicated by its high enrichment value. Although involving fewer genes, the TNF-, Notch-, and PPAR-signaling pathways demonstrate greater upregulation compared to retrograde endocannabinoid signaling, as indicated by their higher -log10(FDR) values.
Formalin-induced inflammatory pain activates pathways rather than suppressing them (Figure 4c). When applied to the hind paw, formalin primarily upregulates metabolic pathways in the S1 cortex, including purine metabolism and protein digestion/absorption. This is supported by positive enrichment and high log10(FDR) values (Figure 4c).
These findings indicate that burn injury induces unique gene expression patterns in the primary somatosensory cortex, distinct from those observed in the formalin-induced inflammatory pain model.
2.5. Impact of Burn Injury on the Components of the Retrograde Endocannabinoid Network in the S1 Cortex
A KEGG graph generated by Pathview was used to visualize the retrograde endocannabinoid signaling pathway (04723 4/17/17, provided by Kanehisa Labs). This pathway involves the release of endocannabinoids, such as anandamide and 2-AG, from postsynaptic neurons. These endocannabinoids then travel retrogradely to presynaptic neurons, where they regulate neurotransmitter release and influence synaptic plasticity, resulting in both short-term and long-term changes [25,26]. By utilizing Pathview, we mapped our experimental RNA-seq data onto this pathway to illustrate the role of these components in the retrograde endocannabinoid signaling relevant to the molecular mechanisms underlying burn injury. Figure 5 overviews the retrograde endocannabinoid network influenced by burn injury.
Despite an overall increase in retrograde endocannabinoid signaling, as shown in Figure 3 and Figure 4a,b, several key genes were downregulated following burn injury, as illustrated in Figure 5. Specifically, the cannabinoid receptor 1 (CB1R) gene, inward-rectifying potassium channels (referred to as GIRK in Figure 5), and certain voltage-gated L-type calcium channels (noted as VGCC in Figure 5) exhibited decreased expression. The observed results may be attributed to multiple factors, including the temporal dynamics and cell type specificity of the components involved in retrograde endocannabinoid signaling. However, exploring these details was beyond the scope of the current study. Further research is necessary to understand these factors and to gain insight into the complex regulatory mechanisms that influence the pain response to burns.
Moreover, our analysis of RNA-seq data using Pathview revealed that synaptic vesicle cycling, crucial for releasing neurotransmitters from presynaptic terminals, was reduced (Figure 5). In contrast, the overall process of oxidative phosphorylation, vital for energy production, was found to be enhanced in the somatosensory cortex following burn injury (Figure 5).
2.6. Contrary to CB1 Receptors, Ggamma14 Fluorescence Intensity in the S1 Cortex and Ggamma14-IR Cell Numbers in Layer 5 of the S1 Cortex Exhibit Significant Changes One-Hour Post-Burn Injury
Peripheral burn injury can affect and overstimulate the retrograde endocannabinoid network in the cortex (see Figure 3 and Figure 4). Despite its overall upregulation, certain key components of this system, such as CB1 receptors (CB1R), exhibit reduced gene expression, while other components, including Gi/o proteins, show upregulation (Figure 5). To validate these observations at the protein level, an immunofluorescent approach targeting CB1R as well as one member of the heterotrimeric G protein complex, Ggamma14, was employed.
Cannabinoid receptors are distributed throughout the pain circuits, extending from peripheral sensory nerve endings to the brain [27]. The CB1R is located in multiple layers of the rat somatosensory cortex, with notable expression in layers 2/3, and 5/6 [28]. These layers exhibit a high density of CB1R-positive fibers, indicating significant endocannabinoid signaling activity in the primary somatosensory cortex [28]. Consistent with this observation, we obtained similar results in mice (Figure 6 a-b). The fluorescence intensity of the CB1R showed a slight reduction one hour after burn injury, decreasing from 1213.4 ± 70.6 to 911.8 ± 80.5 across the total thickness of the contralateral S1 cortex; however, it did not reach statistically significant levels compared to the control group (5-8 sections from 2 animals per group; Figure 6c). Supplementary Figure S1 illustrates that the fluorescence intensity of the CB1R did not exhibit significant variations across various layers of the S1 cortex.
Ggamma14, also known as G protein subunit gamma 14 (Gγ14), is encoded by the GNGT2 gene and functions as a component of the G protein complex involved in cellular signaling [29,30]. While direct studies linking G gamma signaling to the endocannabinoid system are limited [31], its role in cellular signaling suggests potential involvement in this pathway. The findings from KEGG Pathview provide additional support for this hypothesis (Figure 5). Representative confocal images of Ggamma14-IR in layers 2/3 and 5 from both a control mouse and a burn-injured mouse are displayed in Figure 6a-b. We observed a significant increase in relative fluorescence intensity in the entire thickness of the contralateral S1 cortex following burn injury compared to the control group (p = 0.043; Figure 7d). Specifically, the Ggamma14 fluorescence intensity increased to 106.1 ± 19.7% on the contralateral side and decreased to 95.8 ± 27% on the ipsilateral side (3-5 sections from 2 animals per group; Figure 7d). Interestingly, a noticeable increase was detected in the number of Ggamma14-IR cells in layer 5 post-burn injury (Figure 6e). Layer 5, with its pyramidal neurons, plays a pivotal role in integrating sensory information and transmitting outputs to diverse cortical and subcortical regions. The percentage of Ggamma14-IR cells significantly increased to 202.5% ± 22 in layer 5 of the contralateral S1 cortex (p = 0.015), while it remained unchanged at 94.8% ± on the ipsilateral side (Figure 7e).
In summary, a decrease in CB1 receptor mRNA was observed in the somatosensory cortex one hour post-burn injury. However, no significant difference was detected at the protein level throughout the observation period. This discrepancy between mRNA and protein levels observed one-hour post-injury is likely attributed to the CB1 receptor’s relatively stable half-life of approximately 24 hours, which enables it to maintain functionality over an extended period. Along with the significant increase in Ggamma14 fluorescence intensity in the full thickness of the contralateral S1 cortex, there was a notable increase in the number of Ggamma14-IR cells, specifically in layer 5 of the S1 cortex. This suggests that pyramidal neurons in this layer may play a crucial role in the functional remodeling induced by burn injury, potentially through G-protein signaling pathways.
3. Discussion
Burn injury (BI) is a chronic condition marked by increased sensitivity and spontaneous pain [1,2,3,4]. Its management is often inadequate due to a limited understanding of the underlying mechanisms. Like other chronic pain conditions [5,6,13,15,16,17,18], BI can cause lasting changes in neural activity in specific brain regions, altering sensory perception and pain experiences. Thus, understanding the molecular mechanisms of pain processing in burn injury is crucial. We recently found that pain models from different origins, like burn injury and formalin application to the hind paw, lead to distinct gene expression patterns in the mouse spinal cord [20]. We hypothesized that burn injury (BI)-induced pain also triggers transcriptomic alterations in the primary somatosensory cortex, which may account for the subsequent pain-related behaviors observed afterward.
To elucidate the pathogenesis and neurobiological mechanisms underlying burn injury-induced pain, we conducted transcriptome-wide RNA sequencing (RNA-seq). This was followed by comprehensive bioinformatic analysis to interpret the extensive data generated, aiming to extract meaningful biological insights into gene expression patterns, regulatory mechanisms, and potential therapeutic targets for non-severe burn injury. Mice in the burn injury group were subjected to a full-thickness burn injury, affecting the entire hind leg up to the knee joint, by immersing them in 60°C hot water for two minutes. This represents a burn of about 5% of the total body surface area (TBSA), consistent with a non-severe burn model [32]. We compared the changes in the gene expression pattern caused by burn injury to those induced by formalin-induced inflammatory pain models. Our hypothesis is that pain models originating from various etiologies induce distinct transcriptomic alterations in the S1 cortex, similar to those observed in the spinal cord [20]. We identified differentially expressed genes (DEGs) and clustered them signaling pathways and functional gene networks to better understand the molecular mechanisms involved in pain processing. KEGG pathway analysis has revealed the involvement of retrograde endocannabinoid signaling in the modulation of burn pain within the S1 cortex of mice. Additionally, two components of retrograde endocannabinoid signaling that had exhibited transcriptional changes following burn injury were validated at the protein level using immunofluorescent staining (the CB1 receptor and Ggamma14).
RNA-seq identified 1116 and 136 DEGs in the S1 cortex of burn injury-injured and formalin-treated mice, respectively, compared to the controls. The number of downregulated genes following burn injury was six times higher than that of upregulated genes (920 versus 196). This significant gene suppression may be attributed to two main factors: first, epigenetic modifications, including DNA methylation [11,20]; and second, the activation of stress responses, which tend to prioritize certain pathways (e.g., stress responses) over normal cellular functions. Gene Ontology (GO) enrichment analysis revealed that burn injury significantly altered GO terms related to neuronal functions, including synaptic plasticity, excitability, and axon guidance. In contrast, formalin treatment notably impacted cell cycle-related processes, protein digestion and absorption as well as the metabolism of phospholipids and carbohydrates. This underscores that different painful modalities induce distinct molecular and biochemical processes, resulting in unique transcriptional patterns in the cortex depending on the modality.
In the S1 cortex of wild-type mice following burn injury (BI), we identified 1116 differentially expressed genes (DEGs) using the criteria of -1 < logFC < 1 compared to untreated mice. Among these, 196 DEGs were upregulated, while 920 were downregulated. In this section, we have focused on the genes that are uniquely influenced by the burn stimulus, with particular emphasis on those exhibiting the most significant alterations in log fold change (logFC).
Significant upregulation was observed in genes linked to oxidative phosphorylation (MT-ATP6), glycosphingolipid biosynthesis (A4GALT), and epithelial cell differentiation (LRG1, TAGLN, RBM46). The MT-ATP6 gene plays a vital role in mitochondrial oxidative phosphorylation and the production of ATP. Notably, MT-ATP6 expression is increased in spinal cord samples from both pain models [20]. However, in the S1 cortex, it is overexpressed exclusively after burn injury, likely due to heightened mitochondrial stress. In contrast, the expression of MT-ATP6 decreased in nociceptive dorsal root ganglion (DRG) neurons following sciatic nerve injury [33]. A4GALT is essential for the biosynthesis of glycosphingolipids, which are vital components of cell membranes and important for cell signaling [34]. Glycosphingolipids are implicated in diseases related to nerve injuries and neuropathic and inflammatory pain [35]. Given the role of A4GALT as previously discussed, its increased expression may influence pain mechanisms associated with burn injuries by modifying cell membrane composition and signaling pathways. The TAGLN gene, involved in tissue repair after spinal cord injury [36], and GPR183, which regulates astrocyte migration and signaling [37], showed increased cortical expression after heat exposure. Similarly, LRG1, which promotes wound healing through angiogenesis in diabetic rats [38], was also elevated. The NMR gene that encodes neuromedin B was upregulated following burn injury. In addition to its role in acute itch induction [39], the NMB/NMB-R system is involved in stress responses and may influence them through neural pathways associated with learning and memory [40].
Burn exposure leads to the downregulation of several key genes, including those involved in semaphorin signaling (SEMA5A, PLXNA4), axon guidance (DCC, ERBB4), cytoskeleton remodeling (PLXNA4), and pathways related to synaptic plasticity and neurotransmission (CBLN1, GRIN2A, GRIK3, BSN). The PLXNA4 gene, which encodes the protein Plexin A4, plays a crucial role in axon guidance by interacting with SEMA3A [41]. Another crucial molecule in neuronal morphogenesis is the transmembrane protein semaphorin-5A, which is regulated by sulfated proteoglycans [42]. Additionally, the DCC gene, which encodes the netrin-1 receptor, is significant to this process as well [43]. These findings align with Hozumi’s study, which demonstrated that chronic pain alters neuronal structure in the spinal cord by influencing genes involved in axon and dendrite growth [44]. Interestingly, DCC is highly expressed in the central nervous system and is associated with the basal lamina of epithelial cells [45]. Elevated levels of DCC may, therefore, enhance the structural integrity of the blood-brain barrier by promoting the proper organization and maintenance of endothelial cells and their tight junctions, as verified by the KEEG pathway analysis. Chronic pain is associated with anatomical changes, such as spine remodeling and synaptic reorganization in the spinal cord [46]. In line with the KEGG pathview shown in Figure 5, our findings suggest that burn injury downregulated genes critical for synaptic plasticity and the regulation of neuronal activity, including GRIK3 and GRIN2A. GRIK3 encodes glutamate receptor 7, which is essential for excitatory synaptic transmission mediated by the neurotransmitter L-glutamate. GRIN2A, which encodes the NMDA receptor subunit 2A, was also downregulated. This receptor is crucial for excitatory postsynaptic currents, long-term potentiation, and learning [47]. Numerous studies have established that the BSN gene (Bassoon), which encodes a scaffold protein in the presynaptic cytomatrix, is crucial for presynaptic function [48,49]. A decrease in BSN levels may indicate reduced synaptic activity and potential downregulation of synaptic transmission. Again, the KEGG Pathview analysis derived from our current RNA-seq data supports this assumption. Conversely, increased axonal bouton turnover was observed in chronic neuropathic pain, indicating heightened synaptic activity [50]. The observed discrepancy between our findings and theirs may be attributed to differences in pathophysiological mechanisms and/or the timing of gene expression analyses in the respective models (acute vs. chronic). ERBB4 transcription is crucial for axon guidance and the activation of the MAPK1/ERK2 and MAPK3/ERK1 signaling pathways, which are essential for cell proliferation, neurite outgrowth, and heat sensation in the spinal cord [51,52,53]. As a result, lower levels of ERBB4 transcription are linked to dysfunction in all of these activities. Overall, the downregulation of these genes resulting from burn injury may cause impaired synaptic function, decreased neural plasticity, and potential deficits in sensory processing.
136 DEGs were detected after formalin treatment in wild-type mice compared to the untreated sample, of which 92 were upregulated, and 44 were downregulated. In this section, we have focused on the genes that are uniquely influenced by formalin injection, with particular emphasis on those exhibiting the most significant alterations in log fold change (logFC).
Significant enrichment of upregulated genes was observed in G protein-mediated cell signaling (PCP2, ARHGEF33, NPR1), transcriptional and translational regulation (IKZF1, EIF4EBP3, ATOH8, CYREN), phospholipid homeostasis (e.g., PLA2G4A, CYP39A1), and glycosaminoglycan (GAG) biosynthesis (CHST14). The transcripts of PCP2 and ARHGEF33, which are involved in G protein-mediated signaling and the regulation of Rho GTPases, respectively, were observed to be upregulated in response to formalin treatment. However, their mRNA levels decreased in the burn injury model. PLA2G4A, which encodes a calcium-dependent phospholipase A2 and is essential for phospholipid hydrolysis and cell membrane remodeling, was elevated following FA. Interestingly, the upregulation of PLA2G4A has been demonstrated to induce neurotoxicity in microglia via a MAPK/ERK-dependent mechanism following systemic inflammation [54]. Upon the application of formalin, we observed an elevated NPR1 expression in the S1 cortex, which encodes natriuretic peptide receptor A. Activation of the presynaptic natriuretic peptide receptor A has been found to reduce CFA-induced thermal hyperalgesia in rats [55], indicating that NPR1 activation has an analgesic effect, at least in the context of CFA-induced pain. Furthermore, this receptor may play a role in sensory processing related to itch perception in the spinal cord of mice [56].
Among the top 15 downregulated genes, several are implicated in glycosaminoglycan metabolism (CHST5) and fatty acid metabolism (NUDT7), while others are associated with the cAMP signaling pathway (KCTD14) or ERK/MAPK signaling (PDGFC, TIRAP). The variation in gene expression related to cholesterol and glycosaminoglycan synthesis, with some genes (PLA2G4A, CYP39A1, CHST14) being upregulated and others (CHST5, NUDT7) downregulated, can be attributed to cell type-dependent metabolic shifts that occur during inflammation. Interestingly, the expression of NUDT7 decreased following formalin injection but increased after burn injury. Moreover, CHRNA5, which encodes the alpha-5 subunit of nicotinic acetylcholine receptors (nAChRs), was also suppressed during inflammatory pain. This receptor facilitates fast signal transmission at synapses [57]. The downregulation of cholinergic receptors like CHRNA5 in the S1 cortex can lead to impaired cholinergic signaling, altered neural plasticity, and increased vulnerability to neurodegeneration, all of which can significantly impact neural function [57]. We also found that PRPH expression, which encodes the protein peripherin ˗ essential for axon elongation and regeneration [58] ˗ decreased in the cortex following formalin treatment.
By comparing the two pain models, we identified 59 DEGs in the S1 cortex that responded similarly to noxious treatments. Of these genes, 42 were upregulated, while 17 were downregulated. In this chapter, we focus on genes that show similar changes in response to painful stimuli (BI and FA), emphasizing those with the most significant alterations in log fold change (logFC).
We observed elevated levels of energy-related metabolic pathway genes (e.g., MT-ATP8, MT-CO2), transcriptional regulatory genes (ZFP459 and 764), and stress-responsive genes (e.g., HBA-A2) in pain models, regardless of their origin. Our findings are consistent with previous research indicating that elevated levels of HBA genes, especially HBA-A2, play a role in the cellular response to social stress in the prefrontal cortex [59] and also in response to spinal cord injury [60]. Additionally, HBA2 has been identified in various brain regions, indicating potential roles that extend beyond its known function in oxygen transport (https://www.informatics.jax.org/marker/MGI:96016). Genes associated with oxidative phosphorylation and mitochondrial homeostasis, specifically components of cytochrome c oxidase (MT-CO2) and the mitochondrial proton-transporting ATP synthase complex (MT-ATP8), exhibited upregulation in both nociceptive conditions. Consistent with this observation, a significant increase in the levels of mt-Co2 and mt-Atp6 has been demonstrated in the tibial nerve injury model, as measured by the mitochondria-specific marker Tomm20 [13]. In contrast to our findings, mitochondrial dysfunction has been shown to significantly contribute to the pathophysiology of neuropathic pain [61]. This dysfunction is characterized by the repression of ATP synthase and a reduction in electron transport chain activity by day 7 following the partial sciatic nerve ligation model. The discrepancies between the two studies may be attributed to the different pain models used and the timing of sample collection post-injury (7 days versus one hour).
Both treatments led to a downregulation of genes associated with G-protein-coupled receptors (HTR2C, BDKRB2), ion channels (P2RX3, KCNJ2), and ECM remodeling (GLB1L, THBS4). HTR2C is the gene responsible for encoding the 5-HT2C receptor, a subtype of serotonin receptor that plays a critical role in the regulation of energy homeostasis [62] as well as in the development of neuropathic pain-related behaviors [63]. BDKRB2 is the gene that encodes the bradykinin receptor B2. Bradykinin, a pro-inflammatory peptide, interacts with its receptors to activate pain pathways, which can result in inflammation and neuropathic conditions as previously reported in models of brachial plexus avulsion [64]. Nociceptive stimuli negatively affected the P2X3 receptor for ATP, which is essential for sensory perception [65], and the KCNJ2 potassium channel, which is critical for neuronal excitability [66]. Glycosaminoglycans (GAGs) generally elicit an anti-inflammatory response in pain conditions, including osteoarthritis [67]. However, genes involved in GAG metabolism, such as GLB1L, were downregulated in the acute pain models studied. Thrombospondin-4 (TSP4), which is encoded by the THBS4 gene, is a key regulator of ECM organization, cell-to-cell and cell-to-matrix interactions [68]. It has been reported that TSP4 expression is elevated in the spinal cord following peripheral nerve injuries, contributing to the development of neuropathic, bone cancer, and inflammatory pain [69,70,71]. In contrast, we found that THBS4 was downregulated in the S1 cortex. Consequently, we hypothesize that reducing TSP4 levels may attenuate this hyperexcitability, potentially acting as a protective mechanism to prevent the transition from acute to chronic pain at the onset of injury.
The findings indicate that exposure to stressful stimuli, such as pain, results in alterations in the expression of specific genes, which may be either upregulated or downregulated. This process contributes to a decrease in overall neural excitability and disrupts the organization of the ECM. Such an adaptive response could serve a neuroprotective function by mitigating excessive neuronal activation, thereby potentially aiding in the prevention of chronic pain associated with nociceptive insults.
Through Gene Ontology Enrichment Analysis (GOEA) of the RNA-seq data, we identified several gene clusters involved in key biological processes. Following burn injury, the upregulated genes were predominantly enriched in mitochondrial functions and processes related to gene expression, including ATP metabolism and ribosome activity. In contrast, many downregulated DEGs were associated with axon guidance, synaptic plasticity, and neurotransmission. In the context of burn injury, KEGG pathway analysis identified significant enrichment in several biological processes. These include synaptic function, axon guidance, serotonergic synapses, and various signaling pathways such as TNF, Notch, and PPAR. Furthermore, the analysis emphasized the significance of the retrograde endocannabinoid signaling pathway. In contrast, formalin application leads to the enrichment of genes involved in protein digestion and absorption, phospholipid and carbohydrate metabolism, and processes associated with the cell cycle. This KEGG-based in silico analysis revealed that different painful sensory modalities activate or suppress various functional gene clusters and signaling networks within the S1 cortex. As a result, we hypothesize that the observed changes in transcription may lead to distinct structural and functional remodeling of the cerebral cortex, depending on the specific etiology of pain.
Retrograde endocannabinoid signaling in the cortex plays a vital role in endogenous pain control [72,73,74]. Nonetheless, the specific involvement of its components in the cortical processing of pain associated with burn injury remains unexplored. When neurons in the cortex are activated by painful stimuli, they release endocannabinoids. These endocannabinoids travel backward across synapses to bind to presynaptic cannabinoid receptors. This binding inhibits the release of both inhibitory and excitatory neurotransmitters, which reduces overall neuronal excitability and alters pain perception [72,73]. CB1R activation reduces chronic pain and modulates sensory processing [75,76]. Moreover, in the CNS, activation of CB1R, which is typically coupled to Gi/o-proteins, inhibits adenylate-cyclase and influences gene expression. The oxidative phosphorylation components of the retrograde endocannabinoid signaling pathway highlight the crucial role of mitochondria in this process, as indicated by the KEGG pathview. In addition to its well-known presence in the plasma membrane, the CB1R is also found in mitochondrial membranes [77,78]. In this location, it reduces mitochondrial respiration and contributes to “depolarization-induced suppression of inhibition” by preventing the release of the inhibitory neurotransmitter GABA from inhibitory terminals. Despite the repression of several key proteins involved in retrograde endocannabinoid signaling following burn injury (e.g., CB1R, CACNA1C), KEGG pathway analysis revealed an overall upregulation of the signaling process. This upregulation can be attributed to the increased expression of components involved in oxidative phosphorylation, highlighting the significant interrelationship between these two processes. Consequently, the inclusion of oxidative phosphorylation components in the retrograde endocannabinoid pathway underscores the connection between endocannabinoid signaling and mitochondrial activity, emphasizing the broader impact of this signaling mechanism on cellular energy processes and, more specifically, neuronal and synaptic function.
To the best of our knowledge, this study represents the first comprehensive analysis of gene expression changes and the associated key signaling pathways in the primary somatosensory cortex of a mouse model subjected to burn injury utilizing whole transcriptome bulk RNA sequencing. However, several limitations inherent to this study must be recognized. Firstly, tissue collection for RNA sequencing was performed 1 hour after the induction of nociceptive stimuli, representing the acute phase of pain models. However, due to animal welfare considerations, it was not feasible to examine changes in gene expression at multiple time points. This ethical limitation restricts our ability to gain a comprehensive understanding of the shift of gene expression profiles and the associated dynamics of energy demand following burn injury. Sampling at different intervals would likely produce varying expression patterns. Secondly, neurons and glial cells likely undergo activity-dependent changes in response to tissue injury and pain, influenced by epigenetic tagging [79,80]. This study did not investigate these changes. Future research should employ single nucleus RNA-seq for detailed genetic profiling of specific cell subpopulations. Thirdly, previous studies have suggested that the ipsilateral and contralateral brain regions may have different roles in pain processing [17,81]. However, in our RNA-seq experiments, we did not analyze the two sides of the brain separately; both hind legs were exposed to nociceptive stimuli. As a result, both sides of the brain were considered contralateral to the stimulus. Additionally, we did not investigate the specific changes in the transcriptome resulting from pain stimuli at the level of cortical columns and layers. Therefore, further research is needed to explore any layer-specific differences. Fourthly, a single bulk RNA-seq run was conducted using pooled biological samples from 12 to 13 animals per group rather than employing multiple replicates. Ultimately, while RNA-seq is an effective preliminary screening method, it is essential to conduct further direct experiments to determine whether the transcriptional changes observed in the S1 cortex are linked to changes in neuronal function.
A two-minute peripheral burn injury triggers molecular reprogramming of the central nervous system, resulting in moderate changes not only in the spinal cord [20] but also in higher supraspinal pain pathway-related regions such as the primary somatosensory area. Interestingly, we observed that the majority of the differentially expressed genes (DEGs) were downregulated in the S1 cortex following burn injury. This downregulation may reflect a protective mechanism to reduce neuronal excitability and prevent excessive activation, thereby mitigating the risk of consequent neuronal damage. Factors such as epigenetic changes (e.g., DNA methylation and histone modifications) and microglial activation are likely to contribute to this downregulation, aiming to achieve neuroprotection [11,20].
4. Materials and Methods
Animals and Ethical Considerations
The experiments were conducted on adult male mice, with approval from the Animal Care and Protection Committee at the University of Debrecen (Approval No.: 15-1/2023/DEMÁB). All procedures adhered to the European Community Council Directives and the IASP Guidelines. The animals were housed individually in a temperature-controlled colony room, maintained on a 12-hour light/dark cycle, and provided with food and water ad libitum. Wild-type CD1 mice were obtained from Charles River Laboratories (Wilmington, MA, USA). A total of 43 wild-type CD1 mice were utilized in this study.
Establishment of Burn Injury Model
For RNA sequencing, both hind legs were simultaneously exposed to a non-severe burn injury under deep anesthesia induced by sodium pentobarbital (Release, 50 mg/kg intraperitoneal). Detailed methodology is available [82,83]. In control cases, the hind legs were immersed in 37°C water for 2 minutes. For immunostaining studies, only the left leg was subjected to the stimulus. A total of 13 wild-type CD1 mice were exposed to burn injuries for transcriptome analysis, while an additional 5 mice were used for immunostaining.
Establishment of Formalin-Induced Inflammatory Pain Model
In the formalin-induced inflammatory pain model, 25 μL of a 5% formaldehyde solution was injected intraplantarly under the plantar pads of both hind legs [84] while the mice were under deep anesthesia (50 mg/kg i.p. sodium pentobarbital). This experiment involved 12 CD1 wild-type mice and was used for transcriptome analysis.
Immunofluorescent Staining
Two control and three burn-injured wild-type mice were used for this experiment. After one hour survival period, the experiment was terminated without the animals regaining consciousness, and they were perfused transcardially with a 4% paraformaldehyde (PFA) solution under deep sodium pentobarbital anesthesia. The bilateral S1 cortical region was dissected, post-fixed in 4% paraformaldehyde (PFA) for 3 hours, and sectioned at 100 µm thickness using a vibrating blade vibratome (VT 1000S; Leica Biosystems, Wetzlar, Germany). Immunofluorescent staining was outlined as described in our previous studies [82,85], with the following modification: antigen retrieval was performed in sodium-citrate buffer (pH 6.0) containing 0.05% Tween 20 at 95°C for 30 minutes. Briefly, the primary antibody was incubated at 4°C for two days, followed by an overnight incubation with the secondary antibody. The primary antibodies used in this study are detailed in Table 1.
Species-specific secondary antibodies conjugated to Alexa Fluor-488, 555, and 647 (Invitrogen, Waltham, MA, USA) were utilized. To identify cell nuclei and layer boundaries, 4′,6-diamidino-2-phenylindole (DAPI) staining was applied based on its density. The sections were subsequently mounted in Hydromount (National Diagnostics, Brandon, FL, USA) and imaged using Olympus FV3000 confocal systems (Tokyo, Japan).
Total RNA Isolation and RNA-Seq Analysis
A total of 38 male wild-type CD1 mice were divided into three groups: a control group (n=13), a burns group (n=13), and a formalin-treated group (n=12). Painful stimuli were applied to the hind legs of the mice. After an hour of survival, the experiment was terminalized by perfusing the mice with ice-cold sterile saline without the animals regaining consciousness. The cortical regions associated with the primary somatosensory cortex were immediately excised and placed into TRIzol™ Reagent (Thermo Fisher Scientific Inc., Waltham, MA, USA) for RNA sequencing analysis as detailed in [20]. The RNA concentration was quantified using a NanoDrop spectrophotometer. RNA samples from individual animals were pooled equal amounts into three treatment-specific groups. The quality of the pooled RNA was then assessed using an Agilent BioAnalyzer and the Eukaryotic Total RNA Nano Kit (Agilent Technologies), following the manufacturer’s protocol.
RNA-seq was performed as described previously by [20]. Briefly, samples with an RNA integrity number (RIN) above 8 were selected for library preparation using the NEBNext® Ultra II RNA Sample Preparation Kit (New England BioLabs, Ipswich, MA, USA). Sequencing was conducted on the Illumina NextSeq 500 instrument (Illumina, San Diego, CA) with single-end 75-cycle runs. Raw .bcl files were converted to fastq files and demultiplexed using Illumina’s bcl2fastq software. The data were aligned and normalized with the STAR aligner (version 2.7.11; [87]) and mapped to the mouse genome (Ensembl, GRCm39). Differentially expressed genes were identified using edgeR (version 3.18). Gene expression data were visualized with R packages VennDiagram and ggplot2 (version 4.3.3), along with SRPLOT [88]. Gene pathway analysis was conducted using Cytoscape (version 3.10.1; [89]) with the built-in String and EnrichmentMap apps. Aligned sequencing data have been deposited into the NCBI SRA database under accession number PRJNA1137110.
Data Analysis
For Gene Ontology KEGG pathway analysis, the false discovery rate (FDR) was used to indicate the correlation strength between the identified genes in our dataset and all the genes described in a given pathway, employing the built-in algorithm of the EnrichmentMap app in Cytoscape. In a KEGG bubble chart, the enrichment was calculated with the following formula: E = (#up - #down)/√#total genes, where E is the enrichment, #up is the number of upregulated genes, #down is the number of the downregulated genes and #total is the number of all genes found in the particular KEGG pathway. It is used to signify the relative importance or significance of a pathway within the context of the analyzed data. All data obtained from immunostaining are presented as the mean ± standard error of the mean (SEM). Statistical analyses were performed with Past4 software using the Mann–Whitney U-test.
5. Conclusions
This study utilized a systematic and unbiased approach to document alterations in gene expression within the primary somatosensory cortex at the early onset of a full-thickness burn injury and an inflammatory pain model. The findings offer novel insights into the distinct molecular mechanisms underlying pain processing across different pain models. These results highlight the complex molecular pathways driving sensory modality-specific responses, providing valuable information for potential therapeutic targets and experimental hypotheses for future research.
While the spinal cord undergoes epigenetic and transcriptomic reorganization to amplify burning pain signals [20], the S1 cortex regulates gene expression downwards to safeguard neurons against overactivation and potential damage. These contrasting responses illustrate the complexity and specificity involved in pain processing mechanisms across different regions of the CNS.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Table S1: List of the top 15 upregulated and downregulated differentially expressed genes (DEGs) in the primary somatosensory cortex (S1) in response to burn injury (BI). Supplementary Table S2: List of the top 15 upregulated and downregulated differentially expressed genes (DEGs) in the primary somatosensory cortex (S1) in response to formalin-induced inflammatory pain (FA). Supplementary Table S3: List of the top 15 genes upregulated or downregulated similarly in the S1 cortex by both burn injury (BI) and formalin-induced inflammatory pain (FA). Supplementary Figure S1: Layer-specific CB1 Receptor fluorescence intensity in the S1 cortex shows no significant change one-hour post-burn injury.
Author Contributions
Conceptualization, AV and ZM; Performance of experiments, VE, AV, ZM; Data analysis, VE, AV, ZM; Original Draft preparation, AV; Review & Editing, ZM. All authors have read and agreed to the submitted version of the manuscript.
Funding
2019-2.1.7-ERA-NET-2021-00039 has been implemented with the support provided by the Ministry of Culture and Innovation of Hungary from the National Research, Development and Innovation Fund, financed under the 2019-2.1.7-ERA-NET funding scheme (This action has received funding from the ERA-NET COFUND /EJP COFUND Programme with co-funding from the European Union Horizon 2020 research and innovation programme). TKP2021-EGA-20 has been implemented with the support provided by the Ministry of Culture and Innovation of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-EGA funding scheme.
Institutional Review Board Statement
The animal study protocol was approved by the University of Debrecen’s Animal Care and Protection Committee (No. 15-1/2023/DEMÁB) for studies involving animals.
Data Availability Statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
Acknowledgments
We thank Mrs. Tímea Nánási M. for her technical animal maintenance and breeding assistance.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Allahham, A.; Rowe, G.; Stevenson, A.; Fear, M.W.; Vallence, A.-M.; Wood, F.M. The Impact of Burn Injury on the Central Nervous System. Burns Trauma 2024, 12, tkad037. [Google Scholar] [CrossRef] [PubMed]
- Vetrichevvel, T.P.; Randall, S.M.; Fear, M.W.; Wood, F.M.; Boyd, J.H.; Duke, J.M. Burn Injury and Long-Term Nervous System Morbidity: A Population-Based Cohort Study. BMJ Open 2016, 6, e012668. [Google Scholar] [CrossRef]
- Xie, C.; Hu, J.; Cheng, Y.; Yao, Z. Researches on Cognitive Sequelae of Burn Injury: Current Status and Advances. Front. Neurosci. 2022, 16, 1026152. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, D.; Zhang, J.; Wang, Y. Pathological Changes in the Brain after Peripheral Burns. Burns Trauma 2023, 11, tkac061. [Google Scholar] [CrossRef] [PubMed]
- Seifert, F.; Maihöfner, C. Central Mechanisms of Experimental and Chronic Neuropathic Pain: Findings from Functional Imaging Studies. Cell. Mol. Life Sci. CMLS 2009, 66, 375–390. [Google Scholar] [CrossRef]
- Bak, M.S.; Park, H.; Kim, S.K. Neural Plasticity in the Brain during Neuropathic Pain. Biomedicines 2021, 9, 624. [Google Scholar] [CrossRef]
- Ong, W.-Y.; Stohler, C.S.; Herr, D.R. Role of the Prefrontal Cortex in Pain Processing. Mol. Neurobiol. 2019, 56, 1137–1166. [Google Scholar] [CrossRef]
- Sun, G.; McCartin, M.; Liu, W.; Zhang, Q.; Kenefati, G.; Chen, Z.S.; Wang, J. Temporal Pain Processing in the Primary Somatosensory Cortex and Anterior Cingulate Cortex. Mol. Brain 2023, 16, 3. [Google Scholar] [CrossRef]
- Quiton, R.L.; Masri, R.; Thompson, S.M.; Keller, A. Abnormal Activity of Primary Somatosensory Cortex in Central Pain Syndrome. J. Neurophysiol. 2010, 104, 1717–1725. [Google Scholar] [CrossRef]
- Timmermann, L.; Ploner, M.; Haucke, K.; Schmitz, F.; Baltissen, R.; Schnitzler, A. Differential Coding of Pain Intensity in the Human Primary and Secondary Somatosensory Cortex. J. Neurophysiol. 2001, 86, 1499–1503. [Google Scholar] [CrossRef]
- Tao, W.; Chen, C.; Wang, Y.; Zhou, W.; Jin, Y.; Mao, Y.; Wang, H.; Wang, L.; Xie, W.; Zhang, X.; et al. MeCP2 Mediates Transgenerational Transmission of Chronic Pain. Prog. Neurobiol. 2020, 189, 101790. [Google Scholar] [CrossRef] [PubMed]
- Orczyk, J.J.; Sethia, R.; Doster, D.; Garraghty, P.E. Transcriptome Response to Infraorbital Nerve Transection in the Gonadally Intact Male Rat Barrel Cortex: RNA-Seq. J. Comp. Neurol. 2016, 524, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Ping, X.; Xie, J.; Yuan, C.; Jin, X. Electroacupuncture Induces Bilateral S1 and ACC Epigenetic Regulation of Genes in a Mouse Model of Neuropathic Pain. Biomedicines 2023, 11, 1030. [Google Scholar] [CrossRef] [PubMed]
- Labus, J.S.; Osadchiy, V.; Hsiao, E.Y.; Tap, J.; Derrien, M.; Gupta, A.; Tillisch, K.; Le Nevé, B.; Grinsvall, C.; Ljungberg, M.; et al. Evidence for an Association of Gut Microbial Clostridia with Brain Functional Connectivity and Gastrointestinal Sensorimotor Function in Patients with Irritable Bowel Syndrome, Based on Tripartite Network Analysis. Microbiome 2019, 7, 45. [Google Scholar] [CrossRef]
- Blom, S.M.; Pfister, J.-P.; Santello, M.; Senn, W.; Nevian, T. Nerve Injury-Induced Neuropathic Pain Causes Disinhibition of the Anterior Cingulate Cortex. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 5754–5764. [Google Scholar] [CrossRef]
- Alvarado, S.; Tajerian, M.; Millecamps, M.; Suderman, M.; Stone, L.S.; Szyf, M. Peripheral Nerve Injury Is Accompanied by Chronic Transcriptome-Wide Changes in the Mouse Prefrontal Cortex. Mol. Pain 2013, 9, 21. [Google Scholar] [CrossRef]
- Zheng, Q.-M.; Zhou, Z.-R.; Hou, X.-Y.; Lv, N.; Zhang, Y.-Q.; Cao, H. Transcriptome Analysis of the Mouse Medial Prefrontal Cortex in a Chronic Constriction Injury Model. Neuromolecular Med. 2023, 25, 375–387. [Google Scholar] [CrossRef]
- Li, H.; Li, X.; Wang, J.; Gao, F.; Wiech, K.; Hu, L.; Kong, Y. Pain-Related Reorganization in the Primary Somatosensory Cortex of Patients with Postherpetic Neuralgia. Hum. Brain Mapp. 2022, 43, 5167–5179. [Google Scholar] [CrossRef]
- Sneddon, L.U. Comparative Physiology of Nociception and Pain. Physiol. Bethesda Md 2018, 33, 63–73. [Google Scholar] [CrossRef]
- Mészár, Z.; Erdei, V.; Szücs, P.; Varga, A. Epigenetic Regulation and Molecular Mechanisms of Burn Injury-Induced Nociception in the Spinal Cord of Mice. Int. J. Mol. Sci. 2024, 25, 8510. [Google Scholar] [CrossRef]
- Lee, G.J.; Kim, Y.J.; Lee, K.; Oh, S.B. Patterns of Brain C-Fos Expression in Response to Feeding Behavior in Acute and Chronic Inflammatory Pain Condition. Neuroreport 2021, 32, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qin, Y.; Zhong, Z.; Meng, L.; Huang, L.; Li, B. Pain Experience Reduces Social Avoidance to Others in Pain: A c-Fos-Based Functional Connectivity Network Study in Mice. Cereb. Cortex N. Y. N 1991 2024, 34, bhae207. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Qin, Y.; Yang, S.; Zhang, H.; Zheng, J.; Wen, S.; Li, W.; Liang, Z.; Zhang, X.; Li, B.; et al. Differences in the Neural Basis and Transcriptomic Patterns in Acute and Persistent Pain-Related Anxiety-like Behaviors. Front. Mol. Neurosci. 2023, 16, 1185243. [Google Scholar] [CrossRef] [PubMed]
- Hjornevik, T.; Leergaard, T.B.; Darine, D.; Moldestad, O.; Dale, A.M.; Willoch, F.; Bjaalie, J.G. Three-Dimensional Atlas System for Mouse and Rat Brain Imaging Data. Front. Neuroinformatics 2007, 1, 4. [Google Scholar] [CrossRef]
- Donvito, G.; Nass, S.R.; Wilkerson, J.L.; Curry, Z.A.; Schurman, L.D.; Kinsey, S.G.; Lichtman, A.H. The Endogenous Cannabinoid System: A Budding Source of Targets for Treating Inflammatory and Neuropathic Pain. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2018, 43, 52–79. [Google Scholar] [CrossRef]
- Wilson, R.I.; Nicoll, R.A. Endocannabinoid Signaling in the Brain. Science 2002, 296, 678–682. [Google Scholar] [CrossRef]
- Manzanares, J.; Julian, M.; Carrascosa, A. Role of the Cannabinoid System in Pain Control and Therapeutic Implications for the Management of Acute and Chronic Pain Episodes. Curr. Neuropharmacol. 2006, 4, 239–257. [Google Scholar] [CrossRef]
- Bodor, A.L.; Katona, I.; Nyíri, G.; Mackie, K.; Ledent, C.; Hájos, N.; Freund, T.F. Endocannabinoid Signaling in Rat Somatosensory Cortex: Laminar Differences and Involvement of Specific Interneuron Types. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 6845–6856. [Google Scholar] [CrossRef]
- Tennakoon, M.; Senarath, K.; Kankanamge, D.; Ratnayake, K.; Wijayaratna, D.; Olupothage, K.; Ubeysinghe, S.; Martins-Cannavino, K.; Hébert, T.E.; Karunarathne, A. Subtype-Dependent Regulation of Gβγ Signalling. Cell. Signal. 2021, 82, 109947. [Google Scholar] [CrossRef]
- Tennakoon, M.; Senarath, K.; Kankanamge, D.; Chadee, D.N.; Karunarathne, A. A Short C-Terminal Peptide in Gγ Regulates Gβγ Signaling Efficacy. Mol. Biol. Cell 2021, 32, 1446–1458. [Google Scholar] [CrossRef]
- Shi, C.; Szczesniak, A.; Mao, L.; Jollimore, C.; Coca-Prados, M.; Hung, O.; Kelly, M.E.M. A3 Adenosine and CB1 Receptors Activate a PKC-Sensitive Cl- Current in Human Nonpigmented Ciliary Epithelial Cells via a G Beta Gamma-Coupled MAPK Signaling Pathway. Br. J. Pharmacol. 2003, 139, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Bruen, K.J.; Campbell, C.A.; Schooler, W.G.; deSerres, S.; Cairns, B.A.; Hultman, C.S.; Meyer, A.A.; Randell, S.H. Real-Time Monitoring of Keratin 5 Expression during Burn Re-Epithelialization. J. Surg. Res. 2004, 120, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-H.; Lin, C.-R.; Cheng, J.-T.; Cheng, J.-K.; Liao, W.-T.; Yang, C.-H. Altered Mitochondrial ATP Synthase Expression in the Rat Dorsal Root Ganglion after Sciatic Nerve Injury and Analgesic Effects of Intrathecal ATP. Cell. Mol. Neurobiol. 2014, 34, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Sonnino, S.; Mauri, L.; Ciampa, M.G.; Prinetti, A. Gangliosides as Regulators of Cell Signaling: Ganglioside-Protein Interactions or Ganglioside-Driven Membrane Organization? J. Neurochem. 2013, 124, 432–435. [Google Scholar] [CrossRef]
- Sántha, P.; Dobos, I.; Kis, G.; Jancsó, G. Role of Gangliosides in Peripheral Pain Mechanisms. Int. J. Mol. Sci. 2020, 21, 1005. [Google Scholar] [CrossRef]
- Xiao, L.; Ma, Z.-L.; Li, X.; Lin, Q.-X.; Que, H.-P.; Liu, S.-J. cDNA Microarray Analysis of Spinal Cord Injury and Regeneration Related Genes in Rat. Sheng Li Xue Bao 2005, 57, 705–713. [Google Scholar]
- Rutkowska, A.; Preuss, I.; Gessier, F.; Sailer, A.W.; Dev, K.K. EBI2 Regulates Intracellular Signaling and Migration in Human Astrocyte. Glia 2015, 63, 341–351. [Google Scholar] [CrossRef]
- Yang, P.; Li, S.; Zhang, H.; Ding, X.; Tan, Q. LRG1 Accelerates Wound Healing in Diabetic Rats by Promoting Angiogenesis via the Wnt/β-Catenin Signaling Pathway. Int. J. Low. Extrem. Wounds 2024, 23, 568–576. [Google Scholar] [CrossRef]
- Kiguchi, N.; Ding, H.; Park, S.H.; Mabry, K.M.; Kishioka, S.; Shiozawa, Y.; Alfonso Romero-Sandoval, E.; Peters, C.M.; Ko, M.-C. Functional Roles of Neuromedin B and Gastrin-Releasing Peptide in Regulating Itch and Pain in the Spinal Cord of Non-Human Primates. Biochem. Pharmacol. 2022, 198, 114972. [Google Scholar] [CrossRef]
- Yamada, K.; Santo-Yamada, Y.; Wada, K. Stress-Induced Impairment of Inhibitory Avoidance Learning in Female Neuromedin B Receptor-Deficient Mice. Physiol. Behav. 2003, 78, 303–309. [Google Scholar] [CrossRef]
- Suto, F.; Ito, K.; Uemura, M.; Shimizu, M.; Shinkawa, Y.; Sanbo, M.; Shinoda, T.; Tsuboi, M.; Takashima, S.; Yagi, T.; et al. Plexin-A4 Mediates Axon-Repulsive Activities of Both Secreted and Transmembrane Semaphorins and Plays Roles in Nerve Fiber Guidance. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 3628–3637. [Google Scholar] [CrossRef] [PubMed]
- Kantor, D.B.; Chivatakarn, O.; Peer, K.L.; Oster, S.F.; Inatani, M.; Hansen, M.J.; Flanagan, J.G.; Yamaguchi, Y.; Sretavan, D.W.; Giger, R.J.; et al. Semaphorin 5A Is a Bifunctional Axon Guidance Cue Regulated by Heparan and Chondroitin Sulfate Proteoglycans. Neuron 2004, 44, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Finci, L.; Zhang, Y.; Meijers, R.; Wang, J.-H. Signaling Mechanism of the Netrin-1 Receptor DCC in Axon Guidance. Prog. Biophys. Mol. Biol. 2015, 118, 153–160. [Google Scholar] [CrossRef]
- Hozumi, T.; Sawai, S.; Jitsuishi, T.; Kitajo, K.; Inage, K.; Eguchi, Y.; Shiga, Y.; Narita, M.; Orita, S.; Ohtori, S.; et al. Gene Expression Profiling of the Spinal Cord at the Chronic Pain Phase Identified CDKL5 as a Candidate Gene for Neural Remodeling. Neurosci. Lett. 2021, 749, 135772. [Google Scholar] [CrossRef]
- Schneiders, F.I.; Maertens, B.; Böse, K.; Li, Y.; Brunken, W.J.; Paulsson, M.; Smyth, N.; Koch, M. Binding of Netrin-4 to Laminin Short Arms Regulates Basement Membrane Assembly. J. Biol. Chem. 2007, 282, 23750–23758. [Google Scholar] [CrossRef]
- Kuner, R.; Flor, H. Structural Plasticity and Reorganisation in Chronic Pain. Nat. Rev. Neurosci. 2016, 18, 20–30. [Google Scholar] [CrossRef]
- Tumdam, R.; Hussein, Y.; Garin-Shkolnik, T.; Stern, S. NMDA Receptors in Neurodevelopmental Disorders: Pathophysiology and Disease Models. Int. J. Mol. Sci. 2024, 25, 12366. [Google Scholar] [CrossRef]
- Dresbach, T.; Hempelmann, A.; Spilker, C.; tom Dieck, S.; Altrock, W.D.; Zuschratter, W.; Garner, C.C.; Gundelfinger, E.D. Functional Regions of the Presynaptic Cytomatrix Protein Bassoon: Significance for Synaptic Targeting and Cytomatrix Anchoring. Mol. Cell. Neurosci. 2003, 23, 279–291. [Google Scholar] [CrossRef]
- Terry-Lorenzo, R.T.; Torres, V.I.; Wagh, D.; Galaz, J.; Swanson, S.K.; Florens, L.; Washburn, M.P.; Waites, C.L.; Gundelfinger, E.D.; Reimer, R.J.; et al. Trio, a Rho Family GEF, Interacts with the Presynaptic Active Zone Proteins Piccolo and Bassoon. PloS One 2016, 11, e0167535. [Google Scholar] [CrossRef]
- Kim, S.K.; Nabekura, J. Rapid Synaptic Remodeling in the Adult Somatosensory Cortex Following Peripheral Nerve Injury and Its Association with Neuropathic Pain. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 5477–5482. [Google Scholar] [CrossRef]
- Vaskovsky, A.; Lupowitz, Z.; Erlich, S.; Pinkas-Kramarski, R. ErbB-4 Activation Promotes Neurite Outgrowth in PC12 Cells. J. Neurochem. 2000, 74, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, W.; Dong, Z.; Xing, G.; Cui, W.; Yao, L.; Zou, W.-J.; Robinson, H.L.; Bian, Y.; Liu, Z.; et al. A Novel Spinal Neuron Connection for Heat Sensation. Neuron 2022, 110, 2315–2333.e6. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Cranfill, S.L.; Luo, W. ErbB4+ Spinal Cord Dorsal Horn Neurons Process Heat Pain. Neuron 2022, 110, 2206–2208. [Google Scholar] [CrossRef] [PubMed]
- Plastira, I.; Bernhart, E.; Joshi, L.; Koyani, C.N.; Strohmaier, H.; Reicher, H.; Malle, E.; Sattler, W. MAPK Signaling Determines Lysophosphatidic Acid (LPA)-Induced Inflammation in Microglia. J. Neuroinflammation 2020, 17, 127. [Google Scholar] [CrossRef]
- Zhang, F.-X.; Liu, X.-J.; Gong, L.-Q.; Yao, J.-R.; Li, K.-C.; Li, Z.-Y.; Lin, L.-B.; Lu, Y.-J.; Xiao, H.-S.; Bao, L.; et al. Inhibition of Inflammatory Pain by Activating B-Type Natriuretic Peptide Signal Pathway in Nociceptive Sensory Neurons. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 10927–10938. [Google Scholar] [CrossRef]
- Fatima, M.; Ren, X.; Pan, H.; Slade, H.F.E.; Asmar, A.J.; Xiong, C.M.; Shi, A.; Xiong, A.E.; Wang, L.; Duan, B. Spinal Somatostatin-Positive Interneurons Transmit Chemical Itch. Pain 2019, 160, 1166–1174. [Google Scholar] [CrossRef]
- Venkatesan, S.; Lambe, E.K. Chrna5 Is Essential for a Rapid and Protected Response to Optogenetic Release of Endogenous Acetylcholine in Prefrontal Cortex. J. Neurosci. Off. J. Soc. Neurosci. 2020, 40, 7255–7268. [Google Scholar] [CrossRef]
- Chadan, S.; Moya, K.L.; Portier, M.M.; Filliatreau, G. Identification of a Peripherin Dimer: Changes during Axonal Development and Regeneration of the Rat Sciatic Nerve. J. Neurochem. 1994, 62, 1894–1905. [Google Scholar] [CrossRef]
- Stankiewicz, A.M.; Goscik, J.; Swiergiel, A.H.; Majewska, A.; Wieczorek, M.; Juszczak, G.R.; Lisowski, P. Social Stress Increases Expression of Hemoglobin Genes in Mouse Prefrontal Cortex. BMC Neurosci. 2014, 15, 130. [Google Scholar] [CrossRef]
- Sugeno, A.; Piao, W.; Yamazaki, M.; Takahashi, K.; Arikawa, K.; Matsunaga, H.; Hosokawa, M.; Tominaga, D.; Goshima, Y.; Takeyama, H.; et al. Cortical Transcriptome Analysis after Spinal Cord Injury Reveals the Regenerative Mechanism of Central Nervous System in CRMP2 Knock-in Mice. Neural Regen. Res. 2021, 16, 1258–1265. [Google Scholar] [CrossRef]
- Lim, T.K.Y.; Rone, M.B.; Lee, S.; Antel, J.P.; Zhang, J. Mitochondrial and Bioenergetic Dysfunction in Trauma-Induced Painful Peripheral Neuropathy. Mol. Pain 2015, 11, 58. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; He, J.; Cui, Z.; Wang, R.; Bao, K.; Huang, Y.; Wang, R.; Liu, T. Central 5-HTR2C in the Control of Metabolic Homeostasis. Front. Endocrinol. 2021, 12, 694204. [Google Scholar] [CrossRef] [PubMed]
- Nakae, A.; Nakai, K.; Tanaka, T.; Hosokawa, K.; Mashimo, T. Serotonin 2C Receptor Alternative Splicing in a Spinal Cord Injury Model. Neurosci. Lett. 2013, 532, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Quintão, N.L.M.; Passos, G.F.; Medeiros, R.; Paszcuk, A.F.; Motta, F.L.; Pesquero, J.B.; Campos, M.M.; Calixto, J.B. Neuropathic Pain-like Behavior after Brachial Plexus Avulsion in Mice: The Relevance of Kinin B1 and B2 Receptors. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 2856–2863. [Google Scholar] [CrossRef]
- Li, M.; Wang, Y.; Banerjee, R.; Marinelli, F.; Silberberg, S.; Faraldo-Gómez, J.D.; Hattori, M.; Swartz, K.J. Molecular Mechanisms of Human P2X3 Receptor Channel Activation and Modulation by Divalent Cation Bound ATP. eLife 2019, 8, e47060. [Google Scholar] [CrossRef]
- Prüss, H.; Derst, C.; Lommel, R.; Veh, R.W. Differential Distribution of Individual Subunits of Strongly Inwardly Rectifying Potassium Channels (Kir2 Family) in Rat Brain. Mol. Brain Res. 2005, 139, 63–79. [Google Scholar] [CrossRef]
- Bishnoi, M.; Jain, A.; Hurkat, P.; Jain, S.K. Chondroitin Sulphate: A Focus on Osteoarthritis. Glycoconj. J. 2016, 33, 693–705. [Google Scholar] [CrossRef]
- Stenina-Adognravi, O.; Plow, E.F. Thrombospondin-4 in Tissue Remodeling. Matrix Biol. J. Int. Soc. Matrix Biol. 2019, 75–76, 300–313. [Google Scholar] [CrossRef]
- Crosby, N.D.; Zaucke, F.; Kras, J.V.; Dong, L.; Luo, Z.D.; Winkelstein, B.A. Thrombospondin-4 and Excitatory Synaptogenesis Promote Spinal Sensitization after Painful Mechanical Joint Injury. Exp. Neurol. 2015, 264, 111–120. [Google Scholar] [CrossRef]
- Düzenli, N.; Ülker, S.; Şengül, G.; Kayhan, B.; Önal, A. Effects of Cyanocobalamin and Its Combination with Morphine on Neuropathic Rats and the Relationship between These Effects and Thrombospondin-4 Expression. Korean J. Pain 2022, 35, 66–77. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, M.; Xu, X.; Gao, Y.; Li, X.; Li, Y.; Su, S.; Xie, X.; Yang, Z.; Ke, C. Thrombospondin 4, a Mediator and Candidate Indicator of Pain. Eur. J. Cell Biol. 2024, 103, 151395. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Kumar, U. Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef] [PubMed]
- Noriega-Prieto, J.A.; Kofuji, P.; Araque, A. Endocannabinoid Signaling in Synaptic Function. Glia 2023, 71, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Urits, I.; Orhurhu, V.; Peck, J.; Orhurhu, M.S.; Giacomazzi, S.; Smoots, D.; Piermarini, C.; Manchikanti, L.; Kaye, A.D.; et al. The Role of the Cannabinoid System in Pain Control: Basic and Clinical Implications. Curr. Pain Headache Rep. 2020, 24, 35. [Google Scholar] [CrossRef]
- Agarwal, N.; Pacher, P.; Tegeder, I.; Amaya, F.; Constantin, C.E.; Brenner, G.J.; Rubino, T.; Michalski, C.W.; Marsicano, G.; Monory, K.; et al. Cannabinoids Mediate Analgesia Largely via Peripheral Type 1 Cannabinoid Receptors in Nociceptors. Nat. Neurosci. 2007, 10, 870–879. [Google Scholar] [CrossRef]
- Bishay, P.; Schmidt, H.; Marian, C.; Häussler, A.; Wijnvoord, N.; Ziebell, S.; Metzner, J.; Koch, M.; Myrczek, T.; Bechmann, I.; et al. R-Flurbiprofen Reduces Neuropathic Pain in Rodents by Restoring Endogenous Cannabinoids. PloS One 2010, 5, e10628. [Google Scholar] [CrossRef]
- Xu, Z.; Lv, X.-A.; Dai, Q.; Ge, Y.-Q.; Xu, J. Acute Upregulation of Neuronal Mitochondrial Type-1 Cannabinoid Receptor and It’s Role in Metabolic Defects and Neuronal Apoptosis after TBI. Mol. Brain 2016, 9, 75. [Google Scholar] [CrossRef]
- Cosentino, L.; Urbinati, C.; Lanzillotta, C.; De Rasmo, D.; Valenti, D.; Pellas, M.; Quattrini, M.C.; Piscitelli, F.; Kostrzewa, M.; Di Domenico, F.; et al. Pharmacological Inhibition of the CB1 Cannabinoid Receptor Restores Abnormal Brain Mitochondrial CB1 Receptor Expression and Rescues Bioenergetic and Cognitive Defects in a Female Mouse Model of Rett Syndrome. Mol. Autism 2024, 15, 39. [Google Scholar] [CrossRef]
- Imai, S.; Ikegami, D.; Yamashita, A.; Shimizu, T.; Narita, M.; Niikura, K.; Furuya, M.; Kobayashi, Y.; Miyashita, K.; Okutsu, D.; et al. Epigenetic Transcriptional Activation of Monocyte Chemotactic Protein 3 Contributes to Long-Lasting Neuropathic Pain. Brain J. Neurol. 2013, 136, 828–843. [Google Scholar] [CrossRef]
- Liang, L.; Tao, Y.-X. Expression of Acetyl-Histone H3 and Acetyl-Histone H4 in Dorsal Root Ganglion and Spinal Dorsal Horn in Rat Chronic Pain Models. Life Sci. 2018, 211, 182–188. [Google Scholar] [CrossRef]
- Wang, G.-Q.; Cen, C.; Li, C.; Cao, S.; Wang, N.; Zhou, Z.; Liu, X.-M.; Xu, Y.; Tian, N.-X.; Zhang, Y.; et al. Deactivation of Excitatory Neurons in the Prelimbic Cortex via Cdk5 Promotes Pain Sensation and Anxiety. Nat. Commun. 2015, 6, 7660. [Google Scholar] [CrossRef] [PubMed]
- Varga, A.; Mészár, Z.; Sivadó, M.; Bácskai, T.; Végh, B.; Kókai, É.; Nagy, I.; Szücs, P. Spinal Excitatory Dynorphinergic Interneurons Contribute to Burn Injury-Induced Nociception Mediated by Phosphorylated Histone 3 at Serine 10 in Rodents. Int. J. Mol. Sci. 2021, 22, 2297. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.M.; Ko, C.W.; Fidalgo, A.R.; Cibelli, M.; Paule, C.C.; Anderson, P.J.; Cruz, C.; Gomba, S.; Matesz, K.; Veress, G.; et al. Severe Burn Injury Induces a Characteristic Activation of Extracellular Signal-Regulated Kinase 1/2 in Spinal Dorsal Horn Neurons. Eur. J. Pain Lond. Engl. 2011, 15, 683–690. [Google Scholar] [CrossRef]
- Tjølsen, A.; Berge, O.-G.; Hunskaar, S.; Rosland, J.H.; Hole, K. The Formalin Test: An Evaluation of the Method. Pain 1992, 51, 5–17. [Google Scholar] [CrossRef]
- Mészár, Z.; Kókai, É.; Varga, R.; Ducza, L.; Papp, T.; Béresová, M.; Nagy, M.; Szücs, P.; Varga, A. CRISPR/Cas9-Based Mutagenesis of Histone H3.1 in Spinal Dynorphinergic Neurons Attenuates Thermal Sensitivity in Mice. Int. J. Mol. Sci. 2022, 23, 3178. [Google Scholar] [CrossRef]
- Echeazarra, L.; Barrondo, S.; García Del Caño, G.; Bonilla-Del Río, I.; Egaña-Huguet, J.; Puente, N.; Aretxabala, X.; Montaña, M.; López de Jesús, M.; González-Burguera, I.; et al. Up-Regulation of CB1 Cannabinoid Receptors Located at Glutamatergic Terminals in the Medial Prefrontal Cortex of the Obese Zucker Rat. Front. Neuroanat. 2022, 16, 1004702. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinforma. Oxf. Engl. 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Tang, D.; Chen, M.; Huang, X.; Zhang, G.; Zeng, L.; Zhang, G.; Wu, S.; Wang, Y. SRplot: A Free Online Platform for Data Visualization and Graphing. PloS One 2023, 18, e0294236. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
Figure 1.
The impact of burn injury on cortical neuronal activity: c-Fos immunoreactivity analysis. (a) Schematic representation illustrating the time scale of this experiment. (b) Schematic drawing of a coronal section of the mouse brain representing the lower limb area of the S1 cortex. (c) Representative immunostaining with an antibody against c-Fos (green) was obtained from four optical sections of a coronal slice of a wild-type mouse brain following burn injury. DAPI counterstained cell nuclei for blue. The term “burn injury-contralateral” specifically refers to the right hemisphere of the brain, which is opposite the burn injury site. After the burn injury, c-Fos immunoreactivity in the contralateral S1 cortex increased along the entire length of the somatosensory cortex. Scale bar, 220 μm. (d) Quantitative analysis of the number of c-Fos-IR nuclei in the S1 cortex following burn injury, relative to the control. The data are presented as mean ± SEM (5-6 slices from 2 animals per group). Asterisk means p < 0.05. (e) Quantitative analysis of c-Fos fluorescence intensity in the S1 cortex following burn injury, relative to the control. The data are presented as mean ± SEM (5-6 slices from 2 animals per group). Asterisk means p < 0.05. (f) Representative triple immunostaining with antibodies against c-Fos, parvalbumin (Pvalb), and calretinin (CR) from layer 2/3 of the contralateral-sided S1 cortex following burn injury. Arrowhead shows a CR-positive interneuron that expresses c-Fos. Scale bar, 30 um. (g) Representative triple immunostaining with antibodies against c-Fos, calbindin (Calb), and a nuclear marker, DAPI, from layer 2/3 of the contralateral-sided S1 cortex following burn injury. The dotted line indicates the boundary between layer 1 and layer 2/3. Arrowheads show co-localizations of c-Fos and Calb. Scale bar, 30 um. Abbreviations: WT, wild-type; PFA, paraformaldehyde; S1, primary somatosensory cortex; MOp, primary motor cortex; V3, third ventricle; IC, internal capsule; LV, lateral ventricle; CC, corpus callosum.
Figure 1.
The impact of burn injury on cortical neuronal activity: c-Fos immunoreactivity analysis. (a) Schematic representation illustrating the time scale of this experiment. (b) Schematic drawing of a coronal section of the mouse brain representing the lower limb area of the S1 cortex. (c) Representative immunostaining with an antibody against c-Fos (green) was obtained from four optical sections of a coronal slice of a wild-type mouse brain following burn injury. DAPI counterstained cell nuclei for blue. The term “burn injury-contralateral” specifically refers to the right hemisphere of the brain, which is opposite the burn injury site. After the burn injury, c-Fos immunoreactivity in the contralateral S1 cortex increased along the entire length of the somatosensory cortex. Scale bar, 220 μm. (d) Quantitative analysis of the number of c-Fos-IR nuclei in the S1 cortex following burn injury, relative to the control. The data are presented as mean ± SEM (5-6 slices from 2 animals per group). Asterisk means p < 0.05. (e) Quantitative analysis of c-Fos fluorescence intensity in the S1 cortex following burn injury, relative to the control. The data are presented as mean ± SEM (5-6 slices from 2 animals per group). Asterisk means p < 0.05. (f) Representative triple immunostaining with antibodies against c-Fos, parvalbumin (Pvalb), and calretinin (CR) from layer 2/3 of the contralateral-sided S1 cortex following burn injury. Arrowhead shows a CR-positive interneuron that expresses c-Fos. Scale bar, 30 um. (g) Representative triple immunostaining with antibodies against c-Fos, calbindin (Calb), and a nuclear marker, DAPI, from layer 2/3 of the contralateral-sided S1 cortex following burn injury. The dotted line indicates the boundary between layer 1 and layer 2/3. Arrowheads show co-localizations of c-Fos and Calb. Scale bar, 30 um. Abbreviations: WT, wild-type; PFA, paraformaldehyde; S1, primary somatosensory cortex; MOp, primary motor cortex; V3, third ventricle; IC, internal capsule; LV, lateral ventricle; CC, corpus callosum.
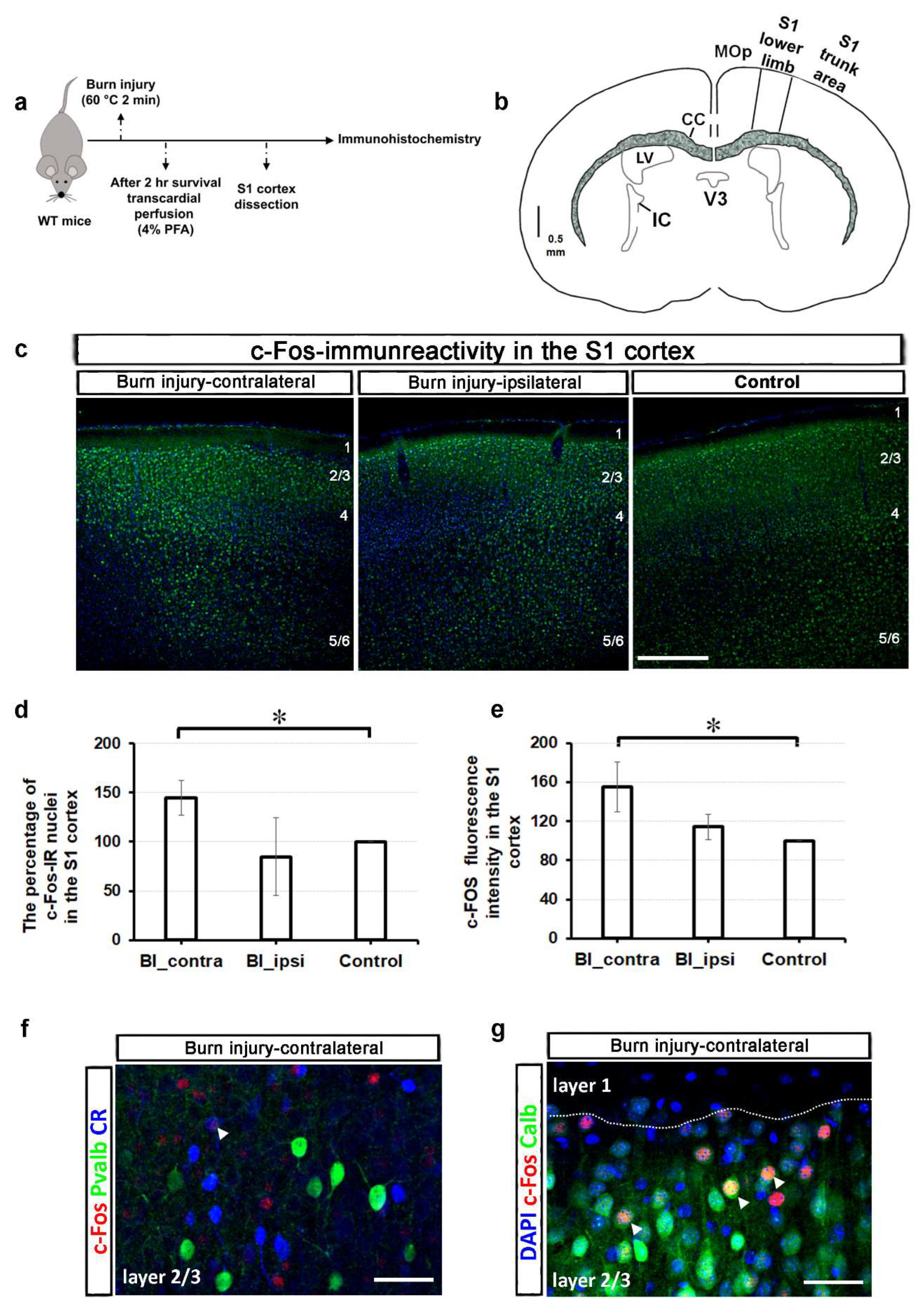
Figure 2.
Distinct transcriptomic profiles of the S1 cortex in two pain models. RNA-seq analysis reveals limited overlap in differentially expressed genes (DEGs) between burn injury and inflammatory pain models. (a) Schematic representation illustrating the time scale of this experiment. (b) The Venn diagram analysis of DEGs in the S1 cortex illustrates the overlap of DEGs (logFC between -1 and 1, relative to untreated samples) between the burn injury model and the formalin-induced inflammatory pain model. The intersections represent DEGs commonly dysregulated in response to these painful conditions. (c) The heatmap of RNA-seq data illustrates the top 15 upregulated differentially expressed genes (DEGs) in the S1 cortex following burn injury (BI, shown in the left panel) or formalin application (FA, seen in the middle panel). The panel on the right lists 15 commonly upregulated DEGs across the pain models (out of the 42 genes shown in the intersection of the above Venn diagram). In each panel, color-coded expression values of identical genes are displayed side by side to compare the two pain models. Blue indicates downregulated genes, while warm colors represent upregulated ones. (d) This heatmap of RNA-seq data illustrates the top 15 downregulated DEGs in the S1 cortex following burn injury (BI, on the left panel) or formalin application (FA, in the middle panel). The right panel lists 15 (out of 17) commonly downregulated DEGs across the pain models. In each panel, the expression values of identical genes are color-coded and displayed side by side to compare the two pain models. Blue indicates downregulated genes, while warm colors indicate upregulated ones. Supplementary Tables S1–S3 detail the molecular functions and biological processes related to the top 15 up- and downregulated DEGs.
Figure 2.
Distinct transcriptomic profiles of the S1 cortex in two pain models. RNA-seq analysis reveals limited overlap in differentially expressed genes (DEGs) between burn injury and inflammatory pain models. (a) Schematic representation illustrating the time scale of this experiment. (b) The Venn diagram analysis of DEGs in the S1 cortex illustrates the overlap of DEGs (logFC between -1 and 1, relative to untreated samples) between the burn injury model and the formalin-induced inflammatory pain model. The intersections represent DEGs commonly dysregulated in response to these painful conditions. (c) The heatmap of RNA-seq data illustrates the top 15 upregulated differentially expressed genes (DEGs) in the S1 cortex following burn injury (BI, shown in the left panel) or formalin application (FA, seen in the middle panel). The panel on the right lists 15 commonly upregulated DEGs across the pain models (out of the 42 genes shown in the intersection of the above Venn diagram). In each panel, color-coded expression values of identical genes are displayed side by side to compare the two pain models. Blue indicates downregulated genes, while warm colors represent upregulated ones. (d) This heatmap of RNA-seq data illustrates the top 15 downregulated DEGs in the S1 cortex following burn injury (BI, on the left panel) or formalin application (FA, in the middle panel). The right panel lists 15 (out of 17) commonly downregulated DEGs across the pain models. In each panel, the expression values of identical genes are color-coded and displayed side by side to compare the two pain models. Blue indicates downregulated genes, while warm colors indicate upregulated ones. Supplementary Tables S1–S3 detail the molecular functions and biological processes related to the top 15 up- and downregulated DEGs.
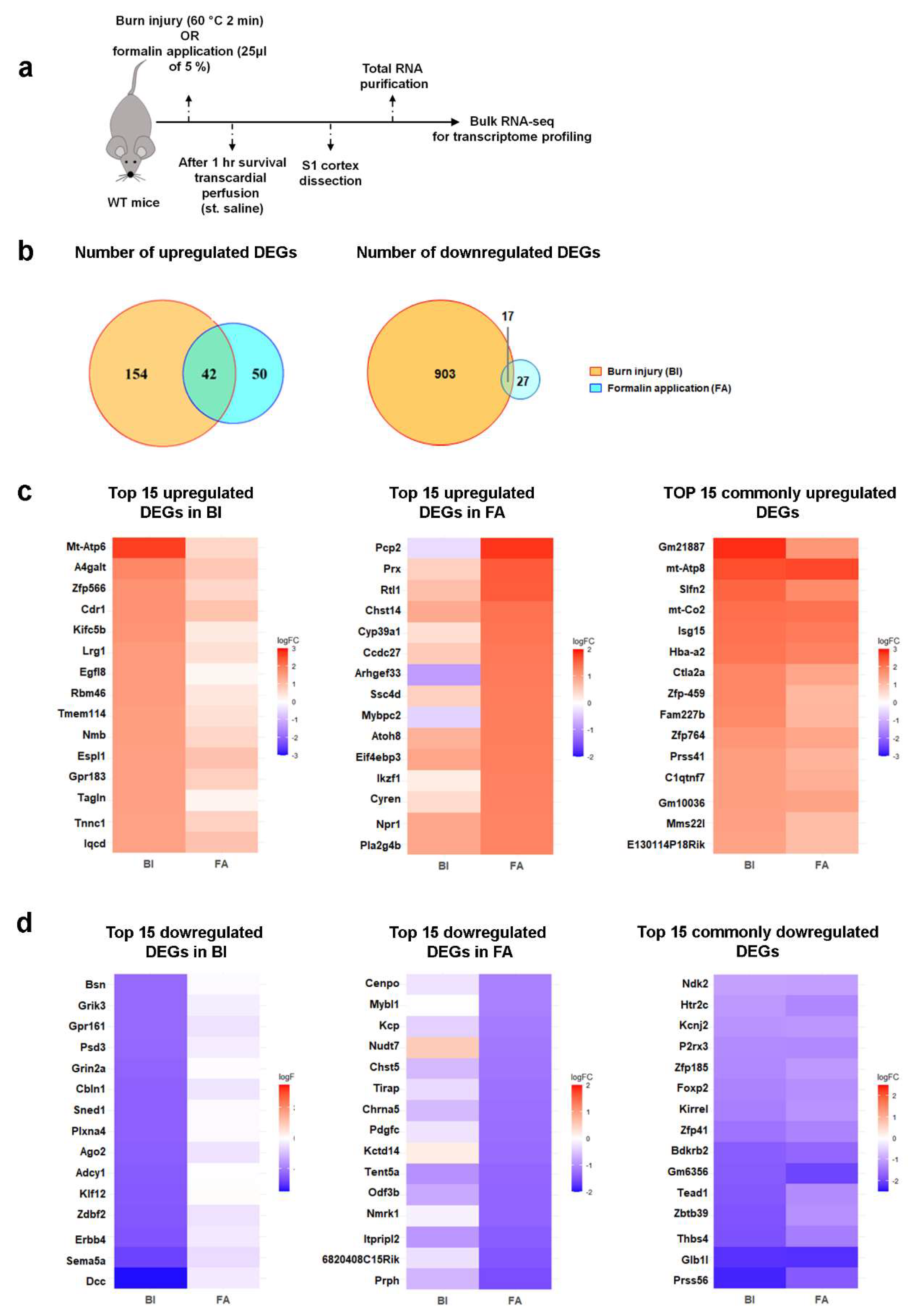
Figure 3.
Gene ontology enrichment analysis (GOEA) reveals distinct molecular signaling pathways activated by thermal injury and formalin-induced inflammatory pain in the S1 cortex. (a) Bar-dot plot illustrates the results of the GO terms and KEGG pathway enrichment analysis of the DEGs in the S1 cortex in response to burn injury, compared to the control. (b) Bar-dot plot illustrates the results of the GO terms and KEGG pathway enrichment analysis of the DEGs in the S1 cortex in response to formalin-induced inflammation, compared to the control. The vertical axis lists the terms, while the horizontal axis displays the transformed false discovery rate (−log10(FDR)). Upregulated terms are depicted in red, and downregulated terms in blue based on the ratio of the numbers of up- and downregulated genes in each entry. Line lengths indicate the significance level, and dots represent the number of genes. Abbreviations: BP, Gene Ontology biological process; CC, Gene Ontology cellular compartment; MF, Gene Ontology molecular function; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Figure 3.
Gene ontology enrichment analysis (GOEA) reveals distinct molecular signaling pathways activated by thermal injury and formalin-induced inflammatory pain in the S1 cortex. (a) Bar-dot plot illustrates the results of the GO terms and KEGG pathway enrichment analysis of the DEGs in the S1 cortex in response to burn injury, compared to the control. (b) Bar-dot plot illustrates the results of the GO terms and KEGG pathway enrichment analysis of the DEGs in the S1 cortex in response to formalin-induced inflammation, compared to the control. The vertical axis lists the terms, while the horizontal axis displays the transformed false discovery rate (−log10(FDR)). Upregulated terms are depicted in red, and downregulated terms in blue based on the ratio of the numbers of up- and downregulated genes in each entry. Line lengths indicate the significance level, and dots represent the number of genes. Abbreviations: BP, Gene Ontology biological process; CC, Gene Ontology cellular compartment; MF, Gene Ontology molecular function; KEGG, Kyoto Encyclopedia of Genes and Genomes.
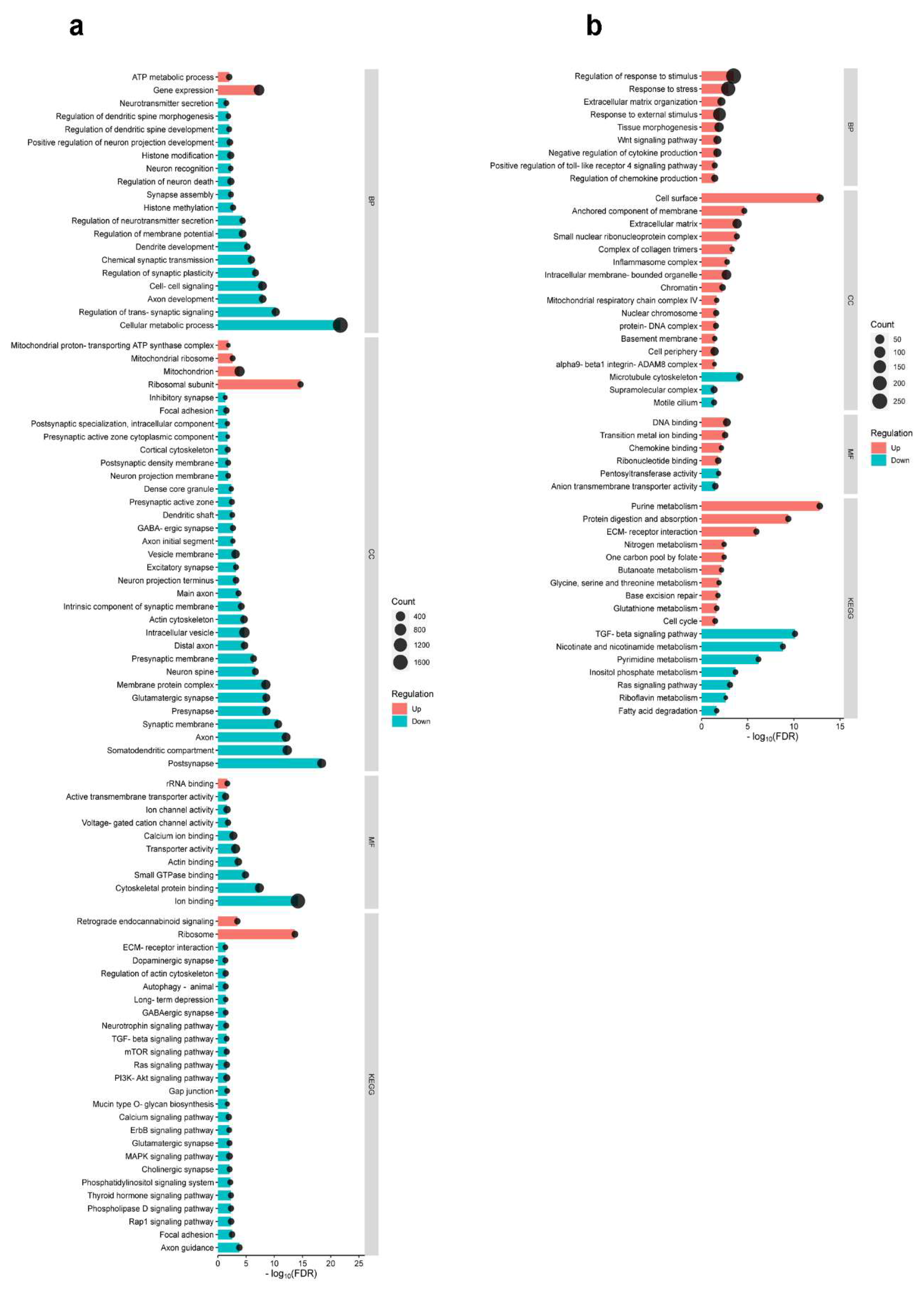
Figure 4.
KEGG pathway analysis identifies distinct pain-related molecular programs in the S1 cortex based on sensory modality. (a) Bubble chart illustrating KEGG pathway enrichment for DEGs related to synaptic and structural functions in burn injury, compared to the control group. (b) Bubble chart depicting KEGG pathway enrichment for DEGs related to signaling processes in burn injury, compared to the control group. (c) Bubble chart illustrating KEGG pathway enrichment for DEGs related to signaling and metabolic processes in formalin treatment compared to the control group. The vertical axis represents KEGG pathway entries, while the horizontal axis shows the ratio of DEGs enriched in each pathway to the total number of genes annotated in that pathway (filtered by a log fold change criterion of -0.5 < log2FC > 0.5). The size of each bubble corresponds to the number of genes associated with a given pathway. The color gradient represents the false discovery rate (FDR) on a log10 scale, indicating statistical significance. The enrichment scale quantitatively expresses the proportion of genes in the given KEGG pathway that are up-, or downregulated.
Figure 4.
KEGG pathway analysis identifies distinct pain-related molecular programs in the S1 cortex based on sensory modality. (a) Bubble chart illustrating KEGG pathway enrichment for DEGs related to synaptic and structural functions in burn injury, compared to the control group. (b) Bubble chart depicting KEGG pathway enrichment for DEGs related to signaling processes in burn injury, compared to the control group. (c) Bubble chart illustrating KEGG pathway enrichment for DEGs related to signaling and metabolic processes in formalin treatment compared to the control group. The vertical axis represents KEGG pathway entries, while the horizontal axis shows the ratio of DEGs enriched in each pathway to the total number of genes annotated in that pathway (filtered by a log fold change criterion of -0.5 < log2FC > 0.5). The size of each bubble corresponds to the number of genes associated with a given pathway. The color gradient represents the false discovery rate (FDR) on a log10 scale, indicating statistical significance. The enrichment scale quantitatively expresses the proportion of genes in the given KEGG pathway that are up-, or downregulated.
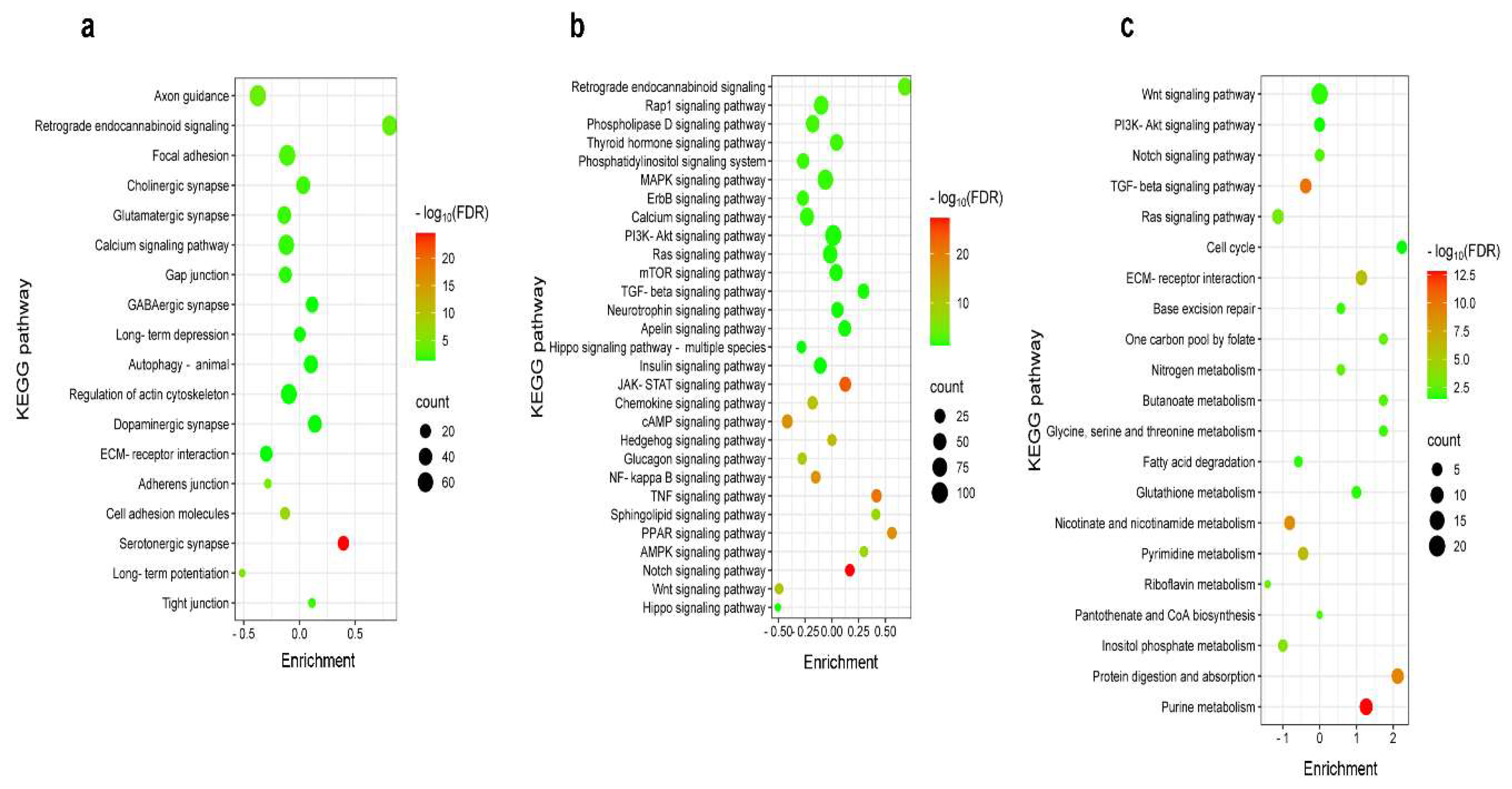
Figure 5.
Identification of key players of burn injury-related retrograde endocannabinoid network. KEGG Pathview analysis of the molecular signature of the retrograde endocannabinoid signaling pathway in burn injury compared to control. This analysis illustrates the pre-and postsynaptic components of retrograde endocannabinoid signaling at both excitatory and inhibitory synapses. The KEGG pathway analysis indicates that burn injury leads to the upregulation or downregulation of multiple components in the retrograde endocannabinoid signaling pathway. Each box represents a certain gene family or a single gene, with color indicating the log2-based fold change. Gene expression levels are shown as higher (red), unchanged (green), or lower (blue) in burn-injured samples compared to non-treated controls. The synaptic vesicle cycle showed downregulation in presynaptic terminals, regardless of the functional nature of the synapse. On the other hand, oxidative phosphorylation was typically upregulated in the axon terminals in response to burn injury. Abbreviations: CB1R, cannabinoid receptor type 1; VGCC, voltage-gated calcium channels; GIRK, inwardly rectifying potassium channels.
Figure 5.
Identification of key players of burn injury-related retrograde endocannabinoid network. KEGG Pathview analysis of the molecular signature of the retrograde endocannabinoid signaling pathway in burn injury compared to control. This analysis illustrates the pre-and postsynaptic components of retrograde endocannabinoid signaling at both excitatory and inhibitory synapses. The KEGG pathway analysis indicates that burn injury leads to the upregulation or downregulation of multiple components in the retrograde endocannabinoid signaling pathway. Each box represents a certain gene family or a single gene, with color indicating the log2-based fold change. Gene expression levels are shown as higher (red), unchanged (green), or lower (blue) in burn-injured samples compared to non-treated controls. The synaptic vesicle cycle showed downregulation in presynaptic terminals, regardless of the functional nature of the synapse. On the other hand, oxidative phosphorylation was typically upregulated in the axon terminals in response to burn injury. Abbreviations: CB1R, cannabinoid receptor type 1; VGCC, voltage-gated calcium channels; GIRK, inwardly rectifying potassium channels.
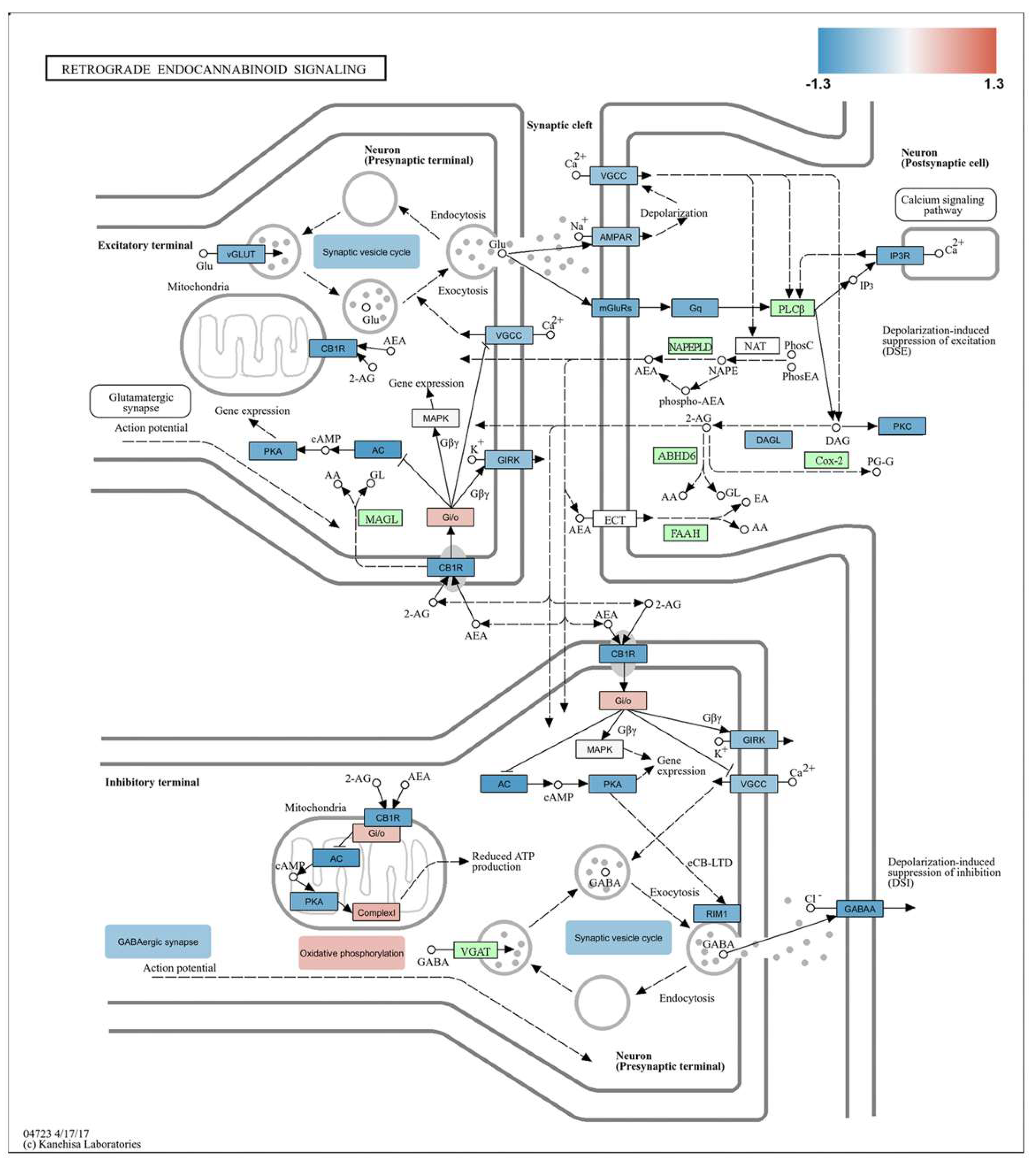
Figure 6.
Validation of RNA-seq datasets using immunofluorescence: increased Ggamma14 fluorescence intensity and a higher number of Ggamma14-IR cells in the S1 cortex post-burn injury. (a) Representative triple immunofluorescent labeling of the S1 cortex in a control mouse (top) and a mouse subjected to burn injury (bottom) is shown, utilizing antibodies against the CB1 receptor (red), Ggamma14 (green), and DAPI (blue) in layer 2/3. Scale bar: 10 µm. (b) Representative triple immunofluorescent labeling of the S1 cortex in a control mouse (top) and a mouse subjected to burn injury (bottom) is shown, utilizing antibodies against the CB1 receptor (red), Ggamma14 (green), and DAPI (blue) in layer 5. Scale bar: 10 µm. (c) Quantitative analysis of CB1 receptor fluorescence intensity throughout the entire thickness of the contralateral S1 cortex, relative to the control. The data are presented as mean ± SEM (n = 5-8 sections from 2 animals per group). (d) Quantitative analysis of fluorescence intensity for Ggamma14-IR throughout the entire thickness of the S1 cortex was normalized to the control. The data are presented as mean ± SEM (n = 3-5 sections from 2 animals per group). An asterisk (*) indicates p < 0.05 when compared to the control group. (e) Quantitative analysis of the percentage of Ggamma14-IR cells in layer 5 of the S1 cortex was normalized to the control value (n = 5 sections/group). The data are presented as mean ± SEM (n = 3-5 sections from 2 animals per group). An asterisk (*) denotes p < 0.05 compared to the control group. The term BI_contra specifically refers to the right hemisphere of the brain, which is opposite the site of the burn injury, while BI_ipsi denotes the left-sided cortical region.
Figure 6.
Validation of RNA-seq datasets using immunofluorescence: increased Ggamma14 fluorescence intensity and a higher number of Ggamma14-IR cells in the S1 cortex post-burn injury. (a) Representative triple immunofluorescent labeling of the S1 cortex in a control mouse (top) and a mouse subjected to burn injury (bottom) is shown, utilizing antibodies against the CB1 receptor (red), Ggamma14 (green), and DAPI (blue) in layer 2/3. Scale bar: 10 µm. (b) Representative triple immunofluorescent labeling of the S1 cortex in a control mouse (top) and a mouse subjected to burn injury (bottom) is shown, utilizing antibodies against the CB1 receptor (red), Ggamma14 (green), and DAPI (blue) in layer 5. Scale bar: 10 µm. (c) Quantitative analysis of CB1 receptor fluorescence intensity throughout the entire thickness of the contralateral S1 cortex, relative to the control. The data are presented as mean ± SEM (n = 5-8 sections from 2 animals per group). (d) Quantitative analysis of fluorescence intensity for Ggamma14-IR throughout the entire thickness of the S1 cortex was normalized to the control. The data are presented as mean ± SEM (n = 3-5 sections from 2 animals per group). An asterisk (*) indicates p < 0.05 when compared to the control group. (e) Quantitative analysis of the percentage of Ggamma14-IR cells in layer 5 of the S1 cortex was normalized to the control value (n = 5 sections/group). The data are presented as mean ± SEM (n = 3-5 sections from 2 animals per group). An asterisk (*) denotes p < 0.05 compared to the control group. The term BI_contra specifically refers to the right hemisphere of the brain, which is opposite the site of the burn injury, while BI_ipsi denotes the left-sided cortical region.
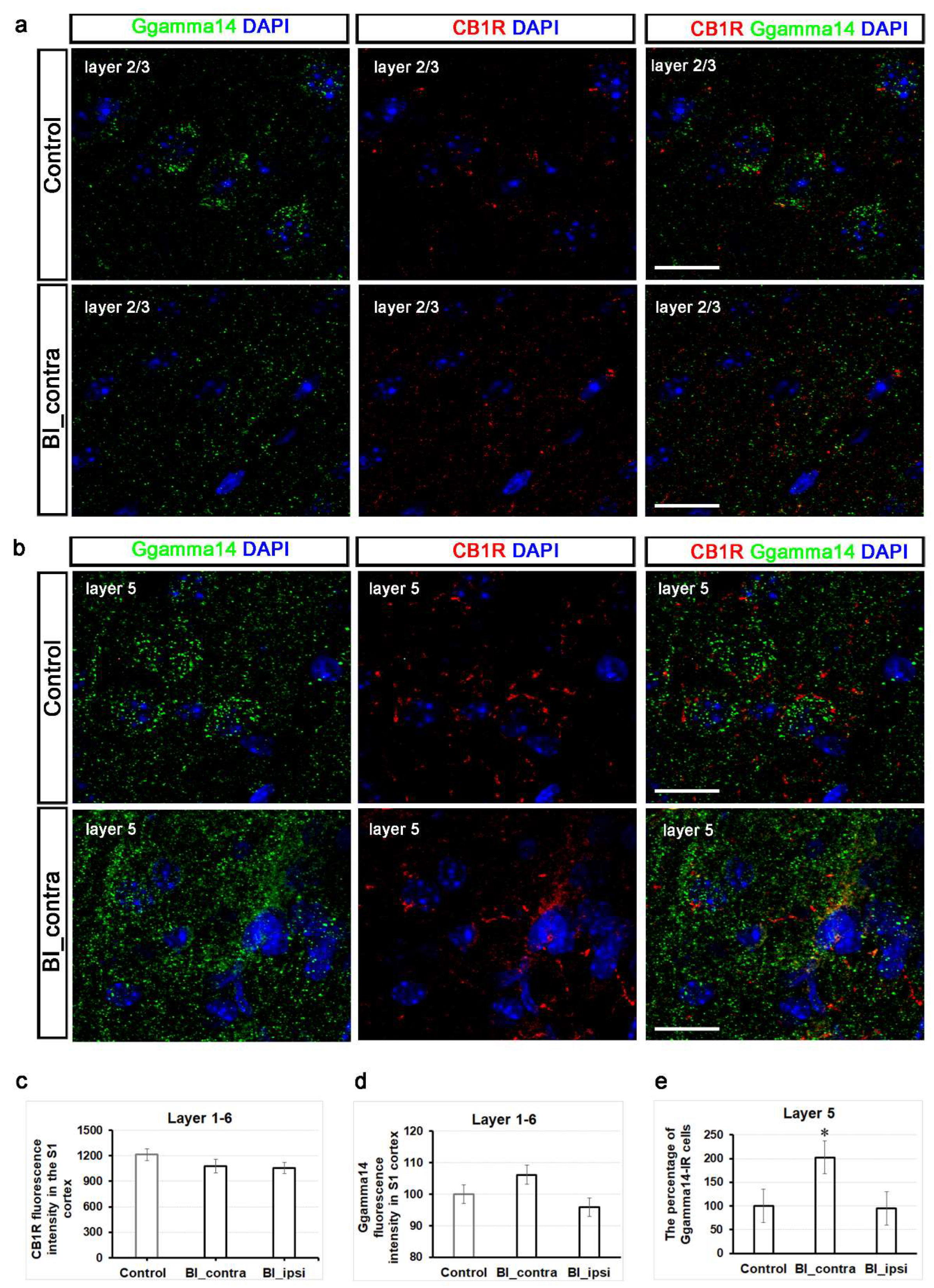

Table 1.
Lists of the primary antibodies used in the study, including their suppliers, catalog numbers, and dilution factors.
Table 1.
Lists of the primary antibodies used in the study, including their suppliers, catalog numbers, and dilution factors.
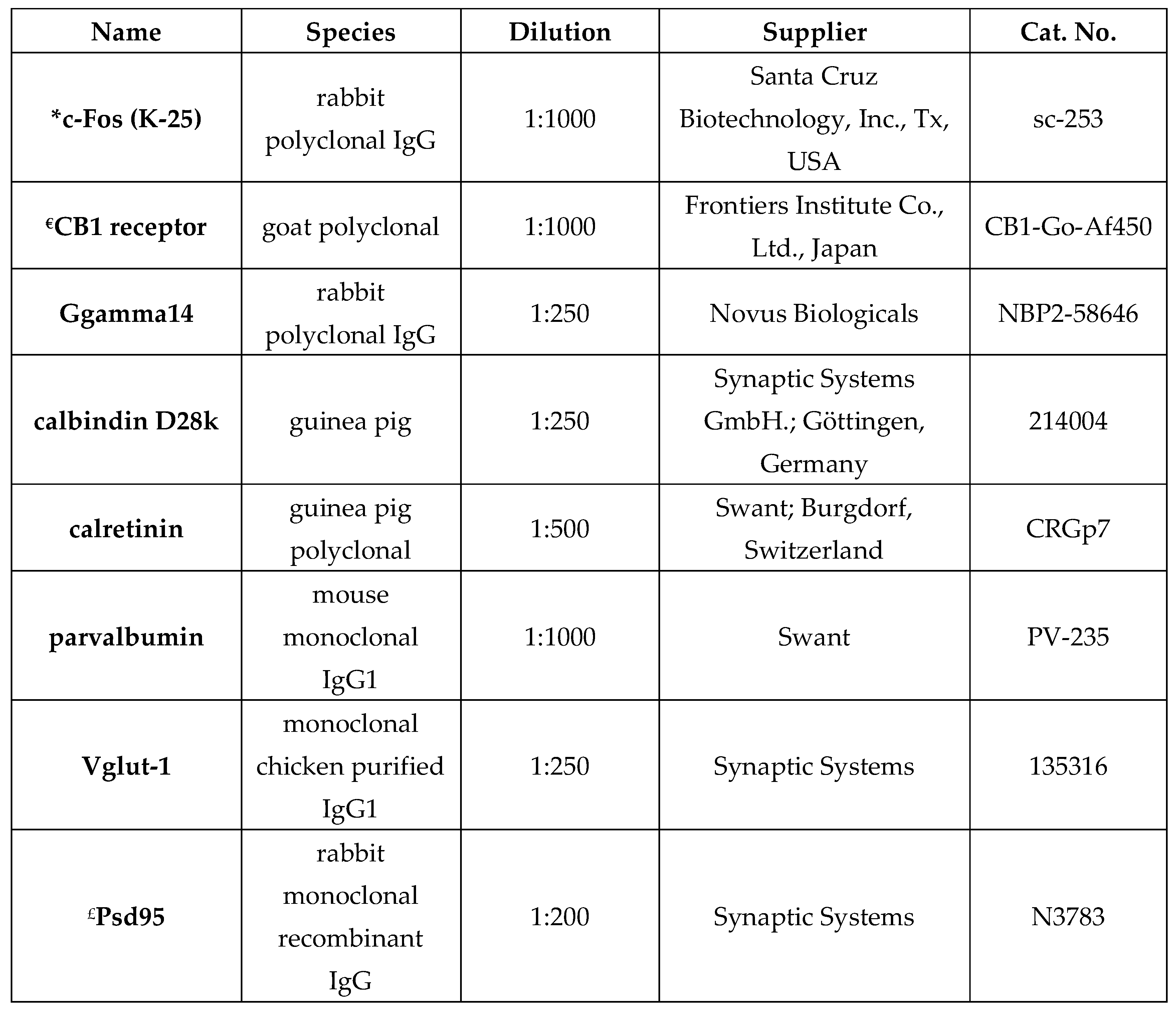
* c-Fos (K-25) has been discontinued and replaced by c-Fos (E-8): sc-166940. € see in [86]. £Psd95 is a component of postsynaptic densities in synapses.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



