
Submitted:
10 February 2025
Posted:
12 February 2025
You are already at the latest version
Abstract
This research investigates the structural, electronic, mechanical, and vibrational properties of double half-Heusler compounds with the generic formula Ti2Pt2ZSb (Z = Al, Ga, and In) using density functional theory calculations. The generalized gradient approximation within the Perdew-Burke-Ernzerhof functional was employed for structural optimization and the hybrid HSE06 functional for electronic properties. Our results demonstrate that these compounds are energetically favorable, dynamically and mechanically stable. Electronic structure calculations reveal that Ti2Pt2AlSb double half-Heusler compound is a non-magnetic semiconductor with an indirect bandgap of 1.49 eV, while Ti2Pt2GaSb and Ti2Pt2InSb are non-magnetic semiconductors with direct bandgaps of 1.40 eV. The alloys exhibit low lattice thermal conductivity (2.35–2.66 W/mK) and high melting temperature (1211–1248 K), making them promising candidates for high-technological applications. Further performed analysis, including phonon dispersion curves, electron localization function (ELF), and Bader charge analysis, provides insights into the bonding character and vibrational properties of these materials.
Keywords:
double half-Heusler alloy
; mechanical properties
; thermal conductivity
; density functional theory
1. Introduction
Heusler alloys, characterized by their diverse transition metal elements (X, Y) and main group elements (Z), have emerged as promising materials for exploring novel electronic properties and multifunctional applications. While full (X2YZ) and half (XYZ) Heusler alloys have been extensively studied [1,2,3,4,5,6,7,8], double Heusler alloys (X′X″Y2Z2, X2Y′Y″Z2, and X2Y2Z′Z″) have garnered significant interest in recent years due to their unique combination of structural symmetry and electronic complexity [9,10]. The term ’double’ emphasizes the doubling of the unit cell in comparison to their single-cell counterparts (XYZ → X2YY′Z2), where Y and Y′ are aliovalent. In the quest for novel materials with tailored electronic properties and multifunctional capabilities, double-half-Heusler (DHH) alloys have emerged as compelling candidates for this role. With applications from spintronics to thermoelectrics, the study of DHH alloys has become a focal point in contemporary materials research.
Anand et al. [9] propose that the thermal conductivity () in DHH compounds is primarily governed by reduced group velocity phonons and constrained by disorder scattering effects, which should reduce compared to contemporary ternary half-Heusler thermoelectric materials, whose efficiency is hindered by their inherently high . Through the synthesis of Ti2FeNiSb2, they have demonstrated and validated that this DHH alloy exhibits markedly lower thermal conductivity than its ternary half-Heusler analog TiCoSb, thus presenting a superior foundation for the enhancement of thermoelectric efficiency [9]. Rached et al. [11] compared the structural, thermoelectric, and elastic properties of identical half (TiXSb, X = Ru, Pt) and DHH alloys (Ti2RuPtSb2). The authors have determined through computational analysis that the Ti2RuPtSb2 compound exhibits pronounced peaks in the absorption coefficient within the ultraviolet (UV) spectral range, suggesting its potential utility as UV filters and UV photodetectors. The investigated materials have also demonstrated promising thermoelectric properties, indicating their suitability as potential candidates for thermoelectric device applications [11,12,13,14,15,16]. In most instances, DHH alloys exhibit semiconductor behavior [17], exemplified by the investigations conducted by Bouhadjer et al. [18]. In their study, Ti2FeNiSb2 and Ti2Ni2InSb alloys were observed, displaying band gap energies of 0.64 and 0.43 eV, respectively.
Ding et al. [19] devised a half-metallic DHH alloy, Mn2FeCoSi2, and conducted an investigation into its electronic structure and magnetism. Additionally, they accurately examined the impact of atomic irregularities, encompassing six varieties of swap disorders and twelve varieties of antisite disorders, on the half-metallic properties of Mn2FeCoSi2 [19]. Transitioning to another study, Douinat et al. [20] explore the valence band contributions in two DHH alloys, Zr2AlBiNi2 and Zr2GaBiNi2. They find that in both alloys, the 3d orbitals of Ni predominantly contribute to the valence band, while the conduction band is primarily influenced by the 4d and 3d orbitals of Zr and Ni, respectively. So beyond thermoelectricity, DHH alloys demonstrate potential in spintronics, offering opportunities for spin injection, manipulation, and detection, which are essential to advancing electronic devices into the next generation [3]. Furthermore, extensive research has demonstrated the potential of double Heusler alloys in cutting-edge applications, including photovoltaics, ultraviolet (UV) sensing, solar cell technologies [21] and transportation [23,24,25].
It should be noted that despite the advantages and large number of DHH alloys that have been extensively studied over the past decade, a considerable number of new compound combinations have not been investigated neither experimentally nor theoretically.
This paper presents a first-time investigation into the structural, electronic, mechanical, and phonon properties of DHH compounds Ti2Pt2ZSb (where Z = Al, Ga, and In). The results obtained should help a better understanding of the physicochemical properties and assess the potential suitability of the aforementioned alloys for advanced technological applications.
2. Computational Methods
The calculations were performed using the Vienna Ab-initio Simulation Package (VASP) package within the density functional theory (DFT) methodology [26,27]. The generalized gradient approximation (GGA) within the Perdew-Burke-Ernzerhof functional (PAW) was utilized to account for the exchange-correlation interaction [28]. A baseline plane wave cutoff of 700 eV was applied in all instances. Integration of the Brillouin zone was achieved through a k-point grid. These parameter choices demonstrated satisfactory convergence in total energy. Convergence tolerance for the calculations was set at a total energy difference within the range of eV/atom. The charge distribution on the ions was investigated employing topological analysis, utilizing the Bader method. The phonon calculations were performed using PhonoPy program [29]. Real-space force constants were calculated using supercell and finite displacement approaches, with supercells for all considered compounds. The elastic stiffness tensor () of considered compounds was calculated using the stress ()–strain () relation and from obtained data all desired mechanical properties were estimated. Lattice thermal conductivity was calculated using a modified Debye–Callaway model, as implemented in AICON [30]. This approach uses the Debye temperature, phonon group velocity, and mode-resolved Gruneisen parameter as input, and it was especially appropriate for bulk crystalline materials.
3. Results
3.1. Structural Properties
Now we present the obtained results on the new DHH compounds.
The selected DHH compounds, Ti2Pt2ZSb (Z = Al, Ga, and In), crystallize in a body-centered tetragonal lattice structure with the space group (No.122). An example of the crystal structure is shown in Figure 1. The X (Ti) and Y (Pt) atoms occupy and sites, while the Z (Al, Ga, and In) and Z′ (Sb) atoms occupy and Wyckoff positions, respectively. The chosen X and Y atoms are transition metals, while the Z atom is an element of the main group. Table 1 provides an overview of the key attributes associated with these alloys.
Stable half-Heusler semiconductor compounds adhere to the 18 valence electron counting (VEC) rule [31]. Alloys that conform to this rule exhibit notable stability and possess intriguing semiconductor properties, making them particularly relevant for various branches of physics.
DHH compounds are distinguished by their formation through the combination of 17 and 19 valence electron half-Heusler compounds, resulting in stable 18 valence electron structures. This transformation from ternary to quaternary compounds enables the creation of a diverse array of new alloys, suitable for a wide range of applications.
In our case, the DHH compound Ti2Pt2AlSb which has 18 valence electrons, is considered to be a combination of the two half-Heuslers: TiPtAl with 17 valence electrons and TiPtSb with 19 valence electrons. This rule also works for the other two Ti2Pt2GaSb and Ti2Pt2InSb double half-Heusler compounds with 18 valence electrons. Accordingly, the chosen compounds should be considered stable. Therefore, further, we investigated the structures for energy, mechanical, and dynamic stability.
Based on the above let us consider whether the selected alloys are energetically favorable. The expected enthalpy of formation without correction for zero-point energy for the studied chemical compounds was calculated.
The formation enthalpy calculation was performed using the following formula:
The enthalpies of the Ti2Pt2AlSb, Ti2Pt2GaSb, and Ti2Pt2InSb alloys are equal to -0.87, -1.01, and -1.06 eV, respectively. The negative values of formation enthalpy indicate that these alloys are stable. As expected from the 18 valence electron counting rule, the studied alloys were found to be energetically stable.
3.2. Vibrational Properties
The vibrational properties of the material are analyzed to describe the dynamic stability. The phonon dispersion curves and phonon density of states of Ti2Pt2ZSb alloys are shown in Figure 2. Results of phonon dispersion curves are displayed in the first Brillouin zone. Since all three compounds have 12 atoms in the primitive cell, there are 36 vibrational modes in the phonon band structure with followed mechanical representation . The first three acoustic modes () represent the movement of the center of mass, while the optical modes () characterize the relative motion between atoms. According to a group theoretical analysis, there are 30 Raman active (), 24 IR active (), and 3 silent () modes for Ti2Pt2ZSb compounds. Calculated vibrational modes at -point are presented in Table 2.
Analyzing the density of phonon states, it can be seen that for all three compounds in the frequency from 0 to 4 THz, the main contribution to acoustic modes corresponds to Pt atoms, while from 4 to 6 THz to Sb atoms. For Ti2Pt2GaSb and Ti2Pt2InSb alloys in the frequency ≈ 5.5–6.2 THz a phonon band gap is observed. Conversely, Ti2Pt2AlSb exhibits two smaller phonon band gaps in the frequencies ≈ 7.3–7.4 THz and ≈ 8.1–8.5 THz. Additionally, a fifth phonon band with frequencies exceeding 9 THz, primarily influenced by the vibrations of the lighter aluminum atoms.
According to the performed calculation of the phonon dispersion, no imaginary modes in the whole Brillouin zone were found, which means that when the system is perturbed, the atoms return to their equilibrium positions. This fact proves that the considered alloys are dynamically stable.
3.3. Elastic Properties
The number of independent elastic constants usually depends on the symmetry of the structure under investigation. In our case, for a tetragonal crystal structure, there are six independent elastic constants as , , , , , . Table 3 contains the calculated elastic constants for all three compounds. A compound that satisfies the Born-Huang condition can be considered mechanically stable [32]. Four principal criteria must be satisfied by our system:
Based on the calculations all three alloys meet the required conditions and could be considered as mechanically stable. Considering the above, the Ti2Pt2ZSb alloys were tested for energy, dynamic, and mechanical stability. They were found to be stable according to all three criteria.
Realizing that the elastic constants and demonstrate compression resistance, while the constants ,, and show shear resistance, from the information in Table 3 the compression resistance of Ti2Pt2ZSb alloys is larger than the resistance to one-dimensional shear deformations. Utilizing the known elastic constants above, the elastic moduli as shear, bulk, Young’s modulus, and Poisson’s ratio can be estimated. Bulk modulus of elasticity B serves as an indicator of a material’s resistance to uniform compression. The shear modulus G characterizes the material’s resistance to shear deformations. The upper and lower boundaries of the actual polycrystalline constants are described by the Voigt [33] and Reuss [34] equations. For a tetragonal crystal, it can be approximated using the Voigt average (,), and the Reuss’s modulus (, ) derived from the elastic constants. Taking into consideration Hill’s recommendations [35,36], the estimation of the volume and shear modulus was the arithmetic mean of the upper and lower limits, and the following equations were used:
The calculated B, G, E, and are presented in Table 4. Upon analyzing the bulk modulus, the Ti2Pt2GaSb alloy exhibits the highest resistance to uniform compression among the three alloys, while Ti2Pt2InSb shows the lowest resistance. Notably, the shear modulus decreases as the ionic radius of the constituent elements increases, implying that larger ionic radii contribute to reduced resistance to shear stress.
Young’s modulus indicates the stiffness of a material. The Young’s modulus equation can be written as follows [37]. The Poisson’s ratio shows the measure of deformation in one direction under the action of stress applied in a perpendicular direction [38]. In our case, for a tetragonal system, it is determined by the following relation [39]. High E values correspond to very stiff materials with strong covalent bonds. It is clear from Table 4 that Young’s modulus decreases with increasing ionic radius, while on the contrary, the Poisson’s ratio increases. These values typically indicate materials with metal compounds and are close to the typical range for metals and ductile materials. Simply put, with an increase in the radius of the Z atom, the stiffness of the material decreases, but at the same time, it maintains a balance between strength and plasticity: the material can deform but retain its shape.
Table 4 also shows values such as fracture toughness, Vickers hardness, and brittleness of the material. Based on our calculations, the Vickers hardness for the Ti2Pt2InSb alloy is the lowest and equals to 4.2 GPa. Ti2Pt2AlSb has the highest value of (5.3 GPa), while the Ti2Pt2GaSb has an intermediate (4.5 GPa).
The calculated values of Ti2Pt2ZSb (Z = Al, Ga, and In) fall below the minimum threshold for hard materials, which is 20 GPa (Figure 3). Therefore, these double half-Heusler alloys could not be classified as hard materials. The ratio gives an idea of the fragility of the material. If the ratio is greater than 1.75, the material is considered to be ductile. If it is less than 1.75, it is considered brittle. In our case, the values are high (), which indicates their ductility. Fracture toughness can be described as the resistance of a material to cracks and fractures when exposed to external forces. As for fracture toughness KIC, this decreases with increasing the ionic radius of the Z atom in Ti2Pt2ZSb alloy [40]. Using the Fine relation [41] for hexagonal and tetragonal structures of metals and intermetallides, we calculated the melting temperature of the alloys and found that it decreases with increasing their stability.
Using the calculated bulk and shear moduli, the percentage of elastic anisotropy for the bulk modulus and shear modulus were calculated. For elastic isotropy, the value corresponds to 0 %, while the maximum possible anisotropy corresponds to a value of 100 %. A comparative analysis of the elastic properties of double half-Heusler alloys and their half-Heusler analogue shows that aliovalent substitution has a negligible effect on the values of the elastic moduli.
3.4. Thermal Conductivity
The lattice thermal conductivity was calculated and illustrated in Figure 4. The calculated values of lattice thermal conductivity (W/(m·K), longitudinal sound velocity (m/s), transverse sound velocity (m/s) and average sound velocity (m/s) of the Ti2Pt2ZSb DHH compounds with TiPtSn are presented in Table 5. The sound velocities for TiPtSn were recalculated using the constants provided in [42]. It can be seen that with increasing temperature, the lattice thermal conductivity for all three double half-Heusler alloys decreases gradually. At 300 K, the lattice thermal conductivity for Ti2Pt2AlSb compound is 2.67 W/(m·K), for Ti2Pt2GaSb is 2.52 W/(m·K), for Ti2Pt2InSb compound 2.35 W/(m·K) respectively. A comparison between the investigated DHH alloys and their analogue TiPtSn indicates that aliovalent substitution of Sn leads to an increase in lattice thermal conductivity by approximately threefold. In the case of sound velocities, their values do not differ significantly.
3.5. Electronic Properties
The electronic properties of the materials were investigated by calculating the density of states and the band structure using density functional theory (DFT). Since it is known that standard DFT functionals greatly underestimate the band gap, the HSE06 hybrid functional [45] was used to estimate the band structure and density of states (DOS). The density of states is an essential factor in characterizing energy levels. The calculated band structure and DOS are shown in Figure 5.
Analysis of the DOS and band structure revealed the presence of a band gap proximate to the Fermi level, with respective values of 1.43 eV (indirect) for the Ti2Pt2AlSb alloy, 1.40 eV (direct) for the Ti2Pt2GaSb and Ti2Pt2InSb alloys respectively. According to the results obtained, the maximum of the valence band (VBM) lies at the -point, and the minimum of the conduction band (CBM) at the M-point. The band profiles are alike for all three compounds. For comparison, the band gap of the 18-valence-electron analogue TiPtSn is reported to be 1.31 eV [44].
The analysis of the three graphs shows that Pt d states make a predominant contribution to the total density of states in the bottom of the valence band from ≈ -5.5 to -2.5 eV, whereas the top of the conduction band from ≈ 1 to 2 eV, the unoccupied Ti d states make the most significant contribution. Furthermore, near the Fermi level, the contribution of indium In p states among the Z atoms is significantly greater than Al p states. Proximal to the Fermi level, at the valence band maximum, contributions from all atomic species are observable, with the occupied d states of Ti exerting the most significant influence.
The results of the electronic characteristics of DHH alloys determine that the chosen alloys exhibit properties of non-magnetic direct bandgap semiconductors.
Furthermore, a Bader analysis was conducted to ascertain the atomic charges and are shown in Table 6. This analysis enables a detailed understanding of the charge distribution within the studied compounds. The Bader charges of Ti atoms decrease slightly as the Z element changes from Al to In. This trend indicates a gradual increase in charge delocalization and a slight reduction in the ionic character of Ti with heavier Z elements.
In case of Pt atoms, increasing negative charge suggests enhanced electron transfer to Pt atoms as the atomic size of the Z element increases, likely due to the influence of bonding and electronegativity differences. Sb atoms show an increasing trend in Bader charges. This trend correlates with an increasing covalent character or electron density redistribution towards Sb as the Z element becomes heavier. The Z element retains a constant Bader charge of (+3e) across all compounds. This uniformity suggests that the Z elements contribute primarily through their stable valence state, with minimal variation in charge transfer dynamics. To illustrate the various interactions and chemical bonds in selected DHH alloys, the electronic localization functions (ELF) were calculated. Figure 6 depict the electronic localization function data for Ti2Pt2AlSb, Ti2Pt2GaSb, and Ti2Pt2InSb. High values of the ELF between atoms indicate regions where electrons tend to pair, reflecting the covalent bonding character between the nearest-neighbor atoms in the compounds. As can be seen from Figure 6, the charge accumulation occurs halfway through each Sb–-Pt vector and the maximum ELF value between Sb and Pt atoms is about 0.4, which is indicative of the ionic bonding. In contrast, the bond between the Z atoms (Al, Ga, In) and Pt atoms predominantly exhibit a covalent nature, with ELF values reaching 0.8. Moreover, as the ionic radius of the Z atom increases (Al – Ga – In), the covalent character of the bond between Pt and Z atoms is enhanced. The comparison of ELF for the selected alloys shows that the nature of the electron density distribution is mainly determined by the nature of the element Z. However, the overall pattern of the electron density distribution remains qualitatively similar for all selected alloys, which indicates the similarity of their electronic structure.
Funding
This study was funded by the Ministry of Science and Higher Education of the Republic of Kazakhstan under the "Zhas Galym" project for 2024–2026 AP22683528 “Computer design of thermoelectric and spintronic materials based on Heusler alloys” and VVK acknowledges Priority-2030 program of NUST MISIS, grant K2-2022-022. The calculations were performed using resources provided by the Novosibirsk State University Supercomputer Center. NES was supported by the state assignment of IGM SB RAS (122041400176-0). The work by TMI was carried out within the state assignment of the Vernadsky Institute of Geochemistry and Analytical Chemistry of the Russian Academy of Sciences (GEOKHI RAS). In addition, M. K and A.I.P. were supported by EUROfusion Enabling Research Project ENR-MAT.02. ISSP-UL- “New dielectric functional materials and interfaces (DFMI) – Theoretical and Experimental analysis.” This work has been carried out within the framework of the EUROfusion Consortium, funded by the European Union via the Euratom Research and Training Programme (Grant Agreement No 101052200 — EUROfusion). Views and opinions ex-pressed are however those of the author(s) only and do not necessarily reflect those of the Euro-pean Union or the European Commission. Neither the European Union nor the European Com-mission can be held responsible for them.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Webster, P.J. Heusler alloys. Contemp. Phys. 1969, 10, 559–577. [Google Scholar] [CrossRef]
- Tavares, S.; Yang, K.; Meyers, M.A. Heusler alloys: Past, properties, new alloys, and prospects. Prog. Mater. Sci. 2023, 132, 101017. [Google Scholar] [CrossRef]
- Elphick, K.; Frost, W.; Samiepour, M.; Kubota, T.; Takanashi, K.; Sukegawa, H.; Mitani, S.; Hirohata, A. Heusler alloys for spintronic devices: Review on recent development and future perspectives. Sci. Technol. Adv. Mater. 2021, 22, 235–271. [Google Scholar] [CrossRef] [PubMed]
- Rogl, G.; Grytsiv, A.; Gürth, M.; Tavassoli, A.; Ebner, C.; Wünschek, A.; Puchegger, S.; Soprunyuk, V.; Schranz, W.; Bauer, E.; et al. Mechanical properties of half-Heusler alloys. Acta Mater. 2016, 107, 178–195. [Google Scholar] [CrossRef]
- Abuova, A.; Merali, N.; Abuova, F.; Khovaylo, V.; Sagatov, N.; Inerbaev, T. Electronic properties and chemical bonding in V2FeSi and Fe2VSi Heusler alloys. Crystals 2022, 12, 1546. [Google Scholar] [CrossRef]
- Abuova, F.; Inerbaev, T.; Abuova, A.; Merali, N.; Soltanbek, N.; Kaptagay, G.; Seredina, M.; Khovaylo, V. Structural, electronic, and magnetic properties of Mn2Co1-xVXZ (Z = Ga, Al) Heusler alloys: An insight from DFT study. Magnetochemistry 2021, 7, 159. [Google Scholar] [CrossRef]
- Casper, F.; Graf, T.; Chadov, S.; Balke, B.; Felser, C. Half-Heusler compounds: Novel materials for energy and spintronic applications. Semicond. Sci. Technol. 2012, 27, 063001. [Google Scholar] [CrossRef]
- Tangirbergen, A.; Amangeldi, N.; Revankar, S.T.; Yergaliuly, G. A review of irradiation-induced hardening in FeCrAl alloy systems for accident-tolerant fuel cladding. Nucl. Eng. Des. 2024, 429, 113659. [Google Scholar] [CrossRef]
- Anand, S.; Wood, M.; Xia, Y.; Wolverton, C.; Snyder, G.J. Double half-Heuslers. Joule 2019, 3, 1226–1238. [Google Scholar] [CrossRef]
- Zeier, W.G.; Schmitt, J.; Hautier, G.; Aydemir, U.; Gibbs, Z.M.; Felser, C.; Snyder, G.J. Engineering half-Heusler thermoelectric materials using Zintl chemistry. Nat. Rev. Mater. 2016, 1, 1–10. [Google Scholar] [CrossRef]
- Rached, Y.; Caid, M.; Merabet, M.; Benalia, S.; Rached, H.; Djoudi, L.; Mokhtari, M.; Rached, D. A comprehensive computational investigations on the physical properties of TiXSb (X: Ru, Pt) half-Heusler alloys and Ti2RuPtSb2 double half-Heusler. Int. J. Quantum Chem. 2022, 122, e26875. [Google Scholar] [CrossRef]
- Hasan, R.; Park, T.; Kim, S.; Kim, H.-S.; Jo, S.; Lee, K.H. Enhanced Thermoelectric Properties of Ti2FeNiSb2 Double Half-Heusler Compound by Sn Doping. Advanced Energy and Sustainability Research 2022, 3, 2100206. [Google Scholar] [CrossRef]
- Hassan, M.A.; El-Khouly, A.; Elsehly, E.M.; Almutib, E.N.; Elshamndy, S.K.; Serhiienko, I.; Argunov, E.V.; Sedegov, A.; Karpenkov, D.; Pashkova, D. Transport and thermoelectric properties of melt spinning synthesized M2FeNiSb2 (M= Ti, Hf) double half-Heusler alloys. Materials Research Bulletin 2023, 164, 112246. [Google Scholar] [CrossRef]
- Charifi, Z.; Baaziz, H.; Uğur, Ş.; Uğur, G. Prediction of the electronic structure, optical and vibrational properties of ScXCo2Sb2 (X= V, Nb, Ta) double half-Heusler alloys: a theoretical study. Indian Journal of Physics 2023, 97, 413–428. [Google Scholar] [CrossRef]
- Boudjelal, M.; Bouhadjer, K.; Matougui, M.; Bentata, S.; Srivastata, V.; Bin-Omran, S.; Khenata, R. Ab initio prediction of the structural, optoelectronic, and thermoelectric properties of double half-Heusler (DHH) ScXRh2Bi2 (X= Nb, Ta) alloys DFT study results. Indian Journal of Physics 2024, 1–14. [Google Scholar] [CrossRef]
- Mekhtiche, M.; Matougui, M.; Houari, M.; Bouadjemi, B.; Lantri, T.; Boudjelal, M.; Bentata, S. Predictive study of the new double half-Heusler compounds Hf2FeNiSb2, Nb2Co2GaSb, and ScNbCo2Sb2, promising candidates for thermoelectric applications. Indian J. Phys. 2024, 1–9. [Google Scholar] [CrossRef]
- Diaf, M.; Righi, H.; Rached, H.; Rached, D.; Beddiaf, R. Ab initio study of the properties of Ti2PdFe (Ru) Sb2 double half-Heusler semiconducting alloys. Journal of Electronic Materials 2023, 52, 6514–6529. [Google Scholar] [CrossRef]
- Bouhadjer, K.; Boudjelal, M.; Matougui, M.; Bentata, S.; Lantri, T.; Batouche, M.; Seddik, T.; Khenata, R.; Bouadjemi, B.; Bin Omran, S.; et al. Structural, optoelectronic, thermodynamic and thermoelectric properties of double half-Heusler (DHH) Ti2FeNiSb2 and Ti2Ni2InSb compounds: A TB-mBJ study. Chin. J. Phys. 2023, 85, 508–523. [Google Scholar] [CrossRef]
- Ding, H.; Li, X.; Feng, Y.; Wu, B. Electronic structure, magnetism and disorder effect in double half-Heusler alloy Mn2FeCoSi2. J. Magn. Magn. Mater. 2022, 555, 169367. [Google Scholar] [CrossRef]
- Douinat, O.; Boucherdoud, A.; Seghier, A.; Houari, M.; Mesbah, S.; Lantri, T.; Bestani, B. Theoretical investigation of the physical, mechanical, and thermal properties of Zr2XBiNi2 (X: Al, Ga) double half-Heusler alloys. Journal of Materials Research 2023, 38, 4509–4521. [Google Scholar] [CrossRef]
- Berarma, K.; Essaoud, S.S.; Al Azar, S.; Al-Reyahi, A.Y.; Mousa, A.A.; Mufleh, A. Computational characterization of structural, optoelectronic, and thermoelectric properties of some double half-Heusler alloys X2FeY′Sb2 (X: Hf, Zr; Y′: Ni, Pd). Phase Transitions 2023, 96, 806–821. [Google Scholar] [CrossRef]
- Surucu, G.; Isik, M.; Candan, A.; Wang, X.; Gullu, H.H. Investigation of structural, electronic, magnetic, and lattice dynamical properties for XCoBi (X: Ti, Zr, Hf) half-Heusler compounds. Physica B: Condensed Matter 2020, 587, 412146. [Google Scholar] [CrossRef]
- Ma, H.; Yang, C.-L.; Wang, M.-S.; Ma, X.-G.; Yi, Y.-G. Effect of M elements (M = Ti, Zr, and Hf) on thermoelectric performance of the half-Heusler compounds MCoBi. J. Phys. D: Appl. Phys. 2019, 52, 255501. [Google Scholar] [CrossRef]
- Zhu, H.; He, R.; Mao, J.; Zhu, Q.; Li, C.; Sun, J.; Ren, W.; Wang, Y.; Liu, Z.; Tang, Z.; et al. Discovery of ZrCoBi based half-Heuslers with high thermoelectric conversion efficiency. Nat. Commun. 2018, 9, 2497. [Google Scholar] [CrossRef] [PubMed]
- Radouan, D.; Besbes, A.; Bestani, B. Investigation on electronic and thermoelectric properties of (P, As, Sb) doped ZrCoBi. East Eur. J. Phys. 2021, 1, 27–33. [Google Scholar]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Fan, T.; Oganov, A.R. AICON: A program for calculating thermal conductivity quickly and accurately. Comput. Phys. Commun. 2020, 251, 107074. [Google Scholar] [CrossRef]
- Anand, S.; Xia, K.; Hegde, V.I.; Aydemir, U.; Kocevski, V.; Zhu, T.; Wolverton, C.; Snyder, G.J. A valence balanced rule for discovery of 18-electron half-Heuslers with defects. Energy Environ. Sci. 2018, 11, 1480–1488. [Google Scholar] [CrossRef]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; Oxford University Press: Oxford, UK, 1996. [Google Scholar]
- Voigt, W. Lehrbuch der Kristallphysik (Textbook of crystal physics). BG Teubner, Leipzig und Berlin 1928.
- Reuss, A.J.Z. Calculation of the flow limits of mixed crystals on the basis of the plasticity of monocrystals. Z. Angew. Math. Mech. 1929, 9, 49–58. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Hill, R. Elastic properties of reinforced solids: some theoretical principles. J. Mech. Phys. Solids 1963, 11, 357–372. [Google Scholar] [CrossRef]
- Chen, H.; Yang, L.; Long, J. First-principles investigation of the elastic, Vickers hardness, and thermodynamic properties of Al–Cu intermetallic compounds. Superlattices Microstruct. 2015, 79, 156–165. [Google Scholar] [CrossRef]
- Frantsevich, I.N. Elastic constants and elastic moduli of metals and insulators. Reference Book; Naukova Dumka: 1982.
- Ranganathan, S.I.; Ostoja-Starzewski, M. Universal elastic anisotropy index. Phys. Rev. Lett. 2008, 101, 055504. [Google Scholar] [CrossRef]
- Niu, H.; Niu, S.; Oganov, A.R. Simple and accurate model of fracture toughness of solids. J. Appl. Phys. 2019, 125, 06. [Google Scholar] [CrossRef]
- Fine, M.E.; Brown, L.D.; Marcus, H.L. Elastic constants versus melting temperature in metals. Scr. Metall. 1984, 18, 951–956. [Google Scholar] [CrossRef]
- Dasmahapatra, A.; Daga, L.E.; Karttunen, A.J.; Maschio, L.; Casassa, S. Key role of defects in thermoelectric performance of TiMSn (M= Ni, Pd, and Pt) half-Heusler alloys. J. Phys. Chem. C 2020, 124, 14997–15006. [Google Scholar] [CrossRef]
- Xi, J.; Dong, Z.; Gao, M.; Luo, J.; Yang, J. Screening of half-Heuslers with temperature-induced band convergence and enhanced thermoelectric properties. arXiv Prepr. 2024, arXiv:2407.00433.
- Gautier, R.; Zhang, X.; Hu, L.; Yu, L.; Lin, Y.; Sunde, T.O.L.; Chon, D.; Poeppelmeier, K.R.; Zunger, A. Prediction and accelerated laboratory discovery of previously unknown 18-electron ABX compounds. Nat. Chem. 2015, 7, 308–316. [Google Scholar] [CrossRef]
- Krukau, Aliaksandr V and Vydrov, Oleg A and Izmaylov, Artur F and Scuseria, Gustavo E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Phys. Chem. C 2006, 125.
Figure 1.
Crystal structure of double half Ti2Pt2ZSb alloys (Z=Al, Ga, In).
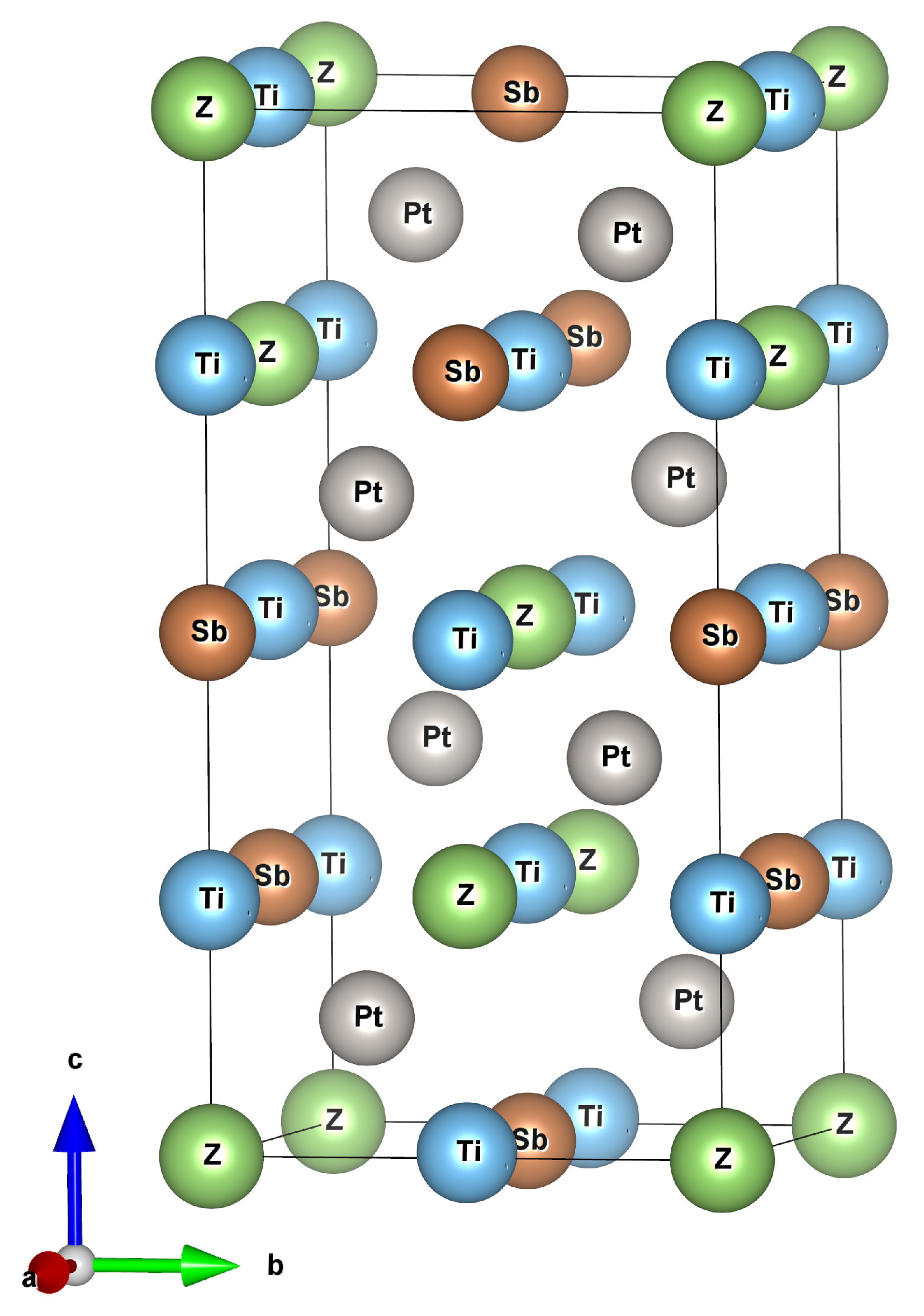
Figure 2.
Calculated phonon dispersion curves and phonon density of states for Ti2Pt2ZSb compounds.
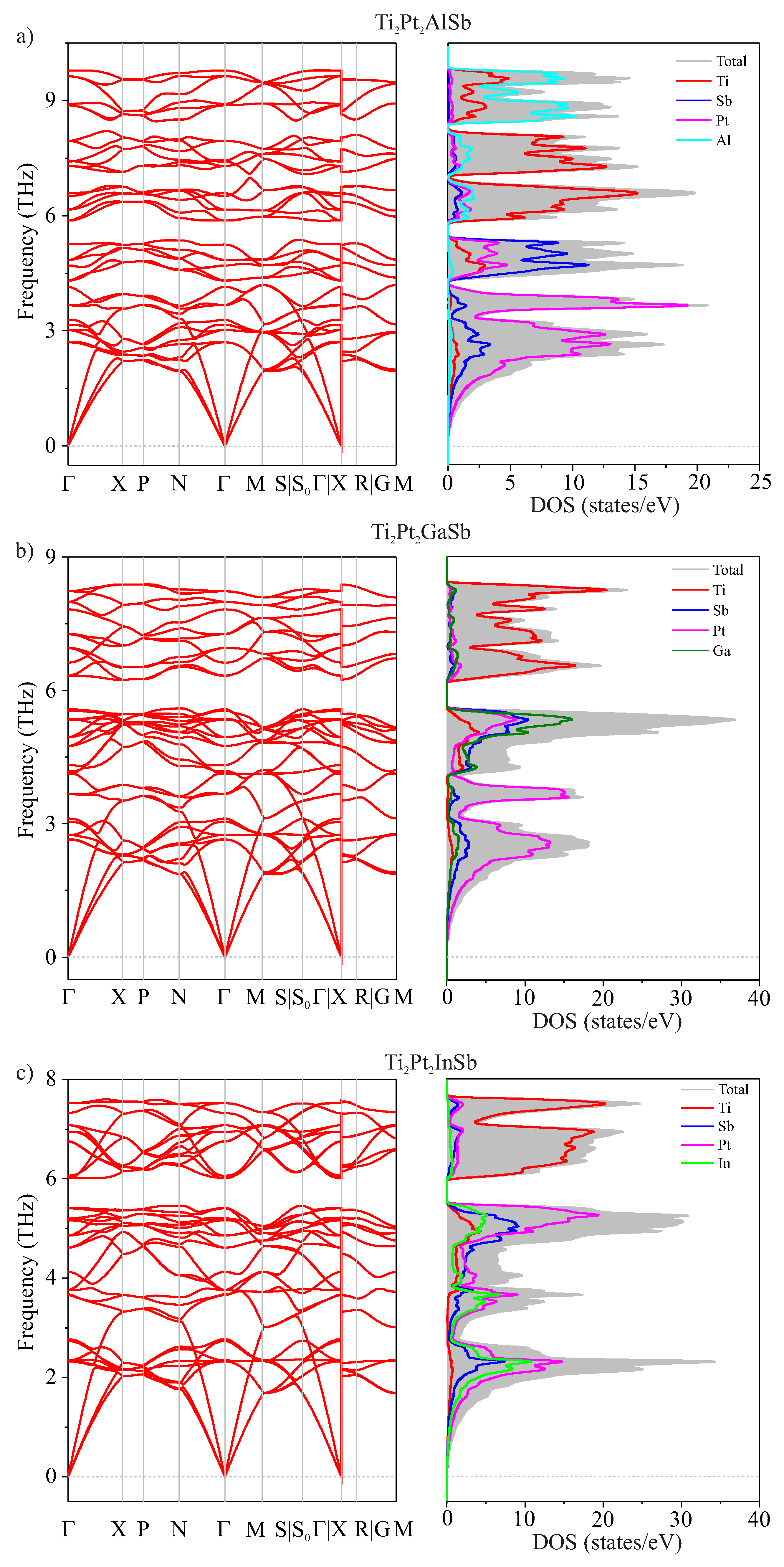
Figure 3.
Comparative analysis of values of hardness (), for Ti2Pt2ZSb compounds.
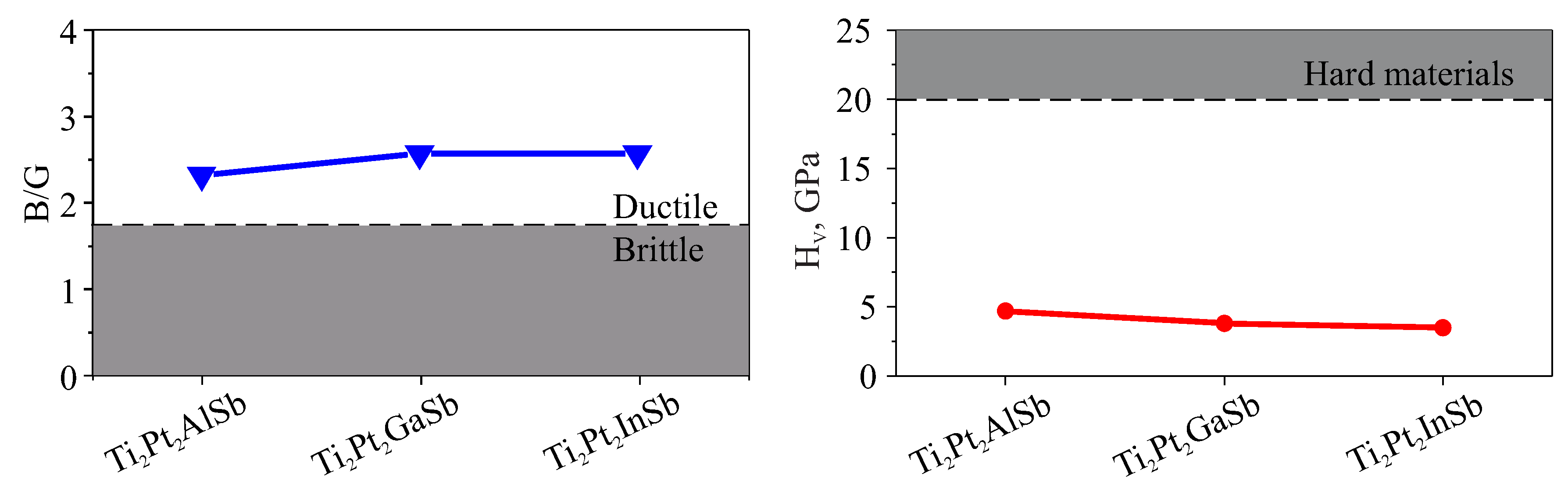
Figure 4.
Lattice thermal conductivity of Ti2Pt2ZSb (Z = Al, Ga, In) at different temperatures ranging from 200 K to 1000 K.
Figure 4.
Lattice thermal conductivity of Ti2Pt2ZSb (Z = Al, Ga, In) at different temperatures ranging from 200 K to 1000 K.
Figure 5.
The density of states and electronic band structure of the Ti2Pt2ZSb double half-Heusler alloys.
Figure 5.
The density of states and electronic band structure of the Ti2Pt2ZSb double half-Heusler alloys.
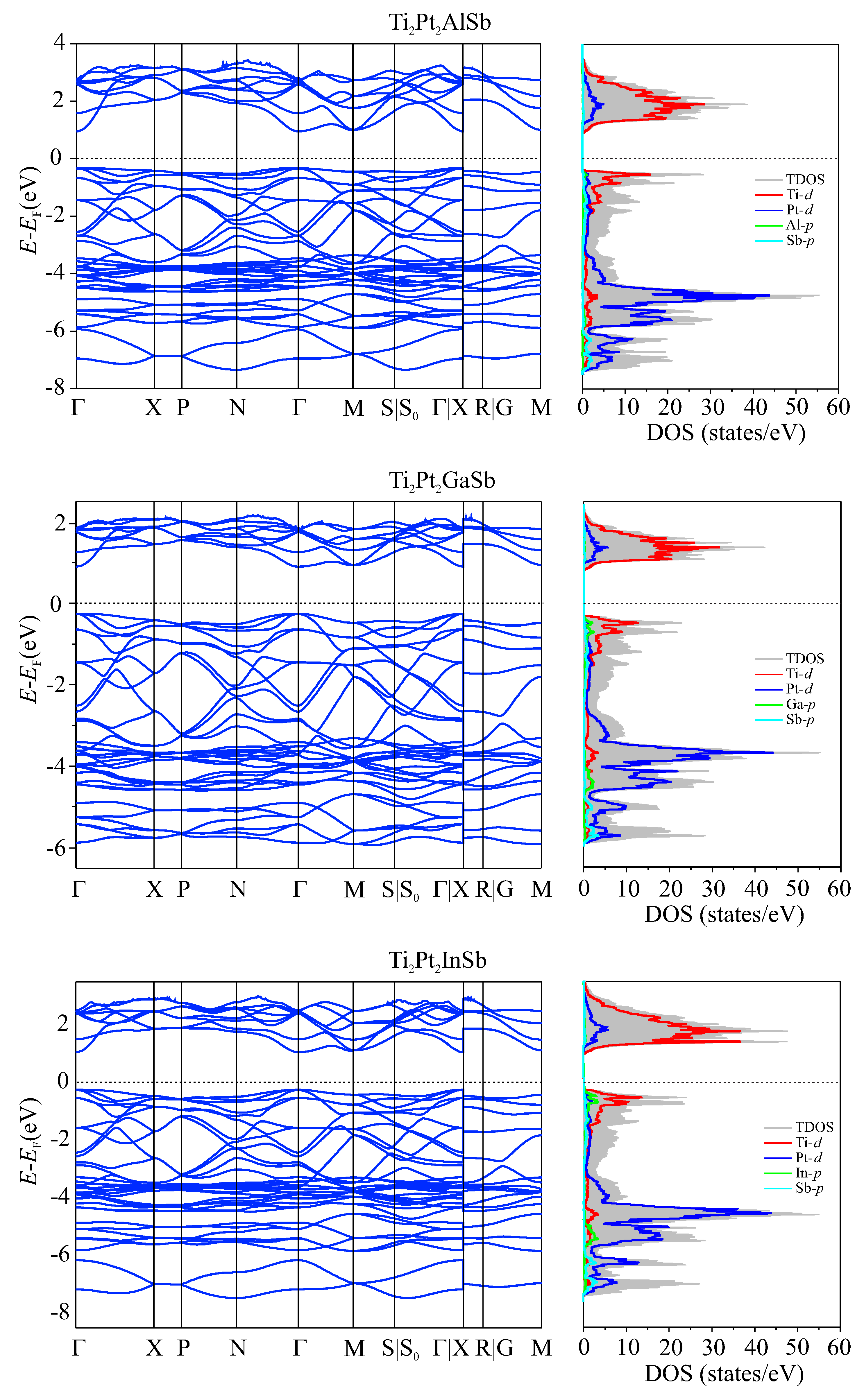
Figure 6.
Calculates ELF of the Ti2Pt2ZSb double half-Heusler alloys in the (110) plane.
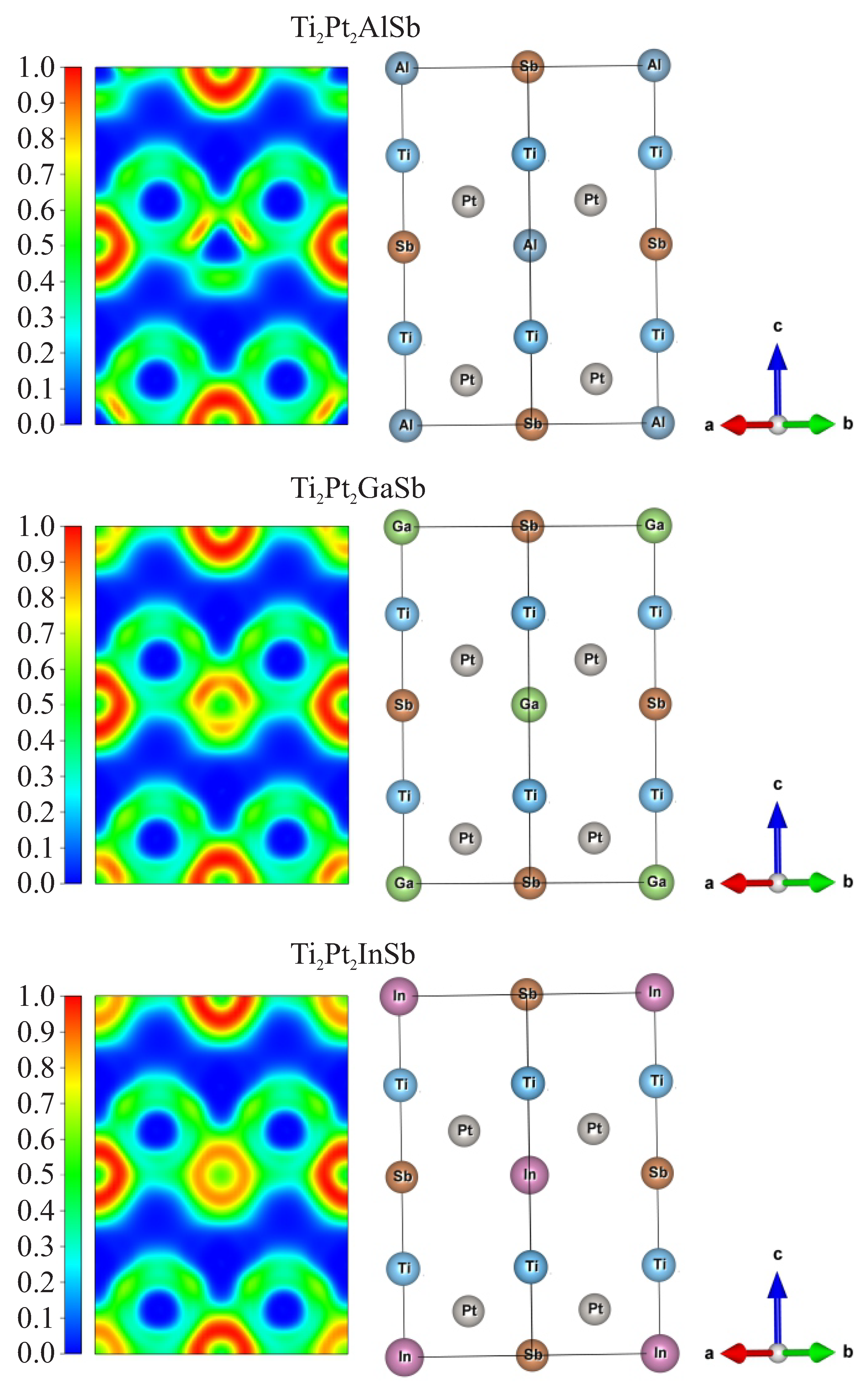

Table 1.
Structural data of considered Ti2Pt2ZSb compounds.
| Compounds | Ti2Pt2AlSb | Ti2Pt2GaSb | Ti2Pt2InSb | |
|---|---|---|---|---|
| Lattice | a = b | 6.120 | 6.121 | 5.905 |
| parameter | c | 12.228 | 12.216 | 11.826 |
| Coordinates | x | y | z | |
| Atoms | Ti | 0 | 0.5 | 0.495 |
| Pt | 0.75 | 0.25 | 0.625 | |
| Al/Ga/In | 0 | 0.5 | 0.25 | |
| Sb | 0 | 0 | 0.5 | |
Table 2.
Calculated optical modes () of Ti2Pt2ZSb (Z = Al, Ga, and In), their activity and corresponding Mulliken symbols.
Table 2.
Calculated optical modes () of Ti2Pt2ZSb (Z = Al, Ga, and In), their activity and corresponding Mulliken symbols.
| Mulliken symbol | Activity | , THz | ||
|---|---|---|---|---|
| Ti2Pt2AlSb | Ti2Pt2GaSb | Ti2Pt2InSb | ||
| E | Raman, IR | 2.700 | 2.644 | 2.322 |
| E | Raman,IR | 3.023 | 2.750 | 2.322 |
| Raman | 3.158 | 3.049 | 2.768 | |
| Raman, IR | 3.287 | 3.116 | 2.732 | |
| Silent | 3.666 | 3.672 | 3.671 | |
| Raman | 3.678 | 3.674 | 3.658 | |
| Silent | 4.147 | 4.192 | 4.125 | |
| E | Raman,IR | 4.312 | 4.127 | 3.759 |
| Raman | 4.704 | 4.318 | 3.763 | |
| E | Raman,IR | 4.848 | 4.749 | 4.615 |
| Raman, IR | 5.251 | 5.358 | 5.163 | |
| E | Raman,IR | 5.874 | 4.949 | 4.862 |
| E | Raman,IR | 6.162 | 5.334 | 5.198 |
| Raman, IR | 6.512 | 5.544 | 5.411 | |
| Raman | 6.587 | 5.572 | 5.406 | |
| E | Raman,IR | 7.295 | 6.330 | 6.010 |
| Raman | 7.414 | 7.269 | 6.750 | |
| Silent | 7.433 | 7.265 | 6.756 | |
| E | Raman,IR | 7.952 | 6.953 | 6.946 |
| E | Raman,IR | 8.874 | 7.991 | 7.079 |
| Raman, IR | 8.914 | 6.625 | 6.059 | |
| E | Raman,IR | 9.634 | 8.231 | 7.323 |
| Raman | 9.783 | 7.824 | 7.523 | |
Table 3.
Calculated elastic constants for Ti2Pt2ZSb compounds (Z = Al, Ga, and In) in GPa.
| Compounds | ||||||
|---|---|---|---|---|---|---|
| Ti2Pt2AlSb | 199.6 | 111.5 | 108.6 | 196.5 | 69.1 | 68 |
| Ti2Pt2GaSb | 194.2 | 115.2 | 113.5 | 191.7 | 66.9 | 66.9 |
| Ti2Pt2InSb | 191.3 | 106.6 | 106.9 | 188.8 | 58.6 | 59 |
Table 4.
Calculated values of the bulk (in GPa), shear (in GPa), Young’s (in GPa) moduli, Poisson’s ratio, ratio, Vickers hardness (in GPa), fracture toughness (in MPa ), bulk and shear anisotropy factors (%), and melting temperature (in K) of Ti2Pt2ZSb (Z=Al, Ga, In) double half Heusler compounds and comparison with TiPtSn [42].
Table 4.
Calculated values of the bulk (in GPa), shear (in GPa), Young’s (in GPa) moduli, Poisson’s ratio, ratio, Vickers hardness (in GPa), fracture toughness (in MPa ), bulk and shear anisotropy factors (%), and melting temperature (in K) of Ti2Pt2ZSb (Z=Al, Ga, In) double half Heusler compounds and comparison with TiPtSn [42].
| Compounds | B | G | E | B/G | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ti2Pt2AlSb | 139 | 58 | 152 | 0.318 | 2.4 | 5.3 | 2.5 | 0.01 | 2.26 | 1248 |
| Ti2Pt2GaSb | 140 | 54 | 144 | 0.329 | 2.6 | 4.5 | 2.4 | 0.01 | 3.25 | 1224 |
| Ti2Pt2InSb | 135 | 51 | 136 | 0.331 | 2.6 | 4.2 | 2.3 | 0.01 | 1.38 | 1211 |
| TiPtSn | 146 | 57 | 150 | 0.329 | 2.5 | 4.7 | – | 0.00 | 1.72 | – |
Table 5.
The calculated lattice thermal conductivity (W/(m·K), longitudinal sound velocity (m/s), transverse sound velocity (m/s) and average sound velocity (m/s) for Ti2Pt2ZSb (Z = Al, Ga, and In) and comparison with TiPtSn.
Table 5.
The calculated lattice thermal conductivity (W/(m·K), longitudinal sound velocity (m/s), transverse sound velocity (m/s) and average sound velocity (m/s) for Ti2Pt2ZSb (Z = Al, Ga, and In) and comparison with TiPtSn.
| Compounds | Ref. | ||||
|---|---|---|---|---|---|
| Ti2Pt2AlSb | 2.67 | 4847 | 2505 | 2911 | this study |
| Ti2Pt2GaSb | 2.52 | 4653 | 2349 | 2732 | this study |
| Ti2Pt2InSb | 2.35 | 4523 | 2273 | 2661 | this study |
| TiPtSn | 9.2 | 4553 | 2569 | 2924 | [43] |
Table 6.
Bader charges of atoms (e) in Ti2Pt2ZSb (Z = Al, Ga, In) alloys.
| Compounds | Ti | Pt | Z | Sb |
|---|---|---|---|---|
| Ti2Pt2AlSb | +1.99 | -3.59 | +3 | +0.20 |
| Ti2Pt2GaSb | +1.98 | -3.62 | +3 | +0.28 |
| Ti2Pt2InSb | +1.94 | -3.63 | +3 | +0.40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



