
Submitted:
04 February 2025
Posted:
05 February 2025
You are already at the latest version
Abstract
A slowly progressive neurological disorder classified as degenerative encephalopathy (DE) occurs in Nova Scotia Duck Tolling Retrievers. The disease is characterized by frequent episodes of pronounced involuntary movements during sleep, cognitive impairment, anxiety, heightened sensitivity to sensory stimuli, and compulsive behaviors. The onset of these signs occurs between 2 months and 5 years of age. Late signs include aggressive behavior, gait abnormalities, and urinary and fecal incontinence. The clinical signs are accompanied by degeneration of the cerebellum, caudate nucleus, and substantia nigra. Next-generation sequencing was used to generate whole-genome sequences from the DNA of affected and unaffected Nova Scotia Duck Tolling Retrievers. The resulting sequences were aligned to the Dog10K_Boxer_Tasha reference genome assembly and the whole genome sequences (WGS) of 334 additonal control canine whole genome sequences generated by the University of Missouri Canine Genetics Laboratory. Analysis of the WGS data identified a missense variant in RB1CC1 exon 22 chromosome 29:4891014, C>T that was uniquely homozygous in the affected dog. This variant predicts a p.G1503R change in the amino acid sequence of RB1CC1. Genotyping of 2,950 Nova Scotia Duck Tolling Retrievers at the variant locus found complete concordance between the disease phenotype and RB1CC1 genotype. RBCC1 is an essential component of a protein complex that mediates the initiation of autophagosome formation. Therefore, it appears likely that the disease results at least in part from impaired autophagy. Consistent with possibility, caudate nucleus neurons of an affected dog were found to contain abnormal lysosomal storage body-like inclusions bodies with heterogenous contents. This disorder could serve as a valuable model to elucidate the mechanisms underlying human diseases associated with impaired autophagy. Identification of the disease-causing DNA sequence variant will enable owners of Nova Scotia Duck Tolling Retrievers to screen their dogs for the RB1CC1 risk variant.
Keywords:
autophagy
; neurodegeneration
; canine
; whole genome sequencing
; lipofuscin
1. Introduction
In 2016 a slowly progressive neurological disorder classified as degenerative encephalopathy was described in Nova Scotia Duck Tolling Retrievers [1]. The most consistent sign is frequent episodes of pronounced involuntary movements during sleep that became progressively more severe over time. Affected dogs also exhibit cognitive impairment, anxiety, heightened sensitivity to sensory stimuli, and compulsive behaviors. As the disease progresses, some affected dogs exhibit aggressive behavior, gait abnormalities, and urinary and fecal incontinence. The severity of signs increases over time. Magnetic resonance imaging of the brains of affected dogs revealed pronounced abnormalities of the caudate nucleus as well as other more subtle abnormalities elsewhere in the brain. Due to the progression of signs, some of the affected dogs were humanely euthanized. Postmortem examination of the brains revealed cavitation lesions in the caudate nucleus. Based on the pattern of inheritance, the disorder appears to be an autosomal recessive trait. Studies were undertaken to identify the molecular genetic basis of this disorder.
2. Materials and Methods
2.1. Identification of Cases and Controls
The hallmark of Nova Scotia Duck Tolling Retriever degenerative encephalopathy is pronounced involuntary movements during sleep [1]. The onset of signs has been reported to occur as early as 2 months of age and as late as 5 years of age [1]. Therefore, for this study cases included dogs that had exhibited the marked and progressive sleep disturbance sign characteristic of this disorder by 5 years of age. Control Nova Scotia Duck Tolling Retrievers consisted of dogs that had not exhibited signs of the disorder by 6 years of age.
2.2. Molecular Genetic Analyses
DNA from affected and unaffected Nova Scotia Duck Tolling Retrievers and unaffected dogs of other breeds was isolated from EDTA anticoagulated blood or FTA Elute cards [2,3] collected with informed consent of the owners. Whole genome sequences were generated from DNA samples from selected dogs by the University of Missouri Genomics Technology Core Facility from Illumina TruSeq PCR-free paired-end libraries. The Burrows-Wheeler Aligner (BWA-mem) was used to map the sequence reads to a canine reference genome assembly (Dog10K_Boxer_Tasha). The reads were then sorted with SAMtools (ver. 1.11), and PCR duplicates were marked with Picard tools (ver. 2.23.8). A modified Genome Analysis Tool Kit (GATK ver. 3.8) best practices pipeline was used for realignment, recalibration, and variant calling. To help identify rare variants in the affected sample sequences, an additional 334 canine whole genome sequences previously generated by the University of Missouri Canine Genetics Laboratory were used as additional unaffected controls. These whole genome sequences have been deposited in the NCBI Sequence Read Archive (SRA) (https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi). The SRA accession numbers for all 336 whole genome sequences used in this analysis are listed in Supplemental Table 1. Variants in samples from the affected dogs were called individually with GATK HaplotypeCaller in the gVCF mode. All sample gVCF files were joined with GATK CombineGVCFs, and jointly genotyped using GATK GenotypeGVCFs. Functional effects of the called variants were predicted with SnpEff software [4] together with Ensembl annotation. SnpSift software was used for filtering low quality variants and extracting annotated variants for the affected samples. Variant reports were generated by tabulating the annotated output to a Microsoft Excel spreadsheet with GATK VariantsToTable. Candidate variants were visually inspected with the Integrative Genomics Viewer (IGV, ver. 2.8.10).
A homozygous missense variant in RB1CC1 exon 22 chromosome 29:4891014, C>T was identified in the whole genome sequence of an affected dog (see Results). This variant was not present in the control dog whole genome sequences. To verify the candidate RB1CC1 variant, Sanger sequencing of the region of RB1CC1 surrounding position 29:4891014 was performed on DNA samples from affected dogs using PCR primers: 5′-TATATGGCTGAACATTGCAGA-3’ and 5′-TCCGAATGCAGAAAATACAAGG--3′. The PCR amplifications were conducted in 30 µL volumes with a DNA Polymerase Kit (Promega) and included an initial denaturation at 950C for 2 min, followed by 40 cycles of denaturation at 950C for 15 s, primer annealing at 600C for 15 s, extension at 720C for 30 s, and a final extension at 720C for 2 min.
An allelic discrimination assay was designed for genotyping individual dogs for the RB1CC1 variant. The PCR primer sequences were 5′-GGGTAGCAGTAAGCCTTTAGGATAATAAAT-3′ and 5′- CGCTCGTCTAGGATGATGAGTAC-3′. The competing probe sequences were 5′-VIC- TTCAGGTGGGAGATTT-NFQ-3′ (reference allele) and 5′-FAM- TTTCAGGTGAGAGATTT-NFQ-3′ (variant allele). These amplifications were conducted in 20 µL volumes with a TaqMan Genotyping Master Mix (Applied Biosystems, Waltham, MA) and included an initial denaturation at 950C for 10 min, followed by 40 cycles of denaturation at 950C for 15 sec, primer annealing at 600C for 1 min, and a final extension at 600C for 30sec on a StepOnePlus Real-Time PCR system (Applied Biosystems).
2.3. Electron Microscopy
Cavitation and astrocytosis of the caudate nucleus is the most pronounced brain lesion in Nova Scotia Duck Tolling Retrievers suffering from degenerative encephalopathy [1]. To determine whether this pathology was accompanied by ultrastructural abnormalities in caudate nucleus neurons, brain tissue was collected from an affected dog that was humanely euthanized due to the progression of disease signs. The caudate nucleus was fixed in 2% glutaraldehyde, 1.2% paraformaldehyde, 120 mM sodium cacodylate, 1 mM CaCl2, pH 7.4 for 7 days. The sample was then rinsed in 170 mM sodium cacodylate, post-fixed with 1% osmium tetroxide, and embedded in Embed 812 resin (Electron Microscopy Sciences cat. no. 14120, 14900). Sections of the sample were cut at thicknesses of 70 to 90 nm and stained with uranyl acetate and lead citrate. The sections were examined with a JEOL JEM-1400 transmission electron microscope equipped with a Gatan digital camera.
3. Results
Whole genome sequences of the affected and unaffected Nova Scotia Duck Tolling Retrievers were generated with a target 30-fold average coverage. The sequence of the affected dog contained 2 uniquely homozygous variants relative to the unaffected dog and the 334 reference whole genome sequences. Among these was a C-to-T transition at position 4,891,014 on chromosome 22 that is predicted to produce a p.G1503R missense variant in the RB1 inducible coiled-coil protein 1 (RB1CC1). The validity of this variant call was confirmed by inspection of aligned reads from the affected dog’s whole genome sequence to the Tasha reference sequence from positions 4,890,982 to 4,891,047 on chromosome 22 using the Integrative-Genomics-Viewer (Figure 1), and by Sanger sequencing (Figure 2).
To assess the concordance between the RB1CC1 29:4891014 genotype and phenotype, a total of 2,950 Nova Scotia Duck Tolling Retrievers were genotyped with the allelic discrimination assay that distinguished between the reference and variant alleles. Of these dogs, 2,590 were homozygous for the reference C allele, 336 were heterozygous, and 24 were homozygous for the T risk allele. None of the heterozygous dogs exhibited disease signs, including the parents of affected dogs. The only potential exception to concordance between genotype and phenotype was one dog that exhibited signs similar to those of dogs with degenerative encephalopathy but was homozygous for the reference allele. Although this dog did exhibit running movements during sleep, it also exhibited sudden-onset bouts of aggression and altered behavior that are not typical of the disorder. Due to these phenotypic differences, this dog was not considered to suffer from DE. Of the 4,114 dogs in the WGS reference cohort, the sequence was reliably called at 29:4891014 in 4,103 dogs. All of these were homozygous for the reference C allele.
The PolyPhen-2 tool (PolyPhen-2: prediction of functional effects of human nsSNPs (harvard.edu) was employed to estimate the functional effect of the RB1CC1 variant on the encoded protein. The PolyPhen-2 score was 0.9999, which predicted that the effect of the variant was very likely to be deleterious.
The amino acid sequence predicted from the proband WGS from p.1470 to p.1503 was aligned to the predicted corresponding RB1CC1 amino acid sequences of 50 mammalian species in the NCBI database. The predicted amino acid sequences were completely conserved across all 50 species except that all but the proband had glycine at the position corresponding to canine p.1503 rather than arginine predicted from the proband sequence.
Neurons of the proband’s caudate nucleus contained large membrane-bounded inclusions with heterogeneous contents similar to those that accumulate in some lysosomal storage disorders [5,6,7] (Figure 3, Figure 4 and Figure 5). Within these inclusion bodies were membrane-like components, lipid droplets, amorphous granular materials, and very electron dense matter. In some planes of section, some of the large inclusion bodies could be seen to be at least partially surrounded by membrane-bounded finger-like cytoplasmic extensions (Figure 4 and Figure 5).
4. Discussion
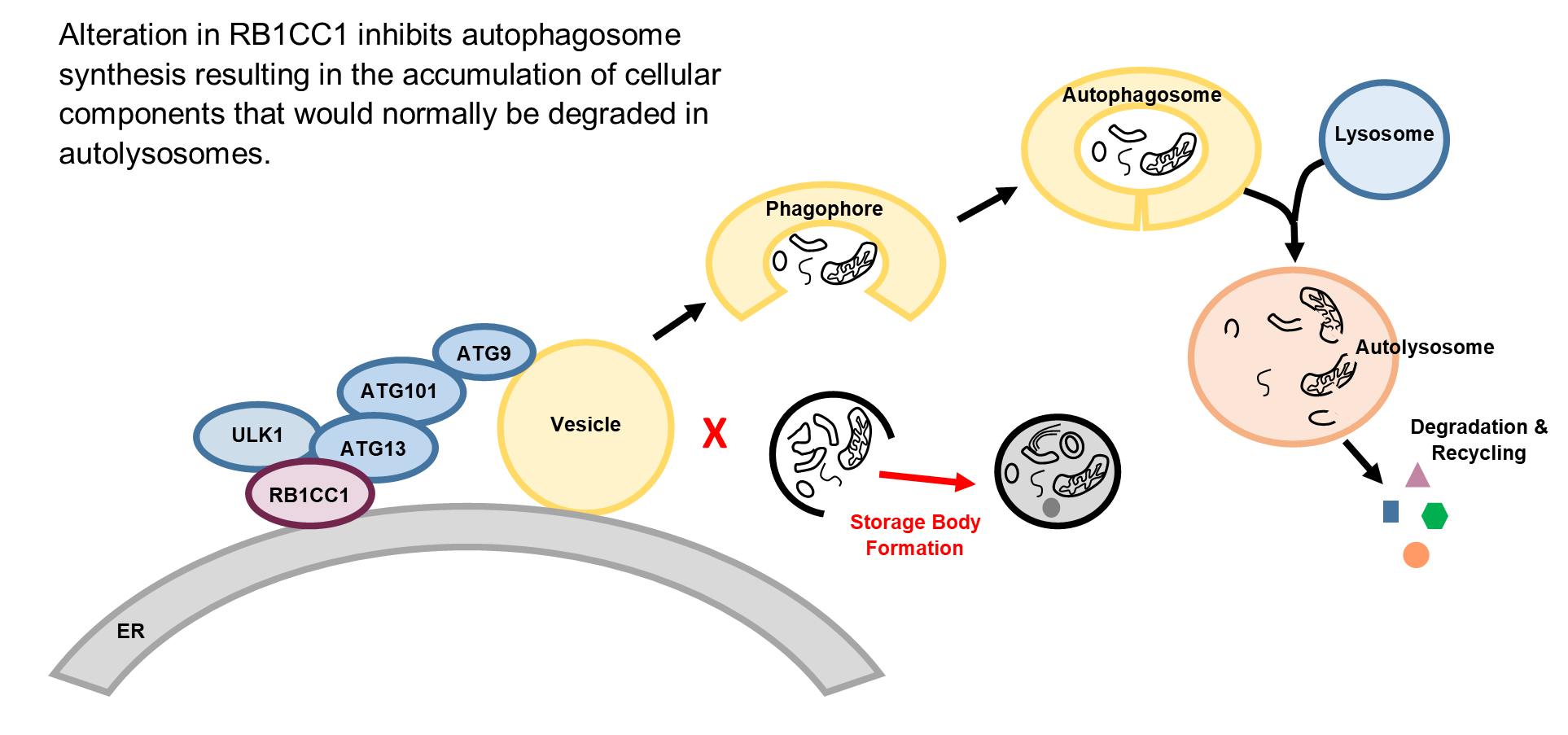
The RB1-inducible coiled-coil 1 protein (RB1CC1; also known as FIP200) plays a central role in macroautophagy, a process by which intracellular components are engulfed into membrane-bounded organelles (autophagosomes) and degraded after fusion of the autophagosomes with lysosomes [8,9,10,11,12,13]. Degradation of damaged cellular components via autophagy is necessary for maintaining cellular homeostasis. The autophagic process is initiated by formation of the ULK protein complex consisting of RB1CC1, ULK1, ATG13 and ATG101 proteins near the endoplasmic reticulum (Figure 6) [8,9,10,11,12,13]. Activation of the ULK complex by intracellular signals starts the process of autophagosome formation. Formation of the double-walled autophagosome membrane is initiated when the activated ULK complex recruits ATG9 protein-containing vesicles to the surface of the endoplasmic reticulum. Many other proteins are involved in subsequent steps of autophagosome formation and function [13].
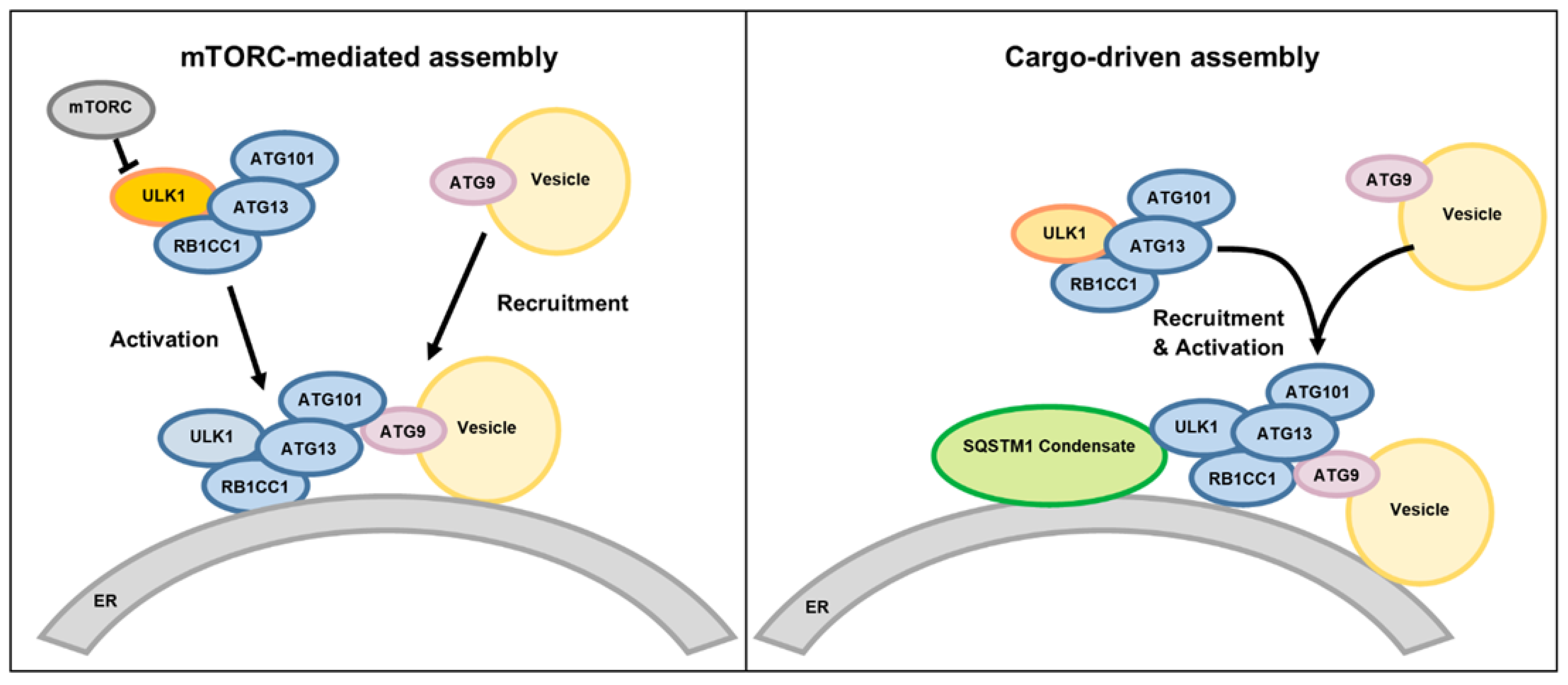
Figure 5.
Alternative mechanisms of macroautophagy initiation. In mTORC-mediated initiation of autophagosome formation, intracellular signaling by factors such as starvation inhibit the mTORC-mediated phosphorylation of ULK1 resulting in dephosphorylation of ULK1 which activates the ULK complex which then binds to the endoplasmic reticulum (ER) and recruits ATG9-containing membrane vesicles. In a cargo-driven process, the SQSTM1 condensate, consisting of a complex of specific proteins and ubiquitinated cargo molecules, binds to the ER and recruits the ULK complex and ATG-containing vesicles to initiate autophagosome formation. (Adapted from [13]).
Figure 5.
Alternative mechanisms of macroautophagy initiation. In mTORC-mediated initiation of autophagosome formation, intracellular signaling by factors such as starvation inhibit the mTORC-mediated phosphorylation of ULK1 resulting in dephosphorylation of ULK1 which activates the ULK complex which then binds to the endoplasmic reticulum (ER) and recruits ATG9-containing membrane vesicles. In a cargo-driven process, the SQSTM1 condensate, consisting of a complex of specific proteins and ubiquitinated cargo molecules, binds to the ER and recruits the ULK complex and ATG-containing vesicles to initiate autophagosome formation. (Adapted from [13]).
Consistent with the conclusion that the canine disorder is the result of the RB1CC1 29:4891014 C>T variant, variants in autophagy genes have been associated with a number of human autosomal recessive hereditary neurodegenerative diseases. Among these are ataxia with developmental delay [14] , mild to severe intellectual disability with ataxia and tremor [15], global developmental delay, seizures and spastic quadriplegia [16], and spinocerebellar ataxia [17]. Cases of schizophrenia with abnormalities in the dentate gyrus have also been associated specifically with RB1CC1 variants [18,19]. The importance of RB1CC1-mediated functions in maintaining normal neurological structure and function is supported by a number of gene-knockout and knock-in studies in mice [20,21,22,23,24,25]. For example, neural-specific deletion of RB1CC1 resulted in cerebellar atrophy associated with axonal degeneration and neuronal death [25]. These data and presence of storage body accumulations in neurons of dogs with the RB1CC1 p.G1503R variant support the conclusion that the neurological disorder in these dogs is the result of impaired autophagy. Variants in genes encoding other proteins involved in the autophagy pathway also result in neurological disorders [26,27].
Autophagosomes are double-membraned vesicles. Defects in the later stages of autophagy result in the intracellular accumulation of these organelles. For example, mutations in TECPR2 that encodes a protein that mediates fusion of autophagosomes with lysosomes, result in accumulation of double-membranes vesicles [27,28,29]. In contrast, the RB1CC1 variant was associated with an accumulation of inclusions surrounded by a single membrane, consistent with impairment in the formation of the normal double-membrane structures. The impaired ability to synthesize autophagosomes (and their precursor phagophores) apparently results in cellular components that would normally be degraded by autophagy accumulating in single-membrane bound lipofuscin-like inclusions. Numerous studies indicate that impaired autophagy is associated with the accumulation of lipofuscin-like intracellular inclusions, similar to that observed in neurons of the affected dog evaluated in this study [30,31,32,33,34,35,36,37]. For example, treatment of retinal pigment epithelial cells in vitro with an inhibitor of autophagy resulted in increased accumulation of lipofuscin-like inclusions [30]. In addition, mice that were homozygous for a null mutation in Per1 (Period 1) exhibited impaired autophagy and a massive accumulation of lipofuscin-like material in the hippocampus [35]. The mechanisms by which autophagy defects result in the accumulation of lipofuscin-like inclusions have not been elucidated. It appears that when damaged intracellular components cannot traverse the autophagosome to autolysosome degradative pathway, they are sequestered in inclusion bodies such as those observed in the caudate nucleus of a dog that was homozygous for the RB1CC1 risk variant. The ultrastructural findings in this study suggest that normal age-related accumulation of lipofuscin may be the result, at least in part, of insufficient degradation of cellular components via autophagy [34,35,38,39,40,41].
The change in RB1CC1 (FIP200) amino acid residue at position 1503 from glycine to arginine likely hinders autophagosome formation by altering the interaction of the protein with other components of the initiation complex. A component of this complex is a condensate of a specific group of proteins with ubiquitinated cargo molecules (SQSTM1 condensate) (Figure 5). It was found that the region of RB1CC1 surrounding residue 1503 is involved in the interaction between RB1CC1 and the SQSTM1 protein complex that initiates cargo-driven autophagosome synthesis [42,43,44]. The replacement of glycine with arginine within this region would be expected to alter this interaction, as well as interactions with other components of the autophagosome initiation complex [45].
Although the accumulation of membrane-bound inclusion bodies in neurons of the proband suggests that the autophagy process was impaired, RB1CC1 has also been found to have other functions in addition to its role in autophagy [46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63]. For example, RB1CC1 has been shown to have autophagy-independent roles in cell adhesion and motility [46], modulating gene expression [49], and immune signaling [47,55]. It is possible that impairment of these functions by the alteration in the RB1CC1 amino acid sequence contributed to the disease pathology in the affected dogs.
A number of studies have demonstrated the importance of RB1CC1 in many tissues in outside of the central nervous system [25,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82]. For example, variants in RB1CC1 have been associated with differences in muscle strength in human subjects [83], and conditional rb1cc1 knockout in mice results in myopathy with the accumulation of inclusion bodies in muscle fibers [84]. Considering the key role that RB1CC1 plays in autophagy and other cellular functions, it is not surprising that multiple tissues are impacted by alterations in the function of this protein. It is possible that some of the signs exhibited by the dogs with the RB1CC1 missense variant were secondary to pathology outside of the central nervous system. For example, extra-neuronal pathology may have played a role in the gait abnormalities and urinary and fecal incontinence that characterized the later stages of the canine disorder. It will be of interest to evaluate additional tissues from affected dogs, particularly skeletal muscle, should they become available.
Based on our findings, owners of Nova Scotia Duck Tolling Retrievers can now screen their dogs for the RB1CC1 degenerative encephalopathy risk variant and avoid breeding dogs that could produce affected offspring.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Acknowledgments
Our thanks to the dog owners who provided us with the samples and health information that were used in this study. Assistance in sample acquisition, processing archiving was provided Liz Hansen, Peggy Ann Eichen, and Timothy Pullian. The research was supported in part by a grant from the Orthopedic Foundation for Animals.
References
- Barker, E.N.; Dawson, L.J.; Rose, J.H.; Van Meervenne, S.; Frykman, O.; Rohdin, C.; Leijon, A.; Soerensen, K.E.; Jarnegren, J.; Johnson, G.C.; et al. Degenerative Encephalopathy in Nova Scotia Duck Tolling Retrievers Presenting with a Rapid Eye Movement Sleep Behavior Disorder. J Vet Intern Med 2016, 30, 1681–1689. [CrossRef]
- Katz, M.L.; Khan, S.; Awano, T.; Shahid, S.A.; Siakotos, A.N.; Johnson, G.S. A Mutation in the CLN8 Gene in English Setter Dogs with Neuronal Ceroid-Lipofuscinosis. Biochem Biophys Res Commun 2005, 327. [CrossRef]
- Zeng, R.; Coates, J.R.; Johnson, G.C.; Hansen, L.; Awano, T.; Kolicheski, A.; Ivansson, E.; Perloski, M.; Lindblad-Toh, K.; O’Brien, D.P.; et al. Breed Distribution of SOD1 Alleles Previously Associated with Canine Degenerative Myelopathy. J Vet Intern Med 2014, 28. [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain W1118; Iso-2; Iso-3. Fly (Austin) 2012, 6, 80–92. [CrossRef]
- Ferreira, C.R.; Gahl, W.A. Lysosomal Storage Diseases. Transl Sci Rare Dis 2017, 2, 1–71. [CrossRef]
- Bullock, G.; Johnson, G.S.; Mhlanga-Mutangadura, T.; Petesch, S.C.; Thompson, S.; Goebbels, S.; Katz, M.L. Lysosomal Storage Disease Associated with a CNP Sequence Variant in Dalmatian Dogs. Gene 2022, 830, 146513. PT - Journal Article. [CrossRef]
- Furuta, A.; Kikuchi, H.; Fujita, H.; Yamada, D.; Fujiwara, Y.; Kabuta, T.; Nishino, I.; Wada, K.; Uchiyama, Y. Property of Lysosomal Storage Disease Associated with Midbrain Pathology in the Central Nervous System of Lamp-2-Deficient Mice. Am J Pathol 2015, 185, 1713–1723. [CrossRef]
- Alers, S.; Wesselborg, S.; Stork, B. ATG13: Just a Companion, or an Executor of the Autophagic Program?. Autophagy 2014, 10, 944–956. [CrossRef]
- Noda, N.N.; Mizushima, N. Atg101: Not Just an Accessory Subunit in the Autophagy-Initiation Complex. Cell Struct Funct 2016, 41, 13–20. [CrossRef]
- Yamashita, S.-I.; Kanki, T. How Autophagy Eats Large Mitochondria: Autophagosome Formation Coupled with Mitochondrial Fragmentation. Autophagy 2017, 13, 980–981. [CrossRef]
- Corona Velazquez, A.F.; Jackson, W.T. So Many Roads: The Multifaceted Regulation of Autophagy Induction. Mol Cell Biol 2018, 38. [CrossRef]
- Yamano, K.; Youle, R.J. Two Different Axes CALCOCO2-RB1CC1 and OPTN-ATG9A Initiate PRKN-Mediated Mitophagy. Autophagy 2020, 16, 2105–2107. [CrossRef]
- Yamamoto, H.; Zhang, S.; Mizushima, N. Autophagy Genes in Biology and Disease. Nat Rev Genet 2023, 24, 382–400. [CrossRef]
- Kim, M.; Sandford, E.; Gatica, D.; Qiu, Y.; Liu, X.; Zheng, Y.; Schulman, B.A.; Xu, J.; Semple, I.; Ro, S.-H.; et al. Mutation in ATG5 Reduces Autophagy and Leads to Ataxia with Developmental Delay. Elife 2016, 5. [CrossRef]
- Collier, J.J.; Guissart, C.; Olahova, M.; Sasorith, S.; Piron-Prunier, F.; Suomi, F.; Zhang, D.; Martinez-Lopez, N.; Leboucq, N.; Bahr, A.; et al. Developmental Consequences of Defective ATG7-Mediated Autophagy in Humans. N Engl J Med 2021, 384, 2406–2417. [CrossRef]
- Almannai, M.; Marafi, D.; Abdel-Salam, G.M.H.; Zaki, M.S.; Duan, R.; Calame, D.; Herman, I.; Levesque, F.; Elbendary, H.M.; Hegazy, I.; et al. El-Hattab-Alkuraya Syndrome Caused by Biallelic WDR45B Pathogenic Variants: Further Delineation of the Phenotype and Genotype. Clin Genet 2022, 101, 530–540. [CrossRef]
- Seidahmed, M.Z.; Hamad, M.H.; AlBakheet, A.; Elmalik, S.A.; AlDrees, A.; Al-Sufayan, J.; Alorainy, I.; Ghozzi, I.M.; Colak, D.; Salih, M.A.; et al. Ancient Founder Mutation in RUBCN: A Second Unrelated Family Confirms Salih Ataxia (SCAR15). BMC Neurol 2020, 20, 207. [CrossRef]
- Guo, A.; Lun, P.; Chen, J.; Li, Q.; Chang, K.; Li, T.; Pan, D.; Zhang, J.; Zhou, J.; Wang, K.; et al. Association Analysis of Risk Genes Identified by SCHEMA with Schizophrenia in the Chinese Han Population. Psychiatr Genet 2022, 32, 188–193. [CrossRef]
- Lappas, A.S.; Ioannou, M.; Christodoulou, N.G. Histopathological Evidence of Cellular Alterations in the Dentate Gyrus Is Associated with Aberrant RB1CC1-ATG16L1 Expression in the Hippocampus among Older Adults with Chronic Schizophrenia: A Pilot Post-Mortem Study. Schizophr Res 2025, 275, 14–24. [CrossRef]
- Yao, J.; Jia, L.; Khan, N.; Lin, C.; Mitter, S.K.; Boulton, M.E.; Dunaief, J.L.; Klionsky, D.J.; Guan, J.-L.; Thompson, D.A.; et al. Deletion of Autophagy Inducer RB1CC1 Results in Degeneration of the Retinal Pigment Epithelium. Autophagy 2015, 11, 939–953. [CrossRef]
- Wang, C.; Chen, S.; Yeo, S.; Karsli-Uzunbas, G.; White, E.; Mizushima, N.; Virgin, H.W.; Guan, J.-L. Elevated P62/SQSTM1 Determines the Fate of Autophagy-Deficient Neural Stem Cells by Increasing Superoxide. J Cell Biol 2016, 212, 545–560. [CrossRef]
- Chen, S.; Wang, C.; Yeo, S.; Liang, C.-C.; Okamoto, T.; Sun, S.; Wen, J.; Guan, J.-L. Distinct Roles of Autophagy-Dependent and -Independent Functions of FIP200 Revealed by Generation and Analysis of a Mutant Knock-in Mouse Model. Genes Dev 2016, 30, 856–869. [CrossRef]
- Zhu, Y.-F.; Yu, R.-H.; Zhou, S.; Tang, P.-P.; Zhang, R.; Wu, Y.-X.; Xu, R.; Wei, J.-M.; Wang, Y.-Y.; Zhang, J.-L.; et al. TAX1BP1 and FIP200 Orchestrate Non-Canonical Autophagy of P62 Aggregates for Mouse Neural Stem Cell Maintenance. Zool Res 2024, 45, 937–950. [CrossRef]
- Wang, C.; Yeo, S.; Haas, M.A.; Guan, J.-L. Autophagy Gene FIP200 in Neural Progenitors Non-Cell Autonomously Controls Differentiation by Regulating Microglia. J Cell Biol 2017, 216, 2581–2596. [CrossRef]
- Liang, C.-C.; Wang, C.; Peng, X.; Gan, B.; Guan, J.-L. Neural-Specific Deletion of FIP200 Leads to Cerebellar Degeneration Caused by Increased Neuronal Death and Axon Degeneration. J Biol Chem 2010, 285, 3499–3509. [CrossRef]
- Deneubourg, C.; Ramm, M.; Smith, L.J.; Baron, O.; Singh, K.; Byrne, S.C.; Duchen, M.R.; Gautel, M.; Eskelinen, E.-L.; Fanto, M.; et al. The Spectrum of Neurodevelopmental, Neuromuscular and Neurodegenerative Disorders Due to Defective Autophagy. Autophagy 2022, 18, 496–517. [CrossRef]
- Hahn, K.; Rohdin, C.; Jagannathan, V.; Wohlsein, P.; Baumgartner, W.; Seehusen, F.; Spitzbarth, I.; Grandon, R.; Drogemuller, C.; Jaderlund, K.H. TECPR2 Associated Neuroaxonal Dystrophy in Spanish Water Dogs. PLoS One 2015, 10, e0141824. [CrossRef]
- Tamim-Yecheskel, B.-C.; Fraiberg, M.; Kokabi, K.; Freud, S.; Shatz, O.; Marvaldi, L.; Subic, N.; Brenner, O.; Tsoory, M.; Eilam-Altstadter, R.; et al. A Tecpr2 Knockout Mouse Exhibits Age-Dependent Neuroaxonal Dystrophy Associated with Autophagosome Accumulation. Autophagy 2021, 17, 3082–3095. [CrossRef]
- Fraiberg, M.; Tamim-Yecheskel, B.-C.; Kokabi, K.; Subic, N.; Heimer, G.; Eck, F.; Nalbach, K.; Behrends, C.; Ben-Zeev, B.; Shatz, O.; et al. Lysosomal Targeting of Autophagosomes by the TECPR Domain of TECPR2. Autophagy 2021, 17, 3096–3108. [CrossRef]
- Lei, L.; Tzekov, R.; Li, H.; McDowell, J.H.; Gao, G.; Smith, W.C.; Tang, S.; Kaushal, S. Inhibition or Stimulation of Autophagy Affects Early Formation of Lipofuscin-Like Autofluorescence in the Retinal Pigment Epithelium Cell. Int J Mol Sci 2017, 18. [CrossRef]
- Liu, A.; Guo, E.; Yang, J.; Yang, Y.; Liu, S.; Jiang, X.; Hu, Q.; Dirsch, O.; Dahmen, U.; Zhang, C.; et al. Young Plasma Reverses Age-Dependent Alterations in Hepatic Function through the Restoration of Autophagy. Aging Cell 2018, 17. [CrossRef]
- Nian, F.-S.; Li, L.-L.; Cheng, C.-Y.; Wu, P.-C.; Lin, Y.-T.; Tang, C.-Y.; Ren, B.-S.; Tai, C.-Y.; Fann, M.-J.; Kao, L.-S.; et al. Rab18 Collaborates with Rab7 to Modulate Lysosomal and Autophagy Activities in the Nervous System: An Overlapping Mechanism for Warburg Micro Syndrome and Charcot-Marie-Tooth Neuropathy Type 2B. Mol Neurobiol 2019, 56, 6095–6105. [CrossRef]
- Sarraf, S.A.; Shah, H. V; Kanfer, G.; Pickrell, A.M.; Holtzclaw, L.A.; Ward, M.E.; Youle, R.J. Loss of TAX1BP1-Directed Autophagy Results in Protein Aggregate Accumulation in the Brain. Mol Cell 2020, 80, 779-795.e10. [CrossRef]
- Li, W.-W.; Wang, H.-J.; Tan, Y.-Z.; Wang, Y.-L.; Yu, S.-N.; Li, Z.-H. Reducing Lipofuscin Accumulation and Cardiomyocytic Senescence of Aging Heart by Enhancing Autophagy. Exp Cell Res 2021, 403, 112585. [CrossRef]
- Borner, J.H.; Rawashdeh, O.; Rami, A. Exacerbated Age-Related Hippocampal Alterations of Microglia Morphology, Beta-Amyloid and Lipofuscin Deposition and Presenilin Overexpression in <ovid:I>Per1</Ovid:I><ovid:Sup>-/-</Ovid:Sup>-Mice. Antioxidants (Basel) 2021, 10. [CrossRef]
- Hyttinen, J.M.T.; Koskela, A.; Blasiak, J.; Kaarniranta, K. Autophagy in Drusen Biogenesis Secondary to Age-Related Macular Degeneration. Acta Ophthalmol 2024, 102, 759–772. [CrossRef]
- Wang, X.-L.; Gao, Y.-X.; Yuan, Q.-Z.; Zhang, M. NLRP3 and Autophagy in Retinal Ganglion Cell Inflammation in Age-Related Macular Degeneration: Potential Therapeutic Implications. Int J Ophthalmol 2024, 17, 1531–1544. [CrossRef]
- Sosulski, M.L.; Gongora, R.; Danchuk, S.; Dong, C.; Luo, F.; Sanchez, C.G. Deregulation of Selective Autophagy during Aging and Pulmonary Fibrosis: The Role of TGFbeta1. Aging Cell 2015, 14, 774–783. [CrossRef]
- Li, W.-W.; Wang, H.-J.; Tan, Y.-Z.; Wang, Y.-L.; Yu, S.-N.; Li, Z.-H. Reducing Lipofuscin Accumulation and Cardiomyocytic Senescence of Aging Heart by Enhancing Autophagy. Exp Cell Res 2021, 403, 112585. [CrossRef]
- Winicki, N.M.; Nanavati, A.P.; Morrell, C.H.; Moen, J.M.; Axsom, J.E.; Krawczyk, M.; Petrashevskaya, N.N.; Beyman, M.G.; Ramirez, C.; Alfaras, I.; et al. A Small Erythropoietin Derived Non-Hematopoietic Peptide Reduces Cardiac Inflammation, Attenuates Age Associated Declines in Heart Function and Prolongs Healthspan. Front Cardiovasc Med 2022, 9, 1096887. [CrossRef]
- Terman, A.; Kurz, T.; Gustafsson, B.; Brunk, U.T. The Involvement of Lysosomes in Myocardial Aging and Disease. Curr Cardiol Rev 2008, 4, 107–115. [CrossRef]
- Turco, E.; Witt, M.; Abert, C.; Bock-Bierbaum, T.; Su, M.-Y.; Trapannone, R.; Sztacho, M.; Danieli, A.; Shi, X.; Zaffagnini, G.; et al. FIP200 Claw Domain Binding to P62 Promotes Autophagosome Formation at Ubiquitin Condensates. Mol Cell 2019, 74, 330-346.e11. [CrossRef]
- Turco, E.; Witt, M.; Abert, C.; Bock-Bierbaum, T.; Su, M.-Y.; Trapannone, R.; Sztacho, M.; Danieli, A.; Shi, X.; Zaffagnini, G.; et al. How RB1CC1/FIP200 Claws Its Way to Autophagic Engulfment of SQSTM1/P62-Ubiquitin Condensates. Autophagy 2019, 15, 1475–1477. [CrossRef]
- Zhang, M.; Wang, Y.; Gong, X.; Wang, Y.; Zhang, Y.; Tang, Y.; Zhou, X.; Liu, H.; Huang, Y.; Zhang, J.; et al. Mechanistic Insights into the Interactions of TAX1BP1 with RB1CC1 and Mammalian ATG8 Family Proteins. Proc Natl Acad Sci U S A 2024, 121, e2315550121. [CrossRef]
- Fu, T.; Zhang, M.; Zhou, Z.; Wu, P.; Peng, C.; Wang, Y.; Gong, X.; Li, Y.; Wang, Y.; Xu, X.; et al. Structural and Biochemical Advances on the Recruitment of the Autophagy-Initiating ULK and TBK1 Complexes by Autophagy Receptor NDP52. Sci Adv 2021, 7. [CrossRef]
- Assar, E.A.; Tumbarello, D.A. Loss of the Essential Autophagy Regulators FIP200 or Atg5 Leads to Distinct Effects on Focal Adhesion Composition and Organization. Front Cell Dev Biol 2020, 8, 733. [CrossRef]
- Yeo, S.K.; Guan, J.-L. Regulation of Immune Checkpoint Blockade Efficacy in Breast Cancer by FIP200: A Canonical-Autophagy-Independent Function. Cell Stress 2020, 4, 216–217. [CrossRef]
- Liu, H.; Wang, C.; Yi, F.; Yeo, S.; Haas, M.; Tang, X.; Guan, J.-L. Non-Canonical Function of FIP200 Is Required for Neural Stem Cell Maintenance and Differentiation by Limiting TBK1 Activation and P62 Aggregate Formation. Sci Rep 2021, 11, 23907. [CrossRef]
- Xue, X.; Ma, L.; Zhang, X.; Xu, X.; Guo, S.; Wang, Y.; Qiu, S.; Cui, J.; Guo, W.; Yu, Y.; et al. Tumour Cells Are Sensitised to Ferroptosis via RB1CC1-Mediated Transcriptional Reprogramming. Clin Transl Med 2022, 12, e747. [CrossRef]
- Yang, Y.; Klionsky, D.J. A Novel Role of ATG9A and RB1CC1/FIP200 in Mediating Cell-Death Checkpoints to Repress TNF Cytotoxicity. Autophagy 2023, 19, 1617–1618. [CrossRef]
- Chen, P.; Duan, Y.; Lu, X.; Chen, L.; Zhang, W.; Wang, H.; Hu, R.; Liu, S. RB1CC1 Functions as a Tumor-Suppressing Gene in Renal Cell Carcinoma via Suppression of PYK2 Activity and Disruption of TAZ-Mediated PDL1 Transcription Activation. Cancer Immunol Immunother 2021, 70, 3261–3275. [CrossRef]
- Wang, L.; Song, K.; Hao, W.; Wu, Y.; Patil, G.; Hua, F.; Sun, Y.; Huang, C.; Ritchey, J.; Jones, C.; et al. FIP200 Restricts RNA Virus Infection by Facilitating RIG-I Activation. Commun Biol 2021, 4, 921. [CrossRef]
- Yang, Y.; Klionsky, D.J. A Novel Role of ATG9A and RB1CC1/FIP200 in Mediating Cell-Death Checkpoints to Repress TNF Cytotoxicity. Autophagy 2023, 19, 1617–1618. [CrossRef]
- Okamoto, T.; Yeo, S.K.; Hao, M.; Copley, M.R.; Haas, M.A.; Chen, S.; Guan, J.-L. FIP200 Suppresses Immune Checkpoint Therapy Responses in Breast Cancers by Limiting AZI2/TBK1/IRF Signaling Independent of Its Canonical Autophagy Function. Cancer Res 2020, 80, 3580–3592. [CrossRef]
- Yeo, S.K.; Wang, C.; Guan, J.-L. Role of FIP200 in Inflammatory Processes beyond Its Canonical Autophagy Function. Biochem Soc Trans 2020, 48, 1599–1607. [CrossRef]
- Goodwin, J.M.; Dowdle, W.E.; DeJesus, R.; Wang, Z.; Bergman, P.; Kobylarz, M.; Lindeman, A.; Xavier, R.J.; McAllister, G.; Nyfeler, B.; et al. Autophagy-Independent Lysosomal Targeting Regulated by ULK1/2-FIP200 and ATG9. Cell Rep 2017, 20, 2341–2356. [CrossRef]
- Yang, Y.; Klionsky, D.J. A Novel Role of ATG9A and RB1CC1/FIP200 in Mediating Cell-Death Checkpoints to Repress TNF Cytotoxicity. Autophagy 2023, 19, 1617–1618. [CrossRef]
- Abbi, S.; Ueda, H.; Zheng, C.; Cooper, L.A.; Zhao, J.; Christopher, R.; Guan, J.-L. Regulation of Focal Adhesion Kinase by a Novel Protein Inhibitor FIP200. Mol Biol Cell 2002, 13, 3178–3191. [CrossRef]
- Melkoumian, Z.K.; Peng, X.; Gan, B.; Wu, X.; Guan, J.-L. Mechanism of Cell Cycle Regulation by FIP200 in Human Breast Cancer Cells. Cancer Res 2005, 65, 6676–6684. [CrossRef]
- Choi, J.D.; Ryu, M.; Ae Park, M.; Jeong, G.; Lee, J.-S. FIP200 Inhibits Beta-Catenin-Mediated Transcription by Promoting APC-Independent Beta-Catenin Ubiquitination. Oncogene 2013, 32, 2421–2432. [CrossRef]
- Wang, C.; Liang, C.-C.; Bian, Z.C.; Zhu, Y.; Guan, J.-L. FIP200 Is Required for Maintenance and Differentiation of Postnatal Neural Stem Cells. Nat Neurosci 2013, 16, 532–542. [CrossRef]
- Watanabe, R.; Chano, T.; Inoue, H.; Isono, T.; Koiwai, O.; Okabe, H. Rb1cc1 Is Critical for Myoblast Differentiation through Rb1 Regulation. Virchows Arch 2005, 447, 643–648. [CrossRef]
- Chano, T.; Saeki, Y.; Serra, M.; Matsumoto, K.; Okabe, H. Preferential Expression of RB1-Inducible Coiled-Coil 1 in Terminal Differentiated Musculoskeletal Cells. Am J Pathol 2002, 161, 359–364. [CrossRef]
- Wei, H.; Gan, B.; Wu, X.; Guan, J.-L. Inactivation of FIP200 Leads to Inflammatory Skin Disorder, but Not Tumorigenesis, in Conditional Knock-out Mouse Models. J Biol Chem 2009, 284, 6004–6013. [CrossRef]
- Liu, F.; Guan, J.-L. FIP200, an Essential Component of Mammalian Autophagy Is Indispensible for Fetal Hematopoiesis. Autophagy 2011, 7, 229–230. [CrossRef]
- Wei, H.; Wei, S.; Gan, B.; Peng, X.; Zou, W.; Guan, J.-L. Suppression of Autophagy by FIP200 Deletion Inhibits Mammary Tumorigenesis. Genes Dev 2011, 25, 1510–1527. [CrossRef]
- Bae, H.; Guan, J.-L. Suppression of Autophagy by FIP200 Deletion Impairs DNA Damage Repair and Increases Cell Death upon Treatments with Anticancer Agents. Mol Cancer Res 2011, 9, 1232–1241. [CrossRef]
- Ma, D.; Molusky, M.M.; Song, J.; Hu, C.-R.; Fang, F.; Rui, C.; Mathew, A. V; Pennathur, S.; Liu, F.; Cheng, J.-X.; et al. Autophagy Deficiency by Hepatic FIP200 Deletion Uncouples Steatosis from Liver Injury in NAFLD. Mol Endocrinol 2013, 27, 1643–1654. [CrossRef]
- Liu, F.; Fang, F.; Yuan, H.; Yang, D.; Chen, Y.; Williams, L.; Goldstein, S.A.; Krebsbach, P.H.; Guan, J.-L. Suppression of Autophagy by FIP200 Deletion Leads to Osteopenia in Mice through the Inhibition of Osteoblast Terminal Differentiation. J Bone Miner Res 2013, 28, 2414–2430. [CrossRef]
- Li, Y.; Gan, C.; Zhang, S.; Zhou, X.; Li, X.; Wei, Y.; Yang, J.; Wu, M. FIP200 Is Involved in Murine Pseudomonas Infection by Regulating HMGB1 Intracellular Translocation. Cell Physiol Biochem 2014, 33, 1733–1744. [CrossRef]
- Yao, J.; Jia, L.; Khan, N.; Lin, C.; Mitter, S.K.; Boulton, M.E.; Dunaief, J.L.; Klionsky, D.J.; Guan, J.-L.; Thompson, D.A.; et al. Deletion of Autophagy Inducer RB1CC1 Results in Degeneration of the Retinal Pigment Epithelium. Autophagy 2015, 11, 939–953. [CrossRef]
- Chen, S.; Wang, C.; Yeo, S.; Liang, C.-C.; Okamoto, T.; Sun, S.; Wen, J.; Guan, J.-L. Distinct Roles of Autophagy-Dependent and -Independent Functions of FIP200 Revealed by Generation and Analysis of a Mutant Knock-in Mouse Model. Genes Dev 2016, 30, 856–869. [CrossRef]
- Wang, C.; Yeo, S.; Haas, M.A.; Guan, J.-L. Autophagy Gene FIP200 in Neural Progenitors Non-Cell Autonomously Controls Differentiation by Regulating Microglia. J Cell Biol 2017, 216, 2581–2596. [CrossRef]
- Wang, B.; Iyengar, R.; Li-Harms, X.; Joo, J.H.; Wright, C.; Lavado, A.; Horner, L.; Yang, M.; Guan, J.-L.; Frase, S.; et al. The Autophagy-Inducing Kinases, ULK1 and ULK2, Regulate Axon Guidance in the Developing Mouse Forebrain via a Noncanonical Pathway. Autophagy 2018, 14, 796–811. [CrossRef]
- Oestreich, A.K.; Chadchan, S.B.; Medvedeva, A.; Lydon, J.P.; Jungheim, E.S.; Moley, K.H.; Kommagani, R. The Autophagy Protein, FIP200 (RB1CC1) Mediates Progesterone Responses Governing Uterine Receptivity and Decidualization . Biol Reprod 2020, 102, 843–851. [CrossRef]
- Li, D.; Vogel, P.; Li-Harms, X.; Wang, B.; Kundu, M. ATG14 and RB1CC1 Play Essential Roles in Maintaining Muscle Homeostasis. Autophagy 2021, 17, 2576–2585. [CrossRef]
- Wang, L.; Song, K.; Hao, W.; Wu, Y.; Patil, G.; Hua, F.; Sun, Y.; Huang, C.; Ritchey, J.; Jones, C.; et al. FIP200 Restricts RNA Virus Infection by Facilitating RIG-I Activation. Commun Biol 2021, 4, 921. [CrossRef]
- Xue, X.; Ma, L.; Zhang, X.; Xu, X.; Guo, S.; Wang, Y.; Qiu, S.; Cui, J.; Guo, W.; Yu, Y.; et al. Tumour Cells Are Sensitised to Ferroptosis via RB1CC1-Mediated Transcriptional Reprogramming. Clin Transl Med 2022, 12, e747. [CrossRef]
- Yang, Y.; White, E. Autophagy in PDGFRA<ovid:Sup>+</Ovid:Sup> Mesenchymal Cells Is Required for Intestinal Homeostasis and Mammalian Survival. Autophagy 2023, 19, 726–728. [CrossRef]
- Yang, F.; Kalantari, S.; Ruan, B.; Sun, S.; Bian, Z.; Guan, J.-L. Autophagy Inhibition Prevents Lymphatic Malformation Progression to Lymphangiosarcoma by Decreasing Osteopontin and Stat3 Signaling. Nat Commun 2023, 14, 978. [CrossRef]
- Zhu, Y.-F.; Yu, R.-H.; Zhou, S.; Tang, P.-P.; Zhang, R.; Wu, Y.-X.; Xu, R.; Wei, J.-M.; Wang, Y.-Y.; Zhang, J.-L.; et al. TAX1BP1 and FIP200 Orchestrate Non-Canonical Autophagy of P62 Aggregates for Mouse Neural Stem Cell Maintenance. Zool Res 2024, 45, 937–950. [CrossRef]
- Li, L.; Wang, G.; Hu, J.-S.; Zhang, G.-Q.; Chen, H.-Z.; Yuan, Y.; Li, Y.-L.; Lv, X.-J.; Tian, F.-Y.; Pan, S.-H.; et al. RB1CC1-Enhanced Autophagy Facilitates PSCs Activation and Pancreatic Fibrogenesis in Chronic Pancreatitis. Cell Death Dis 2018, 9, 952. [CrossRef]
- Huang, Y.; Bodnar, D.; Chen, C.-Y.; Sanchez-Andrade, G.; Sanderson, M.; Shi, J.; Meilleur, K.G.; Hurles, M.E.; Gerety, S.S.; Tsai, E.A.; et al. Rare Genetic Variants Impact Muscle Strength. Nat Commun 2023, 14, 3449. [CrossRef]
- Li, D.; Vogel, P.; Li-Harms, X.; Wang, B.; Kundu, M. ATG14 and RB1CC1 Play Essential Roles in Maintaining Muscle Homeostasis. Autophagy 2021, 17, 2576–2585. [CrossRef]
Figure 1.
Screenshot of the proband’s whole genome sequence reads aligned to the reference sequence in the vicinity of position on 4,891,014 chromosome 22 (red bar at top), as viewed with the Integrative Genomics Viewer. The variant T is highlighted in red. The reference DNA and mRNA sequences are shown along with the predicted protein amino acid sequence. The codon change from GGA to AGA predicts an amino acid change from glycine (G) to arginine (R).
Figure 1.
Screenshot of the proband’s whole genome sequence reads aligned to the reference sequence in the vicinity of position on 4,891,014 chromosome 22 (red bar at top), as viewed with the Integrative Genomics Viewer. The variant T is highlighted in red. The reference DNA and mRNA sequences are shown along with the predicted protein amino acid sequence. The codon change from GGA to AGA predicts an amino acid change from glycine (G) to arginine (R).
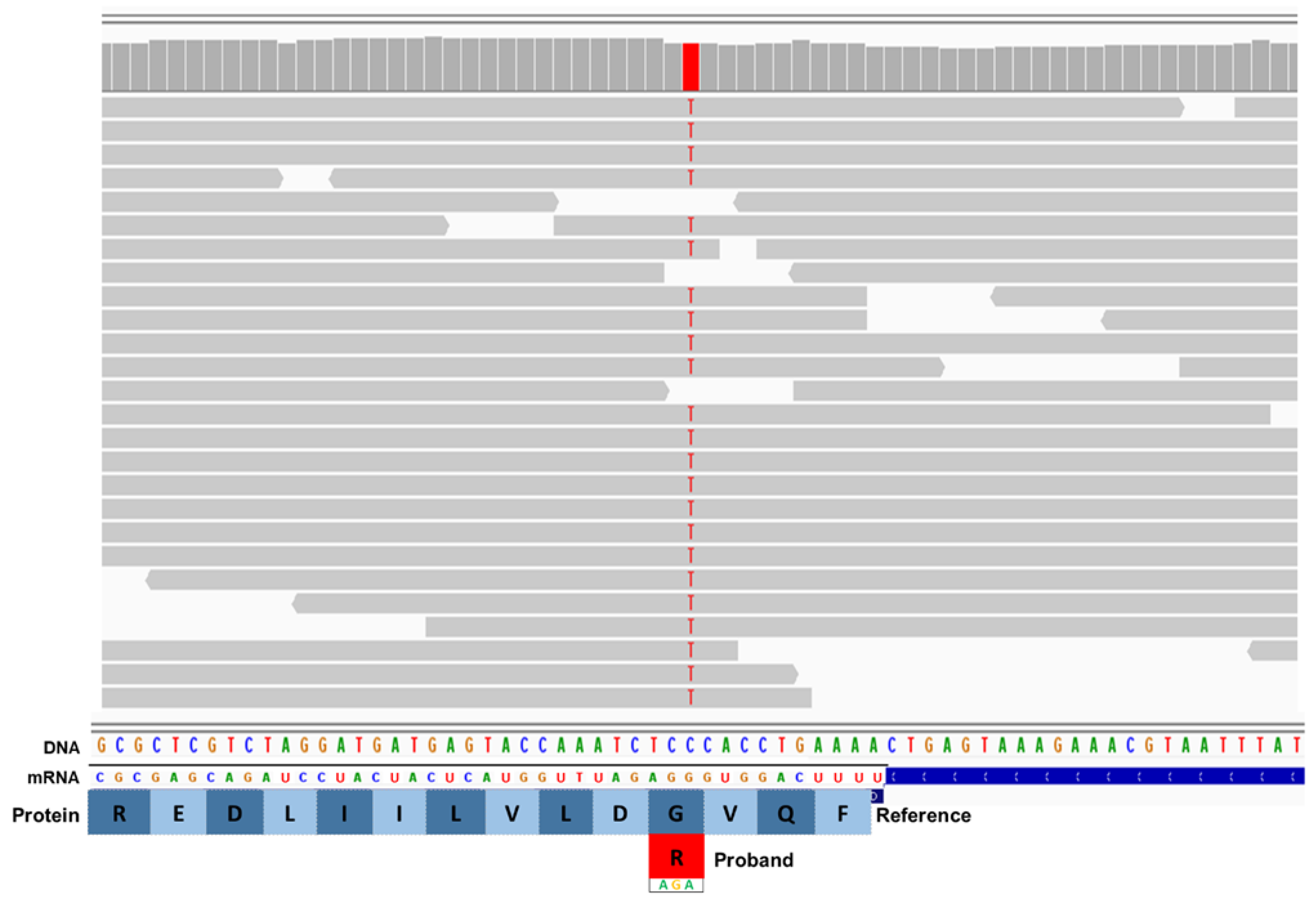
Figure 2.
Automated Sanger sequence electrophoretograms of DNA from an unaffected dog (top line) and proband (bottom line). The codon for RB1CC1 p.1503 is GGA in the unaffected dog and AGA in the affected dog, confirming the finding from whole genome sequence analysis.
Figure 2.
Automated Sanger sequence electrophoretograms of DNA from an unaffected dog (top line) and proband (bottom line). The codon for RB1CC1 p.1503 is GGA in the unaffected dog and AGA in the affected dog, confirming the finding from whole genome sequence analysis.
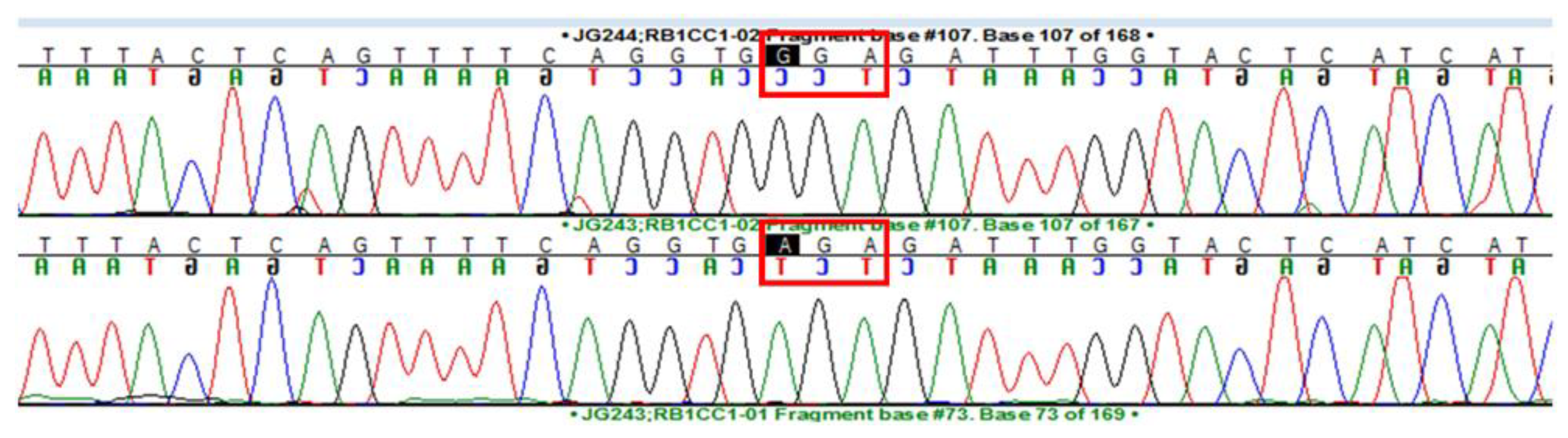
Figure 3.
Electron micrograph of a disease-specific inclusion body (IB) in a caudate nucleus neuron of the proband. The contents of the inclusion body are quite heterogeneous in appearance.
Figure 3.
Electron micrograph of a disease-specific inclusion body (IB) in a caudate nucleus neuron of the proband. The contents of the inclusion body are quite heterogeneous in appearance.
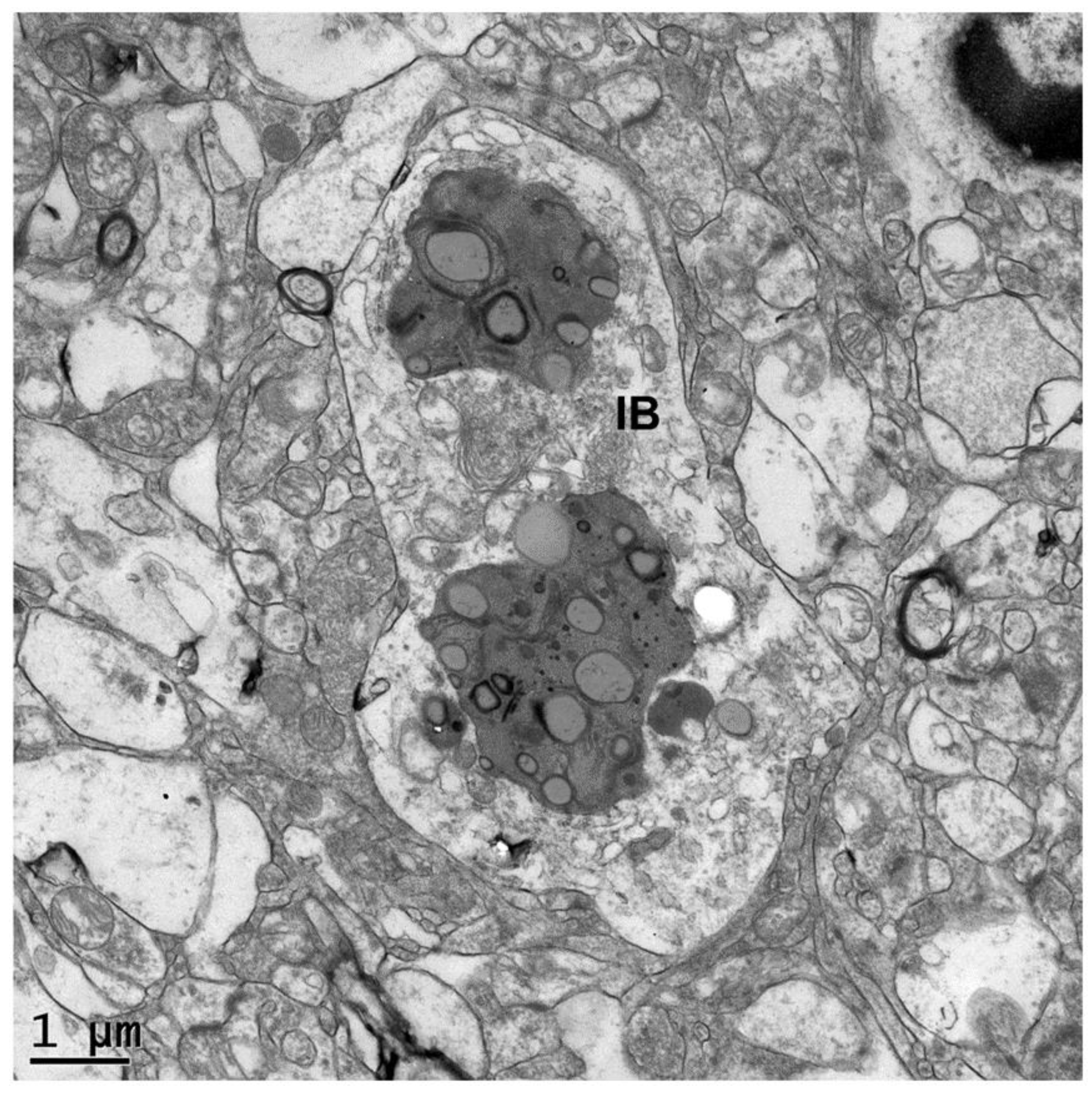
Figure 4.
Electron micrograph of a disease-specific inclusion body in a caudate nucleus neuron of the proband. Finger-like membrane-bounded cytoplasmic extensions partially engulf the inclusion body (red arrows).
Figure 4.
Electron micrograph of a disease-specific inclusion body in a caudate nucleus neuron of the proband. Finger-like membrane-bounded cytoplasmic extensions partially engulf the inclusion body (red arrows).
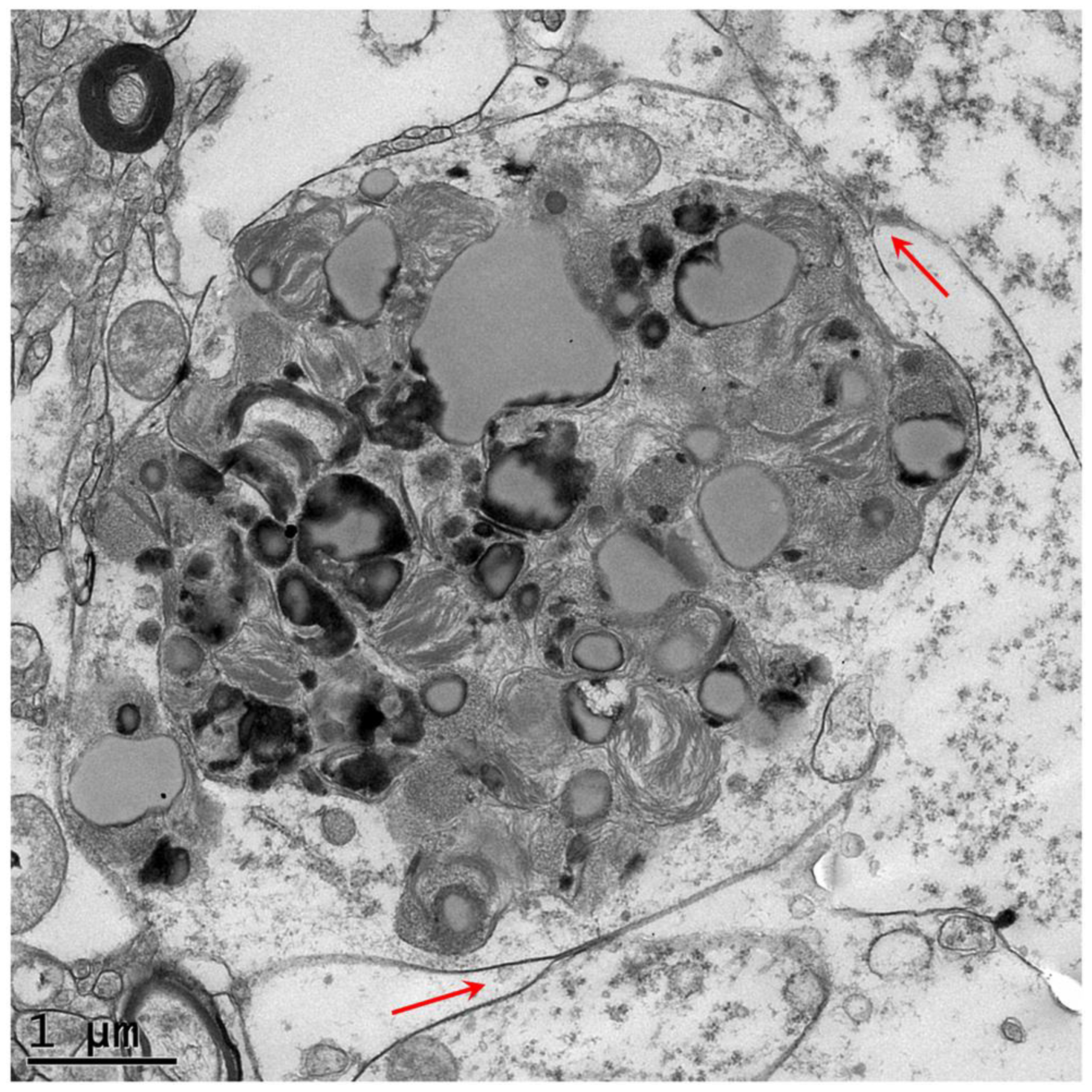
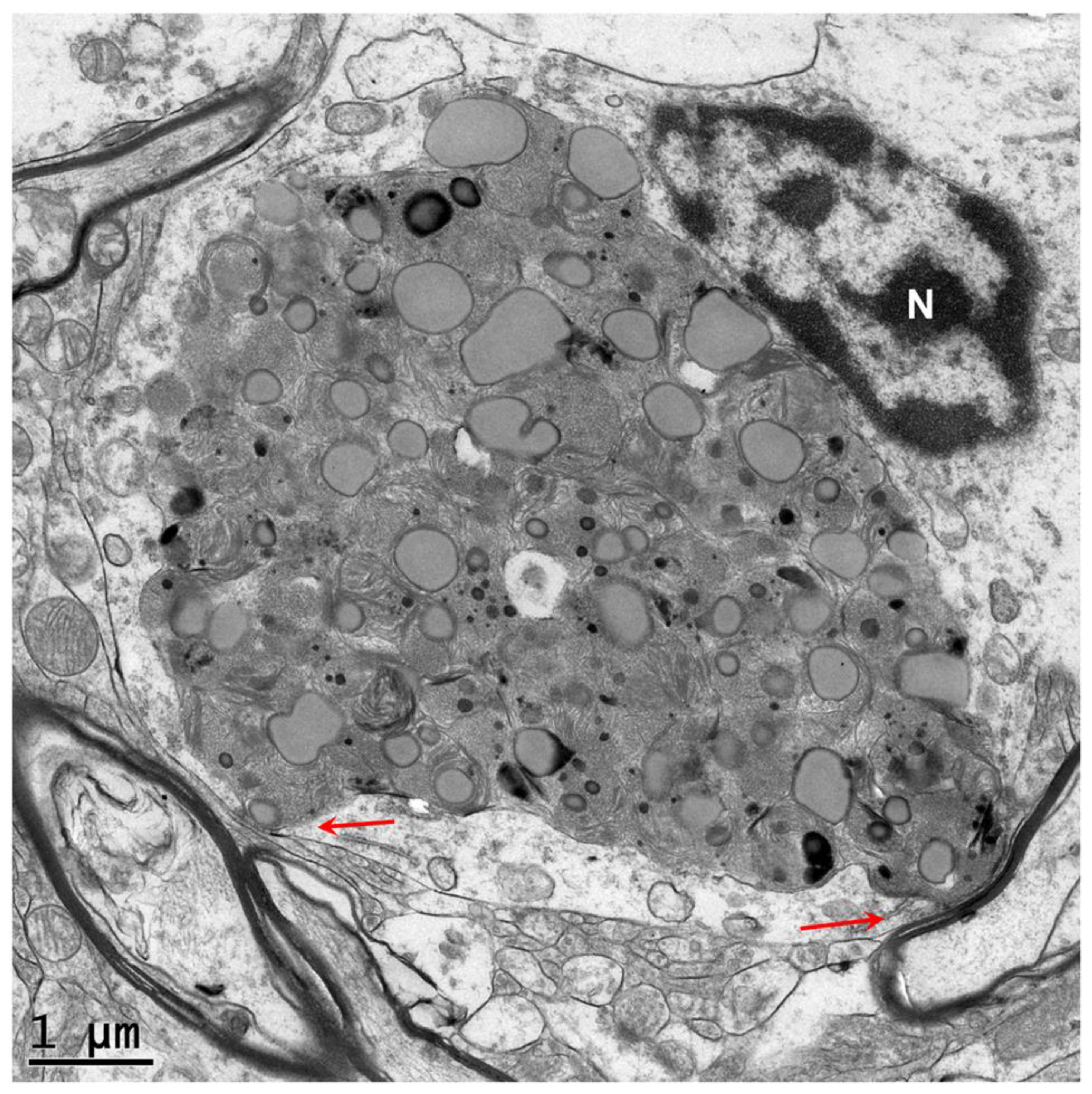
Figure 5.
Electron micrograph of a disease-specific inclusion body in a caudate nucleus neuron of the proband. A membrane-bounded structure abuts the inclusion body (red arrows).
Figure 5.
Electron micrograph of a disease-specific inclusion body in a caudate nucleus neuron of the proband. A membrane-bounded structure abuts the inclusion body (red arrows).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



