
Submitted:
31 December 2024
Posted:
02 January 2025
You are already at the latest version
Abstract
Cm-p5, as well as its cyclic monomer and dimeric analogues are known for their antifungal, antibacterial, antiviral, and antibiofilm activities. Previously, our cyclization method generated a mixture of peptides that were difficult to separate, which was then improved by a selective synthesis of the parallel dimer and its differentiation from the antiparallel by comparing the retention times in RP-HPLC. Here, we developed a more reliable identification method, comprising chymotrypsin proteolytic degradation and sequencing of the different fragments by ESI-MSMS, for the Cm-p5 dimer identification. Also, we improved our cyclization methods to specifically produce higher amounts of the desired cyclic variant, either cyclic monomer or dimer. We show that liquid phase oxidation with 20% DMSO or iodine oxidation affords only the cyclic analogue. However, the on-resin oxidation with iodine showed more efficacy and efficiency. Also, cyclization in liquid phase renders the antiparallel dimer in high EtOH or peptide concentration, indicating a kinetic control. On the other hand, the parallel dimer was preferentially produced in 5% of TFE and low peptide concentration without the formation of the cyclic analogue indicating a thermodynamic control. In summary, we report that chymotryptic digestion combined with ESI-MS and MS/MS allows an unambiguous differentiation of Cm-p5 dimers. Here we demontrate demonstrate a more selective and efficient synthesis yielding in high amounts of the cyclic analogue and the dimers of Cm-p5.
Keywords:
antimicrobial peptide
; SPPS
; cyclization
; disulfide bond
; dimer
; peptide sequencing
1. Introduction
Antimicrobial peptides (AMPs) constitute a fundamental part of the innate immune system among all organisms [1,2]. These molecules are broad-spectrum antibiotics that exhibit fundamental structural differences among prokaryotic and eukaryotic cells, acting selectively against potential pathogens [3]. It is widely accepted that some of these compounds adopt amphipathic conformations that interact strongly with biological membranes, provoking metabolic destabilization of the prokaryotic cell [4]. Consequently, the search for natural and synthetic AMPs is an active research area of interest [5]. Nevertheless, one important drawback of the use of AMPs and other peptidic compounds for medicinal purposes has been their low metabolic stability [6]. Nature and synthetic chemists have usually addressed this limitation using covalent modification strategies to reduce proteolytic degradation, such as N-alkylation, cyclization and bioisosteric replacements [7,8,9]. In particular, cyclization has the advantage of reducing the conformational space of the peptide, thus increasing the proportion of the active conformation and improving the interactions with the molecular target [9]. Many modern trustworthy methodologies for peptide cyclization have been established in the last years [10,11]. However, the more classical macrolactamization, intermolecular disulfide bond formation, Grubbs metathesis, and azide-alkyne click reaction remain the more widely used [12,13]. Cyclization by the formation of disulfide bonds between the native cysteine residues is a powerful strategy that has the extra advantage of having high chemoselectivity and taking place in metal-free and mild reaction conditions [14,15]. In addition, a disulfide bond in the peptide structure influences the conformation and proteolytic stability, and enhances the pharmacological properties [16,17].
In 2015, Otero et al. reported the characterization of the novel Cm-p5 peptide (SRSELIVHQRLF-NH2), a derivative of Cm-p1, isolated from the coastal mollusk Cenchritis muricatus, with relevant antifungal activity [18]. Recently, several aminoacid replacements and truncations were explored, and the residues relevant to the biological activity were confirmed to rationally understand structure-activity relationships. Furthermore, three analogues of Cm-p5 with diverse and promising biological activities were developed [19]. It turned out that the cyclic Cm-p5 version showed increased antifungal activity. In contrast, the antiparallel dimer showed antibacterial activity even against virulent tuberculosis and multi-resistant bacteria isolates, and the parallel dimer showed antiviral activity [20]. Later on, it was additionally proved that all the derivatives inhibited the formation of Candida auris biofilms at semi-inhibitory concentrations, even arresting the growth of mature biofilms [21]. Cm-p5, the cyclic or the dimers does not show cytotoxicity against normal eukaryotic cells [19,20].
The cyclic version of Cm-p5 was prepared by replacing Glu and His residues in the sequence with Cys residues, followed by intramolecular disulfide formation in diluted conditions (0.5 mg/ml) (Figure S1). Those positions were chosen because we demonstrated that Glu and His residues are relevant for maintaining the optimal conformation required for the antifungal activity. This folding constraint is probably mediated by a salt bridge interaction between these residues, stabilizing the amphipathic helicoidal structure capable of interacting with microbial membranes [19].
During cyclization of the lineal precursor CysCysCm-p5, two collateral products were obtained, generating a mixture of three peptides, difficult to separate by RP-HPLC (Figure S1, zoom to Figure 18 (48 h)-Supp. Inf. Morales et al. ACS-Omega, 2019, [19]). Molecular mass measurement identified these collateral products as the parallel and antiparallel dimers of CysCysCm-p5 mediated by two intermolecular disulfide bonds [19]. The selective synthesis of one of them, the parallel, using an orthogonal protection strategy allowed differentiation of both dimers by comparing their retention times in RP-HPLC analysis. Here, we report that combining chymotrypsin digestion and ESI-MSMS analysis allows an unambiguous differentiation of the parallel and antiparallel dimers [22].
To go deeper into the biological applications of these derivatives, a reliable synthetic approach to obtain cyclic peptide, as well as parallel and antiparallel dimers with high purity and yield must be available. The success of any cyclization process relies on the fine tuning of experimental conditions such as temperature, dilution, number of equivalents, solvents, etc. [10,11,12,13,14,15]. This work presents the formation of the cyclic Cm-p5 monomer by using liquid oxidation with DMSO or iodine and on-resin cyclization, which is more efficacious and efficient. The selective synthesis of the antiparallel dimer usually requires an orthogonal protection scheme that is more expensive [23,24]. This aspect is important to establish a feasible production process for this promising analogue. We overcame this limitation by using highly concentrated ethanol or peptide, which allowed the exclusive formation of the antiparallel dimer. On the other hand, the parallel dimer was mainly produced at low concentrations of peptide or TFE without the formation of the cyclic monomer analogue. Future investigations will allow a deeper understanding of the chemical-physical behavior of these reactions.
2. Results
2.1. Differentiating Cyclic Parallel and Antiparallel Dimers of Cm-p5 by Chymotryptic Digestion and ESI-MSMS Analysis
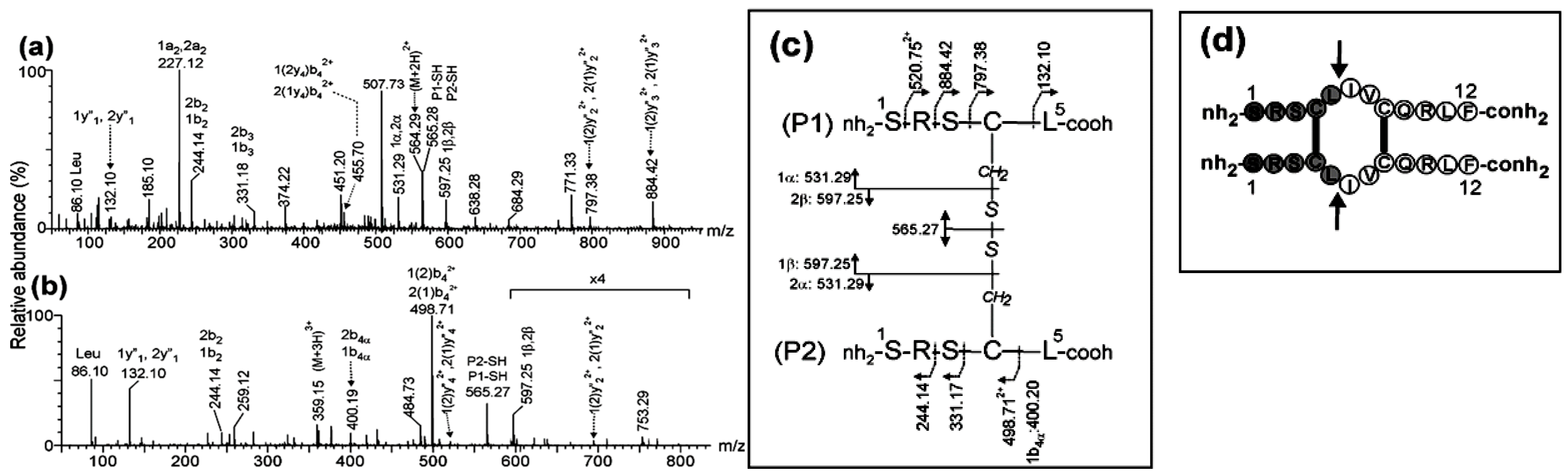
The ESI-MS analysis of the chymotrypsin digestion of dimer that eluted in peak 2 (Figure S1) shows two intense signals detected at m/z 394.22 (3+) and m/z 590.82 (2+). These m/z values agree with the expected ones for the (M+3H)3+ and (M+2H)2+ protonated peptides (1Ser-Leu5)-S-S-(6Ile-Arg10) and (1Ser-Leu5)-S-S-(6Ile-Phe12-oh) linked by an intermolecular disulfide bond between Cys4-Cys8 and with the C-terminal of the original linear peptide deamidated also by chymotrypsin. This result suggests that peak 2 contains the antiparallel dimer.
To confirm this assignment, the MS/MS spectra of the triply-and doubly-charged precursor ions detected at m/z 394.22 and m/z 590.82, respectively were acquired (Figure 1a-b).The singly-charged fragment ions detected at m/z 565.27 and m/z 618.34 (Figure 1a) were assigned to P1-SH and P2-SH due to the symmetric cleavage of the intermolecular disulfide bond between Cys4 and Cys8 producing the reduced peptides 1Ser-Leu5and 6Ile-Arg10, respectively.
Several signals in Figure 1a-b were produced by the asymmetric cleavage of the disulfide bond and assigned to 1α and 1β fragment ions (m/z 531.29 (1+) and m/z 597.25 (1+)) of the P1 peptide, and 2α and 2β fragment ions (m/z 584.35 (1+) and m/z 650.31 (1+)) of the P2 peptide (Figure 1c) with Cys4 and Cys8 residues modified as dehydroalanine and disulfohydryl cysteine [25]. In addition, further fragmentation of the backbone of 1α, 2α and 2β fragment ions were detected at m/z 400.19 (1+), 372.20 (1+) and 438.16 (1+) and assigned to 1b4α, 2y3α and 2y3β ions, respectively. In P1 peptide, the mass difference of 718.30 Da between the ions detected at m/z 850.40 (1+) and 132.10 (1+) in Figure 1c and assigned as 1(2)y2 and 1y1, respectively, agrees with the expected (718.33 Da) for Cys4 in peptide 1Ser-Leu5 linked by an intermolecular disulfide bond to Cys8 in the 6Ile-Arg10 peptide. The same conclusion is reached when calculating the mass difference (718.33 Da) between the complementary ions at m/z 525.75 (2+) and 331.17 (1+) and assigned as 1(2)b4 and 1b3, respectively (the ion corresponding to the fragment of m/z 525.75 (2+) appear with hydroxylated C-terminus at 534.25 (2+)). A similar result was obtained in P2 peptide since the mass difference of 665.28 Da between the fragment ions assigned as 2(1)y3 and 2y2 or the complementary backbone ions 2(1)b3 and 2b2 agrees with the expected (665.24 Da) for Cys8 in peptide 6Ile-Arg10 linked by an intermolecular disulfide bond to Cys4 in the 1Ser-Leu5peptide.
The ESI-MS analysis of the chymotrypsin digestion of the dimer eluted in peak 1 (Figure S1), showed two signals at m/z 564.29 (2+), m/z 376.51 (3+) and m/z 617.35 (2+), m/z 411.98 (3+). These m/z values were tentatively assigned to the homodimer peptides (1Ser-Leu5)-S-S-(1Ser-Leu5) and (6Ile-Arg10)-S-S-(6Ile-Arg10) forming two different disulfide bonds between Cys4-Cys4 and Cys8-Cys8, respectively (Table S1). This result suggests the parallel dimer (Figure 2d) eluted in peak 1. To confirm this assignment, the MS/MS spectra of both homodimer were acquired (Figure 2 and Figure 3).
The MS/MS spectra of the ions detected at m/z 564.29 (2+) and m/z 376.51 (3+) (appear as 359.15 (3+) due to the loss of OH) assigned to the (1Ser-Leu5)-S-S-(1Ser-Leu5) homodimer peptide is shown in Figure 2(a-b). A signal detected at m/z 565.27 (1+) corresponds to the symmetric cleavage of the intermolecular disulfide bond producing the reduced 1Ser-Leu5 peptide (P1-SH or P2-SH, Figure 2c). In addition, two single-charged signals detected at m/z 531.29 and m/z 597.25 were assigned to [1α or 2α] and [1β or 2β] produced by the asymmetric cleavage of the disulfide bond containing the Cys4 modified as dehydroalanine and disulfohydryl cysteine. The mass difference of 665.28 Da between the ions detected at m/z 797.38 (1+) and 132.10 (1+) and assigned as 1(2)y2 or 2(1)y2 and 1y1 or 2y1, respectively, agreed with the expected (665.26 Da) for Cys4 linked to the 1Ser-Leu5 peptide by an intermolecular disulfide bond. The same evidence was obtained through the analysis of bn ions. All these results confirmed the presence of the homodimer (1Ser-Leu5)-S-S-(1Ser-Leu5). The remaining assignments of other fragment ions observed in the MS/MS spectra are summarized in Figure 2c.
Figure 2.
ESI-MS/MS analysis of the precursor ions (a) (M+2H)2+ at m/z 564.29 and (b) (M+3H)3+ at m/z 376.51 (359.15) corresponding to a chymotryptic peptide derived from the parallel dimer of CysCys-Cm-p5. (c) The fragmentation scheme shows the assignment for the fragment ions. (d) Structural desing with solid arrows indicate the chymotrypsin cleavage sites. Connecting lines between Cys residues indicate two intermolecular disulfide bonds between Cys4-Cys4 and Cys8-Cys8. The nomenclature of fragment ions in the MS/MS spectra agrees with the proposed by Morman et al. [25].
Figure 2.
ESI-MS/MS analysis of the precursor ions (a) (M+2H)2+ at m/z 564.29 and (b) (M+3H)3+ at m/z 376.51 (359.15) corresponding to a chymotryptic peptide derived from the parallel dimer of CysCys-Cm-p5. (c) The fragmentation scheme shows the assignment for the fragment ions. (d) Structural desing with solid arrows indicate the chymotrypsin cleavage sites. Connecting lines between Cys residues indicate two intermolecular disulfide bonds between Cys4-Cys4 and Cys8-Cys8. The nomenclature of fragment ions in the MS/MS spectra agrees with the proposed by Morman et al. [25].
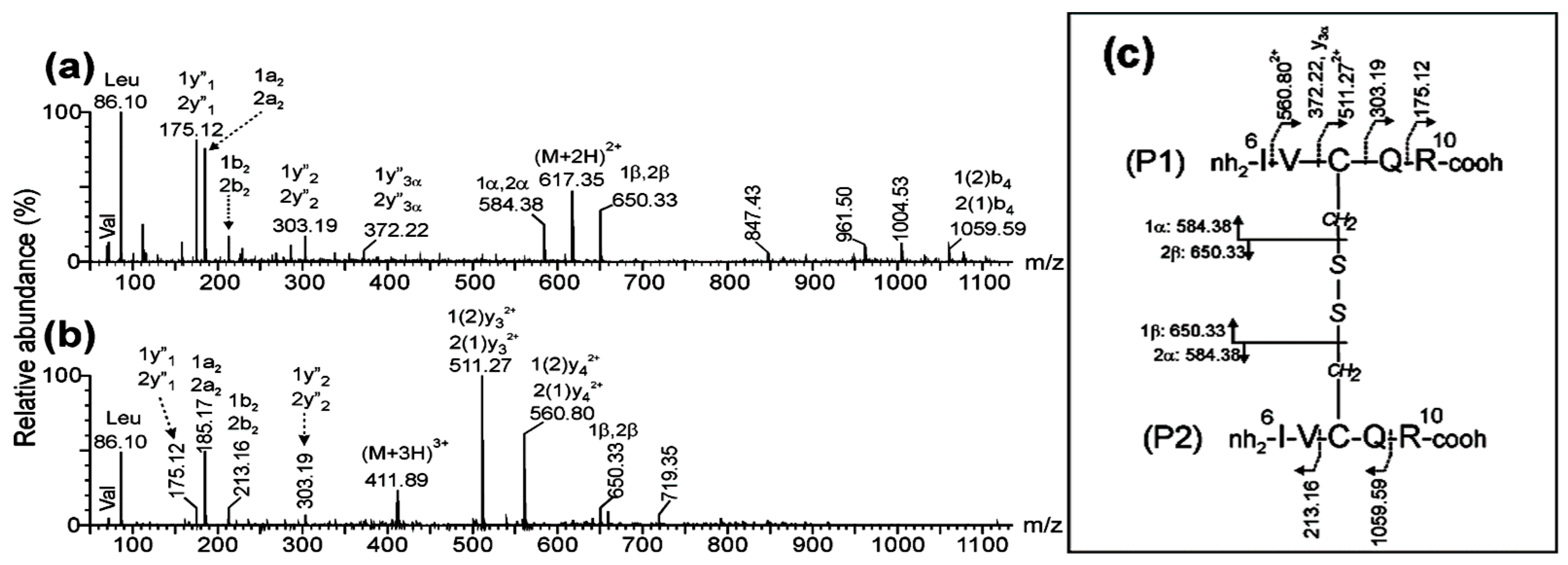
The MS/MS spectra of the precursor ions detected at m/z 617.35 (2+) and m/z 411.89 (3+) (Figure 3a-b) also support the assignment of the (6Ile-Arg10)-S-S-(6Ile-Arg10) homodimer peptide (Figure 3c) derived from the parallel dimer of CysCysCm-p5. The signals at m/z 584.38 (1+) and 650.33 (1+) were assigned to the asymmetric cleavage of the disulfide bond to yield the peptide (6Ile-Arg10) with the Cys8 transformed as dehydroalanine [1α or 2α] and disulfohydryl cysteine [1β or 2β] residues, respectively. The mass difference of 718.35 Da between the 1(2)y3 (m/z 511.27, 2+) and 1y2 (m/z 303.19, 1+) fragment ions agreed with the expected (718.33 Da) for Cys8 linked to the 6Ile-Arg10 peptide by an intermolecular disulfide bond.
Figure 3.
ESI-MS/MS analysis of the precursor ions (a) (M+2H)2+ at m/z 617.35 and (b) (M+3H)3+ at m/z 411.89 corresponding to a chymotryptic peptide derived from the parallel dimer of CysCys-Cm-p5. (c) Fragmentation scheme of peptide and summary of the assignment for fragment ions.
Figure 3.
ESI-MS/MS analysis of the precursor ions (a) (M+2H)2+ at m/z 617.35 and (b) (M+3H)3+ at m/z 411.89 corresponding to a chymotryptic peptide derived from the parallel dimer of CysCys-Cm-p5. (c) Fragmentation scheme of peptide and summary of the assignment for fragment ions.
2.2. Selective Synthesis of the Cyclic Monomer CysCysCm-p5ss
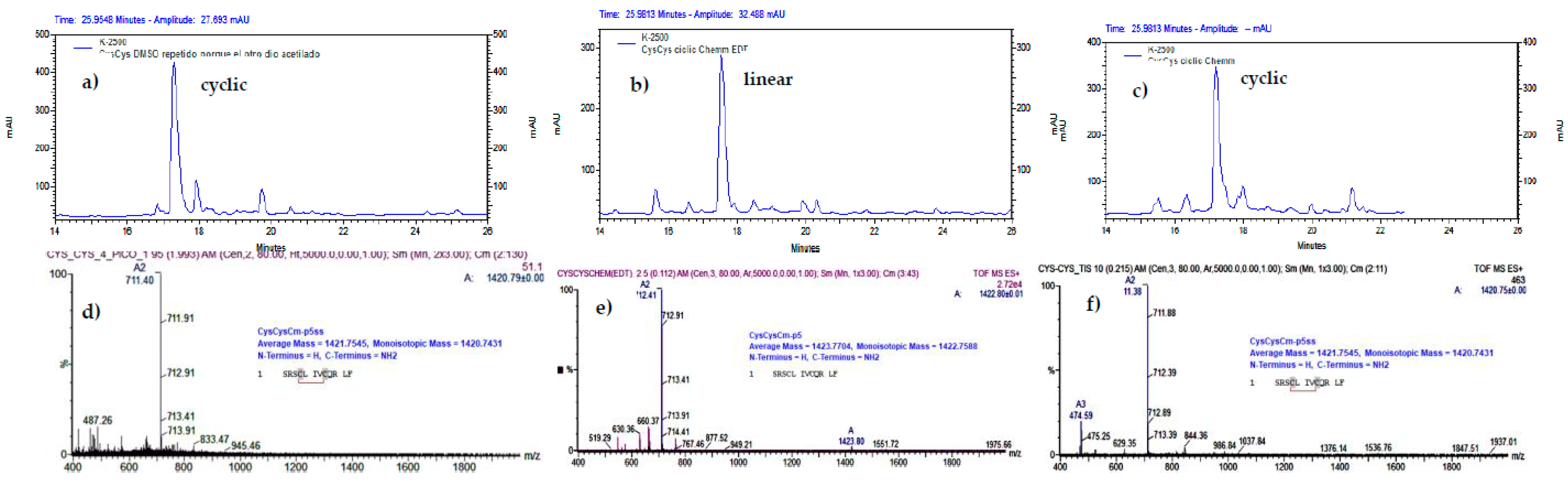
The DMSO (20%) oxidation at pH = 3 and 1 mg/ml of CysCysCm-p5 produced only the cyclic monomer with an intramolecular disulfide bond between Cys4 and Cys8 (Figure 4). On-resin oxidation of CysCysCm-p5 with DMSO (35%) in Chemmatrix resin with 0.7 mmol/g of substitution produced the cyclic peptide only if EDT was eliminated from the cleavage mixture (Figure 4 and Scheme 1). However, in these experimental conditions, the peptide yield was very poor (34%). The retention time changed from 19 min to 17.7 min (the original retention time was 19 min, Figure S1) due to a reduction of the tubing length between the column and the HPLC UV detector.
To increase the yield, the resin substitution was decreased to 0.41mmol/g but in this case, the same cleavage cocktail without EDT (TIS 2.5%/H2O 2.5%) reduced the disulfide bond. We tried to reproduce the previous result by increasing the DMSO concentration up to 50% and the reaction time to 12 h, but the results were almost the same; suggesting that TIS in the cleavage cocktail reduced the intramolecular disulfide bond. In an additional experiment, where the Chemmatrix resin was replaced with a highly substituted Knorr-MBHA matrix (1.11 mmol/g), the cyclization was not possible (Figure S5).
To check the efficiency of the on-resin oxidation by DMSO we did a minicleavage with TFA 10%/TIS 1% in DCM that guarantees the maintenance of lateral chain protecting groups. We also tested the behavior of other oxidation methods, such as I2/DMF or O2/NH3. The RP-HPLC profiles of the cleaved peptides are shown in Figure S6 and eluted in one peak at higher retention times with the mass of cyclic and protected peptide (except the Trt protecting group of Gln).
We also evaluated two other conditions with reduced content of TIS, varying water, in Chemmatrix resin, 0.41 mmol/g (Figure S7), partially preventing the disulfide bond reduction, in the case of lower amount water. However, a compound that reduced the peptide solubility remained attached to it and change retention time to 23.8 min. This compound was also the cyclic peptide (lower-center ESI-MS panel, Figure S7), but with a different counterion or hydrophobic adduct that modified its retention time and could not be replaced during the column equilibrium step nor the purification. ESI-MS measurement of the solid peptide showed a signal with a delta mass of 97 Da, corresponding to HSO4-or H2PO4-counterions (lower-right ESI-MS panel, Figure S7).
We also tested TIS or PhSiH3 as one of the scavenger but without water, in Chemmatrix resin of 0.2 mmol/g and DMSO/HCl oxidation. The results corroborated that besides a lower substitution, the elimination of water protects the cyclic product (sum of cyclic and counterion) and that PhSiH3 is more efficient than TIS in reducing the disulfide bond in those conditions and also avoids the formation of adducts with counterions (Figure S8).
The result of the cleavage of peptides in Figure S6 with TFA 94-95% is shown in Figure S8 (lower panels). After DMSO or O2 oxidation, the cleavage with 1% TIS, without water, reduced the disulfide bond by 25-30%. However, the disulfide bond formed by I2 oxidation resisted the cleavage; besides, some dimers were formed. Diastereomers are formed by Lewis-acid promoted Cys racemization, especially when the Trt protecting group is eliminated before oxidation.
Final experiments were conducted to test the use of iodine as a better oxidation method but in the inexpensive Knorr-MBHA resin. Testing the on-resin cyclization with I2 in MBHA 0.46 mmol/g, 0.2 g, the cyclic monomer was protected, and 100 mg of crude was obtained (62%). In 0.13 g of MBHA resin, 1.11 mmol/g, and using iodine oxidation, only 80 mg (32%) of crude was produced, indicating that an increased substitution leads to a lower yield. At both substitutions, the quantity of the dimer increased while racemization remained constant (Figure 5). Aiming to increase the dimers we used MBHA resin with a highest substitution (1.35 mmol/g) and changed the solvent during the iodine oxidation, but surprisingly dimers was reduced. Unfortunately, the yield with this resin was reduced dramatically and is it not recommended for this synthesis.
2.3. Selective Synthesis of the Antiparallel Dimer (CysCysCm-p5)2ss-ss
For the synthesis of the antiparallel dimer by the one-pot formation of two disulfides with iodine and Acm protected CysAcmCysCm-p5, the cyclic peptide was produced quantitatively (Scheme S3 and Figure S9). We observe that helical inducer solvent showed interesting results on the chemoselectivity for one dimer depending on the experimental conditions. For example, in pure water, the parallel was favored, but the cyclic was also produced, and insoluble material remained. Experiments with TFE showed that conditions of extreme helical formation, TFE 90% or pure, favor the antiparallel (Figure 6).
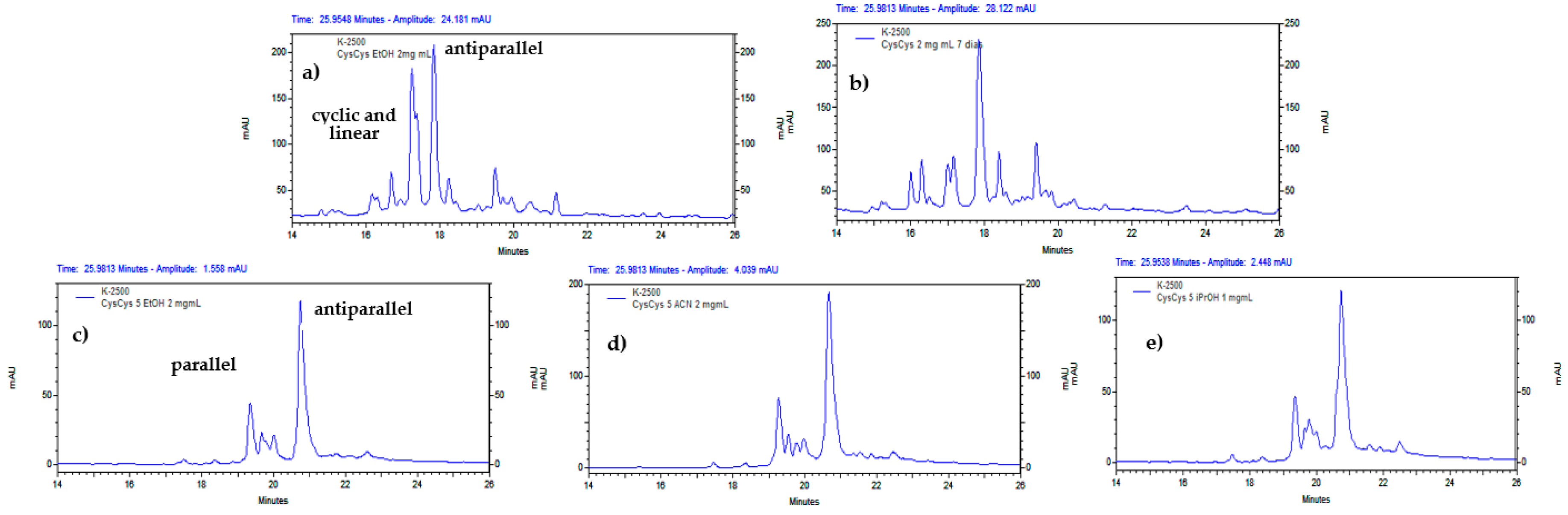
Screening several EtOH concentrations for cyclization conditions improved the result obtained with TFE regarding of the use of a greener solvent. Indeed, 81% EtOH generated almost exclusively the antiparallel dimer at a peptide concentration of 1mg/ml (Figure 7). On the other hand, 90% TFE did not induce the formation of the antiparallel only, probably due to its higher polarity. It was also observed that the reaction time for the consumption of the starting material increases with the concentration of EtOH (Figure 8).
Finally, several experiments at 2 mg/ml of peptide showed shorter reaction times to yield mostly the antiparallel dimer in 5% EtOH or other organic solvents (Figure 8). Notably, at 1 mg/ml or more, insoluble material was present during cyclization, even in the presence of organic solvent. However, for the RP-HPLC analysis, the aliquots were solubilized by dilution to compare the chromatographic profiles and to ensure the completion of the reaction until the acyclic peptide was consumed.
Figure 8.
Analytical RP-HPLC profile of the cyclization of CysCysCm-p5 at 2 mg/ml: a) 90% EtOH 6 h, b) 90% EtOH 24 h, c) 5% EtOH, d) 5% ACN, e) 5% iPrOH (12 h).
Figure 8.
Analytical RP-HPLC profile of the cyclization of CysCysCm-p5 at 2 mg/ml: a) 90% EtOH 6 h, b) 90% EtOH 24 h, c) 5% EtOH, d) 5% ACN, e) 5% iPrOH (12 h).
2.4. Selective Synthesis of the Parallel Dimer (CysCysCm-p5)2(ss)2
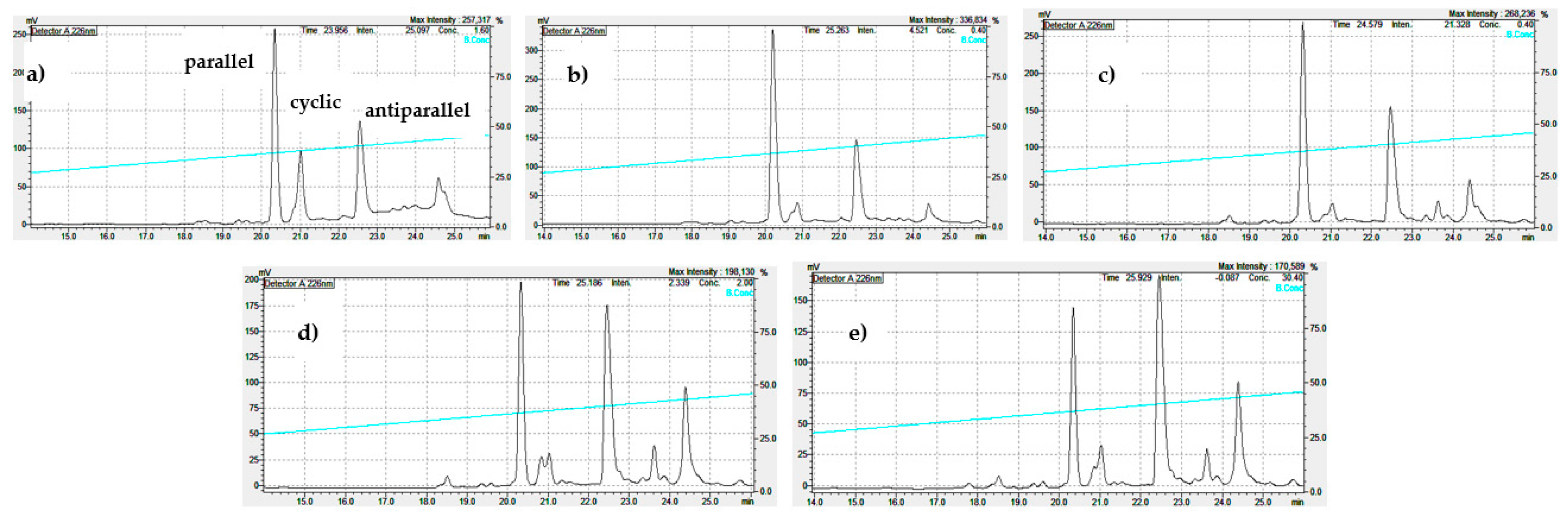
As shown in Figure 6a and Figure 7a-b, the reaction of the linear monomer at 1 mg/ml in pure water or between 20-50% EtOH yields mainly the parallel dimer. The results shown in Figure S10, a-c indicates that at 0.5 mg/ml peptide concentration, this tendency is even more pronounced, being the low peptide concentration and 5% TFE or 20-25% EtOH selected conditions for the synthesis of the parallel dimer.
Figure 9.
Analytical RP-HPLC profile (Shimatsu system) of the cyclization of the linear CysCysCm-p5 at 0.5 mg/ml and different EtOH concentration: a) 5% EtOH, b) 25% EtOH, c) 50% EtOH, d) 75% EtOH, e) 90% EtOH, pH = 8, 12-24 h.
Figure 9.
Analytical RP-HPLC profile (Shimatsu system) of the cyclization of the linear CysCysCm-p5 at 0.5 mg/ml and different EtOH concentration: a) 5% EtOH, b) 25% EtOH, c) 50% EtOH, d) 75% EtOH, e) 90% EtOH, pH = 8, 12-24 h.
3. Discussion
3.1. Differentiating Cyclic Parallel and Antiparallel Dimers of Cm-p5 by ESI-MS/MS
One goal of the present investigation was the development of an MS-based method for the unambiguous differentiation between the parallel and the antiparallel dimer. In our previous work, we used an orthogonal protection scheme to unambiguously synthesize the parallel dimer and the comparison of its retention time in the RP-HPLC analysis, allowing the two dimers to be differentiated [19]. Identifying the dimers only by their retention times is useful during routine quality control within a well-known synthesis process. However, the RP-HPLC method may not be reliable when synthesis conditions are changed since unknown contaminants generated during the new reaction might change retention times of the expected dimers. Therefore, we envisioned a more reliable method based on the combination of proteolytic degradation and ESI-MS/MS analysis. This MS-based approach was also useful to differentiate the parallel and antiparallel dimers generated by air oxidation of the anticancer peptide CIGB-300 [22].
Parallel and antiparallel dimers have identical elemental composition and cannot be differentiated based on mass accuracy by ESI-MS analysis. However, ESI-MS analysis of the proteolysis allowed their differentiation. The proteolytic digestion of the antiparallel dimer yields a unique chymotryptic peptide with the Cys4-Cys8 linked by a disulfide bond, while in the case of the parallel dimer chymotrypsin generates two different homodimer chymotryptic peptides with Cys4-Cys4 and Cys8-Cys8 linked by an intermolecular disulfide bond (Figure S2). Despite of the wider cleavage specificity of Chymotrypsin (mainly cleaves at Trp, Tyr, Phe, and Leu) it was the protease of choice due to the presence of a cleavage site (Leu5) inside the cyclic sequence constrained by the two intermolecular disulfide bonds present in both dimers.
In addition to the differentiation provided by ESI-MS analysis of the proteolytic digestions, ESI-MS/MS analysis confirmed these results. The symmetric and asymmetric cleavages of the disulfide bond, as well as the mass differences of the backbone fragment ions generated by collision-induced dissociation, allowed an unambiguous identification of the molecular masses of the disulfide-linked peptides as well as the connectivity of cysteine residues (Figure 1, Figure 2 and Figure 3) [25].
3.2. Selective Synthesis of the Cyclic Monomer CysCysCm-p5ss
Previous to the present study, dimers and cyclic of Cm-p5 were produced as a mixture during the liquid phase cyclization at 0.5 mg/ml, pH = 8 in water/ACN (50:50). The separation of both analogues was challenging, even though several RP-HPLC gradients were evaluated. The lack of selectivity in the synthesis of these molecules led us to study different cyclization conditions to reach this goal.
Beginning with the cyclic analogue of Cm-p5 (CysCysCm-p5ss), we demonstrated that only extreme dilution could avoid dimer formation; however, such a condition is not applicable in large scale production. Considering that the presence of chaotropic agents would favor the cyclization because helicoidal secondary structures are destroyed, favoring twisted conformations in which thiols are closer, we performed the DMSO oxidation at pH = 3, and only the cyclic peptide was produced at the same concentration (even at 1 mg/ml) that with oxygen at pH = 8 (Figure 4).
It is known that the reactivity of thiol to oxidation is related to the presence of the thiolate anion at pH = 8, a kinetic factor that implies that small quantities of free sulfur catalyze reaction in a different pathway [26]. In these sense, dimer or cyclic peptides have the same kinetic factor allowing their formation, except for the necessity that exist aggregation to be able to form dimers or oligomers. Dimers follow a second-order kinetic law reaction with respect to peptides, and helical or extended conformation should favor them. At pH = 8, some populations of Cys are as thiolate ions (Scheme S1), decreasing the net positive charge of the peptide and allowing some aggregation that, together with the increase in the reactivity of thiolate, favor the dimerization because of the facial amphiphilicity of the molecule. The characteristic facial amphiphilicity of this molecule in the helical conformation determines the joining of the hydrophobic face of two molecules while the hydrophilic face containing four positive Arg residues covers the aggregate and avoids the formation of trimmer, tetramer, and larger oligomers. On the other hand, at pH = 3, in the presence of 20% DMSO, CysCysCm-p5 is totally protonated (two Arg residues, two Cys residues and free N-terminus), avoiding aggregation, which together with the chaotropic effects of DMSO that favor twisted conformation, eliminate dimerization. Intramolecular cyclization follows a first-order kinetic law with respect to peptides, and twisted conformation should favor it. The DMSO oxidation avoids aggregation and favors the formation of beta-hairpin structures that facilitate intramolecular cyclization.
Taking into account that DMSO oxidation could need an intermediate step of concentration/DMSO-elimination when compared with air oxidation, we pay attention to a recently reported on resin simultaneous Cys deprotection/cyclization of Atosiban using I2/DMF in polystyrene resin [23]. On-resin peptide cyclization should be favorable by three factors: i. pseudo dilution condition of low substitution of resins, ii. impossibility of aggregates formation or iii. undefined secondary structure of protected peptides. On-resin supported CysCysCm-p5 have protected Arg, Ser, and Gln residues, and defined secondary structures are difficultly present, but random coil and twisted chain conformations could favor the approximation of the intramolecular thiol groups. In principle, the on-resin oxidation using DMSO/water or ACN-THF/water mixtures should follow a similar behavior to that of I2/DMF; besides, the former comprises one step (I2 also eliminates Trt protection of Cys) and cyclization in water mixtures could be done only in Chemmatrix resin. DMSO and DMF are chaotropic agents and favor disordered structures such as twisted chains, and also prevent aggregation. Chemmatrix also swells more than MBHA and maintains peptide chains further away, preventing dimers formation. Because I2 has the drawback of intense color and the possibility of Lewis-acid-catalyzed Cys racemization, we prefer to start with DMSO oxidation, which is faster than O2 oxidation (Scheme 1).
With the aim of developing a solid phase cyclization method, we synthesized the complete sequence of CysCysCm-p5 in Chemmatrix resin (0.7 mmol/g) and deprotected the Cys by several washing with TFA 1%/TIS 1% in DCM. After that, the on-resin cyclization was carried out with 35% DMSO and HCl 0.37%, pH = 3 in water for 3 h (Scheme 1). Hydrochloric acid maximizes the oxidation potential of DMSO because HX/DMSO combination rapidly oxidizes the peptide coming from the usual basic media of the Fmoc/tBu protocol. The cleavage of the peptide from the resin should be done without EDT because, once the disulfide bond is formed, EDT reduces it. The results are shown in Figure 4 and indicate that the dimer formation is prevented in these conditions.
However, we observed a poor yield of crude peptide, a known drawback of Chemmatrix resin. In contrast to the usual 50% yield, we obtained only 38 mg (34% yield) with cleavage mixture without EDT and 54 mg (48% yield) with EDT (111 mg, 0.15 g of resin for each experiment) (Exp. 1-2, Table 1). The lower yield in the first case is due to the loss of cyclic peptide due to its higher solubility in ether. Also, in our experience with peptide protocols, resins of high substitution are not normally used, among other things, mainly because of the poor yield.
In the case of cyclization in Chemmatrix resin of minor substitution, besides the peptide was correctly oxidized (Figure S6), we could only say that the cause of reduction is the ratio TIS/peptide because almost the same volume of cleavage mixture is employed. The application of the two-stage procedure for detachment/deprotection of Rink amide resin corroborates the idea that TIS is responsible for disulfide reduction during cleavage once formed during oxidation. This reduction was already reported for the solid phase cyclization of Atosiban where it was shown that TIS and water quantities in the cleavage cocktail must be modulated [23].
The reduction of a disulfide bond by TIS will depend on TIS and water equivalents, resin substitution, and quantity of protecting groups and linker that generate carbocations. These factors should be regulated peptide to peptide by calculating the available TIS equivalents. Chemmatrix resin swells so much in TFA and needs at least 15 ml/g of cleavage mixture for substitution between 0.41-0.7 mmol/g because the swelling is almost the same, but, evidently for 0.41 mmol/g the excess of TIS is sufficient for scavenging of carbocations and also reduce disulfide.
In the experiments with lower TIS and water in the cleavage mixture, it was not possible to protect the disulfide bond from being reduced by TIS, and the formation of a counterion of +97 Da (H3PO4 or H2SO4 counterions) is not desirable. Phosphoric acid is not present in our samples and has pKa1 = 2.15, meaning it do not replace TFA counterions. In the case of sulfuric acid, it should be generated by Pbf abnormal cleavage and has pKa = -10, much stronger than TFA (pKa = 0.23), so it could replace it. A plausible explanation for this experimental fact is that at reduced H2O equivalents, Pbf could be scavenged only by TIS, which kinetically tends to attack the C-S bond, generating a sulfonated Arg (SO3 = 80 Da) that is finally converted in HSO4- (97 Da) after hydrolysis. The use of 1% of TIS (3.55 eq) and excess of water (3.5%) corroborate its influence in disulfide reduction via TIS probably by reaction with other cations (tBu, linker, Pbf sulfocation) (Scheme S2), increasing the effective quantity of TIS available for disulfide reduction.
A final test showed that the total elimination of water from the cleavage mixture was unsuccessful in protecting the disulfide bond. It is important to note that TFA absorbs at least 1% of humidity from the ambient. The more water there is, the more hydronium is formed, which is the only way TFA can dissociate as its autoprotolysis (constant of 4 × 10-14 [27]) is unlikely to occur. The initial result with Chemmatrix was impossible to reproduce even in high substituted Knorr-MBHA (Exp. 18, Table 1). Apparently, the resin matrix is involved in the reduction of disulfide bond by TIS; for example, polyethylene glycol should trap more water by hydrogen bridge interactions.
In the case of iodine, the cyclic product was protected from TIS action by residual I2, preventing almost completely the reduction of the disulfide bond besides forming dimers. It was observed that I2 causes racemization of Cys due to its Lewis acid character if the Trt of Cys is eliminated previously. In an experiment with a resin of 0.2 mmol/g and direct I2 deprotection of Trt, dimers and diasteromers were minimized (Figure S8).
In addition to the good results, developing the same iodine oxidation method in the cheaper resin MBHA is desirable. In two experiments with MBHA resin, the use of iodine was confirmed as a better oxidation method, even in 2% water/2% TIS as a cleavage mixture; besides, increased dimers and reduced yield at 1.11 mmol/g was observed (Figure 5). Surprisingly, at 1.11 mmolg MBHA-resin substitution, the quantity of dimers related to 0.46 mmol/g was the same.
At this point, the results indicate that DMSO oxidation in liquid phase and iodine oxidation in MBHA resin are the best methods for producing the cyclic analogue of Cm-p5. Reduction of the disulfide bond by TIS depends on oxidation conditions, TIS and water equivalents, the volume of cleavage cocktail, resin type, and substitution, and the quantity of protecting groups and linkers that generate carbocation, which is difficult to control unless it occurs in highly substituted Chemmatrix resins.
In Table 1, we summarized all the evaluated experimental conditions. The combination of I2 oxidation and a low substituted Knorr-MBHA resin was the best choice to protect the disulfide bond during cleavage. In addition, this reaction was carried out with good yield, in one pot and using a yellow solvent, such as THF.
Aiming to produce the dimers by the double oxidation of CysAcmCysCm-p5 at 5 mg/ml, we developed another method for the synthesis of the cyclic (Scheme S3, Table 1). In this reaction, 5 eq of iodine per Acm group were added to eliminate it and activate the sulfur atom. Subsequently, a sulfur atom from another Cys(Acm) or a free Cys nucleophilically replaces the iodine in the activated sulfur, generating the disulfide bond. As each molecule of peptide has a free Cys and a protected Cys(Acm), we expected that at a high peptide concentration (even 5 mg/mL), once the sulfur atom was activated, the free Cys of another peptidic molecule replaced the iodine and eventually formed the antiparallel dimer by intercalation of both sequences. Unfortunately, the intramolecular attack of the free thiol to the activated sulfur in the same molecule was favored in that case, indicating that the experimental conditions led the reaction through a different path due to factors other than concentration. For example, the HAc solution should favor the formation of a twisted/beta-peptide structure which brings the intramolecular Cys residues together to form a disulfide bond and prevents aggregation. We also tried the reaction in THF to induce the formation of a helicoidal structure, but the cyclic monomer was always formed. Nonetheless, this cyclization method is interesting because higher peptide concentrations (5 mg/ml) can be used compared the process under DMSO conditions (1 mg/ml peptide); however, it requires the use of the more expensive Acm protection, iodine and acetic acid.

Table 1.
Summary of experimental conditions, the purity and the yield of cyclic product obtained by different methods.
Table 1.
Summary of experimental conditions, the purity and the yield of cyclic product obtained by different methods.
| Exp. | Chemmatrix | Oxidation | Cleavage mixture | Cyclic | Linear | Racemic | Yield | Dimers |
|---|---|---|---|---|---|---|---|---|
| 1 | 0.7 mmol/g, 0.15 g | 40% DMSO, 3 h | TFA/EDT 2.5%/TIS 1%/H2O 2.5% | 0% | >95% | - | 48% | - |
| 2 | TFA/TIS 2.5%/H2O 2.5% | >95% | - | - | 34% | - | ||
| 3 | 0.41 mmol/g, 0.1 g | 40% DMSO, 6 h | TFA/TIS 2.5%/H2O 2.5% | 10% | >80% | - | 45% | - |
| 4 | 50% DMSO, 12 h | 25% | >70% | - | 47% | |||
| 5 | 0.49 mmol/g, 0.2 g | 35% DMSO, 3 h | TFA/TIS 2%/H2O 2% | 0% | >95% | 35% | ||
| 6 | 0.41 mmol/g | 35% DMSO, 3 h | TFA 10% | >98% | - | - | - | - |
| 7 | I2/DMF-Trt, 30 min | |||||||
| 8 | O2, 6 h | |||||||
| 9 | 0.41 mmol/g, 0.2 g | 35% DMSO, 3 h | TFA/TIS 1%/H2O 3.5% | 25% | 75% | 43% | - | |
| TFA/TIS 1%/H2O 1% | 50% | 50% | 50% | - | ||||
| 10 | 0.41 mmol/g, 0.1 g | 35% DMSO, 3 h | TFA/TIS 1% | 68% | 27% | 35% | - | |
| 11 | I2/DMF-Trt, 30 min | 80% | - | 10% | 32% | - | ||
| 12 | O2, 6 h | 71% | 25% | 37% | - | |||
| 13 | 0.2 mmol/g, 0.1 g | I2/DMF, 30 min | 70% | - | 5% | |||
| 14 | 40% DMSO, 3 h | 61% | 37% | - | - | - | ||
| 15 | TFA/PhSiH3 1% | 48% | 47% | |||||
| MBHA | ||||||||
| 16 | 0.46 mmol/g, 0.2 g | I2/DMF, 30 min | TFA/TIS 2%/H2O 2% | 43% | - | 9.7% | 62% | 27% |
| 17 | 1.11 mmol/g, 0.13 g | I2/DMF, 30 min | 41% | - | 10% | 32% | 34% | |
| 18 | 1.11 mmol/g, 0.17 g | 60% DMSO, 3 h | TFA/TIS 1.5%/H2O 1.5% | 28% | 43% | 1.8% | 57% | 22% |
| 19 | 1.35 mmol/g, 0.15 g | I2/THF, 30 min | 57% | - | 9.8% | 30% | 6.7% | |
| 20 | 1.35 mmol/g, 0.15 g | I2/DMF, 30 min | 56% | - | 7.6% | 26% | 9% |
3.3. Selective Synthesis of the Dimers
Generally, dimers or oligomers are formed due to various factors such as concentration, solvent mixture, and tendency to aggregation [22,28]. However, in the case of peptides, the favored secondary structure could play a determining role [29]. The concentration must be optimized from case to case, but in our experience, disulfide formation of peptides can be done at 0.3-0.5 mg/ml without major disadvantages related to dimer formation. The tendency of CysCysCm-p5 to dimerize (at 0.5 mg/ml) was partially explained previously based on the distance between thiol groups determined by molecular dynamic simulation of the helicoidal conformation of the acyclic peptide and the different behavior of HcyHcyCm-p5 during cyclization [19]. In principle, the results indicate that the cyclization rate is limited by the energetic barrier needed to distort the helical structure because thiol groups are farther-away than the covalent distance of a sulfur-sulfur bond. This behavior could imply that the helical conformation is favored in the solvent mixtures used for air oxidation (50% ACN in water). In principle, more water favors the formation of beta, twisted and random coil structures, whereas non solvating or non chaotropic organic solvents favor helicoidal structures. Beta and random coil structures should also favor aggregation because of the availability of CO-NH motifs for hydrogen bond formation. Cm-p5 helical form should only interact facially due to its amphipathicity, conferred by the hydrophobic regions where Cys are located. The polar positive region surrounding the aggregate, while the hydrophobic phases are approached internally may be the reason why polymerization does not occur.
If the above were true, more water or chaotropic agent in the oxidation mixture (solvent with fewer tendencies to favor helical structure, DMF, DMSO, or salts like GnCl) would favor the cyclization and more non-solvating organic solvent would favor the dimerization. For example, it is known that DMSO is a stronger solvating agent and destroys helical structures, favoring beta structures [30]. The experimental result of Figure 4, may confirm in part that hypothesis because the use of DMSO as oxidant favors only the cyclic analogue of Cm-p5.
The results of the cyclization in water or with variable quantity of TFE corroborate these ideas because cyclic was favored by water (strong solvating inducer solvent reduce aggregation) besides parallel was also formed (apolar regions of peptide suffer some grade of hydrophobic collapse) (Figure 6). As said before, pH has a dominant role in dimer formation, only explained in terms of favoring aggregation at pH = 8. Once fixed this pH and in the presence of helical inducer solvent (aggregation inducer solvent), dimers would compete in the reaction mixture. Apparently, more polar solvent favors the parallel, however, pure water doesn’t produce the parallel only, may be an indication of kinetic control for antiparallel, favored in pure TFE. Previous computational modeling reported demonstrates that parallel dimer is more thermodynamically stable [19].
TFE is expensive hence we decided to study the cyclization at different concentration of EtOH (peptide at 1 mg/ml), green solvent compared to ACN or TFE (Figure 7). These results were more conclusive and showed that above 50% EtOH the antiparallel is favored and below 50% the parallel. The use of 81% EtOH generate almost the antiparallel dimer (better that with TFE 90%). The scale up of this reaction with 15 mg of peptide but directly in 90% EtOH improves the method besides an increase in reaction time to 15 h. The formation of antiparallel at 2 mg/ml in 90% EtOH besides slower, indicates that is favored by concentration. By that reason at 2 mg/ml of peptide but with 5% EtOH, ACN or iPrOH, antiparallel is favored and no acyclic material appears after 6 h (Figure 8), water increased the rate and antiparallel is kinetically favored.
Even though we already have developed a synthetic method of the parallel based on the mono dimerization maintaining one Cys protected with Acm in two stages, a direct method starting from the linear peptide with two free Cys is desirable. As has been showed before, in several conditions like pH = 8 and organic solvent below 50%, the parallel is favored (Figure 8), especially at 22% EtOH. In pure water parallel is favored but also the cyclic indicating that pH allows aggregation and dimerization but helicoidal induction eliminates the cyclic. The chemoselectivity to the parallel in water was improved by dissolving first in DMF 5% at 1mg/ml because this reaction always present insoluble material and DMF have a powerful capability of dissolving peptides. Also, in TFE 5%, parallel is preferred formed in more quantity than in EtOH 5%, probably due to its major polar character. In several of these experiments (Table 2), parallel should be obtained in a modest 50-60% but the best is 25% EtOH and 0.5 mg/ml in with acyclic or cyclic peptide are minimal what favors subsequent purification.
Table 2.
Resume of condition and relative percent of linear, cyclic and dimers obtained by different methods.
Table 2.
Resume of condition and relative percent of linear, cyclic and dimers obtained by different methods.
| Exp. | Solvent mixture | Time | Parallel | Acyclic | Cyclic | Antiparallel |
|---|---|---|---|---|---|---|
| CysCysCm-p5 at 0.5 mg/ml | ||||||
| 1 | ACN 50% | 30% | - | - | 40% | |
| 2 | MeOH 50% | 40% | 10% | 10% | 25% | |
| 3 | H2O | 40% | - | 25% | 10% | |
| 4 | EtOH 5% | 60% | - | - | 39% | |
| 5 | EtOH 25% | 67% | - | - | 32% | |
| 6 | EtOH 50% | 57% | - | - | 43% | |
| 7 | EtOH 75% | 44% | - | - | 55% | |
| 8 | EtOH 90% | 36% | - | - | 64% | |
| CysCysCm-p5 at 1 mg/ml | ||||||
| 10 | EtOH 22% | 45% | - | 10% | 31% | |
| 11 | EtOH 45% | 41% | - | 12% | 28% | |
| 12 | EtOH 67% | 28% | - | 13% | 45% | |
| 13 | EtOH 81% | 12% | 4% | 11% | 51% | |
| 14 | DMF 5% | 40% | - | 10% | 35% | |
| 15 | TFE 5% | 6 h | 30% | 30% | 10% | 20% |
| 16 | TFE 5% | 12 h | 50% | 5% | 10% | 20% |
| 17 | TFE 90% | 30% | - | 10% | 50% | |
| CysCysCm-p5 at 2 mg/ml | ||||||
| 18 | EtOH 90% | 8% | 5% | 7% | 68% | |
| 19 | EtOH 5% | 10% | 5% | 5% | 73% | |
| 20 | ACN 5% | 10% | 5% | 5% | 71% | |
| 21 | iPrOH 5% | 10% | 5% | 5% | 70% | |
4. Materials and Methods
4.1. Peptide Characterization
4.1.1. Analytical RP-HPLC
Analytical RP-HPLC was performed on an WellChrom HPLC (Knauer, Germany) using the EZChrom-Elite® chromatography software ChromGate v3.1 from Agilent (USA) using a Zorbax RP-C18 (5 μm, 4.6 × 150 mm) column, at flow rate of 0.8 ml/min and UV detection at 226 nm. Mobile phase with gradient of 5-60% B in 35 min, Solvent A: 0.1% TFA in H2O; solvent B: 0.1% TFA in CH3CN. Chromatograms were recorded at 226 nm and processed by the UNICORN 4.11 (GE Healthcare USA) software package.
4.1.2. ESI-MS
Low-energy ESI-MS and MS/MS spectra were obtained by using a hybrid quadrupole time-of-flight QTOF-2™ instrument (Waters, Milford, MA, USA) fitted with a nanospray ion source in positive ion mode. Peptide solutions were collected from RP-HPLC and dried, were dissolved in 1 ml of 60% (v/v) ACN/water solution containing 0.2% formic acid and loaded into a metal-coated borosilicate capillary nanotip (Proxeon, Denmark), inserted into the Z-spray nanoflow electrospray ion source, and slightly pressured with nitrogen to guarantee their stable spray during measurement. The capillary and cone voltage were set to 900 to 1200 and 35 volts, respectively. Mass spectra were acquired in the m/z range of 400–2000 Th. Electrospray ionization mass spectrometry spectra was processed using the MassLynx v4.1 program (Micromass, UK). Argon was used as a collision gas and appropriate collision energy was selected for each peptide to obtain informative MS/MS spectra.
4.1.3. Proteolytic Digestion and Desalting of Chymotryptic Peptides
The two dried fractions of dimers were dissolved in 20 μL of 100 mM Tris-HCl pH 8 containing 1 μg of chymotrypsin (Worthington, USA) and incubated for 8 h at 37 °C. The digestion was acidified with 5% formic acid (v/v) and the chymotryptic peptides were absorbed on ZipTip-C18 (Millipore, USA), previously equilibrated in 0.2% formic acid (v/v) solution. ZipTips were washed with 0.2% formic acid solution and the desalted peptides were eluted in 4.0 μL of 60% ACN containing 0.2% formic acid in water. Samples of intact peptides and proteolytic digestion were directly infused through borosilicate nano-capillaries into the mass spectrometer.
4.2. Peptide Synthesis
All reagents and solvents were obtained from commercial suppliers and used without further purification. Rink-MBHA-Polystyrene and Rink-Chemmatrix resins were prepared by reaction of Fmoc-Rink-OH linker with commercial MBHA-Polystyrene or AM-Chemmatrix resins following controlled acetylation after desired substitution respectively. All the Fmoc-amino acids, DIEA, I2, Ascorbic Acid, Ac2O and resins were purchased from Merck. Organic solvents (analytical grade) (DMF, DCM, MeOH, Et2O and ACN) were purchased from Merck. RP-HPLC quality ACN and ultrapure water quality were used for RP-HPLC analysis and purification. All reactions were carried in polypropylene plastic syringes (10 ml) fitted with polypropylene frits at room temperature with mechanical shaking and excess of solvent and reagent was eliminated by filtration. Side chain of amino acids was protected with Pbf for Arg, Trt for Gln, Cys and His, tBu for Ser and Glu and also Acm for Cys.
Solid phase peptide synthesis was carried out using Fmoc/tBu chemistry on Rink amide resin based in polystyrene or PEG (Chemmatrix). Fmoc removal was achieved with 20% piperidine in DMF (2 × 10 min), and the subsequent aminoacids were added using the following coupling conditions: Fmoc-Aa-OH/DIC/Oxyma (4 eq each) in DMF, after negative ninhydrin test (approximately 30 min). Between the different steps, the resin was washed with DMF (4 × 1 min). After the last coupling reaction, Fmoc is eliminated and peptide-resin was washed subsequently with DMF (4 × 1 min), MeOH (4 × 1 min) and Et2O (4 × 1 min). Peptide-resin is dried in desiccator during 24 h and finally is frizzed at -20 °C by 30 min before cleavage. Crude peptides were obtained as a mainly peak as ascertained by analytical RP-HPLC. The experimental molecular mass corresponded with the theoretically calculated monoisotopic mass for each peptide.
4.2.1. Peptide Cleavage
Cleavage from the resin and global deprotection: for Cys containing peptide was employed the cleavage cocktail: TFA/TIS/EDT/H2O (94:1:2.5:2.5, 3 ml/g of peptide-resin in the case of MBHA-polystyrene or 5 ml/g of peptide-resin in the case of Chemmatrix) was added to the frizzing peptide-resin and left for 2 h with shaking. Cleavage mixture was filtered and added over cold Et2O (40 ml/g of peptide-resin, -70 °C), mixed in vortex and centrifuged. The supernatant was discarded and the operation was repeated two more times. The solid residue was re-dissolved in H2O/ACN (7:3), frizzed at -70 °C and lyophilized (LABCONCO, EUA).
4.2.2. Two-Stage Procedure for Detachment/Deprotection of Rink Amide Resin
Wash the peptide-resin with 10% TFA/1% TIS in DCM, 3 × 5 min. Remove the excess TFA/DCM under reduced pressure. The solid residue was re-dissolved in H2O/ACN (7:3), frizzed at -70 °C and lyophilized (LABCONCO, EUA).
4.2.3. Liquid Phase Peptide Cyclization
Cyclization was conducted by dissolving the peptides crude (approx. 150 mg) in H2O (0.1% TFA)/DMSO (4:1) at 1 mg/ml in a necked flask of 300 ml. The reaction was stirred 6 h (or until consumption of the staring material, checked by RP-HPLC and ESI-MS. The mixture was diluted 10-fold and DMSO removed by absortion-desortion of peptide in LiChroprep RP-18 matrix (25-40 μm). The 40% ACN eluate was lyophilized (LABCONCO, EUA) and purified as described before.
4.2.4. On Resin Peptide Cyclization with DMSO, O2 or I2
On-resin DMSO cyclization was conducted by adding DMSO (35% for Chemmatrix-50% for MBHA resin) followed by water to the peptide-resin (5 ml/g total volume). To acidulate the mixture to pH = 3, 150 ul of HCl (aq.) (35%) was added and reaction was mechanically stirred during 3 h. Finally the peptide-resin was washed and cleaved as described before.
On-resin oxygen cyclization was conducted by swelling the peptide-resin with THF, addition of THF/H2O (2:1) (5 ml/g) and NH3 25% to reach pH = 8-9. Reaction was mechanically stirred overnight and finally the resin was washed and prepared for the cleavage as described before.
On-resin cyclization with I2 was done by adding 3 eq of iodine dissolved in DMF to the peptide resin (5 ml/g), allowing reacting with mechanical stirring during 30 min. Finally the resin was washed and prepared for the cleavage as described before.
4.2.5. Concentrated Iodine Oxidation of Acm Containing Peptide
The peptide containing a free Cys and Cys-Acm was dissolved in HAc (5mg/ml) and 5eq of I2 in MeOH was added dropwise until there is a persistent brown color. Water was added, 20% of the volume of HAc used and the mixture stirred for 1h.Iodine was quenched by adding 1M aq. ascorbic acid drop-wise until the mixture is colorless, and concentrates by evaporation under reduced pressure to approximately one third of the original volume and analyzed by RP-HPLC.
4.2.5. Low Scale Experiments with EtOH
At 0.5 mg/ml: In a 1.5 ml eppendorf, dissolve 0.5 mg of peptide in the necessary water with 1% TFA, sonicate during 1 min, Add absolute EtOH to reach 1 ml of final volume, 10-20 ul of 25% NH3 or until reach pH = 8-9 and shake overnight or until reaction was completed.
At 1 mg/ml: In a 1.5 ml eppendorf, dissolve 1 mg of peptide in the necessary water with 1% TFA, sonicate during 1 min, add 90% natural grade EtOH to reach 1 ml at the final volume, 10-20 ul of 25% NH3 or until reach pH = 8-9 and shake overnight or until reaction was completed.
4.2.6. Dimerization Scale-Up to Afford the Antiparallel Dimer
At 1 mg/ml (90% EtOH): Suspend 15 mg of crude peptide in 15 ml of Natural Ethanol (Cuba Ron S.A/Lawton Havana Distillery)/Jesús Rabí Distillery) and sonicate during 1 min. Add 150 ul of 25% NH3 or until pH = 8-9. Close the reaction vessel (50 ml corning) and shake overnight or until reaction was completed.
At 2 mg/ml (5% EtOH): Suspend 30 mg of crude peptide in 13.5 ml water with 1% TFA and sonicate during 1 min. Add 1.5 ml of absolute EtOH to reach 15 ml of final volume, 70 ul of 25% NH3 or until reach pH = 8-9. Close the reaction vessel (50 ml corning) and shake overnight or until reaction was completed.
4.2.7. Dimerization Scale-Up to Afford Parallel Dimer
At 0.5 mg/ml (25% EtOH): Suspend 7 mg of crude peptide in 11.25 ml of water with 1% TFA and sonicate during 1 min. Add 3.74 ml of absolute EtOH and 70 ul of 25% NH3 or until reach pH = 9. Close the reaction vessel (50 ml corning) and shake overnight or until reaction was completed.
5. Conclusions
The selective syntheses of dimers and cyclic analogues of Cm-p5 have a notable importance because each of these molecules had shown potent but variable biological activities like antifungal, antibacterial, antibiofilm and antiviral. Also, Cm-p5 and its derivatives are short peptides easily covalently modifiable and none of them showed cytotoxicity or hemolytic. To summarize, ESI-MS and -MS/MS analysis in combination with the chymotryptic digestion allowed an unambiguous differentiation of the parallel and antiparallel dimers of antifungal peptide Cm-p5.
Improved methods of preparation of the antifungal Cm-p5 cyclic monomer by liquid phase or on-resin oxidation were developed. In terms of efficiency and effectivity, DMSO oxidation has the disadvantage of additional steps of cyclization, concentration by absortion-desortion and lyophilization. On-resin iodine oxidation has the disadvantage of formation of low quantity of dimers but is advantageous in terms of time and operative steps that are most efficiently and robust (yield, time, cost, applicability to other peptides). Future research would be directed to the application of these methods to other peptides with several covalent modifications, for example, lipidated peptides have limited solubility in water and major tendency to aggregation and if the DMSO/water oxidation is a problem, the on-resin cyclization should be the preferred method.
Linear CysCysCm-p5 helix does not cycle between i, i+4 because of structural constrains and need to have turns generated by the solvent to close the thiol groups. At pH = 8 dimers are favored because of the presence of thiolates and apparently major populations of helicoidal secondary structures that together favor aggregation and makes the cyclization difficult. At pH = 3 the cyclic form is favored because the molecules could not aggregate due to a total positive charge and DMSO destroys the helix. At pH = 8, polar solvent or more water favor parallel and cyclic forms, more organic solvent and concentration favor antiparallel forms, but at 3 mg/ml the parallel form is the preferred one. A complex reaction coordinates kinetic and thermodynamic control and is governing this reaction but, due to the extension of these results, theoretical calculations and several physic-chemical characterizations and explanation using DC and DLS should be part of future research.
Finally, this work represents the first deep study of on-resin cyclization conditions and its dependence of type of resin, substitution, oxidation method and cleavage mixture and the protection of disulfide by iodine. This is the first time that a not orthogonal and selective synthesis of peptide dimers is reported indicating the importance of the solvent and pH induction of the secondary structure and aggregation. It is notable that dimeric but not always covalently bonded helix exist in proteins, toxins, chemokines and defensins, etc. Taking into account the new method of synthesis, these kinds of covalent dimers arise as an interesting structural motif that should be object of revision and generalization as structural building block of bioorganic and medicinal chemistry.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org.
Author Contributions
Conceptualization, F.M.V., L.A.E.; methodology, F.M.V., L.A.E.; investigation, M.G., F.M.V., E.D.P.; resources, H.G.P., L.J.G., L.S. and A.O.G.; writing—original draft preparation, F.M.V., L.A.E., G.O., E.D.P.; writing—review and editing, L.J.G., L.S., A.O.G.; supervision, A.R., O.F., F.R., H.G.P.; project administration, L.S., H.G.P.; funding acquisition, L.S., A.O.G., H.G.P. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the German Research Society (DFG) project CRC1279 (number 316249678). We are also grateful to DAAD-Germany for facilitating the financial support of the German Ministry for Foreign Affairs via the program Global Health and Pandemic Prevention Centers (project 57592717-GLACIER).
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank to the Center for Protein Studies, Faculty of Biology, University of Havana, 25 Str. and I Str., 10400, La Habana, Cuba and Core Facility for Functional Peptidomimetics, Ulm Peptide Pharmaceuticals (U-PEP), University Ulm, Faculty of Medicine, Ulm University 89081 Ulm, Germany for the previous done biological test
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The antimicrobial peptides and their potential clinical applications. Am. J. Translat. Res. 2019, 11, 3919–3931. [Google Scholar] [PubMed] [PubMed Central]
- Kościuczuk, E.M.; Lisowski, P.; Jarczak, J.; Strzałkowska, N.; Jóźwik, A.; Horbańczuk, J.; Krzyżewski, J.; Zwierzchowski, L.; E., B. Cathelicidins: family of antimicrobial peptides. A review. Mol. Biol. Rep. 2012, 39, 10957–10970. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.J.; Gallo, R.L. Antimicrobial peptides. Curr. Biol. 2016, 26, R14–R19. [Google Scholar] [CrossRef]
- Bechinger, B.; Gorr, S.U. Antimicrobial Peptides: Mechanisms of Action and Resistance. J. Dent. Res. 2017, 96, 254–260. [Google Scholar] [CrossRef]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef]
- Otvos, L.; Wade, J.D. Current challenges in peptide-based drug discovery. Front. Chem. 2014, 6, 62. [Google Scholar] [CrossRef]
- Chatterjee, J.; Gilon, C.; Hoffman, A.; Kessler, H. N-Methylation of Peptides: A New Perspective in Medicinal Chemistry. Acc. Chem. Res. 2008, 41, 1331–1342. [Google Scholar] [CrossRef]
- Meanwell, N.A. The Influence of Bioisosteres in Drug Design: Tactical Applications to Address Developability Problems. In Tactics in Contemporary Drug Design. Springer: Berlin, Heidelberg 2013, 9, 283–381. [Google Scholar] [CrossRef]
- Jing, X.; Jin, K. A gold mine for drug discovery: Strategies to develop cyclic peptides into therapies. Med. Res. Rev. 2019, 40, 753–810. [Google Scholar] [CrossRef]
- Reguera, L.; Rivera, D.G. Multicomponent Reaction Toolbox for Peptide Macrocyclization and Stapling. Chem. Rev. 2019, 119, 9836–9860. [Google Scholar] [CrossRef]
- Rivera, D.G.; Ojeda-Carralero, G.M.; Reguera, L.; Van der Eycken, E.V. Peptide macrocyclization by transition metal catalysis. Chem. Soc. Rev. 2020, 49, 2039–2059. [Google Scholar] [CrossRef] [PubMed]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Chow, H.Y.; Zhang, Y.; Matheson, E.; Li, X. Ligation Technologies for the Synthesis of Cyclic Peptides. Chem. Rev. 2019, 119, 9971–10001. [Google Scholar] [CrossRef] [PubMed]
- Shinbara, K.; Liu, W.; van Neer, R.H.P.; Katoh, T.; Suga, H. Methodologies for Backbone Macrocyclic Peptide Synthesis Compatible With Screening Technologies. Front. Chem 2020, 8, 447. [Google Scholar] [CrossRef] [PubMed]
- Gang, D.; Kim, D.W.; Park, H.S. Cyclic Peptides: Promising Scaffolds for Biopharmaceuticals. Genes 2018, 9, 557. [Google Scholar] [CrossRef]
- Khoo, K.K.; Norton, R.S.; Hughes, A.B. Role of Disulfide Bond in Peptide and Protein Conformation. In Amino Acids, Peptides and Proteins in Organic Chemistry: Analysis and Function of Amino Acids and Peptides. Wiley-VCH Verlag GmbH & Co. KGaA 2011, 5, 395–417. [Google Scholar]
- Góngora-Benítez, M.; Tulla-Puche, J.; Albericio, F. Multifaceted Roles of Disulfide Bonds. Peptides as Therapeutics. Chem. Rev. 2014, 114, 901–926. [Google Scholar] [CrossRef]
- López-Abarrategui, C.; McBeth, C.; Zhen-Yu, J.S.; Heffron, G.; García, M.; Alba-Menéndez, A.; Migliolo, L.; Reyes-Acosta, O.; Campos-Dias, S.; Brandt, W.; Porzel, A.; Wessjohann, L.; Starnbach, M.; Franco, O.L.; Otero-González, A.J. Cm-p5: an antifungal hydrophilic peptide derived from the coastal mollusk Cenchritis muricatus (Gastropoda: Littorinidae). FASEB Journal 2015, 29, 3315–3325. [Google Scholar] [CrossRef]
- Morales-Vicente, F.E.; González-García, M.; Díaz Pico, E.; Moreno-Castillo, E.; Garay, H.E.; Rosi, P.E.; Jimenez, A.M.; Campos-Delgado, J.A.; Rivera, D.G.; Chinea, G.; Pietro, R.C.L.R.; Stenger, S.; Spellerberg, B.; Kubiczek, D.; Bodenberger, N.; Dietz, S.; Rosenau, F.; Paixao, M.E.; Standker, L.; Otero-González, A.J. Design of a Helical-Stabilized, Cyclic, and Nontoxic Analogue of the Peptide Cm-p5 with Improved Antifungal Activity. ACS Omega 2019, 4, 19081–19095. [Google Scholar] [CrossRef]
- González-Garcia, M.; Morales-Vicente, F.; Diáz-Pico, E.; Garay, H.; Rivera, D.G.; Grieshober, M.; Olari, L.R.; Grob, R.; Conzelmann, C.; Kruger, F.; Zech, F.; Prelli, C.B.; Muller, J.A.; Zelikin, A.; Raber, H.; Kubiczek, D.; Rosenau, F.; Munch, J.; Stenger, S.; Spellerberg, B.; Franco, O.L.; Rodriguez Alfonso, A.A.; Standker, L.; Otero-González, A.J. Antimicrobial activity of cyclic-monomeric and dimeric derivatives of the snail-derived peptide Cm-p5 against viral and multidrug-resistant bacterial strains. Biomolecules 2021, 11, 745. [Google Scholar] [CrossRef]
- Kubiczek, D.; Raber, H.; Gonzalez-García, M.; Morales-Vicente, F.; Staendker, L.; Otero-Gonzalez, A.J.; Rosenau, F. Derivates of the Antifungal Peptide Cm-p5 Inhibit Development of Candida auris Biofilms In Vitro. Antibiotics 2020, 9, 363. [Google Scholar] [CrossRef] [PubMed]
- Garay, H.; Espinosa, L.A.; Perera, Y.; Sánchez, A.; Diago, D.; Perea, S.E.; Besada, V.; Reyes, O.; González, L.J. Characterization of low-abundance species in the active pharmaceutical ingredient of CIGB-300: A clinical-grade anticancer synthetic peptide. J. Pep. Sci. 2018, 24, e3081. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hansen, L.; Badalassi, F. Investigation of On-Resin Disulfide Formation for Large-Scale Manufacturing of Cyclic Peptides: A Case Study. Org. Process Res. Dev. 2020, 24, 1281–1293. [Google Scholar] [CrossRef]
- Postma, T.M.; Albericio, F. Disulfide Formation Strategies in Peptide Synthesis. Eur. J. Org. Chem. 2014, 3519–3530. [Google Scholar] [CrossRef]
- Mormann, M.; Eble, J.; Schwoppe, C.; Mesters, R.M.; Berdel, W.E.; Peter-Katalinic, J.; Pohlentz, G. Fragmentation of intra-peptide disulfide bonds of proteolytic peptides by nanoESI collision induced dissociation. Anal Bioanal Chem. 2008, 392, 831–838. [Google Scholar] [CrossRef]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radical Biology and Medicine 2014, 80, 148–157. [Google Scholar] [CrossRef]
- Harriss, M.G.; Milne, J.B. The trifluoracetic acid solvent system. Part III. The acid, HB(OOCCF3)4 and the solvent autoprotolysis constant. Can. J. Chem. 1971, 49, 3612. [Google Scholar] [CrossRef]
- Cabodevilla, J.F.; Odriozola, L.; Santiago, E.; Martínez-Irujo, J.J. Factors affecting the dimerization of the p66 form of HIV-1 reverse transcriptase. Eur. J. Biochemistry 2001, 268, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.P.; Morejón, M.C.; Rivera, D.G.; Wessjohann, L.A. Peptide macrocyclization assisted by traceless turn inducers derived from Ugi peptide ligation with cleavable and resin-linked amines. Org. Lett. 2017, 19, 4022–4025. [Google Scholar] [CrossRef]
- Tuncer, S.; Gurbanov, R.; Sheraj, I.; Solel, E.; Esenturk, O.; Banerjee, S. Low dose dimethyl sulfoxide driven gross molecular changes have the potential to interfere with various cellular processes. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
Figure 1.
ESI-MS/MS analysis of the precursor ions (a) (M+3H)3+ at m/z 394.22 and (b) (M+2H)2+ at m/z 590.82 corresponding to the chymotryptic peptide derived from antiparallel dimer of CysCys-Cm-p5. (c) Fragmentation scheme of peptide and assignment for fragment ions. (d) Structural desing with solid arrows indicates the chymotrypsin cleavage sites. Connecting lines between Cys residues indicate two intermolecular disulfide bonds between Cys4-Cys8. The nomenclature of fragment ions in the MS/MS spectra agrees with the proposed by Morman et al. [25].
Figure 1.
ESI-MS/MS analysis of the precursor ions (a) (M+3H)3+ at m/z 394.22 and (b) (M+2H)2+ at m/z 590.82 corresponding to the chymotryptic peptide derived from antiparallel dimer of CysCys-Cm-p5. (c) Fragmentation scheme of peptide and assignment for fragment ions. (d) Structural desing with solid arrows indicates the chymotrypsin cleavage sites. Connecting lines between Cys residues indicate two intermolecular disulfide bonds between Cys4-Cys8. The nomenclature of fragment ions in the MS/MS spectra agrees with the proposed by Morman et al. [25].
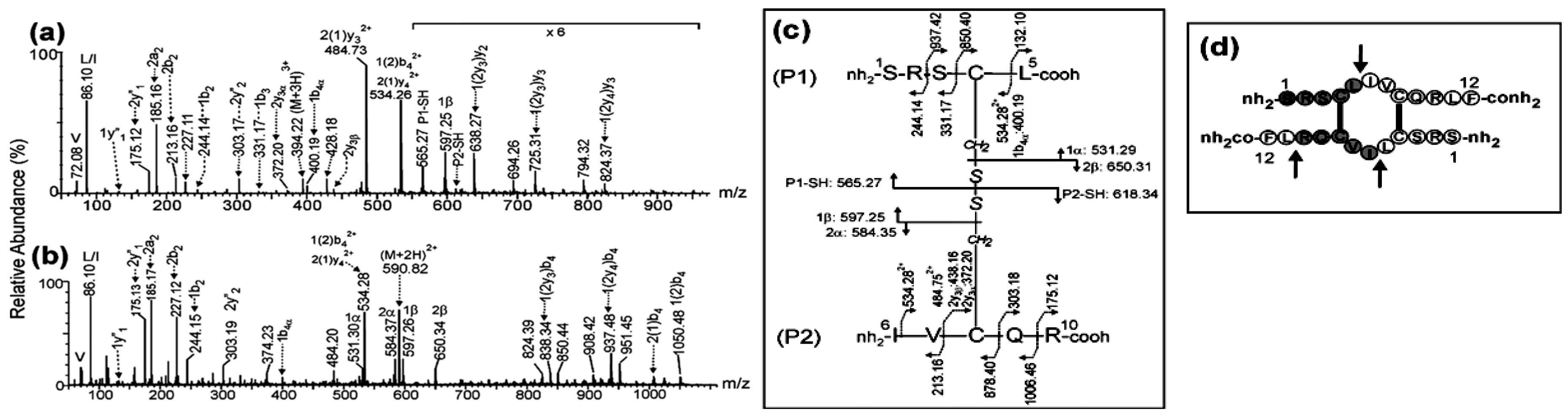
Figure 4.
Analytical RP-HPLC profile (Knauer system) and ESI-MS analysis of crude peptide CysCysCm-p5 after the liquid phase cyclization (1 mg/ml) with DMSO (20%) at pH = 3 (a, d) and after the on resin cyclization with DMSO (35%), pH = 3, 3 h, and cleavage with a mixture containing EDT (b, e) or not (c, f). The m/z 711.40 (2+) and m/z 712.41 (2+) correspond to the expected m/z values for the (M+2H)2+ precursor ions of the linear and disulfide containing CysCysCm-p5, respectively.
Figure 4.
Analytical RP-HPLC profile (Knauer system) and ESI-MS analysis of crude peptide CysCysCm-p5 after the liquid phase cyclization (1 mg/ml) with DMSO (20%) at pH = 3 (a, d) and after the on resin cyclization with DMSO (35%), pH = 3, 3 h, and cleavage with a mixture containing EDT (b, e) or not (c, f). The m/z 711.40 (2+) and m/z 712.41 (2+) correspond to the expected m/z values for the (M+2H)2+ precursor ions of the linear and disulfide containing CysCysCm-p5, respectively.
Scheme 1.
On-resin synthesis and cyclization of CysCysCm-p5.
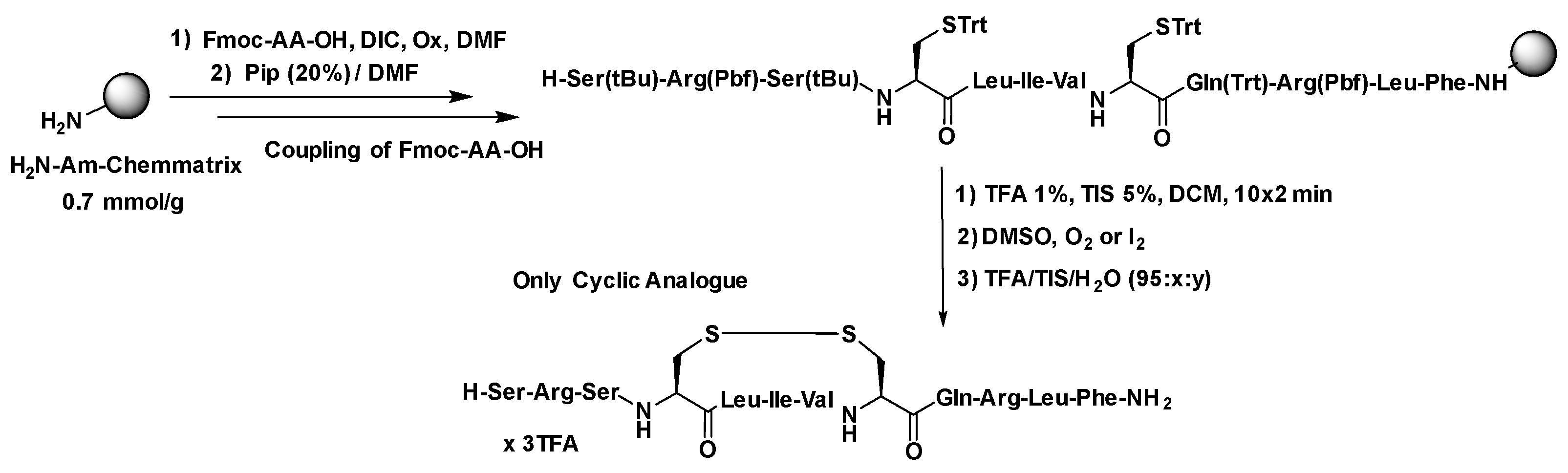
Figure 5.
Analytical RP-HPLC profiles (Shimatsu new) of CysCysCm-p5 oxydation with I2 in Rink-MBHA resin of: a) 0.46 mmol/g, b) 1.11 mmol/g, c) 1.35 mmol/g (I2/DMF), d) 1.35 mmol/g, (I2/THF).
Figure 5.
Analytical RP-HPLC profiles (Shimatsu new) of CysCysCm-p5 oxydation with I2 in Rink-MBHA resin of: a) 0.46 mmol/g, b) 1.11 mmol/g, c) 1.35 mmol/g (I2/DMF), d) 1.35 mmol/g, (I2/THF).
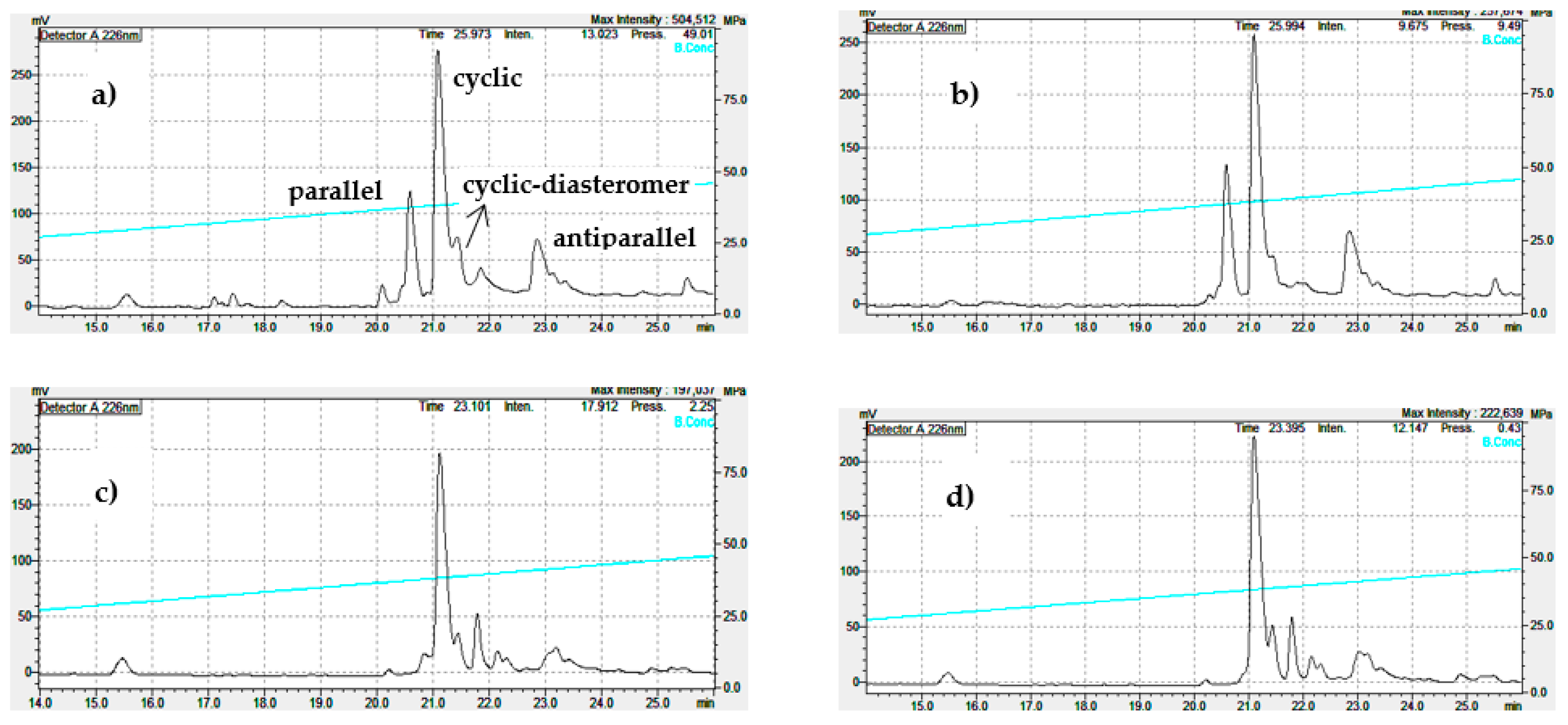
Figure 6.
Analytical RP-HPLC profile (Knauer HPLC system) after the liquid phase air oxidation/cyclization (0.5 mg/ml) in water (a), 90% TFE (b), 100% TFE (c) during 12 h.
Figure 6.
Analytical RP-HPLC profile (Knauer HPLC system) after the liquid phase air oxidation/cyclization (0.5 mg/ml) in water (a), 90% TFE (b), 100% TFE (c) during 12 h.
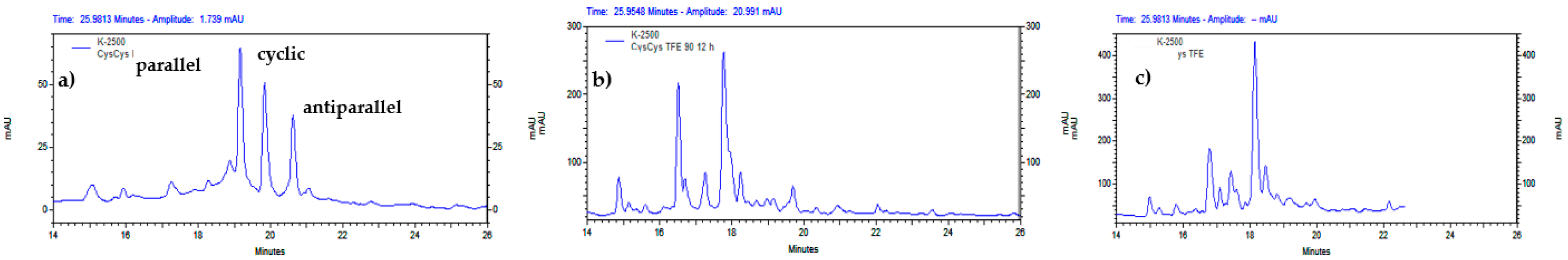
Figure 7.
Analytical RP-HPLC profile of the dimerization of CysCysCm-p5 at different EtOH concentration: a) 22% EtOH, b) 45% EtOH, c) 67% EtOH, d) 81% EtOH, pH = 8, 12-24 h, 1 mg/ml. The percentual values over each chromatogram indicate the integrated relation between the parallel dimer, cyclic monomer and antiparallel dimer. The m/z 711.40 (4+) and m/z 948.20 (3+) correspond to the expected m/z values for the (M+4H)4+ and (M+3H)3+ precursor ions of the parallel (e) and antiparallel (f) dimers of CysCysCm-p5, respectively.
Figure 7.
Analytical RP-HPLC profile of the dimerization of CysCysCm-p5 at different EtOH concentration: a) 22% EtOH, b) 45% EtOH, c) 67% EtOH, d) 81% EtOH, pH = 8, 12-24 h, 1 mg/ml. The percentual values over each chromatogram indicate the integrated relation between the parallel dimer, cyclic monomer and antiparallel dimer. The m/z 711.40 (4+) and m/z 948.20 (3+) correspond to the expected m/z values for the (M+4H)4+ and (M+3H)3+ precursor ions of the parallel (e) and antiparallel (f) dimers of CysCysCm-p5, respectively.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



