
Submitted:
27 December 2024
Posted:
30 December 2024
You are already at the latest version
Abstract
Among neurotrophins, which are composed of nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin-4 (NT-4/5), BDNF has been extensively studied for its physiological role in cell survival and synaptic regulation in the central nervous system (CNS) neurons. BDNF binds to TrkB (a tyrosine kinase) with high affinity and the resulting downstream intracellular signaling cascades play crucial roles in determining cell fate, including neuronal differentiation and maturation of the CNS neurons. It has been well demonstrated that the downregulation/dysregulation of the BDNF/TrkB system is implicated in the pathogenesis of neurologic and psychiatric disorders, such as Alzheimer’s disease (AD) and depression. Interestingly, the effects of BDNF mimetic compounds including flavonoids, small molecules which can activate TrkB-mediated signaling, has been extensively investigated as potential therapeutic strategies for brain diseases, given that p75NTR, a common neurotrophin receptor, also contributes to cell death under a variety of pathological conditions such as neurodegeneration. The present review shows recent advances in the molecular mechanisms underlying the BDNF/TrkB system in neuronal survival and plasticity, providing critical insights into the potential therapeutic impact of BDNF mimetics in pathophysiology of brain diseases.
Keywords:
BDNF
; TrkB
; depression
; schizophrenia
; Alzheimer’s disease
; Parkinson's disease
; Huntington’s disease
; BDNF mimetics
1. Introduction
A growing body of evidence has demonstrated critical importance of BDNF and its receptor in the development and function of brains. BDNF, which belongs to neurotrophins, has multiple roles in both the PNS and CNS neurons [1,2]. Trks, tyrosine protein kinase receptors for neurotrophins, also consist of TrkA, TrkB, and TrkC, and mainly regulate the cell proliferation, differentiation, maturation, and synaptic function in the PNS and CNS neurons when specific neurotrophin binds to them, respectively. It is well known that NGF, the first identified neurotrophin, binds to TrkA with high affinity. BDNF, which is expressed in the variety of brain regions including the cerebral cortex and hippocampus contributing to learning and memory function of brain, also has a high affinity TrkB receptor in the same way [1-3]. Similarly, NT-3 binds to TrkC, and NT-4 does to TrkB, although all the members of the neurotrophin family interacts with the p75 neurotrophin receptor (p75NTR), which has no catalytic activity, with low affinity [4].
Firstly, all the members including BDNF is translated as a precursor (named a proneurotrophin), which binds to the p75NTR with high affinity, regulating neurite outgrowth, cell survival and/or death via interacting with varieties of co-receptors or adaptor proteins [5-7]. On the other hand, small mature neurotrophins after receiving the subsequent cleaving process (mature NGF, mature BDNF, and so on, see Figure 1) preferentially interact with Trks [3,4].
Importantly, evidence indicates critical contribution of BDNF in the development and maintenance of brain. The roles of BDNF in the CNS are very important for brain functions, thus, dysregulation of BDNF function has been suggested to be involved in cognitive impairment observed in the various mental disorders including depression [3].Therefore, it has been demonstrated that upregulation of BDNF/TrkB system is effective against the cognitive impairment of mental disorders. In this review, we show current studies concerning upregulation of BDNF by natural compounds and influence of BDNF mimetics in behaviors of in vivo and in vitro models for mental disorders.
AD, which is recognized as the most common neurodegenerative disorder, shows abnormal accumulation of amyloid-beta (Aβ) and neurofibrillary tangles in which hyperphosphorylated tau protein is aggregated in the brain tissue, and exhibits significant cognitive decline. A variety of pharmacological interventions aiming at deprivation of Aβ and tau proteins have carried out, however, limited success in treatment for progression of the disease [8]. Recently, as one of the alternative therapeutic targets going over the amyloid-centric approach, facilitation of the BDNF/TrkB system is considered as a critical target to improve disease progression of AD [9]. Parkinson’s disease (PD), which is also a neurodegenerative disorder, displays bradykinesia, rigidity, and tremors, in addition to non-motor symptoms impacting quality of life, exhibits the loss of dopaminergic neurons within the substantia nigra pars compacta, resulting in depletion of striatal dopamine levels. In the PD pathogenesis, it has been also demonstrated that the downregulation of BDNF/TrkB system could contribute to dopaminergic neuronal degeneration [10]. Taken together, how to enhance BDNF/TrkB system is promising target for therapeutic development in these brain diseases. Similarly, it has been demonstrated that changed neurotrophic support by BDNF/TrkB is involved in pathogenesis of Huntington’s disease (HD), which exhibits significant neurodegeneration, particularly in the striatum and cortex [11].
Recent evidence has demonstrated that natural compounds which increase endogenous expression of BDNF and/or TrkB, and BDNF mimetic compounds that are small molecules passing blood-brain barrier (BBB), look promising to treat brain diseases including mental and neurological disorders. Here we show recent evidence in regulation of BDNF/TrkB system by small molecules using in vivo and in vitro disease models, to take critical insights developing the potential therapeutic approach to mental and neurological disorders.
2. Biological Roles of BDNF/TrkB System and Its Downstream Intracellular Signaling
There are three main intracellular signaling pathways triggered after TrkB activation. BDNF can stimulate the phospholipase Cγ (PLCγ), which is involved in Ca2+ homeostasis regulated by BDNF, the mitogen-activated protein kinase (MAPK) contributing to a variety of neuronal aspects including synaptic plasticity, and the phosphatidylinositol 3 kinase (PI3K)/Akt maintaining cell survival, and guanosine triphosphate hydrolases of Ras homolog gene family pathways via the TrkB activation [12-14].
We previously reported that enhanced intracellular Ca2+ concentration by BDNF is important for release of glutamate, an excitatory ammino acid transmitter, which is mainly dependent on the PLCγ pathway [15]. Furthermore, we also found a significant downregulation of the PLCγ pathway in brain neurons obtained from low-weight birth pups produced by intrauterine growth retardation (IUGR) [16]. Importantly, it is well accepted that the activation of Ca2+-calmodulin-dependent kinases (CaMKs) and protein kinase C, and generation of 1,2-diacylglycerol are regulated via activating PLCγ pathway [13,17].
The family of MAPK, which is a serine-threonine kinase, is composed of p38, c-Jun NH2-terminal kinase (JNK, or stress-activated protein kinase), and ERK, and has multiple roles in the mammalian cells. In the many kinds of cell population including the CNS neurons, MAPKs contribute to cell proliferation, differentiation, survival, maturation, and synaptic function [18-20]. Especially, ERK regulates the expression of synaptic proteins stimulated by BDNF. We previously reported that attenuated ERK levels after glucocorticoid (stress hormone) exposure resulted in downregulation of BDNF-enhanced expression of synaptic proteins [21]. Interestingly, recent evidence suggests that SPROUTY2, a critical regulatory molecule for signaling of neurotrophic factors including BDNF, is possibly an effective therapeutic target for neurodevelopmental disorders, because it regulates a variety of neuronal aspects including cell death and axon outgrowth via affecting ERK signaling pathway (see [22]).
Importantly, the mammalian target of rapamycin (mTOR), which is a key molecule for the cellular homeostasis, functions as a downstream molecule of the PI3K/Akt signaling pathway, leading to facilitating cell differentiation and survival [23-26], therefore, the PI3K/Akt/mTOR pathway also exerts a critical role in BDNF/TrkB-mediated neuronal survival in the CNS neurons. Furthermore, evidence demonstrates that Akt/cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB) pathway, which induces the upregulation of BDNF, is promising therapeutic target in the neurodegenerative diseases including AD [27]. In addition to ERK signaling, these PI3K/Akt/ mTOR and Akt/CREB pathways are also suggested to be involved in the regulation of the apoptosis observed in the neurodegenerative diseases [28].
3. BDNF/TrkB System and Antidepressant Effects of Natural Compounds in Depression Models
As mentioned above, a growing body of evidence has demonstrated that altered expression and/or function of BDNF/TrkB system are closely correlated with major depressive disorder (MDD) [29]. At the current time, although the pathogenesis of depression is not fully understood, it has been suggested that signal pathways affected by BDNF and its receptors (TrkB and p75NTR) have a role in the pathogenesis of depression [30]. Since patients with depressive disorders exhibit cognitive impairments [31], decreased activation of BDNF/TrkB system, which regulates neuronal function, has been considered as one of the major mechanism underlying the pathophysiology of depressive disorders [32]. Importantly, it is well known that the overactivation of hypothalamic-pituitary-adrenal (HPA) axis induces abnormally increased glucocorticoids (the stress hormones), which influence the BDNF/TrkB system [32]. A variety of environmental challenges affect the status of the HPA axis, leading to changed blood levels of glucocorticoids, which in turn influence synaptic function and BDNF-mediated neuronal aspects [32,33].
When searching novel antidepressant drugs, Bupleurum, which is the traditional Chinese medicine to treat liver illness including its inflammation, fibrosis, and cancer, is promising candidate [34]. It has been shown that rutin, puerarin, saikosaponin A, saikosaponin D, and quercetin, which are ingredients obtained from Bupleurum, have antidepressant effects through different mechanisms including regulation of neurotransmitters, the NMDA receptor system, the BDNF/TrkB system, and so on [34]. 18β-glycyrrhetinic acid (18β-GA) in Xiaoyaosan (XYS), which is a traditional Chinese medicine and used for the treatment of depression, has been identified as a primary active compound [35]. Using the chronic social defeat stress (CSDS) model, it was revealed that an injection of XYS showed antidepressant-like effects with activation of BDNF transcription in the medial prefrontal cortex of CSDS mice. Furthermore, 18β-GA was identified as the primary product in the mouse brain after XYS administration, also found to interact with ERK, leading to activation of ERK, and to induce BDNF transcription via activating nuclear factor-erythroid factor 2-related factor 2 (Nrf2), CREB, and c-Jun [35]. Many studies show neuroprotective properties of phytocannabinoids and a relationship between cannabis and psychiatric conditions, including depression, has been discussed [36]. Interestingly, a recent study examined the biological effect of both natural tetrahydro-cannabiorcol (trans-(-)-Δ9-THCO) and unnatural Δ9-tetrahydrocannabiorcol (trans-(+)-Δ9-THCO), focusing on the levels of BDNF as an indicator for potential in neuropathologies [37]. Both THCO enantiomers significantly reversed cell death caused by CORT (corticosterone), one of the glucocorticoids, in the SH-SY5Y cells, and increased the expression of BDNF simultaneously. It was also shown that unnatural trans-(+)-Δ9-THCO has better activity compared with that by natural enantiomer [37]. It has been demonstrated that the nuclear factor erythroid 2-related factor 2-antioxidant responsive element (Nrf2-ARE) is involved in cellular defense system under the oxidative stress [38]. Interestingly, an activation and/or nuclear translocation of Nrf2 are triggered via PI3K/Akt pathway stimulated by BDNF/TrkB system. Notably, recent evidence shows that indole-3-carbinol and its metabolite, diindolylmethane, exert neuroprotective effects via activating Akt, leading to the antioxidant action, by inhibiting the Nrf2-kelch-like ECH-associated protein 1 (Keap1) complex [38]. Recently, NMC-4 (new macamide compound-4) has been synthesized and analyzed [39]. Using depression rat model caused by chronic unpredictable mild stress, antidepressant action of NMC-4 with significant improvement of reduced BDNF/PI3K/Akt pathways in the cerebral cortex of depression rats. Further, increased levels of IL- 1β, IL- 6, and TNF-α in the cortex of depression animals were all downregulated by NMC-4 application [39]. Recently, the antidepressant effect of ononin, which is a flavonoid compound, was investigated using a depressive rat models induced by chronic mild stress (CMS) [40]. It was revealed that significantly decreased depression-like behaviors assessed with sucrose consumption test, open field test, and tail suspension test (TST), were achieved after ononin treatment [40]. Markedly, the treatment with ononin increased the protein levels of neurotrophic factors including BDNF, GDNF, and NGF in the hippocampus and frontal cortex of the depressive rats [40]. Zeng et al. (2024) examined the antidepressant effects of Rosmarinic acid (RA), which is one of the polyphenol compounds found in a variety of herbs [41]. They made depression-like behaviors in male C57BL/6 J mice by chronic CORT intraperitoneally injection and performed daily oral application of RA for 21 consecutive days. They found that depression-like and anxious behaviors assessed with forced swimming test (FST), tail suspension test (TST), splash test (ST), novelty-suppressed feeding test (NSFT), and sucrose preference test (SPT) were improved by the application of RA [41]. Furthermore, it was revealed that hippocampal expression of pGR (glucocorticoid receptor), HSP90 (heat shock protein 90), FKBP51 (FK506 binding protein 51), and SGK-1 (recombinant serum/glucocorticoid regulated kinase 1) was decreased by treatment with RA while a density of Nissl bodies and number of dendritic spines in the hippocampus were increased after RA treatment. Interestingly, expressions of BDNF, pTrkB, and pCREB were upregulated by RA [41].
Antineuroinflammatory and/or antidepressant effects of oleacein (OC), a rare compound obtained from olives, has been found [42]. In SH-SY5Y cells, the upregulation of BDNF was induced by OC application although the BDNF upregulation was canceled in the presence of an inhibitor for PI3K, or MEK (an upper molecule for ERK). Interestingly, an administration of OC significantly improved depression-like behaviors of model mice caused by lipopolysaccharide (LPS). In addition, OC also decreased mRNA levels of hippocampal pro-inflammatory cytokines including Il6, Il1β, and Tnfα in LPS mice [42].
It has been recognized that high-molecular weight proteins including neurotrophins are not able to cross the BBB, thus small molecules which mimic actions of neurotrophins has been searched. Fukuyama et al. (2020) have summarized chemical constituents of neurotrophic compounds derived from Garcinia subelliptica, Magnolia ovobata, Phytolacca americana, Illicium species, and Viburnum species, and showed their neurite outgrowth and neuroprotective effects using cellular systems including PC12, primary cortical neurons, and MEB5 cells [43].
These results indicate significant upregulation of BDNF by a variety of natural compounds, resulting in antidepressant-like effects in vivo and in vitro depression models. The interventions concerning a mechanism underlying an upregulation of BDNF expression by these compounds could provide beneficial platform to seek another drugs candidate.
4. BDNF Mimetics and Their Antidepressant Effects in Depression Models
It was reported that impaired learning and memory, and exploratory behavior detected in depression rats after receiving unavoidable electric foot-shock were corrected by GSB-106, N-monosuccinyl-L-seryl-L-lysine hexamethylenediamide, which is a dipeptide mimetic of the BDNF loop 4 [44]. To analyze the effectiveness of GSB-106 against the depression, an animal study using depression mouse model caused by 28-day social defeat stress with 21-day oral administration of GSB-106 after the stress was performed [45]. A decreased locomotor activity and an impaired sucrose preference were restored by the administration of GSB-106. Remarkably, antidepressant-like activity of the GSB-106 assessed with FST was canceled by K252A, a blocker for Trk receptors, and by U73122, an inhibitor for PLC, suggesting probable contribution of TrkB/PLCγ signaling in effects of GSB-106 [45]. Furthermore, male rats exhibited depressive-like behavior and impaired memory after transient middle cerebral artery occlusion (MCAO), although GSB-106 administration completely reversed these depressive-like behavior and memory impairment [46]. Moreover, it was revealed that the decreased content of CREB in the hippocampus of MCAO animals was also repaired by GSB-106 administration [46].
5. A Variety of Mechanisms Under the Influence of BDNF in Depression Models
Tiliwaerde et al. (2024) recently showed that GW043, which is a novel NMDA receptor modulator, has antidepressant effect using animal model [47]. They found that an activation of NMDAR and hippocampal long-term potentiation (LTP) were induced by GW043 in mice. Furthermore, depressive behaviors of rats caused by chronic unpredictable mildstress (CUMS) were improved by GW043 treatment. Downregulation of BDNF and phosphorylation of mTOR in the hippocampus and prefrontal cortex of CUMS rats was reversed by GW043 [47]. Growing evidence has demonstrated that possible involvement of histone deacetylase 5 (HDAC5) in the pathogenesis of MDD. Meng et al. (2024) performed the overexpression of HDAC5 with adenoviral vectors in the dentate gyrus of mice and found that depressive-like behaviors were evoked by the overexpression. Importantly, in addition to improvement of depressive-like behaviors, the downregulated BDNF expression (mRNA and protein) in the hippocampus of mice overexpressing HDAC5 was restored by application of TMP269, a class IIA histone deacetylase inhibitor [48].
Recently, effects of polygonatum sibiricum polysaccharides (PSPs) on microglia has been shown [49]. The depressive-like behaviors (with SPT and FST) observed in chronic restraint stress (CRS) mice were significantly improved by PSP treatment. In the prefrontal cortex of the depressive mice, it was revealed that PSP decreased activation of microglial, and upregulated microglial BDNF levels in the prefrontal cortex and hippocampal neurogenesis, suggesting an involvement of regulation of microglial in the effects of PSP [49]. Valenza et al. (2024) reported the responses of astrocytes and microglia to acute stress. In rat model of post-traumatic stress disorder, acute inescapable footshock caused prompt astrocytes and microglia response, NF-κB pathway activation, an increase in levels of IL-18 and TNF-α in the prefrontal cortex of vulnerable rats. Furthermore, significantly changed levels of glial proteins (S100B, CD11b, and CX43), and trophic factors (BDNF and FGF2) were also confirmed [50].
Interestingly, a recent study reported that patients with MDD displayed lower levels of BDNF in plasma astrocyte-derived extracellular vesicles (ADEVs) than that from healthy controls at baseline [51]. Importantly, BDNF levels in ADEVs were more stable than that in plasma, suggesting ADEVs is more suitable for biomarkers than plasma in depression [51].
6. Relationship Between BDNF/TrkB System and Schizophrenia
Zou et al. (2024) performed a network meta-analysis on different psychiatric disorders, and found that peripheral BDNF concentration in patients with MDD, bipolar disorder, obsessive-compulsive disorder, panic disorder, or schizophrenia is lower compared with that in controls although patients with PTSD shows an increased BDNF levels, suggesting BDNF is a valuable biomarker for diagnosis of mental disorders [52].
Schizophrenia, a severe and highly heritable psychiatric disorder, also exhibits abnormality in thinking and emotions, and has been suggested to be associated with altered BDNF function. Importantly, using search terms: (schizophrenia) AND (BDNF gene polymorphism) to get articles published in the last 5 years, Farcas et al. (2023) conducted a systematic search on Val66Met polymorphism of BDNF gene and showed that the Met/Met genotype is associated with higher positive scores of Positive and Negative Syndrome Scale (PANSS), reflecting disease severity, and with a history of suicide attempts [53]. Moreover, Met/Met homozygous individuals exhibit lower levels of peripheral BDNF and worse cognitive ability [53]. Indeed, the resent systematic review of the literature on the differences in baseline levels of BDNF and a Val66Met polymorphism of BDNF in bipolar disorder and schizophrenia among responders and non-responders, presents that higher levels of baseline BDNF and the BDNF Val/Va genotype may be present in responders to both pharmacological and non-pharmacological treatments [54].
Chen et al. (2022) reported schizophrenia-like behaviors in Bdnf-e6-/- mice (promoter VI mutant mice) when exposed to postnatal stress although stress or the deficiency of Bdnf promoter alone did not cause behavioral abnormalities. These behavioral abnormalities were improved by RU-486 (an antagonist for corticosterone), or Ab4B19 (an agonistic antibody for TrkB), suggesting both environmental and genetic factors contributing in the pathogenesis of schizophrenia [55]. In addition to BDNF gene, the expression of antisense RNA for BDNF gene (BDNF-AS) has been also demonstrated to be involved in the vulnerability to psychiatric disorders, including depression, schizophrenia, and bipolar disorder [56]. As expected, a lot of studies suggest significant connections among specific polymorphisms of BDNF-AS gene and these psychiatric disorders [56], reconfirming an importance of BDNF expression in the pathogenesis of psychiatric disorders including schizophrenia because of its role in regulation of BDNF expression. The recent cross-sectional comparative study (208 healthy control subjects, 123 patients with mild cognitive impairment (MCI), and 123 patients with schizophrenia) also reported a lower concentration of plasma BDNF levels in patients with schizophrenia and MCI compared with that in healthy controls [57]. It was revealed that cognitive functions (estimated by mini mental state examination) of patients with schizophrenia and individuals with MCI were lower than that in control, and the lower cognitive activity showed a significant association with decreased plasma BDNF levels [57].
As shown, increasing evidence demonstrates that restoring BDNF/TrkB system also could be effective to improve symptoms of schizophrenia.
7. Contribution of BDNF/TrkB System in Antipsychotic Effects of Natural Compounds in Schizophrenia Models
As we will discuss in Section 8, 7,8-dihydroxyflavone (7,8 DHF) has been expensively studied using a wide variety of brain disease models. Even in regard to schizophrenia models, effects of 7,8 DHF have been examined. Han et al. (2016) had showed possible involvement of downregulation of BDNF/TrkB system in abnormal neurodevelopment of offspring produced by polyriboinosinic-polyribocytidylic acid [poly(I:C)] [58]. In their system, adult offspring from pregnant animals which received poly(I:C) exhibited decreased BDNF/TrkB signaling and immunoreactivity of parvalbumin in the prefrontal cortex and hippocampus, in addition to PPI deficits, although supplementation of 7,8 DHF revered all these abnormalities, including the downregulation of BDNF/TrkB signaling, reduced PV immunoreactivity, and abnormal behavior [58]. Using animal maternal immune activation (MIA) model which was induced with polycytidylic acid (Poly-I:C) infection during pregnancy, schizophrenia-like behaviors of the offspring was examined [59]. Fetal brains from MIA dams showed increased IL-6, and downregulation of Ntrk2 and glutamatergic/GABAergic neuronal markers. As expected, adult offspring from MIA dams exhibited anxiety-like behaviors. Interestingly, 7,8-DHF applied through drinking water for MIA dams could not inhibit negative influence of MIA on behavioral disturbances of adult offspring although 7-8 DHF treatment reversed the expression of some glutamatergic (Grm5) and GABAergic (Gabra1) genes, implying an importance of the timing of potential interventional treatment [59]. When exposed to Poly I:C, cultured hippocampal neurons exhibited reduced cell viability and increased lipoperoxidation, and showed inflammatory responses including upregulation of NFkB canonical pathway and decreased expression of BDNF [60]. Interestingly, omega-3 polyunsaturated fatty acids (n3 PUFAs) and clozapine, an atypical antipsychotic, inhibited the pro-inflammatory responses although only n3 PUFAs prevented both neuronal cell death and the depletion of BDNF expression caused by exposure to Poly I:C, [60]. Using an animal model for intrauterine growth retardation (IUGR) caused by maternal administration of thromboxane A2, we also found that significant downregulation of TrkB in the cortex of IUGR rats at birth [16]. Interestingly, glutamate release triggered by BDNF was decreased in cultured IUGR cortical neurons where the decreased TrkB was still maintained, which were reversed after the transfection of human TrkB. It was reported that downregulation of BDNF/TrkB system, deficits in learning ability and hippocampal synaptic plasticity of the schizophrenia-like animals were also rescued by 7,8-DHF (Yang et al., 2014). MK-801-treated rats exhibited poor working learning ability (MWM test), decreased expression of BDNF, and deficits in hippocampal LTP although enhanced activation of TrkB, ERK1/2, CaMKII, CREB and GluR1, with an improved synaptic plasticity and learning ability were observed after the chronic 7,8-DHF treatment [61]. Using schizophrenia mouse model induced by ketamine, an NMDA antagonist, effects of polyphenolic flavonoid, silymarin on disease-like symptoms was examined [62]. Preventive and curative effects of silymarin against hyperlocomotion, stereotypy, memory, and social impairments were observed and significant downregulation of BDNF, glutathione, superoxide-dismutase, and catalase in the prefrontal cortex, hippocampus, and striatum of mice received the ketamine administration were all restored by silymarin [62]. Khalid et al. (2024) examined antipsychotic effects of esculetin, a derivative of natural coumarin, against schizophrenia-like behaviors of mice received the chronic administration of ketamine [63]. They found improvements in behavioral symptoms, oxidative stress, and status of neuroinflammation caused by ketamine. Further, they observed restoration of BDNF, which was decreased in the hippocampus, cortex and striata by ketamine [63].
Ordinarily, both fingolimod (also named as FTY-720) and siponimod, which are modulators for a sphingosine 1-phosphate receptor (S1PR), have been recognized as effective drugs to inhibit progression and relapse of multiple sclerosis [64-66]. Recently, to search the molecular target for the improvement of decreased cognitive function of schizophrenia, Li et al. (2024) performed bioinformatics and computational analyses focusing pharmacological mechanisms underlying neuroprotective effects by both fingolimod and siponimod [67]. Interestingly, the regulations of the MAPK and PI3K/Akt signaling pathways involving TNF, IL1B, IL6, INS, BCL2, AKT1, and BDNF by fingolimod while the MAPK signaling pathways involving TNF, AKT1, and CASP3 (Caspase-3) by siponimod have been demonstrated [67].
As genetic variation in CACNA1C (for the alpha-1 subunit of CaV1.2 L-type voltage-dependent Ca2+ channels is demonstrated as a risk factor for both bipolar disorder and schizophrenia, using a Cacna1c+/− rat model [68], Tigaret et al. (2024) examined cognitive and synaptic phenotypes of the animal models and found synaptic and cognitive abnormalities in the rats, which was also rescued by LM22B-10, an agonist for TrkB/TrkC receptor [69]. Recently, it was reported that LT-102, a novel potentiator of AMPAR, enhanced the hippocampal LTP and restored cognitive deficits in schizophrenia-like animal model induced by phencyclidine [70]. LT-102 binds to AMPAR's GluA2 subunit, and induced upregulation of BDNF protein and the phosphorylation levels of GluA1 [70].
As shown in these sections concerning psychiatric disorders, boosting BDNF function through its specific receptor TrkB is indeed effective to improve symptoms of depression and schizophrenia judging from results using in vivo and in vitro models (see Figure 2). Further, changed levels of biomarkers, including BDNF and/or downstream signaling molecules, for mental disorders, are clearly suggestive of the promising potential of BDNF mimetics although further information concerning actions of BDNF mimetics, including the duration of its activity for TrkB stimulation and side effects at the cellular level, should be approached before clinical applications as excess BDNF/TrkB activation may cause side reactions, resulting in abnormal synaptic plasticity.
8. The Therapeutic Potential of BDNF Mimetics in Alzheimer’s Disease (AD)
8.1. The Role of BDNF in AD
AD, a progressive neurodegenerative disorder, continues to pose an enormous challenge to public health, affecting millions globally. Current treatment modalities predominantly address symptomatic relief but fail to effectively halt or reverse the disease's underlying pathophysiological processes. AD is characterized by the accumulation of amyloid-beta (Aβ) plaques and neurofibrillary tangles, leading to widespread neuronal dysfunction and cognitive decline. Over the past few decades, despite significant research investments, pharmacological interventions targeting Aβ and/or tau proteins have yielded limited success in modifying disease progression [8]. Thus, there is a growing need to explore alternative therapeutic targets that go beyond the conventional amyloid-centric approach. Among these, BDNF has emerged as a key player in maintaining neuronal integrity and synaptic function, positioning it as a critical target for therapeutic development in neurodegenerative diseases like AD [9].
BDNF belongs to the neurotrophin family, a group of growth factors which are essential for the development and maintenance of the CNS. As mentioned above, it regulates key aspects of brain function, including synaptic plasticity like LTP and neuronal survival—all of which are vital for learning and memory processes. Several studies have demonstrated that BDNF levels are significantly reduced in the brains of AD patients, particularly in regions associated with memory, such as the hippocampus [71,72]. This decline is thought to exacerbate neuronal loss and cognitive decline, making BDNF signaling a critical therapeutic target (Figure 3). However, delivering BDNF itself as a treatment is challenging due to its large size and poor BBB permeability [73]. Consequently, the development of BDNF mimetics, which can circumvent these limitations, has garnered increasing attention.
BDNF exerts its effects primarily by binding to TrkB receptor, triggering downstream signaling pathways that promote neuronal survival, differentiation, and synaptic modulation [9]. As shown in Figure 1, activation of TrkB initiates several intracellular cascades, including the PI3K/Akt, MAPK, and PLCγ pathways, all of which are implicated in cellular survival, synaptic growth, and plasticity. In AD, disruption of these pathways contributes to neurodegeneration [9]. Reduced levels of BDNF impair TrkB signaling, leading to synaptic loss and neuronal atrophy, which are hallmarks of AD pathology.
Given that synaptic dysfunction is one of the earliest signs of AD, strategies aimed at enhancing TrkB signaling could provide significant therapeutic benefits. While direct administration of BDNF has shown neuroprotective effects in preclinical models [73-75], its therapeutic application in humans remains impractical due to delivery challenges and short half-life. As a result, BDNF mimetics, which are designed to replicate the neurotrophic and neuroprotective functions of BDNF by activating TrkB, offer an attractive alternative for AD treatment.
8.2. Challenges in Direct BDNF Therapy
The biological hurdles to directly using BDNF as a therapeutic agent for AD were recognized early on. Despite its promising neuroprotective properties, BDNF is a large protein (27-kDa noncovalently linked homodimer) with poor pharmacokinetic properties when administered systemically [73]. Intranasal delivery of BDNF, while bypassing the BBB, still faces challenges related to achieving therapeutic concentrations in the brain [76]. Furthermore, the short half-life of BDNF in circulation, combined with the need for repeated dosing, makes it a less viable long-term treatment option [77]. In addition, there are concerns regarding the specificity and safety of direct BDNF therapy. High concentrations of exogenous BDNF may lead to non-specific activation of not only TrkB but also other receptors, such as p75NTR, which may induce pro-apoptotic signals under certain pathological conditions [9]. These limitations highlighted the need to develop alternative strategies, specifically the use of small molecule mimetics that can selectively target TrkB receptors while overcoming the pharmacological constraints associated with the BDNF protein.
8.3. Small Molecule BDNF Mimetics: Early Efforts
The first generation of BDNF mimetics focused on small molecules with an ability to activate TrkB, either by mimicking BDNF binding or by inducing receptor dimerization and autophosphorylation. One of the most studied early candidates is 7,8-DHF, a flavonoid that has demonstrated significant neuroprotective effects in various animal models of neurodegeneration [78]. Discovered through high-throughput screening, 7,8-DHF was identified as a small molecule which is capable of crossing the BBB and selectively activating TrkB [79]. In preclinical studies, 7,8-DHF has been shown to exert multiple beneficial effects, including the restoration of synaptic plasticity, reduction in Aβ deposition, and mitigation of cognitive deficits in AD models [80-82]. By activating TrkB, 7,8-DHF triggers downstream signaling cascades such as the PI3K/Akt, MAPK/ERK, and PLCγ pathways, thereby promoting neuronal survival, reducing apoptosis, and enhancing synaptic strength—processes that are severely compromised in AD [82]. Its ability to cross the BBB and engage TrkB with relatively high specificity made it an attractive candidate for further development. Another example of the first generation of BDNF mimetics is LM22A-4, a small molecule that was specifically designed to target the extracellular domain of TrkB, thereby selectively activating TrkB without engaging the p75NTR or other neurotrophic receptors [83]. LM22A-4 demonstrated promising neuroprotective properties in animal models, including the promotion of neuronal survival, enhanced synaptic plasticity, and improved cognitive outcomes in rodent models of neurodegeneration including AD [84,85].However, despite these promising preclinical results, 7,8-DHF and LM22A-4 face some limitations in their pharmacokinetic properties, including rapid metabolism and poor oral bioavailability [78]. Efforts have been made to improve their stability and effectiveness, leading to the synthesis of structural analogs with enhanced pharmacological profiles. Despite these challenges, the first generation of BDNF mimetics remain foundational molecules in the field of BDNF mimetic development, demonstrating the feasibility of small molecule approaches to induce TrkB activation.
8.4. Next-Generation BDNF Mimetics: Improved Selectivity and Potency
The limitations of first-generation mimetics such as 7,8-DHF and LM22A-4 spurred the development of second-generation compounds with improved pharmacokinetics, BBB permeability, and receptor selectivity. For example, Chen et al. (2018) developed a prodrug of 7,8-DHF named R13, by modifying the catechol group with a carbamate moiety, which resulted in significantly improved drug-like characteristics [86]. Chronic oral administration of R13 activated TrkB signaling and reduced Aβ deposition in 5xFAD mice, thereby inhibiting the pathological cleavage of APP and Tau mediated by δ-secretase. Furthermore, R13 mitigated hippocampal synapse loss and alleviated memory impairments in a dose-dependent manner [86]. Importantly, R13 appears to exhibit a more favorable pharmacokinetic profile compared to 7,8-DHF, with better stability and specificity for TrkB. Another study further optimized 7,8-DHF, producing a highly stable CF3CN derivative that demonstrates TrkB agonist activity in cellular assays with an EC50 of approximately 26.4 nM [87]. Chronic oral administration of CF3CN exhibited promising therapeutic effects in the 5xFAD mouse model [87]. PTXBD4-3, another second-generation small-molecule TrkB agonist derived from LMA22A-4, activated TrkB signaling following intraperitoneal and chronic administration of relatively low doses into MeCP2 mutant mice (model mice of Rett syndrome, a severe neurodevelopmental disorder). This treatment reversed respiratory and motor control deficits in MeCP2 mice [88].
Beyond small molecules, the development of BDNF mimetic peptides has gained traction in recent years. Peptide-based mimetics are designed to interact directly with the ligand-binding domain of TrkB, effectively replicating BDNF’s action but in a smaller, more manageable form. For example, the peptide GSB-214 was engineered to selectively activate TrkB and promote neurotrophic signaling in animal models [89]. Importantly, GSB-214 has shown promise in preventing memory impairment in AD model rats [90].Peptide-based approaches have certain advantages, including high specificity for the target receptor and minimal off-target effects. However, they are often limited by poor oral bioavailability and susceptibility to enzymatic degradation. Researchers are actively working on overcoming these obstacles by modifying peptide structures, enhancing their stability, and developing novel delivery methods to improve their therapeutic potential.
8.5. Preclinical and Clinical Development
While the majority of research on BDNF mimetics remains in the preclinical stage, there has been significant progress in moving promising candidates toward a clinical trial. For instance, a literature mentioned that R13 had entered a phase 1 clinical trial as a potential treatment for AD, though the result from the trial is still pending [91]. Ongoing preclinical studies continue to evaluate the safety, efficacy, and optimal dosing of second-generation mimetics as well as peptide-based compounds. A key challenge in the clinical development of BDNF mimetics is demonstrating their long-term efficacy and safety in human patients. Neurodegenerative diseases like AD progress slowly, making it essential to assess whether these mimetics can provide sustained neuroprotective effects over extended periods. Moreover, the potential for TrkB overactivation, which could lead to unwanted side effects such as abnormal cell proliferation, must be carefully monitored in clinical studies.
9. The Therapeutic Potential of BDNF Mimetics in Parkinson's Disease (PD) and Huntington’s Disease (HD)
9.1. The Role of BDNF in PD
PD is a progressive neurodegenerative disorder marked by motor symptoms such as bradykinesia, rigidity, and tremors, as well as a range of non-motor symptoms that severely impact quality of life. Central to PD pathology is the loss of dopaminergic neurons within the substantia nigra pars compacta, leading to the depletion of dopamine in the striatum. While genetic and environmental factors contribute to PD etiology, a key area of interest has been the neurotrophic support mechanisms, especially the role of BDNF. BDNF is known for its critical role in neuronal survival, differentiation, and plasticity. However, BDNF levels and signaling are significantly altered in PD, suggesting that impaired BDNF function could exacerbate dopaminergic neuronal degeneration [10].
BDNF supports dopaminergic neuron health through activation of the TrkB receptor, promoting signaling pathways that counteract cellular stress and apoptosis [92]. In PD, both endogenous BDNF expression and TrkB receptor activation disappeared in the substantia nigra, resulting in reducing neuroprotection and allowing oxidative stress and mitochondrial dysfunction to induce neuronal death (Figure 3) [93]. Studies using animal models of PD have shown that BDNF administration can restore dopaminergic neuron populations and improve motor function, underscoring its neuroprotective potential [94,95]. Additionally, BDNF’s role in synaptic plasticity is crucial in maintaining functional neural circuits that are compromised by the progressive neuronal loss in PD [3].
BDNF’s involvement in PD is further complicated by various mechanisms contributing to its dysregulation. Genetic factors, including single nucleotide polymorphism in the BDNF gene, have been linked to reduced BDNF expression in PD patients [96]. Additionally, epigenetic factors, such as DNA methylation and histone modification, can alter BDNF expression [97]. The relationship between alpha-synuclein, a protein implicated in PD pathology, and BDNF signaling has also been of interest. Studies suggest that alpha-synuclein accumulation may impair BDNF trafficking and reduce its availability to dopaminergic neurons [93,98]. Moreover, inflammatory processes common in PD can interfere with BDNF expression and signaling, further hindering its neuroprotective functions [99].
The potential to harness BDNF for therapeutic interventions in PD is promising but presents numerous challenges. Direct BDNF administration has shown positive effects in preclinical models [94,95], yet its therapeutic application in humans is limited by the protein’s poor BBB permeability and rapid degradation. Novel delivery methods, such as nanocarriers, and small molecules that mimic BDNF activity, are under investigation [3]. Small molecules that activate TrkB or modulate endogenous BDNF levels represent another promising area for PD therapeutics, although safety and efficacy in human trials remain to be fully established.
9.2. BDNF Mimetics in PD
One of the most well-studied BDNF mimetics in PD is also 7,8-DHF. Preclinical studies have demonstrated that 7,8-DHF can prevent dopaminergic neuron loss, reduce oxidative stress, and improve motor function in animal models of PD [100-102]. For example, treatment with 7,8-DHF in rodent models of PD resulted in significant preservation of dopaminergic neurons in the substantia nigra and increased levels of striatal dopamine, translating into improved motor performance [103]. The neuroprotective effects of 7,8-DHF are attributed to its ability to activate key survival pathways, including the PI3K/Akt and ERK pathways, which are essential for reducing neuronal apoptosis and promoting synaptic plasticity [3]. Additionally, 7,8-DHF has been found to attenuate neuroinflammation, which is increasingly recognized as a contributing factor in PD pathology. By reducing microglial activation and the release of pro-inflammatory cytokines [104], 7,8-DHF may offer a dual benefit by protecting neurons and reducing neuroinflammatory damage. Other small-molecule TrkB agonists are also being actively investigated due to their potential for oral administration and ability to cross the BBB [105]. CF3CN has shown efficacy in promoting TrkB activation, inhibiting δ-secretase, and increasing TH-positive dopaminergic neurons in MPTP-induced human SNCA transgenic PD mice [106]. These agents are attractive candidates because they can be synthesized with structural modifications that enhance their specificity for TrkB, thereby reducing the risk of side effects associated with non-specific neurotrophin receptor activation.
Beyond small-molecule TrkB agonists, other classes of BDNF mimetics are being explored, including peptidomimetics—short peptides or peptide-like molecules designed to mimic specific regions of the BDNF protein that interact with the TrkB. These peptidomimetics are engineered for enhanced receptor specificity, minimizing off-target effects and reducing the risk of adverse reactions. Indeed, the peptide compound GSB-214 has demonstrated TrkB agonist activity, promoting neuroprotection and ameliorating motor dysfunction in 6-OHDA-induced rat model of PD [107].
9.3. The Role of BDNF in HD
HD is a hereditary neurodegenerative disorder characterized by progressive motor dysfunction, cognitive decline, and psychiatric disturbances. This disease is caused by an expansion of CAG trinucleotide repeats in the huntingtin (HTT) gene, leading to the production of a mutant huntingtin protein (mHTT) that is prone to aggregation. These aggregates are toxic to neurons, particularly in the striatum and cortex, resulting in widespread neuronal dysfunction and cell death. Despite the well-established genetic basis of HD, the precise molecular mechanisms that contribute to neurodegeneration remain incompletely understood. However, evidence increasingly points to deficiencies in neurotrophic support, especially by BDNF, as a crucial factor in HD pathophysiology [11].
The hippocampus, cortex, and striatum—regions critically affected in HD—are particularly reliant on BDNF signaling for their function and structural integrity [11]. However, in HD, there is a marked reduction in BDNF levels, which has been implicated as a significant contributor to disease progression (Figure 3) [108,109]. The reduction in BDNF levels in HD is thought to occur through multiple mechanisms. One of the primary contributors is the dysregulation of transcriptional machinery caused by mHTT. The transcriptional repressor element-1 silencing transcription factor (REST) functions as a corepressor, inhibiting the expression of its target genes, including BDNF. The wild-type Htt protein, located in the cytosol, interacts with REST, thereby preventing its translocation into the nucleus and consequently promoting the expression of its target genes through derepression. In contrast, mHtt exhibits a reduced affinity for REST, which facilitates REST's nuclear localization and results in the suppression of its target genes, such as BDNF [110,111]. As a result, the synthesis of BDNF is significantly impaired. Furthermore, mHTT has been shown to disrupt axonal transport, thereby hindering the delivery of BDNF from the cortical neurons, where it is synthesized, to the striatal neurons, where it is required for cellular health [112]. This impaired transport mechanism exacerbates the striatal atrophy observed in HD patients. Additionally, BDNF signaling is compromised at the receptor level in HD (Figure 3). A previous study has demonstrated that TrkB receptor expression is decreased in the brains of HD patients [113], which further impairs the neurotrophic support needed to counteract neurodegenerative processes. This reduction in both BDNF availability and TrkB receptor activity creates a vicious cycle of neuronal vulnerability, synaptic dysfunction, and cell death. The downstream signaling cascades activated by TrkB, PI3K/Akt and MAPK pathways, are essential for promoting cell survival and neuroprotection. In HD, the downregulation of BDNF/TrkB system disrupts these protective pathways, contributing to the accelerated neurodegeneration observed in affected brain regions.
The interplay between BDNF deficiency and HD pathology is further supported by animal model studies. Importantly, genetic or pharmacological interventions that increase BDNF levels have been shown to alleviate some of the motor and cognitive deficits in these models [114-116], suggesting that enhancing BDNF signaling could be a viable therapeutic strategy. For instance, overexpression of BDNF in HD mouse models has been reported to delay disease onset and improve motor function, providing compelling evidence for the neuroprotective role of BDNF [114].
Collectively, these findings underscore the critical role of BDNF in the pathogenesis of HD. The loss of BDNF and the resulting impairments in TrkB-mediated signaling contribute to neuronal dysfunction, synaptic deficits, and neurodegeneration, which are hallmarks of HD. The therapeutic potential of strategies aimed at restoring BDNF levels or mimicking its activity has garnered significant interest. Given the central role of BDNF in neuronal survival and plasticity, targeted interventions to enhance the BDNF/TrkB system could provide a promising avenue for slowing disease progression and ameliorating symptoms in HD patients.
9.4. BDNF Mimetics in HD
Several classes of BDNF mimetics have been developed, each with distinct mechanisms of action aimed at either directly activating TrkB or enhancing TrkB-mediated signaling indirectly. As expected, 7,8-DHF has shown considerable promise in preclinical studies in HD model mice [117-119]. By binding to TrkB, 7,8-DHF activates PI3K/Akt and MAPK pathways, enhancing neuronal survival and plasticity while also promoting synaptic function in HD [117-119]. Another mimetic, LM22A-4, has also demonstrated efficacy in mitigating HD pathology [120]. In R6/2 HD mice, systemic administration of LM22A-4 led to the activation of TrkB, along with the activation of downstream pathways, including Akt, PLCγ, and CREB. Additionally, it reduced intranuclear HTT aggregates within the striatum and cortex, suppressed microglial activation in the striatum, and protected striatal neurons from degeneration associated with HD [83]. Moreover, LM22A-4 preserved dendritic spine density and enhanced motor function in both R6/2 and BACHD mouse models [83].
In addition to directly targeting TrkB, some small molecules function via modulating endogenous BDNF levels. Compounds that enhance the expression or release of BDNF from neurons can potentially address the underlying deficit observed in HD. HDAC inhibitors, for example, have been shown to increase BDNF levels by promoting chromatin relaxation, thereby facilitating gene transcription [121,122]. The use of HDAC inhibitors like suberoylanilide hydroxamic acid (SAHA) and CKD-504 in HD models has demonstrated neuroprotective effects, partly attributable to increased BDNF expression [123,124]. Furthermore, certain natural compounds have been identified as potential BDNF mimetics due to their ability to enhance TrkB-mediated signaling indirectly. For example, curcumin, an active ingredient in turmeric, has shown neuroprotective properties in R6/2 HD mice, partly by upregulating BDNF expression and activating TrkB signaling pathways [125]. The compound not only possesses neurotrophic activity but also exhibits anti-inflammatory and antioxidant properties, which may further mitigate the neurodegenerative processes in HD [126].
10. Conclusion and Future Directions
The exploration of BDNF mimetics as therapeutic agents has opened new avenues for the treatment of neuropsychiatric and neurodegenerative disorders. The dysregulation of BDNF/TrkB system is a shared pathological hallmark across a wide range of conditions, including depression, schizophrenia, AD, PD, and HD. Consequently, restoring TrkB signaling has emerged as a promising strategy for mitigating the neuropsychiatric or neurodegenerative processes that are characteristic of these disorders.
However, despite encouraging preclinical evidence, there are several challenges to translating the therapeutic potential of BDNF mimetics into effective clinical treatments. One of the most significant barriers is the pharmacokinetic profile of current BDNF mimetics. For example, while 7,8-DHF has shown efficacy in animal models, its bioavailability and stability in humans remain uncertain. Ensuring that these compounds can achieve therapeutic concentrations in the brain without rapid degradation or off-target effects is crucial. Additionally, the ability of BDNF mimetics to effectively penetrate the BBB in human patients remains a significant hurdle. Advances in drug delivery systems, such as nanoparticle carriers, or prodrug formulations, may enhance the bioavailability of these compounds, thereby increasing their therapeutic potential.
Moreover, a deeper understanding of disease-specific alterations in BDNF/TrkB system is essential to optimize the use of BDNF mimetics. The patterns of BDNF dysregulation in AD, PD, and HD are complex and may vary across disease stages and brain regions. Precision medicine approaches that tailor BDNF-based therapies to the specific molecular profiles of individual patients could enhance the efficacy of these treatments. This would involve the integration of biomarkers such as cerebrospinal fluid BDNF levels, neuroimaging of TrkB receptor expression, or genetic profiling to predict patient responses to BDNF mimetics.
It is also crucial to consider the potential benefits of combination therapies. Given the multifactorial nature of neuropsychiatric and neurodegenerative disorders, targeting BDNF/TrkB system alone may not be sufficient to achieve robust clinical benefits. Combining BDNF mimetics with established therapies, such as selective serotonin reuptake inhibitors (SSRIs) for depression or anti-Aβ treatment for AD, could enhance therapeutic outcomes. Synergistic effects might be achieved by simultaneously addressing neurotransmitter imbalances, neuroinflammation, and oxidative stress alongside neurotrophic support. Furthermore, interventions like exercise, cognitive training, or dietary modifications known to naturally upregulate BDNF expression could complement the pharmacological effects of BDNF mimetics, offering a more holistic treatment strategy.
In conclusion, BDNF mimetics represent a promising frontier in the treatment of neuropsychiatric and neurodegenerative disorders, offering the potential to address the neurotrophic deficits that underlie these conditions. While significant challenges remain, advances in drug delivery systems, selective receptor targeting, and personalized medicine approaches could pave the way for the successful translation of BDNF mimetics into clinical practice. Future research must continue to unravel the complexities of TrkB signaling in the brain and develop innovative strategies to harness its therapeutic potential. If these challenges can be overcome, BDNF mimetics may offer not just symptomatic relief but also disease-modifying benefits, ultimately improving the quality of life for patients suffering from these debilitating conditions.
Author Contributions
TN and RK wrote and edited the paper. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the Grant-in-Aid for Scientific Research (C) (JSPS KAKENHI 24K09645) (TN) of the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and from the Takeda Science Foundation (TN). This study was also supported by Kawano Masanori Memorial Public Interest Incorporated Foundation for Promotion of Pediatrics (Grant Number: 35-18) (RK), and the Grant-in-Aid for Scientific Research (JSPS KAKENHI 23K07772) (RK).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or reported in this review article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Alfonsetti, M.; d'Angelo, M.; Castelli, V. Neurotrophic factor-based pharmacological approaches in neurological disorders. Neural Regen Res 2023, 18, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D. Neurotrophic Factors: An Overview. Methods Mol Biol 2018, 1727, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Kajihara, R. Involvement of brain-derived neurotrophic factor signaling in the pathogenesis of stress-related brain diseases. Front Mol Neurosci 2023, 16, 1247422. [Google Scholar] [CrossRef]
- Ali, N.H.; Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Alnaaim, S.A.; Saad, H.M.; Batiha, G.E. The Molecular Pathway of p75 Neurotrophin Receptor (p75NTR) in Parkinson's Disease: The Way of New Inroads. Mol Neurobiol 2024, 61, 2469–2480. [Google Scholar] [CrossRef] [PubMed]
- Teng, H.K.; Teng, K.K.; Lee, R.; Wright, S.; Tevar, S.; Almeida, R.D.; Kermani, P.; Torkin, R.; Chen, Z.Y.; Lee, F.S.; et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci 2005, 25, 5455–5463. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Harte-Hargrove, L.C.; Siao, C.J.; Marinic, T.; Clarke, R.; Ma, Q.; Jing, D.; Lafrancois, J.J.; Bath, K.G.; Mark, W.; et al. proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. Cell Rep 2014, 7, 796–806. [Google Scholar] [CrossRef]
- Meeker, R.B.; Williams, K.S. The p75 neurotrophin receptor: at the crossroad of neural repair and death. Neural Regen Res 2015, 10, 721–725. [Google Scholar] [CrossRef]
- Boxer, A.L.; Sperling, R. Accelerating Alzheimer's therapeutic development: The past and future of clinical trials. Cell 2023, 186, 4757–4772. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Kajihara, R. An Interaction between Brain-Derived Neurotrophic Factor and Stress-Related Glucocorticoids in the Pathophysiology of Alzheimer's Disease. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef]
- Palasz, E.; Wysocka, A.; Gasiorowska, A.; Chalimoniuk, M.; Niewiadomski, W.; Niewiadomska, G. BDNF as a Promising Therapeutic Agent in Parkinson's Disease. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Kim, A.; Lalonde, K.; Truesdell, A.; Gomes Welter, P.; Brocardo, P.S.; Rosenstock, T.R.; Gil-Mohapel, J. New Avenues for the Treatment of Huntington's Disease. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem 2003, 72, 609–642. [Google Scholar] [CrossRef]
- Minichiello, L. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci 2009, 10, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Moya-Alvarado, G.; Gonzalez-Billaut, C.; Bronfman, F.C. Cellular and molecular mechanisms regulating neuronal growth by brain-derived neurotrophic factor. Cytoskeleton (Hoboken) 2016, 73, 612–628. [Google Scholar] [CrossRef]
- Numakawa, T.; Kumamaru, E.; Adachi, N.; Yagasaki, Y.; Izumi, A.; Kunugi, H. Glucocorticoid receptor interaction with TrkB promotes BDNF-triggered PLC-gamma signaling for glutamate release via a glutamate transporter. Proc Natl Acad Sci U S A 2009, 106, 647–652. [Google Scholar] [CrossRef]
- Numakawa, T.; Matsumoto, T.; Ooshima, Y.; Chiba, S.; Furuta, M.; Izumi, A.; Ninomiya-Baba, M.; Odaka, H.; Hashido, K.; Adachi, N.; et al. Impairments in brain-derived neurotrophic factor-induced glutamate release in cultured cortical neurons derived from rats with intrauterine growth retardation: possible involvement of suppression of TrkB/phospholipase C-γ activation. Neurochem Res 2014, 39, 785–792. [Google Scholar] [CrossRef]
- Alcántara, S.; Frisén, J.; del Río, J.A.; Soriano, E.; Barbacid, M.; Silos-Santiago, I. TrkB signaling is required for postnatal survival of CNS neurons and protects hippocampal and motor neurons from axotomy-induced cell death. J Neurosci 1997, 17, 3623–3633. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Compromised MAPK signaling in human diseases: an update. Arch Toxicol 2015, 89, 867–882. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Lahair, M.M.; Franklin, R.A. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid Redox Signal 2006, 8, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Suzuki, S.; Kumamaru, E.; Adachi, N.; Richards, M.; Kunugi, H. BDNF function and intracellular signaling in neurons. Histol Histopathol 2010, 25, 237–258. [Google Scholar] [CrossRef] [PubMed]
- Kumamaru, E.; Numakawa, T.; Adachi, N.; Kunugi, H. Glucocorticoid suppresses BDNF-stimulated MAPK/ERK pathway via inhibiting interaction of Shp2 with TrkB. FEBS Lett 2011, 585, 3224–3228. [Google Scholar] [CrossRef] [PubMed]
- Puranik, N.; Jung, H.; Song, M. SPROUTY2, a Negative Feedback Regulator of Receptor Tyrosine Kinase Signaling, Associated with Neurodevelopmental Disorders: Current Knowledge and Future Perspectives. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef] [PubMed]
- Panwar, V.; Singh, A.; Bhatt, M.; Tonk, R.K.; Azizov, S.; Raza, A.S.; Sengupta, S.; Kumar, D.; Garg, M. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct Target Ther 2023, 8, 375. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Datta, S.R.; Greenberg, M.E. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol 2001, 11, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Nito, C.; Kamada, H.; Nishi, T.; Chan, P.H. Activation of the Akt/GSK3beta signaling pathway mediates survival of vulnerable hippocampal neurons after transient global cerebral ischemia in rats. J Cereb Blood Flow Metab 2006, 26, 1479–1489. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Zarneshan, S.N.; Fakhri, S.; Khan, H. Targeting Akt/CREB/BDNF signaling pathway by ginsenosides in neurodegenerative diseases: A mechanistic approach. Pharmacol Res 2022, 177, 106099. [Google Scholar] [CrossRef]
- Kumari, S.; Dhapola, R.; Reddy, D.H. Apoptosis in Alzheimer's disease: insight into the signaling pathways and therapeutic avenues. Apoptosis 2023, 28, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Autry, A.E. Function of brain-derived neurotrophic factor in the hypothalamus: Implications for depression pathology. Front Mol Neurosci 2022, 15, 1028223. [Google Scholar] [CrossRef]
- Fan, Y.; Luan, X.; Wang, X.; Li, H.; Zhao, H.; Li, S.; Li, X.; Qiu, Z. Exploring the Association between BDNF related Signaling Pathways and Depression: A Literature Review. Brain Res Bull 2024. [Google Scholar] [CrossRef] [PubMed]
- Airaksinen, E.; Larsson, M.; Lundberg, I.; Forsell, Y. Cognitive functions in depressive disorders: evidence from a population-based study. Psychol Med 2004, 34, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Richards, M.; Nakajima, S.; Adachi, N.; Furuta, M.; Odaka, H.; Kunugi, H. The role of brain-derived neurotrophic factor in comorbid depression: possible linkage with steroid hormones, cytokines, and nutrition. Front Psychiatry 2014, 5, 136. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Huang, T.; Zhang, M.; Zhang, Y.; Zeng, Y.; Chen, X. Glucocorticoid receptor signaling in the brain and its involvement in cognitive function. Neural Regen Res 2025, 20, 2520–2537. [Google Scholar] [CrossRef] [PubMed]
- Ran, S.; Peng, R.; Guo, Q.; Cui, J.; Chen, G.; Wang, Z. Bupleurum in Treatment of Depression Disorder: A Comprehensive Review. Pharmaceuticals (Basel) 2024, 17. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Mo, X.; He, L.; Ma, Q.; Cai, L.; Zheng, Y.; Huang, L.; Lin, X.; Wu, M.; Ding, W.; et al. The role of BDNF transcription in the antidepressant-like effects of 18β-glycyrrhetinic acid in a chronic social defeat stress model. Phytomedicine 2024, 132, 155332. [Google Scholar] [CrossRef]
- Langlois, C.; Potvin, S.; Khullar, A.; Tourjman, S.V. Down and High: Reflections Regarding Depression and Cannabis. Front Psychiatry 2021, 12, 625158. [Google Scholar] [CrossRef]
- Anand, R.; Anand, L.K.; Rashid, N.; Painuli, R.; Malik, F.; Singh, P.P. Synthesis and Evaluation of Natural and Unnatural Tetrahydrocannabiorcol for Its Potential Use in Neuropathologies. J Nat Prod 2024, 87, 167–175. [Google Scholar] [CrossRef]
- Singh, A.A.; Yadav, D.; Khan, F.; Song, M. Indole-3-Carbinol and Its Derivatives as Neuroprotective Modulators. Brain Sci 2024, 14. [Google Scholar] [CrossRef]
- Chen, Y.; Gu, H.; Ye, D.; Li, Y.; Chen, Y.; Qiao, H.; Huang, Y.; Tao, R.; Yu, S.; Zhang, J.; et al. NMC-4 Ameliorates Depression-Like Behavior and Neuroinflammation Caused by Chronic Unpredictable Mild Stress. Chem Biol Drug Des 2024, 104, e14626. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Ganesan, K.; Wang, Y.; Zhang, Z.; Liu, Y.; Wang, J.; Yang, F.; Zheng, Y. Ononin ameliorates depression-like behaviors by regulating BDNF-TrkB-CREB signaling in vitro and in vivo. J Ethnopharmacol 2024, 320, 117375. [Google Scholar] [CrossRef]
- Zeng, J.; Xie, Z.; Chen, L.; Peng, X.; Luan, F.; Hu, J.; Xie, H.; Liu, R.; Zeng, N. Rosmarinic acid alleviate CORT-induced depressive-like behavior by promoting neurogenesis and regulating BDNF/TrkB/PI3K signaling axis. Biomed Pharmacother 2024, 170, 115994. [Google Scholar] [CrossRef]
- Wakasugi, D.; Kondo, S.; Ferdousi, F.; Mizuno, S.; Yada, A.; Tominaga, K.; Takahashi, S.; Isoda, H. A rare olive compound oleacein functions as a TrkB agonist and mitigates neuroinflammation both in vitro and in vivo. Cell Commun Signal 2024, 22, 309. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, Y.; Kubo, M.; Harada, K. The search for, and chemistry and mechanism of, neurotrophic natural products. J Nat Med 2020, 74, 648–671. [Google Scholar] [CrossRef] [PubMed]
- Garibova, T.L.; Kraineva, V.A.; Kotel'nikova, S.O.; Povarnina, P.Y.; Gudasheva, T.A.; Seredenin, S.B. Behavioral Effects of Dimeric Dipeptide BDNF Mimetic GSB-106 in a Rat Model of Depressive-Like State. Bull Exp Biol Med 2020, 169, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Gudasheva, T.A.; Tallerova, A.V.; Mezhlumyan, A.G.; Antipova, T.A.; Logvinov, I.O.; Firsova, Y.N.; Povarnina, P.Y.; Seredenin, S.B. Low-Molecular Weight BDNF Mimetic, Dimeric Dipeptide GSB-106, Reverses Depressive Symptoms in Mouse Chronic Social Defeat Stress. Biomolecules 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Povarnina, P.Y.; Antipova, T.A.; Logvinov, I.O.; Gudasheva, T.A.; Seredenin, S.B. Сhronically Administered BDNF Dipeptide Mimetic GSB-106 Prevents the Depressive-like Behavior and Memory Impairments after Transient Middle Cerebral Artery Occlusion in Rats. Curr Pharm Des 2023, 29, 126–132. [Google Scholar] [CrossRef]
- Tiliwaerde, M.; Gao, N.; Yang, Y.; Jin, Z. A novel NMDA receptor modulator: the antidepressant effect and mechanism of GW043. CNS Neurosci Ther 2024, 30, e14598. [Google Scholar] [CrossRef]
- Meng, Y.; Xiao, L.; Liu, R.; Du, J.; Liu, N.; Yu, J.; Li, Y.; Lu, G. Antidepressant effect and mechanism of TMP269 on stress-induced depressive-like behavior in mice. Biochem Pharmacol 2024, 225, 116320. [Google Scholar] [CrossRef]
- Yuan, Z.Y.; Zhang, X.; Yu, Z.Z.; Wang, X.Y.; Zeng, Z.H.; Wei, M.X.; Qiu, M.T.; Wang, J.; Cheng, J.; Yi, L.T. Polygonatum sibiricum Polysaccharides Alleviate Depressive-like Symptoms in Chronic Restraint Stress-Induced Mice via Microglial Regulation in Prefrontal Cortex. Polymers (Basel) 2024, 16. [Google Scholar] [CrossRef]
- Valenza, M.; Facchinetti, R.; Torazza, C.; Ciarla, C.; Bronzuoli, M.R.; Balbi, M.; Bonanno, G.; Popoli, M.; Steardo, L.; Milanese, M.; et al. Molecular signatures of astrocytes and microglia maladaptive responses to acute stress are rescued by a single administration of ketamine in a rodent model of PTSD. Transl Psychiatry 2024, 14, 209. [Google Scholar] [CrossRef]
- Li, K.; Wang, K.; Xu, S.X.; Xie, X.H.; Tang, Y.; Zhang, L.; Liu, Z. Investigating Neuroplasticity Changes Reflected by BDNF Levels in Astrocyte-Derived Extracellular Vesicles in Patients with Depression. Int J Nanomedicine 2024, 19, 8971–8985. [Google Scholar] [CrossRef]
- Zou, Y.; Zhang, Y.; Tu, M.; Ye, Y.; Li, M.; Ran, R.; Zou, Z. Brain-derived neurotrophic factor levels across psychiatric disorders: A systemic review and network meta-analysis. Prog Neuropsychopharmacol Biol Psychiatry 2024, 131, 110954. [Google Scholar] [CrossRef]
- Farcas, A.; Hindmarch, C.; Iftene, F. BDNF gene Val66Met polymorphisms as a predictor for clinical presentation in schizophrenia - recent findings. Front Psychiatry 2023, 14, 1234220. [Google Scholar] [CrossRef] [PubMed]
- Liberona, A.; Jones, N.; Zúñiga, K.; Garrido, V.; Zelada, M.I.; Silva, H.; Nieto, R.R. Brain-Derived Neurotrophic Factor (BDNF) as a Predictor of Treatment Response in Schizophrenia and Bipolar Disorder: A Systematic Review. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, S.; Zhang, T.; Yang, F.; Lu, B. Corticosterone antagonist or TrkB agonist attenuates schizophrenia-like behavior in a mouse model combining Bdnf-e6 deficiency and developmental stress. iScience 2022, 25, 104609. [Google Scholar] [CrossRef] [PubMed]
- Shkundin, A.; Halaris, A. Associations of BDNF/BDNF-AS SNPs with Depression, Schizophrenia, and Bipolar Disorder. J Pers Med 2023, 13. [Google Scholar] [CrossRef]
- Gredicak, M.; Nikolac Perkovic, M.; Nedic Erjavec, G.; Uzun, S.; Kozumplik, O.; Svob Strac, D.; Pivac, N. Association between reduced plasma BDNF concentration and MMSE scores in both chronic schizophrenia and mild cognitive impairment. Prog Neuropsychopharmacol Biol Psychiatry 2024, 134, 111086. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Zhang, J.C.; Yao, W.; Yang, C.; Ishima, T.; Ren, Q.; Ma, M.; Dong, C.; Huang, X.F.; Hashimoto, K. Intake of 7,8-Dihydroxyflavone During Juvenile and Adolescent Stages Prevents Onset of Psychosis in Adult Offspring After Maternal Immune Activation. Sci Rep 2016, 6, 36087. [Google Scholar] [CrossRef]
- Gillespie, B.; Dunn, A.; Sundram, S.; Hill, R.A. Investigating 7,8-Dihydroxyflavone to combat maternal immune activation effects on offspring gene expression and behaviour. Prog Neuropsychopharmacol Biol Psychiatry 2024, 134, 111078. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, B.M.M.; Chaves Filho, A.J.M.; Costa, D.; de Menezes, A.T.; da Fonseca, A.C.C.; Gama, C.S.; Moura Neto, V.; de Lucena, D.F.; Vale, M.L.; Macêdo, D.S. N-3 polyunsaturated fatty acids and clozapine abrogates poly I: C-induced immune alterations in primary hippocampal neurons. Prog Neuropsychopharmacol Biol Psychiatry 2019, 90, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.J.; Li, Y.K.; Wang, W.; Wan, J.G.; Yu, B.; Wang, M.Z.; Hu, B. Small-molecule TrkB agonist 7,8-dihydroxyflavone reverses cognitive and synaptic plasticity deficits in a rat model of schizophrenia. Pharmacol Biochem Behav 2014, 122, 30–36. [Google Scholar] [CrossRef]
- Ben-Azu, B.; Fokoua, A.R.; Annafi, O.S.; Adebayo, O.G.; Del Re, E.C.; Okuchukwu, N.; Aregbesola, G.J.; Ejenavi, A.C.; Isiwele, D.M.; Efezino, A.J.; et al. Effective action of silymarin against ketamine-induced schizophrenia in male mice: Insight into the biochemical and molecular mechanisms of action. J Psychiatr Res 2024, 179, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Khalid, I.; Saleem, U.; Ahmad, B.; Hawwal, M.F.; Mothana, R.A. NMDA receptor modulation by Esculetin: Investigating behavioral, biochemical and neurochemical effects in schizophrenic mice model. Saudi Pharm J 2024, 32, 101994. [Google Scholar] [CrossRef]
- Mandala, S.; Hajdu, R.; Bergstrom, J.; Quackenbush, E.; Xie, J.; Milligan, J.; Thornton, R.; Shei, G.J.; Card, D.; Keohane, C.; et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 2002, 296, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.A.; Chun, J. Mechanisms of fingolimod's efficacy and adverse effects in multiple sclerosis. Ann Neurol 2011, 69, 759–777. [Google Scholar] [CrossRef] [PubMed]
- Shirani, A.; Okuda, D.T.; Stüve, O. Therapeutic Advances and Future Prospects in Progressive Forms of Multiple Sclerosis. Neurotherapeutics 2016, 13, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhuo, C.; Ma, X.; Li, R.; Chen, X.; Li, Y.; Zhang, Q.; Yang, L.; Wang, L. Exploring the molecular targets of fingolimod and siponimod for treating the impaired cognition of schizophrenia using network pharmacology and molecular docking. Schizophrenia (Heidelb) 2024, 10, 80. [Google Scholar] [CrossRef]
- Sykes, L.; Haddon, J.; Lancaster, T.M.; Sykes, A.; Azzouni, K.; Ihssen, N.; Moon, A.L.; Lin, T.E.; Linden, D.E.; Owen, M.J.; et al. Genetic Variation in the Psychiatric Risk Gene CACNA1C Modulates Reversal Learning Across Species. Schizophr Bull 2019, 45, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Tigaret, C.M.; Lin, T.E.; Morrell, E.R.; Sykes, L.; Moon, A.L.; O'Donovan, M.C.; Owen, M.J.; Wilkinson, L.S.; Jones, M.W.; Thomas, K.L.; et al. Neurotrophin receptor activation rescues cognitive and synaptic abnormalities caused by hemizygosity of the psychiatric risk gene Cacna1c. Mol Psychiatry 2021, 26, 1748–1760. [Google Scholar] [CrossRef]
- Qi, X.; Yu, X.; Wei, L.; Jiang, H.; Dong, J.; Li, H.; Wei, Y.; Zhao, L.; Deng, W.; Guo, W.; et al. Novel α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor (AMPAR) potentiator LT-102: A promising therapeutic agent for treating cognitive impairment associated with schizophrenia. CNS Neurosci Ther 2024, 30, e14713. [Google Scholar] [CrossRef]
- Amidfar, M.; de Oliveira, J.; Kucharska, E.; Budni, J.; Kim, Y.K. The role of CREB and BDNF in neurobiology and treatment of Alzheimer's disease. Life Sci 2020, 257, 118020. [Google Scholar] [CrossRef]
- Du, Y.; Wu, H.T.; Qin, X.Y.; Cao, C.; Liu, Y.; Cao, Z.Z.; Cheng, Y. Postmortem Brain, Cerebrospinal Fluid, and Blood Neurotrophic Factor Levels in Alzheimer's Disease: A Systematic Review and Meta-Analysis. J Mol Neurosci 2018, 65, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wei, X.X.; Chang, L.S.; Dong, L.; Wang, Y.L.; Li, N.N. Ultrasound Combined With Microbubbles Loading BDNF Retrovirus to Open BloodBrain Barrier for Treatment of Alzheimer's Disease. Front Pharmacol 2021, 12, 615104. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, A.H.; Merrill, D.A.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med 2009, 15, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, A.H.; Mateling, M.; Kovacs, I.; Wang, L.; Eggert, S.; Rockenstein, E.; Koo, E.H.; Masliah, E.; Tuszynski, M.H. Early BDNF treatment ameliorates cell loss in the entorhinal cortex of APP transgenic mice. J Neurosci 2013, 33, 15596–15602. [Google Scholar] [CrossRef] [PubMed]
- Braschi, C.; Capsoni, S.; Narducci, R.; Poli, A.; Sansevero, G.; Brandi, R.; Maffei, L.; Cattaneo, A.; Berardi, N. Intranasal delivery of BDNF rescues memory deficits in AD11 mice and reduces brain microgliosis. Aging Clin Exp Res 2021, 33, 1223–1238. [Google Scholar] [CrossRef] [PubMed]
- Thorne, R.G.; Frey, W.H. , 2nd. Delivery of neurotrophic factors to the central nervous system: pharmacokinetic considerations. Clin Pharmacokinet 2001, 40, 907–946. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhu, G. 7,8-Dihydroxyflavone and Neuropsychiatric Disorders: A Translational Perspective from the Mechanism to Drug Development. Curr Neuropharmacol 2022, 20, 1479–1497. [Google Scholar] [CrossRef]
- Jang, S.W.; Liu, X.; Yepes, M.; Shepherd, K.R.; Miller, G.W.; Liu, Y.; Wilson, W.D.; Xiao, G.; Blanchi, B.; Sun, Y.E.; et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci U S A 2010, 107, 2687–2692. [Google Scholar] [CrossRef]
- Akhtar, A.; Dhaliwal, J.; Sah, S.P. 7,8-Dihydroxyflavone improves cognitive functions in ICV-STZ rat model of sporadic Alzheimer's disease by reversing oxidative stress, mitochondrial dysfunction, and insulin resistance. Psychopharmacology (Berl) 2021, 238, 1991–2009. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Jeong, Y.J.; Kang, E.J.; Kang, B.S.; Lee, S.H.; Kim, Y.J.; Kang, S.S.; Suh, S.W.; Ahn, E.H. GAP-43 closely interacts with BDNF in hippocampal neurons and is associated with Alzheimer's disease progression. Front Mol Neurosci 2023, 16, 1150399. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, X.; Schroeder, J.P.; Chan, C.B.; Song, M.; Yu, S.P.; Weinshenker, D.; Ye, K. 7,8-dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology 2014, 39, 638–650. [Google Scholar] [CrossRef]
- Simmons, D.A.; Belichenko, N.P.; Yang, T.; Condon, C.; Monbureau, M.; Shamloo, M.; Jing, D.; Massa, S.M.; Longo, F.M. A small molecule TrkB ligand reduces motor impairment and neuropathology in R6/2 and BACHD mouse models of Huntington's disease. J Neurosci 2013, 33, 18712–18727. [Google Scholar] [CrossRef]
- Korkmaz, O.T. Can Brain-derived Neurotrophic Factor (BDNF) Mimetics be a Way Out for Neurodegenerative Diseases? Curr Pharm Des 2023, 29, 246–250. [Google Scholar] [CrossRef]
- Massa, S.M.; Yang, T.; Xie, Y.; Shi, J.; Bilgen, M.; Joyce, J.N.; Nehama, D.; Rajadas, J.; Longo, F.M. Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J Clin Invest 2010, 120, 1774–1785. [Google Scholar] [CrossRef]
- Chen, C.; Wang, Z.; Zhang, Z.; Liu, X.; Kang, S.S.; Zhang, Y.; Ye, K. The prodrug of 7,8-dihydroxyflavone development and therapeutic efficacy for treating Alzheimer's disease. Proc Natl Acad Sci U S A 2018, 115, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ahn, E.H.; Liu, X.; Wang, Z.H.; Luo, S.; Liao, J.; Ye, K. Optimized TrkB Agonist Ameliorates Alzheimer's Disease Pathologies and Improves Cognitive Functions via Inhibiting Delta-Secretase. ACS Chem Neurosci 2021, 12, 2448–2461. [Google Scholar] [CrossRef]
- Adams, I.; Yang, T.; Longo, F.M.; Katz, D.M. Restoration of motor learning in a mouse model of Rett syndrome following long-term treatment with a novel small-molecule activator of TrkB. Dis Model Mech 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Gascon, S.; Jann, J.; Langlois-Blais, C.; Plourde, M.; Lavoie, C.; Faucheux, N. Peptides Derived from Growth Factors to Treat Alzheimer's Disease. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Povarnina, P.Y.; Volkova, A.A.; Vorontsova, O.N.; Kamensky, A.A.; Gudasheva, T.A.; Seredenin, S.B. A Low-Molecular-Weight BDNF Mimetic, Dipeptide GSB-214, Prevents Memory Impairment in Rat Models of Alzheimer's Disease. Acta Naturae 2022, 14, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ahn, E.H.; Kang, S.S.; Liu, X.; Alam, A.; Ye, K. Gut dysbiosis contributes to amyloid pathology, associated with C/EBPβ/AEP signaling activation in Alzheimer's disease mouse model. Sci Adv 2020, 6, eaba0466. [Google Scholar] [CrossRef] [PubMed]
- Hyman, C.; Hofer, M.; Barde, Y.A.; Juhasz, M.; Yancopoulos, G.D.; Squinto, S.P.; Lindsay, R.M. BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature 1991, 350, 230–232. [Google Scholar] [CrossRef]
- Jin, W. Regulation of BDNF-TrkB Signaling and Potential Therapeutic Strategies for Parkinson's Disease. J Clin Med 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.H.; Leem, E.; Jeon, M.T.; Jeong, K.H.; Park, J.W.; Jung, U.J.; Kholodilov, N.; Burke, R.E.; Jin, B.K.; Kim, S.R. Induction of GDNF and BDNF by hRheb(S16H) transduction of SNpc neurons: neuroprotective mechanisms of hRheb(S16H) in a model of Parkinson's disease. Mol Neurobiol 2015, 51, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Guillin, O.; Diaz, J.; Carroll, P.; Griffon, N.; Schwartz, J.C.; Sokoloff, P. BDNF controls dopamine D3 receptor expression and triggers behavioural sensitization. Nature 2001, 411, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Przybylska, I.; Marusiak, J.; Toczyłowska, B.; Stępień, A.; Brodacki, B.; Langfort, J.; Chalimoniuk, M. Association between the Val66Met (rs6265) polymorphism of the brain-derived neurotrophic factor (BDNF) gene, BDNF protein level in the blood and the risk of developing early-onset Parkinson's disease. Acta Neurobiol Exp (Wars) 2024, 84, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Wang, Y.; Sun, Z. The Relationships Between Stress, Mental Disorders, and Epigenetic Regulation of BDNF. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Gamble, K.L.; Schultheiss, C.E.; Riddle, D.M.; West, A.B.; Lee, V.M. Formation of α-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol Biol Cell 2014, 25, 4010–4023. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.C.; Yao, W.; Hashimoto, K. Brain-derived Neurotrophic Factor (BDNF)-TrkB Signaling in Inflammation-related Depression and Potential Therapeutic Targets. Curr Neuropharmacol 2016, 14, 721–731. [Google Scholar] [CrossRef]
- Zuo, L.; Dai, C.; Yi, L.; Dong, Z. 7,8-dihydroxyflavone ameliorates motor deficits via regulating autophagy in MPTP-induced mouse model of Parkinson's disease. Cell Death Discov 2021, 7, 254. [Google Scholar] [CrossRef] [PubMed]
- Massaquoi, M.S.; Liguore, W.A.; Churchill, M.J.; Moore, C.; Melrose, H.L.; Meshul, C.K. Gait Deficits and Loss of Striatal Tyrosine Hydroxlase/Trk-B are Restored Following 7,8-Dihydroxyflavone Treatment in a Progressive MPTP Mouse Model of Parkinson's Disease. Neuroscience 2020, 433, 53–71. [Google Scholar] [CrossRef]
- Mohankumar, T.; Chandramohan, V.; Lalithamba, H.S.; Jayaraj, R.L.; Kumaradhas, P.; Sivanandam, M.; Hunday, G.; Vijayakumar, R.; Balakrishnan, R.; Manimaran, D.; et al. Design and Molecular dynamic Investigations of 7,8-Dihydroxyflavone Derivatives as Potential Neuroprotective Agents Against Alpha-synuclein. Sci Rep 2020, 10, 599. [Google Scholar] [CrossRef]
- Li, X.H.; Dai, C.F.; Chen, L.; Zhou, W.T.; Han, H.L.; Dong, Z.F. 7,8-dihydroxyflavone Ameliorates Motor Deficits Via Suppressing α-synuclein Expression and Oxidative Stress in the MPTP-induced Mouse Model of Parkinson's Disease. CNS Neurosci Ther 2016, 22, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Park, C.; Hwang, H.J.; Kim, B.W.; Kim, G.Y.; Kim, C.M.; Kim, N.D.; Choi, Y.H. 7,8-Dihydroxyflavone attenuates the release of pro-inflammatory mediators and cytokines in lipopolysaccharide-stimulated BV2 microglial cells through the suppression of the NF-κB and MAPK signaling pathways. Int J Mol Med 2014, 33, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.H.; Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Alexiou, A.; Papadakis, M.; AlAseeri, A.A.; Alruwaili, M.; Saad, H.M.; Batiha, G.E. BDNF/TrkB activators in Parkinson's disease: A new therapeutic strategy. J Cell Mol Med 2024, 28, e18368. [Google Scholar] [CrossRef]
- Kang, S.S.; Wu, Z.; Liu, X.; Edgington-Mitchell, L.; Ye, K. Treating Parkinson's Disease via Activation of BDNF/TrkB Signaling Pathways and Inhibition of Delta-Secretase. Neurotherapeutics 2022, 19, 1283–1297. [Google Scholar] [CrossRef]
- Firouzan, B.; Iravanpour, F.; Abbaszadeh, F.; Akparov, V.; Zaringhalam, J.; Ghasemi, R.; Maghsoudi, N. Dipeptide mimetic of BDNF ameliorates motor dysfunction and striatal apoptosis in 6-OHDA-induced Parkinson's rat model: Considering Akt and MAPKs signaling. Behav Brain Res 2023, 452, 114585. [Google Scholar] [CrossRef]
- Ciammola, A.; Sassone, J.; Cannella, M.; Calza, S.; Poletti, B.; Frati, L.; Squitieri, F.; Silani, V. Low brain-derived neurotrophic factor (BDNF) levels in serum of Huntington's disease patients. Am J Med Genet B Neuropsychiatr Genet 2007, 144b, 574–577. [Google Scholar] [CrossRef] [PubMed]
- Krzysztoń-Russjan, J.; Zielonka, D.; Jackiewicz, J.; Kuśmirek, S.; Bubko, I.; Klimberg, A.; Marcinkowski, J.T.; Anuszewska, E.L. A study of molecular changes relating to energy metabolism and cellular stress in people with Huntington's disease: looking for biomarkers. J Bioenerg Biomembr 2013, 45, 71–85. [Google Scholar] [CrossRef]
- Bithell, A.; Johnson, R.; Buckley, N.J. Transcriptional dysregulation of coding and non-coding genes in cellular models of Huntington's disease. Biochem Soc Trans 2009, 37, 1270–1275. [Google Scholar] [CrossRef]
- Kumar, A.; Vaish, M.; Ratan, R.R. Transcriptional dysregulation in Huntington's disease: a failure of adaptive transcriptional homeostasis. Drug Discov Today 2014, 19, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, E.; Zuccato, C.; Tartari, M. Normal huntingtin function: an alternative approach to Huntington's disease. Nat Rev Neurosci 2005, 6, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Ginés, S.; Bosch, M.; Marco, S.; Gavaldà, N.; Díaz-Hernández, M.; Lucas, J.J.; Canals, J.M.; Alberch, J. Reduced expression of the TrkB receptor in Huntington's disease mouse models and in human brain. Eur J Neurosci 2006, 23, 649–658. [Google Scholar] [CrossRef]
- Cho, S.R.; Benraiss, A.; Chmielnicki, E.; Samdani, A.; Economides, A.; Goldman, S.A. Induction of neostriatal neurogenesis slows disease progression in a transgenic murine model of Huntington disease. J Clin Invest 2007, 117, 2889–2902. [Google Scholar] [CrossRef]
- Lenoir, S.; Lahaye, R.A.; Vitet, H.; Scaramuzzino, C.; Virlogeux, A.; Capellano, L.; Genoux, A.; Gershoni-Emek, N.; Geva, M.; Hayden, M.R.; et al. Pridopidine rescues BDNF/TrkB trafficking dynamics and synapse homeostasis in a Huntington disease brain-on-a-chip model. Neurobiol Dis 2022, 173, 105857. [Google Scholar] [CrossRef] [PubMed]
- Speidell, A.; Bin Abid, N.; Yano, H. Brain-Derived Neurotrophic Factor Dysregulation as an Essential Pathological Feature in Huntington's Disease: Mechanisms and Potential Therapeutics. Biomedicines 2023, 11. [Google Scholar] [CrossRef]
- Ahmed, S.; Kwatra, M.; Gawali, B.; Panda, S.R.; Naidu, V.G.M. Potential role of TrkB agonist in neuronal survival by promoting CREB/BDNF and PI3K/Akt signaling in vitro and in vivo model of 3-nitropropionic acid (3-NP)-induced neuronal death. Apoptosis 2021, 26, 52–70. [Google Scholar] [CrossRef]
- García-Díaz Barriga, G.; Giralt, A.; Anglada-Huguet, M.; Gaja-Capdevila, N.; Orlandi, J.G.; Soriano, J.; Canals, J.M.; Alberch, J. 7,8-dihydroxyflavone ameliorates cognitive and motor deficits in a Huntington's disease mouse model through specific activation of the PLCγ1 pathway. Hum Mol Genet 2017, 26, 3144–3160. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Peng, Q.; Liu, X.; Jin, J.; Hou, Z.; Zhang, J.; Mori, S.; Ross, C.A.; Ye, K.; Duan, W. Small-molecule TrkB receptor agonists improve motor function and extend survival in a mouse model of Huntington's disease. Hum Mol Genet 2013, 22, 2462–2470. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.A. Modulating Neurotrophin Receptor Signaling as a Therapeutic Strategy for Huntington's Disease. J Huntingtons Dis 2017, 6, 303–325. [Google Scholar] [CrossRef]
- Sada, N.; Fujita, Y.; Mizuta, N.; Ueno, M.; Furukawa, T.; Yamashita, T. Inhibition of HDAC increases BDNF expression and promotes neuronal rewiring and functional recovery after brain injury. Cell Death Dis 2020, 11, 655. [Google Scholar] [CrossRef] [PubMed]
- Sartor, G.C.; Malvezzi, A.M.; Kumar, A.; Andrade, N.S.; Wiedner, H.J.; Vilca, S.J.; Janczura, K.J.; Bagheri, A.; Al-Ali, H.; Powell, S.K.; et al. Enhancement of BDNF Expression and Memory by HDAC Inhibition Requires BET Bromodomain Reader Proteins. J Neurosci 2019, 39, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Mielcarek, M.; Benn, C.L.; Franklin, S.A.; Smith, D.L.; Woodman, B.; Marks, P.A.; Bates, G.P. SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington's disease. PLoS One 2011, 6, e27746. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Choi, J.; Sim, H.R.; Kim, J.; Jun, J.H.; Kyung, J.; Ha, N.; Kim, S.; Ryu, K.H.; Chung, S.S.; et al. A novel HDAC6 inhibitor, CKD-504, is effective in treating preclinical models of huntington's disease. BMB Rep 2023, 56, 178–183. [Google Scholar] [CrossRef]
- Elifani, F.; Amico, E.; Pepe, G.; Capocci, L.; Castaldo, S.; Rosa, P.; Montano, E.; Pollice, A.; Madonna, M.; Filosa, S.; et al. Curcumin dietary supplementation ameliorates disease phenotype in an animal model of Huntington's disease. Hum Mol Genet 2019, 28, 4012–4021. [Google Scholar] [CrossRef]
- Kaur, K.; Al-Khazaleh, A.K.; Bhuyan, D.J.; Li, F.; Li, C.G. A Review of Recent Curcumin Analogues and Their Antioxidant, Anti-Inflammatory, and Anticancer Activities. Antioxidants (Basel) 2024, 13. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Neurotrophins and their receptors in the CNS neurons. Brain-derived neurotrophic factor (BDNF), nerve growth factor (NGF, the firstly identified one), neurotrophin-3 (NT-3), and neurotrophin-4 (NT-4) are produced firstly as precursor pro-forms (proneurotrophins), respectively. Proneurotrophins bind to p75NTR (a common receptors for all neurotrophin) with a high affinity. Sortilin, a co-receptor, is also critical for the binding with pro-forms, resulting in neuronal cell death. After receiving proteolytic cleavage, mature BDNF produced from pro-forms, binds to TrkB, a high affinity receptor for mature BDNF and NT-4, and triggers downstream intracellular signaling including MAPK-, PLCγ-, and PI3K/Akt-pathways, leading to cell differentiation and survival, in addition to neurite outgrowth. It is well known that the BDNF/TrkB system, which has multiple roles in neural cell aspects, is closely associated with synaptic function, and learning and memory function of brain. NGF binds to TrkA with a high affinity, similarly, NT-3 to TrkC.
Figure 1.
Neurotrophins and their receptors in the CNS neurons. Brain-derived neurotrophic factor (BDNF), nerve growth factor (NGF, the firstly identified one), neurotrophin-3 (NT-3), and neurotrophin-4 (NT-4) are produced firstly as precursor pro-forms (proneurotrophins), respectively. Proneurotrophins bind to p75NTR (a common receptors for all neurotrophin) with a high affinity. Sortilin, a co-receptor, is also critical for the binding with pro-forms, resulting in neuronal cell death. After receiving proteolytic cleavage, mature BDNF produced from pro-forms, binds to TrkB, a high affinity receptor for mature BDNF and NT-4, and triggers downstream intracellular signaling including MAPK-, PLCγ-, and PI3K/Akt-pathways, leading to cell differentiation and survival, in addition to neurite outgrowth. It is well known that the BDNF/TrkB system, which has multiple roles in neural cell aspects, is closely associated with synaptic function, and learning and memory function of brain. NGF binds to TrkA with a high affinity, similarly, NT-3 to TrkC.
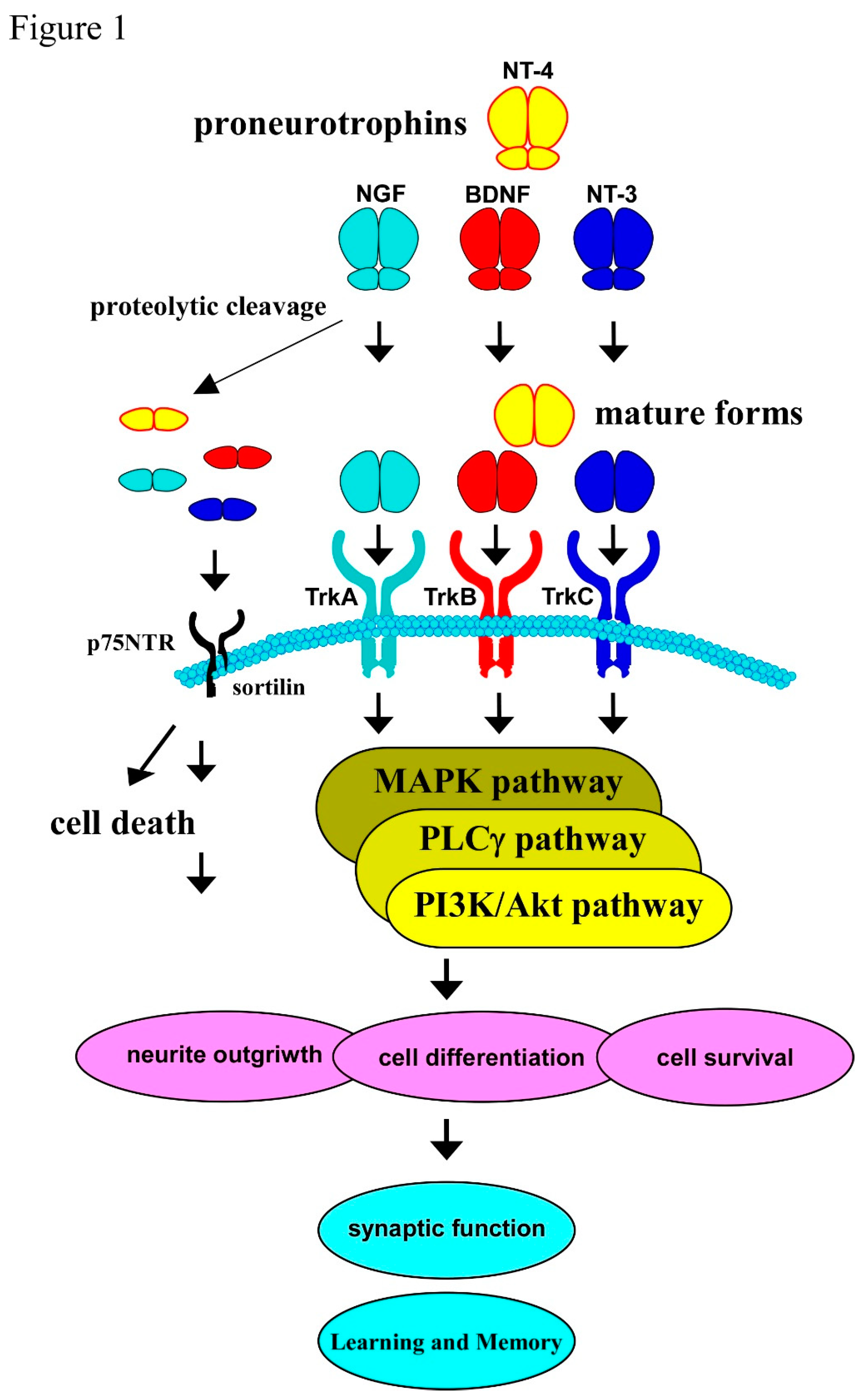
Figure 2.
BDNF mimetics, natural compounds and BDNF/TrkB system. It is possible that the downregulation of BDNF/TrkB system is involved in impaired neuronal function associated with psychiatric disorders, including depression and schizophrenia. Thus, BDNF mimetics, which stimulate TrkB, and natural compounds which can increase the expression of endogenous BDNF (and or TrkB), could improve an activation status of intracellular signaling (MAPK-, PLCγ-, and PI3K/Akt-pathways), leading to potentiation of synaptic function, and learning and memory in the patients.
Figure 2.
BDNF mimetics, natural compounds and BDNF/TrkB system. It is possible that the downregulation of BDNF/TrkB system is involved in impaired neuronal function associated with psychiatric disorders, including depression and schizophrenia. Thus, BDNF mimetics, which stimulate TrkB, and natural compounds which can increase the expression of endogenous BDNF (and or TrkB), could improve an activation status of intracellular signaling (MAPK-, PLCγ-, and PI3K/Akt-pathways), leading to potentiation of synaptic function, and learning and memory in the patients.
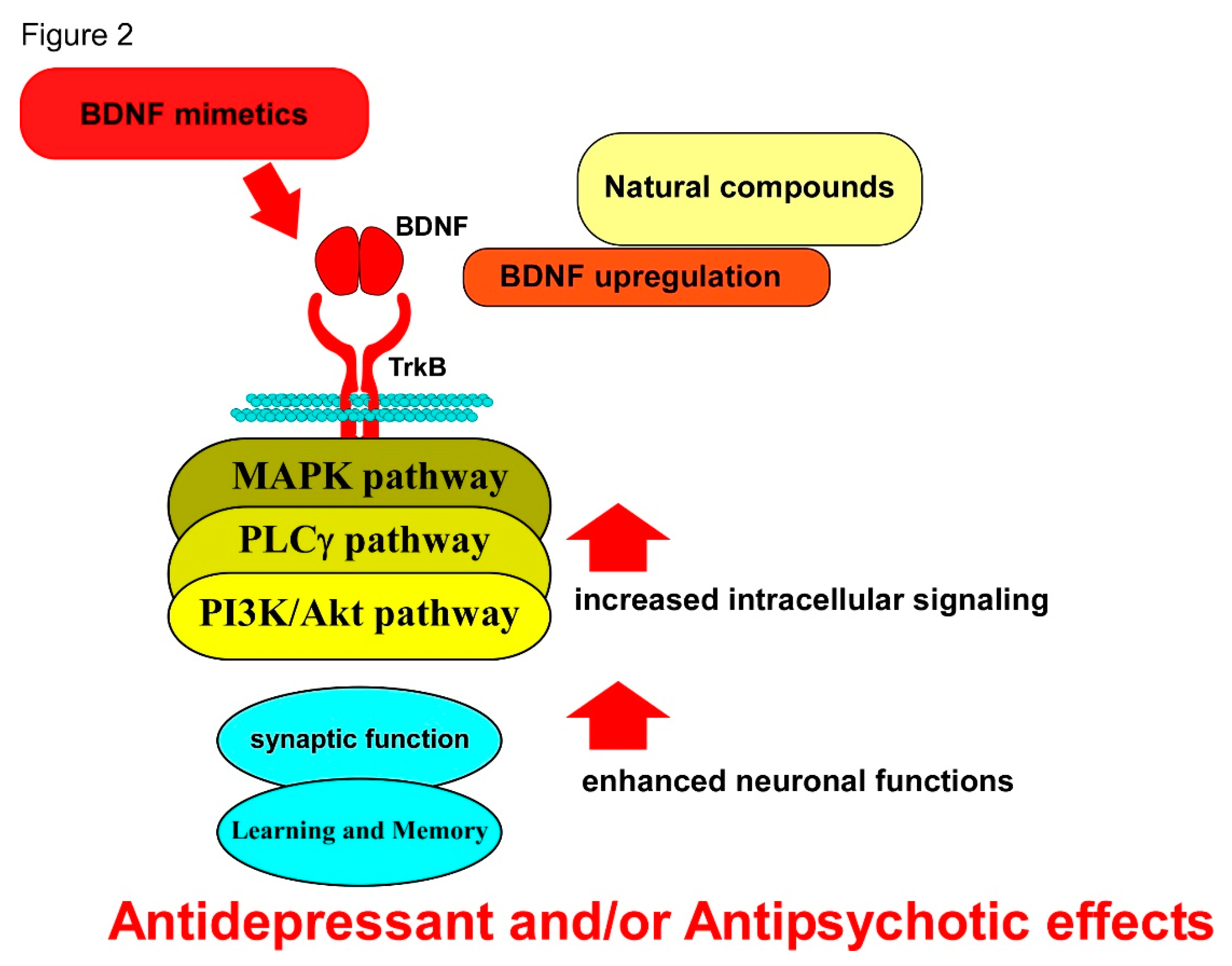
Figure 3.
The role of the BDNF/TrkB system in neurodegenerative diseases and the therapeutic potential of BDNF mimetics for these conditions. BDNF exerts its effects primarily by binding to TrkB receptor, initiating downstream signaling pathways such as PI3K-Akt, MAPK-ERK, and PLCγ, which are critical for neuroprotection, synaptic development, and plasticity. In neurodegenerative disorders such as AD, PD and HD, reduced levels of BDNF and/or TrkB impair intracellular signaling, leading to synaptic loss and neuronal atrophy—key features of neurodegeneration. BDNF mimetics, which are designed to replicate the neurotrophic and neuroprotective functions of BDNF by activating TrkB, offer an attractive alternative for the treatment.
Figure 3.
The role of the BDNF/TrkB system in neurodegenerative diseases and the therapeutic potential of BDNF mimetics for these conditions. BDNF exerts its effects primarily by binding to TrkB receptor, initiating downstream signaling pathways such as PI3K-Akt, MAPK-ERK, and PLCγ, which are critical for neuroprotection, synaptic development, and plasticity. In neurodegenerative disorders such as AD, PD and HD, reduced levels of BDNF and/or TrkB impair intracellular signaling, leading to synaptic loss and neuronal atrophy—key features of neurodegeneration. BDNF mimetics, which are designed to replicate the neurotrophic and neuroprotective functions of BDNF by activating TrkB, offer an attractive alternative for the treatment.
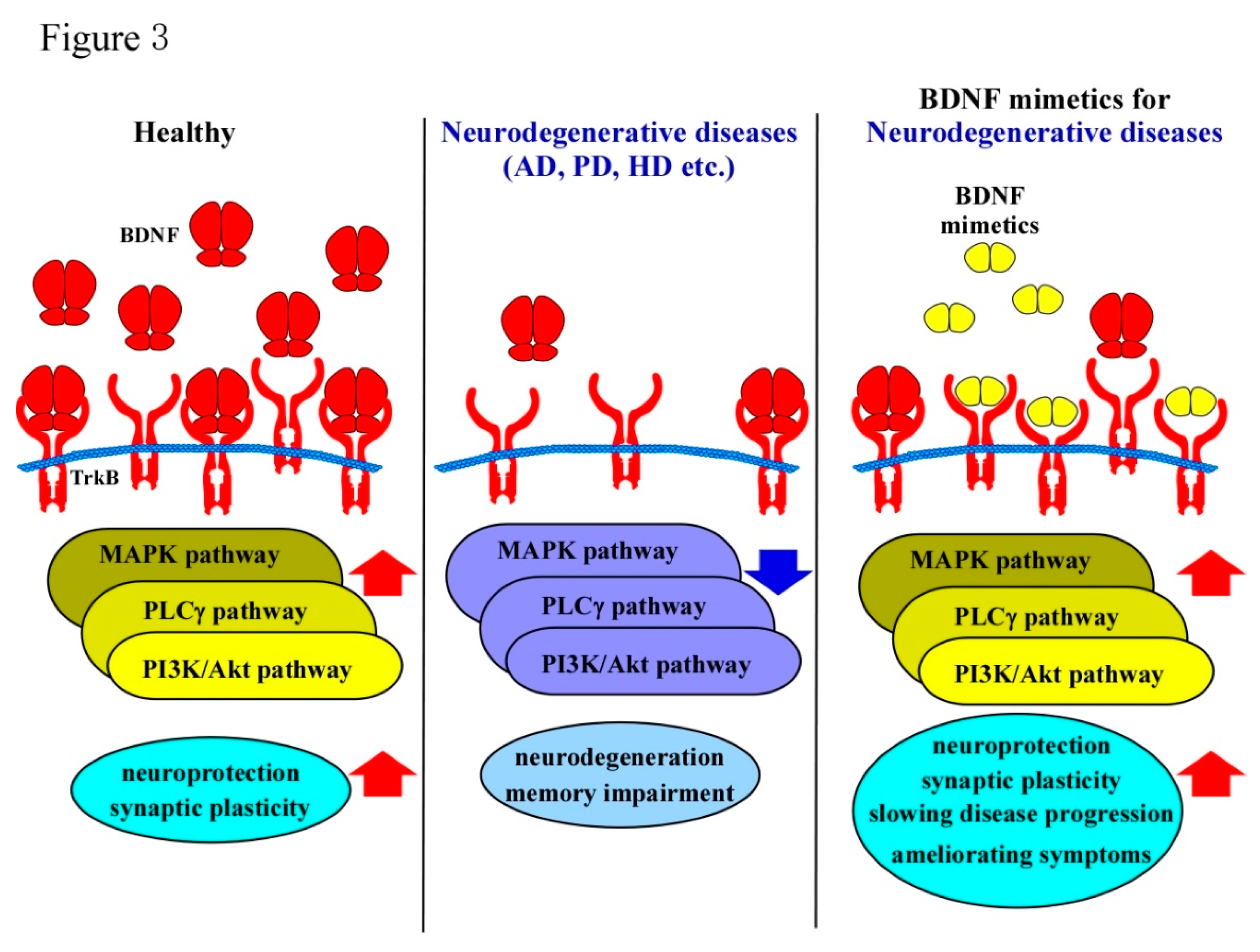
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



