
Submitted:
22 December 2024
Posted:
24 December 2024
You are already at the latest version
Abstract
4-Chloro-5-chlorosulfonylsalicylic acid [C7H4Cl2O5S] is a derivative of salicylic acid and a diuretic agent. Its ability to form polymorphs through recrystallization from various solvents was demonstrated. As a result, four polymorphs were successfully obtained and analyzed using single-crystal X-ray diffraction and powder X-ray diffraction. The solid-state properties were investigated by evaluating crystal lattice energies and intermolecular interactions.
Keywords:
polymorphism
; 4-chloro-5-chlorosulfonylsalicylic acid
; salicylic acid derivative
1. Introduction
Salicylic acid is a naturally occurring compound and a plant hormone [1], which is currently widely used in various applications, such as for treating skin conditions [2,3,4,5,6], as a food preservative, and for its antiseptic and bactericidal properties [7].
The derivatives of salicylic acid have been employed in the synthesis of various pharmaceutical active ingredients. The title compound (Figure 1) is a diuretic agent that is used as a reagent in the synthesis of the diuretic drug xipamide [8,9].
The CSD database and the literature report a limited number of structures of salicylic acid derivatives with various substitutions, including studies on polymorphism [10,11,12], solid-state crystal packing, and hydrogen bonding investigations [13,14]. However, the title compound has not yet been studied in this context.
The current research aims to obtain polymorphs of this compound, primarily based on the functional groups it contains: carbonyl (C=O) and carboxyl (R-COOH). These groups have an increased affinity for participating in the formation of new hydrogen bonds and supramolecular synthons, leading to the formation of polymorphs. Polymorphism refers to the potential of a chemical compound to adopt different molecular self-arrangements from a crystallographic point of view [15,16]. The pursuit of new polymorphs of a pharmaceutical compound with biological activity is driven by the desire to obtain crystalline forms with improved solubility, bioavailability, and melting points [17].
We report the identification of four polymorphs of the diuretic drug 4-chloro-5-chlorosulfonyl salicylic acid. Their crystal structures were elucidated using single-crystal X-ray diffraction and further investigated by powder X-ray diffraction. Solid-state analysis was performed using Hirshfeld surfaces, and lattice energy was evaluated.
2. Materials and Methods
2.1. Crystallization Experiments
The sample of 4-chloro-5-chlorosulfonylsalicylic acid was purchased from Microsin, Bucharest, and the solvents were purchased from Merck, Germany.
Single crystals suitable for X-ray diffraction analysis were prepared through high-throughput screening for new solid crystalline forms using the Crissy XL light crystallization platform, which can handle up to 24 experiments simultaneously.
The solvents and various solvent mixtures were added in three steps, resulting in a final solution volume of 1 mL with the sample completely dissolved. Crystals suitable for X-ray analysis were grown by slow evaporation as follows:
(i) The first polymorph (denoted Polymorph 1) was obtained from a methanol solution;
(ii) The second polymorph (denoted Polymorph 2) was obtained from an acetonitrile solution;
(iii) The third polymorph (denoted Polymorph 3) was obtained from a 1:1 mixture of ethanol and acetone;
(iv) The fourth polymorph (denoted Polymorph 4) was obtained from a tetrahydrofuran solution.
2.2. Single Crystal X-Ray Diffraction and Refinement
Suitable single crystals of the polymorphs were coated in oil and placed on a fine nylon loop on the goniometer of a SuperNova diffractometer, which operates at 50 kV and 0.8 mA. The diffractometer is equipped with dual Cu and Mo micro-sources and an Eos CCD detector. Data collection and correction for Lorentz, polarization, and absorption effects were accomplished using CrysAlis PRO software [18].
The structures were treated and solved in Olex2 software [19], using the ShelXL solution program with Direct Methods for Polymorphs 1, 2, and 3, which were solved by direct methods with SHELXS [20], while Polymorph 4 was solved using the SHELXT program [21] with Intrinsic Phasing. The structures were further refined using SHELXL [22] by Least Squares minimization.
Hydrogen atoms bound to carbon atoms were treated as riding with the isotropic displacement parameter of their parent atom (e.g., Uiso(H) = 1.2Ueq(C) for tertiary CH groups [C-H = 0.98 Å]). Hydrogen atoms attached to hydroxyl O-H groups were located from Fourier maps and refined with O-H = 0.82 Å.
2.3. Powder X-Ray Diffraction Analysis
Powder X-ray diffraction data were collected using a Bruker D8 Advance diffractometer, equipped with a Cu tube (CuKα1, λ = 1.54056 Å) operating at 40 kV and 40 mA. The system included a Ge (111) monochromator in the incident beam to isolate CuKa1 radiation and a LYNXEYE detector. Data were acquired at a scan rate of 0.02°/s in the 2ϴ range of 5-40֯.
2.4. Computational Chemistry
The intermolecular interactions were examined in terms of both their nature and magnitudes for all asymmetric units and molecules positioned at distances equal to or shorter than the sum of their van der Waals radii. Interaction energies were calculated pairwise by considering four distinct energy components: electrostatic (Eele), polarization (Epol), dispersion (Edis), and exchange repulsion (Erep) [23,24]. These calculations were carried out using the [B3LYP/6-31G(d,p)] function in the CrystalExplorer software [23]. The scaling factors for the CE-B3LYP model are kele = 1.057, kpol = 0.740, kdisp = 0.871, and krep = 0.618. Additionally, C-H and O-H bond lengths were adjusted to the normalized distances used in neutron diffraction, with C-H = 1.083 Å and O-H = 0.993 Å [25].
The evaluation of crystal lattice energies was performed using the CrystalExplorer [23] software based on the evaluation of a molecular cluster with a radius of 45 Å.
3. Results
3.1. Descriptions of the Crystal Structures
Table 1 presents the detailed crystallographic and refinement data of the analyzed crystals.
3.1.1. Polymorph 1
The starting form of 4-chloro-5-chlorosulfonylsalicylic acid crystallizes centrosymmetrically in the monoclinic C2/c space group with one molecule in the asymmetric unit (Figure 2a). In the crystal, the molecules are linked via mutual O1-H1···O2 carboxyl···carboxyl hydrogen bonds (1.656 Å), which bridge molecular dimers and form a R22(8) graph set motif. Along the b-axis direction, the molecules are linked via C4-H4···O4 interactions (2.441 Å) between the phenyl ring and the O4 oxygen of the sulfonyl group, and are located in the same plane. The molecular layers are further connected via O3-H3···O5 (2.485 Å) hydrogen bonds between the hydroxyl group and the O5 sulfonyl oxygen. The crystal packing perspective of polymorph 1 is depicted in Figure 2b.
3.1.2. Polymorph 2
Polymorph 2 was found to crystallize centrosymmetrically with one molecule in the asymmetric unit (Figure 3a), but it belongs to the triclinic P-1 space group. Its packing similarities are high, as it once again forms R22(8) molecular synthons that link molecular dimers via strong O1-H1···O2 carboxyl···carboxyl hydrogen bonds (1.695 Å). Other similarities include the presence of C4-H4···O4 interactions (2.303 Å) between the phenyl ring and the O4 sulfonyl oxygen, which occur this time in the direction of the a-axis and connect diuretic molecules located in the same plane. The adjacent molecular layers are further connected via O3-H3···O5 (2.383 Å) hydrogen bonds between the hydroxyl group and the O5 sulfonyl oxygen. Polymorph 2 is packed along the a-axis, as depicted in Figure 3b.
3.1.3. Polymorph 3
Polymorph 3 crystallized centrosymmetrically and belongs to the monoclinic P21/c crystal system with one molecule in the asymmetric unit (Figure 4a). The structure is characterized by reciprocal O1-H1···O2 carboxyl···carboxyl hydrogen bonds (1.708 Å), which bind together to form a dimer that exhibits an R22(8) graph set motif. The difference in crystal packing compared to polymorphs 1 and 2 lies in the π···π interactions, which occur between the C4···C4 phenyl rings at a distance of 3.393 Å, leading to the formation of molecular layers located in the same plane (Figure 4b)
3.1.4. Polymorph 4
By X-ray single crystal analysis, it was found that, similarly to polymorph 3, the fourth polymorph investigated crystallized in the centrosymmetric P21/c space group of the monoclinic crystal system, with one diuretic molecule in the asymmetric unit (Figure 5a). As in the case of the other crystals, polymorph 4 consists of molecular dimers that form R22(8) motifs based on the formation of reciprocal O1-H1···O2 carboxyl···carboxyl hydrogen bonds (1.689 Å). The molecules are linked in the direction of the c-axis via O3-H3···O5 (2.6 Å) hydrogen bonds between the O1 hydroxyl atom and the O5 oxygen of the sulfonyl group, forming infinite chains that depict a sinusoidal-like shape (Figure 5b).
The molecular packing similarity analysis for the four polymorphs was performed using the Mercury software package [26], which calculated positional differences between a cluster of 15 molecules in each crystal [27]. The parameters were set to include only molecules with a positional variation within 20% for both distances and angles, while molecules with deviations greater than 20% were excluded. Hydrogen atom positions were omitted, and inversions were allowed during the comparison. The results show that polymorphs 1 and 2 have the highest packing similarity, with five out of 15 molecules overlapping and an RMS deviation of 0.348. Polymorphs 2 and 4 also exhibit a good overlap for three of the 15 molecules, with an RMS deviation of 0.106, while the remaining molecules align well only for the molecular pairs that form the R22(8) synthons. The molecular overlap is displayed in Figure S1 (Supplementary Material).
The analysis of the four crystal structures reveals several key structural features as follows:
(i) All polymorphs crystallized in centrosymmetric space groups: monoclinic C2/c for polymorph 1, triclinic P-1 for polymorph 2, and monoclinic P21/c for polymorphs 3 and 4.
(ii) The polymorphs are characterized by asymmetric units consisting of single diuretic molecules.
(iii) All four polymorphs exhibit molecular dimers based on the formation of R22(8) homosynthons via strong O-H···O mutual hydrogen bonds.
(iv) In terms of molecular layout, polymorph 1 differs from polymorphs 2, 3, and 4 due to the twisted chlorosulfonyl group.
3.2. Powder X-Ray Diffraction
X-ray diffraction analysis was performed to assess the structural homogeneity and purity of the prepared samples, as well as to confirm that the analyzed single crystals are representative of the entire bulk powder sample. Comparisons between the simulated diffraction patterns (generated from the CIF files) and the experimental patterns are shown in Figure S2 (Supporting Information). A good match is observed in all paired comparisons, except for some diffraction intensities that are lower in the experimental patterns, which can be attributed to the preferred orientation of the crystallites.
3.3. Hirshfeld Surface Analysis
The Hirshfeld surface analysis was carried out to gain a deeper understanding of the contact interactions in the crystal structures of the polymorphs. The analysis reveals both similarities and differences among the polymorphs in terms of distinct contact features (Table 2), as demonstrated by the corresponding dnorm-surface mappings in Figure 6.
The color code (red, white, and blue) can be interpreted as follows: red areas represent distances shorter than the sum of the van der Waals (∑vdW) radii, indicating strong interactions; white areas correspond to distances near or equal to ∑vdW, signifying weak contacts; and blue areas represent longer distances. For each 3D Hirshfeld surface, a corresponding 2D fingerprint plot was generated (Figure S3 and Figure S4, Supporting Information).
The most obvious similarity observed in all four polymorphs is the presence of deep paired red areas that correspond to intermolecular O1-H1···O2 contacts. The deep red color suggests that these interactions are strong, which is also evident from the separation distances, which are significantly shorter compared to the sum of the van der Waals radii (Table 2). Polymorph 1 shows a contact distance for the O1-H1···O2 of 1.666 Å, in polymorph 2 the distance is 1.705 Å, in polymorph 3 it is 1.718 Å, and in polymorph 4 it is 1.698 Å, all of which are considerably shorter than the ∑vdW of 2.72 Å.
Other similarities were found in polymorphs 1, 2, and 3 in terms of the O3-H3···O5 contact (2.490 Å vs ∑vdW = 2.72 Å in polymorph 1), (2.387 Å vs ∑vdW = 2.72 Å in polymorph 2), and (2.606 Å vs ∑vdW = 2.72 Å in polymorph 3), which can be seen as two faint red spots on the surfaces in polymorphs 1 and 2, while in polymorph 3 they appear as white patches since they are very close to the ∑vdW.
The presence of the C4-H4···O4 hydrogen bond between the phenyl ring and the chlorosulfonyl functional group is seen as a faint red spot in both structures of polymorphs 1 and 2 (2.445 Å vs ∑vdW = 2.72 Å in polymorph 1) and (2.308 Å vs ∑vdW = 2.72 Å in polymorph 2), but it is not present in polymorphs 3 and 4.
A unique feature that can be observed in polymorph 3, but is lacking in the others, is the white patches indicative of π···π interactions, which are very close to the van der Waals sum limit in separation distance (3.393 Å vs ∑vdW = 3.40 Å). Another interaction that can be distinguished in polymorphs 3 and 4 are the C-H···Cl interactions, characterized by white spots on the Hirshfeld surfaces (2.949 Å vs ∑vdW = 2.95 Å) and (2.925 Å vs ∑vdW = 2.95 Å).
Other non-traditional intermolecular interactions identified by faint red spots and white patches on the Hirshfeld surfaces include O···C, C···Cl, O···Cl, and Cl···Cl contacts.
The breakdown of various close contacts for the polymorphs was carried out by analyzing the fingerprint plots and the di and de distances (Figure S3 and S4, Supporting Information).
As a first similarity, all fingerprint diagrams are paw-like in shape and symmetrical, due to the fact that the crystals are characterized by a single molecule in the asymmetric unit. All four crystals are dominated by H···O/O···H contacts, which represent 27.2% in polymorph 1, 31.0% in polymorph 2, 25.3% in polymorph 3, and 23.6% in polymorph 4. These are distinguished on the fingerprint plots as two sharp protruding spikes, suggesting that the O-H···O interactions play an important role in cohesion.
The second contribution to the fingerprint plots is from Cl···O/O···Cl contacts, which account for 16.8% in polymorph 1, 14.7% in polymorph 2, and 20.6% in both polymorphs 3 and 4. The third contribution is from H···Cl/Cl···H contacts, which appear as symmetrical wings on the plots of polymorphs 2, 3, and 4, while in polymorph 1, they are less prominent. A common feature seen in all crystals is the lack of prominent H···H spikes on the fingerprint plots; these contacts account for only 7.2% in polymorph 1, 4.3% in polymorph 2, 3.3% in polymorph 3, and 4.7% in polymorph 4.
Moreover, the C···H/H···C contacts exhibit a similar pattern in all polymorphs and are characterized by distances longer than the ∑vdW, accounting for 5.8% in polymorph 1, 4.3% in polymorph 2, 6.1% in polymorph 3, and a higher percentage of 9.1% in polymorph 4.
The Hirshfeld and fingerprint plots analysis concludes that the hydrogen bonding and other van der Waals interactions are governing the crystal cohesion.
The Hirshfeld surfaces were also mapped with the electrostatic potential (Figure S5, Supporting Information), highlighting the complementarity of charges between various interactions found on the Hirshfeld surfaces. The electrostatic potential mappings reveal that the identified donor and acceptor atoms are charge-complemented and attract each other electrostatically.
3.4. Intermolecular Interaction Energies
The identified interactions were analyzed through interaction energy calculations to assess the relative strength of specific contacts within the respective crystals for a cluster with molecules located at distances up to 3.8 Å from the central molecule. The results are presented interactively in Figure S6 (Supporting Information). Table 3 presents a summary of the most relevant interactions.
In all four polymorphs, the strongest interactions are similar and occur within the formation of eight-membered R22(8) homosynthons via the mutual O1-H1···O2 hydrogen bonds. Thus, polymorph 1 displays a value of -59.8 kJ/mol, polymorph 2 shows -59.5 kJ/mol, polymorph 3 exhibits -55.9 kJ/mol, and polymorph 4 shows -58.1 kJ/mol. All these intermolecular interactions are primarily driven by the dominant electrostatic term, which was found to be -115.0 kJ/mol in polymorph 1 and -101.0 kJ/mol in polymorph 3.
Even though the separation distances between stacked molecular layers are longer than the ∑vdW radii, the dispersion energy is significant enough for their combined contributions to become important. Note that polarization forces are the least significant in all polymorphs.
Energy framework simulations were conducted to assess the impact of overall interaction forces on the packing behavior of the four polymorph crystals. As depicted in Figure 7, the molecular packing of the polymorphs is primarily stabilized by electrostatic forces (illustrated by red bars), particularly the strong O1-H1···O2 hydrogen bond interactions, along with the long-range molecular stacking interactions that account for the dispersion forces (green bars), even though their separation distances are shorter than to the ∑vdW only in Polymorph 3.
3.5. Crystal Lattice Energies in Polymorphs
To assess the relative stability of the polymorphs, lattice energy calculations were performed using the CrystalExplorer21 [23] software and the results are presented in Table 4. The data indicate that the stability is relatively similar among all polymorphs. Analyzing the intermolecular interaction energies between neighboring molecules (Table 3 and Figure S6) shows that, although the dispersion terms are not very large, they accumulate and become the primary attractive force governing the crystal cohesion. Polymorph 3 shows the magnitude of the dispersion energy which is enhanced by the stacking molecules found with separation distances shorter than the ∑vdW.
In terms of attraction, the Coulomb interaction has the second-largest contribution, which can be explained by the hydrogen bonds involved in the formation of R22(8) synthons. The Coulomb energy is relatively close in value to the dispersion terms and does not differ significantly between polymorphs.
The evaluation of the lattice energies is consistent with the polymorph densities and the packing index [28], with the exception of polymorph 4, which has a higher density and packing index than the other polymorphs, but slightly lower lattice energy than polymorph 3.
4. Conclusions
The crystal structures of four polymorphs of the diuretic agent 4-chloro-5-chlorosulfonylsalicylic acid have been determined and reported. All crystals belong to centrosymmetric space groups: three belong to the monoclinic crystal system, with one being C2/c and two P21/c, while one belongs to the triclinic P-1 space group.
The asymmetric unit is comprised by one diuretic molecule in all polymorphs which are found to build molecular dimmers based on the R22(8) homosynthons which are driven by strong mutual carboxyl···carboxyl O1-H1···O2 hydrogen bonds.
The analysis of the nature and strength of intermolecular interactions reveals that cohesion within the crystal lattices is primarily driven by dispersion forces associated with the stacking of molecular layers, complemented by strong electrostatic forces resulting from the presence of O-H···O hydrogen bonds, while polarization energies play a lesser role
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: X-ray powder diffraction analysis comparison between experimental (Exp) and simulate (Sim) diffraction patterns: Polymorph 1 (a); Polymorph 2(b); Polymorph 3 (c); Polymorph 4 (d); Figure S2: Comparison between packing similarities between polymorphs; Figure S3: Fingerprint plots of the polymorphs; Figure S4: Breakdown of the fingerprint plots in individual contributions; Figure S5: Front and back views of the electrostatic potential mapped on the Hirshfeld surfaces: Polymorph 1 (-0.065 to 0.244) (a); Polymorph 2 (-0.069 to 0.228) (b); Polymorph 3 (0.0613 to 0.2231) (c); Polymorph 4 (-0.0568 to 0.2295) (d); Figure S6: Intermolecular interaction energies for a molecular cluster with a radius of 3.8 Å: Polymorph 1 (a); Polymorph 2 (b); Polymorph 3 (c) and polymorph 4 (d).
CIF files of the structures were deposited by the Cambridge Crystallographic Data Centre: 2410826 (Polymorph 1); 2410827 (Polymorph 2); 2410828 (Polymorph 3); 2410829 (Polymorph 4). The copies can be obtained free of charge on written application to CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK (fax:+44 1223 336033); or by access to http://www.ccdc.cam.ac.uk.
Author Contributions
Conceptualization, A.T. and G.B.; methodology, G.B.; software, A.T.; validation, A.T., M.O.M and G.B.; formal analysis, G.B. and M.O.M; investigation, M.O.M.; resources, G.B.; data curation, A.T.; writing—original draft preparation, A.T. and G.B.; writing—review and editing, A.T.; visualization, G.B.; supervision, G.B.; project administration, G.B.; funding acquisition, A.T.. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by MINISTRY OF RESEARCH, INNOVATION AND DIGITALIZATION through the "Nucleu" Programe within the National Plan for Research, Development and Innovation 2022-2027, project PN 23 24 01 01.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- War, A.R.; Paulraj, M.G.; War, M.Y.; Ignacimuthu, S. Role of Salicylic Acid in Induction of Plant Defense System in Chickpea ( Cicer Arietinum L.). Plant Signaling & Behavior 2011, 6, 1787–1792. [Google Scholar] [CrossRef]
- Madan, R.K.; Levitt, J. A Review of Toxicity from Topical Salicylic Acid Preparations. Journal of the American Academy of Dermatology 2014, 70, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Bilal, H.; Xiao, Y.; Khan, M.N.; Chen, J.; Wang, Q.; Zeng, Y.; Lin, X. Stabilization of Acne Vulgaris-Associated Microbial Dysbiosis with 2% Supramolecular Salicylic Acid. Pharmaceuticals 2023, 16, 87. [Google Scholar] [CrossRef]
- Andrýsková, N.; Motyčka, J.; Babincová, M.; Babinec, P.; Šimaljaková, M. Computational Design of a Novel Dithranol–Salicylic Acid Antipsoriatic Prodrug for Esterase-Activated Topical Drug Delivery. Applied Sciences 2024, 14, 1094. [Google Scholar] [CrossRef]
- Măgerușan, Șoimița E.; Hancu, G.; Rusu, A. A Comprehensive Bibliographic Review Concerning the Efficacy of Organic Acids for Chemical Peels Treating Acne Vulgaris. Molecules 2023, 28, 7219. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, Y.H. Exploring Acne Treatments: From Pathophysiological Mechanisms to Emerging Therapies. IJMS 2024, 25, 5302. [Google Scholar] [CrossRef]
- Song, X.; Li, R.; Zhang, Q.; He, S.; Wang, Y. Antibacterial Effect and Possible Mechanism of Salicylic Acid Microcapsules against Escherichia Coli and Staphylococcus Aureus. IJERPH 2022, 19, 12761. [Google Scholar] [CrossRef] [PubMed]
- Lebedev, A.A.; Bazhmina, M.Yu.; Smirnov, V.A. Synthesis and Diuretic Activity of 4-Chlorosalicylic Acid Derivatives. Pharm Chem J 1987, 21, 641–645. [Google Scholar] [CrossRef]
- Smith, R.L.; Woltersdorf, O.W.; Cragoe, E.J. Chapter 8: Diuretics. In Annual Reports in Medicinal Chemistry; Elsevier, 1976; Vol. 11, pp. 71–79 ISBN 978-0-12-040511-4.
- Braun, D.E.; Karamertzanis, P.G.; Arlin, J.-B.; Florence, A.J.; Kahlenberg, V.; Tocher, D.A.; Griesser, U.J.; Price, S.L. Solid-State Forms of β-Resorcylic Acid: How Exhaustive Should a Polymorph Screen Be? Crystal Growth & Design 2011, 11, 210–220. [Google Scholar] [CrossRef]
- Sarma, B.; Sanphui, P.; Nangia, A. Polymorphism in Isomeric Dihydroxybenzoic Acids. Crystal Growth & Design 2010, 10, 2388–2399. [Google Scholar] [CrossRef]
- Turza, A.; Borodi, G.; Miclaus, M.O.; Kacso, I. Structural Studies of the Diuretic Compound 4-Chloro Salicylic Acid-5-Sulfonamide. Journal of Molecular Structure 2020, 1212, 128154. [Google Scholar] [CrossRef]
- Montis, R.; Hursthouse, M.B. Surprisingly Complex Supramolecular Behaviour in the Crystal Structures of a Family of Mono-Substituted Salicylic Acids. CrystEngComm 2012, 14, 5242. [Google Scholar] [CrossRef]
- Alexandru, T.; Maria, M.O.; Liviu, Z.; Maria, D.; Irina, K.; Gheorghe, B. New Solid Forms of the Diuretic Compound 4-Chloro Salicylic Acid-5-Sulfonamide. Journal of Molecular Structure 2021, 1241, 130682. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorphism in Molecular Crystals; IUCr monographs on crystallography; Second edition.; Oxford University Press: Oxford ; New York, 2020; ISBN 978-0-19-965544-1.
- Vippagunta, S.R.; Brittain, H.G.; Grant, D.J.W. Crystalline Solids. Advanced Drug Delivery Reviews 2001, 48, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Chistyakov, D.; Sergeev, G. The Polymorphism of Drugs: New Approaches to the Synthesis of Nanostructured Polymorphs. Pharmaceutics 2020, 12, 34. [Google Scholar] [CrossRef]
- CrysAlis PRO 2018.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2 : A Complete Structure Solution, Refinement and Analysis Program. J Appl Crystallogr 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr A Found Crystallogr 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT – Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr A Found Adv 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr C Struct Chem 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer : A Program for Hirshfeld Surface Analysis, Visualization and Quantitative Analysis of Molecular Crystals. J Appl Crystallogr 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer Model Energies and Energy Frameworks: Extension to Metal Coordination Compounds, Organic Salts, Solvates and Open-Shell Systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef]
- Hahn, T. International Tables for Crystallography; 5th ed.; Kluwer academic publ: Dordrecht, 2002; ISBN 978-0-7923-6590-7.
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0 : From Visualization to Analysis, Design and Prediction. J Appl Crystallogr 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Chisholm, J.A.; Motherwell, S. COMPACK : A Program for Identifying Crystal Structure Similarity Using Distances. J Appl Crystallogr 2005, 38, 228–231. [Google Scholar] [CrossRef]
- Spek, A.L. checkCIF validation ALERTS: What they mean and how to respond. Acta Crystallogr. Sect. E Crystallogr. Commun. 2020, 76, 1–11. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Chemical diagram of 4-chloro-5-chlorosulfonyl salicylic acid polymorphs.

Figure 2.
Asymmetric unit of polymorph 1 presenting the atoms as thermal ellipsoids at 50% probability (a); crystal packing of polymorph 1 seen along ob-axis (b).
Figure 2.
Asymmetric unit of polymorph 1 presenting the atoms as thermal ellipsoids at 50% probability (a); crystal packing of polymorph 1 seen along ob-axis (b).

Figure 3.
Asymmetric unit of polymorph 2 presenting the atoms as thermal ellipsoids at 50% probability (a); crystal packing of polymorph 1 seen along oa-axis (b).
Figure 3.
Asymmetric unit of polymorph 2 presenting the atoms as thermal ellipsoids at 50% probability (a); crystal packing of polymorph 1 seen along oa-axis (b).

Figure 4.
Asymmetric unit of polymorph 3 presenting the atoms as thermal ellipsoids at 50% probability (a); crystal packing of polymorph 1 seen along ob-axis (b).
Figure 4.
Asymmetric unit of polymorph 3 presenting the atoms as thermal ellipsoids at 50% probability (a); crystal packing of polymorph 1 seen along ob-axis (b).

Figure 5.
Asymmetric unit of polymorph 4 presenting the atoms as thermal ellipsoids at 50% probability (a); crystal packing of polymorph 1 seen along ob-axis (b).
Figure 5.
Asymmetric unit of polymorph 4 presenting the atoms as thermal ellipsoids at 50% probability (a); crystal packing of polymorph 1 seen along ob-axis (b).

Figure 6.
Front and back views of Hirshfeld surfaces of polymorphs mapped with dnorm in a range of -0.734 to 1.275 arbitrary units for polymorph 1 (a); -0.703 to 1.393 arbitrary units for polymorph 2; -0.693 to 1.125 arbitrary units for polymorph 3 and -0.708 to 0.961 arbitrary units for polymorph 4 (d).
Figure 6.
Front and back views of Hirshfeld surfaces of polymorphs mapped with dnorm in a range of -0.734 to 1.275 arbitrary units for polymorph 1 (a); -0.703 to 1.393 arbitrary units for polymorph 2; -0.693 to 1.125 arbitrary units for polymorph 3 and -0.708 to 0.961 arbitrary units for polymorph 4 (d).

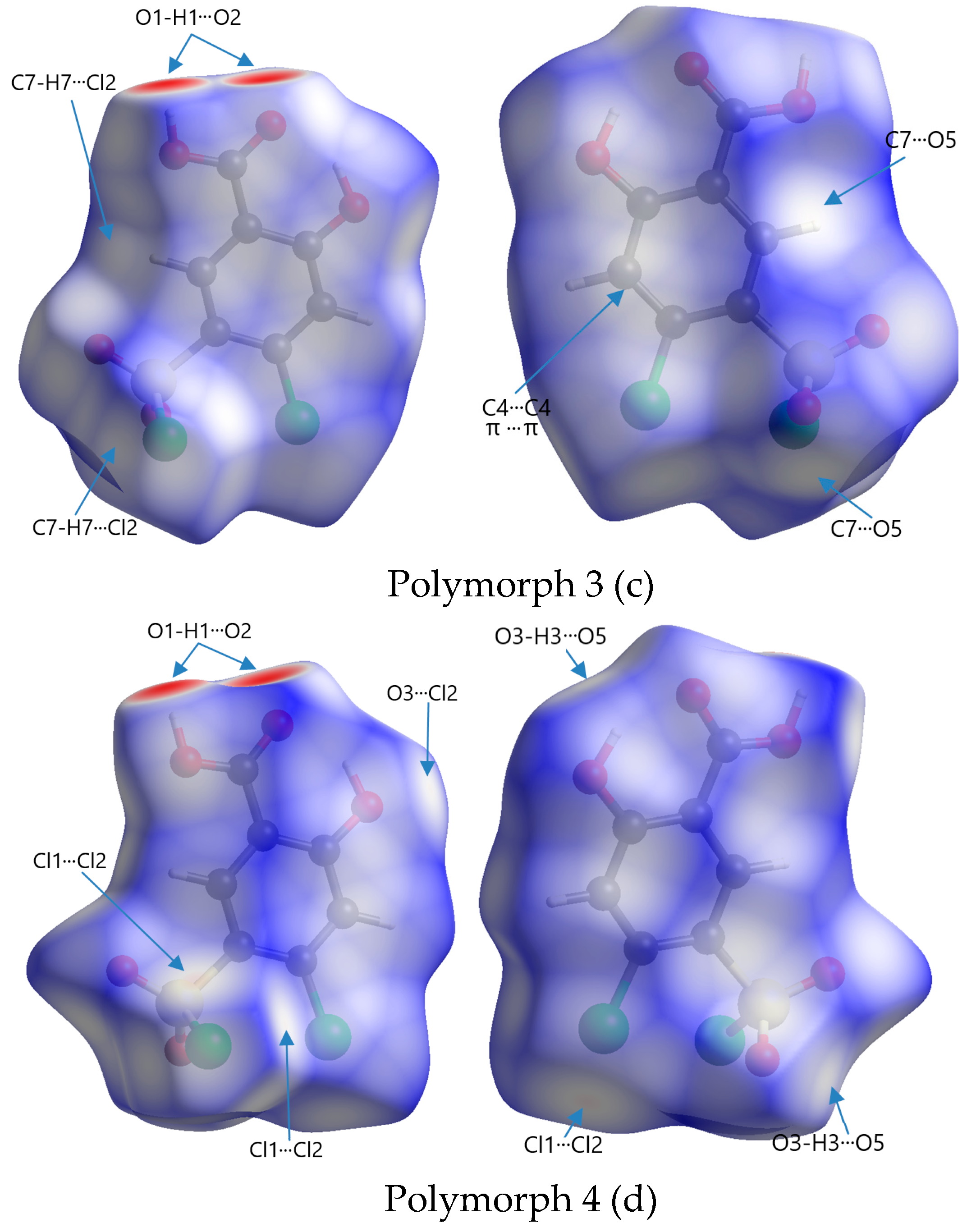
Figure 7.
Crystal packing views over a domain of 2x2x2 unit cells considering a scale factor of 100 and a energy cut-off of 8kJ/mol illustrating of the electrostatic energy (red), dispersion energy (green), and the energy frameworks (blue) for the polymorphs. The relative magnitude of interaction energies is proportional to the cylindrical radius.
Figure 7.
Crystal packing views over a domain of 2x2x2 unit cells considering a scale factor of 100 and a energy cut-off of 8kJ/mol illustrating of the electrostatic energy (red), dispersion energy (green), and the energy frameworks (blue) for the polymorphs. The relative magnitude of interaction energies is proportional to the cylindrical radius.


Table 1.
Crystallographic and refinement details of analyzed polymorphs.
| Identification code | Polymorph 1 | Polymorph 2 | Polymorph 3 | Polymorph 4 |
| Empirical formula | C7H4Cl2O5S | C7H4Cl2O5S | C7H4Cl2O5S | C7H4Cl2O5S |
| Formula weight | 271.06 | 271.06 | 271.06 | 271.06 |
| Temperature/K | 293(2) | 293(2) | 293(2) | 293(2) |
| Crystal system | monoclinic | triclinic | monoclinic | monoclinic |
| Space group | C2/c | P-1 | P21/c | P21/c |
| a/Å | 16.1353(7) | 7.6935(7) | 7.4495(3) | 5.9570(2) |
| b/Å | 7.3277(3) | 8.1150(7) | 8.5652(2) | 9.6985(3) |
| c/Å | 17.1273(7) | 8.7459(9) | 15.4974(5) | 16.8231(5) |
| α/° | 90 | 81.717(8) | 90 | 90 |
| β/° | 95.571(4) | 68.786(9) | 97.386(3) | 95.364(3) |
| γ/° | 90 | 79.410(8) | 90 | 90 |
| Volume/Å3 | 2015.48(15) | 498.57(9) | 980.63(6) | 967.68(5) |
| Z’ | 1 | 1 | 1 | 1 |
| Z | 8 | 2 | 4 | 4 |
| ρcalcg/cm3 | 1.787 | 1.806 | 1.836 | 1.861 |
| μ/mm-1 | 7.781 | 0.856 | 0.870 | 8.103 |
| F(000) | 1088.0 | 272.0 | 544.0 | 544.0 |
| Radiation | CuKα (λ = 1.54184) | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | Cu Kα (λ = 1.54184) |
| 2Θ range for data collection/° | 10.378 to 142.836 | 6.862 to 58.076 | 7.124 to 58.136 | 10.54 to 142.178 |
| Index ranges | -15 ≤ h ≤ 19, -8 ≤ k ≤ 4, -18 ≤ l ≤ 20 | -9 ≤ h ≤ 9, -11 ≤ k ≤ 9, -11 ≤ l ≤ 10 | -9 ≤ h ≤ 9, -11 ≤ k ≤ 11, -20 ≤ l ≤ 20 | -7 ≤ h ≤ 7, -11 ≤ k ≤ 11, -20 ≤ l ≤ 12 |
| Reflections collected | 3114 | 3305 | 13001 | 3406 |
| Independent reflections | 1909 [Rint = 0.0140, Rsigma = 0.0230] | 2147 [Rint = 0.0372, Rsigma = 0.0643] | 2384 [Rint = 0.0458, Rsigma = 0.0372] | 1829 [Rint = 0.0206, Rsigma = 0.0301] |
| Data/restraints/parameters | 1909/0/138 | 2147/4/144 | 2384/2/144 | 1829/0/139 |
| Goodness-of-fit on F2 | 1.050 | 1.072 | 1.078 | 1.049 |
| Final R indexes [I>=2σ (I)] | R1 = 0.0344, wR2 = 0.0886 | R1 = 0.0565, wR2 = 0.1320 | R1 = 0.0628, wR2 = 0.1714 | R1 = 0.0359, wR2 = 0.0970 |
| Final R indexes [all data] | R1 = 0.0384, wR2 = 0.0926 | R1 = 0.0783, wR2 = 0.1656 | R1 = 0.0967, wR2 = 0.1967 | R1 = 0.0406, wR2 = 0.1032 |
| Largest diff. peak/hole / e Å-3 | 0.32/-0.48 | 0.42/-0.62 | 0.72/-0.67 | 0.35/-0.33 |
Table 2.
Normalized contact distances of intermolecular interactions of polymorphs shorter than the sum of van der Waals radii.
Table 2.
Normalized contact distances of intermolecular interactions of polymorphs shorter than the sum of van der Waals radii.
| Contact | dnorm distance (Å) | ∑vdW (Å) | Δ(∑vdW-dnorm distance) | Symmetry operation |
| Polymorph 1 | ||||
| O1-H1···O2 | 1.666 | 2.72 | 0.944 | ½-x,1/2-y,-z |
| O3-H3···O5 | 2.490 | 2.72 | 0.12 | x, 1-y, -1/2+z |
| C4-H4···O4 | 2.445 | 2.72 | 0.165 | x, 1+y, z |
| O4···C1 | 2.904 | 3.22 | 0.316 | ½-x, ½+y, 1/2-z |
| O4···C2 | 3.129 | 3.22 | 0.091 | ½-x, ½+y, 1/2-z |
| C1···Cl2 | 3.409 | 3.45 | 0.041 | 1-x, y, ½-y |
| Polymorph 2 | ||||
| O1-H1···O2 | 1.705 | 2.72 | 0.905 | 1-x, 1-y, -1-z |
| O3-H3···O5 | 2.387 | 2.72 | 0.223 | 1+x, y, -1+z |
| C4-H4···O4 | 2.308 | 2.72 | 0.302 | 1+x, y, z |
| Cl2···O3 | 3.249 | 3.27 | 0.021 | 1-x, 2-y, -z |
| Polymorph 3 | ||||
| O1-H1···O2 | 1.718 | 2.72 | 0.892 | -x, -y, -z |
| C4···C4 (π ···π) | 3.393 | 3.40 | 0.007 | 1-x, 1-y, -z |
| C7-H7···Cl2 | 2.949 | 2.95 | 0.109 | -x, -1/2+y, ½-z |
| C7···O5 | 3.213 | 3.22 | 0.007 | 1-x, -1/2+y, ½-z |
| Polymorph 4 | ||||
| O1-H1···O2 | 1.698 | 2.72 | 0.912 | -x, 2-y, 1-z |
| O3-H3···O5 | 2.606 | 2.72 | 0.004 | x, 1.5-y, ½+z |
| C7-H7···Cl2 | 2.925 | 2.95 | 0.025 | -x, ½+y, ½-z |
| Cl1···Cl2 | 3.425 | 3.50 | 0.075 | 1+x, y, z |
| O3···Cl2 | 3.258 | 3.27 | 0.012 | 1-x, 1-y, 1-z |
Table 3.
Strength and nature of intermolecular interactions for some relevant contacts.
| Crystal | Contact | Eele | Epol | Edisp | Erep | Etot |
| Polymorph 1 | O1-H1···O2 | -115.0 | -19.9 | -11.5 | 87.6 | -59.8 |
| O3-H3···O5 | -2.5 | -0.9 | -7.4 | 3.1 | -7.6 | |
| C4-H4···O4 | -4.7 | -1.4 | -11.5 | 7.1 | -10.5 | |
| O4 ···C1 O4 ···C2 |
-16.1 | -2.7 | -24.9 | 10.6 | -33.1 | |
| Cl2 ···C1 | -8.9 | -0.7 | -39.0 | 19.8 | -28.8 | |
| Polymorph 2 | O1-H1···O2 | -104.9 | -17.6 | -11.6 | 74.6 | -59.5 |
| O3-H3···O5 | -5.3 | -1.0 | -7.2 | 4.5 | -9.0 | |
| C4-H4···O4 | -6.9 | -0.9 | -8.4 | 6.6 | -9.6 | |
| Cl2···O3 | -6.1 | -0.6 | -29.5 | 14.9 | -21.3 | |
| Polymorph 3 | O1-H1···O2 | -101.0 | -16.6 | -11.1 | 72.8 | -55.9 |
| C4···C4 (π ···π) | -2.7 | -0.8 | -34.1 | 14.4 | -23.2 | |
| C7-H7···Cl2 | -4.1 | -1.1 | -13.5 | 0 | -18.7 | |
| C7···O5 | -7.6 | -2.3 | -11.9 | 4.5 | -17.3 | |
| Polymorph 4 | O1-H1···O2 | -107.4 | -19.1 | -11.5 | 79.9 | -58.1 |
| O3-H3···O5 | -3.7 | -0.7 | -6.8 | 2.8 | -8.4 | |
| C7-H7···Cl2 | -12.8 | -1.4 | -11.2 | 7.4 | -18.0 | |
| Cl1···Cl2 | 4.3 | -1.0 | -18.6 | 9.4 | -5.9 | |
| O3 ···Cl2 | -2.8 | -0.7 | -33.0 | 15.0 | -21.5 |
Table 4.
Comparison of lattice energy with the corresponding energy components for the polymorphs.
| Structure | Ecoul (kJ/mol) | Epol (kJ/mol) | Edisp (kJ/mol) | Erep (kJ/mol) | Elatt (kJ/mol) | Density (g/cm3) | Packing index % |
| Polymorph 1 | -88.7 | -9.2 | -87.9 | 79.4 | -106.4 | 1.787 | 68.5 |
| Polymorph 2 | -81.4 | -6.0 | -98.0 | 77.1 | -108.3 | 1.806 | 69.3 |
| Polymorph 3 | -76.6 | -10.2 | -101.2 | 73.4 | -114.6 | 1.836 | 70.8 |
| Polymorph 4 | -82.2 | -10.3 | -97.3 | 80.5 | -109.3 | 1.861 | 71.7 |
Ecoul: the Coulombic term; Epol: the polarization term; Edisp: the dispersion term; Erep: the repulsion term; Elatt: crystal lattice energy
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



