
Submitted:
20 December 2024
Posted:
23 December 2024
You are already at the latest version
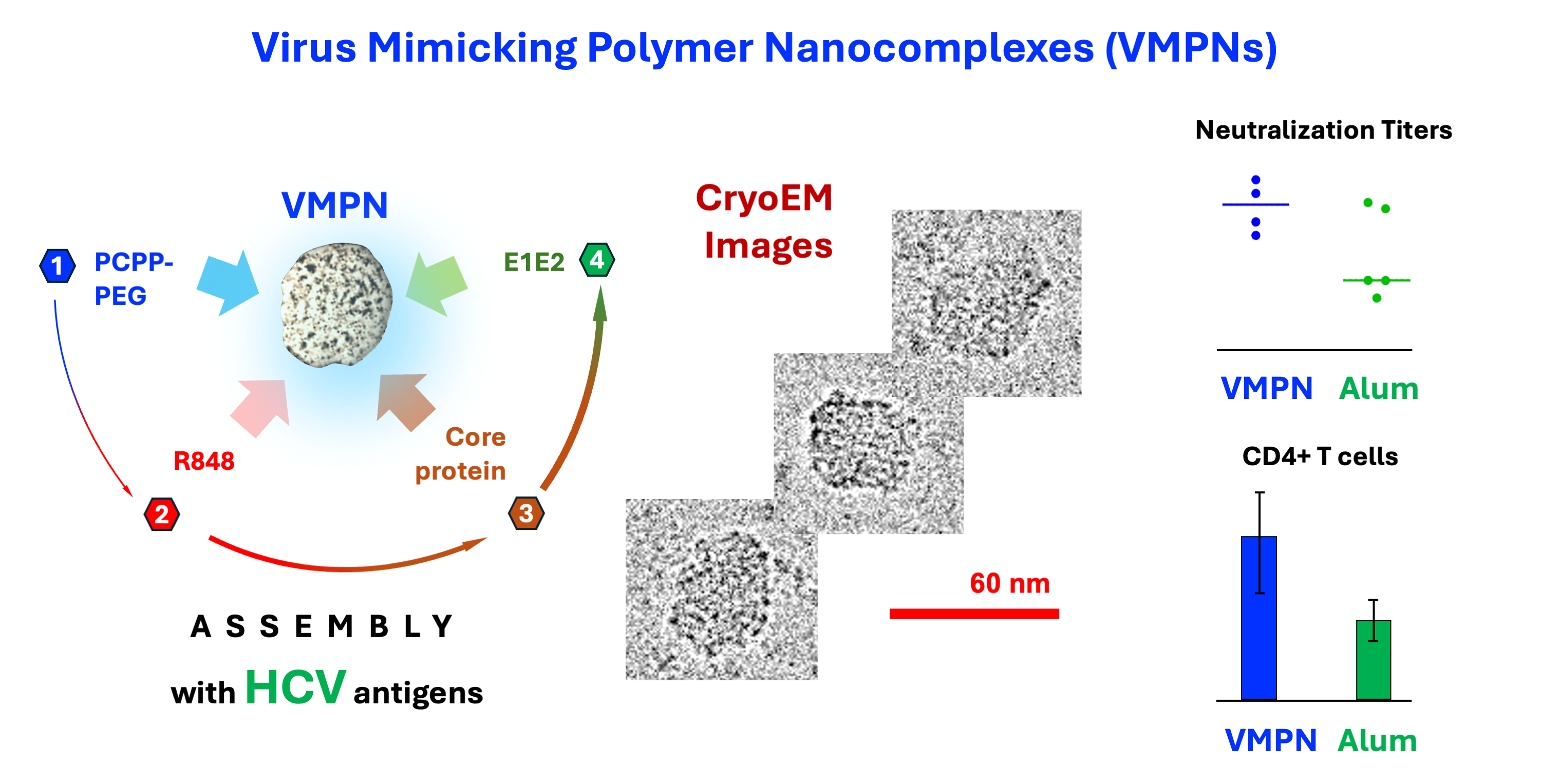
Abstract
Hepatitis C virus (HCV) is a major public health concern, and the development of an effective HCV vaccine plays an important role in an effort to prevent new infections. Supramolecular co-assembly and co-presentation of the HCV envelope E1E2 heterodimer complex and core protein presents an attractive vaccine design strategy for achieving effective humoral and cellular immunity. With this objective, the two antigens were non-covalently assembled with a TLR 7/8 agonist into virus-mimicking polymer nanocomplexes (VMPNs) using biodegradable synthetic polyphosphazene delivery vehicle. The resulting assemblies were characterized using dynamic light scattering and asymmetric flow field flow fractionation methods and directly visualized in their vitrified state by cryogenic electron microscopy. The in vivo superiority of VMPNs over the individual components and an Alum formulated vaccine manifests in higher neutralizing antibody titers, promotion of a balanced IgG response, and induction of CD4+ T cell responses to core proteins. Aqueous-based spontaneous co-assembly of antigens and immunopotentiating molecules enabled by a synthetic biodegradable carrier offers a simple and effective pathway to the development of polymer-based supramolecular vaccine systems.
Keywords:
polyphosphazenes
; polymer nanocomplexes
; vaccine delivery
; Hepatitis C virus
; immunoadjuvants
; supramolecular assembly
; resiquimod
; PEGylated polymer
1. Introduction
Hepatitis C virus (HCV) is a major public health concern and has chronically infected over 50 million people worldwide with nearly one million new infections every year [1]. HCV is the leading cause of liver cancer in North America, Europe, and Japan with high infection rates in Southeast Asia and other parts of the world [2,3]. Although highly effective direct-acting antivirals were introduced that can nearly eliminate the virus, the prevalence of HCV infection still remains high due to the limited availability to patients in low and middle-income countries, and the inability to provide protection against reinfection [4,5,6]. This has prompted the World Health Organization (WHO) to announce a global hepatitis strategy, which aims to reduce new infections from all types of hepatitis viruses by 90% and associated deaths by 65% by 2030 [7]. Development of an effective HCV vaccine will play a major role in achievement of this WHO mandate to address this major public health problem.
HCV is an enveloped, positive strand RNA virus that contains three structural proteins: core and two envelope glycoproteins, E1 and E2, and seven non-structural proteins [8,9]. The E1 and E2 glycoproteins are transmembrane proteins that form a membrane-anchored heterodimer complex, mbE1E2, which interacts with host cell receptors and is critical for viral entry into the cell. The E1E2 complex has been a prime subject of vaccine studies pursuing the induction of neutralizing antibodies [10]. More recently, a secreted E1E2 heterodimer (sE1E2) has been developed that maintains that natural conformational structure of mbE1E2 as a promising vaccine candidate [11,12,13].
A major challenge for HCV vaccine development is that the virus exhibits considerable genetic diversity with eight known genotypes and more than one hundred subtypes identified thus far [14]. Given this diversity, induction of broadly reactive immune responses that target genetically conserved regions of the viral genome will likely be required for protection against HCV. Data from both chimpanzee and human studies suggest that HCV-specific CD4+ and CD8+ T cells are important in the control of primary and secondary infections ([10,15]. In addition, clearance of HCV infection is also associated with early development of serum antibodies capable of blocking infection of multiple heterologous strains [16,17,18]. For these reasons, an effective vaccine must be capable of inducing both humoral and cellular responses. In view of this, the HCV core protein is more conserved across HCV genotypes compared to E1E2 and has been shown to be effective as an antigen in HCV prototype vaccines providing strong cellular immune responses [19,20,21]. Therefore, vaccine designs combining the E1E2 heterodimer along with the HCV core protein are highly attractive in context of inducing both B cell and T cell responses [22].
Virus-like particles (VLPs) represent a promising vaccine strategy due to their ability to mimic the structure of natural viruses without transmission of infectious material. Several VLP-based vaccines have been licensed for commercial use targeting hepatitis B virus and human-papillomavirus [23]. It has been shown that VLPs are effective in stimulating both arms of the immune system, humoral and cellular responses. Several expression systems have been used for HCV VLP production including mammalian and insect cells by co-expression of one or more of HCV structural proteins such as E1E2 and core [24,25,26]. These studies showed promising results in preclinical model systems for induction of potent humoral and cellular responses [26,27,28,29,30,31,32]. However, several limitations exist in the scale-up manufacturing of these VLP-based vaccines including contamination with residual host cell or viral components, bioproduction yields, and/or uniformity of the particles thereby posing a challenge in VLP purification and vaccine development [25].
Due to these limitations, attempts to co-immobilize core and E1E2 proteins using synthetic delivery carriers have been undertaken in the past. For instance, “immune stimulating complexes” (ISCOMs), which are composed of cholesterol, phospholipid, and saponin showed significant potential. These components can form cage-like pentagonal dodecahedral structures (ISCOMATRIX) upon mixing, which were shown to be efficient in adsorbing core protein via electrostatic interactions and eliciting cellular responses in macaques [33]. Further studies demonstrated that such core-adsorbed ISCOMATRIX vehicles can also serve as an efficient adjuvant for an E1E2 antigen in mice [33]. The described formulation approach, however, leads to a dramatic increase in the dimensions of complexes – from 40 nm for INSCOMATRIX to micrometer-sized particulates for antigen loaded formulations. Although significant cellular responses were detected in macaques with ISCOMATRIX-core formulation, the exact mechanism remained unclear. Authors note that internalization of micrometer-sized particulates by phagocytosis typically results to their delivery directly to lysosomes. This leads to antigen presentation of the Ag through the MHC class II pathway, which is primarily involved in humoral immune responses [33]. Furthermore, the evaluation of ISCOMATRIX - HCV core vaccine in Phase I clinical study showed no indication of dose response with CD8+ T cell responses detected only in two of the eight participants receiving the highest vaccine dose [34].
Polymer-based nanoparticulates present an alternative approach to virus mimicry-based vaccine delivery [35,36,37,38,39,40,41,42,43]. Synthetic polyphosphazene macromolecules offer a compelling choice as water-soluble vehicles for the delivery of vaccine antigens with their in vivo performance, which was validated in multiple animal models and clinical [44,45]. The ability of these biodegradable ionic polymers to spontaneously self-assemble with antigenic proteins in aqueous solutions is one of the key factors responsible for their immunopotentiating activity. The resulting nano-sized protein-polymer complexes are stable at near physiological conditions, retain excellent water-solubility and are capable of displaying antigenic molecules in a multimeric form [46,47,48,49]. Poly(dicarboxylatophenoxy)phosphazene (PCPP) is an advanced representative of this class of macromolecules, which demonstrated excellent safety profile and dose-dependent immunoadjuvant effect in clinical trials .
In present study, we introduce a novel approach in which we co-assembled two HCV antigens - sE1E2 and core protein, with a small molecule TLR7/8 agonist - R848 (resiquimod) [50,51,52,53] using a lightly PEGylated version of PCPP macromolecule as an interconnecting delivery system. The resulting nano-sized complexes were directly visualized in their vitrified state by cryogenic electron microscopy (cryoEM), showed monomodal size distribution by dynamic light scattering (DLS) and confirmed to comprise antigenic and adjuvant components by asymmetric flow field flow fractionation (AF4) method. We refer to these nano-sized polymer-protein complexes as Virus-Mimicking Polymer Nano-assemblies (VMPNs). In murine studies, we demonstrate that these VMPNs can be used as a vaccine candidate that demonstrate in vivo superiority over the individual components by inducing higher neutralizing antibody titers, promote a balanced IgG class, and are capable of inducing CD4+ T cell responses to core proteins.
2. Materials and Methods
2.1. Plasmid Construction
For the expression sE1E2SZ.H445P protein the human codon-optimized cDNA sequence was synthesized by GenScript and cloned into pcDNA3.1(+) vector as described previously [12]. Plasmid expressing HCV Core protein was kindly provided by Dr. Mariuzza, (Institute for Bioscience and Biotechnology Research, University of Maryland, Rockville, MD)
2.2. Protein Expression and Purification
Transient expression of recombinant sE1E2SZ.H445P was performed in human Expi239 cells using Expi293 Expression system by following manufacturer’s instructions (Thermo Fisher Scientific, Carlsbad, CA). Briefly, Expi293 cells (ATCC, Manassas, VA) were cultured in Expi293 Expression medium in the shaker incubator at 37 oC, with 120 rpm and 8% carbon dioxide. After cells reached density 2x106 cells/mL, cells were cotransfected with sE1E2SZ.H445P and furin constructs (kindly provided by Dr. Yuxing Li, Institute for Bioscience and Biotechnology Research, University of Maryland, Rockville, MD) at a 2:1 ratio. Culture supernatant of sE1E2SZ.H445P was harvested after 72 h, clarified by centrifugation and filtered through 0.22 μM filter (Pall Corporation, Port Washington, NY). Protein was then purified by sequential HisTrap Ni2+ -NTA and Superdex200 size-exclusion chromatography (Cytiva, Uppsala, Sweden) as described previously [11].
HCV Core protein was produced in E.coli cells, purified and refolded from inclusion bodies. For expression, the HCV Core construct was transformed into BL21(DE3) competent cells. A single colony from a fresh transformation was inoculated into 0.5 L of Luria-Bertani (LB) broth (Fisher Scientific, Hampton, NH) medium containing 100 μg/mL ampicillin. The culture was allowed to grow at 37 °C until it reached an OD600 of 0.6-0.7 and induced with Isoprophyl β-D-1-thiogalactopyranoside (IPTG) (Gold Biotechnology, St. Louis, MO) at a final concentration of 1 mM. After incubation for 3-3.5 h the bacteria were harvested by centrifugation at 7000 rpm. The cell pellet was resuspended in 75-100 mL of 100 mM Tris-HCl pH 8, 150 mM NaCl, 1 mM EDTA, 5% Triton X100 (Sigma-Aldrich, St. Louis, MO) and sonicated for 5 minutes at ~50 kHz on ice. Inclusion bodies were centrifuged at 8000 rpm at 4 °C for 15 minutes. Next, the pellet was resuspended in 75-100 mL of 100 mM Tris-HCl pH 8, 150 mM NaCl, 1 mM EDTA, and further sonicated for 5 min at ~50 kHz on ice. Inclusion bodies were again centrifuged at 8000 rpm at 4°C for 15 min. The remaining pellet was resuspended in 10-15 mL of 100 mM Tris-HCl pH 8, 8 M Urea (Fisher Scientific, Fairlawn, NJ), and placed on a nutating mixer at 4°C overnight. The dissolved inclusion bodies were centrifuged at 25,000 rpm for 1 h and the remaining pellet was dissolved in 6 M Guanidinium chloride (Fisher Scientific, Fairlawn, NJ) and centrifuged again. Solubilized inclusion bodies were refolded by adding dropwise to the refolding buffer 0.4 M Arginine-HCl (Sigma-Aldrich, St. Louis, MO), 100 mM Tris-HCl pH 8.0, 3.7 mM Cystamine (Sigma-Aldrich, St. Louis, MO), 6.6 mM Cysteamine (Sigma-Aldrich, St. Louis, MO) and 5 mM EDTA and stirred at 4 °C for 72 hrs. The mixture was then concentrated and dialyzed in PBS buffer before loading onto a Superdex 200 increase 10/300 column (Cytiva, Uppsala, Sweden). The peak from the size exclusion column was loaded onto Pierce high-capacity endotoxin removal resin (Thermo Scientific, Rockford, IL) to remove endotoxins. Refolded HCV Core was aliquoted and stored at -80°C.
Purified sE1E2SZ.H445P and HCV Core proteins were separated by precast, 4-20% Mini-Protean TGX stain-free gels (Bio-Rad, Hercules, CA) on a Mini-Protean Tetra cell electrophoresis instrument (Bio-Rad, Hercules, CA). For Western blot (Figure S1, Supplementary Materials), protein samples were transferred onto Trans-Blot Turbo Mini nitrocellulose membranes (Bio-Rad, Hercules, CA) and probed with anti-HCV E2 mAb HCV.1 at 5 μg/ml, anti-HCV E1 mAb H-111 at 10 μg/ml followed by detection with a secondary goat anti-human IgG-HRP conjugate (Invitrogen) at a 1:10 000 dilution and the Western ECL substrate (Bio-Rad Laboratories, Inc., Hercules, CA). HCV Core sample was probed with anti-HCV Core C7-50 mAb (Invitrogen, Rockford, IL) at 1μg/ml and detected with goat anti-mouse IgG-HRP conjugate (Abcam Inc., Waltham, MA) at 1:10,000 dilution.
2.3. Synthesis of PCPP-PEG
A copolymer of PCPP containing 2% (mol) of 5 kDa PEG chains (PCPP-PEG) was synthesized using macromolecular substitution of polydichlorophosphazene (Supplementary Materials, Figure S2). Polymer structure and composition were confirmed by 1H NMR (400 MHz, D2O): δ [ppm] 7.3 (br, 2H, =CH2-); 6.4 (br, 2H, =CH2-]); 3.6 (br, 4H, -CH2-CH2-O). Molecular weight (510,000 g/mol) was determined by size exclusion chromatography using Agilent 1260 Infinity II Binary LC instrument (G7112B binary pump, G7167A Multisampler, G7116A multicolumn thermostat, G7117C diode array detector). TSKgel GMPW size exclusion column (Tosoh Bioscience, LLC, Japan), poly(ethylene oxide) molecular weight standards (American Polymer Standards Corporation, Mentor, OH) and 0.1xPBS with 10% acetonitrile (mobile phase) were employed.
2.4. Preparation of Virus-Mimicking Polymer Nanocomplexes (VMPNs)
In a typical procedure, PCPP-PEG based VMPNs was prepared as follows. In a typical procedure, PCPP-PEG based VMPNs was prepared as follows. PCPP-PEG (0.5 ml of 4 mg/mL in PBS) was mixed with R848 (0.5 mL 0.8 mg/mL in DI water, pH 6.2) to bind a cationic small-molecule TLR agonist with negatively charged carboxylic acid groups of the polymer. The resulting formulation was mixed with the solution of a core protein (1 mL of 0.02, 0.2 or 0.5 mg/mL) and then 2 mL of 0.25 mg/mL E1E2 protein were added under mixing by pipetting.
2.5. Physico-Chemical Characterization
Dynamic Light Scattering (DLS) analysis was performed using a Malvern Zetasizer Nano ZS (Malvern Instruments Ltd., Worcestershire, UK) using a 532 nm laser with a back-scattering detector angle of 173°. Data recorded and processed using Malvern Zetasizer Software 7.10.
The Asymmetric flow field flow fractionation (AF4) analysis was conducted using AF2000 AT instrument (Postnova Analytics GmbH, Germany). A regenerated cellulose membrane with molecular mass cutoff of 10 kDa (Postnova, Germany) was used in a separation cartridge and PBS, pH 7.4 (filtered through 0.1 μm pore size) was employed as an eluent.
2.6. CryoEM Visualization of Complexes
An aliquot of polyphosphazene formulation (3.0 μL) was deposited on holey carbon film transmission electron microscopy (TEM) grids (Q3100CR1.3-2 nm, Electron Microscopy Sciences, Hatfield, PA; plasma-treated using glow discharge PELCO EasiGlow (Ted Pella Inc., Redding, CA) for 15 s at 10 mA). The grids were then double blotted for 2 s at 12 C on a Vitrobot (Vitrobot Mark IV, FEI, Hillsboro, OR) leaving a thin layer stretched over the grid holes and vitrified in liquid ethane. The samples were then transferred to a 200 kV Talos Arctica electron microscope (FEI, Hillsboro, OR) equipped with a FEI Falcon3EC direct electron detector. Imaging was performed at a temperature near 90 K with an acceleration voltage of 200 kV. The data were collected using EPU software and processed in CryoSPARC 4.2.1 (Structura Biotechnology Inc., Toronto, Canada). ImageJ software was later used to tune the contrast and brightness of the acquired images.
2.7. Immunization
Thirty-five female Balb/c mice aged 6-8 weeks (Jackson Labs, ME) were randomly distributed into seven groups of five mice each. The mice were immunized with PCPP-PEG (50µg)-R848 (20µg) based VMPN formulations containing 25µg of HCV envelope - sE1mE2.SZ_H445P antigen alone (Group 1) or combination of 25µg sE1mE2.SZ_H445P and HCV core protein, with the core protein included at 1µg (Group 2), 10µg (Group 3) or 25 µg (Group 4), respectively. The mice in remaining groups received 25µg sE1mE2.SZ_H445P and 10 µg core protein formulated in PCPP-PEG alone (50µg PCPP-PEG, Group 5) or Alum (35µg Alum, Group 6) or no adjuvant (control Group 7), respectively (see Table 2). The mice were immunized by intraperitoneal route. The immunization regimen comprised prime immunization on day 0, followed by boosts on days 14, 28, and day 42, respectively. Blood samples were collected prior to each vaccination on days 0 (prebleed), 14, 28, and 42, and a terminal bleeding on day 56. The blood samples were processed for serum by centrifugation and stored at −80 °C until analysis was performed. For memory T cell assays, mice were given an additional boost on Day 136 and then euthanized 7 days later for analysis. Mice were observed daily for mortality and signs of morbidity and weighed biweekly. One mouse (#15) from Group 3 was found dead overnight on day 15 following boost 1, and was deemed to have resulted from injury from intraperitoneal immunization. As post immunization for all mice in the study there was no signs of morbidity, hunched posture, ruffled fur, lethargy, diarrhea, and/or loss of >20% body weight associated with the administration of vaccine formulations.
2.8. Enzyme-Linked Immunosorbent Assay (ELISA)
ELISAs were performed in 96-well plates (MaxiSorp, Thermo Fisher Scientific, Waltham, MA) coated overnight with 5 μg/ml Galanthus Nivalis Lectin (Vector Laboratories, Newark, CA at 4°C. The next day, plates were washed with PBS containing 0.05% Tween 20 and coated with 200 ng per well sE1E2SZ.H445P antigen at 4°C overnight. Next day plates were washed and blocked with blocking buffer 1xPBS, 2% dry milk, 5% fetal bovine serum (FBS, GIBCO, Grand Island, NY, for 1h at room temperature. Serially diluted mice sera samples were added to the plates and incubated for another hour. The binding of HCV E1E2-spesific antibodies was detected by 1:5000 dilution of HRP-conjugated goat anti-mouse IgG (H+L), IgG1 and IgG2a secondary antibody (Southern Biotech, Homewood, AL) with TMB substrates (Bio-Rad, Hercules, CA). Absorbance values at 450 nm (SpectraMax M3 microplate reader, Molecular Devices, San Jose, CA) were used to determine endpoint titers, which were calculated by curve fitting in GraphPad Prism software (Dotatics, San Diego, CA) and defined as four times the highest absorbance value of preimmune sera.
2.9. HCV Pseudoparticles (HCVpp) Neutralization Assay
Huh7, a human hepatoma cell line, was grown and maintained in a DMEM media supplemented with 10% heat-inactivated FBS (GIBCO) and 5% Penicillin-Streptomycin (10,000 U/mL) (Thermo Fisher Scientific, Waltham, MA) and used as the target cell line for neutralization assays [54]. Huh7 cells were seeded onto 96-well plates at a density of 1 × 104 per well to test sera for neutralization. The next day, the pseudoparticles (HCVpp) were incubated with defined serial dilutions of heat-inactivated serum for 1 h at 37 °C and added to each well. After the plates were incubated in a CO2 incubator at 37 °C for 5 to 6 h, the mixtures were replaced with fresh medium and then incubated for 72 h. After incubation, 100 μL Bright-Glo (Promega, Fitchburg, WI) was added to each well for 2 min at room temperature. The luciferase activity was measured using a FLUOstar Omega plate reader (BMG Labtech, Ortenberg, Germany) and stored with the MARS software (Friday Harbor, WA). The ID50 was calculated as the serum dilution that caused a 50% reduction in relative light units (RLU) compared with HCVpp in the control wells. All values were calculated using a dose-response curve fit with nonlinear regression in GraphPad Prism. All experiments involving the use of HCVpp were performed under biosafety level 2 conditions.
2.10. Intracellular Staining for Cytokines and Low Cytometry Analysis
Single cell suspensions from lymph nodes and spleens of immunized animals were incubated with specific antigen or 100 ng/ml Phorbol 12-myristate 13-acetate (PMA; Millipore Sigma, MO) and 1 µg/ml Ionomycin (Millipore Sigma, MO) in complete RPMI (Thermo Fisher Scientific, MA) for 2 h at 37°C. At this time, cells were treated with 10 µg/ml Brefeldin A (eBioscience, CA) for an additional 4 hours at 37°C. For staining, Fc receptor blocking (using a cocktail of 2.4G2 (BioXcell, CA) and chrompure IgG (Jackson labs, ME)) was performed for 15 minutes at 4°C in FACS buffer (1X PBS supplemented with 0.2% bovine serum albumin ,BSA, 0.01% azide). Surface staining was performed for 30 minutes at 4°C in FACS buffer using the antibodies listed. Cells were washed once with FACS buffer, fixed and permeabilized in BD Cytofix/Cytoperm solution (BD Biosciences, CA) for 20 minutes at 4°C, followed by an overnight incubation at 4°C in 1X Fixation/Permeabilization Solution (eBioscience, CA). Intracellular staining was performed using the antibodies described above in 1X eBioscience Permeabilization Buffer for 1 hour at 4°C. Stained cells were washed twice with 1X Permeabilization Buffer (eBioscience, CA) followed by an additional two washes with FACS buffer. Cells were analyzed on the BD LSR-II cytometer and all data was analyzed using FlowJo (TreeStar Inc., Ashland, OR).
2.11. Statistical Analysis
The differences among group % neutralizations were statistically analyzed using the nonparametric Kruskal–Wallis test with Dunn’s multiple comparisons test. P < 0.05 was considered significant. All statistical analyses were performed using GraphPad Prism software (GraphPad Software, Boston, MA).
3. Results
3.1. Macromolecular Design of a Polyphosphazene Delivery Vehicle and its Optimization
From the biophysical standpoint, the optimal design of polyphosphazene delivery vehicle, which is capable of carrying multiple proteins and small molecules, has to satisfy at least two main criteria. First, the system should enable nano-scale dimensions of the resulting assemblies and prevent uncontrolled agglomeration. Second, sufficient avidity between components should be provided. Initially, the formulation was undertaken using a clinical stage polyphosphazene immunoadjuvant – PCPP (Figure 1a). However, DLS study of molecular interactions of this polymer with core protein revealed a severe dose dependent agglomeration in the system (Figure 1b.c). This problem was alleviated through the synthesis of a structural analog of PCPP, which contains 2% (mol) of graft PEG chains (Figure 1d). Such structural modification of the polymer using short chains of PEG allowed for a complete prevention of agglomeration (Figure 1c,e). Furthermore, PCPP-PEG copolymer showed superior resistance to agglomeration caused by acidic and high ionic strength environment (Supplementary Materials, Figure S3). No agglomeration of PCPP-PEG was detected upon addition of E1E2 antigen or R848 to PCPP-PEG in doses intended for in vivo experiments.
3.2. Assembly of VMPNs Containing Ternary E1E2 – core protein – TLR7/8 Agonist
The nano-assembly of four-component VMPNs was conducted through the sequential addition of R848, E1E2 and core protein to the aqueous solution of PCPP-PEG (Figure 2a). PCPP-PEG, which contains negatively charged carboxylic acid moieties, was first modified with positively charged small molecule, R848 to afford its binding to the polymer as a counterion (Supplementary Materials, Figure S4), as described previously [55]. Binding of sequentially added core protein – E1E2 to PCPP-PEG scaffold modified with R848 was demonstrated by AF4 analysis. This method allows for the separation and detection of analyte components on the basis of their molecular and supramolecular sizes [56,57] and has been previously successfully applied to polyphosphazene systems [47,58]. Figure 2b shows AF4 fractogram of VMPNs (solid line at approximately, peak at 14 min) contrasted with PCEP-PEG – R848 system (dashed line) and a mixture of E1E2 – core proteins (peak at 10 min). The binding of proteins to the polymer scaffold is demonstrated by a complete disappearance of protein peaks (10 min) in the VMPN and change in a peak profile compared to PCPP-PEG – R848 (dashed line, peak at 14 min). The latter indicates a modified character of interactions with analytical membrane of AF4, which is caused by binding of proteins and the formation of VMPN. The hydrodynamic diameter of VMPN was determined by DLS and roughly corresponded to the dimensions of the protein-free polyphosphazene scaffold (about 70 nm). The formulation shows unimodal distribution and is completely free of agglomerates.
3.3. Direct Visualization of VMPNs by cryoEM
Cryogenic Electron Microscopy (cryo-EM) is one of the most advanced techniques for visualizing biomacromolecules, such as proteins. It provides microscopy images of biomolecules embedded in vitreous, glass-like ice revealing the real space architecture of macromolecules and their nanoassemblies in their native environment [59,60,61,62]. Although, the visualization of synthetic polymer chains has been challenging due to the low electron-optical contrast of the backbone and their inherent flexibility [63,64,65], the technique was proven to be successful for the analysis of their hierarchical structures, such as micellar assemblies [66,67,68,69]. CryoEM images of E1E2 and core protein-loaded VMPNs are shown in Figure 3. The VMPN assemblies can be described as compact spherical nanocomplexes of somewhat irregular shape. Both, the shape, diameter and size distribution of VMPNs are reminiscent of HCV virions visualized using the same technique [70] and are strikingly different from single chains of PCPP macromolecules [46]. The overall dimensions of VMPNs seen from cryoEM images are in line with those determined by DLS (approximately 60-70 nm, Figure 2c) and are slightly larger than those of HCV virions (approximately 50 nm) [70].
3.4. Immunization Studies
The immunogenicity of VMPNs was evaluated in Balb/c mice. Three animal groups were immunized with VMPNs containing polyphosphazene-R848 system (PPZ-R) assembled with E1E2 (25 μg) and various doses of core protein (1, 10 and 25 μg) (Table 1, groups 2-4). These groups were compared with animals received VMPNs without a core protein and R848-free VMPNs containing 10 μg of core protein (groups 1 and 5, respectively). The results were also benchmarked against those in animals immunized with a mixture of E1E2-core proteins adsorbed on Alum or formulated without a delivery vehicle (groups 6 and 7, respectively). The immunization schedule included prime and three boosts.
3.5. Evaluation of Serological Responses and Homologous and Heterologous Neutralization
Figure 4a shows total IgG titers are influenced by both the addition of R848 and core protein, with both leading to an increase in overall titers. Moreover, all of the PPZ formulations elicit higher titers than antigens formulated with Alum or without any adjuvant. Isotype-specific analysis of IgG1 and IgG2a titers shows that, while the IgG1 titers do not vary appreciably between groups (Figure 4b, upper panel), there is a marked disparity in the IgG2a titers (Figure 4b, lower panel), which act as a proxy measure for the strength of the cellular immune response. Among these groups, the differences that achieve statistical significance in an uncorrected pairwise Kruskal-Wallis test are PPZ-R(sE1E2-C10) versus Alum(sE1E2-C10), PPZ-R(sE1E2-C25) versus sE1E2-C10 (P < 0.05) and PPZ-R(sE1E2-C25) vs. Alum(sE1E2-C10) (P < 0.01).
Presence of core protein did not have a dose-dependent effect on IgG2a levels, but inclusion of R848 did appear to enhance IgG2a levels. Remarkably, the Alum-formulation barely elicited any IgG2a at all, and in fact performed worse in this regard than the no adjuvant group (Figure 4b, lower panel). As a result, the immune response was skewed such that for the Alum group the IgG2a/IgG1 ratio is near zero whereas the PPZ groups, and in particular the PPZ-R groups, show a much more balanced immune response with ratios of 0.1 or higher (Figure 4c). Based on these data, we analyzed the ability of mice immunized with sE1E2 and 10 μg of core protein to neutralize HCV pseudoviruses from the homologous GT1a (H77) strain (Figure 5 and Figure S6) and heterologous strains (Table 2 and Figure S5). Three heterologous strains were chosen, one from GT1b (UKNP1.18.1), one from GT2a (J6), and one from GT2b (UKNP2.5.1). The PPZ-R group performs the best in neutralizing the homologous pseudovirus, with a mean ID50 that is more than three times that of the other two groups. This difference achieves statistical significance for PPZ-R versus PPZ, but not versus Alum (likely due to data spread). Heterologous neutralization was generally weak and the data are inconclusive, with each group neutralizing one of the three heterologous pseudoviruses better than the other two groups.
3.6. Evaluation of Memory VMPN T-Cell Responses to sE1E2 and Core Antigens
Antigen-specific memory T cells primed by a vaccine must persist for a long time in animals to provide enhanced protection during a subsequent re-challenge. Typically, effector T cells (marked by higher levels of the surface marker CD44) are formed in the first weeks after immunization but the persistence of antigen-specific T cells with such a phenotype beyond 3-4 months of challenge is evidence of generation of durable immunological memory. Accordingly, to evaluate the presence of long-term memory T cell responses generated by the VMPNs in mice, we further boosted immunized mice at Day 136 and isolated single cell suspensions 7 days later. Effector cytokine production in an antigen-specific manner is a strong correlate of such memory T cells [71]. The data in Figure 6 compare the responses of isolated splenocytes by measuring intracellular IFNγ upon restimulation with E1E2 or Core proteins (with PMA/Ionomycin used as a positive control polyclonal stimulator). Cells were analyzed by gating on the live CD4+ population (gating strategy discussed in Figure S7, Supplementary Materials). CD4 T cells from the PPZ-R vaccinate mice, re-stimulated with core protein, show a significant increase in the proportion of IFNγ producers within the CD44-hi (Effector/Memory phenotype) subset compared to those from PPZ or Alum immunizations. At this time-point, no significant response to E1E2 proteins were observed; in previous studies, lymphoproliferative responses tend to be higher for core than for E1E2 [29].
4. Discussion
We previously reported the supramolecular assembly of PCPP with the toll-like receptor 7/8 agonist, R848, that could significantly enhance immune stimulation in vitro and in vivo to the HCV E2 protein [55]. In the present study, we expanded on this assembly approach and analyzed the antigenic compositions and immunogenicity of VMPNs comprised of the HCV sE1E2 envelope heterodimer and core protein. The secreted sE1E2 heterodimer is a molecular mimic of the membrane-associated form that can be uniformly produced and shown to induce robust, cross-neutralizing antibody responses in mice [12]. The core protein is the most highly conserved HCV protein among the different viral genotypes, and cellular immune responses to core protein have been associated with improved responses to antiviral treatment and disease outcome. Therefore, we view our synthetic VMPNs incorporating sE1E2 and core as a chemically-defined VLP-type platform designed to elicit both humoral and cellular immunity to further advance development of an HCV vaccine.
PCPP – is a clinical stage macromolecule that has demonstrated promising immunoadjuvant properties in clinical settings [72,73,74,75]. Similarly to another ionic adjuvant – Alum (Alhydrogel), PCPP has been shown to bind (adsorb) antigenic macromolecules non-covalently to realize its immunopotentiating activity [76,77,78]. However, in contrast to Alum, which can only exist in a three-dimensional physical state as a hydrogel [79,80,81], PCPP is inherently water-soluble. This important physical feature is also maintained for the majority of its nano-scale complexes with antigenic proteins, which makes PCPP highly attractive from the formulation standpoint [44]. Yet, our attempts to utilize PCPP as a carrier for all three payload molecules – sE1E2 and core proteins along with R848 TLR7/8 agonist, resulted in the undesirable and uncontrolled phase separation (Figure 1b). To alleviate this issue, we synthesized a new polyphosphazene derivative of PCPP, which contained 2% (mol) of relatively short (5 kDa) graft poly(ethylene glycol) (PEG) chains. The presence of a small PEG content in PCPP did not detrimentally affect the ability of the polymer to efficiently bind antigenic and immunoadjuvant components, which was proven by AF4 experiments (Figure 2b). However, such light PEGylation allowed for a dramatically improved solution behavior of the resulting nanocomplexes (Figure 2c). Moreover, immunization experiments confirmed that a new PCPP-PEG macromolecule was effective in facilitating immune responses in animals. It can be expected that PCPP-PEG can be a potent alternative to PCPP in cases when antigen-polymer interactions are sufficiently strong to cause charge neutralization and undesirable phase separation.
The simplicity of the supramolecular assembly of all payload components in aqueous solutions is extremely attractive from the formulation standpoint as it basically involves sequential addition of the components. It links all formulation components together and results in monomodal size distribution in a nano-scale range. AF4 and DLS methods allow full in process control of the assembly (Figure 2).
CryoEM visualization approach facilitated by high-contrast properties of polyphosphazene backbone [46] reveals morphological features of VMPN assemblies. As mentioned above, the shape and size characteristics of VMPNs resemble actual HCV virions when imaged in their vitrified state [70]. It is also important to note that the co-immobilization of TLR7/8 agonist on VMPNs is critical for achieving both high neutralization titers and cellular responses (Figure 5 and Figure 6). As R848 was noted to be somewhat ineffective as vaccine adjuvant in vivo due to its rapid dissociation from the antigen [50,51,52,53], the effect can be attributed to its complexation with polyphosphazene carrier (Supplementary Materials, Figure S4) [55].
In vivo evaluation of PCPP-PEG complexed with R848 (referred to as PPZ-R) and formulated with sE1E2 in the absence and presence of increasing amounts of core (1, 10, and 25 mcg) was performed in Balb/c mice. These formulations were compared to Alhydrogel (Alum) and no adjuvant controls (Table 1). As shown in Figure 4a, increasing amounts of core in the VMPN assemblies in the presence of R848 (PPZ-R (sE1E2-C1), PPZ-R (sE1E2-C10), PPZ-R (sE1E2-C25)) resulted in an elevation in the anti-E1E2 IgG endpoint titers. Although there was no apparent significant differences among these groups, it is noteworthy that increasing the amount of core did not disrupt the biophysical properties of the PPZ-R assembled VMPNs as presented to the immune system. Moreover, this is also reflected in the IgG1 titers (Figure 2b, upper panel) in which there is no significant difference among these groups. Lastly, the PPZ-R group performed the best in neutralizing the homologous pseudovirus (H77C, Figure 5) in which there is a significant difference in the mean ID50 that is more than three times that of the other two groups. This difference is likely due to the elevated total IgG response but there was also a more uniform clustering of ID50 values around the mean in mice immunized with the PPZ-R adjuvant versus the PPZ and Alum groups.
Effective vaccines against infectious agents require elicitation of both humoral and cellular responses to engender protective immunity, along with persistent memory responses [82]. This has led to the design of tailored immunoadjuvant systems that can drive specific alterations of the immune response according to specific chemical pattern of a pathogen, e.g., double-stranded RNA of viral origin for TLR3, bacterial lipopolysaccharide (LPS) for TLR4, singled stranded RNA for TLR7/8, unmethylated CpG motifs found in bacterial DNA or viruses for TRL9, and so forth [83]. To that end, achieving functionally appropriate type of immunity, such as T helper 1 (Th1) mediated immunity, is highly desirable. We have shown previously that the supramolecular assembly of the R848 TRL7/8 agonist with PCPP drive can drive and modulate IgG1 and IgG2 isotypes in mice that are commonly used on surrogate markers of Th2 and Th1 responses, respectively [84]. In this study, we show that inclusion of R848 (PPZ-R) significantly enhances the IgG2a responses (Figure 2b, lower panel) and the resulting IgG2a/IgG1 ratio was significantly higher than in the absence of R848. Moreover, the IgG2a/IgG1 ratio was near to zero for the Alum group in which the IgG2a response was negligible. This is further supported by the ability to increase generation of memory T cells, a highly desirable features of optimal vaccine adjuvants. CD4 T cells play a critical role in the maintenance of functional CD8 responses as well as formation of memory B cells [85]. Their function is typically evaluated by the production of cytokines, such as gamma interferon (IFN-γ) and tumor necrosis factor alpha (TNF-α) with multiple cytokine producing cells considered to be superior. As shown in Figure 6, CD4 T cells from the PPZ-R vaccinated mice, re-stimulated with core protein, show a significant increase in the proportion of IFNγ producers within the CD44-hi (memory phenotype) cell population compared to those from PPZ or Alum immunizations. Although we did not see a similar response to sE1E2 under these conditions, it has been shown that the E1E2 protein exhibits immunosuppressive properties thereby making it a more challenging immunogen [86,87]. These results demonstrate generation of antigen-specific memory responses in mice immunized with PPZ-R adjuvanted antigens in the VMPN assemblies, and correlate with data on IgG isotype profiles confirming that addiction of PPZ-R favors Th1 responses. Overall, demonstration that synthetic VMPNs can be assembled via simple and highly controlled formulation approaches and display potent immunostimulating activity in vivo, suggest that these bifunctional polymer-based supramolecular constructs can serve as a viable approach for parenteral vaccine applications.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Table S1: Binding affinities HCV envelope antigens using human mAbs. Figure S1: Characterization of sE1E2.SZ-H445P and HCV core antigens. Figure S2: Synthesis of PCPP-PEG. Figure S3: DLS studies demonstrating superior environmental stability of PCPP-PEG over PCPP. Figure S4: Ionic binding of R848 to a PCPP-PEG side chain. Figure S5: Breadth of neutralization against 4 core genotypes with HCVpp. Figure S6: Breadth of neutralization of individual mouse sera against H77C (GT1a). Figure S7: Gating strategy.
Author Contributions
Conceptualization, T.R.F. and A.K.A.; methodology, E.A.T., N.J.S., T.R.F. and A.K.A.; validation, A.M., S.J., L.K. and R.H.; formal analysis, A.M., S.J., L.K. and E.A.T.; investigation, A.M., S.J., L.K., R.H. and K.G.; resources, N.J.S., T.R.F., A.K.A.; data curation, A.M., S.J., L.K. and R.H.; writing—original draft preparation, T.R.F., E.A.T., N.J.S. and A.K.A.; writing—review and editing, T.R.F., E.A.T., N.J.S. and A.K.A.; visualization, R.H., N.J.S. and A.K.A.; supervision, T.R.F., N.J.S., A.K.A.; project administration, T.R.F., N.J.S. and A.K.A.; funding acquisition, T.R.F., N.J.S. and A.K.A. All authors have read and agreed to the published version of the manuscript.
Funding
Please add: This research was funded by the National Institutes of Health, NIAID grants R01 AI132212 (T.R.F., E.A.T., A.K.A.) and R01 AI168048 (T.R.F., E.A.T., A.K.A.).
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Maryland Medical School (protocol number: 1022019).
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Acknowledgments
We thank Dr. Yuxing Li and Dr. Mariuzza (University of Maryland) for kindly providing furin constructs and plasmid expressing HCV Core protein, respectively. Authors are also grateful to Yunus S. Abdul (University of Maryland) for help with coordinating the animal studies.
Conflicts of Interest
Thomas R. Fuerst and Alexander K. Andrianov are shareholders of NeuImmune, Inc. – the company, which develops HCV vaccine. This company had no role in the design of the study, collection, analyses, or interpretation of data, in the writing of the manuscript and in the decision to publish the results. These potential conflicts of interest have been disclosed and are managed by the University of Maryland. The other authors have no conflicts of interest to report.
References
- WHO Hepatitis C Key Facts. https://www/who.int/en/news-room/fact-sheets/detail/hepatitis-c (April 9),.
- Fiehn, F.; Beisel, C.; Binder, M. Hepatitis C virus and hepatocellular carcinoma: carcinogenesis in the era of direct-acting antivirals. Curr Opin Virol 2024, 67, 101423. [Google Scholar] [CrossRef] [PubMed]
- Costa, G.L.; Sautto, G.A. Exploring T-Cell Immunity to Hepatitis C Virus: Insights from Different Vaccine and Antigen Presentation Strategies. Vaccines 2024, 12. [Google Scholar] [CrossRef] [PubMed]
- Yukawa, Y.; Tamori, A.; Iio, E.; Ogawa, S.; Yoshida, K.; Uchida-Kobayashi, S.; Enomoto, M.; Tanaka, Y.; Kawada, N. Hepatitis C virus recurrence in two patients who achieved sustained viral response with interferon-free direct-acting antiviral therapy: reinfection or relapse? Clinical journal of gastroenterology 2019, 12, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Berenguer, J.; Gil-Martin, A.; Jarrin, I.; Montes, M.L.; Dominguez, L.; Aldamiz-Echevarria, T.; Tellez, M.J.; Santos, I.; Troya, J.; Losa, J.E.; et al. Reinfection by hepatitis C virus following effective all-oral direct-acting antiviral drug therapy in HIV/hepatitis C virus coinfected individuals. AIDS 2019, 33, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Rossi, C.; Butt, Z.A.; Wong, S.; Buxton, J.A.; Islam, N.; Yu, A.; Darvishian, M.; Gilbert, M.; Wong, J.; Chapinal, N.; et al. Hepatitis C virus reinfection after successful treatment with direct-acting antiviral therapy in a large population-based cohort. Journal of hepatology 2018, 69, 1007–1014. [Google Scholar] [CrossRef]
- Who, Global Hepatitis Report 2024. World Health Organization: 2024.
- Bartenschlager, R.; Ahlborn-Laake, L.; Yasargil, K.; Mous, J.; Jacobsen, H. Substrate determinants for cleavage in cis and in trans by the hepatitis C virus NS3 proteinase. J Virol 1995, 69, 198–205. [Google Scholar] [CrossRef]
- Grakoui, A.; Wychowski, C.; Lin, C.; Feinstone, S.M.; Rice, C.M. Expression and identification of hepatitis C virus polyprotein cleavage products. J Virol 1993, 67, 1385–95. [Google Scholar] [CrossRef]
- Cox, A.L. Challenges and Promise of a Hepatitis C Virus Vaccine. Cold Spring Harb Perspect Med 2020, 10, (2).
- Guest, J.D.; Wang, R.; Elkholy, K.H.; Chagas, A.; Chao, K.L.; Cleveland, T.E. t.; Kim, Y.C.; Keck, Z.Y.; Marin, A.; Yunus, A.S.; et al. Design of a native-like secreted form of the hepatitis C virus E1E2 heterodimer. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar] [CrossRef]
- Wang, R.; Suzuki, S.; Guest, J.D.; Heller, B.; Almeda, M.; Andrianov, A.K.; Marin, A.; Mariuzza, R.A.; Keck, Z.Y.; Foung, S.K.H.; et al. Induction of broadly neutralizing antibodies using a secreted form of the hepatitis C virus E1E2 heterodimer as a vaccine candidate. Proc Natl Acad Sci U S A 2022, 119, e2112008119. [Google Scholar] [CrossRef]
- Toth, E.A.; Chagas, A.; Pierce, B.G.; Fuerst, T.R. Structural and Biophysical Characterization of the HCV E1E2 Heterodimer for Vaccine Development. Viruses 2021, 13. [Google Scholar] [CrossRef]
- Borgia, S.M.; Hedskog, C.; Parhy, B.; Hyland, R.H.; Stamm, L.M.; Brainard, D.M.; Subramanian, M.G.; McHutchison, J.G.; Mo, H.; Svarovskaia, E.; et al. Identification of a novel hepatitis C virus genotype from Punjab, India: Expanding classification of hepatitis C virus into 8 genotypes. Journal of Infectious Diseases 2018, 218, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Duggal, P.; Thio, C.L.; Wojcik, G.L.; Goedert, J.J.; Mangia, A.; Latanich, R.; Kim, A.Y.; Lauer, G.M.; Chung, R.T.; Peters, M.G.; et al. Genome-wide association study of spontaneous resolution of hepatitis C virus infection: data from multiple cohorts. Annals of internal medicine 2013, 158, 235–45. [Google Scholar] [CrossRef] [PubMed]
- Pestka, J.M.; Zeisel, M.B.; Blaser, E.; Schurmann, P.; Bartosch, B.; Cosset, F.L.; Patel, A.H.; Meisel, H.; Baumert, J.; Viazov, S.; et al. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc Natl Acad Sci U S A 2007, 104, 6025–30. [Google Scholar] [CrossRef] [PubMed]
- Osburn, W.O.; Snider, A.E.; Wells, B.L.; Latanich, R.; Bailey, J.R.; Thomas, D.L.; Cox, A.L.; Ray, S.C. Clearance of hepatitis C infection is associated with the early appearance of broad neutralizing antibody responses. Hepatology 2014, 59, 2140–51. [Google Scholar] [CrossRef] [PubMed]
- Logvinoff, C.; Major, M.E.; Oldach, D.; Heyward, S.; Talal, A.; Balfe, P.; Feinstone, S.M.; Alter, H.; Rice, C.M.; McKeating, J.A. Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc Natl Acad Sci U S A 2004, 101, 10149–54. [Google Scholar] [CrossRef]
- Huang, X.-j.; Lü, X.; Lei, Y.-f.; Yang, J.; Yao, M.; Lan, H.-y.; Zhang, J.-m.; Jia, Z.-s.; Yin, W.; Xu, Z.-k. Cellular immunogenicity of a multi-epitope peptide vaccine candidate based on hepatitis C virus NS5A, NS4B and core proteins in HHD-2 mice. J. Virol. Methods 2013, 189, 47–52. [Google Scholar] [CrossRef]
- Martínez-Donato, G.; Piniella, B.; Aguilar, D.; Olivera, S.; Pérez, A.; Castañedo, Y.; Alvarez-Lajonchere, L.; Dueñas-Carrera, S.; Lee, J.W.; Burr, N.; et al. Protective T Cell and Antibody Immune Responses against Hepatitis C Virus Achieved Using a Biopolyester-Bead-Based Vaccine Delivery System. Clinical and Vaccine Immunology 2016, 23, 370–378. [Google Scholar] [CrossRef]
- Roohvand, F.; Aghasadeghi, M.-R.; Sadat, S.M.; Budkowska, A.; Khabiri, A.-R. HCV core protein immunization with Montanide/CpG elicits strong Th1/Th2 and long-lived CTL responses. Biochem. Biophys. Res. Commun. 2007, 354, 641–649. [Google Scholar] [CrossRef]
- Guest, J.D.; Pierce, B.G. Structure-Based and Rational Design of a Hepatitis C Virus Vaccine. Viruses 2021, 13, 837. [Google Scholar] [CrossRef]
- Fuenmayor, J.; Godia, F.; Cervera, L. Production of virus-like particles for vaccines. N Biotechnol 2017, (Pt B), 174–180. [Google Scholar] [CrossRef]
- Baumert, T.F.; Ito, S.; Wong, D.T.; Liang, T.J. Hepatitis C virus structural proteins assemble into viruslike particles in insect cells. J Virol 1998, 72, 3827–36. [Google Scholar] [CrossRef] [PubMed]
- Masavuli, M.G.; Wijesundara, D.K.; Torresi, J.; Gowans, E.J.; Grubor-Bauk, B. Preclinical Development and Production of Virus-Like Particles As Vaccine Candidates for Hepatitis C. Frontiers in microbiology 2017, 8, 2413. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, F.; Rostami, S.; Meshkat, Z. Progress in the development of vaccines for hepatitis C virus infection. World journal of gastroenterology 2015, 21, 11984–2002. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Escobar, E.; Roingeard, P.; Beaumont, E. Current Hepatitis C Vaccine Candidates Based on the Induction of Neutralizing Antibodies. Viruses, 2023; 15. [Google Scholar]
- Christiansen, D.; Earnest-Silveira, L.; Grubor-Bauk, B.; Wijesundara, D.K.; Boo, I.; Ramsland, P.A.; Vincan, E.; Drummer, H.E.; Gowans, E.J.; Torresi, J. Pre-clinical evaluation of a quadrivalent HCV VLP vaccine in pigs following microneedle delivery. Sci Rep 2019, 9, 9251. [Google Scholar] [CrossRef]
- Lechmann, M.; Murata, K.; Satoi, J.; Vergalla, J.; Baumert, T.F.; Liang, T.J. Hepatitis C virus-like particles induce virus-specific humoral and cellular immune responses in mice. Hepatology 2001, 34, 417–23. [Google Scholar] [CrossRef]
- Jeong, S.H.; Qiao, M.; Nascimbeni, M.; Hu, Z.; Rehermann, B.; Murthy, K.; Liang, T.J. Immunization with hepatitis C virus-like particles induces humoral and cellular immune responses in nonhuman primates. J Virol 2004, 78, 6995–7003. [Google Scholar] [CrossRef]
- Earnest-Silveira, L.; Christiansen, D.; Herrmann, S.; Ralph, S.A.; Das, S.; Gowans, E.J.; Torresi, J. Large scale production of a mammalian cell derived quadrivalent hepatitis C virus like particle vaccine. J Virol Methods 2016, 236, 87–92. [Google Scholar] [CrossRef]
- Earnest-Silveira, L.; Chua, B.; Chin, R.; Christiansen, D.; Johnson, D.; Herrmann, S.; Ralph, S.A.; Vercauteren, K.; Mesalam, A.; Meuleman, P.; et al. Characterization of a hepatitis C virus-like particle vaccine produced in a human hepatocyte-derived cell line. J Gen Virol 2016, 97, 1865–76. [Google Scholar] [CrossRef]
- Polakos, N.K.; Drane, D.; Cox, J.; Ng, P.; Selby, M.J.; Chien, D.; O'Hagan, D.T.; Houghton, M.; Paliard, X. Characterization of hepatitis C virus core-specific immune responses primed in rhesus macaques by a nonclassical ISCOM vaccine. J. Immunol. 2001, 166, 3589–98. [Google Scholar] [CrossRef]
- Drane, D.; Maraskovsky, E.; Gibson, R.; Mitchell, S.; Barnden, M.; Moskwa, A.; Shaw, D.; Gervase, B.; Coates, S.; Houghton, M.; et al. Priming of CD4+ and CD8+ T cell responses using a HCV core ISCOMATRIX™ vaccine: A phase I study in healthy volunteers. Hum. Vaccines 2009, 5, 151–157. [Google Scholar] [CrossRef]
- Eshaghi, B.; Fofana, J.; Nodder, S.B.; Gummuluru, S.; Reinhard, B.M. Virus-Mimicking Polymer Nanoparticles Targeting CD169+ Macrophages as Long-Acting Nanocarriers for Combination Antiretrovirals. ACS Appl. Mater. Interfaces 2022, 14, 2488–2500. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-Y.; Li, X.; Wang, Z.-G.; Liu, S.-L. Virus-mimicking nanosystems: from design to biomedical applications. Chem. Soc. Rev. 2023, 52, 8481–8499. [Google Scholar] [CrossRef] [PubMed]
- Somiya, M.; Kuroda, S. i. Development of a virus-mimicking nanocarrier for drug delivery systems: The bio-nanocapsule. Adv. Drug Delivery Rev. 2015, 95, 77–89. [Google Scholar] [CrossRef]
- Li, X.; Liu, S.; Yin, P.; Chen, K. Enhanced immune responses by virus-mimetic polymeric nanostructures against infectious diseases. Front. Immunol. 2022, 12, 804416. [Google Scholar] [CrossRef]
- Lou, B.; De Beuckelaer, A.; Boonstra, E.; Li, D.; De Geest, B.G.; De Koker, S.; Mastrobattista, E.; Hennink, W.E. Modular core-shell polymeric nanoparticles mimicking viral structures for vaccination. J. Controlled Release 2019, 293, 48–62. [Google Scholar] [CrossRef]
- Somiya, M.; Liu, Q.; Kuroda, S. i. Current progress of virus-mimicking nanocarriers for drug delivery. Nanotheranostics 2017, 1, 415. [Google Scholar] [CrossRef]
- van Rijn, P.; Schirhagl, R. Viruses, Artificial Viruses and Virus-Based Structures for Biomedical Applications. Adv. Healthcare Mater. 2016, 5, 1386–1400. [Google Scholar] [CrossRef]
- Liu, C.; Xu, H.; Sun, Y.; Zhang, X.; Cheng, H.; Mao, S. Design of Virus-Mimicking Polyelectrolyte Complexes for Enhanced Oral Insulin Delivery. J. Pharm. Sci. 2019, 108, 3408–3415. [Google Scholar] [CrossRef]
- Lee, C.; Jeong, J.; Lee, T.; Zhang, W.; Xu, L.; Choi, J.E.; Park, J.H.; Song, J.K.; Jang, S.; Eom, C.-Y.; et al. Virus-mimetic polymer nanoparticles displaying hemagglutinin as an adjuvant-free influenza vaccine. Biomaterials 2018, 183, 234–242. [Google Scholar] [CrossRef]
- Andrianov, A.K.; Langer, R. Polyphosphazene immunoadjuvants: Historical perspective and recent advances. J. Controlled Release 2021, 329, 299–315. [Google Scholar] [CrossRef]
- Chand, D.J.; Magiri, R.B.; Wilson, H.L.; Mutwiri, G.K. Polyphosphazenes as Adjuvants for Animal Vaccines and Other Medical Applications. Front. Bioeng. Biotechnol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Hlushko, R.; Pozharski, E.; Prabhu, V.M.; Andrianov, A.K. Directly visualizing individual polyorganophosphazenes and their single-chain complexes with proteins. Commun. Mater. 2024, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- Marin, A.; Kethanapalli, S.H.; Andrianov, A.K. Immunopotentiating Polyphosphazene Delivery Systems: Supramolecular Self-Assembly and Stability in the Presence of Plasma Proteins. Mol. Pharm. 2024, 21, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Leyva-Grado, V.H.; Marin, A.; Hlushko, R.; Yunus, A.S.; Promeneur, D.; Luckay, A.; Lazaro, G.G.; Hamm, S.; Dimitrov, A.S.; Broder, C.C.; et al. Nano-Assembled Polyphosphazene Delivery System Enables Effective Intranasal Immunization with Nipah Virus Subunit Vaccine. ACS Applied Bio Materials 2024, 7, 4133–4141. [Google Scholar] [CrossRef]
- Andrianov, A.K.; Marin, A.; Fuerst, T.R. Molecular-Level Interactions of Polyphosphazene Immunoadjuvants and Their Potential Role in Antigen Presentation and Cell Stimulation. Biomacromolecules 2016, 17, 3732–3742. [Google Scholar] [CrossRef]
- Vasilakos, J.P.; Tomai, M.A. The use of Toll-like receptor 7/8 agonists as vaccine adjuvants. Expert Rev. Vaccines 2013, 12, 809–819. [Google Scholar] [CrossRef]
- Tomai, M.A.; Vasilakos, J.P. TLR-7 and -8 agonists as vaccine adjuvants. Expert Rev. Vaccines 2011, 10, 405–407. [Google Scholar] [CrossRef]
- Bhagchandani, S.; Johnson, J.A.; Irvine, D.J. Evolution of Toll-like receptor 7/8 agonist therapeutics and their delivery approaches: From antiviral formulations to vaccine adjuvants. Adv. Drug Delivery Rev. 2021, 175, 113803. [Google Scholar] [CrossRef]
- Dowling, D.J. Recent Advances in the Discovery and Delivery of TLR7/8 Agonists as Vaccine Adjuvants. ImmunoHorizons 2018, 2, 185–197. [Google Scholar] [CrossRef]
- Midgard, H.; Bjøro, B.; Mæland, A.; Konopski, Z.; Kileng, H.; Damås, J.K.; Paulsen, J.; Heggelund, L.; Sandvei, P.K.; Ringstad, J.O.; et al. Hepatitis C reinfection after sustained virological response. J. Hepatol. 2016, 64, 1020–1026. [Google Scholar] [CrossRef]
- Andrianov, A.K.; Marin, A.; Wang, R.; Karauzum, H.; Chowdhury, A.; Agnihotri, P.; Yunus, *!!! REPLACE !!!*; Abdul, *!!! REPLACE !!!*; Mariuzza, R.A.; Fuerst, T.R. Supramolecular assembly of Toll-like receptor 7/8 agonist into multimeric water-soluble constructs enables superior immune stimulation in vitro and in vivo. ACS Appl. Bio Mater. 2020, 3, 3187–3195. [Google Scholar] [CrossRef] [PubMed]
- Messaud, F.A.; Sanderson, R.D.; Runyon, J.R.; Otte, T.; Pasch, H.; Williams, S.K.R. An overview on field-flow fractionation techniques and their applications in the separation and characterization of polymers. Prog. Polym. Sci. 2009, 34, 351–368. [Google Scholar] [CrossRef]
- Pitkänen, L.; Striegel, A.M. AF4/MALS/QELS/DRI characterization of regular star polymers and their “span analogs”. Analyst 2014, 139, 5843–5851. [Google Scholar] [CrossRef] [PubMed]
- Andrianov, A.K.; Marin, A.; Wang, R.; Chowdhury, A.; Agnihotri, P.; Yunus, A.S.; Pierce, B.G.; Mariuzza, R.A.; Fuerst, T.R. In Vivo and In Vitro Potency of Polyphosphazene Immunoadjuvants with Hepatitis C Virus Antigen and the Role of Their Supramolecular Assembly. Mol. Pharm. 2021, 18, 726–734. [Google Scholar] [CrossRef]
- Weissenberger, G.; Henderikx, R.J.M.; Peters, P.J. Understanding the invisible hands of sample preparation for cryo-EM. Nat. Methods 2021, 18, 463–471. [Google Scholar] [CrossRef]
- Yip, K.M.; Fischer, N.; Paknia, E.; Chari, A.; Stark, H. Atomic-resolution protein structure determination by cryo-EM. Nature 2020, 587, 157–161. [Google Scholar] [CrossRef]
- Cheng, Y. Single-particle cryo-EM - How did it get here and where will it go. Science 2018, 361, 876–880. [Google Scholar] [CrossRef]
- Gopal, A.; Zhou, Z.H.; Knobler, C.M.; Gelbart, W.M. Visualizing large RNA molecules in solution. RNA 2012, 18, 284–99. [Google Scholar] [CrossRef]
- Lyu, Z.; Yao, L.; Chen, W.; Kalutantirige, F.C.; Chen, Q. Electron Microscopy Studies of Soft Nanomaterials. Chem. Rev. 2023, 123, 4051–4145. [Google Scholar] [CrossRef]
- Wang, F.; Gnewou, O.; Solemanifar, A.; Conticello, V.P.; Egelman, E.H. Cryo-EM of Helical Polymers. Chem. Rev. 2022, 122, 14055–14065. [Google Scholar] [CrossRef]
- Mai, D.J.; Schroeder, C.M. 100th Anniversary of Macromolecular Science Viewpoint: Single-Molecule Studies of Synthetic Polymers. ACS Macro Lett. 2020, 9, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Parry, A.L.; Bomans, P.H.H.; Holder, S.J.; Sommerdijk, N.A.J.M.; Biagini, S.C.G. Cryo Electron Tomography Reveals Confined Complex Morphologies of Tripeptide-Containing Amphiphilic Double-Comb Diblock Copolymers. Angew. Chem. Int. Ed. 2008, 47, 8859–8862. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ting, J.M.; Tirrell, M.V. Mechanism of Dissociation Kinetics in Polyelectrolyte Complex Micelles. Macromolecules 2020, 53, 102–111. [Google Scholar] [CrossRef]
- Lueckheide, M.; Vieregg, J.R.; Bologna, A.J.; Leon, L.; Tirrell, M.V. Structure–Property Relationships of Oligonucleotide Polyelectrolyte Complex Micelles. Nano Letters 2018, 18, 7111–7117. [Google Scholar] [CrossRef] [PubMed]
- Marras, A.E.; Vieregg, J.R.; Ting, J.M.; Rubien, J.D.; Tirrell, M.V. Polyelectrolyte Complexation of Oligonucleotides by Charged Hydrophobic—Neutral Hydrophilic Block Copolymers. Polymers 2019, 11, 83. [Google Scholar] [CrossRef]
- Yu, X.; Qiao, M.; Atanasov, I.; Hu, Z.; Kato, T.; Liang, T.J.; Zhou, Z.H. Cryo-electron microscopy and three-dimensional reconstructions of hepatitis C virus particles. Virology 2007, 367, 126–134. [Google Scholar] [CrossRef]
- Seder, R.A.; Darrah, P.A.; Roederer, M. T-cell quality in memory and protection: implications for vaccine design. Nature reviews. Immunology 2008, 8, 247–58. [Google Scholar] [CrossRef]
- Bouveret Le Cam, N.N.; Ronco, J.; Francon, A.; Blondeau, C.; Fanget, B. Adjuvants for influenza vaccine. Res. Immunol. 1998, 149, 19–23. [Google Scholar] [CrossRef]
- Ison, M.G.; Mills, J.; Openshaw, P.; Zambon, M.; Osterhaus, A.; Hayden, F. Current research on respiratory viral infections: Fourth International Symposium. Antiviral Res. 2002, 55, 227–278. [Google Scholar] [CrossRef]
- O'Connell, R.J.; Excler, J.-L.; Polonis, V.R.; Ratto-Kim, S.; Cox, J.; Jagodzinski, L.L.; Liu, M.; Wieczorek, L.; McNeil, J.G.; El-Habib, R. Safety and Immunogenicity of a randomized Phase I prime-boost trial with ALVAC-HIV (vCP205) and Oligomeric gp160 MN/LAI-2 Adjuvanted in Alum or Polyphosphazene. J. Infect. Dis. 2016, 213, 1946–1954. [Google Scholar] [CrossRef]
- O'Connell, R.; Polonis, V.; Ratto-Kim, S.; Cox, J.; Jagodzinski, L.; Malia, J.; Michael, N.; Excler, J.; Robb, M.; Kim, J. Safety and immunogenicity of a randomized phase I prime-boost trial with ALVAC-HIV (vCP205) and gp160 MN/LAI-2 adjuvanted in alum or polyphosphazene. Retrovirology 2012, 2 (Suppl 2)), O50. [Google Scholar] [CrossRef]
- Laera, D.; HogenEsch, H.; O’Hagan, D.T. Aluminum Adjuvants—‘Back to the Future’. Pharmaceutics 2023, 15, 1884. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; He, P.; Guo, D.; Chen, K.; Hu, Z.; Zou, Y. Research Progress of Aluminum Phosphate Adjuvants and Their Action Mechanisms. Pharmaceutics 2023, 15, 1756. [Google Scholar] [CrossRef] [PubMed]
- Jully, V.; Mathot, F.; Moniotte, N.; Préat, V.; Lemoine, D. Mechanisms of Antigen Adsorption Onto an Aluminum-Hydroxide Adjuvant Evaluated by High-Throughput Screening. J. Pharm. Sci. 2016, 105, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Liu, G. Nano alum: A new solution to the new challenge. Hum. Vaccines Immunother. 2022, 18, 2060667. [Google Scholar] [CrossRef]
- Kurzątkowski, W.; Kartoğlu, Ü.; Górska, P.; Główka, M.; Woźnica, K.; Zasada, A.A.; Szczepańska, G.; Trykowski, G.; Gniadek, M.; Donten, M. Physical and chemical changes in Alhydrogel™ damaged by freezing. Vaccine 2018, 36, 6902–6910. [Google Scholar] [CrossRef]
- Mardliyati, E.; Hawa Syaifie, P.; El Muttaqien, S.; Ria Setyawati, D. Nanoscale alum-based adjuvants: Current status and future prospects. Materials Today: Proceedings.
- Reed, S.G.; Orr, M.T.; Fox, C.B. Key roles of adjuvants in modern vaccines. Nat Med 2013, 19, 1597–608. [Google Scholar] [CrossRef]
- Kayesh, M.E.H.; Kohara, M.; Tsukiyama-Kohara, K. TLR agonists as vaccine adjuvants in the prevention of viral infections: an overview. Frontiers in microbiology 2023, 14, 1249718. [Google Scholar] [CrossRef]
- Andrianov, A.K.; Marin, A.; Wang, R.; Karauzum, H.; Chowdhury, A.; Agnihotri, P.; Yunus, A.S.; Mariuzza, R.A.; Fuerst, T.R. Supramolecular Assembly of Toll-like Receptor 7/8 Agonist into Multimeric Water-Soluble Constructs Enables Superior Immune Stimulation In Vitro and In Vivo. ACS Applied Bio Materials 2020, 3, 3187–3195. [Google Scholar] [CrossRef]
- Kannanganat, S.; Ibegbu, C.; Chennareddi, L.; Robinson, H.L.; Amara, R.R. Multiple-cytokine-producing antiviral CD4 T cells are functionally superior to single-cytokine-producing cells. J Virol 2007, 81, 8468–76. [Google Scholar] [CrossRef]
- Pierce, B.G.; Felbinger, N.; Metcalf, M.; Toth, E.A.; Ofek, G.; Fuerst, T.R. Hepatitis C Virus E1E2 Structure, Diversity, and Implications for Vaccine Development. Viruses 2024, 16. [Google Scholar] [CrossRef]
- Vijayamahantesh, V.; Patra, T.; Meyer, K.; Alameh, M.G.; Reagan, E.K.; Weissman, D.; Ray, R. Modified E2 Glycoprotein of Hepatitis C Virus Enhances Proinflammatory Cytokines and Protective Immune Response. J Virol 2022, 96, e0052322. [Google Scholar] [CrossRef]
Figure 1.
Selection of polyphosphazene delivery vehicle. (a) Chemical structure and schematic presentation of PCPP, (b) DLS profiles of PCPP and its complexes with core protein, (c) degree of aggregation determined by DLS as a function of core protein concentration for PCPP and PCPP-PEG, (d) chemical structure and schematic presentation of PCPP-PEG and (e) DLS profiles of PCPP-PEG and its complexes with core protein (25 g E1E2, 50 g PCPP or PCPP-PEG, PBS, pH 7.4).
Figure 1.
Selection of polyphosphazene delivery vehicle. (a) Chemical structure and schematic presentation of PCPP, (b) DLS profiles of PCPP and its complexes with core protein, (c) degree of aggregation determined by DLS as a function of core protein concentration for PCPP and PCPP-PEG, (d) chemical structure and schematic presentation of PCPP-PEG and (e) DLS profiles of PCPP-PEG and its complexes with core protein (25 g E1E2, 50 g PCPP or PCPP-PEG, PBS, pH 7.4).
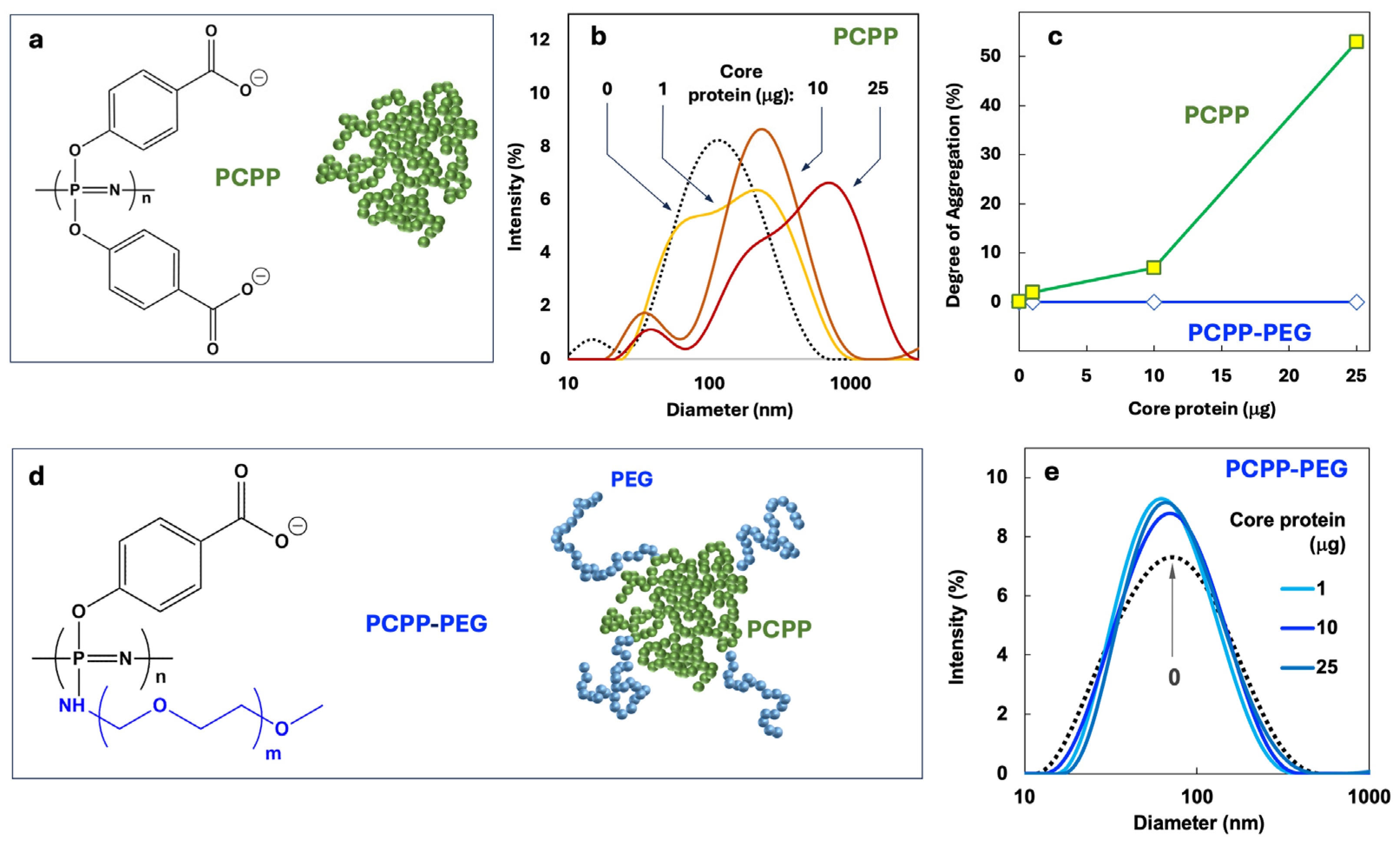
Figure 2.
Preparation and characterization of VMPNs. (a) Schematic diagram of VMPN nano-assembly showing sequential steps of component addition, (b) AF4 profiles showing protein-polymer binding and (c) DLS profiles of VMPNs contrasted with protein-free polymer.
Figure 2.
Preparation and characterization of VMPNs. (a) Schematic diagram of VMPN nano-assembly showing sequential steps of component addition, (b) AF4 profiles showing protein-polymer binding and (c) DLS profiles of VMPNs contrasted with protein-free polymer.
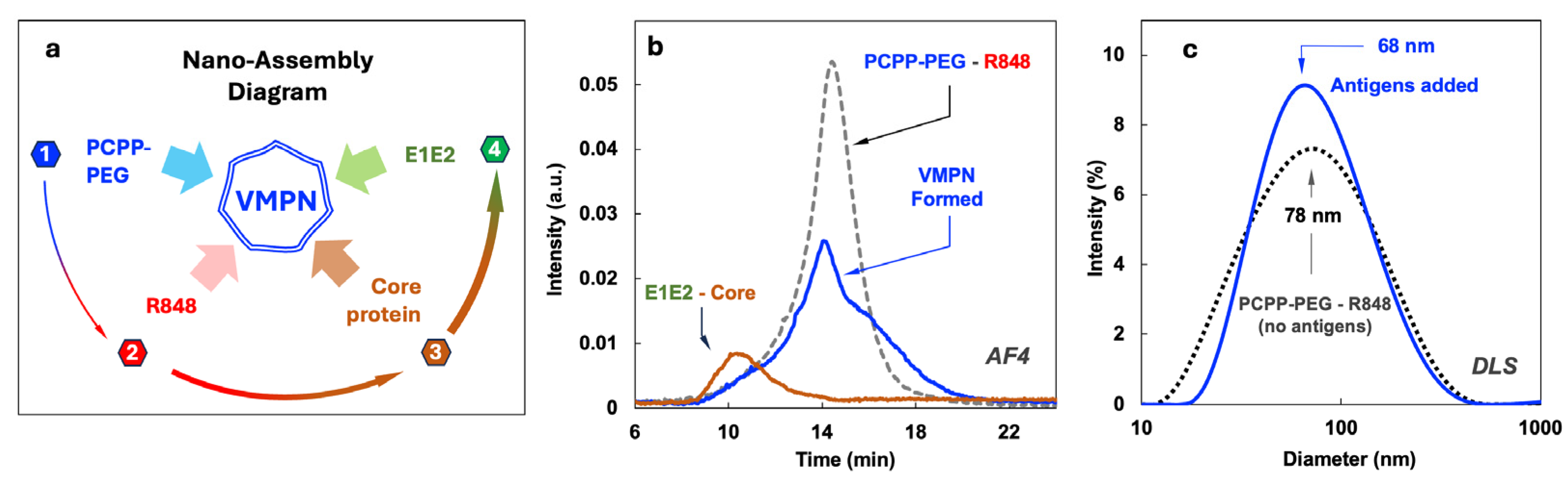
Figure 3.
CryoEM images of VMPNs in their vitrified state (50 mM phosphate buffer, pH 7.4).
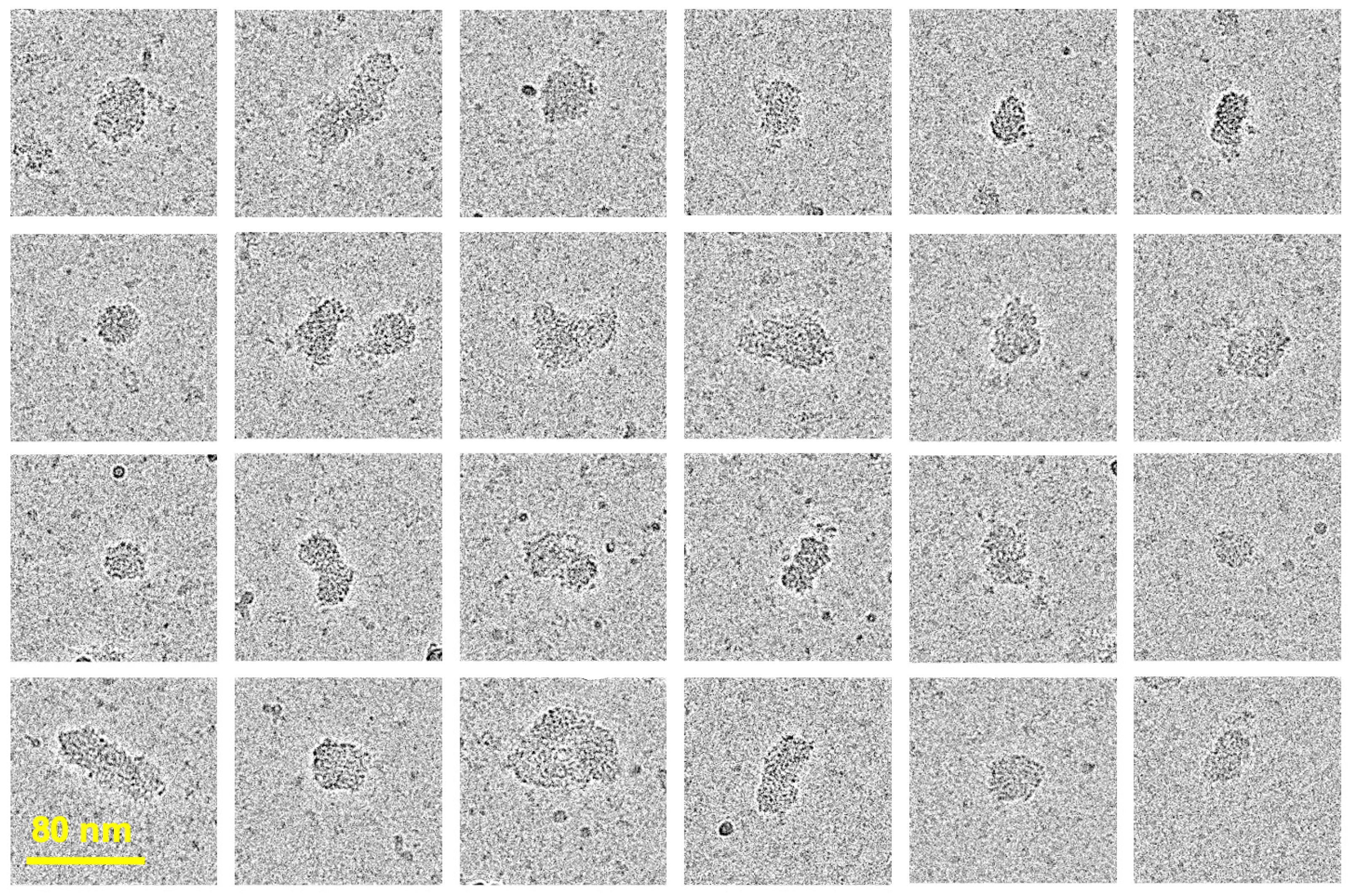
Figure 4.
Immunogenicity assessment by ELISA of antibodies induced in immunized mice at day 56 after immunization. (a) Total IgG titers, (b) IgG1 and IgG2a titers, and (c) the ratio of IgG2a to IgG1 for each immunogen. Pooled sera from the indicated groups were analyzed. Endpoint titers were calculated by curve fitting in GraphPad software with endpoint optical density defined as four times the highest absorbance value of day 0 sera.
Figure 4.
Immunogenicity assessment by ELISA of antibodies induced in immunized mice at day 56 after immunization. (a) Total IgG titers, (b) IgG1 and IgG2a titers, and (c) the ratio of IgG2a to IgG1 for each immunogen. Pooled sera from the indicated groups were analyzed. Endpoint titers were calculated by curve fitting in GraphPad software with endpoint optical density defined as four times the highest absorbance value of day 0 sera.

Figure 5.
Potency of neutralization of individual mouse sera against H77C (GT1a). Individual immunized mice sera were assessed for neutralization activities at day 56 and day 0. Serum dilutions were performed as three-fold dilutions starting at 1:150 for HCVpp neutralization. The experiment was performed in duplicate and the geometric mean of each group is denoted. P values were calculated using Kruskal-Wallis analysis of variance with Dunn’s multiple comparison test, and significant P values are shown (*P < 0.05).
Figure 5.
Potency of neutralization of individual mouse sera against H77C (GT1a). Individual immunized mice sera were assessed for neutralization activities at day 56 and day 0. Serum dilutions were performed as three-fold dilutions starting at 1:150 for HCVpp neutralization. The experiment was performed in duplicate and the geometric mean of each group is denoted. P values were calculated using Kruskal-Wallis analysis of variance with Dunn’s multiple comparison test, and significant P values are shown (*P < 0.05).
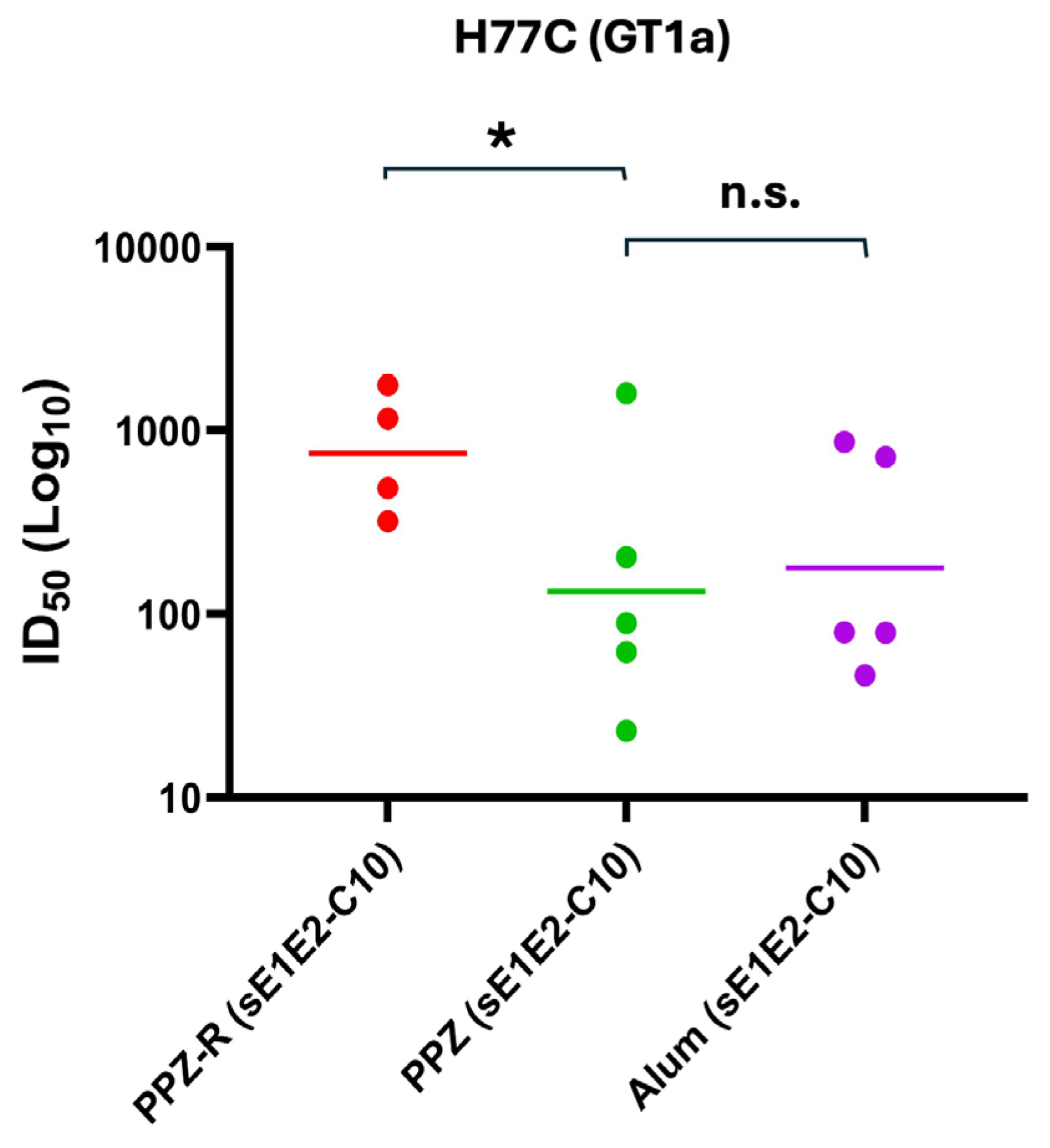
Figure 6.
T cell responses to PPZ-R adjuvanted HCV antigens. (a) Representative Intracellular Cytokine Staining FACS plots showing PPZ-R, PPZ or Alum vaccinated mice, stimulated as labelled on each column. Plots are gated on CD4+ T cells and (b) quantitation of the total percentage of IFNγ-producing CD4+ CD44-hi T cells each condition, after stimulation with core or E1E2 recombinant proteins.
Figure 6.
T cell responses to PPZ-R adjuvanted HCV antigens. (a) Representative Intracellular Cytokine Staining FACS plots showing PPZ-R, PPZ or Alum vaccinated mice, stimulated as labelled on each column. Plots are gated on CD4+ T cells and (b) quantitation of the total percentage of IFNγ-producing CD4+ CD44-hi T cells each condition, after stimulation with core or E1E2 recombinant proteins.
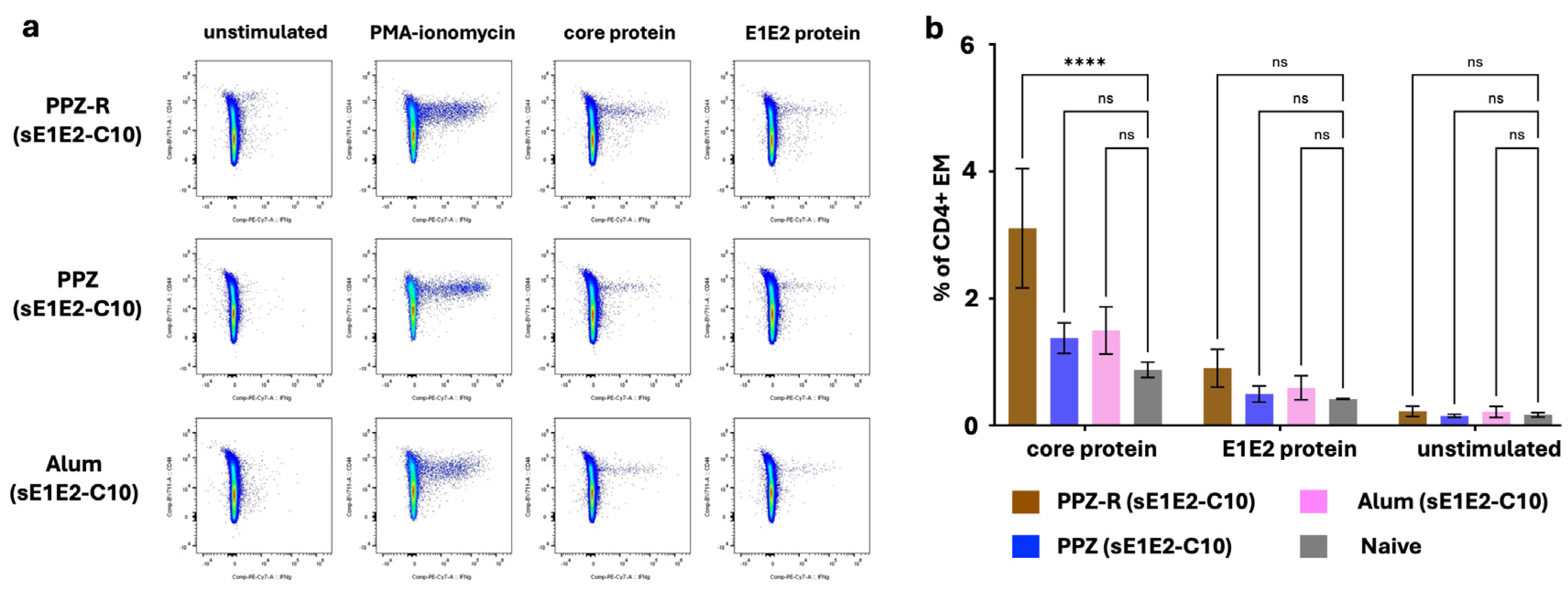

Table 1.
Immunization groups.
| Group | sE1E2-H445P (µg) |
Core (µg) |
Vehicle | Reference | ||
|---|---|---|---|---|---|---|
| PCPP-PEG (µg) |
R848 (µg) |
Alum (µg) |
||||
| 1 | 25 | - | 100 | 20 | - | PPZ-R (sE1E2) |
| 2 | 25 | 1 | 100 | 20 | - | PPZ-R (sE1E2-C1) |
| 3 | 25 | 10 | 100 | 20 | - | PPZ-R (sE1E2-C10) |
| 4 | 25 | 25 | 100 | 20 | - | PPZ-R (sE1E2-C25) |
| 5 | 25 | 10 | 100 | 20 | - | PPZ (sE1E2-C10) |
| 6 | 25 | 10 | - | - | 35 | Alum (sE1E2-C10) |
| 7 | 25 | 10 | - | - | - | (sE1E2-C10) |
Table 2.
Homologous and heterologous neutralization serum ID50 values.
| HCVpp * | PPZ-R (sE1E2-C10) | PPZ (sE1E2-C10) | Alum (sE1E2-C10) |
|---|---|---|---|
| GT1a | 587.9 | 173.3 | 184.4 |
| GT1b | 36.7 | 65.6 | 72.4 |
| GT2a | 125.9 | 113.7 | 102.0 |
| GT2b | 73.4 | 109.9 | 63.4 |
* Please see Section 3.5 for more details on genotypes tested here.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



