
Submitted:
15 August 2025
Posted:
18 August 2025
You are already at the latest version
Abstract
Background: The quest for well-defined immunoadjuvants remains one of the highest priorities for the successful development of effective vaccines. Combination adjuvants, which are designed to integrate both the ability to activate a variety of immune mechanisms and synergistically improve delivery of vaccine components, are well-positioned to address the unmet needs. The development of preventive vaccine against hepatitis C virus (HCV) – a major public health concern, is a particular instance in which choice of the immunoadjuvant is of utmost importance. Methods: We assembled a lipid A Toll-like receptor 4 (TLR4) agonist BECC438 and TLR7/8 agonist resiquimod (R848) on a polyphosphazene macromolecule (PCPP) to create a nanoscale immunoadjuvant-vaccine delivery system: PCPP-R+BECC438. This aqueous-based system was formulated with HCV sE2 antigen, and the resulting vaccine candidate was evaluated in vivo for the ability to induce immune responses. Results: Co-assembly of adjuvants resulted in a visually clear aqueous system of nanoscale dimensions, monomodal size distribution and entropy driven interactions between components. Intramuscular immunization of mice with HCV sE2 antigen formulated in polyphosphazene-based nano-system induced ten-fold higher IgG and IgG2a titers than the antigen adjuvanted with BECC438 alone. PCPP-R+BECC438 formulated HCV sE2 also produced statistically significant improvements in IgG2a/IgG1 ratio and more robust HCVpp neutralization ID50 titers than control formulations. Conclusions: Polyphosphazene-assembled adjuvant nano-system promotes in vivo immune responses of enhanced quantity and quality of antibodies with increased potency of HCV neutralization.
Keywords:
Hepatitis C virus
; vaccine adjuvants
; polyphosphazenes
; vaccine delivery
; Toll-like receptor agonist
; intermolecular interactions
1. Introduction
Immunoadjuvants are key components in vaccine formulations by enhancing and modulating immune responses to vaccines [1,2,3,4,5]. Immunization using purified protein antigens alone often elicits weak antibody responses and minimal T cell activation, requiring multiple booster doses to provide protection. Therefore, generation of a toolbox of adjuvants with well-defined profiles, which can be applied to new vaccines against diverse pathogens, is one of the highest priorities in the field. According to their mechanism of action, adjuvants are commonly classified as immunostimulating molecules, vaccine delivery systems, or a combination of both. Immunostimulating molecules mainly activate the innate immune receptors, including Toll-like receptors (TLRs) or Nucleotide-binding and oligomerization domain (NOD-like) receptors, generating signals that drive the proper activation of downstream adaptive immune responses. Vaccine delivery systems are designed to improve stability and presentation of the vaccine to the immune system. Combination adjuvants integrate both components that act synergistically by activating a variety of immune mechanisms and improving their delivery. TLR4 agonists, such as lipid A-based molecules, are among the most potent immunostimulating compounds [6].
Monophosphoryl lipid A (MPL), known for enhancing Th1 humoral and cell-mediated immune responses to protein antigens, is currently being used in numerous vaccine trials and was approved by the Food and Drug Administration (FDA) as part of Shingrix and Cervarix vaccines [7,8]. MPL is produced from Salmonella minnesota R595 lipid A using a multi-step detoxification process. Instead of the chemical treatments employed in the generation of MPL, a different approach was undertaken in the synthesis of MPL-like molecules termed Bacterial Enzymatic Combinatorial Chemistry (BECC). Using heterologous expression of various acyltransferases, phosphatases, deacylases, and glycosyltransferases in different bacterial backgrounds, we have been able to generate numerous lipid A structures, such as BECC438 with altered proinflammatory responses in humans and murine cells [9]. BECC lipid A molecules have proven potent adjuvant capabilities and, due to their lipophilic nature, present opportunities for innovative formulation strategies to improve their stability in aqueous dispersions [10]. These potent adjuvants can benefit from delivery vehicles, such as water-soluble polymers, emulsions, liposomes, and nanoparticles, for optimal formulation stability, effective display of TLR4 ligands, and association with vaccine antigen.
Polyphosphazenes immunoadjuvants are synthetic biodegradable macromolecules, which are composed of an inorganic phosphorus-nitrogen backbone and organic side groups containing anionic functionalities [11]. Polyphosphazenes are known to display intrinsic immunostimulatory activity both in vitro and in vivo [12,13,14]. However, perhaps even more important feature of these macromolecules is to act as vaccine delivery vehicles. This enabling functionality is largely supported by their remarkable ability to spontaneously self-assemble with vaccine antigens in aqueous solutions [15,16]. The resulting supramolecular complexes are of nano-scale dimensions and may be viewed as virus-mimicking structures in terms of their size and ability to present protein antigens [17]. Poly[di(carboxylatophenoxy)phosphazene] (PCPP) is an advanced polyphosphazene adjuvant, which has demonstrated safety and potency in several clinical trials [18,19,20,21,22]. In addition to antigen carrying capacity, PCPP can effectively bind Toll-like receptor (TLR) agonists, such as resiquimod, via counterion-polyelectrolyte interactions to both extend their half-life and provide association with the antigen [23]. The capacity of this macromolecule to bind to and transport other immunostimulating molecules remains understudied. Hepatitis C virus (HCV) is a major public health concern that infects more than 50 million people worldwide and is a leading cause of liver disease and cancer [24,25,26]. HCV infection is known as the “silent epidemic” because many people with the disease do not know they are infected. In the United States, more than 3.2 million Americans are infected, and yet as many as 75% are unaware that they carry the virus [27]. Highly effective direct-acting antivirals (DAAs) are available that can nearly eliminate the virus in HCV-infected individuals. However, these DAAs cannot provide protection against reinfection and associated disease later in life, and they have limited availability to people in low- and middle-income countries [28,29,30]. Therefore, the World Health Organization (WHO) has established a global strategy with a primary goal of reducing new HCV infections by 90% and associated deaths by 65% by the year 2030 [31]. Development of an effective vaccine is a critical step in addressing this major public health problem.
The genetic diversity of HCV is a major challenge for vaccine development, with eight known genotypes and more than one hundred subtypes identified to date [32]. The diversity of the virus is due to the lack of a proof-reading function of the virally encoded RNA-dependent RNA polymerase, leading to the extremely high sequence variability. As a result, the amino acid median sequence divergence within and between genotypes is approximately 23% and 33%, respectively [33]. Given this diversity, a prophylactic vaccine must be capable of inducing broadly reactive immune responses that can provide protection against a global pool of circulating viruses. Moreover, data from both chimpanzee and human studies have shown that induction of HCV-specific CD4+ and CD8+ T cells are important in the control of infection [34,35]. For these reasons, an effective vaccine must be capable of inducing both humoral and cellular responses in which the choice of the immunoadjuvant and delivery system is of utmost importance.
In this study, we combined BECC438, a lipid A TLR4 agonist, with PCPP or PCPP-resiquimod (PCPP-R) to derive binary adjuvants PCPP+BECC438 and PCPP-R+BECC438, respectively, that remain in an aqueous state. Here, PCPP scaffold served as the solubilizing excipient for BECC438, and stability was imparted via lyophilization, such that upon resuspension in PBS, the binary adjuvant remained visually clear and retained a nano-scale monomodal size distribution similar to PCPP. We tested the immunostimulatory effects of the binary adjuvant in vitro and in mice with HCV-sE2 antigen. Using immortalized mouse macrophages, the BECC438 component of the binary adjuvant retained immunostimulatory activity implying no antagonistic effect from complexing with PCPP, and at higher concentrations the binary adjuvant showed somewhat superior activity implying a possibility for dose-dependent synergy with further ratio optimizations. Intramuscular immunization of mice with HCV sE2 antigen formulated in PCPP-R+BECC438 (or PCPP-R) produced 10-fold higher IgG titers and IgG2a titers than BECC438 alone. PCPP-R+BECC438 also produced statistically significant differences in IgG2a/IgG1 ratio and more robust HCVpp neutralization ID50 titers that were two to three times higher than BECC438 and PCPP-R, respectively. Our experiments demonstrate that a combination of the novel BECC438 adjuvant with a water-soluble PCPP-based delivery system led to enhanced quantity and quality of antibodies with increased potency of HCV neutralization.
2. Materials and Methods
2.1. Protein Expression and Purification
The protein sE2 was transiently expressed using the Expi293 Expression System and the protocol provided by the manufacturer (ThermoFisher). The cells were cultured in a shaking incubator at 37 °C, 120 rpm, and 8% CO2 in Expi293 medium until reaching a cell density of 2.0 × 106 cells/mL. The transfection mixture contained a predetermined amount of plasmid, ExpiFectamine™ 293 Reagent and other transfection enhancers. The sE2 supernatant was harvested 72 hours after transfection then centrifuged in order to remove the suspended cells at 10,000rpm for 10 minutes and filtered using 0.22 μm filters. The protein was then purified from the supernatant using the sequential HisTrap Ni2+-NTA and Superdex 200 size-exclusion chromatography as described previously [36,37].
2.2. Preparation of BECC438
BECC438b was made and resuspended as previously described [9]. Briefly, BECC436 was grown in shaking culture at 26 °C for 18 h, during which time the culture reached an OD600 of 1.0–1.4. After pelleting bacteria from the liquid culture, lipooligosaccharide (LOS) was extracted from the pellet as previously described using a double hot phenol method [38]. Mass spectrometry was used to confirm the extracted lipid A structures (Bruker Microflex MALDI TOF, norharmane matrix, negative ion mode). The resulting LOS was designated BECC438b (b indicating biologically derived).
2.3. Preparation of PCPP-BECC438 Formulations
Aqueous-based formulations of PCPP and BECC438 were prepared at PCPP and BECC438 concentrations of 0.5 and 0.25 mg/mL, respectively. Lyophilized powder of BECC438 was suspended in endotoxin-free deionized water containing 0.02% triethanolamine (TEA) (Millipore Sigma, St. Louis, MO) to make a 0.5 mg/mL stock solution. The dispersion was vortexed and then sonicated in a Branson CPX2800H ultrasonic cleaner (Emerson Electric Co., St. Louis, MO) for approximately 10 min at room temperature. The obtained micellar solution of BECC438 was mixed at 1:1 (v/v) ratio with 1 mg/mL aqueous solution of PCPP (pH 7.4). Formulations were then lyophilized and re-suspended to a desirable concentration in a phosphate-buffered saline (PBS) (Thermo-Fisher Scientific, Grand Island, NY) at pH 7.4.
2.3. Dynamic Light Scattering (DLS) and Isothermal Titration Calorimetry (ITC) Analysis
DLS characterization of BECC438, PCPP and their lyophilized formulation re-dissolved in PBS (pH 7.4) was conducted using a Malvern Zetasizer Nano ZS instrument with data analysis carried out with version 7.10 Zetasizer Software (Malvern Instruments Ltd., Worcestershire, UK).
ITC measurements were conducted using a Nano ITC SV instrument (TA Instruments, Waters, New Castle, DE, USA) in an aqueous solution (50 mM phosphate buffer, pH 7.5) at 25 °C. BECC438 (2.0 mg/mL) was dispersed in an aqueous phase and placed into an isothermal cell of the instrument. Titration was carried out using a 250 μL syringe rotating at 200 rpm (5.0 mg/mL PCPP, 300 s delay between 8 μL injections). A heat release curve (μJ/s vs s) was integrated using NanoAnalyze software, version 3.12.5 (TA Instruments, Waters, New Castle, DE, USA).
2.4. In Vitro Evaluation of Immunostimulating Activity
In vitro evaluation was conducted using engineered mouse macrophages - RAW BLUE cells (InvivoGen, San Diego, CA) assays. Cells were maintained in culture in Dulbecco’s modified eagle medium containing glucose and L-glutamine (Thermo-Fisher Scientific, Grand Island, NY) supplemented with 10% fetal bovine serum (10%), penicillin streptomycin (1%) (Thermo-Fisher Scientific, Grand Island, NY) and normocin (100 µg/ml) (InvivoGen, San Diego, CA). The experiment was initiated by adding ternary formulations containing PCPP, BECC438 and E2 antigen in culture to an equal volume of RAW BLUE cells in 96 well plates (120,000 cells/well) and incubating them at 37 °C in carbon dioxide (5%) for 20 h. The level of the RAW BLUE cell activation was assessed spectrophotometrically by the conversion of SEAP substrate - p-nitrophenylphosphate (Millipore Sigma, St. Louis, MO). Culture supernatant was added to the reagent at one-to-ten ratio by volume. The absorbance was read at 405 nm using a ThermoScientific SpectraMax plate reader (Molecular Devices, San Jose, CA). All experiments were performed in triplicate.
2.5. In Vivo Studies
Six- to eight-week-old, Balb/c mice (Jackson Laboratories, ME, USA) were immunized intramuscularly (IM) in the hind, caudal thigh on day 0 (prime), followed by three boosts (Days 14, 28, and 42). Fifty µL of vaccines (sE2-PCPP-R, sE2-BECC438, sE2-PCPP-R+BECC438, sE2-no adjuvant) were used for each vaccination dose. The control group was immunized with sterile PBS. Terminal bleeds were performed on day 56 to obtain serum. Animals were housed in individually vented cages and had access to food and water ad libitum. All animal procedures were reviewed and approved by the University of Maryland, Baltimore Institutional Animal Care and Use Committee.
2.6. Enzyme-Linked Immunosorbent Assay (ELISA)
ELISAs were performed to determine the HCV E2 antibody binding profile of the immunized mice sera. The plates were initially coated with 5 µg/mL Galanthus Nivalis Lectin (GNL, Vector Laboratories) and then incubated at 4 °C overnight. The subsequent day, the plates were first washed with PBST (1x PBS with Tween 20 at 0.05% v:v) and blocked with a blocking buffer (1x PBS/2% dry milk/ 5% FBS), and then the sE2 antigen was immobilized at a concentration of 2 μg/mL, with incubation overnight at 4 °C. The plates were then blocked in the same manner as above. Sera was initially diluted 1:100, followed by seven consecutive 5-fold serial dilutions, and then incubated on the plate for 1 hour at room temperature. The antibodies were detected by a 1:5,000 dilution of HRP-conjugated anti-mouse IgG secondary antibody (Abcam) with TMB substrates (Bio-Rad Laboratories). The data was collected on a SpectraMax M3 microplate reader by measuring the absorbance at 450 nm. The endpoint titers were defined as four times the highest signal elicited by the preimmune sera and determined using a curve fitting program in GraphPad Prism software version 10 (Dotatics, San Diego, CA, USA).
2.7. HCV Pseudoparticles (HCVpp) Neutralization Assay
The neutralizing ability of the antibodies induced by immunization of the mice was assessed with HCV pseudoparticles (HCVpp). Huh7 cells derived from human hepatoma tissue were grown in DMEM 10% FBS and 1% Pen/Strep and were seeded onto a white 96-well plate (Corning) at 1.5 x 104 per well and were incubated in a CO2 incubator at 37 °C overnight. The next day, heat-inactivated sera or HmAbs were serially diluted and then incubated in HCVpp for 1 hour. After one hour, the mixture is transferred to the plates and then incubated for 5-6 hours. At the end of the incubation period, the HCVpp and antibody mixture is replaced with media. Seventy-two hours later, the plates are developed with 100 μL Bright-Glo (Promega) per well for 2 minutes at room temperature. The FLUOstar Omega plate reader (BMG Labtech), was used to detect luminescence and the data is retrieved from the Mars software. The percent neutralization is calculated relative to a Relative Luminescence Units (RLU) control which is the HCVpp containing PBS in place of the serum. The RLU sample is the mixture containing HCVpp as well as the serum. The calculation is as follows 100*(1-(RLU sample/ RLU control). The ID50, which is the dilution of sera that achieved 50% neutralization was determined with nonlinear regression curve fitting with GraphPad Prism software version 10 (Dotatics, San Diego, CA, USA). Neutralization assays involving pseudoparticles were performed under BSL-2 conditions.
2.8. Statistical Analysis
The P values between groups IgG2a/IgG1 were determined using the nonparametric Kruskal–Wallis analysis of variance with Dunn’s multiple comparison test. p < 0.05 was considered significant. Statistical analyses were performed using GraphPad Prism software version 10 (Dotatics, San Diego, CA, USA).
3. Results
3.1. PCPP Enables Water-Soluble Formulations of BECC438
The approach to the development of PCPP-enabled nano-scale formulations of BECC438 involved a two-step process. First, a micellar dispersion of lipid A molecules was prepared by mixing BECC438b with deionized water, adding triethanolamine, agitating and sonicating the mixture (Figure 1a, step (I)). The sub-micron size dispersion of BECC438 (Figure 1b) was then admixed to an aqueous solution of PCPP as a solubilizing excipient, and the resulting formulation was stabilized and dehydrated by lyophilization (Figure 1b, step (II)). Analysis of the resuspended lyophilized material in PBS (pH 7.4) by DLS revealed a monomodal size distribution in a nano-scale range, which resembled the profile of unmodified PCPP (Figure 1b). The formulation was visually clear and did not contain any submicron or micron-sized aggregates as detected by DLS.
PCPP and BECC438 were studied for potential interactions in an aqueous phase using an isothermal titration calorimetry (ITC) method. Figure 2a shows raw ITC signals of PCPP addition to BECC438 (red curve and points) as well as the heat of PCPP dilution (blue curve and points). As seen from the figure, polymer dilution generates signals that are below the baseline, indicating that this is an exothermic process. In contrast, the addition of PCPP to BECC438 results in an upward trend at the beginning of titration, which reveals a weakly endothermic reaction. For further data analysis, the heats of dilution were subtracted from the heats of adsorption (Figure 2b). The resulting curve shows a positive heat of absorbance, which suggests entropy-driven interactions between PCPP and BECC438.
In vitro assessment of formulations was conducted using engineered mouse macrophages (RAW BLUE cells). These cells are derived from mouse RAW 264.7 macrophages and harbor a secreted embryonic alkaline phosphatase (SEAP) reporter construct. SEAP is inducible by NF-kB and AP-1 transcription factors that are downstream of several pattern recognition receptors (PRRs) of the innate immune system. The analysis of immunostimulatory activity of PCPP-BECC438 combination in RAW BLUE cells showed that although PCPP was capable of augmenting the activity of the formulation partner, the activity of the binary formulation was mainly determined by BECC438 component (Figure 3). No adverse effect of PCPP on the immunostimulatory effect of lipid A molecule was detected. Notably, at high dilutions, PCPP-BECC438 appears to show a superior activity compared to individual components.
3.2. In Vivo Studies
Based on the results of immunoactivation profiles and DLS study, a combination PCPP-BECC438 adjuvant system was selected for in vivo studies with one adjustment to the study design. PCPP was used in its salt form with the TLR7/8 agonist resiquimod (R848). Previous studies demonstrated that although R848 was not effective in improving immune responses to HCV E2 secreted antigen (sE2) in mice due to poor PK/PD profile, PCPP can convert it into a multimeric form, and such modified (PCPP-R) form resulted in superior performance compared to PCPP alone [23]. The addition of R848 did not affect the homogeneity of formulation or its DLS profiles. The design of in vivo studies is shown in Table 1.
3.3. Evaluation of Serological Responses and Neutralization
To assess the immune response, mice were immunized with a formulation of sE2 antigen (refer to Table 1) in which the prime consisted of 50 μg sE2A and three boosts of 10 μg sE2 two weeks apart. A terminal bleed was taken at day 56 after the initial immunization. As shown in Figure 4a, the total IgG titers for both sE2-PCPP-R and sE2-PCPP-R+BECC438 groups are about 10-fold higher than those of BECC438 alone or the adjuvant-sE2 group. Similarly, the isotype-specific analysis of IgG1 (T helper 1, Th1) and IgG2a (T helper 2, Th2, a surrogate marker for the strength of the cellular immune response) shows that in comparison to other groups, the sE2-PCPP-R and sE2-PCPP-R+BECC438 have superior IgG1 titers (Figure 4b) and a significantly higher IgG2a titer (Figure 4c). Since BECC438 and the no-adjuvant group showed negligible Ig2a antibodies, the elicitation of IgG2a isotype antibodies can be largely attributed to PCPP-R component (Figure 4c). A further analysis shows that the combination adjuvant PCPP-R+BECC438 achieves a statistically significant difference in the ratio of IgG2a/IgG1 over sE2-PCPP-R (1.8-fold) and sE2-BECC438 (30-fold) using an uncorrected pairwise Kruskal-Wallis test of sE2-PCPP-R+BECC438 versus sE2-BECC438 (p<0.05) (Figure 4d). Thus, inclusion of BECC438 in the PCPP-R+BECC438 helped establish a more balanced Th1/Th2 response.
We next analyzed the serum from individual mice in each group for its ability to neutralize HCV pseudoparticles (HCVpp) made from the homologous GT1a (H77) strain from which the sE2 antigen was derived (Figure 5). Our results show that in comparison to the non-adjuvant group (sE2 only), individual mice in all adjuvant groups (PCPP-R, BECC-438, and PCPP-R+BECC438) produced neutralizing responses as measured by ID50 values (inhibitory dilution at which 50 percent neutralization is attained) with some mice in the binary adjuvant group showing higher titers over individual mice in other groups (Figure 5a). As shown in Figure 5b, the mean HCVpp ID50 values show that the titers for PCPP-R+BECC438 group is indeed 2-3 times higher than BECC438 or PCPP-R group, respectively. Although not statistically significant, the neutralization responses indicate a potential synergy for the binary adjuvant with HCV sE2 antigen.
4. Discussion
Binary formulations of lipids and polymers are widely exploited in pharmaceutical sciences [39]. The disordered amorphous phase created by the polymer can improve the solubility of sophisticated dosage forms of poorly soluble active pharmaceutical ingredients (APIs), such as lipids. To that end, the formation of non-covalent bonds between the polymer and API is important, as such interactions inhibit the potential for nucleation and crystallization of a lipophilic molecule [39]. Furthermore, polymers impart barriers, both through steric and electrostatic stabilization of a suspension, thereby preventing undesirable aggregation [40,41]. Nanosizing—an important pharmaceutical technique for reducing the size of colloidal suspensions of APIs commonly relies on the stabilizing effect of polymers. The resulting polymer-based nanosuspensions are then converted to a solid dosage form using lyophilization or other dehydrating techniques [39,40,41]. Aqueous formulations of hydrophobic lipid A molecules - BECC438 (Figure 1a), can typically be described as sub-micron to micron size micellar dispersions with excipient-dependent size distribution and stability. In contrast, PCPP is a large synthetic macromolecule, which forms true solutions in water with the hydrated polymer coil reaching hydrodynamic diameters in the range of 60-80 nm. Due to the presence of carboxylic acid moieties and aromatic rings in the PCPP structure (Figure 1a), this macromolecule is capable of forming supramolecular assemblies and complexes with various substrates via hydrogen bonds and hydrophobic interactions [15]. ITC results confirm the presence of interactions in the PCPP - BEC438 system. The biphasic pattern of titration, which involves exothermic and endothermic processes (Figure 2b), suggests two distinct stages in the mechanism of interactions between the polymer and the BECC438 dispersion. In the beginning, the process is exothermic, i.e., enthalpy-driven, which suggests bonding of PCPP macromolecules to the polar surface of BECC438 micelles. Since the heat of interactions is relatively weak, it can be assumed that this process is dominated by the formation of hydrogen bonds and/or hydrophobic interactions. Further saturation of the system with PCPP manifests in endothermic changes. This may be attributed to a breakdown of large BECC438 micelles with the re-assembly of small adjuvant molecules along polymer chains. As this endothermic process must be driven by entropy, the results suggest that BECC438 molecules associated with flexible PCPP chains are less constrained in their mobility compared to the same molecules physically entrapped in large micelles. In this context, PCPP acts not only as a vaccine delivery vehicle due to its binding of HCV vaccine antigens demonstrated previously [15,17], but also as an efficient vehicle for displaying BECC438 lipid molecules. Therefore, PCPP-enabled BECC438 formulations, which are obtained in a straightforward production process (Figure 1a), can be described as a nano-scale lipid-polymer system with a monomodal size distribution profile.
Development of effective vaccines has shown that there is an increasingly important dependence on the choice of adjuvants to stimulate the desired immune responses. Moreover, the choice of adjuvants can be further divided into two main categories: delivery systems and immune stimulators. Research efforts on the development of HCV vaccine candidates have largely focused on the search for potent immunoadjuvants in which side-by-side comparisons are relatively uncommon [42]. Alum (Alhydrogel), along with various emulsion-based systems such as MF59 [43], remains the most popular choice in such studies. Polyphosphazene adjuvants used in the present study combine delivery vehicle characteristics, which are realized through the association of polymer with the antigen, with the capability of inducing a local proinflammatory response leading to the recruitment of immune cells to the site of injection [11]. Effective vaccines against infectious agents require the elicitation of both humoral and cellular responses to engender protective immunity, along with persistent memory responses [44]. This has led to the design of tailored immunoadjuvant systems that can give specific alterations of the immune response according to the specific chemical pattern of a pathogen, e.g., double-stranded RNA of viral origin for TLR3, bacterial lipopolysaccharide (LPS) for TLR4, singled-stranded RNA for TLR7/8, unmethylated CpG motifs found in bacterial DNA or viruses for TRL9, and so forth [45]. To that end, achieving a functionally appropriate type of immunity, such as Th1-mediated immunity, is highly desirable. In this regard, we previously reported the supramolecular assembly of PCPP with the TLR7/8 agonist, R848 (PCPP-R) that could significantly enhance immune stimulation in vivo to the HCV E2 protein [23]. In this study, we further augmented this approach by incorporating the rationally designed BECC438 TLR4 agonist with PCPP-R to derive a novel binary adjuvant (PCPP-R+BECC438) and used the HCV sE2 antigen to compare immune responses against its individual components PCPP-R and BECC438 in BALB/c mice (Table 1).
As shown in Figure 4a, immunization of mice with the binary adjuvant or the PCPP-R alone formulation produced an overall 10-fold higher anti-E2 IgG response compared to BECC438 alone or the no-adjuvant group. The magnitude of anti-E2 IgG titers remained similar between the PCPP-R and binary adjuvant groups (Figure 4a). Since PCPP-R is known to induce high IgG2a titers with HCV-sE2 [23], and IgG2a and IgG1 isotypes are commonly used as surrogate markers of Th1 and Th2 responses in mice, respectively [46,47,48], we measured the impact of combining BECC438 with PCPP-R adjuvant. We determined that the binary adjuvant produced a statistically significant 1.8-fold higher IgG2a/IgG1 ratio than PCPP-R alone, indicating that the binary adjuvant further improves the immune response to favor Th1 type immunity (Figure 4d). In HCV neutralization assays, all three adjuvants produced modest to high neutralization titers; however, individual mice in binary adjuvant group showed higher geometrical mean titers than the other groups such that the neutralization ID50 titer from the binary adjuvant group was 2-3 fold higher than BECC438 or PCPP-R group, respectively (Figure 5b), implying a potential synergy from combining PCPP-R+BECC438 as a single binary adjuvant.
Overall, our results provide a simple method that utilizes PCPP scaffold to present multiple TLR agonists—a Lipid A-TLR4 agonist (BECC438) and small molecule—Resiquimod—TLR7/8 as a as single-stable adjuvant formulation. Such a combination adjuvant should help vaccine antigens with inherently poor immunogenicity such as HCV-envelope antigen. The aqueous formulation should ameliorate the injection site reactions associated with use lipid containing adjuvants (MF59, AS03) in vaccines [49]. Additionally, unlike lipid/oil emulsion adjuvants that suffer from phase separation from antigen upon longer term storage [50,51], PCPP remains amenable to lyophilization and, upon resuspension can potentially maintain the stable-complex with antigen and TLR-agonists while maintaining the desired particle size. Therefore, our approach should help hold the antigen and TLR agonist(s) in close proximity for optimal immune stimulation and facilitate stable long-term storage of the prepared vaccine formulation.
Author Contributions
Conceptualization, A.K.A., R.K.E. and T.R.F.; methodology, A.K.A., R.K.E. and T.R.F.; formal analysis, A.M., A.C., R.H., S.J. and L.K.; investigation, A.M., A.C., R.H., S.D., S.J., L.K. and F.M.; resources, A.K.A., R.K.E. and T.R.F.; writing—original draft preparation, A.K.A., R.K.E. and T.R.F.; writing—review and editing, A.K.A., R.K.E., E.A.T. and T.R.F.; supervision, A.K.A., R.K.E., E.A.T., and T.R.F.; project administration, A.K.A., R.K.E. and T.R.F.; funding acquisition, A.K.A., R.K.E., E.A.T., and T.R.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Institutes of Health, NIAID grants R01 AI132212 (T.R.F., E.A.T., A.K.A.), R01 AI168048 (T.R.F., E.A.T., A.K.A.) and HHS-NIH-NIAID-BAA2017 (R.K.E.).
Institutional Review Board Statement
The animal study protocol was approved by the University of Maryland, Baltimore Institutional Animal Care and Use Committee (Protocol # 0921009, expiry: 09.17.2024 BAA-NIAID-75N93022R00009 and HHS-NIH-NIAID-BAA2017).
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Conflicts of Interest
Thomas R. Fuerst and Alexander K. Andrianov are shareholders of NeuImmune, Inc.—the company that develops HCV vaccine. Robert K Ernst is the cofounder of TollereBio—the company that has licensed the BECC adjuvant platform. All companies had no role in the design of the study, collection, analysis, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results. These potential conflicts of interest have been disclosed and are managed by the University of Maryland, College Park, and the University of Maryland- Baltimore. The other authors have no conflicts of interest to report.
Abbreviations
The following abbreviations are used in this manuscript:
| PCPP | Poly[di(carboxylatophenoxy)phosphazene] |
| BECC | Bacterial Enzymatic Combinatorial Chemistry |
| DLS | Dynamic light scattering |
| ITC…. | Isothermal titration calorimetry |
| Dz | z-average hydrodynamic diameter |
| pdi | polydispersity index |
| TLR | Toll-like receptor |
| R848 | Resiquimod |
| PCPP-R | PCPP containing resiquimod as a counterion |
| API | Active pharmaceutical ingredient |
| SEAP | Secreted embryonic alkaline phosphatase |
| ELISA | Enzyme-linked immunosorbent assay |
References
- Reed, S.G.; Orr, M.T.; Fox, C.B. Key roles of adjuvants in modern vaccines. Nat. Med. 2013, 19, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- Brito, L.A.; Malyala, P.; O’Hagan, D.T. Vaccine adjuvant formulations: A pharmaceutical perspective. Semin. Immunol. 2013, 25, 130–145. [Google Scholar] [CrossRef]
- Petrovsky, N.; Heinzel, S.; Honda, Y.; Lyons, A.B. New-Age Vaccine Adjuvants: Friend or Foe? New-Age 2007. [Google Scholar]
- Pulendran, B.; S. Arunachalam, P.; O’Hagan, D.T. Emerging concepts in the science of vaccine adjuvants. Nat. Rev. Drug. Discov. 2021, 20, 454–475. [Google Scholar] [CrossRef]
- Nanishi, E.; Dowling, D.J.; Levy, O. Toward precision adjuvants: optimizing science and safety. Curr. Opin. Pediatr. 2020, 32, 125–138. [Google Scholar] [CrossRef]
- Persing, D.H.; Coler, R.N.; Lacy, M.J.; Johnson, D.A.; Baldridge, J.R.; Hershberg, R.M.; Reed, S.G. Taking toll: lipid A mimetics as adjuvants and immunomodulators. Trends Microbiol. 2002, 10, s32–s37. [Google Scholar] [CrossRef]
- Garçon, N.; Chomez, P.; Van Mechelen, M. GlaxoSmithKline Adjuvant Systems in vaccines: concepts, achievements and perspectives. Expert review of vaccines 2007, 6, 723–739. [Google Scholar] [CrossRef]
- Catanese, M.T.; Ansuini, H.; Graziani, R.; Huby, T.; Moreau, M.; Ball, J.K.; Paonessa, G.; Rice, C.M.; Cortese, R.; Vitelli, A.; et al. Role of scavenger receptor class B type I in hepatitis C virus entry: kinetics and molecular determinants. J Virol 2010, 84, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Gregg, K.A.; Harberts, E.; Gardner, F.M.; Pelletier, M.R.; Cayatte, C.; Yu, L.; McCarthy, M.P.; Marshall, J.D.; Ernst, R.K. Rationally designed TLR4 ligands for vaccine adjuvant discovery. MBio 2017, 8, e00492–00417. [Google Scholar] [CrossRef]
- Gregg, K.A.; Harberts, E.; Gardner, F.M.; Pelletier, M.R.; Cayatte, C.; Yu, L.; McCarthy, M.P.; Marshall, J.D.; Ernst, R.K. A lipid A-based TLR4 mimetic effectively adjuvants a Yersinia pestis rF-V1 subunit vaccine in a murine challenge model. Vaccine 2018, 36, 4023–4031. [Google Scholar] [CrossRef] [PubMed]
- Andrianov, A.K.; Langer, R. Polyphosphazene immunoadjuvants: Historical perspective and recent advances. J. Controlled Release 2021, 329, 299–315. [Google Scholar] [CrossRef]
- Palmer, C.D.; Ninković, J.; Prokopowicz, Z.M.; Mancuso, C.J.; Marin, A.; Andrianov, A.K.; Dowling, D.J.; Levy, O. The effect of stable macromolecular complexes of ionic polyphosphazene on HIV Gag antigen and on activation of human dendritic cells and presentation to T-cells. Biomaterials 2014, 35, 8876–8886. [Google Scholar] [CrossRef]
- Awate, S.; Wilson, H.L.; Singh, B.; Babiuk, L.A.; Mutwiri, G. The adjuvant PCEP induces recruitment of myeloid and lymphoid cells at the injection site and draining lymph node. Vaccine 2014, 32, 2420–2427. [Google Scholar] [CrossRef]
- Awate, S.; Wilson, H.L.; Lai, K.; Babiuk, L.A.; Mutwiri, G. Activation of adjuvant core response genes by the novel adjuvant PCEP. Mol. Immunol. 2012, 51, 292–303. [Google Scholar] [CrossRef]
- Andrianov, A.K.; Marin, A.; Fuerst, T.R. Molecular-Level Interactions of Polyphosphazene Immunoadjuvants and Their Potential Role in Antigen Presentation and Cell Stimulation. Biomacromolecules 2016, 17, 3732–3742. [Google Scholar] [CrossRef] [PubMed]
- Andrianov, A.K.; Marin, A.; Wang, R.; Chowdhury, A.; Agnihotri, P.; Yunus, A.S.; Pierce, B.G.; Mariuzza, R.A.; Fuerst, T.R. In Vivo and In Vitro Potency of Polyphosphazene Immunoadjuvants with Hepatitis C Virus Antigen and the Role of Their Supramolecular Assembly. Mol. Pharm. 2021, 18, 726–734. [Google Scholar] [CrossRef]
- Fuerst, T.R.; Marin, A.; Jeong, S.; Kulakova, L.; Hlushko, R.; Gorga, K.; Toth, E.A.; Singh, N.J.; Andrianov, A.K. Virus-Mimicking Polymer Nanocomplexes Co-Assembling HCV E1E2 and Core Proteins with TLR 7/8 Agonist—Synthesis, Characterization, and In Vivo Activity. J. Funct. Biomater. 2025, 16, 34. [Google Scholar] [CrossRef]
- Bouveret Le Cam, N.N.; Ronco, J.; Francon, A.; Blondeau, C.; Fanget, B. Adjuvants for influenza vaccine. Res. Immunol. 1998, 149, 19–23. [Google Scholar] [CrossRef]
- Ison, M.G.; Mills, J.; Openshaw, P.; Zambon, M.; Osterhaus, A.; Hayden, F. Current research on respiratory viral infections: Fourth International Symposium. Antiviral Res. 2002, 55, 227–278. [Google Scholar] [CrossRef] [PubMed]
- Thongcharoen, P.; Suriyanon, V.; Paris, R.M.; Khamboonruang, C.; de Souza, M.S.; Ratto-Kim, S.; Karnasuta, C.; Polonis, V.R.; Baglyos, L.; El Habib, R. A Phase 1/2 Comparative Vaccine Trial of the Safety and Immunogenicity of a CRF01_AE (Subtype E) Candidate Vaccine: ALVAC-HIV (vCP1521) Prime With Oligomeric gp160 (92TH023/LAI-DID) or Bivalent gp120 (CM235/SF2) Boost. J. Acquired Immune Defic. Syndr. 2007, 46, 48–55. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.J.; Excler, J.-L.; Polonis, V.R.; Ratto-Kim, S.; Cox, J.; Jagodzinski, L.L.; Liu, M.; Wieczorek, L.; McNeil, J.G.; El-Habib, R. Safety and Immunogenicity of a randomized Phase I prime-boost trial with ALVAC-HIV (vCP205) and Oligomeric gp160 MN/LAI-2 Adjuvanted in Alum or Polyphosphazene. J. Infect. Dis. 2016, 213, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.; Polonis, V.; Ratto-Kim, S.; Cox, J.; Jagodzinski, L.; Malia, J.; Michael, N.; Excler, J.; Robb, M.; Kim, J. Safety and immunogenicity of a randomized phase I prime-boost trial with ALVAC-HIV (vCP205) and gp160 MN/LAI-2 adjuvanted in alum or polyphosphazene. Retrovirology 2012, 9, O50. [Google Scholar] [CrossRef]
- Andrianov, A.K.; Marin, A.; Wang, R.; Karauzum, H.; Chowdhury, A.; Agnihotri, P.; Yunus, A.S.; Mariuzza, R.A.; Fuerst, T.R. Supramolecular assembly of Toll-like receptor 7/8 agonist into multimeric water-soluble constructs enables superior immune stimulation in vitro and in vivo. ACS Appl. Bio Mater. 2020, 3, 3187–3195. [Google Scholar] [CrossRef]
- WHO Hepatitis C Key Facts. Available online: https://www/who.int/en/news-room/fact-sheets/detail/hepatitis-c (accessed on 9 April 2025).
- Fiehn, F.; Beisel, C.; Binder, M. Hepatitis C virus and hepatocellular carcinoma: carcinogenesis in the era of direct-acting antivirals. Curr Opin Virol 2024, 67, 101423. [Google Scholar] [CrossRef]
- Caniglia, E.C.; Cain, L.E.; Sabin, C.A.; Robins, J.M.; Logan, R.; Abgrall, S.; Mugavero, M.J.; Hernandez-Diaz, S.; Meyer, L.; Seng, R.; et al. Comparison of dynamic monitoring strategies based on CD4 cell counts in virally suppressed, HIV-positive individuals on combination antiretroviral therapy in high-income countries: a prospective, observational study. Lancet HIV 2017, 4, e251–e259. [Google Scholar] [CrossRef]
- Hepatitis - The Silent Epidemic. May 20, 2020. 20 May.
- Yukawa, Y.; Tamori, A.; Iio, E.; Ogawa, S.; Yoshida, K.; Uchida-Kobayashi, S.; Enomoto, M.; Tanaka, Y.; Kawada, N. Hepatitis C virus recurrence in two patients who achieved sustained viral response with interferon-free direct-acting antiviral therapy: reinfection or relapse? Clinical journal of gastroenterology 2019, 12, 598–602. [Google Scholar] [CrossRef]
- Berenguer, J.; Gil-Martin, A.; Jarrin, I.; Montes, M.L.; Dominguez, L.; Aldamiz-Echevarria, T.; Tellez, M.J.; Santos, I.; Troya, J.; Losa, J.E.; et al. Reinfection by hepatitis C virus following effective all-oral direct-acting antiviral drug therapy in HIV/hepatitis C virus coinfected individuals. AIDS 2019, 33, 685–689. [Google Scholar] [CrossRef]
- Rossi, C.; Butt, Z.A.; Wong, S.; Buxton, J.A.; Islam, N.; Yu, A.; Darvishian, M.; Gilbert, M.; Wong, J.; Chapinal, N.; et al. Hepatitis C virus reinfection after successful treatment with direct-acting antiviral therapy in a large population-based cohort. Journal of hepatology 2018, 69, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Hepatitis Report 2024; World Health Organization, 2024. [Google Scholar]
- Borgia, S.M.; Hedskog, C.; Parhy, B.; Hyland, R.H.; Stamm, L.M.; Brainard, D.M.; Subramanian, M.G.; McHutchison, J.G.; Mo, H.; Svarovskaia, E.; et al. Identification of a novel hepatitis C virus genotype from Punjab, India: Expanding classification of hepatitis C virus into 8 genotypes. Journal of Infectious Diseases 2018, 218, 1722–1729. [Google Scholar] [CrossRef]
- Fuerst, T.R.; Pierce, B.G.; Keck, Z.Y.; Foung, S.K.H. Designing a B Cell-Based Vaccine against a Highly Variable Hepatitis C Virus. Frontiers in microbiology 2017, 8, 2692. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.L. Challenges and Promise of a Hepatitis C Virus Vaccine. Cold Spring Harb Perspect Med 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Duggal, P.; Thio, C.L.; Wojcik, G.L.; Goedert, J.J.; Mangia, A.; Latanich, R.; Kim, A.Y.; Lauer, G.M.; Chung, R.T.; Peters, M.G.; et al. Genome-wide association study of spontaneous resolution of hepatitis C virus infection: data from multiple cohorts. Annals of internal medicine 2013, 158, 235–245. [Google Scholar] [CrossRef]
- Urbanowicz, R.A.; Wang, R.; Schiel, J.E.; Keck, Z.-y.; Kerzic, M.C.; Lau, P.; Rangarajan, S.; Garagusi, K.J.; Tan, L.; Guest, J.D.; et al. Antigenicity and Immunogenicity of Differentially Glycosylated Hepatitis C Virus E2 Envelope Proteins Expressed in Mammalian and Insect Cells. J. Virol. 2019, 93, e01403–01418. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Keck, Z.-Y.; Wang, R.; Lau, P.; Garagusi, K.; Elkholy, K.; Toth, E.A.; Urbanowicz, R.A.; Guest, J.D.; Agnihotri, P.; et al. Structure-based design of hepatitis C virus E2 glycoprotein improves serum binding and cross-neutralization. J. Virol. 2020, 94, e00704–00720. [Google Scholar] [CrossRef]
- Zacharia, A.; Harberts, E.; Valencia, S.M.; Myers, B.; Sanders, C.; Jain, A.; Larson, N.R.; Middaugh, C.R.; Picking, W.D.; Difilippantonio, S.; et al. Optimization of RG1-VLP vaccine performance in mice with novel TLR4 agonists. Vaccine 2021, 39, 292–302. [Google Scholar] [CrossRef]
- Siepmann, J.; Faham, A.; Clas, S.-D.; Boyd, B.J.; Jannin, V.; Bernkop-Schnürch, A.; Zhao, H.; Lecommandoux, S.; Evans, J.C.; Allen, C.; et al. Lipids and polymers in pharmaceutical technology: Lifelong companions. Int. J. Pharm. 2019, 558, 128–142. [Google Scholar] [CrossRef]
- Kesisoglou, F.; Panmai, S.; Wu, Y. Nanosizing — Oral formulation development and biopharmaceutical evaluation. Adv. Drug Delivery Rev. 2007, 59, 631–644. [Google Scholar] [CrossRef]
- Heurtault, B.; Saulnier, P.; Pech, B.; Proust, J.-E.; Benoit, J.-P. Physico-chemical stability of colloidal lipid particles. Biomaterials 2003, 24, 4283–4300. [Google Scholar] [CrossRef]
- Andrianov, A.K.; Fuerst, T.R. Immunopotentiating and Delivery Systems for HCV Vaccines. Viruses 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Landi, A.; Law, J.; Hockman, D.; Logan, M.; Crawford, K.; Chen, C.; Kundu, J.; Ebensen, T.; Guzman, C.A.; Deschatelets, L.; et al. Superior immunogenicity of HCV envelope glycoproteins when adjuvanted with cyclic-di-AMP, a STING activator or archaeosomes. Vaccine 2017, 35, 6949–6956. [Google Scholar] [CrossRef]
- Reed, S.G.; Orr, M.T.; Fox, C.B. Key roles of adjuvants in modern vaccines. Nat Med 2013, 19, 1597–1608. [Google Scholar] [CrossRef]
- Kayesh, M.E.H.; Kohara, M.; Tsukiyama-Kohara, K. TLR agonists as vaccine adjuvants in the prevention of viral infections: an overview. Frontiers in microbiology 2023, 14, 1249718. [Google Scholar] [CrossRef] [PubMed]
- Weissenberger, G.; Henderikx, R.J.M.; Peters, P.J. Understanding the invisible hands of sample preparation for cryo-EM. Nat Methods 2021, 18, 463–471. [Google Scholar] [CrossRef]
- Yip, K.M.; Fischer, N.; Paknia, E.; Chari, A.; Stark, H. Atomic-resolution protein structure determination by cryo-EM. Nature 2020, 587, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y. Single-particle cryo-EM-How did it get here and where will it go. Science 2018, 361, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Petrovsky, N. Comparative Safety of Vaccine Adjuvants: A Summary of Current Evidence and Future Needs. Drug Saf 2015, 38, 1059–1074. [Google Scholar] [CrossRef]
- Iyer, V.; Cayatte, C.; Guzman, B.; Schneider-Ohrum, K.; Matuszak, R.; Snell, A.; Rajani, G.M.; McCarthy, M.P.; Muralidhara, B. Impact of formulation and particle size on stability and immunogenicity of oil-in-water emulsion adjuvants. Hum Vaccin Immunother 2015, 11, 1853–1864. [Google Scholar] [CrossRef]
- Iyer, V.; Cayatte, C.; Marshall, J.D.; Sun, J.; Schneider-Ohrum, K.; Maynard, S.K.; Rajani, G.M.; Bennett, A.S.; Remmele, R.L., Jr.; Bishop, S.M.; et al. Feasibility of Freeze-Drying Oil-in-Water Emulsion Adjuvants and Subunit Proteins to Enable Single-Vial Vaccine Drug Products. J Pharm Sci 2017, 106, 1490–1498. [Google Scholar] [CrossRef]
Figure 1.
Preparation and characterization of PCPP-BECC438 formulations. (a) Flowchart of a multi-step preparation of aggregate-free PCPP-BECC438 formulations and (b) DLS profiles of BECC438, PCPP and their formulation obtained using the process shown in panel a (0.25 mg/mL BECC438, 0.5 mg/mL PCPP, PBS, pH 7.4, z-average hydrodynamic diameter (Dz) and polydispersity index (pdi) of lyophilized and redissolved PCPP-BECC438 formulations are shown).
Figure 1.
Preparation and characterization of PCPP-BECC438 formulations. (a) Flowchart of a multi-step preparation of aggregate-free PCPP-BECC438 formulations and (b) DLS profiles of BECC438, PCPP and their formulation obtained using the process shown in panel a (0.25 mg/mL BECC438, 0.5 mg/mL PCPP, PBS, pH 7.4, z-average hydrodynamic diameter (Dz) and polydispersity index (pdi) of lyophilized and redissolved PCPP-BECC438 formulations are shown).
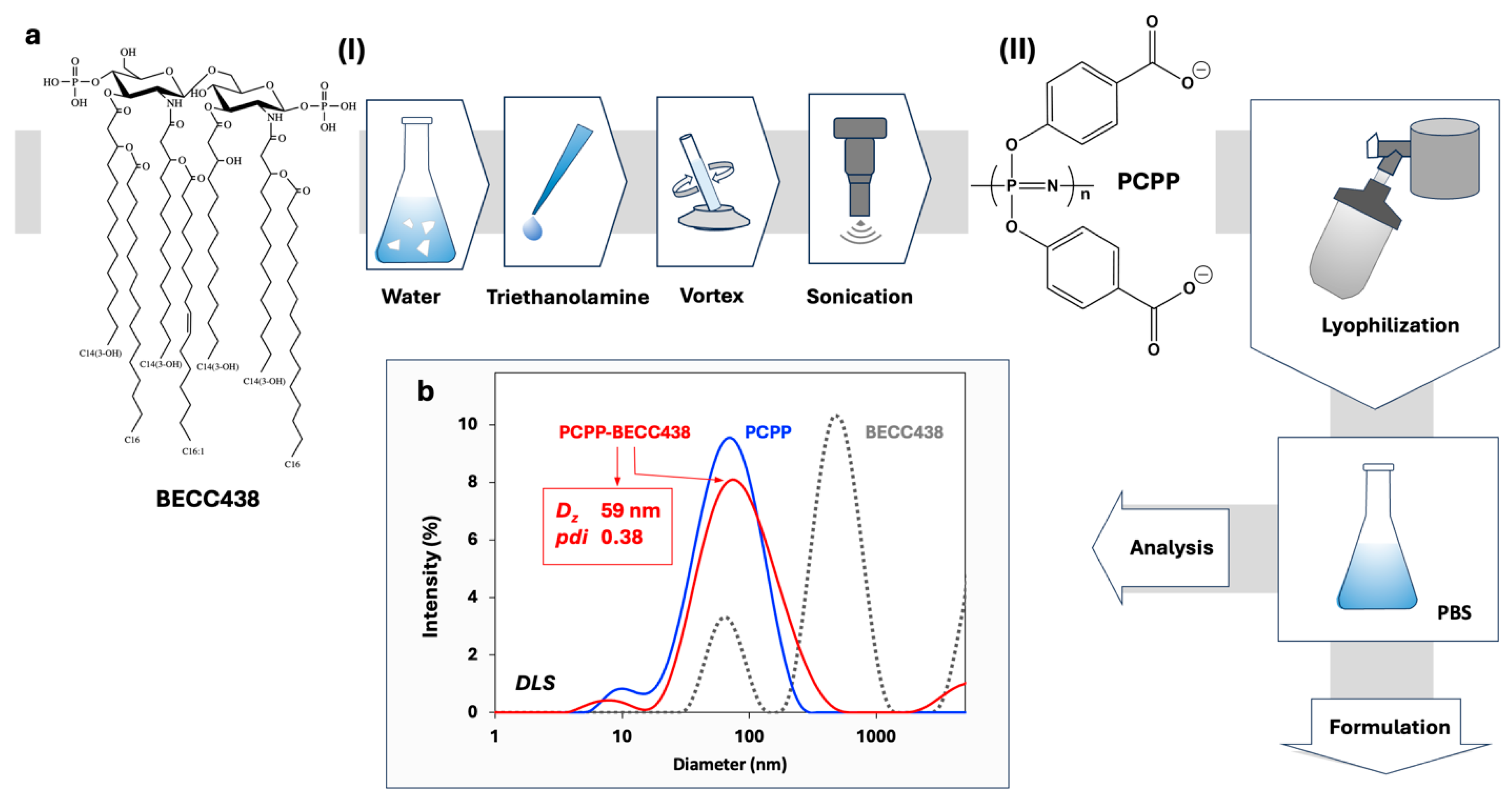
Figure 2.
(a) ITC data of PCPP interaction with BECC438 and the corresponding heats of dilution of PCPP (50 mM phosphate buffer, pH = 7.4, 25 °C) and (b) binding isotherm corrected for the heat of dilution.
Figure 2.
(a) ITC data of PCPP interaction with BECC438 and the corresponding heats of dilution of PCPP (50 mM phosphate buffer, pH = 7.4, 25 °C) and (b) binding isotherm corrected for the heat of dilution.
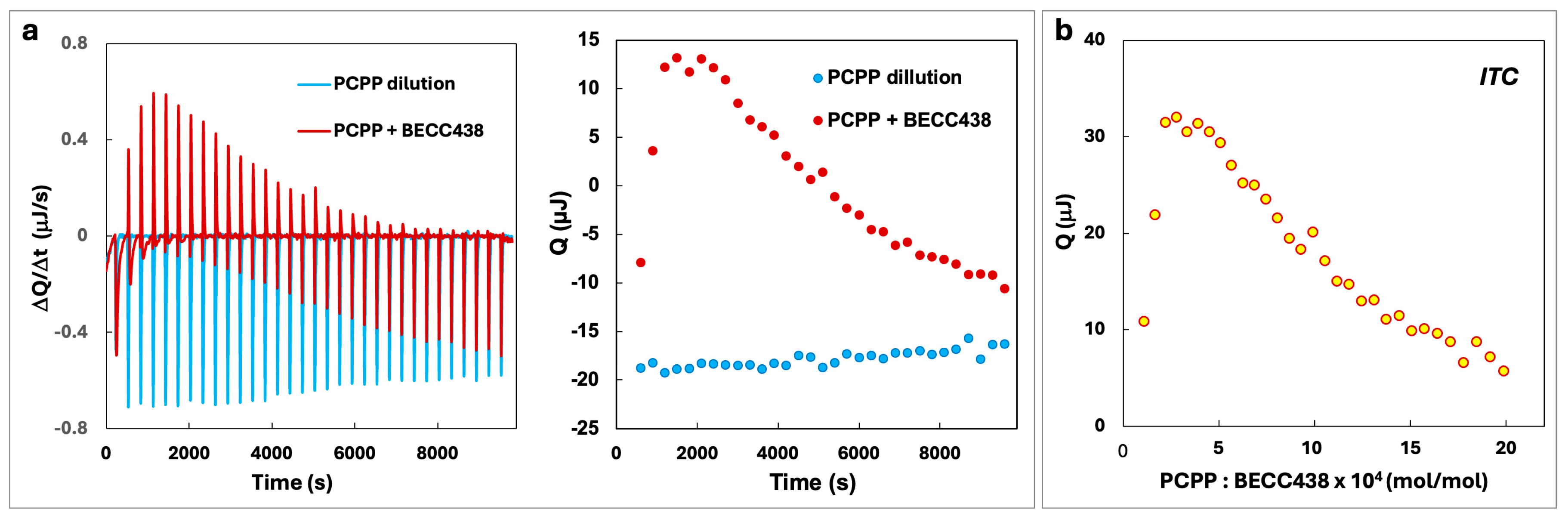
Figure 3.
Immunoactivation of RAW Blue cells by the PCPP-BECC438, BECC438 and PCPP formulations as measured by the spectrophotometric monitoring of SEAP substrate hydrolysis (all formulations contain HCV E2 antigen at 1:1 HCV E2-PCPP ratio, the absorbance measured at 405 nm, n=3, error bars - standard error).
Figure 3.
Immunoactivation of RAW Blue cells by the PCPP-BECC438, BECC438 and PCPP formulations as measured by the spectrophotometric monitoring of SEAP substrate hydrolysis (all formulations contain HCV E2 antigen at 1:1 HCV E2-PCPP ratio, the absorbance measured at 405 nm, n=3, error bars - standard error).
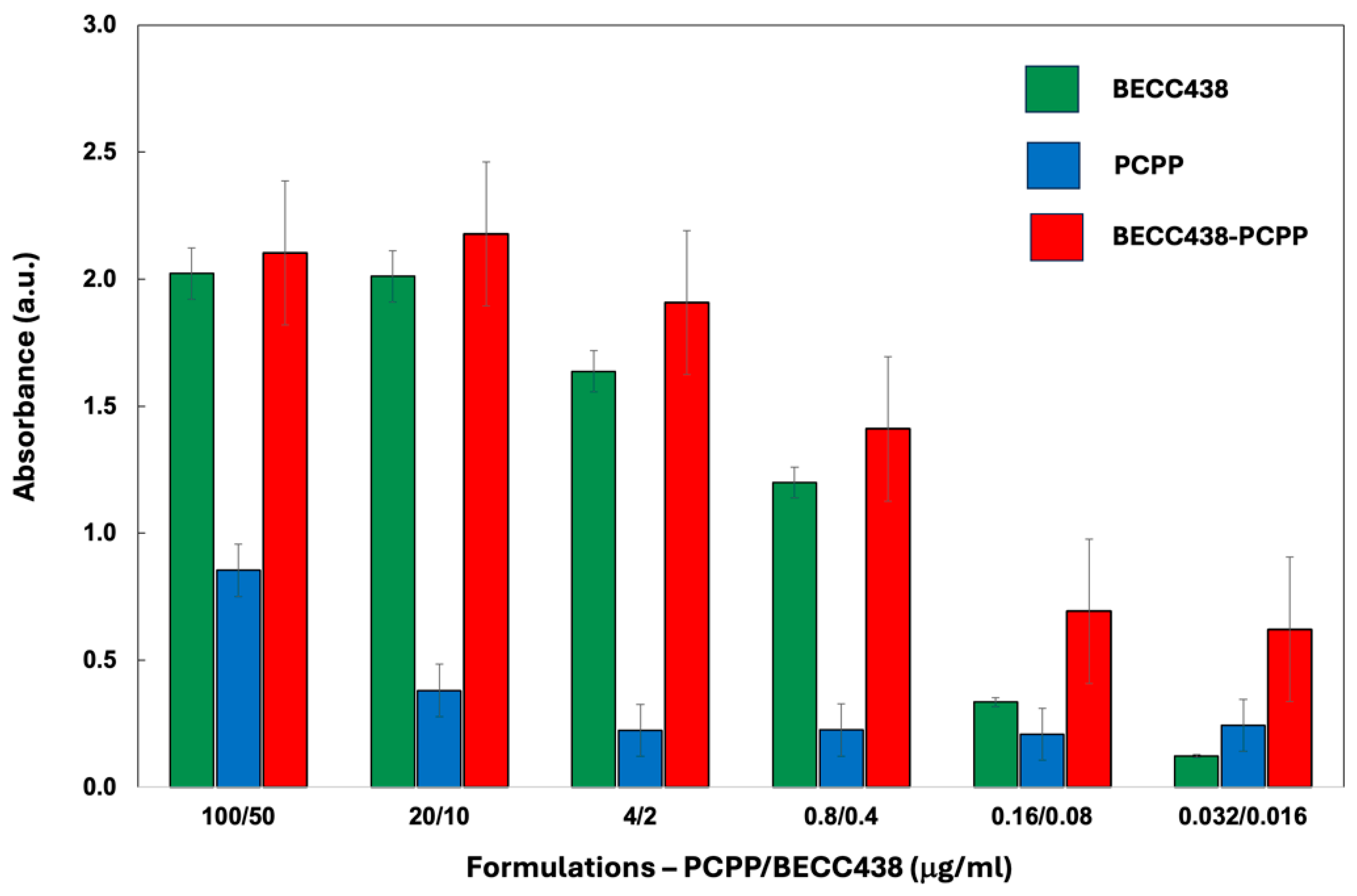
Figure 4.
Immunogenicity assessment of PPZ-R and BECC combination formulations by ELISA of antibodies induced in mice at day 56 after immunization. (a) Total IgG titers, (b) Total IgG1 titers, (c) Total IgG2 titers, and (d) the ratio of IgG2a to IgG1 for each immunogen. Individual mouse sera from the indicated groups were analyzed. Endpoint titers were calculated by curve fitting in GraphPad software (version 10) with endpoint optical density defined as four times the highest absorbance value of day 0 sera.
Figure 4.
Immunogenicity assessment of PPZ-R and BECC combination formulations by ELISA of antibodies induced in mice at day 56 after immunization. (a) Total IgG titers, (b) Total IgG1 titers, (c) Total IgG2 titers, and (d) the ratio of IgG2a to IgG1 for each immunogen. Individual mouse sera from the indicated groups were analyzed. Endpoint titers were calculated by curve fitting in GraphPad software (version 10) with endpoint optical density defined as four times the highest absorbance value of day 0 sera.
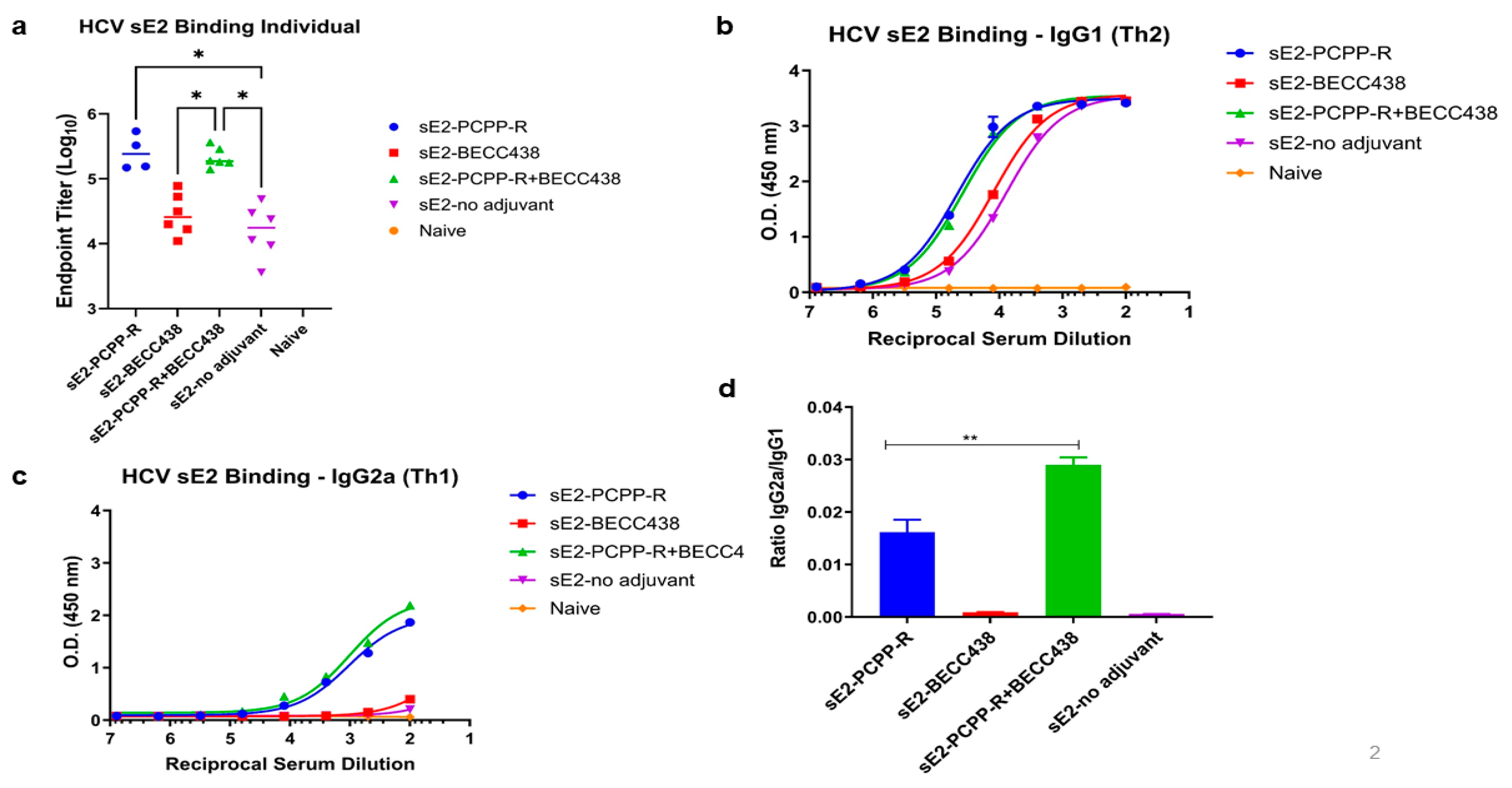
Figure 5.
Potency of neutralization of individual mouse sera against H77C (GT1a) isolate, comparing PCPP-R and BECC438 formulations. (a) Individual immunized mouse sera were assessed for neutralization activities at day 56 and day 0. Serum dilutions were performed as three-fold dilutions starting at 1:150 for HCVpp neutralization. (b) Inhibition dilutions values corresponding to 50 percent neutralization (ID50) values for each group.
Figure 5.
Potency of neutralization of individual mouse sera against H77C (GT1a) isolate, comparing PCPP-R and BECC438 formulations. (a) Individual immunized mouse sera were assessed for neutralization activities at day 56 and day 0. Serum dilutions were performed as three-fold dilutions starting at 1:150 for HCVpp neutralization. (b) Inhibition dilutions values corresponding to 50 percent neutralization (ID50) values for each group.
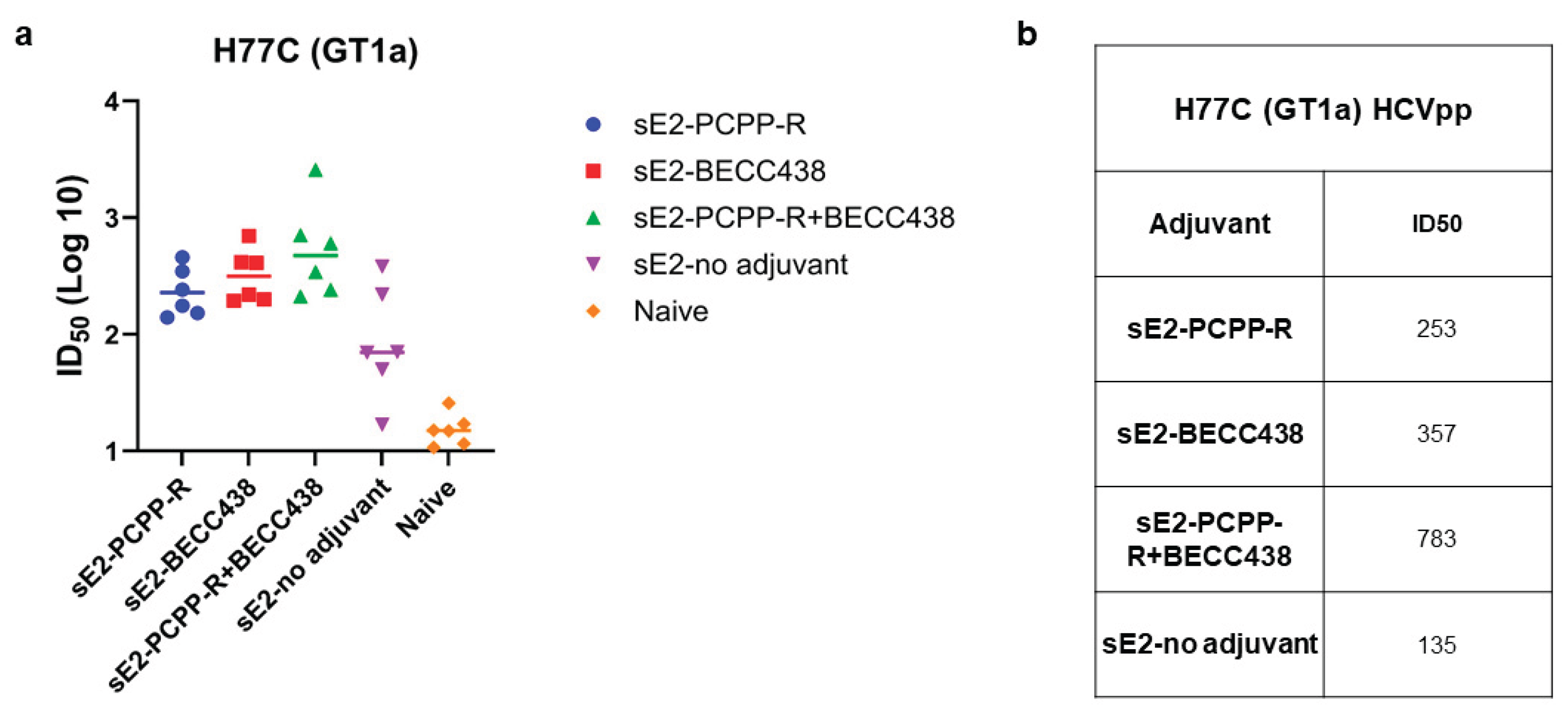

Table 1.
The Design of In Vivo Studies.
| Group | Label | sE2 FL (µg) | PCPP-R (µg) | BECC438 (µg) |
|---|---|---|---|---|
| 1 | sE2-PCPP-R | 50 | 50 | 0 |
| 2 | sE2-BECC438 | 50 | 0 | 25 |
| 3 | sE2-PCPP-R+BECC438 | 50 | 50 | 25 |
| 4 | sE2-no adjuvant | 50 | 0 | 0 |
| 5 | Naive | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



