Submitted:
13 December 2024
Posted:
16 December 2024
You are already at the latest version
Abstract
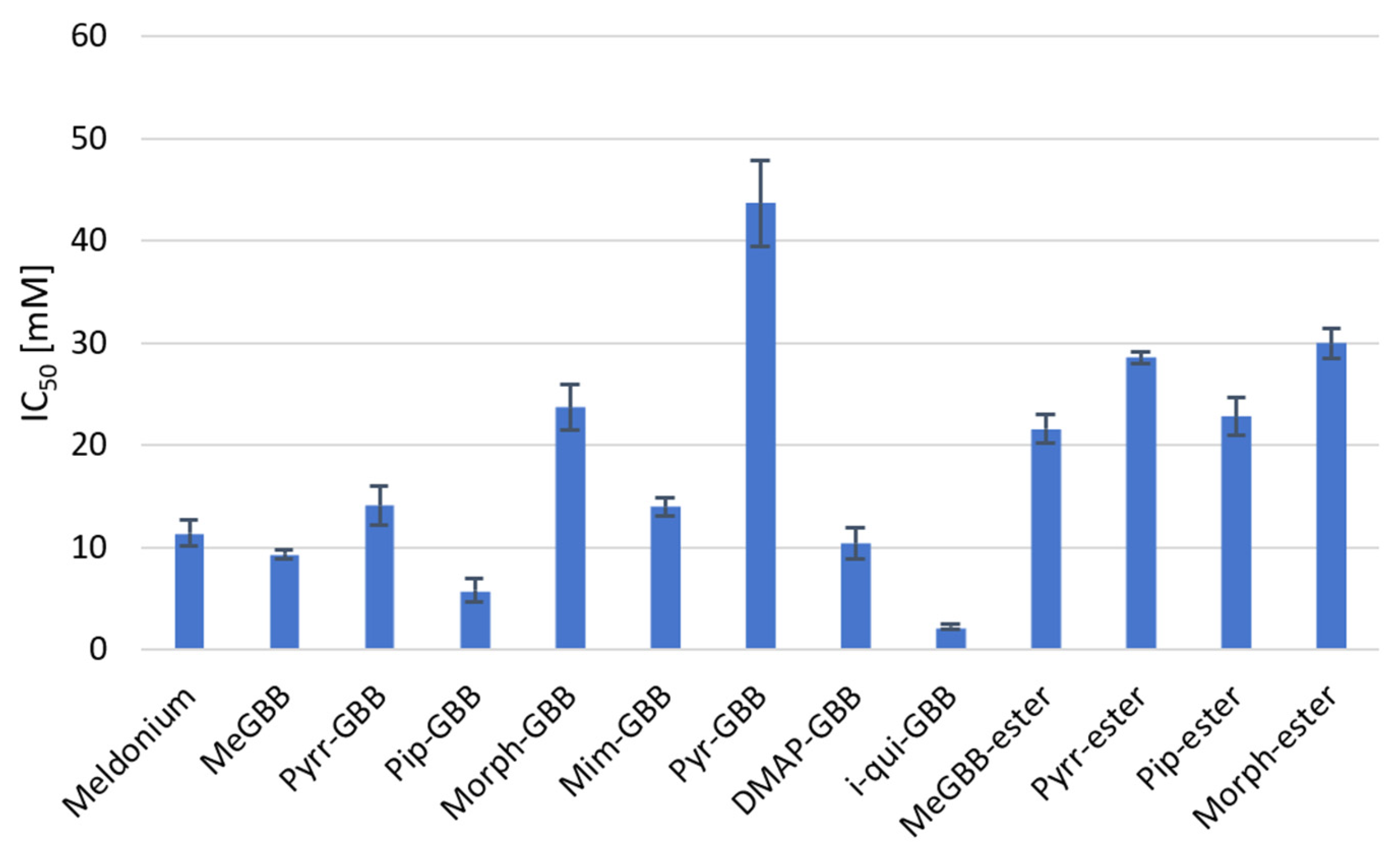
This study examined heterocyclic gamma-butyrobetaine (GBB) analogs as metabolic modulators through rational design, docking, synthesis, and in vitro analyses. The compounds inhibited carnitine acetyltransferase (CAT) and possibly other enzymes in the carnitine transferase family, showing inhibitory potential in the low millimolar range (IC50 = 2.24–43.6 mM), with some more active than the well-known drug Meldonium (IC50 = 11.39 mM). Key findings include that bulky and hydrophobic substituent at the gamma-position enhances inhibition, while esterification and increased polarity reduce it. The most active compound was identified as a reversible competitive inhibitor of CAT, with a Ki value of 3.5 mM, similar to Meldonium’s Ki of 1.63 mM. These results indicate that heterocyclic GBB analogs are potential candidates for regulating metabolic processes and for treating conditions such as ischemic diseases, diabetes, and certain cancers.
Keywords:
1. Introduction
2. Results and Discussion
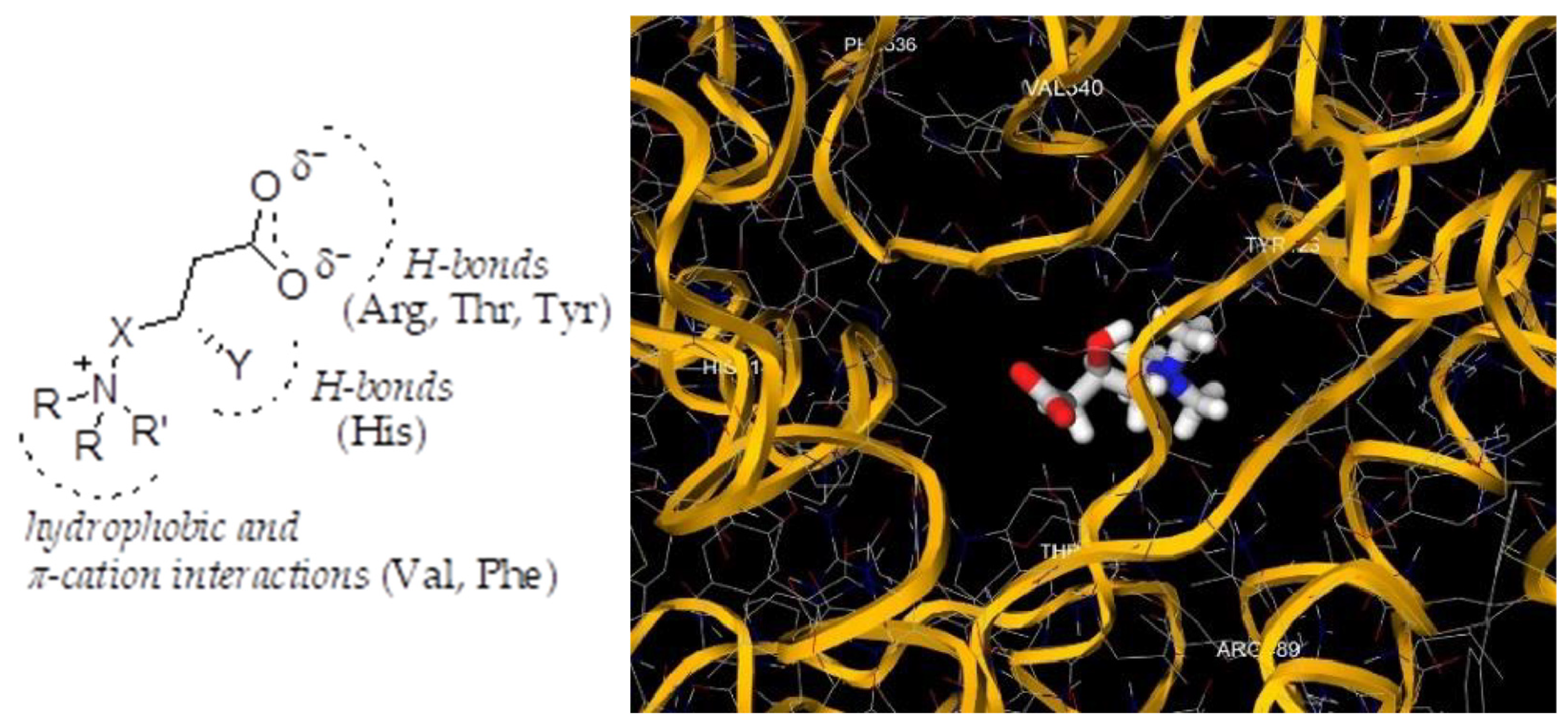
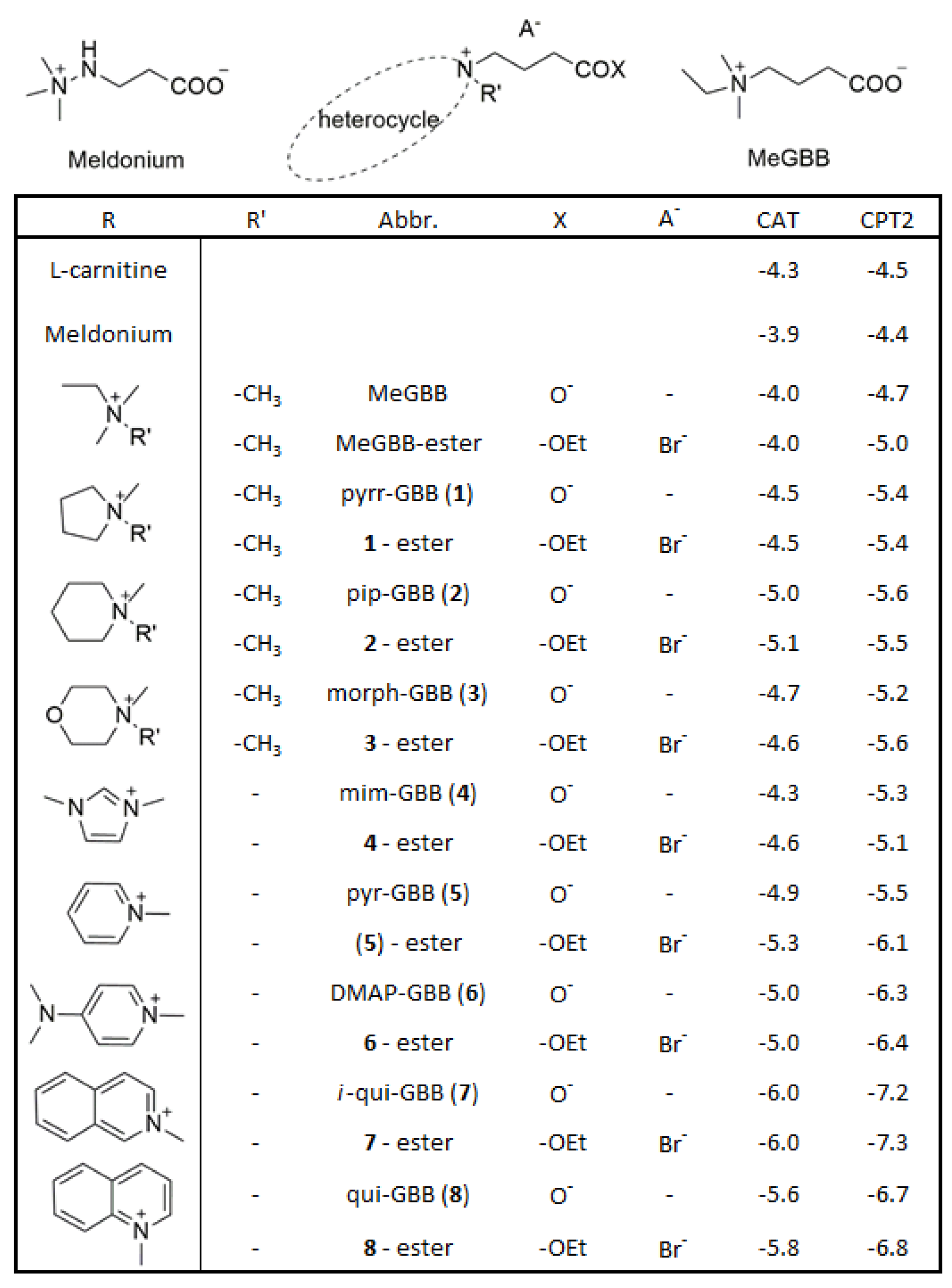
2.1. Rational Design
2.2. Synthesis and Characterization
2.3. Biological Assessment
3. Materials and Methods
3.1. General
3.2. Synthesis
3.2.1. Synthesis of Bromide Salts of Heterocyclic Gamma-Butyrobetaine Ethyl Esters
3.2.2. Synthesis of Heterocyclic Gamma-Butyrobetaines
3.3. In-Vitro Studies
3.3.1. IC50 Determination
3.3.2. Enzyme Kinetics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Foley, J. Rationale and application of fatty acid oxidation inhibitors in treatment of diabetes mellitus. Diabetes Care. 1992, 15, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Keung, W.; Samokhvalov, V.; Wang, W.; Lopaschuk, G. Role of fatty acid uptake and fatty acid beta-oxidation in mediating insulin resistance in heart and skeletal muscle. Biochim. Biophys. Acta. 2010, 1801, 1–22. [Google Scholar] [CrossRef]
- Hue, L.; Taegtmeyer, H. The Randle cycle revisited: a new head for an old hat. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E578–591. [Google Scholar] [CrossRef]
- Ramsay, R.; Arduini, A. The carnitine acyltransferases and their role in modulating acyl-CoA pools. Arch. Biochem. Biophys. 1993, 302, 307–314. [Google Scholar] [CrossRef]
- Fritz, I.; Yue, K. Long-chain carnitine acyltransferase and the role of acylcarnitine derivatives in the catalytic increase of fatty acid oxidation induced by carnitine. J. Lipid Res. 1963, 4, 279–288. [Google Scholar] [CrossRef]
- Wang, M.; Wang, K.; Liao, X.; Hu, H.; Chen, L.; Meng, L.; Gao, W.; Li, Q. Carnitine Palmitoyltransferase System: A New Target for Anti-Inflammatory and Anticancer Therapy? Front. Pharmacol. 2021, 12, 760581. [Google Scholar] [CrossRef]
- Jacques, F.; Rippa, S.; Perrin, Y. Physiology of L-carnitine in plants in light of the knowledge in animals and microorganisms. Plant. Sci. 2018, 274, 432–440. [Google Scholar] [CrossRef]
- Govindasamy, L.; Kukar, T.; Lian, W.; Pedersen, B.; Gu, Y.; Agbandje-McKenna, M.; Jin, S.; McKenna, R.; Wu, D. Structural and mutational characterization of L-carnitine binding to human carnitine acetyltransferase. J. Struct. Biol. 2004, 146, 416–424. [Google Scholar] [CrossRef]
- Jogl, G.; Tong, L. Crystal structure of carnitine acetyltransferase and implications for the catalytic mechanism and fatty acid transport. Cell. 2003, 112, 113–122. [Google Scholar] [CrossRef]
- Bonnefont, J.; Djouadi, F.; Prip-Buus, C.; Gobin, S.; Munnich, A.; Bastin, J. Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol. Aspects. Med. 2004, 25, 495–520. [Google Scholar] [CrossRef]
- Sierra, A.; Gratacós, E.; Carrasco, P.; Clotet, J.; Ureña, J.; Serra, D.; Asins, G.; Hegardt, F.; Casals, N. CPT1c is localized in endoplasmic reticulum of neurons and has carnitine palmitoyltransferase activity. J. Biol. Chem. 2008, 283, 6878–6885. [Google Scholar] [CrossRef]
- Wu, D.; Govindasamy, L.; Lian, W.; Gu, Y.; Kukar, T.; Agbandje-McKenna, M.; McKenna, R. Structure of human carnitine acetyltransferase. Molecular basis for fatty acyl transfer. J. Biol. Chem. 2003, 278, 13159–13165. [Google Scholar] [CrossRef]
- Ramsay, R.; Gandour, R.; van der Leij, F. Molecular enzymology of carnitine transfer and transport. Biochim. Biophys. Acta. 2001, 1546, 21–43. [Google Scholar] [CrossRef]
- Randle, P.; Garland, P.; Hales, C.; Newsholme, E. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963, 1, 785–789. [Google Scholar] [CrossRef]
- Nechaeva, G.; Zheltikova, E. Effects of Meldonium in Early Postmyocardial Infarction Period. Kardiologiia. 2015, 55, 35–42. [Google Scholar] [CrossRef]
- Liamina, N.; Kotel’nikova, E.; Karpova, É.; Biziaeva, E.; Senchikhin, V.; Lipchanskaia, T. Cardioprotective capabilities of drug meldonium in secondary prevention after percutaneous coronary intervention in patients with documented myocardial ischemia. Kardiologiia. 2014, 54, 60–65. [Google Scholar] [CrossRef]
- Lopaschuk, G.; Wall, S.; Olley, P.; Davies, N. Etomoxir, a carnitine palmitoyltransferase I inhibitor, protects hearts from fatty acid-induced ischemic injury independent of changes in long chain acylcarnitine. Circ. Res. 1988, 63, 1036–1043. [Google Scholar] [CrossRef]
- Hübinger, A.; Knode, O.; Susanto, F.; Reinauer, H.; Gries, F. Effects of the carnitine-acyltransferase inhibitor etomoxir on insulin sensitivity, energy expenditure and substrate oxidation in NIDDM. Horm. Metab. Res. 1997, 29, 436–439. [Google Scholar] [CrossRef]
- Ratheiser, K.; Schneeweiss, B.; Waldhäusl, W.; Fasching, P.; Korn, A.; Nowotny, P.; Rohac, M.; Wolf, H. Inhibition by etomoxir of carnitine palmitoyltransferase I reduces hepatic glucose production and plasma lipids in non-insulin-dependent diabetes mellitus. Metabolism. 1991, 40, 1185–1190. [Google Scholar] [CrossRef]
- Liepinsh, E.; Vilskersts, R.; Zvejniece, L.; Svalbe, B.; Skapare, E.; Kuka, J.; Cirule, H.; Grinberga, S.; Kalvinsh, I.; Dambrova, M. Protective effects of mildronate in an experimental model of type 2 diabetes in Goto-Kakizaki rats. Br. J. Pharmacol. 2009, 157, 1549–1556. [Google Scholar] [CrossRef]
- Liepinsh, E.; Skapare, E.; Svalbe, B.; Makrecka, M.; Cirule, H.; Dambrova, M. Anti-diabetic effects of mildronate alone or in combination with metformin in obese Zucker rats. Eur. J. Pharmacol. 2011, 658, 277–283. [Google Scholar] [CrossRef]
- Anderson, R. Carnitine Palmitoyltransferase: A Viable Target for the Treatment of NIDDM? Curr. Pharm. Des. 1998, 4, 1–15. [Google Scholar] [CrossRef]
- Giannessi, F.; Chiodi, P.; Marzi, M.; Minetti, P.; Pessotto, P.; De Angelis, F.; Tassoni, E.; Conti, R.; Giorgi, F.; Mabilia, M.; Dell’Uomo, N.; Muck, S.; Tinti, M.; Carminati, P.; Arduini, A. Reversible carnitine palmitoyltransferase inhibitors with broad chemical diversity as potential antidiabetic agents. J. Med. Chem. 2001, 44, 2383–2386. [Google Scholar] [CrossRef]
- Wagman, A.; Nuss, J. Current therapies and emerging targets for the treatment of diabetes. Curr. Pharm. Des. 2001, 7, 417–450. [Google Scholar] [CrossRef]
- Đurašević, S.; Stojković, M.; Bogdanović, L.; Pavlović, S.; Borković-Mitić, S.; Grigorov, I.; Bogojević, D.; Jasnić, N.; Tosti, T.; Đurović, S.; Đorđević, J.; Todorović, Z. The Effects of Meldonium on the Renal Acute Ischemia/Reperfusion Injury in Rats. Int. J. Mol. Sci. 2019, 20, 5747. [Google Scholar] [CrossRef]
- Đurašević, S.; Stojković, M.; Sopta, J. The effects of meldonium on the acute ischemia/reperfusion liver injury in rats. Sci. Rep. 2021, 11, 1305. [Google Scholar] [CrossRef]
- Mørkholt, A.; Wiborg, O.; Nieland, J. Blocking of carnitine palmitoyl transferase 1 potently reduces stress-induced depression in rat highlighting a pivotal role of lipid metabolism. Sci. Rep. 2017, 7, 2158. [Google Scholar] [CrossRef]
- Shriver, L.; Manchester, M. Inhibition of fatty acid metabolism ameliorates disease activity in an animal model of multiple sclerosis. Sci. Rep. 2011, 1, 79. [Google Scholar] [CrossRef]
- Mørkholt, A.; Oklinski, M.; Larsen, A.; Bockermann, R.; Issazadeh-Navikas, S.; Nieland, J.; Kwon, T.; Corthals, A.; Nielsen, S.; Nieland, J. Pharmacological inhibition of carnitine palmitoyl transferase 1 inhibits and reverses experimental autoimmune encephalitis in rodents. PLoS One. 2020, 15, e0234493. [Google Scholar] [CrossRef]
- Beitnere, U.; van Groen, T.; Kumar, A.; Jansone, B.; Klusa, V.; Kadish, I. Mildronate improves cognition and reduces amyloid-β pathology in transgenic Alzheimer’s disease mice. J. Neurosci. Res. 2014, 92, 338–346. [Google Scholar] [CrossRef]
- Otsubo, C.; Bharathi, S.; Uppala, R.; Ilkayeva, O.; Wang, D.; McHugh, K.; Zou, Y.; Wang, J.; Alcorn, J.; Zuo, Y.; Hirschey, M.; Goetzman, E. Long-chain Acylcarnitines Reduce Lung Function by Inhibiting Pulmonary Surfactant. J. Biol. Chem. 2015, 290, 23897–23904. [Google Scholar] [CrossRef]
- Sainero-Alcolado, L.; Liaño-Pons, J.; Ruiz-Pérez, M.; Arsenian-Henriksson, M. Targeting mitochondrial metabolism for precision medicine in cancer. Cell. Death. Differ. 2022, 29, 1304–1317. [Google Scholar] [CrossRef]
- Melone, M.; Valentino, A.; Margarucci, S.; Galderisi, U.; Giordano, A.; Peluso, G. ; The carnitine system and cancer metabolic plasticity. Cell Death Dis. 2018, 9, 228. [Google Scholar] [CrossRef]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; Andreeff, M. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Invest. 2010, 120, 142–156. [Google Scholar] [CrossRef]
- Slebe, F.; Rojo, F.; Vinaixa, M.; García-Rocha, M.; Testoni, G.; Guiu, M.; Planet, E.; Samino, S.; Arenas, E.; Beltran, A.; Rovira, A.; Lluch, A.; Salvatella, X.; Yanes, O.; Albanell, J.; Guinovart, J.; Gomis, R. FoxA and LIPG endothelial lipase control the uptake of extracellular lipids for breast cancer growth. Nat. Commun. 2016, 7, 11199. [Google Scholar] [CrossRef]
- Kant, S.; Kesarwani, P.; Prabhu, A.; Graham, S.; Buelow, K.; Nakano, I.; Chinnaiyan, P. Enhanced fatty acid oxidation provides glioblastoma cells metabolic plasticity to accommodate to its dynamiC-Nutrient microenvironment. Cell Death Dis. 2020, 11, 253. [Google Scholar] [CrossRef]
- Ricciardi, M.; Mirabilii, S.; Allegretti, M.; Licchetta, R.; Calarco, A.; Torrisi, M.; Foà, R.; Nicolai, R.; Peluso, G.; Tafuri, A. Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias. Blood. 2015, 126, 1925–1929. [Google Scholar] [CrossRef]
- Gugiatti, E.; Tenca, C.; Ravera, S.; Fabbi, M.; Ghiotto, F.; Mazzarello, A.; Bagnara, D.; Reverberi, D.; Zarcone, D.; Cutrona, G.; Ibatici, A.; Ciccone, E.; Darzynkiewicz, Z.; Fais, F.; Bruno, S. A reversible carnitine palmitoyltransferase (CPT1) inhibitor offsets the proliferation of chronic lymphocytic leukemia cells. Haematol. 2018, 103, 531–536. [Google Scholar] [CrossRef]
- Yao, C.; Liu, G.; Wang, R.; Moon, S.; Gross, R.; Patti, G. Identifying off-target effects of etomoxir reveals that carnitine palmitoyltransferase I is essential for cancer cell proliferation independent of β-oxidation. PLoS Biol. 2018, 16, 2003782. [Google Scholar] [CrossRef]
- Pacilli, A.; Calienni, M.; Margarucci, S.; D’Apolito, M.; Petillo, O.; Rocchi, L.; Pasquinelli, G.; Nicolai, R.; Koverech, A.; Calvani, M.; Peluso, G.; Montanaro, L. Carnitine-acyltransferase system inhibition, cancer cell death, and prevention of myc-induced lymphomagenesis. J. Nat. Cancer Inst. 2013, 105, 489–498. [Google Scholar] [CrossRef]
- Cheng, S.; Wang, G.; Wang, Y.; Cai, L.; Qian, K.; Ju, L.; Liu, X.; Xiao, Y.; Wang, X. Fatty acid oxidation inhibitor etomoxir suppresses tumor progression and induces cell cycle arrest via PPARγ-mediated pathway in bladder cancer. Clin. Sci. (Lond). 2019, 133, 1745–1758. [Google Scholar] [CrossRef]
- Simkhovich, B.; Shutenko, Z.; Meirena, D.; Khagi, K.; Mezapuķe, R.; Molodchina, T.; Kalviņs, I.; Lukevics, E. 3-(2,2,2-Trimethylhydrazinium)propionate (THP)—a novel gamma-butyrobetaine hydroxylase inhibitor with cardioprotective properties. Biochem. Pharmacol. 1988, 37, 195–202. [Google Scholar] [CrossRef]
- Berlato, D.; Bairros, A. Meldonium: Pharmacological, toxicological and analytical aspects. Toxicol. Res. Appl. 2020, 4, 239784732091514. [Google Scholar] [CrossRef]
- Jaudzems, K.; Kuka, J.; Gutsaits, A.; Zinovjevs, K.; Kalvinsh, I.; Liepinsh, E.; Liepinsh, E.; Dambrova, M. Inhibition of carnitine acetyltransferase by mildronate, a regulator of energy metabolism. J. Enzyme Inhib. Med. Chem. 2009, 24, 1269–1275. [Google Scholar] [CrossRef]
- Kruszynska, Y.; Sherratt, H. Glucose kinetics during acute and chronic treatment of rats with 2[6(4-chloro-phenoxy)hexyl]oxirane-2-carboxylate, etomoxir. Biochem. Pharmacol. 1987, 36, 3917–3921. [Google Scholar] [CrossRef]
- Gerondaes, P.; Alberti, K.; Agius, L. Interactions of inhibitors of carnitine palmitoyltransferase I and fibrates in cultured hepatocytes. Biochem. J. 1988, 253, 169–173. [Google Scholar] [CrossRef]
- Tars, K.; Leitans, J.; Kazaks, A.; Zelencova, D.; Liepinsh, E.; Kuka, J.; Makrecka, M.; Lola, D.; Andrianovs, V.; Gustina, D.; Grinberga, S.; Liepinsh, E.; Kalvinsh, I.; Dambrova, M.; Loza, E.; Pugovics, O. Targeting carnitine biosynthesis: discovery of new inhibitors against γ-butyrobetaine hydroxylase. J. Med. Chem. 2014, 57, 2213–2236. [Google Scholar] [CrossRef]
- Dambrova, M.; Makrecka-Kuka, M.; Vilskersts, R.; Makarova, E.; Kuka, J.; Liepinsh, E. Pharmacological effects of meldonium: Biochemical mechanisms and biomarkers of cardiometabolic activity. Pharmacol. Res. 2016, 113, 771–780. [Google Scholar] [CrossRef]
- Grube, M.; Meyer zu Schwabedissen, H.; Präger, D.; Haney, J.; Möritz, K.; Meissner, K.; Rosskopf, D.; Eckel, L.; Böhm, M.; Jedlitschky, G.; Kroemer, H. Uptake of cardiovascular drugs into the human heart: expression, regulation, and function of the carnitine transporter OCTN2 (SLC22A5). Circulation. 2006, 113, 1114–1122. [Google Scholar] [CrossRef]
- Schmitz, F.; Rösen, P.; Reinauer, H. Improvement of myocardial function and metabolism in diabetic rats by the carnitine palmitoyl transferase inhibitor Etomoxir. Horm. Metab. Res. 1995, 27, 515–522. [Google Scholar] [CrossRef]
- Morillas, M.; Clotet, J.; Rubí, B.; Serra, D.; Ariño, J.; Hegardt, F.; Asins, G. Inhibition by etomoxir of rat liver carnitine octanoyltransferase is produced through the co-ordinate interaction with two histidine residues. Biochem. J. 2000, 351, 495–502. [Google Scholar] [CrossRef]
- Lilly, K.; Chung, C.; Kerner, J.; VanRenterghem, R.; Bieber, L. Effect of etomoxiryl-CoA on different carnitine acyltransferases. Biochem. Pharmacol. 1992, 43, 353–61. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, R.; Guo, L.; Ghassemi, S.; Snyder, N.; Worth, A.; Weng, L.; Kam, Y.; Philipson, B.; Trefely, S.; Nunez-Cruz, S.; Blair, I.; June, C.; Milone, M. The CPT1a inhibitor, etomoxir induces severe oxidative stress at commonly used concentrations. Sci. Rep. 2018, 8, 6289. [Google Scholar] [CrossRef]
- Holubarsch, C.; Rohrbach, M.; Karrasch, M.; Boehm, E.; Polonski, L.; Ponikowski, P.; Rhein, S. A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: the ERGO (etomoxir for the recovery of glucose oxidation) study. Clin. Sci. (Lond.) 2007, 113, 205–212. [Google Scholar] [CrossRef]
- Johnson, T.; Kocher, H.; Anderson, R.; Nemecek, G. Cloning, sequencing and heterologous expression of a cDNA encoding pigeon liver carnitine acetyltransferase. Biochem. J. 1995, 305, 439–444. [Google Scholar] [CrossRef]
- Chase, J. Carnitine acetyltransferase from pigeon breast muscle: [EC 2. 3.1.7 Acetyl-CoA: carnitine O-acetyltransferase. 1969, 13, 387–393. [Google Scholar]
- Mcule. Available online: http://mcule.com.
- Szafran, M.; Dega-Szafran, Z.; Nowak-Wydra, B.; Pietrzak, M. Differences in proton–proton coupling constants of N+–CH2–CH2 protons of some betaines, N+–(CH2)2-3–COO−, and their complexes in aqueous solution. J. Mol. Struct. 2001, 563–564, 555–564. [Google Scholar] [CrossRef]
- Chevalier, A.; Zhang, Y.; Khdour, O.; Hecht, S. Selective Functionalization of Antimycin A Through an N-Transacylation Reaction. Org. Lett. 2016, 18, 2395–2398. [Google Scholar] [CrossRef]
- Kuroda, K.; Satria, H.; Miyamura, K.; Tsuge, Y.; Ninomiya, K.; Takahashi, K. Design of Wall-Destructive but Membrane-Compatible Solvents. J. Am. Chem. Soc. 2017, 139, 16052–16055. [Google Scholar] [CrossRef]
- Dega-Szafran, Z.; Dulewicz, E.; Szafran, M. 1H and 13C-NMR spectra of betaines, >N+(CH2)nCOO−, and their hydrogen halides. Additivity rules for carbon-13 chemical shifts. 2000, 38, 43–50. [Google Scholar]
- Jadhav, S.; Ganvir, V.; Shinde, Y.; Revankar, S.; Thakre, S.; Singh, M. Carboxylate functionalized imidazolium-based zwitterions as benign and sustainable solvent for cellulose dissolution: Synthesis and characterization. J. Mol. Liq. 2021, 344, 117724. [Google Scholar] [CrossRef]
- Marquis, N.; Fritz, I. Enzymological determination of free carnitine concentrations in rat tissues. J. Lipid. Res. 1964, 5, 184–187. [Google Scholar] [CrossRef]
- Dega-Szafran, Z.; Przybylak, R. Synthesis, IR and NMR studies of zwitterionic ω-(1-pyrrolidine)alkanocarboxylic acids and their N-methyl derivatives. J. Mol. Struct. 1997, 436-437, 107–121. [Google Scholar] [CrossRef]
- Barczynski, P.; Dega-Szafran, Z.; Dulewicz, E.; Petryna, M. Aqueous basicity and proton affinity of zwitterionic omega-(N-methylpiperidine)-alkanocarboxylates and omega-(N-piperidine)-alkanocarboxylic acids. Pol. J. Chem. 2000, 74, 1149–1161. [Google Scholar]
- Lukeš, R.; Pliml, J. Die reduktion von Pyridinbasen durch Ameisensäure VIII. Über die Reduktion von N-(Äthoxycarbonylalkyl)-pyridiniumformiaten. Collect. Czech. Chem. Commun. 1956, 21, 1602–1606. [Google Scholar] [CrossRef]


| AA sequence | AA position | AA sequence | AA position | AA sequence | AA position | |
| Human (CAT)8 | TYESASLRMFHLGRTD | 430-445 | GEAFDRHLLGL | 492-502 | DCVMFFGPVVP | 540-550 |
| Mouse (CAT)8 | TYESASLRMFHLGRTD | 452-467 | GEAFDRHLLGL | 514-524 | DCVMFFGPVVP | 562-572 |
| Mouse (CAT)57 | TYESASLRMFHLGRTD | 422-437 | GEAFDRHLLGL | 484-494 | DCVMFFGPVVP | 532-542 |
| Pigeon (CAT)55 | TYESASLRMFRLGRTD | 453-468 | GNAIDRHLLGL | 514-524 | DCVMCFGPVVP | 562-572 |
| Human (COT)8 | CYETAMTRHFYHGRTE | 438-453 | GKGFDRHLLGL | 500-510 | GYLRVQGVVVP | 548-558 |
| Human (CPT1)8 | TYEASMTRLFREGRTE | 588-603 | GSGIDRHLFCL | 650-660 | SSGGGFGPVAP | 706-716 |
| Human (CPT2)13 | TYESCSTAAFKHGRTE | 485-500 | GQGFDRHLFAL | 549-559 | VNLGGFAPVVS | 598-608 |
| Rat (CPT2)57 | TYESCSTAAFKHGRTE | 454-469 | GQGFDRHLFAL | 518-528 | VSLGGFAPVVP | 566-576 |
| Type of inhibition | R2 | AIC | Sy.x |
|---|---|---|---|
| Competitive | 0,99382 | -1710,565 | 5.215e-9 |
| Noncompetitive | 0,97754 | -1652,521 | 9.938e-9 |
| Noncompetitive (partial) | 0.97754 | -1,649.983 | 1.006e-8 |
| Uncompetitive | 0,95923 | -1625,683 | 1.339e-8 |
| Uncompetitive (partial) | 0.95922 | -1,623.139 | 1.355e-8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



