Submitted:
07 July 2025
Posted:
07 July 2025
You are already at the latest version
Abstract
This study explores a series of 3,4-dihydroisocoumarins as potential inhibitors of fatty acid oxidation through rational design, molecular docking, synthesis, and in vitro evaluation. The compounds studied were designed as structural analogs of the natural substrates of carnitine acetyltransferase (CAT) and other enzymes in the carnitine transferase family, which play a crucial role in fatty acid metabolism. Comparative in vitro analyses revealed that the presence of an alkyl substituent at the 3rd position of the heterocyclic core, along with its chain length, significantly influences inhibitory activity, yielding IC50 values in the micromolar range. Kinetic studies of one of the most potent compounds—cis- and trans-3-decyl-6,7-dimethoxy-3,4-dihydroisocoumarin-4- carboxylic acids—demonstrated mixed inhibition of CAT, with Ki values of 130 μM and 380 μM, respectively. These findings underscore the therapeutic potential of the compounds under investigation in modulating fatty acid catabolism, with possible applications in treating metabolic disorders.
Keywords:
Carnitine acyltransferase
; fatty acid oxidation
; isocoumarins
; metabolism
; enzyme inhibition
1. Introduction
Carnitine acyltransferases (CTs) are a family of enzymes essential for energy production in human and animal cells. They regulate fatty acid oxidation by catalyzing the reversible transfer of acyl groups between L-carnitine and coenzyme A (CoA). This enzyme family comprises three members—carnitine acetyltransferase (CAT), carnitine octanoyltransferase (COT), and carnitine palmitoyltransferase (CPT)—each responsible for transporting fatty acids of varying chain lengths across cellular compartments, including the cytosol, mitochondria, and peroxisomes. [1,2,3,4,5] Given their critical role in fatty acid metabolism, inhibiting CTs activity has emerged as a promising therapeutic strategy for various chronic diseases linked to excessive fatty acid breakdown. These conditions include cardiovascular diseases [6,7], diabetes [8], kidney and liver diseases [9,10], psychiatric disorders [11], neurodegenerative diseases [12], certain cancers [13,14,15,16,17,18,19,20].
As part of an ongoing project focused on synthesizing potential inhibitors of CTs [21], we directed our attention to a class of natural compounds—3,4-dihydroisocoumarins, which are prominent due to their broad spectrum of biological activities [22,23,24,25,26,27,28,29,30,31,32,33,34]. When appropriately substituted, these compounds can be regarded as molecular hybrids that integrate structural fragments of the natural substrates of carnitine acyltransferases – the L-carnitine fragment and the fatty acid residue (see Figure 1). Moreover, they resemble the structure of well-known CTs inhibitors, such as Clomoxir [35,36], Etomoxir [37,38], Meldonium [39], and Hemiacetylcarnitine [40], which additionally suggests their potential.
In the present study, we employed a rational design and preliminary molecular docking, and synthesized a series of 3,4-dihydroisocoumarin derivatives with a carboxylic group at the 4th position and varied substituents (alkyl or aryl) in the 3rd position of the heterocyclic moiety. We further assessed their inhibitory potential towards CAT as a model enzyme. To the best of our knowledge, this investigation is the first to explore the possibility of such compounds serving as metabolic modulators by inhibiting CAT.
2. Results
2.1. Rational Design
As can be seen from Figure 1, the target 3-substituted 3,4-dihydroisocoumarin-4-carboxylic acids exhibit structural similarity with the native substrates of CTs. The free carboxyl group and the oxygen atom in the β-position relative to it (given in red) resemble the L-carnitine molecule, while the alkyl substituent at C3 (given in blue) those of the fatty acid’s hydrocarbon chain. Furthermore, the presence of a lactone ring would lead to reduced reactivity and increased selectivity. The latter suggests their lower toxicity compared to that demonstrated by the well-known CT inhibitors Etomoxir and Clomoxir, which contain a highly reactive oxirane ring [35,36,37,38].
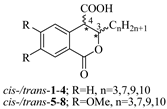
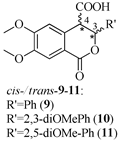
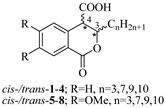
Based on the above analysis, we hypothesized that the target compounds may interact with both active sites of CAT—specifically for L-carnitine and fatty acids—thereby leading to more effective inhibition by simultaneously blocking two regions within its active center. To test this hypothesis, we designed hypothetical structures of 3,4-dihydroisocoumarin-4-carboxylic acids with different substituents at the 3rd position of the benzopyranone skeleton (see Table 1). We chose propyl, heptyl, nonyl, and decyl as alkyl groups to evaluate the impact of the alkyl chain length as a key factor, and included some aryl-substituted compounds (phenyl, 2,3-dimethoxyphenyl, and 2,5-dimethoxyphenyl) to study the effect of substituent type. We also added methoxy groups at the 6th and 7th positions, hypothesizing that this might result in additional hydrogen bonding interactions or influence others, such as π-π stacking or π-cation interactions. Relying on this rationale, we believed that our compounds would be more effective inhibitors of CAT than established ones like Meldonium [39], which target only one site in the enzyme's active center, or Etomoxir and Clomoxir [41], which are suicidal inhibitors causing hepatotoxicity.
To investigate our hypothesis, we conducted a theoretical study using the freely accessible online platform Mcule [42]. We used the structures for CAT from mouse (Mus musculus, PDB identifier: 1NDI) [2] and CPT2 from rat (Rattus norvegicus, PDB identifier: 2FW3) [43]. Our goal was to compare the binding affinities of the hypothetical inhibitors with those of the native substrates—L-carnitine and acetyl-CoA, as well as L-carnitine and Palmitoyl-CoA—along with the positive controls Meldonium and Etomoxir, which target CAT and CPT2, respectively. Additionally, we examined the impact of stereochemistry by analyzing the four possible stereoisomers for each compound.
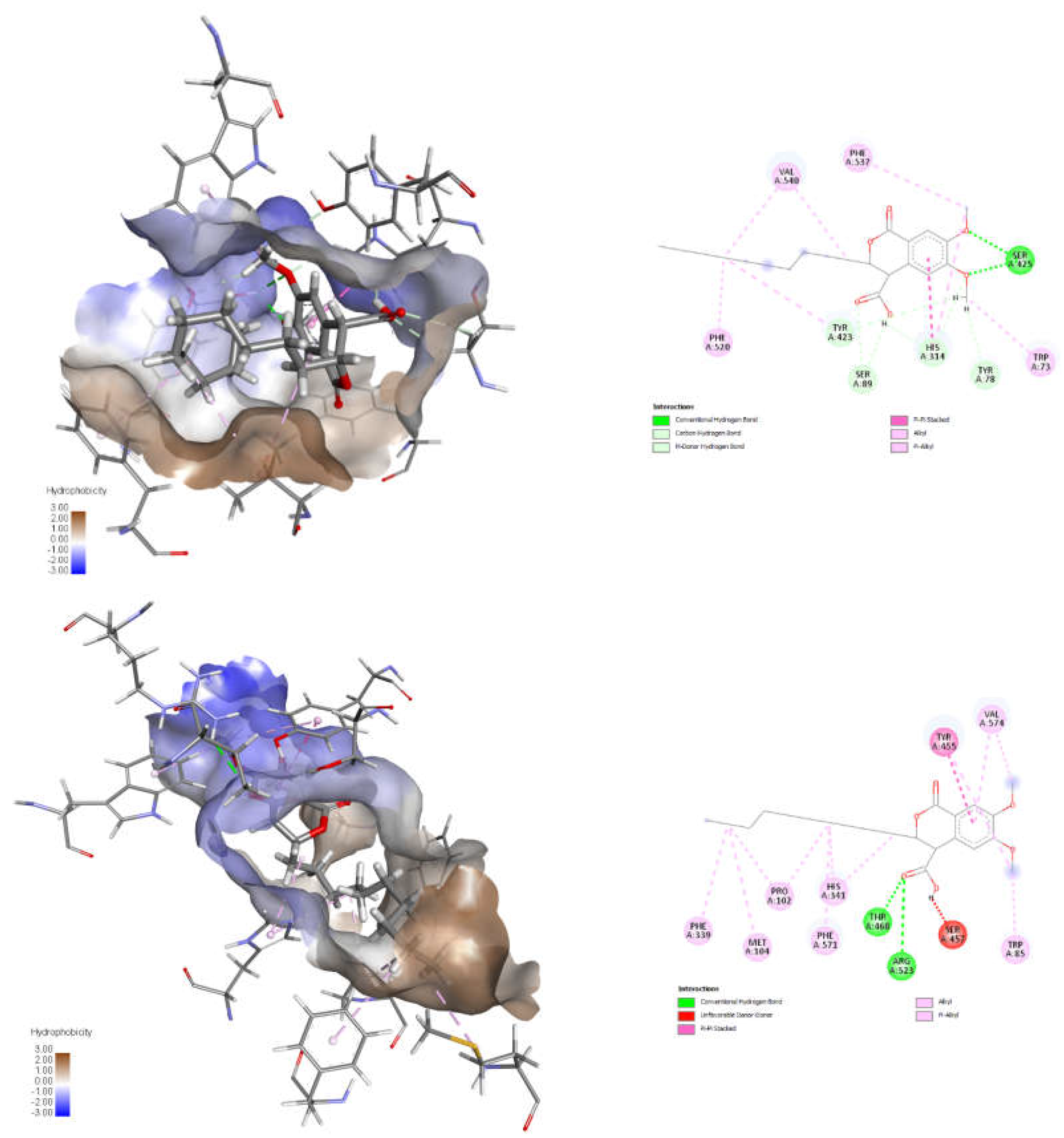
The results of the docking studies and the general structure of the compounds are presented in Table 1 and Table 2, and 3D and 2D visualization of the poses for one of the most promising inhibitors with respect to CAT (Mus musculus, PDB identifier: 1NDI) and CPT2 (Rattus Norvegicus, PDB identifier: 2FW3) – trans-8, in Figure 2.
The most important finding evident from Table 1 is that all compounds exhibit a greater binding affinity than the natural substrates and positive controls, suggesting they may have a strong inhibitory effect. This also provides preliminary support for the hypothesis mentioned earlier, as both substrates and standards bind to a single site within the enzyme's active center. In contrast, the studied compounds are expected to bind to two different sites simultaneously. It is also clear that these compounds have a higher binding affinity for CPT2 than for CAT, emphasizing the role of hydrophobic interactions and the specificity of the hydrophobic pocket that accommodates the alkyl group of fatty acids. Further exploration of this phenomenon was pursued through additional analysis and visualization of the automatically generated docking poses for compound trans-8. This was done because of its relevance, which stems from the conformational flexibility in both the pyranonone ring and the alkyl chain. As shown in Figure 2, the alkyl chain at C3 adopts a straight conformation in CPT2 and a folded one in CAT, matching the size of the hydrophobic pocket, which affects enzyme selectivity based on the chain length of the fatty acid residue. Following this discussion, we consider these initial theoretical results as a strong foundation for future experiments.
2.2. Synthesis and Characterization
Scheme 1 outlines the synthesis of the target 3-substituted-3,4-dihydroisocoumarin-4-carboxylic acids. We employed the straightforward method previously reported by Bogdanov and Palamareva [44] for synthesizing 3-aryl-3,4-dihydroisocoumarin-4-carboxylic acids from homophthalic anhydride and aromatic aldehydes, which we had also recently adapted for synthesizing 3-alkyl-substituted derivatives, particularly cis- and trans-5–8 [45].
During the reaction, two new stereogenic centers form at atoms C-3 and C-4. This results in σ-diastereomerism in these compounds, with the cis and trans arrangement of the substituents relative to the benzopyranone ring system. In the majority of cases, the resulting diastereoisomeric mixtures were successfully separated by flash chromatography, and the products were isolated in pure, crystalline form. Because of the similar behavior of the two diastereoisomers, 3-propyl derivatives 1 and 5, as well as the 2,5-dimethoxyphenyl substituted ones (compd. 11), were isolated, characterized, and tested as isomeric mixtures. Compound 1 was isolated as two mixtures, M1-1 and M2-1, with the cis isomer making up 60% and 35% of each mixture, respectively. Compound 5 was obtained as two mixtures, M1-5 and M2-5, with the cis isomer comprising 90% and 30% of each mixture, respectively. The cis isomer of compound 11 was successfully isolated, but the trans isomer was obtained as a mixture (M-11) with a 60/40 cis/trans ratio.
The structure of the synthesized compounds was unambiguously determined using various spectral techniques, including 1H, 13C, DEPT-135 NMR, and HRMS analysis. The interpretation of spectral data aligns with literature data [44,45,46,47,48]. For the 6,7-dimethoxy-substituted analogues, the spectral data include two singlets for the aromatic protons H-8 and H-5, two singlets for the methoxy groups, and multiplet signals for the methyl and methylene groups in the alkyl chain at C-3. Additional signals reflecting differences in configuration and allowing the determination of substances as cis and trans diastereomers are clearly distinguishable multiplets for H-3 and doublets for H-4. The ratios between cis and trans isomers in the case of racemic mixtures were determined from the integrals of signals for the protons H-3 and H-4. All compounds demonstrate conformational flexibility; however, a comprehensive discussion of this subject exceeds the scope of this study (see Refs. 45 and 46 for additional details). Spectral data are included in the Supplementary Materials.
2.3. Biological Assessment
To assess the inhibitory potential of the synthesized compounds against CAT, we conducted an initial screening at three concentrations: 100 μM, 250 μM, and 1000 μM. Readings were taken within the first minute after the reaction started, and inhibition was measured using a kinetic method, as described in the Experimental section. The results are summarized and presented as a percentage of inhibition in Figure 3 and Figure 4 for the 3-alkyl-3,4-dihydroisocoumarin-4-carboxylic acids and the 3-alkyl/3-aryl-6,7-dimethoxy-3,4-dihydroisocoumarin-4-carboxylic acids, respectively.
As shown in Figure 3 and Figure 4, all tested compounds exhibit promising inhibitory properties, with some demonstrating significant effects at micromolar concentrations. These findings align with the preliminary docking studies and confirm that activity depends on both the substituent type and alkyl chain length, supporting our initial hypothesis. At the highest tested concentration (1 mM), the nonyl and decyl derivatives induce near-complete inhibition, whereas propyl, heptyl, and their aromatic analogues show lower activity. Notably, the inhibitory potency appears independent of stereochemistry and substitution pattern, as evidenced by the similar effects demonstrated by the cis- and trans-diastereomeric pairs and methoxy-substituted and unsubstituted derivatives at positions 6 and 7.
The most potent inhibitors in this series were the cis- and trans-3-decyl-3,4-dihydroisocoumarin-4-carboxylic acids, with IC₅₀ values of approximately 250 μM—an order of magnitude lower than the positive control, Meldonium (IC₅₀ = 11.4 mM, Ref. 21).
Notably, theoretical calculations suggested that 3-aryl-substituted isocoumarins have a stronger affinity for the enzyme's active site than their 3-alkyl-substituted counterparts. However, experimental results showed significantly reduced activity when a conjugated system is present at the 3-position. This decrease in activity most likely occurs because the inhibitor cannot form additional π-π interactions with the enzyme's hydrophobic pocket, which is crucial for stabilizing binding.
Given carnitine acetyltransferase’s high affinity for short-chain fatty acids (C₂–C₄), the observed correlation between increasing hydrocarbon chain length and enhanced inhibitory activity suggests an uncompetitive or mixed-type inhibition mechanism. To validate this hypothesis, we conducted additional kinetic studies as detailed in the Experimental section on the most active diastereomeric pair cis-8 and trans-8. These experiments allowed us to identify the inhibition mechanism and calculate Km, Vmax, and Ki. We used SigmaPlot version 12.5 (Systat Software Inc., San Jose, CA, USA), which includes modules for regression analysis and various inhibition models. This software also lets us choose the mechanism of inhibition with the highest correlation factors (coefficient of determination (R2) and Akaike information coefficient (AIC) and the lowest Sy,x value. The numerical results are summarized in Table 3, while the corresponding Lineweaver–Burk and Michaelis–Menten kinetic plots are available in the Supplementary Materials. Our findings indicate that cis-8 and trans-8 are mixed-type of inhibitors with Ki values of 130 μM and 377 μM, respectively. The trans isomer demonstrated a cooperative effect (α = 0.21, where α < 1), indicating an increased inhibitory activity in the presence of the substrate. Conversely, the cis isomer functions as an allosteric inhibitor, attaching to a different site on the enzyme and inducing a conformational change that reduces the enzyme's affinity for its substrate.
The results obtained demonstrate the possibility of the studied compounds to be applied as metabolic modulators and their potential for the treatment of systemic inflammatory processes, ischemic disease, diabetes and some types of cancer. It would be of interest to study the individual enantiomers, which is also the subject of upcoming research.
3. Materials and Methods
3.1. General
All chemicals and CAT (isolated from pigeon breast muscle, ammonium sulfate suspension, CAS Number: 9029-90-7) used in this study were purchased from Sigma-Aldrich (FOT, Sofia, Bulgaria). The organic solvents were of analytical grade and were used without further purification. Molecular docking was performed with the freely available online platform Mcule (Mcule Inc., Palo Alto, CA, USA; http://mcule.com). NMR spectra were recorded on a Bruker Avance III HD (Bruker BioSpin GmbH, Rheinstetten, Germany, 500 MHz and 126 MHz for 1H and 13C, respectively) DMSO as a solvent. The chemicals shifts (δ) are given in ppm and J values are reported in Hz. TLC was performed on pre-coated 0.2 mm aluminum plates with silica gel 60 with fluorescence indicator (Alugram® SIL G/UV254, Macherey-Nagel, Merck, Darmstadt, Germany). Column chromatography was performed on Horizon High Performance FLASH chromatography system (HPFC) with cartridges filled with Silica gel 60 [particle size – 0.06–0.2 mm (70–230 mesh), MACHEREY-NAGEL, Düren, Germany]. Biological assessment was performed on ELISA Reader Biotek 800TS (Biotek Instruments, Inc, ELTA90, Sofia, Bulgaria). High-Resolution Mass Spectra (HRMS) were obtained on a Shimadzu LCMS-9050 (Shimadzu Handels GmbH., Korneuburg, Austria).
3.2. Synthesis
The corresponding aldehyde (1 equiv.) was added to a solution of 1.1 equiv. homophthalic anhydride for the series of (±)-3-alkyl-3,4-dihydro-1-oxo-1H-isochromene-4-carboxylic acids or 6,7-dimethoxyhomophtalic anhydride (1.1 equiv.) for the series of (±)-3-alkyl/aryl-3,4-dihydro-6,7-dimethoxy-1-oxo-1H-isochromene-4-carboxylic acids in 10 mL dry chloroform and DMAP (1 equiv.) was added. The resulting mixture was stirred for 1 h at r.t. (22–23 ◦C). At the end of the reaction (TLC monitoring), the obtained carboxylic acids were extracted with 10% NaHCO3 and the aqueous layer was acidified (pH = 3) with 18% HCl and extracted with EtOAc. The organic layer was dried with Na2SO4. The solvent was evaporated and the diastereoisomers of the product were isolated via column chromatography (mobile phase: petroleum ether/EtOAc = 1/1 + formic acid).
3.2.1. cis- and trans-(±)-3-propyl-3,4-dihydro-1-oxo-1H-isochromene-4-carboxylic Acids (1)
Homophtalic anhydride (2.00 g, 12.0 mmol) reacted with butanal (0.80 g, 11.0 mmol) in the presence of 1.30 g (11.0 mmol) DMAP to give white crystals of 1 (1.62 g, 63% yield). After purification two mixtures – M1-1 and M2-1, were acquired with percentages of the cis isomer of 60% and 35%, respectively:
1H-NMR (500 MHz, DMSO):
cis diastereomer: δ = 4.73–4.65 (1H, m, 3-CH), 3.98 (1H, d, 3J3,4 = 3.1 Hz, 4-CH)
trans diastereomer: δ = 4.90 (1H, dt, J = 8.8, 4.4 Hz, 3-CH), 4.07 (1H, d, 3J3,4 = 4.0 Hz, 4-CH)
other signals for both diastereomers: δ = 12.99 (1H, s, COOH), 7.98–7.91 (1H, m, 8-CH), 7.72–7.61 (1H, m, 6-CH), 7.51 (1H, t, J = 7.6 Hz, 7-CH), 7.44 (1H, t, J = 7.1 Hz, 5-CH), 1.83–1.64 (1H, m, 1’-CH2), 1,64-1,33 (3H, m, 1’-CH2, 2’-CH2), 0.94 (3H, t, 3J2’,3’ = 7.4 Hz, 3’-CH3).
HRMS (ESI) m/z, calculated for [M-H]- C13H13O4-: 233.08193, found [M-H]-: 233.08096.
3.2.2. cis- and trans-(±)-3-Heptyl-3,4-dihydro-1-oxo-1H-isochromene-4-carboxylic Acids (2)
Homophtalic anhydride (2.00 g, 12.0 mmol) reacted with octanal (1.40 g, 11.0 mmol) in the presence of 1.30 g (11.0 mmol) DMAP to give white crystals of 2 (2.70 g, 85% yield). After purification and separation, cis and trans isomers were acquired:
cis-2, m.p. = 105–109 °C (from CH2Cl2: petroleum ether, b.p. = 35–60 °C); Rf = 0.41 (EtOAc: CH2Cl2 = 2:3); 1H-NMR (500 MHz, DMSO): δ = 12.95 (1H, s, COOH), 7.95 (1H, d, J = 7.7 Hz, 8-CH), 7.64 (1H, t, J = 7.5 Hz, 5-CH), 7.51 (1H, t, J = 7.6 Hz, 7-CH), 7.45 (1H, d, J = 7.6 Hz, 5-CH), 4.67 (1H, td, 3J3,1′ = 6.8, 3J3,4 = 3.1 Hz, 3-CH), 3.99 (1H, d, 3J3,4 = 3.0 Hz, 4-CH), 1.81–1.70 (2H, m, 1′-CH2), 1.56–1.39 (2H, m, 2′-CH2), 1.38–1.19 (8H, m, 3′–6′-CH2), 0.87 (3H, t, 3J6′,7′ = 6.6 Hz, 7′-CH3). 13C NMR (126 MHz, DMSO): δ = 170.57 (C, C=O, COOH), 164.34 (C, 1C), 138.25 (C, 8aC), 133.63 (CH, 8C), 129.49 (CH, 6C), 128.49 (CH, 7C), 127.71 (CH, 5C), 125.25 (C, 4aC), 78.41 (CH, 3C), 46.68 (CH, 4C), 32.23 (CH2), 31.17 (CH2), 28.68 (CH2), 28.57 (CH2), 24.69 (CH2), 22.08 (CH2), 13.96 (CH3, 7′-CH3).
HRMS (ESI) m/z, calculated for [M-H]- C17H21O4-: 289.14453, found [M-H]-: 289.14390.
trans-2, m.p = 126–127 °C (from CH2Cl2: petroleum ether, b.p. = 35–60 °C); Rf = 0.38 (EtOAc: CH2Cl2 = 2:3); 1H-NMR (500 MHz, DMSO): δ = 13.20 (1H, s, COOH), 7.95 (1H, d, J = 7.7 Hz, 8-CH), 7.68 (1H, t, J = 7.5 Hz, 6-CH), 7.50 (1H, t, J = 7.6 Hz, 7-CH), 7.43 (1H, d, J = 7.6 Hz, 5-CH), 4.95–4.82 (1H, m, 3-CH), 4.07 (1H, d, 3J3,4 = 4.1 Hz, 4-CH), 1.63–1.47 (2H, m, 1′-CH2), 1.46–1.30 (2H, m, 2′-CH2), 1.29–1.03 (8H, m, 3′–6′-CH2), 0.83 (3H, t, 3J6′,7′ = 6.8 Hz, 7′-CH3). 13C-NMR (126 MHz, DMSO): δ = 171.79 (C, C=O, COOH), 163.09 (C, 1C), 136.47 (C, 8aC), 134.09 (CH, 8C), 129.15 (CH, 6C), 128.63 (CH, 7C), 128.37 (CH, 5C), 124.45 (C, 4aC), 79.06 (CH, 3C), 47.04 (CH, 4C), 33.06 (CH2), 31.10 (CH2), 28.44 (CH2), 24.69 (CH2), 22.04 (CH2), 13.92 (CH3, 7′-CH3).
HRMS (ESI) m/z, calculated for [M-H]- C19H25O6-: 289.14453, found [M-H]-: 289.14493.
3.2.3 cis- and trans-(±)-3,4-Dihydro-3-nonyl-1-oxo-1H-isochromene-4-carboxylic Acids (3)
Homophtalic anhydride (2.00 g, 12.0 mmol) reacted with decanal (1.72 g, 11.0 mmol) in the presence of 1.30 g (11.0 mmol) DMAP to give white crystals of 3 (2.00 g, 56% yield). After purification and separation, cis and trans isomers were acquired:
cis-3, m.p. = 133–135 °C (from CH2Cl2: petroleum ether, b.p. = 35–60 °C); Rf = 0.42 (EtOAc: CH2Cl2 = 2:3); 1H-NMR (500 MHz, DMSO): δ = 12.95 (1H, s, COOH), 7.95 (1H, d, J = 7.7 Hz, 8-CH), 7.64 (1H, t, J = 7.5 Hz, 6-CH), 7.51 (1H, t, J = 7.6 Hz, 7-CH), 7.44 (1H, d, J = 7.6 Hz, 5-CH), 4.67 (1H, td, 3J3,1′ = 6.8, 3J3,4 = 3.1 Hz, 3-CH), 3.99 (1H, d, 3J3,4 = 3.1 Hz, 4-CH), 1.82–1.71 (2H, m, 1′-CH2), 1.61–1.41 (2H, m, 2′-CH2), 1.41–1.11 (12H, m, 3′–8′-CH2), 0.86 (3H, t, 3J8′,9′ = 6.8 Hz, 9′-CH3). 13C-NMR (126 MHz, DMSO): δ = 170.57 (C, C=O, COOH), 164.33 (C, 1C), 138.25 (C, 8aC), 133.63 (CH, 8C), 129.49 (CH, 6C), 128.49 (CH, 7C), 127.71 (CH, 5C), 125.25 (C, 4aC), 78.41 (CH, 3C), 46.69 (CH, 4C), 32.23 (CH2), 31.31 (CH2), 28.91 (CH2), 28.72 (CH2), 28.70 (CH2), 24.69 (CH2), 22.11 (CH2), 13.96 (CH3, 9′-CH3).
HRMS (ESI) m/z, calculated for [M-H]- C19H25O4-: 317.17583, found [M-H]-: 317.17582.
trans-3, m.p = 110–111 °C (from CH2Cl2: petroleum ether, b.p. = 35–60 °C); Rf = 0.39 (EtOAc: CH2Cl2 = 2:3); 1H-NMR (500 MHz, DMSO): δ = 13.20 (1H, s, COOH), 7.95 (1H, d, J = 7.7 Hz, 8-CH), 7.68 (1H, t, J = 7.5 Hz, 6-CH), 7.50 (1H, t, J = 7.6 Hz, 7-CH), 7.43 (1H, d, J = 7.6 Hz, 5-CH), 4.92–4.83 (1H, m, 3-CH), 4.07 (1H, d, 3J3,4 = 4.1 Hz, 4-CH), 1.63–1.46 (2H, m, 1′-CH2), 1.47–1.30 (2H, m, 2′-CH2), 1.30–1.12 (12H, m, 3′–8′-CH2), 0.84 (3H, t, 3J8′,9′ = 6.8 Hz, 9′-CH3). 13C-NMR (126 MHz, DMSO): δ = 172.24 (C, C=O, COOH), 163.54 (C, 1C), 136.94 (C, 8aC), 134.54 (CH, 8C), 129.60 (CH, 6C), 129.08 (CH, 7C), 128.81 (CH, 5C), 124.92 (C, 4aC), 79.53 (CH, 3C), 47.53 (CH, 4C), 33.54 (CH2), 31.72 (CH2), 29.29 (CH2), 29.26 (CH2) 29.12 (CH2), 28.94 (CH2), 25.15 (CH2), 22.54 (CH2) 14.39 (CH3, 9′-CH3).
HRMS (ESI) m/z, calculated for [M-H]- C19H25O4-: 317.17583, found [M-H]-: 317.17582.
3.2.4. cis- and trans-(±)-3-Decyl-3,4-dihydro-1-oxo-1H-isochromene-4-carboxylic Acids (4)
Homophtalic anhydride (2.00 g, 12.0 mmol) reacted with undecanal (1.90 g, 11.0 mmol) in the presence of 1.30 g (11.0 mmol) DMAP to give white crystals of 4 (2.20 g, 60% yield). After purification and separation, cis and trans isomers were acquired:
cis-4, m.p. = 129–131 °C (from CH2Cl2: petroleum ether, b.p. = 35–60 °C); Rf = 0.43 (EtOAc: CH2Cl2 = 2:3); 1H-NMR (500 MHz, DMSO): δ = 7.93 (1H, d, J = 7.9 Hz, 8-CH), 7.65 (1H, t, J = 7.5 Hz, 6-CH), 7.50 (1H, t, J = 7.6 Hz, 7-CH), 7.43 (1H, d, J = 7.5 Hz, 5-CH), 4.70–4.60 (1H, m, 3-CH), 3.97 (1H, d, 3J3,4 = 2.9 Hz, 4-CH), 1.81–1.69 (2H, m, 1′-CH2), 1.57–1.37 (2H, m, 2′-CH2), 1.35–1.18 (14H, m, 3′–9′-CH2), 0.86 (3H, t, 3J9′,10′ = 6.7 Hz, 10′-CH3). 13C-NMR (126 MHz, DMSO): δ = 170.53 (C, C=O, COOH), 164.38 (C, 1C), 136.80 (C, 8aC), 133.58 (CH, 8C), 129.45 (CH, 6C), 128.39 (CH, 7C), 127.68 (CH, 5C), 125.26 (C, 4aC), 78.47 (CH, 3C), 46.87 (CH, 4C), 32.24 (CH2), 31.29 (CH2), 28.99 (CH2), 28.95 (CH2), 28.91 (CH2), 28.71 (CH2), 24.70 (CH2), 22.09 (CH2), 13.96 (CH3, 10′-CH3).
HRMS (ESI) m/z, calculated for [M-H]- C20H21O4-: 289.14453, found [M-H]-: 289.14390.
trans-3, m.p = 110–111 °C (from CH2Cl2: petroleum ether, b.p. = 35–60 °C); Rf = 0.39 (EtOAc: CH2Cl2 = 2:3); 1H-NMR (500 MHz, DMSO): δ = 13.20 (1H, s, COOH), 7.94 (1H, d, J = 7.7 Hz, 8-CH), 7.68 (1H, t, J = 7.5 Hz, 6-CH), 7.50 (1H, t, J = 7.6 Hz, 7-CH), 7.43 (1H, d, J = 7.6 Hz, 5-CH), 4.92–4.83 (1H, m, 3-CH), 4.07 (1H, d, 3J3,4 = 4.1 Hz, 4-CH), 1.63–1.46 (2H, m, 1′-CH2), 1.47–1.30 (2H, m, 2′-CH2), 1.30–1.12 (12H, m, 3′–9′-CH2), 0.84 (3H, t, 3J8′,9′ = 6.8 Hz, 10′-CH3). 013C-NMR (126 MHz, DMSO): δ = 172.25 (C, C=O, COOH), 163.54 (C, 1C), 136.94 (C, 8aC), 134.55 (CH, 8C), 129.60 (CH, 6C), 129.09 (CH, 7C), 128.82 (CH, 5C), 124.92 (C, 4aC), 79.53 (CH, 3C), 47.52 (CH, 4C), 33.54 (CH2), 31.74 (CH2), 29.42 (CH2), 29.34 (CH2), 29.25 (CH2), 29.14 (CH2), 28.94 (CH2), 25.15 (CH2), 22.55 (CH2) 14.40 (CH3, 10′-CH3).
HRMS (ESI) m/z, calculated for [M-H]- C19H25O6-: 317.17583, found [M-H]-: 317.17582.
3.2.5. cis- and trans-(±)-3-propyl-3,4-dihydro-6,7-dimethoxy-1-oxo-1H-isochromene-4-caboxylic Acids (5)
6,7-Dimethoxyhomophtalic anhydride (2.00 g, 9.00 mmol) reacted with butanal (0.59 g, 8.18 mmol) in the presence of 1.00 g (8.18 mmol) DMAP to give white crystals of 5 (1.32 g, 56% yield). After purification two mixtures – M1-5 and M2-5, were acquired with percentages of the cis isomer of 90% and 30%, respectively:
1H-NMR (500 MHz, DMSO):
cis diastereomer: δ = 4.68–4.58 (1H, m, 3-CH), 3.87-3.78 (1H, m, 4-CH − the signal overlaps with the signals of 6-OCH3 and 7-OCH3)
trans diastereomer: δ = 4.91–4.85 (1H, m, 3-CH), 3.93 (1H, d, 3J3,4 = 3.1 Hz, 4-CH)
other signals for both diastereomers: δ = 12.88 (1H, s, COOH), 7.40 (1H, s, 8-CH), 7.02 (1H, s, 5-CH), 3.87-3.78 (6H, m, 6-OCH3, 7-OCH3), 1.81-1.34 (4H, m, 1’-CH2, 2’-CH2), 0.95 (3H, t, 3J2’,3’ = 7.4 Hz, 3’-CH3).
HRMS (ESI) m/z, calculated for [M-H]- C15H17O6-: 293.10306, found [M-H]-: 293.10158.
3.2.6. cis- and trans-(±)-3-Heptyl-3,4-dihydro-6,7-dimethoxy-1-oxo-1H-isochromene-4-carboxylic Acids (6)
6,7-Dimethoxyhomophtalic anhydride (2.00 g, 9.00 mmol) reacted with octanal (1.05 g, 8.18 mmol) in the presence of 1.00 g (8.18 mmol) DMAP to give white crystals of 6 (2.63 g, 92% yield). After purification and separation, cis and trans isomers were acquired:
cis-6, m.p. = 132–134 °C (from CH2Cl2: petroleum ether, b.p. = 35–60 °C); Rf = 0.37 (EtOAc: CH2Cl2 = 2:3); 1H-NMR (500 MHz, DMSO): δ = 12.86 (1H, s, COOH), 7.39 (1H, s, 8-CH), 7.02 (1H, s, 5-CH), 4.65–4.58 (1H, td, 3J3,1′ = 7.0, 3J3,4 = 3.3 Hz, 3-CH), 3.86 (1H, d, 3J3,4 = 3.2 Hz, 4-CH), 3.84 (3H, s, 7-OCH3), 3.81 (3H, s, 6-OCH3), 1.81–1.68 (2H, m, 1′-CH2), 1.55–1.37 (2H, m, 2′-CH2), 1.37–1.20 (8H, m, 3′–6′-CH2), 0.87 (3H, t, 3J6′,7′ = 6.8 Hz, 7′-CH3). 13C-NMR (126 MHz, DMSO): δ = 170.69 (C, C=O, COOH), 164.26 (C, 1C), 153.10 (C, 6C), 148.59 (C, 7C), 132.52 (C, 4aC), 117.19 (C, 8aC), 111.24 (CH, 8C), 110.14 (CH, 5C), 78.41 (CH, 3C), 55.97 (CH3, 6-OCH3), 55.68 (CH3, 7-OCH3), 46.41 (CH, 4C), 32.27 (CH2), 31.16 (CH2), 28.69 (CH2), 28.57 (CH2), 24.69 (CH2), 22.08 (CH2), 13.96 (7′-CH3).
HRMS (ESI) m/z, calculated for [M-H]- C19H25O6-: 349.16566, found [M-H]-: 349.16430.
trans-6, m.p = 134–136 °C (from CH2Cl2: petroleum ether, b.p. = 35–60 °C); Rf = 0.33 (EtOAc: CH2Cl2 = 2:3); 1H-NMR (500 MHz, DMSO): δ = 13.10 (1H, s, COOH), 7.37 (1H, s, 8-CH), 6.99 (1H, s, 5-CH), 4.88–4.81 (1H, ddd, 3J3,1′ = 8.5, 3J3,1′ = 5.2, 3J3,4 = 3.3 Hz, 3-CH), 3.93 (1H, d, 3J3,4 = 3.3 Hz, 4-CH), 3.84 (3H, s, 7-OCH3), 3.81 (3H, s, 6-OCH3), 1.64–1.46 (2H, m, 1′-CH2), 1.45–1.30 (2H, m, 2′-CH2), 1.30–1.15 (8H, m, 3′–6′-CH2), 0.84 (3H, t, 3J6′,7′ = 6.9 Hz, 7′-CH3). 13C-NMR (126 MHz, DMSO): δ = 172.49 (C, C=O, COOH), 163.30 (C, 1C), 153.88 (C, 6C), 148.94 (C, 7C), 130.92 (C, 4aC), 116.98 (C, 8aC), 111.66 (CH, 8C), 111.33 (CH, 5C), 79.56 (CH, 3C), 56.34 (CH3, 6-OCH3), 56.10 (CH3, 7-OCH3), 47.01 (CH, 4C), 33.60 (CH2), 31.58 (CH2), 28.94 (CH2), 28.91 (CH2), 25.32 (CH2), 22.50 (CH2), 14.39 (CH3, 7′-CH3).
HRMS (ESI) m/z, calculated for [M-H]- C19H25O6-: 349.16566, found [M-H]-: 349.16366.
3.2.7. cis- and trans-(±)-3,4-Dihydro-6,7-dimethoxy-3-nonyl-1-oxo-1H-isochromene-4-carboxylic Acids (7)
6,7-dimethoxyhomophtalic anhydride (1.29 g, 5.8 mmol) reacted with decanal (0.83 g, 5.3 mmol) in the presence of 0.646 g (5.31 mmol) DMAP to give white crystals of 7 (1.71 g, 85% yield). After purification and separation, cis and trans isomers were acquired:
cis-7, m.p. = 137–139 °C (from CH2Cl2: petroleum ether, b.p. = 35–60 °C); Rf = 0.39 (EtOAc: CH2Cl2 = 2:3); 1H-NMR (500 MHz, DMSO): δ = 12.85 (1H, s, COOH), 7.39 (1H, s, 8-CH), 7.02 (1H, s, 5-CH), 4.66–4.56 (1H, td, 3J3,1′ = 6.9, 3J3,4 = 3.3 Hz, 3-CH), 3.85 (1H, d, 3J3,4 = 3.2 Hz, 4-CH), 3.84 (3H, s, 7-OCH3), 3.81 (3H, s, 6-OCH3), 1.81–1.67 (2H, m, 1′-CH2), 1.56–1.38 (2H, m, 2′-CH2), 1.38–1.19 (12H, m, 3′–8′-CH2), 0.86 (3H, t, 3J8′,9′ = 6.9 Hz, 9′-CH3). 13C-NMR (126 MHz, DMSO): 171.15 (C, C=O, COOH), 164.72 (C, 1C), 153.57 (C, 6C), 149.06 (C, 7C), 132.98 (C, 4aC), 117.65 (C, 8aC), 111.70 (CH, 8C), 110.60 (CH, 5C), 78.87 (CH, 3C), 56.42 (CH3, 6-OCH3), 56.13 (CH3, 7-OCH3), 46.88 (CH, 4C), 32.74 (CH2), 31.77 (CH2), 29.40 (CH2), 29.37 (CH2), 29.20 (CH2), 29.16 (CH2), 25.16 (CH2), 22.57 (CH2), 14.43 (CH3, 9′-CH3).
HRMS (ESI) m/z, calculated for [M-H]- C21H29O6-: 377.19696, found [M-H]-: 377.19537;
trans-7, m.p. = 140–143 °C (from CH2Cl2: petroleum ether, 35–60 °C); Rf = 0.35 (EtOAc: CH2Cl2 = 2:3); 1H-NMR (500 MHz, DMSO): δ = 13.05 (1H, s, COOH), 7.38 (1H, s, 8-CH), 6.99 (1H, s, 5-CH), 4.89–4.80 (1H, ddd, 3J3,1′ = 8.5, 3J3,1′ = 5.2, 3J3,4 = 3.3 Hz, 3-CH), 3.92 (1H, d, 3J3,4 = 3.3 Hz, 4-CH), 3.84 (3H, s, 7-OCH3), 3.81 (3H, s, 6-OCH3), 1.64–1.46 (2H, m, 1′-CH2), 1.44–1.30 (2H, m, 2′-CH2), 1.28–1.17 (12H, m, 3′–8′-CH2), 0.84 (3H, t, 3J8′,9′ = 6.9 Hz, 9′-CH3). 13C-NMR (126 MHz, DMSO): 172.48 (C, C=O, COOH), 163.28 (C, 1C), 153.88 (C, 6C), 148.95 (C, 7C), 130.90 (C, 4aC), 116.98 (C, 8aC), 111.66 (CH, 8C), 111.32 (CH, 5C), 79.55 (CH, 3C), 56.33 (CH3, 6-OCH3), 56.09 (CH3, 7-OCH3), 47.00 (CH, 4C), 33.60 (CH2), 31.72 (CH2), 29.30 (CH2), 29.28 (CH2), 29.12 (CH2), 28.95 (CH2), 25.30 (CH2), 22.54 (CH2), 14.39 (CH3, 9′-CH3).
HRMS (ESI) m/z, calculated for [M-H]- C21H29O6-: 377.19696, found [M-H]-: 377.19482.
3.2.8. cis- and trans-(±)-3-Decyl-3,4-dihydro-6,7-dimethoxy-1-oxo-1H-isochromene-4-carboxylic Acids (8)
6,7-dimethoxyhomophtalic anhydride (0.611 g, 2.80 mmol) reacted with undecanal (0.426 g, 2.50 mmol) in the presence of 0.306 g (2.50 mmol) DMAP to give white crystals of 8 (0.76 g, 77% yield). After purification and separation, cis and trans isomers were acquired:
cis-8, m.p. = 143–145 °C (from CH2Cl2: petroleum ether, b.p. = 35–60 °C); Rf = 0.41 (EtOAc: CH2Cl2 = 2:3); 1H-NMR (500 MHz, DMSO): δ = 12.86 (1H, s, COOH), 7.38 (1H, s, 8-CH), 7.02 (1H, s, 5-CH), 4.66–4.55 (1H, td, 3J3,1′ = 7.0, 3J3,4 = 3.3 Hz, 3-CH), 3.85 (1H, d, J = 3.2 Hz, 4-CH), 3.85 (3H, s, 7-OCH3), 3.82 (3H, s, 6-OCH3), 1.80–1.69 (2H, m, 1′-CH2), 1.56–1.38 (2H, m, 2′-CH2), 1.37–1.17 (14H, m, 3′–9′-CH2), 0.86 (3H, t, 3J9′,10′ = 6.9 Hz, 10′-CH3). 13C-NMR (126 MHz, DMSO): 171.15 (C, C=O, COOH), 164.71 (C, 1C), 153.57 (C, 6C), 149.06 (C, 7C), 132.97 (C, 4aC), 117.65 (C, 8aC), 111.70 (CH, 8C), 110.59 (CH, 5C), 78.87 (CH, 3C), 56.42 (CH3, 6-OCH3), 56.13 (CH3, 7-OCH3), 46.88 (CH, 4C), 32.74 (CH2), 31.77 (CH2), 29.47 (CH2), 29.42 (CH2), 29.40 (CH2), 29.20 (CH2), 25.17 (CH2), 22.57 (CH2), 14.42 (CH3, 10′-CH3).
HRMS (ESI) m/z, calculated for [M-H]- C22H31O6-: 391.21261, found [M-H]-: 391.21064.
trans-8, m.p. = 148–150 °C (from CH2Cl2: petroleum ether, b.p. = 35–60 °C); Rf = 0.38 (EtOAc: CH2Cl2 = 2:3); 1H-NMR (500 MHz, DMSO): δ = 13.06 (1H, s, COOH), 7.37 (1H, s, 8-CH), 6.99 (1H, s, 5-CH), 4.89–4.79 (1H, ddd, 3J3,1′ = 8.5, 3J3,1′ = 5.2, 3J3,4 = 3.3 Hz, 3-CH), 3.92 (1H, d, J = 3.3 Hz, 4-CH), 3.84 (3H, s, 7-OCH3), 3.81 (3H, s, 6-OCH3), 1.63–1.44 (2H, m, 1′-CH2), 1.44–1.30 (2H, m, 2′-CH2), 1.30–1.14 (14H, m, 3′–9′-CH2), 0.84 (3H, t, 3J9′,10′ = 6.9 Hz, 10′-CH3). 13C-NMR (126 MHz, DMSO): 172.48 (C, C=O, COOH), 163.28 (C, 1C), 153.88 (C, 6C), 148.95 (C, 7C), 130.90 (C, 4aC), 116.98 (C, 8aC), 111.66 (CH, 8C), 111.32 (CH, 5C), 79.55 (CH, 3C), 56.33 (CH3, 6-OCH3), 56.09 (CH3, 7-OCH3), 47.00 (CH, 4C), 33.60 (CH2), 31.74 (CH2), 29.42 (CH2), 29.35 (CH2), 29.28 (CH2), 29.14 (CH2), 28.95(CH2), 25.31 (CH2), 22.55 (CH2), 14.40 (CH3, 10′-CH3).
HRMS (ESI) m/z, calculated for [M-H]- C22H31O6-: 391.21261, found [M-H]-: 391.21098.
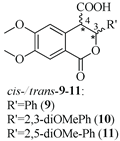
3.2.9. cis- and trans-(±)-3,4-Dihydro-6,7-dimethoxy-1-oxo-3-phenyl-1H-isochromene-4-carboxylic Acids (9)
6,7-dimethoxyhomophtalic anhydride (2.00 g, 9.00 mmol) reacted with benzaldehyde (0.832 g, 7.84 mmol) in the presence of 0.958 g (7.84 mmol) DMAP to give white crystals of 9 (2.41 g, 92% yield). After purification and separation, cis and trans isomers were acquired:
cis-9, m.p. = 191–193 °C (from CH2Cl2: petroleum ether, b.p. = 35–60 °C); Rf = 0.42 (EtOAc: CH2Cl2 = 2:3); 1H-NMR (500 MHz, DMSO): δ = 12.58 (1H, s, COOH), 7.53–7.47 (3H, m, 8-CH, 2’-CH, 6’-CH), 7.43 (2H, t, J = 7.5, 3’-CH, 5’-CH), 7.39–7.34 (1H, m, 4’-CH), 7.06 (1H, s, 5-CH), 5.91 (1H, d, 3J3,4 = 3.6 Hz, 3-CH), 4.17 (1H, d, 3J3,4 = 3.6 Hz, 4-CH), 3.87 (3H, s, OCH3), 3.85 (3H, s, OCH3). 13C-NMR (126 MHz, DMSO): 170.12 (C, C=O, COOH), 164.06 (C, 1C), 153.34 (C, 6C), 148.82 (C, 7C), 137.23 (C, 1’C), 132.05 (C, 4aC), 128.24 (CH, 2’C, 6’C), 125.79 (CH, 3’C, 5’C), 117.00 (C, 8aC), 111.44 (CH, 8C), 110.01 (CH, 5C), 78.72 (CH, 3C), 56.05 (CH3, OCH3), 55.74 (CH3, OCH3), 49.11 (CH, 4C).
HRMS (ESI) m/z, calculated for [M-H]- C18H15O6-: 327.08741, found [M-H]-: 327.08598.
trans-9, m.p. = 208–210 °C (from CH2Cl2: petroleum ether, b.p. = 35–60 °C); Rf = 0.39 (EtOAc: CH2Cl2 = 2:3); 1H-NMR (500 MHz, DMSO): δ = 13.23 (1H, s, COOH), 7.40 (1H, s, 8-CH), 7.38–7.28 (5H, m, 2’–6’-CH), 6.93 (1H, s, 5-CH), 5.99 (1H, d, 3J3,4 = 5.0 Hz, 3-CH), 4.49 (1H, d, 3J3,4 = 5.1 Hz, 4-CH), 3.81 (3H, s, OCH3), 3.81 (3H, s, OCH3). 13C-NMR (126 MHz, DMSO): 171.45 (C, C=O, COOH), 163.22 (C, 1C), 153.64 (C, 6C), 148.60 (C, 7C), 138.04 (C, 1’C), 130.55 (C, 4aC), 128.54 (CH, 2’C, 6’C), 128.34 (CH, 4’C), 126.47 (CH, 3’C, 5’C), 116.63 (C, 8aC), 110.88 (CH, 8C), 110.31 (CH, 5C), 79.62 (CH, 3C), 55.87 (CH3, OCH3), 55.65 (CH3, OCH3), 47.94 (CH, 4C).
HRMS (ESI) m/z, calculated for [M-H]- C18H15O6-: 327.08741, found [M-H]-: 327.08581.
3.2.10. cis- and trans-(±)-3,4-Dihydro-3-(2,3-dimethoxyphenyl)-6,7-dimethoxy-1-oxo-1H-isochromene-4-carboxylic Acids (10)
6,7-dimethoxyhomophtalic anhydride (2.00 g, 9.00 mmol) reacted with 2,3-dimethoxybenzaldehyde (1.302 g, 7.84 mmol) in the presence of 0.958 g (7.84 mmol) DMAP to give white crystals of 10 (2.81 g, 91% yield). After purification and separation, cis and trans isomers were acquired:
cis-10, m.p. = 215–217 °C (from CH2Cl2: petroleum ether, b.p. = 35–60 °C); Rf = 0.41 (EtOAc: CH2Cl2 = 2:3); 1H-NMR (500 MHz, DMSO): δ = 12.57 (1H, s, COOH), 7.48 (1H, s, 8-CH), 7.16–7.05 (4H, m, 5-H, 4’- 6’-CH), 6.01 (1H, d, 3J3,4 = 3.6, 3-CH), 4.05 (1H, d, 3J3,4 = 3.6 Hz, 4-CH), 3.91–3.79 (12H, m, 6-OCH3, 7-OCH3, 2’-OCH3, 3’-OCH3). 13C-NMR (126 MHz, DMSO): 170.06 (C, C=O, COOH), 164.10 (C, 1C), 153.45 (C), 151.76 (C), 148.83 (C), 145.07 (C), 131.93 (C), 130.20 (C), 123.76 (CH), 118.16 (CH), 117.00 (C), 112.88 (CH), 111.44 (CH3, OCH3), 110.14 (CH), 74.80 (CH, 3C), 60.35 (CH3, OCH3), 56.08 (CH3, OCH3), 55.75 (CH3, OCH3), 55.71 (CH3, OCH3), 47.94 (CH, 4C).
HRMS (ESI) m/z, calculated for [M-H]- C20H19O8-: 387.10854, found [M-H]-: 387.10692.
trans-10, m.p. = 208–210 °C (from CH2Cl2: petroleum ether, b.p. = 35–60 °C); Rf = 0.36 (EtOAc: CH2Cl2 = 2:3); 1H-NMR (500 MHz, DMSO): δ = 13.21 (1H, s, COOH), 7.44 (1H, s, 8-CH), 7.07–6.90 (2H, m, 4’-CH, 6’-CH), 6.94 (1H, s, 5-CH), 6.66 (1H, dd, J = 7.7, 1.4 Hz, 5’-CH), 6.18 (1H, d, 3J3,4 = 4.9 Hz, 3-CH), 4.39 (1H, d, 3J3,4 = 5.0 Hz, 4-CH), 3.90-3.71 (12H, m, 6-OCH3, 7-OCH3, 2’-OCH3, 3’-OCH3). 13C-NMR (126 MHz, DMSO): 171.97 (C, C=O, COOH), 163.84 (C, 1C), 154.13 (C), 152.87 (C), 149.09 (C), 146.45 (C), 131.52 (C), 130.94 (C), 124.34 (CH), 118.98 (CH), 116.91 (C), 113.77 (CH), 111.31 (CH), 110.93 (CH), 75.98 (CH), 60.85 (CH3, OCH3), 56.34 (CH3, OCH3), 56.20 (CH3, OCH3), 56.14 (CH3, OCH3) 47.63 (CH, 4C).
HRMS (ESI) m/z, calculated for [M-H]- C18H15O6-: 327.08741, found [M-H]-: 387.10689.
3.2.11. cis- and trans-(±)-3,4-Dihydro-3-(2,5-dimethoxyphenyl)-6,7-dimethoxy-1-oxo-1H-isochromene-4-carboxylic Acids (11)
6,7-dimethoxyhomophtalic anhydride (2.00 g, 9.00 mmol) reacted with 2,5-dimethoxybenzaldehyde (1.302 g, 7.84 mmol) in the presence of 0.958 g (7.84 mmol) DMAP to give white crystals of 11 (2.80 g, 90% yield). After purification and separation, cis isomer and mixture of cis and trans isomers – M-11 in ratio 60%/40% were acquired:
cis-11, m.p. = 210–212 °C (from CH2Cl2: petroleum ether, b.p. = 35–60 °C); Rf = 0.40 (EtOAc: CH2Cl2 = 2:3); 1H-NMR (500 MHz, DMSO): δ = 12.57 (1H, s, COOH), 7.47 (1H, s, 8-CH), 7.11 (1H, s, 5-H), 7.05–6.99 (2H, m, 3’-CH, 6’-CH), 6.92 (1H, dd, 3J3,4 = 8.9, 3.2 Hz, 4’-CH), 5.95 (1H, d, 3J3,4 = 3.5 Hz, 3-CH), 4.11 (1H, d, 3J3,4 = 3.5 Hz, 4-CH), 3.87–3.73 (12H, m, 6-OCH3, 7-OCH3, 2’-OCH3, 5’-OCH3). 13C-NMR (126 MHz, DMSO): 170.05 (C, C=O, COOH), 164.04 (C, 1C), 153.45 (C), 153.03 (C), 149.53 (C), 148.81 (C), 131.80 (C), 125.83 (C), 116.95 (C), 113.42 (CH), 112.67 (CH), 111.83 (CH), 111.45 (CH), 110.17 (CH), 74.38 (CH, 3C), 59.76 (CH3, OCH3), 56.08 (CH3, OCH3), 55.73 (CH3, OCH3), 55.43 (CH3, OCH3), 46.98 (CH, 4C).
HRMS (ESI) m/z, calculated for [M-H]- C20H19O8-: 387.10854, found [M-H]-: 387.10685.
trans diastereomer: Rf = 0.37 (EtOAc: CH2Cl2 = 2:3); 1H-NMR (500 MHz, DMSO): δ = 6.17 (1H, d, 3J3,4 = 3.8 Hz, 3-CH), 4.36 (1H, d, 3J3,4 = 3.9 Hz, 4-CH).
HRMS (ESI) m/z, calculated for [M-H]- C20H19O8-: 387.10854, found [M-H]-: 387.10677.
3.3. In Vitro Studies
To determine the effect of the synthesized compounds on CAT activity, a modified procedure for spectrophotometric determination of L-carnitine using Ellman's reagent [49] was used. In all analyses, an enzyme isolated from pigeon breast muscle (Sigma Aldrich) was used in the form of an ammonium sulfate suspension with an activity of 71 U/mg protein. One unit of enzyme catalyzes the conversion of 1 μmol of L-carnitine and acetyl-CoA into acetylcarnitine and free CoA in 1 min. Phosphate buffer (0.5 M, pH = 7.6) is used to dilute the enzyme to obtain stock solution with concentration 24 U/mL.
Ellman's reagent was prepared immediately before each measurement by dissolving 25mg DTNB in 5 mL 1 mM solution of Na2EDTA in phosphate buffer (0.5 M, pH = 7.6). Stock solutions of acetyl-CoA and L-carnitine were prepared in deionized water with concentration 348.0 μM and 303.4, respectively. An aqueous solution of tris(hydroxymethyl)aminomethane (TRIS) with pH = 7.6 and 1 M concentration was used as buffer solution. Test compounds were dissolved and diluted in phosphate buffer (0.5 M, pH = 7.6) to the desired stock concentration. The working volume of the reaction is 300 μl and contains 50 μl of each of the six components (DTNB, TRIS, acetyl-CoA, CAT, L-carnitine and inhibitor) solutions with concentrations as described below. In the control samples, the inhibitor solution is replaced with phosphate buffer (0.5 M, pH = 7.6). The incubation time for all components without L-carnitine is 5 min at 37 ˚C. The reaction was started by adding L-carnitine, and its progress was monitored by reading the change in absorbance at 405 nm in kinetic mode. The concentrations of the components in the final volume were respectively: c (DTNB) = 114 μM, c (TRIS) = 100 mM, c (acetyl-CoA) = 58 μM, c (CAT) = 4 U/mL, c (L-carnitine) = 50.56 μM. The time for reading the results was the first minute after starting the reaction.
To establish the mechanism of inhibition of the most active compounds, extensive kinetic studies were carried out and the obtained data on the initial rate of the reaction, v0, were used to construct the v0 vs. [S] (Michaelis-Menten) and 1/v0 vs. [S] (Lainweaver-Burke) relationships. For this purpose, five solutions with different concentrations of the inhibitor and five solutions with different concentrations of L-carnitine were prepared. The concentrations of the remaining components in the final volume were c (DTNB) = 114μM, c (TRIS) = 100 mM, c (acetyl-CoA) = 58 μM and c (CAT) = 4 U/mL, respectively. The time for reading the results is every six seconds for three minutes after the start of the reaction, which corresponds to the end of the linear interval. The initial velocity v0 was determined using the method proposed by Baici [50], by calculating the cut-off of the v vs. t (time) dependence for the linear interval of each of the reactions. Graphical dependencies are provided in the Supplementary Materials (Figures S1, S2 and S3).
3.4. Molecular Docking
Binding energies (kcal/mol) for the compounds studied herein were automatically computed using the 1-Click docking tool available on the Mcule online platform [https:// mcule.com (accessed on 27 January 2025, Mcule Inc., Palo Alto, CA, USA)] [42]. This online platform incorporates an embedded version of AutoDock Vina for docking operations [51] and utilizes AutoDock tools to prepare both ligand and target for the docking process [52]. Detailed instructions on using the 1-Click docking tool, including a tutorial, can be found at https://mcule.com/apps/1-click-docking/ (accessed on 27 January 2025). The target proteins were selected from the Mcule database—CAT isolated from mouse (Mus musculus, PDB identifier: 1NDI) [2] and CPT2 isolated from rat (Rattus norvegicus, PDB identifier: 2FW3) [43]. The x, y, and z coordinates for the binding center of CAT corresponded to His314 (30.133, 34.746, 90.272), while these for the CPT2 were default values (18.0041, 6.6764, 32.0234). We considered protein molecules as rigid structures without water molecules, while the ligands were flexible and optimized by the 1-Click docking algorithm. The pose for each compound was generated, and the results for the binding energy (lower binding energy indicating the higher binding affinity) are listed in Table 1 and Table 2. The poses of the natural substrate L-carnitine and one of the most active compounds – trans-8 with CAT (Mus musculus, PDB identifier: 1NDI) [2] were further evaluated and visualized using BIOVIA Discovery Studio Visualizer v24.1.0.23298 [53]. The poses demonstrating the highest binding affinity and the specific interactions within the active center are shown in Figure 4.
4. Conclusions
We investigated 3-alkyl-3,4-dihydroisocoumarin-4-carboxylic acids as potential inhibitors of carnitine acetyltransferase (CAT) using a structure-based rational approach. Structure-activity relationship studies and docking simulations showed that these compounds bind more effectively to the enzyme’s active site than natural substrates or known inhibitors, indicating a potential impact on fatty acid β-oxidation. Eleven diastereomeric pairs were synthesized, purified, and tested in vitro, demonstrating CAT inhibitory activity with IC50 values between 100 μM and 1 mM. Notably, some derivatives outperformed the reference inhibitor Meldonium (IC50 = 11.4 mM). The presence and length of a hydrophobic alkyl group at the 3rd position of the benzopyranone moiety were crucial for activity. Kinetic analyses identified the most potent compounds (cis-8, and trans-8) as mixed inhibitors. Given their demonstrated inhibitory potential, the compounds studied herein are promising metabolic modulators for the treatment of inflammation, ischemic diseases, diabetes, and certain types of cancer.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/doi/s1, Michaelis-Menten and Lineweaver-Burk plots for different types of inihibition; 1H- and 13C-NMR spectra of all compounds described in the script.
Author Contributions
S.S. and M.G.B. contributed equally to this article. All authors have read and agreed to the published version of the manuscript
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the supplementary material. Further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Govindasamy, L.; Kukar, T.; Lian, W.; Pedersen, B.; Gu, Y.; Agbandje-McKenna, M.; Jin, S.; McKenna, R.; Wu, D. Structural and mutational characterization of L-carnitine binding to human carnitine acetyltransferase. J. Struct. Biol. 2004, 146, 416–424. [Google Scholar] [CrossRef]
- Jogl, G.; Tong, L. Crystal structure of carnitine acetyltransferase and implications for the catalytic mechanism and fatty acid transport. Cell 2003, 112, 113–122. [Google Scholar] [CrossRef]
- Bonnefont, J.; Djouadi, F.; Prip-Buus, C.; Gobin, S.; Munnich, A.; Bastin, J. Carnitine palmitoyltransferases 1 and 2: Biochemical, molecular and medical aspects. Mol. Aspects Med. 2004, 25, 495–520. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.; Gratacós, E.; Carrasco, P.; Clotet, J.; Ureña, J.; Serra, D.; Asins, G.; Hegardt, F.; Casals, N. CPT1c is localized in endoplasmic reticulum of neurons and has carnitine palmitoyltransferase activity. J. Biol. Chem. 2008, 283, 6878–6885. [Google Scholar] [CrossRef]
- Wu, D.; Govindasamy, L.; Lian, W.; Gu, Y.; Kukar, T.; Agbandje-McKenna, M.; McKenna, R. Structure of human carnitine acetyltransferase. Molecular basis for fatty acyl transfer. J. Biol. Chem. 2003, 278, 13159–13165. [Google Scholar] [CrossRef] [PubMed]
- Nechaeva, G. Zheltikova, E. Effects of Meldonium in early postmyocardial infarction period. Kardiologiia 2015, 55, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Liamina, N. Kotel'nikova, E., Karpova, É., Biziaeva, E., Senchikhin, V., Lipchanskaia, T. Cardioprotective capabilities of drug meldonium in secondary prevention after percutaneous coronary intervention in patients with documented myocardial ischemia. Kardiologiia 2014, 54, 60–65. [Google Scholar] [CrossRef]
- Keung, W. Ussher, J. R., Jaswal, J. S., Raubenheimer, M., Lam, V. H., Wagg, C. S., Lopaschuk, G. D. Inhibition of carnitine palmitoyltransferase-1 activity alleviates insulin resistance in diet-induced obese mice. Diabetes 2013, 62, 711–720. [Google Scholar] [CrossRef]
- Đurašević, S. Stojković, M., Bogdanović, L., Pavlović, S., Borković-Mitić, S., Grigorov, I., Bogojević, D., Jasnić, N., Tosti, T., Đurović, S., Đorđević, J., Todorović, Z. The Effects of Meldonium on the Renal Acute Ischemia/Reperfusion Injury in Rats. Int. J. Mol. Sci. 2019, 20, 5747. [Google Scholar] [CrossRef]
- Đurašević, S. Stojković, M., Sopta, J., Pavlović, S., Borković-Mitić, S., Ivanović, A., Jasnić, N., Tosti, T., Đurović, S., Đorđević, J., Todorović, Z. The effects of meldonium on the acute ischemia/reperfusion liver injury in rats. Sci. rep. 2021, 11, 1305. [Google Scholar]
- Mørkholt, A. Wiborg, O., Nieland, J. Blocking of carnitine palmitoyl transferase 1 potently reduces stress-induced depression in rat highlighting a pivotal role of lipid metabolism. Sci. Rep. 2017; 7, 2158. [Google Scholar]
- Trabjerg, M. Andersen, D., Huntjens, P., Mørk, K., Warming, N., Kullab, U., Skjønnemand, M., Oklinski, M., Oklinski, K., Bolther, L., Kroese, L., Pritchard, C., Huijbers, I., Corthals, A., Søndergaard, M., Kjeldal, H., Pedersen, C., Nieland, J. Inhibition of carnitine palmitoyl-transferase 1 is a potential target in a mouse model of Parkinson's disease. NPJ Parkinson's Dis. 2023, 9, 6. [Google Scholar]
- Gregory, K. Elliott, G., Robertson, H., Kumar, A., Wanless, E., Webber, G., Craig, V., Andersson, G., Page, A. Mitochondrial and metabolic alterations in cancer cells. Eur. J. Cell Biol. 2022, 101, 151225. [Google Scholar]
- Liu, Y. Chen, S., Liu, Z., Lu, Y., Xia, G., Liu, H., He, L., She, Z. Bioactive Metabolites from Mangrove Endophytic Fungus Aspergillus sp. 16-5B. Mar. drugs, 2015; 13, 3091–3102. [Google Scholar]
- Jariwala, N. Mehta, G., Bhatt, V., Hussein, S., Parker, K., Yunus, N., Parker, J., Guo, J., Gatza, M. CPT1A and fatty acid β-oxidation are essential for tumor cell growth and survival in hormone receptor-positive breast cancer. NAR cancer, 2021; zcab035. [Google Scholar]
- Wang, X. Yang, C., Huang, C., Wang, W. Dysfunction of the carnitine cycle in tumor progression. Heliyon 2024, 10, e35961. [Google Scholar] [CrossRef] [PubMed]
- Monaco, M. Fatty acid metabolism in breast cancer subtypes. Oncotarget 2017, 8, 29487–29500. [Google Scholar] [CrossRef]
- Maher, M., Diesch. Diesch, J., Casquero, R., Buschbeck, M. Epigenetic-Transcriptional Regulation of Fatty Acid Metabolism and Its Alterations in Leukaemia. Front. Genet. 2018, 9, 405. [Google Scholar] [CrossRef]
- Zhu, Y. Wang, Y., Li, Y., Li, Z., Kong, W., Zhao, X., Chen, S., Yan, L., Wang, L., Tong, Y., Shao, Y. Carnitine palmitoyltransferase 1A promotes mitochondrial fission and regulates autophagy by enhancing MFF succinylation in ovarian cancer. Commun. Biol. 2023, 6, 618. [Google Scholar] [CrossRef]
- Ma, L. Chen, C., Zhao, C., Li, T., Ma, L., Jiang, J., Duan, Z., Si, Q., Chuang, T. H., Xiang, R., & Luo, Y. Targeting carnitine palmitoyl transferase 1A (CPT1A) induces ferroptosis and synergizes with immunotherapy in lung cancer. Sign. Transduct. Targ. Ther. 2024, 9–64. [Google Scholar]
- Stoyanova, S. Bogdanov, M. G. Rational Design, Synthesis, and In Vitro Activity of Heterocyclic Gamma-Butyrobetaines as Potential Carnitine Acetyltransferase Inhibitors. Molecules 2025, 30, 735. [Google Scholar] [CrossRef]
- Saeed, A. Isocoumarins, miraculous natural products blessed with diverse pharmacological activities. Eur. J. Med. Chem. 2016, 116, 290–317. [Google Scholar] [CrossRef]
- Barry, R. Isocoumarins. Developments since 1950. Chem. Rev. 1964, 64, 229–260. [Google Scholar] [CrossRef]
- Napolitano, E. The synthesis of isocoumarins over the last decade. A review. Org. Prep. Proced. Int. 1997, 29, 631–664. [Google Scholar] [CrossRef]
- Noor, A. O. Almasri, D. M., Bagalagel, A. A., Abdallah, H. M., Mohamed, S. G. A., Mohamed, G., Ibrahim, S. Naturally Occurring Isocoumarins Derivatives from Endophytic Fungi: Sources, Isolation, Structural Characterization, Biosynthesis, and Biological Activities. Molecules 2020, 25, 395. [Google Scholar] [CrossRef]
- Hussain, H. Jabeen, F., Krohn, K., Al-Harrasi, A., Ahmad, M., Mabood, F., Shah, A., Badshah, A., Ur Rehman, N., Green, I., Ali, I., Draeger, S., Schulz, B. Antimicrobial activity of two mellein derivatives isolated from an endophytic fungus. Med. Chem. Res. 2015, 24, 2111–2114. [Google Scholar] [CrossRef]
- Hussain, H. Krohn, K., Draeger, S., Meier, K., Schulz, B. Bioactive chemical constituents of a sterile endophytic fungus from Meliotus dentatus. Rec. Nat. Prod. 2009, 3, 114–117. [Google Scholar]
- Zhao, M. Yuan, L., Guo, D., Ye, Y., Da-Wa, Z., Wang, X., Ma, F. W., Chen, L., Gu, Y., Ding, L., Zhou, Y. (2018). Bioactive halogenated dihydroisocoumarins produced by the endophytic fungus Lachnum palmae isolated from Przewalskia tangutica. Phytochem. 2018, 148, 97–103. [Google Scholar] [CrossRef]
- Orfali, R. Perveen, S., AlAjmI, M., Ghaffar, S., Rehman, M., AlanzI, A., Gamea, S., Essa Khwayri, M. Antimicrobial Activity of Dihydroisocoumarin Isolated from Wadi Lajab Sediment-Derived Fungus Penicillium chrysogenum: In Vitro and In Silico Study. Molecules 2022, 27, 3630. [Google Scholar] [CrossRef]
- Aly, A. H. Edrada-Ebel, R., Wray, V., Müller, W. E., Kozytska, S., Hentschel, U., Proksch, P., & Ebel, R. Bioactive metabolites from the endophytic fungus Ampelomyces sp. isolated from the medicinal plant Urospermum picroides. Phytochem. 2008, 69, 1716–1725. [Google Scholar]
- Furuta, T. Fukuyama, Y., Asakawa, Y. Polygonolide, an isocoumarin from Polygonum hydropiper possessing anti-inflammatory activity. Phytochem. 1986, 25, 517–520. [Google Scholar] [CrossRef]
- Ju, Z. Lin, X., Lu, X., Tu, Z., Wang, J., Kaliyaperumal, K., Liu, J., Tian, Y., Xu, S., Liu, Y. Botryoisocoumarin A, a new COX-2 inhibitor from the mangrove Kandelia candel endophytic fungus Botryosphaeria sp. KcF6. J. Antibiot. 2015, 68, 653–656. [Google Scholar] [CrossRef]
- Koopklang, K. Choodej, S., Hantanong, S., Intayot, R., Jungsuttiwong, S., Insumran, Y., Ngamrojanavanich, N., Pudhom, K. Anti-Inflammatory Properties of Oxygenated Isocoumarins and Xanthone from Thai Mangrove-Associated Endophytic Fungus Setosphaeria rostrata. Molecules 2024, 29, 603. [Google Scholar] [CrossRef]
- Sukandar, E. Kaennakam, S., Raab, P., Nöst, X., Rassamee, K., Bauer, R., Siripong, P., Ersam, T., Tip-pyang, S., Chavasiri, W. Cytotoxic and Anti-Inflammatory Activities of Dihydroisocoumarin and Xanthone Derivatives from Garcinia picrorhiza. Molecules 2021, 26, 6626. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, K. Sherratt, St., Turnbull, D. (1984). Inhibition of hepatic and skeletal muscle carnitine palmitoyltransferase I by 2[5(4-chlorophenyl)pentyl]-oxirane-2-carbonyl-CoA. Biochem. Soc. Trans., 12(4): 688–689.
- Wolf, H. Eistetter, K., Ludwig, G. (1982). Phenylalkyloxirane carboxylic acids, a new class of hypoglycaemic substances: hypoglycaemic and hypoketonaemic effects of sodium 2-[5-(4-chlorophenyl)-pentyl]-oxirane-2-carboxylate (B 807-27) in fasted animals. Diabetologia., 22(6):456-63.
- Lilly, K. Chung, C., Kerner, J., VanRenterghem, R., Bieber, L. (1992). Effect of etomoxiryl-CoA on different carnitine acyltransferases. Biochem. Pharmacol., 43(2):353-61.
- Selby, P. Sherratt, H. (1989). Substituted 2-oxiranecarboxylic acids: a new group of candidate hypoglycaemic drugs. Trends Pharmacol. Sci., 10(12):495-500.
- Jaudzems, K. Kuka, J., Gutsaits, A., Zinovjevs, K., Kalvinsh, I., Liepinsh, E., Liepinsh, E., Dambrova, M. Inhibition of carnitine acetyltransferase by mildronate, a regulator of energy metabolism. J. Enzyme Inhib. Med. Chem. 2009, 24, 1269–75. [Google Scholar] [CrossRef] [PubMed]
- Gandour, R. Colucci, W., Stelly, T., Brady, P., Brady, L. (1986). Active-site probes of carnitine acyltransferases. Inhibition of carnitine acetyltransferase by hemiacetylcarnitinium, a reaction intermediate analogue. Biochem. Biophys. Res. Commun., 138:735-741.
- O’Connor, R.; Guo, L.; Ghassemi, S.; Snyder, N.; Worth, A.; Weng, L.; Kam, Y.; Philipson, B.; Trefely, S.; Nunez-Cruz, S.; et al. The CPT1a inhibitor, etomoxir induces severe oxidative stress at commonly used concentrations. Sci. Rep. 2018, 8, 6289. [Google Scholar] [CrossRef]
- Mcule. Available online: http://mcule.com (accessed on 27 January 2025).
- Rufer, A. Thoma, R., Benz, J., Stihle, M., Gsell, B., De Roo, E., Banner, D., Mueller, F., Chomienne, O., Hennig, M. (2006). The crystal structure of carnitine palmitoyltransferase 2 and implications for diabetes treatment. Structure 2006, 14, 713–23. [Google Scholar] [CrossRef]
- Bogdanov, M. Palamareva, M. Cis/trans-Isochromanones. DMAP induced cycloaddition of homophthalic anhydride and aldehydes. Tetrahedron 2004, 60, 2525–2530. [Google Scholar] [CrossRef]
- Stoyanova, S.; Bogdanov, M.G. Synthesis and Characterization of cis-/trans-(±)-3-Alkyl-3,4-dihydro-6,7-dimethoxy-1-oxo-1H-isochromene-4-carboxylic Acids. Molbank 2025, 2025, M1988. [Google Scholar] [CrossRef]
- Bogdanov, M.; Todorov, I.; Manolova, P.; Cheshmedzhieva, D.; Palamareva, M. Configuration and conformational equilibrium of (±)-trans-1-oxo-3-thiophen-2-yl-isochroman-4-carboxylic acid methyl ester. Tetrahedron Lett. 2004, 45, 8383–8386. [Google Scholar] [CrossRef]
- Miliovsky, M.; Svinyarov, I.; Mitrev, Y.; Evstatieva, Y.; Nikolova, D.; Chochkova, M.; Bogdanov, M. A novel one-pot synthesis and preliminary biological activity evaluation of cis-restricted polyhydroxy stilbenes incorporating protocatechuic acid and cinnamic acid fragments. Eur. J. Med. Chem. 2013, 66, 185–192. [Google Scholar] [CrossRef]
- Miliovsky, M.; Svinyarov, I.; Prokopova, E.; Batovska, D.; Stoyanov, S.; Bogdanov, M. Synthesis and Antioxidant Activity of Polyhydroxylated trans-Restricted 2-Arylcinnamic Acids. Molecules 2015, 20, 2555–2575. [Google Scholar] [CrossRef]
- Marquis, N. Fritz, I. Enzymological determination of free carnitine concentrations in rat tissues. J Lipid Res. 1964, 5, 184–7. [Google Scholar] [CrossRef]
- Baici, A. Kinetics of Enzyme-Modifier Interactions; 1st ed.; Springer: Vienna, 2015. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock 4 and AutoDock Tools 4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA Discovery Studio Visualizer. Available online: https://discover.3ds.com/discovery-studio-visualizer-download (accessed on 27 January 2025.).
Figure 1.
Structure of Clomoxir-CoA and Extomoxir-CoA, Meldonium and Hemiacetylcarnitine.
Figure 2.
3D and 2D visualizations of docking poses for the 3R,4S isomer of trans-8 and the specific interactions in the active site of CAT (top) and CPT2 (bottom). The hydrocarbon residue in the 3rd position preferentially locates in the hydrophobic pocket according to its size specificity.
Figure 2.
3D and 2D visualizations of docking poses for the 3R,4S isomer of trans-8 and the specific interactions in the active site of CAT (top) and CPT2 (bottom). The hydrocarbon residue in the 3rd position preferentially locates in the hydrophobic pocket according to its size specificity.
Scheme 1.
Synthesis of cis- and trans-3,4-dihydroisocoumarin-4-carboxylic acids 1–11 and their derivatives.
Scheme 1.
Synthesis of cis- and trans-3,4-dihydroisocoumarin-4-carboxylic acids 1–11 and their derivatives.
Figure 3.
CAT inhibition (%) in the presence of (±)-3-alkyl-3,4-dihydro-3,4-dihydroisocoumarin-4-carboxylic acids.
Figure 3.
CAT inhibition (%) in the presence of (±)-3-alkyl-3,4-dihydro-3,4-dihydroisocoumarin-4-carboxylic acids.
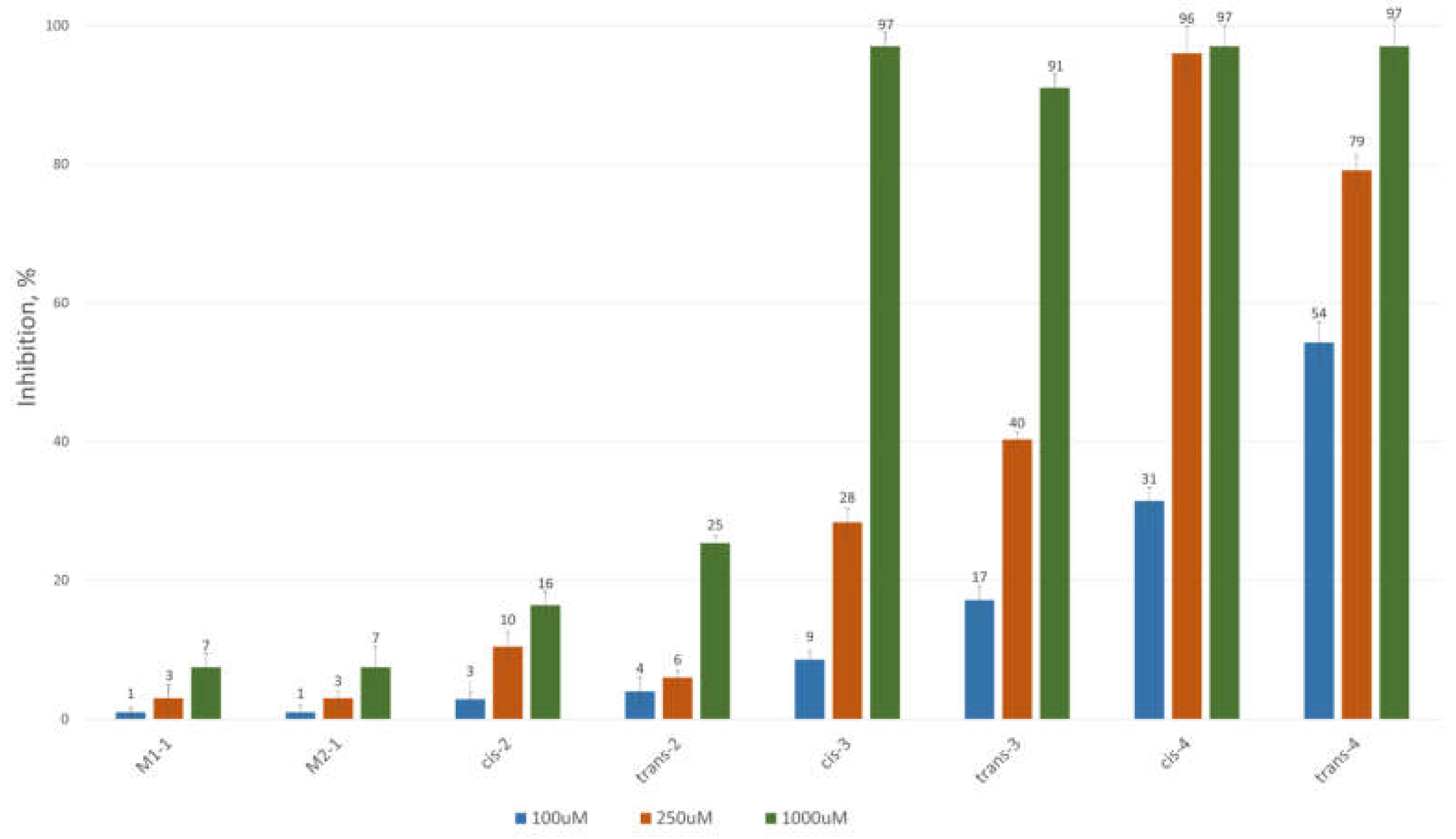
Figure 4.
CAT inhibition (%) in the presence of (±)-3-alkyl/aryl-3,4-dihydro-6,7-dimethoxy-3,4-dihydroisocoumarin-4-carboxylic acids.
Figure 4.
CAT inhibition (%) in the presence of (±)-3-alkyl/aryl-3,4-dihydro-6,7-dimethoxy-3,4-dihydroisocoumarin-4-carboxylic acids.
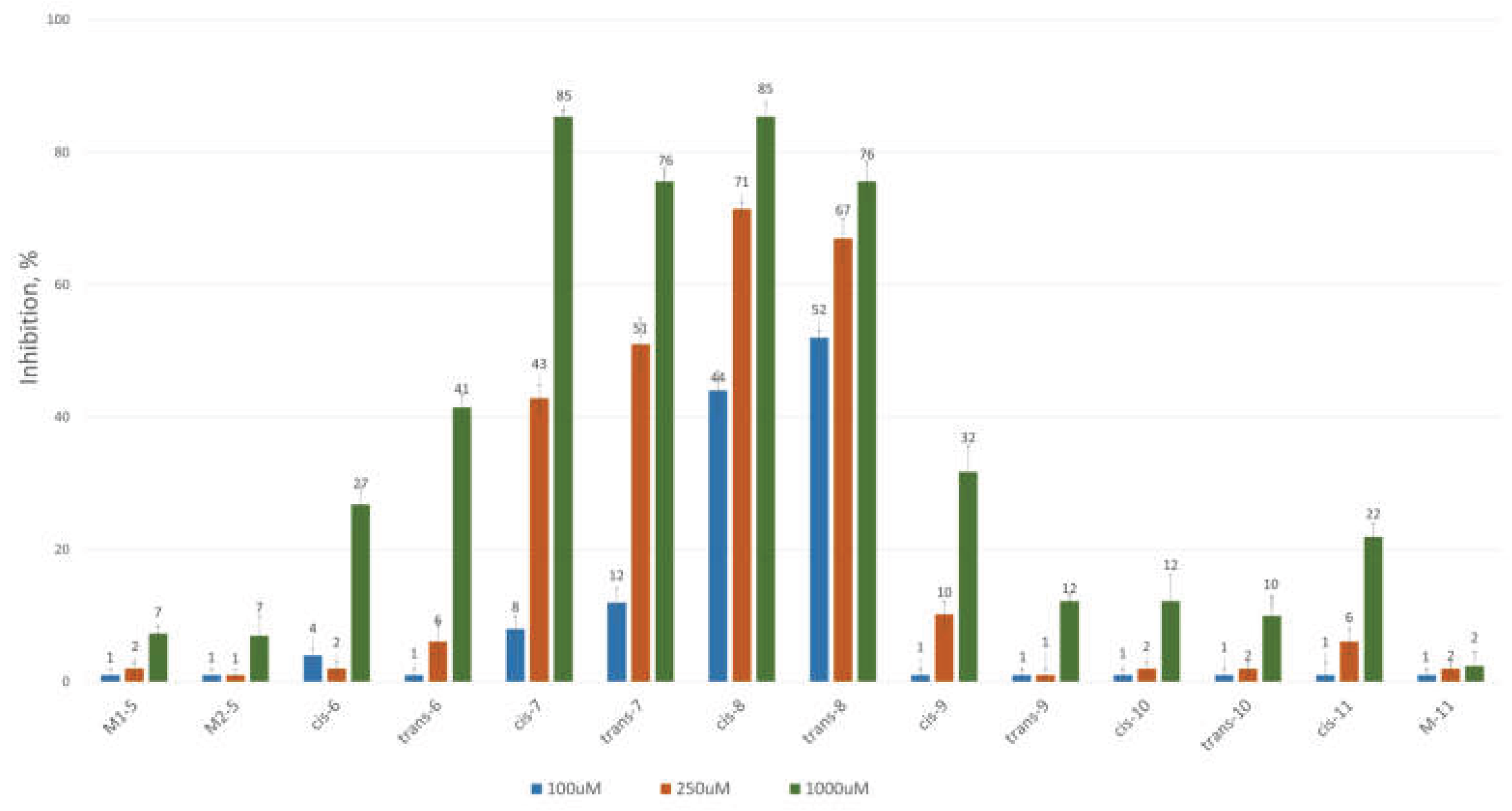

Table 1.
Numbering, substitution pattern and theoretically calculated affinity of the studied compounds 1-11. Lower value denotes a higher binding affinity of the inhibitor to the active cite of CAT isolated from mouse (Mus musculus, PDB identifier: 1NDI).
Table 1.
Numbering, substitution pattern and theoretically calculated affinity of the studied compounds 1-11. Lower value denotes a higher binding affinity of the inhibitor to the active cite of CAT isolated from mouse (Mus musculus, PDB identifier: 1NDI).
 |
 |
|||
| Compd. | Binding Energy (kcal/mol) | |||
| (3R,4R) | (3S,4S) | (3R,4S) | (3S,4R) | |
| 1 | -5.9 (cis) | -6.5 (cis) | -6.2 (trans) | -6.1 (trans) |
| 2 | -6.2 (cis) | -6.5 (cis) | -6.5 (trans) | -5.5 (trans) |
| 3 | -6.4 (cis) | -6.4 (cis) | -6.3 (trans) | -5.5 (trans) |
| 4 | -6.2 (cis) | -5.8 (cis) | -6.1 (trans) | -6.5 (trans) |
| 5 | -6.5 (cis) | -6.0 (cis) | -5.9 (trans) | -5.9 (trans) |
| 6 | -6.6 (cis) | -5.7 (cis) | -6.4 (trans) | -6.1 (trans) |
| 7 | -6.2 (cis) | -5.6 (cis) | -6.0 (trans) | -5.9 (trans) |
| 8 | -6.0 (cis) | -5.6 (cis) | -5.4 (trans) | -6.0 (trans) |
| 9 | -6.6 (trans) | -6.9 (trans) | -6.8 (cis) | -6.9 (cis) |
| 10 | -7.0 (trans) | -7.0 (trans) | -6.4 (cis) | -6.8 (cis) |
| 11 | -6.8 (trans) | -7.0 (trans) | -6.7 (cis) | -6.4 (cis) |
| Compd. | ||||
| L-carnitine | -4.5 | |||
| acetyl-CoA | -5.8 | |||
| Meldonium | -4.5 | |||
Table 2.
Numbering, substitution pattern and theoretically calculated affinity of the studied compounds 1-11. Lower value denotes a higher binding affinity of the inhibitor to the active cite of CPT2 isolated from rat (Rattus Norvegicus, PDB identifier: 2FW3).
Table 2.
Numbering, substitution pattern and theoretically calculated affinity of the studied compounds 1-11. Lower value denotes a higher binding affinity of the inhibitor to the active cite of CPT2 isolated from rat (Rattus Norvegicus, PDB identifier: 2FW3).
 |
 |
|||
| Compd. | Binding Energy (kcal/mol) | |||
| (3R,4R) | (3S,4S) | (3R,4S) | (3S,4R) | |
| 1 | -7.6 (cis) | -7.4 (cis) | -7.2 (trans) | -7.3 (trans) |
| 2 | -6.0 (cis) | -8.4 (cis) | -7.0 (trans) | -7.6 (trans) |
| 3 | -8.2 (cis) | -7.3 (cis) | -7.5 (trans) | -7.9 (trans) |
| 4 | -7.5 (cis) | -7.6 (cis) | -7.6 (trans) | -8.1 (trans) |
| 5 | -7.4 (cis) | -7.4 (cis) | -7.2 (trans) | -7.4 (trans) |
| 6 | -7.1 (cis) | -7.1 (cis) | -7.8 (trans) | -7.0 (trans) |
| 7 | -7.7 (cis) | -7.2 (cis) | -7.5 (trans) | -7.1 (trans) |
| 8 | -7.3 (cis) | -7.7 (cis) | -7.3 (trans) | -7.7 (trans) |
| 9 | -8.6 (trans) | -8.7 (trans) | -8.1 (cis) | -8.6 (cis) |
| 10 | -8.3 (trans) | -8.3 (trans) | -8.3 (cis) | -8.3 (cis) |
| 11 | -8.5 (trans) | -8.3 (trans) | -8.3 (cis) | -8.4 (cis) |
| Compd. | ||||
| L-carnitine | -4.5 | |||
| palmitoyl-CoA | -4.6 | |||
| Meldonium | -4.4 | |||
| Etomoxir | -7.2 | |||
| Compd. |
Type of Inhibition |
α | R2 | AIC | Sy,x |
| cis-8 | mixed | 0.97 | 0.98027 | -1562.764 | 2.987 × 10-9 |
| non-competitive | 1 | 0.98027 | -1565.380 | 2.947 × 10-9 | |
| uncompetitive | - | 0.97141 | -1550.555 | 3.547 × 10-9 | |
| competitive | - | 0.93830 | -1519.782 | 5.211 × 10-9 | |
| trans-8 | mixed | 0.21 | 0.97682 | -1548.364 | 3.576 × 10-9 |
| uncompetitive | - | 0.97551 | -1548.795 | 3.626 × 10-9 | |
| non-competitive | 1 | 0.97122 | -1542.332 | 3.931 × 10-9 | |
| competitive | - | 0.89542 | -1490.723 | 7.493 × 10-9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



