
Submitted:
09 December 2024
Posted:
11 December 2024
You are already at the latest version
Abstract
Fucosidosis is a rare lysosomal storage disease caused by α-L-fucosidase deficiency following mutation in the FUCA1 gene, in which fucose-bound glycoconjugates accumulate in the lysosomes of different tissues of the body. Fucosidosis is characterized by rapid mental and motor loss, along with growth retardation, coarse facial features, hepatosplenomegaly, telangiectasis or an-giokeratomas, epilepsy, inguinal hernia, and dysostosis multiplex. Patients usually die at an early age. Treatment of fucosidosis is a great challenge, and there is currently no definitive effective treatment. Hematopoietic cell transplantation studies are ongoing in the treatment of fucosidosis. However, early diagnosis of this disease and treatment can be effective. In addition, the body's immune system decreases due to chemotherapy applied after transplantation, leaving the body vulnerable to microbes and infections, and the risk of death is high with this treatment. In another treatment method, gene therapy, the use of retroviral vectors is promising due to their easy inte-gration, high cell efficiency and safety. In another treatment approach, enzyme replacement ther-apy, preclinical studies are ongoing for fucosidosis, but the blood-brain barrier is a major obstacle in lysosomal storage diseases affecting the central nervous system. Early diagnosis is im-portant in fucosidosis, a rare disease, due to the delay in diagnosis of patients identified so far and the rapid progression of the disease. In addition, enzyme replacement therapy, which carries fewer risks, is promising.
Keywords:
α-L-fucosidase
; FUCA1
; autosomal recessive
; lysosomal storage disorders
; rare disease
; enzyme deficiency
1. Introduction
α-L-fucosidase deficiency is observed after a mutation in the FUCA1 gene. Deficiency of this enzyme causes accumulation of α-L-fucose-linked glycoconjugates in tissue lysosomes of different organs [1]. Fucosidosis is a lysosomal storage disease (LSD) characterized by accumulation of these sugars in α-L-fucosidase deficiency. This autosomal recessive and the rare disease negatively affects many other systems, especially the central nervous system. In particular, it appears to cause rapid loss of mental and motor functions, causing irreversible damage [2]. Fucose-linked conjugates accumulated in lysosomes can inhibit the activity of hydrolases by changing the substrate binding properties of other hydrolases or by affecting the stability of enzymes [3]. This situation of the activity of other hydrolases in lysosomes prevents the separation of fucose-linked glycoconjugates into monomers and causes their storage. In studies examining the hydrolysis of glycoproteins showed that fucose must first be removed for the activity of the N-glycanase aspartylglucosaminidase, which hydrolyzes the bond between N-linked glycans and peptides [4]. However, substrate accumulation is observed in α-L-fucosidase deficiency and lysosomal dysfunction leads to necrosis and apoptosis of cells (Figure 1) [5].
1.1. History of the Disease
The first case of fucosidosis was seen in a study conducted by Durand et al. in 1966. In this case seen in two siblings, the clinical picture of the 3-year study reported gradually increasing memory loss, mental disorders and degeneration in motor functions. As a result of histochemical studies, they observed the accumulation of a material consisting of a mixture of mucopolysaccharides and lipids in various organs and tissues of the body, including the central nervous system. Along with the abnormal mitochondria, the liver biopsy result showed a swollen appearance in balloon or pseudo-gargoyle cells. Although an idea was obtained about the composition of the accumulated material as well as the clinical picture obtained in this study, its definitive chemical definition could not be made with the histochemical techniques available at that time. It was also seen to be different from other lysosomal storage diseases [6].
In another study conducted by Van Hoof and Hers in 1968, the activity of enzymes found in different tissue lysosomes of patients with Hurler syndrome or related diseases was analyzed. The results showed that α-L-fucosidase enzyme showed excessive increase in lysosomes of 2 out of 5 patients, but this enzyme was not active in the brains and livers of 3 patients. At the same time, attention was drawn to the significant increase in the amount of mucopolysaccharides seen in Hurler syndrome in the tissues of these 3 patients. Later, mucopolysaccharides were isolated from the livers of these 3 patients and their molecular structures were examined by chromatographic analysis. They reported that there was a large accumulation of fucose derivatives in the structures of these monosaccharides. The clinical report of these patients was given by Durand and colleagues, who had previously reported the accumulation of 2 unusual glycolipids in the liver. In the first study conducted by Durand and colleagues, the deficiency of α-L-fucosidase enzyme was thus identified retrospectively. Van Hoof and Hers later proposed to name this new lysosomal storage disease mucopolysaccharidosis F (F for fucosidosis) because of the similar histological findings and symptoms seen in Hurler syndrome [7]. Later, Durand and his colleagues continued their studies on this case and confirmed that the substances accumulated in the tissues were due to the deficiency of α-L-fucosidase and that fucose accumulated in all tissues in its pathogenesis. They proposed the term ‘fucosidosis’ for the inborn error of carbohydrate metabolism and included sphingolipids and polysaccharides containing fucose under this name. Thus, a new lysosomal storage disease continued to be named in this way in the literature [8].
Later, Kousseff et al. stated that there may be two different types of fucosidosis. Although α-L-fucosidase activity is deficient in both types, rapidly progressive psychomotor and neurological deterioration was observed in type I fucosidosis. It was also stated that patients died in early childhood (before approximately 5 years of age). In type II fucosidosis, psychomotor retardation and neurological deterioration were observed that progressed more slowly than in type I. In this type, where longer survival was observed, evidence of lysosomal storage was also found in angiokeratomas, vascular endothelium and skin fibroblasts [9].
In addition to the description of 58 patients in the literature in 1989 from 105 previously published reports and international survey studies, Willems et al. prepared a review in 1991 by examining a total of 77 patients, 19 of which had not been previously reported. The age of death was recorded in 44 patients in the review report. The number of patients who died before the age of 10 was recorded as 19 (43%), while the number of patients who died after the age of 20 was recorded as 18 (41%). The number of patients who died between the ages of 10 and 20 was determined as 7 (16%), indicating a bimodal distribution. It was suggested that the clinical variability among the patients was determined by at least 4 different mutations, but the description of more fucosidosis patients caused clinical gaps between type I and type II. It was later thought that these clinical variability was not only due to different allelic mutations in FUCA1, but additional factors also contributed to the phenotype of the fucosidosis patient. According to the distribution and incidence report of the disease presented by the same review, since only 77 patients were identified in a comprehensive screening in North America, Western Europe, Japan and Australia, the incidence of fucosidosis was determined to be extremely low. While Italians and the Mexican-Indian population in New Mexico and Colorado in the USA showed a high incidence, fucosidosis disease was not detected only in the Australian continent. The high incidence of fucosidosis patients (20), the majority of whom were of Italian origin, was explained by the high consanguineous marriage and founder effect found in the Italian population [2]. Fucosidosis is a rare disease with an estimated incidence of less than 1 in 200,000 live births worldwide, and recent studies on the disease have identified a high incidence in Cuba [10,11,12].
2. Other Lysosomal Storage Diseases
Lysosomal storage diseases are characterized by the accumulation of macromolecules in lysosomes as a result of deficiency of lysosomal hydrolases. In these diseases, which are generally inherited as autosomal recessive, sphingolipids, mucopolysaccharides, glycolipids and oligosaccharides accumulate in different tissues and lysosomes of the body, resulting in excessive activation of the inflammatory response and cell death. Lysosomal storage diseases are categorized according to the substance that cannot be hydrolyzed and stored as a result of enzyme deficiency [13]. For example, in the glycogen storage disease type II known as Pompe disease, carbohydrates accumulate in the glycogen structure in cell lysosomes as a result of lysosomal acid α-glycosidase deficiency. Cholesteryl ester storage disease and Wolman disease are characterized by the storage of neutral lipids such as cholesteryl ester and triglycerides in lysosomes as a result of acid cholesteryl hydrolase deficiency. Sphingolipidoses known as Fabry, Tay-Sachs, Niemann-Pick, Krabbe and Gaucher diseases and mucolipidoses known as Pseudo-Hurler Polydystrophy disease are disease groups in which glycolipids are stored due to lysosomal enzyme deficiencies. Mannosidosis, aspartoglycosidosis, galactosylceramidosis and Schindler disease are included in the glycoproteinosis group and are characterized by the storage of N-glycans in lysosomes as a result of the deficiency of the enzyme that breaks down glycans. Fucosidosis disease, such as α- and β-mannosidosis, sialidosis and aspartylglucosaminuria diseases, are diseases named according to non-hydrolyzed sugar and in which both glycolipids and glycoproteins are stored [14].
In lysosomal storage diseases, progressive neurological deterioration together with delayed motor and cognitive activities, seizures, and visual disturbances are common symptoms. These symptoms appear in late infancy. Gene therapy, hematopoietic stem cell transplantation and enzyme replacement therapy are clinically applicable for only a small portion of these disease groups [15]. After the initial discovery of fucosidosis, clinical observations have been confused with other lysosomal storage diseases. Angiokeratoma, which is among the symptoms of fucosidosis, is seen in GM1 gangliosidosis, sialidosis type II, Schindler neuroaxonal dystrophy syndrome, Kanzaki disease, galactosialidosis, and β-mannosidosis, but is not distinctive for Fabry disease. For this reason, advanced biochemical and other molecular analyses were needed for a complete diagnosis of fucosidosis disease [6,14].
3. Symptoms and Clinical Features
In α-L-fucosidase deficiency, the inability to degradation fucosylated glycolipids and glycoproteins causes many clinical symptoms with the accumulation of these glycoconjugates in various tissues and organs such as the brain, liver, bone and skin. The occurrence of significant neurodegeneration in the course of the disease leads to significant deterioration in the mental and physical functions of the patients. In addition to these, growth retardation, coarse facial features, hepatosplenomegaly, telangiectasis or angiokeratomas, epilepsy, recurrent respiratory tract infections, inguinal hernia and dysostosis multiplex are observed (Table 1). These clinical features of fucosidosis are similar to the features of other lysosomal storage disorders such as mucopolysaccharidosis and mucolipidosis [16]. The majority of patients with fucosidosis die at an early age due to neurological deterioration or respiratory tract infections [1].
3.1. Motor and Mental Function
Fucosidosis causes serious deterioration in mental and motor functions. In addition to neurological disorders such as difficulty in standing, loss of walking ability or frequent falls, equinovarus deformity in the feet, difficulty in sitting, and deterioration in language skills, patients are also described with flexion contractures and spastic quadraparesis with increased tendon reflexes. However, in rare cases, permanent incontinence, epileptic seizures or asymmetric mild-moderate sensorineural hearing loss and eustachian tube dysfunction can be observed [1,17,18].
In the neuroimaging results of the patients, changes in white matter and gray matter were identified. In particular, the prominent white matter abnormalities of the globus pallidus in fucosidosis are considered to be a specific finding for fucosidosis, helping to distinguish it from other neurometabolic disorders. The globi pallidi and substantia nigra have low signal intensity on T2/FLAIR (Fluid Attenuated Inversion Recovery) sequences but high signal intensity on T1 sequences [1,24,25,26].
3.2. Facial and Physical Features
In fucosidosis, a coarse facial appearance is generally accepted as a common symptom. Along with a wide nasal bridge, an abnormal distance between the normally symmetrical paired organs known as hypertelorism is seen. In cases of malformations, the increase in height and weight stops after a certain age and growth retardation is observed [1,2,19].
3.3. Recurrent Respiratory Tract Infections
Generally, clinical findings include recurrent upper respiratory tract infections, pulmonary emphysema, difficulty breathing during sleep, and otitis media. High sweat electrolytes and recurrent respiratory tract infections are also seen in cystic fibrosis. Fucose residues accumulated due to enzyme deficiency seen in fucosidosis, and sialic acid affect the viscoelasticity of mucus. Although there are connections between fucosidosis and cystic fibrosis, such as mucus-secreting ciliated epithelial areas and recurrent infections, the immune systems of fucosidosis are not directly affected, so this symptom can be eliminated by mucus cleaning in fucosidosis [1,20].
3.4. Dysostosis Multiplex
Some patients with fucosidosis have abnormalities in ossification. Bone anomalies called dysostosis multiplex include cervical platyspondylosis, wide ribs or deformities, scoliosis and hunchback deformity of the lumbar vertebrae, and odontoid processes [1].
3.5. Dermatological Abnormalities
The symptoms of fucosidosis include abnormalities on the skin. Skin lesions are usually seen as red or purple raised dots on the lower abdomen and genital area. Angiokeratoma corporis diffusum is more likely to be seen on the skin depending on age and although not pathognomonic, it raises suspicion of the diagnosis of fucosidosis. Angiokeratomas, which cover the whole body, are punctate deposits of capillaries within the papillary dermis [1]. Angiokeratomas are more likely to be seen in patients over 20 years of age than in patients under 10 years of age. The cause of this skin abnormality is unclear, but one view is that it is a condition in which undegradable glycolipids and glycoproteins accumulate in endothelial cells, leading to apoptosis of these cells and subsequent regeneration of the cells, resulting in capillaries [21]. Telangiectasias are known as permanent capillary dilations, and some patients have only telangiectasias without angiokeratomas. In addition to these skin conditions seen in fucosidosis, skin thickness, acrocyanosis, keratinocyte differentiation, hyperhidrosis, hypohidrosis, and transverse nail bands are also seen [1].
3.6. Organomegaly
Organomegaly, known as abnormal growth of internal organs in fucosidosis disease, has been recorded in some cases of fucosidosis disease. The most common of these and non-progressive is hepatospenomegaly, where both the liver and spleen grow simultaneously. Liver balloon or pseudogargoyle cells give a clear and swollen appearance as a result of periodic acid-schiff stain analysis. At the same time, hepatic cells containing stored ceramide tetra and pentahexoid are observed in liver biopsies [1,22,23].
3.7. Ophthalmological Abnormalities
Although severe visual impairment is rarely seen in fucosidosis, it has been shown that storage material accumulates in conjunctival, retinal and skin vessels. In addition, histological evaluation of conjunctival endothelial cells revealed clear vacuoles with a reticular structure similar to those seen in mucopolysaccharidoses and dark inclusions with dense granular material. In addition, mild corneal clouding, strabismus and congestive papilledema have been reported in some cases [1].
3.8. Clinical Features
Fucosidosis is divided into 2 types according to the age of onset and clinical features. However, since fucosidosis is a rare disease, the diagnosis of patients described so far has been delayed and the clinical findings have varied, and the current view is that the severity of the clinical disorder is a single disorder with a wide range of symptoms rather than two distinct types [10,21]. Clinical symptoms of type I fucosidosis begin early (approximately 6 months of age). Neurological deterioration progresses rapidly and patients usually die between the ages of 5 and 10. Type II progresses more slowly and therefore symptoms appear before the age of 2. Although patients rarely survive to the age of 30, they usually die in the second decade of life. The slower progression of neurological deterioration and the milder course of the disease have been considered features that distinguish it from type I. In addition, the fact that angiokeratomas usually occur with age and that type II patients are more likely to survive into adulthood has led to the symptoms of angiokeratomas being suggestive of type II [1,10,21].
In the review conducted by Willems et al. in 1991 on 77 patients reported in 18 different countries, 95% mental disorder, 87% motor disorder, 78% growth retardation, 79% coarse facial appearance, 58% dystosis multiplex, 78% recurrent infections, 52% angiokeratoma, 38% seizures and 44% organomegaly were recorded [2]. In 2019, Wali et al. identified 89 cases of fucosidosis reported from 26 different countries/ethnic origins since Willems et al.'s review in their case report and literature review. Of these, 75 patients with sufficient clinical data were taken as the basis to determine the clinical spectrum. According to the results, 50-60% mental disorder, 50-60% motor disorder, 30-40% growth retardation, 60-70% coarse facial appearance, 50-60% dystosis multiplex, 30-40% recurrent infections, 40-50% angiokeratoma, 10-20% seizures and 30-40% organomegaly were observed [27]. When the clinical finding percentages of these two studies were examined, growth retardation, especially together with mental and motor disorders, was considered to be one of the important findings of the disease in fucosidosis. However, it was suggested that the inconsistencies observed in the results of both reports could be affected by inadequate or unsystematic collection of data or improved supportive treatments. In 2022, a patient with a new homozygous pathogenic variant and atypical clinical findings was described in a case report by Şanlı and Uysal and in addition to this case, they summarized the clinical and molecular features of 13 Turkish patients reported and presented in the literature. In the clinical findings of the Turkish patients, 100% mental disorder, 100% motor disorder, 90% growth retardation, 100% coarse facial appearance, 54% dystosis multiplex, 63% recurrent infections, 54% angiokeratoma, 27% seizures and 27% organomegaly were recorded and the findings of this study were found to be more consistent with the report of Willems et al. [19].
4. Molecular Perspective
The FUCA1 gene is located on the short arm (p) of chromosome 1. It contains eight exons spanning 23 kb and encodes an α-L-fucosidase of 461 amino acids, of which 22 amino acids are the signal peptide and 439 amino acids are the mature protein [1,28]. The enzyme is a homotetramer of approximately 50–60 kDa that hydrolyzes fucosyl bonds by acting on natural oligosaccharide and glycosphingolipid substrates, resulting from changes in N-glycosylation and proteolytic processing [2].
Homozygous or heterozygous mutations occurring in the germline of the FUCA1 gene cause α-L-fucosidase deficiency. In the deficiency of the enzyme, fucose-containing glycolipids and glycoproteins accumulate in various tissues and urine as a result of incomplete degradation of N- and O-glycosylproteins [28]. In HGMD (Human Gene Mutation Database) for fucosidosis, a rare lysosomal storage disease transmitted as an autosomal recessive trait, 36 biallelic pathogenic variants have been reported so far. 6 missense and 11 nonsense substitutions, along with 8 small and 5 large deletions have been described. In addition, only three splice site variants have been described, including one small deletion, one complete deletion, and one stop-loss mutation. At the same time, since the genotype-phenotype correlation is not well defined, there is significant clinical variability in symptoms [1].
5. Biochemical and Molecular Diagnosis
Clinical symptoms seen in fucosidosis may be the first step for the suspicion of the disease. However, the common symptoms seen with other lysosomal storage diseases are insufficient for the definitive diagnosis of this disease. The continuously changing signal intensity and hypomyelination in the globus pallidus seen in neuroimaging results raise high suspicion for the diagnosis of fucosidosis [29]. At the same time, excessive amounts of glycopeptides are seen in the urine of patients with fucosidosis. In biochemical screenings performed for the diagnosis of fucosidosis, enzyme activity is measured in serum and plasma. However, the definitive diagnosis of the disease is proven by the analysis of mutations occurring in the FUCA1 gene [30]. Individuals with distinct phenotypes can be diagnosed with FUCA1 gene-targeted tests, but fucosidosis has a wide clinical presentation. Therefore, comprehensive tests such as chromosomal microarray analysis or array comparative genomic hybridization (aCGH) analysis are recommended for patients with inadequate clinical diagnosis. However, prenatal diagnosis is important. Since fucosidosis is inherited as an autosomal recessive trait, biochemical tests are not recommended for prenatal diagnosis. Because mutation carriers still show partial protein expression. Therefore, if there is a family member affected by FUCA1 pathogenic variants, prenatal testing or molecular genetic tests such as preimplantation genetic testing (PGT-M) are used in subsequent pregnancies [1].
6. Treatment and Therapeutic Options
Since the first case of the disease was seen in 1966, studies have been carried out to define the disease, determine the clinical appearance and examine it at the molecular level. However, due to the fact that it is a rare disease and few cases are seen or the cases that come are diagnosed with advanced disease and the disease progresses rapidly, studies on the treatment of the disease are slow. The treatment of fucosidosis is a great difficulty and there is currently no definitive effective treatment. Therefore, symptomatic treatments are applied to improve the quality of life. The multidisciplinary team that applies these treatments consists of physiotherapists, orthopedists, cardiologists, ophthalmologists and neurologists [19]. Since neurological symptoms and respiratory tract infections are the most important cause of early death in patients with fucosidosis, this problem needs to be treated early. Abnormally growing tissues that appear in the upper respiratory tract and protrude from the mucous membrane cause nasal polyps. This situation negatively affects the flow of passing air and causes problems such as nasal congestion, difficulty in smelling, and sleep apnea in the patient. Nasal polyps that do not go away on their own can be removed surgically. In the lower respiratory tract, albuterol, nebulized beclomethasone, and fluticasone inhalers can be used to relieve swelling and inflammation in the walls of the airways in the lungs. The airway clearance system is used to displace mucus in the bronchial walls from the small airways to the large airways to prevent atelectasis and pneumonia. Seizures, which are among the other symptoms, can be controlled with anti-epileptic treatment. However, involuntary contractions seen in patients with dystonia are prevented for a while with baclofen or by preventing the release of acetylcholine, a toxin produced by the Colostridium botulinum bacteria that carries the muscle contraction command. In addition to these treatments, physical massage therapy can also be applied [25].
6.1. Animal Models
6.1.1. Canine
Fucosidosis due to deficiency of α-L-fucosidase enzyme has been observed in Springer Spaniels in Australia and the United Kingdom. The mutant fucosidosis gene was transferred from England to Australia via dogs imported from English show clubs. Sick Springer Spaniels develop normally for 1 year and appear superior to healthy puppies. For this reason, they are frequently selected as show candidates. In 1971, an English Springer Spaniel was presented to Hartley and his team as a case study. Later, in 1978 and 1980, two dogs aged 2-3 years in Australia were observed to show progressive neuropathological signs. It was known that these two dogs were closely related to a dog previously imported from England to Australia. In a study published by Hartley et al. in 1982, they stated that these dogs had loss of myelin and Purkinje cells in the central nervous system and that most of their neurons showed severe cytoplasmic vacuolization. They suggested that some of these vacuoles may contain oligosaccharide-like material [31]. In 1983, one of the puppies of these dogs was 12 months old and the other was 19 months old, and the same symptoms began to appear. The earliest clinical sign of canine fucosidosis is the failure to walk on the forelimbs with the hind legs held high. Later, tremors and numbness are observed, along with difficulty swallowing. Blindness, deafness, and progressive ataxia are among the symptoms of canine fucosidosis. Springer Spaniels die in a lethargic state between the ages of 3 and 5. Since canine fucosidosis is closely related to the symptoms of human fucosidosis, it is used as a potential animal model [32]. So far, the canine model has been used as a model for Hematopoietic stem cell and enzyme replacement therapy treatments of fucosidosis [2].
6.1.2. Mouse
For therapeutic research of fucosidosis treatment, the canine model has contributed greatly to the development of approaches such as bone marrow transplantation and intracisternal enzyme replacement therapy by crossing the blood-brain barrier. In order to easily establish more diagnostic and treatment strategies, a new knockout mouse model with symptoms similar to human fucosidosis was developed by Wolf et al. in 2016. While creating the knockout model, the Escherichia coli neomycin phosphotransferase I (nptI) gene was inserted into the exon 1 region of the FUCA1 gene, which encodes lysosomal α-L-fucosidase. With analyses of enzyme levels, genomic and transcript studies, FUCA1 deficiency in the mouse model was definitely confirmed, and it was shown that the mice were completely devoid of α-L-fucosidase activity in all tissue homogenates tested. As a result, storage vacuoles were observed in the internal organs of FUCA1-deficient mice and control littermates. Mice with normal weight and growth up to 6 months of age were indistinguishable from wild-type mice. Later, knockout mice showed impaired motor function. Mice subjected to motor and cognitive testing showed sensorimotor fragmentation and impaired spatial learning and fear memory. To prevent unnecessary suffering of the animals due to the severe phenotype, knockout mice were euthanized at 9–11 months of age [33].
This mouse model reflects the type II form of human fucosidosis despite severe neuropathology. Unlike the canine model, although the mouse model shows histological changes in many regions of the central nervous system and contains storage materials, it lacks demyelination due to loss of oligodendrocytes in the central nervous system [34]. However, the paucity of data on patient cases of the rare disease fucosidosis makes studies on the treatment of the disease difficult. Compared to the dog model, cost-reducing factors such as easier reproduction, a defined genetic background, shorter life span and much lower body weight provide convenience. In addition, it has been shown that producing sufficient amounts of recombinant enzyme for high-dose and long-term treatments in mice is much more economical and feasible than the dog model.
6.2. Hematopoietic Cell Transplantation
Hematopoietic cell transplantation, such as bone marrow or umbilical cord blood transplantation, is a frequently used treatment method for lysosomal storage diseases. After transplantation, some cells that cross the blood-brain barrier can differentiate into microglia and secrete lysosomal enzymes [35].
Hematopoietic stem cell transplantation studies have been conducted on a fucosidosis canine model that best represents human fucosidosis. The first transplantation to fucosidosis Springer spaniels was successfully performed by Taylor et al. The results showed that α-L-fucosidase enzyme activity returned to normal in many tissues and especially in neural tissues behind the blood-brain barrier. With this improvement in enzyme activity, improvement was observed in peripheral nerve and visceral lesions and central nervous system pathology. However, they emphasized that treatment together with early diagnosis of this disease, not after the onset of clinical symptoms, may be effective [36,37]. Hematopoietic stem cell transplantation was first performed in humans in 1995 on an 8-month-old boy by A Vellodi et al. Although his development was normal, an MRI scan was performed on the boy after symptoms of the disease were seen in his older brother. After the abnormalities were observed, a volunteer donor was found for stem cell transplantation. Although mild neurodevelopmental delay continued to be observed eighteen months after transplantation, his older brother showed much greater developmental delay at the same age [38]. When the 4-year follow-up period of the fucosidosis case first treated with bone marrow transplantation was examined, it was seen that the enzymatic activity gradually increased and psychomotor development improved as observed in the MRI scan results [39]. In another study, a patient diagnosed with fucosidosis was treated with unrelated donor umbilical cord blood transplantation. After transplantation, the patient's enzymatic activity was shown to return to normal and his neurological symptoms improved. Umbilical cord blood transplantation is considered as a new approach to treat fucosidosis patients who do not have a suitable bone marrow donor [18].
In conclusion, hematopoietic stem cell transplantation is promising as an effective method for the treatment of the rare disease fucosidosis after its early diagnosis. The economic feasibility of this treatment method as a single intervention makes it an attractive option [40]. However, this treatment method is not effective when individuals are diagnosed with advanced disease. In addition, finding a suitable donor for bone marrow transplantation is difficult due to the presence of more than one child affected by fucosidosis in the family [27]. The body's immune system is weakened due to chemotherapy applied after transplantation, leaving the body vulnerable to microbes and infections. For this reason, the risk of death is seen as high with this treatment method [35]. Gene therapy is also seen as one of the basic research methods in the future. However, hematopoietic stem cell transplantation treatment method studies are considered as a treatment method that has reached the clinical stage to improve the quality of life of patients affected by fucosidosis [16].
6.3. Gene Therapy
In hematopoietic stem cell transplants, daughter cells that produce the missing enzyme can pass to the central nervous system, differentiate there and secrete the lysosomal enzyme [41]. In another treatment method, gene therapy, hematopoietic stem cells are taken from the patient and genetically modified. The use of retroviral vectors for gene correction is gaining importance in genetic disease treatment studies because they contain complex transcriptional units and transfer stem cells with high efficiency. In addition, the integration of these vectors is safer than other vectors [42].
In a study, a full-length cDNA clone encoding the lysosomal hydrolase α-L-fucosidase was cloned into a retroviral vector. It was used to efficiently transfer the α-L-fucosidase gene into both human and canine fucosidosis fibroblasts in vitro. The results resulted in the correction of the characteristic α-L-fucosidase enzyme deficiency. Therefore, these retroviral constructs need to be developed using animal models for gene therapy studies of fucosidosis disease. Gene therapy is considered as one of the future research methods.
6.4. Enzyme Replacement Therapy
The use of enzymes as biopharmaceuticals continues to gain importance in biotechnological studies. Enzyme replacement has become a widely used therapy method, especially in lysosomal storage diseases where enzymatic activity is impaired or deficient [43]. Enzyme replacement therapy is a treatment method based on replacing an enzyme that is inadequately produced or absent in the body at regular intervals throughout life. Although its safety and efficacy were initially debated, it was developed as the first effective treatment for type I Gaucher disease by Brady et al. in the early 1990s. Later, this treatment method became available for other lysosomal storage diseases such as Fabry disease, mucopolysaccharidosis type I, Pompe disease, and Neimann Pick B disease, and clinical studies gained momentum [44].
The enzymes to be used in this treatment method can be isolated from animals, plants or microorganisms, but in order to improve tissue targeting, the closest forms of enzymes to mammals need to be determined. Therefore, recombinant production and characterization of modified, highly purified enzymes are important. Microbial-derived enzymes have advantages over plant and animal-derived enzymes due to their high catalytic activity, stability, ease of isolation, high yield, no undesirable by-product formation and easy economic applicability. They are also important potential therapeutic agents due to their suitability for nanocarrier encapsulation, fusion proteins, PEGylation and other genetic manipulation methods [45].
Enzyme replacement therapy is ongoing in preclinical studies for fucosidosis, but the blood-brain barrier is a major obstacle in lysosomal storage diseases affecting the central nervous system. In order to overcome this problem, for the first time in a study, dogs with fucosidosis were injected with canine α-L-fucosidase intracisternal enzyme infusion method at regular intervals for 3 months starting from 8 weeks of age. After treatment, α-L-fucosidase reached all central nervous system regions, although it was found to be higher in areas close to the injection site (39-73% normal) and lower in deep brain structures (2.6-5.5% normal). However, it was observed that fucosyl-linked oligosaccharide accumulation was significantly reduced, especially in the spinal cord and brainstem, liver, bone marrow and lymph nodes. At the same time, partial improvement in neuropathology was recorded. No antibody formation or inflammatory reaction was observed in plasma and cerebrospinal fluid after the injections. Although this treatment method is not completely curative at this dose and treatment frequency, it has been shown that the enzyme is well tolerated and safe when delivered directly to the central nervous system. It has also been concluded that early treatment reduces substrate storage. Therefore, enzyme replacement therapy is promising for the treatment of fucosidosis [1]. In a study using a new knockout mouse model by Wolf et al in 2016, an expression system was established for the production of human α-L-fucosidase in CHO-K1 cells. The purified enzyme was administered intravenously to mouse models. It was observed that the enzyme was effectively taken to the visceral organs. As a preliminary result, promising data were presented from the new knockout mouse model for therapeutic studies of human fucosidosis disease in the development of enzyme replacement therapy [46].
7. Conclusions
Fucosidosis is a rare lysosomal storage disease caused by α-L-fucosidase deficiency following a mutation in the FUCA1 gene. In lysosomal storage disease, which is usually inherited as an autosomal recessive trait, sphingolipids, mucopolysaccharides, glycolipids, and oligosaccharides accumulate in the lysosomes of different tissues of the body. Lysosomal storage diseases are usually categorized according to the storage material that cannot be hydrolyzed. However, fucosidosis is named according to the nonhydrolyzable sugar, and both fucosylated glycolipids and fucosylated glycoproteins are stored in the lysosomes. Fucose-linked glycoconjugates negatively affect the activity of hydrolases by changing the substrate binding properties of other hydrolases in the lysosomes or by affecting the stability of the enzymes. Therefore, in order for fucose-linked glycoconjugates to be separated into their monomers, fucose must first be removed. This dysregularity in lysosomes triggers oxidative stress, cell damage and the subsequent onset of an inflammatory response. Dysfunction seen in lysosomes can lead to cell death. Especially microglia, known as the protective cells of the brain, contain a large number of lysosomes in their cytoplasm. This dysfunction in lysosomes, which can cause cell death, greatly affects the central nervous system. Fucosidosis was initially divided into 2 types according to the age of onset and clinical features. However, since fucosidosis is a rare disease, the diagnosis of patients identified so far has been delayed and their clinical findings have varied, and the current view is that the severity of the clinical disorder has been referred to as a single disorder with a wide range of symptoms rather than two separate types. Clinical symptoms of fucosidosis, called type I, begin early (approximately at 6 months of age). In type I, where neurological deterioration progresses rapidly, patients usually die between the ages of 5-10. Type II progresses more slowly and therefore symptoms appear before the age of 2. The slower progression of neurological deterioration and the mild course of the disease is considered to be a feature that distinguishes it from type I. Although patients rarely live to the age of 30, the majority die at an early age due to neurological deterioration or respiratory tract infections, usually in the second decade of their lives. In addition to the psychomotor regression and loss of mental functions seen in fucosidosis, symptoms such as growth retardation, coarse facial features, hepatosplenomegaly, telangiectasis or angiokeratomas, epilepsy, inguinal hernia and dysostosis multiplex are also seen. The clinical symptoms seen in fucosidosis can be the first step for the suspicion of the disease, but the common symptoms seen with other lysosomal storage diseases are insufficient for the definitive diagnosis of this disease. The definitive diagnosis of the disease is proven by the analysis of mutations in the FUCA1 gene. Although studies have been carried out since the first case of the disease was seen in 1966, such as defining the disease, determining the clinical appearance and examining it at the molecular level, studies on the treatment of the disease have been slow due to the fact that it is a rare disease and that only a small number of cases are seen or that the cases that come are diagnosed with advanced disease or that the disease progresses rapidly. The treatment of fucosidosis is a great difficulty and there is currently no definitive effective treatment. Therefore, symptomatic treatments are applied to improve the quality of life. Hematopoietic cell transplantation studies are continuing in the treatment of fucosidosis. The results of the study conducted on the canine model, which is a preserved form of the disease, showed that α-L-fucosidase enzyme activity returned to normal in many tissues and especially in the neural tissues behind the blood-brain barrier, and even with this improvement in enzyme activity, improvement was observed in peripheral nerve and visceral lesions and central nervous system pathology. However, they emphasized that it would be effective to diagnose this disease at an early age and apply treatment together with the onset of clinical symptoms. This method was first applied to humans in 1995 on an 8-month-old boy. When the 4-year follow-up period of the fucosidosis case treated for the first time with bone marrow transplantation is examined, it is seen that the enzymatic activity gradually increased and the psychomotor development improved as observed in the MRI scan results. This treatment method promises to be an effective treatment method. The economic feasibility of this treatment method as a one-time intervention makes it an attractive option. However, this treatment method is not effective since individuals are diagnosed with advanced disease. In addition, since there is more than one child affected by fucosidosis in the family, finding a suitable donor for bone marrow transplantation makes this treatment method difficult. The body's immune system decreases due to chemotherapy applied after transplantation, making the body open to microbes and infections. For this reason, the risk of death is seen as high in this treatment method. Nevertheless, hematopoietic stem cell transplantation treatment method studies are considered as a treatment method that has reached the clinical stage to improve the quality of life of patients affected by fucosidosis. In another treatment method, gene therapy, hematopoietic stem cells are taken from the patient and genetically modified. The use of retroviral vectors for gene correction is gaining importance in genetic disease treatment studies because they contain complex transcriptional units and transfer stem cells with high efficiency. In addition, the integration of these vectors is safer than other vectors. Therefore, these retroviral structures need to be developed using animal models in gene therapy studies of fucosidosis disease. Gene therapy is considered as one of the research methods in the future. Another treatment approach, enzyme replacement therapy, is ongoing preclinical studies for fucosidosis, but the blood-brain barrier is a major obstacle in lysosomal storage diseases affecting the central nervous system. In order to overcome this problem, in a first study, dogs with fucosidosis were injected with the enzyme at regular intervals for 3 months using the canine α-L-fucosidase intracisternal enzyme infusion method starting from the age of 8 weeks. Although this treatment method was not completely curative at this dose and treatment frequency, it showed that the enzyme delivered directly to the central nervous system was well tolerated and safe. It was also concluded that early treatment reduced substrate storage. Therefore, ERT is promising for the treatment of fucosidosis.
Author Contributions
Conceptualization, S.K., and M.B.; writing—original draft preparation, S.K., M.B., and B.P.; writing—review and editing, S.K.; visualization, B.P.; supervision, S.K., and M.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Stepien, K.M.; Ciara, E.; Jezela-Stanek, A. Fucosidosis—Clinical Manifestation, Long-Term Outcomes, and Genetic Profile—Review and Case Series. Genes (Basel) 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Willems, P.J.; Gatti, R.; Darby, J.K.; Romeo, G.; Durand, P.; Dumon, J.E.; O’Brien, J.S. Fucosidosis Revisited: A Review of 77 Patients. Am J Med Genet 1991, 38. [Google Scholar] [CrossRef]
- Parc, A. Le; Karav, S.; Bell, J.M.L.N.D.M.; Frese, S.A.; Liu, Y.; Mills, D.A.; Block, D.E.; Barile, D. A Novel Endo-β-N-Acetylglucosaminidase Releases Specific N-Glycans Depending on Different Reaction Conditions. Biotechnol Prog 2015. [CrossRef]
- Winchester, B. Lysosomal Metabolism of Glycoproteins. Glycobiology 2005, 15. [Google Scholar] [CrossRef] [PubMed]
- Pekdemir, B.; Karav, S. Exploring the Diverse Biological Significance and Roles of Fucosylated Oligosaccharides. Front Mol Biosci 2024, 11, 1403727. [Google Scholar] [CrossRef] [PubMed]
- DURAND, P. A NEW MUCOPOLYSACCHARIDE LIPIDSTORAGE DISEASE? The Lancet 1966, 288. [Google Scholar] [CrossRef]
- Van Hoof, F.; Hers, H.G. Mucopolysaccharidosis by Absence of Alpha-Fucosidase. Lancet 1968, 1. [Google Scholar] [CrossRef]
- Durand, P.; Borrone, C.; Cella, G. Della Fucosidosis. J Pediatr 1969, 75. [Google Scholar] [CrossRef]
- Kousseff, B.G.; Beratis, N.G.; Strauss, L.; Brill, P.W.; Rosenfield, R.E.; Kaplan, B.; Hirschhorn, K. Fucosidosis Type 2. Pediatrics 1976, 57. [Google Scholar] [CrossRef]
- Gowda, V.K.; Srinivasan, V.M.; Vegda, H.; Bhat, M. Fucosidosis with Pathogenic Variant in FUCA1 Gene. Indian J Pediatr 2020, 87. [Google Scholar] [CrossRef]
- Tamayo Chang, V.J.; Morales Peralta, E.; Santana Hernández, E.E.; Lantigua Cruz, A.; Collazo Mesa, T.; Lardoeyt Ferrer, R. Epidemiological and Population Genetic Characterization of Fucosidosis in Holguin Province, Cuba. Salud, Ciencia y Tecnología - Serie de Conferencias 2024, 3, 978–978. [Google Scholar] [CrossRef]
- Willems, P.J.; Seo, H.C.; Coucke, P.; Tonlorenzi, R.; O’Brien, J.S. Spectrum of Mutations in Fucosidosis. European Journal of Human Genetics 1999, 7. [Google Scholar] [CrossRef]
- Khatiwada, B.; Pokharel, A. Lysosomal Storage Disease. Journal of Nepal Medical Association 2009, 48, 242–245. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Gahl, W.A. Lysosomal Storage Diseases. Transl Sci Rare Dis 2017, 2, 1–71. [Google Scholar] [CrossRef] [PubMed]
- Burlina, A.P.; Manara, R.; Gueraldi, D. Lysosomal Storage Diseases. 2024, 204, 147–172. [CrossRef]
- Mao, S.J.; Zhao, J.; Shen, Z.; Zou, C.C. An Unusual Presentation of Fucosidosis in a Chinese Boy: A Case Report and Literature Review (Childhood Fucosidosis). BMC Pediatr 2022, 22. [Google Scholar] [CrossRef] [PubMed]
- Domin, A.; Zabek, T.; Kwiatkowska, A.; Szmatola, T.; Deregowska, A.; Lewinska, A.; Mazur, A.; Wnuk, M. The Identification of a Novel Fucosidosis-Associated Fuca1 Mutation: A Case of a 5-Year-Old Polish Girl with Two Additional Rare Chromosomal Aberrations and Affected Dna Methylation Patterns. Genes (Basel) 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Liu, S.; Jiang, H.; Lin, Y.; Shao, Y.; Hu, H.; Zhao, X.; Liu, H.; Huang, Y.; Liu, L. Brain Abnormalities in Fucosidosis: Transplantation or Supportive Therapy? Metab Brain Dis 2017, 32. [Google Scholar] [CrossRef] [PubMed]
- Şanlı, M.E.; Uysal, S. Fucosidosis: Clinical and Molecular Findings of Turkish Patients. Turkish Journal of Pediatrics 2022, 64. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B.K.; Macleod, P.M.; Sturgess, J.; King, M. Recurrent Respiratory Infections in a Child with Fucosidosis: Is the Mucus Too Thin for Effective Transport? Pediatr Pulmonol 1991, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, M.; Hong, S.; Tang, T.; Zhuang, J.; Huang, H. Fucosidosis in a Chinese Boy: A Case Report and Literature Review. Journal of International Medical Research 2020, 48. [Google Scholar] [CrossRef]
- Terespolsky, D. Evolution of the Neuroimaging Changes in Fueosidosis Type II. J Inherit Metab Dis 1996, 19. [Google Scholar] [CrossRef]
- Ediz, S.S.; Aralasmak, A.; Yilmaz, T.F.; Toprak, H.; Yesil, G.; Alkan, A. MRI and MRS Findings in Fucosidosis; a Rare Lysosomal Storage Disease. Brain Dev 2016, 38. [Google Scholar] [CrossRef]
- Zubarioglu, T.; Kiykim, E.; Zeybek, C.A.; Cansever, M.S.; Benbir, G.; Aydin, A.; Yalcinkaya, C. Clinical and Neuroradiological Approach to Fucosidosis in a Child with Atypical Presentation. Ann Indian Acad Neurol 2015, 18. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Dhaliwal, A.S.; Raynes, H.; Naidich, T.P.; Kaufman, D.M. Diagnosis and Supportive Management of Fucosidosis: A Case Report. Cureus 2019. [CrossRef] [PubMed]
- BoAli, A.; Tlili-Graiess, K.; AlHashem, A.; Tabarki, B. Neuroregression, Coarse Features, and Oligosaccharides in Urines. Neurosciences 2017, 22. [Google Scholar] [CrossRef]
- Wali, G.; Wali, G.M.; Sue, C.M.; Kumar, K.R. A Novel Homozygous Mutation in the FUCA1 Gene Highlighting Fucosidosis as a Cause of Dystonia: Case Report and Literature Review. Neuropediatrics 2019, 50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, S.; Liu, H.; Wang, X.; Wang, X.; Du, N.; Liu, H.; Duan, H. Identification of a Novel Homozygous Loss-of-Function Mutation in FUCA1 Gene Causing Severe Fucosidosis: A Case Report. Journal of International Medical Research 2021, 49. [Google Scholar] [CrossRef] [PubMed]
- Meena, A.K.; Wander, A.; Manikandan, S.; Peer, S.; Bansal, A. Magnetic Resonance Imaging Pattern Recognition in Fucosidosis. Indian J Pediatr 2024. [CrossRef]
- Wynne, E.; Wynne, K.; Cleary, M.; Brogan, P.A. Fucosidosis Mimicking Juvenile Idiopathic Arthritis. Rheumatol Adv Pract 2018, 2. [Google Scholar] [CrossRef] [PubMed]
- Hartley, W.J.; Canfield, P.J.; Donnelly, T.M. A Suspected New Canine Storage Disease. Acta Neuropathol 1982, 56. [Google Scholar] [CrossRef] [PubMed]
- Kelly, W.R.; Clague, A.E.; Barns, R.J.; Bate, M.J.; MacKay, B.M. Canine α-l-Fucosidosis: A Storage Disease of Springer Spaniels. Acta Neuropathol 1983, 60. [Google Scholar] [CrossRef] [PubMed]
- Wolf, H.; Damme, M.; Stroobants, S.; D’Hooge, R.; Beck, H.C.; Hermans-Borgmeyer, I.; Lüllmann-Rauch, R.; Dierks, T.; Lubke, T. A Mouse Model for Fucosidosis Recapitulates Storage Pathology and Neurological Features of the Milder Form of the Human Disease. DMM Disease Models and Mechanisms 2016, 9. [Google Scholar] [CrossRef]
- Stroobants, S.; Wolf, H.; Callaerts-Vegh, Z.; Dierks, T.; Lübke, T.; D’Hooge, R. Sensorimotor and Neurocognitive Dysfunctions Parallel Early Telencephalic Neuropathology in Fucosidosis Mice. Front Behav Neurosci 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Massaro, G.; Geard, A.F.; Liu, W.; Coombe-tennant, O.; Waddington, S.N.; Baruteau, J.; Gissen, P.; Rahim, A.A. Gene Therapy for Lysosomal Storage Disorders: Ongoing Studies and Clinical Development. Biomolecules 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.M.; Farrow, B.R.H.; Stewart, G.J.; Healy, P.J.; Tiver, K. The Clinical Effects of Lysosomal Enzyme Replacement by Bone Marrow Transplantation after Total Lymphoid Irradiation on Neurologic Disease in Fucosidase Deficient Dogs. Transplant Proc 1988, 20. [Google Scholar]
- Taylor, R.M.; Farrow, B.R.H.; Stewart, G.J. Amelioration of Clinical Disease Following Bone Marrow Transplantation in Fucosidase-Deficient Dogs. Am J Med Genet 1992, 42. [Google Scholar] [CrossRef]
- Vellodi, A.; Cragg, H.; Winchester, B.; Young, E.; Young, J.; Downie, C.J.C.; Hoare, R.D.; Stocks, R.; Banerjee, G.K. Allogeneic Bone Marrow Transplantation for Fucosidosis. Bone Marrow Transplant 1995, 15, 153–158. [Google Scholar]
- Miano, M.; Lanino, E.; Gatti, R.; Morreale, G.; Fondelli, P.; Celle, M.E.; Stroppiano, M.; Crescenzi, F.; Dini, G. Four Year Follow-up of a Case of Fucosidosis Treated with Unrelated Donor Bone Marrow Transplantation. Bone Marrow Transplant 2001, 27. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Gupta, A.K.; Gupta, N.; Meena, J.P.; Seth, R.; Kabra, M. Hematopoietic Stem Cell Transplantation for Storage Disorders: Present Status. Indian J Pediatr 2024, 91, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Occhiodoro, T.; Hopwood, J.J.; Phillip Morris, C.; Anson, D.S. Correction of α-L-Fucosidase Deficiency in Fucosidosis Fibroblasts by Retroviral Vector-Mediated Gene Transfer. Hum Gene Ther 1992, 3. [Google Scholar] [CrossRef]
- Cavazzana, M.; Bushman, F.D.; Miccio, A.; André-Schmutz, I.; Six, E. Gene Therapy Targeting Haematopoietic Stem Cells for Inherited Diseases: Progress and Challenges. Nat Rev Drug Discov 2019, 18. [Google Scholar] [CrossRef]
- Li, M. Enzyme Replacement Therapy: A Review and Its Role in Treating Lysosomal Storage Diseases. Pediatr Ann 2018, 47. [Google Scholar] [CrossRef] [PubMed]
- Desnick, R.J. Enzyme Replacement and Enhancement Therapies for Lysosomal Diseases. J Inherit Metab Dis 2004, 27. [Google Scholar] [CrossRef] [PubMed]
- Vachher, M.; Sen, A.; Kapila, R.; Nigam, A. Microbial Therapeutic Enzymes: A Promising Area of Biopharmaceuticals. Curr Res Biotechnol 2021, 3. [Google Scholar] [CrossRef]
- Wolf, H. The Lysosomal Storage Disease Fucosidosis : Towards Enzyme Replacement Therapy. Dissertation 2016.
Figure 1.
Fucosidosis is a lysosomal storage disease characterized by a deficiency of α-L-fucosidase. Fucose-linked conjugates accumulated in lysosomes can inhibit the activity of hydrolases and dysfunction of lysosomes lead to cell death (Created by Biorender).
Figure 1.
Fucosidosis is a lysosomal storage disease characterized by a deficiency of α-L-fucosidase. Fucose-linked conjugates accumulated in lysosomes can inhibit the activity of hydrolases and dysfunction of lysosomes lead to cell death (Created by Biorender).
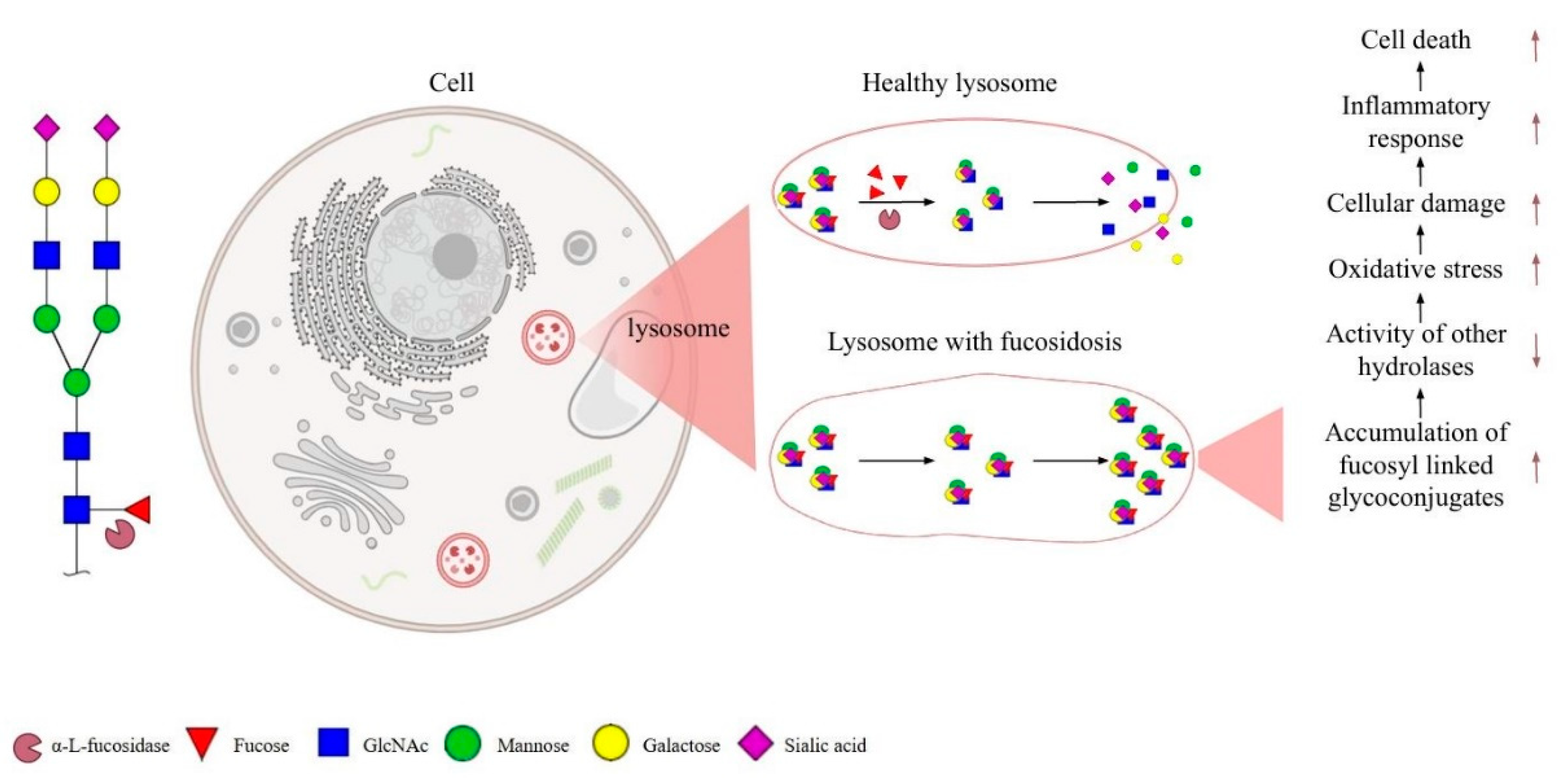

Table 1.
Clinical features of fucosidosis.
| Symptoms | Clinical features | References |
| Motor and mental dysfunction | Loss of walking ability, difficulty sitting and speaking, seizures, impairment of cognitive functions. | [1,17,18] |
| Facial and physical features | Coarse facies, wide nose bridge, growth retardation. | [1,2,19] |
| Recurrent respiratory infections | Emphysema, sleep apnea, loss of smell ability. | [1,20] |
| Radiologic abnormalities | Cervical platyspondyly, wide ribs or deformities, scoliosis and humpback deformity of the lumbar vertebrae, short odontoid processes. | [1] |
| Dermatological abnormalities | Angiokeratoma corporis diffusum, telenjiektaziler, skin thickness, acrocyanosis, keratinocyte differentiation, hyperhidrosis, hypohidrosis and transverse nail bands. | [1,21] |
| Organomegaly | The most common and non-progressive is hepatospenomegaly, in which both the liver and spleen enlarge at the same time. | [1,22,23] |
| Ophthalmological abnormalities | Accumulation of storage material in conjunctival, retinal and skin vessels, mild clouding of the corneas, strabismus and congestive papilledema. | [1] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



