
Submitted:
04 December 2024
Posted:
04 December 2024
You are already at the latest version
Abstract
The Arabidopsis transcription factor WUSCHEL-Related Homeobox 14 (AtWOX14) has versatile roles in plant growth and development. However, its biochemical specificity of DNA-binding and genome-wide regulatory targets remain uncharacterized. With the recently developed 5mC-incorporation strategy[1], this study performed SELEX and DAP-seq for AtWOX14 both in the presence and absence of cytosine methylation, and systematically curated 65 motif models and identified 51,039 genomic binding sites for AtWOX14. Overall, 5mC represses DNA binding of AtWOX14 monomers but facilitates binding of its dimers, and the methylation effect on a cytosine’s affinity to AtWOX14 is positional dependent. Notably, we found that the most preferred homodimeric configuration of AtWOX14 has changed from ER1 to ER0 upon methylation. Such change has the potential to rewire the regulatory network downstream of AtWOX14, as suggested by the GO analyses and the strength changes of the DAP-seq peaks upon methylation. Therefore, this work comprehensively illustrates the specificity and targets of AtWOX14, and reports a yet unrecognized effect of DNA methylation on transcription factor binding.
Keywords:
WUSCHEL-related homeobox 14
; DNA binding specificity
; SELEX
; DNA methylation
; Transcription factor
; Dimeric spacing
1. Introduction
The homeobox (HB) superfamily, defined by a conserved homeodomain (HD) of 60–66 amino acids, is widely distributed across plants and animals [2]. HB superfamily members serve as key regulators of growth and development. Plant HB superfamily TFs are involved in various processes, such as embryo patterning [3], organ development (root, shoot, floral meristems) [4], vascular development, and stress responses [5,6,7,8]. Although the HB superfamily is shared in eukaryotes, the HB TFs belonging to the WUSCHEL (WOX) family are plant-specific. WOX TFs are phylogenetically categorized into the modern, intermediate, and ancient clades based on evolutional evidences and species distributions [9]. The Arabidopsis TF — AtWOX14 (WUSCHEL-related homeobox 14) is a member of the ancient clade. AtWOX14 plays critical roles in diverse developmental processes, including bud regeneration in callus [10], lateral root and stamen development [11], vascular cell differentiation [12], lignification [13], and the accumulation of bioactive gibberellins in the stem of inflorescence [14].
To regulate the expression of target genes, transcription factors (TFs) selectively bind to specific DNA sequences. This binding process can interact with chromatin accessibility [15], histone modification [16], TF-TF interaction (cooperative or competitive) [17], as well as DNA methylation [18,19]. DNA methylation typically inhibits TF binding, for example, plant WRKY-family TFs show reduced DNA-binding affinities upon cytosine methylation [20]. However, a few TFs do enhance their binding affinity to methylated DNA [21,22,23]. For example, a comprehensive survey of 542 TFs reported that TFs critical for early embryonic development (e.g., POU5F1 and HOXB13) preferentially bind methylated DNA, highlighting a dual role of methylation in modulating TF-DNA interactions [21]. TFs frequently bind DNA as dimers, for instance, ARF forms homodimers upon binding [24], and bZIP10 and bZIP53 can heterodimerize [17]. While the previous studies systematically addressed the impact of methylation on nucleotide specificity of TFs [21], there are limited insights into how 5mC affects the configuration of dimeric binding of TFs.
Given the critical roles of AtWOX14 in plant growth and development, this study aims to thoroughly clarify its DNA-binding specificity and regulatory targets, and how these are influenced by DNA-methylation. In the absence and presence of methylation, altogether we curated 4 monomeric and 63 dimeric binding models for AtWOX14, and validated the ER0/1 dimeric binding model with EMSA. Methylation has globally changed the binding specificity of AtWOX14, not only the sequence specificity, but notably, also the configurational specificity (the spacing between the monomers) of dimerization. The specificity changes upon methylation also dictate the binding strength of AtWOX14 to its genomic targets — either being enhanced or inhibited dependent on the sequence of the cis-regulatory elements (CREs) of AtWOX14, suggesting that the methylation effect on AtWOX14’s regulatory activity can be complex.
For AtWOX14, here we use SELEX to profile the biochemical specificity, and DAP-seq to identify the genome-wide regulatory targets. This is because the randomness and complexity of SELEX input library ensure unbiased and sensitive discovery of a TF’s binding models [25,26], while DAP-seq uses genomic DNA to directly interrogate the binding sites and target genes of the TF [23]. These two methods complement each other and are preferably combined. Also, in contrast to animals whereby the majority of methylation happens on the CG dinucleotides, in plants methylation can occur in all of the CG, CHG, and CHH contexts [27]. Taking this into consideration, in this study we replaced all Cs in the SELEX/DAP-seq library with 5mCs when examining the methylation effects, using a recently developed protocol [1].
2. Results
2.1. Methylation Inhibits Monomeric But Enhances Dimeric Binding of AtWOX14
To unravel the original DNA-binding specificity of AtWOX14, we first performed SELEX without methylation (Figure 1A). Recombinant AtWOX14 protein is incubated with a 101-bp dsDNA library containing a randomized region. The ligands with high affinities to AtWOX14 were then purified together with AtWOX14, and amplified for the next cycle. To examine the impact of methylation on the specificity of AtWOX14. The SELEX process is also modified to incorporate 5mCs instead of Cs during the PCR amplification [1], generating a fully methylated DNA library in each cycle (Figure 1A). After 5 cycles, the enriched DNA pools were sequenced and analyzed.
We first confirmed that SELEX has enriched binding signals of AtWOX14. First, specific 10-mer subsequences were enriched over SELEX cycles (Figure 1B). Such enrichment is exponential because a straight-line is observed after taking the logarithm (Figure S1). Enrichment-based mutual information (E-MI) analysis [28,29] has also validated the presence of TF-binding signals in the enriched library (Figure 1C).
We next investigated the impact of methylation on the preferred kmers of AtWOX14 (Figure 1D). Interestingly, when we compare all 8-mer subsequences between methylated and normal libraries, the most abundant 8-mers are more enriched in the SELEX library (Figure 1D, left). In contrast, when all 12-mer subsequences were compared, the most abundant 12-mers are enriched in the Methyl-SELEX library (Figure 1D, right). We further performed motif discovery with 8-mers and 12-mers as seeds (using Autoseed [30]), and found that 8-mers give monomeric AtWOX14 motifs, while 12-mers give dimeric motifs. Therefore, it can be concluded that methylation has inhibited the monomeric binding of AtWOX14, while enhancing its dimeric binding.
2.2. Methylation Changes the Preferred Homodimeric Configuration of AtWOX14
While methylation is known to affect TFs’ nucleotide specificity and affinity [21,23], how the incorporation of 5mC affects TFs’ dimeric configuration is yet to be explored. We noticed that dependent on methylation, the most enriched dimeric motifs are with different configurations regarding the spacings of the half-sites (Figure 1D, right). Therefore, we next de novo identified all dimeric (also monomeric) binding motifs of WOX14 (Figure 2A, B) with the reported method [30], and compared their enrichments between the SELEX and the Methyl-SELEX libraries.
The identified dimeric motifs covered all possible relative orientations of monomers (direct repeats: DR, inverted repeats: IR, or everted repeats: ER). By defining the half-site as “YAATYA”, we are able to calculate the spacing between the half-sites, and name each dimeric motif by combining the relative orientation and the spacing (name labeled to the left of each motif, Figure 2A, B).
From the normal SELEX library, we identified 34 motif models, including 2 monomeric and 32 homodimeric motifs (Figure 1C). In general, the dimeric motifs with narrower spacings displayed greater enrichments and higher information contents (IC) (Figure 1C). The ER1 motif exhibited the highest IC and enrichment, followed by ER0, DR3-5, and IR6-8 (Figure 1C). An important signature of the functional TF motifs is that they are enriched around transcription start sites (TSSs). Accordingly, we found that the identified high-IC dimeric motifs all enriched upstream of TSSs (Figure 1D). However, we noticed that the dimeric models show higher enrichments at TSSs than the monomeric models. This consists with the stronger binding signals of dimeric models in SELEX (Figure 1C), and potentially also because the higher transactivating activity of the dimeric CREs are more strongly selected for during the evolutional history. To date, the reported motifs (e.g., in public databases [31,32,33]) of WOX TFs are mainly monomeric. Here we show that the dimeric motifs are more enriched in SELEX and around the TSSs, revealing that WOX14 has a strong preference to bind as homodimers.
In the presence of methylation, we identified 2 monomeric and 29 dimeric motifs, including 8 IR, 13 DR, and 8 ER motifs (Figure 2D). Consistent with the SELEX results (Figure 1C), dimeric motifs show higher enrichments and ICs compared to the monomeric motifs, further supporting the preference of WOX14 to bind as a dimer. The high-IC motifs are also enriched around TSSs (Figure 2D). Interestingly, the most enriched dimeric motif has switched from ER0 to ER1 (Figure 2B), the motif-based enrichment analysis here is also consistent with the 12-mer based analysis (Figure 1D, right), confirming the methylation effect on the homodimeric configuration of AtWOX14.
2.3. The Targets of ER0 and ER1 Are of Different Physiological Functions
We next focused on the high-affinity dimeric configurations ER0 and ER1 to investigate their specificity changes to DNA methylation. Positional comparison revealed that methylation affected the affinity of cytosines at positions 1, 5, 6, 7, and 11 (Figure 3A). Interestingly, the affinity of cytosines is not universally reduced upon methylation. Whereas the cytosines at pos1 of ER0/1 and pos11 of ER1 show decreased affinities when methylated (most preferred base changed from G to A, or C to T), the methylation effects on cytosines at the boundaries of the half-sites (pos5/6 of ER0, corresponding to pos5/7 of ER1) are more complex (Figure 3A) — their affinity increases in ER0, but decreases in ER1. This suggests that the effects of methylation on cytosine affinity can depend on the spacing of the TF dimer, which was also previously unrecognized. Similar to the dimeric motifs, the effect of methylation on cytosines of the monomeric motifs is also positional-dependent.
We further validated the dimeric binding of ER0 and ER1 to WOX14 by electrophoretic mobility shift assays (EMSA). Recombinant WOX14 protein is incubated with DNA ligands containing ER0/ER1 consensus, and also the consensus of the monomeric motif M1 as a control (Figure 3B). The results show that all ligands can bind WOX14 monomer; however, only ER0 and ER1 ligands can bind dimers of WOX14 at high protein concentrations (Figure 3B). This indicates that ER0 and ER1 are genuine homodimeric CREs of WOX14, and also suggests that the protein-level dimerization of AtWOX14 is weak, a proper CRE element on DNA is necessary to facilitate its dimerization. This could have explained why the dimeric configuration of AtWOX14 is sensitive to DNA methylation.
To explore the potential biological roles of ER0 and ER1 CREs, we conducted Gene Ontology (GO) analysis of their target genes (Figure 3C) using motif matches in the promoter region. We found that there is almost no overlap between the biological processes regulated by ER0 and ER1. Considering only the reported functions for AtWOX14 [10,11,12,14], genes downstream of ER0 were enriched in biological processes such as cell wall organization, cell wall modification, lignin metabolism and biosynthesis, gibberellin metabolism, and meristem initiation (Figure 3C), which were associated with vascular cell lignification. In contrast, genes associated with ER1 were predominantly involved in cell fate determination and auxin transport. Therefore, DNA methylation could facilitate WOX14 to utilize different dimeric binding modes (ER0 vs. ER1) and regulate distinct physiological processes.
2.4. Methylation Effects on Genome-Wide Binding Targets of AtWOX14
To explore the impact of methylation on genomic binding sites of AtWOX14, we generated native (DAP-seq), unmethylated (ampDAP-seq), and fully methylated (Methyl-ampDAP-seq) DAP-seq libraries. Similarly to Methyl-SELEX, Methyl-ampDAP was performed by amplifying genomic DNA with 5mCs instead of Cs during PCR. The Venn diagram (Figure 4A) revealed that the libraries of DAP and ampDAP are more similar to each other, while Methyl-ampDAP has identified much more (12,138) unique peaks. The similarity between DAP and ampDAP is potentially explained by the relative scarcity of CHH methylation in the genome.
Motif discovery of the DAP-seq libraries using Homer [34] has confirmed that the peak sites of all libraries contained the monomer motif of WOX14 (Figure 4B). These motifs are consistent with the motifs derived in SELEX (Figure 2A, B). From the DAP libraries, we also confirmed that ER0 CREs were preferentially bound under methylated conditions (Figure 4C-E), because: (1) there are more peaks containing ER0 in Methyl-ampDAP than DAP/ampDAP libraries (Figure 4C); (2) the intensity of ER0 peaks is also higher than ER1 peaks in the Methyl-ampDAP but not in the DAP/ampDAP libraries (Figure 4C). In contrast to ER1, while the number of peaks containing ER0 is similar across all the DAP libraries (Figure 4C), the intensity of ER0 peaks is weaker than ER1 peaks only in the ampDAP library (Figure 4D). We next examined the individual binding sites of AtWOX14, and found that, the ER0 site near AT4G35410 showed the highest occupancy upon methylation (Figure 4C, left), whereas the ER1 site upstream of AT1G60545 showed the highest occupancy in the absence of methylation (Figure 4C, right). The ER0 site within the promoter of AT1G15640 harbors a sequence almost the same as the consensus of ER0 motif (Figure 4F), we also validated with EMSA for its capability to bind AtWOX14 dimer (Figure 4D).
Overall, the analysis of DAP-seq libraries also confirmed that upon methylation, AtWOX14 has switched its preferred dimeric mode from ER1 to ER0.
3. Discussion
This study performed SELEX and DAP-seq for AtWOX14 both in the presence and absence of cytosine methylation, and systematically studied how methylation affects the specificity and genome-wide binding sites of AtWOX14. The most interesting finding is that the preferred dimeric configuration has switched from ER0 to ER1 upon DNA methylation (Figure 4E). ER0 and ER1 were also found to regulate target genes with different functions. Therefore, it is reasonable to hypothesize that when genome-wide methylation level is low, AtWOX14 will regulate gibberellin synthesis and accumulation, thereby promoting the lignification of vascular cell [14]. However, as genome-wide methylation level increases, for example, when the level of methyl-transferase DRM increases or when the RdDM pathway is activated [35], AtWOX14 could rebalance between the ER0 and ER1 sites and emphasize more on the ER0 targets, which are involved in cell differentiation, and cell fate determination processes [10,11] (Figure 4E). Consequently, the methylation level may serve as a switch to toggle between different downstream regulatory networks of AtWOX14.
This mechanism can potentially be extrapolated to other TFs that bind as homodimers but not protein-level homodimers. This is because in general, protein-level dimerization of TFs depends on amino acids outside the DNA-binding domain (DBD) [36], which tends to be far away from the DNA and thus unlikely affected by DNA methylations [37]. However, if the protein-level interaction between TF monomers is weak, then the homodimeric binding can be heavily mediated by the DNA scaffold. In this case, it is conceivable that the chemical modifications on DNA will impose more prominent effects on the preferred dimeric configurations. Notably, dimeric binding mediated by DNA scaffold is not a rare case but represents a prevalent phenomenon, because a high-throughput study has revealed that many TFs only show a small interaction surface between the two monomers when docked to the high-affinity dimeric CRE sequence [38]. The solved TF-DNA structures also suggest that TF dimers can form purely dependent on DNA, with two monomers spaced at >10 Å (e.g., PDB: 1HJB) [39]. Dimeric binding without direct contacts can be induced by the allosteric effect [40], or if a TF alters DNA shape upon binding.
When addressing the methylation effect on a TF’s binding affinity and specificity, the previous work mainly relied on comparing between the DAP and ampDAP libraries. This approach artfully made use of the sporadic 5mCs present in the native genome, to interrogate how methylation changes the affinity of each C in a known motif model of the TF [20]. However, this method suffers from the inability to de novo build binding models in the presence of 5mC. Also, because the non-CG methylation sites on the genome are relatively sparse [41], one can easily overfit the methylation effect with limited data points. In this work, we adopted the recently developed Methyl-SELEX method [1]to prepare input DNA ligands with 100% 5mC, and extrapolated the protocol to set up Methyl-ampDAP. These methods will largely facilitate our understanding on the methylation effects of TF binding.
Consistent with previous researches [21,42], we found that the methylation effect on a cytosine’s affinity to AtWOX14 is positional dependent (Figure 3A). But interestingly, we also found that 5mC overall represses DNA binding of AtWOX14 monomers but facilitates the binding of its dimers (Figure 1D). Thus, the methylation effect is also dependent on the binding mode of a TF.
In summary, this work comprehensively analyzed the influence of DNA methylation on DNA-binding of AtWOX14. It is also interesting to explore whether the mechanistic insights revealed by this work — DNA methylation can rewire the regulatory network by changing the preferred homodimeric configuration of a TF — can be extrapolated to other TFs.
4. Materials and Methods
4.1. Cloning and Protein Expression
The full-length CDS sequence of WOX14 was cloned into the bacterial expression vector pETG20A-His using the In-Fusion Kit (Vazyme, C112). This resulted in the generation of an N-terminal His-tagged fusion protein construct of WOX14. The construct was then transformed into Rosetta 2 (DE3) pLysS strains and induced overnight at 17°C to obtain the recombinant WOX14 protein. After overnight incubation, the bacteria were collected by centrifugation and resuspended in 150 µL of Buffer A (50 mM Tris, 300 mM NaCl, 10 mM imidazole, pH 7.5). The bacterial suspension was then subjected to freeze at -80°C overnight. The frozen bacteria were lysed at room temperature for 2 h using lysis buffer (0.5 mg/ml lysozyme, 2 mg/ml DNase I, 1 mM PMSF). Following lysis, His-tag Ni Sepharose (Sangon, C600332) was added and incubated with the lysate for 1 h. The protein-bead complexes were washed twice with Buffer A and Buffer B (50mM Tris, 300 mM NaCl, 50 mM Imidazole, pH 7.5) using a 10 kD ultra-centrifugal filter (Sigma, UFC8010) to remove contaminants. Finally, the recombinant WOX14 protein was eluted from the beads using Buffer C (50mM Tris, 300 mM NaCl, 500 mM Imidazole, pH 7.5).
4.2. SELEX
SELEX (Systematic Evolution of Ligands by Exponential Enrichment) was performed following previously described protocols [43]. The input SELEX library was designed to include a central random region of 11 bp and flanking regions that were compatible with Illumina sequencing (TruSeq adapter sequences). 6 µl fusion proteins were incubated with 6 µl of SELEX library (30 ng/µl) and 30 µl of TCAPT buffer (140 mM KCl, 5 mM NaCl, 1 mM MgCl2, 3 µM ZnSO4, 100 µM EGTA, 10 mM Tris, pH 8, 0.1% Tween) at 25 °C for 1 h. The DNA-protein complexes were enriched with His-Tag Beads (Cytiva, 17371222), followed by washing away unbound DNA ligands using the HydroSpeed plate washer (Tecan, 30190101). Subsequently, the bound DNA ligands were amplified by PCR (for Methyl-SELEX, PCR using the 5-methyl-dCTP solution) with amplification primers
(forward primer 5′-CCCTACACGACGCTCTTCC-3′,
reverse primer 5′-CAGACGTGTGCTCTTCCG-3′), and the resulting PCR products were used as the input library for the next cycle. After 5 cycles, the enriched DNA ligands were amplified with the PE primers
(forward primer5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTAC ACGACGCTCTTCC-3′,
reverse primer 5′-CAAGCAGAAGACGGCATACGAGAT-barcode-GTGACTGGA GTTCAGACGTGTGCTCTTCCG-3′) and Illumina sequenced.
4.3. Methyl-SELEX
Methyl-SELEX process was derived from SELEX with the addition of a DNA methylation step [1]. Specifically, the input SELEX library was amplified by PCR using 5-methyl-dCTP to generate the methylated library which was then incubated with the protein. This methylated library was incubated with 6 µl recombinant WOX14 protein and 30 µl of TCAPT buffer at 25 °C for 1 h. Methylated DNA ligands bound to WOX14 were separated from the unbound sequences through a washing step, and subsequently amplified again by PCR using 5-methyl-dCTP. The bound DNA ligands amplified by PCR were sequenced for analysis and used as input for the next cycle.
4.4. Data Analyses of SELEX
The raw sequencing data generated from the Illumina NovaSeq 6000 platform was first demultiplexed according to the unique i7 index of each sample. In the pre-processing stage, the low-quality reads and PCR duplicates were removed, the adaptors were trimmed, and then the paired-end reads were merged. To de novo discover the binding motifs of WOX14 from the pre-processed SELEX reads, the Autoseed algorithm was used with parameters “-40N <Background sequence> <Signal sequence> 1 8 10 0.35 - 40 100” [30]. The previously described Enrichment-based Mutual Information (E-MI) analysis is used to evaluate the signal strength of transcription factors in the SELEX library [28,29]. The information content (IC) of motifs also was calculated. To calculate motif enrichments, motif matching analysis was performed using motifmatchr [44]. Gene Ontology (GO) analyses were conducted using the R package clusterProfiler [45].
4.5. Electrophoretic Mobility Shift Assays
The protein-DNA binding buffer was prepared with 10 mM Tris, 50 mM NaCl, 1 mM MgCl2, 4% glycerol, and 0.5 mM EDTA. Double-stranded DNA probes, either monomeric or dimeric, were synthesized and diluted to a concentration of 50nM for use in EMSA. The probe sequences used were as follows:
M1: ATGCTAGCTCCATCTGTATTGATTGTTTATGGCGGTGACGTACT;
ER0: ATGCTAGCTCCATCTGTGATTGCAATCAATGGCGGTGACGTACT;
ER1: ATGCTAGCTCCATCTGTGATTGACAATCAATGGCGGTGACGTACT;
ER0-AT1G15640: AATAAGATGTACCCAATGATTGCAATGATGAGCCCAATGA GTGC.
The purified WOX14 protein was then diluted into various gradients and incubated with 30 ng of the DNA probe in a 20 µl protein-DNA binding buffer at 25°C for 40 minutes. The resulting reaction mixture was loaded onto a 6% native PAGE gel and run in 0.5× TBE buffer at 4°C for 90 minutes at 110 V. The gel was subsequently stained with Gel-Blue (UElandy, S2019L) and imaged using a Bio-Rad scanner (Bio-Rad, 1708195EDU).
4.6. DAP and Data Pre-Processing
The ampDAP, methyl-ampDAP, and DAP protocols followed the previously described workflow [46]. To construct the DAP library, genomic DNA (gDNA) was extracted from 7-day-old Arabidopsis seeding and fragmented using the Covaris S2 sonicator. The fragmented DNA (~200bp) was then purified with DNA clean beads (Vazyme, N411) and ligated with adapters using ligation kits (Vazyme, N203, N204). For the input library of amp-DAP, the gDNA was further amplified with specific primers to obtain completely unmethylated gDNA. For the input library of methyl-amp-DAP, the gDNA was amplified with specific primers and 5-methyl-dCTP to obtain the fully methylated gDNA. Next, 6 µl of His-tagged WOX14 protein and ~200 ng of the input DAP library were incubated within30 µl of TCAPT buffer for 1 h. The protein-DNA complexes were pulled down using His-Tag Beads and washed with a HydroSpeed plate washer (Tecan, 30190101). The gDNA bound to WOX14 was amplified by PCR and sequenced on an Illumina NovaSeq 6000. For methyl-ampDAP, PCR amplification of gDNA using the 5-methyl-dCTP solution.
Raw reads were demultiplexed, and sequences with low quality or adapters were removed before aligning to the TAIR10 version of the Arabidopsis genome (https:// www.arabidopsis.org/download_files/Genes/TAIR10_genome_release/TAIR10_chromosome_files/TAIR10_chr_all.fas.gz) using BWA v.0.7.17 [47]. Genome annotations were obtained from the Araport11 files (https://www.arabidopsis.org/download/list?dir=Gene s%2FAraport11_genome_release). Peak calling was performed with MACS3v.3.0.0a7 [48]. Motif discovery was performed using Homer(http://homer.ucsd.edu/homer/) [34].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1 The frequencies of subsequence (10-mers) after taking the logarithm in each cycle of WOX14 SELEX and Methyl-SELEX.
Author Contributions
H.G and F.Z. designed the research. D.J., X.Z., L.L., T.L., H.C., N.M., L.F., P.T., P.L., and F.M. collected the data. D.J., H.G, F.Z. and X.Z. analyzed the data. F.Z., H.G, D.J. and X.Z. wrote the manuscript. All authors read and approved the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China [32370582,32170554]; Natural Science Foundation of Fujian [2022J06017]; Major Special Project of Fujian [2021NZ029009].
Data Availability Statement
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in China National Center for Bioinformation under the accession number PRJCA032646.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Morgunova, E. et al. Interfacial water confers transcription factors with dinucleotide specificity. 2023.10.03.560647. (2023). Preprint at. [CrossRef]
- Gehring, W. J. et al. The structure of the homeodomain and its functional implications. Trends Genetics 6, 323–329 (1990). [CrossRef]
- Chandler, J., Nardmann, J. & Werr, W. Plant development revolves around axes. Trends in Plant Science 13, 78–84 (2008). [CrossRef]
- Byrne, M. E. Shoot meristem function and leaf polarity: the role of class III HD-ZIP genes. PLoS Genetics 2, e89 (2006). [CrossRef]
- Ito, M., Sato, Y. & Matsuoka, M. Involvement of homeobox genes in early body plan of monocot. International Review of Cytology 218, 1–35 (2002).
- Chan, R. L., Gago, G. M., Palena, C. M. & Gonzalez, D. H. Homeoboxes in plant development. Biochimica et Biophysica Acta 1442, 1–19 (1998).
- Hake, S. et al. The role of knox genes in plant development. Annual Reviews Cell and Developmental Biology 20, 125–151 (2004).
- Hay, A., Craft, J. & Tsiantis, M. Plant hormones and homeoboxes: bridging the gap? Bioessays 26, 395–404 (2004).
- Graaff, E. van der, Laux, T. & Rensing, S. A. The WUS homeobox-containing (WOX) protein family. Genome Biology 10, 248 (2009). [CrossRef]
- Wang, J. et al. WUS-RELATED HOMEOBOX 14 boosts de novo plant shoot regeneration. Plant Physiology 192, 748 (2023). [CrossRef]
- Deveaux, Y. et al. Genes of the most conserved WOX clade in plants affect root and flower development in Arabidopsis. BMC Evolutionary Biology 8, 291 (2008). [CrossRef]
- Etchells, J. P., Provost, C. M., Mishra, L. & Turner, S. R. WOX4 and WOX14 act downstream of the PXY receptor kinase to regulate plant vascular proliferation independently of any role in vascular organisation. Development 140, 2224–2234 (2013). [CrossRef]
- Smit, M. E. et al. A PXY-Mediated Transcriptional Network Integrates Signaling Mechanisms to Control Vascular Development in Arabidopsis. The Plant Cell 32, 319 (2019). [CrossRef]
- Denis, E. et al. WOX14 promotes bioactive gibberellin synthesis and vascular cell differentiation in Arabidopsis. The Plant Journal 90, 560–572 (2017). [CrossRef]
- Bell, O., Tiwari, V. K., Thomä, N. H. & Schübeler, D. Determinants and dynamics of genome accessibility. Nature Reviews Genetics 12, 554–564 (2011). [CrossRef]
- Li, Z., Li, D., Li, Y., Guo, X. & Yang, R. Deciphering the regulatory code of histone modifications in plants. Journal of Genetics and Genomics 49, 1064–1067 (2022). [CrossRef]
- Li, M. et al. Double DAP-seq uncovered synergistic DNA binding of interacting bZIP transcription factors. Nature Commuications 14, 2600 (2023). [CrossRef]
- Héberlé, É. & Bardet, A. F. Sensitivity of transcription factors to DNA methylation. Essays in Biochemistry 63, 727 (2019). [CrossRef]
- Zhang, Y. et al. Transposable elements orchestrate subgenome-convergent and -divergent transcription in common wheat. Nature Communications 13, 6940 (2022).
- Charvin, M. et al. Single-cytosine methylation at W-boxes repels binding of WRKY transcription factors through steric hindrance. Plant Physiology 192, 77–84 (2023). [CrossRef]
- Yin, Y. et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356, eaaj2239 (2017). [CrossRef]
- Hu, S. et al. DNA methylation presents distinct binding sites for human transcription factors. eLife 2, e00726 (2013). [CrossRef]
- O’Malley, R. C. et al. Cistrome and Epicistrome Features Shape the Regulatory DNA Landscape. Cell 165, 1280–1292 (2016). [CrossRef]
- Boer, D. R. et al. Structural Basis for DNA Binding Specificity by the Auxin-Dependent ARF Transcription Factors. Cell 156, 577–589 (2014). [CrossRef]
- Yan, J. et al. Systematic Analysis of Transcription Factors Binding to Noncoding Variants. Nature 591, 147–151 (2021).
- Jolma, A. et al. Binding specificities of human RNA-binding proteins toward structured and linear RNA sequences. Genome Research 30, 962–973 (2020). [CrossRef]
- Law, J. A. & Jacobsen, S. E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nature Reviews Genetics 11, 204–220 (2010). [CrossRef]
- Zhu, F. et al. The interaction landscape between transcription factors and the nucleosome. Nature 562, 76–81 (2018). [CrossRef]
- Wen, C. et al. Sea-ATI unravels novel vocabularies of plant active cistrome. Nucleic Acids Research 51, 11568–11583 (2023). [CrossRef]
- Nitta, K. R. et al. Conservation of transcription factor binding specificities across 600 million years of bilateria evolution. eLife 4, e04837 (2015). [CrossRef]
- Chow, C. et al. PlantPAN3.0: a new and updated resource for reconstructing transcriptional regulatory networks from ChIP-seq experiments in plants. Nucleic Acids Research 47, D1155–D1163 (2019).
- Jin, J. et al. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res 45, D1040–D1045 (2017). [CrossRef]
- Bryne, J. C. et al. JASPAR, the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic Acids Research 36, D102–D106 (2008). [CrossRef]
- Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular Cell 38, 576–589 (2010). [CrossRef]
- Zhang, H. & Zhu, J.-K. Epigenetic gene regulation in plants and its potential applications in crop improvement. Nature Reviews Molecular Cell Biology (2024). [CrossRef]
- Fontana, M. et al. Cooperative action of separate interaction domains promotes high-affinity DNA binding of Arabidopsis thaliana ARF transcription factors. Proceedings of the National Academy of Sciences of the United States of America. 120, e2219916120 (2023). [CrossRef]
- Nanao, M. H. et al. Structural basis for oligomerization of auxin transcriptional regulators. Nature Communications 5, 3617 (2014). [CrossRef]
- Jolma, A. et al. DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature 527, 384–388 (2015). [CrossRef]
- Tahirov, T. H. et al. Structural Analyses of DNA Recognition by the AML1/Runx-1 Runt Domain and Its Allosteric Control by CBFβ. Cell 104, 755–767 (2001).
- Kim, S. et al. Probing Allostery Through DNA. Science 339, 816–819 (2013). [CrossRef]
- Niederhuth, C. E. Widespread natural variation of DNA methylation within angiosperms. Genome Biology 17, 194 (2016). [CrossRef]
- Kribelbauer, J. F. et al. Quantitative analysis of the DNA methylation sensitivity of transcription factor complexes. Cell Rep 19, 2383–2395 (2017). [CrossRef]
- Jolma, A. et al. DNA-Binding Specificities of Human Transcription Factors. Cell 152, 327–339 (2013). [CrossRef]
- Korhonen, J., Martinmäki, P., Pizzi, C., Rastas, P. & Ukkonen, E. MOODS: fast search for position weight matrix matches in DNA sequences. Bioinformatics 25, 3181–3182 (2009). [CrossRef]
- Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb) 2, 100141 (2021). [CrossRef]
- Bartlett, A. et al. Mapping genome-wide transcription-factor binding sites using DAP-seq. Nature Protocols 12, 1659–1672 (2017). [CrossRef]
- Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [CrossRef]
- Zhang, Y. et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biology 9, R137 (2008).
Figure 1.
The Binding Specificity of WOX14 in the Presence and Absence of Methylation. (A) Schematic representation of the SELEX and Methyl-SELEX workflow; (B) The frequencies of subsequence (10-mers) in each cycle of WOX14 SELEX. In addition to the bound sequences (top 50 enriched sequences, red), 50 randomly selected sequences (blue) were also visualized; (C) E-MI signal near the hypotenuse of the triangle becomes stronger than elsewhere, a pattern indicating enrichment of TF signals; (D) Enrichment comparison of 8-mers and 12-mers between SELEX and Methyl-SELEX libraries.
Figure 1.
The Binding Specificity of WOX14 in the Presence and Absence of Methylation. (A) Schematic representation of the SELEX and Methyl-SELEX workflow; (B) The frequencies of subsequence (10-mers) in each cycle of WOX14 SELEX. In addition to the bound sequences (top 50 enriched sequences, red), 50 randomly selected sequences (blue) were also visualized; (C) E-MI signal near the hypotenuse of the triangle becomes stronger than elsewhere, a pattern indicating enrichment of TF signals; (D) Enrichment comparison of 8-mers and 12-mers between SELEX and Methyl-SELEX libraries.
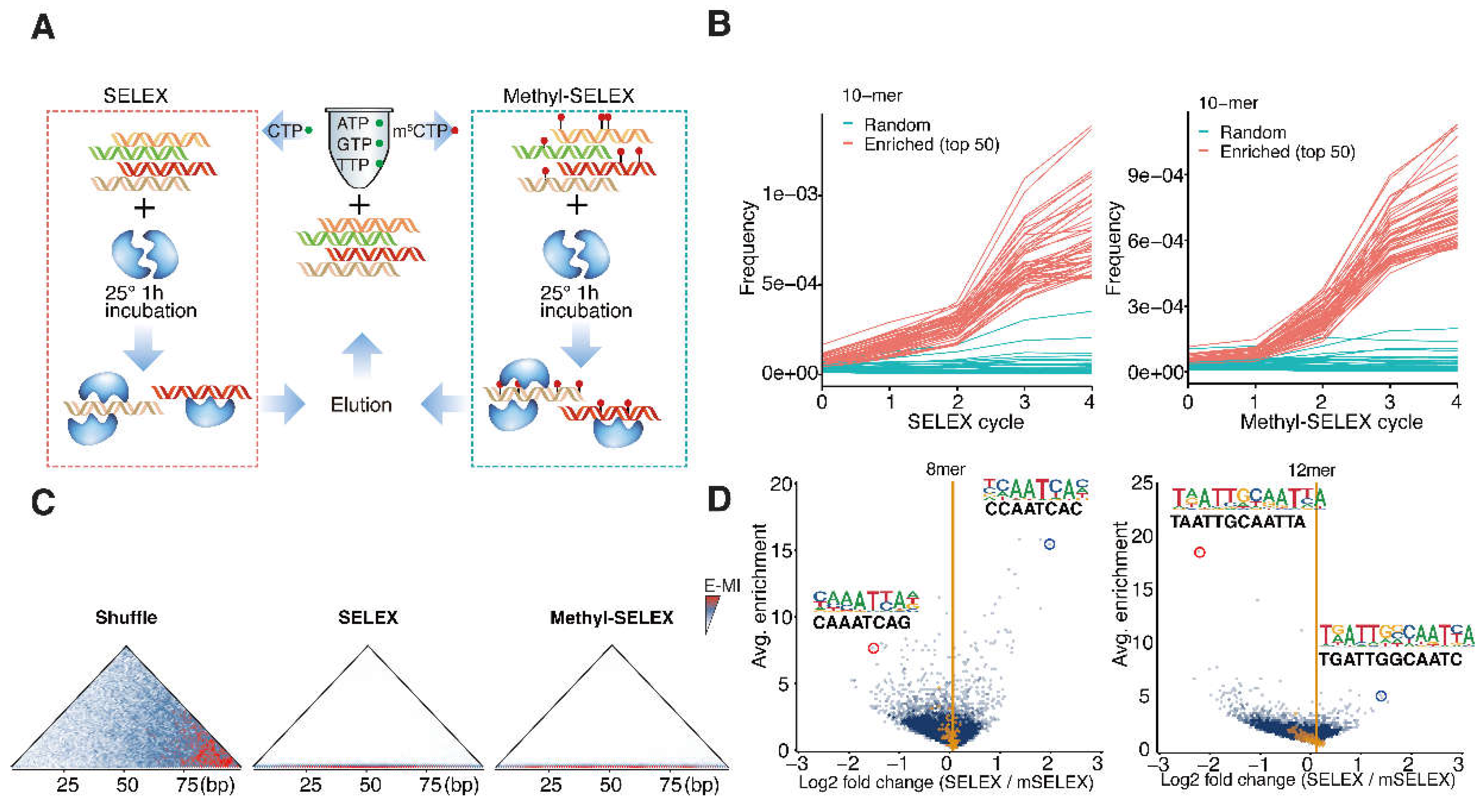
Figure 2.
The Binding Models of WOX14 in the Presence and Absence of Methylation. De novo discovered motifs in the SELEX (A) and Methyl-SELEX (B) libraries are shown, with their information content (blue squares) and enrichment (red squares) indicated to the left. By defining the half-site as “AATYA”, the spacing regions of the dimeric motifs are colored grey. Distributions of the discovered motifs (mean IC > 0.3) around TSSs of A. thaliana genes were evaluated by the density of motif matches, and visualized both for the SELEX motifs (C) and Methyl-SELEX motifs (D).
Figure 2.
The Binding Models of WOX14 in the Presence and Absence of Methylation. De novo discovered motifs in the SELEX (A) and Methyl-SELEX (B) libraries are shown, with their information content (blue squares) and enrichment (red squares) indicated to the left. By defining the half-site as “AATYA”, the spacing regions of the dimeric motifs are colored grey. Distributions of the discovered motifs (mean IC > 0.3) around TSSs of A. thaliana genes were evaluated by the density of motif matches, and visualized both for the SELEX motifs (C) and Methyl-SELEX motifs (D).
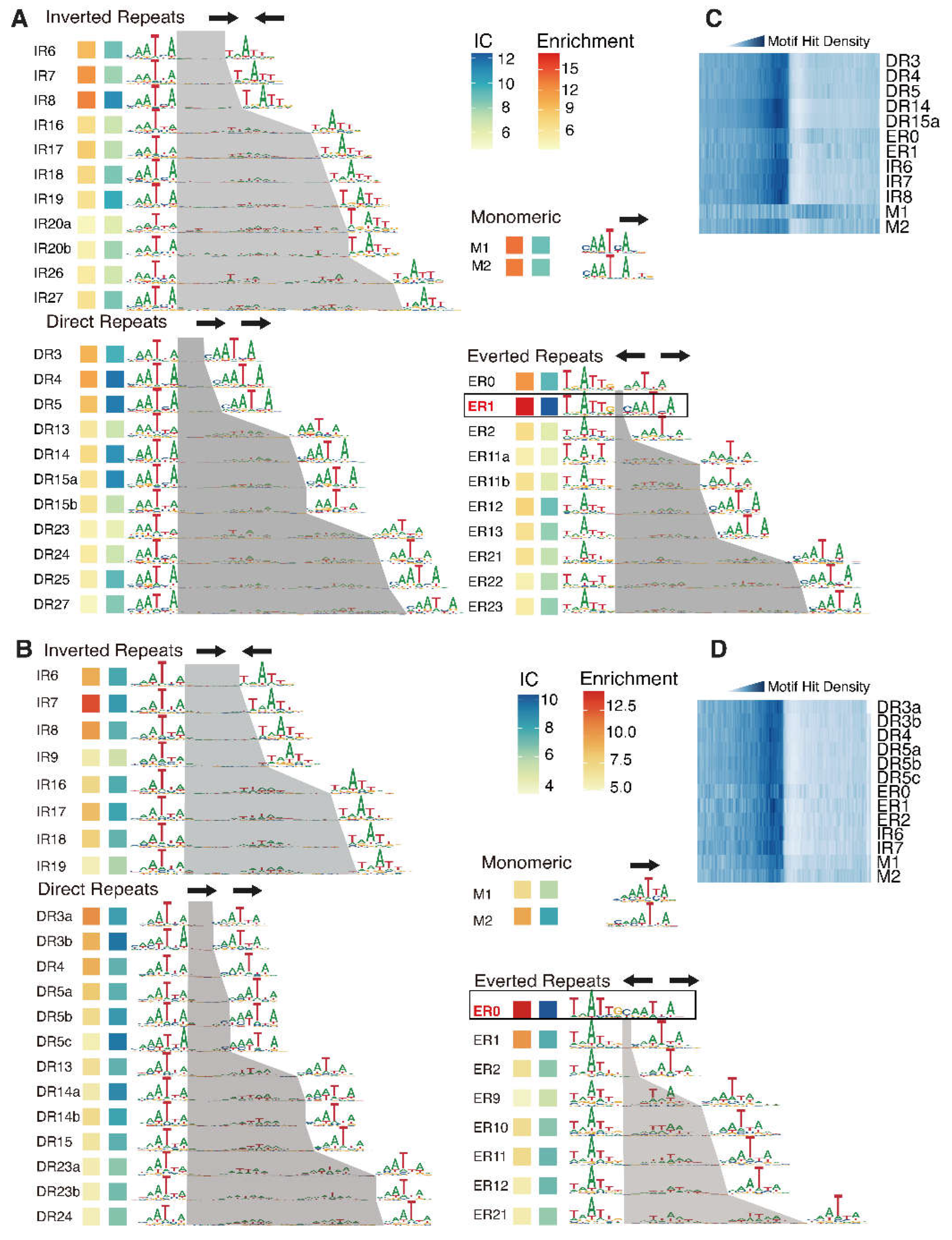
Figure 3.
ER0 and ER1 Regulate Different Physiological Functions. (A) Comparison of the methylated motifs of ER0 and ER1 with their unmethylated counterparts. Cytosine affinity can either increase (“+”) or decrease (“-”) after methylation; (B) Electrophoretic mobility shift assay (EMSA) of WOX14 binding to monomer, ER0, and ER1 consensus. The concentrations of WOX14 are increased from left to right (50, 100, and 200nM); (C) The GO enrichment of the target genes of ER0 and ER1. Arrows indicate the GO terms related to the development of vascular tissues and plant growth.
Figure 3.
ER0 and ER1 Regulate Different Physiological Functions. (A) Comparison of the methylated motifs of ER0 and ER1 with their unmethylated counterparts. Cytosine affinity can either increase (“+”) or decrease (“-”) after methylation; (B) Electrophoretic mobility shift assay (EMSA) of WOX14 binding to monomer, ER0, and ER1 consensus. The concentrations of WOX14 are increased from left to right (50, 100, and 200nM); (C) The GO enrichment of the target genes of ER0 and ER1. Arrows indicate the GO terms related to the development of vascular tissues and plant growth.
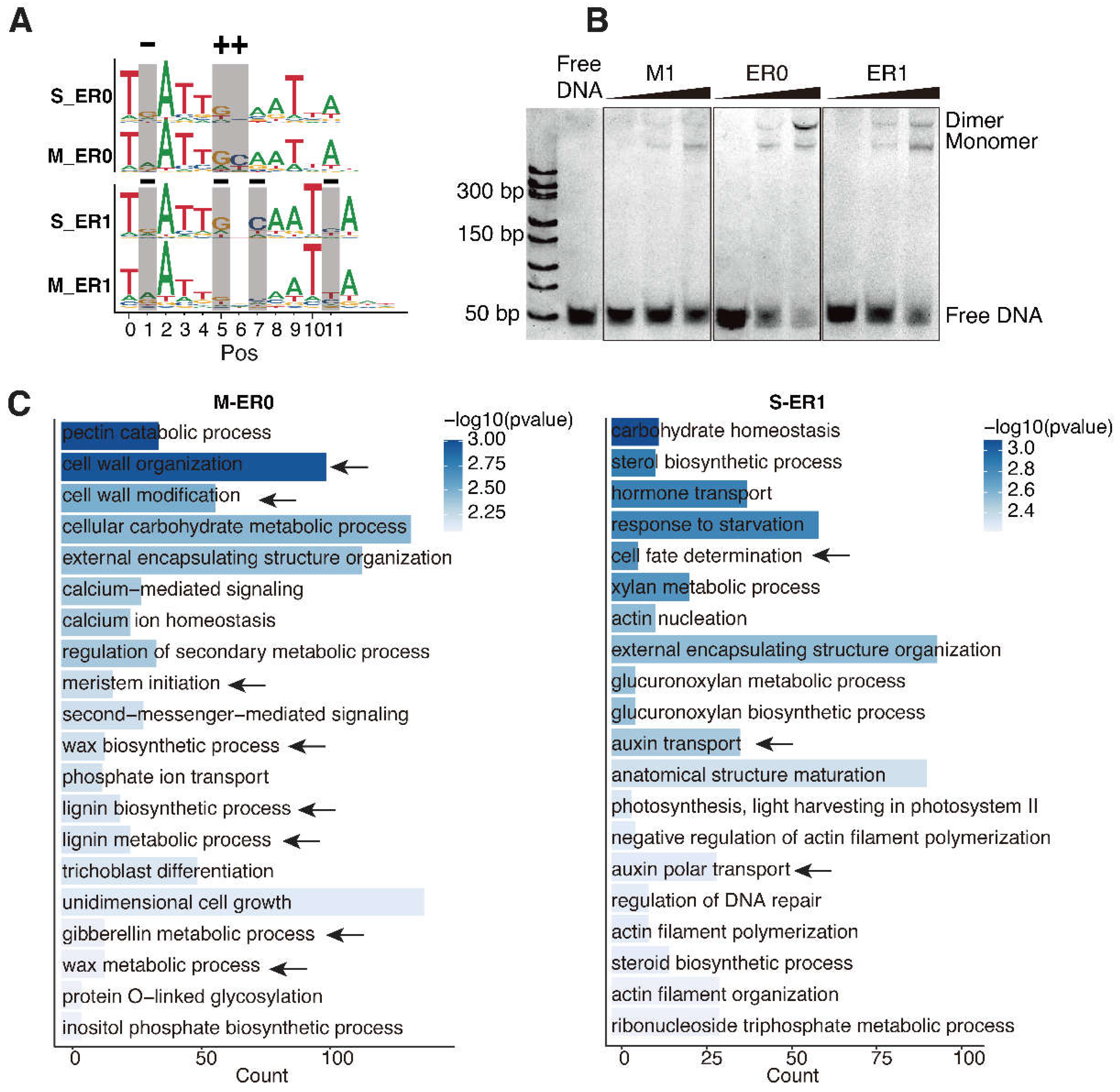
Figure 4.
The Genome-wide Binding of AtWOX14 in the Presence and Absence of Methylation. (A) Venn diagram comparing peaks identified in DAP, ampDAP, and Methyl-ampDAP; (B) PWM models of the most enriched motifs identified from DAP, ampDAP, and Methyl-ampDAP peaks using Homer; (C) Proportion of the top 3,000 peaks that contain each motif; (D) Intensities of peaks that contain ER0 or ER1 motif; (E) Normalized coverage tracks of Methyl-ampDAP, DAP, and ampDAP near the ER0/ER1 target sites. The ER0/ER1 CRE sequences are displayed above, with their genomic positions marked by gray vertical line, and mismatches to the consensus marked by red rectangles; (F) The EMSA (right) showing the dimeric binding of WOX14 protein to the ER0 CRE sequence within the promoter of AT1G15840 (left). The concentrations of WOX14 are 25, 50, 100, and 200nM, from left to right.
Figure 4.
The Genome-wide Binding of AtWOX14 in the Presence and Absence of Methylation. (A) Venn diagram comparing peaks identified in DAP, ampDAP, and Methyl-ampDAP; (B) PWM models of the most enriched motifs identified from DAP, ampDAP, and Methyl-ampDAP peaks using Homer; (C) Proportion of the top 3,000 peaks that contain each motif; (D) Intensities of peaks that contain ER0 or ER1 motif; (E) Normalized coverage tracks of Methyl-ampDAP, DAP, and ampDAP near the ER0/ER1 target sites. The ER0/ER1 CRE sequences are displayed above, with their genomic positions marked by gray vertical line, and mismatches to the consensus marked by red rectangles; (F) The EMSA (right) showing the dimeric binding of WOX14 protein to the ER0 CRE sequence within the promoter of AT1G15840 (left). The concentrations of WOX14 are 25, 50, 100, and 200nM, from left to right.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



