
Submitted:
02 December 2024
Posted:
03 December 2024
You are already at the latest version
Abstract
Spodoptera frugiperda is a globally significant migratory agricultural pest that requires proactive monitoring. Understanding the molecular mechanisms underlying the interactions between pathogenic microorganisms and S. frugiperda is crucial for enhancing the effectiveness of microbial control agents against this pest. This study used transcriptome sequencing and molecular biology techniques on S. frugiperda larvae infected by bacteria and fungi to investigate the composition and molecular regulatory mechanisms of its immune system. A total of 655 immune-related genes were identified, including pattern recognition receptors, signal transduction factors, immune effectors, and immune regulators. Following microbial infection, most immune-related genes showed an upregulated expression trend. Phylogenetic analysis revealed that the immune gene repertoire of S. frugiperda is relatively conserved. The expression changes of immune recognition receptors PGRP and βGRP were validated using qPCR, confirming that these genes were significantly upregulated under specific pathogenic infections. This study elucidates the immune transcriptome of S. frugiperda in response to various pathogenic microorganisms, providing valuable insights for improving the effectiveness of existing microbial agents and developing new highly efficient and specific biopesticides.
Keywords:
Spodoptera frugiperda
; Pathogenic microorganisms
; Transcriptome
; Induced expression
; Immune genes
1. Introduction
Spodoptera frugiperda, commonly known as the fall armyworm, is a highly migratory agricultural pest of major significance, globally recognized by the Food and Agriculture Organization (FAO) as a severe threat. It is characterized by a broad host range, strong migratory ability, rapid spread, high reproductive rate, and significant resistance to pesticides, posing substantial risks to agricultural production in many countries [1]. Currently, chemical pesticides are the primary method for controlling S. frugiperda. However, excessive use of chemical pesticides has led to increased pest resistance and resurgence, disrupted the natural balance of ecosystems, and caused severe environmental pollution [2]. Biological control, especially the use of microbial biopesticides, has emerged as a promising sustainable strategy for managing S. frugiperda [3,4]. Among the insecticides recommended by China's Ministry of Agriculture and Rural Affairs for emergency control of S. frugiperda, six are biopesticides. These include five pathogenic microorganism-based products and one viral product: Mamestra brassicae nucleopolyhedrovirus, Bacillus thuringiensis (Bt), Metarhizium anisopliae, Beauveria bassiana (Bb), and Empedobacter brevis. Among these, B. thuringiensis is the most widely used and successful microbial insecticide, known for its high specificity, safety, and lack of residues [5].
Microorganisms can cause the death of S. frugiperda, but the pest simultaneously activates immune responses to resist and eliminate the invading pathogens [6,7]. Through long-term co-evolution, insects have developed a highly efficient and comprehensive innate immune system to defend against microbial and parasitic infections. Bai Yaoyu and colleagues discovered that injecting Escherichia coli (Ec) affects the cellular immune functions of S. frugiperda [8], while the invasion of Steinernema carpocapsae, a nematode species, leads to a "decrease-increase-decrease" trend in the hemolymph phenoloxidase activity of S. frugiperda larvae [9]. Pathogenic microbial invasion triggers a series of humoral and cellular immune responses in the host, stimulating the production of melanin and various antimicrobial peptides. The robust immune system of the host is one of the most critical factors limiting the "high virulence" of pathogens. Currently, research on the immune functions of S. frugiperda remains in its early stages. Therefore, an in-depth investigation into the composition and molecular regulatory mechanisms of the immune system of S. frugiperda is crucial for improving the efficacy of pathogenic microorganisms and developing novel, specific-target, and highly efficient biopesticides.
This study focuses on transcriptome sequencing of S. frugiperda infected by different pathogenic microorganisms, identifying immune-related genes through differential expression analysis, and conducting bioinformatic analyses and expression validation of these genes. The aim is to uncover the molecular mechanisms underlying the interaction between pathogenic microorganisms and S. frugiperda, thereby providing new methods and theoretical foundations for utilizing pathogens in the biological control of S. frugiperda.
2. Materials and Methods
2.1. Test Insects
The S. frugiperda used in this experiment was collected from Fengyang County, Chuzhou City, Anhui Province (117.56°E, 32.86°N) and propagated for multiple generations indoors to establish an experimental population. The larvae of S. frugiperda were reared with artificial feed in an insectary at 27 ± 1°C, relative humidity of 80% ± 10%, and a photoperiod of 14L:10D.
The bacteria used in this experiment were Gram-positive bacteria Staphylococcus aureus (SA) and B. thuringiensis (Bt), Gram-negative bacteria E. coli (Ec), and fungus B. bassiana (Bb). The bacterial strains were purchased from the Institute of Microbiology, Chinese Academy of Sciences. The bacteria were activated and cultured at 37°C and 200 rpm until the OD600 reached approximately 1. The cultures were centrifuged at 5000 g for 5 minutes at 4°C, and the supernatant was discarded. The bacteria were washed three times with sterile PBS (7.7 mmol/L Na2HPO4, 2.65 mmol/L NaH2PO4, 150 mmol/L NaCl, pH 6.4), adjusted to a concentration of 1×10^6 cells/mL, sterilized at 121°C under high pressure for 20 minutes, and stored for future use. B. bassiana was provided by Professor Hu Fei from the Institute of Plant Protection and Agro-Product Safety, Anhui Academy of Agricultural Sciences. It was cultured on PDA medium at 27°C, collected in enzyme-free EP tubes, shaken thoroughly with sterile water (containing 0.05% Tween-80), filtered with sterile cotton, counted using a hemocytometer, and diluted to 1×10^6 cells/mL for future use.
2.2. Injection Experiment
Healthy fourth-instar S. frugiperda larvae of consistent size were selected and divided into a control group, PBS injection group, S. aureus injection group, E. coli injection group, B. thuringiensis injection group, and B. bassiana injection group, with 15 larvae per group and three replicates for each treatment. The injection experiment was performed according to the method of Sun & Bai (2020) [10]. The larvae were anesthetized on ice for 5 minutes, and 5 µL of inactivated bacterial suspension (approximately 3.0×10^6 cells/mL) was injected into the proleg using a microsyringe, with an equal amount of PBS as the control. The injection site was surface sterilized with 70% alcohol, and the larvae were reared in an artificial climate chamber with artificial feed at 27 ± 1°C, relative humidity of 80% ± 10%, and a photoperiod of 14L:10D for 24 hours, then stored in liquid nitrogen for future use.
2.3. Total RNA Extraction and Sequencing
Total RNA was extracted from the whole S. frugiperda using the Trizol method. The quality of the RNA was assessed by 1% agarose gel electrophoresis, and its concentration and purity were evaluated with a Nanodrop 2000 (OD260/280 and OD260/230 ratios). The integrity of RNA was accurately assessed with an Agilent 2100 Bioanalyzer to ensure sample quality. Qualified RNA samples were used for transcriptome sequencing on the Illumina NovaSeq 2000 platform.
2.4. Gene Expression Quantification and Differential Analysis
The raw sequencing data were processed by removing adapter sequences, low-quality reads, and reads with uncertain base information, resulting in high-quality clean reads. The reads were assembled using Trinity (Trinity-v2.5.1) software. The assembly quality of Trinity.fasta, unigene.fasta, and cluster.fasta was assessed using BUSCO software, and the accuracy and completeness of the assembly were evaluated based on the GC content and unigene sequence integrity. Gene expression levels were quantified by FPKM (Fragments Per Kilobase of transcript per Million fragments mapped). Differential gene expression analysis was performed using DESeq2 from edgeR 3.8.6, with differentially expressed genes selected based on an FDR < 0.01 and fold change (FC) greater than 2. GO functional enrichment analysis was performed using TopGO, and KEGG pathway analysis was conducted using ClusterProfiler.
2.5. Identification and Analysis of Immune-Related Genes
The S. frugiperda unigenes served as the original database, and immune sequences from other known insect model species were used as a reference database. Immune pathway-related gene sequences were screened from the S. frugiperda transcriptome. CD-search (http://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi) and SMART (http://smart.embl-heidelberg.de/) were used to examine conserved domains in candidate genes of non-redundant protein families. Signal peptides and transmembrane domains of different protein families were verified using SignalP 4.1 (http://www.cbs.dtu.dk/services/SignalP) and TMHMM (http://www.cbs.dtu.dk/services/TMHMM/). Heatmaps of the FPKM values of differentially expressed genes across treatments were generated using TBtools, and MEGA 11.0 was used to align homologous immune proteins of S. frugiperda with those of other insects. A phylogenetic tree of S. frugiperda immune family proteins and homologous proteins from other insects was constructed using the maximum likelihood method, and FigTree1.4.4 was used for visualization to clarify the evolutionary relationship of immune proteins between S. frugiperda and other insects.
2.6. qPCR Verification of S. frugiperda Immune Receptor Genes
The gene sequences were obtained from NCBI, and primers were designed online (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). RPL3 and RPL18 [11,12] were used as reference genes. The primer sequences for candidate genes are shown in Table 1. High-quality RNA samples were reverse transcribed into cDNA using the TransScript One-step gDNA Removal and cDNA Synthesis SuperMix kit. The qPCR reaction (20 µL) consisted of 1 µL cDNA template, 1 µL of each forward and reverse primer, 1 µL ROX Reference Dye II, 10 µL TB Green Premix Ex Taq, and 6 µL ddH2O. The program was set according to RR420 A as follows: 95°C for 30 s, 95°C for 5 s, 60°C for 30 s, 40 cycles, followed by 95°C for 15 s, 60°C for 1 min, and 95°C for 15 s. Gene expression was calculated using the 2^−ΔΔCt method [13], and data were analyzed using Prismchs (Table 1).
3. Results
3.1. Transcriptome Sequencing Data Statistics and Analysis
S. frugiperda larvae through transcriptome sequencing and analysis. Each sample had more than 6.72 Gb of Clean Data. The Q30 was above 95.72%, Q20 above 98.462%, the GC content ranged from 45.15% to 46.7%, and the error rate was 0.1% (Table 2)
3.2. Differentially Expressed Gene Analysis
A total of 10,453 differentially expressed genes (DEGs) were identified after treating S. frugiperda with pathogenic microorganisms. Comparative analysis of DEGs with the control group revealed that the Bb-treated group exhibited significantly more DEGs than other treatment groups, with a total of 3,593 DEGs. Among these, 2,044 genes were upregulated, and 1,549 genes were downregulated.
In the SA-treated group, 2,419 DEGs were identified, including 1,734 upregulated genes and 685 downregulated genes. The Bt-treated group showed 2,571 DEGs, with 1,474 genes upregulated and 1,097 genes downregulated. Lastly, the Ec-treated group had 1,870 DEGs, with 994 genes upregulated and 876 genes downregulated (Figure 1).
3.3. GO Functional Enrichment Analysis
GO functional enrichment analysis was performed on differentially expressed genes induced by pathogenic microorganisms in fourth-instar larvae of S. frugiperda. The results showed that the differential genes were mainly distributed in Biological Process (BP), Cellular Component (CC), and Molecular Function (MF) (Figure 3). In the Bb treatment, 707 unigenes were annotated to Biological Process, 125 to Cellular Component, and 1198 to Molecular Function. In the Bt treatment, 477 unigenes were annotated to Biological Process, 88 to Cellular Component, and 686 to Molecular Function. In the Ec treatment, 481 unigenes were annotated to Biological Process, 78 to Cellular Component, and 712 to Molecular Function. In the SA treatment, 368 unigenes were annotated to Biological Process, 97 to Cellular Component, and 617 to Molecular Function. These results indicate that the level of changes in differentially expressed genes varies after infection of S. frugiperda fourth-instar larvae by different pathogenic microorganisms. However, most changes are concentrated in Molecular Function, followed by Biological Process, with relatively fewer differentially expressed genes in Cellular Component.
3.4. KEGG Pathway Enrichment Analysis
Differentially expressed genes (DEGs) (P < 0.05) induced by microbial treatments in S. frugiperda were analyzed using KEGG pathway enrichment. The results showed that in the Bb-treated group, among 3,593 DEGs, KEGG analysis identified Carbon metabolism as the most significantly enriched pathway, containing the highest number of differentially expressed genes. In the Bt-treated group, out of 2,571 DEGs, significantly enriched KEGG pathways included Biosynthesis of cofactors, Lysosome, Neuroactive ligand-receptor interaction, and Carbon metabolism. In the Ec-treated group, among 1,870 DEGs, significantly enriched pathways included Peroxisome, Lysosome, and Neuroactive ligand-receptor interaction. In the SA-treated group, among 2,419 DEGs, significantly enriched pathways were Biosynthesis of amino acids, Carbon metabolism, and Motor proteins.
3.5. Screening of Immune-Related Genes
By comparing the immune-related gene sequences of known model insects, 655 immune-related genes of various categories were identified from the transcriptome sequences of S. frugiperda (see Appendix 1). Based on their functions, these genes were categorized into four major groups: pattern recognition receptors, immune effectors, signal transduction factors, and immune regulatory factors. Signal transduction factors include components of the IMD, Toll, JAK/STAT, JNK, RNA interference, and autophagy immune pathways.
Specifically, the study identified 98 pattern recognition receptors belonging to 12 gene families, accounting for 14.96% of the total immune-related genes. The serine protease inhibitor family with clip domains and the serine protease family were found to have 20 and 61 members, respectively, which regulate the amplification and attenuation of extracellular immune signals. These immune regulatory factors account for 12.37% of the total immune-related genes.
In the IMD, Toll, JAK/STAT, JNK, RNA interference, and autophagy immune signal transduction pathways of S. frugiperda, 212 components responsible for signal transduction were identified, representing 32.37% of the total. Immune effectors in insects, such as antimicrobial peptides, lysozymes, melanin, and antioxidative molecules, were also identified. A total of 264 immune effectors, accounting for 40.30% of the total immune-related genes, were identified in this study.
As shown in Figure 4, when S. frugiperda is infected by pathogenic microorganisms, its pattern recognition receptors, immune effectors, signal transduction factors, and immune regulatory factors are all activated. Among these, genes related to immune effectors constitute the largest proportion, indicating that through long-term biological evolution, S. frugiperda has developed a relatively complete innate immune system.
3.6. Analysis of Immune-Related Genes
The immune gene families of S. frugiperda are complex, with functional differences observed among genes within the same family. This study conducted phylogenetic and expression variation analyses of immune genes in comparison with known model species to better understand the functions and expression trends of immune-related genes in S. frugiperda.
3.6.1. Pattern Recognition Receptors
Insects rely on unique pattern-recognition receptors (PRRs) to detect pathogen-associated molecular patterns (PAMPs) on the surfaces of microorganisms. In the transcriptome of S. frugiperda, 12 types of PRRs were identified (see Appendix 1). Among them, Integrin was the most abundant, with 27 unigenes, followed by ubiquitin-conjugating enzyme (20), DSCAM (11), PGRP (9), SR (9), Galectin (8), Vitellogenin (5), βGRP (3), C-type lectin (2), ApoLp (2), TEP (1), and Croquemort (1). The transcription levels of PRR genes varied significantly under different pathogenic microbial infections (Figure 5). Specifically, SR-1 was consistently and significantly downregulated following all pathogen treatments. In contrast, PGRP-1, -3, -4, ubiquitin-conjugating enzyme, DSCAM-2, βGRP-1, and CTL were significantly upregulated under all pathogenic conditions. Meanwhile, βGRP-2, Vitellogenin-1, and ApoLp exhibited an overall downregulation trend. These findings suggest that different PRR genes may play distinct roles in immune pathways during the recognition and response to pathogenic microorganisms.
The most notable feature of the PGRP (Peptidoglycan Recognition Protein) family is the presence of a T4 bacteriophage lysozyme domain [14]. In insects, PGRPs are classified into three types based on molecular weight: short (S), intermediate (I), and long (L) [15].
The short type (S), with a molecular weight of approximately 20–25 kD, typically contains signal peptides, lacks transmembrane domains, and functions as small, secreted extracellular proteins. The intermediate type (I) has a molecular weight of approximately 40–45 kD. The long type (L), generally exceeding 90 kD in molecular weight, can be further subdivided into two subtypes: intracellular proteins lacking both signal peptides and transmembrane domains, and transmembrane proteins containing signal peptides and transmembrane domains, which exist as transmembrane proteins [16].
In the transcriptome Unigene data of S. frugiperda, nine PGRP family genes were identified and named PGRP-L1, L2, L3, LB, LB1, LB2, LE2, S1, S2 based on sequence characteristics and multiple sequence alignments. As shown in Figure 6 (b), the amino acid sequences of PGRP-S1 and PGRP-S2 contain signal peptides, suggesting that they are likely secreted extracellular proteins of the short type and may participate in melanization upon receiving signals. PGRP-L1 and PGRP-LB1 lack signal peptides and transmembrane domains, indicating they function as intracellular proteins. PGRP-L2, L3, LB2, and LE2 contain transmembrane domains, suggesting that they may act as transmembrane proteins to activate immune signaling pathways. Notably, PGRP-L3 contains two transmembrane domains, indicating a potentially broader role in immune recognition compared to other proteins.
Phylogenetic tree analysis revealed that the PGRP gene family of S. frugiperda is closely related to those of lepidopteran insects such as Bombyx mori and Helicoverpa armigera. The results of the phylogenetic analysis align well with previous nomenclature based on sequence characteristics, with L-type PGRPs clustering together and S-type PGRPs forming their own distinct clade.
βGRP, also known as GNBP (Gram-negative binding protein), contains two main conserved domains: an N-terminal β-1,3-glucan recognition domain and a C-terminal β-1,3-glucan recognition domain lacking catalytic residues. These domains are responsible for recognizing the cell wall polysaccharides of Gram-negative bacteria or β-1,3-glucans in fungi [17,18].
In the transcriptome data of S. frugiperda, three homologous βGRP genes were identified, all of which contained signal peptide sequences at their N-terminal ends. This indicates their ability to be secreted into the hemolymph to perform pathogen recognition functions. Phylogenetic analysis revealed that βGRP1 of S. frugiperda clustered with HamGRP-2 from H. armigera, while βGRP2 clustered with BmorGRP-1 and BmorGRP-2 from B. mori. Previous studies have demonstrated that BmorGRP-1 can bind to fungi and bacteria, activating phenoloxidase-mediated melanization [19]. Thus, βGRP2 is likely a key recognition receptor in the immune signaling pathways for pathogenic microorganisms. Meanwhile, βGRP3 clustered with the βGRP family of Aedes aegypti, suggesting evolutionary similarity.
3.6.2. Immune Regulatory Factors
Regulatory factors in insect plasma include serine proteases, their non-catalytic homologs (serine protease homologs, SPH), and serine protease inhibitors (serpins). Serine proteases (SPs), one of the largest protein families in insects, amplify invading immune signals through proteolytic cascade reactions, especially those containing clip domains [20]. Serpins, as inhibitors of SPs, attenuate immune signals and provide feedback regulation. Members of a protein superfamily, serpins often form covalent complexes with SPs, blocking SP cascades to precisely regulate the prophenoloxidase cascade and the Toll pathway [21].
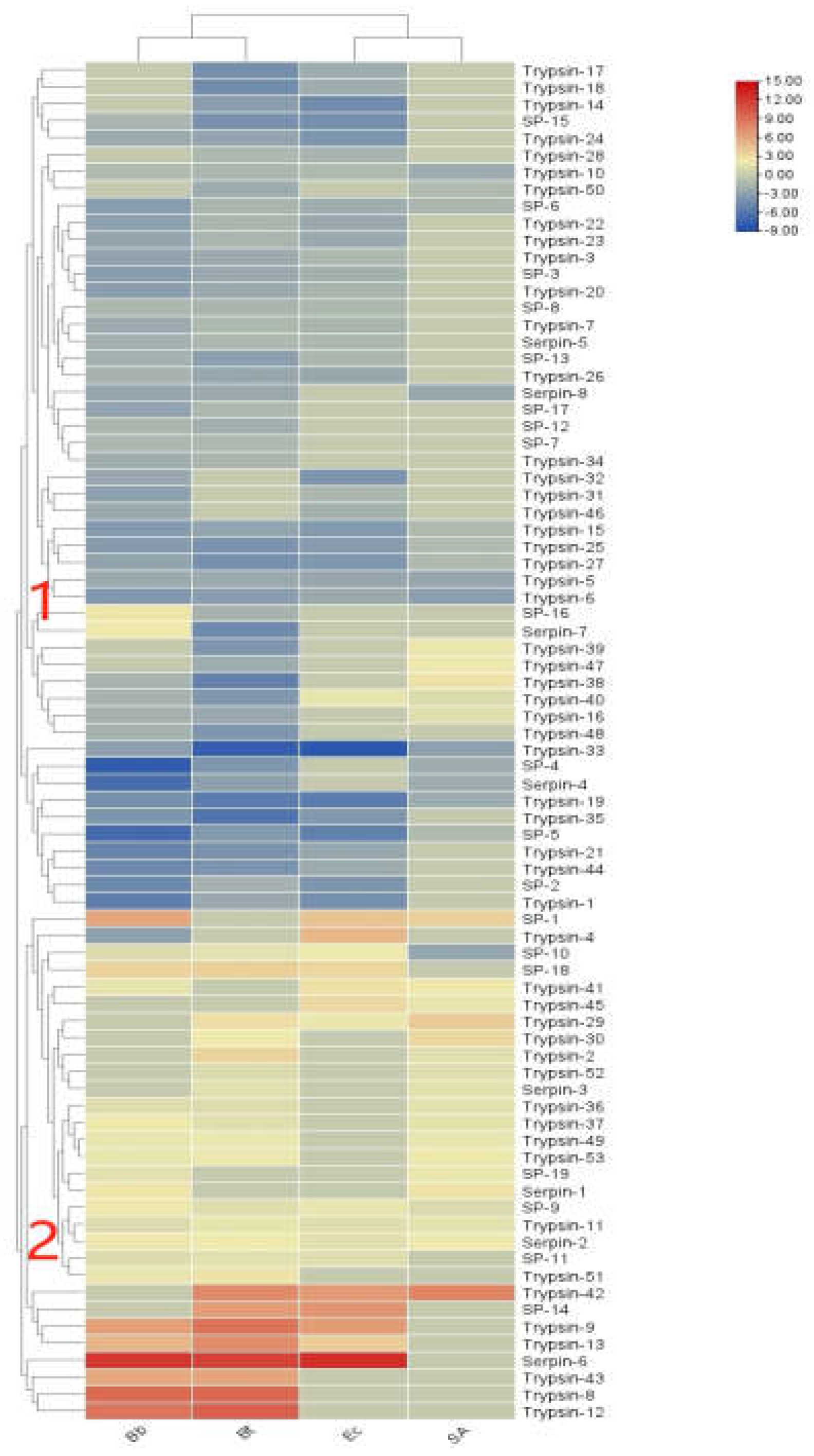
In the transcriptome of S. frugiperda, 61 serine protease (SP) genes and 20 serine protease inhibitor serpin genes were identified (see Appendix 1). As shown in Figure 9, the expression levels of immune regulators in S. frugiperda exhibited varied transcriptional responses following infection by different pathogenic microorganisms. Overall, they can be divided into two clusters. Cluster 1 showed a general downregulation trend after infection with various pathogens, likely due to the involvement of serpin genes. In contrast, Cluster 2 exhibited a general upregulation trend, indicating that amplification of immune signaling in response to different pathogens requires the coordinated participation of multiple serine proteases, serine protease inhibitors, and trypsins, which collectively regulate immune signal transmission.
Figure 8.
Cluster heat map of differential expression of immunomodulatory factors in S. frugiperda infected by pathogenic microorganisms.
Figure 8.
Cluster heat map of differential expression of immunomodulatory factors in S. frugiperda infected by pathogenic microorganisms.
Phylogenetic tree analysis revealed that the serine proteases of S. frugiperda are closely related to those of other lepidopteran insects, including B. mori, Ostrinia furnacalis, and H. armigera. Among these, SfurSP-59 clustered with HarmSP-6. Previous research by Xiong et al. demonstrated that cSP6 in H. armigera is a key activator of prophenoloxidase, suggesting that SfurSP-59 may have a similar function and play a role in the activation of melanization [21].
The serpins of S. frugiperda also showed close phylogenetic relationships with those of lepidopteran and hymenopteran insects. Among the identified serpin genes, seven (SPN genes) possess complete domains and signal peptide sequences, indicating their extracellular functionality. Studies have shown that MsSerpin-6 in Manduca sexta can block upstream signals in the prophenoloxidase cascade [22].
In the phylogenetic tree, Sfruserpin-3 clustered with MsSerpin-6, suggesting that Sfruserpin-3 may have a similar function. This indicates that Sfruserpin-3 could participate in the precise regulation of melanization and the Toll signaling pathway in S. frugiperda.
3.6.3. Signal Transduction Factors
The innate immune signaling pathways in the model insect Drosophila melanogaster include four primary pathways: Toll, Imd, JAK/STAT, and JNK4. Among these, the Toll and Imd pathways have been gradually confirmed in many lepidopteran insects [23,24]. The Toll signaling pathway in insects is evolutionarily highly conserved and primarily defends against fungi and Gram-positive bacteria. In S. frugiperda, the Toll signaling pathway is relatively complete, encompassing genes encoding the extracellular cytokine Spätzle, transmembrane receptors such as Toll proteins, tolloid-like proteins, and toll-like receptors, as well as intracellular signaling components Tube, myeloid differentiation factor 88, Pelle kinase, the inhibitor molecule Cactus, Cactin, Pellino, and the NF-κB transcription factor dorSAl.
A total of 20 Toll genes were identified in S. frugiperda. As shown in Figure 12(a), these Toll genes cluster closely with those of other lepidopteran insects, such as B. mori and H. armigera. The red-highlighted region of the phylogenetic tree shows that all Toll sequences of S. frugiperda form a unique homolog cluster, suggesting that these genes underwent a certain degree of variation after species differentiation.
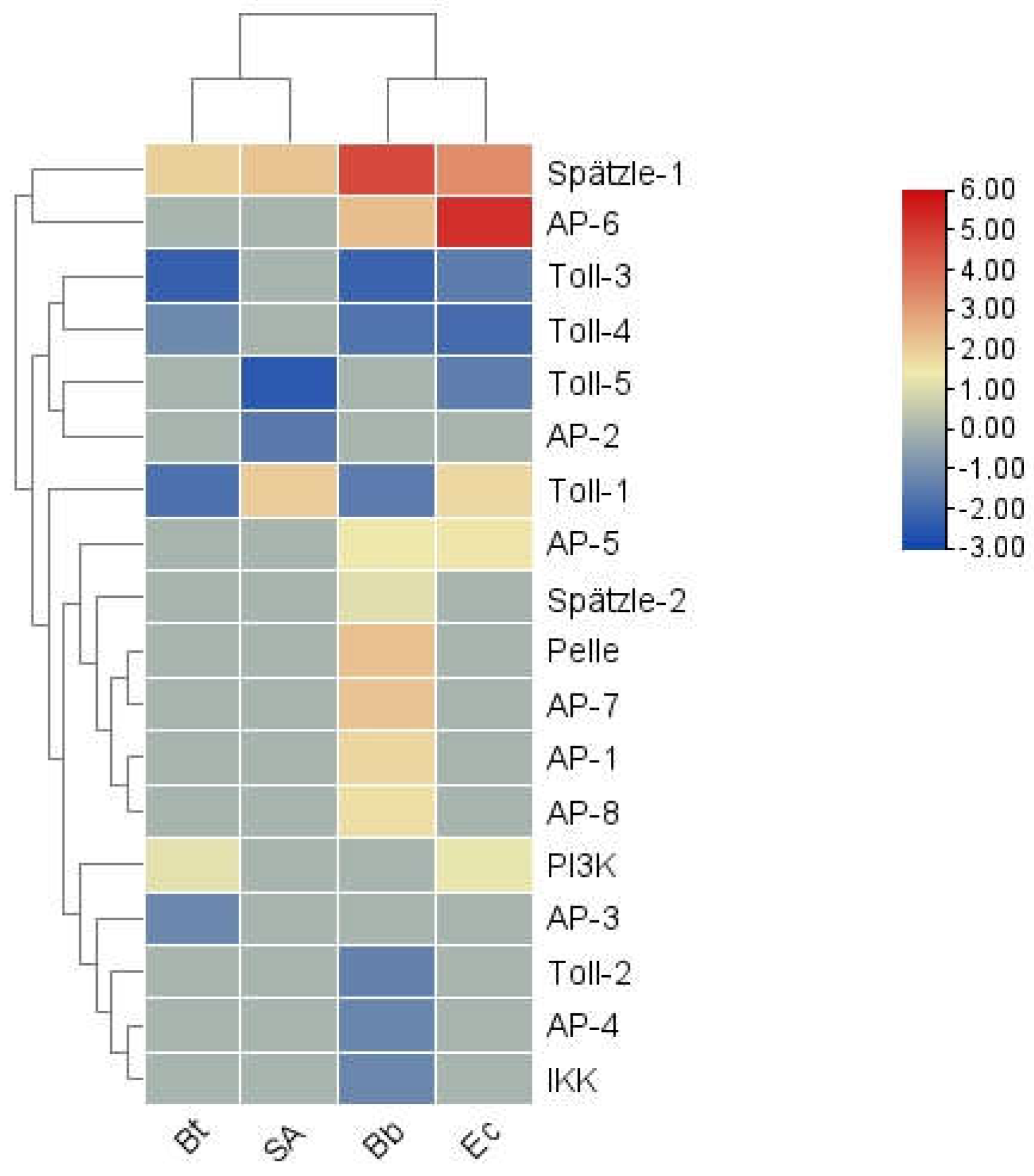
Figure 11.
Cluster heat map of differential expression of signal transduction factors in S. frugiperda infected by pathogenic microorganisms.
Figure 11.
Cluster heat map of differential expression of signal transduction factors in S. frugiperda infected by pathogenic microorganisms.
Toll receptors in insects do not directly detect foreign substances; instead, they function as receptors for the cytokine Spätzle. In S. frugiperda, five Spz genes were identified. Studies have shown that activating Bmorspz1 in B. mori increases the mRNA levels of antimicrobial peptides [25], and BmorSpz4 can activate intracellular Toll signaling to enhance the host's immunity against external infections [26]. Given the homology between SfruSpz2 in S. frugiperda and BmorSpz4 in B. mori, it is hypothesized that SfruSpz2 may act as a cytokine binding to Toll receptors in S. frugiperda, regulating Toll pathway activity and influencing the host's immune response.
The IMD pathway is a critical pathway for defending against Gram-negative bacteria. In S. frugiperda, key regulatory genes identified in this pathway include 1 TAK1, 2 IKKs, 1 Sickie, 1 Akirin, and 6 Cullins (see Appendix 1).
The JNK and JAK-STAT signaling pathways are also essential immune defense mechanisms in insects. In the transcriptome data of S. frugiperda, the core genes of the JAK-STAT pathway were identified, including those encoding JAK kinase (Hopscotch) and STAT factors, as well as negative regulatory genes SOCS and PIAS.
In terms of expression levels, the signal transduction factor genes were generally downregulated, with the exception of Spätzle-1 and AP6, which exhibited upregulated trends under certain pathogenic infections. Overall, the four innate immune signaling pathways are interconnected, working in concert to transduce danger signals and ultimately stimulate the production of immune effector molecules.
3.6.4. Immune Effector Factors
Microbial induction can lead to the production of numerous effectors, which are small molecular weight proteins. In the transcriptome of S. frugiperda, a total of 264 effector factor genes were identified (see Appendix 1). Common effectors include antimicrobial peptides (AMPs), melanin mediated by prophenoloxidase (PPO), lysozymes (Lys), and reactive oxygen species (ROS) [27,28].
In S. frugiperda, five types of AMP genes were identified: 2 unigenes of drosomycin, 5 of attacin, 2 of defensin, 1 of holotricin, 1 of cecropin, and 2 of anionic antimicrobial peptides. Additionally, the transcriptome revealed 7 unigenes of lysozyme, 11 of chitinase, 18 of heat shock proteins (HSP), and 39 of actin.
Based on expression patterns, immune effector factors were categorized into three clusters:Cluster 1 and Cluster 2 showed an overall downregulation trend following infection by various pathogenic microorganisms, with Cluster 2 displaying high expression levels, suggesting a role as negative regulatory components.Cluster 3 exhibited overall high expression levels, functioning as positive regulatory components.
As shown in Figure 14, S. frugiperda contains four gloverin genes, which are antimicrobial peptides specific to lepidopteran species. In the phylogenetic tree, these genes cluster with those of H. armigera. Most lysozyme genes also cluster with those of H. armigera, indicating a close evolutionary relationship with other lepidopteran species. In contrast, attacin forms a distinct clade in the phylogenetic tree, suggesting considerable evolutionary divergence of antimicrobial peptides among different insect species.
3.7. Effects of Pathogenic Microbial Infection on the Expression of PGRP and βGRP Immune Genes in S. frugiperda
The studies above demonstrate that during pathogenic microbial infections, various immune genes in S. frugiperda are activated to combat different types of pathogens. To further validate these findings, the immune gene familie peptidoglycan recognition proteins (PGRPs) and β-glucan recognition proteins (βGRPs), which play critical roles in initiating innate immunity, were selected for validation analysis.
Infections by three types of bacteria induced varying degrees of expression in both long-form (L) and short-form (S) PGRP genes in the fourth instar larvae of S. frugiperda. Among the long-form genes, the expression levels of PGRP-LE2, LB1, L1, L2, and L3 decreased in the order of Ec > Bt > SA, with Ec treatment inducing expression levels 6.88, 60.36, 529.78, 5.04, and 4.57 times higher than the control (CK), respectively. The expression of PGRP-LE2 under SA treatment was significantly lower than the control. PGRP-LB and LB2 exhibited the highest expression levels under Bt treatment, with levels 9.55 and 43.22 times higher than the control, respectively. However, PGRP-LB showed no significant differences in expression between Bt, SA, and the control.
For short-form genes, PGRP-S1 and S2 exhibited the highest expression levels under Bt treatment. Notably, the expression of PGRP-S1 under Bt treatment was significantly higher than the control, reaching 2700.55 times the control level. Conversely, the expression of PGRP-S2 under Ec treatment was significantly lower than the control.
As pattern recognition receptors, some PGRPs recognize and bind to external pathogens, subsequently activating the Toll and Imd pathways. Lysine-type PGN primarily activates the Toll pathway, while DAP-type PGN activates the Imd pathway [29].
These findings suggest that PGRP-LE2, LB1, L1, L2, and L3 genes may be involved in the activation of the Imd pathway by Gram-positive bacteria such as S. aureus and B. thuringiensis. Conversely, PGRP-S1 and S2 genes may activate the Toll pathway in response to Gram-negative bacteria such as E. coli.
As shown in Figure 16, induction by the fungus B. bassiana did not result in a significant difference in the expression of the pattern recognition receptor gene βGRP-3 compared to the control. However, the expression levels of βGRP-1 and βGRP-2 increased significantly, reaching 32.48 and 274.52 times the levels of the control group, respectively. These findings suggest that βGRP-1 and βGRP-2 are critical binding proteins in S. frugiperda in response to induction by B. bassiana.
4. Discussion
With the continuous advancements in next-generation sequencing, insect immunogenomics research has garnered increasing attention, expanding its focus beyond model insects such as Drosophila and Bombyx mori. In recent years, innate immunity in insects has become a research hotspot. The components of the innate immune systems of many insects have been gradually elucidated. For example, in 2013, the immune system of the tobacco hornworm Manduca sexta was characterized, identifying 232 immune-related genes [30]. In 2014, Liu et al. identified 190 immune-related genes in the Asian corn borer Ostrinia furnacalis [29]. In 2015, the immune systems of the agricultural pests Helicoverpa armigera and Plutella xylostella were analyzed, with 233 and 149 immune genes identified, respectively [21,31]. In 2018, the immune system of the invasive pest Dendroctonus valens was revealed by Xu Letian's research group at Hubei University, identifying 185 immune-related genes [32]. As a globally significant migratory agricultural pest under surveillance, the fall armyworm (Spodoptera frugiperda) remains poorly understood in terms of its innate immune system. However, the widespread use of Bacillus thuringiensis (Bt)-based biopesticides in pest control poses a high risk of resistance development. By targeting the host immune response to pathogens and reducing its innate immunity, novel green and effective pest management strategies could be developed.
To this end, this study employed transcriptome sequencing to comprehensively analyze the composition and dynamic changes of immune genes in fall armyworm larvae under different bacterial and fungal infections. A total of 655 immune-related genes were identified, including four major categories: pattern recognition receptors, immune effectors, signal transduction factors, and immune regulators.
This study analyzed these four categories of immune factors in fall armyworm, showing that the immune gene repertoire of this species is relatively conserved, possessing a complete set of components from pathogen recognition to the production of effectors, without significant loss. This suggests that S. frugiperda has a well-developed innate immune defense system. However, some insects exhibit component loss during evolution, such as the absence of the JAK/STAT pathway ligand hopscotch protein in the genome of B. mori [17] and the absence of the Toll pathway ligand MyD88 in the transcriptome of P. xylostella [31]. The systematic identification and analysis of immune gene families in S. frugiperda not only provide insights into their evolutionary history but also establish a theoretical foundation for further functional analysis of these immune genes.
Among pattern recognition receptors, different genes exhibited varied trends in response to pathogens, suggesting their involvement in distinct pathways for combating microbial infections. Phylogenetic analysis and expression validation revealed that S. frugiperda PGRP-L2 clustered with Drosophila PGRP-LD, which is associated with maintaining gut microbial homeostasis [29]. Previous studies have shown that BmPGRP-S1 and HaPGRP-A can activate the phenoloxidase cascade, while DmPGRP-LC mediates the regulation of the Drosophila Imd pathway and initiates phagocytosis and Imd signaling by recognizing Gram-negative bacteria [33,34,35]. It is hypothesized that S. frugiperda PGRP-L1 and PGRP-S1 may share similar functions. In the transcriptome database, three βGRP genes were identified in S. frugiperda. Studies have shown that B. mori βGRP1 can bind bacterial cell wall polysaccharides and β-1,3-glucan, activating PO-mediated melanization [36]. It is inferred that S. frugiperda βGRP2, which shares conserved glucan-binding domains at its N-terminus with B. mori βGRP1, might serve as a key receptor for bacterial detection and activation of downstream immune signaling pathways.
Immune regulators in S. frugiperda mainly include serine proteases and serine protease inhibitors, whose expression levels varied in response to pathogenic infections but function cooperatively in immune signaling. Previous studies found high similarity between H. armigera cSP6 and M. sexta PAP3 [19], the latter being critical for PPO activation. It is hypothesized that S. frugiperda SP-59 has similar functions in melanization activation. Serine protease inhibitors provide feedback regulation to attenuate immune signaling. Phylogenetic analysis indicated that S. frugiperda Serpin-3 might share similar functions with M. sexta Serpin-6, regulating melanization and Toll signaling pathways [21]. Signal transduction factors were mainly identified in the Toll, Imd, JAK/STAT, and JNK pathways. Toll is the primary signaling pathway in insects for defense against fungi and Gram-positive bacteria. Over evolutionary time, it has formed unique homologs, with Spätzle cytokines in S. frugiperda modulating Toll pathway activity to influence host immunity.
Most immune effectors showed increased expression following pathogen infection. Phylogenetic analysis of gloverin, an antimicrobial peptide unique to Lepidoptera, showed that S. frugiperda gloverin clustered with H. armigera, indicating functional similarity. Attacin may have undergone significant variation during evolution, while lysozyme shares close evolutionary relationships within Lepidoptera. These adaptations likely enhance host resilience in complex and dynamic environments.
As two major classes of pattern recognition receptors initiating immune responses, expression analysis showed significant upregulation of PGRP-S1 and S2 following infection with Staphylococcus aureus and B. thuringiensis, while infections with Escherichia coli led to increased expression of PGRP-LE2, LB1, L1, L2, and L3. These findings suggest that PGRP genes are involved in pathway regulation and activation. Based on structural analysis, it is inferred that PGRP-S1 and S2 are likely involved in Toll pathway activation, while PGRP-LE2, LB1, L1, L2, and L3 participate in Imd pathway activation [37,38,39]. Additionally, infection with Beauveria bassiana significantly upregulated βGRP-1 and βGRP-2 expression, indicating their role in activating PO-mediated melanization [19].
This study has clarified the immune transcriptome of S. frugiperda against different microbial pathogens. Future work will focus on identifying highly responsive and differentially expressed PGRP and βGRP genes during pathogen infections. These genes could serve as targets for developing novel insecticides, providing a valuable reference for enhancing the efficacy of existing microbial agents and designing new bio-insecticides with specific molecular targets.
Author Contributions
Conceptualization: ZX W, F H; Methodology: ZX W, Y T, Q Z; Data curation: ZX W; Formal analysis: GJ Y, F L; Investigation: Y T, Y W, WS W; Project administration: ZX W; Resources; Supervision: F H;ZX W; Validation: ZX W; Visualization: XY Z, XC L; Writing—original draft: Y T, Q Z; Funding acquisition: ZX W, F H; Writing—review and editing: ZX W,F H.
Funding
This research was partially supported by the following grants: Key Projects of Natural Science Research project for Anhui Universities (2022AH051654). National Key Research and Development Program(2021YFD1400701)
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on reasonable request from the corresponding author.
Acknowledgments
We would like to thank Fei Hu for providing Beauveria bassiana.
Conflicts of Interest
The authors declare no conflicts of interest.
Appendix 1. Summary of main immune genes of S. frugiperda.
| Gene Functions | Gene | Encoded Protein | Protein Function | Number | |
|
Pattern Recognition Receptors |
Ubiquitin-conjugating enzyme | Ubiquitin-conjugating enzyme | Immune recognition | 20 | |
| DSCAM | Down syndrome cell adhesion molecule | Immune recognition,phagocytosis | 11 | ||
| βGRP | β-1,3-glucan recognition protein | Immune recognition | 3 | ||
| PGRP | Peptidoglycan recognition protein | Immune recognition | 9 | ||
| C-type lectin | C-type lectin | Immune recognition, phagocytosis, melanization | 2 | ||
| TEP | Thioester-containing protein | Immune recognition, phagocytosis | 1 | ||
| Galectin | Galectin | Immune recognition, phagocytosis | 8 | ||
| ApoLp | Apolipoprotein family member | Immune recognition, apoptosis, phagocytosis | 2 | ||
| SR | Scavenger receptor | Immune recognition, phagocytosis | 9 | ||
| Croquemort | Scavenger receptor | Immune recognition | 1 | ||
| Integrin | Integrin | Immune recognition, encapsulation, phagocytosis | 27 | ||
| Vitellogenin | Vitellogenin | Immune recognition, phagocytosis | 5 | ||
|
Signal Transduction and Regulation |
Regulators |
SP | Serine protease | Activates Toll signaling pathway, melanization | 61 |
| Serpin | Serine protease inhibitor | Regulates Toll signaling pathway, melanization | 20 | ||
| Trypsin | Trypsin | Melanization, activates Toll signaling pathway | 107 | ||
| Elastase | Elastase | Melanization, activates Toll signaling pathway | 5 | ||
|
Toll Signaling Pathway |
Spätzle | Spätzle-like protein | Activates Toll signaling pathway | 5 | |
| Toll | Toll protein | Activates Toll signaling pathway | 20 | ||
| MyD88 | Cytoplasmic protein | Activates NF-κB transcription factor | 1 | ||
| Pelle | Cytoplasmic protein | Activates NF-κB transcription factor | 1 | ||
| Cactus | Inhibitor | Synthesis of antimicrobial peptides | 1 | ||
| DorSAl | NF-κB transcription factor | Synthesis of antimicrobial peptides | 2 | ||
| Cactin | Regulator | Regulates Toll signaling pathway | 1 | ||
| Pellino | Regulator | Regulates Toll signaling pathway | 1 | ||
|
IMD Signaling Pathway |
TAK1 | TGF-βactivated kinase | Activates IMD and JNK signaling pathways | 1 | |
| IKK | Protein kinase | Activates NF-κB transcription factor | 2 | ||
| Sickie | Inhibitor | Regulates IMD signaling pathway | 1 | ||
| Akirin | Nuclear protein | Immune modulation | 1 | ||
| Cullin | SCF complex subunit | Regulates IMD signaling pathway | 6 | ||
|
JAK-STAT Signaling Pathway |
SOCS | Suppressor of cytokine signaling | Regulates JAK-STAT signaling pathway | 3 | |
| Hop | JAK tyrosine kinase signaling molecule | Activates JAK-STAT signaling pathway | 1 | ||
| PIAS | Protein inhibitors of activated STATs | Synthesis of antimicrobial peptides | 1 | ||
| STAT | Signal transducer and activator of transcription | Synthesis of antimicrobial peptides | 2 | ||
|
MAPK-JNK -p38 Signaling Pathway |
activating protein | Transcription factor complex | Switch for initiating gene transcription | 39 | |
| MKK4 | MAPKK4 kinase | Activates JNK pathway | 1 | ||
| Eiger | Receptor protein | Activates JNK pathway | 1 | ||
| Wengen | Receptor protein | Activates JNK pathway | 1 | ||
| RNA Interference |
Vig | Argonaut homolog gene | Forms RISC complex | 1 | |
| Ago-2 | RNA-binding protein | Participates in RNA interference, antiviral response | 3 | ||
|
Autophagy |
PI3K | Phosphoinositide 3-kinase | Autophagy signal transduction | 2 | |
| TOR | Target of rapamycin | Regulates autophagy | 1 | ||
| Dronc | Caspase | Initiates apoptosis | 1 | ||
|
Immune Effectors |
Lysozyme | Lysozyme | Degrades peptidoglycan, antimicrobial | 7 | |
| Attacin | Antimicrobial peptide | Pathogen inhibition | 5 | ||
| Defense | Defensin | Pathogen inhibition | 2 | ||
| Cecropin | Cecropin | Pathogen inhibition | 1 | ||
| Holotricin | Antimicrobial peptide | Pathogen inhibition | 1 | ||
| Anionic antimicrobial peptide | Anionic antimicrobial peptide | Pathogen inhibition | 2 | ||
| Chitinase | Chitinase | Degrades chitin, antimicrobial | 11 | ||
| Heat Shock Protein | Heat Shock Protein | Stress response, antiviral reaction | 18 | ||
| Actin | Actin | Stress response, antiviral reaction | 39 | ||
| phenoloxidase | Phenoloxidase | Melanization | 15 | ||
| Hexamerin | Hexamerin | Melanization | 3 | ||
| Arylphorin | Arylphorin | Melanization | 2 | ||
| Nitric Oxide Synthase | Nitric Oxide Synthase | Generates free radicals | 4 | ||
| NADPH oxidase | NADPH Oxidase | Generates free radicals | 2 | ||
| Peroxidase | Peroxidase | Generates free radicals | 18 | ||
| Glutathione S-transferase | Glutathione S-transferase | Regulates free radicals | 24 | ||
| SOD | Superoxide Dismutase (SOD) | Regulates free radicals | 6 | ||
| Catalase | Catalase | Regulates free radicals | 20 | ||
| Thioredoxin | Thioredoxin | Regulates free radicals | 17 | ||
| gloverin | Gloverin | Inhibits pathogens | 2 | ||
| lebocin | Antimicrobial Peptide | Inhibits pathogens | 2 | ||
| cadherin | Cadherin | Signal transmission | 15 | ||
| Lysine | Lysozyme | Degrades peptidoglycan, antimicrobial | 48 | ||
References
- Wang, L.; Chen, K.H.; Zhong, G.H. Progress for occurrence and management and the strategy of the fall armyworm Spodoptera frugiperda (Smith). Journal of Environmental Entomology,2019, 41(3): 479-487.
- Rebeca G. M., David M. S., Blanco C. A. Field-Evolved Resistance of the Fall Armyworm (Lepidoptera: Noctuidae) to Synthetic Insecticides in Puerto Rico and Mexico. Journal of Economic Entomology, 2019(2): 792-802.
- Hu, F.; Xu, T.T.; Su, X.Y. Control Efficacy of Bacillus thuringiensis Tiny Microgranules on Maize Lepidopteran Pests. Chinese Journal of Biological Control, 2022, Doi: 10.16409/j.cnki.2095-039x.
- Xu, Y.D.; Wei, H.S.; Shi, J.W. Comparison of virulence of three Beauveria bassiana strains against fall armyworm Spodoptera frugiperda. Journal of Plant Protection, 2020, 47(4): 867-874.
- Hu, F.; Xu, T.T.; Hu, B.J. Control Efficacy of Biopesticide Bacillus thuringiensis G033A Combined with Reduced Low Dose Chemical Pesticides on Spodoptera frugiperda. Chinese Journal of Biological Control,2021, 37(6): 1103-1110.
- Alaka, S.; Shasank, S.S.; Behera, A. Antimicrobial Peptides Derived from Insects Offer a Novel Therapeutic Option to Combat Biofilm: A Review[J]. Frontiers in Microbiology, 2021, 12.
- Yi, H.Y.; Chowdhury, M.; Huang, Y.D. Insect antimicrobial peptides and their applications. Applied Microbiology and Biotechnology, 2014, 98(13): 5807-5822.
- Bai, Y.Y.; Sun, J.Q. Immunological and stress responses of fall armyworm Spodoptera frugiperda larvae to injecting Escherichia coli. Journal of Plant Protection,2020, 47(4): 859-866.
- Li, E.T.; Lu, Q.H.; Zhang, D.F. Effects of infection of the entomopathogenic nematode Steinernema carpocapsae All on the innate immune response in Spodoptera frugiperda (Lepidoptera: Noctuidae) larvae. Acta Entomologica Sinica, 2022, 65(12):1623-1635. DOI: 10.16380/j.kcxb.2022.12.008.
- Sun, J.Q.; Bai, Y.Y. Predator-induced stress influences fall army-worm immune response to inoculating bacteria. Journal of Inver-tebrate Pathology, 2020, 172: 107352.
- Shu, B.S.; Huang, Y.T.; Yu, X.Y. Expression Stability of Reference Genes in Larvae of Spodoptera frugipferda Under Azadirachtin Stress by Real-Time Quantitative PCR Analysis. Guangdong Agricultural Sciences, 2024, 51(8): 21-30..
- Wu, T.Y.; Zhao, Y.; Wang, Z.Y.; Song, Q.S.; Wang, Z.X.; Xu, Q.W.; Wang, Y.; Wang, L.; Zhang, Y.; Feng, C. β-1,3-glucan recognition protein 3 activates the prophenoloxidase system in response to bacterial infection in Ostrinia furnacalis guenée. Dev. Comp. Immunol. 2018, 79,31-43..
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR. Methods 2002, 25,402–408.
- Dziarski, R.; Gupta, D. The peptidoglycan recognition proteins (PGRPs) [J]. Genome Biology, 2006, 7(8): 232.
- Werner, T.; Liu, G.; Kang, D. A family of peptidoglycan recognition proteins in the fruit fly Drosophila melanogaster. Proceedings of the National Academy of Sciences of the United StatesofAmerica, 2000,97(25): 13772-13777.
- Lemaitre, B.; Hoffmann, J. The host defense of Drosophila Melanogaster. Annual Review of Immunology, 2007,25: 697-743.
- Tanaka, H.; Ishibashi, J.; Fujita, K. A genome-wide analysis of genes and gene families involved in innate immunity of Bombyx mori [J]. Insect Biochem Mol Biol, 2008, 38(12): 1087-110.
- Meng, Q.; Yu, H.Y.; Zhang, H. Transcriptomic insight into the immune defenses in the ghost moth, Hepialus xiaojinensis, during an Ophiocordyceps sinensis fungal infection [J]. Insect Biochem Molec, 2015, 64: 1-15.
- Ochiai, M.; Ashida, M. A pattern-recognition protein for beta-1,3-glucan. The binding domain and the cDNA cloning of beta-1,3-glucan recognition protein from the silkworm, Bombyx mori [J]. J Biol Chem, 2000, 275(7): 4995-5002.
- Christophides, G.K.; Zdobnov, E.; Barillas-Mury, C. Immunity-related genes and gene families in Anopheles gambiae [J]. Science, 2002, 298(5591): 159-65.
- Xiong, G. H.; Xing, L.S.; Lin, Z. High throughput profiling of the cotton bollworm Helicoverpa armigera immunotranscriptome during the fungal and bacterial infections [J]. BMC Genomics, 2015, 16(1): 321.
- Zou, Z.; Jiang, H. Gene structure and expression profile of Manduca sexta prophenoloxidase-activating proteinase-3 (PAP-3), an immune protein containing two clip domains. Insect Mol Biol. 2005 Aug;14(4): 433-42.
- Meng, Q.; Yu, H.Y.; Zhang, H. Transcriptomic insight into the immune defenses in the ghost moth, Hepialus xiaojinensis, during an Ophiocordyceps sinensis fungal infection [J]. Insect Biochem Molec, 2015, 64:1-15.
- Liu, Y.; Shen, D.; Zhou, F. Identification of immunity-related genes in Ostrinia furnacalis against entomopathogenic fungi by RNA-seq analysis [J]. PLoS One, 2014, 9(1): e86436.
- Wang, Y.; Cheng, T.; Rayaprolu, S.; Zou, Z.; Xia, Q.; Xiang, Z.; Jiang, H. Proteolytic activation of pro-spätzle is required for the induced transcription of antimicrobial peptide genes in lepidopteran insects. Dev. Comp. Immunol, 2007, 31, 1002–1012.
- Huang, X.Y. Discovery of a new splicing isomer of silkworm BmSpatzle4 and its immune response to different microorganisms in the body wall [D]. Jiangsu University of Science and Technology, 2019.
- Lu, D.; Geng, D.; Hou, C.; Huang, Y.; Qin, G.; Guo, X. Bombyx mori cecropin A has a high antifungal activity to entomopathogenic fungus Beauveria bassiana [J]. Gene, 2016, 583(1): 29-35.
- Yi, H.; Chowdhury, M.; Huang, Y.; Yu, X. Insect antimicrobial peptides and their applications[J]. Applied Microbiology & Biotechnology, 2014, 98(13): 5807-5822.
- Lu, Y.; Su, F.; Li, Q.; Zhang, J.; Li, Y.; Tang, T.; Hu, Q.; Yu, X.Q. Pattern recognition receptors in Drosophila immune responses. Developmental and Comparative Immunology, 2020, 102: 103468.
- Gunaratna, R.T.; Jiang, H. A comprehensive analysis of the Manduca sexta immunotranscriptome [J]. Dev Comp Immunol, 2013, 39(4): 388-98.
- Xia, X.; Yu, L.; Xue, M. Genome-wide characterization and expression profiling of immune genes in the diamondback moth, Plutella xylostella (L.) [J]. Sci Rep, 2015, 5: 9877.
- Xu, L.; Zhang, Y.; Zhang, S. Comparative analysis of the immune system of an invasive bark beetle, Dendroctonus Bombyx mori, infected by an entomopathogenic fungus [J]. Dev Comp Immunol, 2018, 88: 65-9.
- Song, X.M.; Wang, M.F.; Dong, L. PGRP-LD mediates A. stephensi vector competency by regulating homeostasis of microbiota-induced peritrophic matrix synthesis [J]. Plos Pathogens, 2018, 14(2): e1006899.
- Yoshida, H.; Kinoshita, K.; Ashida, M. Purification of a peptidoglycan recognition protein from hemolymph of the silkworm, Bombyx mori [J]. J Biol Chem, 1996, 271(23): 13854-60.
- Chang, C.; Chelliah, Y.; Borek, D.; Mengin-Lecreulx, D.; Deisenhofer, J. Structure of tracheal cytotoxin in complex with a heterodimeric pattern-recognition receptor[J]. Science, 2006, 311(5768): 1761-1764.
- Choe, K.M.; Werner, T.; Stoven, S. Requirement for a peptidoglycan recognition protein (PGRP) in Relish activation and antibacterial immune responses in Drosophila [J]. Science, 2002, 296(5566): 359-62.
- Wang, X.H.; 2018. Mechanism of peptidoglycan recognition protein RfPGRP-S1 in the maintenance of intestinal flora homeostasis in red palm weevil [D]. Fujian Agriculture and Forestry University.
- Wang, X.H. PGRP-S1 Downregulates the Intestinal Immunity to Maintain the Homeostasis of Gut Microbiota in Rhynchophorus ferrugineus Olivier. Fujian Agriculture and Forestry University, 2018.
- Werner, T.; Borge-Renberg, K.; Mellroth, P.; Steiner, H.; Dan, H. Functional diversity of the Drosophila PGRP-LC gene cluster in the response to lipopolysaccharide and peptidoglycan. J. Biol. Chem. 2003, 278,26319–26322.
Figure 1.
Differentially expressed gene (DEG) volcano plot of S. frugiperda treated with pathogenic microorganisms.
Figure 1.
Differentially expressed gene (DEG) volcano plot of S. frugiperda treated with pathogenic microorganisms.
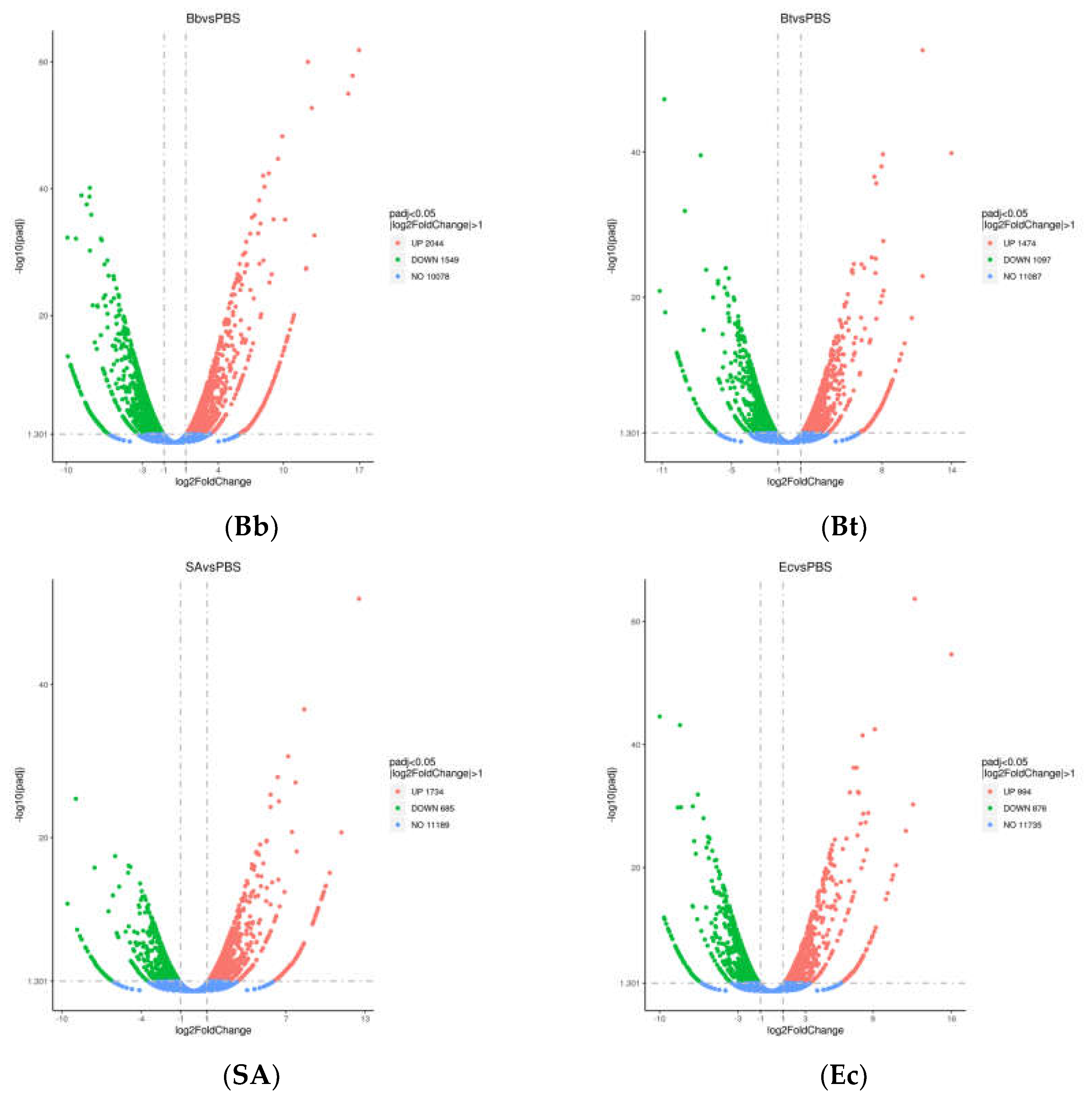
Figure 2.
Functional Classification of Unigenes in Telenomus remus Nixon.
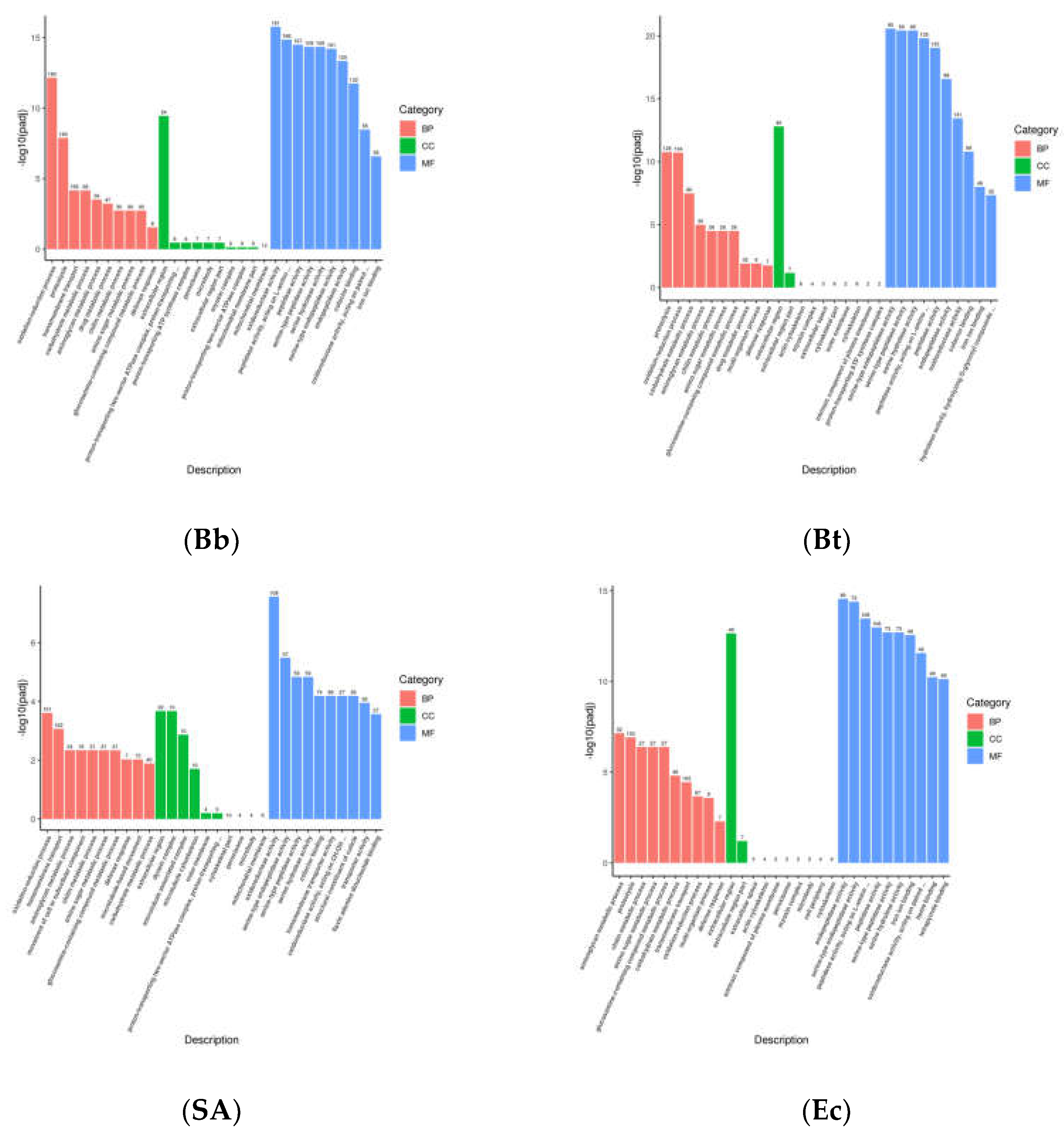
Figure 3.
KEGG pathway enrichment analysis of differentially expressed genes in S. frugiperda treated with pathogenic microorganisms.
Figure 3.
KEGG pathway enrichment analysis of differentially expressed genes in S. frugiperda treated with pathogenic microorganisms.
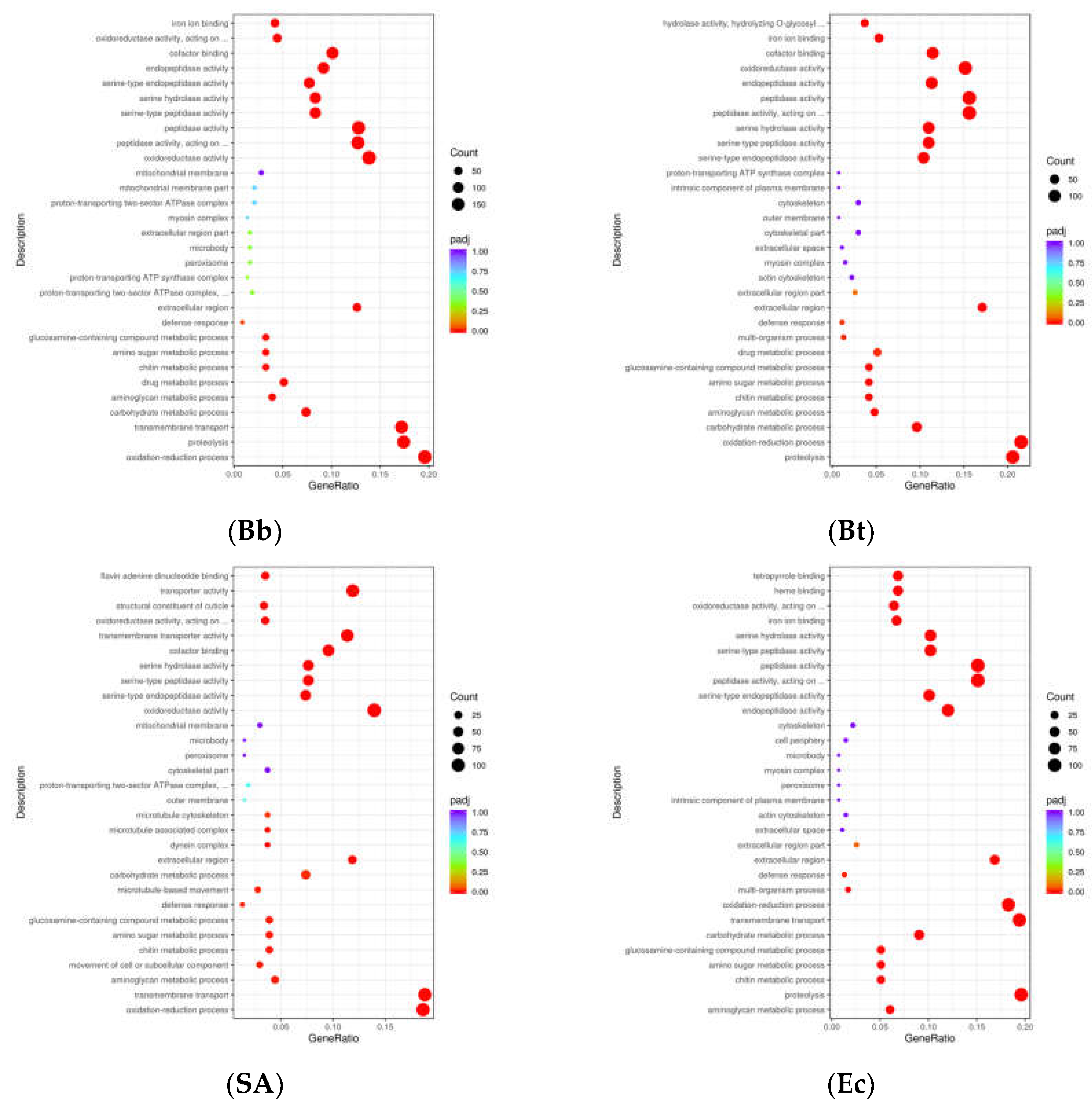
Figure 4.
Functional distribution of immune-related genes in S. frugiperda.
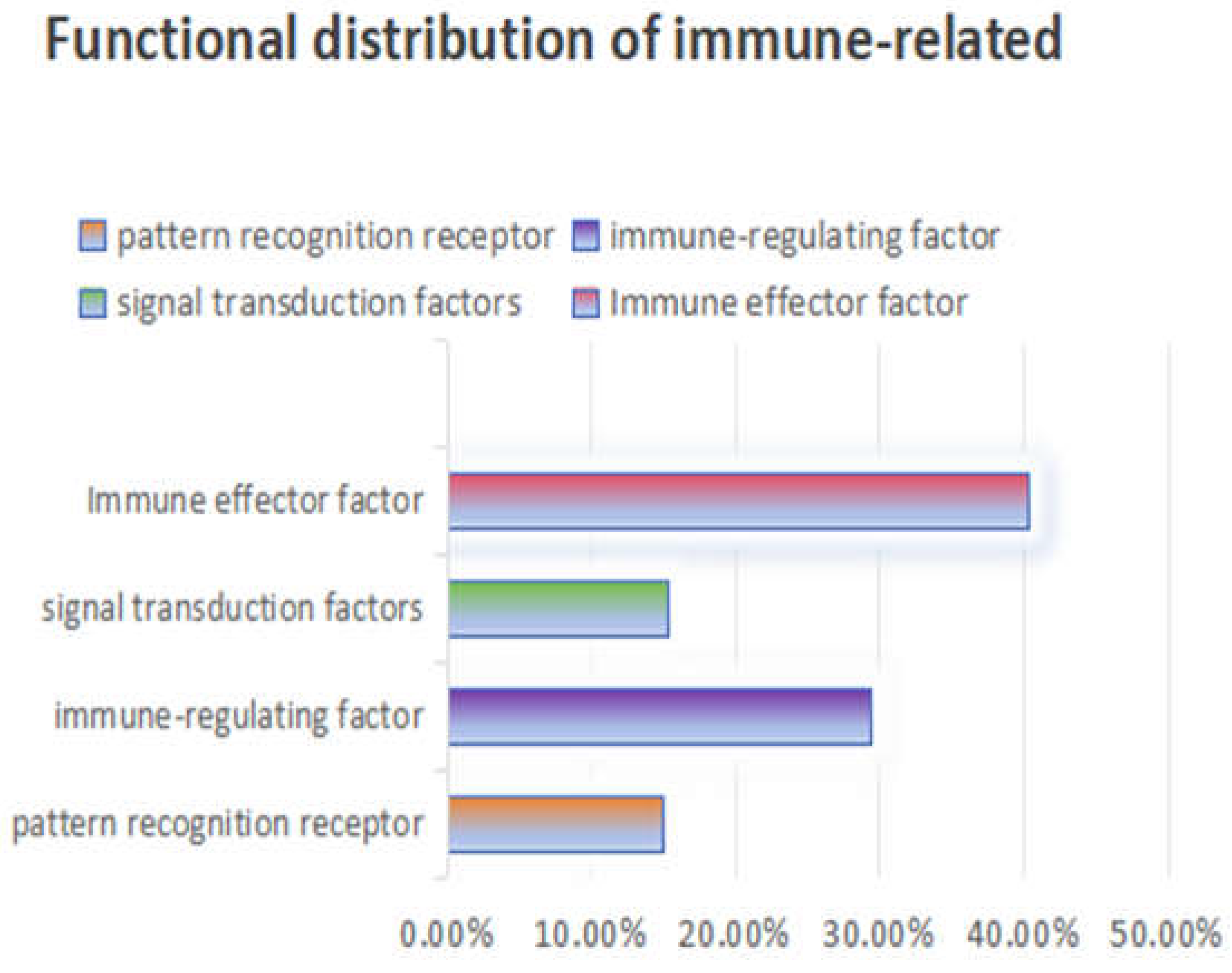
Figure 5.
Cluster heat map of differential expression of pattern recognition receptors in S. frugiperda infected by pathogenic microorganisms.
Figure 5.
Cluster heat map of differential expression of pattern recognition receptors in S. frugiperda infected by pathogenic microorganisms.
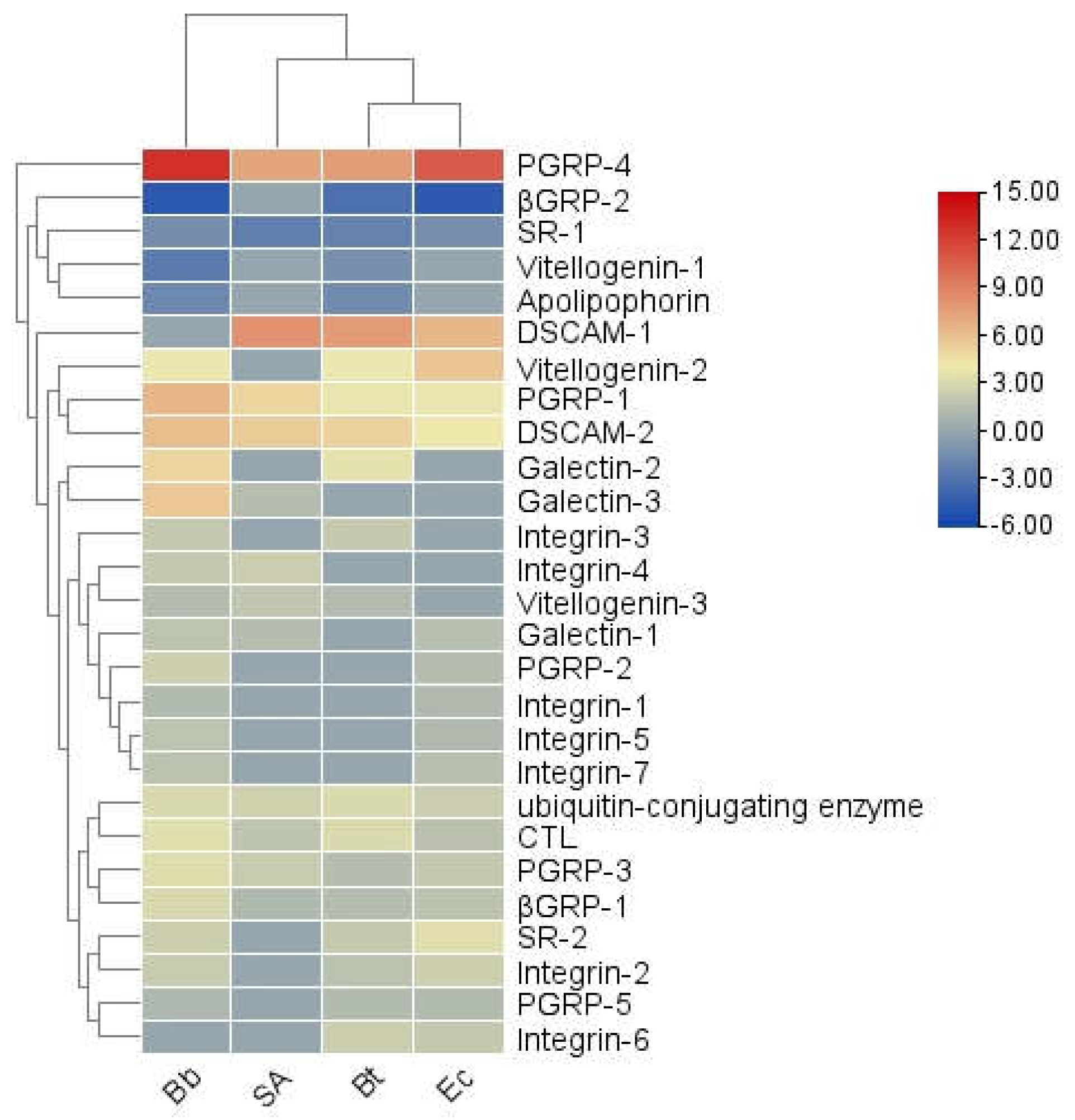
Figure 6.
(a) Sequence structure of PGRP protein, (b) Phylogenetic tree of PGRP family, in which the PGRP family of S.frugiperda was marked with red fonts. Note: Insects included in the phylogenetic tree Aedes aegypti; Drosophila melanogaster; Bombyx mori; Musca domestica; Helicoverpa armigera.
Figure 6.
(a) Sequence structure of PGRP protein, (b) Phylogenetic tree of PGRP family, in which the PGRP family of S.frugiperda was marked with red fonts. Note: Insects included in the phylogenetic tree Aedes aegypti; Drosophila melanogaster; Bombyx mori; Musca domestica; Helicoverpa armigera.
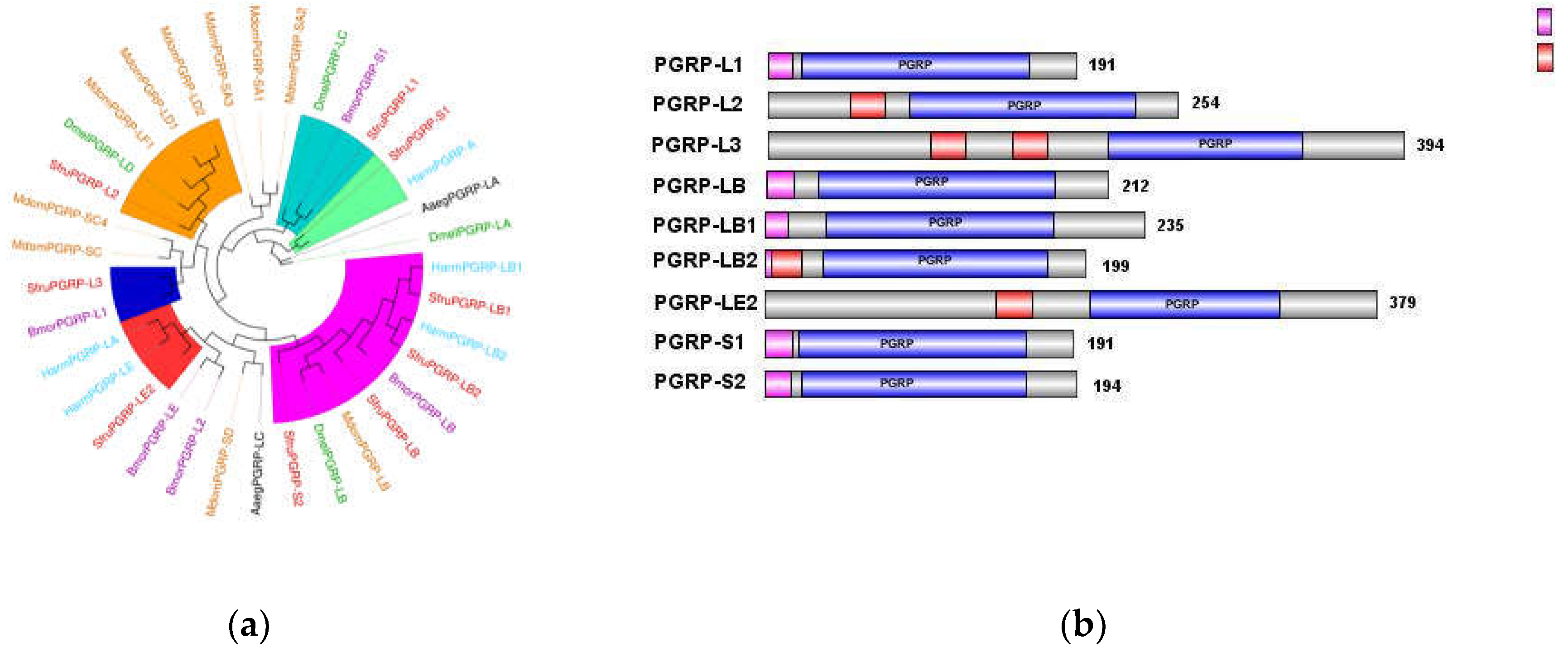
Figure 7.
(a) Sequence structure of βGRP protein, (b) Phylogenetic tree of βGRP family, in which the βGRP family of S. frugiperda was marked with red fonts. Note: Insects included in the phylogenetic tree (a) Aedes aegypti; Spodoptera litura; Helicoverpa armigera; Bombyx mori; Ostrinia furnacalis.
Figure 7.
(a) Sequence structure of βGRP protein, (b) Phylogenetic tree of βGRP family, in which the βGRP family of S. frugiperda was marked with red fonts. Note: Insects included in the phylogenetic tree (a) Aedes aegypti; Spodoptera litura; Helicoverpa armigera; Bombyx mori; Ostrinia furnacalis.

Figure 9.
SP family phylogenetic tree, in which the SP family of S. frugiperda is marked with red font. Note: Insects included in the phylogenetic tree Helicoverpa armigera; Bombyx mori; Musca domestica; Ostrinia furnacalis; Drosophila melanogaster.
Figure 9.
SP family phylogenetic tree, in which the SP family of S. frugiperda is marked with red font. Note: Insects included in the phylogenetic tree Helicoverpa armigera; Bombyx mori; Musca domestica; Ostrinia furnacalis; Drosophila melanogaster.
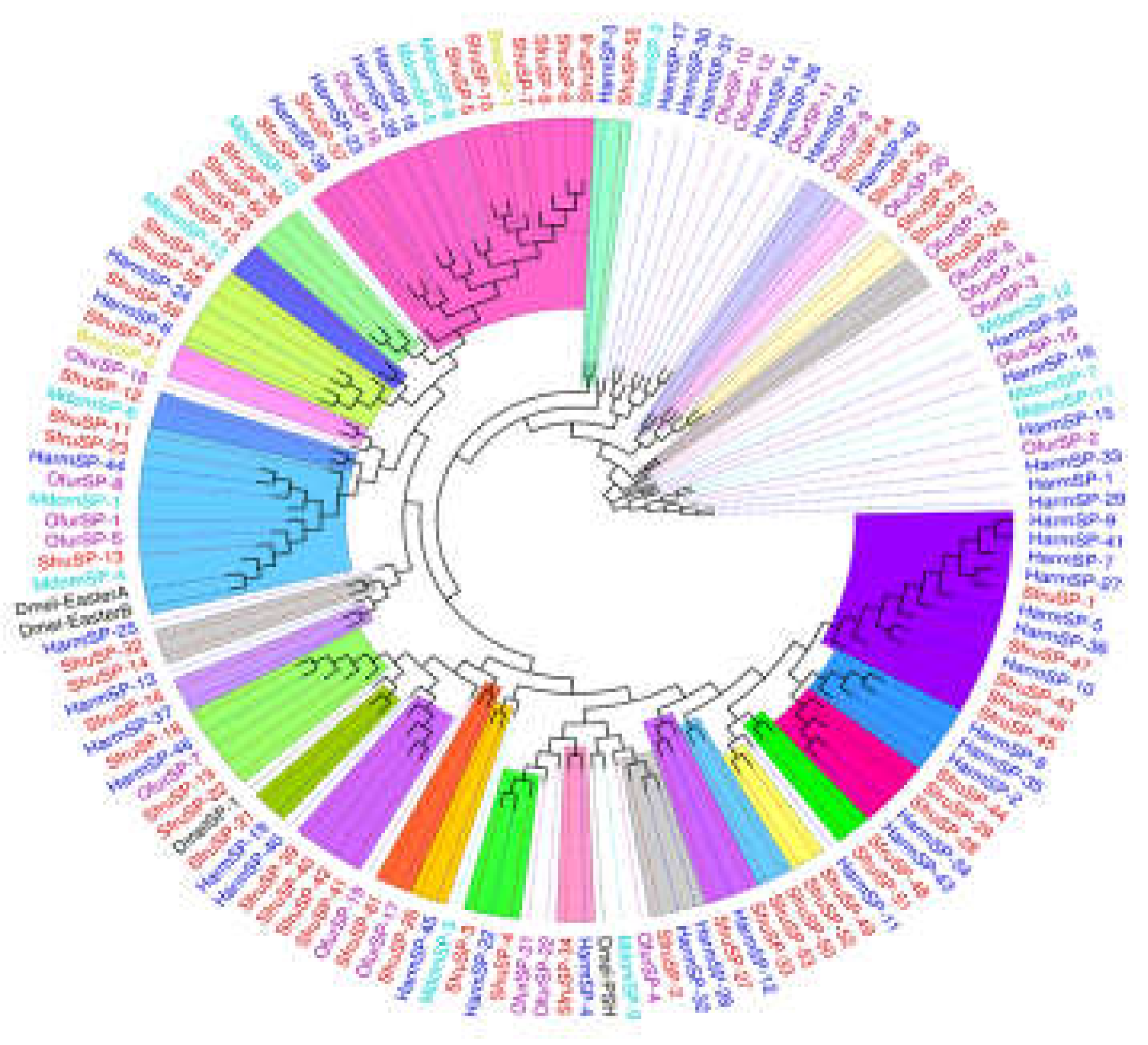
Figure 10.
(a) Sequence structure of Serpin protein, (b) Phylogenetic tree of Serpin family, in which the Serpin family of S. frugiperda was marked with red fonts. Note: Insects included in the phylogenetic tree Bombyx mori; Aedes aegypti; Spodoptera litura; Ostrinia furnacalis; Helicoverpa armigera; Manduca sexta.
Figure 10.
(a) Sequence structure of Serpin protein, (b) Phylogenetic tree of Serpin family, in which the Serpin family of S. frugiperda was marked with red fonts. Note: Insects included in the phylogenetic tree Bombyx mori; Aedes aegypti; Spodoptera litura; Ostrinia furnacalis; Helicoverpa armigera; Manduca sexta.
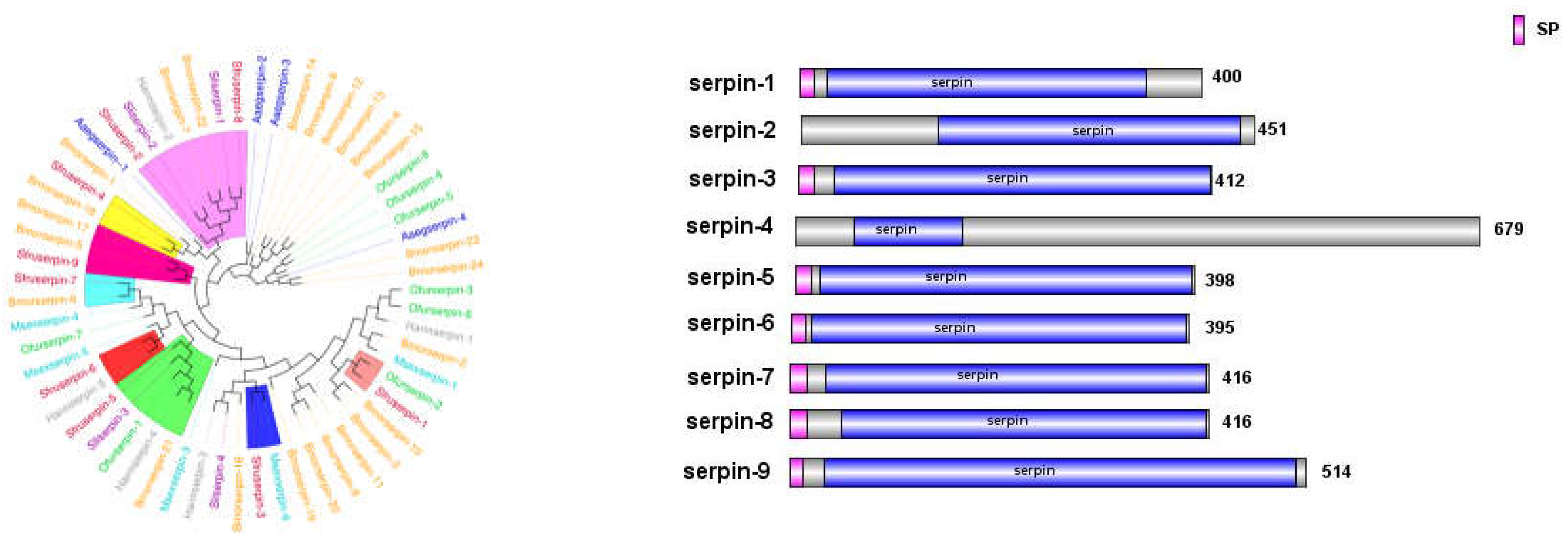
Figure 12.
(a) The phylogenetic tree of the Toll family, (b) The phylogenetic tree of the Spz family, in which the S. frugiperda related family is marked with red fonts. Note: Insects included in the phylogenetic tree (a) Musca domestica; Ostrinia furnacalis; Spodoptera litura; Helicoverpa armigera; Bombyx mori. (b) Musca domestica; Spodoptera litura; Helicoverpa armigera; Bombyx mori; Ostrinia furnacalis.
Figure 12.
(a) The phylogenetic tree of the Toll family, (b) The phylogenetic tree of the Spz family, in which the S. frugiperda related family is marked with red fonts. Note: Insects included in the phylogenetic tree (a) Musca domestica; Ostrinia furnacalis; Spodoptera litura; Helicoverpa armigera; Bombyx mori. (b) Musca domestica; Spodoptera litura; Helicoverpa armigera; Bombyx mori; Ostrinia furnacalis.
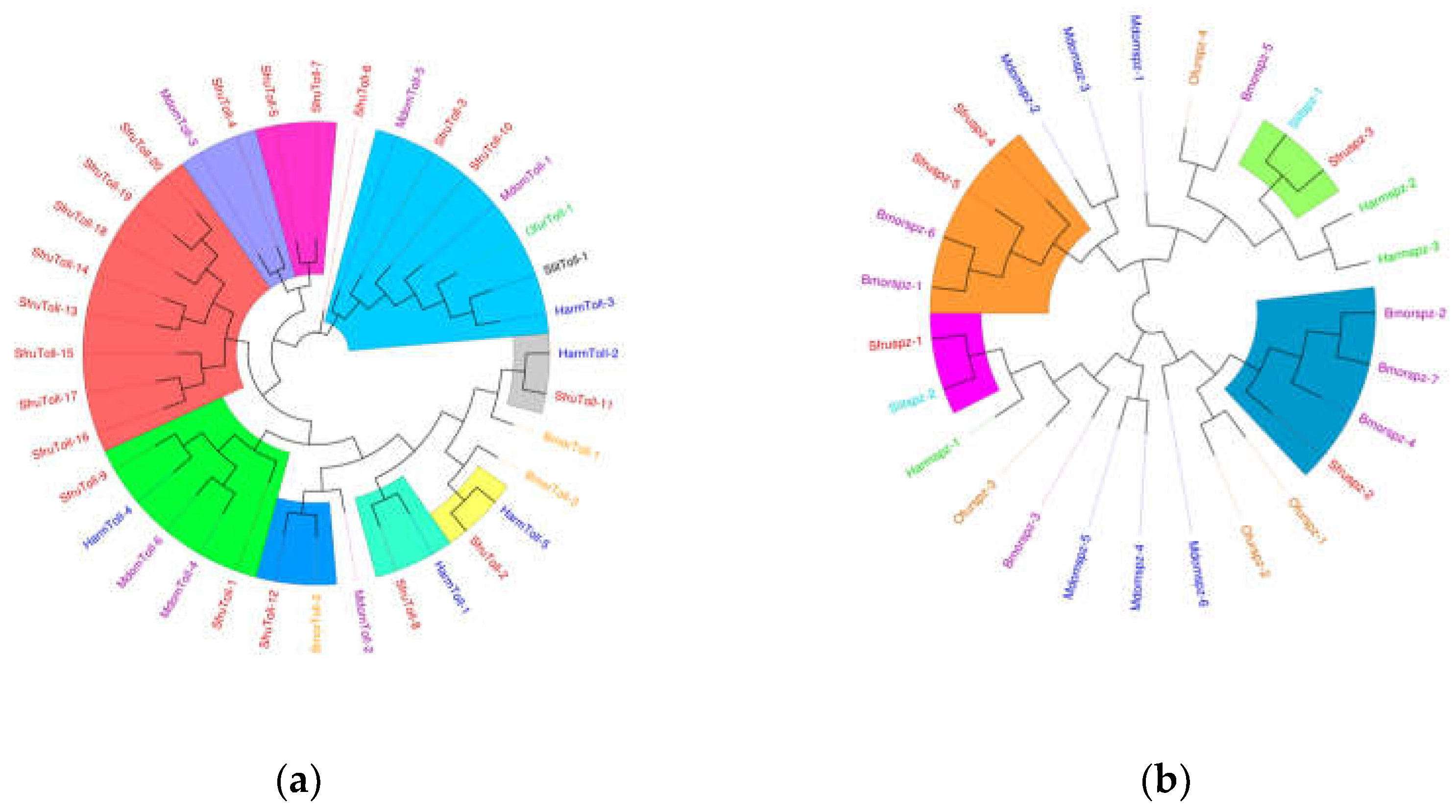
Figure 13.
Cluster heat map of differential expression of immune effectors in S.frugiperda infected by pathogenic microorganisms.
Figure 13.
Cluster heat map of differential expression of immune effectors in S.frugiperda infected by pathogenic microorganisms.
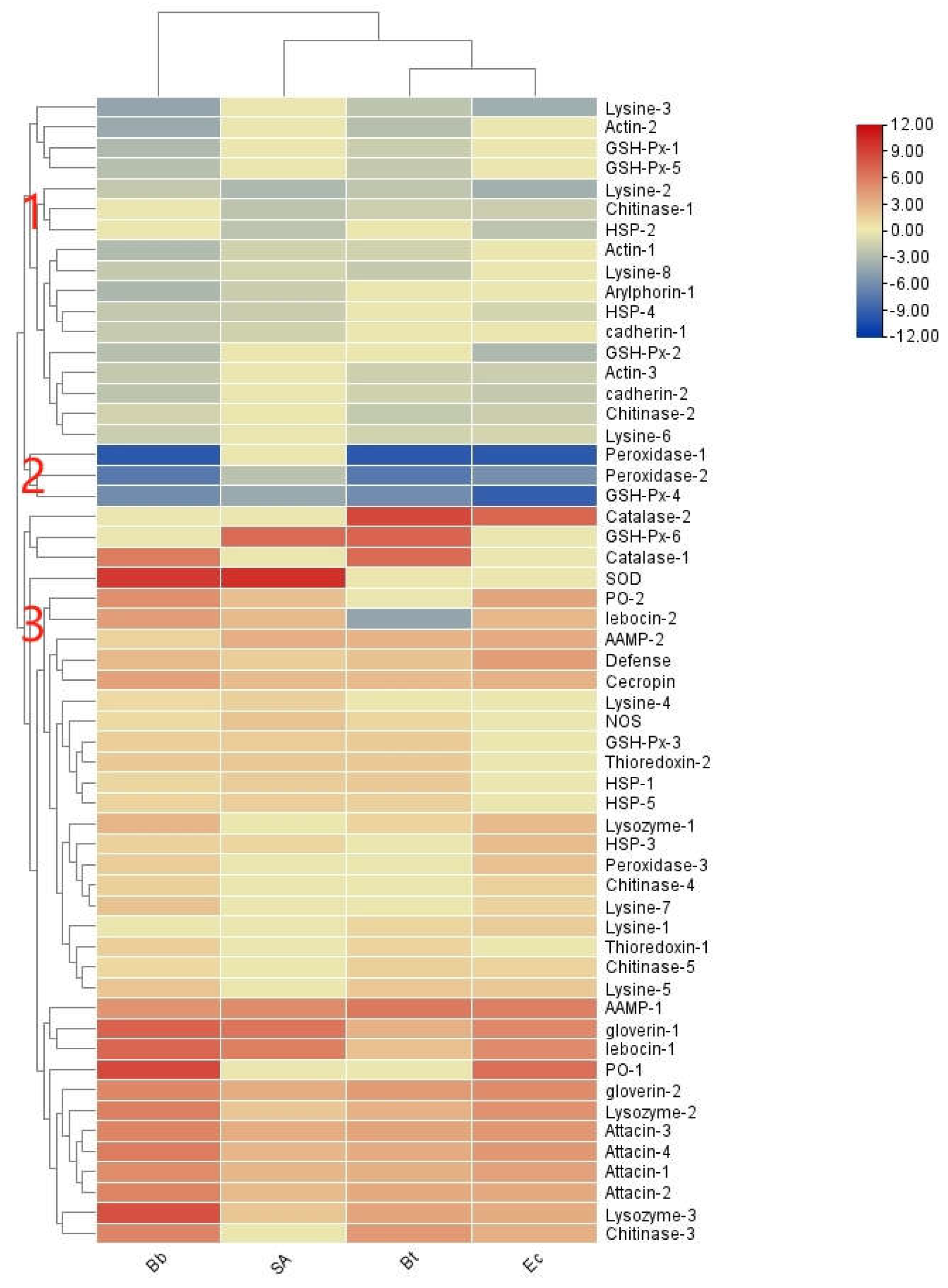
Figure 14.
Phylogenetic analysis of antimicrobial peptides in S. frugiperda. Note: (a) Drosophila melanogaster; Musca domestica; Helicoverpa armigera; Ostrinia furnacalis. (b).
Figure 14.
Phylogenetic analysis of antimicrobial peptides in S. frugiperda. Note: (a) Drosophila melanogaster; Musca domestica; Helicoverpa armigera; Ostrinia furnacalis. (b).
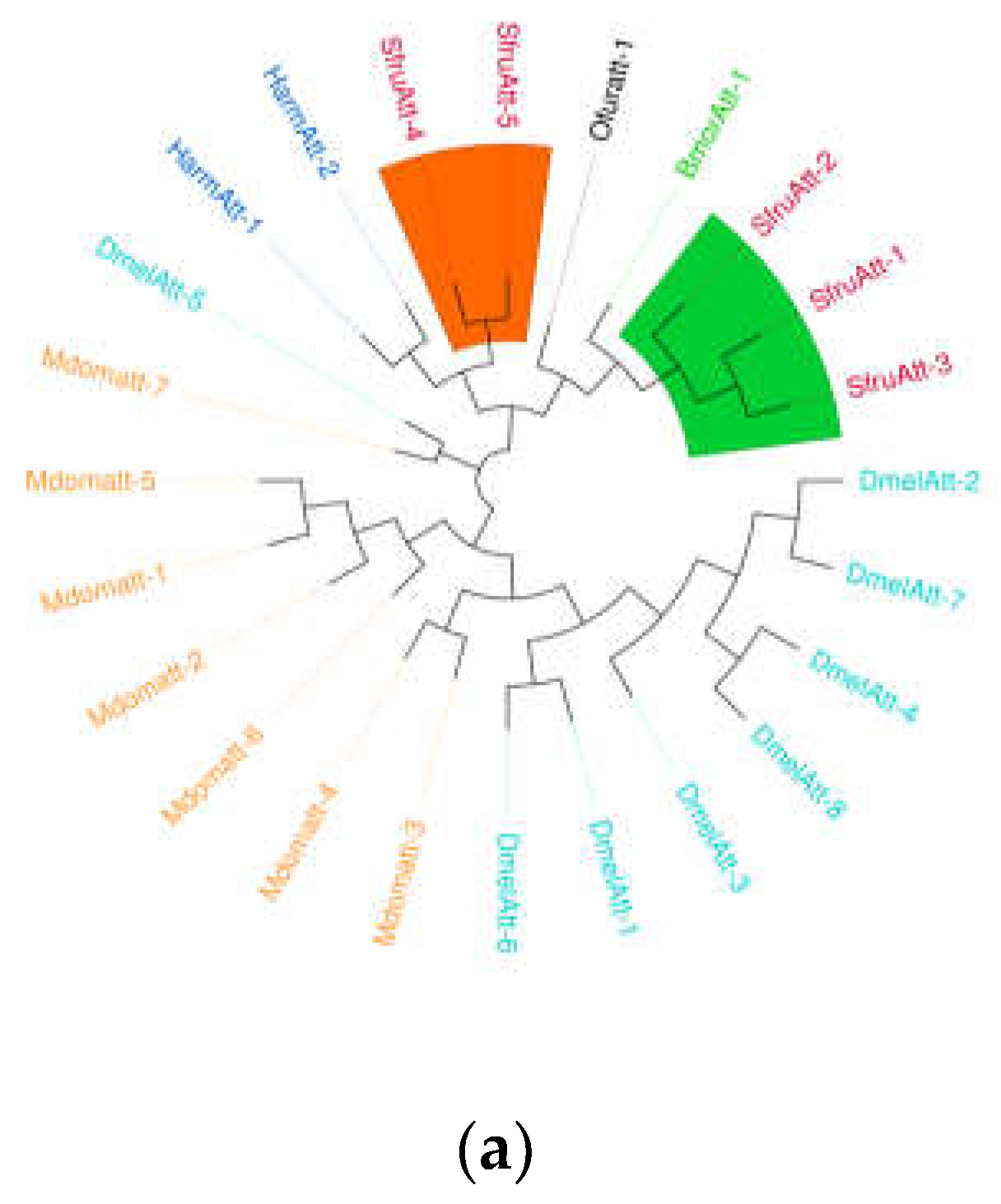
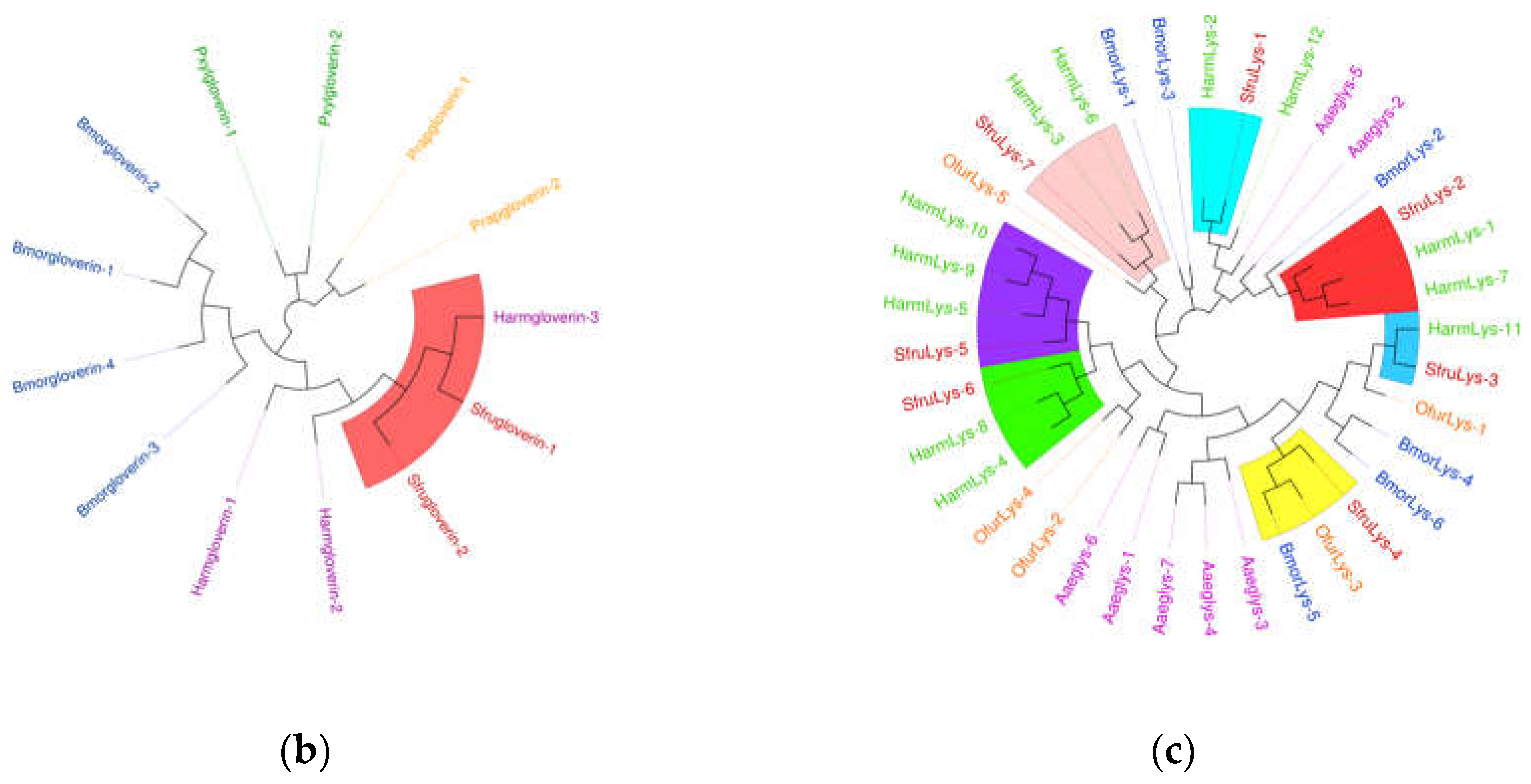
Figure 15.
Expression level of PGRP gene in S. frugiperda infected by pathogenic microorganisms. Note: There is a significant difference in gene expression between the ' * ' representative and the control in the figure. (*: FDR < 0.05, **: FDR < 0.01, ***: FDR<0.001, ****: FDR<0.0001), There was no significant difference in gene expression between ' ns ' and control.
Figure 15.
Expression level of PGRP gene in S. frugiperda infected by pathogenic microorganisms. Note: There is a significant difference in gene expression between the ' * ' representative and the control in the figure. (*: FDR < 0.05, **: FDR < 0.01, ***: FDR<0.001, ****: FDR<0.0001), There was no significant difference in gene expression between ' ns ' and control.
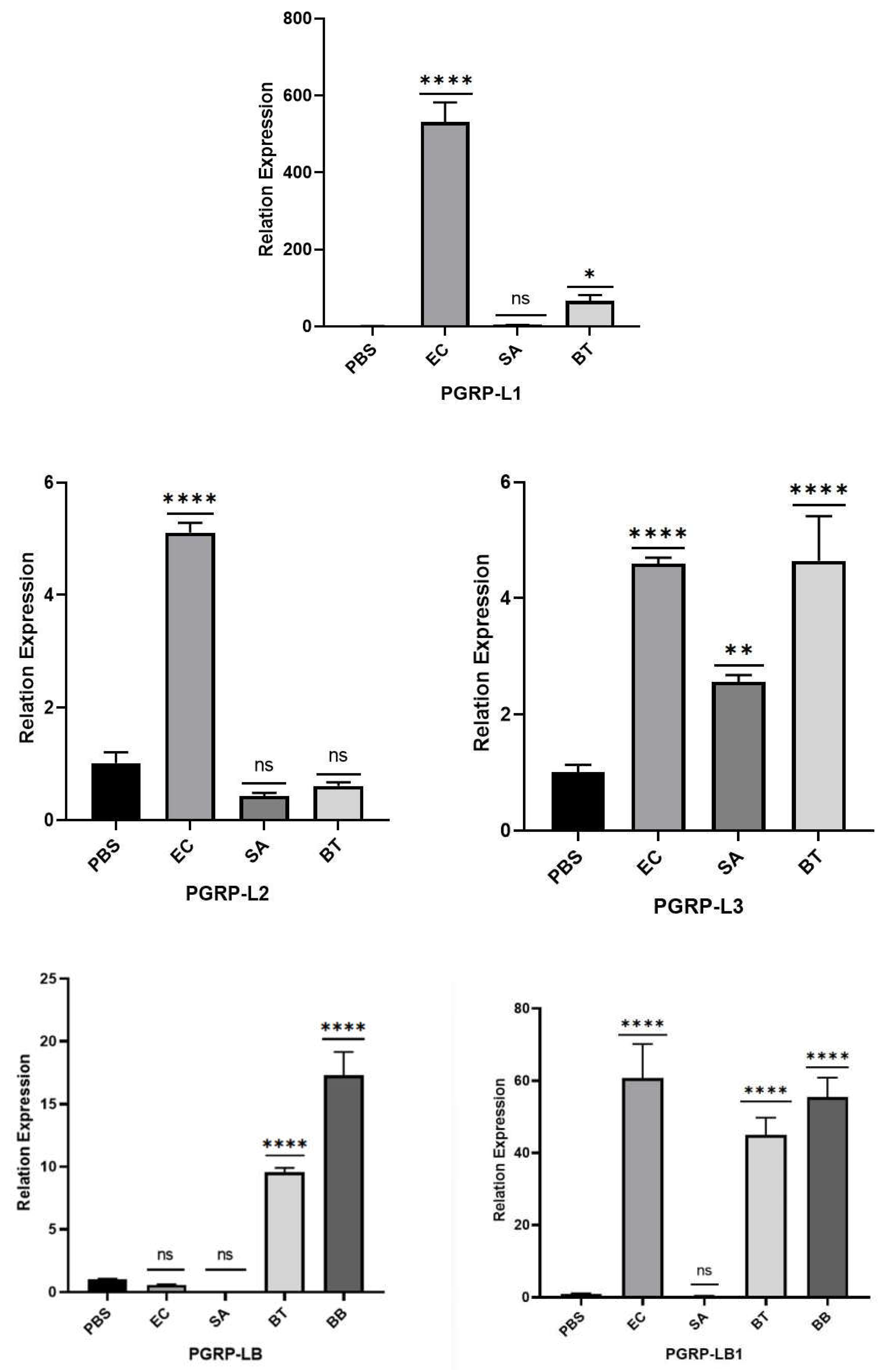

Figure 16.
Expression level of βGRP gene in S. frugiperda infected by pathogenic microorganisms. Note: There is a significant difference in gene expression between the ' * ' representative and the control in the figure. (****: FDR<0.0001), There was no significant difference in gene expression between ' ns ' and control.
Figure 16.
Expression level of βGRP gene in S. frugiperda infected by pathogenic microorganisms. Note: There is a significant difference in gene expression between the ' * ' representative and the control in the figure. (****: FDR<0.0001), There was no significant difference in gene expression between ' ns ' and control.
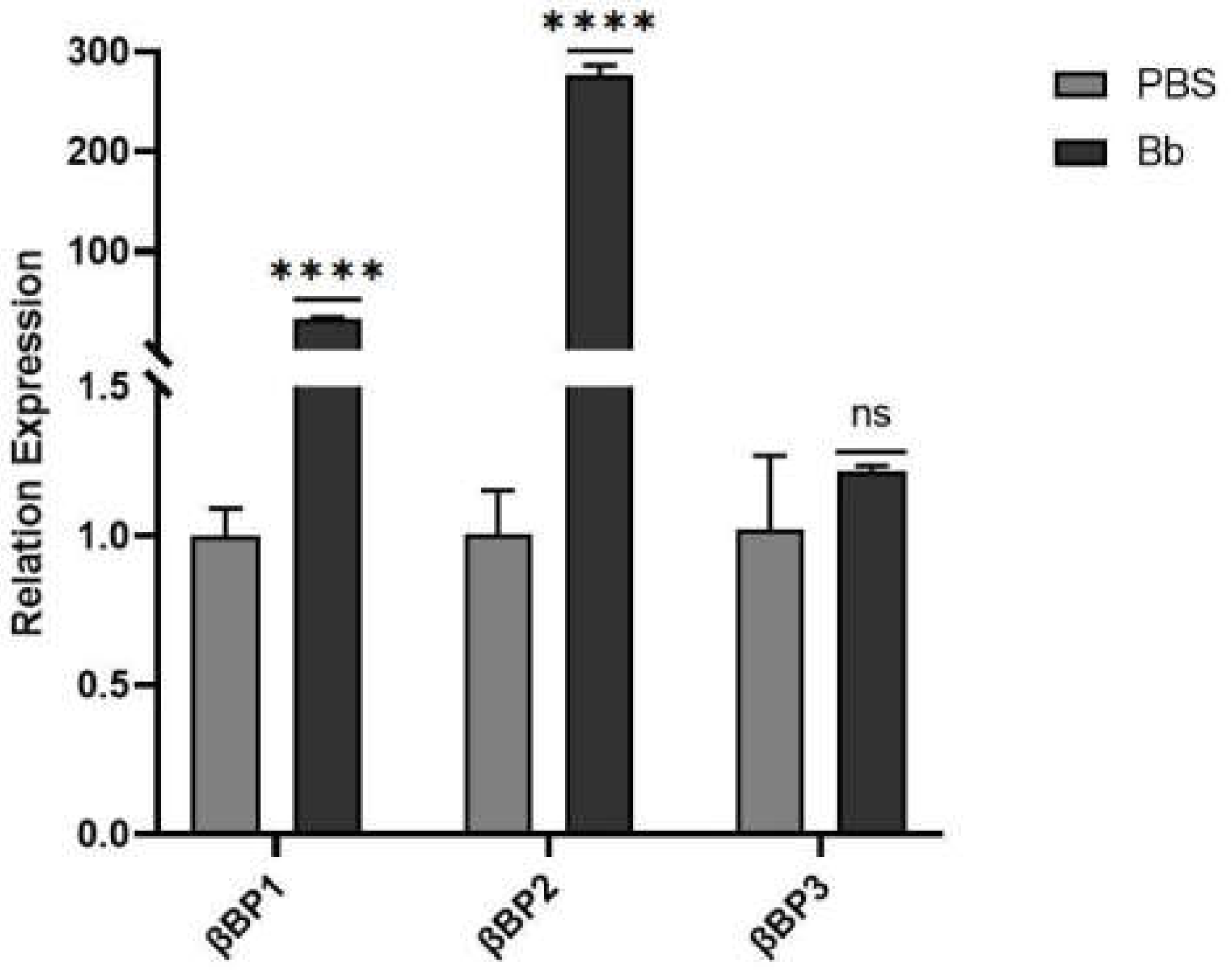

Table 1.
qRT-PCR primer information.
| Primer | Forward primer | Reverse primer |
|---|---|---|
| PGRP-LE2 | ATTTCGCACACTGCTACCGA | TGGACTGAGAGTAGACGCCA |
| PGRP-LB | CAAGGAAGACTGCTCAGCGA | AGGCAGTTCCAGGACATTCG |
| PGRP-LB1 | GCACGCGCTACATTTCAACA | TTGAAGAGTGCGTCTCCTGG |
| PGRP-LB2 | AGACCGCCTAATGGTTCGAC | AGCCAAGCTTCACTCCAGTC |
| PGRP-L1 | AGCAGCCAATGGAATCAGGA | GAGAGCTGACTATGGGCCAC |
| PGRP-L2 | GTCAGCTTGCTCCTGGTGAT | ATCGTTCCGTTCCCGTTTGA |
| PGRP-L3 | GAATTGCGCAGCTGAGATGG | CAAGCTCGACACCCTTGTCT |
| PGRP-S1 | AAATGGGGACTGTGGCGTAG | CGTATACTTTGCCGTTGCCG |
| PGRP-S2 | TTGTGTCGAGGATCGGTTGG | CTCATACACTGTCCCCTGGC |
| βGRP1 | GAAGTGCTCCAACCGAAGGA | CGAATATGGTTTGGCCTGCG |
| βGRP2 | CCCTGGAGAACCGGACTTTC | AGGTGATGATCGGTGGGAGA |
| βGRP3 | GTTAGCCGGAGTATTGGCGA | AATCTCCGGCGGGCATTTTA |
| PRL18 | GCCAAGACCGTTCTGCTGC | CGCTCGTGTCCCTTAGTGC |
| PRL3 | CCAAGGGTAAAGGATACAAAGGTG | TCATTCACCGTTGCCCGT |
Table 2.
Evaluation statistics of transcriptome data of Spodoptera frugiperda larvae treated with pathogenic microorganisms.
Table 2.
Evaluation statistics of transcriptome data of Spodoptera frugiperda larvae treated with pathogenic microorganisms.
| SAmple | Raw reads | Raw bases | Clean bases | Error rate(%) | Q20(%) | Q30(%) | GC pct(%) |
|---|---|---|---|---|---|---|---|
| PBS | 54529714 | 8180000000 | 7540000000 | 0.01 | 98.49 | 95.76 | 45.95 |
| Ec | 48893730 | 7330000000 | 6940000000 | 0.01 | 98.67 | 96.11 | 46.7 |
| Bt | 48788450 | 7320000000 | 6890000000 | 0.01 | 98.46 | 95.72 | 45.24 |
| SA | 51879208 | 7780000000 | 7460000000 | 0.01 | 98.67 | 96.2 | 45.3 |
| Bb | 46291118 | 6940000000 | 6720000000 | 0.01 | 98.56 | 95.96 | 45.15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



