
Submitted:
30 November 2024
Posted:
02 December 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The SARS-CoV-2 induced disease, COVID-19 is a worldwide public health concern with a rapid transmission, pathogenicity, and mortality rate. To elucidate the viral mutational changes and screening the emergence of new variants of concern, we conducted this study in Bangladesh. The viral RNA genome was sequenced using Illumina COVID Seq protocol and genomic data processing and evaluation was performed in DRAGEN COVID Lineage software. Variant analysis revealed that the number of Delta, Omicron and Mauritius variants identified were 88%, 8%, and 4%, respectively. All of the 25 samples had 23403A>G (D614G, S gene), 3037 C>T (nsp3) and 14408 C>T (nsp12) mutations, where 23403 A>G was responsible for increased transmission. Omicron had the highest number of unique mutations in spike protein (i.e., substitutions, deletions and insertions) which may explain its more transmissibility and immune evading ability than Delta. A total of 779 mutations were identified where 691 substitution, 85 deletion and 3 insertion mutations were observed. To sum up, our study will enrich the genomic database of SARS-CoV-2, aiding treatment strategy along with understanding the virus’s preferences in both mutation type and site for predicting newly emerged virus’s survival strategy and thus for being prepared to counteract them.
Keywords:
SARS-CoV-2
; COVID-19
; Omicron
; Delta
; Mauritius
; Mutation
; Bangladesh
1. Introduction
The novel severe acute respiratory syndrome corona virus 2 (SARS-CoV-2) that introduced itself as a global health threat, was first reported on 30 December 2019 in Wuhan city of Hubei province in China [1] and its associated disease is known as Corona Virus Disease 2019 (COVID-19) [2]. The devastating accelerated geographic spread of COVID-19 resembles the SARS and MERS outbreak which also caused pandemics [3,4]. According to the WHO, the number of total infected individuals is above 776 million and death count is beyond 7 million. In Bangladesh, the first three COVID-19 cases were reported officially on 8th March 2020 having a travel history from Italy when COVID-19 had already spread in that region [5]. In Bangladesh, 2,051,201 were infected with SARS-CoV-2 and over 29000 people died from COVID-19 as per information provided by the Government designated institution [6].
The spherical pleomorphic viral particle, coronavirus possesses a longest non-segmented RNA genome of 26 to 32kb in length which is also a positive sense single-strand in nature with a 5′ cap and 3′ poly-adenylation [7,8,9,10]. The genome includes 14 functional open reading frames (ORFs) comprising of genes for 16 non-structural (nsp1-nsp16), 4 structural and 9 accessory proteins along with 2 non-coding regions at both of the ends [11,12]. ORF1a and ORF1b encoded 16 NSPs are essential for viral RNA synthesis while accessory proteins gave selective facilities in host cell for its pathogenicity [13,14]. This virus is made of 4 types of structural proteins that are vital for viral assembly i.e., spike (S), membrane (M), envelope (E) and nucleocapsid protein (N) and among them, the spike protein is the most significant one for attachment, fusion and entry into the host cell [15,16]. The S, M and E proteins are embedded in the envelope and N protein forms the core structure. Spike protein is a trimeric protein where its monomer comprises 2 distinct functional subunits, S1 and S2. These subunits are necessary for attachment and membrane fusion, respectively, for entry into the host cell [17]. The viral genome is encapsulated with the nucleocapsid protein that forms a helical ribonucleoprotein (RNP) complex [18]. Moreover, envelope and membrane proteins form the envelope and the membrane of the virus, respectively, and thus the membrane protein gives distinct shape to the virus [19,20].
The COVID-19 that emerged through a zoonotic transmission event [20], is an acute disease without any carrier status and has continued to spread rapidly throughout the world [21]. Viral infection occurs due to human-to-human transmission by having contact with the droplets of saliva that come out while talking, coughing, and sneezing from a close distance or due to direct contact with oral, nasal, and eye mucous membranes [22,23,24]. The incubation period of the SARS-CoV-2 virus ranges from 2 to 14 days and symptoms are similar to those of influenza with some other clinical manifestations i.e., fever, dry cough, myalgia, headache, nausea or vomiting, chills, diarrhea, conjunctival congestion, shortness of breath, sore throat, and fatigue [25,26,27]. Being infected with SARS-CoV-2, the mortality rate was observed to increase with age where the rate increases significantly above 80 years of age, and other comorbidities that were believed to play role in increasing mortality rate are heart disease, diabetes, chronic lung or kidney disease and other socio-demographic factors. Moreover, an increased risk of being infected was observed to be associated with hypertension, diabetes, and coronary heart disease [28,29]. Real-time Polymerase Chain Reaction (RT-PCR) and RT-LAMP (reverse-transcriptase-loop mediated isothermal amplification) techniques were used to detect viral RNA in laboratory [30].
After the first wave of COVID-19, several variants were observed when compared to the reference genome SARS-CoV-2 (GenBank). Viruses generally acquire alteration of their genetic constitution over time, giving rise to new variants, more specifically said Variant of Interest (VOI) or Variant of Concern (VOC) which have increased transmissibility and ability to escape immunity and/or have severe pathogenicity. Concerning variants are often referred to by using the terms variant, strain, and lineage interchangeably or by country in which they were first identified. The term “emerging variant” is used to label a new variant that appears in a population [31]. The havoc caused by SARS-CoV-2 started to decrease after April 2023 and was at the lower point near April 2024 in Italy, Greece, Romania, Northern Ireland, Poland, and United Kingdom of Great Britain though the graph exhibits that the number is on the rise again. But at the same time the number of COVID-19 cases were higher during April 2024 in China and New Zealand.
To control SARS-CoV-2 by intervention, vaccination is the vital key and evidence has proved that the risk of death, hospitalization, mild/moderate/severe disease, and incident infection was reducing due to different type of vaccine administration [32,33]. A big threat is posed when variants of concern (VOCs) emerge which perturbs the attainment of herd immunity (HI) by changing the effectiveness of vaccine plus herd immunity threshold (HIT) and this was evident from the incidence of Pfizer-BioNTech, and AstraZeneca vaccines being less effective against the beta variant compared to the wild type one. The new mutant strains of SARS-CoV-2 with higher infecting ability and immune evading characteristics leads to new wave of COVID infection and death. So far, the alpha, beta, gamma, Delta, Omicron variants emerged having changes in viral behavior, infectivity and pathogenicity i.e., with the ability of evading the immune system by altering their receptor binding domain of S protein [34]. For this reason, regular sequencing for surveillance of VOCs is a must to inform global vaccination programs [35]. However, everyday a noteworthy number of datasets of SARS-CoV-2-virus whole genome sequence are being deposited to genomic databases from different parts of the world whereas the number is very low in case of SARS-CoV-2 virus that caused great havoc in Bangladesh. Keeping in mind the necessity of surveillance of new variants of concern, we sequenced the genome of SARS-CoV-2 from 25 COVID-19 patients via Next Generation Sequencing (NGS) in Bangladesh and evaluated their clade, lineage, signature mutation, and mutational spectra.
2. Materials and Methods
2.1. Study Subjects Selection
The Institutional Review Board (IRB) of BSMMU under the declaration-of-Helsinki-ethical-principles provided formal approval (No 3506, date 28.06.2021) to conduct the present study. This cross-sectional descriptive study was performed in the Genomic Research Laboratory in the Anatomy Department, Bangabandhu Sheikh Mujib Medical University (BSMMU), Bangladesh. A consortium was formed with the Laboratory of Neuroscience and Neurogenetics, Department of Biochemistry and Molecular Biology, University of Dhaka, Bangladesh; Department of Physiology and Molecular Biology, Bangladesh University of Health Sciences, Dhaka, Bangladesh; The National Institute of Laboratory Medicine & Referral Center (NILMRC), Dhaka, Bangladesh; and Dhaka Medical College, Dhaka, Bangladesh. The time frame of the study is March 2021 to February 2022. After having consent, 25 patients with the COVID-19 who met the exclusion and inclusion criteria were selected. From the patients’ selection check list and data collection questionnaire the socio demographic, and comorbidity data of the patients were collected.
2.2. Ethical Implication
In this research, all the patients were treated equally expressing due respect. They were informed about the aim and possible benefits of the variants’ characterization before asking for their written consent in a prescribed consent form. It was also made it clear to the patients that their RNA will only be used for the research purpose and they can withdraw their name anytime during the study. Moreover, to avoid anonymity and to ensure safeguarding confidentiality each of the COVID-19 patient was given a unique ID number.
2.3. Isolation of RNA
Genomic RNA was extracted from nasopharyngeal swab sample of selected COVID-19 patients using ReliaPrep™ (Promega, USA) RNA extraction kit. Micro centrifuge tube with 200μl nasopharyngeal swab was treated sequentially with the enzyme proteinase K, cell lysis buffer, isopropanol and wash buffer where proteinase k, and cell-lysis buffer were used for cell lysis and protein denaturation of nuclease along with other proteins and to assist in removal of these proteins from the RNA extraction specimen following centrifugation. In addition, Isopropanol was used for precipitation of RNA that forms a gel like pellet.
2.4. Synthesis and Amplification of cDNA
Nano Drop spectrophotometer was used for concentration measurement and checking the quality of genomic RNA. Then using random hexamers, the isolated RNAs were annealed to prepare for cDNA generation. Reverse Transcriptase reversely transcribed RNA fragments that were primed with random hexamers. Thus, the first strand of cDNA was synthesized. The cDNA amplification was performed using two sets of COVID Seq primers in 2 separate PCR reactions. This reaction includes IPM HT (Illumina PCR Mix HT), CPP1 HT (COVID Seq Primer Pool 1HT), and CPP2 HT (COVID Seq Primer Pool 2HT).
2.5. Amplification of Tagmented Amplicons and Library Preparation
Tagmentation of PCR amplicons with adapter sequence was facilitated using EBLTS-HT (Enrichment BLT HT), and TB1 HT (Tagmentation Buffer 1 HT). This step executes fragmentation as well as tagging of PCR amplicons with adapter sequences. In post tagmentation clean up step, adapter-tagged amplicons were washed with ST2 HT (Stop Tagment Buffer 2 HT), and TWB HT (Tagmentation Wash Buffer HT) before performing their PCR amplification. The tagmented amplicons were amplified using a PCR program that includes EPM HT (Enhanced PCR Mix HT), Index adapters, and sequences that were required for sequencing clue. Libraries from all the 96 well sample plate are then combined and placed in a 1.7 ml tube followed by binding of optimal size libraries to the magnetic bead using ITB (Illumina Tune Beads). Afterwards, RSB (Resuspension Buffer HT) washed away the too large or small fragments. Thus, pooling and cleaning of libraries were accomplished. Qubit dsDNA HS Assay kit was employed for analysis of 2 µl library pool. Standard range of libraries were maintained and or else, RSB was used for 10x dilution and then analyzed once again. The average library size was 400bp and the diluted normalized concentration was 4nM. As per library preparation documentation, for denaturation and dilution, libraries standard normalization method was applied. Libraries were diluted to the starting concentration (according to the manufacturer guidelines) for the sequencing system.
2.6. Loading of the Libraries to the MiSeq Flow Cell and Sequencer Running
The denatured and diluted libraries were loaded into the reagent cartridge and then the sequencing run was set up. For evaluation of sequencing reads of RNA libraries, the Illumina DRAGEN COVID Seq Test Pipeline was used. At first for starting the run setup steps, sequence option was clicked on from the screen of software interface which was followed by selection of the run setup option. The reagent cartridge was loaded and the run parameters were reviewed and results were checked by pre-run. After selection of Start button, run was observed using either Local Run Manager or Sequencing Analysis Viewer (SAV) from MCS interface. Finally, the libraries were loaded to the MiSeq flow cell and the system was run. After running, data was automatically uploaded into BaseSpace Sequence hub by base calling within 12 to 13 hours. FASTQ files were uploaded and analyzed in DRAGEN COVID Lineage software which helps in assembling SNV with reference SARS-CoV-2 genome. One Excel and another FASTA file were generated in BaseSpace whereas the Excel file include clade and lineage files. The SARS-CoV-2 variant evaluation was performed by using COVID-Seq Lineage software.
2.7. Sequence Data Processing and Evaluation
The raw output was the BCL formatted file which was eventually converted into FASTA format after several steps. For the detection and surveillance of viral pathogens, DRAGEN RNA Pathogen Detection Pipeline was used. To detect viral pathogens coverage and k-mer-based approaches were exploited. Reads were aligned to a SARS-CoV-2 reference genome and generated a consensus genome sequence. Lineage/clade analysis was performed using Pangolin. DRAGEN RNA Pathogen Detection Pipeline encompasses Human and viral references which is a combination of reference sequences of the human genome (hg38) with selected reference sequences of the virus. Moreover, this pipeline consists of an Integrated FASTA generation feature which can be uploaded to public databases i.e., in our case the GenBank Database. The accession number of the 25 virus genomes are OM019149, OM019148, OM019138, OM019145, OM019139, OM019150, OM019143, OM019146, OM090130, OM090137, OM090136, OM090135, OM090140, OM019153, OM019152, OM019154, OM019140, OM019147, OM019141, OM019155, OM090139, OM277491, OM277497, OM277498, and OM019151. The resulting sequences of our study were compared with the GenBank of NCBI (National Centre for Biotechnology Information) database. Furthermore, MEGA X software automatically translated the codons. Thereafter, the NCBI Entrez search engine aided in the comparison of translated codons with the reference sequence.
2.8. Statistical Analysis and Graph Construction
SPSS (Statistical Package for the Social Sciences, version 23) was used for statistical and mutational analysis i.e., determining the mutation count and percentage of specific mutation (i.e., D614G) falling in a specific category of mutation (i.e., substitution mutation) observed in the viral genome from COVID-19 patients and also for the data of socio-demographic and disease condition-related characteristics. In this study, pie charts and lollipop plots were constructed for better visualization of the data using R studio and SRPlot, respectively [36].
3. Results
3.1. Socio-Demographic and Disease-Related Data of COVID-19 Patients
The present study includes twenty-five adult COVID-19 positive patients where 48% were male and 52% patients were female. Most of the patients (84%) had no family history of COVID-19. The quarantine period was maintained by 48% of the patients. Moreover, among the twenty-five patients, 12% had comorbidities like hypertension but only 4% had a history of hypertension with asthma. Other comorbidities like chronic kidney disease, diabetes mellitus, and chronic obstructive pulmonary disease conditions did not present. Substantially, only 12% of people had 2nd time positive history of COVID-19 (Table 1).
Additionally, 4% of patients remain positive for a long duration. Most of the people had no specific long traveling history. They move only short distances such as from home to working place and some people had a history of social gatherings, such as invitations or religious Festival (Table. 1). Fifty two percent patients were vaccinated by two doses. Eight percent of patients had taken only 1st dose of COVID-19 vaccine. The rest of the forty percent of patients didn’t take a vaccine against SARS-CoV-2 (Figure 1).
3.2. SARS-CoV-2 Variants, Clades and Lineage Evaluation
Omicron, Delta, and Mauritius variants were detected in the present study. Among twenty fife patients, twenty-two of the COVID-19 patients were Delta variant carriers, two were Omicron carriers and only one patient was identified to be infected with the Mauritius variant of SARS-CoV-2 (Figure 2).
Evaluation of the viral clade demonstrated that 21A clade and 21J clade of the Delta variants were comprising 80% and 8% of all the variants, respectively. In addition, the four percent 20B clade was established for the Mauritius variant, whereas 20A clade and 21K clade the Omicron variants fall in the 8% of all the variants (Table 2).
Lineage analysis of the SARS-CoV-2 variants revealed that in the case of Delta variants, 28% were under B.1.617.2 lineage, 24% were under AY.4 lineages, 8% were under AY.131 lineage, 8%) were under AY.26 lineage, and rest of the Delta variants come from a different category of lineages like AY.122, AY.122.1, AY.29, AY.30 and AY.39 (Table 3). Mauritius variant was under B.1.1.318 (4%). The 2 Omicron variants were under BA.1 lineage (8%) (Table 3).
3.3. Substitution, and Insertion-Deletion Mutation Analysis
Study on different types of mutations in three of the SARS-CoV-2 variants unveiled that in Omicron variants, the number of maximum and minimum substitutions were 53 (highest among all three variants) and 41, respectively; while the maximum and minimum number of deletion loci were observed to be 6 and 6 with their maximum created gaps of 45bp and 39 bp, accordingly (Table 4). In addition, 2 insertion mutations of 9 nucleotide base length were identified in 2 Omicron variants.
However, Delta variants, on the other side exhibited maximum and minimum substitutions of 45 and 14, respectively. Whereas for deletion mutations the number of deletion loci were 4 and 3, with maximum created gaps of 18bp and 13bp, in accordance. Noticeably, no insertion mutation was seen in the Delta variants (Table 4). Moreover, in case of the Mauritius variant, the numbers of substitution, deletion, and insertion mutations were 36, 5 (35bp gap), and 1 (3bp in length), respectively. Using Next Generation Sequencing of the whole viral genome of the extracted RNA of twenty-five COVID-19 patients, a total of 779 mutations were identified of which 691 substitution, 85 deletion, and 3 insertion mutations were observed.
3.4. Comprehensive Investigation of Nucleotide Substitution Mutations
The most common substitution mutations were 23403A>G (S gene), 3037C>T (ORF1a (nsp3) gene), and 14408C>T (ORF1b (nsp12) gene) which mutations were present in all of the twenty-five samples (Table 5). Whereas, substitution mutation 26767T>C (M gene) was found in 23 samples, and 15451G>A (ORF1b (nsp12)), 25469C>T (ORF3a), and 23604C>G (S gene) were identified in 22 viral genomes.
These aforementioned mutations were detected in 21 (88%) samples and the other mutations encompass the 5′ Leader Sequence, ORF1a, ORF1b, S, N, ORF7b, ORF7a, and the 3′ end region (Table 5). Mutations that were detected in at least 4 samples are shown in Table 5 and other minimal count mutations are given in Supplementary Table S1.
The most common type of alteration of base pair was C>T being around 38.48% in all the variants which is followed by G>T (18.23%), A>G (11.52%), G>A (8.61%), and T>C (8.39%) in order and all of these aforementioned base changes are transition mutations except G>T (Table 6).
A comparison of the 3 variants’ viral genome unveiled that in all these 3 variants transition of nucleotide bases was way higher than transversion mutation and the percentages of transition and transversion mutation were 67% and 33% in order (Table 7).
3.5. Extensive Analysis of Nucleotide Deletion Mutations
In-depth analysis of deletion mutations revealed that 22029-22035bp(S), 28248-28254bp (ORF8), 28274bp (N), and 29750-29752bp (non-coding) deletions were identified in 20 Delta variant samples of 21A clade and the last one was observed in only one sample (Table 8).
Besides, deletion in 22029-22034bp (S gene), 28248-28253bp (ORF8), and 28271bp (non-coding region) regions were noted in both 21J Delta variants samples except 21992-21994bp deletion mutation, which was found only in one sample. Additionally, most of the Delta variants had 15bp deletions. On the contrary, in case of Omicron variants most of the deletions were unique and these were different from Delta or Mauritius variants i.e., deletion at 6513-6516/6513-6515bp (ORF1a) was observed only in Omicron variant and deletion at around 11287bp (ORF1a) locus was noticed in both Omicron and Mauritius variant but not in Delta variant. The 2 Omicron had 45bp (6513-6516 (ORF1a), 11287-11296 (ORF1a), 21766-21772 (S), 21987-21996 (S), 22194-22197 (S), and 28363-28372 (N)) and 39bp (6513-6515 (ORF1a), 11285-11293 (ORF1a), 21765-21770 (S), 21987-21995 (S), 22194-22196 (S), 28362-28370 (N)) deletions at 6 deletion loci. However, 35bp deletions at 5 deletion sites (11288-11297 (ORF1a), 21994-21997(S), 27887-27902, (ORF7b, ORF8), 28254 (ORF8), 28896-28899 (N)) were revealed in Mauritius variant that also had unique deletions at 27887-27902bp covering both ORF7b, ORF8 genes (Table 8).
3.6. Detailed Evaluation of Insertion Mutations
Two insertion mutations of 9bp length in the spike glycoprotein region were seen in two of the Omicron variants and they are 22204:GAGCCAGAA and 22206: GCCAGAAGA (Table 9). In the case of the Mauritius variant, an insertion mutation of 3bp (28250:CTG) in length was observed at the ORF8 region and no insertion mutation was seen in case of the Delta variant.
3.7. Missense Mutations Analysis in SARS-CoV-2 Viral Genome
Comparison of MEGA X translated codons with reference genomes facilitated the unraveling of amino acid substitutions. The amino acid substitution (missense/nonsense) mutations that were obtained after analysis and were found in at least 3 viral genomic sequences (Table 10).
On the contrary, mutations that were least common and found in less than 3 samples are shown in Supplementary Table S2. S:D614G missense mutation was the most frequent one and was present in all of the study subjects. Then M:I82T was most frequent in order being present in 24 samples and ORF3a:S26L, S:P681R, and ORF1b:G662S were the 3rd most frequent mutations being present in 22 samples. Moreover, ORF1b:P314L, N:M1X, ORF9b:T60A, S:T19R and S:E156G missense mutations were found in at least 20 samples. Other mutations are presented in the Table 10.
Besides, to visualize the mutational spectra of S, N proteins, and OR1a, ORF1b polyproteins lollipop plots of amino acid substitution and deletion mutations in all of the 3 variants are given in Figure 3, Figure 4, Figure 5, and Figure 6, accordingly. The mutations that were observed in more than 1 variant were in bold letter and black in color, and the mutations that play important roles in viral pathogenesis and transmission are in bold font and red color.
Furthermore, mutations that were present in most of the sample of a specific variant are bolded, blue in color and have larger lollipop in size. In all these Figures (Figure 3, Figure 4, Figure 5 and Figure 6) panel a), panel b) and panel c) represent Delta, Omicron and Mauritius variants respectively. From the Figure 3, it is clear that in Spike protein of the Omicron variant, most of the unique mutations as well as deletion and substitution mutation were noticed. Substitution mutation T19R, E156G, L452R, T478K, D614G, P681R, D950N were observed in most of the Delta variant and T95I, as well as D614G were found in all the 3 variants. Moreover, P681 was substituted with R (Arginine) in Delta but with H (Histidine) in Omicron and Mauritius variants while A67 was substituted with V (Valine) in Omicron but it was substituted with T (Threonine) in Mauritius variant. In addition, D796 was substituted with Y (Tyrosine) in Omicron and with H in Mauritius variant. Furthermore, G142D, T478K, and H655Y were found both in Delta and Omicron variants. Interestingly, E484 was substituted with Q, A, and K in Delta, Omicron and Mauritius variants, respectively.
However, in Nucleocapsid protein (N) of Delta variant no amino acid deletion was observed and compared to the other two variants most of the mutations were in Delta variant (Figure 4). Interestingly, only R203 was found to be substituted in all these 3 variants but it was substituted to M (Methionine) in Delta while it was substituted to K (Lysine) in Omicron and Mauritius variants. In the Omicron and Mauritius variants common mutation was G204R. Besides, M1X, D63G, R203M, G215C, and D377Y were the most frequent mutations in Delta variant samples.
However, regarding ORF1a, in the case of Delta mutations encompassed the whole polyprotein region and the other 2 variants showed fewer mutations in the N terminal regions (Figure 5). In Delta variants K261N, P309L, A1306S, P1640L, H2092Y, P2287S, V2930L, A3209V, T3255I, T3646A, and V3718A were the most frequent mutations. Moreover, while comparing the variants, only L3606F was observed to be common in all of the 3 variants and A3209V was observed in the Delta and Mauritius variants whereas T3255I was found in both Delta and Omicron variants.
From our investigation, we found that ORF1b protein did not have mutations at all after 2473th amino acid residue and downright from the N terminal domain of NSP4 (Figure 6). However, P314L was observed in all the 3 variants and in some samples of Delta variant, P314 was substituted with F (Phenylalanine) while V2371 was substituted with L (Leu) in Delta variant which was substituted to M (Methionine) in the Mauritius variant. In addition, P314L, G662S, P1000L, and A1918V were most frequent mutations in the Delta variant viral genomes.
3.8. Amino Acid Deletion Mutation Analysis
Deletion mutation analysis of Omicron Variant genome revealed that N:E31-,N:R32-,N:S33-, ORF1a:S3675-, ORF1a:G3676-, ORF9b:N28-, ORF9b:A29-, S:H69-, S:V70-, S:V143-, S:Y144- and ORF1a:S2083-, ORF1a:L2084-, ORF1a:L3674-, ORF1a:F3677-, ORF9b:E27-, ORF9b:V30-, S:G142-, S:Y145-, S:N211-, S:L212- deletions occur in nucleocapsid, non-structural proteins and spike protein encoding regions (Table 11).
However, in Mauritius variant there were 7 amino acid deletion mutations (N:R209-, ORF1a:S3675-, ORF1a:G3676-, ORF1a:F3677-, ORF8:M1-, ORF8:K2-, and S:Y144-) and they are located in 1 nucleocapsid, 5 non-structural protein and 1 spike protein encoding regions (Table 11). On the other side, 6 amino acid deletion mutations were observed in spike protein and non-structural protein encoding regions of Delta variant and they are ORF8:D119-, ORF8:F120-, S:F156-, S:F157-, S:R158-, and S:Y144- (Table 11). Figure 3 suggests that Y144- deletion mutation occurs in S protein of all the 3 variants. In ORF1a protein, it was observed that S3675-, G3676-, and E3677- were common mutations for Omicron and Mauritius variants (Figure 5). From Figure 6, we noticed that in ORF1b protein, none of the variants had deletion mutation.
3.9. Proteins with the Highest Mutation Count
The amino acid substitution and deletion mutation count in different protein of the SARS-CoV-2 virus as observed in the 25 samples (Figure 7). Clearly the figure states that in order, Spike protein (S), ORF1a as well as ORF1b and Nucleocapsid (N) protein were highly mutated by substitution mutations. Moreover, ORF8 and S proteins have the highest amino acid deletion mutation count. On the other side, none of the SARS-CoV-2 variants acquired deletion mutation at the ORF1b, ORF3a, E, M, ORF7a, ORF7b gene regions.
4. Discussion
The rapid geographical spread of the novel coronavirus disease, COVID-19, that commenced at the end of 2019 leaving a pernicious and traumatic impact in every aspect of human life, spread drastically and rapidly around the whole world posing a global health threat and even now, it is prevailing and circulating in different regions of the world taking away more lives [37]. The secret of this viral particle being uncontrollable altogether is believed to be hidden in its threatening potential of attaining genetic diversity due to random recombinations and mutations of different categories like synonymous, non-synonymous mutations (substitution) as well as insertion-deletion mutations [38]. Like other RNA viruses, for fitness, survival and possibly for it’s pathogenicity SARS-CoV-2 goes through genetic alterations to create genetic diversity. During replication of the viral RNA, random nucleotide sequence error occurs that may or may not alter the protein sequence. If it does not result in protein sequence alteration, it may have regulatory impact [37]. However, despite low fidelity of RNA-dependent RNA polymerase [39], SARS-CoV-2 has 3′ to 5′ exonuclease activity that performs the proofreading job which explains a slower mutation attainment of this viral particle [40].
Therefore, to combat this continual threat and havoc evoked by this virus, analysis of genomic surveillance, mutational spectrum, viral behavior and its related pathogenesis, as well as infectibility is crucial for adopting preventive strategies like development of therapeutics as well as vaccines of different categories [39,41]. Potential thrapeutic strategies include use of RNA interference [42] as weapon to combat SARS-CoV-2 i.e., siRNA-based therapeutics [43], miRNAs as antiviral therapy [44], and circRNAs as antiviral targets [45]. Moreover, recent startling techniques for vaccine development, that are under evaluation includes DNA/RNA vaccines and Epitope base vaccines [46,47,48]. Recurrent mutations occur in RNA viruses and SARS-CoV-2 is not different from this and so, designing and developing the treatment therapeutics and vaccines for controlling the viral catastrophe entirely, is difficult [49,50,51].
Socio-demographic and disease condition related data of COVID-19 patients in Table 1 shows that among the 25 COVID positive patients 48% (n=12) were male and 52% (n=13) were female and none of them had chronic kidney disease, diabetes mellitus, chronic obstructive pulmonary disease or long-distance travelling history whereas our previous research on COVID-19 conducted in vast population reported that 47.20% and 3.27% of the COVID-19 positive patients had Diabetes Mellitus and chronic renal disease, respectively [52]. In this study, the gender distribution of COVID-19 contrasts our previous study which depicts that male subjects (42%) were prone to SARS-CoV-2 virus infection compared to female and that research work was conducted in a larger population comprising 8480 participants [52]. From several analysis it was found that people suffering from diabetes, chronic obstructive pulmonary disease or chronic kidney disease before being infected with SARS-CoV-2 had poor disease progression, needed extra treatment, higher hospitalization rate and had 4 times higher mortality rate if they were chronic obstructive pulmonary disease patients [53]. Among the study participants, 48% maintained quarantine period while 16% had positive family history of COVID infection. However, 12% of the study subjects undergo reinfection with SARS-CoV-2 and 4% of them were long term COVID-19 positive though 60% of them were vaccinated (Figure 1). Of most interest, 12% had hypertension and 4% had asthma with hypertension whereas our previous investigation in lager size of population (n=8480) revealed that 46.57% and 18.22% of the COVID-19 patients had history of hypertension and asthma, respectively [52]. Moreover, patients with asthma were reported to have severe disadvantage with higher risk of being infected and suffered much more than the others [53].
Lineage and clade analysis of SARS-CoV-2 variants in Bangladesh demonstrated that 88% (n=22) of the study subjects were carrier of Delta variants (Figure 2) whereas 20 of the 22 Delta variants fall in 21A clade and the other 2 are of 21J clade (Table 2). However, lineage evaluation by Pangolin revealed that the Delta variants evolved from 9 lineages (i.e., AY.122, AY.122.1, AY.131, AY.26, AY.29, AY.30, AY.39, AY.4, B.1.617.2) where 7 (28%) and 6 (24%) of the Delta variants were from B.1.617.2 and AY.4 lineages respectively (Table 3). Besides, 2 (8%) were from AY.131 and other 2 (8%) of the Delta variants were from AY26 lineage and the rest of the variants were from AY.122, AY.122.1, AY.29, AY.30 and AY.39 lineages each comprising of 4% of all the 25 viral entities. The disappearance of one variant may be due to a lack of adaptability while the emergence of the other can be demonstrated from the example of Delta B.1.617.1 and B.1.617.2 variants. In Maharashtra, these 2 variants had proportions of 55-60% and 10-60%, respectively, within the period of February to March 2021 [54]. However, in Bangladesh, the SARS-CoV-2 sample collected from July to August 2021 manifested that B.1.617.1 became rare in our population while the second one (B.1.617.2) predominantly prevails among the other 8 lineages with a proportion of 28%. However, the most prevalent clade of Omicron in different countries is claimed to be BA.1, among the 3 clades (i.e., BA.1.1, BA.2, and BA.3), which also goes the same in case of our observation in Bangladesh [55]. Hence, 2 (8%) of the Omicron variants follow the BA.1 lineage and one of them is of 21K clade and the other is of 20A clade. After the emergence of Mauritius in January, 2021 it spread at a high rate of eleven to seventy-five percent within the time of March to May of that year [56]. However, in August 2021, Mauritius was identified in this present study with 4% (n=1) of incidence which is lower than the estimated incidence in Gabon. The lineage of this variant in our country was identified to be B.1.318, which belongs to the 20B clade. However, in the present study, Delta, Mauritius, and Omicron were found except alpha, beta, and gamma. It might be due to the spatial and temporal travelling of variants of the SARS-CoV-2 virus.
In this cross-sectional descriptive study, the maximum number of substitution mutations in Delta variant was 45, while the minimum number of this type of mutation was 14 (Table 4). On the contrary, the Delta variant that first emerged in India in Oct 2020, had 21 non-synonymous mutations, and 5 synonymous mutations. Nevertheless, in place of only 1 deletion in the Indian Delta variant, the Delta variant from our country contains a maximum of 4 (creating a gap of 18bp) and a minimum of 3 deletion (13bp gap) which implies that at most 4x increased number of deletions occur within this time frame of Delta traveling from India to Bangladesh (Table 4). Similar to the Indian Delta variant, the spike protein in this study also had a deletion at 156/157 position (Table 11) [55]. Unlike the Indian variant, the Bangladeshi variant had an amino acid deletion at Y144, and R158 in S protein (Table 11). However, none of the Delta variants from our country was found to have insertion mutation (Table 9).
The first emergence of Omicron (another name: GRA) was documented in November 2021 in several countries around the world. Though the number of mutations that Omicron carries is much higher than previous VOCs, it shares some common mutations with the previous variants. Notwithstanding, the first documented Omicron had 45 nonsynonymous mutations, in our genomic analysis we found that it contains maximum 53 and minimum 41 substitution mutations (Table 4). Whereas in place of 7 deletions we have seen Bangladeshi Omicrons harboring maximum 6 (45bp gap created) and minimum 6 (39bp gap) deletion loci (Table 4) [55]. Additionally, 2 Omicron variants acquired 9bp insertion mutation in the spike protein region (Table 4, Table 9). In our study Mauritius, on the other hand, was observed to have 36 substitution mutation, 5 (35bp gap) deletions and 1 insertion (3bp) mutations where the number of mutations is higher than the previously recorded one (Table 4) [56,57,58].
The sequence of the viral genetic makeup was published by GenBank in January 2020 (accession no. MN908947.3). So far, the most frequent mutations of this viral genome are 23403A>G (D614G, in S protein), 241C>T (in 5′ UTR region), 3037C>T (in nsp3 of ORF1a), and 14408C>T (in nsp12 of ORF1b) [59,60]. However, in Bangladesh, the most commonly occurring mutations are detected to be 28881G>A (Nucleocapsid protein (N) region), 23403A>G, 28882G>A (N), 28883G>C (N), 1163A>T (ORF1a), and 14408C>T (ORF1ab) [5]. From our analysis, a total of 691 substitutions mutations were observed, and among the identified mutations, A23403G, C3037T, C14408T, G15451A, C25469T, T26767C, C23604G, A28461G, and C21618G, were the utmost frequent substitutions being present in at least 20 of the 25 viral genomes (Table 5). D614G is one of the ten hotspot mutations that occurs at the frequency of 0.10 and was found to be associated with viral infectivity, greater transmissibility as well as higher fatality rate [61,62]. SARS-CoV-2 packaging and titers may be impaired due to mutations at non-coding region like 5′ UTR i.e., 241C>T, and 210G>T [11,60]. Mutational spectra analysis of Asian and European SARS-CoV-2 demonstrated that 23403A>G, 3037C>T, and 28144T>C (L84S, ORF8), mutations occur together with mutations 241C>T which variants have higher infectability [63]. Similarly, in the present study, a co-occurrence of 241C>T with 23403A>G, 3037C>T, and 28144T>C mutations was seen. It was noticeable that, 3037C>T and 23403A>G, and 14408C>T mutation had been seen in most of the cases (Table 5). Moreover, mutation 241C>T tends to coincide with mutation 210G>T most of the time among these twenty-five adult COVID-19 Bangladeshi patients which should be underscored for further evaluating the molecular and biological aspect of viral behavior.
It was reported that host cellular enzymes modify host RNAs as well as viral RNAs like methylation of adenosine and deamination (by ADAR and APOBEC enzymes) of cytidine to uracil and adenosine to inosine. Deamination results in C to T and A to G change in the sequence which explains the fact that C-T and A-G mismatch occurs during multiple sequence alignment with wild type reference genome of SARS-CoV-2. As a result, the deamination of cytidine and adenosine were held accountable for 65% of the C>T synonymous substitutions as well as 22% of the A>G substitution mutations [37,64]. In our study we also found that the frequency of C>T (38.48%) mutation was much higher than that of G>T (18.23%), A>G (11.52%) mutation (Table 6). It’s also mention-worthy that, so far, in our study, we observed that transition (T>C or G>A) mutations occur more often than transversion mutation (i.e., T>G or A>C) and the percentage of transition as well as transversion mutations were 67% and 33%, accordingly (Table 7). As mentioned earlier, the possible reason behind this can be the increased rate of deamination of cytidine to uracil, and adenosine to inosine. However, further study can be conducted to explain this phenomenon as well.
As presented in Table 8, deletions observed in the Delta variant genome include 22029-22035 (S), 28248-28254 (ORF8), 28274 (N), as well as 29750-29752 (non-coding) in case of 21A clade and 21992-21994 (S), 22029-22034 (S), 28248-28253(ORF8), 28271 (non-coding) in case of 21J clade, who reside in the Spike (S) protein-encoding gene, in Nucleocapsid encoding gene and in ORF8 as well as in a non-coding region. Research suggests that deletion at the S gene can pose many more challenges because it can upsurge the ability of spike protein attachment with the ACE2 protein of the host cell and therefore increase transmissibility or even pathogenicity [54]. All the aforementioned deletions of the 21J clade were present in both of the variants except the deletion at 21992-21994 which was seen only in a sample (accession number OM277498) which gives a hint that the later one may gather more advantageous mutations and evolve as a new variant of interest. On the other hand, most of the deletions in the Omicron genome were unique and were not found in Delta or Mauritius variants. Moreover, the Omicron variant’s 20A clade contains deletions at 6 loci i.e., 6513-6516 (ORF1a), 11287-11296 (ORF1a), 21766-21772 (S), 21987-21996 (S), 22194-22197 (S), and 28363-28372 (N) among them deletion at 2 loci of ORF1a were not seen in the Delta variants though deletion at around 11.3kbp site was seen in the Mauritius variant. However, though at around the same loci, 21K clade of Omicron variant contain a little different range of deletion varying in 1bp or 2bp and they are 6513-6515 (ORF1a), 11285-11293 (ORF1a), 21765-21770 (S), 21987-21995 (S), 22194-22196 (S), and 28362-28370 (N) deletions residing in ORF1a, Spike glycoprotein and Nucleocapsid gene regions. Deletion at S gene site may facilitate viral transmission, infectivity, pathogenicity, and/or vaccine efficacy. However, Mauritius variant includes 11288-11297 (ORF1a), 21994-21997(S), 27887-27902 (ORF7b, ORF8), 28254 (ORF8), and 28896-28899 (N) deletion mutations which also have unique deletions at ORF7b, ORF1a sites along with deletions at S, N and ORF8 genes. To sum up, Omicron variant has more diverse deletions at vulnerable site S gene, then the Mauritius variant and at last least number of deletions were encompassed by the Delta variant.
While looking for insertion mutations in our investigation, we found that 2 insertion mutations of 9bp length in the spike glycoprotein region was seen in 2 of the Omicron variants, 22206: GCCAGAAGA (in 20A clade) and 22204:GAGCCAGAA (in 21K clade) (Table 9). In the case of the Mauritius variant, a 3bp (28250:CTG) insertion mutation was observed in the ORF8 region.
To have a bird’s eye view of the mutational spectra of S, N proteins, and OR1a, ORF1b polyproteins lollipop plots of amino acid substitution (red lollipop) and deletion (blue lollipop) mutations in all of the 3 variants are given in Figure 3, Figure 4, Figure 5, and Figure 6, accordingly. From the Figure 3, it is clear that the most significant one- Spike protein of the Omicron variant, most of the unique mutations as well as deletion and substitution mutation were noticed. Substitution mutation T19R, T95I, E156G, L452R, T478K, D614G, P681R, D950N were observed in most of the Delta variant and T95I, as well as D614G were found in all the 3 variants. Moreover, P681 was substituted with R (Arginine) in Delta but with H (Histidine) in Omicron and Mauritius variants while A67 was substituted with V (Valine) in Omicron but it was substituted with T (Threonine) Mauritius variant which refers to a phylogenetic deviation point. In addition, D796 was substituted with Y (Tyrosine) in Omicron and with H in Mauritius variant while E484 was substituted with Q (Glutamine), A (Alanine), and K (Lysine) in the Delta, Omicron and Mauritius variants, respectively. Furthermore, G142D and H655Y were found both in Delta and Omicron variants. Besides, in the case of Omicron variant, most of the substitution mutations were concentrated to the Receptor Binding Domain (RBD) while deletion mutations were found to be clustered to the N-terminal region of the S1 functional subunit of spike glycoprotein.
However, in Nucleocapsid protein (N) of Delta variant no amino acid deletion was observed as we have seen in the nucleotide deletion section only a single bp was deleted at 28274 locus which eventually didn’t result in AA deletion. In addition, compared to the other two variants most of the mutations were in the Delta variant (Figure 4). Interestingly, only R203 was found to be substituted in all these 3 variants but it was substituted to M (Methionine) in Delta while it was substituted to K (Lysine) in Omicron and Mauritius variants. In the Omicron and Mauritius variants common mutation was G204R. Besides, M1X, D63G, R203M, G215C, and D377Y were the most frequent mutations in Delta variant samples.
Regarding ORF1a, in the case of Delta mutations encompassed the whole polyprotein region and the other 2 variants showed fewer mutations in the N terminal regions from which angle we can give a thought for further research to dive into figuring out their changes in viral behavior (Figure 5). In Delta variants K261N, P309L, A1306S, P1640L, H2092Y, P2287S, V2930L, A3209V, T3255I, T3646A, and V3718A were the most frequent mutations. Moreover, while comparing the variants, only L3606F was observed to be common in all of the 3 variants and A3209V was observed in the Delta and Mauritius variants whereas T3255I was found in both Delta and Omicron variants. However, Though S3675-, G3676-, and E3677- deletion mutations were found to happen in the Omicron and Mauritius variants, no deletion mutation was found in the Delta variant’s ORF1a polyprotein.
From our investigation, it was revealed that ORF1b protein did not have mutations at all after 2473th amino acid residue, in other words downright from the N terminal domain of NSP4 in all these 3 variants (Figure 6). In the case of the Delta variants P314L, G662S, P1000L, and A1918V were most frequent mutations in the viral genomes. When comparing the variants of Delta, Omicron and Mauritius, it was observed that P314L was present in all the 3 variants and in some samples of Delta variant, in place of L (Leu) P314 was substituted with F (Phenylalanine) which might indicate the emergence of a new strain of the Delta variant. Moreover, V2371 was substituted with L (Leu) in Delta variant which was substituted to M (Methionine) in the Mauritius variant that kind of differential substitutions rise new research interest on the fact that if they can give more advantages i.e., post translational modification to the later one [65]. In a nutshell, it can be said that the mutational spectrum analysis gives clear insights into the significant mutational variations and gradual changes of the viral genome until it evolves into a new strain or variant of interests.
Recent research implies that the unique and characteristic spike protein mutations of Delta variants include P681R, T19R, T478K, L452R, D614G, and D950N as well as deletion at the part of the N-terminal domain region, Δ157-158. Higher transmissibility of Delta variant can be demonstrated from the adoption of 6 point mutations in spike protein gene over time and the noteworthy among them are P681R and L452R as well as the aforementioned deletion at Δ157-158, residing in the Receptor Binding Domain and altogether they were found to be associated with increase in binding to the ACE2 protein and antibody neutralization for evading the immune system [54]. The point mutation at the S1-S2 cleavage site of spike protein, P681R found in Delta variants in our study (Figure 3 (a)), gives an assumption that this deletion might be associated with its strain’s higher replication ability resulting in higher viral load and increasing transmission [66]. In our investigation, 30 signature/unique mutations were found in the spike protein of Omicron variants among the twenty-five adult COVID-19 patients (Figure 3). Due to the various unique deleterious alterations especially in the Receptor Binding Domain (RBD) of the spike glycoprotein, where in place of 4 unique alterations detected in the Delta variant Omicron contains 12 unique variants that are absent in the Delta (Figure 3 (a), and 3 (b)), it is believed that the Omicron variant intensified the health concern by posing an increased risk of reinfection, reduced treatment response that threatens the treatment strategy, and host immunity evasion [67,68,69].
Regarding Mauritius variant, Cherian et al. hypothesized that this variant possesses some unique mutations that are common in this clade like P618R, L452R, and E484Q in the S gene region which might be associated with the rapid infections and transmission in the province of eastern Maharashtra [54]. Similarly, in this present study Y144-, D614G, D796H, E484K, P681H, and T95I were identified in the Spike protein of Mauritius variant (Figure 3(c)). Nevertheless, in the Republic of Gabon this variant was reported as carrying P681H, E484K, D614G, D796H, and T95I, as well as deletion at Y144- where they proposed that D614G might have a role in the rapid transmission of this VOI. Furthermore, it was stated that E484K and Y144- have an impact in decreasing vaccine efficacy.
It was documented that the genes of ORF1ab open reading frame i.e., nsp1-3, nsp12, and nsp15, as well as the spike protein gene, S, and ORF8 gene were reported to have remarkably more mutations than other genes [37]. On the other hand, in present study ORF1a, ORF1b, ORF3a, and nucleocapsid gene were detected to acquire comparatively significant number of substitution mutations than the other genes (Figure 7). Moreover, analysis on deletion mutation unraveled that most of the aa deletion mutation was acquired by the variants at S and ORF8 genes while no aa deletion mutation was observed in ORF1b, ORF3a, E, M, ORF7a, and ORF7b proteins (Figure 7).
To conclude, it can be said that the results presented here in this investigation can provide new insights into the mutational spectra of SARS-CoV-2 viral genome and can help in constructing genetic database of COVID-19 of Bangladeshi adult COVID-19 patients based on the course of period from the very beginning of this disease incidence in Bangladesh till this date and genetic surveillance should be continued as this SARS-CoV-2 has yet not been eradicated from our country or many parts of this world. The remarkable and significant findings of our investigation on this novel viral genome shaded light on the demand for understanding the molecular basis of SARS-CoV-2 genetics isolated from Bangladeshi COVID-19 patients and might have value in the near future in the field of diagnosis of the disease in medicinal treatment, development of vaccines and decision making in its management. Moreover, by exploring the evolution of mutational spectra of SARS-CoV-2 Variants of Concern or Variants of Interest from different global regions at different time frames, researchers can have an understanding of the preference of virus’s genomic mutation types and preference of mutation sites that the virus tends to acquire for increasing their adaptation strategy, transmissibility, survival, pathogenicity, and for growing resistance against drugs that ultimately leads to the emergence of newly evolved viral strain with their extraordinary advantages over the old one and in this project our evaluated information on viral genome can add a new dimension in unraveling the challenges that SARS-CoV-2 imposed.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Substitution and Amino Acid substitution mutations are presented in the Supplementary materials. Table S1. Nucleotide substitution mutations in 3 different SARS-CoV-2 variants enlisted with their genomic annotations observed in at most 3 samples. Table S2. List of amino acid substitution mutations and annotated genes with their number of occurrences which were found in the least number of samples.
Author Contributions
Conceptualization: LAB, MH; Methodology: LAB, AS, MH, NA, SKS, ZH; Software: AS, LAB, NA; Validation: AS, LAB; Formal Analysis: AS, LAB, MH, NNN; Investigation: AS, LAB, MH, NA; Resources: LAB, MH, SKS, ZH; Data Curation: AS, LAB, MH, NA, NNN; Writing—Original Draft Preparation: AS, LAB, MH, NA, NNN; Writing—Review and Editing: LAB, MH, NNN, SKS, ZH; Visualization: LAB, MH, NNN; Supervision: LAB, MH, SKS, ZH; Project Administration: LAB; All authors have read and approved the final work.
Funding
Integrated Health Science Research and Development Fund, the Ministry of Health, Government of Bangladesh;.
Data Availability Statement
All the data are accessible using https://www.ncbi.nlm.nih.gov/ website by the following accession numbers: OM019149, OM019148, OM019138, OM019145, OM019139, OM019150, OM019143, OM019146, OM090130, OM090137, OM090136, OM090135, OM090140, OM019153, OM019152, OM019154, OM019140, OM019147, OM019141, OM019155, OM090139, OM277491, OM277497, OM277498, and OM019151. 10.6084/m9.figshare.27263484.
Acknowledgments
We would like to acknowledge the Integrated Health Science Research and Development Fund activities from the Ministry of Health, Government of Bangladesh for funding support of this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sharma, A.; Tiwari, S.; Deb, M. K.; Marty, J. L. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): a global pandemic and treatment strategies. International Journal of Antimicrobial Agents 2020, 56. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, S. M.; Yu, X. H.; Tang, S. L.; Tang, C. K. Coronavirus disease 2019 (COVID-19): current status and future perspectives. Int J Antimicrob Agents 2020, 55. [Google Scholar] [CrossRef] [PubMed]
- Gabutti, G.; d’Anchera, E.; Sandri, F.; Savio, M.; Stefanati, A. Coronavirus: update related to the current outbreak of COVID-19. Infectious diseases and therapy 2020, 9, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Umakanthan, S.; Sahu, P.; Ranade, A. V.; Bukelo, M. M.; Rao, J. S.; Abrahao-Machado, L. F.; Dahal, S.; Kumar, H.; Kv, D. Origin, transmission, diagnosis and management of coronavirus disease 2019 (COVID-19). Postgrad Med J 2020, 96, 753–758. [Google Scholar] [CrossRef]
- Siam, M. H. B.; Hasan, M. M.; Tashrif, S. M.; Rahaman Khan, M. H.; Raheem, E.; Hossain, M. S. Insights into the first seven-months of COVID-19 pandemic in Bangladesh: lessons learned from a high-risk country. Heliyon 2021, 7. [Google Scholar] [CrossRef]
- COVID-19 Dynamic Dashboard for Bangladesh. (DGHS), D. G. o. H. S., Ed.; 2024.
- Fehr, A. R.; Perlman, S. Coronaviruses: an overview of their replication and pathogenesis. Coronaviruses: methods and protocols 2015, 1-23.
- Hartog, N.; Faber, W.; Frisch, A.; Bauss, J.; Bupp, C. P.; Rajasekaran, S.; Prokop, J. W. SARS-CoV-2 infection: molecular mechanisms of severe outcomes to suggest therapeutics. Expert review of proteomics 2021, 18, 105–118. [Google Scholar] [CrossRef]
- Millet, J. K.; Whittaker, G. R. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus research 2015, 202, 120–134. [Google Scholar] [CrossRef]
- Wang, N.; Shang, J.; Jiang, S.; Du, L. Subunit vaccines against emerging pathogenic human coronaviruses. Frontiers in microbiology 2020, 11, 298. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. The lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Ziebuhr, J. Molecular biology of severe acute respiratory syndrome coronavirus. Current Opinion in Microbiology 2004, 7, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Bartlam, M.; Xu, Y.; Rao, Z. Structural proteomics of the SARS coronavirus: a model response to emerging infectious diseases. Journal of Structural and Functional Genomics 2007, 8, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Zheng, Q.; Zhang, H.; Niu, Y.; Lou, Y.; Wang, H. The SARS-CoV-2 spike glycoprotein biosynthesis, structure, function, and antigenicity: implications for the design of spike-based vaccine immunogens. Frontiers in immunology 2020, 11, 576622. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J. W.; Kim, V. N.; Chang, H. The architecture of SARS-CoV-2 transcriptome. Cell 2020, 181, 914–921. [Google Scholar] [CrossRef]
- Liu, S.; Xiao, G.; Chen, Y.; He, Y.; Niu, J.; Escalante, C. R.; Xiong, H.; Farmar, J.; Debnath, A. K.; Tien, P. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. The Lancet 2004, 363, 938–947. [Google Scholar] [CrossRef]
- Ke, Z.; Oton, J.; Qu, K.; Cortese, M.; Zila, V.; McKeane, L.; Nakane, T.; Zivanov, J.; Neufeldt, C. J.; Cerikan, B. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 2020, 588, 498–502. [Google Scholar] [CrossRef]
- Schoeman, D.; Fielding, B. C. Coronavirus envelope protein: current knowledge. Virology journal 2019, 16, 1–22. [Google Scholar] [CrossRef]
- Li, J.; Lai, S.; Gao, G. F.; Shi, W. The emergence, genomic diversity and global spread of SARS-CoV-2. Nature 2021, 600, 408–418. [Google Scholar] [CrossRef]
- Wu, Y. C.; Chen, C. S.; Chan, Y. J. The outbreak of COVID-19: An overview. J Chin Med Assoc 2020, 83, 217–220. [Google Scholar] [CrossRef]
- El Baz, S.; Draoui, A.; Echchakery, M.; del Rey, N. L.-G.; Chgoura, K. Spread of COVID-19 and Its Main Modes of Transmission. In Handbook of Research on Pathophysiology and Strategies for the Management of COVID-19, IGI Global, 2022; pp 78-95.
- Tang, B.; Bragazzi, N. L.; Li, Q.; Tang, S.; Xiao, Y.; Wu, J. An updated estimation of the risk of transmission of the novel coronavirus (2019-nCov). Infectious disease modelling 2020, 5, 248–255. [Google Scholar] [CrossRef]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K. S.; Lau, E. H.; Wong, J. Y. Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia. New England journal of medicine 2020, 382, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Ruan, F.; Huang, M.; Liang, L.; Huang, H.; Hong, Z.; Yu, J.; Kang, M.; Song, Y.; Xia, J. SARS-CoV-2 viral load in upper respiratory specimens of infected patients. New England journal of medicine 2020, 382, 1177–1179. [Google Scholar] [CrossRef] [PubMed]
- Singhal, T. A review of coronavirus disease-2019 (COVID-19). The indian journal of pediatrics 2020, 87, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; Zumla, A.; Locatelli, F.; Ippolito, G.; Kroemer, G. Coronavirus infections: Epidemiological, clinical and immunological features and hypotheses. Cell stress 2020, 4. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; Zhang, Y. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA internal medicine 2020, 180, 934–943. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. The lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Teymouri, M.; Mollazadeh, S.; Mortazavi, H.; Naderi Ghale-noie, Z.; Keyvani, V.; Aghababaei, F.; Hamblin, M. R.; Abbaszadeh-Goudarzi, G.; Pourghadamyari, H.; Hashemian, S. M. R.; et al. Recent advances and challenges of RT-PCR tests for the diagnosis of COVID-19. Pathology—Research and Practice 2021, 221, 153443. [Google Scholar] [CrossRef]
- Mascola, J. R.; Graham, B. S.; Fauci, A. S. SARS-CoV-2 viral variants—tackling a moving target. Jama 2021, 325, 1261–1262. [Google Scholar] [CrossRef]
- Rella, S. A.; Kulikova, Y. A.; Dermitzakis, E. T.; Kondrashov, F. A. Rates of SARS-CoV-2 transmission and vaccination impact the fate of vaccine-resistant strains. Scientific Reports 2021, 11. [Google Scholar] [CrossRef]
- for Immunization, N. C. Science Brief: SARS-CoV-2 Infection-induced and Vaccine-induced Immunity. In CDC COVID-19 Science Briefs [Internet], Centers for Disease Control and Prevention (US), 2021.
- Forchette, L.; Sebastian, W.; Liu, T. A comprehensive review of COVID-19 virology, vaccines, variants, and therapeutics. Current medical science 2021, 1–15. [Google Scholar] [CrossRef]
- Iftekhar, E. N.; Priesemann, V.; Balling, R.; Bauer, S.; Beutels, P.; Valdez, A. C.; Cuschieri, S.; Czypionka, T.; Dumpis, U.; Glaab, E. A look into the future of the COVID-19 pandemic in Europe: an expert consultation. The Lancet Regional Health–Europe 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, M.; Huang, X.; Zhang, G.; Zeng, L.; Zhang, G.; Wu, S.; Wang, Y. SRplot: A free online platform for data visualization and graphing. PLOS ONE 2023, 18. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, A.; Mirzazadeh, A.; Tavakolpour, S. Genetics and genomics of SARS-CoV-2: A review of the literature with the special focus on genetic diversity and SARS-CoV-2 genome detection. Genomics 2021, 113, 1221–1232. [Google Scholar] [CrossRef]
- Chen, Z.-w.; Li, Z.; Li, H.; Ren, H.; Hu, P. Global genetic diversity patterns and transmissions of SARS-CoV-2. medRxiv 2020, 2020.2005. 2005.20091413.
- Rehman, S. U.; Shafique, L.; Ihsan, A.; Liu, Q. Evolutionary trajectory for the emergence of novel coronavirus SARS-CoV-2. Pathogens 2020, 9. [Google Scholar] [CrossRef]
- Minskaia, E.; Hertzig, T.; Gorbalenya, A. E.; Campanacci, V.; Cambillau, C.; Canard, B.; Ziebuhr, J. Discovery of an RNA virus 3′→ 5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proceedings of the National Academy of Sciences 2006, 103, 5108–5113. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E. Mechanisms of viral emergence. Veterinary research 2010, 41. [Google Scholar] [CrossRef]
- Qureshi, A.; Tantray, V. G.; Kirmani, A. R.; Ahangar, A. G. A review on current status of antiviral siRNA. Reviews in Medical Virology 2018, 28. [Google Scholar] [CrossRef]
- Ghosh, S.; Firdous, S. M.; Nath, A. siRNA could be a potential therapy for COVID-19. EXCLI journal 2020, 19, 528. [Google Scholar]
- Demirci, M. D. S.; Adan, A. Computational analysis of microRNA-mediated interactions in SARS-CoV-2 infection. PeerJ 2020, 8, e9369. [Google Scholar] [CrossRef]
- Zhang, X.; Chu, H.; Wen, L.; Shuai, H.; Yang, D.; Wang, Y.; Hou, Y.; Zhu, Z.; Yuan, S.; Yin, F. Competing endogenous RNA network profiling reveals novel host dependency factors required for MERS-CoV propagation. Emerging microbes & infections 2020, 9, 733–746. [Google Scholar]
- Bhattacharya, M.; Sharma, A. R.; Patra, P.; Ghosh, P.; Sharma, G.; Patra, B. C.; Lee, S. S.; Chakraborty, C. Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-COV-2): Immunoinformatics approach. Journal of medical virology 2020, 92, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Kream, R. M.; Stefano, G. B. An evidence based perspective on mRNA-SARS-CoV-2 vaccine development. Medical science monitor: international medical journal of experimental and clinical research 2020, 26, e924700–924701. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-y.; Kong, W.-p.; Huang, Y.; Roberts, A.; Murphy, B. R.; Subbarao, K.; Nabel, G. J. A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature 2004, 428, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Parvez, M. S. A.; Rahman, M. M.; Morshed, M. N.; Rahman, D.; Anwar, S.; Hosen, M. J. Genetic analysis of SARS-CoV-2 isolates collected from Bangladesh: Insights into the origin, mutational spectrum and possible pathomechanism. Comput Biol Chem 2021, 90, 107413. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y. J.; Wei, C. L.; Ee, A. L.; Vega, V. B.; Thoreau, H.; Su, S. T.; Chia, J. M.; Ng, P.; Chiu, K. P.; Lim, L.; et al. Comparative full-length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection. Lancet 2003, 361, 1779–1785. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Wang, X.; Yuan, X.; Xiao, G.; Wang, C.; Deng, T.; Yuan, Q.; Xiao, X. The epidemiology and clinical information about COVID-19. Eur J Clin Microbiol Infect Dis 2020, 39, 1011–1019. [Google Scholar] [CrossRef]
- Hossain, M.; Mannan, R.; Islam, S.; Banu, L. A.; Jamee, A. R.; Hassan, Z.; Elias, S. M.; Das, S. K.; Azad Khan, A. K. Unveiling the occurrence of COVID-19 in a diverse Bangladeshi population during the pandemic. Frontiers in Public Health 2024, 12, Original. [Google Scholar] [CrossRef]
- Sanyaolu, A.; Okorie, C.; Marinkovic, A.; Patidar, R.; Younis, K.; Desai, P.; Hosein, Z.; Padda, I.; Mangat, J.; Altaf, M. Comorbidity and its Impact on Patients with COVID-19. SN Compr Clin Med 2020, 2, 1069–1076. [Google Scholar] [CrossRef]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9. [Google Scholar] [CrossRef]
- McLean, G.; Kamil, J.; Lee, B.; Moore, P.; Schulz, T. F.; Muik, A.; Sahin, U.; Türeci, Ö.; Pather, S. The Impact of Evolving SARS-CoV-2 Mutations and Variants on COVID-19 Vaccines. mBio 2022, 13. [Google Scholar] [CrossRef]
- Manouana, G. P.; Nzamba Maloum, M.; Bikangui, R.; Oye Bingono, S. O.; Ondo Nguema, G.; Honkpehedji, J. Y.; Rossatanga, E. G.; Zoa-Assoumou, S.; Pallerla, S. R.; Rachakonda, S.; et al. Emergence of B.1.1.318 SARS-CoV-2 viral lineage and high incidence of alpha B.1.1.7 variant of concern in the Republic of Gabon. Int J Infect Dis 2022, 114, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Ramuth, M.; Amoako, D.; Scheepers, C.; Wilkinson, E.; Giovanetti, M.; Lessells, R.; Giandhari, J.; Ismail, A.; Martin, D.; et al. A Novel and Expanding SARS-CoV-2 Variant, B.1.1.318, dominates infections in Mauritius; 2021. [CrossRef]
- Olawoye, I. B.; Oluniyi, P. E.; Oguzie, J. U.; Uwanibe, J. N.; Kayode, T. A.; Olumade, T. J.; Ajogbasile, F. V.; Parker, E.; Eromon, P. E.; Abechi, P.; et al. Emergence and spread of two SARS-CoV-2 variants of interest in Nigeria. Nature Communications 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, D.; Giorgi, F. M. Geographic and genomic distribution of SARS-CoV-2 mutations. Frontiers in microbiology 2020, 11, 1800. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T.; Platt, D.; Parida, L. Variant analysis of SARS-CoV-2 genomes. Bulletin of the World Health Organization 2020, 98. [Google Scholar] [CrossRef]
- Laamarti, M.; Alouane, T.; Kartti, S.; Chemao-Elfihri, M.; Hakmi, M.; Essabbar, A.; Laamarti, M.; Hlali, H.; Bendani, H.; Boumajdi, N. Large scale genomic analysis of 3067 SARS-CoV-2 genomes reveals a clonal geo-distribution and a rich genetic variations of hotspots mutations. PloS one 2020, 15. [Google Scholar] [CrossRef]
- Cardozo, T. SARS-CoV-2 viral spike G614 mutation exhibits higher case fatality rate. Int J Clin Pract 2020, 74. [Google Scholar]
- Yin, C. Genotyping coronavirus SARS-CoV-2: methods and implications. Genomics 2020, 112, 3588–3596. [Google Scholar] [CrossRef]
- Li, Y.; Yang, X.; Wang, N.; Wang, H.; Yin, B.; Yang, X.; Jiang, W. The divergence between SARS-CoV-2 and RaTG13 might be overestimated due to the extensive RNA modification. Future Virology 2020, 15, 341–347. [Google Scholar] [CrossRef]
- Fung, T. S.; Liu, D. X. Post-translational modifications of coronavirus proteins: roles and function. Future Virol 2018, 13, 405–430. [Google Scholar] [CrossRef]
- Lopez Bernal, J.; Andrews, N.; Gower, C.; Gallagher, E.; Simmons, R.; Thelwall, S.; Stowe, J.; Tessier, E.; Groves, N.; Dabrera, G. Effectiveness of Covid-19 vaccines against the B. 1.617. 2 (Delta) variant. New England Journal of Medicine 2021, 385, 585–594. [Google Scholar] [CrossRef]
- SARS-CoV-2, B. 1.1.529 (Omicron) Variant—United States, December 1-8, 2021. MMWR Morb Mortal Wkly Rep 2021, 70, 1731–1734. [Google Scholar] [CrossRef]
- Choudhary, O. P.; Dhawan, M. Omicron variant (B. 1.1. 529) of SARS-CoV-2: threat assessment and plan of action. International Journal of Surgery 2022, 97, 106187. [Google Scholar] [CrossRef] [PubMed]
- Abulsoud, A. I.; El-Husseiny, H. M.; El-Husseiny, A. A.; El-Mahdy, H. A.; Ismail, A.; Elkhawaga, S. Y.; Khidr, E. G.; Fathi, D.; Mady, E. A.; Najda, A.; et al. Mutations in SARS-CoV-2: Insights on structure, variants, vaccines, and biomedical interventions. Biomed Pharmacother 2023, 157, 113977. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Vaccination status of COVID-19 patients.
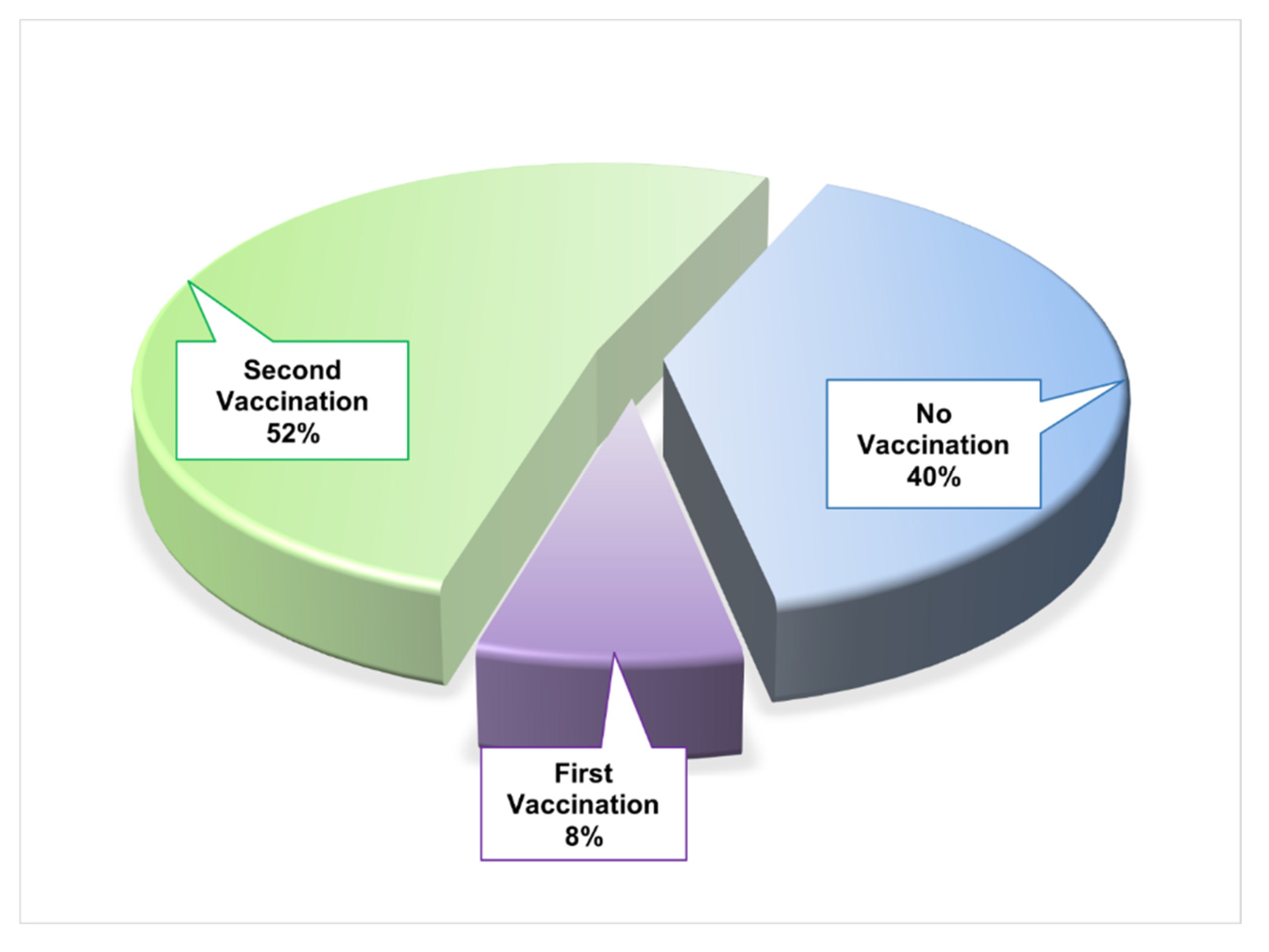
Figure 2.
Distribution of SARS-CoV-2 variants in COVID-19 patients.
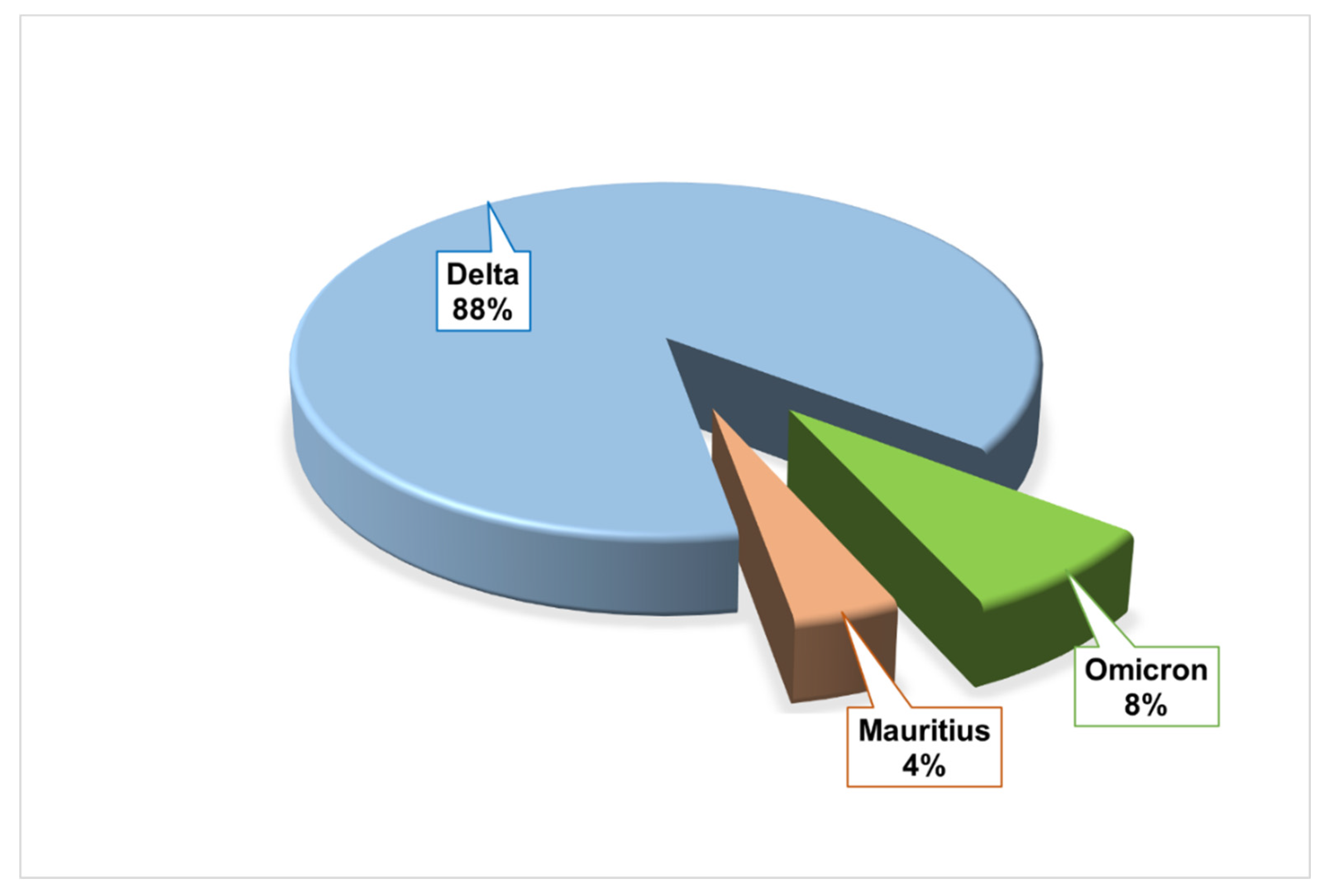
Figure 3.
Needle Plot for the visual representation of amino acid (AA) substitution (red lollipop) and deletion (blue lollipop) mutations in different domains and motifs of Spike glycoprotein (S protein) of the Delta (a), Omicron (b), and Mauritius (c) variants of SARS-CoV-2 virus. Bold and Blue font was used to refer to the most frequent mutations in a specific variant’s samples, while red color represents remarkable variants, and sometimes, the red colored mutations are common for 3 variants. On the other hand, black and bold font represent common mutations for any of the 2 variants.
Figure 3.
Needle Plot for the visual representation of amino acid (AA) substitution (red lollipop) and deletion (blue lollipop) mutations in different domains and motifs of Spike glycoprotein (S protein) of the Delta (a), Omicron (b), and Mauritius (c) variants of SARS-CoV-2 virus. Bold and Blue font was used to refer to the most frequent mutations in a specific variant’s samples, while red color represents remarkable variants, and sometimes, the red colored mutations are common for 3 variants. On the other hand, black and bold font represent common mutations for any of the 2 variants.
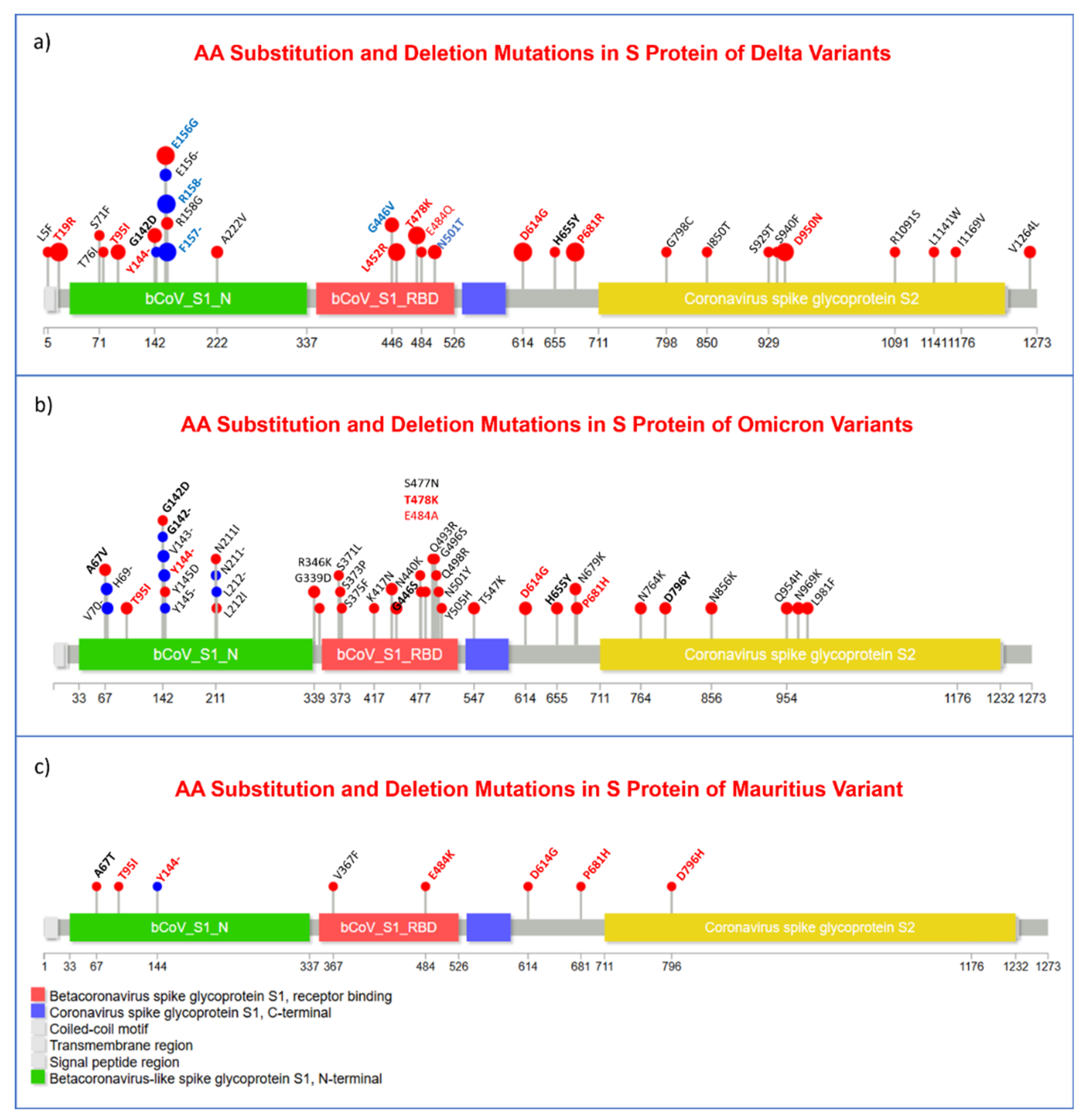
Figure 4.
Lollipop plot representing the amino acid (AA) substitution and deletion mutations in nucleocapsid protein (N protein) of Delta (a), Omicron (b), and Mauritius (c) variants where the red lollipop represents the aa substitution mutation and deletion mutations are symbolized by the blue one. No deletion mutation was observed in the nucleocapsid protein of the Delta variant. Regarding the annotation of mutations red-bold font represents the signature of important mutations reported in previous studies while blue-bold font represents mutations that were found in a specific variant in highest frequency. Black-bold font was used for referring to mutations that were present in 2 variants at least.
Figure 4.
Lollipop plot representing the amino acid (AA) substitution and deletion mutations in nucleocapsid protein (N protein) of Delta (a), Omicron (b), and Mauritius (c) variants where the red lollipop represents the aa substitution mutation and deletion mutations are symbolized by the blue one. No deletion mutation was observed in the nucleocapsid protein of the Delta variant. Regarding the annotation of mutations red-bold font represents the signature of important mutations reported in previous studies while blue-bold font represents mutations that were found in a specific variant in highest frequency. Black-bold font was used for referring to mutations that were present in 2 variants at least.

Figure 5.
Substitution (red lollipop) and deletion (blue lollipop) mutations in ORF1a polyprotein region of the 3 variants, Delta (a), Omicron (b), and Mauritius (c) are shown in this Needle plot. Annotation of mutations were done based on the following font style- black-bold font refers to mutations being identified in at least 2 variants; blue-bold font refers to mutations that were observed in highest frequency in a specific variant, red-bold font indicates the noteworthy mutations and, in some cases mutations that were found in the 3 variants.
Figure 5.
Substitution (red lollipop) and deletion (blue lollipop) mutations in ORF1a polyprotein region of the 3 variants, Delta (a), Omicron (b), and Mauritius (c) are shown in this Needle plot. Annotation of mutations were done based on the following font style- black-bold font refers to mutations being identified in at least 2 variants; blue-bold font refers to mutations that were observed in highest frequency in a specific variant, red-bold font indicates the noteworthy mutations and, in some cases mutations that were found in the 3 variants.
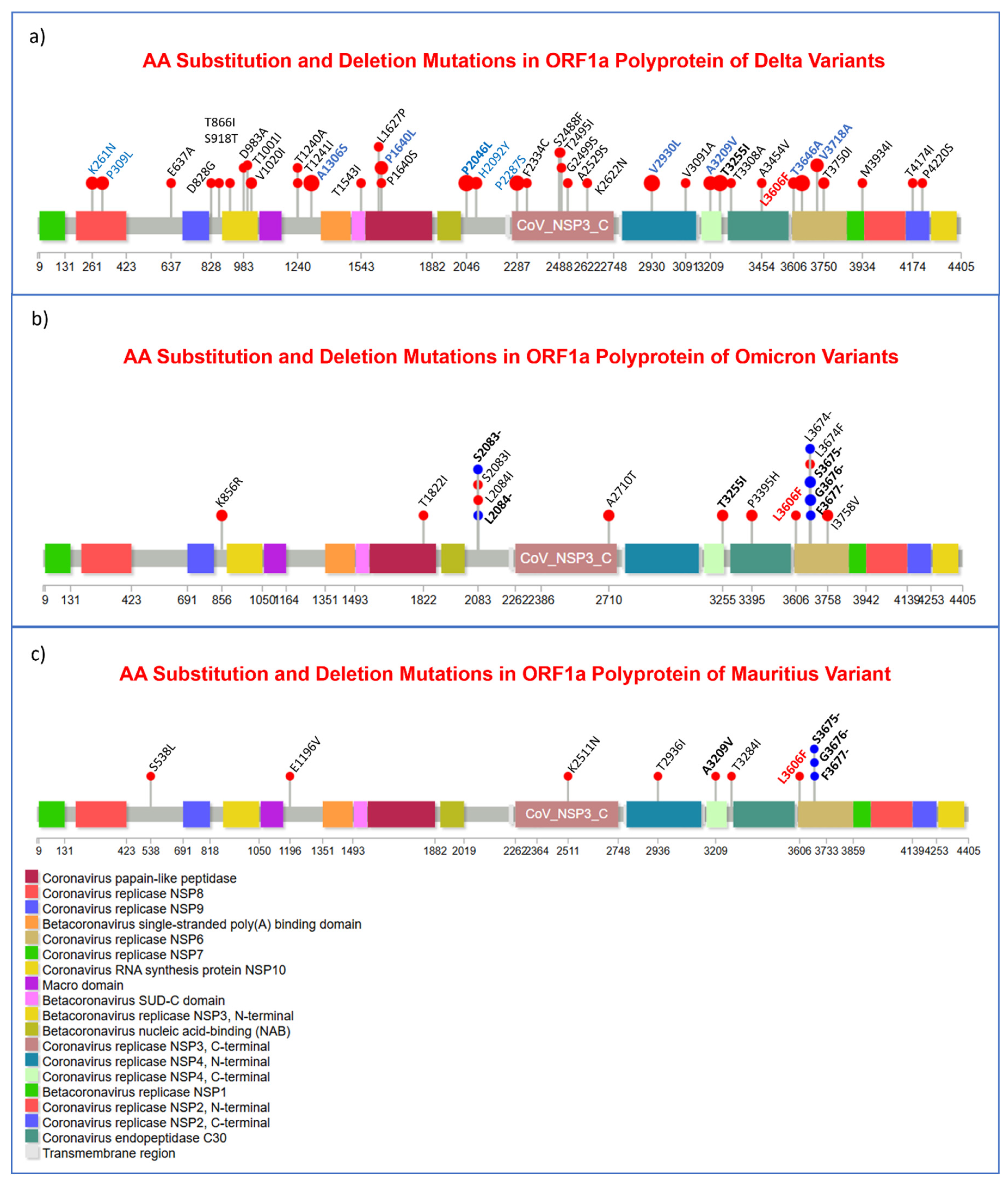
Figure 6.
Exhibition of substitution mutations in ORF1b polyprotein region in Delta (a), Omicron (b), and Mauritius (c) viral genomes. No amino acid deletion was observed in this region of ORF1b gene. Here the red lollipop presents the aa substitution mutation and deletion mutations are symbolized by the blue one. For annotation of the mutations, black-bold font refers to mutations that were present in 2 variants, whereas, the blue-bold one represents mutations that were present in most of the samples of a specific variant and red-bold implies to the remarkable and in some cases, it refers to the mutations that were present in all the 3 variants. .
Figure 6.
Exhibition of substitution mutations in ORF1b polyprotein region in Delta (a), Omicron (b), and Mauritius (c) viral genomes. No amino acid deletion was observed in this region of ORF1b gene. Here the red lollipop presents the aa substitution mutation and deletion mutations are symbolized by the blue one. For annotation of the mutations, black-bold font refers to mutations that were present in 2 variants, whereas, the blue-bold one represents mutations that were present in most of the samples of a specific variant and red-bold implies to the remarkable and in some cases, it refers to the mutations that were present in all the 3 variants. .
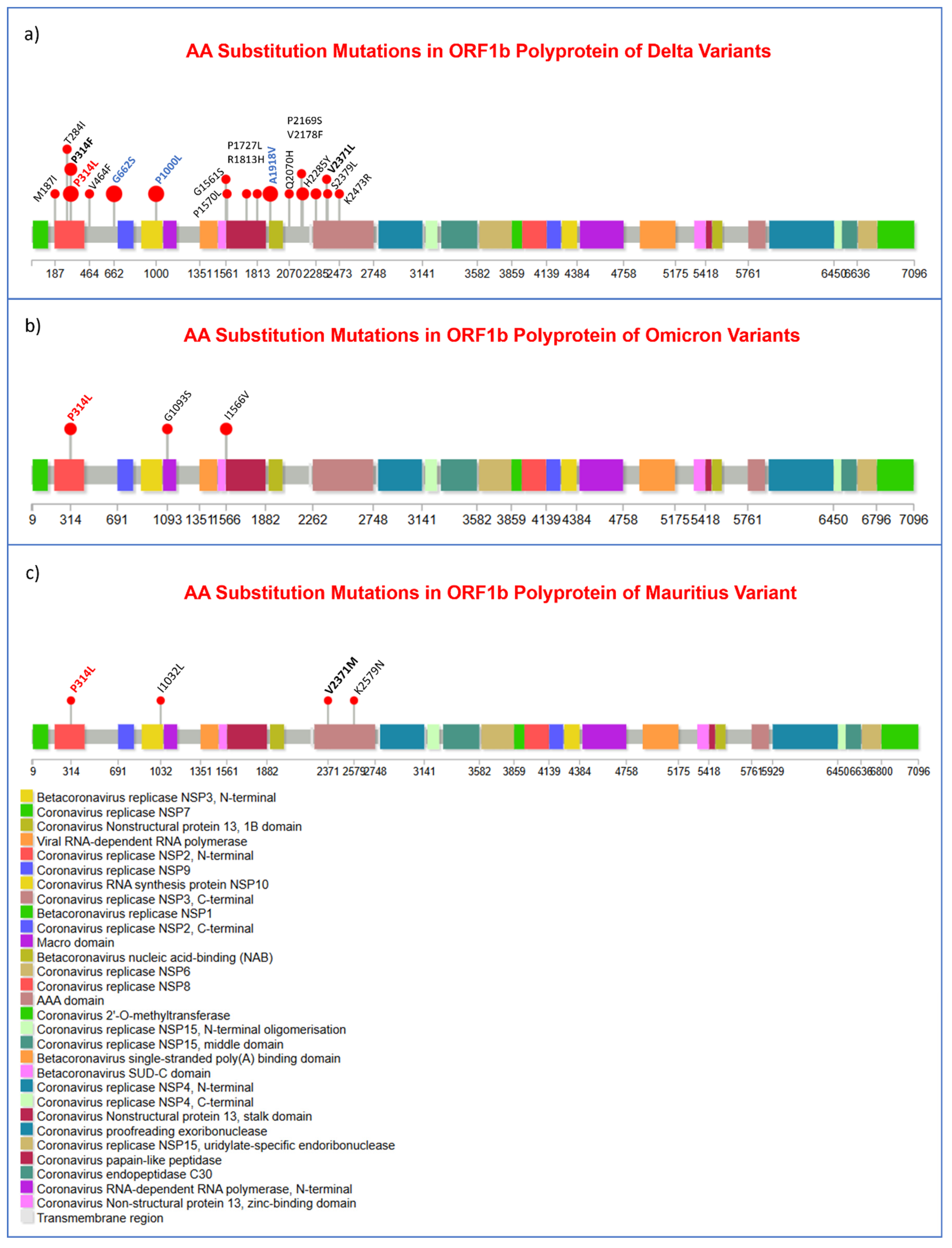
Figure 7.
Amino acid (AA) substitution and AA deletion mutation number in different proteins of viral genomes.
Figure 7.
Amino acid (AA) substitution and AA deletion mutation number in different proteins of viral genomes.
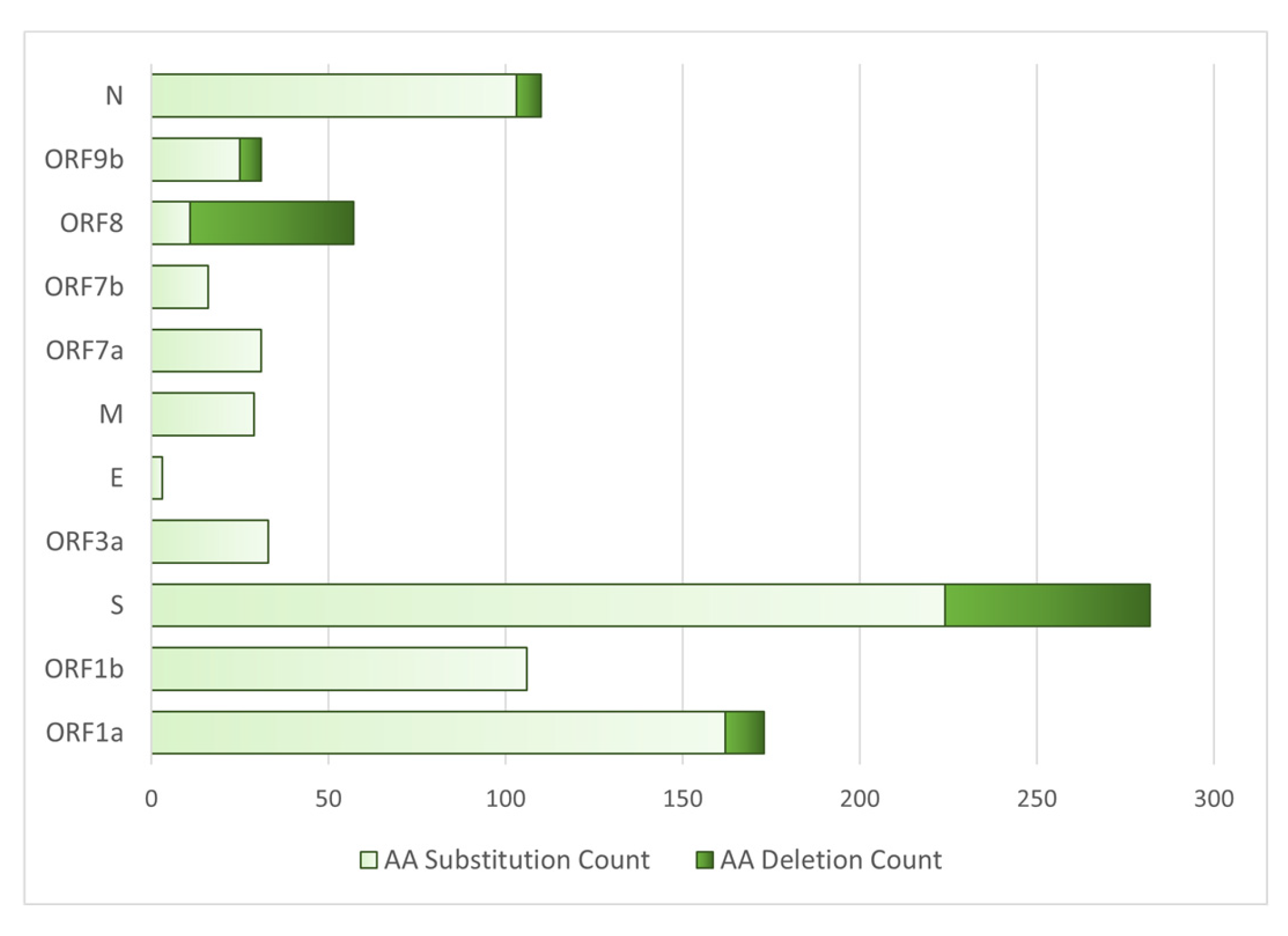

Table 1.
Frequency of sociodemographic and comorbidity of the COVID-19 patients.
| Socio-demography and Comorbidity | Number | Percentage (%) |
|---|---|---|
| Male | 12 | 48 |
| Female | 13 | 52 |
| Family history of COVID-19 Infection | 4 | 16 |
| Maintained quarantine period | 12 | 48 |
| Hypertension | 3 | 12 |
| Hypertension with asthma | 1 | 4 |
| Chronic kidney disease (CKD) | 0 | 0 |
| Diabetes mellitus | 0 | 0 |
| Chronic obstructive pulmonary disease (COPD) | 0 | 0 |
| Re-infected with SARS-CoV-2 | 3 | 12 |
| Vaccinated | 15 | 60 |
| Long duration of COVID-19 positive | 1 | 4 |
| History of long-distance traveling | 0 | 0 |
Table 2.
Frequency of identified clades of SARS-CoV-2.
| Variant | Clade | Number | Percentage (%) |
|---|---|---|---|
| Delta | 21A | 20 | 80 |
| 21J | 2 | 8 | |
| Omicron | 20A | 1 | 4 |
| 21K | 1 | 4 | |
| Mauritius | 20B | 1 | 4 |
Table 3.
Frequency of lineage distribution of SARS-CoV-2 variants.
| Variant | Lineage | Number | Percentage (%) |
|---|---|---|---|
| Delta | B.1.617.2 | 7 | 28 |
| AY.4 | 6 | 24 | |
| AY.131 | 2 | 8 | |
| AY.26 | 2 | 8 | |
| AY.29 | 1 | 4 | |
| AY.30 | 1 | 4 | |
| AY.39 | 1 | 4 | |
| AY.122 | 1 | 4 | |
| AY.122.1 | 1 | 4 | |
| Omicron | BA.1 | 2 | 8 |
| Mauritius | B.1.1.318 | 1 | 4 |
Table 4.
The types of mutations (in numbers) observed in each COVID-19 patient.
| Mutation | Variant | ||||
|---|---|---|---|---|---|
| Omicron | Delta | Mauritius | |||
| Maximum | Minimum | Maximum | Minimum | ||
| Substitution | 53 | 41 | 45 | 14 | 36 |
| Deletion sites with gap in bp | 6 (45) | 6 (39) | 4 (18) | 3(13) | 5 (35) |
| Insertion sites with length in bp | 1 (9) | 0 | - | - | 1 (3) |
Table 5.
List of substitution mutations with their mapped gene/genomic regions in SARS-CoV-2.
| Genomic region | Substitution | Number of Mutation | Genomic region | Substitution | Number of Mutation |
|---|---|---|---|---|---|
| ORF1a | C3037T | 25 | S | C23604G | 22 |
| C10029T | 16 | C21618G | 20 | ||
| G4181T | 15 | G24410A | 17 | ||
| C6402T | 15 | C22995A | 16 | ||
| C8986T | 15 | T22917G | 14 | ||
| G9053T | 15 | C21846T | 9 | ||
| A11201G | 14 | G21987A | 5 | ||
| A11332G | 14 | G22899T | 5 | ||
| C7124T | 10 | A23064C | 4 | ||
| C9891T | 7 | ORF3a | C25469T | 22 | |
| C5184T | 6 | G26104T | 4 | ||
| T12946C | 6 | M | T26767C | 23 | |
| C1191T | 5 | ORF7a | C27752T | 14 | |
| C1267T | 5 | T27638C | 13 | ||
| T11418C | 5 | ORF7b | C27874T | 15 | |
| G1048T | 4 | ORF8 | C28054G | 5 | |
| A2560G | 4 | N | A28461G | 21 | |
| G11083T | 4 | G28881T | 19 | ||
| ORF1b | C14408T | 25 | G29402T | 15 | |
| G15451A | 22 | G28916T | 14 | ||
| C16466T | 19 | T29014C | 4 | ||
| C19220T | 15 | 5′ Leader Sequence | C241T | 16 | |
| C14407T | 5 | G210T | 14 | ||
| G19999T | 5 | near 3′ end | G29742T | 14 | |
| S | A23403G | 25 | G29688T | 5 |
Table 6.
Nucleotide base changes of substitution mutation in the SARS-CoV-2 viral genome.
| Nucleotide base change | Number | Percentage (%) |
|---|---|---|
| C>T | 344 | 38.48 |
| G>T | 163 | 18.23 |
| A>G | 103 | 11.52 |
| G>A | 77 | 8.61 |
| T>C | 75 | 8.39 |
| C>G | 48 | 5.37 |
| C>A | 33 | 3.69 |
| T>G | 22 | 2.46 |
| A>C | 10 | 1.12 |
| A>T | 7 | 0.78 |
| G>C | 6 | 0.67 |
| T>A | 6 | 0.67 |
Table 7.
Comparison of frequency of transition (i.e., T>C, G>A) and transversion (i.e., T>A, G>C) mutation in SARS-CoV-2.
Table 7.
Comparison of frequency of transition (i.e., T>C, G>A) and transversion (i.e., T>A, G>C) mutation in SARS-CoV-2.
| Mutation | Base Change | Number | Percentage (%) |
|---|---|---|---|
| Transition | Purine>Purine/ Pyrimidine>Pyrimidine |
599 | 67 |
| Transversion | Purine>Pyrimidine/ Pyrimidine>Purine |
295 | 33 |
Table 8.
Deletion mutations in the SARS-CoV-2 viral genome from distinctive clades along with their genomic annotation.
Table 8.
Deletion mutations in the SARS-CoV-2 viral genome from distinctive clades along with their genomic annotation.
| Deletion (bp) of variant (gene) | ||||
|---|---|---|---|---|
| Delta | Omicron | Mauritius | ||
| 21A | 21J | 20A | 21K | 20B |
| 22029-22035 (S) | 21992-21994 (S) | 6513-6516 (ORF1a) | 6513-6515 (ORF1a) | 11288-11297 (ORF1a) |
| 28248-28254 (ORF8) | 22029-22034 (S) | 11287-11296 (ORF1a) | 11285-11293 (ORF1a) | 21994-21997(S) |
| 28274 (N) | 28248-28253 (ORF8) |
21766-21772 (S) |
21765-21770 (S) | 27887-27902 (ORF7b, ORF8) |
| 29750-29752 (Non-coding) | 28271 (Non-coding) |
21987-21996 (S) | 21987-21995 (S) | 28254 (ORF8) |
| 22194-22197 (S) |
22194-22196 (S) | 28896-28899 (N) | ||
| 28363-28372 (N) | 28362-28370 (N) | |||
Table 9.
Insertion mutation and their genomic locations in different types of SARS-CoV-2 viral genome.
Table 9.
Insertion mutation and their genomic locations in different types of SARS-CoV-2 viral genome.
| Variant (Clade) |
Insertion position with inserted sequence | Length (bp) | Mapped gene |
|---|---|---|---|
| Omicron (21K) |
22204:GAGCCAGAA | 9 | S |
| Omicron (20A) |
22206:GCCAGAAGA | 9 | S |
| Mauritius (20B) |
28250:CTG | 3 | ORF8 |
Table 10.
Amino acid substitution in SARS-CoV-2 viral genome.
| Genomic region | Amino Acid Substitutions | Number of Mutations | Genomic region | Amino Acid Substitutions | Number of Mutations |
|---|---|---|---|---|---|
| ORF1a | T3255I | 16 | S | D950N | 17 |
| A1306S | 15 | T478K | 16 | ||
| P2046L | 15 | L452R | 14 | ||
| V2930L | 15 | T95I | 9 | ||
| T3646A | 14 | G142D | 6 | ||
| P2287S | 10 | G446V | 5 | ||
| A3209V | 7 | N501T | 4 | ||
| P1640L | 5 | H655Y | 3 | ||
| P309L | 5 | P681H | 3 | ||
| V3718A | 5 | ORF3a | S26L | 22 | |
| K261N | 4 | D238Y | 4 | ||
| L3606F | 4 | M | I82T | 24 | |
| H2092Y | 3 | ORF7a | T120I | 14 | |
| ORF1b | G662S | 22 | V82A | 13 | |
| P314L | 20 | L116F | 3 | ||
| P1000L | 19 | ORF7b | T40I | 15 | |
| A1918V | 15 | ORF8 | S54* | 5 | |
| P314F | 5 | ORF9b | T60A | 21 | |
| V2178F | 5 | N | M1X | 20 | |
| S | D614G | 25 | D63G | 19 | |
| P681R | 22 | R203M | 19 | ||
| E156G | 20 | D377Y | 15 | ||
| T19R | 20 | G215C | 14 |
Table 11.
Amino acid deletion mutations of SARS-CoV-2 viral genome.
| Gene | Deletion Mutations |
Number of Mutations |
|---|---|---|
| ORF1a | S3675- | 3 |
| G3676- | 3 | |
| F3677- | 2 | |
| L2084- | 1 | |
| S2083- | 1 | |
| L3674- | 1 | |
| S | F157- | 22 |
| R158- | 20 | |
| E156- | 2 | |
| H69- | 2 | |
| V70- | 2 | |
| V143- | 2 | |
| Y144- | 4 | |
| Y145- | 1 | |
| L212- | 1 | |
| G142- | 1 | |
| N211- | 1 | |
| ORF8 | D119- | 22 |
| F120- | 22 | |
| M1- | 1 | |
| K2- | 1 | |
| ORF9b | N28- | 2 |
| A29- | 2 | |
| V30- | 1 | |
| E27- | 1 | |
| N | E31- | 2 |
| R32- | 2 | |
| S33- | 2 | |
| R209- | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



