
Submitted:
17 October 2025
Posted:
20 October 2025
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The Delta variant of SARS-CoV-2 virus, one of the alarming variants of concern (VOC) with a distinct mutation characteristic, was immensely detrimental and a significant cause of the prolonged pandemic waves. This study intends to analyze the genetic characteristics of the predominant Delta variant detected from Acute Febrile Illness (AFI) patients in Ethiopia. Nasopharyngeal samples were collected from AFI patients in four hospitals during February 2021 to June 2022 and tested for SARS-CoV-2 by using RT-qPCR. Of 101 positive samples, 48 stored specimens were re-tested, and 26 with sufficient RNA quality (Ct < 30) were sequenced using whole-genome sequencing to identify variants of concern, specific virus lineages and mutation features. Delta variants (21J clade) found predominant among all the sequenced SARS-CoV-2 isolate (80.8%, 21/26). AY.120 (46.2%) and B.1.617.2 (26.9%) were the predominant sub lineages of the delta variant. Omicron (21k, Pango BA.1.1/BA.1.17/ BA.1) and Alpha (20I, Pango B.1.1.7) variants accounted for 11.5% and 7.7% of the total sequenced samples. Phylogenetic analysis showed evidence of local transmission and possible multiple introductions of SARS-CoV-2 VOCs in Ethiopia. The number of mutations increases dramatically from Alpha (~35 avg) to Delta (~42 avg) to Omicron (~56 avg). The Delta variant revealed a spike mutation on L452R and T478K and P681R, and characterized by the double deletion E156-F157- in Spike protein. The findings observed in this study is indicative of a gradual change in the genetic coding of the virus underscoring the importance of ongoing genomic surveillance to track the evolution and spread of SARS-CoV-2 and other emerging virus.
Keywords:
SARS-CoV-2
; mutation
; delta variant
; genetic characteristics
; Ethiopia
Introduction
SARS-CoV-2, an RNA virus, has a high mutation rate due to the error-prone nature of viral RNA-dependent RNA polymerase (RdRp) and recombination events, allowing for rapid evolution and adaptation [1,2,3]. Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), Delta (B.1.617.2), and Omicron (B.1.1.529) are some of the variants of concern (VOCs) that have emerged since SARS-CoV-2 was first discovered in Wuhan, China [1,4,5]. These VOCs all have unique mutations in the spike (S) protein and other genomic regions [6,7,8,9,10,11]. Enhanced binding affinity to the human angiotensin-converting enzyme 2 (ACE2) receptor, greater transmissibility, and immune evasion have all been linked to mutations in the receptor-binding domain (RBD) of the S protein [3,12]. Furthermore, genomic insertions and deletions have been noted, which have been linked to antigenic drift and changed viral fitness [13,14].
Due to its increased transmissibility and capacity for immune evasion, the SARS-CoV-2 Delta variant (B.1.617.2), which was initially discovered in India in late 2020, swiftly rose to prominence as a dominant strain worldwide [1,4,14]. Delta variant was responsible for more than 90% of sequenced cases in multiple countries by the middle of 2021, causing major COVID-19 outbreaks that put a pressure on healthcare systems around the globe [1,4,5,14,15].
SARS-CoV-2 genetic characterization has been crucial for understanding the mutation characteristics, tracking viral transmission, determining toxicity, and influencing public health actions like as vaccine development [16]. There are limitations in publications on the genetic evolution and mutation patterns of SARS-CoV-2 variants in Ethiopia, despite international genomic surveillance efforts, emphasizes the necessity of localized research to guide public health actions [17,18]. This study intends to analyze the genetic characteristics of the predominance Delta variant in Ethiopia aiming to address existing knowledge gap and contributing advance worldwide comprehension of SARS-CoV-2 development in low- and middle-income environments.
Methods and Material
Ethical Clearance
Ethical clearance for this study was obtained from EPHI’s Scientific and Ethical Review Office (EPHI-IRB-254-2020; MM No. 065) and the Institutional Review Board of Addis Ababa University (IRB/19/025). Written informed consent was obtained from each participant prior to the enrollment in the study for an interview and sample collection.
Study Population and Specimen Collection
Genetic analysis of SARS-CoV-2 was conducted using repository nasopharyngeal samples collected from AFI patients in a cross-sectional study during February 2021 to June 2022 in four hospitals (St. Paul Hospital, Jimma University Hospital, Hiwot Fana Hospital and Gonder University Hospital) [19]. The nasopharyngeal swabs were collected from enrolled cases using a sterile COPAN brand universal transport medium containing 1-3ml Viral Transport Media. The collected sample was vortexed and aliquoted in two cryovials for molecular testing at the National Influenza Center (NIC), Ethiopian Public Health Institute (EPHI). The samples were stored at 4°C until transported by trained postal service officers to the NIC at EPHI. EPHI’s NIC (laboratory) identified respiratory viruses from nasopharyngeal swabs.
A total of 737 samples were collected, of which 101 (13.7%) tested positive for SARS-COV-2 by real-time PCR. From these, 48 stored positive samples were randomly selected and re-tested for SARS-CoV-2, and 26 samples with cycle threshold (Ct) values <30 were prepared for the Whole-genome sequencing. The enrolled cases were Inpatients and Outpatients presented at the selected facilities, and who met the case definition criteria for AFI with a predefined inclusion and exclusion criteria.
Viral Load Determination and cDNA Synthesis
Nucleic acid was extracted from the swabs using the MagaBio plus Virus RNA Purification Kit II by MGISP-NE32 automated extractor. Real-time PCR was conducted on an ABI 7500FAST system (Life Technologies, Carlsbad, CA USA) using primers provided by CDC International Reagent Resource (CDC-IRR), a biological reagent repository established to provide better access to laboratory reagents. Samples with Ct values of 30 or less were deemed uniformly suggestive of a substantial viral load for the purposes of genome sequencing, eligible for whole-genome sequencing, and suitable for downstream analysis.
Whole-Genome Sequencing and Assembly
Sequencing of the 26 SARS-CoV-2 genomes were conducted by using Oxford Nanopore Technology's MinION platform, combining the Rapid Barcoding Kit 96 (SQK-RBK110.96) with the Midnight RT PCR Expansion (EXP-MRT001). First, reverse transcription of the extracted viral RNA from 26 positive samples were performed using LunaScript RT SuperMix, followed by a tiled PCR amplification step designed to generate overlapping ~1.2 kb amplicons that cover the entire viral genome. This step utilizes two separate primer pools (Pool A and Pool B) from the Midnight kit, which have been optimized for balanced amplification. The resulting amplicons from both pools were combined, and unique rapid barcodes were ligated to each sample, enabling multiplexed sequencing. The generated barcoded library was then purified using AMPure XP bead-based clean-up, quantified, and prepared for loading by combining with sequencing buffer and loading beads.
The prepared library was loaded onto a primed R9.4.1 flow cell (FLO-MIN106) for sequencing, which was controlled by the MinKNOW software for real-time data acquisition and base calling. Critical settings in MinKNOW, such as enabling barcoding and setting specific barcode filtering scores, were applied for accurate demultiplexing. Following sequencing, downstream analysis was performed using the wf-artic bioinformatics workflow, which leverages the ARTIC pipeline for consensus genome generation, variant calling, and phylogenetic analysis. SARS-CoV-2 raw reads obtained from Oxford Nanopore sequencing machine were assembled by using Genome Detective Platform Version 2.15.1 (https://www.genomedetective.com/) to generate consensus genomes. Genome coverage of consensus genomes > 95% was used for the downstream analysis.
Variant Analysis
The consensus genomes generated by Genome Detective were analyzed for variants of SARS-COV-2 using Web based (https://clades.nextstrain.org/) and CLI Nextclade (version v3.8.2) tools. Sequence quality checks, viral genome alignment, mutation identification, clade assignment, and phylogenetic placement were performed using CLI Nextclade (version v3.8.2). Pangolin lineage assignments (for Pango nomenclature) were also performed using the Web based tool called Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN) (https://pangolin.cog-uk.io/). Consensus genome that contains less than 3000 missing or ambiguous (N’s) nucleotide was used for variant analysis.
Phylogenetic Analysis
For constructing phylogenetic trees and analyzing evolutionary relationships, the CLI Augur pipeline (version 23.1) was employed. The maximum likelihood (ML) method, using the General Time Reversible (GTR) model, was applied for tree building and ancestral state reconstruction. Visualization of the phylogenetic trees generated by the Augur pipeline was done using Auspice.us (https://auspice.us/).
Statistical Analysis and Visualization
Python scripts were created to process the CSV output obtained from NextClade pipeline. Descriptive statistics were used to summarize and highlight the main features of the dataset. The findings were presented through graphs and tables. For the analysis of the mutational profile, a subset of amino acid substitutions and deletion with frequencies above 75% were examined.
Results
Socio-Demographic and Clinical Characteristics
The randomly selected SARS-CoV-2 positive samples for genetic analysis (n = 48 participants consented) was composed of patients with a median age of 43 years old (range: 19-90 years old), where 54% were male and 46% were females (Table 1, below). Sudden onset of fever (>380C measured axillary body temperature) observed in all the participants, Cough (83%), Sore Throat (65%), Headache (71%) and Muscle ache (63%) were the most commonly reported symptoms among participants. Most of participants (90%) in this study were inpatients admitted with a complaint of acute febrile illness.
Assembly and Sequencing Metrics of SARS-CoV-2 Raw Read Sequences
The Genome Detective platform, which generated the consensus sequence of 26 isolates and provided metrics like the number of reads, depth of coverage, nucleotide identity, amino acid identity, and genome coverage. The results showed that the 26 isolates had high sequencing depth, with an average depth of coverage ranging from 279 to 1014; the nucleotide identity and amino acid identity were consistently high across all isolates, with an average of 99.8% and 99.7%, respectively. The average genome coverage was 99.4%, confirming that the data were suitable for downstream genomic analysis. (Table 2).
SARS-CoV-2 Genetic Diversity over the Study Period
In the study period the analysis of genetic diversity showed the predominance of Delta variants or 21J clade (80.8%, 21/26) among all the sequenced SARS-CoV-2 isolate according to WHO and Next strain naming system. The Delta variant was also sub-classified as AY.120 (46.2%) and B.1.617.2 (26.9%) under the Pangolin Lineage naming systems. Omicron and Alpha (Pango B.1.1.7) variants accounted for 11.5% and 7.7% of the total sequenced samples.

Table 3.
The frequency and percentage of SARS-CoV-2 variants in three naming systems (WHO, Pangolin and Next strain).
Table 3.
The frequency and percentage of SARS-CoV-2 variants in three naming systems (WHO, Pangolin and Next strain).
| WHO Variant N (%) | Pangolin Lineage N (%) | Next strain Clade N (%) | |||
|---|---|---|---|---|---|
| Alpha | 2 (7.7) | B.1.1.7 | 2 (7.7) | 20I | 2 (7.7) |
| Delta | 21 (80.8) | AY.120 | 12 (46.2) | 21J | 21 (80.8) |
| B.1.617.2 | 7 (26.9) | ||||
| AY.32 | 2 (7.7) | ||||
| Omicron | 3 (11.5) | BA.1.1 | 1 (3.8) | 21K | 3 (11.5) |
| BA.1.17 | 1 (3.8) | ||||
| BA.1 | 1 (3.8) | ||||
Patterns of SARS-CoV-2 Genetic Variation During the Study
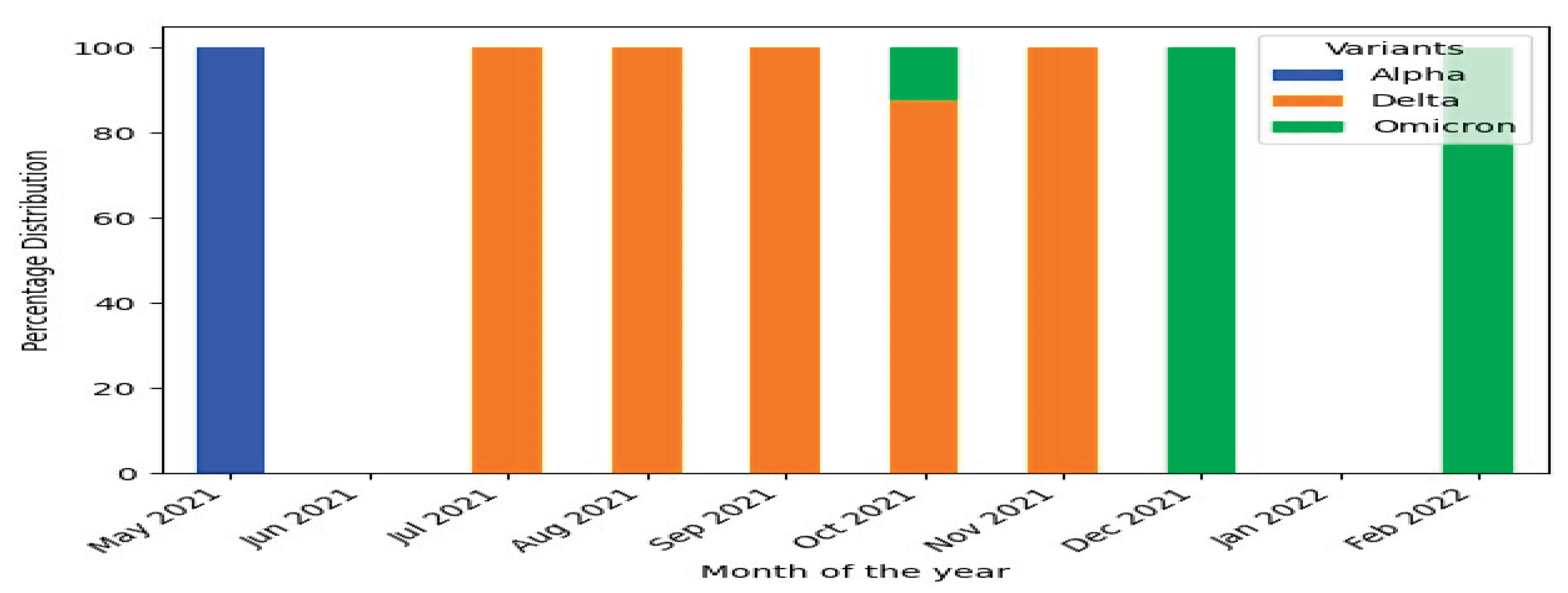
The analysis of the temporal trends in the prevalence of the three SARS-CoV-2 variants during the study period showed that there was Alpha variants in May 2021 with low prevalence. The Delta variants emerged as the predominant variant starting from July 2021 and remained dominant until November 2021. The Omicron variants detected for the first time in October 2021 and observed again on December 2021 and February 2022.
Figure 1.
Distribution of SARS-CoV-2 WHO variants (alpha, delta and omicron) by month during the study period (2021-2022).
Figure 1.
Distribution of SARS-CoV-2 WHO variants (alpha, delta and omicron) by month during the study period (2021-2022).
Nucleotide and Amino Acid Mutation Analysis in SARS-CoV-2 Genes
The mutational analysis reveals a clear evolutionary progression from Alpha to Omicron, characterized by a significant increase in genetic changes, particularly in the Spike protein, which enhances the virus's ability to evade immunity and infect cells. The number of mutations increases from Alpha (~35 avg) to Delta (~42 avg) to Omicron (~56 avg). The alpha variant featured a spike mutation on N501Y (improved cell entry) and P681H (enhanced infectivity). The Delta variant revealed a spike mutation on L452R and T478K (strong immune evasion) and P681R (high infectivity), and characterized by the double deletion E156-F157- in Spike. The Omicron variant contains over 30 changes in Spike alone. Key mutations include G339D, S371L, N440K, S477N, T478K, E484A, Q498R, N501Y (all in the receptor-binding domain, allowing it to evade most existing antibodies), Table 4.
Phylogenetic Analysis
The phylogenetic tree depicted on Figure 2 below shows 3 major clusters (clades) of SARS-CoV-2 variants based on the known variant classification system and distinct branching patterns. Genetically more closely related consensus sequences are grouped in to the same cluster and each cluster forms a distinct branch or set of branches on the tree. On this figure at the bottom left, there is a reference genome, which is the original SARS-CoV-2 isolated in late 2019. All the current SARS-CoV-2 variants are descended from this initial strain. The first cluster, cluster 1, was the smallest (n=2), containing only two SARS-CoV-2 genomes that are Alpha variant. The 2nd cluster consist of Omicron (n=3), which is composed of entirely Omicron variants. The 3rd clade, occupying the upper part of the tree, comprised Delta variants (n=21) that have many sub-lineages within it (Figure 3).
When we see the common ancestral relationships of the variants in the three cluster, the phylogenetic tree shows that Cluster 1 (Alpha variant) is the older group, branching off earlier from the common ancestor of all sequences (i.e from the Wuhan). The Delta variant shows several sub-branches, indicating ongoing evolution and diversification within this variant. The Omicron variant, while small in number, shows the greatest genetic divergence from the root. This shows Omicron's many mutations compared to earlier variants (Figure 2).
Discussion
In this study, SARS-CoV-2 genomes was characterized on nasopharyngeal specimens collected from patients with AFI between February 2021 to June 2022 in four hospitals in Ethiopia. It was found that the Delta variant (lineage B.1.617.2 and AY.120) was predominant among specimens with high viral load, accompanied by characteristic spike and non-spike protein mutations; a smaller proportion of Alpha and Omicron variants were also detected. The high depth and coverage of sequencing enabled reliable variant calling and phylogenetic placement, allowing us to observe diversification of Delta, including emergence of local sub-lineage signatures.
The sequencing metrics found in this study show high-quality SARS-CoV-2 genomic data, with consistently high nucleotide and amino acid identities across all isolates. The nucleotide identity averaged 99.8%, whereas the amino acid identity was 99.7%, showing little sequence divergence from the reference genome. These findings are consistent with recent investigations from East Africa, which have revealed similar high sequence identities ranging from 99.68% to 99.92% and amino acid identities of around 99.94% [17,18,20].
This dataset has a depth of coverage ranging from 279 to 1,014, which guaranteed accurate variant calling and sequence assembly. Similar to earlier research conducted in Africa, where sequencing depths of more than 500× have been documented for SARS-CoV-2 genomes, this degree of coverage supports the precision of consensus sequences and mutation identification. The degree of coverage is similarly comparable to earlier studies conducted in France (coverage range: 279–1,014×) and Northeast Ohio (average coverage: 1,081×; range: 223–2,235×) [21,22]. Averaging 99.4%, genome coverage was also consistently high, with very little differences between isolates. The somewhat decreased genome coverage (96.0% and 95.7%) in some isolates could be due to a combination of factors such as sequencing depth, RNA quality, or the difficulty of sequencing particular genomic areas.
The depth of coverage in this dataset ranging from 279 to 1,014, this ensured a reliable variant calling and sequence assembly. This degree of coverage supports the accuracy of consensus sequences and mutation detection and is comparable to earlier research conducted in Africa, where sequencing depths of more than 500× have been documented for SARS-CoV-2 genomes. The degree of coverage is similarly comparable to other studies conducted in North-east Ohio (average coverage: 1,081×; range: 223–2,235×) and France (range: 279–1,014×) [21,22]. Furthermore, the average genome coverage was 99.4%, with very slight differences amongst isolates. It's possible that the somewhat decreased genome coverage of some isolates (96.0% and 95.7%, for example) was caused by issues with RNA quality, sequencing depth, or the difficulty of sequencing particular genomic areas.
Majority of the sequenced isolates (21/26) in this study were the Delta variant (21J clade). Perhaps this illustrates that the isolates found in this study within a particular timeframe were not considered to be the real variations in Ethiopia during the epidemic. This result was consistent with the GISAID data analysis (up to July 2021), which showed that the Delta variant's prevalence ranged from 67.6% to 98.3% of all circulating variations in Africa ([21]. Before Omicron surpassed it in early 2022, Delta first appeared in the spring of 2021, gained dominance in the middle of the year, and continued to be significantly higher until the end of 2021 [21,23]. Furthermore, Africa's third COVID wave was caused by Delta, which also accounted for more than one-third of all infections on the continent; [21,24]. Compared to earlier variants, Delta has a little increase in immune evasion and is more transmissible, which leads to its increased prevalence[6,25].
AY.120 and B.1.617.2 were the two most common sub-lineages within the Delta variant, accounting for 46.2% and 26.9% of the 26 sequenced samples, respectively. According to a comparative study carried out in England, the increase in AY.120 detections suggested an estimated rate of spatial growth of 6.56 (6.16–6.97), and there were 18,400 detections during the same study period, supporting the predominance observation of sub-lineage AY.120 in this study [26]. This indicates that during the middle of 2021, a Delta infection wave evolving both locally and internationally included the sub-lineages AY.120 and B.1.617.2. In 74 nations, including the UK (85%), India (3%), USA (3%), Germany (2%), and France (1%), AY.120 sub-lineage detections were reported, with a global prevalence of 1% [27]. Furthermore, AY.120 sub-lineage reported in 74 countries including in UK (85%), India (3%), USA (3%), Germany (2%), and France (1%) with a 1% global prevalence [27].
Although a large number of AY.120 lineages have been identified in multiple countries, relatively little information has been published regarding the epidemiological and genetic characteristics of these lineages. Crucially, there is no indication of either a progressive contextual effect, such as rising vaccination rates or natural immunity, that would contribute to the predominance of this sub-lineage, or biological selection pressure favoring the growth advantage of emerging lineages ([26]. However, the core Delta mutations in the spike protein (L452R, T478K, P681R, and others) that are known to increase transmissibility and decrease vaccine susceptibility may be shared by AY.120 because it is a member of the larger Delta clade [28]. No discernible variations in clinical severity were reported in studies on comparable AY lineages (e.g., AY.28, AY.104, AY.3, AY.122); however, AY.120 is not included in these data [29]. Given the results that have been reported, it would appear that this merits more research.
Delta variant sub-lineage B.1.617.2, the other prominent sub-lineage found in this study, was initially discovered in Maharashtra, India, in October 2020, and was designated as a Variant of Concern (VOC) by the WHO on May 11, 2021 [1,4,5,14,30,31]). Because of its high transmissibility and shorter transmission cycles, the Delta variety showed a remarkable global growth. By June 15, 2021, the U.S. CDC reported that B.1.617.2 had been found in at least 66 countries and had spread to over 130 countries [1,5,6,14,25], According to several studies, it is 40–60% more transmissible than Alpha; R₀ is roughly 5 compared to 2.7 for wild-type [5,14,31].
Numerous distinguishing mutations, especially in the spike (S) protein, which increase transmissibility and impact immunological interactions, are present in delta variant sub-lineage B.1.617.2. Similar to the finding in this study, few other studies reported that the most prevalent key spike mutations are T19R, G142D, Δ157 158, R158G, L452R, T478K, D614G, P681R, and D950N [32]. L452R raises the affinity for ACE2 binding and could aid in avoiding the CD8 T-cell response. T478K, which is found in RBD, helps the immune system evade antibodies. P681R increases the efficiency of viral entry by enhancing spike cleavage at the furin site (S1/S2) [11,33,34]. Other mutations that may affect viral replication, assembly, and immunological interactions include D950N in S2, as well as modifications in ORF1a/b, M, N, ORF3a, ORF7a/b, and ORF8 [30].
The Delta variant's L452R and T478K spike mutations worked in concert to prevent neutralizing antibody binding, providing considerable immune evasion against immunity caused by infection and vaccination. At the same time, the P681R mutation significantly improved the effectiveness of spike protein cleavage, which is necessary for viral entry, resulting in significantly higher viral loads and infectivity. The E156-F157 deletion, which is thought to form a glycan shield and add another layer of antibody avoidance, was added to these alterations. The rapid global dominance of the Delta variety was supported by this potent combination of increased transmissibility and robust immune evasion, which were driven by these precise molecular changes [35].
Compared to earlier variants, Delta variant is linked to more severe illness outcomes as reported by some studies. Data from Public Health England & Scotland indicated that the hospitalization was 2× riskier than Alpha. Ct values are lower (less than 30 more frequently), indicating higher infectivity; the viral load is approximately 1,000 times greater than that of ancestral strains [30]. Perhaps there is lack of data indicating the severity of this variant in Ethiopia, studies conducted in other countries has indicated that Delta infection increases the probability of hospitalization by 120%, ICU admission by 287 percent, and fatality by 137% when compared to non-VOC strains.
Clustering patterns suggestive of several importation and local transmission episodes were found in the phylogenetic analyses conducted for this study. Similarly, studies in Zambia [36], that used phylogenetic analysis revealed indications of local transmission and potential numerous introductions of SARS-CoV-2 variants from various European and African countries corroborate this observation. These results highlight the interdependence of SARS-CoV-2 transmission networks as well as the part that worldwide travel and trade play in the virus's propagation [36,37].
Limitations of the Study
This study presented the genetic features of Delta variant detected from AFI patients in Ethiopia where scientific data on genetic characteristics were scares. The AY.120 strain of delta variant presented in this study were not thoroughly studied globally and very few publications were available, the findings on this study may contribute on providing the genetic features of this variant for the global database. Also, it might give an insight on mutation features of this delta variant in the low-middle incoming countries. However, the number of isolates sequenced in this study are few (n= 26). However, the findings can provide a valuable insight on genetic information of the circulating SARS-CoV-2 like other published studies with few numbers of samples such as in Guinea (n=19), Nigeria (n=34) and Zhengzhou, China (n=5); [38,39,40]. Association b/n the epidemiological factors and the identified variants not done in this study due to the few numbers of sample size.
Conclusion
Delta variant sub-lineage AY.120 and B.1.617.2 were the predominant strain of SARS-CoV-2 virus circulating in Ethiopia during the study period. The mutational analysis reveals a clear evolutionary progression, characterized by a significant increase in genetic changes, particularly in the Spike protein. These findings provide partial but valuable insights into the genetic diversity of SARS-CoV-2 in Ethiopia and highlight the importance of continuous genomic surveillance and expanded sequencing efforts to track the spread and evolution of emerging variants.
Author Contributions
MT conceptualized, designed, and drafted the manuscript. DS, AT, WS, MG, AA, AH, AA, MW, AH, NT, TK, NK performed article searching, data extraction, and quality assessment. MT, DS, WS & NT conducted data analysis and wrote the manuscript. AT, MG, AA, AH, AA, MW, AH, TK, NK reviewed the final manuscript. All authors read and approved the final manuscript.
Funding
The author(s) declare that financial support was not received for the research and/or publication of this article.
Institutional Review Board Statement
The studies involving humans were approved by Ethiopian Public Health Institute IRB AND Addis Ababa University’s Aklilu Lemma Institute of Pathobiology IRB. The studies were conducted in accordance with the local legislation and institutional re-quirements. Written informed consent for participation in this study was provided by the participants.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to express our gratitude to all study sites (hospitals), Ethiopian Public Health Institute’s National Influenza center and the personnel that operate there for their crucial contribution. We are grateful to the United States Centers for Disease Control and Prevention (CDC) for their technical support and provision of lab supplies and reagents.
Author Disclaimer
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the U.S. Centers for Disease Control and Prevention.
References
- World Health Organization. Tracking SARS-CoV-2 variants, https://www.who.int/en/activities/tracking-SARS-CoV-2-variants, 2021.
- Domingo E, García-Crespo C, Lobo-Vega R, Perales C. Mutation rates, mutation frequencies, and proofreading-repair activities in rna virus genetics. Vol. 13, Viruses. MDPI; 2021. [CrossRef]
- Wu A, Peng Y, Huang B, Ding X, Wang X, Niu P, et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Vol. 27, Cell Host and Microbe. Cell Press; 2020. p. 325–8. [CrossRef]
- Centers for Disease Control and Prevention. SARS-CoV-2 Variant Classifications and Definitions, https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html. 2021;
- GISAID) GI o. SAID. Tracking of Variants, https://www.gisaid.org/hcov19-284 variants (2021). 2021;
- CDC. Delta variant: what we know about the science. Atlanta, GA: US Department of Health and Human Services, CDC; 2021. 2021.
- Tao K, Tzou PL, Nouhin J, Gupta RK, de Oliveira T, Kosakovsky Pond SL, et al. The biological and clinical significance of emerging SARS-CoV-2 variants. Vol. 22, Nature Reviews Genetics. Nature Research; 2021. p. 757–73. [CrossRef]
- World Health Organization Regional Office for Africa. Guidance on tools for detection and surveillance of SARS CoV-2 Variants Interim Guidance, Geneva: World Health Organization; 2021. Version 1.0. 2021.
- Eur Centre Dis Prev Control. Ba A, Changed VO, Added VO, Ba A, Replaced O. Changes to list of SARS-CoV-2 variants of concern, variants of interest, and variants under monitoring. 2022.
- World Health Organization. *Tracking SARS-CoV-2 variants*. 2023.
- Bhattacharya M, Chatterjee S, Sharma AR, Lee SS, Chakraborty C. Delta variant (B.1.617.2) of SARS-CoV-2: current understanding of infection, transmission, immune escape, and mutational landscape. Vol. 68, Folia Microbiologica. Springer Science and Business Media B.V.; 2023. p. 17–28. [CrossRef]
- Planas D, Veyer D, Baidaliuk A, Staropoli I, Guivel-Benhassine F, Rajah MM, et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature. 2021 Aug 12;596(7871):276–80. [CrossRef]
- McBroome J, Thornlow B, Hinrichs AS, Kramer A, De Maio N, Goldman N, et al. A Daily-Updated Database and Tools for Comprehensive SARSCoV-2 Mutation-Annotated Trees. Mol Biol Evol. 2021;38(12):5819–24. [CrossRef]
- National Collaborating Centre for Infectious Diseases Concern https://nccid.ca/covid-19-variants. Updates on COVID-19 Variants of. 2021.
- Mlcochova P, Kemp SA, Dhar MS, Papa G, Meng B, Ferreira IATM, et al. SARS-CoV-2 B.1.617.2 Delta variant replication and immune evasion. Nature. 2021 Nov 4;599(7883):114–9. [CrossRef]
- Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, Harrison EM, et al. SARS-CoV-2 variants, spike mutations and immune escape. Vol. 19, Nature Reviews Microbiology. Nature Research; 2021. p. 409–24. [CrossRef]
- Aga AM, Mulugeta D, Gebreegziabxier A, Zeleke GT, Girmay AM, Tura GB, et al. Genome diversity of SARS-CoV-2 lineages associated with vaccination breakthrough infections in Addis Ababa, Ethiopia. BMC Infect Dis. 2025 Dec 1;25(1). [CrossRef]
- Hailu Alemayehu D, Adnew B, Alemu F, Abeje Tefera D, Seyoum T, Tesfaye Beyene G, et al. Whole-Genome Sequences of SARS-CoV-2 Isolates from Ethiopian Patients GENOME SEQUENCES. 2025. [CrossRef]
- Chekol MT, Sugerman D, Tayachew A, Mekuria Z, Tesfaye N, Alemu A, et al. Clinical and epidemiological characteristics of influenza and SARS-CoV-2 virus among patients with acute febrile illness in selected sites of Ethiopia 2021–2022. Front Public Health. 2025;13.
- Kia P, Katagirya E, Kakembo FE, Adera DA, Nsubuga ML, Yiga F, et al. Genomic characterization of SARS-CoV-2 from Uganda using MinION nanopore sequencing. Sci Rep. 2023 Dec 1;13(1). [CrossRef]
- Chan WS, Lam YM, Law JHY, Chan TL, Ma ESK, Tang BSF. Geographical prevalence of SARS-CoV-2 variants, August 2020 to July 2021. Sci Rep. 2022 Dec 1;12(1). [CrossRef]
- Nicot F, Trémeaux P, Latour J, Jeanne N, Ranger N, Raymond S, et al. Whole-genome sequencing of SARS-CoV-2: Comparison of target capture and amplicon single molecule real-time sequencing protocols. J Med Virol. 2023 Jan 1;95(1).
- Cohen C, Kleynhans J, von Gottberg A, McMorrow ML, Wolter N, Bhiman JN, et al. Characteristics of infections with ancestral, Beta and Delta variants of SARS-CoV-2 in the PHIRST-C community cohort study, South Africa, 2020-2021. BMC Infect Dis. 2024 Dec 1;24(1). [CrossRef]
- Tegally H, San JE, Cotten M, Moir M, Tegomoh B, Mboowa G, et al. The evolving SARS-CoV-2 epidemic in Africa: Insights from rapidly expanding genomic surveillance. Science (1979). 2022 Oct 7;378(6615). [CrossRef]
- CDC. Science brief: COVID-19 vaccines and vaccination. Atlanta, GA: US Department of Health and Human Services, CDC; 2021. 2021.
- Smallman-Raynor MR, Cliff AD, Robson SC, Connor TR, Loman NJ, Golubchik T, et al. Spatial growth rate of emerging SARS-CoV-2 lineages in England, September 2020-December 2021. Epidemiol Infect. 2022 Jul 20;150.
- Gangavarapu K, Latif AA, Mullen JL, Alkuzweny M, Hufbauer E, Tsueng G, et al. Outbreak.info genomic reports: scalable and dynamic surveillance of SARS-CoV-2 variants and mutations. Nat Methods. 2023 Apr 1;20(4):512–22. [CrossRef]
- Taboada B, Zárate S, García-López R, Muñoz-Medina JE, Sanchez-Flores A, Herrera-Estrella A, et al. Dominance of Three Sublineages of the SARS-CoV-2 Delta Variant in Mexico. Viruses. 2022 Jun 1;14(6). [CrossRef]
- Ranasinghe D, Jayathilaka D, Jeewandara C, Gunasinghe D, Ariyaratne D, Jayadas TTP, et al. Molecular Epidemiology of AY.28 and AY.104 Delta Sub-lineages in Sri Lanka. Front Public Health. 2022 Jun 21;10. [CrossRef]
- Tulimilli SR V., Dallavalasa S, Basavaraju CG, Kumar Rao V, Chikkahonnaiah P, Madhunapantula SR V., et al. Variants of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) and Vaccine Effectiveness. Vol. 10, Vaccines. MDPI; 2022.
- Alhamlan FS, Al-Qahtani AA. SARS-CoV-2 Variants: Genetic Insights, Epidemiological Tracking, and Implications for Vaccine Strategies. Vol. 26, International Journal of Molecular Sciences. Multidisciplinary Digital Publishing Institute (MDPI); 2025. [CrossRef]
- Zhan Y, Yin H, Yin JY. B.1.617.2 (Delta) Variant of SARS-CoV-2: Features, transmission and potential strategies. Vol. 18, International Journal of Biological Sciences. Ivyspring International Publisher; 2022. p. 1844–51. [CrossRef]
- Shayan S, Jamaran S, Askandar RH, Rahimi A, Elahi A, Farshadfar C, et al. The SARS-Cov-2 Proliferation Blocked by a Novel and Potent Main Protease Inhibitor via Computer-aided Drug Design. Iranian Journal of Pharmaceutical Research. 2021 Jun 1;20(3):399–418. [CrossRef]
- Hoteit R, Yassine HM. Biological Properties of SARS-CoV-2 Variants: Epidemiological Impact and Clinical Consequences. Vol. 10, Vaccines. MDPI; 2022. [CrossRef]
- Carabelli AM, Peacock TP, Thorne LG, Harvey WT, Hughes J, de Silva TI, et al. SARS-CoV-2 variant biology: immune escape, transmission and fitness. Vol. 21, Nature Reviews Microbiology. Nature Research; 2023. p. 162–77. [CrossRef]
- Katowa B, Kalonda A, Mubemba B, Matoba J, Shempela DM, Sikalima J, et al. Genomic Surveillance of SARS-CoV-2 in the Southern Province of Zambia: Detection and Characterization of Alpha, Beta, Delta, and Omicron Variants of Concern. Viruses. 2022 Sep 1;14(9). [CrossRef]
- G/meskel W, Desta K, Diriba R, Belachew M, Evans M, Cantarelli V, et al. SARS-CoV-2 variant typing using real-time reverse transcription-polymerase chain reaction–based assays in Addis Ababa, Ethiopia. IJID Regions. 2024 Jun 1;11. [CrossRef]
- Sow MS, Togo J, Simons LM, Diallo ST, Magassouba ML, Keita MB, et al. Genomic characterization of SARS-CoV-2 in Guinea, West Africa. PLoS One. 2024 Mar 1;19(3 March). [CrossRef]
- Shaibu JO, Onwuamah CK, James AB, Okwuraiwe AP, Amoo OS, Salu OB, et al. Full-length genomic sanger sequencing and phylogenetic analysis of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) in Nigeria. PLoS One. 2021 Jan 1;16(1 January). [CrossRef]
- Wang Y, Chen D, Zhu C, Zhao Z, Gao S, Gou J, et al. Genetic Surveillance of Five SARS-CoV-2 Clinical Samples in Henan Province Using Nanopore Sequencing. Front Immunol. 2022 Apr 4;13. [CrossRef]
Figure 2.
Phylogenetic tree visualized using Auspice.us and showing the evolutionary relationships of SARS-CoV-2 variants. (The branch lengths represent genetic divergence between variants).
Figure 2.
Phylogenetic tree visualized using Auspice.us and showing the evolutionary relationships of SARS-CoV-2 variants. (The branch lengths represent genetic divergence between variants).
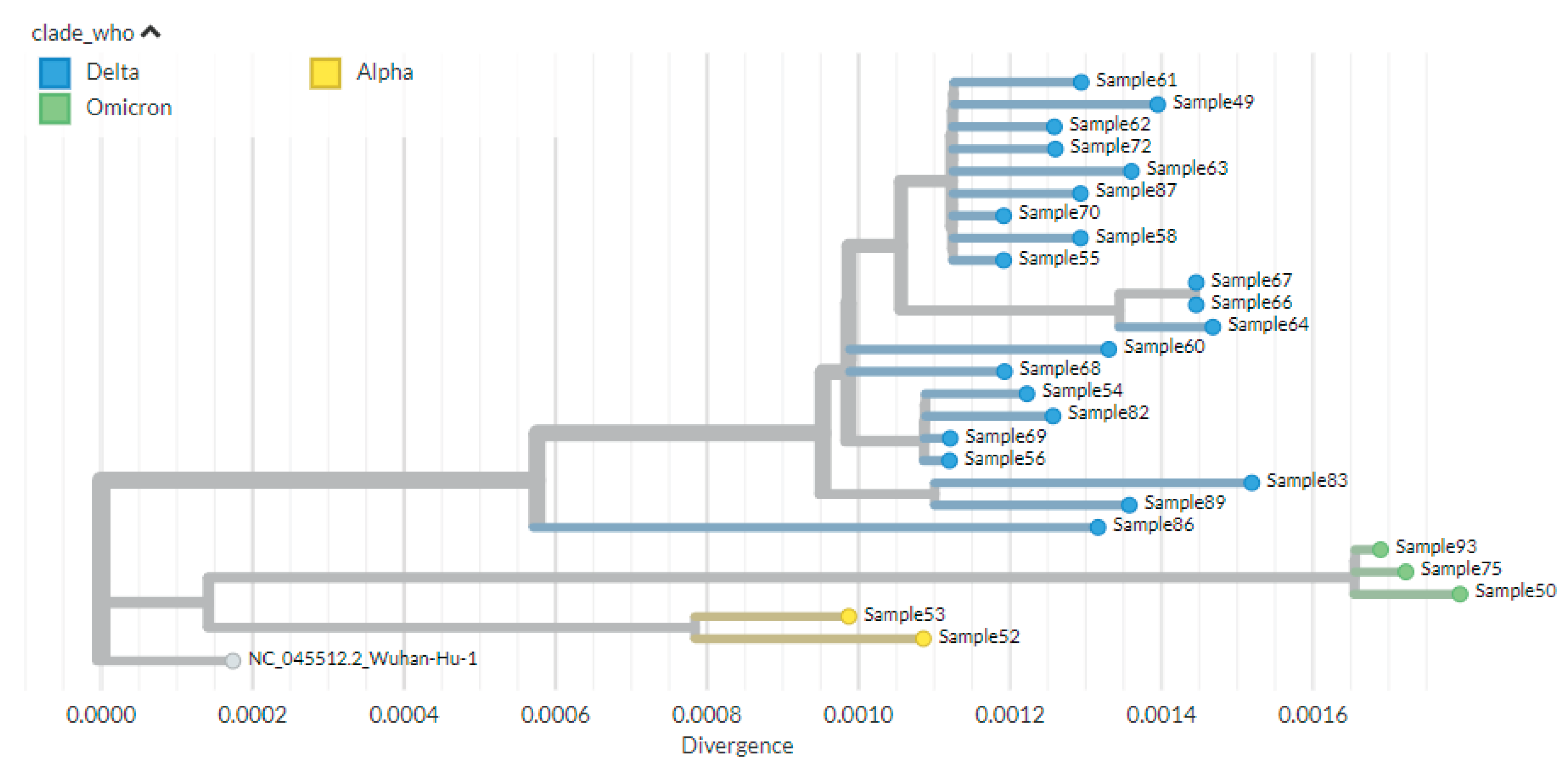
Table 1.
Socio-demographic and clinical characteristics of the study population (n = 48 participants).
Table 1.
Socio-demographic and clinical characteristics of the study population (n = 48 participants).
| Characteristics | Frequency (n=48) | Percentage | |
|---|---|---|---|
| Sex | Female | 22 | 46% |
| Male | 26 | 54% | |
| Age | 15-24 Years old | 3 | 6% |
| 25-44 Years old | 22 | 46% | |
| 45-64 years old | 8 | 17% | |
| 65+ | 15 | 31% | |
| Symptoms | Fever | 48 | 100% |
| Cough | 40 | 83% | |
| Sore Throat | 31 | 65% | |
| Difficulty of breathing | 27 | 56% | |
| Headache | 34 | 71% | |
| Muscle ache | 30 | 63% | |
| Arthralgia | 16 | 33% | |
| Admission Status | Inpatient | 43 | 90% |
| Outpatient | 5 | 10% | |
Table 2.
Sequencing depth and accuracy for SARS-CoV-2 sequence according to Genome Detective Platform.
Table 2.
Sequencing depth and accuracy for SARS-CoV-2 sequence according to Genome Detective Platform.
| Barcode | # Reads | Depth of Coverage | Nucleotide Identity (%) | Amino Acid Identity (%) | Genome Coverage (%) |
|---|---|---|---|---|---|
| 49 | 27604 | 485 | 99.8 | 99.7 | 99.4 |
| 50 | 27620 | 502 | 99.8 | 99.6 | 96.0 |
| 52 | 55585 | 1014 | 99.9 | 99.9 | 99.4 |
| 53 | 15574 | 279 | 99.9 | 99.8 | 99.4 |
| 54 | 28642 | 486 | 99.9 | 99.7 | 99.4 |
| 55 | 42675 | 769 | 99.9 | 99.7 | 99.4 |
| 56 | 51054 | 915 | 99.9 | 99.7 | 99.4 |
| 58 | 27377 | 485 | 99.9 | 99.7 | 99.4 |
| 60 | 51101 | 931 | 99.9 | 99.7 | 97.7 |
| 61 | 43354 | 771 | 99.9 | 99.7 | 99.4 |
| 62 | 27608 | 495 | 99.9 | 99.7 | 99.4 |
| 63 | 55379 | 979 | 99.8 | 99.7 | 99.4 |
| 64 | 53559 | 959 | 99.8 | 99.7 | 99.4 |
| 66 | 26429 | 466 | 99.8 | 99.7 | 99.2 |
| 67 | 53016 | 937 | 99.8 | 99.7 | 99.4 |
| 68 | 37235 | 666 | 99.9 | 99.7 | 98.2 |
| 69 | 26743 | 467 | 99.9 | 99.7 | 97.5 |
| 70 | 33827 | 595 | 99.9 | 99.7 | 99.4 |
| 72 | 35043 | 610 | 99.9 | 99.7 | 99.2 |
| 75 | 37569 | 646 | 99.8 | 99.6 | 99.4 |
| 82 | 46077 | 841 | 99.9 | 99.7 | 99.4 |
| 83 | 17440 | 293 | 99.8 | 99.7 | 95.7 |
| 86 | 23716 | 430 | 99.9 | 99.7 | 99.4 |
| 87 | 37558 | 662 | 99.9 | 99.7 | 99.4 |
| 89 | 21772 | 360 | 99.8 | 99.7 | 99.4 |
| 93 | 54886 | 949 | 99.8 | 99.7 | 99.4 |
Table 4.
Mutation Characteristics of SARS-CoV-2 variants by Sub-lineages.
| Variant (Sub-lineage) | Protein | High-Frequency Substitutions (>75%) | High-Frequency Deletions (>75%) |
|---|---|---|---|
| Alpha (B.1.1.7) | ORF1a | T1001I, A1708D, P2287S | S3675-, G3676-, F3677- |
| ORF1b | P314L, G662S | ||
| ORF8 | Q27, R52I, K68, Y73C | ||
| N | D3L, R203K, G204R, S235F | ||
| S | N501Y, D614G, P681H | H69-, V70-, Y144- | |
| Delta (B.1.617.2) | ORF1a | A1306S, P2046L, P2287S, V2930L, T3255I, T3646A | |
| ORF1b | P314L, G662S, P1000L, A1918V | ||
| ORF3a | S26L | ||
| ORF7a | V82A, T120I | ||
| ORF7b | T40I | ||
| ORF9b | T60A | ||
| M | I82T | ||
| N | D63G, R203M, G215C, D377Y | ||
| S | T19R, G142D, R158G, L452R, T478K, D614G, P681R, D950N | E156-, F157- | |
| Delta (AY.32) | ORF1a | A1306S, P2046L, P2287S, V2930L, T3255I, T3646A | |
| ORF1b | P314L, G662S, P1000L, A1918V, T2376I, R2613C | ||
| ORF3a | S26L | ||
| ORF7a | V82A, T120I | ||
| ORF7b | T40I | ||
| ORF9b | T60A | ||
| M | I82T | ||
| N | D63G, R203M, G215C, D377Y | ||
| S | T19R, G142D, R158G, L452R, T478K, D614G, P681R, D950N | E156-, F157- | |
| Delta (AY.120) | ORF1a | V28I, A1306S, P2046L, S2048F, P2287S, V2930L, T3255I, T3646A | |
| ORF1b | P314L, G662S, P1000L, A1918V, A2306T | ||
| ORF3a | S26L | ||
| ORF7a | V82A, T120I | ||
| ORF7b | T40I | ||
| ORF9b | T60A | ||
| M | I82T | ||
| N | D63G, R203M, G215C, D377Y | ||
| S | T19R, T95I, G142D, R158G, L452R, T478K, D614G, P681R, D950N | E156-, F157- | |
| Omicron (BA.1; BA.1.1, BA.1.17) | ORF1a | K856R, A2710T, T3255I, P3395H, I3758V | L3674-, S3675-, G3676- |
| ORF1b | P314L, I1566V | ||
| ORF9b | P10S | E27-, N28-, A29- | |
| M | D3G, Q19E, A63T | ||
| N | P13L, R203K, G204R | E31-, R32-, S33- | |
| S | A67V, T95I, G142D, Y145D, L212I, G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H, T547K, D614G, H655Y, N679K, P681H, N764K, D796Y, N856K, Q954H, N969K, L981F | H69-, V70-, G142-, V143-, Y144-, N211- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



