
Submitted:
26 November 2024
Posted:
27 November 2024
You are already at the latest version
Abstract
Simple Summary: DNA damage response (DDR) is a multi-factor network that is responsible for the removal of DNA lesions, thus enabling cells to function normally. An imbalance between the generation of reactive oxygen species and their removal by defense mechanisms is known as oxidative stress. Previous studies demonstrated that deregulation of the DDR network and redox imbalance are implicated in the onset and progression of several diseases, including cancer, as well as in the outcome of chemotherapy. In this study, we found that DDR-associated parameters and the intracellular redox status display significant differences among lung cancer patients at baseline and correlate with the clinical responses to subsequent platinum-based therapy. The exploitation of these results might lead to the identification of new therapeutic targets, the design of effective and sensitive biomarkers and the development of new therapeutic regimens for the treatment of this devastating malignancy.

Keywords:
DNA damage response (DDR)
; lung cancer
; PBMCs
; platinum-based chemotherapy
; clinical response
; redox status
; oxidative stress
; apurinic/apyrimidinic (AP) sites
; Nucleotide Excision Repair (NER)
; Interstrand Cross-links Repair (ICL/R)
1. Introduction
Lung cancer remains one of the most-diagnosed malignant diseases over decades, characterized by high mortality rates, with over 2 million cases per year rising worldwide [1]. Based on the histology of the cancer cells, lung cancer can be categorized into two types, small cell lung cancer (SCLC) that comprises about 15% of lung cancers and non-small cell lung cancer (NSCLC) that accounts for 85% of all cases and can be further subdivided into three histological types: adenocarcinoma, large cell carcinoma and squamous cell carcinoma [2]. Despite considerable progress in understanding, diagnosing and treating the disease, further advances seem obligatory. One major challenge appears to be the identification of accurate predictive biomarkers that can be utilized in the clinic to improve treatment design [3].
Lung cancer has early been related to cigarette smoking and many carcinogenic compounds have been identified in tobacco ever since [4]. These substances induce DNA damage contributing to genomic instability associated with the high mutational burden of lung cancer cells. Nevertheless, a significant percentage of patients who develop lung cancer have not ever used tobacco, implying further DNA damaging factors such as environmental pollution, radiation, exposure to industrial hazardous agents etc. pose as risk factors, interplaying with genetic predisposition [5,6].
The human genome is constantly exposed to multiple exogenous (genotoxic chemicals, UV light, ionizing radiation, etc.) and endogenous DNA damaging factors (e.g., oxidation, alkylation, hydrolysis, mismatch of DNA bases) [7,8,9]. Genotoxic stress may also emerge from several cellular processes, such as replication and transcription [10,11]. Specifically, the generation and response to Reactive Oxygen Species (ROS) remain among the most well-studied genotoxic mechanisms, as cancer cells are frequently characterized by impaired regulation of ROS. Several pathways are involved in ROS regulation as their function is critical for cell signaling and metabolism. Disrupted ROS levels lead to pathological outcomes and disease development [12,13], while redox status has been highlighted as critical for both cancer progression and chemotherapy response [14]. Particularly in lung cancer, evidence suggests that oxidative stress is critical for the onset and progression of the disease, as lungs are more susceptible due to their exposure to oxygen and blood circulation [15]. Moreover, ROS are capable of directly inducing DNA lesions, including oxidized purines and pyrimidines, single-strand breaks (SSBs), double-strand breaks (DSBs) and abasic (AP; apurinic/apyrimidinic) sites [16,17,18]. Specifically, AP-sites are common DNA lesions that may occur both spontaneously, due to oxidative stress, and as intermediates of DNA repair pathways, such as Base Excision Repair (BER) [19]. Repair of AP-sites includes their cleavage and SSBs formation that may result in DSBs during DNA replication. As such, AP-sites levels have been suggested as a possible biomarker for oxidative stress and BER capacity, while it has been proposed that could even predict survival in resected NSCLC patients [19]. Moreover, a key factor of BER, the Apurinic/apyrimidinic endonuclease 1 (APE1), has been underlined as a therapeutic target in NSCLC, as its inhibition resulted in excessive DNA damage and augmented tumor cell death in vitro and in vivo [20].
Interestingly, genotoxic drugs like platinum-based compounds have been reported to induce oxidative stress-related cytotoxic effects, either by directly generating ROS or by blocking the antioxidant system [21,22,23]. As a result, redox status appears to be crucial for response to chemotherapy-based treatment [14,24,25,26]. Glutathione, a key antioxidant factor, has been found to react with cisplatin and regulate resistance to this drug [26,27,28,29,30]. GSH, the reduced form of glutathione, binds and deactivates cisplatin molecules, thus preventing them from reaching the DNA and forming adducts [28,29]. In parallel, GSH reacts with the cisplatin-induced ROS, interfering with ROS-mediated cytotoxicity [29]. In particular, cisplatin cytotoxicity depends on the glutathione levels and the expression of the nuclear factor erythroid 2-related factor 2 (NRF2), which controls the transcription of glutathione components in lung cancer [29] and other tumor cells [31].
To confront the above challenges and ensure genomic stability, cells have developed a complex system of molecules and pathways, commonly known as the DNA damage response (DDR) network, including sensors of the lesion sites, cell cycle kinases, signaling cascades and effector proteins that maintain genomic integrity [32,33]. Disruption of the function of DDR network contributes to genomic instability and is involved in tumorigenesis. Furthermore, recent data have shown that DDR strongly impacts the immune system suggesting crucial therapeutic implications [34,35]. Specifically, defects in repair mechanisms may lead to the accumulation of cytosolic DNA resulting in stimulation of innate immune response and/or genomic mutations. These effects increase tumor mutational burden and levels of MHC-presented neoantigens, thus potentiating anti-tumor immune response [36,37,38].
Lung cancer is currently commonly treated with chemotherapy, molecular-targeted therapy, immunotherapy, radiation therapy and surgery [1,2]. The standard of care for most advanced NSCLC-patients includes platinum-based chemotherapy [39], thanks to its cytotoxicity. These drugs function by inducing the formation of DNA monoadducts that are almost exclusively repaired by nucleotide excision repair (NER), and interstrand cross-links (ICL), which are repaired by NER, translesion synthesis, Fanconi anemia pathway, homologous recombination (HR) and nonhomologous end-joining (NHEJ) [18].
In this study, we tested the hypothesis that redox status and DDR-related parameters of lung cancer patients at baseline correlate with therapeutic benefit from subsequent platinum-based treatment. Towards this, we evaluated GSH/GSSG ratio (a reliable estimation of cellular redox status), apurinic/apyrimidinic sites, and several DDR parameters, including the endogenous/baseline DNA damage, the efficiencies of critical DNA repair mechanisms and the apoptosis rates in normal and lung cancer cell lines, as well as in peripheral blood mononuclear cells (PBMCs) from healthy controls and lung cancer patients at baseline.
2. Materials and Methods
2.1. Patients
A total of 32 lung cancer patients were included in this study: seventeen (n = 17) patients with partial response to therapy (PR; 3 females/14 males; median age, 66 years; range, 49–82) , five (n = 5) with stable disease (SD; 2 females/3 males; median age, 68 years; range, 65–76) and ten (n = 10) with progressive disease (PD; 3 females/7 males; median age, 68.5 years; range, 62–81) (Table 1). Twenty (n=20) healthy individuals were also included as controls (HC; 8 females/12 males; median age 61.4 years; range, 41–82). PBMCs were isolated from freshly drawn peripheral blood and purified using the standard Ficoll gradient centrifugation, as previously described [40]. Cells were resuspended in freezing medium (90% Fetal Bovine Serum, 10% Dimethyl sulfoxide) and stored at −80°C until further processing. The study was approved by the Institutional Review Board of Soteria Hospital (No. 15627/11.6.2020 and 25950/10.10.2022), and all subjects provided informed consent. The study was conducted according to the Declaration of Helsinki.
2.2. Cell Lines
Human 1BR3hT cells (immortalized normal skin fibroblasts; provided by Dr. Maria Fousteri, Institute for Fundamental Biomedical Research, BSRC "Alexander Fleming") and H1299 cells (epithelial-like non-small-cell lung carcinoma; provided by Prof. Athanassios Kotsinas, National and Kapodistrian University of Athens) were maintained in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Human A549 cells (non-small-cell lung carcinoma; provided by Dr. Panagiotis Georgiadis, National Hellenic Research Foundation) were maintained in DMEM/Ham’s F12 (1:1) medium supplemented with 10% FBS and 1% penicillin-streptomycin. Human WS1 cells (normal skin fibroblasts; provided by Dr. Panagiotis Georgiadis, National Hellenic Research Foundation) were maintained in DMEM, supplemented with 1% non-essential amino acids, 10% FBS and 1% penicillin-streptomycin.
2.3. Measurement of Nucleotide Excision Repair (NER) – Alkaline Comet Assay
To assess the NER capacity of cell lines or PBMCs, cells were irradiated with ultraviolet C (UVC) (100 J/m2 and 5 J/m2 respectively), incubated in the appropriate culture medium for 0-6h at 37°C, harvested and stored in freezing medium at -80°C. DNA damage was measured using alkaline comet assay as follows: an appropriate cell suspension was diluted in 120µl of 1% low-melting point agarose and 70µl were loaded onto glass slides pre-coated with 1% standard agarose and covered with coverslip. Slides were then left to dry at 4°C for 30min. Next, slides were covered with an alkaline lysis buffer (NaCl 2.5 M, EDTA 0.1 M, Tris 0.01 M; pH=10 and 1% Triton X-100) for 2h at 4°C. After lysis, slides were placed in an electrophoresis tank. Electrophoresis was performed at 25V, 225mA for 30min in the comet assay tank at 4°C. Afterward, slides were washed in neutralizing buffer (0.4 M Tris; pH=7.5) and, subsequently, deionized H2O, and left to dry overnight. After staining with SYBR™ Gold nucleic acid gels stain (Thermo Fisher Scientific, #S11494), slides were photographed with a 10x microscopy lens under UV light. Comet parameters (Olive Tail Moment-OTM) were analyzed by Image J Analysis/Open Comet v1.3.1 (https://cometbio.org/). For each sample, 2 gels were scored and the average OTM value of 150 cells was calculated.
2.4. Measurement of the Interstrand Cross-Links Repair
Cell lines or PBMCs were treated with cisplatin (25μg/ml and 5μg/ml respectively) for 3h at 37°C in the appropriate culture medium, incubated in drug-free medium for 0-24h at 37°C, harvested and stored in freezing medium at -80°C. The ICL repair was measured using Southern blot analysis, as described previously [41].
2.5. GSH/GSSG Ratio and Abasic Sites
The GSH/GSSG ratio was measured using a luminescence-based system that detects and quantifies total glutathione (GSH+GSSG), oxidized glutathione (GSSG) and the GSH/GSSG ratio, according to manufacturer’s protocol (GSH/GSSG-Glo Assay, #V6612, Promega). Abasic sites were evaluated using the OxiSelect Oxidative DNA Damage Quantitation Kit (Cell Biolabs; #STA-324) according to the manufacturer’s protocol.
2.6. Apoptosis Rates
PBMCs were incubated with variable cisplatin doses (0-150 μg/ml) for 3h at 37oC in complete RPMI-1640 medium and then cultured in cisplatin-free medium for 24h. Apoptosis rates were measured using the Cell Death Detection ELISA PLUS kit (Roche Diagnostics Corp., #11.774.425.001, Mannheim, Germany). Experiments were performed as described by the manufacturer.
2.7. Western Blot Analysis
Cell lysates were prepared in RIPA Lysis Buffer System (Santa Cruz Biotechnology, #sc-24948) according to the manufacturer’s protocol. Protein electrophoresis was performed in 4-20% FastGene PAGE Gels (Nippon Genetics, #PG-S420) with MOPS buffer (Nippon Genetics, #PG-MOPS10). Subsequently, the proteins were transferred to nitrocellulose membranes (GE Healthcare, Amersham Protran 0.45μm, #10600002), which were incubated for 1h at room temperature with 5% nonfat dry milk in Tris-buffered saline-Tween 20 (TBST) followed by incubation with primary antibodies (Cell Signaling Technology; γH2AΧ, #80312; β-tubulin, #15115l; β-actin, #3700) diluted in either nonfat dry milk or BSA (5%) in TBST overnight at 4°C. Then, the membranes were washed with TBST and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Cell Signaling Technology; anti-mouse IgG: HRP, #7076S; anti-rabbit IgG: HRP, #7074S) for 1h at room temperature. After washing with TBST, the bound antibody complexes were visualized using the Pierce™ ECL Western Blotting Substrate (Thermo Scientific, #32106) and BioRad Gel Doc XR Imaging Systems.
2.8. Statistical Analysis
Unpaired t test with Welch's correction was applied for p-value determination. The results were of statistical significance when P < 0.05. All statistical analyses and graph design were carried out with GraphPad Prism 8.0.1. The mean ± SD was used to present the data.
3. Results
3.1. DDR-Associated Parameters in Lung Cancer Cell Lines
DDR-related signals were analyzed in two lung cancer cell lines (A549, H1299) and two normal fibroblast cell lines (WS1, 1BR3hT). For all parameters examined, similar results were obtained for the cell lines of each group. First, the endogenous/baseline DNA damage was evaluated using alkaline comet assay, which measures SSBs and/or DSBs. As seen in Figure 1A, the endogenous/baseline DNA damage was found to be significantly higher in lung cancer cell lines than in normal ones (P < 0.001), showing accumulation of DNA lesions in malignant cells when there is no known exogenous genotoxic attack. To further investigate the formation of the endogenous/baseline DNA damage, we measured intracellular factors that lead to the formation of SSBs and DSBs, such as redox dysregulation and AP-sites. Interestingly, the GSH/GSSG ratio was found decreased, while AP-sites were augmented in cancer cells, compared with the normal ones (all P < 0.001; Figure 1B, C).
Then, the efficiency of NER was evaluated. That is, all cell lines were irradiated with 100 J/m2 UVC, which induces cyclobutane pyrimidine dimers (CPDs) and 6–4 photoproducts (6-4PPs), DNA lesions that are repaired by the NER pathway [42], and the DNA damage was measured using alkaline comet assay (Figure 2A). Significant differences in the efficiencies of NER were found between malignant and normal cell lines. Indeed, lung cancer cell lines showed reduced NER efficiency compared with normal cells (Figure 2B), resulting in higher UVC-induced DNA damage accumulation in malignant cells, expressed as the Area Under the Curve (AUC) for DNA damage during the whole experiment (0-6h) (P < 0.001; Figure 2C). Moreover, in both lung cancer and normal cell lines we found that UVC irradiation reduced the GSH/GSSG ratio and increased AP-sites (P < 0.05; Figure 2D, E). Although kinetic patterns of UVC-induced AP-sites showed no significant differences between lung cancer and normal cells (Figure 2E), augmented accumulation of AP-sites was found in the lung cancer cell lines (P < 0.001; Figure 2F), due to increased levels of endogenous/baseline AP-sites in these cells. Together, these data suggest that the increased endogenous/baseline levels of DNA damage found in malignant cells may result, at least partly, from disruption of redox homeostasis and the subsequent formation of AP-sites.
To study the efficiency of the ICL repair, cell lines were treated with 25μg/ml cisplatin for 3h and the kinetics of ICL repair was followed for up to 24h after treatment. Significant differences in the cisplatin-induced ICL burden were found between malignant and normal cell lines, with lung cancer cells showing slightly higher ICL repair capacity (P < 0.05; Figure 3A). In addition, in both lung cancer and normal cell lines, cisplatin treatment resulted in reduction of GSH/GSSG ratio (Figure 3B) and higher levels of AP-sites (Figure 3C); maximal effect on both factors analyzed was observed at the end of the 3-h cisplatin treatment (time-point, T0). Importantly, significant differences were observed in the GSH/GSSG ratio and AP-sites kinetic patterns after cisplatin treatment between lung cancer and normal cells, with the malignant cells returning to baseline levels much faster than normal cells. Moreover, robustly higher total amounts of AP-sites expressed as AUC were found in malignant than in normal cell lines (Figure 3D). Next, the cisplatin-induced phosphorylation of H2AX at the serine residue 139, as a marker of DSBs, was also evaluated. In all cell lines analyzed, maximal levels of γH2AX were observed at the 24h time-point (Figure 3E).
3.2. DDR Signals in PBMCs from Lung Cancer Patients
To test the hypothesis that DDR-associated signals and redox status are implicated in the response to platinum-based chemotherapy, changes in the DDR parameters and the GSH/GSSG ratios were evaluated in PBMCs from 20 healthy controls and 32 lung cancer patients at baseline (17 responders and 15 non-responders to subsequent platinum-based chemotherapy).
Firstly, factors implicated in the formation of DNA damage were evaluated. In line with our previous data [43] and the cell lines’ results, compared with PBMCs from healthy individuals, patients’ cells exhibited significantly lower GSH/GSSG ratio (Figure 4A) and higher burden of AP-sites (Figure 4B) at baseline (all P < 0.001). Interestingly, responders to subsequent chemotherapy were characterized by significantly lower baseline GSH/GSSG ratio and higher baseline levels of AP-sites compared to non-responders (all P < 0.001; Figure 4A, B). In addition, the lowest doses of cisplatin required for the induction of apoptosis at 24h were significantly higher in PBMCs from patients at baseline compared with healthy controls (all P < 0.001), indicating that patients’ PBMCs exhibited significantly decreased apoptosis rates (Figure 4C). In particular, non-responders at baseline exhibited significantly lower apoptotic rates than responders, as their samples required the highest cisplatin dose for apoptosis induction (all P < 0.001; Figure 4C).
Subsequently, NER efficiency was analyzed in PBMCs following irradiation with 5 J/m2 UVC. In line with our previous data [43] and the cell lines’ results, significantly lower NER capacity was observed in lung cancer patients compared with healthy controls, resulting in higher accumulation of NER-repaired lesions in patients’ PBMCs (P < 0.01). Intriguingly, patients who responded to subsequent platinum chemotherapy showed significantly lower rates of NER compared with both non-responders and healthy controls, resulting in significantly higher DNA damage burden in responders’ cells (all P < 0.001; Figure 5A, B). Non-responders exhibit similar DNA damage levels to healthy controls, suggesting that impaired NER efficiency might be crucial to chemotherapy response.
In addition, we found that, in a 6-hour time frame after UVC irradiation, patients’ samples were characterized by lower GSH/GSSG ratio and higher UVC-induced AP-sites than healthy controls, with responders showing the lowest GSH/GSSG ratio and the highest levels of AP-sites (all P < 0.01; Figure 5C, D, E).
Next, to investigate the ICL repair efficiency, PBMCs were treated ex vivo with 5μg/ml cisplatin for 3h and the ICL levels were analyzed up to 24h after treatment. We found that lung cancer patients showed significantly lower ICL repair capacity than healthy controls, as depicted by higher DNA damage burden after cisplatin treatment (P < 0.001). In line with the NER capacity, responders’ cells showed much lower ICL repair capacity than non-responders, resulting in significantly higher accumulation of ICLs in responders’ PBMCs (P < 0.001, Figure 6A, B). Once again, non-responders show DNA damage levels equivalent to the ones of healthy controls. The GSH/GSSG ratio and AP-sites within 24 hours after cisplatin treatment were also evaluated. Significantly lower GSH/GSSG ratio and higher levels of AP-sites were obtained in patients’ samples compared with healthy controls, with responders presenting significantly diminished GSH/GSSG ratio and augmented levels of AP-sites compared to non-responders (all P < 0.001; Figure 6C, D, E).
4. Discussion
The development of drug resistance poses a significant challenge to platinum-based chemotherapy, a crucial treatment regimen employed in the therapeutic management of several malignancies, including lung cancer [44]. Cisplatin kills cancer cells by creating intra- and inter-strand DNA cross-links that cause genotoxic stress and cytotoxicity. Oxidative stress and ROS generation are also linked to cisplatin's cytotoxic effects [21,24,25,26,27]. Therefore, in this study we tested the hypothesis that redox status and DDR-related signals measured in PBMCs from lung cancer patients might correlate with therapeutic response to platinum-based therapy.
Firstly, the redox status, expressed as the GSH/GSSG ratio was assessed. In line with previous studies showing that in cancer cells a number of variables, such as hypoxia, aerobic glycolysis, and oncogene activation, disrupt the redox balance and lead to ROS accumulation, we found that PBMCs from lung cancer patients showed lower GSH/GSSG ratio than healthy controls. Prior research has demonstrated a robust association between lung cancer and redox imbalance [45]. In fact, patients with lung cancer had higher levels of oxidative stress biomarkers, including 8-oxodG and malondialdehyde, as well as reduced levels of antioxidative biomarkers, such as red cell superoxide dismutase and glutathione peroxidase activities [46]. Of note, chronic inflammation of the lung tissue is known to be associated with lung cancer [47]. Interestingly, cigarette smoke is known to increase inflammation by raising the number of inflammatory immune cells in the airways and causing the release of proinflammatory cytokines including GMCSF, TNF-α, IL-1, IL-6 and IL-8 [48]. Furthermore, DNA damage can also result in elevated ROS, which can further exacerbate oxidative damage, creating a vicious cycle and raising the burden of DNA damage. Given that the oxidative damage caused by ROS within cells leads to modifications of DNA bases, the greater numbers of AP-sites observed in the lung cancer patients examined herein may be explained by increased oxidative stress.
Importantly, significant differences in the redox status were found between patients sensitive or resistant to subsequent platinum-based therapy. Indeed, we found that PBMCs from responders are characterized by lower baseline and cisplatin-induced GSH/GSSG ratios, compared with non-responders patients. Previous studies have shown that the increase in cellular GSH plays a crucial role in cisplatin resistance because of its detoxification effect [49]. That is, before attaching to DNA, many platinum molecules form a Pt(GS)2 conjugate with GSH, which is subsequently removed from the cell. Even though this conjugation may deplete the antioxidant reservoir of the cells and result in oxidative stress, high levels of GSH lower the quantity of reactive cisplatin, thus limiting its anticancer effectiveness [50]. Therefore, it would be expected that an increase in GSH synthesis in cancer cells will cause resistance to platinum-based regimens.
Since the generation of ROS following treatment with many anticancer drugs may augment the treatment efficacy, there is growing interest in combining ROS-inducing agents with chemotherapy [51,52,53]. Indeed, pro-oxidative anticancer drugs, including curcumin and its derivatives, Choline Tetrathiomolybdate (ATN-224), 15-Deoxy-Delta-12,14-prostaglandin J2 (15d-PGJ2), 2-Methoxyoestradiol and carnosol are in various stages of research and development [54]. On the other hand, oxidative stress disrupts cellular processes, including the regulation of cell cycle, apoptosis, and DNA repair mechanisms that are essential for antineoplastic agents to exert their maximum cytotoxicity on cancer cells [55]. This results in the increased lipid peroxidation products, the decrease in blood plasma's capacity to trap radicals, the reduction of the plasma levels of beta-carotene, vitamin C and vitamin E, the induction of oxidative DNA damage, as well as the decrease in tissue glutathione levels after chemotherapy [56]. Thus, oxidative stress can also adversely affect normal tissues that undergo rapid proliferation, such as the heart, liver, lungs, kidneys, and gastrointestinal system [57]. Moreover, other adverse events, such as tumor cells adaptation to oxidative stress and cell cycle changes by oxidative stress, decrease the effectiveness of chemotherapy and induce cancer metastasis and recurrence [58]. Interestingly, combination treatment including antioxidants to reduce the side effects of chemotherapy might potentially decrease the efficacy of anticancer agents [59].
Since cisplatin cytotoxicity is mainly due to its ability to cause DNA damage, DNA repair capacity is expected to be one of most important cisplatin-resistance mechanisms. In this study, we found that lung cancer patients exhibited decreased NER and ICL repair capacities, compared with healthy controls. NER pathway is an important repair mechanism, as it is responsible for the repair of a variety of DNA lesions caused by multiple factors such as UV light, ionizing irradiation, ROS and chemotherapeutic drugs, including cisplatin [60]. Evidence has suggested that this pathway may be inhibited in lung cells exposed to tobacco smoke [61], with a recent study showing correlations between NER mutations and smoking status in NSCLC patients [62]. Specifically, an ERCC1 genetic polymorphism was found to be increased in heavy smokers NSCLC patients [62]. ERCC1 is known to be indispensable for NER pathway and it has been previously implicated with chemoresistance in lung adenocarcinoma [63]. Moreover, our previous study has shown deregulation of the genes encoding for the NER-related molecular components of the heterodimer DDB complex (DDB1 and DDB2) in lung cancer patients [45]. In line with the NER results, herein we found that PBMCs derived from lung cancer patients are characterized by decreased ICL repair capacities. In addition, we found reduced apoptosis rates in lung cancer patients. These results are in accordance with previous data showing that the absence of apoptotic regulation prolongs the life of cancer cells and provides more time for mutations to accumulate, which might enhance invasiveness as the tumor grows, deregulate differentiation pathways and promote angiogenesis [43]. Corresponding results on most DDR-related parameters were also obtained in cell lines experiments, thus further validating the broad applicability of our results. Interestingly, lung cancer cell lines additionally showed higher endogenous/baseline DNA damage (both single- and double strand breaks) compared to normal fibroblasts, partly due to the elevated levels of oxidative stress that were found in malignant cell lines.
Importantly, significant differences in the NER and ICL repair capacities were observed between lung patients sensitive or resistant to subsequent platinum-based therapy. Indeed, following treatment with UVC or cisplatin, limited lesion accumulation in non-responders’ PBMCs was found, probably emerging from excessive DNA repair activity of both NER and ICL repair mechanisms, resulting in strong resistance to apoptosis in non-responders samples. These results are in line with previous studies in solid tumors, including lung cancer [64,65,66,67], head and neck cancer [68,69,70,71], ovarian cancer [45,72,73], testicular cancer [72] and colorectal cancer [22], as well as in hematologic malignancies [74,75,76,77,78]. These results suggest that it might be possible to predict chemotherapy outcomes by measuring DDR signals in PBMCs derived from cancer patients.
5. Conclusions
In order to protect against genotoxic effects, cells have developed a number of genome-protection mechanisms, which collectively referred to as DNA damage response network. Interestingly, dysregulation of this system has been linked to the onset and progression of multiple diseases, such as cancer, as well as the response to therapies that cause damage to DNA. In addition, multiple diseases, such as cancer, are caused by disruption of redox homeostasis, which is necessary for human health, with oxidative stress also playing a crucial role in the cytotoxicity of platinum drugs. Therefore, herein we investigated the relationship between the therapeutic benefit of platinum-based regimens, the redox status and the DDR-related signals of PBMCs derived from patients with lung cancer at baseline. We found that redox status expressed as the GSH/GSSG ratio, the apurinic/apyrimidinic sites, the DNA repair capacity of critical DNA repair mechanisms, namely NER and ICL repair, and the apoptosis rates display significant differences among patients and correlate with the clinical responses to platinum-based therapy. These findings might be exploited as tools to design novel non-invasive predictive biomarkers and might contribute to the identification of lung cancer patients who are more likely to benefit from this type of therapy.
Author Contributions
Conceptualization, Dimitra Mavroeidi, Dimitra Stefanou, Konstantinos Syrigos and Vassilis Souliotis; Data curation, Dimitra Mavroeidi and Vassilis Souliotis; Formal analysis, Dimitra Mavroeidi and Vassilis Souliotis; Funding acquisition, Dimitra Mavroeidi, Dimitra Stefanou and Konstantinos Syrigos; Investigation, Dimitra Mavroeidi, Anastasia Georganta, Dimitra Stefanou and Vassilis Souliotis; Project administration, Vassilis Souliotis; Resources, Vassilis Souliotis; Supervision, Konstantinos Syrigos and Vassilis Souliotis; Validation, Dimitra Mavroeidi, Dimitra Stefanou, Christina Papanikolaou, Konstantinos Syrigos and Vassilis Souliotis; Visualization, Dimitra Mavroeidi and Vassilis Souliotis; Writing – original draft, Dimitra Mavroeidi and Vassilis Souliotis; Writing – review & editing, Dimitra Mavroeidi, Anastasia Georganta, Dimitra Stefanou, Christina Papanikolaou, Konstantinos Syrigos and Vassilis Souliotis. All authors will be updated at each stage of manuscript processing, including submission, revision, and revision reminder, via emails from our system or the assigned Assistant Editor.
Funding
This work was supported by the Hellenic Society of Medical Oncology (HeSMO) (8177/10-02-2021 and 9597/19-6-2024 Research Grants).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board of Sotiria Hospital (Protocol Nr. 15627/11.6.2020 and 25950/10.10.2022).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available by specific request to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Thai, A.A.; Solomon, B.J.; Sequist, L.V.; Gainor, J.F.; Heist, R.S. Lung Cancer. The Lancet 2021, 398, 535–554. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yan, B.; He, S. Advances and Challenges in the Treatment of Lung Cancer. Biomedicine & Pharmacotherapy 2023, 169, 115891. [Google Scholar] [CrossRef]
- Sears, C.R.; Mazzone, P.J. Biomarkers in Lung Cancer. Clinics in Chest Medicine 2020, 41, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hecht, S.S. Carcinogenic Components of Tobacco and Tobacco Smoke: A 2022 Update. Food and Chemical Toxicology 2022, 165, 113179. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Rosell, R.; Cardona, A.F.; Martín, C.; Zatarain-Barrón, Z.L.; Arrieta, O. Lung Cancer in Never Smokers: The Role of Different Risk Factors Other than Tobacco Smoking. Critical Reviews in Oncology/Hematology 2020, 148, 102895. [Google Scholar] [CrossRef]
- Laguna, J.C.; García-Pardo, M.; Alessi, J.; Barrios, C.; Singh, N.; Al-Shamsi, H.O.; Loong, H.; Ferriol, M.; Recondo, G.; Mezquita, L. Geographic Differences in Lung Cancer: Focus on Carcinogens, Genetic Predisposition, and Molecular Epidemiology. Ther Adv Med Oncol 2024, 16, 17588359241231260. [Google Scholar] [CrossRef]
- Cadet, J.; Wagner, J.R. DNA Base Damage by Reactive Oxygen Species, Oxidizing Agents, and UV Radiation. Cold Spring Harbor Perspectives in Biology 2013, 5, a012559–a012559. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair, and Mutagenesis. Environ and Mol Mutagen 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Van Houten, B.; Santa-Gonzalez, G.A.; Camargo, M. DNA Repair after Oxidative Stress: Current Challenges. Current Opinion in Toxicology 2018, 7, 9–16. [Google Scholar] [CrossRef]
- Ganai, R.A.; Johansson, E. DNA Replication—A Matter of Fidelity. Molecular Cell 2016, 62, 745–755. [Google Scholar] [CrossRef]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; Vousden, K.H. The Role of ROS in Tumour Development and Progression. Nat Rev Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Lian, G. ROS and Diseases: Role in Metabolism and Energy Supply. Mol Cell Biochem 2020, 467, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Simon, M.C. Glutathione Metabolism in Cancer Progression and Treatment Resistance. Journal of Cell Biology 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Kim, S.R.; Lee, Y.C. Impact of Oxidative Stress on Lung Diseases. Respirology 2009, 14, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M. Oxidatively Induced DNA Damage: Mechanisms, Repair and Disease. Cancer Letters 2012, 327, 26–47. [Google Scholar] [CrossRef]
- Renaudin, X. Reactive Oxygen Species and DNA Damage Response in Cancer. In International Review of Cell and Molecular Biology; Elsevier, 2021; Vol. 364, pp. 139–161 ISBN 978-0-323-85561-7.
- Souliotis, V.L.; Vlachogiannis, N.I.; Pappa, M.; Argyriou, A.; Ntouros, P.A.; Sfikakis, P.P. DNA Damage Response and Oxidative Stress in Systemic Autoimmunity. IJMS 2019, 21, 55. [Google Scholar] [CrossRef]
- Zhao, H.; Shen, J.; Deininger, P.; Hunt, J.D. Abasic Sites and Survival in Resected Patients with Non-Small Cell Lung Cancer. Cancer Letters 2007, 246, 47–53. [Google Scholar] [CrossRef]
- Long, K.; Gu, L.; Li, L.; Zhang, Z.; Li, E.; Zhang, Y.; He, L.; Pan, F.; Guo, Z.; Hu, Z. Small-Molecule Inhibition of APE1 Induces Apoptosis, Pyroptosis, and Necroptosis in Non-Small Cell Lung Cancer. Cell Death Dis 2021, 12, 503. [Google Scholar] [CrossRef]
- Berndtsson, M.; Hägg, M.; Panaretakis, T.; Havelka, A.M.; Shoshan, M.C.; Linder, S. Acute Apoptosis by Cisplatin Requires Induction of Reactive Oxygen Species but Is Not Associated with Damage to Nuclear DNA. Intl Journal of Cancer 2007, 120, 175–180. [Google Scholar] [CrossRef]
- Chen, W.; Lian, W.; Yuan, Y.; Li, M. The Synergistic Effects of Oxaliplatin and Piperlongumine on Colorectal Cancer Are Mediated by Oxidative Stress. Cell Death Dis 2019, 10, 600. [Google Scholar] [CrossRef]
- Van Loenhout, J.; Peeters, M.; Bogaerts, A.; Smits, E.; Deben, C. Oxidative Stress-Inducing Anticancer Therapies: Taking a Closer Look at Their Immunomodulating Effects. Antioxidants 2020, 9, 1188. [Google Scholar] [CrossRef] [PubMed]
- Cadoni, E.; Valletta, E.; Caddeo, G.; Isaia, F.; Cabiddu, M.G.; Vascellari, S.; Pivetta, T. Competitive Reactions among Glutathione, Cisplatin and Copper-Phenanthroline Complexes. Journal of Inorganic Biochemistry 2017, 173, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Jamali, B.; Nakhjavani, M.; Hosseinzadeh, L.; Amidi, S.; Nikounezhad, N.; Shirazi, F.H. Intracellular GSH Alterations and Its Relationship to Level of Resistance Following Exposure to Cisplatin in Cancer Cells. 2015.
- Li, S.; Li, C.; Jin, S.; Liu, J.; Xue, X.; Eltahan, A.S.; Sun, J.; Tan, J.; Dong, J.; Liang, X.-J. Overcoming Resistance to Cisplatin by Inhibition of Glutathione S-Transferases (GSTs) with Ethacraplatin Micelles in Vitro and in Vivo. Biomaterials 2017, 144, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.W.; Kuo, M.T. Role of Glutathione in the Regulation of Cisplatin Resistance in Cancer Chemotherapy. Metal-Based Drugs 2010, 2010, 1–7. [Google Scholar] [CrossRef]
- Kasherman, Y.; Sturup, S.; Gibson, D. Is Glutathione the Major Cellular Target of Cisplatin? A Study of the Interactions of Cisplatin with Cancer Cell Extracts. J. Med. Chem. 2009, 52, 4319–4328. [Google Scholar] [CrossRef]
- Silva, M.M.; Rocha, C.R.R.; Kinker, G.S.; Pelegrini, A.L.; Menck, C.F.M. The Balance between NRF2/GSH Antioxidant Mediated Pathway and DNA Repair Modulates Cisplatin Resistance in Lung Cancer Cells. Sci Rep 2019, 9, 17639. [Google Scholar] [CrossRef]
- Vascellari, S.; Valletta, E.; Perra, D.; Pinna, E.; Serra, A.; Isaia, F.; Pani, A.; Pivetta, T. Cisplatin, Glutathione and the Third Wheel: A Copper-(1,10-Phenanthroline) Complex Modulates Cisplatin–GSH Interactions from Antagonism to Synergism in Cancer Cells Resistant to Cisplatin. RSC Adv. 2019, 9, 5362–5376. [Google Scholar] [CrossRef]
- Hannon Barroeta, P.; O’Sullivan, M.J.; Zisterer, D.M. The Role of the Nrf2/GSH Antioxidant System in Cisplatin Resistance in Malignant Rhabdoid Tumours. J Cancer Res Clin Oncol 2023, 149, 8379–8391. [Google Scholar] [CrossRef]
- Black, H.; Mills, K. The DNA Damage Repair Response. COO 2020, 03. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-Damage Response in Human Biology and Disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Hsiehchen, D.; Hsieh, A.; Samstein, R.M.; Lu, T.; Beg, M.S.; Gerber, D.E.; Wang, T.; Morris, L.G.T.; Zhu, H. DNA Repair Gene Mutations as Predictors of Immune Checkpoint Inhibitor Response beyond Tumor Mutation Burden. Cell Reports Medicine 2020, 1, 100034. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Nowsheen, S.; Deng, M. DNA Repair Deficiency Regulates Immunity Response in Cancers: Molecular Mechanism and Approaches for Combining Immunotherapy. Cancers 2023, 15, 1619. [Google Scholar] [CrossRef] [PubMed]
- Härtlova, A.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kröger, A.; Nilsson, J.A.; et al. DNA Damage Primes the Type I Interferon System via the Cytosolic DNA Sensor STING to Promote Anti-Microbial Innate Immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS–STING Pathway in Cancer. Cancer Discovery 2020, 10, 26–39. [Google Scholar] [CrossRef]
- Pilger, D.; Seymour, L.W.; Jackson, S.P. Interfaces between Cellular Responses to DNA Damage and Cancer Immunotherapy. Genes Dev. 2021, 35, 602–618. [Google Scholar] [CrossRef]
- Rossi, A.; Di Maio, M. Platinum-Based Chemotherapy in Advanced Non-Small-Cell Lung Cancer: Optimal Number of Treatment Cycles. Expert Review of Anticancer Therapy 2016, 16, 653–660. [Google Scholar] [CrossRef]
- Pappa, M.; Ntouros, P.A.; Papanikolaou, C.; Sfikakis, P.P.; Souliotis, V.L.; Tektonidou, M.G. Augmented Oxidative Stress, Accumulation of DNA Damage and Impaired DNA Repair Mechanisms in Thrombotic Primary Antiphospholipid Syndrome. Clinical Immunology 2023, 254, 109693. [Google Scholar] [CrossRef]
- Souliotis, V.L.; Dimopoulos, M.A.; Episkopou, H.G.; Kyrtopoulos, S.A.; Sfikakis, P.P. Preferential in Vivo DNA Repair of Melphalan-Induced Damage in Human Genes Is Greatly Affected by the Local Chromatin Structure. DNA Repair 2006, 5, 972–985. [Google Scholar] [CrossRef]
- Scharer, O.D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harbor Perspectives in Biology 2013, 5, a012609–a012609. [Google Scholar] [CrossRef]
- Stefanou, D.T.; Kouvela, M.; Stellas, D.; Voutetakis, K.; Papadodima, O.; Syrigos, K.; Souliotis, V.L. Oxidative Stress and Deregulated DNA Damage Response Network in Lung Cancer Patients. Biomedicines 2022, 10, 1248. [Google Scholar] [CrossRef] [PubMed]
- Shahlaei, M. Platinum-Based Drugs in Cancer Treatment: Expanding Horizons and Overcoming Resistance. Journal of Molecular Structure 2023, 1301, 137366. [Google Scholar] [CrossRef]
- Stefanou, D.T.; Souliotis, V.L.; Zakopoulou, R.; Liontos, M.; Bamias, A. DNA Damage Repair: Predictor of Platinum Efficacy in Ovarian Cancer? Biomedicines 2021, 10, 82. [Google Scholar] [CrossRef] [PubMed]
- Peddireddy, V.; Siva Prasad, B.; Gundimeda, S.D.; Penagaluru, P.R.; Mundluru, H.P. Assessment of 8-Oxo-7, 8-Dihydro-2′-Deoxyguanosine and Malondialdehyde Levels as Oxidative Stress Markers and Antioxidant Status in Non-Small Cell Lung Cancer. Biomarkers 2012, 17, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Golubnitschaja, O.; Zhan, X. Chronic Inflammation: Key Player and Biomarker-Set to Predict and Prevent Cancer Development and Progression Based on Individualized Patient Profiles. EPMA Journal 2019, 10, 365–381. [Google Scholar] [CrossRef]
- Lu, L.-M.; Zavitz, C.C.J.; Chen, B.; Kianpour, S.; Wan, Y.; Stämpfli, M.R. Cigarette Smoke Impairs NK Cell-Dependent Tumor Immune Surveillance. The Journal of Immunology 2007, 178, 936–943. [Google Scholar] [CrossRef]
- Han, Y.; Yin, W.; Li, J.; Zhao, H.; Zha, Z.; Ke, W.; Wang, Y.; He, C.; Ge, Z. Intracellular Glutathione-Depleting Polymeric Micelles for Cisplatin Prodrug Delivery to Overcome Cisplatin Resistance of Cancers. Journal of Controlled Release 2018, 273, 30–39. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular Mechanisms of Cisplatin Resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef]
- Li, X.; Hou, Y.; Zhao, J.; Li, J.; Wang, S.; Fang, J. Combination of Chemotherapy and Oxidative Stress to Enhance Cancer Cell Apoptosis. Chem. Sci. 2020, 11, 3215–3222. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxidative Medicine and Cellular Longevity 2019, 2019, 1–12. [Google Scholar] [CrossRef]
- Liu, T.-I.; Lu, T.-Y.; Yang, Y.-C.; Chang, S.-H.; Chen, H.-H.; Lu, I.-L.; Sabu, A.; Chiu, H.-C. New Combination Treatment from ROS-Induced Sensitized Radiotherapy with Nanophototherapeutics to Fully Eradicate Orthotopic Breast Cancer and Inhibit Metastasis. Biomaterials 2020, 257, 120229. [Google Scholar] [CrossRef]
- Nizami, Z.N.; Aburawi, H.E.; Semlali, A.; Muhammad, K.; Iratni, R. Oxidative Stress Inducers in Cancer Therapy: Preclinical and Clinical Evidence. Antioxidants 2023, 12, 1159. [Google Scholar] [CrossRef]
- Iqbal, M.J.; Kabeer, A.; Abbas, Z.; Siddiqui, H.A.; Calina, D.; Sharifi-Rad, J.; Cho, W.C. Interplay of Oxidative Stress, Cellular Communication and Signaling Pathways in Cancer. Cell Commun Signal 2024, 22, 7. [Google Scholar] [CrossRef]
- Chiang, F.-F.; Huang, S.-C.; Yu, P.-T.; Chao, T.-H.; Huang, Y.-C. Oxidative Stress Induced by Chemotherapy: Evaluation of Glutathione and Its Related Antioxidant Enzyme Dynamics in Patients with Colorectal Cancer. Nutrients 2023, 15, 5104. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.S.; Bae, C.; Wang, J.; Lee, K.-H.; Hankerd, K.M.; Kim, H.K.; Chung, J.M.; La, J.-H. Peripheral and Central Oxidative Stress in Chemotherapy-Induced Neuropathic Pain. Mol Pain 2019, 15, 1744806919840098. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zuo, J.; Li, B.; Chen, R.; Luo, K.; Xiang, X.; Lu, S.; Huang, C.; Liu, L.; Tang, J.; et al. Drug-Induced Oxidative Stress in Cancer Treatments: Angel or Devil? Redox Biology 2023, 63, 102754. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Zhou, L.; Huang, Z.; Li, B.; Nice, E.C.; Xu, J.; Huang, C. Antioxidant Therapy in Cancer: Rationale and Progress. Antioxidants 2022, 11, 1128. [Google Scholar] [CrossRef] [PubMed]
- Petruseva, I.O.; Evdokimov, A.N.; Lavrik, O.I. Molecular Mechanism of Global Genome Nucleotide Excision Repair. Acta Naturae 2014, 6, 23–34. [Google Scholar] [CrossRef]
- Holcomb, N.; Goswami, M.; Han, S.G.; Clark, S.; Orren, D.K.; Gairola, C.G.; Mellon, I. Exposure of Human Lung Cells to Tobacco Smoke Condensate Inhibits the Nucleotide Excision Repair Pathway. PLoS ONE 2016, 11, e0158858. [Google Scholar] [CrossRef]
- Enjo-Barreiro, J.R.; Ruano-Ravina, A.; Pérez-Ríos, M.; Kelsey, K.; Varela-Lema, L.; Torres-Durán, M.; Parente-Lamelas, I.; Provencio-Pulla, M.; Vidal-García, I.; Piñeiro-Lamas, M.; et al. Radon, Tobacco Exposure and Non-Small Cell Lung Cancer Risk Related to BER and NER Genetic Polymorphisms. Archivos de Bronconeumología 2022, 58, 311–322. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, L.; Liu, L.; Zheng, Y.; Zhang, Y.; Yang, S.; Shi, R.; Wang, S. Chemosensitizing Effect of shRNA-Mediated ERCC1 Silencing on a Xuanwei Lung Adenocarcinoma Cell Line and Its Clinical Significance. Oncology Reports 2017, 37, 1989–1997. [Google Scholar] [CrossRef] [PubMed]
- Fikrova, P.; Stetina, R.; Hrnciarik, M.; Hrnciarikova, D.; Hronek, M.; Zadak, Z. DNA Crosslinks, DNA Damage and Repair in Peripheral Blood Lymphocytes of Non-Small Cell Lung Cancer Patients Treated with Platinum Derivatives. Oncology Reports 2014, 31, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Lobachevsky, P.N.; Bucknell, N.W.; Mason, J.; Russo, D.; Yin, X.; Selbie, L.; Ball, D.L.; Kron, T.; Hofman, M.; Siva, S.; et al. Monitoring DNA Damage and Repair in Peripheral Blood Mononuclear Cells of Lung Cancer Radiotherapy Patients. Cancers 2020, 12, 2517. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Li, F.; Sun, N.; Shukui, Q.; Baoan, C.; Jifeng, F.; Lu, C.; Zuhong, L.; Hongyan, C.; YuanDong, C.; et al. Polymorphisms in XRCC1 and XPG and Response to Platinum-Based Chemotherapy in Advanced Non-Small Cell Lung Cancer Patients. Lung Cancer 2009, 65, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Ren, S.; Zhou, S.; Zhang, L.; Su, C.; Zhang, Z.; Deng, Q.; Zhang, J. Predictive Effects of ERCC1 and XRCC3 SNP on Efficacy of Platinum-Based Chemotherapy in Advanced NSCLC Patients. Japanese Journal of Clinical Oncology 2010, 40, 954–960. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, H.; Han, P.; Gao, F.; Dahlstrom, K.R.; Li, G.; Owzar, K.; Zevallos, J.P.; Sturgis, E.M.; Wei, Q. Apoptotic Capacity and Risk of Squamous Cell Carcinoma of the Head and Neck. European Journal of Cancer 2017, 72, 166–176. [Google Scholar] [CrossRef]
- Raturi, V.P.; Wu, C.; Mohammad, S.; Hojo, H.; Bei, Y.; Nakamura, M.; Okumura, M.; Rachi, T.; Singh, R.; Gupta, R.; et al. Could Excision Repair Cross-complementing Group-1 mRNA Expression from Peripheral Blood Lymphocytes Predict Locoregional Failure with Cisplatin Chemoradiation for Locally Advanced Laryngeal Cancer? Asia-Pac J Clncl Oncology 2020, 16. [Google Scholar] [CrossRef]
- Borgmann, K.; Röper, B.; El-Awady, R.A.; Brackrock, S.; Bigalke, M.; Dörk, T.; Alberti, W.; Dikomey, E.; Dahm-Daphi, J. Indicators of Late Normal Tissue Response after Radiotherapy for Head and Neck Cancer: Fibroblasts, Lymphocytes, Genetics, DNA Repair, and Chromosome Aberrations. Radiotherapy and Oncology 2002, 64, 141–152. [Google Scholar] [CrossRef]
- Quintela-Fandino, M.; Hitt, R.; Medina, P.P.; Gamarra, S.; Manso, L.; Cortes-Funes, H.; Sanchez-Cespedes, M. DNA-Repair Gene Polymorphisms Predict Favorable Clinical Outcome Among Patients With Advanced Squamous Cell Carcinoma of the Head and Neck Treated With Cisplatin-Based Induction Chemotherapy. JCO 2006, 24, 4333–4339. [Google Scholar] [CrossRef]
- Reed, E.; Yuspa, S.H.; Zwelling, L.A.; Ozols, R.F.; Poirier, M.C. Quantitation of Cis-Diamminedichloroplatinum 11 (Cisplatin)-DNA-Lntrastrand Adducts in Testicular and Ovarian Cancer Patients Receiving Cisplatin Chemotherapy.
- Stefanou, D.T.; Bamias, A.; Episkopou, H.; Kyrtopoulos, S.A.; Likka, M.; Kalampokas, T.; Photiou, S.; Gavalas, N.; Sfikakis, P.P.; Dimopoulos, M.A.; et al. Aberrant DNA Damage Response Pathways May Predict the Outcome of Platinum Chemotherapy in Ovarian Cancer. PLoS ONE 2015, 10, e0117654. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Souliotis, V.L.; Anagnostopoulos, A.; Bamia, C.; Pouli, A.; Baltadakis, I.; Terpos, E.; Kyrtopoulos, S.A.; Sfikakis, P.P. Melphalan-Induced DNA Damage in Vitro as a Predictor for Clinical Outcome in Multiple Myeloma. Haematologica 2007, 92, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Gkotzamanidou, M.; Terpos, E.; Bamia, C.; Munshi, N.C.; Dimopoulos, M.A.; Souliotis, V.L. DNA Repair of Myeloma Plasma Cells Correlates with Clinical Outcome: The Effect of the Nonhomologous End-Joining Inhibitor SCR7. Blood 2016, 128, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- Gkotzamanidou, M.; Sfikakis, P.P.; Kyrtopoulos, S.A.; Bamia, C.; Dimopoulos, M.A.; Souliotis, V.L. Chromatin Structure, Transcriptional Activity and DNA Repair Efficiency Affect the Outcome of Chemotherapy in Multiple Myeloma. Br J Cancer 2014, 111, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Gkotzamanidou, M.; Terpos, E.; Dimopoulos, M.A.; Souliotis, V.L. The Combination of Panobinostat and Melphalan for the Treatment of Patients with Multiple Myeloma. IJMS 2022, 23, 15671. [Google Scholar] [CrossRef]
- Stefanou, D.T.; Episkopou, H.; Kyrtopoulos, S.A.; Bamias, A.; Gkotzamanidou, M.; Bamia, C.; Liakou, C.; Bekyrou, M.; Sfikakis, P.P.; Dimopoulos, M.; et al. Development and Validation of a PCR-based Assay for the Selection of Patients More Likely to Benefit from Therapeutic Treatment with Alkylating Drugs. Brit J Clinical Pharma 2012, 74, 842–853. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
DDR-associated parameters in cell lines at baseline. (A) Bar charts showing the endogenous/baseline DNA damage in normal and lung cancer cell lines measured by comet assay. (B) Redox status expressed by the GSH/GSSG ratio at untreated cell lines. (C) Baseline AP-sites levels for all cell lines. Error bars represent SD; ***P < 0.001. The experiments shown were based on a minimum of three independent repeats.
Figure 1.
DDR-associated parameters in cell lines at baseline. (A) Bar charts showing the endogenous/baseline DNA damage in normal and lung cancer cell lines measured by comet assay. (B) Redox status expressed by the GSH/GSSG ratio at untreated cell lines. (C) Baseline AP-sites levels for all cell lines. Error bars represent SD; ***P < 0.001. The experiments shown were based on a minimum of three independent repeats.
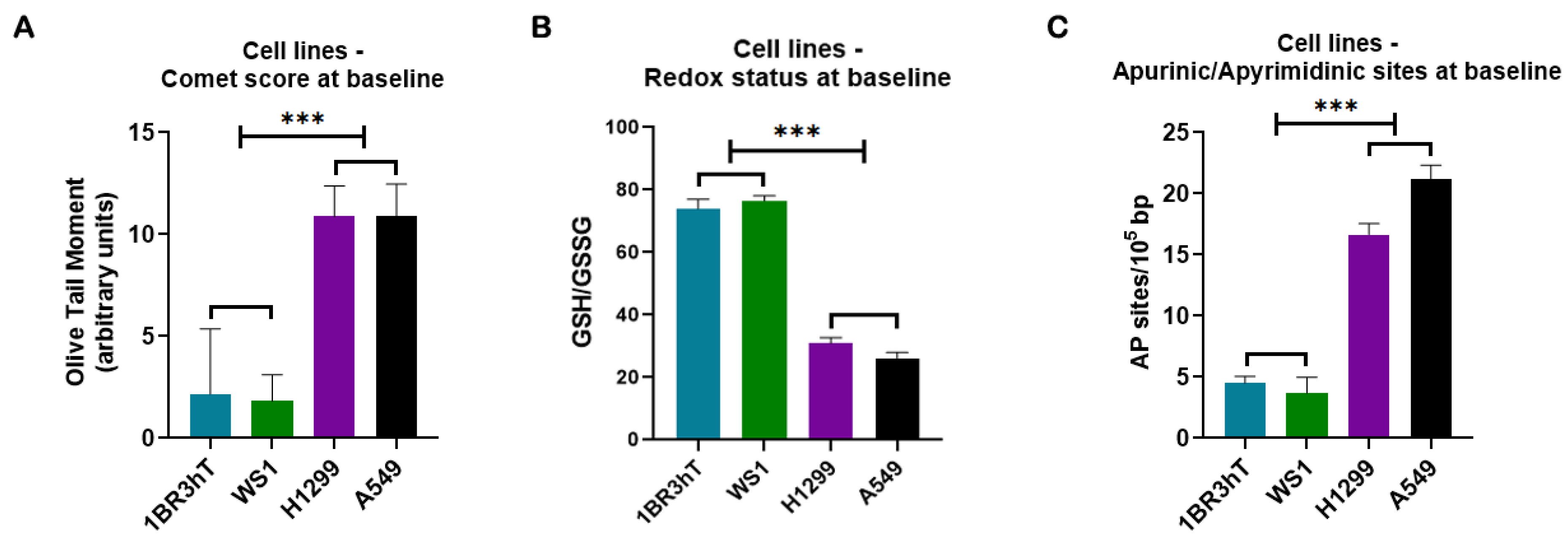
Figure 2.
DNA damage response signals in cell lines after UVC irradiation. (A) Alkaline comet assay images of A549 lung cancer cell line at baseline and at different time-points after UVC irradiation. (B) The kinetics of UVC-induced NER-repaired adducts using alkaline comet assay and (C) total amounts of DNA damage expressed as AUC for DNA damage during the whole experiment (0–6h). (D) Redox status and (E) AP-sites at different time-points after UVC irradiation. (F) Total amounts of AP-sites expressed as AUC. Error bars represent SD; *P < 0.05, **P < 0.01, ***P < 0.001. The experiments shown were based on a minimum of three independent repeats.
Figure 2.
DNA damage response signals in cell lines after UVC irradiation. (A) Alkaline comet assay images of A549 lung cancer cell line at baseline and at different time-points after UVC irradiation. (B) The kinetics of UVC-induced NER-repaired adducts using alkaline comet assay and (C) total amounts of DNA damage expressed as AUC for DNA damage during the whole experiment (0–6h). (D) Redox status and (E) AP-sites at different time-points after UVC irradiation. (F) Total amounts of AP-sites expressed as AUC. Error bars represent SD; *P < 0.05, **P < 0.01, ***P < 0.001. The experiments shown were based on a minimum of three independent repeats.
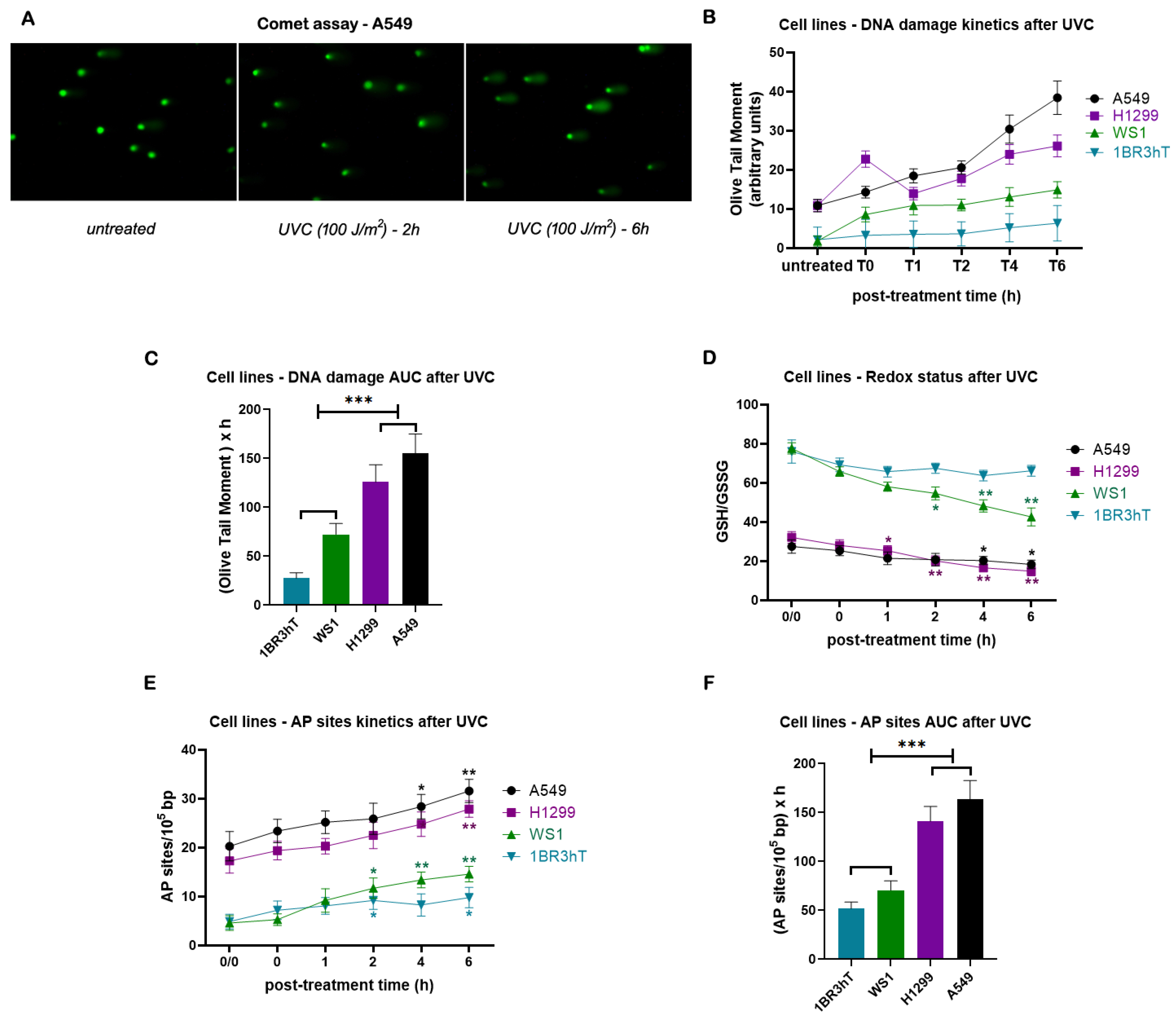
Figure 3.
DDR-associated parameters in cell lines following cisplatin treatment. (A) The total amounts of cisplatin-induced ICLs expressed as AUC for DNA damage. (B) Redox status and (C) AP-sites at different time-points after cisplatin treatment. (D) Total amounts of AP-sites expressed as AUC. (E) Western blots showing the amounts of γH2AΧ at different time-point after cisplatin treatment. β-tubulin and β-actin were used as loading controls. Error bars represent SD; *P < 0.05, **P < 0.01, ***P < 0.001. The experiments shown were based on a minimum of three independent repeats.
Figure 3.
DDR-associated parameters in cell lines following cisplatin treatment. (A) The total amounts of cisplatin-induced ICLs expressed as AUC for DNA damage. (B) Redox status and (C) AP-sites at different time-points after cisplatin treatment. (D) Total amounts of AP-sites expressed as AUC. (E) Western blots showing the amounts of γH2AΧ at different time-point after cisplatin treatment. β-tubulin and β-actin were used as loading controls. Error bars represent SD; *P < 0.05, **P < 0.01, ***P < 0.001. The experiments shown were based on a minimum of three independent repeats.
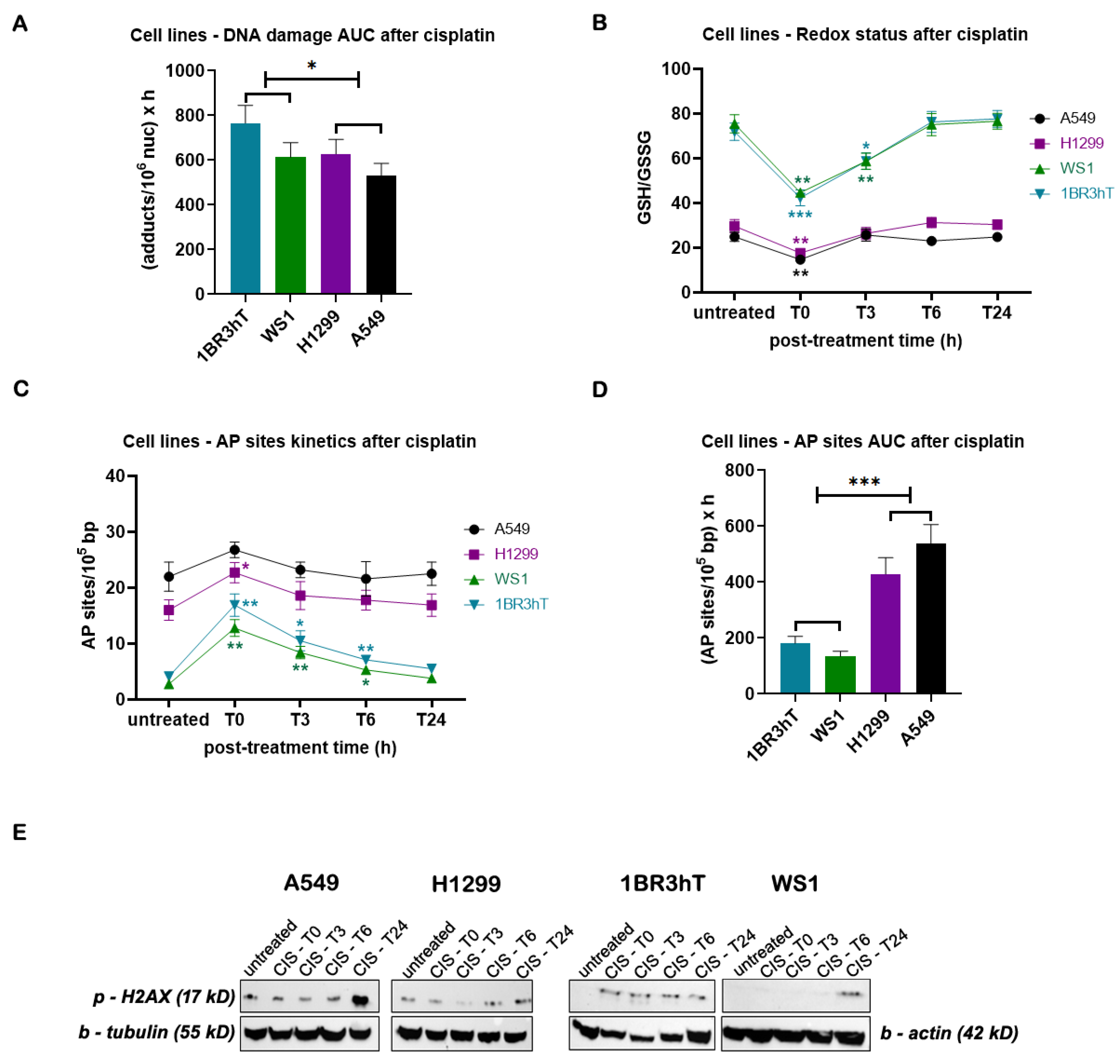
Figure 4.
DDR-associated parameters in PBMCs at baseline (A) Bar charts showing the endogenous/baseline redox status expressed by the GSH/GSSG ratio at PBMCs from healthy controls (HC) and patients, responders (R) and non-responders (NR) to subsequent chemotherapy-based treatment. (B) Baseline AP-sites levels for PBMCs. (C) Apoptosis rates at baseline for PBMCs. Error bars represent SD; *P < 0.05, **P < 0.01, ***P < 0.001. The experiments shown were based on a minimum of three independent repeats.
Figure 4.
DDR-associated parameters in PBMCs at baseline (A) Bar charts showing the endogenous/baseline redox status expressed by the GSH/GSSG ratio at PBMCs from healthy controls (HC) and patients, responders (R) and non-responders (NR) to subsequent chemotherapy-based treatment. (B) Baseline AP-sites levels for PBMCs. (C) Apoptosis rates at baseline for PBMCs. Error bars represent SD; *P < 0.05, **P < 0.01, ***P < 0.001. The experiments shown were based on a minimum of three independent repeats.
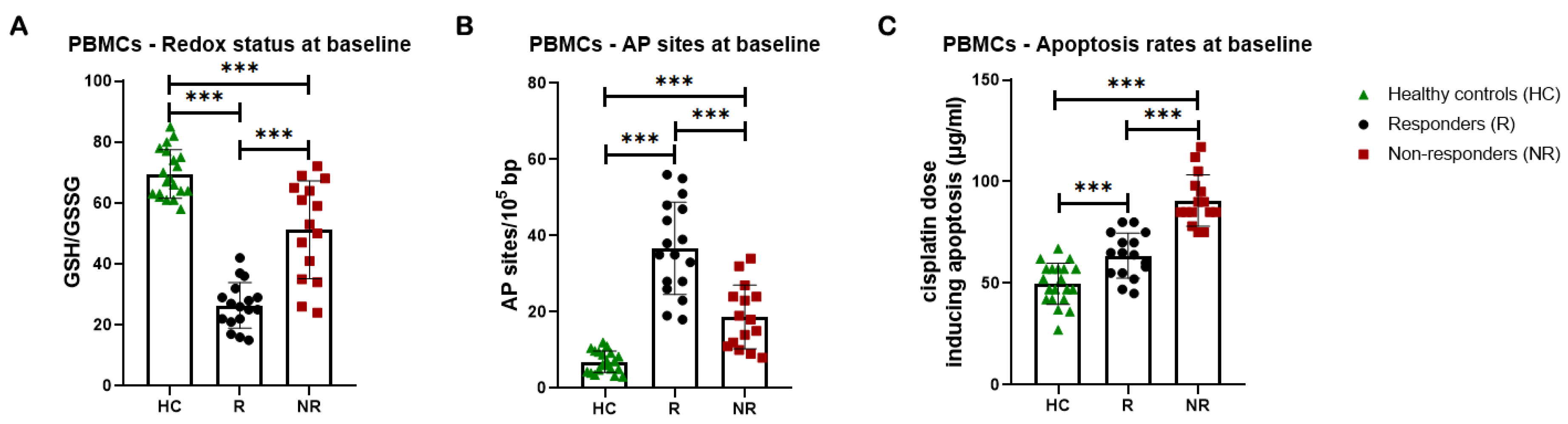
Figure 5.
DDR signals in PBMCs after UVC irradiation. (A) The kinetics of UVC-induced DNA lesions using alkaline comet assay and (B) total amounts of DNA lesions expressed as AUC in PBMCs from healthy controls and lung cancer patients. (C) Redox status and (D) AP-sites at different time-points after UVC irradiation of PBMCs. (E) Total amounts of AP-sites expressed as AUC. Error bars represent SD; *P < 0.05, **P < 0.01, ***P < 0.001. The experiments shown were based on a minimum of three independent repeats.
Figure 5.
DDR signals in PBMCs after UVC irradiation. (A) The kinetics of UVC-induced DNA lesions using alkaline comet assay and (B) total amounts of DNA lesions expressed as AUC in PBMCs from healthy controls and lung cancer patients. (C) Redox status and (D) AP-sites at different time-points after UVC irradiation of PBMCs. (E) Total amounts of AP-sites expressed as AUC. Error bars represent SD; *P < 0.05, **P < 0.01, ***P < 0.001. The experiments shown were based on a minimum of three independent repeats.
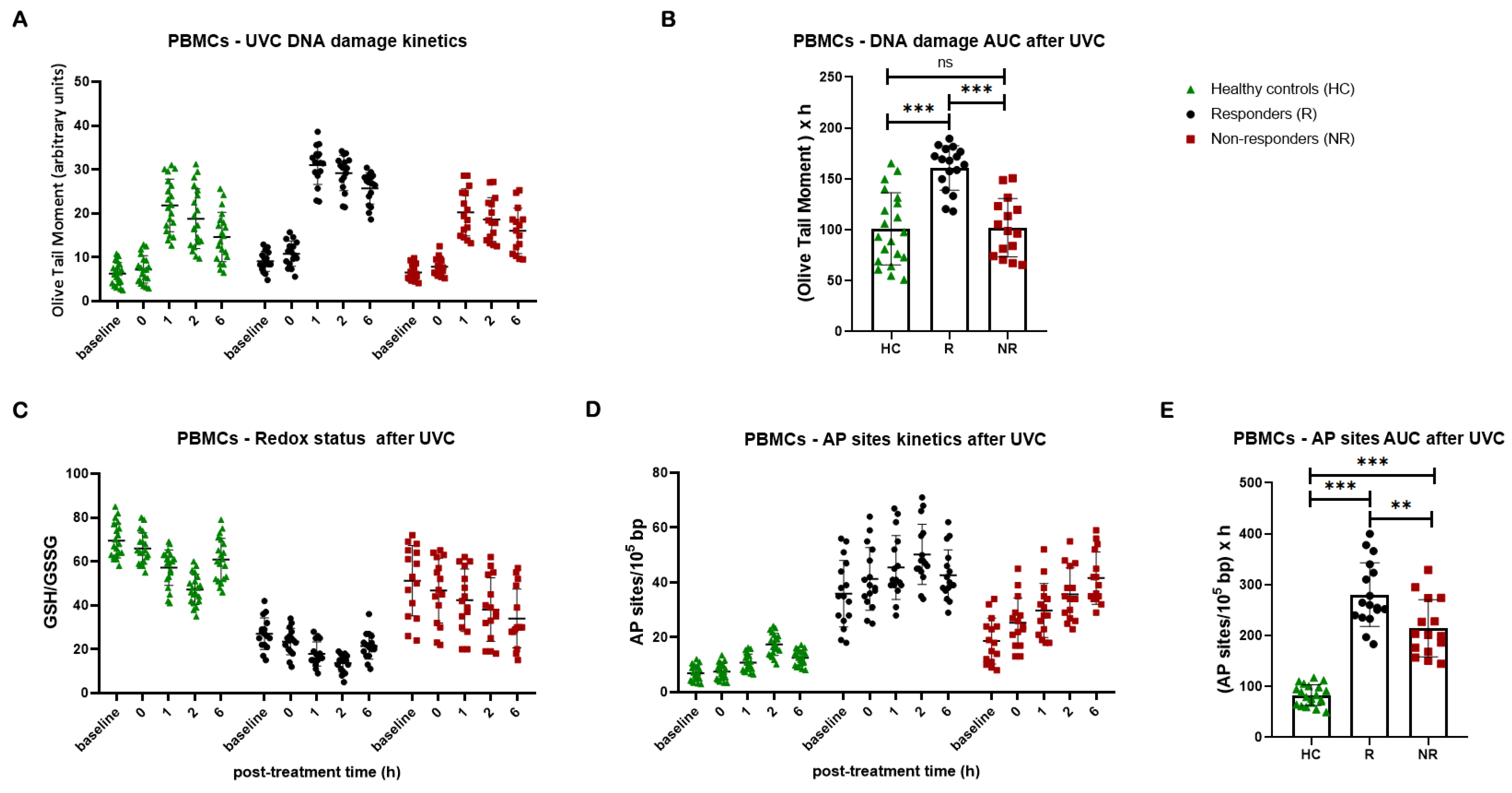
Figure 6.
DNA damage response parameters in PBMCs after the ex vivo cisplatin treatment. (A) The kinetics of cisplatin-induced ICLs and (B) total amounts of ICLs expressed as AUC for DNA damage in PBMCs from healthy controls and lung cancer patients. (C) Redox status and (D) AP-sites at baseline and after cisplatin treatment. (E) Total amounts of AP-sites expressed as AUC in PBMCs. Error bars represent SD; *P < 0.05, **P < 0.01, ***P < 0.001. The experiments shown were based on a minimum of three independent repeats.
Figure 6.
DNA damage response parameters in PBMCs after the ex vivo cisplatin treatment. (A) The kinetics of cisplatin-induced ICLs and (B) total amounts of ICLs expressed as AUC for DNA damage in PBMCs from healthy controls and lung cancer patients. (C) Redox status and (D) AP-sites at baseline and after cisplatin treatment. (E) Total amounts of AP-sites expressed as AUC in PBMCs. Error bars represent SD; *P < 0.05, **P < 0.01, ***P < 0.001. The experiments shown were based on a minimum of three independent repeats.
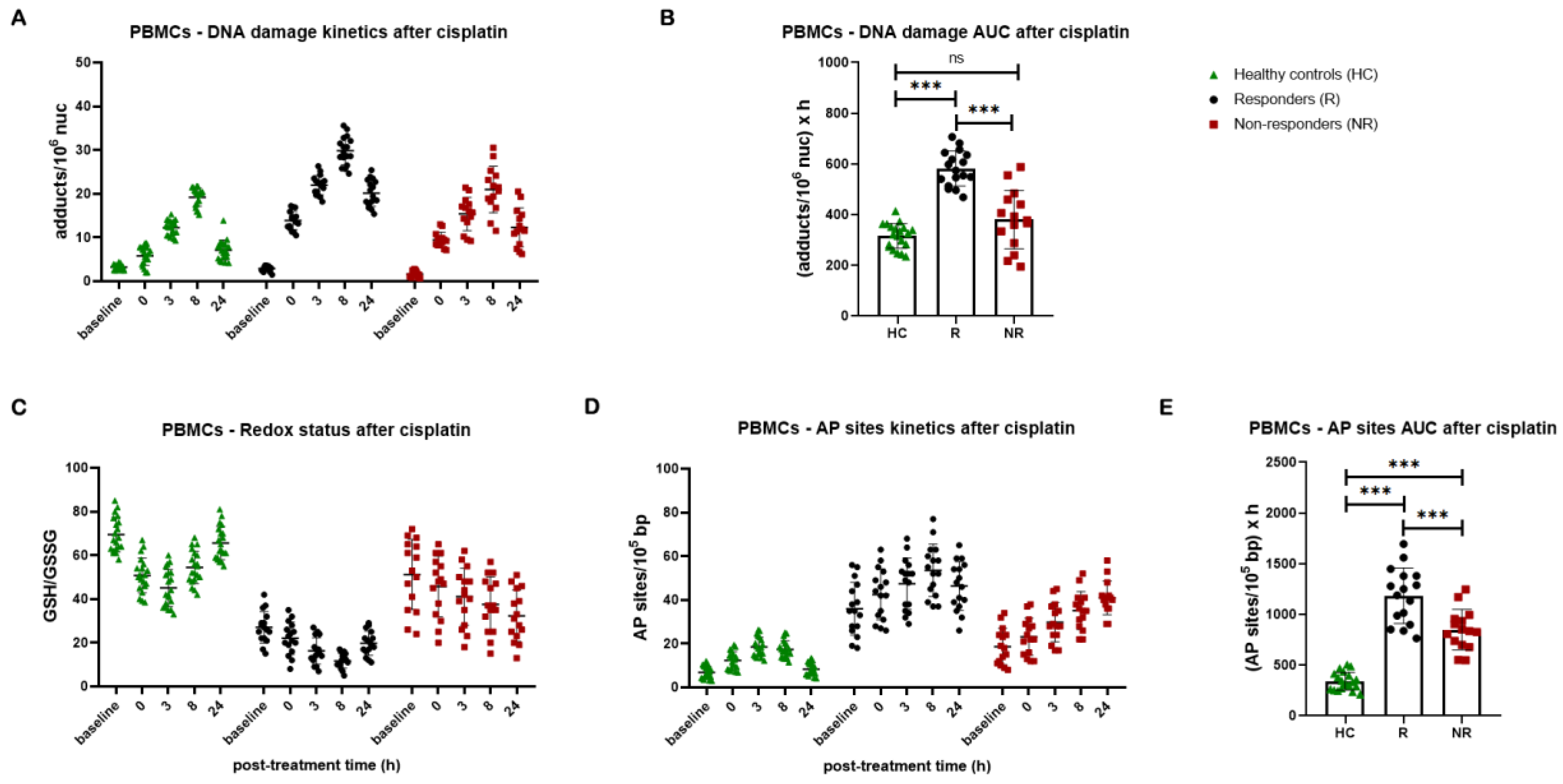

Table 1.
Patients and disease characteristics.
| Patients (N=32) | |||
|---|---|---|---|
| Characteristic | N | Years | % of Total |
| Sex | |||
| Male | 24 | - | 75 |
| Female | 8 | - | 25 |
| Age | |||
| Median | - | 67.5 | - |
| Range | - | 49-82 | - |
| Histology | |||
| squamous | 10 | - | 31,3 |
| Non-squamous | 17 | - | 53,1 |
| Small cell | 5 | - | 15,6 |
| Stage | |||
| I-II / LD | 4 | - | 12,5 |
| III | 7 | - | 21,9 |
| IV | 21 | - | 65,6 |
| Smoking | |||
| Never | 3 | - | 9,3 |
| Current | 3 | - | 9,3 |
| Former | 24 | - | 75 |
| PD-L1 expression | |||
| <1% | 7 | - | 21,9 |
| 1-50% | 7 | - | 21,9 |
| >50% | 7 | - | 21,9 |
| Therapy | |||
| Chemotherapy | 18 | - | 56,2 |
| Chemotherapy – Immunotherapy combination | 14 | - | 43,8 |
| Response | |||
| PR | 17 | - | 53,1 |
| SD | 5 | - | 15,6 |
| PD | 10 | - | 31,3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



