Submitted:
25 November 2024
Posted:
26 November 2024
You are already at the latest version
Abstract
The PD1/PD-L1 axis plays an important immunosuppressive role during the T cell mediated immune response, which is essential for the physiological homeostasis of the immune system. The biology of the immunological microenvironment is extremely complex and crucial for the development of treatment strategies for immunotherapy. Characterization of the immunological, genomic or transcriptomic landscape of cancer patients could allow discrimination between responders and non-responders to anti-PD-1/PD-L1 therapy. Immune Checkpoint Inhibitor (ICI) therapy has shown remarkable efficacy in a variety of malignancies in landmark trials and has fundamentally changed cancer therapy. Current research focuses on strategies to maximize patient selection for therapy, clarify mechanisms of resistance, improve existing biomarkers, including PD-L1 expression and tumor mutational burden (TMB), and discover new biomarkers. In this review, we focus on the function of the PD-1/PD-L1 signaling pathway and discuss the immunological, genomic, epigenetic and transcriptomic landscape in cancer patients receiving anti-PD-1/PD-L1 therapy. Finally, we provide an overview of the clinical trials testing the efficacy of antibodies against PD-1/PD-L1.
Keywords:
PD1/PD-L1
; Immune Checkpoint Inhibitor
; immunological landscape
; genomic landscape
; transcriptomic landscape
1. Introduction
Immuno-oncology is an evolving field of drug development that involves agents that harness the patient’s own immune system to fight cancer by counteracting the mechanisms by which the tumor evades the immune system [1]. Key effector cells, such as CD8+ effector T cells, are components of the immune system that recognize and eliminate cancer cells through the expression of tumor-specific antigens (neoantigens) [2]. The activation of T cells is regulated by immune checkpoints, that effectively limit and control an ongoing immune response to prevent autoimmunity. However, tumor cells (TCs) manage to evade the immune system by disrupting immune checkpoints. Overexpression of PD-1 ligands (programmed death-ligand 1, PD-L1) by TCs binds programmed cell death protein (PD-1) on T cells and inhibits their activity. Immune checkpoint inhibitors (ICIs) against PD-1/PD-L1 can release the brakes and lead to the activation of T cells [3].
The tumor microenvironment (TME) plays an essential role in tumor survival and function and mediates pro-tumorigenic effects through metabolic reprogramming [4]. The composition of the TME varies by tumor type, but characteristic features include cellular components and the extracellular matrix (ECM) that promote tumor growth, angiogenesis, invasion and metastasis [5]. M2-like tumor-associated macrophages (TAMs) [6] and cancer-associated fibroblasts (CAFs) [7] are essential components of the TME that facilitate tumor metastasis and enhance tumor drug resistance. Modulation of TAMs represents a novel therapeutic strategy and could be achieved by reducing or altering the function of TAMs [8]. Targeted therapies with CAFs include chemotherapy, immunotherapy and functional modification or reprogramming. However, many limitations such as the heterogeneity and plasticity of CAFs and the lack of specific target markers for CAFs may hinder the development of effective anti-CAF therapy [9].
Genomic landscapes have revealed great heterogeneity in different human cancers as demonstrated by genome-wide sequencing studies [10]. However, the identification of somatic mutations and copy number alterations has been associated with potential response to immunotherapy [11]. Many independent studies have reported that tumor mutational burden (TMB) is a predictive biomarker for treatment success with ICIs in various tumor types [12,13,14]. On the other hand, cancer patients with low TMB may also have a good response to ICI monotherapies [15]. Different genomic landscapes have been found in tumors with different PD-L1 combined positive score (CPS) [16], and it has also been reported that TMB and PD-L1 are independent biomarkers [17]. In this review, we focus on the function of the PD-1/PD-L1 signaling pathway and discuss immunological, genomic, epigenetic and transcriptomic landscape in cancer patients receiving anti-PD-1/PD-L1 therapy. Finally, an overview of the clinical trials testing the efficacy of antibodies against PD-1/PD-L1 is provided.
2. Function of the PD-1/PD-L1 Signaling Pathway
2.1. T Cell Response and Maintenance of Self-Tolerance
The PD1/PD-L1 axis plays an important immunosuppressive role during the T-cell-mediated immune response, which is essential for the physiological homeostasis of the immune system and the maintenance of self-tolerance [18]. T cell activation is a multistep process that begins with the recognition of antigen by the T cell receptor (TCR) bound to major histocompatibility molecules (MHC) class I or II on the surface of antigen-presenting cells (APCs). This interaction forms a TCR-antigen-MHC complex that is supported by the CD4 or CD8 coreceptors [19]. Shortly after T cell activation, PD1 expression is induced on their surface, which prevents their overactivation and establishes an activation threshold [20]. T cells that cannot be further activated were initially termed “exhausted” [21]. This exhausted PD1-positive population has been shown to contain two subpopulations, both pre-exhausted and terminally exhausted cells, only the former of which can resume function after the PD1 blockade. This state of exhaustion is epigenetically determined, especially in terminally exhausted T cells [22]. It is evident that the downregulation of T cell activation by PD1 and the subsequent suppression of the T cell-mediated immune response has important physiological effects on the prevention of autoimmunity, the maintenance of self-tolerance and the maintenance of immune homeostasis [23]. Specifically, in regulatory T cells (Tregs), inhibitory PD-1 signaling, independent of Foxp3-mediated regulatory cell function, controls immune tolerance and the development of autoimmunity [24].
2.2. Effects of PD-1 and PD-L1 on Signal Transduction Pathways
Several signaling pathways have been reported to be affected by PD-1/PD-L1. Although there are still unresolved questions about which are the primary direct targets of PD1, between TCR and CD28, and the exclusivity of SHP2 in mediating PD1-derived inhibitory signaling, it is well established that PD1 targets multiple downstream signaling pathways [3,25]. These include members of the TCR signalosome ZAP70 and CD3zeta and downstream PKCtheta signaling [26], the Ras/mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K)/Akt and mammalian target of rapamycin (mTOR) [27,28] including PTEN (phosphatase and tensin homolog) activation mediated the PD1 [29].
2.3. PD1 Activation
The PD1 receptor is expressed on the surface of antigen-stimulated T and B cells and has two main ligands, PD-L1 and PD-L2, which have different expression patterns. Thus, PD-L1 is expressed in various cell types, including both normal, hematopoietic and non-hematopoietic cells as well as cancer cells, whereas PD-L2 expression is mainly restricted to hematopoietic cells such as dendritic cells (DCs), macrophages, B cells, mast cells and activated T cells, but also in tumor and stromal cells [20,30]. Although the expression of PD-L2 in head and neck tumors can be associated with an enhanced PD1-mediated T cell response [30], PD-L2 is not considered a significant immunotherapy target in the clinic compared to PD-L1, the predominant inhibitory ligand of PD1 [20]. Activation of PD1 and subsequent inhibitory signaling occurs after binding of PD-L1 and PD-L2. The availability of PD-L1 can influence PD1 activation, as PD-L1 can also bind to the co-stimulatory molecule CD80. The binding affinity of PD-L1 to CD80 is slightly weaker than the binding affinity for PD1, whereas CD80 binds CD28 with a threefold stronger binding affinity [31,32].
2.4. PD1 Expression
Apart from activated T cells, PD1 can be expressed on the surface of natural killer (NK) cells, B cells, macrophages and dendritic cells [33,34]. PD1 is encoded by the programmed cell death 1 (PDCD1) gene, whose transcription is induced by the TCR and NOTCH signaling pathways as well as various cytokines through the downstream activation of different transcription factors such as NFATc1, RBPJκ, STAT3/4/5, FOXO1, AP-1 and NF-κΒ [35]. PD1 expression can be epigenetically regulated via both DNA methylation and histone modification [36]. Thus, PD1 expression in naïve CD8+ T cells is caused by hypermethylation of CRB and CR-C regulatory elements, whereas PD1 overexpression, which is associated with the phenotype of exhausted T cells, is induced by demethylation of the PD1 promoter [37,38]. In addition, PD1 expression can be regulated by post-translational modifications such as fucosylation and ubiquitination [39,40].
2.5. PD-L1 Expression
PD-L1 expression, reflecting a microenvironment rich in inflammatory cells, is considered a biomarker, albeit not a perfect one, for a favorable clinical outcome when administering ICIs [41]. PD-L1 expression can be regulated by gene amplification. For example, an increased copy number of the genetic locus encoding CD274, the gene responsible for PD-L1 protein synthesis, was found in patients with Hodgkin’s lymphoma and small-cell lung cancer (SCLC) [42,43]. Both extrinsic signals related to the immune response and intrinsic signals of the oncogenic pathway are involved in the transcriptional regulation of PD-L1 expression. For example, interferon gamma (IFNγ) produced by activated T cells and NK cells strongly triggers of PD-L1 expression in the tumor microenvironment. Mutations in IFNGR1/2 or JAK1/2, components of the IFNγ signaling cascade, are a common cause of acquired and primary resistance to ICIs blockade therapies [44,45]. In addition, IFNγ targets such as PTPN2, APLNR, and the chromatin remodeling complex PBAF have been identified by genetic screens as putative enhancers of ICIs in preclinical cancer models [46,47,48]. Apart from IFNγ, PD-L1 expression in various cancer cells can be induced by different immunostimulatory cytokines such as IL-6, INF-α and -β and TNF or immunosuppressive cytokines such as IL-10 and TGFβ [46]. PD-L1 expression is regulated in both cancer and immune cells by a variety of intrinsic such as activation of oncogenic EGFR, PI3K/AKT, MAPK and STAT3 signaling pathways and extrinsic stimuli such as various immunostimulatory cytokines [49]. PD-L1 expression can be regulated at post-transcriptional and post-translational levels. In the first case, various oncogenic microRNAs directly interfere with PD-L1 mRNA stability [50]. Regulation at the post-translational level includes modifications such as phosphorylation, glycosylation, ubiquitination and palmitoylation, which influence the stabilization of the PD-L1 protein [51,52].
3. Landscape of Anti-PD-1/PD-L1 Therapy
3.1. Immune Landscape
The biology of the immunological microenvironment is highly complex and plays a crucial role in developing treatment strategies for immunotherapy (Figure 1) [53]. Characterizing this microenvironment and understanding the interactions between stromal and tumor cells is essential for identifying novel neoantigens [54]. Additionally, the definition of a tumor's immunological subtype is key to predicting disease progression, often more so than the specific characteristics of individual cancer types. Identifying these neoantigenic immune targets can help elicit a durable immune response [53].
Characterization of the immune landscape of patients with metastatic melanoma has shown that it is possible to differentiate between responders and non-responders to anti-PD-1 monotherapy [55]. More specifically, in the responder group, high PD-L1 expression was found in macrophages at the tumor-stroma interface, followed by a rapid decrease in expression within the tumor, with even higher PD-L1 expression in the vicinity of cytotoxic T cells, whereas in non-responders, PD-L1 expression and cytotoxic T cell activation follow a more arbitrary spatial regulation [55]. Tumors can be classified into "hot", "intermediate" and "cold" groups according to their different immune profiles, which is highly consistent with the corresponding CD8+ T cell infiltration status [56]. Effective anti-PD-1 immunotherapy has also been associated with the presence of polyclonal CD8+ T cells in the tumor with a limited number of mutations with lower TCR polyclonality [57]. CD8+ T cells are characterized by a highly expressed CD161, while its ligand LLT1 (CLEC2D) is expressed in TCs, that the LLT1/CD161 distribution pattern is characterized by a high proportion of LLT1+ TCs and a low proportion of CD161+ CD8+ T cells and is associated with a higher risk of lymph node metastasis [58]. In MSI-H/mismatch repair-deficient (dMMR) metastatic colorectal cancer (mCRC), the accumulation of more CD8+ T cells in patients receiving first-line anti-PD-1 monotherapy is related to the sensitivity of the therapy [59]. On the other hand, the attenuation of CD8+ T cell function and the increase in immunosuppressive myeloid-derived suppressor cells (MDSCs) in metabolic dysfunction-associated steatohepatitis-related hepatocellular carcinoma (MASH-HCC) could be caused by squalene epoxidase (SQLE), which promotes an impaired antitumor response to anti-PD-1 therapy [60]. In addition, TREM2+ TAMs play an important role in suppressing CD8+ T cells, while their deficiency inhibits tumor growth in vivo HCC models by increasing the antitumor activity of CD8+ T cells [61]. Moreover, in resistant pancreatic tumors in orthotopic pancreatic cancer (PaC) mouse models, the simultaneous presence of more depleted effector CD8+ T cells and increased M2-like TAMs with a reduced capacity for antigen presentation, has been reported [62].
Treg cells suppress anticancer immunity and contribute to immune evasion, tumor progression, and metastasis [63]. In experiments performed on mouse models of melanoma to analyze the intratumoral immune microenvironment, mice that did not respond to anti-PD1 therapy had a significantly higher population of Tregs that shifted the CD8+:Treg ratio in an age-dependent manner. Depletion of Tregs in melanoma using anti-CD25 was shown to reverse the response to anti-PD1 in non-responders [64], while in mice bearing a mammary tumor, neoadjuvant immune checkpoint blockade (ICB) was shown to remodel the intratumoral immune landscape characterized by an increase in CD8+ T cells and NK cells and sustained T cell activation [65]. The spatial distribution of T cells in the TME can predict the response to ICIs [66]. In renal cell carcinoma, there is a potential interaction between CD8+CD39+PD-1+ T cells and Foxp3+PD-1+ Treg cells due to the proximity between the cells, forming a spatial niche that is more specialized for immunosuppression under PD-1 blockade, which is more evident in metastatic lesions [67].
Computational algorithms have also revealed important information about tumor immune complexes in different cancer types [68]. In particular, immune cell infiltration (ICI) clusters and gene clusters have been associated with different immune subtypes and survival outcomes in different cancer types [69,70]. In melanoma, a high ICI score indicated activated immune properties and a better prognosis and was enriched with immune pathways and highly expressed immune signature genes [69]. ICI score was evaluated in HCC. It was found that in the high ICI score group, activation of the Wnt/β-catenin pathway was significantly enriched and expression of immune checkpoint genes was increased, while the low ICI score group was characterized by increased TMB and enrichment of metabolic-related pathways [71]. In lung adenocarcinoma (LUAD), a prognostic signature of tumor microenvironment (LATPS) consisting of UBE2T, KRT6A, IRX2, and CD3D has shown that low LATPS is characterized by an immune phenotype, including increased amount of ICI, tumor immune system dysfunction, and increased activity of immune-related pathways, leading to better benefit from immunotherapy [72]. Similarly, in cervical cancer, an immune-related gene prognostic index (IRGPI) of the tumor immunological microenvironment (TIME) was able to predict the composition of immune cell subtypes such as macrophages T cells CD8 and T cells CD4, TIL exclusion and T cell dysfunction [73]. In colon cancer, high IRGPI levels have been associated with cell adhesion molecules (CAMs) and chemokine signaling pathways, high macrophage M1 infiltration, suppressed immunity, more aggressive colon cancer phenotype, and lower therapeutic benefit of ICIs treatment [74]. A NAD+ metabolism-related gene signature (NMRGS) has been used to establish a comprehensive risk model for glioma patients associated with a more immunosuppressive microenvironment and better therapeutic response to ICIs therapy [75]. In head and neck squamous cell carcinoma (HNSCC), a novel immune signature (IMS) derived from a combined cohort identified hub genes and gene signatures for activated CD8+ T cells correlated with the TIME that can predict response to immunotherapy, prognosis, immune infiltration, and clinical features [76].
In non-small cell lung cancer (NSCLC), a cold tumor immunological microenvironment, i.e. lower infiltration of CD3+ T cells and lower concentration of various immune signatures, correlated with a high chromosomal copy number variant (CNV) burden and was associated with a lower benefit of ICIs, whereas a greater proximity of CD8+GZB+ T cells to malignant cells influenced the benefit of ICIs therapy [77]. In addition, the macrophage landscape plays an important role in the prognosis of NSCLC patients. Patients with low infiltration of M2 macrophages have better overall (OS) and disease-free survival (DFS) [78]. B- cell infiltration, is also an important factor in the sustained benefit of PD-1-based immunotherapy, as demonstrated in biopsy samples from HNSCC prior to treatment [79]. In addition, B-cell concentration in blood mononuclear cells (PBMCs) correlated strongly with tumor B-cell concentration in the tumor and showed a high predictive value for response to ICB [80]. A longitudinal study of the immunologic landscape in peripheral blood samples from NSCLC patients showed the association of immune cells with their correlating cytokines. CD8+ and CD8+CD101hiTIM3+ (CCT T) were detected in low proportions of circulating cells from responders to immune ICB therapy and CCT T cells secreted higher levels of cytokines [81]. Following ICI administration CD8+central memory T cells accumulate and tumor-infiltrating lymphocytes (TILs) develop an effector-like phenotype over time [82]. In the MC38 tumor model, the TIL landscape changes in response to checkpoint blockade, as a switch from an NKT-driven TNFα response to a T cell-driven IFN-γ response has occurred in the tumor microenvironment [83].
Fibroblasts may also play an important role in progression and the immune landscape, as has been reported for pancreatic ductal adenocarcinoma (PDAC). The functional heterogeneity of CAFs in PDAC differentially regulate cancer-associated signaling pathways and the accumulation of regulatory T cells [84]. Infiltration with inflammatory CAFs is also important for the immune-mesenchymal like (IML) subtype, which is associated with ECM activities and immune responses [85]. CAFs and macrophages may also contribute most to CD8 T cell exclusion and immunosuppression by indicating activin A-mediated transcriptional reprogramming toward extracellular matrix remodeling, which polarizes the TME toward immunosuppression [86].
3.2. Genomic Landscape
The identification of potential genetic markers that could be associated with the benefit of immunotherapy is very important for the treatment of cancer patients receiving immunotherapy (Figure 1) [11,87]. However, genomic instability could be a barrier to improved sensitivity to ICIs [88]. The integration of high-throughput technologies such as next generation sequencing (NGS), data extraction and analysis using the cancer genome atlas (TCGA) database has enabled the prediction of genomic data from numerous samples of different cancer types [11] or genetic predispositions related to the immune system [89]. However, the heterogeneity of mutations in tumors can pose a major challenge for personalized medicine and biomarker development [90].
TMB has been postulated as a general determinant of ICIs-dependent tumor rejection that depends on tissue and treatment context. A specific association between TMB and improved treatment outcomes with anti-PD-1/L1 therapies has been demonstrated [12,13,91]. Interaction effects between co-occurring genomic alterations on the efficacy of ICIs in NSCLC have been reported [92]. Large genomic data from 1,846 patients with NSCLC showed that 44% of tumors had a targetable oncogenic alteration, with epidermal growth factor receptor (EGFR) being the most common. An exploratory analysis showed that the impact of TMB on survival with ICIs treatment was associated with improved OS [93]. In addition, relatively high levels of TMB or DNA damage repair (DDR) alterations were found in esophageal squamous cell carcinoma (ESCC) patients prior to administration of neoadjuvant chemoimmunotherapy, suggesting that these different types of alterations are associated with therapeutic response [94]. Moreover, deficit mutations in checkpoint kinase 2 (CHEK2), an important gene in the DDR, enhance the anti-tumor effect of anti-PD-1 therapy, as shown in mice with MC38 and B16 tumors, by affecting the tumor immunological microenvironment [95].
Ataxia-telangiectasia mutations (ATM) may also define a specific subset of NSCLC associated with kirsten rat sarcoma virus (KRAS) mutations, increased TMB, decreased tumor protein P53 (TP53) and EGFR coexistence, and potentially increased sensitivity to combined therapy of ICIs and chemotherapy [96]. Transcription activator BRG1 (SMARCA4)-mutated tumors are also very common recurrent alterations in NSCLC and are characterized by higher TMB and low or negative PD-L1, while the use of ICIs is associated with significantly improved survival [97]. In addition, PD-L1 expression and TMB have been shown to be independent biomarkers related to response rate to PD-1/PD-L1 inhibitors and could be generally used to categorize the immunological subtypes of cancer [17].
Analysis of intra-patient stability of TMB from tissue biopsies at different time points in advanced cancers has shown that TMB remains stable between tumor biopsies regardless of the time interval between sampling, but certain tumor types such as breast cancer (BC), colorectal carcinoma, and glioma may experience an increase in TMB over time [98]. The landscape of frameshift mutations (FS) may also be important as a predictive biomarker for ICIs in combination with TMB, especially in tumors with low TMB, as FS have been found in a high proportion of patients with low TMB (<10 mut/Mb) [15]. Similarly, patients with low TMB might benefit from ICIs when T-cell immunity is already present, without the need to distinguish between high and low TMB tumours [99].
The complexity of genomic analysis emphasizes the discovery potential of integrative analysis in large, well-curated, cancer-specific cohorts [100]. A pan-cancer study of 283,050 patient samples from multiple tumor types found that CD274 (PD-L1) gene rearrangements may be important for ICI-prone cancers when other genomic alterations are present [101]. In addition, PD-L1-positive tumors (score ≥ 1) are associated with significantly higher exonic TMB, leading to enrichment of the PD-L1 pathway and other immune-responsive pathways [102]. Recently, analysis of a cohort of 2504 patients from a broad spectrum of cancer types presented a new tumor classification system that includes 11 mutation-based genes and has the potential to support ICIs treatment decisions [14].
On the other hand, the evolving mutational landscape may be related to acquired resistance to immune checkpoint blockade. Some of these mutations encode tumor neoantigens that can be recognized by T cells and could be exploited for the development of patient-specific immunotherapy approaches [103]. NSCLC patients with acquired resistance to EGFR-TKIs (tyrosine kinase inhibitors) could achieve superior efficacy of anti-PD-1/PD-L1 blockers, as specific T cell responses could be achieved with high-value neoantigens generated from EGFR driver mutations [104]. It has been shown that a better understanding of the presentation of neoantigens by the tumor could be achieved if human leukocyte antigen-I loss of heterozygosity (HLA-I LOH) has the potential to refine TMB as a biomarker for response to checkpoint inhibitors [105]. Furthermore, PTEN loss in NSCLC tumors could lead to the development of an immunosuppressive microenvironment resulting in resistance to anti-PD-1 therapy, which can be overcome by targeting the immunosuppression mediated by PTEN loss [106].
3.3. Epigenetic Landscape
Epigenetic mechanisms underlie many aspects of antitumor immunity. The targeted use of epigenetic modifiers to remodel the immunological microenvironment holds great potential as an integral component of cancer treatment. Epigenetic reprogramming of immune cells underlies the development of tumor-reactive CD8+ T cells and mediates the dysfunctional state of T cells, differentiation into MDSCs, while Treg cells possess a unique set of transcriptional and epigenetic features [107]. Understanding the epigenetic mechanisms in immune cells and their impact on immunologic responses in TIME would enable the development of novel combinatorial treatments/biomarkers for correct treatment regimens and improved efficacy of immunotherapy (Figure 1) [108,109].
A risk model based on three m5C regulator-related genes in rectal adenocarcinoma (READ) patients was established and showed that high-risk patients had enrichment of cancer markers and were less sensitive to immunotherapy [110]. In addition, an m6A-related scoring system was developed to quantify the m6A modification pattern of individual samples using multiple data sets. Unsupervised clustering of 56 m6A regulator-related genes identified three distinct m6A regulator-related patterns, highlighting their important role in shaping TME diversity and clinical/biological features of gastric cancer [111].
METTL3 (Methyltransferase 3, N6-Adenosine-Methyltransferase Complex Catalytic Subunit) plays a key role in a variety of cancers, either dependent or independent of its m6A RNA methyltransferase activity [112]. Recently, M2-TAMs were shown to downregulate METTL3 in thyroid cancer cells and promote immunosuppressive TME through the transfer of extracellular vesicles (EVs), reversing the immunosuppressive effect of M2-TAMs that mediates resistance to anti-PD-1 therapy in thyroid cancer [113]. Following an unbiased clustered regularly interspaced short palindromic repeats - associated protein 9 (CRISPR–Cas9) epigenome screen, the tripartite motif containing 28 - SET domain bifurcated histone lysine methyltransferase 1 (TRIM28-SETDB1) complex was identified as a regulator of PD-L1 expression. In a cohort of anti–PD-1 treated melanoma patients, TRIM28 was significantly less expressed in responders compared to non-responders, while loss of SEATB1 increased infiltration of effector CD8+ T cells into the tumor microenvironment, a prerequisite for response to ICB [114].
3.4. Transcriptomic Landscape
Technologies such as RNA sequencing have identified new key factors and cellular subpopulations that create a microenvironment receptive to immunotherapy. The transcriptome at the single cell level of innate and adaptive intra-tumoral immune cells has been studied in mouse models and in testing the efficacy of different types of chemo-immunotherapies [115]. Differentially expressed genes can reveal important biological signaling pathways in cancer patients undergoing anti-PD1 therapy (Figure 1) [116]. Transcriptome profiling can also reveal changes in gene expression during disease progression and clusters of genes based on relative expression changes between different stages [117].
An analysis of gene expression in lymphocytes within the tumor microenvironment co-expressing with the pan T-cell marker CD3 epsilon subunit (CD3E) in 9,601 human tumors from 31 cancer types was reported. Targets with a high CD3E correlation and a relatively low PDCD1 correlation were identified, indicating the presence of T cells with low PD-1 expression and thus promising targets for therapy [118]. RNA-Seq data from TCGA data of patients with lung adenocarcinoma have revealed the gene landscape of PD-L1 and the status of B-cell infiltration, offering potential targets for future drug development [119]. Similarly, in melanoma cancer, six synthetic viability (SV) gene pairs were constructed as signatures to predict the clinical utility of ICIs [120].
Genetic mutations are also associated with RNA signatures, as demonstrated for myeloid inflammation in intrahepatic cholangiocarcinoma (iCCA). A negative feedback mechanism involves upregulation of interleukin-1 receptor antagonist (IL1RN)-201/203 due to alternative splicing, which exerts important anti-inflammatory effects in KRAS-mutated iCCA that are significantly associated with a better response to anti-PD-1 immunotherapy [121]. Transmembrane protein 92 (TMEM92) mRNA expression has been identified as an immune resistance and prognostic marker in pancreatic cancer. More specifically, in patients with high expression of TMEM92, increased TMB is consistent with frequent mutations of KRAS and TP53 [122].
Transcriptomic data have shown that angiogenesis and immune activities are exclusively enriched in the Alveolar Soft Part Sarcoma Chromosome Region, Candidate 1 - Transcription factor E3 (ASPSCR1-TFE3) in renal cell carcinoma (rRCC), suggesting the superior clinical outcomes of ICIs and TKI combination therapy [123]. The stem cell landscape was also investigated in CRC using data from 1,467 CRC samples. Three stem cell-related subtypes were characterized, with CRC patients with higher stem cell levels having a poorer prognosis, more immunosuppressive components in the TME and a lower response to immunotherapies [124].
4. Antibodies Targeting PD-1/PD-L1
ICIs have shown remarkable efficacy in a variety of malignancies in landmark trials and have fundamentally changed cancer therapy [125,126]. Over the past decade, several monoclonal antibodies targeting PD-1/PD-L1 have been shown to prolong survival and have been incorporated into routine clinical practice [1].
Although this novel approach of manipulating the immune system with monoclonal antibodies to generate an immune response was introduced by Chambers et al. in the 1990s [127], Hodi et al. were the first to publish the results of a landmark phase III trial showing a dramatic improvement in OS with the anti-Cytotoxic T-lymphocyte associated protein 4 (CTLA4) antibody ipilimumab in metastatic melanoma [128]. Since then, major melanoma treatment trials have shown a dramatic improvement in survival with anti-PD-1 inhibitors (Table 1) [129]. Recently, the final ten-year results of the CheckMate-067 trial were published [130]. CheckMate-067 is a landmark phase III trial that evaluated the combination of nivolumab and ipilimumab versus nivolumab monotherapy and ipilimumab monotherapy in metastatic melanoma. The median OS for patients receiving the combination was 71.9 months, compared with 36.9 months for nivolumab monotherapy and 19.9 months for ipilimumab monotherapy. After three years, 96% of patients who were still alive and showed no signs of disease progression were on the combination therapy. 97% were treated with nivolumab alone and 88% with ipilimumab alone [130]. Since 2014, the anti-PD-1 antibodies pembrolizumab and nivolumab have been included in the melanoma treatment algorithm as standard treatment for all patients [131].
In addition, anti-PD-1/PD-L1 immunotherapy represents a major breakthrough in the treatment of NSCLC [139]. KEYNOTE-189 is a landmark phase III trial that investigated the efficacy of pembrolizumab in combination with chemotherapy in metastatic NSCLC. The combination therapy resulted in a median OS of 22 months compared to 10.7 months with chemotherapy alone and was approved by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) as first-line treatment for metastatic NSCLC without actionable oncogenic alterations (AGAs) [132]. On the other hand, the IMpower150 study investigated the efficacy of the monoclonal anti-PD-L1 antibody atezolizumab in conjunction with the monoclonal anti-vascular endothelial growth factor (VEGF) antibody bevacizumab and chemotherapy. With a hazard ratio of 0.78, the combination treatment led to a median OS time of 19.2 months compared to 14.7 months in the control group (Table 2) [140]. In locally advanced stage III NSCLC, the PACIFIC study investigated the monoclonal antibody durvalumab as a consolidation treatment after chemoradiation. Durvalumab showed a remarkable improvement in progression free survival (PFS) (17.2 months compared to 5.6 months) and OS and has established itself as standard treatment in patients who respond to chemotherapy or have stable disease [141].
For recurrent or metastatic (R/M) HNSCC, pembrolizumab and nivolumab were established as first- and second-line treatments, based on the results of the KEYNOTE 048 and CheckMate 141 trials. KEYNOTE-048 showed that pembrolizumab was superior to chemotherapy in patients with PD-L1-positive HNSCC [133]. On the other hand, the CheckMate-141 trial showed that nivolumab prolonged survival compared to standard chemotherapy or cetuximab in patients with platinum-resistant R/M disease [134]. These results have established ICIs as a fundamental component of the therapeutic algorithm for head and neck cancer [146].
In gastrointestinal cancer, predictive biomarkers such as MSI, PD-L1 expression, mutations in KRAS and B-Raf proto-oncogene, serine/threonine kinase (BRAF) genes, and expression of receptor tyrosine-protein kinase erbB-2 (HER2) are crucial for treatment selection in first-line tretment. In MSI-H/dMMR mCRC, pembrolizumab has established itself as first-line treatment after the results of the Keynote-177 trial, showed the superiority of pembrolizumab compared to chemotherapy [135]. In gastric cancer, the Keynote-811 trial showed that pembrolizumab in combination with trastuzumab and chemotherapy improved OS in HER2-positive patients, with the greatest benefit observed in patients whose tumors expressed PD-L1 (CPS ≥1) [147]. In patients with advanced gastric or esophageal cancer, the CheckMate-649 trial showed that nivolumab in combination with chemotherapy provided a significant survival benefit [136]. In addition, immunotherapy has shown great success in the treatment of liver cancer, particularly in HCC and cholangiocarcinoma. In patients with advanced HCC, the Imbrave 050 study found a statistically significant difference in OS compared to sorafenib (20.2 months vs. 15.5 months) [145]. In addition, the HIMALAYA study investigated durvalumab in combination with a single dose of tremelimumab and durvalumab as monotherapy in comparison with sorafenib. This showed an improvement in the survival rate: 25% of patients were still alive after 4 years with the combination, compared to 15% with sorafenib [142]. For patients with advanced cholangiocarcinoma, TOPAZ-1 was the first study to show a survival benefit from the addition of durvalumab to standard chemotherapy [143].
RCC is considered a traditionally immunogenic tumor that was treated several years ago with first-generation immunotherapy (cytokines, interleukins, interferon). In intermediate- and high-risk populations, the CheckMate 214 trial showed that nivolumab/ipilimumab significantly improved OS and PFS compared to sunitinib [148]. On the other hand, the combination of pembrolizumab and axitinib resulted in a median PFS of 15.7 months and an objective response rate of 60% [137]. In addition, the CheckMate 9ER study showed a significant improvement in survival in patients receiving nivolumab-cabozantinib versus sunitinib [149]. In addition, pembrolizumab was included in the treatment algorithm for patients with early-stage disease. The Keynote-564 trial demonstrated an extension of DFS at 24 months in high-risk renal cell carcinoma after nephrectomy [150].
Recent important research on advanced urothelial carcinoma has shown improved outcomes in initial treatment. Three important trials—CheckMate 901, EV-302, and JAVELIN—have significantly improved the treatment of bladder cancer. The CheckMate 901 trial found that nivolumab in combination with chemotherapy resulted in a median OS of 21.7 months [151]. Most importantly, the combination of enfortumab vedotin and pembrolizumab resulted in an excellent OS of 31.5 months and a PFS of 12.5 months (EV-302/Keynote-A39 study) [138]. Maintenance immunotherapy with avelumab in patients without disease progression after platinum-based chemotherapy resulted in a significant benefit in OS and PFS benefit in the JAVELIN 100 trial [144].
In the past, advanced gynecologic malignancies, including uterine and cervical cancer, were treated with conventional chemotherapy. The new molecular classification of endometrial cancer enables personalized therapy. Based on the Keynote-775 and GARNET trials, immunotherapy—including pembrolizumab and lenvatinib—has improved survival and progression-free survival in patients with advanced endometrial cancer [152].
Immunotherapy has achieved encouraging results in the treatment of triple-negative breast cancer (TNBC). In particular, in patients with high PD-L1 expression, the KEYNOTE-355 study showed that pembrolizumab in combination with chemotherapy significantly extended survival. In patients with a PD-L1 CPS ≥10, the combination of pembrolizumab plus chemotherapy resulted in a median PFS of 9.7 months, compared to 5.6 months for chemotherapy alone [153].
5. Future Perspectives and Conclusions
The PD1/PD-L1 axis acts as a brake on the immune response and prevents overactivity, while T cells are the effector actors of the immune system that attack and destroy damaged cells such as virus-infected and cancer cells [154]. Expression of PD-L1 by tumor cells often activates PD1 signaling and allows the tumor to evade immune surveillance. Novel ICB and combination therapies to block the PD-L1/PD-1 pathway are leading to improved anti-tumor efficacy in patients with advanced cancers [1]. Characterizing the molecular landscape of the immunotherapy response by delineating activated cell states or cellular interactions may elucidate the molecular mechanisms of tumor response [55,81]. Even in patients who initially respond effectively to immunotherapy, the disease progresses over a period of 5 years. Resistance to immunotherapy is an evolving area of research that is challenging due to the complex and dynamic interplay between TCs and the immune system [155]. Key mechanisms of resistance include low TMB and heterogeneity of antigen presentation, alterations in the tumor microenvironment, and host-related factors [156]. Current research is focused on strategies to maximize patient selection for therapy, clarify mechanisms of resistance, improve existing biomarkers, including PD-L1 expression and TMB, and discover new biomarkers [87,99].
In summary, anti-PD-1 and anti-PD-L1 antibodies have led to prolonged response and improved survival rates in a variety of malignant tumors. Ongoing research and clinical developments have established immuno-therapeutics as indispensable components of modern oncology.
Author Contributions
Conceptualization, A.S.; investigation, A.S., C.A., I.K.; writing—original draft preparation, A.S., C.A., I.K.; writing—review and editing, A.S., A.P., E.L., A.P.; supervision, A.S.; All authors have read and agreed to the published version of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| Full Name | Abbreviation | Full Name | Abbreviation |
| Programmed death-ligand 1 | PD-L1 | Epidermal growth factor receptor | EGFR |
| Programmed cell death protein | PD1 | DNA damage repair | DDR |
| Immune checkpoint inhibitors | ICIs | Esophageal squamous cell carcinoma | ESCC patients |
| Tumor microenvironment | TME | checkpoint kinase 2 | CHEK2 |
| Extracellular matrix | ECM | Ataxia-telangiectasia mutations | ATM |
| Tumor-associated macrophages | TAMs | Kirsten rat sarcoma virus | KRAS |
| Cancer-associated fibroblasts | CAFs | Tumor Protein P53 | TP53 |
| Tumor mutational burden | TMB | Transcription activator BRG1 | SMARCA4 |
| Regulatory T cells | Tregs | Breast cancer | BC |
| Ras/mitogen-activated protein kinase | MAPK | Frameshift mutations | FS |
| Phosphoinositide 3-kinase | PI3K | Tyrosine kinase inhibitors | TKIs |
| Mammalian target of rapamycin | mTOR | Human leukocyte antigen-I loss of heterozygosity | HLA-I LOH |
| Dendritic cells | DC | Phosphatase and tensin homolog | PTEN |
| tumor cells | TCs | Rectal adenocarcinoma | READ |
| Mismatch repair-deficient | dMMR | Methyltransferase 3, N6-adenosine-methyltransferase complex catalytic subunit | METTL3 |
| myeloid-derived suppressor cells | MDSCs | Extracellular vesicles | EVs |
| Steatohepatitis -related hepatocellular carcinoma | MASH-HCC | Clustered regularly interspaced short palindromic repeats- associated protein 9 | CRISPR–Cas9 |
| Squalene epoxidase | SQLE | Tripartite Motif Containing 28- SET Domain Bifurcated Histone Lysine Methyltransferase 1 | TRIM28-SETDB1 |
| Metastatic Colorectal Cancer | mCRC | CD3 Epsilon Subunit | CD3E |
| Hepatocellular cancer | HCC | Programmed cell death protein 1 | PDCD1 |
| Pancreatic cancer | PaC | Synthetic viability | SV |
| Immune checkpoint blockade | ICB | Intrahepatic cholangiocarcinoma | iCCA |
| Natural killer | NK | Interleukin-1 receptor antagonist | IL1RN |
| Immune cell infiltration | ICI | Transmembrane protein 92 | TMEM92 |
| Lung adenocarcioma | LUAD | Renal cell carcinoma | rRCC |
| Tumor microenvironment prognostic signature | LATPS | Alveolar Soft Part Sarcoma Chromosome Region, Candidate 1 - Transcription factor E3 | ASPSCR1-TFE3 |
| Immune -related gene prognostic index | IRGPI | Cytotoxic T-lymphocyte associated protein 4 | CTLA4 |
| Tumor immunological microenvironment | TIME | Food and drugs administration | FDA |
| NAD+ metabolism-related gene signature | NMRGS | European medicines agency | EMA |
| Non-small cell lung cancer | NSCLC | Actionable oncogenic alterations | AGAs |
| Head and neck squamous cell carcinoma | HNSCC | Vascular endothelial growth factor | VEGF |
| Pancreatic ductal adenocarcinoma | PDAC | Recurrent or metastatic | R/M |
| Immune -mesenchymal like | IML | B-Raf proto-oncogene, serine/threonine kinase | BRAF |
| Overall Survival | OS | Receptor tyrosine-protein kinase erbB-2 | HER2 |
| Disease-free survival | DFS | Progression Free Survival | PFS |
| Next-generation sequencing | NGS | Triple Negative Breast Cancer | TNBC |
| The Cancer Genome Atlas | TCGA | Small-cell lung cancer | SCLC |
| Interferon gamma | IFΝγ | Immune cell infiltration | ICI |
| Cell adhesion molecules | CAMs | Blood mononuclear cells | PBMCs |
References
- Robert, C. A Decade of Immune-Checkpoint Inhibitors in Cancer Therapy. Nat Commun 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T Cells in Cancer and Cancer Immunotherapy. Br J Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Wang, Q.; Strauss, L.; Boussiotis, V.A. Revisiting the PD-1 Pathway. Sci Adv 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Nwabo Kamdje, A.H.; Seke Etet, P.F.; Simo Tagne, R.; Vecchio, L.; Lukong, K.E.; Krampera, M. Tumor Microenvironment Uses a Reversible Reprogramming of Mesenchymal Stromal Cells to Mediate Pro-Tumorigenic Effects. Front Cell Dev Biol 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The Tumor Microenvironment. Curr Biol 2020, 30, R921–R925. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, J.; Chen, Z.; Luo, J.; Guo, W.; Sun, L.; Lin, L. Targeting M2-like Tumor-Associated Macrophages Is a Potential Therapeutic Approach to Overcome Antitumor Drug Resistance. NPJ Precis Oncol 2024, 8. [Google Scholar] [CrossRef]
- Zhang, H.; Yue, X.; Chen, Z.; Liu, C.; Wu, W.; Zhang, N.; Liu, Z.; Yang, L.; Jiang, Q.; Cheng, Q.; et al. Define Cancer-Associated Fibroblasts (CAFs) in the Tumor Microenvironment: New Opportunities in Cancer Immunotherapy and Advances in Clinical Trials. Mol Cancer 2023, 22. [Google Scholar] [CrossRef]
- Anderson, N.R.; Minutolo, N.G.; Gill, S.; Klichinsky, M. Macrophage-Based Approaches for Cancer Immunotherapy. Cancer Res 2021, 81, 1201–1208. [Google Scholar] [CrossRef]
- Glabman, R.A.; Choyke, P.L.; Sato, N. Cancer-Associated Fibroblasts: Tumorigenicity and Targeting for Cancer Therapy. Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Ock, C.Y.; Hwang, J.E.; Keam, B.; Kim, S.B.; Shim, J.J.; Jang, H.J.; Park, S.; Sohn, B.H.; Cha, M.; Ajani, J.A.; et al. Genomic Landscape Associated with Potential Response to Anti-CTLA-4 Treatment in Cancers. Nat Commun 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Muquith, M.; Espinoza, M.; Elliott, A.; Xiu, J.; Seeber, A.; El-Deiry, W.; Antonarakis, E.S.; Graff, S.L.; Hall, M.J.; Borghaei, H.; et al. Tissue-Specific Thresholds of Mutation Burden Associated with Anti-PD-1/L1 Therapy Benefit and Prognosis in Microsatellite-Stable Cancers. Nat Cancer 2024, 5, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Cohen, P.R.; Boichard, A.; Frampton, G.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Genomic Landscape of Advanced Basal Cell Carcinoma: Implications for Precision Treatment with Targeted and Immune Therapies. Oncoimmunology 2017, 7. [Google Scholar] [CrossRef]
- Long, J.; Wang, D.; Wang, A.; Chen, P.; Lin, Y.; Bian, J.; Yang, X.; Zheng, M.; Zhang, H.; Zheng, Y.; et al. A Mutation-Based Gene Set Predicts Survival Benefit after Immunotherapy across Multiple Cancers and Reveals the Immune Response Landscape. Genome Med 2022, 14. [Google Scholar] [CrossRef]
- Florou, V.; Floudas, C.S.; Maoz, A.; Naqash, A.R.; Norton, C.; Tan, A.C.; Sokol, E.S.; Frampton, G.; Soares, H.P.; Puri, S.; et al. Real-World Pan-Cancer Landscape of Frameshift Mutations and Their Role in Predicting Responses to Immune Checkpoint Inhibitors in Cancers with Low Tumor Mutational Burden. J Immunother Cancer 2023, 11. [Google Scholar] [CrossRef]
- Wang, J.Y.; Xiu, J.; Baca, Y.; Arai, H.; Battaglin, F.; Kawanishi, N.; Soni, S.; Zhang, W.; Millstein, J.; Shields, A.F.; et al. Distinct Genomic Landscapes of Gastroesophageal Adenocarcinoma Depending on PD-L1 Expression Identify Mutations in RAS-MAPK Pathway and TP53 as Potential Predictors of Immunotherapy Efficacy. Ann Oncol 2021, 32, 906–916. [Google Scholar] [CrossRef]
- Yarchoan, M.; Albacker, L.A.; Hopkins, A.C.; Montesion, M.; Murugesan, K.; Vithayathil, T.T.; Zaidi, N.; Azad, N.S.; Laheru, D.A.; Frampton, G.M.; et al. PD-L1 Expression and Tumor Mutational Burden Are Independent Biomarkers in Most Cancers. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, Y.; Miao, Q.; Chen, Y. The Therapeutic Potential of PD-1/PD-L1 Pathway on Immune-Related Diseases: Based on the Innate and Adaptive Immune Components. Biomed Pharmacother 2023, 167. [Google Scholar] [CrossRef]
- Hwang, J.R.; Byeon, Y.; Kim, D.; Park, S.G. Recent Insights of T Cell Receptor-Mediated Signaling Pathways for T Cell Activation and Development. Exp Mol Med 2020, 52, 750–761. [Google Scholar] [CrossRef]
- Chamoto, K.; Yaguchi, T.; Tajima, M.; Honjo, T. Insights from a 30-Year Journey: Function, Regulation and Therapeutic Modulation of PD1. Nat Rev Immunol 2023, 23, 682–695. [Google Scholar] [CrossRef]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring Function in Exhausted CD8 T Cells during Chronic Viral Infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining “T Cell Exhaustion. ” Nat Rev Immunol 2019, 19, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, C.; Luong, G.; Sun, Y. A Snapshot of the PD-1/PD-L1 Pathway. J Cancer 2021, 12, 2735–2746. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chikuma, S.; Hori, S.; Fagarasan, S.; Honjo, T. Nonoverlapping Roles of PD-1 and FoxP3 in Maintaining Immune Tolerance in a Novel Autoimmune Pancreatitis Mouse Model. Proc Natl Acad Sci U S A 2016, 113, 8490–8495. [Google Scholar] [CrossRef]
- Xu, X.; Hou, B.; Fulzele, A.; Masubuchi, T.; Zhao, Y.; Wu, Z.; Hu, Y.; Jiang, Y.; Ma, Y.; Wang, H.; et al. PD-1 and BTLA Regulate T Cell Signaling Differentially and Only Partially through SHP1 and SHP2. J Cell Biol 2020, 219. [Google Scholar] [CrossRef]
- Sheppard, K.A.; Fitz, L.J.; Lee, J.M.; Benander, C.; George, J.A.; Wooters, J.; Qiu, Y.; Jussif, J.M.; Carter, L.L.; Wood, C.R.; et al. PD-1 Inhibits T-Cell Receptor Induced Phosphorylation of the ZAP70/CD3ζ Signalosome and Downstream Signaling to PKCθ. FEBS Lett 2004, 574, 37–41. [Google Scholar] [CrossRef]
- Kleffel, S.; Posch, C.; Barthel, S.R.; Mueller, H.; Schlapbach, C.; Guenova, E.; Elco, C.P.; Lee, N.; Juneja, V.R.; Zhan, Q.; et al. Melanoma Cell-Intrinsic PD-1 Receptor Functions Promote Tumor Growth. Cell 2015, 162, 1242–1256. [Google Scholar] [CrossRef]
- Patsoukis, N.; Brown, J.; Petkova, V.; Liu, F.; Li, L.; Boussiotis, V.A. Selective Effects of PD-1 on Akt and Ras Pathways Regulate Molecular Components of the Cell Cycle and Inhibit T Cell Proliferation. Sci Signal 2012, 5. [Google Scholar] [CrossRef]
- Patsoukis, N.; Li, L.; Sari, D.; Petkova, V.; Boussiotis, V.A. PD-1 Increases PTEN Phosphatase Activity While Decreasing PTEN Protein Stability by Inhibiting Casein Kinase 2. Mol Cell Biol 2013, 33, 3091–3098. [Google Scholar] [CrossRef]
- Yearley, J.H.; Gibson, C.; Yu, N.; Moon, C.; Murphy, E.; Juco, J.; Lunceford, J.; Cheng, J.; Chow, L.Q.M.; Seiwert, T.Y.; et al. PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer. Clin Cancer Res 2017, 23, 3158–3167. [Google Scholar] [CrossRef]
- Van Der Merwe, P.A.; Bodian, D.L.; Daenke, S.; Linsley, P.; Davis, S.J. CD80 (B7-1) Binds Both CD28 and CTLA-4 with a Low Affinity and Very Fast Kinetics. J Exp Med 1997, 185, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Butte, M.J.; Peña-Cruz, V.; Kim, M.J.; Freeman, G.J.; Sharpe, A.H. Interaction of Human PD-L1 and B7-1. Mol Immunol 2008, 45, 3567–3572. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yu, Y.; Yang, S.; Zeng, B.; Zhang, Z.; Jiao, G.; Zhang, Y.; Cai, L.; Yang, R. Regulation of Arginase I Activity and Expression by Both PD-1 and CTLA-4 on the Myeloid-Derived Suppressor Cells. Cancer Immunol Immunother 2009, 58, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Petrovas, C.; Casazza, J.P.; Brenchley, J.M.; Price, D.A.; Gostick, E.; Adams, W.C.; Precopio, M.L.; Schacker, T.; Roederer, M.; Douek, D.C.; et al. PD-1 Is a Regulator of Virus-Specific CD8+ T Cell Survival in HIV Infection. J Exp Med 2006, 203, 2281–2292. [Google Scholar] [CrossRef]
- Oestreich, K.J.; Yoon, H.; Ahmed, R.; Boss, J.M. NFATc1 Regulates PD-1 Expression upon T Cell Activation. J Immunol 2008, 181, 4832–4839. [Google Scholar] [CrossRef]
- Bally, A.P.R.; Austin, J.W.; Boss, J.M. Genetic and Epigenetic Regulation of PD-1 Expression. J Immunol 2016, 196, 2431–2437. [Google Scholar] [CrossRef]
- Youngblood, B.; Noto, A.; Porichis, F.; Akondy, R.S.; Ndhlovu, Z.M.; Austin, J.W.; Bordi, R.; Procopio, F.A.; Miura, T.; Allen, T.M.; et al. Cutting Edge: Prolonged Exposure to HIV Reinforces a Poised Epigenetic Program for PD-1 Expression in Virus-Specific CD8 T Cells. J Immunol 2013, 191, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, B.; Oestreich, K.J.; Ha, S.J.; Duraiswamy, J.; Akondy, R.S.; West, E.E.; Wei, Z.; Lu, P.; Austin, J.W.; Riley, J.L.; et al. Chronic Virus Infection Enforces Demethylation of the Locus That Encodes PD-1 in Antigen-Specific CD8(+) T Cells. Immunity 2011, 35, 400–412. [Google Scholar] [CrossRef]
- Zhang, N.; Li, M.; Xu, X.; Zhang, Y.; Liu, Y.; Zhao, M.; Li, P.; Chen, J.; Fukuda, T.; Gu, J.; et al. Loss of Core Fucosylation Enhances the Anticancer Activity of Cytotoxic T Lymphocytes by Increasing PD-1 Degradation. Eur J Immunol 2020, 50, 1820–1833. [Google Scholar] [CrossRef]
- Meng, X.; Liu, X.; Guo, X.; Jiang, S.; Chen, T.; Hu, Z.; Liu, H.; Bai, Y.; Xue, M.; Hu, R.; et al. FBXO38 Mediates PD-1 Ubiquitination and Regulates Anti-Tumour Immunity of T Cells. Nature 2018, 564, 130–135. [Google Scholar] [CrossRef]
- Ribas, A.; Hu-Lieskovan, S. What Does PD-L1 Positive or Negative Mean? J Exp Med 2016, 213, 2835–2840. [Google Scholar] [CrossRef]
- George, J.; Saito, M.; Tsuta, K.; Iwakawa, R.; Shiraishi, K.; Scheel, A.H.; Uchida, S.; Watanabe, S.I.; Nishikawa, R.; Noguchi, M.; et al. Genomic Amplification of CD274 (PD-L1) in Small-Cell Lung Cancer. Clin Cancer Res 2017, 23, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Roemer, M.G.M.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J Clin Oncol 2016, 34, 2690–2697. [Google Scholar] [CrossRef]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov 2017, 7, 188–201. [Google Scholar] [CrossRef]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e9. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Kobayashi, A.; Jiang, P.; De Andrade, L.F.; Tay, R.E.; Luoma, A.M.; Tsoucas, D.; Qiu, X.; Lim, K.; Rao, P.; et al. A Major Chromatin Regulator Determines Resistance of Tumor Cells to T Cell-Mediated Killing. Science 2018, 359, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N.; et al. Identification of Essential Genes for Cancer Immunotherapy. Nature 2017, 548, 537–542. [Google Scholar] [CrossRef]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; La Fleur, M.W.; Juneja, V.R.; et al. In Vivo CRISPR Screening Identifies Ptpn2 as a Cancer Immunotherapy Target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef]
- Yi, M.; Niu, M.; Xu, L.; Luo, S.; Wu, K. Regulation of PD-L1 Expression in the Tumor Microenvironment. J Hematol Oncol 2021, 14. [Google Scholar] [CrossRef]
- Yi, M.; Xu, L.; Jiao, Y.; Luo, S.; Li, A.; Wu, K. The Role of Cancer-Derived MicroRNAs in Cancer Immune Escape. J Hematol Oncol 2020, 13. [Google Scholar] [CrossRef]
- Kataoka, K.; Shiraishi, Y.; Takeda, Y.; Sakata, S.; Matsumoto, M.; Nagano, S.; Maeda, T.; Nagata, Y.; Kitanaka, A.; Mizuno, S.; et al. Aberrant PD-L1 Expression through 3’-UTR Disruption in Multiple Cancers. Nature 2016, 534, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Hsu, J.M.; Yang, W.H.; Hung, M.C. Mechanisms Regulating PD-L1 Expression in Cancers and Associated Opportunities for Novel Small-Molecule Therapeutics. Nat Rev Clin Oncol 2022, 19, 287–305. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Apollonio, B.; Ioannou, N.; Papazoglou, D.; Ramsay, A.G. Understanding the Immune-Stroma Microenvironment in B Cell Malignancies for Effective Immunotherapy. Front Oncol 2021, 11. [Google Scholar] [CrossRef]
- Antoranz, A.; Van Herck, Y.; Bolognesi, M.M.; Lynch, S.M.; Rahman, A.; Gallagher, W.M.; Boecxstaens, V.; Marine, J.C.; Cattoretti, G.; van den Oord, J.J.; et al. Mapping the Immune Landscape in Metastatic Melanoma Reveals Localized Cell-Cell Interactions That Predict Immunotherapy Response. Cancer Res 2022, 82, 3275–3290. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tang, B.; Fu, J.; Zhu, X.; Xie, W.; Wang, N.; Ding, Z.; Song, Z.; Yang, Y.; Xu, G.; et al. High-Plex Spatial Transcriptomic Profiling Reveals Distinct Immune Components and the HLA Class I/DNMT3A/CD8 Modulatory Axis in Mismatch Repair-Deficient Endometrial Cancer. Cellular oncology (Dordrecht, Netherlands) 2024, 47, 573–585. [Google Scholar] [CrossRef]
- Puig-Saus, C.; Sennino, B.; Peng, S.; Wang, C.L.; Pan, Z.; Yuen, B.; Purandare, B.; An, D.; Quach, B.B.; Nguyen, D.; et al. Neoantigen-Targeted CD8+ T Cell Responses with PD-1 Blockade Therapy. Nature 2023, 615, 697–704. [Google Scholar] [CrossRef]
- Hu, X.; Dong, Y.; Xie, S.; Song, Y.; Yu, C.; He, Y.; Wang, Z.; Hu, Q.; Ni, Y.; Ding, L. Immune Checkpoint CD161/LLT1-Associated Immunological Landscape and Diagnostic Value in Oral Squamous Cell Carcinoma. J Pathol Clin Res 2024, 10. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zhang, X.; Liu, X.; Cai, X.; Shen, T.; Pan, D.; Liang, R.; Ding, R.; Hu, R.; Dong, J.; et al. Single-Cell Sequencing Reveals the Immune Microenvironment Landscape Related to Anti-PD-1 Resistance in Metastatic Colorectal Cancer with High Microsatellite Instability. BMC Med 2023, 21. [Google Scholar] [CrossRef]
- Wen, J.; Zhang, X.; Wong, C.C.; Zhang, Y.; Pan, Y.; Zhou, Y.; Cheung, A.H.K.; Liu, Y.; Ji, F.; Kang, X.; et al. Targeting Squalene Epoxidase Restores Anti-PD-1 Efficacy in Metabolic Dysfunction-Associated Steatohepatitis-Induced Hepatocellular Carcinoma. Gut 2024. [Google Scholar] [CrossRef]
- Tan, J.; Fan, W.; Liu, T.; Zhu, B.; Liu, Y.; Wang, S.; Wu, J.; Liu, J.; Zou, F.; Wei, J.; et al. TREM2+ Macrophages Suppress CD8+ T-Cell Infiltration after Transarterial Chemoembolisation in Hepatocellular Carcinoma. J Hepatol 2023, 79, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Jiang, Y.; Huang, Y.; Wang, Q.; Kaifi, J.T.; Kimchi, E.T.; Chabu, C.Y.; Liu, Z.; Joshi, T.; Li, G. Single-Cell RNA Sequencing to Characterize the Response of Pancreatic Cancer to Anti-PD-1 Immunotherapy. Transl Oncol 2022, 15. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T Cells in Cancer Immunosuppression - Implications for Anticancer Therapy. Nat Rev Clin Oncol 2019, 16, 356–371. [Google Scholar] [CrossRef]
- Kugel, C.H.; Douglass, S.M.; Webster, M.R.; Kaur, A.; Liu, Q.; Yin, X.; Weiss, S.A.; Darvishian, F.; Al-Rohil, R.N.; Ndoye, A.; et al. Age Correlates with Response to Anti-PD1, Reflecting Age-Related Differences in Intratumoral Effector and Regulatory T-Cell Populations. Clin Cancer Res 2018, 24, 5347–5356. [Google Scholar] [CrossRef] [PubMed]
- Blomberg, O.S.; Kos, K.; Spagnuolo, L.; Isaeva, O.I.; Garner, H.; Wellenstein, M.D.; Bakker, N.; Duits, D.E.M.; Kersten, K.; Klarenbeek, S.; et al. Neoadjuvant Immune Checkpoint Blockade Triggers Persistent and Systemic Treg Activation Which Blunts Therapeutic Efficacy against Metastatic Spread of Breast Tumors. Oncoimmunology 2023, 12. [Google Scholar] [CrossRef]
- Yin, Y.; Sakakibara, R.; Honda, T.; Kirimura, S.; Daroonpan, P.; Kobayashi, M.; Ando, K.; Ujiie, H.; Kato, T.; Kaga, K.; et al. High Density and Proximity of CD8+ T Cells to Tumor Cells Are Correlated with Better Response to Nivolumab Treatment in Metastatic Pleural Mesothelioma. Thorac Cancer 2023, 14, 1991–2000. [Google Scholar] [CrossRef]
- Murakami, T.; Tanaka, N.; Takamatsu, K.; Hakozaki, K.; Fukumoto, K.; Masuda, T.; Mikami, S.; Shinojima, T.; Kakimi, K.; Tsunoda, T.; et al. Multiplexed Single-Cell Pathology Reveals the Association of CD8 T-Cell Heterogeneity with Prognostic Outcomes in Renal Cell Carcinoma. Cancer Immunol Immunother 2021, 70, 3001–3013. [Google Scholar] [CrossRef]
- Peng, M. Immune Landscape of Distinct Subtypes in Urothelial Carcinoma Based on Immune Gene Profile. Front Immunol 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yang, X.; Wu, X. Tumor Immune Microenvironment Characterization Identifies Prognosis and Immunotherapy-Related Gene Signatures in Melanoma. Front Immunol 2021, 12. [Google Scholar] [CrossRef]
- Sui, Z.; Wu, X.; Du, L.; Wang, H.; Yuan, L.; Zhang, J. V.; Yu, Z. Characterization of the Immune Cell Infiltration Landscape in Esophageal Squamous Cell Carcinoma. Front Oncol 2022, 12. [Google Scholar] [CrossRef]
- Guo, Y.; Yang, J.; Ren, K.; Tian, X.; Gao, H.; Tian, X.; Zhang, X.; Kan, Q. The Heterogeneity of Immune Cell Infiltration Landscape and Its Immunotherapeutic Implications in Hepatocellular Carcinoma. Front Immunol 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yuan, L.; Huang, W.; Liao, L.; Zhu, X.; Wang, X.; Li, J.; Liang, W.; Wu, Y.; Liu, X.; et al. LATPS, a Novel Prognostic Signature Based on Tumor Microenvironment of Lung Adenocarcinoma to Better Predict Survival and Immunotherapy Response. Front Immunol 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, H.; Feng, Y.; Liu, X.; Wang, Y.; Liu, Y.; Li, H.; Zhang, Y. Decoding the Immune Landscape: A Comprehensive Analysis of Immune-Associated Biomarkers in Cervical Carcinoma and Their Implications for Immunotherapy Strategies. Front Genet 2024, 15. [Google Scholar] [CrossRef]
- Wang, Z.; Song, J.; Azami, N.L.B.; Sun, M. Identification of a Novel Immune Landscape Signature for Predicting Prognosis and Response of Colon Cancer to Immunotherapy. Front Immunol 2022, 13. [Google Scholar] [CrossRef]
- Jiang, C.; Zhou, Y.; Yan, L.; Zheng, J.; Wang, X.; Li, J.; Jiang, X. A Prognostic NAD+ Metabolism-Related Gene Signature for Predicting Response to Immune Checkpoint Inhibitor in Glioma. Front Oncol 2023, 13. [Google Scholar] [CrossRef]
- Wang, Q.; Zhao, Y.; Wang, F.; Tan, G. A Novel Immune Signature Predicts Immunotherapy Responsiveness and Reveals the Landscape of the Tumor Immune Microenvironment in Head and Neck Squamous Cell Carcinoma. Front Genet 2022, 13. [Google Scholar] [CrossRef]
- Parra, E.R.; Zhang, J.; Duose, D.Y.; Gonzalez-Kozlova, E.; Redman, M.W.; Chen, H.; Manyam, G.C.; Kumar, G.; Zhang, J.; Song, X.; et al. Multi-Omics Analysis Reveals Immune Features Associated with Immunotherapy Benefit in Patients with Squamous Cell Lung Cancer from Phase III Lung-MAP S1400I Trial. Clin Cancer Res 2024, 30, 1655–1668. [Google Scholar] [CrossRef]
- Cao, L.; Che, X.; Qiu, X.; Li, Z.; Yang, B.; Wang, S.; Hou, K.; Fan, Y.; Qu, X.; Liu, Y. M2 Macrophage Infiltration into Tumor Islets Leads to Poor Prognosis in Non-Small-Cell Lung Cancer. Cancer Manag Res 2019, 11, 6125–6138. [Google Scholar] [CrossRef] [PubMed]
- Gavrielatou, N.; Fortis, E.; Spathis, A.; Anastasiou, M.; Economopoulou, P.; Foukas, G.R.P.; Lelegiannis, I.M.; Rusakiewicz, S.; Vathiotis, I.; Aung, T.N.; et al. B-Cell Infiltration Is Associated with Survival Outcomes Following Programmed Cell Death Protein 1 Inhibition in Head and Neck Squamous Cell Carcinoma. Ann Oncol 2024, 35, 340–350. [Google Scholar] [CrossRef]
- Chang, T.-G.; Spathis, A.; Schäffer, A.A.; Gavrielatou, N.; Kuo, F.; Jia, D.; Mukherjee, S.; Sievers, C.; Economopoulou, P.; Anastasiou, M.; et al. Tumor and Blood B Cell Abundance Outperforms Established Immune Checkpoint Blockade Response Prediction Signatures in Head and Neck Cancer. Ann Oncol 2024. [Google Scholar] [CrossRef]
- Leung, E.L.H.; Li, R.Z.; Fan, X.X.; Wang, L.Y.; Wang, Y.; Jiang, Z.; Huang, J.; Pan, H.D.; Fan, Y.; Xu, H.; et al. Longitudinal High-Dimensional Analysis Identifies Immune Features Associating with Response to Anti-PD-1 Immunotherapy. Nat Commun 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Araujo, B. de Lima, V.; Borch, A.; Hansen, M.; Draghi, A.; Spanggaard, I.; Rohrberg, K.; Reker Hadrup, S.; Lassen, U.; Svane, I.M. Common Phenotypic Dynamics of Tumor-Infiltrating Lymphocytes across Different Histologies upon Checkpoint Inhibition: Impact on Clinical Outcome. Cytotherapy 2020, 22, 204–213. [Google Scholar] [CrossRef]
- Aoyama, S.; Nakagawa, R.; Nemoto, S.; Perez-Villarroel, P.; Mulé, J.J.; Mailloux, A.W. Checkpoint Blockade Accelerates a Novel Switch from an NKT-Driven TNFα Response toward a T Cell Driven IFN-γ Response within the Tumor Microenvironment. J Immunother Cancer 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- McAndrews, K.M.; Chen, Y.; Darpolor, J.K.; Zheng, X.; Yang, S.; Carstens, J.L.; Li, B.; Wang, H.; Miyake, T.; De Sampaio, P.C.; et al. Identification of Functional Heterogeneity of Carcinoma-Associated Fibroblasts with Distinct IL6-Mediated Therapy Resistance in Pancreatic Cancer. Cancer Discov 2022, 12, 1580–1597. [Google Scholar] [CrossRef]
- Li, L.; Shen, L.; Wu, H.; Li, M.; Chen, L.; Zhou, Q.; Ma, J.; Huai, C.; Zhou, W.; Wei, M.; et al. An Integrated Analysis Identifies Six Molecular Subtypes of Pancreatic Ductal Adenocarcinoma Revealing Cellular and Molecular Landscape. Carcinogenesis 2023, 44, 726–740. [Google Scholar] [CrossRef]
- Pich-Bavastro, C.; Yerly, L.; DiDomizio, J.; Tissot-Renaud, S.; Gilliet, M.; Kuonen, F. Activin A-Mediated Polarization of Cancer-Associated Fibroblasts and Macrophages Confers Resistance to Checkpoint Immunotherapy in Skin Cancer. Clin Cancer Res 2023, 29, 3498–3513. [Google Scholar] [CrossRef]
- Tunger, A.; Sommer, U.; Wehner, R.; Kubasch, A.S.; Grimm, M.O.; Bachmann, M.P.; Platzbecker, U.; Bornhäuser, M.; Baretton, G.; Schmitz, M. The Evolving Landscape of Biomarkers for Anti-PD-1 or Anti-PD-L1 Therapy. J Clin Med 2019, 8. [Google Scholar] [CrossRef]
- Landen, C.N.; Molinero, L.; Hamidi, H.; Sehouli, J.; Miller, A.; Moore, K.N.; Taskiran, C.; Bookman, M.; Lindemann, K.; Anderson, C.; et al. Influence of Genomic Landscape on Cancer Immunotherapy for Newly Diagnosed Ovarian Cancer: Biomarker Analyses from the IMagyn050 Randomized Clinical Trial. Clin Cancer Res 2023, 29, 1698–1707. [Google Scholar] [CrossRef]
- Roshan-Zamir, M.; Khademolhosseini, A.; Rajalingam, K.; Ghaderi, A.; Rajalingam, R. The Genomic Landscape of the Immune System in Lung Cancer: Present Insights and Continuing Investigations. Front Genet 2024, 15. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N Engl J Med 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer Immunology. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non-Small Cell Lung Cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, J.; Xu, Y.; Cai, S.; Li, T.; Wang, G.; Li, C.; Zhao, L.; Hu, Y. Co-Occurring Genomic Alterations and Immunotherapy Efficacy in NSCLC. NPJ Precis Oncol 2022, 6. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, N.J.; Lavery, J.A.; Brown, S.; de Bruijn, I.; Jee, J.; Tran, T.N.; Rizvi, H.; Arbour, K.C.; Whiting, K.; Shen, R.; et al. The GENIE BPC NSCLC Cohort: A Real-World Repository Integrating Standardized Clinical and Genomic Data for 1,846 Patients with Non-Small Cell Lung Cancer. Clin Cancer Res 2023, 29, 3418–3428. [Google Scholar] [CrossRef]
- Zhang, H.; Wen, H.; Zhu, Q.; Zhang, Y.; Xu, F.; Ma, T.; Guo, Y.; Lu, C.; Zhao, X.; Ji, Y.; et al. Genomic Profiling and Associated B Cell Lineages Delineate the Efficacy of Neoadjuvant Anti-PD-1-Based Therapy in Oesophageal Squamous Cell Carcinoma. EBioMedicine 2024, 100. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Gao, Y.; Jiang, S.; Cui, Y.; Xie, Y.; Kang, Z.; Chen, Y.X.; Sun, D.; Fang, J.Y. CHEK2 Deficiency Increase the Response to PD-1 Inhibitors by Affecting the Tumor Immune Microenvironment. Cancer Lett 2024, 588. [Google Scholar] [CrossRef]
- Vokes, N.I.; Cobo, A.G.; Fernandez-Chas, M.; Molkentine, D.; Treviño, S.; Druker, V.; Qian, Y.; Patel, S.; Schmidt, S.; Hong, L.; et al. ATM Mutations Associate with Distinct Co-Mutational Patterns and Therapeutic Vulnerabilities in NSCLC. Clin Cancer Res 2023, 29, 4958–4972. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, A.J.; Bandlamudi, C.; Lavery, J.A.; Montecalvo, J.; Namakydoust, A.; Rizvi, H.; Egger, J.; Concepcion, C.P.; Paul, S.; Arcila, M.E.; et al. The Genomic Landscape of SMARCA4 Alterations and Associations with Outcomes in Patients with Lung Cancer. Clin Cancer Res 2020, 26, 5701–5708. [Google Scholar] [CrossRef]
- Pham, T. V.; Goodman, A.M.; Sivakumar, S.; Frampton, G.; Kurzrock, R. Intra-Patient Stability of Tumor Mutational Burden from Tissue Biopsies at Different Time Points in Advanced Cancers. Genome Med 2021, 13. [Google Scholar] [CrossRef]
- Georgoulias, G.; Zaravinos, A. Genomic Landscape of the Immunogenicity Regulation in Skin Melanomas with Diverse Tumor Mutation Burden. Front Immunol 2022, 13. [Google Scholar] [CrossRef]
- Ravi, A.; Hellmann, M.D.; Arniella, M.B.; Holton, M.; Freeman, S.S.; Naranbhai, V.; Stewart, C.; Leshchiner, I.; Kim, J.; Akiyama, Y.; et al. Genomic and Transcriptomic Analysis of Checkpoint Blockade Response in Advanced Non-Small Cell Lung Cancer. Nat Genet 2023, 55, 807–819. [Google Scholar] [CrossRef]
- Kelly, A.D.; Murugesan, K.; Kuang, Z.; Montesion, M.; Ross, J.S.; Albacker, L.A.; Huang, R.S.P.; Lin, D.I.; Demirci, U.; Creeden, J. Pan-Cancer Landscape of CD274 (PD-L1) Rearrangements in 283,050 Patient Samples, Its Correlation with PD-L1 Protein Expression, and Immunotherapy Response. J Immunother Cancer 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Hong, J.Y.; Lee, J.; Kwon, G.Y.; Jeong, B.C.; Park, S.H. Genomic Sequencing for Bladder Urothelial Carcinoma and Its Clinical Implications for Immunotherapy. Cancer Res Treat 2022, 54, 894–906. [Google Scholar] [CrossRef]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov 2017, 7, 264–276. [Google Scholar] [CrossRef]
- Wu, D.; Liu, Y.; Li, X.; Liu, Y.; Yang, Q.; Liu, Y.; Wu, J.; Tian, C.; Zeng, Y.; Zhao, Z.; et al. Identification of Clonal Neoantigens Derived From Driver Mutations in an EGFR-Mutated Lung Cancer Patient Benefitting From Anti-PD-1. Front Immunol 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Montesion, M.; Murugesan, K.; Jin, D.X.; Sharaf, R.; Sanchez, N.; Guria, A.; Minker, M.; Li, G.; Fisher, V.; Sokol, E.S.; et al. Somatic HLA Class I Loss Is a Widespread Mechanism of Immune Evasion Which Refines the Use of Tumor Mutational Burden as a Biomarker of Checkpoint Inhibitor Response. Cancer Discov 2021, 11, 282–292. [Google Scholar] [CrossRef]
- Exposito, F.; Redrado, M.; Houry, M.; Hastings, K.; Molero-Abraham, M.; Lozano, T.; Solorzano, J.L.; Sanz-Ortega, J.; Adradas, V.; Amat, R.; et al. PTEN Loss Confers Resistance to Anti-PD-1 Therapy in Non-Small Cell Lung Cancer by Increasing Tumor Infiltration of Regulatory T Cells. Cancer Res 2023, 83, 2513–2526. [Google Scholar] [CrossRef]
- Tien, F.M.; Lu, H.H.; Lin, S.Y.; Tsai, H.C. Epigenetic Remodeling of the Immune Landscape in Cancer: Therapeutic Hurdles and Opportunities. J Biomed Sci 2023, 30. [Google Scholar] [CrossRef] [PubMed]
- Keshari, S.; Barrodia, P.; Singh, A.K. Epigenetic Perspective of Immunotherapy for Cancers. Cells 2023, 12. [Google Scholar] [CrossRef]
- Yang, J.; Xu, J.; Wang, W.; Zhang, B.; Yu, X.; Shi, S. Epigenetic Regulation in the Tumor Microenvironment: Molecular Mechanisms and Therapeutic Targets. Signal Transduct Target Ther 2023, 8. [Google Scholar] [CrossRef]
- Zhang, R.; Gan, W.; Zong, J.; Hou, Y.; Zhou, M.; Yan, Z.; Li, T.; Lv, S.; Zeng, Z.; Wang, W.; et al. Developing an M5C Regulator-Mediated RNA Methylation Modification Signature to Predict Prognosis and Immunotherapy Efficacy in Rectal Cancer. Front Immunol 2023, 14. [Google Scholar] [CrossRef]
- Peng, B.; Lin, Y.; Yi, G.; Lin, M.; Xiao, Y.; Qiu, Y.; Yao, W.; Zhou, X.; Liu, Z. Comprehensive Landscape of M6A Regulator-Related Gene Patterns and Tumor Microenvironment Infiltration Characterization in Gastric Cancer. Sci Rep 2024, 14. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Huang, W.; Li, Y.; Weng, H. Roles of METTL3 in Cancer: Mechanisms and Therapeutic Targeting. J Hematol Oncol 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Ning, J.; Hou, X.; Hao, J.; Zhang, W.; Shi, Y.; Huang, Y.; Ruan, X.; Zheng, X.; Gao, M. METTL3 Inhibition Induced by M2 Macrophage-Derived Extracellular Vesicles Drives Anti-PD-1 Therapy Resistance via M6A-CD70-Mediated Immune Suppression in Thyroid Cancer. Cell Death Differ 2023, 30, 2265–2279. [Google Scholar] [CrossRef]
- Lin, J.; Guo, D.; Liu, H.; Zhou, W.; Wang, C.; Müller, I.; Kossenkov, A. V.; Drapkin, R.; Bitler, B.G.; Helin, K.; et al. The SETDB1-TRIM28 Complex Suppresses Antitumor Immunity. Cancer Immunol Res 2021, 9, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Carpen, L.; Falvo, P.; Orecchioni, S.; Mitola, G.; Hillje, R.; Mazzara, S.; Mancuso, P.; Pileri, S.; Raveane, A.; Bertolini, F. A Single-Cell Transcriptomic Landscape of Innate and Adaptive Intratumoral Immunity in Triple Negative Breast Cancer during Chemo- and Immunotherapies. Cell Death Discov 2022, 8. [Google Scholar] [CrossRef] [PubMed]
- Amato, C.M.; Hintzsche, J.D.; Wells, K.; Applegate, A.; Gorden, N.T.; Vorwald, V.M.; Tobin, R.P.; Nassar, K.; Shellman, Y.G.; Kim, J.; et al. Pre-Treatment Mutational and Transcriptomic Landscape of Responding Metastatic Melanoma Patients to Anti-PD1 Immunotherapy. Cancers (Basel) 2020, 12, 1–15. [Google Scholar] [CrossRef]
- Lee, Y.M.; Hsu, C.L.; Chen, Y.H.; Ou, D.L.; Hsu, C.; Tan, C.T. Genomic and Transcriptomic Landscape of an Oral Squamous Cell Carcinoma Mouse Model for Immunotherapy. Cancer Immunol Res 2023, 11, 1553–1567. [Google Scholar] [CrossRef]
- Gaffney, S.G.; Perry, E.B.; Chen, P.M.; Greenstein, A.; Kaech, S.M.; Townsend, J.P. The Landscape of Novel and Complementary Targets for Immunotherapy: An Analysis of Gene Expression in the Tumor Microenvironment. Oncotarget 2019, 10, 4532–4545. [Google Scholar] [CrossRef]
- Ho, K.H.; Chang, C.J.; Huang, T.W.; Shih, C.M.; Liu, A.J.; Chen, P.H.; Cheng, K.T.; Chen, K.C. Gene Landscape and Correlation between B-Cell Infiltration and Programmed Death Ligand 1 Expression in Lung Adenocarcinoma Patients from The Cancer Genome Atlas Data Set. PLoS One 2018, 13. [Google Scholar] [CrossRef]
- Liu, M.; Dong, Q.; Chen, B.; Liu, K.; Zhao, Z.; Wang, Y.; Zhuang, S.; Han, H.; Shi, X.; Jin, Z.; et al. Synthetic Viability Induces Resistance to Immune Checkpoint Inhibitors in Cancer Cells. Br J Cancer 2023, 129, 1339–1349. [Google Scholar] [CrossRef]
- Zhang, M.; Huang, Y.; Pan, J.; Sang, C.; Lin, Y.; Dong, L.; Shen, X.; Wu, Y.; Song, G.; Ji, S.; et al. An Inflammatory Checkpoint Generated by IL1RN Splicing Offers Therapeutic Opportunity for KRAS-Mutant Intrahepatic Cholangiocarcinoma. Cancer Discov 2023, 13, 2248–2269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wan, X.; Lv, M.; Li, C.; Chu, Q.; Wang, G. TMEM92 Acts as an Immune-Resistance and Prognostic Marker in Pancreatic Cancer from the Perspective of Predictive, Preventive, and Personalized Medicine. EPMA J 2022, 13, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Tang, Y.; Hu, X.; Yin, X.; Chen, Y.; Chen, J.; Liu, H.; Liu, H.; Liang, J.; Zhang, X.; et al. Patients with ASPSCR1-TFE3 Fusion Achieve Better Response to ICI Based Combination Therapy among TFE3-Rearranged Renal Cell Carcinoma. Mol Cancer 2024, 23. [Google Scholar] [CrossRef]
- Zheng, H.; Liu, H.; Li, H.; Dou, W.; Wang, J.; Zhang, J.; Liu, T.; Wu, Y.; Liu, Y.; Wang, X. Characterization of Stem Cell Landscape and Identification of Stemness-Relevant Prognostic Gene Signature to Aid Immunotherapy in Colorectal Cancer. Stem Cell Res Ther 2022, 13. [Google Scholar] [CrossRef]
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat Rev Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Patnaik, A.; Kang, S.P.; Rasco, D.; Papadopoulos, K.P.; Elassaiss-Schaap, J.; Beeram, M.; Drengler, R.; Chen, C.; Smith, L.; Espino, G.; et al. Phase I Study of Pembrolizumab (MK-3475; Anti-PD-1 Monoclonal Antibody) in Patients with Advanced Solid Tumors. Clin Cancer Res 2015, 21, 4286–4293. [Google Scholar] [CrossRef] [PubMed]
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. CTLA-4-Mediated Inhibition in Regulation of T Cell Responses: Mechanisms and Manipulation in Tumor Immunotherapy. Annu Rev Immunol 2001, 19, 565–594. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N Engl J Med 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G. V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- J, L.; FS, H.; JD, W. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. New England Journal of Medicine 2015, 373, 1270–1271. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N Engl J Med 2018, 379, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulières, D.; Tahara, M.; de Castro, G.; Psyrri, A.; Basté, N.; Neupane, P.; Bratland, Å.; et al. Pembrolizumab Alone or with Chemotherapy versus Cetuximab with Chemotherapy for Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (KEYNOTE-048): A Randomised, Open-Label, Phase 3 Study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Blumenschein, G.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N Engl J Med 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Campos Bragagnoli, A.; et al. First-Line Nivolumab plus Chemotherapy versus Chemotherapy Alone for Advanced Gastric, Gastro-Oesophageal Junction, and Oesophageal Adenocarcinoma (CheckMate 649): A Randomised, Open-Label, Phase 3 Trial. Lancet 2021, 398, 27–40. [Google Scholar] [CrossRef]
- Plimack, E.R.; Powles, T.; Stus, V.; Gafanov, R.; Nosov, D.; Waddell, T.; Alekseev, B.; Pouliot, F.; Melichar, B.; Soulières, D.; et al. Pembrolizumab Plus Axitinib Versus Sunitinib as First-Line Treatment of Advanced Renal Cell Carcinoma: 43-Month Follow-up of the Phase 3 KEYNOTE-426 Study. Eur Urol 2023, 84, 449–454. [Google Scholar] [CrossRef]
- Brave, M.H.; Maguire, W.F.; Weinstock, C.; Zhang, H.; Gao, X.; Li, F.; Yu, J.; Fu, W.; Zhao, H.; Pierce, W.F.; et al. FDA Approval Summary: Enfortumab Vedotin plus Pembrolizumab for Locally Advanced or Metastatic Urothelial Carcinoma. Clin Cancer Res 2024, 30. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the Treatment of Non-Small-Cell Lung Cancer. N Engl J Med 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N Engl J Med 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N Engl J Med 2017, 377, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Sangro, B.; Chan, S.L.; Kelley, R.K.; Lau, G.; Kudo, M.; Sukeepaisarnjaroen, W.; Yarchoan, M.; De Toni, E.N.; Furuse, J.; Kang, Y.K.; et al. Four-Year Overall Survival Update from the Phase III HIMALAYA Study of Tremelimumab plus Durvalumab in Unresectable Hepatocellular Carcinoma. Ann Oncol 2024, 35, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; He, A.R.; Bouattour, M.; Okusaka, T.; Qin, S.; Chen, L.T.; Kitano, M.; Lee, C. kun; Kim, J.W.; Chen, M.H.; et al. Durvalumab or Placebo plus Gemcitabine and Cisplatin in Participants with Advanced Biliary Tract Cancer (TOPAZ-1): Updated Overall Survival from a Randomised Phase 3 Study. Lancet Gastroenterol Hepatol 2024, 9, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Park, S.H.; Caserta, C.; Valderrama, B.P.; Gurney, H.; Ullén, A.; Loriot, Y.; Sridhar, S.S.; Sternberg, C.N.; Bellmunt, J.; et al. Avelumab First-Line Maintenance for Advanced Urothelial Carcinoma: Results From the JAVELIN Bladder 100 Trial After ≥2 Years of Follow-Up. J Clin Oncol 2023, 41, 3486–3492. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Bell, R.B.; Bifulco, C.B.; Burtness, B.; Gillison, M.L.; Harrington, K.J.; Le, Q.T.; Lee, N.Y.; Leidner, R.; Lewis, R.L.; et al. The Society for Immunotherapy of Cancer Consensus Statement on Immunotherapy for the Treatment of Squamous Cell Carcinoma of the Head and Neck (HNSCC). J Immunother Cancer 2019, 7. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Kawazoe, A.; Bai, Y.; Xu, J.; Lonardi, S.; Metges, J.P.; Yanez, P.; Wyrwicz, L.S.; Shen, L.; Ostapenko, Y.; et al. Pembrolizumab plus Trastuzumab and Chemotherapy for HER2-Positive Gastric or Gastro-Oesophageal Junction Adenocarcinoma: Interim Analyses from the Phase 3 KEYNOTE-811 Randomised Placebo-Controlled Trial. Lancet 2023, 402, 2197–2208. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Powles, T.; Burotto, M.; Escudier, B.; Bourlon, M.T.; Zurawski, B.; Oyervides Juárez, V.M.; Hsieh, J.J.; Basso, U.; Shah, A.Y.; et al. Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med 2021, 384, 829–841. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Tomczak, P.; Park, S.H.; Venugopal, B.; Ferguson, T.; Chang, Y.-H.; Hajek, J.; Symeonides, S.N.; Lee, J.L.; Sarwar, N.; et al. Adjuvant Pembrolizumab after Nephrectomy in Renal-Cell Carcinoma. N Engl J Med 2021, 385, 683–694. [Google Scholar] [CrossRef]
- van der Heijden, M.S.; Sonpavde, G.; Powles, T.; Necchi, A.; Burotto, M.; Schenker, M.; Sade, J.P.; Bamias, A.; Beuzeboc, P.; Bedke, J.; et al. Nivolumab plus Gemcitabine-Cisplatin in Advanced Urothelial Carcinoma. N Engl J Med 2023, 389, 1778–1789. [Google Scholar] [CrossRef] [PubMed]
- Makker, V.; Colombo, N.; Casado Herráez, A.; Santin, A.D.; Colomba, E.; Miller, D.S.; Fujiwara, K.; Pignata, S.; Baron-Hay, S.; Ray-Coquard, I.; et al. Lenvatinib plus Pembrolizumab for Advanced Endometrial Cancer. N Engl J Med 2022, 386, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Cescon, D.W.; Schmid, P.; Rugo, H.S.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Perez-Garcia, J.; Iwata, H.; et al. Health-Related Quality of Life with Pembrolizumab plus Chemotherapy vs Placebo plus Chemotherapy for Advanced Triple-Negative Breast Cancer: KEYNOTE-355. J Natl Cancer Inst 2024, 116, 717–727. [Google Scholar] [CrossRef] [PubMed]
- PD-1/PD-L1 Pathway: Current Researches in Cancer - PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/32266087/ (accessed on 19 November 2024).
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Weiss, S.A.; Sznol, M. Resistance Mechanisms to Checkpoint Inhibitors. Curr Opin Immunol 2021, 69, 47–55. [Google Scholar] [CrossRef]
Figure 1.
Immunological, genomic, epigenetic and transcriptomic landscape in cancer patients receiving anti-PD-1/PD-L1 therapy.
Figure 1.
Immunological, genomic, epigenetic and transcriptomic landscape in cancer patients receiving anti-PD-1/PD-L1 therapy.
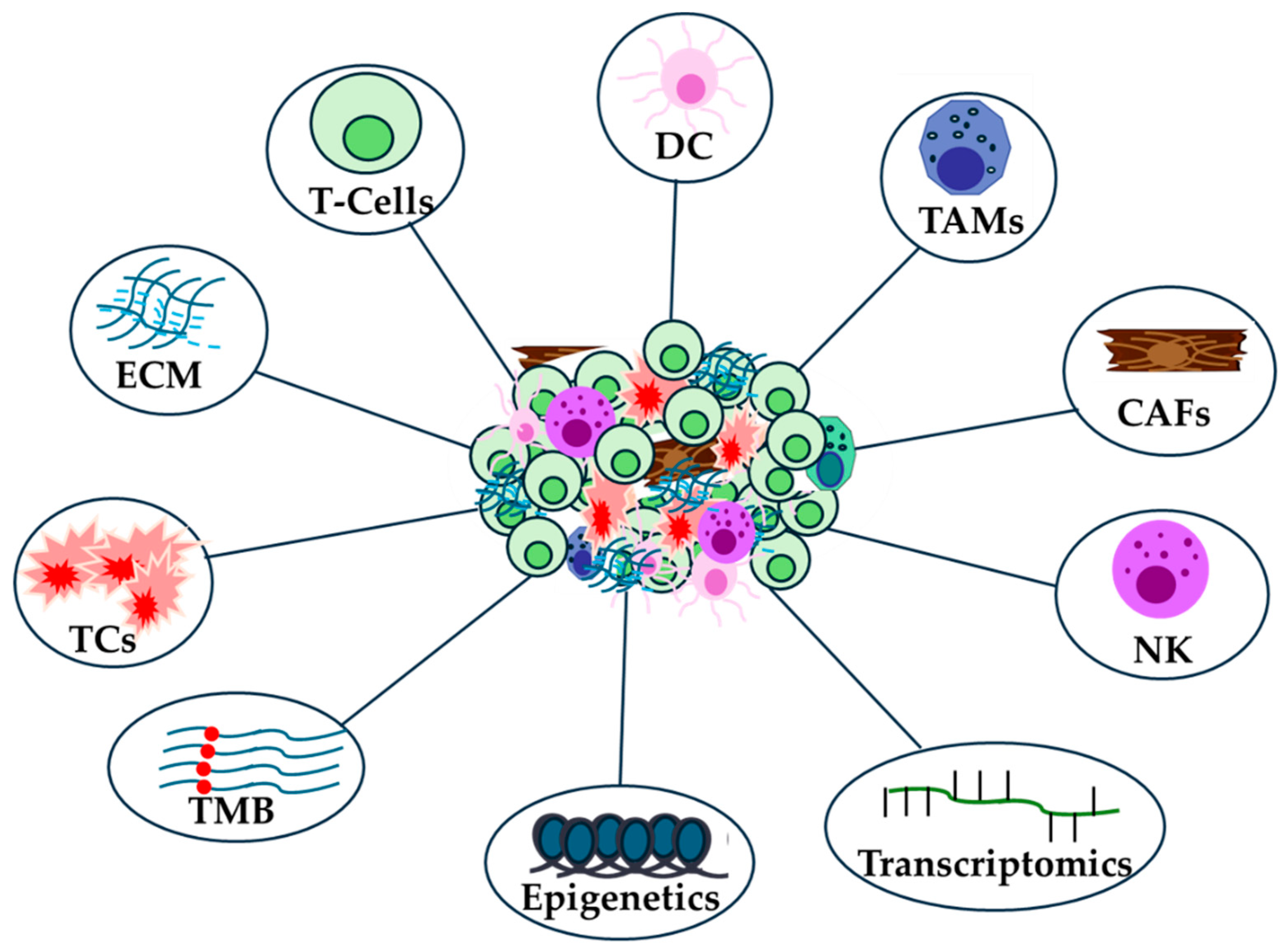

Table 1.
Anti PD-1/PD-L1 inhibitors.
| Clinical Trial (Phase III) |
Drug Name (PD-1/PD-L1) | Cancer Type |
Primary End Points | Response Rate (%) | Sample Size | FDA Approval (Yes/No) | Ref |
|---|---|---|---|---|---|---|---|
| CheckMate-067 | Nivolumab + Ipilimumab Vs Nivolumab Vs Ipilimumab | Melanoma | PFS, OS | 58 | 945 | Yes | [130] |
| Keynote-189 | Pembrolizumab + Chemotherapy Vs Chemotherapy + Placebo | NSCLC (Non-squamous) |
OS, PFS | 48.3 | 616 | Yes | [132] |
| Keynote-048 | Pembrolizumab + Chemotherapy Vs Pembrolizumab Vs Chemotherapy + Cetuximab | Head and Neck Squamous Cell Carcinoma (HNSCC) | PFS, OS | 16.9 | 882 | Yes | [133] |
| CheckMate-141 | Nivolumab Vs Chemotherapy | HNSCC | OS | 13.3 | 361 | Yes | [134] |
| Keynote-177 | Pembrolizumab Vs Chemotherapy + Bevacizumab or cetuximab | Colorectal Cancer (MSI-H/dMMR) | PFS, OS | 43.8 | 307 | Yes | [135] |
| CheckMate-649 | Nivolumab + Chemotherapy Vs Chemotherapy | Gastric/ Esophageal Cancer |
OS, PFS | 47.4 | 1581 | Yes | [136] |
| CheckMate-214 | Nivolumab + Ipilimumab Vs Sunitinib | Renal Cell Carcinoma | OS, PFS, ORR | 42 | 1096 | Yes | [137] |
| EV-302 | Pembrolizumab + Enfortumab vedotin Vs Chemotherapy | Urothelial Cancer 1st Line | PFS, OS | 44 | 608 | Yes | [138] |
Table 2.
Anti PD-L1 inhibitors.
| Clinical Trial (Phase III) | Drug Name (PD-1/PD-L1) | Cancer Type |
Primary End Points | Response Rate (%) | Sample Size | FDA Approval (Yes/No) | Ref |
|---|---|---|---|---|---|---|---|
| IMpower150 | Atezolizumab + Bevacizumab + Chemotherapy Vs Bevacizumab + Chemotherapy | NSCLC 1st Line | PFS, OS | 63 | 1202 | Yes | [140] |
| PACIFIC | Durvalumab Vs placebo after concurrent Chemoradiotherapy | NSCLC (Stage III) |
OS, PFS | 28.4 | 713 | Yes | [141] |
| HIMALAYA | Durvalumab + Tremelimumab (STRIDE) Vs Durvalumab Vs Sorafenib | Hepatocellular Carcinoma (HCC) | OS for STRIDE Vs Sorafenib | 20.1 | 1171 | Yes | [142] |
| TOPAZ-1 | Durvalumab + Chemotherapy Vs Chemotherapy + placebo | Biliary Tract Cancer | OS | 26.7 | 685 | Yes | [143] |
| JAVELIN 100 | Avelumab Vs Best Supportive Care as maintenance Therapy after 4-6 cycles Chemotherapy | Urothelial Cancer | OS | 53.2 | 700 | Yes | [144] |
| IMbrave050 | Atezolizumab + Bevacizumab Vs Active Surveillance | Hepatocellular Carcinoma (HCC) | 33 | 662 | Yes | [145] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



