
Submitted:
15 January 2023
Posted:
18 January 2023
You are already at the latest version
Abstract
Immune checkpoint inhibitors (ICI) are currently use in a wide range of tumors, but only 20-40% of patients achieve clinical benefit. Aim of our study was to find predictive biomarkers of ICI treatment. We analyzed by immunohistochemistry various cell subsets, including CD3+ cells, CD8+ cells, CD68+ cells, CD20+ cells, FoxP3+ cells, and molecules as LAG-3, IDO1, TGfβ. Comprehensive genomic profiles were analyzed. Correlation of various biomarkers with efficacy of ICI treatment in patients with advanced solid tumors was evaluated. We evaluated 56 patients treated with ICI monotherapy. Longer median progression-free survival (PFS) was found in tumors negative for nuclear FoxP3 (P = 0.002, HR 0.14) and in TMB-high tumors (P = 0.024, HR 0.38). Longer overall survival (OS) was found in patient with intraepithelial CD8 negativity (P = 0.045, HR 0.47). In malignant melanoma CD68 negativity, FoxP3 negativity and PDL TPS ≥ 1 was associated with longer PFS. In NSCLC FoxP3 was associated with longer PFS and OS. We found that absence of expression of several biomarkers such as CD68 and FoxP3 is associated with better survival. TMB-high and PD-L1 expression not universally but in certain disease could predict response.
Keywords:
Immune checkpoint inhibitors
; Anti-tumor immunity
; Predictive biomarker
; Malignant melanoma
; NSCLC
1. Introduction
The immune checkpoint inhibitors proved their efficacy in various tumor types, for example, in malignant melanoma, lung, kidney, bladder, breast, head and neck, esophageal, stomach cancer, MSI-high tumors etc. The mechanism of action is to stimulate the patient's immune system and produce immune-based cytotoxicity against the tumor. Although effective in a broad spectrum of tumors, only 20-40% of patients will achieve clinical benefit. Therefore, it is crucial to seek for potential biomarkers that could help to predict the response to immunotherapy treatment. The aim of our project is to describe tumor tissue, its microenvironment and immunoprofile and to correlate it with the response to treatment.
Only a few predictive biomarkers are currently in use in clinical practice. Programmed cell death ligand 1 (PD-L1) expression predicts response to immune checkpoint inhibitors (ICIs) in lung [1], gastric and esophageal [2,3], and breast cancer [4,5]. In other tumor types like malignant melanoma or renal cell carcinoma, ICIs work regardless of PD-L1 expression. Moreover, the use of PD-L1 expression as a universal predictor of response to immunotherapy treatment is burdened by inconsistencies in the use of particular immunohistochemical assays, discriminant values and interobserver concordance. A very strong predictive biomarker for ICI treatment efficacy is microsatellite instability status (MSI). Tumors with high microsatellite instability or deficiency in mismatch repair proteins (MMRd) are significantly sensitive to immunotherapy [6]. Defects resulting in microsatellite instability can potentially produce neoantigens that can be recognized as non-self by the immune system, especially T lymphocytes, and stimulate an immune response against the tumor. Indication of ICIs in MSI high tumors, regardless of the tumor histology, underlines its efficacy as a tumor-agnostic therapy. In certain tumor types like mismatch repair-deficient locally advanced rectal cancer, it could revolutionize treatment algorithms and could potentially lead to the omission of surgery [7]. Another promising predictive factor across multiple tumor types could be tumor mutational burden (TMB). TMB is formed by the increase of non-synonymous single nucleotide variants that lead to enhanced neoantigens formation. Tumor types typically sensitive to ICIs, like lung cancer, bladder cancer, and malignant melanoma, are cancers harboring higher mutational burden. Overall higher TMB could lead to higher ICIs efficacy but setting the optimal cut-off (by the number of non-synonymous mutations per megabase) is very problematic. Different studies used specific panels to estimate TMB and direct comparison of the results is difficult. Optimal TMB cut-off value to discriminate responders may vary significantly between cancer types, and it seems it is not a single independent factor contributing to ICIs sensitivity [8,9].
The tumor microenvironment plays a crucial role in transforming the effect of immunotherapy into tumor regression. Tumor-infiltrating CD8 positive T cells predict ICIs efficacy in many cancer types [10,11]. However, in certain patients, the CD8 T cells positivity does not clearly predict the response to treatment. This may be explained by different subtypes of CD8 T cells - central, effector, stem-like, and tissue resident memory cells [12]. Furthermore, not only functional status (dysfunction, exhaustion) but differences in spatial distribution are contributing to T cell antitumor effector function [13]. CD68 is highly expressed in tissue associated macrophages (TAM), promotes phagocytosis, and mediates the recruitment and activation of macrophages. High expression levels of CD68 in tumors correlated with an adverse prognosis in various tumor types [14]. CD68 expression is associated with ICI resistance, but the results in different tumor types are conflicting [15,16]. Another immune cell population potentially contributing to ICI therapy failure are regulatory lymphocytes (Tregs). Tregs are functioning as central mediators of immune function [17]. Expression of the transcription factor Forkhead box protein P3 (FoxP3) that is highly expressed in Tregs may have utility as a predictor of response to ICIs. B cells express many pro- and anti-inflammatory factors and are essential for tumor inflammation. Studies suggest that B cell infiltration may represent a predictor for response to ICI but can also contribute to immune-related adverse events [18,19]. Expression of other immune inhibitory markers like Lymphocyte-activation gene 3 (LAG-3) [20], T-cell immunoglobulin mucin-3 (TIM-3) [21], and indoleamine 2, 3-dioxygenase 1 (IDO1) [22] has been associated with resistance to ICI therapy. Anti-LAG-3 drug relatlimab in combination with nivolumab already proved its efficacy in patients with malignant melanoma in phase 2-3 trial and is more effective than nivolumab monotherapy [23]. IDO1 inhibitor epacadostat showed promising results in early clinical trials but failed to prove efficacy in a phase 3 trial in advanced melanoma (ECHO-301/Keynote-252 study) [24]. Another signaling contributing to ICI resistance can be driven by TGFβ. Co-administration of TGFβ-blocking and anti-PD-L1 antibodies reduced TGFβ signaling in stromal cells, facilitated T cell penetration into the center of the tumor, and activated anti-tumor immunity and tumor regression. This suggests that TGFβ participates in the tumor microenvironment and restricts T cell infiltration [25].
The aim of our prospective study was to describe different populations of immune cells, including CD3+ cells, CD8+ cells, CD68+ cells, CD20+ cells, FoxP3+ cells, expression of molecules such as LAG-3, IDO1, TGfβ, and analyze comprehensive genomic profiles, and evaluate their contribution to the efficacy of ICIs treatment in patients with advanced solid tumors.
2. Materials and Methods
We prospectively enrolled patients with advanced or metastatic disease treated with anti PD-1 immune checkpoint inhibitor nivolumab from 2017 to 2021 at Masaryk Memorial Cancer Institute in Brno, Czech Republic. Inclusion criteria were: advanced/metastatic disease, measurable disease by RECIST criteria, planned treatment with anti-PD-1 checkpoint inhibitor monotherapy, expected survival of more than 3 months, ability to understand and the willingness to sign a written informed consent document. Informed consent was obtained from each participating subject. The study was approved by the Institutional Ethic Committee of Masaryk Memorial Cancer Institute, reference number 2017/1890/MOU, June 27, 2017.
Immunohistochemistry (IHC) staining was performed using formalin-fixed and paraffin-embedded tissue specimens. The mismatch repair (MMR) protein status was determined by using monoclonal antibodies provided by DAKO: MLH-1 (clone ES05), MSH-2 (clone FE11), MSH-6 (clone EP49) and PMS-2 (clone EP51). Null expression in at least one of the MMR proteins with positive control in non-tumor cells was considered a deficiency. For staining for CD3, we used antibody clone SP7 by DCS - Innovative Diagnostik-Systeme (Hamburg, Germany), for CD8 clone SP16 by Thermo Scientific (Fremont, CA, USA). We evaluated stromal and intraepithelial infiltration separately. For stromal characteristics, we assessed the percentage of tumor stroma formed by lymphocytes (the sample was considered positive if at least 10% of cells demonstrated positive staining by immunohistochemistry for CD3 or CD8). For intraepithelial evaluation, we estimated the average number of lymphocytes in direct contact with tumor cells per area of one high magnification, counted manually (the sample was considered positive if at least 10 cells were positive for CD3 or CD8 staining). For CD20 staining, we used antibody clone L26 by DAKO (Stockholm, Sweden), membrane positivity was evaluated and scored on a qualitative scale from 0 to 3 (score 0 and 1 was considered negative, score 2 a 3 as positive). For CD68 staining clone KP1 by DAKO was used, plasmatic positivity was scored from 0 to 3. For PD-L1 staining, we used antibody clone 22C3 by DACO and scores using CPS and TPS used in clinical practice. For PD1 staining clone NAT105 by Abcam (Cambridge, UK) was used, membrane positivity was scored from 0 to 3. LAG-3 staining was performed by antibody LAG-3 clone BLR028F by Novus Biologicals (Littleton, CO. USA), nuclear positivity was scored from 0 to 3. For IDO1 staining, we used antibody clone 1 A3 by LifeSpan BioSciences (Seattle, WA, USA), plasmatic positivity was scored from 0 to 3. For TGFβ, we used antibody clone EPR21143 by Abcam. For TGFβ, we evaluated immune cells and tumor cells separately and scored on a semiqualitative scale from 0 to 3. For FoxP3 antibody clone SP97 (by Abcam) was used, nuclear positivity was evaluated and estimated the number of lymphocytes with nuclear positivity per area of one high power field (objective 40x, ocular 10x/22 mm) of view and scored as positive if more than 10 lymphocytes were stained positive. The immunohistochemistry assessment was performed by pathologist highly experienced in the evaluation of tissue analysis and blinded to the patient's characteristics or treatment outcomes.
Comprehensive genomic profiling was performed on the patient with available tissue of sufficient quality. Tumor tissue dissected from FFPE blocks was used for mutational analysis. DNA was isolated using the QIAamp DNA FFPE Tissue Kit (Qiagen, Manchester, UK). The sequencing library was prepared using the TruSight Oncology 500 DNA kit according to the recommended protocol. Sequencing was performed on an Illumina NextSeq500 instrument using pair-end sequencing (2x101 bp). Mutations, MSI status and TMB were determined using the tumor-only workflow of TSO500 (Illumina Inc., San Diego, CA, USA). For the TMB status cut-off 10 muts/Mb was used
Factors evaluated in association with progression-free survival (PFS) and overall survival (OS) included age at the onset of checkpoint inhibitors treatment, type of cancer, line of therapy, baseline laboratory values – white blood cell count, absolute neutrophil count, absolute monocyte count, absolute lymphocyte count, neutrophil to lymphocyte ratio (NLR), C-reactive protein (CRP) level, lactate dehydrogenase (LDH) level, drug use (metformin, proton pump inhibitors), treatment toxicity, immunohistochemistry expression of different markers – MMR status, CD3 stromal expression, CD3 intraepithelial expression, CD8 stromal expression, CD8 intraepithelial expression, CD20, CD68, PDL-1, PD1, LAG3, IDO1, TGFβ in immune cell expression, TGFβ in tumor cells, and FoxP3 nuclear expression, TMB. Response to therapy was evaluated by using RECIST criteria version 1.1. PFS was defined as the time from the beginning of checkpoint inhibitors therapy to the first documented objective disease progression or death. OS was defined as the time from the beginning of checkpoint inhibitors therapy to death due to any cause. Clinical benefit from treatment was defined as achieving complete or partial response or at least six months of stable disease. Frequency analysis and summary statistics were used to characterize the sample data set. Survival curves were estimated using the Kaplan-Meier method. A log-rank test was used to test the difference between survival curves (PFS or OS) for different factors. All point estimates include 95% confidence intervals (CIs). Fisher's exact or Chi-squared tests were used to establish the significance of the association between categorical variables. The Cox proportional hazard model was used to calculate hazard ratios (HR). All statistical analyses were performed employing MedCalc version 20.1.4 and a significance level of 0.05.
3. Results
3.1. Patient Characteristics
68 patients were enrolled in our study, and 56 were evaluable for our analysis. Patients' characteristic is summarized in Table 1. The median age was 67 years, 14 patients were female (25%). Histological types of tumors were: 31 patients with malignant melanoma, 15 patients with non-small lung cancer (NSCLC), 7 patients with renal carcinoma, 1 patient with colorectal cancer, 1 patient with bladder carcinoma, 1 patient with yolk sack tumor. Most of the patients started treatment with ICI as a first-line of treatment (36 patients, 64.3%), as a second-line 14 patients (25%), and third or later line 6 patients (10.7%).
Median follow-up was 36.2 months. In 45 patients (80.4%), disease progression was identified, and 22 patients (39.3%) were still alive. Median progression-free survival (PFS) in the whole group was 8.0 months (95% CI 5.1 – 15.1). There was no statistically significant difference in PFS according to tumor type. Median PFS for malignant melanoma was 11.0 months (95% CI 4.7 – 24.5), lung carcinoma 10.4 months (95% CI 2.3 – 19.2), renal cell carcinoma 8.4 months (95% CI 0.5 – 21.3), and for a group of other tumor types 6.0 months (95% CI 0.9 – 7.6). Median overall survival (OS) was 24.8 months (95% CI 15.3 – 37.3). We found a statistically significant difference in OS according to tumor type, The median OS was 27.4 months (95% CI 18.0 – 31.1) for malignant melanoma, 15.3 months (95% CI 7.3 – 23.2) for lung carcinoma, 37.3 months (95% CI 2.5 – NA) for renal cell carcinoma, and 9.3 months (95% CI 1.4 – 10.3) for a group of other types, p = 0.001. There was no survival difference in PFS or OS according to the treatment line. The best response to treatment was complete response in 10 patients (17.9%), partial response in 17 (30.4%), stable disease in 3 patients (5.4%) and disease progression in 24 (42.9%), in 2 patients treatment effect was not evaluable due to treatment toxicity. 27 patients achieved clinical benefit from treatment (48.2%).
Comprehensive genomic profiling was performed on 37 patients. High TMB was found in 21 patients (56.8%), median muts/Mb was 14.9. Two patients had tumors with microsatellite instability. Pathogenic NRAS variant was found in 12 patients (32.4%), BRAF in 14 (37.8%, in malignant melanoma patients only), TP53 in 10 (27.0%), KRAS in 3 (8%), ARID1A in 3 (8%), other pathogenic variants were less common.
3.3. Correlation with Clinical Parameters
We found no correlation between clinical parameters (age, sex, type of tumor, metastatic sites, line of treatment, using metformin, proton pump inhibitors) and IHC expression of different markers. Also, we found no correlation between baseline laboratory parameters (LDH, CRP, levels of leukocytes, neutrophils, monocytes, and lymphocytes) and IHC expression. IHC expressions of different markers were not associated with the type of tumor except for the expression of TGFβ IC, which was predominantly negative in lung carcinoma (p = 0.048).
3.4. Correlation with Survival Parameters
We found no correlation between clinical parameters (age, sex, metastatic sites, line of treatment, patients using metformin, proton pump inhibitors) and survival parameters. We found no correlation between baseline laboratory parameters (LDH, CRP, level of leukocytes, neutrophils, monocytes, and lymphocytes) and survival parameters. Clinical benefit was significantly associated with immune related adverse events (p < 0.001).
For PFS, we found that patients with FoxP3 nuclear negativity achieved longer PFS (14.8 months vs 3.9 months, p = 0.002, HR 0.14, 95% CI 0.04 – 0.48)(Figure 2A). High TMB was associated with longer PFS (15.1 vs 4.4 months, p = 0.024, HR 0.38; 95% CI 0.17 – 0.88)(Figure 2B). Also, TMB above the median (15 and more muts/Mb) was associated with longer PFS (15.1 vs. 6.1 months, p = 0.016, HR 0.38; 95% CI 0.17 – 0.83). There is a trend toward longer PFS in tumors with CD68 negativity (p = 0.059) and PD-L1 TPS 1 and more (p = 0.09). No other factors were associated with PFS.
Clinical benefit was associated with FoxP3 negativity (p = 0.017), with a favorable trend for patients with TPS 1 and more positivity (p = 0.080).
For OS, we found that patient CD8 IEL negativity had longer survival (37.3 vs 19.8 months, p = 0.045, HR 0.47, 95% CI 0.22 – 0.98). We found a trend toward longer OS in patients with LAG-3 negative tumors (p = 0.101), FOXP3 nuclear negativity (p = 0.118), NRAS mutated tumors (p = 0.086), but the results were not statistically significant. Results for PFS and OS are summarized in Table 3.
We analyzed different parameters according to tumor type separately for malignant melanoma and NSCLC (other tumor types had a low number of patients for valid statistical analysis). In patients with malignant melanoma, we found that longer PFS was achieved in patients with FoxP3 nuclear negativity (17.0 vs 3.9 months, p = 0.047, HR 0.19, 95% CI 0.04 – 0.97)(Figure 3A), CD68 negativity (15.1 vs 3.0 months, p = 0.033, HR 0.16, 95% CI 0.03 – 0.87), PD-L1 TPS 1 and more (NR vs 4.8 months, p = 0.006, HR 0.26, 95% CI 0.10 – 0.67), TMB above the median (24.5 vs 4.7 months, p = 0.029, HR 0.32, 95% CI 0.12 – 0.89), a trend toward longer PFS was found in patients with high TMB (P = 0.055) and PD-L1 CPS 1 and more (p = 0.057). No factors were associated with longer OS in malignant melanoma, the trend for longer OS was found in tumors with PD-L1 TPS 1 and more (p = 0.056). Results for malignant melanoma are summarized in Table 3.
In patients with lung carcinoma, longer PFS was found in tumors with FoxP3 nuclear negativity (14.8 vs 1.8 months, p = 0.005, HR 0.05, 95% CI 0.01 – 0.41)(Figure 3B). A trend to longer PFS was found in tumors with CD8 IEL negativity (p = 0.076) and CD68 negativity (p = 0.091). Better OS was associated with FoxP3 nuclear negativity (21.9 vs 7.3 months, p = 0.035, HR 0.16, 95% CI 0.02 – 0.88), and a trend toward longer OS was found in tumors with TGFβ IC negativity (p = 0.066) and high TMB (p = 0.063). Results for NSCLC are summarized in Table 4.
We examined the co-expression of FoxP3 and CD68. There was no correlation between FoxP3 and CD68 expression. When we analyzed patients who were FoxP3 and/or CD68 positive (13 patients) with patients negative for both markers (40 patients), we found significantly prolonged PFS in negative patients for the whole group (median PFS 14.8 vs 3.9 months, p < 0.001, HR 0.21, 95% CI 0.09 – 0.52), and also for patients with malignant melanoma (median PFS 17.0 vs 3.9 months, p < 0.008, HR 0.11, 95% CI 0.02 – 0.56) and NSCLC (median PFS 18.6 vs 2.3 months, p < 0.001, HR 0.04, 95% CI 0.01 – 0.24) separately. No association was found with OS. Combined FoxP3 and CD68 negativity were also significantly associated with treatment benefit (p = 0.021).
4. Discussion
Our study we found different factors associated with response to ICI therapy in advanced solid tumors. The immune system is a network of different effectors, mediators and regulatory cells, in combination with the secretion and degradation of various signaling molecules with stimulating or inhibiting effects. This is creating a complex system forming a tumor microenvironment. By using ICIs therapy, we tilt this balance in the direction of triggering a cascade of events, the result of which is cytotoxic cellular activity and tumor regression. Identifying predictive biomarkers suitable for clinical practice is essential to stratify patients with the greatest benefit from ICIs and to prevent the application of potentially toxic therapy to patients who will benefit minimally from the treatment. This issue is too complex to assume that we will have one or a few markers suitable for all types of immunotherapies across the broad spectrum of different tumor types.
We found that patients with FoxP3 negativity achieved longer PFS when treated with ICIs. The predictive value was confirmed for the whole set and also separately for malignant melanoma and NSCLC, respectively. FoxP3 is a transcription factor that is expressed in Treg cells, a subset of CD4+ T-cells. In contrast to other CD4+ T-cells that enhance local immune function, Tregs maintain immune homeostasis and self-tolerance by suppressing immune activity by regulating other immune cell subsets. They contribute to tumor development and progression by suppressing T effector cell functions [26]. The prognostic negative value of FoxP3 expression was confirmed in several metanalyses, including patients with malignant melanoma and NSCLC [27,28]. In our study, Treg absence was associated with a better response to ICIs used in standard clinical practice. Study with ICI treatment in melanoma and other tumors suggest that ICI increase tumor infiltration of CD4+ and CD8+ cells but without significantly changing or depleting FoxP3+ cells [29]. High infiltration of Tregs was found in patients treated with ICI who achieved disease hyperprogression [30]. Inhibition of FoxP3 augmented antitumor immunity and provides a therapeutic benefit in cancer models [31]. In our study, we found that patients with -negative FoxP3 expression achieved longer PFS and clinical benefit but no OS, suggesting that FoxP3 is a predictive factor for treatment with ICIs. This finding should support the therapeutic approach where targeting the Treg cells population in combination with ICIs could be a potentially effective treatment for an ICIs resistant tumor.
CD68 is overexpressed in tumor associated macrophages. Macrophages' roles in cancer are complex. Activated macrophages are often classified as pro-inflammatory M1 macrophages or anti-inflammatory M2 macrophages. M2-like macrophages support angiogenesis and express immunosuppressive molecules such as IL10, PD-L1, and TGFβ, favoring tumor growth and contributing to tumor aggressiveness [32]. TAMs seem to promote tumor progression in cutaneous melanoma. In particular, CD68+ TAMs and their abundance in the tumor were associated with poor prognostic factors, CD68 expression is associated with worse clinical outcome in patients with various cancers and ICI therapy [14,15,33]. We identified this marker as a negative predictor of the effectiveness of ICI therapy, especially in malignant melanoma, and confirmed previous findings. Combining two independent biomarkers, FoxP3 (Tregs) and CD68 (TAMs), we can select a specific group of patients who benefited from ICIs treatment most and confirm its predictive value for treatment benefit and survival.
We confirmed the predictive value of other known factors. In our study, TMB high (cut/off 10 or more muts/Mb) predicted longer PFS to ICI therapy, but we are aware that establishing a proper threshold for TMB is challenging. TMB high tumors are more sensitive to ICI treatment by producing neoantigens which could attract the immune system. The results of studies and analyses evaluating the effect of TMB on the efficacy of ICI therapy have yielded conflicting results. Although FDA had approved pembrolizumab for the treatment of adult and pediatric patients with unresectable or metastatic solid tumors that are TMB high, it sparked heated debate in the oncology community if it is not too broad characteristic. It is clear that not all patients with TMB 10 or more mutations per megabase profit from ICIs [9,35]. TMB itself results from a broad spectrum of cellular processes that lead to hypermutated status, and it is important to consider the cause of the high TMB and not only the absolute threshold. For example, in colon cancer, patients with high TMB benefit from ICIs treatment only when tumors also harbor microsatellite instability or mutation of polymerase epsilon (POLE) [34]. Tumor type specific approach with different cut-offs could lead to better efficacy prediction [35]. Microsatellite instability is a more robust predictor of response to ICIs, however, even in this group, there are patients who are primarily resistant to therapy [36]. Treatment strategies to overcome this resistance may include the use of dual ICI blockade, the use of ICI therapy earlier in the course of disease treatment or the influence of the tumor microenvironment in favor of the immune response.
A similar situation exists in the area of PD-L1 positivity. PD-L1 positivity was associated with better survival in patients with lung cancer [1], gastric and esophageal cancer [2,3], and breast cancer [4,5]. PD-L1 predictive efficacy depends on the used assay, different thresholds and tumor type specificity ,making it not a universal predictor. In a study with malignant melanoma, PD-L1 tumor cell positivity was predictive of response to ICIs [37]. In the pivotal phase 3 trial in metastatic melanoma, PD-L1 positivity on tumor cells was associated with better treatment response, although responses were also seen in PD-L1 negative tumors [38]. We confirmed these results and found that patients in our study with PD-L1 TPS ≥ 1 had a greater therapeutic benefit, outcome mostly influenced by the malignant melanoma group.
We couldn't confirm the positive role of CD8+ cells in cancer immunotherapy. Patients with CD8 stromal positivity achieved longer PFS, but the results were not statistically significant. We found that not only the presence of CD8+ cells itself but also their spatial differences could play a role in the tumor microenvironment [10,13]. In our study, we did not distinguish between different subtypes of infiltrating CD8+ T cells. Contrary to stromal CD8+ cells, intraepithelial positivity of CD8 was associated with a worse prognosis. Taking into account the different functional states of CD8+ cells, intraepithelial cells could present a population of dysfunctional or exhausted cells unable to trigger anti-tumor cell toxicity.
We are aware of the certain limitation of our study. Although our study was prospective and patients were uniformly treated with ICI monotherapy, it was limited to a relatively low number of enrolled patients. Our results were mainly driven by two diagnoses – malignant melanoma and NSCLC and thus limited our predictive results to only these types of cancer. Patients, especially in the non-melanoma group, were pretreated, which may have influenced the identification and validation of different predictive biomarkers taking into account the dynamics of changes in cancer immunity interpersonally and intrapersonally. A limited number of markers could limit or simplify the view into a complex anti-tumor microenvironment.
5. Conclusions
In summary, we evaluated the correlation between the expression of various clinical and immune-related factors in advanced solid tumors treated with ICI monotherapy. We found that the absence of expression of several biomarkers in the tumor microenvironment, such as CD68 and FoxP3, or its combination, is associated with better survival and could predict response to ICI treatment. We confirmed that other biomarkers, such as high TMB and PD-L1 expression, although not universally but in certain diseases, could predict response.
Author Contributions
Conceptualization, P.G., S.B. and R.V.; Data curation, P.G., S.B., P.F., P.M., D.N., I.K.; Formal analysis, P.G. and I.S.; Funding acquisition, I.K. and R.V.; Investigation, P.G., S.B., P.F., P.M., D.N., I.K.; Methodology, P.G., S.B., P.F. and I.S.; Project administration, P.G. and S.B.; Supervision, I.K. and R.V.; Writing—original draft, P.G., S.B. and P.F.; Writing—review and editing, all authors. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Ministry of Health of the Czech Republic, grant nr. NV18-03-00339. This research was supported by the project National Institute for Cancer Research (Programme EXCELES, ID Project No. LX22NPO5102) - Funded by the European Union - Next Generation EU, and by MH CZ - DRO (MMCI, 00209805).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board and Ethics Committee of Masaryk Memorial Cancer Institute, reference number 2017/1890/MOU; 27 June 2017.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study. Written informed consent has been obtained from the patient(s) to publish this paper.
Data Availability Statement
Data will be made available from the corresponding author on reasonable request.
Conflicts of Interest
P.G. received honoraria from Roche and Servier unrelated to this project. The other authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Five-Year Outcomes With Pembrolizumab Versus Chemotherapy for Metastatic Non–Small-Cell Lung Cancer With PD-L1 Tumor Proportion Score ≥ 50%. J. Clin. Oncol. 2021, 39, 2339–2349. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Campos Bragagnoli, A.; et al. First-Line Nivolumab plus Chemotherapy versus Chemotherapy Alone for Advanced Gastric, Gastro-Oesophageal Junction, and Oesophageal Adenocarcinoma (CheckMate 649): A Randomised, Open-Label, Phase 3 Trial. The Lancet 2021, 398, 27–40. [Google Scholar] [CrossRef]
- Doki, Y.; Ajani, J.A.; Kato, K.; Xu, J.; Wyrwicz, L.; Motoyama, S.; Ogata, T.; Kawakami, H.; Hsu, C.-H.; Adenis, A.; et al. Nivolumab Combination Therapy in Advanced Esophageal Squamous-Cell Carcinoma. N. Engl. J. Med. 2022, 386, 449–462. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Cortes, J.; Rugo, H.S.; Cescon, D.W.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Perez-Garcia, J.; Iwata, H.; et al. Pembrolizumab plus Chemotherapy in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2022, 387, 217–226. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Cercek, A.; Lumish, M.; Sinopoli, J.; Weiss, J.; Shia, J.; Lamendola-Essel, M.; El Dika, I.H.; Segal, N.; Shcherba, M.; Sugarman, R.; et al. PD-1 Blockade in Mismatch Repair–Deficient, Locally Advanced Rectal Cancer. N. Engl. J. Med. 2022, 386, 2363–2376. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-Tumor Genomic Biomarkers for PD-1 Checkpoint Blockade–Based Immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef]
- McGrail, D.J.; Pilié, P.G.; Rashid, N.U.; Voorwerk, L.; Slagter, M.; Kok, M.; Jonasch, E.; Khasraw, M.; Heimberger, A.B.; Lim, B.; et al. High Tumor Mutation Burden Fails to Predict Immune Checkpoint Blockade Response across All Cancer Types. Ann. Oncol. 2021, 32, 661–672. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 Blockade Induces Responses by Inhibiting Adaptive Immune Resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Jin, Y.; Tan, A.; Feng, J.; Xu, Z.; Wang, P.; Ruan, P.; Luo, R.; Weng, Y.; Peng, M. Prognostic Impact of Memory CD8(+) T Cells on Immunotherapy in Human Cancers: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 698076. [Google Scholar] [CrossRef]
- Han, J.; Khatwani, N.; Searles, T.G.; Turk, M.J.; Angeles, C.V. Memory CD8+ T Cell Responses to Cancer. Semin. Immunol. 2020, 49, 101435. [Google Scholar] [CrossRef]
- Egelston, C.A.; Avalos, C.; Tu, T.Y.; Rosario, A.; Wang, R.; Solomon, S.; Srinivasan, G.; Nelson, M.S.; Huang, Y.; Lim, M.H.; et al. Resident Memory CD8+ T Cells within Cancer Islands Mediate Survival in Breast Cancer Patients. JCI Insight 2019, 4, e130000. [Google Scholar] [CrossRef]
- Zhang, J.; Li, S.; Liu, F.; Yang, K. Role of CD68 in Tumor Immunity and Prognosis Prediction in Pan-Cancer. Sci. Rep. 2022, 12, 7844. [Google Scholar] [CrossRef]
- Kato, S.; Okamura, R.; Kumaki, Y.; Ikeda, S.; Nikanjam, M.; Eskander, R.; Goodman, A.; Lee, S.; Glenn, S.T.; Dressman, D.; et al. Expression of TIM3/VISTA Checkpoints and the CD68 Macrophage-Associated Marker Correlates with Anti-PD1/PDL1 Resistance: Implications of Immunogram Heterogeneity. OncoImmunology 2020, 9, 1708065. [Google Scholar] [CrossRef]
- Kazama, A.; Bilim, V.; Tasaki, M.; Anraku, T.; Kuroki, H.; Shirono, Y.; Murata, M.; Hiruma, K.; Tomita, Y. Tumor-Infiltrating Immune Cell Status Predicts Successful Response to Immune Checkpoint Inhibitors in Renal Cell Carcinoma. Sci. Rep. 2022, 12, 20386. [Google Scholar] [CrossRef]
- Kumagai, S.; Togashi, Y.; Kamada, T.; Sugiyama, E.; Nishinakamura, H.; Takeuchi, Y.; Vitaly, K.; Itahashi, K.; Maeda, Y.; Matsui, S.; et al. The PD-1 Expression Balance between Effector and Regulatory T Cells Predicts the Clinical Efficacy of PD-1 Blockade Therapies. Nat. Immunol. 2020, 21, 1346–1358. [Google Scholar] [CrossRef]
- Willsmore, Z.N.; Harris, R.J.; Crescioli, S.; Hussein, K.; Kakkassery, H.; Thapa, D.; Cheung, A.; Chauhan, J.; Bax, H.J.; Chenoweth, A.; et al. B Cells in Patients With Melanoma: Implications for Treatment With Checkpoint Inhibitor Antibodies. Front. Immunol. 2021, 11, 622442. [Google Scholar] [CrossRef]
- Griss, J.; Bauer, W.; Wagner, C.; Simon, M.; Chen, M.; Grabmeier-Pfistershammer, K.; Maurer-Granofszky, M.; Roka, F.; Penz, T.; Bock, C.; et al. B Cells Sustain Inflammation and Predict Response to Immune Checkpoint Blockade in Human Melanoma. Nat. Commun. 2019, 10, 4186. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive Resistance to Therapeutic PD-1 Blockade Is Associated with Upregulation of Alternative Immune Checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Acharya, N.; Sabatos-Peyton, C.; Anderson, A.C. Tim-3 Finds Its Place in the Cancer Immunotherapy Landscape. J. Immunother. Cancer 2020, 8, e000911. [Google Scholar] [CrossRef]
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 Pathway in Cancer: From Bench to Bedside. J. Hematol. Oncol.J Hematol Oncol 2018, 11, 100. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus Pembrolizumab versus Placebo plus Pembrolizumab in Patients with Unresectable or Metastatic Melanoma (ECHO-301/KEYNOTE-252): A Phase 3, Randomised, Double-Blind Study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel III, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Saleh, R.; Elkord, E. Treg-Mediated Acquired Resistance to Immune Checkpoint Inhibitors. Cancer Lett. 2019, 457, 168–179. [Google Scholar] [CrossRef]
- Shang, B.; Liu, Y.; Jiang, S.; Liu, Y. Prognostic Value of Tumor-Infiltrating FoxP3+ Regulatory T Cells in Cancers: A Systematic Review and Meta-Analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef]
- Zhao, S.; Jiang, T.; Zhang, L.; Yang, H.; Liu, X.; Jia, Y.; Zhou, C. Clinicopathological and Prognostic Significance of Regulatory T Cells in Patients with Non-Small Cell Lung Cancer: A Systematic Review with Meta-Analysis. Oncotarget 2016, 7, 36065–36073. [Google Scholar] [CrossRef]
- Sharma, A.; Subudhi, S.K.; Blando, J.; Scutti, J.; Vence, L.; Wargo, J.; Allison, J.P.; Ribas, A.; Sharma, P. Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3+ Regulatory T Cells (Tregs) in Human Cancers. Clin. Cancer Res. 2019, 25, 1233–1238. [Google Scholar] [CrossRef]
- Kamada, T.; Togashi, Y.; Tay, C.; Ha, D.; Sasaki, A.; Nakamura, Y.; Sato, E.; Fukuoka, S.; Tada, Y.; Tanaka, A.; et al. PD-1 + Regulatory T Cells Amplified by PD-1 Blockade Promote Hyperprogression of Cancer. Proc. Natl. Acad. Sci. 2019, 116, 9999–10008. [Google Scholar] [CrossRef]
- Revenko, A.; Carnevalli, L.S.; Sinclair, C.; Johnson, B.; Peter, A.; Taylor, M.; Hettrick, L.; Chapman, M.; Klein, S.; Solanki, A.; et al. Direct Targeting of FOXP3 in Tregs with AZD8701, a Novel Antisense Oligonucleotide to Relieve Immunosuppression in Cancer. J. Immunother. Cancer 2022, 10, e003892. [Google Scholar] [CrossRef]
- van Dalen, F.; van Stevendaal, M.; Fennemann, F.; Verdoes, M.; Ilina, O. Molecular Repolarisation of Tumour-Associated Macrophages. Molecules 2018, 24, 9. [Google Scholar] [CrossRef] [PubMed]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In Vivo Imaging Reveals a Tumor-Associated Macrophage–Mediated Resistance Pathway in Anti–PD-1 Therapy. Sci. Transl. Med. 2017, 9, eaal3604. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, B.; Foote, M.B.; Maron, S.B.; Diplas, B.H.; Lu, S.; Argilés, G.; Cercek, A.; Diaz, L.A. The Spectrum of Benefit from Checkpoint Blockade in Hypermutated Tumors. N. Engl. J. Med. 2021, 384, 1168–1170. [Google Scholar] [CrossRef] [PubMed]
- Valero, C.; Lee, M.; Hoen, D.; Zehir, A.; Berger, M.F.; Seshan, V.E.; Chan, T.A.; Morris, L.G.T. Response Rates to Anti–PD-1 Immunotherapy in Microsatellite-Stable Solid Tumors With 10 or More Mutations per Megabase. JAMA Oncol. 2021, 7, 739. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A.; Shiu, K.-K.; Kim, T.-W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab versus Chemotherapy for Microsatellite Instability-High or Mismatch Repair-Deficient Metastatic Colorectal Cancer (KEYNOTE-177): Final Analysis of a Randomised, Open-Label, Phase 3 Study. Lancet Oncol. 2022, 23, 659–670. [Google Scholar] [CrossRef]
- Morrison, C.; Pabla, S.; Conroy, J.M.; Nesline, M.K.; Glenn, S.T.; Dressman, D.; Papanicolau-Sengos, A.; Burgher, B.; Andreas, J.; Giamo, V.; et al. Predicting Response to Checkpoint Inhibitors in Melanoma beyond PD-L1 and Mutational Burden. J. Immunother. Cancer 2018, 6, 32. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
Figure 1.
Immunohistochemical staining. (A) CD8 intraepithelial expression low. (B) CD8 intraepithelial expression high. (C) Negative expression of CD68. (D) Positive expression of CD68. (E) Negative expression of FoxP3. (F) Positive expression of FoxP3.
Figure 1.
Immunohistochemical staining. (A) CD8 intraepithelial expression low. (B) CD8 intraepithelial expression high. (C) Negative expression of CD68. (D) Positive expression of CD68. (E) Negative expression of FoxP3. (F) Positive expression of FoxP3.
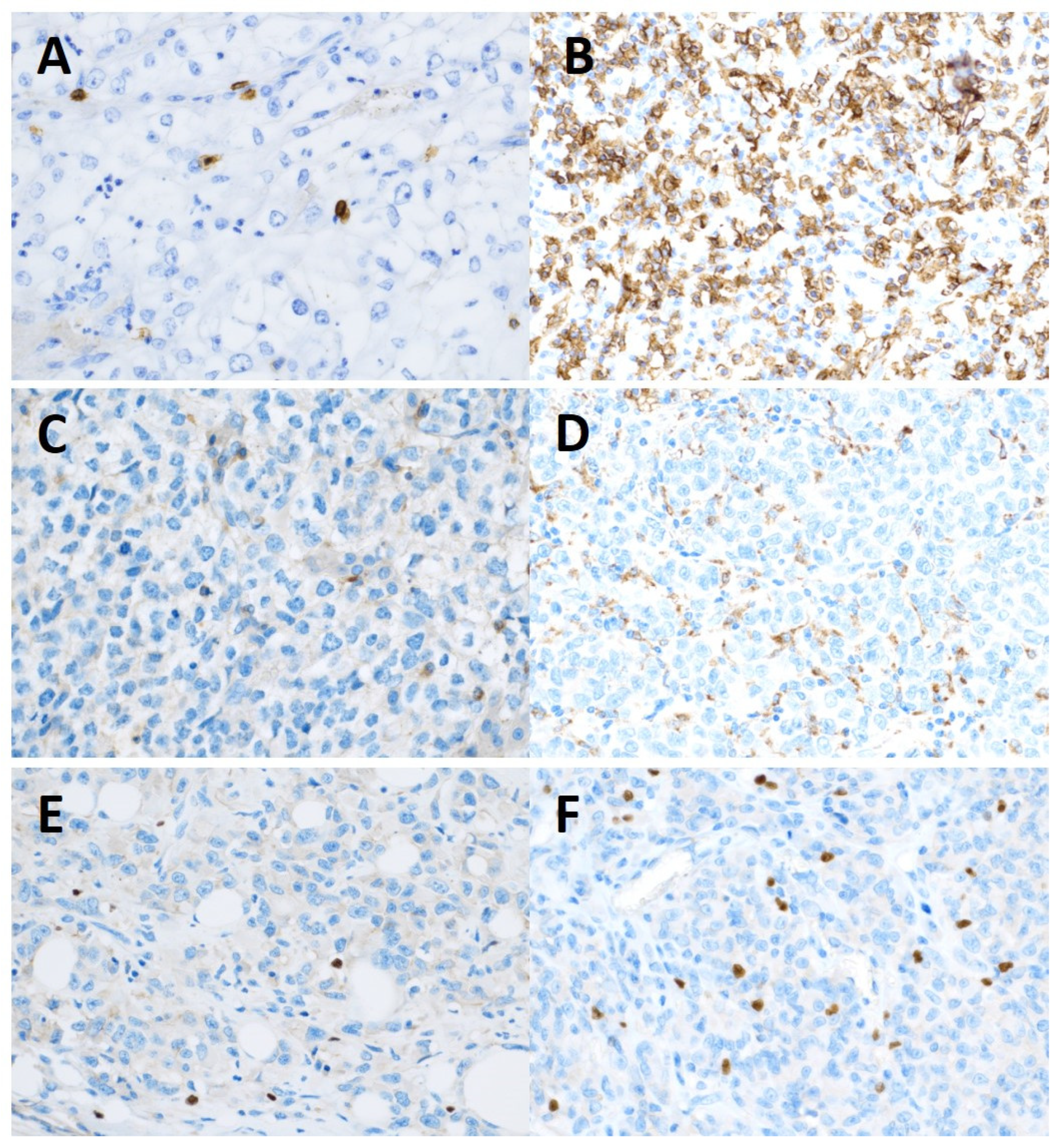
Figure 2.
Kaplan Kaplan-Meier analysis of progression-free survival in the whole group according to (A) FoxP3 expression, median PFS 14.8 months vs 3.9 months, p = 0.002; (B) tumor mutational burden (cut-off 10 muts/Mb), median PFS 15.1 vs 4.4 months, p = 0.024.
Figure 2.
Kaplan Kaplan-Meier analysis of progression-free survival in the whole group according to (A) FoxP3 expression, median PFS 14.8 months vs 3.9 months, p = 0.002; (B) tumor mutational burden (cut-off 10 muts/Mb), median PFS 15.1 vs 4.4 months, p = 0.024.
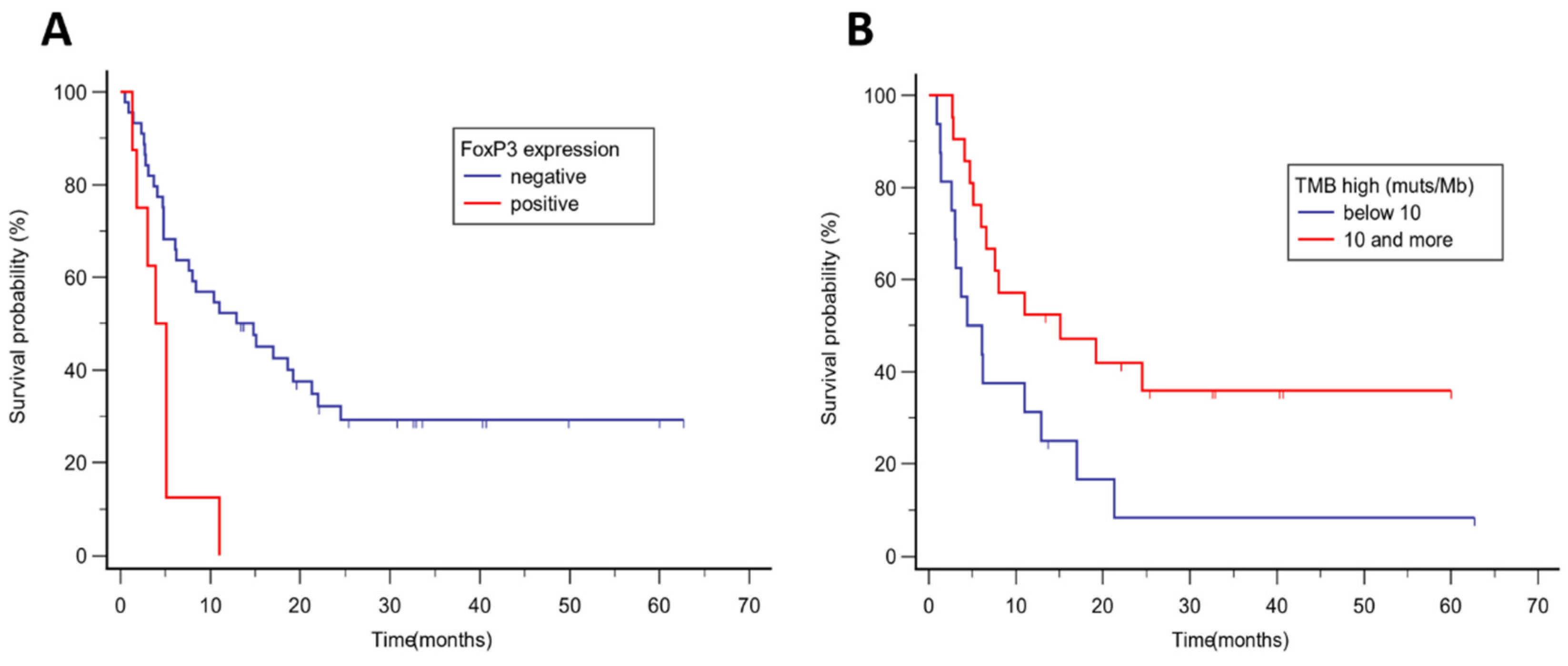
Figure 3.
Kaplan Kaplan-Meier anal_sis of progression-free survival according to FoxP3 expression in (A) malignant melanoma, media_n PFS was 17.0 vs 3.9 months, p = 0.047; (B) NSCLC, median PFS was 14.8 vs 1.8 months, p = 0.005.
Figure 3.
Kaplan Kaplan-Meier anal_sis of progression-free survival according to FoxP3 expression in (A) malignant melanoma, media_n PFS was 17.0 vs 3.9 months, p = 0.047; (B) NSCLC, median PFS was 14.8 vs 1.8 months, p = 0.005.
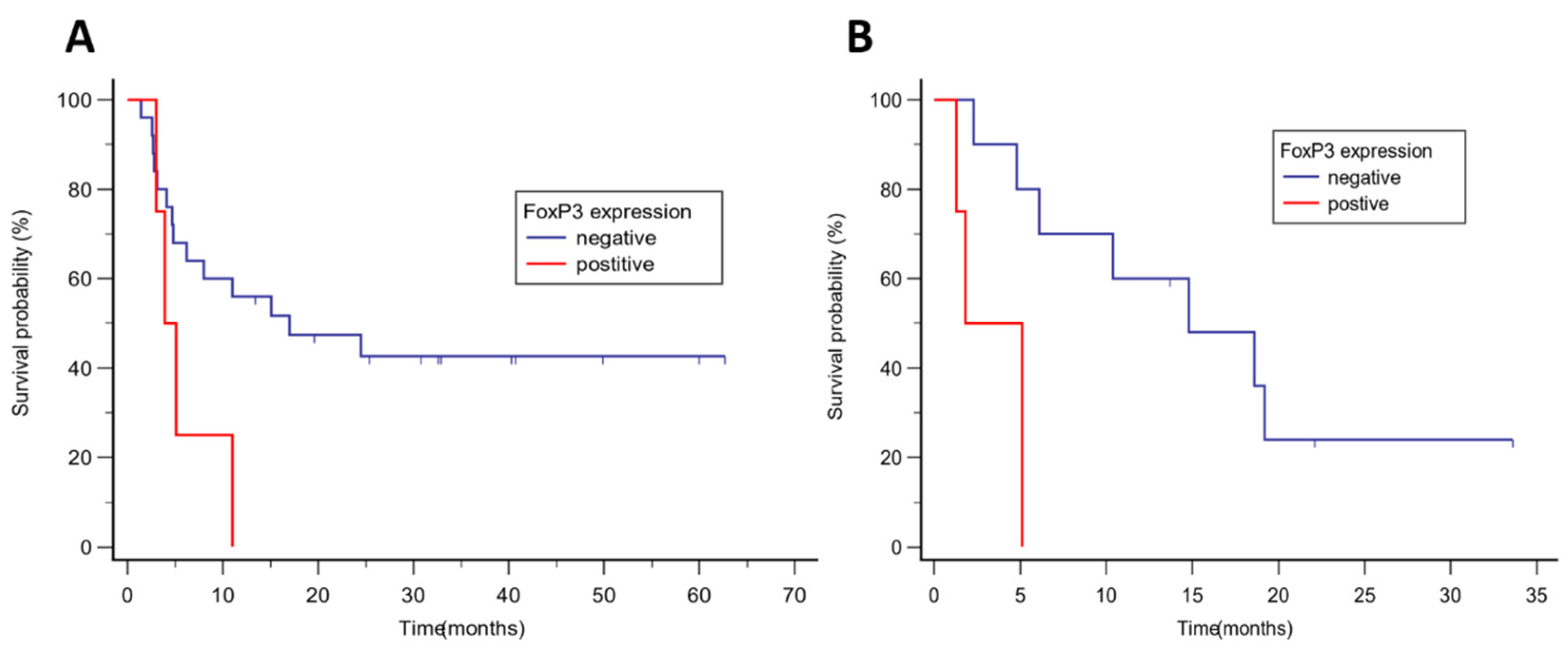
Figure 4.
Kaplan Kaplan-Meier analysis of progression-free survival according to CD68 expression .-in (A) malignant melanoma, median PFS was 15.1 vs 3.0 months, p = 0.033; (B) NSCLC, median PFS was 10.4 vs 2.3 months, p = 0.091.
Figure 4.
Kaplan Kaplan-Meier analysis of progression-free survival according to CD68 expression .-in (A) malignant melanoma, median PFS was 15.1 vs 3.0 months, p = 0.033; (B) NSCLC, median PFS was 10.4 vs 2.3 months, p = 0.091.
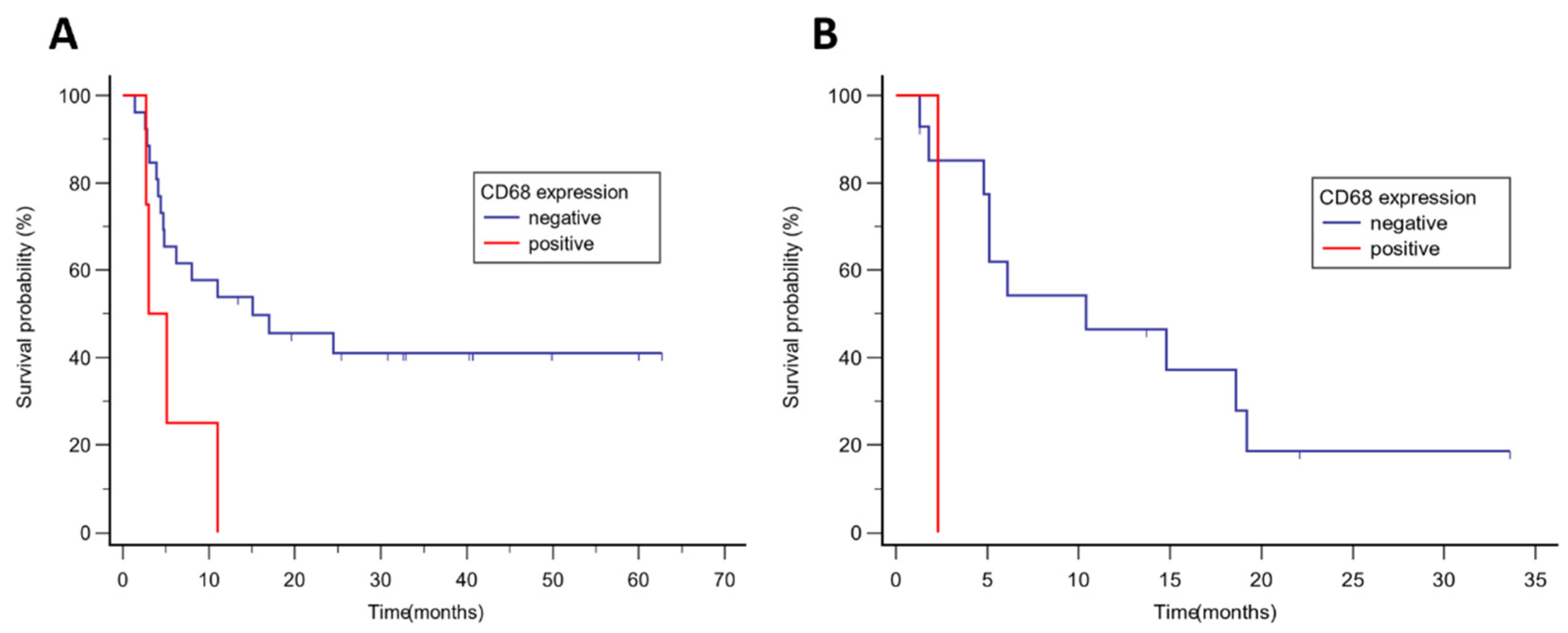

Table 1.
Patient characteristics.
| Characteristic | Patients No. | % |
|---|---|---|
| Age | ||
| median | 67 | |
| range | 37–85 | |
| Sex | ||
| Woman | 14 | 25.0 |
| Men | 42 | 75.0 |
| Cancer type | ||
| Malignant melanoma | 31 | 55.4 |
| NSCLC | 15 | 26.8 |
| Renal cell carcinoma | 7 | 12.5 |
| Colorectal carcinoma | 1 | 1.8 |
| Bladder cancer | 1 | 1.8 |
| Yolk sack tumor | 1 | 1.8 |
| Line of treatment | ||
| 1st line | 36 | 64.3 |
| 2nd line | 14 | 25.0 |
| 3rd or later line | 6 | 10.7 |
| Best overall response | ||
| Complete response | 10 | 17.9 |
| Partial response | 17 | 30.4 |
| Stable disease | 3 | 5.4 |
| Disease progression | 24 | 42.9 |
| ORR | 27 | 48.2 |
| Clinical benefit rate | 27 | 48.2 |
| Survival parameters (median) | ||
| PFS | 8.0 months (95% CI 5.1 – 15.1) | |
| OS | 24.8 months (95% CI 15.3 – 37.3) | |
Abbreviation: NSCLC, non-small cell lung cancer; ORR, overall response rate; PFS, progression-free survival; OS, overall survival.
Table 2.
Immunohistochemical expression of different markers.
| Marker | Negative | Positive | NA | |||
|---|---|---|---|---|---|---|
| N | % | N | % | N | % | |
| MMR deficiency | 52 | 92.9 | 2 | 3.6 | 2 | 3.6 |
| CD3 IEL | 22 | 39.3 | 32 | 57.1 | 2 | 3.6 |
| CD3 stromal | 11 | 19.6 | 42 | 75.0 | 3 | 5.4 |
| CD8 IEL | 29 | 51.8 | 23 | 41.1 | 4 | 7.1 |
| CD8 stromal | 17 | 30.4 | 35 | 62.5 | 4 | 7.1 |
| CD20 | 47 | 83.9 | 7 | 12.5 | 2 | 3.6 |
| CD68 | 46 | 82.1 | 8 | 14.3 | 2 | 3.6 |
| FoxP3 | 45 | 80.4 | 8 | 14.3 | 3 | 5.4 |
| IDO1 | 41 | 73.2 | 12 | 21.4 | 3 | 5.4 |
| LAG-3 | 39 | 69.6 | 15 | 26.8 | 2 | 3.6 |
| TGFβ IC | 32 | 57.1 | 22 | 39.3 | 2 | 3.6 |
| TGFβ TC | 48 | 85.7 | 6 | 12.0 | 2 | 3.6 |
| PD1 | 40 | 71.4 | 14 | 25.0 | 2 | 3.6 |
| PD-L1 CPS ≥1 | 13 | 23.2 | 36 | 64.3 | 7 | 12.5 |
| PD-L1 CPS ≥ 10 | 28 | 50.0 | 21 | 37.5 | 7 | 12.5 |
| PD-L1 CPS ≥ 50 | 37 | 66.1 | 12 | 21.4 | 7 | 12.5 |
| PD-L1 TPS ≥ 1 | 30 | 53.6 | 20 | 35.7 | 6 | 10.7 |
| PD-L1 TPS ≥ 10 | 36 | 64.3 | 14 | 25.0 | 6 | 10.7 |
| PD-L1 TPS ≥ 50 | 41 | 73.2 | 9 | 16.1 | 6 | 10.7 |
Abbreviation: NA, not available; MMR, mismatch repair; IEL, intraepithelial; IC, immune cell; TC tumor cell; CPS, combined positive score; TPS, tumor proportion score.
Table 3.
Association of different markers with survival parameters in the whole group of patients.
| Median PFS in months | Median OS in months | |||||
|---|---|---|---|---|---|---|
| Marker/value | negative | positive | p-value | negative | positive | p-value |
| CD3 IEL | 8.0 | 8.4 | 0.165 | 31.1 | 23.2 | 0.253 |
| CD3 stromal | 5.1 | 11.0 | 0.569 | 18.5 | 25.4 | 0.686 |
| CD8 IEL | 11.0 | 5.1 | 0.190 | 37.8 | 19.8 | 0.045 |
| CD8 stromal | 11.0 | 7.6 | 0.540 | 15.3 | 25.4 | 0.788 |
| CD20 | 8.4 | 11.0 | 0.565 | 26.4 | 24.8 | 0.432 |
| CD68 | 10.4 | 3.0 | 0.059 | 25.4 | 14.9 | 0.837 |
| FoxP3 | 14.8 | 3.9 | 0.002 | 26.4 | 8.3 | 0.118 |
| IDO1 | 8.0 | 11.0 | 0.814 | 21.9 | 24.8 | 0.876 |
| LAG-3 | 11.0 | 5.1 | 0.524 | 31.1 | 23.2 | 0.101 |
| TGFβ IC | 8.0 | 11.0 | 0.736 | 21.9 | 30.0 | 0.345 |
| TGFβ TC | 10.4 | 2.7 | 0.847 | 26.4 | 9.7 | 0.471 |
| PD1 | 8.40 | 5.1 | 0.662 | 27.4 | 23.2 | 0.265 |
| PD-L1 CPS ≥1 | 8.0 | 10.4 | 0.544 | 31.1 | 18.5 | 0.217 |
| PD-L1 CPS ≥ 10 | 8.0 | 10.4 | 0.936 | 19.8 | 23.2 | 0.820 |
| PD-L1 CPS ≥ 50 | 10.4 | 6.1 | 0.892 | 24.8 | 15.3 | 0.794 |
| PD-L1 TPS ≥ 1 | 6.2 | 11.0 | 0.097 | 18.5 | 25.4 | 0.194 |
| PD-L1 TPS ≥ 10 | 8.4 | 6.1 | 0.776 | 24.8 | 15.3 | 0.878 |
| PD-L1 TPS ≥ 50 | 8.4 | 5.1 | 0.778 | 24.8 | 21.9 | 0.881 |
| TMB high | 4.4 | 15.1 | 0.024 | 18.0 | 25.4 | 0.600 |
| TMB above median | 6.1 | 15.1 | 0.016 | 27.4 | 24.8 | 0.871 |
Abbreviation: MMR, mismatch repair; IEL, intraepithelial; IC, immune cell; TC tumor cell; CPS, combined positive score; TPS, tumor proportion score; TMB, tumor mutational burden.
Table 3.
Association of different markers with survival parameters in patients with malignant melanoma.
Table 3.
Association of different markers with survival parameters in patients with malignant melanoma.
| Malignant melanoma | ||||||
|---|---|---|---|---|---|---|
| Median PFS in months | Median OS in months | |||||
| Marker/value | negative | positive | p-value | negative | positive | p-value |
| CD3 IEL | NR | 6.6 | 0.195 | NR | 24.8 | 0.125 |
| CD3 stromal | 8.0 | 15.1 | 0.692 | 31.1 | 25.4 | 0.834 |
| CD8 IEL | 11.0 | 5.1 | 0.474 | NR | 19.8 | 0.093 |
| CD8 stromal | 11.0 | 8.0 | 0.789 | 13.4 | 27.4 | 0.539 |
| CD20 | 8.0 | 11.0 | 0.973 | 31.1 | 24.8 | 0.417 |
| CD68 | 15.1 | 3.0 | 0.033 | 27.4 | 14.9 | 0.969 |
| FoxP3 | 17.0 | 3.9 | 0.047 | 27.4 | 8.3 | 0.852 |
| IDO1 | 8.0 | 15.1 | 0.524 | 26.4 | NR | 0.180 |
| LAG-3 | 11.0 | 15.1 | 0.610 | 31.1 | 25.4 | 0.771 |
| TGFβ IC | 4.8 | 15.1 | 0.376 | 26.4 | NR | 0.366 |
| TGFβ TC | 11.0 | 2.7 | 0.921 | 27.4 | 14.9 | 0.991 |
| PD1 | 11.0 | 5.1 | 0.843 | 31.1 | 24.8 | 0.501 |
| PD-L1 CPS ≥1 | 6.2 | 17 | 0.057 | 27.4 | 25.4 | 0.794 |
| PD-L1 CPS ≥ 10 | 8.0 | NR | 0.197 | 26.4 | NR | 0.529 |
| PD-L1 CPS ≥ 50 | 8.0 | 11.0 | 0.409 | 25.4 | NR | 0.240 |
| PD-L1 TPS ≥ 1 | 4.8 | NR | 0.006 | 19.8 | NR | 0.056 |
| PD-L1 TPS ≥ 10 | 8.0 | NR | 0.268 | 25.4 | NR | 0.468 |
| PD-L1 TPS ≥ 50 | 8.0 | NR | 0.268 | 25.4 | NR | 0.468 |
| TMB high | 4.4 | 15.4 | 0.055 | 27.4 | 26.4 | 0.915 |
| TMB above median | 4.7 | 24.5 | 0.029 | NR | 25.4 | 0.440 |
Abbreviation: MMR, mismatch repair; IEL, intraepithelial; IC, immune cell; TC tumor cell; CPS, combined positive score; TPS, tumor proportion score; TMB, tumor mutational burden.
Table 4.
Association of different markers with survival parameters in patients with NSCLC.
| NSCLC | ||||||
|---|---|---|---|---|---|---|
| Median PFS in months | Median OS in months | |||||
| Marker/value | negative | positive | P-value | negative | positive | P-value |
| CD3 IEL | 6.1 | 10.4 | 0.759 | 13.7 | 15.3 | 0.980 |
| CD3 stromal | 4.8 | 14.8 | 0.321 | 13.7 | 21.9 | 0.258 |
| CD8 IEL | 10.4 | 4.8 | 0.076 | 18.7 | 9.3 | 0.123 |
| CD8 stromal | 10.4 | 5.1 | 0.129 | 15.3 | 11.4 | 0.423 |
| CD20 | 6.1 | 1.3 | 0.503 | 15.3 | 2.7 | 0.701 |
| CD68 | 10.4 | 2.3 | 0.091 | 15.3 | 8.5 | 0.182 |
| FoxP3 | 14.8 | 1.8 | 0.005 | 21.9 | 7.3 | 0.035 |
| IDO1 | 6.1 | 1.3 | 0.750 | 15.3 | 2.7 | 0.601 |
| LAG-3 | 10.4 | 1.8 | 0.135 | 18.5 | 9.3 | 0.293 |
| TGFβ IC | 10.4 | 1.3 | 0.100 | 18.5 | 2.7 | 0.066 |
| TGFβ TC* | NA | 10.4 | NA | NA | 15.3 | NA |
| PD1 | 6.4 | 14.8 | 0.784 | 13.7 | 21.9 | 0.693 |
| PD-L1 CPS ≥1 | NR | 6.1 | 0.161 | NR | 13.7 | 0.222 |
| PD-L1 CPS ≥ 10 | 5.1 | 6.1 | 0.431 | 13.7 | 15.3 | 0.553 |
| PD-L1 CPS ≥ 50 | 10.4 | 4.8 | 0.410 | 18.4 | 11.4 | 0.437 |
| PD-L1 TPS ≥ 1 | 19.2 | 6.1 | 0.279 | 13.7 | 15.3 | 0.712 |
| PD-L1 TPS ≥ 10 | 19.2 | 6.1 | 0.279 | 13.7 | 15.3 | 0.712 |
| PD-L1 TPS ≥ 50 | 10.4 | 4.8 | 0.531 | 13.7 | 15.3 | 0.649 |
| TMB high | 6.1 | 19.2 | 0.199 | 11.4 | 23.2 | 0.063 |
| TMB above median | 6.1 | NR | 0.219 | 11.4 | NR | 0.299 |
Abbreviation: MMR, mismatch repair; IEL, intraepithelial; IC, immune cell; TC tumor cell; CPS, combined positive score; TPS, tumor proportion score; TMB, tumor mutational burden; NA, non-available. *no patient has TGFβ TC negativity.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



