
Submitted:
25 November 2024
Posted:
26 November 2024
You are already at the latest version
Abstract
Malignant peripheral nerve sheath tumor (MPNST) is a rare but aggressive soft-tissue sarcoma characterized by poor response to therapy. The primary treatment remains surgical resection with negative margins. Nonetheless, in the setting of neurofibromatosis type 1 (NF1), the five-year survival rate is at 20-50% with recurrence occurring in up to 50% of individuals. For patients with metastatic and unresectable disease, current treatment options include cytotoxic chemotherapy, which offers minimal benefit, and most patients die within five years of diagnosis. Despite advances in targeted therapy focusing on inhibiting Ras signaling and its downstream effectors, clinical trials report minimal clinical benefit, highlighting the need to explore alternative pathways in MPNST pathogenesis.
Here we discuss the role of the E3 ubiquitin ligase, UBR5, in cancer progression and immune modulation across various malignancies including breast, lung and ovarian cancer. We focus on mechanisms by which UBR5 contributes to tumorigenesis focusing on its influence on tumor microenvironment and immune modulation. We discuss evidence implicating UBR5 in immune evasion and highlight its potential as a therapeutic target to enhance the efficacy of immune checkpoint blockade (ICB) therapy in MPNST, a tumor typically characterized by an immune cold microenvironment. We outline current immune-based strategies and challenges in MPNST management, ongoing efforts to shift the immune landscape in MPNST, and ultimately, we suggest that targeting UBR5 could be a novel strategy to potentiate ICB therapy-mediated anti-tumor immune response and clinical outcomes, particularly in MPNST patients with inoperable or metastatic disease.
Keywords:
Neurofibromatosis Type 1 (NF1)
; MPNST
; Ubiquitin Proteosome System (UPS)
; UBR5
; Ubiquitin
; HECT E3 ligases
; Immune checkpoint blockade (ICB)
; Immunogenicity
; Immune evasion
; Immune “cold”
; Immune “hot
Introduction
Malignant peripheral nerve sheath tumor (MPNST) is a rare but aggressive form of cancer, making up 5–10% of all soft-tissue sarcomas[1,2]. The prevalence of MPNST is one in 100,000 in the general population and affects both genders equally[1]. Approximately 50% of MPNST patients have Neurofibromatosis type 1 (NF1), while 10% have a history of radiation exposure, and the remaining cases are sporadic [1,3,4] . The average age of onset is between 30 and 50 years, but MPNST occurs around a decade earlier in patients with NF1[1]. One of the most prevalent cancer predisposition syndromes, NF1 affects about 1 in 2500 individuals worldwide and 8–13% of those with NF1 develop MPNST in young adulthood, often arising from benign tumors called plexiform neurofibromas (PN)[1,4,5].
Despite advancements in treatment, managing MPNST remains challenging. Surgical resection with wide negative margins remains the only curative treatment[2,6]. Unfortunately, factors such as tumor size, location, and metastasis complicate this approach[6,7]. The 5-year overall survival rate for NF1-associated MPNST ranges from 20% to 50%[5,7]. For patients with recurrent, unresectable, or metastatic disease, treatment options are limited and outcomes are poor[6,7].
Clinical trials targeting signaling pathways involved in MPNST pathogenesis have not yielded significant clinical benefits, highlighting gaps in the understanding of the genetic and molecular underpinnings of this disease[2,6,8].
NF1-MPNST cases are characterized by NF1 gene inactivation and loss of the NF1 encoded tumor suppressor protein, neurofibromin[2,6,8]. A negative regulator of Ras signaling, neurofibromin acts as a Ras-GTPase activating protein[2]. Therefore, the loss of neurofibromin leads to uncontrolled Ras activation and unregulated downstream signaling of pathways such as RAS/MEK/ERK and PI3K/AKT[2,9]. Nonetheless, given that only 10-13% of NF1 patients develop MPNST, biallelic loss of NF1 alone is not sufficient to drive MPNST malignant transformation. Evidence shows that additional genetic alterations, in the tumor suppressors CDKN2A, TP53 and EED/SUZ12, are necessary for the transformation from benign PN to MPNST[6,10,11,12].
Given the limited efficacy of current regimens, there is growing interest in developing targeted therapies for MPNST. The focus has been on inhibiting Ras signaling or downstream pathways with the use of MEK, and mTOR inhibitors. Unfortunately, none of these trials have reported clinical benefit for MPNST patients [2,6,8]. As such, there is a dire need for new therapeutic strategies and a deeper understanding of the molecular drivers of MPNST and the impact of the microenvironment on MPNST progression[6,13,14]. There is growing appreciation for the role of immune, vascular, and stromal cells that form the MPNST microenvironment[14,15].
Immune checkpoint blockade (ICB) has transformed treatment in immune-inflamed cancers such as melanoma, by providing durable anti-tumor immune response[16,17,18]. The use of ICB in MPNST is overwhelmingly understudied[17]. The success of ICB is dependent on an immune cell-rich tumor microenvironment (TME)[16,17], a characteristic, MPNST typically lack, making them "cold" non-inflamed tumors [17,19]. As such, MPNST are unlikely to elicit robust immune response following ICB[17]. Despite isolated reports of ICB response in individual MPNST patients[20,21,22], this lack of immunogenicity poses a significant challenge for use of ICB in MPNST management[17]. Ubiquitin Protein Ligase E3 Component N-Recognin 5 (UBR5) is an E3 ubiquitin ligase commonly amplified and overexpressed in many cancers[23,24]. In cancer, UBR5 is involved in various cellular processes, including the cell cycle, DNA damage response, and the transcriptional machinery[25,26,27,28,29,30]. Overexpression of UBR5 is common in many cancer types, where it often promotes tumor development through mechanisms including but not limited to tumor suppressor degradation[23,31,32]. Similarly, UBR5 has been shown to influence the TME and immune response in Triple Negative Breast Cancer (TNBC) and ovarian cancer (OC). In a murine model of TNBC, genetic knockdown of UBR5 was reported to facilitate antigen processing and presentation, triggering specific immune responses against tumors[33,34]. UBR5 also increased INFγ mediated PDL1 transactivation, leading to immune evasion [33]. In a murine model of OC, UBR5 enhanced cancer growth and metastasis through the recruitment of immunosuppressive tumor-associated macrophages (TAMs) [35].
We have previously reported chromosome 8 (Chr8) gain to be a frequent and critical copy number alteration in MPNST and is associated with poorer overall survival and MPNST pathogenesis[36]. UBR5, one of the genes within the 8q arm[36], is often characterized by genetic alterations, particularly gene amplification in soft tissue sarcoma, including MPNST[36,37][TCGA]. Evidence from studies into the role of UBR5 in cancers such as TNBC and OC points to UBR5 playing a key regulatory role in cancer biology and immune response[33,34,35]. While there is currently no published evidence linking UBR5 to MPNST pathogenesis, by examining evidence of tumorigenic and immunoregulatory roles of UBR5 in cancer, this review will explore mechanisms by which UBR5 mediates immune regulation in cancer and potential therapeutic implications of these mechanisms in MPNST.
1. Ubiquitin Proteasomal System and UBR5 Structure
Ubiquitin proteasomal system (UPS), a fundamental post-translational modification in eukaryotes, attaches ubiquitin (Ub) to protein substrates thereby regulating various cellular processes such as protein degradation, signal transduction, apoptosis, immune response, and DNA repair[24]. While ubiquitination is commonly linked with protein degradation via the proteasome, ubiquitination also regulates protein levels, interactions, and localization[38,39]. UPS orchestrates this regulation, through a cascade of three enzymes: Ub-activating enzyme E1, Ub-conjugating E2, and E3 ligases[38,39]. E3 enzymes, such as UBR5, interact directly with substrates hence imparting substrate specificity to ubiquitination[40,41].
UBR5, a single polypeptide chain homologous to E6AP C terminus, belongs to the HECT-type E3 ubiquitin ligase family and is vital for mammalian embryonic development[24]. UBR5 is one of several E3 ligases encoded in the human genome. E3 ligases are classified into Ring between ring (RBR), the really interesting new gene (RING), and the homologous to E6AP C-terminus (HECT) family[24,26]. The modular structure of UBR5 comprises multiple domains, including a ubiquitin-associated (UBA) domain, a zinc-fingerlike ubiquitin-recognin (UBR) box, and a MLLE/ PABP-interacting motif 2 (PAM2) binding domain adjacent to the HECT domain[26,42]. Structural studies outline the HECT domain to be crucial for catalyzing ubiquitin transfer onto substrates. Notably, UBR5 is unique among N-end rule pathway E3 ligases for containing both an N-degron recognition UBR box and a HECT domain[26]. The N-degron recognition UBR box recognizes and degrades proteins based on the nature of their N-terminal amino acid residue[31]. Unlike other UBR-box containing E3 ligases, UBR5 is the only one containing a HECT domain which regulates proteins by forming a thio-ester linkage between Ub and the conserved catalytic cysteine residue within the HECT domain[31]. Consequently, transferring Ub from E3 ligase to target proteins[26,31,42].
Ubiquitin chain topology, including length and linkage type, dictates protein fate. For instance, branched ubiquitin chains, such as K11/K48, serve as potent signals for proteasomal degradation[38,39,43]. UBR5 plays a key role in assembling K11-K48 linked branched Ub chains, regulating the degradation of various substrates[24,40,41]. Structural analysis reveals that UBR5 specifically assembles K11-K48-linked branched Ub chains, including by branching chains preformed at K11- or K63-linked chains[45]. UBR5 might collaborate with other E3 ligases to synthesize branched K48 linkages off preexisting ubiquitin chains. One such instance is UBR5, along with UBR4, assemble K11/K48 branched ubiquitin chains, facilitating the rapid degradation of aggregation-prone proteins like 73Q-huntingtin (HTT) and C9orf7[38,45].
2. Functional Roles of UBR5
UBR5, a homologue of hyperplastic discs (HYD), plays critical tumor suppressor roles in Drosophila melanogaster, regulating cell proliferation during development[46]. Cells harboring mutated HYD failed to control proliferation, leading to tumorigenesis. Mutations in HYD/UBR5 have been shown to lead to developmental abnormalities, including adult sterility secondary to germ cell defects[46].
In diseases like mantle cell lymphoma and Huntington’s disease, UBR5 plays crucial roles in regulating normal cellular functions. For example, in mantle cell lymphoma, mutations that disrupt the HECT domain of UBR5 impair normal B cell maturation and differentiation, potentially contributing to the transformation of cells into malignancies [43,47]. Similarly, in Huntington’s disease, although the mutant huntingtin protein (HTT) is typically overexpressed, studies using induced pluripotent stem cells (iPSCs) have shown that elevated levels of UBR5 can promote the degradation of the mutant HTT protein. This suggests that UBR5 might help to reduce the toxic effects of mutant HTT in disease models, offering potential insights into therapeutic strategies [43,45,46].
1. Role of UBR5 in DNA Damage Response (DDR)
UBR5 and ATMIN Interaction
Zhang et al. demonstrated that upon irradiation, UBR5 ubiquitinates ATMIN, an ATM interactor, thus promoting ATM phosphorylation at DNA damage sites allowing for efficient DNA repair initiation. UBR5 mediated ATM activation at damage sites, favors cell survival after irradiation, driving radio-resistance. UBR5's role in regulating the DNA damage response highlights its potential as a therapeutic target for enhancing response to DNA damage inducing agents[24,48,49].
UBR5 and TopBP1 Interaction
Previous studies have identified an interaction between UBR5 and DNA repair protein TopBP1. In vitro ubiquitination assays demonstrated that UBR5 ubiquitinates TopBP1 in the presence of the E2 enzyme UBCH4. TopBP1, a target for ubiquitination by UBR5, colocalizes with BRCA1 at stalled DNA replication forks, suggesting a role for UBR5 in regulating DNA replication and repair processes[50]. Additionally, UBR5-mediated ubiquitination of TopBP1 contributes to the activation of the DNA damage checkpoint kinase CHK2, further implicating UBR5 in DDR pathways[24,51]. Marcia munoz et al demonstrated that UBR5 was necessary for CHK2 mediated G1/S and intra S phase DNA damage checkpoint activation and for the maintenance of G2/M arrest after double strand DNA breaks. Defective checkpoint activation in UBR5-depleted cells led to error prone DNA synthesis, premature mitotic entry, accumulation of polyploid cells, and eventual cell death via mitotic catastrophe[52].
UBR5 and TRIP12 Interaction
UBR5 collaborates with TRIP12 to suppress RNF168 mediated chromatin ubiquitination hence preventing the spread of DNA damage signals away from the initial site of damage. TRIP12 and UBR5 maintain the nuclear pool of RNF168, which functions to ubiquitinate a limited fraction of chromatin near double-stranded DNA breaks. The absence of TRIP12 and UBR5 results in the hyperaccumulation of RNF168, leading to aberrant chromatin ubiquitination and impaired DNA repair processes[24,53]. This interaction highlights the coordinated regulation of chromatin dynamics by UBR5 during the DNA damage response.
UBR5 and Replication Fork Components Interaction
UBR5 additionally interacts with components of the replication fork, including the trans-lesion synthesis (TLS) polymerase, polη, to prevent erroneous recruitment of polη via ubiquitylated H2A. Depletion of UBR5 leads to erroneous polη recruitment and accumulation and consequent replication problems, such as slower S-phase progression and the accumulation of single-stranded DNA[54]. polη mediated DNA replication inefficiency secondary to UBR5 loss, emphasizes the significant role UBR5 plays in maintaining replication fork integrity and ensuring accurate DNA replication.
In summary, UBR5 plays diverse roles in both normal cellular functions and tumorigenesis. The unique interaction between UBR5 and key DNA damage associated substrates, such as ATMIN, TopBP1, and replication fork components, highlights the significance of UBR5 in regulating DNA repair processes and maintaining genomic stability. A thorough review of the role of UBR5 in genomic stability is discussed by Shearer. F et al [24].There’s evidence that genomic instability and mutagenesis augment the number and diversity of tumor neoantigens, antigen presentation and consequent, anti-tumor immune responses [55,56,57]. It’s therefore fair to hypothesize that UBR5 deficiency in tumor cells may result in DNA repair deficiencies and consequent genomic instability. As such, further investigation into the intricacies of UBR5 functions and interactions may provide insights into novel therapeutic avenues for targeting the DDR and immune system.
2. UBR5 in Metastasis and Therapeutic Resistance
UBR5 has emerged as a critical player in cancer progression, metastasis, anti-apoptosis, and therapeutic resistance. The interactions of UBR5 with a diversity of substrates, underpin its multifaceted activities that contribute to cancer cell survival and resistance to chemo and radiotherapeutics.
UBR5 in Therapeutic Resistance and Anti-Apoptosis
Overexpression of UBR5 in tumors seems advantageous in the evasion of the cytotoxic effects of DNA-damaging agents. UBR5 drives this evasion by augmenting DNA repair machinery, cellular checkpoints, and the anti-apoptotic response. This is evidenced by UBR5 mediated degradation of the proapoptotic modulator of apoptosis protein 1 (MOAP-1) and modulator of apoptosis protein 1 (MOIP-1) in a HECT domain dependent manner. UBR5 mediated degradation of MOAP-1 and MOIP-1 confers cisplatin resistance in ovarian cancer, illustrating the capacity for UBR5 to enhance cancer cell survival following chemotherapy[58]. Additionally, Matsuura et al., 2017 reported a UBR5 mediated transcriptional activation of the anti-apoptotic protein Mcl-1 in a manner that was independent of its E3 ubiquitin ligase activity. In this context, UBR5 influences apoptosis associated proteins, Mcl-1, MOAP-1, and MOIP-1, through both ubiquitin ligase-dependent and -independent mechanisms. That UBR5 regulates Bcl-2 family proteins and their regulators underscores UBR5's complex role in regulating apoptotic pathways and cancer cell survival. An additional instance in which UBR5 regulates the apoptotic machinery, is evidenced by UBR5 mediated degradation of the proapoptotic regulator TXNIP through K48/K63-branched ubiquitin chains [59].
In an ERa+ breast cancer model, UBR5 overexpression induces tamoxifen resistance by upregulating β-catenin expression and activity. WNT/β-catenin signaling is known to drive acquisition of stemness in various cancers. In a ubiquitin ligase dependent manner, UBR5 led to β-catenin stabilization and consequent β-catenin driven therapeutic resistance. UBR5 knockdown, conversely, decreased β-catenin nuclear accumulation without affecting β-catenin mRNA expression, suggesting a UBR5 mediated post-translational regulation of β-catenin [60]. The inhibition of UBR5/ β-catenin signaling re-sensitized tamoxifen resistant breast cancer in vivo. Similarly, UBR5 mediated β-catenin stabilization has also been described in a TNBC model of breast cancer [35].
In addition to modulating cancer stemness, UBR5 also modulates the levels of other critical proteins involved in cancer progression. UBR5 suppresses MYC expression levels through ubiquitination. MYC is a master regulator of malignant growth, but excess MYC protein levels, primes both normal and cancer cells for apoptosis. In tumor cells, UBR5 fine-tunes MYC protein levels, balancing cell growth and survival, thus contributing to therapeutic resistance by protecting breast tumors from MYC mediated apoptosis-priming [61]. Concordantly, UBR5 depletion in cells treated with replication inhibitors, resulted in MYC mediated apoptosis.
UBR5 in Metastasis
The influence of UBR5 extends to metastasis and cancer cell growth. For instance, UBR5-mediated ubiquitination of the tumor suppressor CDC73 impacts lung colonization of metastatic cells. UBR5 deficiency led to CDC73 upregulation, resulting in decreased metastatic tumor growth and increased sensitivity to apoptosis[62]. This relationship implicates UBR5 in the metastatic process and highlights its role in regulating apoptosis and cell proliferation pathways [48,49,63]. Additionally, Ziqi Yu et al observed upregulation of Trp53 mRNA following Ubr5 deletion. Interestingly, this was reversed by silencing Cdc73, indicating, a role for UBR5 in regulating the p53 pathway in part by degrading the tumor suppressor CDC73[62].
In TNBC, UBR5 abrogation impairs angiogenesis within tumors, increases apoptosis, and induces growth arrest. Absence of UBR5 disrupted mesenchymal-to- epithelial transition (MET) by deregulating E-cadherin expression, thus reducing the ability of micromestases to establish within secondary organs.[34]. Mei Song et al elucidated further the mechanism by which UBR5 loss alters MET: UBR5−/- cells displayed strongly reduced levels of E-cadherin, ID1 and ID3 mRNA and proteins all of which are critical regulators of mesenchymal–epithelial transition (MET) in metastatic colonization. UBR5 directly or indirectly regulated transcription of Id1 and Id3 genes in a manner independent of its E3 ubiquitin ligase activity[35].
Furthermore, UBR5 promotes pancreatic cancer cell migration and invasion by degrading CAPZA1, a member of the F-actin capping protein family, in an E3 ubiquitin ligase-dependent manner. This degradation leads to F-actin accumulation and cytoskeletal remodeling, essential for cancer cell motility and invasion. The process highlights the role of UBR5 in regulating actin dynamics and promoting metastatic behavior [64].
In cervical cancer, UBR5 negatively regulates TIP60, a lysine acetyltransferase involved in transcription, DNA damage response, and apoptosis. UBR5-mediated destabilization of TIP60, promoted HPV E6 oncoprotein mediated tumorigenesis [65].
Finally, in gastric cancer, UBR5 binds and ubiquitinates the tumor suppressor gastrokine 1 (GKN1), reducing its stability and promoting cancer cell growth[66]. Similarly, in colorectal cancer, UBR5 increases ubiquitination and degradation of ECRG4, a tumor suppressor, thus facilitating cell proliferation and survival[67]. These interactions illustrate the critical role UBR5 plays in deregulating tumor suppressors and enhancing oncogenic pathways. In glioma, the tumor suppressor ECRG4 is downregulated. ECRG4 inhibits the activity of NF-kB, suppressing, the invasion, proliferation, and migration of glioma cells[64]. Qiang Wu et al reported UBR5 upregulation in glioma tissues and cells where it promoted the migration and invasion of glioma cells by regulating the ECRG4/NF-kB pathway. Consequently, UBR5 knockdown promoted the expression of ECRG4 and reduced the phospho-activation of NF-kB and phosphorylation of IkBa, a negative regulator of NF-kB[68].
In summary, UBR5 interacts with key proteins such as MOAP-1, MOIP-1, β-catenin, TXNIP, MYC, CDC73, CAPZA1, TIP60, GKN1, and ECRG4, NF-kB, highlighting its pivotal roles in promoting metastasis, therapeutic resistance, and cancer cell survival. Understanding these interactions provides critical insights into the potential of UBR5 as a therapeutic target in cancer treatment.
3. Role of UBR5 in Immune Modulation
Although Chr8 has been reported in MPNST, the precise role played by the genes housed within this chromosome, including UBR5, is not fully understood. UBR5 has been implicated in various other cancers, underscoring its key role in tumorigenesis. Emerging evidence indicates that the tumorigenic activities of UBR5 are, in part, dependent on its modulation of the immune system. This is not surprising as components of the UPS such as E3 ligases and deubiquitinases have been discussed as modulators of both cellular and non-cellular components in the tumor microenvironment [69,70,71].
In a mouse model OC, the loss of Ubr5 was found to enhance chemotherapy response and augment immune response to anti-PD-1 and CAR T cell therapy [35]. This model demonstrated that Ubr5 facilitated OC growth and metastasis by maintaining an immunosuppressive tumor microenvironment (TME). Ubr5 drove growth and immunosuppression through the recruitment and activation of Tumor-Associated Macrophages (TAMs) via paracrine signaling involving CCL2 and CSF-1. UBR5 mediated recruitment of TAMs contributed to an immunosuppressive TME that supports tumor growth and metastasis. Additionally, Ubr5 was essential for sustaining β-catenin signaling, which promoted cancer stemness and distant metastasis independently of TAMs, pointing to a tumor autonomous process. The role of Ubr5 in modulating immune response is highlighted by the observed success of immune checkpoint blockade (ICB) and CAR T cell therapy in this OC mouse model following the loss of Ubr5. This success is particularly noteworthy given the historically limited efficacy of immunotherapy in OC[72,73], underscoring the critical interplay between Ubr5 and the immune system in regulating tumor growth and Immunotherapy response.
In a murine model of Triple-Negative Breast Cancer (TNBC), Ubr5 also enhanced tumor growth in a manner dependent on the immune system. In Ubr5-deficient tumors, there was a notable increase in antigen presentation, the percentage of CD8+ T cells and interferon-gamma (IFN-γ) producing CD4+ T cells in the spleen, as well as an increased infiltration of polymorphonuclear leukocytes in UBR5 knockout tumors. Conversely, Ubr5 wild-type (WT) tumors exhibited a higher number of regulatory T cells (Tregs) in the spleen and TDLN and a greater infiltration of mononuclear leukocytes was observed[34]. The proportion of tumor-infiltrating CD4 + and CD8 + T cells was increased in Ubr5−/- tumor bearing mice compared to control tumors. Furthermore, infiltrating CD8 + T cells exhibited enhanced granzyme B expression, indicating a more active cytolytic state of the CD8+ T cells in Ubr5-deleted tumors [35]. The infiltration of polymorphonuclear leukocytes in Ubr5-/- tumors was associated with reduced angiogenesis, suggesting a potential mechanism by which Ubr5 promotes tumor growth through immune modulation. Additionally, Ubr5 depletion in tumor-bearing mice reduced surface levels of PD-L1 on TAMs suggesting a role for Ubr5 in immune evasion[35].
The molecular mechanisms by which UBR5 regulates immune activation warrants further investigation. It is hypothesized that UBR5 may interact with key signaling pathways and transcription factors involved in immune cell function and tumor-immune interactions. For instance, the role of UBR5 in β-catenin signaling suggests a possible link to the Wnt/β-catenin pathway, which is known to play a role in immune evasion and cancer stemness [35,74]. Additionally, the involvement of UBR5 in CCL2/CSF-1 signaling as reported in the TNBC model [35] indicates a broader impact of UBR5 on the cytokine milieu within the TME, influencing the recruitment and activation of various immune cell populations.
In another murine model of TNBC, UBR5 was found to regulate IFN-γ-mediated PD-L1 expression to promote tumor growth[33]. Tumor cells commonly exploit the PD-L1/PD-1 interaction to evade immune surveillance, a phenomenon associated with poor prognosis across various cancers[75]. PD-L1 expression can be induced by several cytokines, including IFN-γ, tumor necrosis factor-α (TNF-α), interleukins (ILs), and epidermal growth factor (EGF), via multiple signaling pathways such as JAK/STAT1/IRF1, NF-κB, PI3K/AKT/mTOR, and JAK/STAT3 [76].
In this TNBC model [33], UBR5 regulated IFN-γ-induced PD-L1 transcription in an E3 ubiquitin ligase independent manner. Through its polyadenylate binding (PABC) domain, UBR5 enhanced PD-L1 transactivation by upregulating the transcription of EIF2AK2 gene that encodes protein kinase RNA-activated (PKR). The consequence was increased transcription of effectors such as, STAT1 and IRF1, downstream of PKR [33]. Further demonstrated in this same TNBC model, dual hairpin targeting of UBR5 and PD-L1 (CD274) resulted in dramatically reduced tumor growth and lung metastasis and significantly extended survival. Along the same lines, UBR5 has been shown to stabilize the WNT/β-catenin signaling, a key driver of cancer stemness [35,61,74].
In melanoma, β-catenin signaling has been shown to mediate resistance to ICB therapy [74,77]. Underscoring the significant role of WNT/β-catenin signaling in immune evasion. Takeuchi et al. reported that NSCLCs that lacked immune cell infiltration into the TME despite high tumor mutational burden (TMB), a marker high immunogenicity, preferentially up-regulated the WNT/β-catenin pathway. In high TMB NSCLC which are highly immunogenic, TMB does not always correlate with PD-L1 expression. In this NSCLC model, a PD-L1 independent immunosuppressive TME that is driven by WNT/β-catenin signaling was observed [78]. These additional pieces of evidence, underscore the role of UBR5 in regulating oncogenic pathways such as WNT/β-catenin to drive tumor immune evasion.
While the role of UBR5 in sarcomas, including MPNST, is understudied, the oncogenic pathways discussed above, through which UBR5 regulates immune escape, have been implicated in MPNST pathogenesis. For instance, signaling downstream of EGFR is known to induce PD-L1 expression and contribute to immune evasion. Similarly, the PI3K-AKT-mTOR and WNT/β-catenin pathways play roles in PD-L1 induction and immune evasion [76]. These signaling pathways are not only crucial drivers of tumorigenesis in MPNST but are also therapeutic targets in MPNST clinical trials, albeit with limited clinical success[6,8,79]. MAPK-ERK2, a downstream target of the Ras signaling implicated in NF1-MPNST pathogenesis, is reported to regulate UBR5 function in cellular signaling through phosphorylation[80,81]. As such, exploring the interplay between UBR5 and these key oncogenic pathways driving MPNST, could uncover mechanisms to enhance therapeutic response, in part, through augmenting anti-tumor immune response.
4. UBR5 in MPNST: Therapeutic Potential
In our lab, we have extensively studied eight NF1-MPNST parental tumors and their PDX counterparts. Our research highlighted pronounced aneuploidy including chr8 gain, in NF1-MPNST compared to the generally diploid PNFs. Using bulk RNA-seq we identified UBR5 among chr8 genes with the highest expression in MPNST[36]. Sarcomas typically exhibit more copy-number aberrations than other cancers, with MPNST showing particularly high CNAs[37,82]. This is a critical discovery, as DNA aneuploidy is recognized as an independent risk factor, for decreased metastasis-free survival in sarcomas, regardless of histologic grade or lymphovascular invasion [37,83,84].
In dissecting MPNST immune profile, Holand et al reported DNA copy number gains including 8q gain which also correlated with immune deficiency in MPNST tumors[37]. Chr8q gain has been reported in Ewing sarcoma and other pediatric soft-tissue sarcomas , suggesting this may be a critical event in multiple sarcoma types, including NF1-MPNST[36]. Moreover, our data linked Chr8 gain with poor OS in soft-tissue sarcoma datasets from TCGA. Chr8q contains numerous cancer-critical genes, including MYC (8q24), which is well-studied across various cancers, including MPNST where upregulation of MYC and or its targets was correlated with tumor recurrence and immune deficiency [37,85]. Additionally, 8q houses other significant cancer-related genes including UBR5 whose role in posttranslational regulation of Myc and Myc mediated apoptosis-priming has been reported [29,61]. Underscoring the significance of Chr8q gain in MPNST, Maren Holand et al classified MPNSTs into two transcriptomic subtypes defined primarily by immune signatures and proliferative processes. “Immune deficient” MPNSTs were more aggressive, characterized by proliferative signatures, and high genomic complexity (CNAs) including 8q gain.
Based on our study, among the significantly upregulated genes within chr8, genetic knockdown of UBR5 significantly impacted MPNST cell survival. Preliminary unpublished findings from our lab revealed that knockdown of UBR5 in MPNST cells led to reduced proliferative capacity, increased apoptotic cell death, and decreased tumor growth in vivo. These results coupled with established role of UBR5 in the pathogenesis of cancers such as ovarian, lung, and breast cancer, highlights potential involvement in MPNST progression.
5. MPNST Immune Architecture: Mechanisms of Immune Evasion
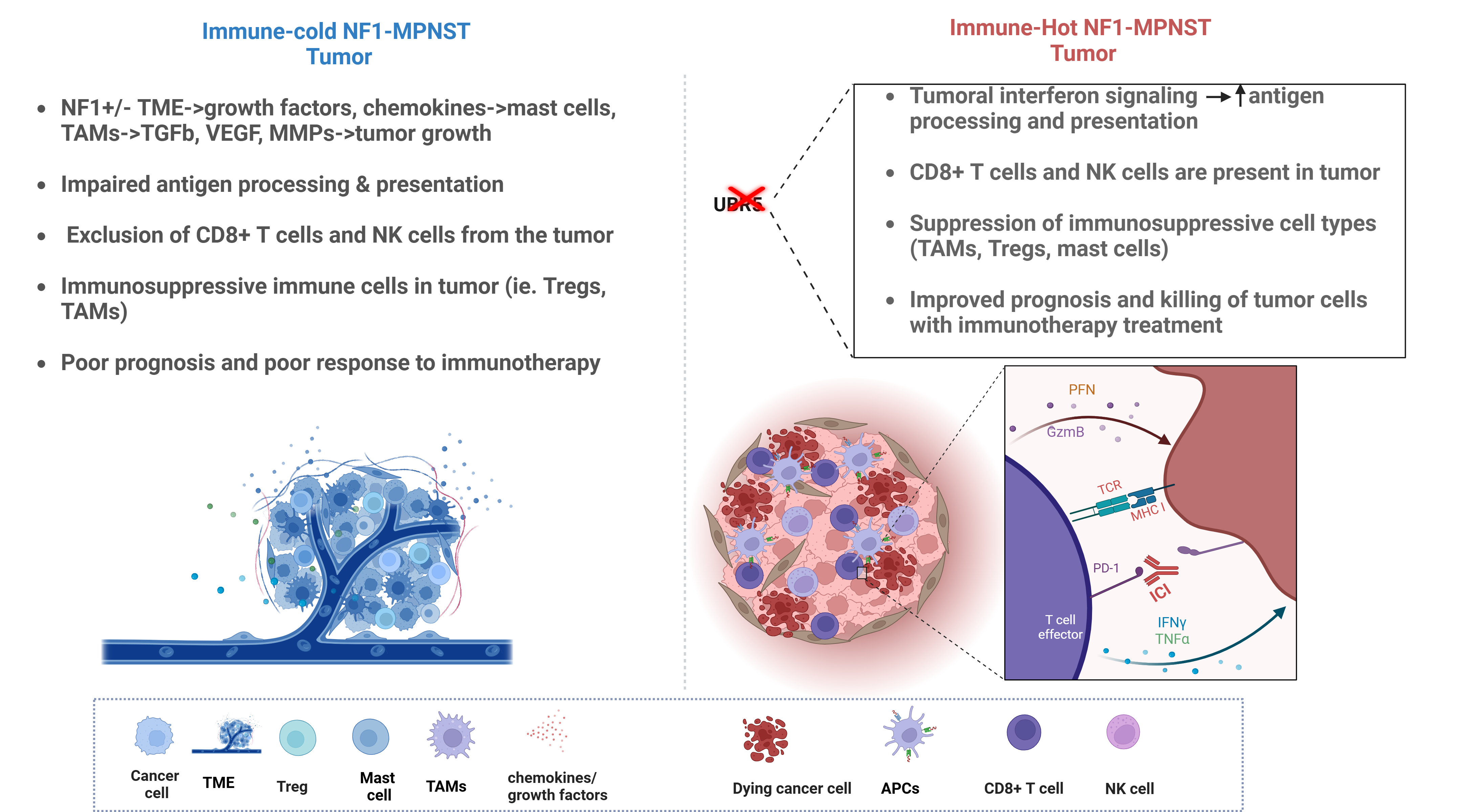
Although immune-based treatment strategies such as ICB, have revolutionized cancer care in malignancies like melanoma and non-small cell lung cancer (NSCLC) [86,87] progress in applying these strategies to MPNST have been notably limited[17,88]. [NF1 patients exhibit germline loss of a single NF1 allele, resulting in NF1 haploinsufficiency (NF1+/−) and hyperactive Ras signaling across all cells in the body including immune cells. Somatic loss of the second NF1 allele in schwann cell precursors triggers plexiform neurofibroma formation. The NF1+/− MPNST TME conditions plays a pivotal role in PN development and malignant transformation through the secretion of growth factors, chemokines, and proinflammatory mediators[14,15,89].
Mast cells are one such immune cell type in the neurofibroma microenvironment, that play a critical role in NF1-associated tumorigenesis. Nf1-/- Schwann cells (SCs) hypersecrete stem cell factor (SCF), the ligand for c-kit, which is essential for mast cell development and survival [13,15]. Nf1 haploinsufficient (Nf1+/− ), mast cells show enhanced migration, proliferation, survival, and degranulation in response to SCF in a Ras/PI3K-dependent manner [14,15]. Among the effector proteins hypersecreted by Nf1+/- mast cells is TGF-beta (TGFβ), promoting increased proliferation and collagen production by Nf1+/- fibroblasts a hallmark of PNF formation[15]. Additionally, mast cells hypersecrete various pro-angiogenic growth factors, including VEGF and metalloproteinases (MMPs), potentially contributing to PNF tumorigenesis [86,87,88]. This tumorigenic TME characteristic has motivated ongoing clinical trials investigating KIT/CSF1R inhibitors, targeting neurofibromas associated with mast cell infiltration[1,14,15]. Similarly, Hirbe et al. and Dodd et al. demonstrated that the Nf1 haploinsufficient TME accelerated MPNST onset in genetically engineered mouse models (GEMMs) of MPNST[11,89]. Unsurprisingly, an enriched myeloid cell population, particularly elevated mast cells, were observed in these tumors. Consistent with these preclinical findings, a phase I clinical trial evaluating a combination strategy targeting mast cells and TAMs in MPNST yielded promising results. The combination of pexidartinib (KIT, CSF1R, and FLT3 inhibitor) and sirolimus, an mTOR inhibitor (NCT02584647), demonstrated efficacy in 12 out of 18 patients with advanced sarcoma, including 5 with MPNST. Median progression-free survival (PFS) and OS in the MPNST cohort were 18.6 weeks and 145.1 weeks, respectively[90].
Despite these advances, other immune strategies such as ICB, ICB combined with targeted therapy, CAR-T cell therapy, and oncolytic viruses are still in their infancy in MPNST [88,91]. Nonetheless, the promotion of tumor formation and growth by mast cell-induced inflammation highlights the involvement of the MPNST TME in tumor pathogenesis and highlights the potential to restore anti-tumor immune function therapeutically. A summary of ongoing clinical evaluating these immune strategies is provided in Table 1.
Emerging clinical data highlight the potential of PD-1/PD-L1 checkpoint blockade in MPNST, particularly in tumors expressing high levels of PD-L1. In clinical case reports, PD-L1 expression was observed in 70-100% of tumor cells in three patients and 2% in one patient, as determined by IHC staining [20,21,22,88,92]. Case reports of these four MPNST patients, who were initially refractory to conventional therapies but achieved complete responses with pembrolizumab or nivolumab, further underscore the potential of ICB in MPNST management.
Discrepancies between the success of these individual responses and broader ICB related clinical outcomes MPNST may be attributed to various factors. Highlighted in these reports, is the need for robust biomarkers to identify which MPNST patients are likely to derive clinical benefit from ICB. Additional factors such as inter- and intra-tumoral heterogeneity, initial treatment prior to ICB, and variable PD-L1 expression levels among patient cohorts may also contribute to this discrepancy [93]. In a study of MPNST transcriptomes pooled from Gene Expression Omnibus (GEO), 30% of MPNST samples had scores predictive of ICB response. This study used a validated predictive score model based on T-cell exclusion and dysfunction signatures to assess the extent of predicted response to ICB. Concomitant with existing evidence of heterogeneity, considerable variability in the behaviors of immune cells even within TME of MPNST tumors with the same CTL score was observed[94].
PD-L1 expression has been considered the best available predictive biomarker to ICB response. However, it has notable limitations, as not all patients with elevated PD-L1 levels exhibit positive response to ICB therapy[95,96]. As such, efforts to identify predictive and prognostic biomarkers aim to enhance our understanding of the complex interplay between cancer and T cell response [93]. In MPNST, potential biomarkers for immunotherapy have not yet been evaluated in large-scale standardized prospective trials. Nonetheless preclinical evidence demonstrates the extent of immune evasion in MPNST. For instance, immune genes such as HLA which encode MHC class I and II, the transcription factor MHC II transactivator (MHC2TA), the transporter associated with antigen processing (TAP1), and the related chaperone CD74, show reduced expression in NF1-associated tumors compared to normal human Schwann cells highlighting downregulation of or impaired antigen presentation processes in MPNST [97].
Biomarkers for future trials in MPNST will need to highlight evidence or lack thereof, of immune activation and intact antigen presentation. Some relevant candidate biomarkers in MPNST include PD-L1copy number gain and expression [97], TILs [88,93], and TMB [88]. Typically, MPNSTs are characterized by low PD-L1 expression, however, reports on PD-L1 expression in MPNST are inconsistent, with some studies reporting high expression and others reporting low PD-L1 expression [17]. Elizabeth Shurell et al. reported 13% of MPNST tissue microarray lesions had at least 5% PD-L1 staining. In these samples, MPNST was characterized by absent PD-1 expression, low PD-L1 expression although higher than benign neurofibroma, and significant CD8+ TIL presence[98]. Similarly, transcriptomic analysis of T cell signatures revealed that a majority of human MPNST (62 out of 73 samples profiled) exhibited an immunologically “cold” phenotype characterized by reduced cytotoxic T cell infiltrates and increased tumor immune dysfunction and exclusion (TIDE) scores[94]. These findings highlight the lack of robust immune cell infiltration in MPNST, that is necessary to elicit robust anti-tumor immune response, emphasizing the challenges to effective application of immunotherapy approaches in this malignancy.
On such basis, MPNST has indeed been described as non-inflamed/immune cold [93,94,98]. Spatial gene expression profiling of human PNSTs across the neurofibroma-MPNST continuum revealed that ANNUBPs exhibited enhanced signatures of antigen presentation and T-cell infiltration, while MPNST was characterized by immune-excluded TME with an increased expression of genes associated with immune exhaustion [37,99]. Interestingly, “benign” neurofibromas contiguous to MPNST, similarly harbored distinct gene expression profiles characterized by signatures of impaired antigen presentation. Of particular interest in this study, is the potential to utilize signatures of impaired antigen presentation to identify precursor lesions histopathologically classified as “benign”, at high risk of undergoing malignant transformation [37,99].
An intriguing question is what oncogenic mechanisms underly this immunosuppressive environment that appears indispensable for the malignant transformation of ANNUBP to MPNST. Changes in immune composition resulting from the loss of the PRC2 complex—a histone-modifying complex primarily responsible for maintaining transcriptional silencing through H3K27 trimethylation (H3K27me3)—represent a plausible hypothesis. Indeed, evidence suggests that loss of the PRC2 complex, a frequent occurrence in MPNST, is linked to reduced antigen presentation and altered immune signatures [4,10,37]. PRC2 loss is also associated with an aggressive disease course, proliferative signatures, and frequent gains of chr8q and triploid genomes in MPNST [4,37]. In a recent study classifying MPNSTs into two transcriptomic subtypes defined primarily by immune signatures and proliferative processes, 8q was reported among the most frequently affected by copy number gain among the “immune deficient” tumors (69% and 66%, respectively). Importantly, identified possible target genes included UBR5, among others on chr8q.
Similarly, PRC2 inactivation, resulted in ICB resistance through the reprograming of chromatin landscape, disrupting chemokine production, impairing antigen presentation and T-cell priming [10]. Consistent with these findings, Cortes-Ciriano et al reported by whole genome sequencing, coupled with transcriptomic and methylation profiling of 95 NF1-related tumors, a significant association between H3K27 trimethylation (H3K27me3) status and immune-phenotype[4]. H3K27me3 loss was strongly correlated with decreased infiltration of immune cells into the TME, downregulation of granzyme expression, and decreased activation of adaptive immunity. H3K27me3 retention on the other hand, was associated with an immune-cell rich phenotype [4].
Tumors with a high tumor mutational burden (TMB) are likely to harbor more neoantigens. As such, albeit some exceptions, higher TMB correlate positively with response to ICB in multiple cancers [18] with a definition of high TMB being >10 mutations per megabase [100]. Soft tissue sarcomas, in general, are reported to have a low TMB of 1–2.5 mutations/Mb [101]. However, in an analysis of 100,000 different cancers, 8.2% of MPNSTs had mutations of more than 20 mutations/Mb [102]. In a separate analysis of whole-exome sequencing data of 12 MPNST patient samples, somatic coding variants per tumor ranged from 7 to 472 with a median value of 63 [103].
While immunotherapy, particularly ICB therapy, has revolutionized the treatment of several malignancies, its efficacy in MPNST remains limited. The immunosuppressive tumor microenvironment driven by NF1 haploinsufficiency, and other pro-tumorigenic factors such as PRC2 loss, WNT/β-catenin, and Ras/PI3K signaling, pose significant challenges to effective immune-based treatments. Promising clinical trials targeting mast cells and TAMs, [(pexidartinib :KIT, CSF1R, and FLT3 inhibitor + sirolimus, an mTOR inhibitor) (NCT02584647)], underscore the potential for combination strategies, yet the immunosuppressive TME and variable response to PD-1/PD-L1 blockade, highlight the need for predictive biomarkers and strategies to augment MPNST immunogenicity. Deeper understanding of the genetic and molecular mechanisms underlying MPNST, including the role of Chr8q gain and UBR5 amplification in shaping the immune architecture and antitumor immune response will be crucial for overcoming immune evasion and improving outcomes in MPNST.
6. Attempts to Enhance MPNST Immunogenicity: Prospects for ICB
Curative treatment for MPNST typically involves surgical resection with negative margins, but this approach is often constrained by factors such as tumor size and proximity to nerves (Ref). For patients with recurrent, unresectable, or metastatic disease, treatment options are limited and outcomes are poor [6,7]. ICB therapy has shown promise in the treatment of inoperable and undruggable malignancies, suggesting potential benefits for MPNST patients [6,7]. As discussed previously, the success of ICB is dependent on an immune-rich TME. Unfortunately, evidence highlights MPNST to be non-inflamed “immune cold” malignancy, characterized by low immune cell infiltration, posing a significant challenge to the use of ICB therapy. Nonetheless, reports of anti-PD-1/PD-L1 success, albeit few, have prompted efforts to increase intratumoral T cell density to enhance MPNST immunogenicity.
A recent study harnessed the cGAS/STING/IFN pathway to augment the innate immune response [17]. The cytosolic DNA–sensing enzyme cGAS binds to dsDNA and initiates a cascade of events leading to the production of type I IFNs and proinflammatory cytokines and chemokines. This cytokine milieu presumably recruits immune cells into the TME. Using a GEMM of MPNST (cisNf1+/– p53+/–), STING agonist reprogramed the TME to enhance T cell infiltration and sensitized tumors to ICB therapy. Treatment of MPNSTs with a STING agonist caused activation of the STING pathway, upregulation of cytokines and chemokines, and infiltration of immune cells, including T cells, into the tumor. Consequently, enhanced tumor growth delay and significant apoptosis was observed upon STING activation followed by ICB therapy compared to STING agonist alone[17].
Other strategies have employed the use of oncolytic viruses. Recent reports have highlighted the potential of viral treatments to enhance immune infiltration in MPNSTs. Ghonime et al. demonstrated that a multimodal oncolytic virus engineered to express EphrinA2—an antigen found in various tumor types—can elicit a strong immune therapeutic response in immune-competent mouse models of both glioma and MPNST [104]. Additionally, a more recent study showed that intratumoral delivery of an inactivated modified vaccinia virus Ankara (MVA) can transform the immune desert environment in MPNSTs, promoting immune infiltration hence rendering tumors more responsive to ICB therapy[105]. In addition to these reports, further preclinical studies evaluating oncolytic virus therapy in MPNST are reviewed here [88].
A 2023 study demonstrated that dual targeting of Ras effector kinases, CDK4/6, and MEK, sensitized MPNST to ICB therapy. Dual inhibition of CDK4/6 and MEK demonstrated synergistic antitumor activity in patient-derived xenografts (PDX) and de novo tumors in immunocompetent mouse models of MPNST. PD-L1 blockade alone delayed tumor growth but caused little tumor regression compared to combination CDK4/6-MEK and PD-L1 inhibition. Resistant tumors were characterized by an immunosuppressive TME with elevated MHC II-low macrophages and increased tumor cell PD-L1 expression. On the other hand, in responsive tumors, CDK4/6-MEK inhibition led to an increase in plasma cells and cytotoxic T cell infiltration compared to resistant tumors[106]. These findings are particularly noteworthy given that tumor infiltrating plasma cells and B cells (TIL-Bs), along with the antibodies they produce are emerging as key components of the immune response against cancer[107]. Notably, plasma cell tumor infiltration has not been reported with the CDK4/6-MEK drug combination or the single agents in any tumor model including MPNST [106] . Emerging evidence suggests that intratumoral plasma cells may serve as a better predictor of ICB response in cancer patients[107,108]. TIL-Bs are believed to use a distinct mode of antigen presentation that fosters the creation and maintenance of immunologically ‘hot’ tumor microenvironments involving T cells, myeloid and natural killer cells. Additionally, by attenuating self-tolerance mechanisms, TIL-Bs minimize the extent of immune editing, a process tumors adopt to evade immune surveillance[109,110,111].
These preclinical studies among others (Table 2) highlight potential avenues to reprogram MPNST immune desert into a more immunologically active TME. The role of UBR5 in immune modulation as already discussed, presents another promising avenue for further research. Evidence from other cancers indicates that UBR5 promotes tumor growth by maintaining an immunosuppressive tumor microenvironment and facilitating immune evasion mediated by WNT/β-catenin, and PD-L1 among other pathways. Targeting UBR5 could enhance the immunogenicity of MPNSTs, improving their response to immune checkpoint blockade. Exploring the interaction between UBR5 and the tumor microenvironment in MPNST will provide valuable insights for developing effective immunotherapeutic strategies for this therapeutically challenging malignancy.
Conclusion/Future Directions
The aggressive and metastatic nature of MPNST coupled with limited effective treatment options, calls for novel therapeutic strategies to improve patient outcomes. To this end, preclinical and clinical studies have focused on the inhibition of the RAS/MEK/ERK and PI3K/AKT/mTOR pathways which play a crucial role in MPNST tumorigenesis and progression, although these trials have resulted in minimal clinical benefit [2,6,8].
There is a growing appreciation for the role of MPNST TME in formation and transformation of PNFs into MPNST. The role of RAS signaling and NF11haploinsufficient TME in PNF formation and malignant transformation has prompted the studies of immune and combinatorial strategies including targeting mast cells and TAMs. Notably, the phase I clinical trial combining pexidartinib with the mTOR inhibitor sirolimus, and the success thereof in MPNST trial patients, support this approach[13,89,90].
Isolated patient cases, albeit limited, have reported success with ICB therapy in patients with elevated tumor PD-L1 expression, suggesting potential for ICB therapy particularly in MPNST patients with inoperable or metastatic disease[20,21,98]. However, the immune cold nature of MPNST, presents a challenge to ICB therapy, emphasizing the need for strategies to enhance MPNST immunogenicity[17,19,93,98,105].
Genetic knockdown of the E3 ubiquitin ligase, UBR5, in preclinical models of cancer, has shown promise in attenuating tumor growth and enhancing anti-tumor immune response through mechanisms including increased antigen processing and presentation, elevated T cell infiltration, reduced TAMs recruitment and reduced PDL-1 mediated immune evasion[33,34,35]. Although further research is needed to substantiate the combined targeting of UBR5 and ICB therapy, the scattered success of ICB therapy coupled with evidence of UBR5’s role in immune evasion, suggest that this combination could address the unmet therapeutic needs of MPNST patients with recurrent, unresectable, or metastatic disease.
Author Contributions
Diana Akinyi Odhiambo (DAO): Conceptualization and writing; Selina Fan (SF): Compiled tables and figures; Angela C. Hirbe, MD, PhD (ACH): Editing and review.
Acknowledgments
DAO is funded by R01NS134815-02 through NINDS.
Conflicts of Interest
ACH has the following COI: Serving on the Advisory Board of Aadi Subsidiary, Inc., engaging in licensing activities with Boehringer Ingelheim GmbH, providing consulting services to Springworks Therapeutic Operating Company, PBC, receiving an honorarium from the American Physician Institute for Advanced Professional Studies, and receiving research funding for Tango.
References
- Yao C, Zhou H, Dong Y, Alhaskawi A, Hasan Abdullah Ezzi S, Wang Z, Lai J, Goutham Kota V, Hasan Abdulla Hasan Abdulla M and Lu H 2023 Malignant Peripheral Nerve Sheath Tumors: Latest Concepts in Disease Pathogenesis and Clinical Management Cancers (Basel) 15 1077. [CrossRef]
- González-Muñoz T, Kim A, Ratner N and Peinado H 2022 The Need for New Treatments Targeting MPNST: The Potential of Strategies Combining MEK Inhibitors with Antiangiogenic Agents Clinical Cancer Research 28 3185–95. [CrossRef]
- Stucky C-C H, Johnson K N, Gray R J, Pockaj B A, Ocal I T, Rose P S and Wasif N 2012 Malignant peripheral nerve sheath tumors (MPNST): the Mayo Clinic experience Ann Surg Oncol 19 878–85. [CrossRef]
- Cortes-Ciriano I, Steele C D, Piculell K, Al-Ibraheemi A, Eulo V, Bui M M, Chatzipli A, Dickson B C, Borcherding D C, Feber A, Galor A, Hart J, Jones K B, Jordan J T, Kim R H, Lindsay D, Miller C, Nishida Y, Proszek P Z, Serrano J, Sundby R T, Szymanski J J, Ullrich N J, Viskochil D, Wang X, Snuderl M, Park P J, Flanagan A M, Hirbe A C, Pillay N and Miller D T 2023 Genomic Patterns of Malignant Peripheral Nerve Sheath Tumor (MPNST) Evolution Correlate with Clinical Outcome and Are Detectable in Cell-Free DNA Cancer Discov 13 654–71. [CrossRef]
- Miettinen M M, Antonescu C R, Fletcher C D M, Kim A, Lazar A J, Quezado M M, Reilly K M, Stemmer-Rachamimov A, Stewart D R, Viskochil D, Widemann B and Perry A 2017 Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1—a consensus overview Human Pathology 67 1–10. [CrossRef]
- Prudner B C, Ball T, Rathore R and Hirbe A C 2020 Diagnosis and management of malignant peripheral nerve sheath tumors: Current practice and future perspectives Neurooncol Adv 2 i40–9. [CrossRef]
- Miller D T, Cortés-Ciriano I, Pillay N, Hirbe A C, Snuderl M, Bui M M, Piculell K, Al-Ibraheemi A, Dickson B C, Hart J, Jones K, Jordan J T, Kim R H, Lindsay D, Nishida Y, Ullrich N J, Wang X, Park P J and Flanagan A M 2020 Genomics of MPNST (GeM) Consortium: Rationale and Study Design for Multi-Omic Characterization of NF1-Associated and Sporadic MPNSTs Genes (Basel) 11 387. [CrossRef]
- Natalie Wu L M and Lu Q R 2019 Therapeutic targets for malignant peripheral nerve sheath tumors Future Neurology 14 FNL7. [CrossRef]
- Wang J, Pollard K, Allen A N, Tomar T, Pijnenburg D, Yao Z, Rodriguez F J and Pratilas C A 2020 Combined Inhibition of SHP2 and MEK Is Effective in Models of NF1-Deficient Malignant Peripheral Nerve Sheath Tumors Cancer Research 80 5367–79. [CrossRef]
- Yan J, Chen Y, Patel A J, Warda S, Lee C J, Nixon B G, Wong E W P, Miranda-Román M A, Yang N, Wang Y, Pachai M R, Sher J, Giff E, Tang F, Khurana E, Singer S, Liu Y, Galbo P M, Maag J L V, Koche R P, Zheng D, Antonescu C R, Deng L, Li M O, Chen Y and Chi P 2022 Tumor-intrinsic PRC2 inactivation drives a context-dependent immune-desert microenvironment and is sensitized by immunogenic viruses J Clin Invest 132. [CrossRef]
- Hirbe A C, Dahiya S, Friedmann-Morvinski D, Verma I M, Clapp D W and Gutmann D H 2016 Spatially- and temporally-controlled postnatal p53 knockdown cooperates with embryonic Schwann cell precursor Nf1 gene loss to promote malignant peripheral nerve sheath tumor formation Oncotarget 7 7403–14. [CrossRef]
- Magallon-Lorenz M, Terribas E, Fernández M, Requena G, Rosas I, Mazuelas H, Uriarte I, Negro A, Castellanos E, Blanco I, DeVries G, Kawashima H, Legius E, Brems H, Mautner V, Kluwe L, Ratner N, Wallace M, Rodriguez J F, Lázaro C, Fletcher J A, Reuss D, Carrió M, Gel B and Serra E 2022 A detailed landscape of genomic alterations in malignant peripheral nerve sheath tumor cell lines challenges the current MPNST diagnosis 2022.05.07.491026. [CrossRef]
- White E E and Rhodes S D 2024 The NF1+/- Immune Microenvironment: Dueling Roles in Neurofibroma Development and Malignant Transformation Cancers 16 994. [CrossRef]
- Staser K, Yang F-C and Clapp D W 2010 Mast cells and the neurofibroma microenvironment Blood 116 157–64. [CrossRef]
- Yang F-C, Ingram D A, Chen S, Zhu Y, Yuan J, Li X, Yang X, Knowles S, Horn W, Li Y, Zhang S, Yang Y, Vakili S T, Yu M, Burns D, Robertson K, Hutchins G, Parada L F and Clapp D W 2008 Nf1-dependent tumors require a microenvironment containing Nf1+/-- and c-kit-dependent bone marrow Cell 135 437–48. [CrossRef]
- Wei S C, Duffy C R and Allison J P 2018 Fundamental Mechanisms of Immune Checkpoint Blockade Therapy Cancer Discovery 8 1069–86. [CrossRef]
- Somatilaka B N, Madana L, Sadek A, Chen Z, Chandrasekaran S, McKay R M and Le L Q 2024 STING activation reprograms the microenvironment to sensitize NF1-related malignant peripheral nerve sheath tumors for immunotherapy J Clin Invest 134. [CrossRef]
- Johnson D B, Frampton G M, Rioth M J, Yusko E, Xu Y, Guo X, Ennis R C, Fabrizio D, Chalmers Z R, Greenbowe J, Ali S M, Balasubramanian S, Sun J X, He Y, Frederick D T, Puzanov I, Balko J M, Cates J M, Ross J S, Sanders C, Robins H, Shyr Y, Miller V A, Stephens P J, Sullivan R J, Sosman J A and Lovly C M 2016 Targeted Next Generation Sequencing Identifies Markers of Response to PD-1 Blockade Cancer Immunol Res 4 959–67. [CrossRef]
- Lee P R, Cohen J E and Fields R D 2006 Immune system evasion by peripheral nerve sheath tumor Neurosci Lett 397 126–9. [CrossRef]
- Özdemir B C, Bohanes P, Bisig B, Missiaglia E, Tsantoulis P, Coukos G, Montemurro M, Homicsko K and Michielin O 2019 Deep Response to Anti-PD-1 Therapy of Metastatic Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheath Tumor With CD274/PD-L1 Amplification JCO Precis Oncol 1–6. [CrossRef]
- Davis L E, Nicholls L A, Babiker H M, Liau J and Mahadevan D 2019 PD-1 Inhibition Achieves a Complete Metabolic Response in a Patient with Malignant Peripheral Nerve Sheath Tumor Cancer Immunology Research 7 1396–400. [CrossRef]
- Payandeh M, Sadeghi M and Sadeghi E 2017 Complete Response to Pembrolizumab in a Patient with Malignant Peripheral Nerve Sheath Tumor: The First Case Reported J App Pharm Sci 7, 182–4. [CrossRef]
- Clancy J L, Henderson M J, Russell A J, Anderson D W, Bova R J, Campbell I G, Choong D Y, Macdonald G A, Mann G J, Nolan T, Brady G, Olopade O I, Woollatt E, Davies M J, Segara D, Hacker N F, Henshall S M, Sutherland R L and Watts C K 2003 EDD, the human orthologue of the hyperplastic discs tumour suppressor gene, is amplified and overexpressed in cancer Oncogene 22 5070–81. [CrossRef]
- Shearer R F, Iconomou M, Watts C K W and Saunders D N 2015 Functional Roles of the E3 Ubiquitin Ligase UBR5 in Cancer Mol Cancer Res 13 1523–32. [CrossRef]
- Wang M, Dai W, Ke Z and Li Y 2020 Functional roles of E3 ubiquitin ligases in gastric cancer (Review) Oncology Letters 20 1–1. [CrossRef]
- Wang F, He Q, Zhan W, Yu Z, Finkin-Groner E, Ma X, Lin G and Li H 2023 Structure of the human UBR5 E3 ubiquitin ligase Structure 31 541-552.e4. [CrossRef]
- Wu B, Song M, Dong Q, Xiang G, Li J, Ma X and Wei F 2022 UBR5 promotes tumor immune evasion through enhancing IFN-γ-induced PDL1 transcription in triple negative breast cancer Theranostics 12 5086–102. [CrossRef]
- Taherbhoy A M and Daniels D L 2023 Harnessing UBR5 for targeted protein degradation of key transcriptional regulators Trends in Pharmacological Sciences 44 758–61. [CrossRef]
- Tsai J M, Aguirre J D, Li Y-D, Brown J, Focht V, Kater L, Kempf G, Sandoval B, Schmitt S, Rutter J C, Galli P, Sandate C R, Cutler J A, Zou C, Donovan K A, Lumpkin R J, Cavadini S, Park P M C, Sievers Q, Hatton C, Ener E, Regalado B D, Sperling M T, Słabicki M, Kim J, Zon R, Zhang Z, Miller P G, Belizaire R, Sperling A S, Fischer E S, Irizarry R, Armstrong S A, Thomä N H and Ebert B L 2023 UBR5 forms ligand-dependent complexes on chromatin to regulate nuclear hormone receptor stability Molecular Cell 83 2753-2767.e10. [CrossRef]
- Mark K G, Kolla S, Aguirre J D, Garshott D M, Schmitt S, Haakonsen D L, Xu C, Kater L, Kempf G, Martínez-González B, Akopian D, See S K, Thomä N H and Rapé M 2023 Orphan quality control shapes network dynamics and gene expression Cell 186 3460-3475.e23. [CrossRef]
- Hu B and Chen S 2024 The role of UBR5 in tumor proliferation and oncotherapy Gene 906 148258. [CrossRef]
- Xiang G, Wang S, Chen L, Song M, Song X, Wang H, Zhou P, Ma X and Yu J 2022 UBR5 targets tumor suppressor CDC73 proteolytically to promote aggressive breast cancer Cell Death Dis 13 1–14. [CrossRef]
- Wu B, Song M, Dong Q, Xiang G, Li J, Ma X and Wei F 2022 UBR5 promotes tumor immune evasion through enhancing IFN-γ-induced PDL1 transcription in triple negative breast cancer Theranostics 12 5086–102. [CrossRef]
- Liao L, Song M, Li X, Tang L, Zhang T, Zhang L, Pan Y, Chouchane L and Ma X 2017 E3 Ubiquitin Ligase UBR5 Drives the Growth and Metastasis of Triple-Negative Breast Cancer Cancer Research 77 2090–101. [CrossRef]
- Song M, Yeku O O, Rafiq S, Purdon T, Dong X, Zhu L, Zhang T, Wang H, Yu Z, Mai J, Shen H, Nixon B, Li M, Brentjens R J and Ma X 2020 Tumor derived UBR5 promotes ovarian cancer growth and metastasis through inducing immunosuppressive macrophages Nat Commun 11 6298. [CrossRef]
- Dehner C, Moon C I, Zhang X, Zhou Z, Miller C, Xu H, Wan X, Yang K, Mashl J, Gosline S J C, Wang Y, Zhang X, Godec A, Jones P A, Dahiya S, Bhatia H, Primeau T, Li S, Pollard K, Rodriguez F J, Ding L, Pratilas C A, Shern J F and Hirbe A C 2021 Chromosome 8 gain is associated with high-grade transformation in MPNST JCI Insight 6. [CrossRef]
- Høland M, Berg K C G, Eilertsen I A, Bjerkehagen B, Kolberg M, Boye K, Lingjærde O C, Guren T K, Mandahl N, Van Den Berg E, Palmerini E, Smeland S, Picci P, Mertens F, Sveen A and Lothe R A 2023 Transcriptomic subtyping of malignant peripheral nerve sheath tumours highlights immune signatures, genomic profiles, patient survival and therapeutic targets eBioMedicine 97 104829. [CrossRef]
- Akizuki Y, Kaypee S, Ohtake F and Ikeda F 2024 The emerging roles of non-canonical ubiquitination in proteostasis and beyond Journal of Cell Biology 223 e202311171. [CrossRef]
- Schnell J D and Hicke L 2003 Non-traditional functions of ubiquitin and ubiquitin-binding proteins J Biol Chem 278 35857–60. [CrossRef]
- Sun A, Tian X, Chen Y, Yang W and Lin Q 2023 Emerging roles of the HECT E3 ubiquitin ligases in gastric cancer Pathol. Oncol. Res. 29 1610931. [CrossRef]
- Wang Y, Argiles-Castillo D, Kane E I, Zhou A and Spratt D E 2020 HECT E3 ubiquitin ligases - emerging insights into their biological roles and disease relevance J Cell Sci 133 jcs228072. [CrossRef]
- Hodáková Z, Grishkovskaya I, Brunner H L, Bolhuis D L, Belačić K, Schleiffer A, Kotisch H, Brown N G and Haselbach D 2023 Cryo-EM structure of the chain-elongating E3 ubiquitin ligase UBR5 The EMBO Journal 42 e113348. [CrossRef]
- Singh S, Ng J and Sivaraman J 2021 Exploring the “Other” subfamily of HECT E3-ligases for therapeutic intervention Pharmacology & Therapeutics 224 107809. [CrossRef]
- Boughton A J, Krueger S and Fushman D 2020 Branching via K11 and K48 Bestows Ubiquitin Chains with a Unique Interdomain Interface and Enhanced Affinity for Proteasomal Subunit Rpn1 Structure 28 29-43.e6. [CrossRef]
- French M E, Koehler C F and Hunter T 2021 Emerging functions of branched ubiquitin chains Cell Discov 7 1–10. [CrossRef]
- Mansfield E, Hersperger E, Biggs J and Shearn A 1994 Genetic and Molecular Analysis of hyperplastic discs, a Gene Whose Product Is Required for Regulation of Cell Proliferation in Drosophila melanogaster Imaginal Discs and Germ Cells Developmental Biology 165 507–26. [CrossRef]
- Swenson S A, Gilbreath T J, Vahle H, Hynes-Smith R W, Graham J H, Law H C-H, Amador C, Woods N T, Green M R and Buckley S M 2020 UBR5 HECT domain mutations identified in mantle cell lymphoma control maturation of B cells Blood 136 299–312. [CrossRef]
- Li C G, Mahon C, Sweeney N M, Verschueren E, Kantamani V, Li D, Hennigs J K, Marciano D P, Diebold I, Abu-Halawa O, Elliott M, Sa S, Guo F, Wang L, Cao A, Guignabert C, Sollier J, Nickel N P, Kaschwich M, Cimprich K A and Rabinovitch M 2019 PPARγ Interaction with UBR5/ATMIN Promotes DNA Repair to Maintain Endothelial Homeostasis Cell Reports 26 1333-1343.e7. [CrossRef]
- Zhang T, Cronshaw J, Kanu N, Snijders A P and Behrens A 2014 UBR5-mediated ubiquitination of ATMIN is required for ionizing radiation-induced ATM signaling and function Proceedings of the National Academy of Sciences 111 12091–6. [CrossRef]
- Henderson M J, Munoz M A, Saunders D N, Clancy J L, Russell A J, Williams B, Pappin D, Khanna K K, Jackson S P, Sutherland R L and Watts C K W 2006 EDD Mediates DNA Damage-induced Activation of CHK2* Journal of Biological Chemistry 281 39990–40000. [CrossRef]
- Honda Y, Tojo M, Matsuzaki K, Anan T, Matsumoto M, Ando M, Saya H and Nakao M 2002 Cooperation of HECT-domain Ubiquitin Ligase hHYD and DNA Topoisomerase II-binding Protein for DNA Damage Response* Journal of Biological Chemistry 277 3599–605. [CrossRef]
- Munoz M A, Saunders D N, Henderson M J, Clancy J L, Russell A J, Lehrbach G, Musgrove E A, Watts C K W and Sutherland R L 2007 The E3 Ubiquitin Ligase EDD Regulates S-Phase and G2/M DNA Damage Checkpoints Cell Cycle 6 3070–7. [CrossRef]
- Gudjonsson T, Altmeyer M, Savic V, Toledo L, Dinant C, Grøfte M, Bartkova J, Poulsen M, Oka Y, Bekker-Jensen S, Mailand N, Neumann B, Heriche J-K, Shearer R, Saunders D, Bartek J, Lukas J and Lukas C 2012 TRIP12 and UBR5 Suppress Spreading of Chromatin Ubiquitylation at Damaged Chromosomes Cell 150 697–709. [CrossRef]
- Cipolla L, Bertoletti F, Maffia A, Liang C-C, Lehmann A R, Cohn M A and Sabbioneda S 2019 UBR5 interacts with the replication fork and protects DNA replication from DNA polymerase η toxicity Nucleic Acids Research 47 11268–83. [CrossRef]
- Mardis E R 2019 Neoantigens and genome instability: impact on immunogenomic phenotypes and immunotherapy response Genome Med 11 71. [CrossRef]
- Chan T A, Yarchoan M, Jaffee E, Swanton C, Quezada S A, Stenzinger A and Peters S 2019 Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic Annals of Oncology 30 44–56. [CrossRef]
- Xu Y, Nowsheen S and Deng M 2023 DNA Repair Deficiency Regulates Immunity Response in Cancers: Molecular Mechanism and Approaches for Combining Immunotherapy Cancers (Basel) 15 1619. [CrossRef]
- Matsuura K, Huang N-J, Cocce K, Zhang L and Kornbluth S 2017 Downregulation of the proapoptotic protein MOAP-1 by the UBR5 ubiquitin ligase and its role in ovarian cancer resistance to cisplatin Oncogene 36 1698–706. [CrossRef]
- Ohtake F, Tsuchiya H, Saeki Y and Tanaka K 2018 K63 ubiquitylation triggers proteasomal degradation by seeding branched ubiquitin chains Proc Natl Acad Sci U S A 115 E1401–8. [CrossRef]
- Yang Y, Zhao J, Mao Y, Lin G, Li F and Jiang Z 2020 UBR5 over-expression contributes to poor prognosis and tamoxifen resistance of ERa+ breast cancer by stabilizing β-catenin Breast Cancer Res Treat 184 699–710. [CrossRef]
- Qiao X, Liu Y, Prada M L, Mohan A K, Gupta A, Jaiswal A, Sharma M, Merisaari J, Haikala H M, Talvinen K, Yetukuri L, Pylvänäinen J W, Klefström J, Kronqvist P, Meinander A, Aittokallio T, Hietakangas V, Eilers M and Westermarck J 2020 UBR5 Is Coamplified with MYC in Breast Tumors and Encodes an Ubiquitin Ligase That Limits MYC-Dependent Apoptosis Cancer Research 80 1414–27. [CrossRef]
- Yu Z, Dong X, Song M, Xu A, He Q, Li H, Ouyang W, Chouchane L and Ma X 2024 Targeting UBR5 inhibits postsurgical breast cancer lung metastases by inducing CDC73 and p53 mediated apoptosis International Journal of Cancer 154 723–37. [CrossRef]
- Bradley A, Zheng H, Ziebarth A, Sakati W, Branham-O’Connor M, Blumer J B, Liu Y, Kistner-Griffin E, Rodriguez-Aguayo C, Lopez-Berestein G, Sood A K, Landen C N and Eblen S T 2014 EDD enhances cell survival and cisplatin resistance and is a therapeutic target for epithelial ovarian cancer Carcinogenesis 35 1100–9. [CrossRef]
- Li J, Zhang W, Gao J, Du M, Li H, Li M, Cong H, Fang Y, Liang Y, Zhao D, Xiang G, Ma X, Yao M, Tu H and Gan Y 2021 E3 Ubiquitin Ligase UBR5 Promotes the Metastasis of Pancreatic Cancer via Destabilizing F-Actin Capping Protein CAPZA1 Front Oncol 11 634167. [CrossRef]
- Subbaiah V K, Zhang Y, Rajagopalan D, Abdullah L N, Yeo-Teh N S L, Tomaić V, Banks L, Myers M P, Chow E K and Jha S 2016 E3 ligase EDD1/UBR5 is utilized by the HPV E6 oncogene to destabilize tumor suppressor TIP60 Oncogene 35 2062–74. [CrossRef]
- Yang M, Jiang N, Cao Q, Ma M and Sun Q 2016 The E3 ligase UBR5 regulates gastric cancer cell growth by destabilizing the tumor suppressor GKN1 Biochemical and Biophysical Research Communications 478 1624–9. [CrossRef]
- Wang J, Zhao X, Jin L, Wu G and Yang Y 2017 UBR5 Contributes to Colorectal Cancer Progression by Destabilizing the Tumor Suppressor ECRG4 Dig Dis Sci 62 2781–9. [CrossRef]
- Wu Q, Liu L, Feng Y, Wang L, Liu X and Li Y 2022 UBR5 promotes migration and invasion of glioma cells by regulating the ECRG4/NF-κB pathway J Biosci 47 45. [CrossRef]
- Hu X, Wang J, Chu M, Liu Y, Wang Z and Zhu X 2021 Emerging Role of Ubiquitination in the Regulation of PD-1/PD-L1 in Cancer Immunotherapy Mol Ther 29 908–19. [CrossRef]
- Layman A A K and Oliver P M 2016 Ubiquitin Ligases and Deubiquitinating Enzymes in CD4+ T Cell Effector Fate Choice and Function The Journal of Immunology 196 3975–82. [CrossRef]
- Li X-M, Zhao Z-Y, Yu X, Xia Q-D, Zhou P, Wang S-G, Wu H-L and Hu J 2023 Exploiting E3 ubiquitin ligases to reeducate the tumor microenvironment for cancer therapy Experimental Hematology & Oncology 12 34. [CrossRef]
- Hu X, Bian C, Zhao X and Yi T 2022 Efficacy evaluation of multi-immunotherapy in ovarian cancer: From bench to bed Front Immunol 13 1034903. [CrossRef]
- Siminiak N, Czepczyński R, Zaborowski M P and Iżycki D 2022 Immunotherapy in Ovarian Cancer Arch Immunol Ther Exp (Warsz) 70 19. [CrossRef]
- Rouzbahani E, Majidpoor J, Najafi S and Mortezaee K 2022 Cancer stem cells in immunoregulation and bypassing anti-checkpoint therapy Biomedicine & Pharmacotherapy 156 113906. [CrossRef]
- Han Y, Liu D and Li L 2020 PD-1/PD-L1 pathway: current researches in cancer Am J Cancer Res 10 727–42.
- Yi M, Niu M, Xu L, Luo S and Wu K 2021 Regulation of PD-L1 expression in the tumor microenvironment J Hematol Oncol 14 10. [CrossRef]
- Liu Q, Aminu B, Roscow O and Zhang W 2021 Targeting the Ubiquitin Signaling Cascade in Tumor Microenvironment for Cancer Therapy International Journal of Molecular Sciences 22 791. [CrossRef]
- Takeuchi Y, Tanegashima T, Sato E, Irie T, Sai A, Itahashi K, Kumagai S, Tada Y, Togashi Y, Koyama S, Akbay E A, Karasaki T, Kataoka K, Funaki S, Shintani Y, Nagatomo I, Kida H, Ishii G, Miyoshi T, Aokage K, Kakimi K, Ogawa S, Okumura M, Eto M, Kumanogoh A, Tsuboi M and Nishikawa H 2021 Highly immunogenic cancer cells require activation of the WNT pathway for immunological escape Science Immunology 6. [CrossRef]
- Knight S W E, Knight T E, Santiago T, Murphy A J and Abdelhafeez A H 2022 Malignant Peripheral Nerve Sheath Tumors—A Comprehensive Review of Pathophysiology, Diagnosis, and Multidisciplinary Management Children (Basel) 9 38. [CrossRef]
- Bethard J R, Zheng H, Roberts L and Eblen S T 2011 Identification of phosphorylation sites on the E3 ubiquitin ligase UBR5/EDD Journal of Proteomics 75 603–9. [CrossRef]
- Eblen S T, Kumar N V, Shah K, Henderson M J, Watts C K W, Shokat K M and Weber M J 2003 Identification of Novel ERK2 Substrates through Use of an Engineered Kinase and ATP Analogs* Journal of Biological Chemistry 278 14926–35. [CrossRef]
- Abeshouse A, Adebamowo C, Adebamowo S N, Akbani R, Akeredolu T, Ally A, Anderson M L, Anur P, Appelbaum E L, Armenia J, Auman J T, Bailey M H, Baker L, Balasundaram M, Balu S, Barthel F P, Bartlett J, Baylin S B, Behera M, Belyaev D, Bennett J, Benz C, Beroukhim R, Birrer M, Bocklage T, Bodenheimer T, Boice L, Bootwalla M S, Bowen J, Bowlby R, Boyd J, Brohl A S, Brooks D, Byers L, Carlsen R, Castro P, Chen H-W, Cherniack A D, Chibon F, Chin L, Cho J, Chuah E, Chudamani S, Cibulskis C, Cooper L A D, Cope L, Cordes M G, Crain D, Curley E, Danilova L, Dao F, Davis I J, Davis L E, Defreitas T, Delman K, Demchok J A, Demetri G D, Demicco E G, Dhalla N, Diao L, Ding L, DiSaia P, Dottino P, Doyle L A, Drill E, Dubina M, Eschbacher J, Fedosenko K, Felau I, Ferguson M L, Frazer S, Fronick C C, Fulidou V, Fulton L A, Fulton R S, Gabriel S B, Gao J, Gao Q, Gardner J, Gastier-Foster J M, Gay C M, Gehlenborg N, Gerken M, Getz G, Godwin A K, Godwin E M, Gordienko E, Grilley-Olson J E, Gutman D A, Gutmann D H, Hayes D N, Hegde A M, Heiman D I, Heins Z, Helsel C, Hepperla A J, Higgins K, Hoadley K A, et al 2017 Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas Cell 171 950-965.e28. [CrossRef]
- Alvegard T A, Berg N O, Baldetorp B, Fernö M, Killander D, Ranstam J, Rydholm A and Akerman M 1990 Cellular DNA content and prognosis of high-grade soft tissue sarcoma: the Scandinavian Sarcoma Group experience J Clin Oncol 8 538–47. [CrossRef]
- Brekke H R, Ribeiro F R, Kolberg M, Agesen T H, Lind G E, Eknaes M, Hall K S, Bjerkehagen B, van den Berg E, Teixeira M R, Mandahl N, Smeland S, Mertens F, Skotheim R I and Lothe R A 2010 Genomic changes in chromosomes 10, 16, and X in malignant peripheral nerve sheath tumors identify a high-risk patient group J Clin Oncol 28 1573–82. [CrossRef]
- Yang J, Ylipää A, Sun Y, Zheng H, Chen K, Nykter M, Trent J, Ratner N, Lev D C and Zhang W 2011 Genomic and Molecular Characterization of Malignant Peripheral Nerve Sheath Tumor Identifies the IGF1R Pathway as a Primary Target for Treatment Clinical Cancer Research 17 7563–73. [CrossRef]
- Hallqvist A, Rohlin A and Raghavan S 2020 Immune checkpoint blockade and biomarkers of clinical response in non–small cell lung cancer Scand J Immunol 92 e12980. [CrossRef]
- Huang A C and Zappasodi R 2022 A decade of checkpoint blockade immunotherapy in melanoma: understanding the molecular basis for immune sensitivity and resistance Nat Immunol 23 660–70. [CrossRef]
- Paudel S N, Hutzen B and Cripe T P 2023 The quest for effective immunotherapies against malignant peripheral nerve sheath tumors: Is there hope? Mol Ther Oncolytics 30 227–37. [CrossRef]
- Dodd R D, Lee C-L, Overton T, Huang W, Eward W C, Luo L, Ma Y, Ingram D R, Torres K E, Cardona D M, Lazar A J and Kirsch D G 2017 NF1+/− hematopoietic cells accelerate malignant peripheral nerve sheath tumor development without altering chemotherapy response Cancer Res 77 4486–97. [CrossRef]
- Manji G A, Van Tine B A, Lee S M, Raufi A G, Pellicciotta I, Hirbe A C, Pradhan J, Chen A, Rabadan R and Schwartz G K 2021 A Phase I Study of the Combination of Pexidartinib and Sirolimus to Target Tumor-Associated Macrophages in Unresectable Sarcoma and Malignant Peripheral Nerve Sheath Tumors Clinical Cancer Research 27 5519–27. [CrossRef]
- Martin E, Lamba N, Flucke U E, Verhoef C, Coert J H, Versleijen-Jonkers Y M H and Desar I M E 2019 Non-cytotoxic systemic treatment in malignant peripheral nerve sheath tumors (MPNST): A systematic review from bench to bedside Critical Reviews in Oncology/Hematology 138 223–32. [CrossRef]
- Larsson A T, Bhatia H, Calizo A, Pollard K, Zhang X, Conniff E, Tibbitts J F, Rono E, Cummins K, Osum S H, Williams K B, Crampton A L, Jubenville T, Schefer D, Yang K, Lyu Y, Pino J C, Bade J, Gross J M, Lisok A, Dehner C A, Chrisinger J S A, He K, Gosline S J C, Pratilas C A, Largaespada D A, Wood D K and Hirbe A C 2023 Ex vivo to in vivo model of malignant peripheral nerve sheath tumors for precision oncology Neuro Oncol 25 2044–57. [CrossRef]
- Haworth K B, Arnold M A, Pierson C R, Choi K, Yeager N D, Ratner N, Roberts R D, Finlay J L and Cripe T P 2017 Immune profiling of NF1-associated tumors reveals histologic subtype distinctions and heterogeneity: implications for immunotherapy Oncotarget 8 82037–48. [CrossRef]
- Bhandarkar A R, Bhandarkar S, Babovic-Vuksanovic D, Parney I F and Spinner R J 2023 Characterizing T-cell dysfunction and exclusion signatures in malignant peripheral nerve sheath tumors reveals susceptibilities to immunotherapy J Neurooncol 164 693–9. [CrossRef]
- Negrao M V, Lam V K, Reuben A, Rubin M L, Landry L L, Roarty E B, Rinsurongkawong W, Lewis J, Roth J A, Swisher S G, Gibbons D L, Wistuba I I, Papadimitrakopoulou V, Glisson B S, Blumenschein G R, Lee J J, Heymach J V and Zhang J 2019 PD-L1 Expression, Tumor Mutational Burden, and Cancer Gene Mutations Are Stronger Predictors of Benefit from Immune Checkpoint Blockade than HLA Class I Genotype in Non–Small Cell Lung Cancer Journal of Thoracic Oncology 14 1021–31. [CrossRef]
- Lei Y, Li X, Huang Q, Zheng X and Liu M 2021 Progress and Challenges of Predictive Biomarkers for Immune Checkpoint Blockade Front Oncol 11 617335. [CrossRef]
- Budczies J, Mechtersheimer G, Denkert C, Klauschen F, Mughal S S, Chudasama P, Bockmayr M, Jöhrens K, Endris V, Lier A, Lasitschka F, Penzel R, Dietel M, Brors B, Gröschel S, Glimm H, Schirmacher P, Renner M, Fröhling S and Stenzinger A 2017 PD-L1 (CD274) copy number gain, expression, and immune cell infiltration as candidate predictors for response to immune checkpoint inhibitors in soft-tissue sarcoma Oncoimmunology 6 e1279777. [CrossRef]
- Shurell E, Singh A S, Crompton J G, Jensen S, Li Y, Dry S, Nelson S, Chmielowski B, Bernthal N, Federman N, Tumeh P and Eilber F C 2016 Characterizing the immune microenvironment of malignant peripheral nerve sheath tumor by PD-L1 expression and presence of CD8+ tumor infiltrating lymphocytes Oncotarget 7 64300–8. [CrossRef]
- Mitchell D K, Burgess B, White E E, Smith A E, Sierra Potchanant E A, Mang H, Hickey B E, Lu Q, Qian S, Bessler W, Li X, Jiang L, Brewster K, Temm C, Horvai A, Albright E A, Fishel M L, Pratilas C A, Angus S P, Clapp D W and Rhodes S D 2024 Spatial Gene-Expression Profiling Unveils Immuno-oncogenic Programs of NF1-Associated Peripheral Nerve Sheath Tumor Progression Clinical Cancer Research OF1–16. [CrossRef]
- Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, Chung H C, Kindler H L, Lopez-Martin J A, Miller W H, Italiano A, Kao S, Piha-Paul S A, Delord J-P, McWilliams R R, Fabrizio D A, Aurora-Garg D, Xu L, Jin F, Norwood K and Bang Y-J 2020 Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study The Lancet Oncology 21 1353–65. [CrossRef]
- Roulleaux Dugage M, Nassif E F, Italiano A and Bahleda R 2021 Improving Immunotherapy Efficacy in Soft-Tissue Sarcomas: A Biomarker Driven and Histotype Tailored Review Front Immunol 12 775761. [CrossRef]
- Chalmers Z R, Connelly C F, Fabrizio D, Gay L, Ali S M, Ennis R, Schrock A, Campbell B, Shlien A, Chmielecki J, Huang F, He Y, Sun J, Tabori U, Kennedy M, Lieber D S, Roels S, White J, Otto G A, Ross J S, Garraway L, Miller V A, Stephens P J and Frampton G M 2017 Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden Genome Medicine 9 34. [CrossRef]
- Brohl A S, Kahen E, Yoder S J, Teer J K and Reed D R 2017 The genomic landscape of malignant peripheral nerve sheath tumors: diverse drivers of Ras pathway activation Sci Rep 7 14992. [CrossRef]
- Ghonime M G, Saini U, Kelly M C, Roth J C, Wang P-Y, Chen C-Y, Miller K, Hernandez-Aguirre I, Kim Y, Mo X, Stanek J R, Cripe T, Mardis E and Cassady K A 2021 Eliciting an immune-mediated antitumor response through oncolytic herpes simplex virus-based shared antigen expression in tumors resistant to viroimmunotherapy J Immunother Cancer 9 e002939. [CrossRef]
- Yan J, Chen Y, Patel A J, Warda S, Nixon B G, Wong E W P, Miranda-Román M A, Lee C J, Yang N, Wang Y, Sher J, Giff E, Tang F, Khurana E, Singer S, Liu Y, Galbo P M, Maag J L, Koche R P, Zheng D, Deng L, Antonescu C R, Li M, Chen Y and Chi P 2022 Tumor-intrinsic PRC2 inactivation drives a context-dependent immune-desert tumor microenvironment and confers resistance to immunotherapy. [CrossRef]
- Kohlmeyer J L, Lingo J J, Kaemmer C A, Scherer A, Warrier A, Voigt E, Raygoza Garay J A, McGivney G R, Brockman Q R, Tang A, Calizo A, Pollard K, Zhang X, Hirbe A C, Pratilas C A, Leidinger M, Breheny P, Chimenti M S, Sieren J C, Monga V, Tanas M R, Meyerholz D K, Darbro B W, Dodd R D and Quelle D E 2023 CDK4/6-MEK Inhibition in MPNSTs Causes Plasma Cell Infiltration, Sensitization to PD-L1 Blockade, and Tumor Regression Clinical Cancer Research 29 3484–97. [CrossRef]
- Laumont C M, Banville A C, Gilardi M, Hollern D P and Nelson B H 2022 Tumour-infiltrating B cells: immunological mechanisms, clinical impact and therapeutic opportunities Nat Rev Cancer 22 414–30. [CrossRef]
- Zhu H, Xu J, Wang W, Zhang B, Liu J, Liang C, Hua J, Meng Q, Yu X and Shi S 2024 Intratumoral CD38+CD19+B cells associate with poor clinical outcomes and immunosuppression in patients with pancreatic ductal adenocarcinoma eBioMedicine 103. [CrossRef]
- Playoust E, Remark R, Vivier E and Milpied P 2023 Germinal center-dependent and -independent immune responses of tumor-infiltrating B cells in human cancers Cell Mol Immunol 20 1040–50. [CrossRef]
- Sanz I, Wei C, Jenks S A, Cashman K S, Tipton C, Woodruff M C, Hom J and Lee F E-H 2019 Challenges and Opportunities for Consistent Classification of Human B Cell and Plasma Cell Populations Front. Immunol. 10. [CrossRef]
- Xu-Monette Z Y, Li Y, Snyder T, Yu T, Lu T, Tzankov A, Visco C, Bhagat G, Qian W, Dybkaer K, Chiu A, Tam W, Zu Y, Hsi E D, Hagemeister F B, Wang Y, Go H, Ponzoni M, Ferreri A J M, Møller M B, Parsons B M, Fan X, van Krieken J H, Piris M A, Winter J N, Au Q, Kirsch I, Zhang M, Shaughnessy J, Xu B and Young K H 2023 Tumor-Infiltrating Normal B Cells Revealed by Immunoglobulin Repertoire Clonotype Analysis Are Highly Prognostic and Crucial for Antitumor Immune Responses in DLBCL Clinical Cancer Research 29 4808–21. [CrossRef]

Table 1.
Immune based strategies in MPNST: Clinical trials.
| No. | Study Name | Phase | Agent | Clinical Trial No. | Modality | Status/Findings | Source |
|---|---|---|---|---|---|---|---|
| 1. | PLX3397 Plus Sirolimus in Unresectable Sarcoma and Malignant Peripheral Nerve Sheath Tumors (PLX3397) | I & II | PLX3397, Sirolimus | NCT02584647 | Small-Molecule Inhibitors | Active, not recruiting; patient enrollment: 43; interventional model: parallel assignment Clinical benefit was observed in 12 of 18 (67%) evaluable subjects with 3 partial responses (all in TGCT) and 9 stable disease. Tissue staining indicated a decreased proportion of activated M2 macrophages within tumor samples with treatment. Manji GA, Van Tine BA, Lee SM, et al. A Phase I Study of the Combination of Pexidartinib and Sirolimus to Target Tumor-Associated Macrophages in Unresectable Sarcoma and Malignant Peripheral Nerve Sheath Tumors. Clin Cancer Res. 2021;27(20):5519-5527. doi:10.1158/1078-0432.CCR-21-1779 |
Paudel SN, Hutzen B, Cripe TP. The quest for effective immunotherapies against malignant peripheral nerve sheath tumors: Is there hope?. Mol Ther Oncolytics. 2023;30:227-237. Published 2023 Jul 31. doi:10.1016/j.omto.2023.07.008 |
| 2. | Vaccine Therapy in Treating Patients With Malignant Peripheral Nerve Sheath Tumor That Is Recurrent or Cannot Be Removed by Surgery | I | Edmonston strain measles virus genetically engineered to express neurofibromatosis type 1 (oncolytic measles virus encoding thyroidal sodium-iodide symporter [MV-NIS]) | NCT02700230 | Oncolytic virus | Completed; patient enrollment: 9; interventional model: single group assignment | Paudel SN, Hutzen B, Cripe TP. The quest for effective immunotherapies against malignant peripheral nerve sheath tumors: Is there hope?. Mol Ther Oncolytics. 2023;30:227-237. Published 2023 Jul 31. doi:10.1016/j.omto.2023.07.008 |
| 3. | Neoadjuvant Nivolumab Plus Ipilimumab for Newly Diagnosed Malignant Peripheral Nerve Sheath Tumor | I | nivolumab and ipilimumab | NCT04465643 | Immune modulators (PD-1 and CTLA-4) | Recruiting; patient enrollment: 18; interventional model: single group assignment | Paudel SN, Hutzen B, Cripe TP. The quest for effective immunotherapies against malignant peripheral nerve sheath tumors: Is there hope?. Mol Ther Oncolytics. 2023;30:227-237. Published 2023 Jul 31. doi:10.1016/j.omto.2023.07.008 |
| 4. | A Study of APG-115 in Combination With Pembrolizumab in Patients With Metastatic Melanomas or Advanced Solid Tumors | Ib/II | APG-115 and pembrolizumab | NCT03611868 | Immune modulator (PD-1) and small molecule inhibitor | Recruiting Interim results show stability in 53% of MPNST cohort when treated for > 4 cycles Somaiah N, Van Tine BA, Chmielowski B, et al. A phase 2 study of alrizomadlin, a novel MDM2/p53 inhibitor, in combination with pembrolizumab for treatment of patients with malignant peripheral nerve sheath tumor (MPNST). Journal of Clinical Oncology. 2023;41(16_suppl):e14627. doi:10.1200/jco.2023.41.16_suppl.e14627 |
Paudel SN, Hutzen B, Cripe TP. The quest for effective immunotherapies against malignant peripheral nerve sheath tumors: Is there hope?. Mol Ther Oncolytics. 2023;30:227-237. Published 2023 Jul 31. doi:10.1016/j.omto.2023.07.008 |
| 5. | B7H3 CAR-T Cell Immunotherapy for Recurrent/Refractory Solid Tumors in Children and Young Adults | I | Biological: second generation 4-1BBζ B7H3-EGFRt-DHFR (B7H3-specific CAR-T cells and CD19 specific CAR-T cells) biological: second generation 4-1BBζ B7H3-EGFRt-DHFR (selected) and a second generation 4-1BBζ CD19-Her2tG | NCT04483778 | CAR-T cells | Active, not recruiting | Paudel SN, Hutzen B, Cripe TP. The quest for effective immunotherapies against malignant peripheral nerve sheath tumors: Is there hope?. Mol Ther Oncolytics. 2023;30:227-237. Published 2023 Jul 31. doi:10.1016/j.omto.2023.07.008 |
| 6. | EGFR806 CAR-T Cell Immunotherapy for Recurrent/Refractory Solid Tumors in Children and Young Adults | I | Biological: second generation 4-1BBζ EGFR806-EGFRt biological: second generation 4-1BBζ EGFR806-EGFRt and a second generation 4 1BBζ CD19-Her2tG | NCT03618381 | CAR-T cells | Recruiting | Paudel SN, Hutzen B, Cripe TP. The quest for effective immunotherapies against malignant peripheral nerve sheath tumors: Is there hope?. Mol Ther Oncolytics. 2023;30:227-237. Published 2023 Jul 31. doi:10.1016/j.omto.2023.07.008 |
| 7. | Donor Stem Cell Transplant After Chemotherapy for the Treatment of Recurrent or Refractory High-Risk Solid Tumors in Pediatric and Adolescent-Young Adults | II | Allogeneic hematopoietic stem cell transplantation | NCT04530487 | Stem cell transfer | Terminated | Paudel SN, Hutzen B, Cripe TP. The quest for effective immunotherapies against malignant peripheral nerve sheath tumors: Is there hope?. Mol Ther Oncolytics. 2023;30:227-237. Published 2023 Jul 31. doi:10.1016/j.omto.2023.07.008 |
| 8. | Nivolumab and Ipilimumab in Treating Patients With Rare Tumors | II | Nivolumab and Ipilimumab | NCT02834013 | Immune modulators (PD-1 and CTLA-4) | Active, not recruiting | Paudel SN, Hutzen B, Cripe TP. The quest for effective immunotherapies against malignant peripheral nerve sheath tumors: Is there hope?. Mol Ther Oncolytics. 2023;30:227-237. Published 2023 Jul 31. doi:10.1016/j.omto.2023.07.008 |
| 9. | A Study of Pembrolizumab in Patients With Malignant Peripheral Nerve Sheath Tumor (MPNST), Not Eligible for Curative Surgery | II | Pembrolizumab | NCT02691026 | Immune modulators | Terminated | |
| 10. | Lorvotuzumab Mertansine in Treating Younger Patients With Relapsed or Refractory Wilms Tumor, Rhabdomyosarcoma, Neuroblastoma, Pleuropulmonary Blastoma, Malignant Peripheral Nerve Sheath Tumor, or Synovial Sarcoma | II | Lorvotuzumab mertansine | NCT02452554 | Antibody-drug conjugates | Clinical activity limited Geller JI, Pressey JG, Smith MA, et al. ADVL1522: A phase 2 study of lorvotuzumab mertansine (IMGN901) in children with relapsed or refractory wilms tumor, rhabdomyosarcoma, neuroblastoma, pleuropulmonary blastoma, malignant peripheral nerve sheath tumor, or synovial sarcoma-A Children's Oncology Group study. Cancer. 2020;126(24):5303-5310. doi:10.1002/cncr.33195 |
|
| 11. | BLESSED: Expanded Access for DeltaRex-G for Advanced Pancreatic Cancer, Sarcoma and Carcinoma of Breast | I/II | DeltaRex-G | NCT04091295 | Tumor targeted gene therapy, human cyclin G1 inhibitor | Available Case report: 14-year-old female with MPNST of parotid gland with lung metastases Previous treatment include: chemotherapies (doxorubicin, ifosfamide, temozolomide, sorafenib), and immunotherapy with interleukin-2. Received intravenous Rexin-G for 2 years. Outcome: no evidence of active disease Kim S, Federman N, Gordon EM, Hall FL, Chawla SP. Rexin-G®, a tumor-targeted retrovector for malignant peripheral nerve sheath tumor: A case report. Mol Clin Oncol. 2017;6(6):861-865. doi:10.3892/mco.2017.1231 |
|
| 12. | HSV1716 in Patients With Non-Central Nervous System (Non-CNS) Solid Tumors | I | HSV1716 | NCT00931931 | Oncolytic virus | Completed | |
| 13. | MASCT-I Combined With Doxorubicin and Ifosfamide for First-line Treatment of Advanced Soft Tissue Sarcoma | II | MASCT-I (dendritic cells and effector T cells injections) with Doxorubicin and Ifosfamide | NCT06277154 | MASCT-I | Not yet recruiting | |
| 14. | Nivolumab and BO-112 Before Surgery for the Treatment of Resectable Soft Tissue Sarcoma | I | BO-112, and BO-112 with nivolumab for patients going through preoperative radiotherapy for definitive surgical resection | NCT04420975 | dsRNA immunotherapy | Active, not recruiting |
Table 2.
Immune based strategies in MPNST: Preclinical studies.
| No. | Study Name | Agent | Modality | Source |
|---|---|---|---|---|
| 1. | Treatment of orthotopic malignant peripheral nerve sheath tumors with oncolytic herpes simplex virus | Oncolytic herpes simplex viruses (oHSVs) | Oncolytic virus | Antoszczyk S, Spyra M, Mautner VF, et al. Treatment of orthotopic malignant peripheral nerve sheath tumors with oncolytic herpes simplex virus. Neuro Oncol. 2014;16(8):1057-1066. doi:10.1093/neuonc/not317 |
| 2. | Aurora A kinase inhibition enhances oncolytic herpes virotherapy through cytotoxic synergy and innate cellular immune modulation | Alisertib and HSV1716 | Oncolytic virus | Currier MA, Sprague L, Rizvi TA, et al. Aurora A kinase inhibition enhances oncolytic herpes virotherapy through cytotoxic synergy and innate cellular immune modulation. Oncotarget. 2017;8(11):17412-17427. doi:10.18632/oncotarget.14885 |
| 3 | Dominant-Negative Fibroblast Growth Factor Receptor Expression Enhances Antitumoral Potency of Oncolytic Herpes Simplex Virus in Neural Tumors | Oncolytic HSV with dominant-negative FGF receptor (dnFGFR) | Oncolytic virus | Liu TC, Zhang T, Fukuhara H, et al. Dominant-negative fibroblast growth factor receptor expression enhances antitumoral potency of oncolytic herpes simplex virus in neural tumors. Clin Cancer Res. 2006;12(22):6791-6799. doi:10.1158/1078-0432.CCR-06-0263 |
| 4. | Oncolytic HSV Armed with Platelet Factor 4, an Antiangiogenic Agent, Shows Enhanced Efficacy | Oncolytic HSV with insertion of transgene platelet factor 4 (PF4) | Oncolytic virus | Liu TC, Zhang T, Fukuhara H, et al. Oncolytic HSV armed with platelet factor 4, an antiangiogenic agent, shows enhanced efficacy. Mol Ther. 2006;14(6):789-797. doi:10.1016/j.ymthe.2006.07.011 |
| 5. | Oncolytic HSV and Erlotinib Inhibit Tumor Growth and Angiogenesis in a Novel Malignant Peripheral Nerve Sheath Tumor Xenograft Model | oHSV mutants (G207 and hrR3) with erlotinib (EGFR inhibitor) | Oncolytic virus | Mahller YY, Vaikunth SS, Currier MA, et al. Oncolytic HSV and erlotinib inhibit tumor growth and angiogenesis in a novel malignant peripheral nerve sheath tumor xenograft model. Mol Ther. 2007;15(2):279-286. doi:10.1038/sj.mt.6300038 |
| 6. | Molecular analysis of human cancer cells infected by an oncolytic HSV-1 reveals multiple upregulated cellular genes and a role for SOCS1 in virus replication | G207 oHSV | Oncolytic virus | Mahller YY, Sakthivel B, Baird WH, et al. Molecular analysis of human cancer cells infected by an oncolytic HSV-1 reveals multiple upregulated cellular genes and a role for SOCS1 in virus replication. Cancer Gene Ther. 2008;15(11):733-741. doi:10.1038/cgt.2008.40 |
| 7. | Molecular engineering and validation of an oncolytic herpes simplex virus type 1 transcriptionally targeted to midkine-positive tumors | oHSV fused with human MDK promoter to the HSV type 1 neurovirulence gene, γ134.5 | Oncolytic virus | Maldonado AR, Klanke C, Jegga AG, et al. Molecular engineering and validation of an oncolytic herpes simplex virus type 1 transcriptionally targeted to midkine-positive tumors. J Gene Med. 2010;12(7):613-623. doi:10.1002/jgm.1479 |
| 8. | Oncolytic measles virus as a novel therapy for malignant peripheral nerve sheath tumors | Engineered MV Edmonston vaccine strain with human sodium iodide symporter | Oncolytic measles virus therapy | Deyle DR, Escobar DZ, Peng KW, Babovic-Vuksanovic D. Oncolytic measles virus as a novel therapy for malignant peripheral nerve sheath tumors. Gene. 2015;565(1):140-145. doi:10.1016/j.gene.2015.04.001 |
| 9. | STING activation reprograms the microenvironment to sensitize NF1-related malignant peripheral nerve sheath tumors for immunotherapy | Activating simulator of IFN genes (STING) signaling with ICB | ICB + tumor microenvironment | Somatilaka BN, Madana L, Sadek A, et al. STING activation reprograms the microenvironment to sensitize NF1-related malignant peripheral nerve sheath tumors for immunotherapy. J Clin Invest. 2024;134(10):e176748. Published 2024 Mar 19. doi:10.1172/JCI176748 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



