
Submitted:
14 October 2024
Posted:
15 October 2024
You are already at the latest version
Abstract
Backgroud
In eukaryotes with a double-stranded linear DNA genome, the loss of terminal DNA during replication is inevitable due to an end-replication problem and telomeres serve as a buffer against DNA loss. Cumulative telomere attrition leads to replicative senescence and the Hayflick limit. Thus, activation of the telomere maintenance mechanism (TMM) is a prerequisite for malignant transformation. We show that extracellular, but not intracellular, signals activate RAD52-dependent alternative lengthening of telomeres (ALT) during malignant transformation of malignant peripheral nerve sheath tumors (MPNSTs).
Methods
We compared neurofibroma (NF, benign) and MPNST occurring in the same patient with type-1 neurofibromatosis, where each NF-MPNST pair shares the same genetic background and differentiation lineage minimizing genetic bias and contrasts only changes related to malignant transformation.
Results
We found that the MEGF11 and the MEGF11-dependent ephrin signaling pathways (EFNA5-EPHPA6-PDZD9/PARD6B) activate the NELL2-PAX7 transcriptional cascade, which sequentially activates RAD52, the recombinase of RAD52-dependent ALT, resulting in telomere elongation, metastasis, and poor prognosis. In contrast, H2AFX, a DNA damage marker and intracellular signal, activates RAD51-dependent ALT to determine the MPNST grade.
Conclusions
We demonstrated the extracellular signals that activate RAD52-dependent ALT and distinguished the biological and clinical manifestations of RAD51 and RAD52-dependent ALT during malignant transformation of MPNSTs.
Keywords:
telomere
; MEGF11
; ephrin
; NELL2
; PAX7
; RAD52‐dependent ALT
Introduction
Telomeres are essential structure on the ends of chromosomes of eukaryotes with double-stranded linear DNA genomes and assist in overcoming end-replication problems[1]. Due to the 5′ to 3′ unidirectionality of DNA polymerization, an Okazaki fragment with an RNA primer is required on lagging strands[2] and DNA loss corresponding to the terminal RNA primer on the lagging strand is inevitable[1], leading to replicative senescence. The cumulative effects of DNA loss in DNA replication are responsible for the Hayflick limit[3]. Telomeres are located at both ends of the chromosomes and serve as a buffer against DNA loss through DNA replication and maintain genomic stability. Cumulative telomere attrition eventually causes the Hayflick limit[4].
Although benign and malignant tumors are clinically distinguished by their metastatic capability, there are distinct differences at the cellular level, which are reflected in the histologic grade. The Fédération Nationale des Centres de Lutte Contre Le Cancer (FNCLCC) grading system consists of three categories: tumor differentiation, mitotic count, and tumor necrosis[5,6]. The mitotic count is the underlying determinant for tumor dedifferentiation and necrosis, and unlimited mitosis is the most fundamental cellular feature of malignant tumors. The activation of the telomere maintenance mechanism (TMM) is essential for overcoming the Hayflick limit and achieving immortalization. The first identified TMM was a type of reverse transcriptase, later called telomerase[7,8]. Human telomerase is composed of TERT, TERC, DKC1, and TEP1. Shelterin, which includes TRF1, TRF2, TPP1, POT1, TIN2, and RAP1 supports telomerase recruitment to telomeres and the protection of telomeres by forming secondary telomeric structures such as T and D-loops[9]. H/ACA snoRNP complexes associated with DKC1, NHP2, NIP10, GAR1, and NAF1 together with hTERT constitute the catalytic core of telomerase[10]. Approximately, 85 to 90% of human cancers exhibit increased telomerase activity, whereas the remaining 10 to 15% lack telomerase activity and adopt alternative lengthening of telomeres (ALT)[11,12,13]. In addition to ALT, which is based on homologous recombination (HR), break-induced replication (BIR)[14,15,16], and mitotic DNA synthesis (MiDAS)[15] have also been reported to be associated mechanisms by which RAD52-dependent ALT overcomes deficient telomerase activity.
Type-1 neurofibromatosis (NF-1) is an autosomal dominant disorder of a tumor suppressor gene, neurofibromin 1, located on 17q11.2, which encodes a GTPase-activating protein involved in the RAS/MAPK pathway[17]. The average global prevalence of NF1 is approximately one in 3,000 individuals, and individuals harboring NF1 have a life expectancy of eight to 21 years inferior to the general population[18]. The most fatal clinical manifestation of NF1 is the malignant peripheral nerve sheath tumor (MPNST), which is malignant transformation of a preexisting neurofibromas. The cumulative lifetime risk for malignant transformation in NF1 patients is estimated to be approximately 8-13%. MPNSTs are rare sarcomas that account for approximately 5-10% of all soft tissue sarcomas. Notably, both telomerase activity and ALT are observed in MPNSTs, instead of ALT replacing telomerase activity[19,20], which indicates that MPNSTs are optimal tumors for the comprehensive analysis of the TMM.
The ALT is a major TMM of malignant tumors originating from the mesenchyme and neuroepithelium. It has been reported that there are two major independent ALT pathways with distinct mechanisms and times of action: RAD51- and RAD52-dependent ALT. RAD51-dependent ALT promotes semiconservative homologous recombination (HR) with high fidelity[21], whereas RAD52-dependent ALT promotes conservative HR as well as breaks-induced replication-related DNA synthesis[22,23]. This conservative DNA synthesis mechanism is observed only in the late phase of the cell cycle, G2/M[14,15]. These findings suggest that RAD51- and RAD52-dependent ALT are regulated independently of each other. Since the molecular mechanisms of ALT share the DNA repair process and DNA damage signals, DNA damage signaling has been characterized as an initiator of ALT. Indeed, the association between the DNA damage response and RAD51-dependent ALT, has been well documented[24,25]. However, the activation signals for RAD52-dependent ALT have not been elucidated and remain an outstanding question in telomere biology.
Cancer is not a simple aggregate of transformed cells but is formed by complex interactions between them and the tumor microenvironment (TME). Currently, genetic variations in protooncogenes and tumor suppressor genes are no longer sufficient to explain oncogenesis even at an early stage. The interactions between transformed cells and the TME, which occur via growth factors and cytokines such as EGF, VEGF, PDGF, and TGF-β secreted by cellular components in the TME, are considered necessary for cancer development and progression[26]. Furthermore, the TME has been reported to play important roles in metastasis as well as oncogenesis and has recently become an anticancer target[26,27,28]. These results suggest that the TME is a repository of extracellular signals related to oncogenesis, some of which can induce of ALT activation.
We hypothesized that extracellular signals beyond intracellular DNA damage signals activate the TMM. New strategies were employed in this study as follows. First, we designed a genetic analysis model conceptually similar to a “subtractive cDNA library”. We focused on MPNSTs associated with NF1 and compared MPNSTs and NFs from the same NF1 patient (Figure 1A). Since MPNSTs associated with NF1 arise from preexisting NFs, each NF-MPNST pair in this model shared the same genetic background and differentiation lineage, minimizing bias due to genetic variation and differentiation. Furthermore, the comparison of each NF-MPNST pair contrasts only malignant transformation-related changes, structurally subtracting changes from the normal cell to the benign tumor (Figure 1B). Second, we designed a combined statistical analysis method in which we analyzed the interaction effects on the telomere length as well as the transcriptional relationships for all the gene pairs, distinguishing the TMM-related transcriptional relationships and filtering out those that are unrelated (Figure 1C). A total of 20 NF-MPNST pairs from 20 NF1 patients were analyzed to evaluate the activation of the TMM during MPNST malignant transformation. Whole transcriptome sequencing (WTS) was performed to profile genome-wide gene expression and whole genome sequencing (WGS) was performed to measure the telomere length.
Materials and Methods
Patients and Samples
We analyzed 20 patients with NF-1 who underwent surgery for both NFs and MPNSTs from November 2000 to July 2017. The clinical characteristics of the patients and tumors (NFs and MPNSTs) are described (Supplementary information 1). Only four NF-MPNST pairs were fresh frozen tissues, and the remaining 17 NF-MPNST pairs were formalin-fixed paraffin-embedded (FFPE) tissues. FFPE samples were prepared as slides for histopathological qualification.
Whole Genome Sequencing (WGS)
The integrity of the genomic DNA was checked by agarose gel electrophoresis and gDNA was quantified using Quant-IT PicoGreen (Invitrogen, Waltham, Massachusetts, USA). The sequencing libraries were prepared according to the instructions of the TruSeq DNA Nano Library Prep Kit. Briefly, genomic DNA (100 ng) was fragmented using adaptive focused acoustic technology (Covaris) and end-repaired to create 5-phosphorylated blunt-ended dsDNA molecules. Following end-repair, the size of the DNA was selected via a bead-based method. DNA fragments were further processed by the addition of a single ‘A’ base, and ligation of TruSeq indexing adapters. The purified libraries were quantified using qPCR according to the qPCR Quantification Protocol Guide (KAPA Library Quantification Kit for Illumina sequencing platforms) and quantified using an Agilent Technologies 2200 TapeStation (Agilent Technologies). Then, paired-end (2×150 bp) sequencing was performed by Macrogen (Seoul, Korea) using the NovaSeq platform (Illumina). WGS was used to measure the telomere length in 38 samples (19 each for NFs and MPNSTs).
Whole Transcriptome Sequencing (WTS)
The total RNA concentration was calculated by Quant-IT RiboGreen (Invitrogen, Waltham, Massachusetts, USA). To determine the DV200 (% of RNA fragments >200 bp) value, samples were run on a TapeStation RNA ScreenTape platform (Agilent). Total RNA (100 ng) was subjected to sequencing library construction using a TruSeq RNA Access Library Prep Kit (Illumina) according to the manufacturer’s protocols. Briefly, total RNA was first fragmented into small pieces using divalent cations at an elevated temperature. The cleaved RNA fragments were copied into first-strand cDNA using SuperScript II reverse transcriptase (Invitrogen, #18064014) and random primers, followed by second-strand cDNA synthesis using DNA Polymerase I, RNase H and dUTP. These cDNA fragments then underwent an end repair process, involving the addition of a single ‘A’ base, and the ligation of adapters. These products were then purified and enriched via PCR to create a cDNA library. All libraries were normalized, and six libraries were pooled into a single hybridization/capture reaction. The pooled libraries were incubated with a cocktail of biotinylated oligos corresponding to coding regions of the genome. The targeted library molecules were captured via hybridized biotinylated oligo probes using streptavidin-conjugated beads. After two rounds of hybridization/capture reactions, the enriched library molecules were subjected to a second round of PCR amplification. The captured libraries were quantified using a KAPA Library Quantification Kits for the Illumina sequencing platforms according to the qPCR Quantification Protocol Guide (KAPA BIOSYSTEMS, #KK4854) and qualified using the TapeStation D1000 ScreenTape platfprm (Agilent Technologies, # 5067-5582). The indexed libraries were then submitted to sequencing on the Illumina NovaSeq platform (Illumina) and paired-end (2×100 bp) sequencing was performed by Macrogen. WTS was used to profile the transcriptomes of 40 samples (20 each for NFs and MPNSTs).
DEG Selection
The raw RNA-seq data (fastq) of all the samples were mapped using STAR (v2.7.3) and quantified by individual genes with a normalized count value, transcripts per million (TPM) using RSEM (v.1.2.31). Noncoding genes, including pseudogenes and long noncoding RNA, were excluded from the analysis and only coding genes were used (20,315 coding genes). The expression value was log2 transformed (log2(TPM+1)), where the entire expression profile was proportionally transformed, and the 0 conversions were corrected by adding 1 to the substitution. By comparing 20 NF-MPNSTs, DEGs between NFs and MPNSTs were selected on the basis of an average fold-change greater than two and a t-test p-value <0.001. Using above filtering criteria, 22 significant DEGs were selected. Statistical testing for each gene was achieved using paired t tests and a p value <0.05 was considered statistically significant.
Measuring the Length of Telomeres
Telomeric reads were calculated using TelomereHunter[57]. The program extracts telomeric reads with nonconsecutive repeats, and the extracted telomeric reads are categorized according to their alignment coordinates and mapping quality. The reads are classified into different telomeric regions. The telomere content is given as the fraction of intratelomeric reads per million reads. The program also accounts for GC bias and GC is used to correct the telomeric content by dividing the intratelomeric reads by the GC number of reads with a GC content between 48% and 52%. In this study, GC-corrected telomeric reads were used.
Statistical Analysis
Statistical analyses were performed using SPSS (SPSS, Inc., Chicago, IL, USA) Transcriptional relationships were analyzed with LR. For each gene pair, transcriptional activation of the B gene by the A gene was defined as the ΔA transcripts during NF-MPNST transformation were correlated with both the ΔB transcripts and the MPNST-B transcripts, where the significance of the LR results between ΔA transcripts and ΔB were confirmed by the Benjamini-Hochberg procedure. The interaction effects of genes A and B on telomere length were also analyzed with a GLM (Figure 1C). Correlations with the histologic grade were evaluated using logistic regression, and MFS and OS were evaluated using Cox regression.
Results
Differentially Expressed Genes during MPNST Malignant Transformation
In this study, 20 NF-MPNST pairs from 20 NF1 patients were analyzed. The patient demographics and tumor characteristics are described in Table 1. The mean latency to MPNST presentation was 36.55 years, and 30.0% of patients were in the metastatic stage at MPNST presentation. The OS rate was 35.0%, and the metastasis rate was 71.4%. In terms of tumors, 50% of MPNSTs were high-grade with an FNCLCC grade of 3. There were no significant differences in tumor sizes (P=0.430). There was a significant difference in the telomere length between NFs (999.96±423.69 intratel_reads*1,000,000/total_reads_with_tel_gc) and MPNSTs (727.32±490.75 intratel_reads*1,000,000/total_reads_with_tel_gc) in the paired t tests (P=0.043), suggesting telomere attrition during malignant transformation (Table 1).
Twenty-two differentially expressed genes (DEGs) were identified from the WTS during MPNST malignant transformation by selecting genes with an average fold-change of greater than twofold. The Z score heatmap is illustrated in Figure 1D. Eleven upregulated genes were mainly classified as related to (1) cell signaling: NELL2 and DLGAP5; (2) cytokinesis: ASPM and BUB1; (3) forkhead box transcription factors: FOXG1 and FOXM1; and (4) transcription factors: SIX1 and SOX11, whereas 11 downregulated genes were mainly classified as related to (1) cell adhesion: CHL1, CDH19, CLDN1, SORBS1, and DMD; and (2) synapse-related: PRIMA1, GRIK3, and SLITKR2. The details of the 22 DEGs are listed in Supplementary Information 1.
Among the upregulated DEGs, BUB1 promotes telomere replication during the S phase in HeLa cells[29]. Although the NELL2-Robo3 complex is involved in the activation of axonal guidance[30], NELL2 is also involved in various cancers, for instance, the NELL2/cdc42-BAF complex in Ewing’s sarcoma cells[31], fat mass and obesity-associated protein (FTO)/E2F1/NELL2 in non-small cell lung cancer (NSCLC)[32], and NELL2/N-cadherin in embryonic carcinoma cells[33]. FOXM1 is highly expressed and a marker for poor prognosis in several cancers[34], including MPNST[35]. SOX11 is also highly expressed in nervous system neoplasms[36]. Among the downregulated DEGs, PRIMA1 has been reported to be a tumor suppressor by restoring mutant p53[37] and inducing tumor cell death[38]. DMD is also a tumor suppressor in sarcomas, hematologic malignancies and nervous system tumors[39]. SORB1 suppression promotes lung adenocarcinoma[40].
NELL2 Activates PAX7
Paired box (PAX) genes encode transcription factors with highly conserved N-terminal DNA binding domains, known as the paired domains, which have been reported to be important in neural development and oncogenesis. Mutations in PAX genes are associated with congenital human diseases related to eye development and deafness, including Aniridia and Peter’s anomaly (PAX6) and Waardenburg syndrome (PAX3), suggesting that PAX genes play a central role in the development of the nervous system[41]. Moreover, PAX genes have been also reported to be involved in oncogenesis[42]. The nine PAX genes are subclassified into four groups according to their homeodomain and octapeptide domain: Group I (PAX1 and PAX9), group II (PAX2, PAX5, and PAX8), group III (PAX3 and PAX7), and group IV (PAX4 and PAX6). Group III contributes to sarcomas (Ewing’s sarcoma and rhabdomyosarcoma) and neural crest-derived tumors[43,44] (Figure 1E).
We hypothesized that NELL2 and PAX genes contribute to the oncogenesis of MPNSTs. We analyzed both the transcriptional relationship and interaction effects on the telomere length for 198 DEG-PAX gene pairs comprising 22 DEGs and nine PAX genes. The transcriptional activation of a PAX gene by a DEG was defined as the significant positive correlation of the ΔDEG transcript with both the ΔPAX transcript and the MPNST-PAX transcript according to linear regression (LR). In addition, to screen for only TMM-related transcriptional activation, the interaction effects of each gene pair on the telomere length (MPNST-telomere) were also analyzed using a generalized linear model (GLM). All 594 P values from the LRs for ΔPAX and MPNST-PAX and the GLM were verified to control the false discovery rate (FDR) by the Benjamini-Hochberg (B-H) procedure (FDR=0.1). Conclusively, transcriptional activation in the TMM activation pathway was defined as significant results for both the LR and GLM and following verification by the B-H procedure (Figure 1C). NELL2 transcriptionally activates PAX7 and SORBS1 transcriptionally activates PAX5 (Figure 2, Supplementary Information 2).
PAX7 Activates RAD52-Dependent ALT and H2AFX Activates RAD51-Dependent ALT
An in-depth search of the literature revealed 100 genes that play important roles in the TMM. These genes were classified into four main categories based on their function in the TMM: (1) telomere machinery, (2) telomere topology, (3) regulatory genes involved in the DNA damage response and cell cycle/checkpoint, and (4) TMM-effector genes. The abbreviations and details for the 100 genes are listed in Supplementary Information 1. To evaluate the downstream regions of PAX7 and PAX5, we analyzed both the transcriptional relationship and interaction effects on the telomere length of 200 pairs comprising these two genes and 100 TMM-related genes. In addition, we evaluated the process of TMM activation caused by DNA damage signals. The DNA damage markers H2AFX, PARP1, P53, and RB1, were also analyzed in the same way as above for 396 pairs comprising these four genes and the remaining 96 TMM-related genes. Finally, all 1,788 P values from the two LRs and GLM for the 596 pairs were verified to control the false discovery rate (FDR) by the Benjamini-Hochberg (B-H) procedure (FDR=0.1). PAX7 activates RAD52 and SLX4IP, whereas PAX5 has no effect on TMM activation. In the DNA damage signaling pathway, H2AFX activates RAD51 and P53 activates FEN1, but those results did not pass verification by B-H procedure (Figure 2, Supplementary Information 3).
MEGF11 Activates the NELL2-PAX7 Transcriptional Cascade
The TME is an emerging field in oncology. Not only genetic and epigenetic changes in transformed cells but also their interactions with the TME are considered necessary for cancer development and progression[26]. We hypothesized that the TME induces the NELL2-PAX7 transcriptional cascade to activate RAD52-dependent ALT during the malignant transformation of MPNSTs. To determine the TME signals that trigger the NELL2-PAX7 cascade, a total of 174 signaling molecules were selected to confirm whether they activated NELL2 and PAX7. The abbreviations and details for the 174 genes are listed in Supplementary Information 1. To increase accuracy, GLM analysis was performed on both the Δtelomere length and MPNST-telomere length and activation of the NELL2-PAX7 cascade by a signaling gene was defined as the activation of both NELL2 and PAX7. All 1,392 P-values were verified by the B-H procedure with an FDR of 0.1. Only MEGF11 was proven to activate the NELL2-PAX7 transcriptional cascade. (Figure 2, Supplementary Information 4). MEGF11 was first reported as a transmembrane protein involved in retinal development[45], but has recently been reported to be related to various cancers, such as triple-negative breast cancer[46,47], Hodgkin lymphoma[48], adrenocortical carcinoma[49], and colorectal cancer[50].
Ephrin Signaling Activates the NELL2-PAX7 Transcriptional Cascade in an MEGF11-Dependent Manner
To determine the downstream targets of MEGF11, we investigated whether MEGF11 interacts with receptor tyrosine kinases (RTKs). A total of 47 RTKs were selected and evaluated in the same manner as mentioned above. The abbreviations and details for the 47 RTKs are listed in Supplementary Information 1. EPHA6 and EPHB6 were proven to interact with MEGF11 under the control conditions with an FDR of 0.1 (Supplementary Information 5). We then investigated whether EPHA6 and EPHB6 activate the NELL2-PAX7 transcriptional cascade. EPHA6 activated both NELL2 and PAX7, whereas EPHB6 activated only PAX7 (Table 2). This finding suggested that EPHA6 is a receptor for ephrin signaling that activates the NELL2-PAX7 transcription cascade. In the next step, we investigated the upstream and downstream regions of EPHA6. Upstream of EPHA6, we assessed whether MEGF11 interacts with ephrin. Among eight ephrins: five ephrin As and three ephrin Bs, EFNA5 was proven to interact with MEGF11 (Supplementary Information 6). Downstream of EPHA6, we assessed the PDZ domain- and SAM domain-containing genes that interact with EPHA6, since EPHA6 has PDZ and SAM binding domain. A total of 137 PDZ domain-containing genes and 55 SAM binding domain-containing genes were evaluated. Two PDZ domain-containing genes, PDZD9 and PARD6B, were proven to interact with both MEGF11 and EPHA6 under the control conditions with an FDR of 0.25 (Supplementary Information 7). Consistent with this, among the 137 PDZ domain containing genes and 55 SAM binding domain containing genes, only PDZD9 and PARD6B activated both NELL2 and PAX7 under the control conditions with an FDR of 0.25 (Supplementary Information 8, Table 2), suggesting that both PDZD9 and PARD6B are associated with EFNA5-EPHA6 signaling. Multiple analyses revealed that all the members of this ephrin signaling pathway, EFNA5-EPHA6-PDZD9 and PARD6B, are dependent on MEGF11 for activating the NELL2-PAX7 cascade (Figure 2, Table 3).
Biological and Clinical Manifestations of RAD51 and RAD52-Dependent ALT
We evaluated and compared the biological and clinical manifestations of RAD51-dependent ALT induced by DNA damage signaling and RAD52-dependent ALT induced by the TME-NELL2-PAX7 cascade. First, we evaluated their effects on telomere length during malignant transformation. The correlations between ΔPAX7, ΔRAD52, ΔH2AFX, and ΔRAD51 and the telomere length: Δtelomere length and MPNST-telomere length were evaluated by LR analysis. RAD52 was the only gene that correlated with the MPNST-telomere length in the simple analysis (B=114.894, P=0.017), suggesting that TME-NELL2-PAX7-induced RAD52-dependent ALT plays a dominant role in telomere maintenance and overcomes replicative senescence and the Hayflick limit, to achieve MPNST malignant transformation (Table 4).
Second, we evaluated the effects of RAD51 and RAD52-dependent ALT on the histologic grade of MPNSTs by logistic regression. H2AFX (OR=2.064, P=0.048) was associated with FNCLCC grade 3 in the simple analysis, but was not independent of RAD51 in the multiple analyses (OR=1.714, P=0.162 and OR=1.309, P=0.336, respectively), indicating that they are involved in the same pathway, whereas no component of the TME-NELL2-PAX7 cascade was associated with the MPNST histologic grade (Table 4).
In addition to archiving telomere maintenance and overcoming the Hayflick limit, acquiring the capacity for distant metastasis is also a prerequisite for malignant transformation. Therefore, we evaluated the effects of RAD51- and RAD52-dependent ALT on metastasis by using Cox regression. PAX7 (HR=2.369, P=0.007), and RAD52 (HR=1.471, P=0.021) were associated with metastasis-free survival (MFS) according to the simple analysis. Only PAX7 was shown to be independent in the multiple analyses (HR=2.132, P=0.037), whereas RAD52, the only factor associated with telomere length, was not found to be independent (HR=1.174, P=0.424). (Table 4).
To evaluate the prognostic effect of factors related to the telomere length, we divided all the factors into two groups according to their changes during malignant transformation: the increased (Δtranscript > 0) and not increased (Δtranscript ≤ 0) groups. Overall survival (OS) and MFS were compared between the two groups using Cox regression. The group with increased telomere length showed inferior OS to the group with not increased telomere length (Figure 3A, Supplementary Information 9, HR=3.809, P=0.038), suggesting that achieving telomere maintenance leads to a poor prognosis. Additionally, the group with increased PAX7 transcript (Figure 3G, Supplementary Information 9, HR=4.896, P=0.046) showed inferior OS to the group with no increase in PAX7 transcript. Among members in the TME-NELL2-PAX7 cascade to activate RAD52-dependent ALT, the groups with increased MEGF11 (Figure 3B, Supplementary Information 9, HR=4.516, P=0.036), PARD6B (Figure 3D, Supplementary Information 9, HR=17.185, P=0.011), PAX7 (Figure 3G, Supplementary Information 9, HR=9.129, P=0.007), and RAD52 (Figure 3H, Supplementary Information 9, HR=8.669, P=0.011) showed inferior MFS to those of the group with no increases in these four genes. On the other hand, H2AFX (Figure 3I, Supplementary Information 9, HR=2.669, P=0.221) and RAD51 (Figure 3J, Supplementary Information 9, HR=2.474, P=0.260) in the DNA damage signaling pathway had no effect on the MFS. These findings strongly suggest that the TME-NELL2-PAX7 transcriptional cascade elongates the telomere length and promotes metastasis.
Discussion
We conducted this study with smart strategies. First; we designed a new analysis model optimized for the study of malignant transformation: we compared NFs and MPNSTs in a single NF1 patient. Since the MPNSTs develop from preexisting NFs; they must have the same genetic background; which minimizes genetic bias and comparisons of the NF-MPNST pairs indicate only the changes that develop during malignant transformation. Second; we designed a combined statistical analysis method: we analyzed the interaction effects on the telomere length and the transcriptional relationship to distinguish only the TMM-related transcriptional relationships and filter out those that are unrelated. Third; we compared NF and MPNST tumor tissues; including the surrounding TME and tumor cells. Since NFs and MPNSTs are rich in tumor stroma and the extracellular matrix; we were able to clearly demonstrate the interactions between the tumors and the TME during malignant transformation.
Using these strategies, we successfully demonstrated that the TME-NELL2-PAX7 cascade activates RAD52-dependent ALT and that DNA damage signaling activates RAD51-dependent ALT during the malignant transformation of MPNSTs. This answers an outstanding question in telomere biology: What signal activates RAD52-dependent ALT? In addition, we also determined the biological and clinical manifestations of RAD51- and RAD52-dependent ALT. RAD52-dependent ALT activated by the TME-NELL2-PAX7 transcriptional cascade elongates the telomere length and promotes metastasis, leading to a poor prognosis of MPNSTs, whereas RAD51-dependent ALT activated by DNA damage signaling determines the histologic grade of MPNSTs. In conclusion, MEGF11 and MERF11-dependent ephrin signaling (EFNA5-EPHA6-PDZD9/PARD6B) activate RAD52-dependent ALT via the NELL2-PAX7 transcriptional cascade, rendering MPNST immortalized and metastatic. Beyond the most well-known roles of the TME in oncogeneses such as tumor immune escape and metastasis, TME also endows cancer cells with immortality by activating RAD52-dependent ALT (Figure 2).
Transcriptional activation is the most universal and fundamental process in gene activation and is involved in numerous signaling pathways and developmental processes. However, although studies of the TERT promoter[51] and transcriptional regulation of the TMM have been attempted[52,53], the transcriptional activation of the TMM is still unknown. No notable TMM activation signals have been identified except for those related to DNA damage. Under these challenging conditions, we have demonstrated, for the first time, a new TME-NELL2-PAX7 transcriptional cascade that activates RAD52-dependent ALT. This discovery could provide a new perspective, deductive insights, and systematic understanding of the overall architecture of the TMM activation process, providing insights into fundamental pathways for TMM activation via transcriptional cascades.
Despite these results, there are some limitations that should be considered. Since this study was based on the changes in mRNA transcripts rather than protein-protein interactions, we were unable to analyze genes with epigenetic changes such as phosphorylation and SUMOylation (small ubiquitin-related modifier). In this study, H2AFX showed no association with ATM and ATR, which encode kinases and convert H2AX to its active form, γH2AX, a DNA damage marker. ATRX/DAXX, which belongs to the SWI/SNF family, suppresses ALT by depositing H3F3A on telomeres and resolving G-quadruplex (G4) loops[54]. ATRX encodes an ATP-dependent helicase that undergoes cell cycle-dependent phosphorylation and DAXX encodes a potent transcriptional repressor that binds to SUMOylated transcription factors. These genes showed no association with the TME-NELL2-PAX7 cascade or with DNA damage signaling in this study. In addition, the SMC5/SMC6 complex and NSMCE2, which are representative examples of SUMOylation regulation in the TMM[55], also showed no significant interactions in this study. Therefore, in this study, the significance of DNA damage signaling (MDC1-ATM/ATR-CHEK1 pathway), the telomere topology (ATRX/DAXX), and telomere recruitment to ALT-associated promyelocytic leukemia nuclear body (APB) (SMC complex and NSMCE2) may have been partly obscured and underestimated for methodological reasons. The findings in this study need to be further validated at the protein-protein interaction level by wet-laboratory research. In addition, our results were derived from analyses between selected TMM-related genes in the literature and database searches[56]. Although most of the prominent TMM-related genes were included in this study, missing associations related to unselected TMM-related genes should be considered.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Supplementary Information 1. Abbreviations of the genes used in this study. Supplementary Information 2. NELL2 activates PAX7 during the malignant transformation of MPNSTs. Supplementary Information 3. PAX7 activates during the malignant transformation of MPNSTs. Supplementary Information 4. MEGF11 activates the NELL2-PAX7 cascade. Supplementary Information 5. MEGF11 interacts with ephrin receptors. Supplementary Information 6. MEGF11 induces ephrin signaling through EFNA5 to EPHA6. Supplementary Information 7. MEGF11-ephrin signaling is transduced to PARD6B and PDZD9. Supplementary Information 8. PARD6B and PDZD9 transduce MEGF11-ephrin signals to the NELL2-PAX7 cascade. Supplementary Information 9. Survival analysis of RAD51 and RAD52-dependent ALT activation pathways.
Author Contributions
Conceptualization, S. H. K.; Methodology, S. H. K.; Formal Analysis, S. H. K., J. L., E. C., H. K., and Y. K.; investigation, J. L., E. C., H. K., Y. K., and S. H. K, Resources, S. H. K. and Y. K., Data Curation, S. H. K., Writing-Original Draft Preparation, S. H. K. and J. L.; Writing-Review & Editing, J. L., E. C., H. K., Y. K., and S. H. K., Visualization, S. H. K., Supervision, S. H. K. and Y. K, Project Administration, S. H. K., Funding Acquisition, S. H. K.; data curation, S. H. K.,. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Bio & Medical Technology Development Program of the NRF funded by the Ministry of Science & ICT (NRF-2017M3A9A7050614 to Y.J.K), the National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST) (2017R1D1A1B03031717 to S. H. K.) and faculty research grant from Yonsei University College of Medicine (6-2016-0101 and 2019-32-0024 to S. H. K.).
Institutional Review Board Statement
This study was done under the Severance Hospital Institutional Review Board-approved protocol (IRB No: 4-2021-0123) and all the patient data in this study were approved by this committee. All methods were carried out in accordance with relevant guidelines and regulations. Informed consent was obtained from participants or their legal guardians.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data is provided within the manuscript or supplementary information files.
Acknowledgments
None.
Conflicts of Interest
The authors have no competing interests to declare. We confirm that neither the manuscript nor any parts of its content are currently under consideration or published in another journal.
References
- Watson, J. D. Origin of Concatemeric T7DNA. Nature New Biology 239, 197-201 (1972). [CrossRef]
- Okazaki, R., Okazaki, T., Sakabe, K., Sugimoto, K. & Sugino, A. Mechanism of DNA chain growth. I. Possible discontinuity and unusual secondary structure of newly synthesized chains. Proceedings of the National Academy of Sciences 59, 598-605 (1968). [CrossRef]
- Hayflick, L. & Moorhead, P. S. The serial cultivation of human diploid cell strains. Exp Cell Res 25, 585-621 (1961). [CrossRef]
- Harley, C. B., Futcher, A. B. & Greider, C. W. Telomeres shorten during ageing of human fibroblasts. Nature 345, 458-460 (1990). [CrossRef]
- Trojani, M. et al. Soft-tissue sarcomas of adults; study of pathological prognostic variables and definition of a histopathological grading system. Int J Cancer 33, 37-42 (1984). [CrossRef]
- Guillou, L. et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. Journal of Clinical Oncology 15, 350-362 (1997). [CrossRef]
- Greider, C. W. & Blackburn, E. H. Identification of a specific telomere terminal transferase activity in tetrahymena extracts. Cell 43, 405-413 (1985). [CrossRef]
- Greider, C. W. & Blackburn, E. H. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 337, 331-337 (1989). [CrossRef]
- Chakravarti, D., Labella, K. A. & Depinho, R. A. Telomeres: history, health, and hallmarks of aging. Cell 184, 306-322 (2021). [CrossRef]
- Ghanim, G. E. et al. Structure of human telomerase holoenzyme with bound telomeric DNA. Nature 593, 449-453 (2021). [CrossRef]
- Cesare, A. J. & Reddel, R. R. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet 11, 319-330 (2010). [CrossRef]
- Shay, J. W. & Wright, W. E. Telomeres and telomerase: three decades of progress. Nat Rev Genet 20, 299-309 (2019). [CrossRef]
- Conomos, D., Pickett, H. A. & Reddel, R. R. Alternative lengthening of telomeres: remodeling the telomere architecture. 3 (2013). [CrossRef]
- Kramara, J., Osia, B. & Malkova, A. Break-Induced Replication: The Where, The Why, and The How. Trends in Genetics 34, 518-531 (2018). [CrossRef]
- Min, J., Wright, W. E. & Shay, J. W. Alternative Lengthening of Telomeres Mediated by Mitotic DNA Synthesis Engages Break-Induced Replication Processes. Mol Cell Biol 37 (2017). [CrossRef]
- Zhang, J. M., Yadav, T., Ouyang, J., Lan, L. & Zou, L. Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Rep 26, 955-968.e953 (2019). [CrossRef]
- Summers, M. A. et al. Skeletal muscle and motor deficits in Neurofibromatosis Type 1. J Musculoskelet Neuronal Interact 15, 161-170 (2015).
- Friedman, J. M. Epidemiology of neurofibromatosis type 1. Am J Med Genet 89, 1-6 (1999).
- Venturini, L. et al. Telomere maintenance mechanisms in malignant peripheral nerve sheath tumors: expression and prognostic relevance. Neuro-Oncology 14, 736-744 (2012). [CrossRef]
- Rodriguez, F. J. et al. Telomere alterations in neurofibromatosis type 1-associated solid tumors. Acta Neuropathologica Communications 7 (2019). [CrossRef]
- Cho, N. W., Dilley, R. L., Lampson, M. A. & Greenberg, R. A. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 159, 108-121 (2014). [CrossRef]
- Bhowmick, R., Minocherhomji, S. & Hickson, I. D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol Cell 64, 1117-1126 (2016). [CrossRef]
- Sotiriou, S. K. et al. Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. Mol Cell 64, 1127-1134 (2016). [CrossRef]
- Hoang, S. M. & O’Sullivan, R. J. Alternative Lengthening of Telomeres: Building Bridges To Connect Chromosome Ends. Trends in Cancer 6, 247-260 (2020). [CrossRef]
- Sobinoff, A. P. & Pickett, H. A. Alternative Lengthening of Telomeres: DNA Repair Pathways Converge. Trends in Genetics 33, 921-932 (2017). [CrossRef]
- Quail, D. F. & Joyce, J. A. Microenvironmental regulation of tumor progression and metastasis. Nat Med 19, 1423-1437 (2013). [CrossRef]
- Anderson, N. M. & Simon, M. C. The tumor microenvironment. Curr Biol 30, R921-r925 (2020). [CrossRef]
- Bader, J. E., Voss, K. & Rathmell, J. C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol Cell 78, 1019-1033 (2020). [CrossRef]
- Li, F. et al. The BUB3-BUB1 Complex Promotes Telomere DNA Replication. Molecular Cell 70, 395-407.e394 (2018). [CrossRef]
- Pak, J. S. et al. NELL2-Robo3 complex structure reveals mechanisms of receptor activation for axon guidance. Nature Communications 11 (2020). [CrossRef]
- Jayabal, P. et al. NELL2-cdc42 signaling regulates BAF complexes and Ewing sarcoma cell growth. Cell Reports 36, 109254 (2021). [CrossRef]
- Wang, Y., Li, M., Zhang, L., Chen, Y. & Zhang, S. m6A demethylase FTO induces NELL2 expression by inhibiting E2F1 m6A modification leading to metastasis of non-small cell lung cancer. Molecular Therapy - Oncolytics 21, 367-376 (2021). [CrossRef]
- Kim, D. H. et al. Neural Epidermal Growth Factor-Like Like Protein 2 (NELL2) Promotes Aggregation of Embryonic Carcinoma P19 Cells by Inducing N-Cadherin Expression. PLoS ONE 9, e85898 (2014). [CrossRef]
- Kalathil, D., John, S. & Nair, A. S. FOXM1 and Cancer: Faulty Cellular Signaling Derails Homeostasis. Front Oncol 10, 626836 (2020). [CrossRef]
- Yu, J. et al. Array-Based Comparative Genomic Hybridization Identifies CDK4 and FOXM1 Alterations as Independent Predictors of Survival in Malignant Peripheral Nerve Sheath Tumor. Clinical Cancer Research 17, 1924-1934 (2011). [CrossRef]
- Yang, Z. et al. SOX11: friend or foe in tumor prevention and carcinogenesis? Therapeutic Advances in Medical Oncology 11, 175883591985344 (2019). [CrossRef]
- Lambert, J. M. R. et al. PRIMA-1 Reactivates Mutant p53 by Covalent Binding to the Core Domain. Cancer Cell 15, 376-388 (2009). [CrossRef]
- Mlakar, V. et al. PRIMA-1MET-induced neuroblastoma cell death is modulated by p53 and mycn through glutathione level. Journal of Experimental & Clinical Cancer Research 38 (2019). [CrossRef]
- Jones, L., Naidoo, M., Machado, L. R. & Anthony, K. The Duchenne muscular dystrophy gene and cancer. Cellular Oncology 44, 19-32 (2021). [CrossRef]
- Wang, C. & Cheng, B. MicroRNA miR-3646 promotes malignancy of lung adenocarcinoma cells by suppressing sorbin and SH3 domain-containing protein 1 via the c-Jun NH2-terminal kinase signaling pathway. Bioengineered 13, 4869-4884 (2022). [CrossRef]
- Dahl, E., Koseki, H. & Balling, R. Pax genes and organogenesis. Bioessays 19, 755-765 (1997). [CrossRef]
- Li, C. G. & Eccles, M. R. PAX Genes in Cancer; Friends or Foes? Front Genet 3, 6 (2012). [CrossRef]
- Robson, E. J., He, S. J. & Eccles, M. R. A PANorama of PAX genes in cancer and development. Nat Rev Cancer 6, 52-62 (2006). [CrossRef]
- Buckingham, M. & Relaix, F. The role of Pax genes in the development of tissues and organs: Pax3 and Pax7 regulate muscle progenitor cell functions. Annu Rev Cell Dev Biol 23, 645-673 (2007). [CrossRef]
- Kay, J. N., Chu, M. W. & Sanes, J. R. MEGF10 and MEGF11 mediate homotypic interactions required for mosaic spacing of retinal neurons. Nature 483, 465-469 (2012). [CrossRef]
- Chiu, J. H. et al. MEGF11 is related to tumour recurrence in triple negative breast cancer via chemokine upregulation. Sci Rep 10, 8060 (2020). [CrossRef]
- Huang, C. P. et al. Overexpression of multiple epidermal growth factor like domains 11 rescues anoikis survival through tumor cells-platelet interaction in triple negative breast Cancer cells. Life Sci 299, 120541 (2022). [CrossRef]
- Osman, Y. et al. Functional multigenic variations associated with hodgkin lymphoma. Int J Lab Hematol 43, 1472-1482 (2021). [CrossRef]
- Fonseca, A. L. et al. Comprehensive DNA methylation analysis of benign and malignant adrenocortical tumors. Genes Chromosomes Cancer 51, 949-960 (2012). [CrossRef]
- Cicek, M. S. et al. Colorectal cancer linkage on chromosomes 4q21, 8q13, 12q24, and 15q22. PLoS One 7, e38175 (2012). [CrossRef]
- Yuan, X., Larsson, C. & Xu, D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: old actors and new players. Oncogene 38, 6172-6183 (2019). [CrossRef]
- Nersisyan, L., Simonyan, A., Binder, H. & Arakelyan, A. Telomere Maintenance Pathway Activity Analysis Enables Tissue- and Gene-Level Inferences. Front Genet 12, 662464 (2021). [CrossRef]
- Nersisyan, L. & Arakelyan, A. A transcriptome and literature guided algorithm for reconstruction of pathways to assess activity of telomere maintenance mechanisms (Cold Spring Harbor Laboratory, 2017).
- Dyer, M. A., Qadeer, Z. A., Valle-Garcia, D. & Bernstein, E. ATRX and DAXX: Mechanisms and Mutations. Cold Spring Harbor Perspectives in Medicine 7, a026567 (2017). [CrossRef]
- Potts, P. R. & Yu, H. The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat Struct Mol Biol 14, 581-590 (2007). [CrossRef]
- Braun, D. M., Chung, I., Kepper, N., Deeg, K. I. & Rippe, K. TelNet - a database for human and yeast genes involved in telomere maintenance. BMC Genetics 19 (2018). [CrossRef]
- Feuerbach, L. et al. TelomereHunter - in silico estimation of telomere content and composition from cancer genomes. BMC Bioinformatics 20, 272 (2019). [CrossRef]
Figure 1.
Schematic illustrations of this study. (A) An example of a T2 sagittal MR image of the thigh showing an NF (blue-circle) and an MPNST (red-circled) in one patient with NF1. (B) Since MPNSTs associated with NF1 arise from preexisting NFs, each NF-MPNST pair shares the same genetic background and differentiation lineage. In each NF-MPNST pair comparison, only the malignant transformation-related changes are determined by structurally subtracting the changes from the normal cell to the benign tumor. (C) A combined statistical analysis method was utilized, where both the interaction effects on the telomere length and the transcriptional relationships for all the gene pairs were analyzed to distinguish only the TMM-related transcriptional relationships and filter out those that were unrelated. (D) A Z score heatmap for 22 DEGs during malignant transformation of MPNSTs. Eleven upregulated DEGs are shown in red, whereas the 11 downregulated DEGs are shown in blue. (E) PAX genes were classified into four groups according to the paired domain, octapeptide, and homeodomain. Each group plays important roles in the development and oncogenesis of various tissues, especially neural tissues. Group III: PAX3 and PAX7 function in the neuromuscular tissue development and the oncogenesis of sarcoma. MR, magnetic resonance; NF, neurofibroma; MPNST, malignant peripheral nerve sheath tumor; NF1, neurofibromatosis 1; TMM, telomere maintenance mechanism; DEG, differentially expressed gene.
Figure 1.
Schematic illustrations of this study. (A) An example of a T2 sagittal MR image of the thigh showing an NF (blue-circle) and an MPNST (red-circled) in one patient with NF1. (B) Since MPNSTs associated with NF1 arise from preexisting NFs, each NF-MPNST pair shares the same genetic background and differentiation lineage. In each NF-MPNST pair comparison, only the malignant transformation-related changes are determined by structurally subtracting the changes from the normal cell to the benign tumor. (C) A combined statistical analysis method was utilized, where both the interaction effects on the telomere length and the transcriptional relationships for all the gene pairs were analyzed to distinguish only the TMM-related transcriptional relationships and filter out those that were unrelated. (D) A Z score heatmap for 22 DEGs during malignant transformation of MPNSTs. Eleven upregulated DEGs are shown in red, whereas the 11 downregulated DEGs are shown in blue. (E) PAX genes were classified into four groups according to the paired domain, octapeptide, and homeodomain. Each group plays important roles in the development and oncogenesis of various tissues, especially neural tissues. Group III: PAX3 and PAX7 function in the neuromuscular tissue development and the oncogenesis of sarcoma. MR, magnetic resonance; NF, neurofibroma; MPNST, malignant peripheral nerve sheath tumor; NF1, neurofibromatosis 1; TMM, telomere maintenance mechanism; DEG, differentially expressed gene.
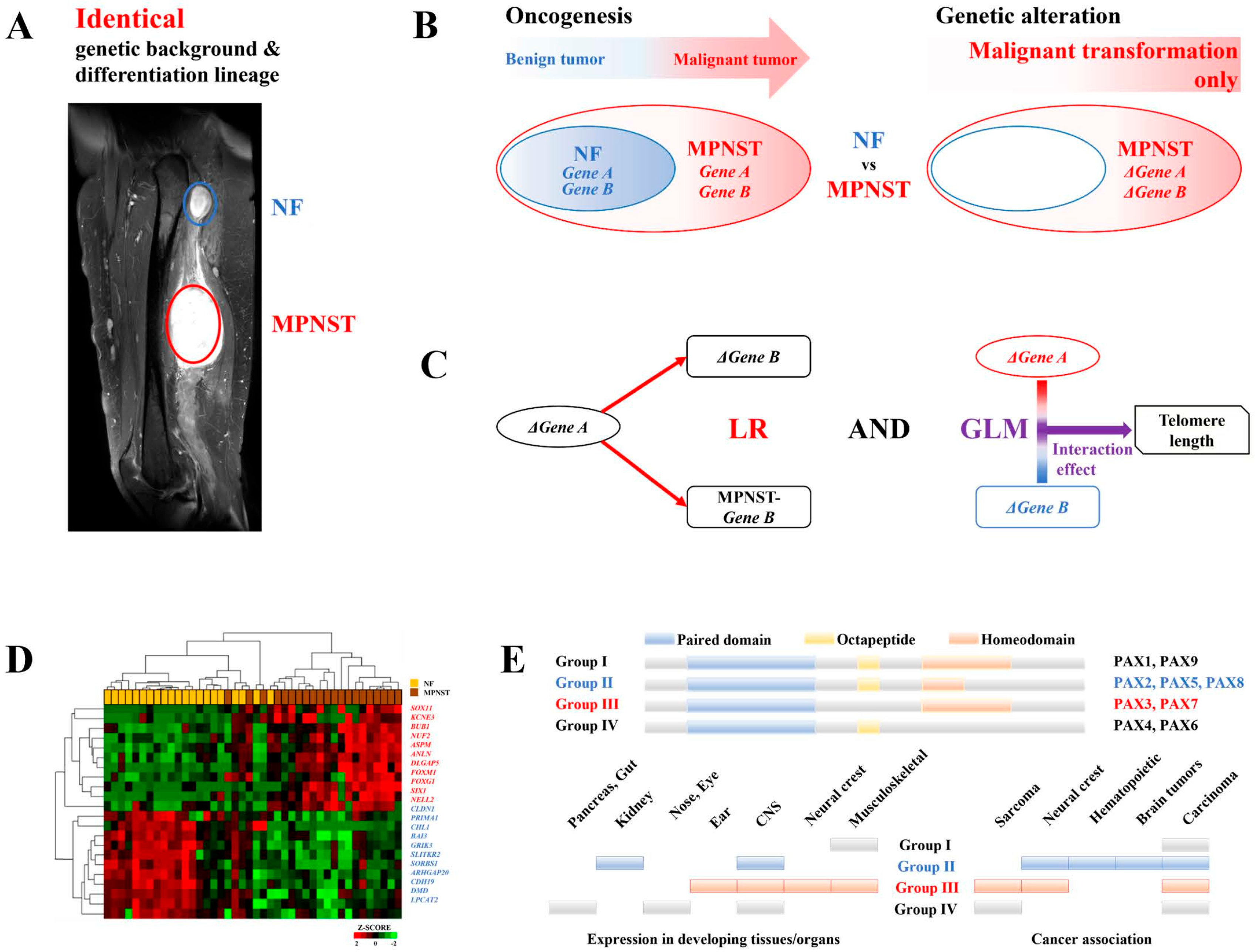
Figure 2.
Summary diagram. RAD52-dependent ALT, which is activated by MEGF11 and the MEGF11-dependent ephrin signaling pathway (EFNA5-EPHA6-PDZD9/PARD6B) through the NELL2-PAX7 transcriptional cascade, increases telomere length and promotes metastasis, leading to a poor prognosis in MPNSTs, whereas RAD51-dependent ALT, which is activated by the DNA damage signal, H2AFX, determines the histologic grade of MPNSTs. TME, tumor microenvironment; MPNST, malignant peripheral nerve sheath tumor; ALT, alternative lengthening of telomeres.
Figure 2.
Summary diagram. RAD52-dependent ALT, which is activated by MEGF11 and the MEGF11-dependent ephrin signaling pathway (EFNA5-EPHA6-PDZD9/PARD6B) through the NELL2-PAX7 transcriptional cascade, increases telomere length and promotes metastasis, leading to a poor prognosis in MPNSTs, whereas RAD51-dependent ALT, which is activated by the DNA damage signal, H2AFX, determines the histologic grade of MPNSTs. TME, tumor microenvironment; MPNST, malignant peripheral nerve sheath tumor; ALT, alternative lengthening of telomeres.
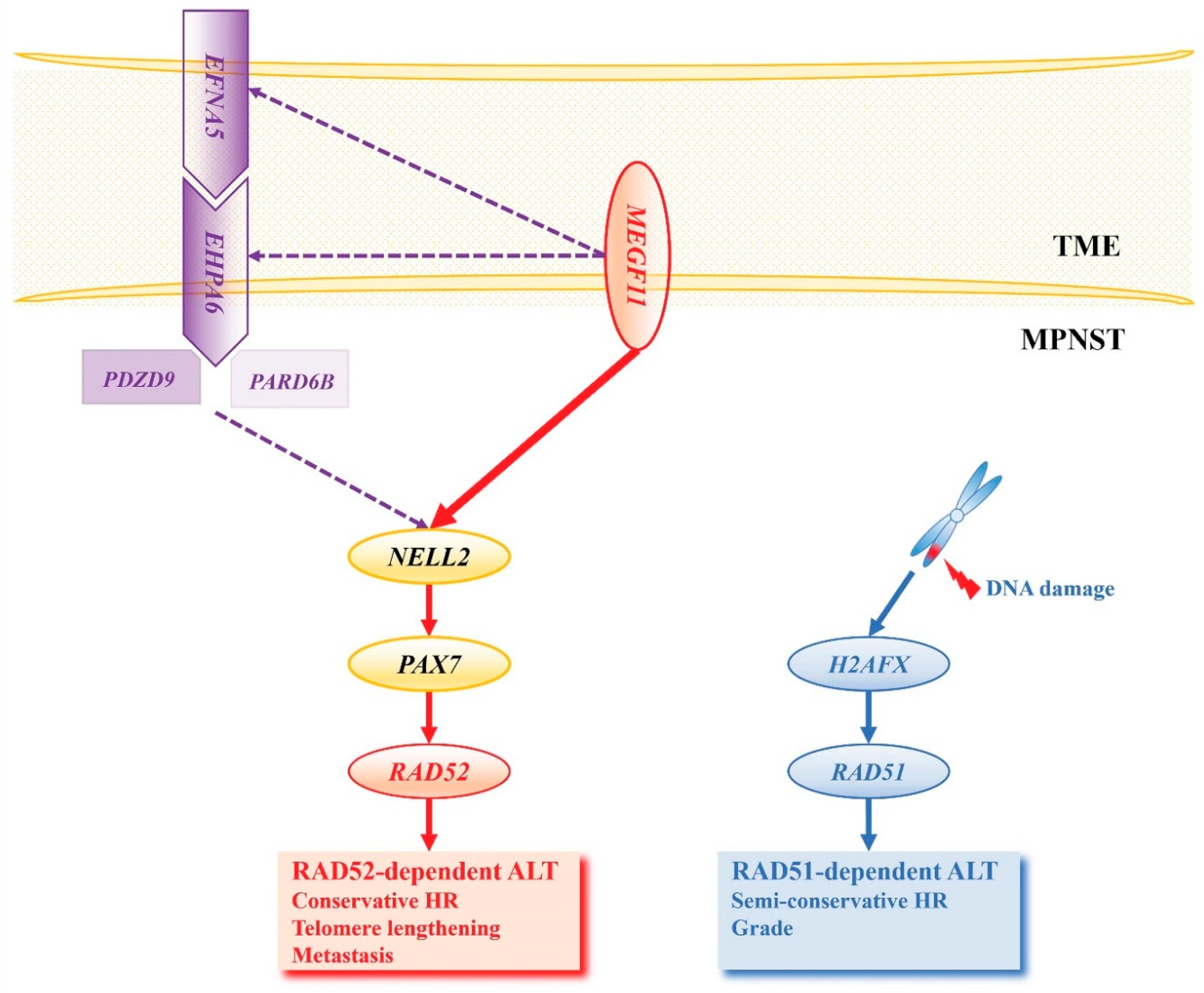
Figure 3.
Survival analysis of the genes involved in the RAD51- and RAD52-dependent ALT activation pathways. MFS and OS were evaluated by Cox regression between the group with increased transcripts (Δfactor > 0) and the group with transcripts that were not increased (Δfactor ≤ 0) during the malignant transformation. (A) Telomere length, (B) MEGF11, (C) EHPA6, (D) PARD6B, (E) PDZD9, (F) NELL2, (G) PAX7, and (H) RAD52 in the TME-NELL2-PAX7 transcriptional cascade were analyzed, and (I) H2AFX and (J) RAD51 in DNA damage signaling were analyzed. The red lines represent the increased group (Δfactor > 0) and the blue lines represent the not increased group (Δfactor ≤ 0). MFS, metastasis-free survival; OS, overall survival; CS, cumulative survival; m, months; TME, tumor microenvironment.
Figure 3.
Survival analysis of the genes involved in the RAD51- and RAD52-dependent ALT activation pathways. MFS and OS were evaluated by Cox regression between the group with increased transcripts (Δfactor > 0) and the group with transcripts that were not increased (Δfactor ≤ 0) during the malignant transformation. (A) Telomere length, (B) MEGF11, (C) EHPA6, (D) PARD6B, (E) PDZD9, (F) NELL2, (G) PAX7, and (H) RAD52 in the TME-NELL2-PAX7 transcriptional cascade were analyzed, and (I) H2AFX and (J) RAD51 in DNA damage signaling were analyzed. The red lines represent the increased group (Δfactor > 0) and the blue lines represent the not increased group (Δfactor ≤ 0). MFS, metastasis-free survival; OS, overall survival; CS, cumulative survival; m, months; TME, tumor microenvironment.

Table 1.
Patient demographics and tumor characteristics.
| NF | MPNST | P | |||
| Patient demographics | |||||
|
Age at diagnosis of MPNST (years, mean±SD)(Min-Max) |
36.55±14.58 (16–71) | - | |||
|
Sex n(%) |
male | 10(50.0) | - | ||
| female | 10(50.0) | - | |||
|
AJCC stage1 n(%) |
I | - | 4(20.0) | - | |
| II & IIIA | 10(50.0) | ||||
|
IIIB & IV (Metastatic) |
6(30.0) | ||||
|
Survival n(%) |
OS2 | - | 7(35.0) | - | |
| DOD | 10(50.0) | ||||
| DOC | 3(15.0) | ||||
|
Metastasis n(%) |
free | - | 4(28.6) | ||
| positive | 10(71.4) | ||||
| Tumor characteristics | |||||
| Size (cm, mean±SD) 3 | 5.31±5.57 | 7.44±3.49 | 0.430 | ||
|
MPNST histologic grade (FNCLCC) n(%) |
1 | - | 4(20.0) | - | |
| 2 | 6(30.0) | ||||
| 3 | 10(50.0) | ||||
|
Location4 n(%) |
visceral | 0(0) | 2(10.0) | 0.625 | |
| axial | 5(25.0) | 5(25.0) | |||
| extremity | 15(75.0) | 13(65.0) | |||
| Telomere length 5 (mean±SD) | 999.96±423.69 | 727.32±490.75 | 0.043 | ||
1 8th edition 2 5 years survival 3 paired t-test 4 Fisher’s exact test 5 paired t-test, measured by TelomereHunter, tel_content = intratel_reads * 1,000,000/total_reads_with_tel_gc Abbreviations: NF, Neurofibroma; MPNST, Malignant Peripheral Nerve Sheath Tumor; SD, Standard Deviation; Min, minimum; Max, maximum; AJCC, American Joint Committee on Cancer; OS, Overall Survival; DOD, Died of Disease; DOC, Died of other cause; FNCLCC, Fédération Nationale des Centres de Lutte Contre Le Cancer.
Table 2.
TME-induced NELL2-PAX7 transcriptional cascades.
| NELL2 | PAX7 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LR | GLM | LR | GLM | ||||||||||||||
| Δ | MPNST | Δtelomere | MPNST-telomere | Δ | MPNST | Δtelomere | MPNST-telomere | ||||||||||
| β (95% CI) |
P | β (95% CI) |
P | β (95% CI) |
P | β (95% CI) |
P | β (95% CI) |
P | β (95% CI) |
P | β (95% CI) |
P | β (95% CI) |
P | ||
| TME | MEGF11 | 0.799 (0.393 to 1.206) |
0.001 | 0.648 (0.330 to 0.967) |
0.000 | 44.551 (19.987 to 69.115) |
0.000 | 45.525 (23.556 to 67.493) |
0.000 | 0.381 (0.188 to 0.575) |
0.001 | 0.383 (0.189 to 0.576) |
0.001 | 89.521 (30.653 to 148.388) |
0.003 | 98.580 (49.698 to 147.462) |
0.000 |
| EFNA5 | 0.106 (-0.421 to 0.633) |
0.676 | 0.283 (-0.115 to 0.681) |
0.152 | 45.446 (17.997 to 72.895) |
0.001 | 38.900 (16.027 to 61.773) |
0.001 | 0.201 (-0.031 to 0.433) |
0.085 | 0.203 (-0.029 to 0.435) |
0.083 | 97.010 (44.392 to 149.628) |
0.000 | 89.744 (48.726 to 130.963) |
0.000 | |
| RTK | EPHA6 | 0.918 (0.217 to 1.620) |
0.013 | 0.808 (0.275 to 1.341) |
0.005 | 57.328 (16.651 to 98.005) |
0.006 | 59.188 (21.406 to 96.970) |
0.002 | 0.499 (0.186 to 0.812) |
0.004 | 0.500 (0.187 to 0.813) |
0.004 | 106.112 (16.647 to 195.577) |
0.020 | 99.886 (15.346 to 184.425) |
0.021 |
| EPHB6 | 0.661 (-0.059 to 1.381) |
0.070 | 0.532 (-0.040 to 1.105) |
0.067 | 89.325 (44.439 to 134.211) |
0.000 | 81.525 (49.131 to 113.918) |
0.000 | 0.439 (0.131 to 0.747) |
0.003 | 0.441 (0.134 to 0.749) |
0.007 | 150.532 (70.277 to 230.787) |
0.000 | 122.220 (57.010 to 17.430) |
0.000 | |
|
PZD proteins |
PARD6B | 0.952 (0.105 to 1.799) |
0.030 | 0.551 (0.077 to 1.025) |
0.025 | 55.943 (13.061 to 98.824) |
0.011 | 68.916 (37.840 to 99.993) |
0.000 | 0.483 (0.088 to 0.878) |
0.019 | 0.485 (0.090 to 0.881) |
0.019 | 187.099 (75.124 to 299.075) |
0.000 | 214.738 (139.247 to 290.230) |
0.000 |
| PDZD9 | 0.919 (0.081 to 1.757) |
0.033 | 0.758 (0.097 to 1.419) |
0.027 | 96.366 (47.433 to 145.300) |
0.000 | 96.710 (67.185 to 126.235) |
0.000 | 0.539 (0.172 to 0.907) |
0.006 | 0.543 (0.175 to 0.910) |
0.006 | 192.099 (107.854 to 276.344) |
0.000 | 175.126 (117.298 to 232.954) |
0.000 | |
Abbreviations: TME, tumor microenvironment; LR, linear regression; GLM, generalized linear model; MPNST, Malignant Peripheral Nerve Sheath Tumor; CI, Confidence Interval, RTK, receptor tyrosine kinase.
Table 3.
MEGF11 and MEGF11-dependent ephrin signaling in the TME-induced NELL2-PAX7 transcriptional cascade.
Table 3.
MEGF11 and MEGF11-dependent ephrin signaling in the TME-induced NELL2-PAX7 transcriptional cascade.
| ΔNELL2 | MPNST-NELL2 | |||||
|---|---|---|---|---|---|---|
| B (95% CI) |
P | B (95% CI) |
P | |||
| Simple analysis | MEGF11 | 0.799 (0.393 to 1.205) |
0.001 | 0.648 (0.330 to 0.967) |
0.000 | |
| EPHA6 | 0.918 (0.217 to 1.620) |
0.013 | 0.808 (0.275 to 1.341) |
0.005 | ||
| PARD6B | 0.952 (0.105 to 1.799) |
0.003 | 55.943 (13.061 to 98.824) |
0.011 | ||
| PDZD9 | 0.919 (0.081 to 1.757) |
0.033 | 0.758 (0.097 to 1.419) |
0.027 | ||
| Multiple analysis | MEGF11 | MEGF11 | 1.059 (0.219 to 1.899) |
0.016 | 0.705 (0.038 to 1.373) |
0.039 |
| EPHA6 | -0.440 (-1.677 to 0.798) |
0.464 | -0.097 (-1.080 to 0.886) |
0.838 | ||
| MEGF11 | 0.697 (0.217 to 1.117) |
0..007 | 0.657 (0.273 to 1.041) |
0.002 | ||
| PARD6B | 0.338 (-0.482 to 1.158) |
0.396 | -0.029 (-0.685 to 0.626) |
0.926 | ||
| MEGF11 | 1.053 (0.346 to 1.760) |
0.006 | 0.836 (0.280 to 1.391) |
0.006 | ||
| PDZD9 | -0.523 (-1.712 to 0.666) |
0.666 | -0.386 (-1.319 to 0.547) |
0.395 | ||
| EHPA6 | EPHA6 | 0.665 (-0.255 to 1.585) |
0.145 | 0.853 (0.138 to 1.567) |
0.022 | |
| PARD6B | 0.460 (-0.605 to 1.526) |
0.375 | -0.081 (-0.909 to 0.746) |
0.839 | ||
| EHPA6 | 0.735 (-0.403 to 1.873) |
0.191 | 0.731 (-0.136 to 1.599) |
0.093 | ||
| PDZD9 | 0.270 (-1.028 to 1.568) |
0.666 | 0.113 (-0.877 to 1.102) |
0.813 | ||
| PARD6B | PARD6B | 0.619 (-0.415 to 1.652) |
0.224 | 0.154 (-0.694 to 1.002) |
0.706 | |
| PDZD9 | 0.566 (-0.451 to 1.583) |
0.257 | 0.670 (-0.165 to 1.504) |
0.109 | ||
Abbreviations: TME, tumor microenvironment; MPNST, Malignant Peripheral Nerve Sheath Tumor.
Table 4.
Biological and clinical manifestations of RAD51- and RAD52-dependent ALT.
| Telomere length1 | MPNST grade2 | Metastasis3 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Δtelomere | MPNST-telomere | Simple analysis | Multiple analysis | Simple analysis | Multiple analysis | ||||||||
| B (95% CI) |
P | B (95% CI) |
P | OR (95% CI) |
P | OR (95% CI) |
P | HR (95% CI) |
P | HR (95% CI) |
P | ||
| Telomere length | Δtelomere | - | - | - | - | 1.250 (0.205 to 7.615) |
0.809 | 1.987 (0.552 to 7.151) |
0.293 | - | - | ||
| MPNST-telomere | - | - | - | - | 1.000 (0.998 to 1.002) |
0.822 | 1.001 (0.999 to 1.002) |
0.427 | - | - | |||
| TME-NELL2-PAX7 cascades | MEGF11 | -0.045 (-0.144 to 0.054) |
0.351 | 46.390 (-48.090 to 140.869) |
0.315 | 1.174 (0.807 to 1.706) |
0.402 | 1.161 (0.920 to 1.464) |
0.208 | ||||
| EHPA6 | -0.069 (-0.216 to 0.078) |
0.334 | 54.320 (-87.270 to 195.911) |
0.429 | 1.355 (0.768 to 2.393) |
0.295 | 1.212 (0.881 to 1.667) |
0.237 | |||||
| PARD6B | 25.699 (-157.424 to 208.822) |
0.771 | 64.970 (-96.681 to 226.621) |
0.408 | 1.317 (0.698 to 2.487) |
0.395 | 1.372 (0.835 to 2.254) |
0.212 | |||||
| PDZD9 | -0.037 (-0.205 to 0.130) |
0.644 | 137.892 (-7.571 to 283.355) |
0.062 | 2.389 (0.839 to 6.797) |
0.103 | 1.394 (0.889 to 2.187) |
0.148 | |||||
| NELL2 | -0.021 (-0.108 to 0.067) |
0.622 | 18.480 (-65.203 to 102.164) |
0.647 | 0.979 (0.718 to 1.333) |
0.891 | 1.174 (0.918 to 1.501) |
0.202 | |||||
| PAX7 | -0.066 (-0.050 to 0.118) |
0.459 | 78.778 (-95.795 to 253.351) |
0.354 | 1.498 (0.740 to 3.033) |
0.262 | 2.369 (1.269 to 4.423) |
0.007 | 2.132 (1.046 to 4.343) |
0.037 | |||
| RAD52 | 0.016 (-0.098 to 0.130) |
0.768 | 114.894 (22.996 to 206.793) |
0.017 | 1.318 (0.829 to 2.097) |
0.243 | 1.471 (1.059 to 2.043) |
0.021 | 1.174 (0.793 to 1.738) |
0.424 | |||
| DNA damage signal | H2AFX | -0.013 (-0.151 to 0.125) |
0.844 | -5.346 (-137.516 to 126.824) |
0.933 | 2.064 (1.007 to 4.229) |
0.048 | 1.714 (0.806 to 3.647) |
0.162 | 1.226 (0.809 to 1.857) |
0.337 | ||
| RAD51 | -0.054 (-0.145 to 0.037) |
0.225 | 19.255 (-71.309 to 109.818) |
0.659 | 1.529 (0.968 to 2.416) |
0.069 | 1.309 (0.757 to 2.266) |
0.336 | 1.312 (0.947 to 1.818) |
0.102 | |||
1 linear regression 2 logistic regression 3 Cox`s regression Abbreviations: ALT, alternative lengthening of telomere; MPNST, Malignant Peripheral Nerve Sheath Tumor; CI, confidence interval; OR, odds ratio; HR, hazard ratio; TME, tumor microenvironment.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



