Submitted:
25 November 2024
Posted:
26 November 2024
You are already at the latest version
Abstract
We have successfully synthesized a 2-oxa androstane derivative, 17β-hydroxy-2-oxa-5α-androstan-3-one (6), and confirmed its structure using NMR spectroscopy and mass spectrometry.
Keywords:
steroids
; oxasteroids
; androgenic-anabolic steroids
; dihydrotestosterone
1. Introduction
Androgenic-anabolic steroids (AAS) have been utilized in clinical practice for many years due to their benefits for structural conditions such as muscle and bone wasting, as well as for various functional disorders (endocrine, metabolic, or infectious). [1,2] A notable representative of AAS, valued for its therapeutic potential, is Oxandrolone [17β-hydroxy-17α-methyl-2-oxa-5α-androstan-3-one (1)]. This compound has a unique chemical structure, characterized by an oxygen atom at C-2 and an additional methyl group at C-17α (Figure 1a). [3] This structural configuration enhances oral bioavailability and results in a high anabolic-to-androgenic ratio (AAR) of 10:1, compared to the well-known endogenous androgen testosterone, which has an AAR of 1:1. [4,5] The AAR is a critical metric for evaluating the relative effects of different steroids on anabolic and androgenic pathways, and it significantly impacts athletic performance and overall health. [5]
Oxandrolone, however, also has drawbacks, including hepatotoxicity due to C-17α-alkylation, which can lead to severe liver conditions such as cholestasis, fibrosis, and even liver cancer. [6] Therefore, it is hypothesized that synthesizing 17α-alkyl-free 2-oxa steroids might offer high androgenic activity and anti-estrogenic effects while reducing liver toxicity. Indeed, such steroids have been researched previously, and their biological potential has been recognized. In 1956, Pappo and Jung [7] reported the synthesis of a series of 2-oxa steroids, including the non-methylated analog of Oxandrolone – 17β-hydroxy-2-oxa-5α-androstan-3-one (6, Figure 1b). They synthesized 6 from 5α-androst-1-ene-3,17-dione (1-androstenedione) using lead tetraacetate in acetic acid, achieving a 50% overall yield after reduction. However, only the melting point, UV absorption spectrum, and specific rotation of the unknown compound (at that time) were reported.
Given the interest in 2-oxa steroids, this communication presents a different synthetic pathway to 17β-hydroxy-2-oxa-5α-androstan-3-one (6) by using 17β-hydroxy-5α-androstan-3-one [(5α-dihydrotestosterone (DHT), 2] as a starting material and employing cheaper reagents in key steps of the synthesis. Furthermore, we unequivocally characterized the structure of compound 6 using NMR and mass spectrometry.
2. Results and Discussion
Several methods are available to introduce an oxygen atom into the steroid skeleton. Hara described one such approach, which employs peroxybenzoic acid in the Bayer-Villiger oxidation of 17β-hydroxy-A-nor-5α-androstan-2-one. [8] This technique enables the synthesis of both 2-oxa and 4-oxaandrostane derivatives in a single-step reaction; however, the starting material is not readily available and must be synthesized in advance. An alternative method, illustrated in Scheme 1, entails the oxidation of α,β-unsaturated ketones to generate secocarbonyl acids, which are subsequently reduced to lactones. [9] This approach enables the synthesis of 2-oxa or 4-oxa derivatives from parent steroids possessing a conjugated double bond in ring A.
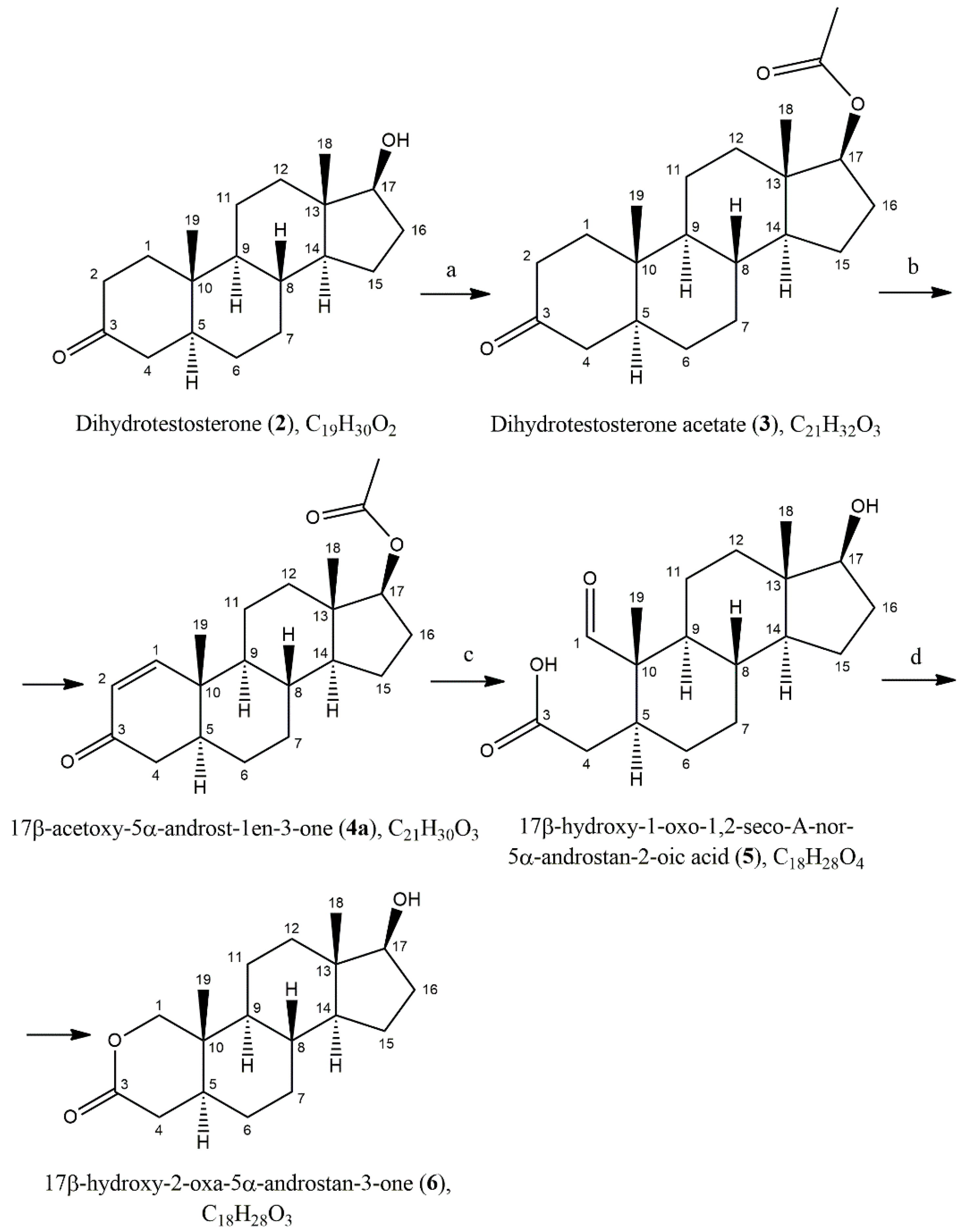
In synthesizing compound 6, we utilized the synthetic method outlined in Scheme 2. Due to the high cost (ranging from €420 to €550 for 25 mg) of 17β-hydroxy-5α-androst-1-en-3-one and its acetylated form (4а), we synthesized the latter from the more affordable precursor, DHT (2). This was achieved through acetylation with acetic anhydride in dry pyridine, followed by dehydrogenation using 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in dry dioxane [10]. The latter transformation results in a mixture of 17β-acetoxy-5α-androst-1-en-3-one (4a) and 17β-acetoxy-androst-1,4-dien-3-one (4b, not shown in Scheme 2). For the second reaction, we observed that the product ratio is influenced by the reaction time and the amount of DDQ used, and that using 1.4 equivalents of DDQ for 24 hours achieved optimal results, yielding a 2:1 ratio of products 4a to 4b.
Various oxidizing agents can oxidize the double bond in ring A. OsO4 is the most selective for producing diols when used in equimolar amounts [11]; however, its high toxicity and cost — approximately €650 per gram — render it an impractical choice for this procedure. Although combining OsO4 in less than equimolar amounts with an excess of NaIO4 has shown some promise, this approach still has limitations. [12,13] An alternative is the ozonation reaction [9,14,15] or oxidation with lead tetraacetate in acetic acid [7]. However, the need for specialized equipment in the first method and the unsatisfactory yields (ca. 50%) in the second made us opt for a combination of KMnO4 and NaIO4 for the one-step production of secocarbonyl acid 5 from compound 4a. This method is based on the conditions established by Lao et al. [16] during their research on 5α-reductase inhibitors and androgen receptor antagonists. We used KMnO4 in amounts less than one equivalent to minimize the risk of overoxidation of the products. Meanwhile, NaIO4 served a dual purpose: it regenerated KMnO4 and oxidized the diol to form the corresponding secocarbonyl acid. In the last step, we obtained the target 2-oxa steroid 6 by reduction of 5 with NaBH4. The structure of all compounds was confirmed using NMR spectroscopy and for the target compound mass spectrometry (HRMS). NMR spectra for all known compounds (3, 4a, 4b, 5) are consistent with those in the literature.
In conclusion, we have successfully synthesized and characterized a known non-alkylated 2-oxa steroid, specifically 17β-hydroxy-2-oxa-5α-androstan-3-one (6), which had not yet undergone spectral characterization. This steroid is believed to possess a high anabolic-to-androgenic ratio and anti-estrogenic effects, all while minimizing the risk of liver toxicity. Our ongoing research aims to assess the in vitro efficacy of this compound and to explore its potential therapeutic benefits for conditions such as estrogen-positive breast cancer, hypogonadism, diabetes, cardiovascular diseases, and osteoporosis.
3. Materials and Methods
3.1. General:
All chemicals used were purchased from Sigma-Aldrich and used without further purification. Column chromatography was performed with Silica gel 60, 0.060-0.2mm. TLC was run on silica gel coated aluminum sheets (Alugram® SIL G/UV254, Macherey-Nagel). NMR spectra were recorded on a Bruker® Avance III 500 MHz spectrometer using TMS as an internal standard. High-Resolution Mass Spectra (HRMS) were obtained on a Shimadzu LCMS-9050. Optical rotation was recorded on a Rudolph Autopol II polarimeter.
3.2. Procedures:
3.2.1. Dihydrotestosterone Acetate (3):
In a round-bottom flask, 5.8 g (20.0 mmol) of 2 was dissolved in 32.0 ml (400 mmol) dry pyridine, and 18.8 ml (200 mmol) acetic anhydride were added. The reaction mixture was stirred at room temperature until the reaction is completed (24h, TLC). The excess pyridine and acetic acid were evaporated under reduced pressure. The residue was then dissolved in ethyl acetate, and washed consecutively with acidified water (pH = 3), 10%Na2CO3 and water to a neutral pH. The organic layer was dried with Na2SO4 an evaporated under reduced pressure. The resulting crystals were recrystallized from ethyl acetate. Dihydrotestosterone acetate (3) was obtained as white crystals (yield 5.5 g, 83%). 1H NMR (500 MHz, CDCl3) δ 4.58 (1H, t, 17-H), 2.43-2.06 (5H, m), 2.01 (3H, s, 17-Acetate-Me), 1.78-1.24 (13H), 1.21-1.12 (1H, m), 1.10-0.98 (4H), 0.97-0.84 (1H, m), 0.84-0.70 (4H). 13C NMR (126 MHz, CDCl3) δ 211.86 (C, C3), 171.18 (C, OC(O)CH3), 82.72 (CH, C17), 53.73, 50.58, 46.62, 44.65, 42.62, 38.50, 38.11, 36.83, 35.72, 35.18, 31.22, 28.77, 27.53, 23.52, 21.17, 20.91, 12.12, 11.47.
3.2.2. 17β-. acetoxy-5α-androst-1en-3-one (4a):
In a round-bottom flask, 5.0 g (15.0 mmol) of 3 was dissolved in 70 ml dry dioxane and 4.8 g (21.0 mmol) DDQ were added. Reaction mixture was refluxed for 24h. After filtration dioxane was evaporated under reduced pressure and ethyl acetate was added. The organic solution was washed with 10%Na2CO3, water to neutral pH and dried with Na2SO4. After evaporation of the solvent, the crude product was purified via column chromatography (mobile phase – benzene/ethyl acetate = 3.5/1.5) to give light yellow crystals of 17β-acetoxy -5α-androst-1en-3-one (4a) in 38% yield (1.9 g). 1H NMR (500 MHz, CDCl3) δ 7.14(1H, d, J = 10.2 Hz, 1-H), 5.84 (1H, d, J = 10.2 Hz, 2-H), 4.64–4.58 (1H, m, 17-H), 2.41–2.32 (1H, m), 2.25–2.12 (2H, m), 2.06–2.03 (3H, s, OC(O)CH3), 1.97–1.89 (1H, m), 1.84–1.60 (5H), 1.57–1.18 (8H), 1.15–1.07 (1H, m), 1.02 (3H, s, 19-Me), 0.83 (3H, s, 18-Me). 13C NMR (126 MHz, CDCl3) δ 200.07 (C, C3), 171.18 (C, OC(O)CH3), 158.22 (CH, C1), 127.47 (CH, C2), 82.55 (CH, C17), 50.70, 49.94, 44.27, 42.76, 40.95, 39.00, 36.76, 35.46, 30.82, 27.53, 27.48, 23.44, 21.17, 20.74, 13.03, 12.25.
3.2.3. 17β-. hydroxy-1-oxo-1,3-seco-A-nor-5α-androstan-2-oic acid (5):
In a round-bottom flask, 1.7 g (5 mmol) of 4a was dissolved in 30 ml isopropyl alcohol and 1.1 g (10 mmol) Na2CO3 dissolved in 10 ml water were added. The mixture was heated to reflux under stirring. A warm solution of 7.7g (36 mmol) NaIO4 and 0.05 g (0.32 mmol) KMnO4 in 100 ml water was added dropwise within 45 min. After the addition, the reaction mixture was heated for 1 hour. The mixture was slowly cooled to room temperature and the inorganic salts were filtered. Isopropyl alcohol was evaporated under reduced pressure, the residual aqueous solution was acidified to pH = 3 with HCl (1:1) and extracted with ethyl acetate. Organic layer was then washed with brine to pH = 5 and dried with Na2SO4. After evaporation, tert-butyl-methyl ether (10 ml) was added to the residual oil and white crystals of 17β-hydroxy-1-oxo-1,3-seco-A-nor-5α-androstan-2-oic acid (5) were isolated within an hour (0.5 g, yield 36%). 1H NMR (500 MHz, DMSO) δ 12.05 (1H, s, COOH), 9.15 (1H, s, 1-H), 4.34 (1H, d), 3.34 (1H, td), 3.23 (1H, s), 2.40 (1H, dt), 2.01–1.92 (1H, m), 1.81–1.66 (3H), 1.55 (3H, dt), 1.40 (1H, ddd), 1.26–1.01 (6H), 0.83 (4H), 0.72 (3H, s, 19-Me), 0.52 (3H, s, 18-Me). 13C NMR (126 MHz, DMSO) δ 207.40 (C, C1), 173.82 (C, COOH), 80.35 (CH, C17), 52.83, 50.65, 46.07, 43.13, 37.25, 36.68, 36.61, 34.04, 30.70, 30.11, 26.63, 23.40, 23.28, 11.67, 8.06.
3.2.4. 17β-. hydroxy-2-oxa-5α-androstan-3-one (6):
In a round-bottom flask, 0.3 g (1 mmol) of 5 was dissolved in 6 ml ethanol and 5 ml 1M NaOH and 0.15 g (3.8 mmol) NaBH4 was added. The reaction mixture was heated to reflux for 2-4 hours (TLC). After complete consumption of the starting material, the reaction mixture was cooled to room temperature, acidified to pH = 3 with HCl (1:1) and stirred for additional 30 min. The acidified solution was then extracted with ethyl acetate and the organic layer was washed with water to neutral pH and dried with Na2SO4. After evaporation the crude product was recrystallized from ethyl acetate. 17β-hydroxy-2-oxa-5α-androstan-3-one (6) was obtained as white crystals (0.26 g, 90% yield), m.p. 201-202°C, [α]D25 = +0.78 (c = 0.5 mg/mL, CHCl3) {Lit. m.p. 198-203°C, [α]D25 = +1.0 (c = 1%, CHCl3) Pappo & Jung [7]} 1H NMR (500 MHz, CDCl3) δ 4.24 (1H, d, J = 10.7Hz, 1-H), 3.94 (1H, d, J = 10.7Hz, 1-H), 3.64 (1H, t, 17-H), 2.52 (1H, dd), 2.22 (1H, dt), 2.10–2.00 (1H, m), 1.87–1.56 (5H), 1.52–1.38 (4H), 1.35–1.15 (3H), 1.12–1.02 (1H, m), 1.00 (3H, s, 19-Me), 0.97–0.78 (3H, m), 0.75 (3H, s, 18-Me). 13C NMR (126 MHz, CDCl3) δ 170.59 (C, C3), 81.68 (CH, C17), 81.02 (CH2, C1), 50.55, 49.75, 42.81, 40.41, 36.24, 34.81, 34.71, 33.79, 30.58, 30.27, 27.08, 23.35, 20.94, 11.07, 10.16. HRMS (ESI) m/z calculated for [M+H]+ C18H29O3+: 293.21112; found: [M+H]+ 293.21104.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, NMR spectra of compounds 2, 3, 4a, 4b, 5 and 6.
Author Contributions
Conceptualization, M.B. and G.D.; methodology, M.B. and S.S.; resources, M.B. and G.D.; data curation, S.S.; writing—original draft preparation, S.S.; writing—review and editing, M.B. and G.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Charles, K. 1976.
- Julius, A. Vida, Androgens and Anabolic Agents: Chemistry and Pharmacology. Academic Press 1969.
- Fox, M.; Minot, A.S.; Liddle, G.W. Oxandrolone: a potent anabolic steroid of novel chemical configuration. J. Clin. Endocrinol. Metab. 1962, 22, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Nutting, E.; Calhoun, D. Estradienes and 2-Oxaestradienes. Potent Oral Anabolic-Androgenic Agents. Endocrinology 1969, 84, 441–442. [Google Scholar] [CrossRef] [PubMed]
- Kam, P.C.A. & Yarrow, M. Anabolic steroid abuse: physiological and anaesthetic considerations. Anaesthesia 2005, 60, 685–692. [Google Scholar] [PubMed]
- Lennon, H. Effects of various 17-alpha-alkyl substitutions and structural modifications of steroids on sulfobromophthalein (BSP) retention in rabbits. Steroids 1966, 7, 157–70. [Google Scholar] [CrossRef] [PubMed]
- Pappo, R.; Jung, C. 2-oxasteroids: a new class of biologically active compounds. Tetrahedron Letters 1962, 9, 365–371. [Google Scholar] [CrossRef]
- Hara, S; Baeyer-Villiger Reaction of 2-Oxo-A-norsteroids. Chem. Pharm. Bull. 1964, 12, 1531. [CrossRef] [PubMed]
- Rogic, M. Reaction of phenylmagnesium bromide with six- and five-membered steroid lactones. Bull. Chem. Soc. Serb. 1964, 29, 57–71. [Google Scholar]
- Turner, A.; Ringold, H. Applications of high-potential quinones. Part I. The mechanism of dehydrogenation of steroidal ketones by 2,3-dichloro-5,6-dicyanobenzoquinone. J. Chem. Soc. 1730. [Google Scholar]
- VanRheenen, V.; Kelly, R.; Cha, D. An improved catalytic OsO4 oxidation of olefins to cis-1,2-glycol using tertiary amine as the oxidant. Tetr. Lett. 1976, 23, 1–73. [Google Scholar]
- Shibata, K.; Takegawa, S.; Koizumi, N.; Yamakoshi, N.; Shimazawa, E. Antiandrogen. I. 2-azapregnane and 2-oxapregnane steroids. Chem. Pharm. Bull. 1992, 40, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Pappo, R.; Allen, D.; Lemieux, R.; Johnson, W.
- Bolt, C. O-hetero-analogues of steroids. Recl. Trav. Chim. Pays-Bas 1951, 70, 940–948. [Google Scholar] [CrossRef]
- Atwater, N.; Ralls, J. 4-Oxasteroid Analogs. J. Am. Chem. Soc. 1960, 82, 2011–2014. [Google Scholar] [CrossRef]
- Lao, K.; Sun, J.; Wang, C.; Wang, Y.; You, Q.; Xiao, H.; Xiang, H. Design, synthesis and biological evaluation of novel 3-oxo-4-oxa-5α-androst-17β-amide derivatives as dual 5α-reductase inhibitors and androgen receptor antagonists. Bioorg. Med. Chem. Lett. 2017, 27, 4212–4217. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structure of 2-oxa steroids. (a) 17β -hydroxy-17α-methyl-2-oxa-5α-androstan-3-one – Oxandrolone (1); (b) 17β-hydroxy-2-oxa-5α-androstan-3-one (6).
Figure 1.
Structure of 2-oxa steroids. (a) 17β -hydroxy-17α-methyl-2-oxa-5α-androstan-3-one – Oxandrolone (1); (b) 17β-hydroxy-2-oxa-5α-androstan-3-one (6).
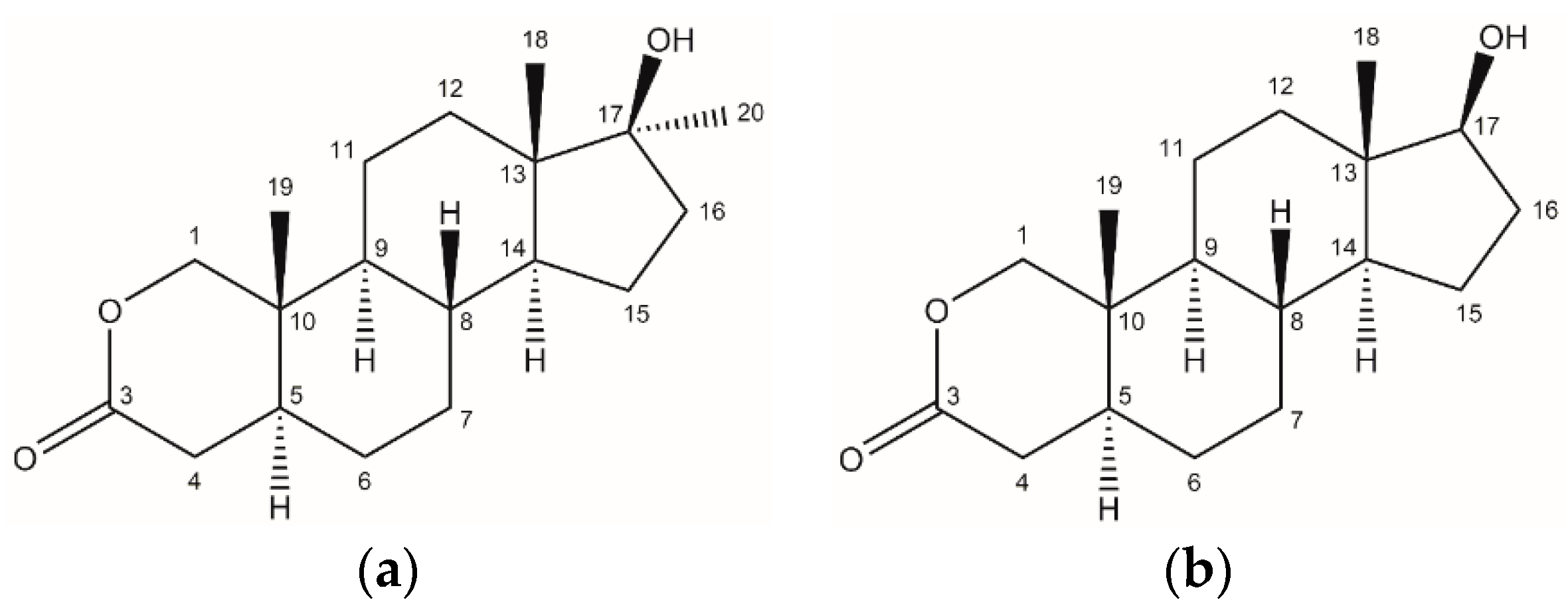
Scheme 1.
General procedure for the synthesis of lactones from the corresponding α,β-unsaturated ketone.
Scheme 1.
General procedure for the synthesis of lactones from the corresponding α,β-unsaturated ketone.
Scheme 2.
Synthesis of 17β-hydroxy-2-oxa-5α-androst-3-one (6). Reaction conditions: a) Ac2O, pyridine, 24h; b) DDQ, dioxane; c) KMnO4/NaIO4, Na2CO3, i-PrOH/H2O; d) NaBH4, NaOH, H2O.
Scheme 2.
Synthesis of 17β-hydroxy-2-oxa-5α-androst-3-one (6). Reaction conditions: a) Ac2O, pyridine, 24h; b) DDQ, dioxane; c) KMnO4/NaIO4, Na2CO3, i-PrOH/H2O; d) NaBH4, NaOH, H2O.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



