Submitted:
24 November 2024
Posted:
25 November 2024
Read the latest preprint version here
Abstract
Cancer therapy evolution has always been about combinations, Timing, and Sequencing. Identifying the optimum combination by clinical trials will take an indeterminate amount of time with the “web” of effective available therapy options today. Additionally, newer targets and effective drugs are added continually, making optimum evidence-based therapy challenging to catch up with. In such a situation, revisiting the foundational aspects of cancer therapy approaches to create actionable trial scaffolds for early and definitive conclusions about the optimum treatment would be appropriate. The trial foundational platform should also allow for the incorporation of effective treatments that will come up in the future. Four fundamental changes exist in cancer development and progression, namely vasculature-metabolic-immune-phenotypic cascades. Hypoxia is the maestro orchestrating interwoven changes through the master manipulator - HIF-1 α. Hypoxia in Cancer is like spider-weaving resistant webs with the potential to overcome every therapy intervention. This review conceptualizes bridging foundational and actionable approaches in cancer therapy to provide a scaffold for stepwise Combinations, Timing, and Sequencing (CTS) strategy with early resolutions in animal/clinical trials. Fundamentally, based on the golden principle of first opportunity, it is the best time to handle and reverse the cancer process’s ramifications. A shift in focus is proposed in the present article from the Tumor Microenvironment (TME) to the Cancer Cell Microenvironment (CCME).
Keywords:
Hypoxia
; HIF-1 α
; vascular-normalization
; clinical trials
; Antiangiogenics
; Chemotherapy
; RT-SBRT
; Immunotherapy
; Nanoparticles
; Vaccines
; CAR-T-Therapy
INTRODUCTION
Cancer therapy evolution has always been about combinations, Timing, and Sequencing. Today, there is a “web of possible combinations” of effective therapies, and identifying the optimum combination through clinical trials will take an indeterminate time. New targets and effective drugs are also continually added, making finding optimum evidence-based therapy like catching up with a moving target.
Angiogenesis is an integral part of the body’s healing and tissue regeneration mechanism, which is an ordinarily well-orchestrated series of events to provide additional oxygen and nutrients to the fast-multiplying healing cells. It is a well-balanced process between growth factors and inhibitors. The unfolding of Angiogenesis involves migration, proliferation, and differentiation of endothelial cells (ECs), forming lumen-bearing tubes with stable branching [1] structure covered by duly formed encasing cells - the pericytes. However, in Cancer, neoangiogenesis (pathological Angiogenesis) or tumor angiogenesis is the process that has gone haywire to a variable degree. Consequently, tumor vasculature ends up as an unstable, chaotic network with the features of tortuous tubes with varying lumen size, functionally leaky due to weak endothelial junctions, supported by a thicker basement membrane topped by deficient pericyte coverage, resulting in a vasculature that is “aberrant” structurally and functionally to a varying degree When the HIF-1 α fails to degrade in cancer initiation due to persistent excessive ROS production, it initiates downstream metabolic and tumor neoangiogenesis changes. The onset of Aberrant Angiogenesis, in turn, stimulates HIF1-α, leading to geometric resistance to flow, inadequate perfusion, tumor progression, and resistance, setting up a vicious cycle [2].
Radioresistance can manifest when the intercapillary distance exceeds 100 microns. With intercapillary distances of more than 140 microns in a breast xenograft, the arterial end of microvessels has Hypoxia. Hypoxia is additionally aggravated by blood flow reduction due to vessel tortuosity, arterio-venous shunt, abnormal red blood cell flux, and tumor microvessel elongation by fast-proliferating cancer cells [3]. The average size of tumor cells can vary from about 20 to 30 microns, depending on the site of origin [4]. Therefore, hypoxia resistance sets in when the tumor size approaches 4 to 7 cell clumps, far ahead of the imaging detection levels. The onset of Hypoxia, which leads to the accumulation of HIF-1α, is the Basic foundational change, a harbinger of the cascade of vascular-metabolic-immunological-phenotypic (VMIP) cancer cell microenvironment (CCME) and Tumor microenvironment (TME) changes.
The first foundational change is the vascular one. The vascular component is the crucial change of the five primary and critical changes in cancer progression - i.e., vascular, metabolic, immunological, mechanical, and phenotypic changes. Because of this, vascular normalization remains a desideratum for retracing the immunological escape of cancer cells and reversing resistant phenotypic changes in the Tumor and its microenvironment (TME) [5].
The second is bypassing the normal glucose metabolism for faster proliferation, switching to lactate production even in the presence of oxygen, described as the Warburg effect. These metabolic changes give an advantage in producing energy even in hypoxic conditions. The cancer cell microenvironment (CCME) also changes to acidic pH, leading to an immunosuppressive milieu [6].
The third foundational alteration is the phenotypic changes leading to increasing therapy resistance. The development of tumor heterogeneity with clones and subclones, each resistant to a particular therapy, results from complex human cell biological evolutionary changes from which the cancer cells inherit the legacy. Extensive intratumoral heterogeneity random event evolutionary biological clonal adoptions limit the cancer progress predictability. Additionally, metastatic clones take another evolutionary dimension [7]. Increasing resistant phenotypes develop with accelerating Hypoxia with each suboptimal therapy intervention. Hypoxia is the maestro orchestrating vasculo-immuno-phenotypic-metabolic changes via the master manipulator - HIF-1 α Hypoxia in Cancer is like spider-weaving resistant webs to overcome every therapy intervention, and the only way to ensure the snuffing of potential evolutionary mutational clones is an upfront appropriate stepwise approach.
The fourthfundamental change is the cancer cell manipulating immune cells in its microenvironment to immune suppressive ones assisted by various cytokines.
The fifth foundational transformation is the cancer tumor developing the physical/mechanical and mechano-biological coupling, signal transduction barriers in its microenvironment (TME), preventing the systemically administered “drug(s)” delivery to the cancer cell microenvironment (CCME), however effective it may be in in-vitro trial settings. Targeting and overcoming these barriers is an essential precondition for “drugs” to reach the cancer cells. The same fate awaits the tumor-infiltrating immune lymphocytes/cells (TILs), preventing them from physically contacting the cancer cells. Additionally, inflammatory dead cell products, tumoral edema, etc., increase interstitial pressure (ISP). Further, increased ISP, in turn, compresses the lymphatics, setting up a vicious cycle of mounting edema impacting the drugs and TILs seeping into the CCME [8].
The sixth factor is the availability of innumerable treatment modalities and possible combinations thereof. Additionally, newer treatment approaches are added continually.
The present review conceptualizes bridging these foundational adaptations in cancer cells and TME with actionable approaches (discussed below) to provide a classification and scaffold for stepwise Combinations, Timing, and Sequencing (CTS) strategy with rapid resolutions with animal/clinical research integration Worldwide.
Part I: Foundational Analysis
A. Basic Change – Hypoxia the “Spider” & HIF -1 α the Spinneret and Tumor-Angiogenesis
Tumor cell hypoxia is a crucial driver of neoangiogenesis primarily through stabilization of HIF-1 α as and when the cancer cells have low oxygen [9]. From then on, HIF -1 α transactivates hundreds of pro-angiogenic genes, including growth factors, and intensifies tumor angiogenesis [1]. Therefore, upregulation of hypoxia-induced HIF-1 α transcription factor is central in the cellular response, having multiple downstream effects on VEGFA, VEGFRs, PlGF, Ang-1/2, and PDGF, TGF-β, etc., which are intimately involved in promoting cell survival, endothelial cell migration, anaerobic metabolism, and metastasis [10].
The core difference between angiogenesis [1] in the healing of tissue essential for restoring normal tissue and Tumor angiogenesis/neoangiogenesis is the malformed, leaky, tortuous, non-functional blood vessels in the latter situation, considered an aberrant angiogenesis (Figure-1{2} & Figure-2 {1,2}). Aberrant Angiogenesis fits into the basic modulation microenvironment by cancer cells bypassing the more intensive normal process, like metabolism, where glycolysis is invoked for energy generation, with quality being sacrificed for growth in quantity. Additionally, these changes facilitate cancer cells’ survival, making them resistant to therapies and normal homeostatic control [11]. Following this, the cancer mass transforms into a bag of varied sub-clonal cells ranging from hypoxic-anoxic and dormant to resistant-proliferating, receptor-negative to receptor-positive stem cells that can evolve mutationally with every therapy intervention. Treatment response can become deceptive, with only susceptible subclones of oxic, receptor/checkpoint-positive, and stem cells in proliferation responding to the treatment. The non-responding cells can widen their resistance, escaping in the web of mutations (Figure 1). The origin of recurrence from the cells in the necrotic wall (probably the most resistant of all cancer cells) is described in the 1960s textbook by Ralston Paterson [12] and holds even today.
The intricate process of aberrant Angiogenesis involves VEGF (Figure 1), Notch, and Ang signaling precipitated and sustained by Hypoxia, orchestrated through an inducible factor (HIF1-α). The VEGF spectrum is primarily made up of VEGF A, B, C, D, and placental growth factor (PlGF), a central driver being VEGF through receptors VEGFR 1, 2, and 3 ending up with intracellular activation of PI3K, PKC, and RAS/RAF/ERK/MAPK pathways [13]. Alternative paths include cyclooxygenase-2 (COX-2) blockade, oligo¬nucleotide complementary to the miRNA competing with the mRNA target, and the inhibition of matrix metal¬loproteinases (MMPs) [13]. Additionally, 18 glycoproteins of the FGF family of growth factors interacting with four transmembrane receptors, FGFR1 to FGFR-4, is also a potent moderator of tumor angiogenesis independent of VEGF signaling [14]. Further, other complementary pathways are described, which depend on critical proteins such as platelet-derived growth factor (PDGF) influencing pericyte recruitment and coverage, fibroblast growth factor (FGF), and various inflammatory mediators of Angiogenesis, etc. [10]. Furthermore, there are stromal pathways where bone marrow-derived cells (BMDCs) are a source of endothelial and pericyte progenitors and pro-angiogenic, tumor-infiltrating immune cells (TILs). Ang-2 increases the expression of the pro-angiogenic genetic phenotype of Tie2+ monocytes/macrophages associated with the blood vessels [10].
2). Complexities of Tumor Angiogenesis: Angiogenesis, a broad term, is the formation of new vasculature with a full-fledged lumen from the existing one. The organization happens either by sprouting of “tip cells” supported by the growth of “stalk cells” for elongation from the side of the blood vessel (sprouting Angiogenesis) by activation of endothelial cells or by intussusceptive Angiogenesis (also known as splitting Angiogenesis) by splitting existing blood-vessel into two. On the other hand, Vasculogenesis is the process where precursor cells differentiate to become endothelial cells. Neo-angiogenesis is the acknowledged characteristic of cancer growth and is considered “aberrant angiogenesis” given its abnormal nature. Thus, Angiogenesis or neo-angiogenesis may end up as “aberrant angiogenesis” or “normal vasculature,” depending on the modulation of intricate pathways. The term neo-vascularization encompasses both neo-angiogenesis and Vasculogenesis [15].
In its quest for easy access to the vasculature, cancer cells adapt to all the available methods enumerated below under the influence of HIF-1 α.
- Vascular Co-option: Solid tumors originating and growing along pre-existing vasculature is a phenomenon of “vessel co-option,” which increases its metastatic potential [13]. Vascular co-option has the advantage of immediate access to vascular supply. Vessel co-option shows inherent resistance to AAGs since co-option is independent of the angiogenic switch and angiogenic growth factors. However, vessel co-option may tend to follow rather than precede AAG. For example, an anti-VEGF antibody in glioblastoma increased vessel co-option [16]. Yet, the co-opted vessels initiate an Ang-2 mediated apoptotic cascade, resulting in regression of the co-opted vessels soon followed by hypoxic tumor cells expressing VEGF, giving rise to further neo-angiogenic response [17].
- Aberrant Angiogenesis: Of the two types of Angiogenesis, sprouting and splitting/ intussusceptive, the latter carries less metabolic demand since no endothelial proliferation is required, and tumors may prefer the latter during AAG therapy. Post-TKI therapy relapse may happen with extensive splitting Angiogenesis [17]. Angiopoietins, Ang-1, Ang-2, and tyrosine kinase receptor (Tie-2) axis in association with vascular endothelial growth factor (VEGF) play a significant role in NAG. Ang-2 initiates NAG in Response to hypoxia/injury. During the normal healing process, these newly formed blood vessels get “matured” and are selectively “sealed” by the Ang-1, and Ang-2 essentially gets restricted at this maturation stage [18]. In Cancer, Ang-2 outstrips Ang-1, leading to deprivation of the maturation process, resulting in progressively immature, thin-walled, tortuous, functionally deficient leaky blood vessels with aggregated endothelial cells covered by defective pericytes low in Ang-1 levels. Ang-2 overexpressed and Ang-1 deficient mice tended to die early of hemorrhage. Ang-2 also facilitates infiltration of myeloid cells, which later polarize into Tumor-Associated Macrophages (TAMS), weakening the local immunity induced through integrin pathway expression of matrix metallopeptidases, promoting invasion and metastases. Hypoxia is the potent inducer of Ang-2. Soluble Tie-2, Ang-2 neutralization antibodies, and small peptides have shown antitumor effectiveness, offering a promising strategy [18].
After anti-VEGF bevacizumab treatment of glioblastoma, recurrence is highly infiltrative, and salvage surgical excision and chemotherapy become ineffective. A study when U87MG glioma–bearing athymic mice were treated with the anti-VEGF agent aflibercept showed that with the extended treatment of 6 weeks, there was enhanced invasion with TEMs migration, a dramatic increase in Ang-2 expression mainly in the invasive periphery, possibly due to increasing VEGF levels but not with a short treatment of 3 weeks. During this period, Ang-1 expression continued to remain low. A similar observation of increased Ang-2 was seen with bevacizumab and not with temozolomide. Further, targeting this Ang2/Tie2 axis to counter the invasive phenotype in AAG therapies prolonged survival in mice [19]. Thus, the literature indicates that the role of Ang-1 is crucial in vascular normalization. However, in a study, stabilizing Ang-1 signals did not contribute to survival [20], showing that studies are required to unravel the complex interaction between Ang-1 and Ang-2.
The role of the other two angiopoietins, Ang-3 and Ang-4, is yet to be understood. In a study, Ang-3 reduced Angiogenesis and pulmonary metastases [18] and is worth exploring.
- c.
- Vasculogenesis: In this type of neoangiogenesis, precursor cells differentiate into endothelial cells. Contribution to Vasculogenesis can come from the differentiation of hemopoiesis (stem) cells or cancer stem cells by direct endothelial differentiation. The latter’s role is emerging as a powerful mechanism for tumor progression. This Vasculogenesis could not be affected by anti-VEGF bevacizumab. Still, it could be inhibited by tyrosine kinase inhibitor sunitinib or the anti-VEGF-receptor-2 neutralizing antibody, suggesting that Vasculogenesis is a VEGF-independent mechanism [21] and targets precisely.
- d.
- Vascular mimicry: There is a process of cancer cells forming vascular-like structures independently of neo-angiogenic blood vessels. Vascular mimicry was shown to get upregulated following treatment with bevacizumab or induction of Hypoxia during the resistance phase [16]. It is proposed that vasculogenic mimicry might depend on cancer stem cells since classical angiogenics has no role in vasculogenic mimicry [17].
3). The Inception of the Antiangiogenesis concept: The manifestation of increased HIF-1 α in terms of metabolic changes in cancer cells would have been more amenable for targeting the carcinogenesis reversal. However, when the “immune escape” [22] breakthrough happens with increased multiplication of cancer cells and goes beyond about three cell layers or a distance of more than 100 µ from the nearest capillary, HIF-1 α shifts the gear to the next level, forming new blood vessels (neoangiogenesis/tumor angiogenesis) (Figure 2 {A, B, C}). Since this is the first foundational and fundamental change that impacts every therapy intervention, it will be dealt with in detail in this review
The introduction of the antiangiogenic (AAG) concept was very innovative and promising; theoretically, depriving oxygen and nutrition to the cancer mass is expected to make the latter wither dramatically. Initial work on this concept in preclinical studies raised immense expectations but generally failed in a clinical situation, with only a modest gain in clinical benefit. Although the clinical role of anti-angiogenics (AAGs) is undisputed today in selected indications, it is necessary to optimize its effects. Following the initial response from the cutting off the feeding vasculature, the resistance almost invariably appears after a while due to a dramatic increase in hypoxic cells. The effect was more dramatic with total “vascular disruption” trials where rebound growth was observed in the viable edge of the cancer mass [23]. This reactive proliferation would induce “evasive resistance” due to the establishment of the alternative route(s) of progression by cancer cells with aggravation of deficiency in nutrient supply [14]. In other words, the therapy approach with the original concept of cutting off the vasculature to the Tumor might help the “spider” to weave additional resistant pathways after a short-term control [24].
4). The Changing Concept of Antiangiogenesis (From Vascular Disruption to Normalization): Enduring responses to AAGs do not happen due to intrinsic “evasive” resistance, or the initial response ends with continued growth while still on treatment due to “acquired” resistance following mutational alteration of the gene encoding a drug target or by modifications in drug uptake and efflux [13]. Tumor growth did not slow down even with extensive pruning of vessels by anti-VEGF [20]. The AAGs, over the long term, may lead to aggravation of tumor hypoxia [25]. High Ang-2 levels correlated with resistance to anti-VEGF therapy, and Ang-2 countered the anti-VEGFR2 treatment-induced initial normalization in mice bearing gliomas [20].
Hypoxia generated ensuing tumor angiogenesis inhibition by vascular disruption is a powerful trigger for setting in the alternative or latent, innumerable pathways that make tumors more aggressive, metastatic, and progressively less sensitive to AAG. The ability of normal and Cancer cell biological systems to reroute through the existing local bypass, activate latent pathways, or adapt to genetic or environmental perturbations is a poorly understood process of adaptive evolution at the molecular level [26]. The development of resistance to AAG can be mainly classified as VEGF and non-VEGF-dependent pathways [10]. Enhanced expression of angiogenic cytokines, such as VEGF and PlGF, or recruitment of pre-metastatic niche EPCs can be induced by AAG [17].
Epithelial-mesenchymal transition in TME also contributes to the aggressive phenotype of surviving tumor cells. The possible mechanisms are the upregulation of a network of alternative angiogenic factors, up-regulation of HIF-1a and over-pruning of blood vessels, TME changes due to expansion and recruitment of myeloid cells, and CAFs, including increased co-option/ mimicry, impediment in the delivery of AAG, etc., promoting further aberrant Angiogenesis, tumor growth, EMT transition, and metastasis [16].
Therefore, an alternative concept of normalization of vasculature for the control of Cancer came into being with worse outcomes for vascular shut-off [27]. RK Jain et al. (2005) popularized the evidence that specific antiangiogenic agents transiently “normalized” the aberrant tumor angiogenesis structure and function, alleviated Hypoxia, improving oxygen and drug delivery. This normalization of the vasculature effect also improved carefully scheduled conventional therapies like chemotherapy and radiotherapy. A better understanding of vascular normalization’s molecular and cellular underpinnings will be the foundation for other therapies [27].
5). Antiangiogenics Cyclicall Administration for sustained vascular normalization: In the first phase, following the evolution of the attempt to cut the blood supply to eliminate Cancer, VEGF became the preferred target since it is considered the prime driver. Soon, it was revealed that the initial response was followed by re-growth due to the development of numerous complementary VEGF and non-VEGF pathways angiogenic pathways [10]. Consequently, to block the vertical and horizontal parallel paths together, combination anti-angiogenics are tried. However, toxicity and subsequent development of resistance became an issue [10]. In clinical situations (unlike animal studies), bevacizumab improved survival only when administered with chemotherapy [17]. However, AAG can decrease the subsequent delivery of cytotoxic drugs by progressively reducing and cutting off the Tumor’s vasculature due to excessive Ang-2 accumulation and pericyte encasement.
In the second phase, a new aspect was introduced: the vascular normalization concept in 2001 by RK Jain et al. [11]. According to this concept, initially, VEGF inhibition altered the aberrant Angiogenesis towards normality by reduced vascular permeability, decreased interstitial pressure, enhanced oxygen perfusion, etc., leading to tumor response. Increased collagenase IV activity restored a thinner basement membrane and improved vascular pericyte coverage by upregulating Ang-1. However, with continued administration, VGFR blockade leads to Ang-2 accumulation, destabilizing blood vessels and compromising the survival benefit of VEGFR-2 inhibition due to increased vascular permeability. This action indicates the negating effects of Ang-2 on vascular normalization of the anti-VEGF blockade, aggravates Hypoxia, and makes cancer cells resistant, which winds up in the selection of more resistant invasive clones [17], contrary to the initial concept of AAG. Additionally, a hypoxic volume is refractory to subsequent radiotherapy and chemotherapy and contributes to cancer cell escape to an aggressive selection of stem-cell-like tumor cells.
However, there is evidence that long-term AAGs, even with continued pruning of vasculature, produce a small survival benefit beyond the progression [17], supporting the original concept of cutting off vasculature for tumor control. A third observation from the literature emerges that the tumor vasculature normalization phenotype becomes re-established upon discontinuing anti-VEGF-targeted therapy [11].
The vascular normalization persisted for at least 28 days after the start of AAGs, the reduction in relative tumor size reversed by day 56, and low permeability persisted till day 112. Therefore, the “vascular normalization time window” lasted at least 28 days. A short survival time in mice precludes validating these observations. Clinically, this phenomenon was demonstrated in patients who required toxicity-associated “drug holidays.” The critical observation was that the normalization phenotype got reversed during drug holidays and returned upon recommencement of therapy, as demonstrated in MRI. From these studies, a reasonable hypothesis emerges to prolong the usefulness of AAGs through strategic “drug holidays” [11]. Potentially, the cyclical administration of AAGs keeps the normalization in a continuum. Therefore, the antiangiogenic “cyclical” administration strategy can be used not only to optimize its effect but also to improve the impact of different treatments due to enhanced oxygen diffusion, the sensitivity of cancer cells, and drug delivery to the Cancer Cell MicroEnvironment (CCME) (Figure 2-{3,4,5,6,7,8}).
6). Mapping the tumor angiogenesis – Vascular Guided Anticancer Therapy: Modern imaging techniques have become essential in developing a model to answer and titrate the appropriate use of the AAG for long-lasting in vivo responses since there are limitations to mice longevity. Dynamic tracking of the “window for vascular normalization” will help determine the optimization of drugs, dosages [28], combinations, and cycles. Indirectly, the vasculature can be evaluated with dynamic contrast-enhanced perfusion magnetic resonance imaging (MRI) or perfusion computed tomography (CT) and PET or both to decide on the period of therapy and drug holidays [17]. CT perfusion study can estimate blood volume, fractional intravascular plasma volume, blood flow, and peak enhancement index. Dynamic contrast-enhanced CT (DCE CT) can evaluate intra-tumoral vascular physiological statuses such as perfusion, permeability surface area, and interstitial space. Using paramagnetic nanoparticles targeting avb3 integrin in positron emission or sonography with gas-filled microbubbles directed against a specific target and optical techniques can also track Angiogenesis directly [17].
Serum level of soluble VEGFR (sFlt1) produced by ECs, Ang 1/Ang 2 ratio, hypoxia-regulated Apelin and its mRNA circulating type IV collagen, Hypoxia correlating TSP-1 [28] will be convenient.
7. Relooking at Terminologies in Tumor Angiogenesis: The abnormal tumor Angiogenesis, as discussed above, has several types of pathological processes like vascular co-option; varied types of aberrant Angiogenesis with ANG-2, Ang-1, and pericyte timeline interplay; Vasculogenesis following precursor cell to endothelial differentiation and vascular mimicry by cancer stem cells [13,16,17,21].
The term antiangiogenics came into existence based on the earlier concept of handling tumor vasculature with the intent to cut off the blood supply and nutrition to starve the cancer mass and reverse the growth. However, subsequently, the strategy changed to normalize the vasculature to reduce Hypoxia [27], making the term antiangiogenic a misnomer in its present clinical applications. Mechanistically, Vasculature-modifying drugs target the Hypoxia and normalize the vasculature for better “drug(s)”/oxygen delivery and cancer cell sensitization to radiotherapy. The more appropriate term would be eu-angiogenics.
In the aberrant type, Angiogenesis is initiated by Ang-2 but leads to immature, ill-formed, thinner vessels in Cancer unless Ang-1 gets accumulated subsequently to complete the process of maturity of blood vessels. The term aberrant Angiogenesis is often used interchangeably with neo-angiogenesis in the description of cancer vasculature, making it difficult to interpret the underlying pathophysiological process at any given time when planning for therapeutic intervention.
In tumor angiogenesis, multiple underlying pathological changes need to be targeted, the final objective of which is to normalize the vasculature. Hence, the present author proposes the terminology eu-angiogenesis to highlight the reversal of abnormal vasculature and differentiate it from the normal angiogenic process. Also, the interventions involved in such an approach can be termed eu-angiogenics. EU-angiogenesis also indicates the intervention process sustaining normal Angiogenesis to prevent them from becoming aberrant or reversing to normalization from aberrant Angiogenesis or a range of combinations thereof. The strategies for synchronizing eu-angiogenic treatment approaches in combinatorial therapies are envisaged in the “implementation of strategies” section.
1) Lessons from long-term anti-angiogenics (AAGs) therapy resistance: In AAGs therapy, aggravation of intratumoral Hypoxia during the vascular regression phase leads to a concomitant increase in HIF-1 α followed by up-regulation of VEGF plays a crucial role in resistance. This upregulation, in turn, results in tumors acquiring more angiogenic and invasive potential, local and metastatic, after the initial response. AAG-induced Hypoxia also leads to downstream changes of over-expression of the tyrosine-protein kinase c-MET, stimulates β1 integrin expression (which in turn interacts with c-MET), decreases adherens junction protein expression affecting pericyte coverage favoring increased invasiveness and colonization [16]. In addition, tumor hypoxia favors angiogenic dormancy, tumor augmentation autophagy (a process of sequestration of dysfunctional cell components and their degradation), glycolysis-induced lactate production in TME aggravating the acidification, Cancer stem cell proliferation, lymph-angiogenesis promoting metastases, etc., all ensuring AAG therapy resistance [1].
Implication: Continuous long-term administration of AAGs can invoke self-resistance and mitigate response to combination therapy schedules when it is Part of the same.
2). Interplay of Ang-1-Ang-2-Tie2 axis: While inhibition of Ang-1 had minimal effect on tumor vasculature, the combined Ang-1 and Ang-2 blockade trims tumor blood vessels without normalization. Contrarily, inhibition of Ang-2 can enhance the interaction of Ang-1/Tie-2. Simultaneous activation of Tie-2 and blockade of Ang-2 in mice in some cancers resulted in more efficient normalization, improved blood perfusion, and reduced lactate acidosis, limiting tumor growth and metastasis. Another study in the mice model demonstrated that dual inhibition of VEGFRs and ANG-2 was more effective than single inhibition due to a widened normalization period [29]. In ovarian xenograft tumors, Ang-1/2 and Tie-2 receptor axis targeting with trebananib, a peptide-Fc fusion protein, reduced Angiogenesis and growth. It improved PFS but not OS in a phase III trial in recurrent ovarian Cancer [30], indicating that blanket Ang-1/2 and Tie2 axis block did not benefit the survival.
Implication: Theoretically, a stratagem synchronizing the Ang-2: Ang-1 optimum balance is paramount. After the initiation of neo-angiogenesis by Ang-2, boosting the accumulation of Ang-1 for thin vessels with tip cell formation in the later phase of neo-angiogenesis would optimize eu-angiogenesis. These harmonized events are the foundation for forming normalized vasculature with a thinner diameter and effective “tip cells,” leading to improved tumor perfusion, reduced interstitial pressure, increased drug delivery, etc.
3). Importance of Pericytes: In the symphony of normal angiogenesis formation, along with the Interplay of the Ang-1-Ang-2-Tie2 axis, the matched role of pericyte completes the swan dance-like steps. Aberrant Angiogenesis has pericytes that are deficient in number and function, the latter due to inadequate vessel coverage. Overall, pericyte depletion disrupts vascular integrity [31]. Pericyte coverage improves Within hours to a day or two. Endothelial cell perfusion function is restored, and vessels are normalized morphologically and functionally within a few days. However, around Day 28 [11], excessive pericyte proliferation and coverage of vessels set in with the return of the Hypoxia. Studies using double blockade with VGFR (endothelial cell targeting) and PDGFR (pericyte targeting) showed improved AAG therapy efficacy. Another study showed that targeting Ang-2 along with pericyte depletion restored vascular stability. In another study, tumor vasculature was enhanced in a breast carcinoma xenograft model using the TGF-β-fibronectin axis, improving pericyte-endothelium association.
Implication: There appears to be a delicate balance between initial deficient coverage and later excessive coverage of pericytes during AAG therapy (the latter coincides with the resistance phase). These observations specify a need for the proper therapy sequencing strategy aimed at initiation by Ang-2, vessel maturation by ang-1, followed by pericyte targeting to reduce its thickening at four weeks of AAG therapy, which can theoretically extend the normalization period required till the completion of combination therapies and cancer elimination.
4) Exploiting the “Normalization Window” (Figure 2 {3,4,5,6,7,8}): A phenomenon is observed in most antiangiogenics therapy where vasculature normalization begins almost immediately and ends by minimum day 28 primarily due to excessive pericyte proliferation - named “normalization window.” Cediranib caused a consistent and dramatic reduction in tumor enhancement within 24 h of therapy along with a decrease in vascular permeability, indicating the start of the normalization window conforms with the onset of normalization following blockade of the VEGF/VEGFR2 pathway in preclinical studies. Upregulation of tumor Ang-1 gene expression typically produced by perivascular cells (PVCs) is the crucial pathway during NW through binding and activating the Tie-2/TEK receptor on ECs. Reorganization of the basement membrane of vessels, which is usually haphazard and thick, is restored to a thinner and more closely associated one during NW commencing in 48–72 hours [11].
This vascular normalization “time window” persisted for at least 28 days, and although relative tumor vessel size was reversed by day 56, interestingly, vascular permeability remained low even till day 112 in the clinical study, not seen in animal studies due to short survival time in mice. Outstandingly, in patients who required “drug holidays” because of toxicity, the reversibility of vascular normalization was also demonstrated in this study with the reversal to normalization phenotype on MRI. Additionally, the cycle gets repeated upon recommencement of therapy [11].
One possible mediator of the escape from the normalization window includes hypoxia-driven bFGF. Temporal changes in circulating FGF2 levels with the VEGF axis inhibition are linked to disease progression seen in glioblastoma [10]. In the U87 xenograft model, one week of short-term VEGF blockade did not increase bFGF levels with a significant reduction of microvessel density, yet in 7 weeks of continued VEGF blockade, bFGF levels increased with microvessel density and tumor cell proliferation [14]. The literature indicates that the normalization process has 3 phases during AAG therapy.
The first normalization phase starts immediately and is known to last about four weeks, bringing down Hypoxia and ensuing improved cancer cell kill. Simultaneously, decreased interstitial pressure further improves oxygenation, reflecting on the cascading effects of TME hostile to the cancer cells. This scenario enhances the effectiveness of combinatorial radiation therapy and Chemotherapy/ immunotherapy [11].
In the following second phase, due to vascular pruning and shrinking of the vasculature, Hypoxia rebounds with resistant phenotypic changes, increasing the invasiveness of Cancer and creating an AAG treatment resistance milieu. The data about subsequent changes are restricted given the short life span of mice. Most reports show that continuing AAG therapy in this stage does not confer significant benefit, letting down the original concept of AAG therapy (that cutting off blood supply would be a strategy to eliminate Cancer). The time between the initiation of AAG and the onset of the second phase is the “normalization window” described by Jain RK et al. [27].
The third phase is substantiated by anecdotal literature. When AAG therapy is discontinued, there will be the return of normalization susceptible phenotypes, and the reintroduction of AAG can set in the process of 2nd “normalization window.” The evidence for restoration of normalization phenotypes comes from MRI studies in patients who were given drug holidays due to toxicity [11]. The present author believes that studying the process of 2nd and possibly further normalization windows is essential to adopt the “cyclical” treatment concept for AAGs.
Implications: Tumor vasculature origin and progression are pretty diverse. The angiopoietins, Ang-1 and Ang-2, ligands of the Tie-2, are known to play an indispensable role in Angiogenesis for recovery from injury. However, their role in the pathophysiology of diseases is quite complex. Primary requirements for any drug action to be effective are that the drug/nanoparticles/immune cells/cell therapies/vaccines should be able to penetrate the cancer cell microenvironment (CCME) by overcoming the physical barriers, should get distributed uniformly within the Tumor, achieve a concentration sufficient to act on cancer cells, and finally be retained in TME enough length of time. However, the vascular pruning action of AAG at the later phase requires strategic dosing schedules and vascular promotion interventions.
C. Foundational Factor 2: Metabolic Changes (Figure 1 {1})
Of the two sub-units of HIF-1, HIF-1α translocated to the cytoplasm, accumulates in the presence of Hypoxia without degradation, and renters the nucleus to integrate with HIF-1β, forming a heterodimer. In turn, the heterodimer promotes the expression of a series of target genes, Glucose Transporters (GLUTs), and glycolysis enzyme Lactate Dehydrogenase A (LDHA), initiating the Warburg effect. LDHA accelerates the conversion of pyruvate to lactate. The anaerobic glycolysis facilitates cancer cell proliferation, and lactate seeping into the TME by creating acidic pH changes produces profound and varied immunosuppressive cell accumulation, favoring genomic mutations and further promoting tumor growth and creation of resistant phenotypic subclones (Figure 1) [9,32]. Theoretically, the abovementioned changes would be reversed with the elimination of Hypoxia.
Widespread intratumor heterogeneity is clonal/subclonal expansion, which gives rise to the theory that Cancer is an evolutionary process that limits the cancer mutational path predictability. The complexity of the deterministic versus stochastic process leads to three fundamental changes in clonal selection, drift, and mutation, which influence cancer evolution, leading to progression, infiltration, metastases, dormancy, recurrence, and driver landscapes [7].
In the history of anticancer therapies, the initial euphoria of targeted therapy results has been belied by an increasing understanding of extensive intratumor genetic heterogeneity, possessing resistance to the drug in a minority of cells in pre-existing subclones. Also, the entire clonal composition cannot be measured precisely [33]. The present author, therefore, hypothesizes that the treatment’s initial core strategy is to prevent such an evolution by effectively targeting Hypoxia (the Spider), which weaves the web of resistant phenotypes, relegating the predictability of subclones to a secondary place since the exact entire clonal composition is not possible to measure. This Hypoxia targeting also increases the therapy sensitivity of the sub-clonal resistant cancer cell population and heralds the long-term control/cure of advanced malignancies.
E. Foundational Factor 4: TME Immunological Changes (Figure 1 {4})
Figure 1 depicts the HIF-1 α domino effects on immunosuppressive domino effects on TME immune cells, including the cytokines and chemokines involved. These changes in cancer cell microenvironments (CCME) are Part of the dynamic immunomodulation process.
Effects on Immune cells: Innate immune cells like mature dendritic cells (mDCs) and M1-like TAMs produce various cytokines such as IFN-α, IL-12, IL-18, TNF, and chemokines such as CXCL9, CXCL10, CCL21 which influences the phenotypic and functional features of tumor vasculature. Adaptive immune cells secrete cytokine IFN-γ, which induces TME vascular normalization. Additionally, mDC, CD8, and TH1 immune cells push macrophage polarization from M2 to M1 phenotype [34].
Endothelial cell effects: Interestingly, CD8+ CTLs and CD4+ T helper34 1 (TH1) cells assist in tumor vessel normalization with vessel phenotype representing high-plump endothelial cells (HEVs) functionally specialized in lymphocyte extravasations by producing IFN-γ in the TME. LTβR signaling pathway generates HEVs in TV with anti-VEGFR2 and anti-PD-L1 combined blockade. STING activation, simulator of IFN genes demonstrated synergism with anti-VEGFR2 and ICIs (either anti-PD-1 or anti-CTLA-4) through type-I IFN signaling activation and the upregulation of genes leading to endothelial-CD8+ CTLs interaction with complete regression of tumors resistant to either of the mono-therapies [34].
Pericyte effects: Depleting CD4+ TH1 cells decreases pericyte coverage and increases distorted vessels. IFN-γ signaling downregulates VEGF-A and simultaneously upregulates CXCL9, CXCL10, CXCL11, etc., with resultant pericyte recruitment along the endothelial cells (Ecs), finally leading to vascular maturation [34].
Immuno-modulatory effects: Immunomodulation is observed in mouse ECs in vitro induced by Interferon-γ (IFN-γ) and TNF in antigen uptake, processing, and presentation process. Also, IFN-γ brings about antigen degradation and antigen loading via ‘immunoproteasome’ [35].
Implication: Designing a combination of therapies to encourage IFN-γ and other cytokine/chemokine accumulation opens possibilities for targeting Hypoxia with added specific immune modulation towards cancer cell elimination.
F: Foundational Factor 5: TME Physical/Mechanical Barrier, Phagocytosis, Interstitial Pressure (Figure 4)
Tumor volume/TME: It comprises a heterogeneous group of cancer cells, vasculature, lymphatics, stromal fibroblasts later transformed to cancer-associated fibroblasts (CAFs), increasingly acidic interstitial fluid, immune modifying cell infiltrates, etc. As the cell mass and interstitial pressure increase, Hypoxia is amplified due to vasculature compression, and increased blood viscosity sets up a vicious cycle that progressively hinders systemically administered therapeutic agent (including nanotherapy particles) concentration in the TME [36]. Enhanced cancer cell-kill leads (e.g., by use of vascular nondisruptive Stereotactic radiotherapy immunogenic dose in the range of 6 Gy to 10 Gy per fraction [37] or Timing of radiotherapy/chemotherapy/ immunotherapy/nanoparticle drugs in the vascular normalization phase of AAG therapy) to decreased interstitial pressure (IP) followed by decompression of vasculature which sets-up a virtues cycle locally.
Fibroblastic physical barrier: Fibroblasts, the engineers of ECM responsible for tissue repair and homeostasis, transform into CAF in cancer promotion. The CAFs cross-link ECM increase its stiffness and, along with tumor cell contractility, induce Hypoxia, aberrant Angiogenesis, and immune cell trapping, setting the stage for cancer progression and metastases [38]. In SBRT, 12 Gy single dose triggered TGF-β release but not with 6 Gy in 1 fraction. An 8 Gy dose per fraction has minimal microvessels and endothelial cell lining damage, and in a study, 10 Gy in 1 fraction is found to be a threshold dose induction of endothelial apoptosis. Since TGF-β is crucial for radiation-induced fibrosis, SBRT-induced fibrosis can be prevented by limiting the dose per fraction below 10 Gy and further reduced by TGF-β blockers to leave an immune stimulatory supple ECM [39].
Professional Phagocytosis: Although phagocytosis is a general phenomenon, those cells that excel are from immune cell lineage, macrophages, neutrophils, and immature dendritic cells, are considered professional. Macrophages, the “big eaters” (which are countered by “don’t eat me” signaling from cancer cells), improve the efficacy of anticancer treatment if promoted. CAR-M has the potential to enhance phagocytosis and reduce interstitial pressure. However, in solid tumors, poor penetration with anomalous unequal distribution of “drugs” due to abnormal-dysfunctional vasculature, the physical barrier of ECM, and increased interstitial pressure (be it chemotherapy/immunotherapy/CAR-T/M/N or any vaccines/Cell therapies) “has always been an insurmountable difficulty.” CAR-M or ones tagged with nanoparticle penetration can be better yet fall short of the full potential. Normalization of the vasculature reduces interstitial pressure, improves oxygen perfusion, overcomes Physical barriers to drug transport, and enhances Tumor Infiltrating Immune cells (TILs), which can be amplified by CAR-M therapy [40]. CD47, CD24, MHC-I, PD-L1, STC-1, and GD2 have emerged as phagocytosis checkpoints, especially CD47, linking innate and adaptive in cancer immunotherapy [41]. This approach can straddle both immunological and interstitial pressure changes.
Implication: One way to further improve AAG’s normalization effect is to complement it with successful concurrent cancer cell killing. This Step can be considered as Vascular promotion, a Phase above the vascular normalization (beyond normalization). This association could be the one reason for positive results when combining AAGs with chemotherapy.
G: Foundational Factor 6: Web of Therapy Options (Figure 5)
Figure 5 shows the web of therapy options available today, and the newer targets are constantly getting added. Each has been found to have a role independently and is suitable for use in combination schedules. How could we decide on the optimum combination of these within a reasonable time?
Part II: Actionable “Block”
Over a hundred years of cancer management, a lesson learned is that searching for a single “magic bullet” has been elusive. Over the decades, improving response and survival has always been achieved through meticulous trials of combinations of available effective therapies. However, with the web of therapy options now available and burgeoning target lists (Figure 6), coming to a consensus about the most effective therapy option is a fast-moving goalpost practically impossible to achieve. This review explores the fast resolution of guidelines using the following strategies.
The above-detailed complex issues require bridging foundational issues with definitive, actionable points. The First Step would be classifying available methods (and the ones going to come up in the future) to fit into a “slot” in the primary mechanisms of eliminating Cancer. From the present author’s perspective, until now, the cancer research community worldwide has treated the situation by exploring the parts of the “elephant in the room” rather than visualizing the “elephant” in its entirety. The following are the fundamental, actionable points to be considered in comprehensively designing Cancer multiarm-multistage (MAMS) research arms, along with Artificial Intelligence and Machine Learning (AIML) algorithms, for fast-tracking the answers. Most of the discussion below revolves around antiangiogenics since they form the foundation of the normalization of the vasculature.
- A.
- Actionable Step 1: Normalization of vasculature & Targeting Hypoxia:
The first prerequisite is overcoming Hypoxia, thus restoring HIF-1 α normalcy. Therefore, the logical approach would be to effectively counter Hypoxia by normalizing perfusion, facilitating the immediate degradation of HIF-1 α on its formation. The other line of attack would be to target HIF-1α. There have been efforts to develop HIF inhibitors, e.g., by dissociating HIF2α and HIF1β dimers, leading to inhibition of transcriptional activation of HIF2. The treatment was successfully tried in multi-treated RCC patients and can be combined with classical antiangiogenic drugs or immunotherapies [1]. However, the best way to target HIF-1α is to simultaneously normalize the oxygenation on one side to restore normal homeostasis and reduce the ROS by reducing cancer inflammation on the other side. Vascular normalization is the sine-qua-non for reversing the cancer process and restoring normal homeostasis [5]. Antiangiogenics and many of the small molecule Tyrosine Kinase Inhibitors (TKIs) have normalization features of the vasculature during the defined period [Goel].
1. Critical Strategy of Cyclic antiangiogenics schedules (Figure 2 {3,4,5,6,7,8} and Figure 6 Sgtep 1): Generally, from day 1, with the start of AAGs, the vascularization to the Tumor improves. However, by day 28, due to the over-proliferation of pericytes, the lumen of the until now normalized vessels start to narrow, leading to an exaggeration of Hypoxia. While targeting aberrant Angiogenesis, identifying a selective therapeutic window is better than targeting pericyte coverage [16]. Continued administration of AAGs at this stage leads to anoxia, taking the tumor mass to palliative control. Stopping the AAG at this stage is expected to reverse this phenotypic-resistant process of vascular obstruction and re-establish vascular normalization for about four weeks. After these four weeks of drug holiday restarting, AAGs initiate the vascular normalization process again [11]. This observation allows us to keep the vasculature in a continuous normalization phase by cyclical administration of four-week AAGs on and off. This strategy makes cancer cells susceptible to radiotherapy/chemotherapy/ targeted drugs/ immunotherapy during their entire course of treatment. The normalization window was identified by fMRI imaging 2–4 days after starting sunitinib with improved perfusion, reduced Hypoxia, and an oxidative shift in redox status of metabolic pyruvate/lactate flux. Fluorodeoxyglucose (FDG)-Positron Emission Tomography (PET), a measure of reduced glucose uptake in cancer cells, is an indicator of vessel normalization [42]. Modern imaging and other techniques for tracking the vascular changes enumerated above, by so-called “vascular-guided anticancer therapy,” will be essential for the trials.
2. Benefits of Moderate/lower dose antiangiogenic therapy: Using low/moderate doses of AAGs is another approach to normalize aberrant tumor microvessels and optimize blood perfusion [43]. Also, low/moderate doses do not transgress into excessive pericyte proliferation. The normalization window is dose and time-dependent, with higher doses leading to pruning of excessive vasculature [44]. Excessive pruning by high-dose AAG results in exaggerated Hypoxia in a shortened normalization window period of four weeks and infiltration by immunosuppressive cells is observed after the normalization window period [29]. Short-term high-dose sunitinib (120 mg/kg per day) increased tumor growth, and low-dosage sunitinib (30 and 60 mg/kg per day) did not stimulate metastasis in mice models [16]. In glioblastoma, low-dose bevacizumab gave better results. Amassed data from other studies also show that low and not high doses facilitate tumor vessel normalization [29]. Ramjiawan et al. showed that lower doses of VEGFR-2 blockade led to increased perfusion and shift to M1-type TAMS and infiltration of CD8+ T-cells, in contrast, to reverse effects with expanded Tregs of a higher dose in breast cancer. In addition, the added cancer vaccine increased IFN-γ+CD8+T-cells and decreased Tregs, resulting in improved overall survival in mice with spontaneous MMTV-PyVT breast cancer [30]. It is not yet clear which dose level is optimal for normalization, but a lower dose of 5 mg/kg of bevacizumab when used in Colorectal Cancer or 15 mg/kg in lung cancer warrants further study [34]. All these studies indicate the potential of combining low-dose AAGs in combinatorial therapies, combining the effect of cell kill with eu-angiogenesis.
- B.
- Actionable Step 2: Vascular Promotion – Beyond Vascular Normalization – Effective Cancer Cell-Kill/Lysis (Figure 6 {Step2})
1. Synchronization (Timing) of combinations: Routinely used Radiotherapy/chemotherapy/ immunotherapy enhances the normalization window of antiangiogenics [11,45] by a more efficient distribution of the drug as well as increased cancer cell lysis and improved immunological reaction (local as well as distant). Sequencing and timing these therapies immediately after initiating AAGs leads to vascular promotion. Theoretically, SBRT with a vascular nondisruptive range (6 to 10 Gy per fraction) can be timed during the normalization window period of AAG as a boost dose to the residual disease in advanced malignancies and oligometastases [46], keeping in mind the possibility of reported increased intra-tumoral bleeding episodes near the critical structures.
Therefore, routine trimodal radiotherapy (including SBRT), chemo-targeted treatment, and immunotherapy promote vascular normalization by killing cancer cells, leading to fewer oxygen-demand cells and decreased pressure on the vasculature. These actions also activate the signals for a fresh network of vascular supply, inducing vascular promotion. Professional phagocytic agents can reinforce these vascular-metabolic-immune phenotypic (VMIP) alterations by reducing interstitial pressure [40]. TGF-β blockers can further reduce interstitial pressure by decreasing inflammation in the extracellular matrix (ECM inflammation) and improving uncollapsed lymphatic functions [47].
2. Fortification and extension of vascular normalization window: Several avenues exist for enhancing and extending vascular normalization to overcome cancer resistance. Some of them are: a). Angiostatic factors such as tumor necrosis factor α (TNFa), thrombospondin-1 (TSP-1), and endostatin improve vascular perfusion. b). Injection of tumor-infiltrating lymphocytes (TILS) and TNF-α to enhance in-vivo vaccination effect. c). LIGHT (also known as herpesvirus entry mediator ligand), activating various intermediaries, can repair abnormal tumor vasculature. d). ABT-510, one of the mimics of TSP-1, promotes normalization and immune modulation without reducing the vascular density. e). Recombinant human endostatin, an angiogenesis inhibitor, to restore vascular homeostasis [28].
The future lies in using miRNAs to detect subclinical disease and target effective treatments, including anticancer vaccines. The miRNAs can also restore the integrity of vasculature, e.g., the use of miR-20b to inhibit the nuclear aggregation of HIF-1α and activation of Transcription 3 (STAT3) [48]. Another group evaluated the suppression of miRNA-153 in preclinical breast cancer studies for HIF-1α & Ang-1 targeting [31]. Studies elucidate the impact of miRNA-140-5p silencing VEGF-A; miRNA-29b inhibiting all steps of Angiogenesis, impacting proliferation, migration, and tube formation by targeting Akt and down-regulating VEGF and c-Myc; overexpression of miRNA-497 reducing VEGF and HIF-1α, etc [31]. Yet another approach is to normalize the extracellular matrix (ECM), especially CAFs, to make them supple [28], facilitating unhindered vascular perfusion of nutrients and therapeutics.
3. Vascular Enhancement – Ang2-Ang1 interplay: Combining AAG and vascular normalizing drugs can extend the former’s effectiveness and the normalization window period. Theoretically, one such combination that has the potential to be explored is a combination of AAG along with appropriately timed Ang-1 to produce more viable vasculature during the window period. Even with prolonged treatment of pancreatic tumors in transgenic mouse models with anti-VEGFR2 antibody, a delay in growth and modest survival benefit was observed due to increased expression of the pro-angiogenic growth factors, Ang-1, Ephrin-A1, Ephrin-A2, and FGF1, FGF2a. Ang-1-Tie pathway results in the maturation or stabilization of blood vessels, which can be blocked by accumulated Ang-2 [16]. Increased expression of factors like Ang-1 antibody immunotherapy indicates the role of appropriately timed vascular maturation agents to reduce Hypoxia, which should be focused on along with synchronized blocking of both VEGF and Ang-2.
4. Gene modification has much potential in sustaining the normalization window. When disrupted, G-protein signaling 5 (Rgs5) could enhance the infiltration of effector immune cells into the tumor parenchyma, resulting in prolonged survival of tumor-bearing mice by an unknown mechanism [29]. The other approach would be to adopt Dual Recombinase Technology to preferentially radiosensitize tumor cells and protect the endothelium, especially during SBRT [24]. During SBRT, a dose range of 6 Gy to 10 Gy per fraction induces immunogenic cell death and vascularization without disruption of endothelium or stimulation of immunosuppressive pathways, retaining the supple ECM [39].
- C.
- Actionable Step 3: TME Immune Promotion – Immune Protection (Figure 6 {Step 3})
Normalization facilitates the delivery to the cancer cell microenvironment (CCME)) of immunotherapy, immunoadjuvants, newer nano-technological targeting, etc., and improves effectiveness. The vascular nondisruptive Immunogenic schedule of SBRT has the added advantage of in vivo therapeutic vaccine generation.
Infiltrating immune cells, stromal cells, and abnormal vascular and lymphatic vessels are the main components of TME, immunologically compromised by Hypoxia, with high interstitial pressure and a low pH [29]. As mentioned above, immunotherapy agents additionally influence vascular promotion, furthering their effectiveness in vascular promotion mechanisms. The NW period improves the immunological milieu of the Tumor. Also, resistant phenotypes get altered to sensitive ones. For example, PDL-1 expression was upregulated in Tumor endothelial cells after AAG treatment, with increased infiltration of CD4+ and CD8+ T cells resulting in improved tumor control following immunotherapy [29]. Combining AAG and immunotherapy agents like bevacizumab, atezolizumab, and apatinib with an anti-PD-L1 antibody could normalize the TME [14,28]. IFN-ϒ secreted by Th1 cells is positively associated with vessel normalization [45]. A virtuous cycle sets in when immunotherapy improves the vasculature, facilitating further infiltration of immune cells.
- D.
Improving survival in Cancer mandates planning to prevent late recurrences and the least or nil long-term side effects. The bedrock of this mandate is leaving a supple ECM after all the combination therapies. Therefore, planning for this Step starts from Actionable Step 1. Two factors significantly fulfill this criterion: ensuring vascular endothelial integrity (especially of the endothelial stem cells) and preventing fibrosis in ECM. When used, the dose per fraction of radiation in SBRT (total dose in regular radiotherapy) and the complex role of TGF-β on ECM play the leading role in ECM suppleness. TGF-β plays a significant role in normal homeostasis and can be responsible for post-therapy tissue inflammation and fibrosis. TGF-β blockers substantially keep the ECM supple, retaining the immunological cross-talk and avoiding breakthroughs in cancer cell dormancy [49]. The consolidation phase elements will include the role of metronomic chemotherapy [50], repurposing drugs [51], senolytics [52,53], lifestyle modifications [54], or a combination thereof, which needs to be considered to complete the cancer reversal process. The critical point in this phase is the yet-to-be-explored intense strategy to eliminate/control minimal surviving cancer cells in dormancy, considering the benefit versus toxicity of anticancer therapies.
Summary of Actionable Points - Optimizing vascular-metabolic-immune-genomic/phenotypic (VMIP) changes and innovations: Exploiting the VMIP interdependence is essential to improving the morphological and immunological tumor response and the possible abscopal effect. Focusing on combinatorial therapy on the component that primarily reduces Hypoxia, which in turn impacts both vascular and metabolic components, accelerates immunological interaction, and sensitizes phenotypically resistant genetically mutated clones of cancer cells, would fulfill the requirements of the VMIP model. For example, VEGFA and ANGPT2 blockade with added bispecific antibody A2 V resulted in enhanced infiltration of CD8+ T cells, leading to increased tumor antigen presentation, promoted perivascular T cell accumulation, and increased the tumor necrosis with normalized residual blood vessels [29].
A sufficient presence of Ang-1 leads to the maturation of vessels with robust “tip cells,” thinner vasculature penetrating deeper into the hypovascular/avascular Part of the Tumor. Theoretically, along with the restriction of Ang-2 at the maturation stage of the vasculature, Ang-1 would produce the eu-angiogenesis, overcoming the “trimming effect.” Timing of Ang-2 blockade and Ang-1 promotion along with AAG would again be an example of an innovative initiative [55].
With immunotherapy dramatically changing the cancer treatment landscape, albeit long-term responses are restricted to a minority. Clinical trials in various cancers continue to evaluate immunotherapy combinations with AAGs. One such combination is PD-1/PD-L1 antibody with an anti-VEGF, with positive studies in several phase III studies. The first objective set in these studies is to circumvent VEGF-mediated immunosuppression by combined VEGF and PD-(L)1 blockade by overcoming inhibition of DC maturation, which facilitates recruitment of T cells to TME with improved perfusion [56]. The next game-changing strategy should convert nonresponding cold nodules to hot ones, exponentially improving immunotherapy responses.
Part III: Bridging the Gap Between Foundational and Actionable Blocks (Figure 7)
The core issues in cancer control and prevention of its recurrence require selecting choice combination therapies as the 1st Step, followed by their Timing and Sequencing strategies (CTS strategy). A comprehensive first-time approach can prevent potential immune escape by resistant cells, a web of genomic mutations, and intransigent recurrence (Figure 2). Cancer cells are derived from the highly complex, designed to survive human cells. Human cells, in turn, have survived adversity and passed on the evolution over millions of years. Due to this, consistent elimination of cancer clones and subclones requires an equally complex but systematic approach. Even studying the breakthroughs in cancer therapy indicates it has always been due to combinations of techniques having different mechanisms of action and overcoming the disadvantage of one by the advantage of another. Survival of cancer cells for one therapy invokes evolutionary-resistant mutations to further lines of therapies with the law of diminishing returns. Hence, if implemented logically, analytically, and systematically, the first-time therapy opportunity is best (Figure 3 {A, B, C, F}).
The overall stratagem should be to reach the therapy choice out of a web of options in the shortest period and with the least expense, with the policy enumerated in this section. Also, it is likely that the therapies, when appropriately “slotted,” will find their place in the treatment program, which otherwise would be discarded as ineffective.
- A.
- Classifications of the available effective Treatments – making sense of the Web of Options (Figure 7).
A combination, Timing, and sequencing (CTS) strategy proposed in this article requires a stepwise approach employing the available therapeutic options with different mechanisms of action to synergize each other. This strategy maximizes the anticancer effect, minimizing the side effects to a great extent. The available and future innovative methods could easily fit into a particular “slot” depending on their mechanism of action in the trial pipeline. The Four primary slots are a). Pre-priming therapy for normalizing the vasculature; b). Priming therapy for vascular promotion; c). Post-priming therapy for immune cycle promotion and immune-memory cell enhancement; and d). Consolidation therapy for cancer reversal. The miscellaneous methods through antigenicity, adjuvanticity, professional phagocytosis, lymphatic function recovery, etc., can be tested in the four particular niches they impact.
The evolution of chemotherapy and radiotherapy (along with surgery) to attain the present-day cure/ control rate also utilized the above CTS strategy steps empirically. The delivery of chemotherapy in cycles is intended to reduce the side effects (established in most situations as cycles of 3 weeks), and the subsequent dose being delivered coincides with the recovery of bone marrow. Weekly schedules were designed to improve the tolerance further and avoid peak peripheral red blood cells and leucocyte drop at the end of 2 weeks. Radiotherapy effectively balanced the side effects versus response in fractionated schedules of 4 to 7 weeks. Ferretti S et al. (2009) tested seven anticancer drugs in 13 experimental tumor models. They showed that most of the anticancer therapeutics significantly lowered interstitial pressure sooner (2 to 3 days) or later (6 to 7 days) and suggested that it was the early marker of tumor response correlating with late changes in tumor volume [57]. Hoffman KE et al., as early as 2007, presented the impact of sequencing chemotherapy drugs in reducing interstitial fluid pressure, improving tumor oxygenation, providing better response, and promoting long-term local control/survival in neoadjuvant therapy of carcinoma breast [58]. Large randomized clinical trials showed significant improvements in survival with chemotherapy and antiangiogenic bevacizumab combination when compared to chemotherapy alone [11], indicating underlying vascular normalization and promotion contribution.
Therefore, underlying this empirical evolution lies the basics of vascular normalization, vascular promotion, and immune promotion. Fundamentally, cancer cells killed by the initial cycles of chemotherapy and initial fractions of radiotherapy reduced the interstitial pressure. Improved oxygenation and drug delivery of subsequent doses enhanced immunological sensitization-susceptibility (certain drugs inducing immunogenic cell death) [59] provided combination therapies take care of repopulating resistant phenotypic cells. Low-dose radiotherapy and SBRT regimen reduced intratumoral pressure and increased intratumorally administered drug delivery and retention [60]. Even with SBRT, the oxygen-utilization rate increased at six to eight fractions, up to 87%, with enhanced response in a schedule of three fractions per week [61].
a). Step 1 - Prepriming Therapy (Figure 6 {Step1}): Mitigating the physical barriers (mechanical TME) for the delivery of” drugs” (can be chemotherapy/immunotherapy/Cell therapy/in vitro vaccines/nano-therapy etc.) first by Pre-Priming therapy with angiogenesis-normalization therapeutics, e.g., multikinase inhibitors.
b). Step 2 - Priming therapy (Figure 6{Step2}) by standard bimodal treatment of ra diation, chemotherapy/targeted drugs (neo-adjuvant) for vascular promotion, and organ preservation oncoplastic surgery for the removal of the core resistant cells before they can “seed” at the distant site.
c). Step 3 - Immune promotion (Figure 6 {Step 3}) using rapidly evolving immune therapy combinations, including various CAR-T cell approaches.
d). Step 4 - Reversing the Cancer Process (Figure 6 {Step4}) by targeting HIF1-α and ROS is in its infancy presently but is likely to take a prominent space with increasing success with the above approaches. The focus is expected to shift from a cancer cell kill approach to reducing the inflammation, which is the fundal reason for the increased ROS and persistent HIF1-α conjoining with HIF1-β in the cell’s nucleus [62].
e). Enhancers of the Above Four (Figure 6 – Modalities to Improve): The rest of the therapies fall into, based on their primary mechanism of action. Primarily, therapies improving antigenicity and adjuvanticity act on immune promotion (Level -3). The reduction of interstitial pressure, e.g. by professional phagocytic agents, acts first on vascular promotion (Level 2) and subsequently on immune promotion (Level 3) by improving lymphatic drainage of Antigen Presenting Cells (APCs).
- TGF-β blockers facilitate the neoantigen-APCs flow to the lymph nodes by restoring lymphatic functions after un-collapsing lymphatic lumens with reduced interstitial pressure [63,64,65]. They also reduce inflammation in the extracellular matrix (ECM), thus retaining the immunological cross-talk [49].
- In-situ therapeutic vaccine - Use of pulsed radiotherapy/stereotactic body radiotherapy [66,67,68] before each cycle of immunotherapy to generate neoantigens specific to the mutation of the time and also fulfilling the booster dose conditions for effective vaccination (like in infective conditions).
- Immune adjuvants enhance immune promotion action by immune-antigenicity-adjuvanticity actions.
- Professional phagocytic agents act at multiple levels. Reduction of the interstitial pressure following the clearance of cancer cell debris induces both vascular and immune promotion.
- Stereotactic Body Radiotherapy (SBRT), including pulsed administration and Nanoparticle therapies, helps at all priming therapies and primarily immune promotion levels.
- Intralesional therapies add value to immunological promotion methods by action of adjuvanticity and antigenicity.
- In vitro-designed Vaccines and Cell therapies will be adjuncts at the immune-promotion level.
- ECM normalizers, not necessarily anticancer therapies, act on Level 4.
- B.
- General Conditions:
- Primary Condition: Any therapy component should not disrupt the tumor/tumor bed vasculature when given with long-term control/curative intent. Generally, ablative therapies leave the tumor bed as a “nidus” for late recurrences with disturbed, rigid ECM, preventing immunological cross-talk [49]. The post-ablative scar is the harbinger of disrupted and deficient vasculature, mitigating subsequent immune cell interactions and drug(s) delivery. Consequently, the resistant dormant cells can get the stimulus for a breakout into proliferation at any time [49].
- The CTS strategy favors, at least in poor prognostic and advanced malignancies, surgical excision after neoadjuvant therapy to reduce the tumor burden and downstage the same as is prevalent in resectable/unresectable locally advanced head & neck cancer [69]. Surgery is essential in the curative program to remove the resistant stem cells, and “Timing” is critical. Also, in the neoadjuvant therapy situation, an optimum immunological cascade may set in before the surgery in the presence of intact drainage lymph nodes [70], requiring further studies. With multimodal chemo-immunotherapy, the infrequent possibility of progression during neoadjuvant therapy can be minimized and should be evaluated for response to treatment often [71].
- C.
- Methodology of Adoption
- Animal/clinical trials should incorporate the classified interventions stepwise as structured above. Adoption of a “Whole-elephant” perspective is required rather than intensely tackling “some part(s) of the elephant” outlook. The CTS strategy enumerated above could be fundamental to the solution of the cancer conundrum. However innovative a treatment may be, in the present-day context and with the experience of the past, there is no way a single/limited approach resolves cancer lesions’ complex evolved architecture.
- Improvise on the way by dropping or adding known effective therapies in a combination of principles enumerated above using adaptive Multi Arm- Multistep (MAMS) methodology by either dropping the losers design or Bayesian design by selecting all promising treatments and their order in treatment effects classified here and prior knowledge of the mechanism of their actions discussed in the present review. Designing the testing models with Artificial Intelligence and Machine learning models is feasible with the analytical approach discussed [72,73].
CONCLUSION
Future research should focus on classifying the “Web of Treatment Options” under the three levels of vascular normalization followed by vascular promotion and immune promotion based on their primary mechanism of action of a particular modality. Once the Cancer becomes sub-clinical, the strategy should be shifted to level four of consolidation therapy with ECM normalization.
Funding
Financial support and sponsorship: None.
Data Availability Statement
Availability of data and materials: Concept paper; Not applicable.
Conflicts of Interest
The author declared no conflicts of interest.
Ethical Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
References
- Montemagno C and Pagès G (2020) Resistance to Antiangiogenic Therapies: A Mechanism Depending on the Time of Exposure to the Drugs. Front. Cell Dev. Biol. 8:584. [CrossRef]
- Multhoff, G., Radons, J., & Vaupel, P. (2014). Critical Role of Aberrant Angiogenesis in the Development of Tumor Hypoxia and Associated Radioresistance. Cancers, 6(2), 813-828. [CrossRef]
- Groebe K, Vaupel P. Evaluation of oxygen diffusion distances in human breast cancer xenografts using tumor-specific in vivo data: role of various mechanisms in the development of tumor hypoxia. Int J Radiat Oncol Biol Phys. 1988 Sep;15(3):691-7. [CrossRef]
- MOORE GE, SANDBERG AA, WATNE AL. The comparative size and structure of tumor cells and clumps in the blood, bone marrow, and tumor imprints. Cancer. 1960 Jan-Feb;13:111-7.. [CrossRef]
- Swamy K. Vascular-Immuno-Phenotypic (VIP) Model for Locally Advanced and Oligo-Metastatic Cancer: A Hypothesis. Med Hypotheses (2021) 152:110618. [CrossRef]
- Liberti MV, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci. 2016 Mar;41(3):211-218. Epub 2016 Jan 5. Erratum in: Trends Biochem Sci. 2016 Mar;41(3):287. Erratum in: Trends Biochem Sci. 2016 Mar;41(3):287. doi: 10.1016/j.tibs.2016.01.004. [CrossRef]
- Lipinski KA, Barber LJ, Davies MN, Ashenden M, Sottoriva A, Gerlinger M. Cancer Evolution and the Limits of Predictability in Precision Cancer Medicine. Trends Cancer. 2016 Jan;2(1):49-63. [CrossRef]
- Zhou H, Wang M, Zhang Y, Su Q, Xie Z, Chen X, Yan R, Li P, Li T, Qin X, Yang H, Wu C, You F, Li S, Liu Y. Functions and clinical significance of mechanical tumor microenvironment: cancer cell sensing, mechanobiology and metastasis. Cancer Commun (Lond). 2022 May;42(5):374-400. [CrossRef]
- Wegiel B, Vuerich M, Daneshmandi Sand Seth P (2018) Metabolic Switch in the Tumor Microenvironment Determines Immune Responses to Anticancer Therapy. Front. Oncol. 8:284. [CrossRef]
- Clarke J M, Hurwitz H I. Understanding and targeting resistance to antiangiogenic therapies. J Gastrointest Oncol 2013;4(3):253-263. [CrossRef]
- Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Normalization of the vasculature for treatment of Cancer and other diseases. Physiol Rev. 2011 Jul;91(3):1071-121. [CrossRef]
- James Ralston Kennedy Paterson. The Treatment of Malignant Disease by Radiotherapy. Williams & Wilkins Company, 1963. Page 45.
- Caporarello N, Lupo G, Olivieri M et al. Classical VEGF, Notch, and Ang signaling in cancer angiogenesis, alternative approaches and future directions (Review). Mol Med Rep 16: 4393-4402, 2017. [CrossRef]
- Zahra, F.T.; Sajib, M. S.; Mikelis, C.M. Role of bFGF in Acquired Resistance upon Anti-VEGF Therapy in Cancer. Cancers 2021, 13, 1422. [CrossRef]
- Al-Ostoot FH, Salah S, Khamees HA, Khanum SA. Tumor angiogenesis: Current challenges and therapeutic opportunities. Cancer Treat Res Commun. 2021;28:100422. [CrossRef] [PubMed]
- Haibe Y, Kreidieh M, El Hajj H et al. Resistance Mechanisms to Antiangiogenic Therapies in Cancer. Front. Oncol. 10:221. [CrossRef]
- Ribatti D. Tumor refractoriness to anti-VEGF therapy. Oncotarget. 2016 Jul 19;7(29):46668-46677. [CrossRef]
- Xiaolan Yu and Fengchun Ye. Role of Angiopoietins in Development of Cancer and Neoplasia Associated with Viral Infection. Cells 2020, 9, 457;. [CrossRef]
- Cortes-Santiago N, Hossain MB, Gabrusiewicz K, Fan X, Gumin J, Marini FC, Alonso MM, Lang F, Yung WK, Fueyo J, Gomez-Manzano C. Soluble Tie2 overrides the heightened invasion induced by antiangiogenesis therapies in gliomas. Oncotarget. 2016 Mar 29;7(13):16146-57. [CrossRef]
- Chae SS, Kamoun WS, Farrar CT, Kirkpatrick ND, Niemeyer E, de Graaf AM, Sorensen AG, Munn LL, Jain RK, Fukumura D. Angiopoietin-2 interferes with anti-VEGFR2-induced vessel normalization and survival benefit in mice bearing gliomas. Clin Cancer Res. 2010 Jul 15;16(14):3618-27. [CrossRef]
- Brossa A, Grange C, Mancuso L, Annaratone L, Satolli MA, Mazzone M, Camussi G, Bussolati B. Sunitinib but not VEGF blockade inhibits cancer stem cell endothelial differentiation. Oncotarget. 2015 May 10;6(13):11295-309. [CrossRef]
- Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. 2007 May;121(1):1-14. [CrossRef]
- Thomas Nielsen; Lise Bentzen; Michael Pedersen; Trine Tramm; Paul F.J.W. Rijken; Johan Bussink; Michael R. Horsman; Leif Østergaard. Combretastatin A-4 Phosphate Affects Tumor Vessel Volume and Size Distribution as Assessed Using MRI-Based Vessel Size Imaging. Clin Cancer Res (2012) 18 (23): 6469–6477. [CrossRef]
- Moding EJ, Castle KD, Perez BA, Oh P, Min HD, Norris H, et al. Tumor cells, but not endothelial cells, mediate the eradication of primary sarcomas by stereotactic body radiation therapy. Sci Trans Med (2015) 7(278):278ra34– 278ra34. [CrossRef]
- Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I, Xu L, Hicklin DJ, Fukumura D, di Tomaso E, Munn LL, Jain RK. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004 Dec;6(6):553-63. [CrossRef]
- Fong SS, Nanchen A, Palsson BO, Sauer U. Latent pathway activation and increased pathway capacity enable Escherichia coli adaptation to loss of key metabolic enzymes. J Biol Chem. 2006 Mar 24;281(12):8024-33. [CrossRef]
- Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005 Jan 7;307(5706):58-62. [CrossRef]
- Yang T, Xiao H, Liu X, Wang Z, Zhang Q, Wei N, Guo X. Vascular Normalization: A New Window Opened for Cancer Therapies. Front Oncol. 2021 Aug 12;11:719836. [CrossRef]
- Li S, Zhang Q, Hong Y. Tumor Vessel Normalization: A Window to Enhancing Cancer Immunotherapy. Technol Cancer Res Treat. 2020 Jan-Dec;19:1533033820980116. [CrossRef]
- Ramjiawan RR, Griffioen AW, Duda DG. Antiangiogenesis for Cancer revisited: Is there a role for combinations with immunotherapy? Angiogenesis. 2017 May;20(2):185-204. [CrossRef]
- Ayoub NM, Jaradat SK, Al-Shami KM, Alkhalifa AE. Targeting Angiogenesis in Breast Cancer: Current Evidence and Future Perspectives of Novel Antiangiogenic Approaches. Front Pharmacol. 2022 Feb 25;13:838133. [CrossRef]
- Weihua Wu, Shimin Zhao, Metabolic changes in Cancer: beyond the Warburg effect, Acta Biochimica et Biophysica Sinica, Volume 45, Issue 1, January 2013, Pages 18–26. [CrossRef]
- Schmitt MW, Loeb LA, Salk JJ. The influence of subclonal resistance mutations on targeted cancer therapy. Nat Rev Clin Oncol. 2016 Jun;13(6):335-47. [CrossRef]
- Lee et al., Lee W S, Yang H, Chon H J and Kim C. Combination of antiangiogenic therapy and immune checkpoint blockade normalize vascular immune cross-talk to potentiate cancer immunity. Experimental & Molecular Medicine (2020) 52:1475–1485 . [CrossRef]
- Amersfoort J, Eelen G, and Carmeliet P. Immunomodulation by endothelial cells — partnering up with the immune system? PERSPECTIVES Nature Reviews. Nat Rev Immunol 2022 Mar 14;1-13. [CrossRef]
- Jain RK, Martin JD, Stylianopoulos T. The role of mechanical forces in tumor growth and therapy. Annu Rev Biomed Eng. 2014 Jul 11;16:321-46. [CrossRef]
- Dewan MZ, Galloway AE, Kawashima N, et al. Fractionated But Not Single-Dose Radiotherapy Induces an ImmuneMediated Abscopal Effect When Combined With Anti-CTLA–4 Antibody. Clin Cancer Res (2009) 15(17):5379–88. [CrossRef]
- Piersma B, Hayward MK, Weaver VM. Fibrosis and cancer: A strained relationship. Biochim Biophys Acta Rev Cancer. 2020 Apr;1873(2):188356. [CrossRef]
- Martinez-Zubiaurre I, Chalmers AJ, Hellevik T. Radiation-induced transformation of immunoregulatory networks in the tumor stroma. Front Immunol (2018) 9:167. [CrossRef]
- Li SY, Guo YL, Tian JW, Zhang HJ, Li RF, Gong P, Yu ZL. Antitumor Strategies by Harnessing the Phagocytosis of Macrophages. Cancers (Basel). 2023 May 11;15(10):2717. [CrossRef]
- Liu, Y., Wang, Y., Yang, Y. et al. Emerging phagocytosis checkpoints in cancer immunotherapy. Sig Transduct Target Ther 8, 104 (2023). [CrossRef]
- Magnussen AL, Mills IG. Vascular normalisation as the stepping stone into tumour microenvironment transformation. Br J Cancer. 2021 Aug;125(3):324-336. [CrossRef]
- Chatterjee S, Wieczorek C, Schöttle J, Siobal M, Hinze Y, Franz T, Florin A, Adamczak J, Heukamp LC, Neumaier B, Ullrich RT. Transient antiangiogenic treatment improves delivery of cytotoxic compounds and therapeutic outcome in lung cancer. Cancer Res. 2014 May 15;74(10):2816-24. [CrossRef]
- Huang Y, Stylianopoulos T, Duda DG, Fukumura D, Jain RK. Benefits of vascular normalization are dose and time-dependent--letter. Cancer Res. 2013 Dec 1;73(23):7144-6. [CrossRef]
- Liu Z, Zhao Q, Zheng Z, Liu S, Meng L, Dong L, Jiang X. Vascular normalization in immunotherapy: A promising mechanisms combined with radiotherapy. Biomed Pharmacother. 2021 Jul;139:111607. [CrossRef]
- Swamy K. Stereotactic Body Radiotherapy Immunological Planning-A Review With a Proposed Theoretical Model. Front Oncol. 2022 Jan 26;12:729250. [CrossRef]
- Ariffin AB, Forde PF, Jahangeer S, Soden DM, Hinchion J. Releasing pressure in tumors: what do we know so far and where do we go from here? A review. Cancer Res. 2014 May 15;74(10):2655-62. [CrossRef]
- Cascio S, D’Andrea A, Ferla R, Surmacz E, Gulotta E, Amodeo V, Bazan V, Gebbia N, Russo A. miR-20b modulates VEGF expression by targeting HIF-1 alpha and STAT3 in MCF-7 breast cancer cells. J Cell Physiol. 2010 Jul;224(1):242-9. [CrossRef]
- Zhao Y, Yu X, Li J. Manipulation of immune‒vascular cross-talk: new strategies towards cancer treatment. Acta Pharm Sin B. 2020 Nov;10(11):2018-2036. [CrossRef]
- Pasquier, E., Kavallaris, M. & André, N. Metronomic chemotherapy: new rationale for new directions. Nat Rev Clin Oncol 7, 455–465 (2010). [CrossRef]
- Xia, Y., Sun, M., Huang, H. et al. Drug repurposing for cancer therapy. Sig Transduct Target Ther 9, 92 (2024). [CrossRef]
- Wyld, L., Bellantuono, I., Tchkonia, T., Morgan, J., Turner, O., Foss, F., George, J., Danson, S., & Kirkland, J. L. (2020). Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers, 12(8), 2134. [CrossRef]
- Kirkland, J.L. Tumor dormancy and disease recurrence. Cancer Metastasis Rev 42, 9–12 (2023). [CrossRef]
- Berrino F. Lifestyle prevention of cancer recurrence: the yin and the yang. Cancer Treat Res. 2014;159:341-51. [CrossRef]
- Biel NM, Siemann DW. Targeting the Angiopoietin-2/Tie-2 axis in conjunction with VEGF signal interference. Cancer Lett. 2016 Oct 1;380(2):525-533. [CrossRef]
- Hack SP, Zhu AX and Wang Y (2020) Augmenting Anticancer Immunity Through Combined Targeting of Angiogenic and PD-1/PD-L1 Pathways: Challenges and Opportunities. Front. Immunol. 11:598877. [CrossRef]
- Ferretti S, Allegrini PR, Becquet MM, McSheehy PM. Tumor interstitial fluid pressure as an early-response marker for anticancer therapeutics. Neoplasia. 2009 Sep;11(9):874-81. [CrossRef]
- K.E. Hoffman ∙ R. Abi-Raad ∙ M. Ancukiewicz ∙ E. Yeh ∙ P. Ryan ∙ L. Schapira ∙ J. Younger ∙ B.L. Smith ∙ I. Kuter ∙ A.G. Taghian. Impact of Interstitial Fluid Pressure, Tumor Oxygenation, and Chemotherapy Drug Sequencing on Response to Neoadjuvant Chemotherapy and Long-Term Local Control in Women Treated for Locally Advanced Breast Cancer. International Journal of Radiation Oncology, Biology, Physics, Volume 69, Issue 3, Supplement S61-S62November 01, 2007. [CrossRef]
- Taghian AG, Abi-Raad R, Assaad SI, Casty A, Ancukiewicz M, Yeh E, Molokhia P, Attia K, Sullivan T, Kuter I, Boucher Y, Powell SN. Paclitaxel decreases the interstitial fluid pressure and improves oxygenation in breast cancers in patients treated with neoadjuvant chemotherapy: clinical implications. J Clin Oncol. 2005 Mar 20;23(9):1951-61. [CrossRef]
- Barsoumian HB, Sheth RA, Ramapriyan R, Hsu E, Gagea M, Crowley K, Sezen D, Williams M, Welsh JW. Radiation Therapy Modulates Tumor Physical Characteristics to Reduce Intratumoral Pressure and Enhance Intratumoral Drug Delivery and Retention. Adv Radiat Oncol. 2022 Dec 5;8(2):101137. [CrossRef]
- Shibamoto Y, Miyakawa A, Otsuka S, Iwata H. Radiobiology of hypofractionated stereotactic radiotherapy: what are the optimal fractionation schedules? J Radiat Res. 2016 Aug;57 Suppl 1(Suppl 1):i76-i82. [CrossRef]
- Ziello JE, Jovin IS, Huang Y. Hypoxia-Inducible Factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J Biol Med. 2007 Jun;80(2):51- 60. [PubMed] [PubMed Central]
- Padera TP, Stoll BR, Tooredman JB, Capen D, di Tomaso E, Jain RK. Pathology: cancer cells compress intratumour vessels. Nature. 2004 Feb 19;427(6976):695. [CrossRef]
- Liao S, Liu J, Lin P, Shi T, Jain RK, Xu L. TGF-beta blockade controls ascites by preventing abnormalization of lymphatic vessels in orthotopic human ovarian carcinoma models. Clin Cancer Res. 2011 Mar 15;17(6):1415-24. [CrossRef]
- Avraham T, Daluvoy S, Zampell J, Yan A, Haviv YS, Rockson SG, Mehrara BJ. Blockade of transforming growth factor-beta1 accelerates lymphatic regeneration during wound repair. Am J Pathol. 2010 Dec;177(6):3202-14. [CrossRef]
- Sezen D, Barsoumian HB, He K, Hu Y, Wang Q, Abana CO, Puebla-Osorio N, Hsu EY, Wasley M, Masrorpour F, Wang J, Cortez MA, Welsh JW. Pulsed radiotherapy to mitigate high tumor burden and generate immune memory. Front Immunol. 2022 Oct 6;13:984318. [CrossRef]
- Moore C, Hsu CC, Chen WM, Chen BPC, Han C, Story M, Aguilera T, Pop LM, Hannan R, Fu YX, Saha D, Timmerman R. Personalized Ultrafractionated Stereotactic Adaptive Radiotherapy (PULSAR) in Preclinical Models Enhances Single-Agent Immune Checkpoint Blockade. Int J Radiat Oncol Biol Phys. 2021 Aug 1;110(5):1306-1316. [CrossRef]
- He K, Barsoumian HB, Sezen D, Puebla-Osorio N, Hsu EY, Verma V, Abana CO, Chen D, Patel RR, Gu M, Cortez MA, Welsh JW. Pulsed Radiation Therapy to Improve Systemic Control of Metastatic Cancer. Front Oncol. 2021 Aug 23;11:737425. [CrossRef]
- Chen Y, Zhong NN, Cao LM, Liu B, Bu LL. Surgical margins in head and neck squamous cell carcinoma: A narrative review. Int J Surg. 2024 Jun 1;110(6):3680-3700. [CrossRef]
- Baniel CC, Heinze CM, Hoefges A, Sumiec EG, Hank JA, Carlson PM, Jin WJ, Patel RB, Sriramaneni RN, Gillies SD, Erbe AK, Schwarz CN, Pieper AA, Rakhmilevich AL, Sondel PM, Morris ZS. In situ Vaccine Plus Checkpoint Blockade Induces Memory Humoral Response. Front Immunol. 2020 Jul 24;11:1610. [CrossRef]
- Caudle AS, Gonzalez-Angulo AM, Hunt KK, Pusztai L, Kuerer HM, Mittendorf EA, Hortobagyi GN, Meric-Bernstam F. Impact of progression during neoadjuvant chemotherapy on surgical management of breast cancer. Ann Surg Oncol. 2011 Apr;18(4):932-8. [CrossRef]
- Wason J, Stallard N, Bowden J, Jennison C. A multi-stage drop-the-losers design for multi-arm clinical trials. Stat Methods Med Res. 2017 Feb;26(1):508-524. [CrossRef]
- Serra A, Mozgunov P, Jaki T. A Bayesian multi-arm multi-stage clinical trial design incorporating information about treatment ordering. Stat Med. 2023 Jul 20;42(16):2841-2854. [CrossRef]
Figure 1.
Hypoxia-HIF-1α Domino Effects - Why is Hypoxia a primary Foundational Factor in cancer initiation and progression? A. The Genesis: Persistently increased ROS (A1) stabilizes HIF-1α and accumulates in the setting of Hypoxia (A2). HIF-1α, reentering the nucleus (A3), forms a transcription complex with the stable HIF-1β sub-unit to form a heterodimer, which binds to DNA motifs (A4) affecting the dozens of homeostatic genes, leading to cascading effects in cancer progression. 1. Shift to Anaerobic Metabolism – Warburg Effect; lactate production altered pH. 2. Vascular endothelial growth factor (VEGF) transcription affects Angiogenesis, finally leading to aberrant neoangiogenesis, aggravating Hypoxia further. 3. Activation of genomic mutations leading to cancer progression, creation of clones and subclones with varied phenotypic resistant population of cancer cells. 4. Accumulation of immunosuppressive cells and cytokines in the TME. B. With the successful treatment, ROS and HIF-1α normalization ensues with the possibility of a cure [Ziello JE et al., Reference 62.] X, Y, Z. Interactive and interdependent vascular-metabolic-immune-phenotypic (VMIP) Cancer Model mandating combination, Timing, and Sequencing (CRS) strategy. Created in BioRender. Swamy, K. (2024) https://BioRender.com/u59r544.
Figure 1.
Hypoxia-HIF-1α Domino Effects - Why is Hypoxia a primary Foundational Factor in cancer initiation and progression? A. The Genesis: Persistently increased ROS (A1) stabilizes HIF-1α and accumulates in the setting of Hypoxia (A2). HIF-1α, reentering the nucleus (A3), forms a transcription complex with the stable HIF-1β sub-unit to form a heterodimer, which binds to DNA motifs (A4) affecting the dozens of homeostatic genes, leading to cascading effects in cancer progression. 1. Shift to Anaerobic Metabolism – Warburg Effect; lactate production altered pH. 2. Vascular endothelial growth factor (VEGF) transcription affects Angiogenesis, finally leading to aberrant neoangiogenesis, aggravating Hypoxia further. 3. Activation of genomic mutations leading to cancer progression, creation of clones and subclones with varied phenotypic resistant population of cancer cells. 4. Accumulation of immunosuppressive cells and cytokines in the TME. B. With the successful treatment, ROS and HIF-1α normalization ensues with the possibility of a cure [Ziello JE et al., Reference 62.] X, Y, Z. Interactive and interdependent vascular-metabolic-immune-phenotypic (VMIP) Cancer Model mandating combination, Timing, and Sequencing (CRS) strategy. Created in BioRender. Swamy, K. (2024) https://BioRender.com/u59r544.
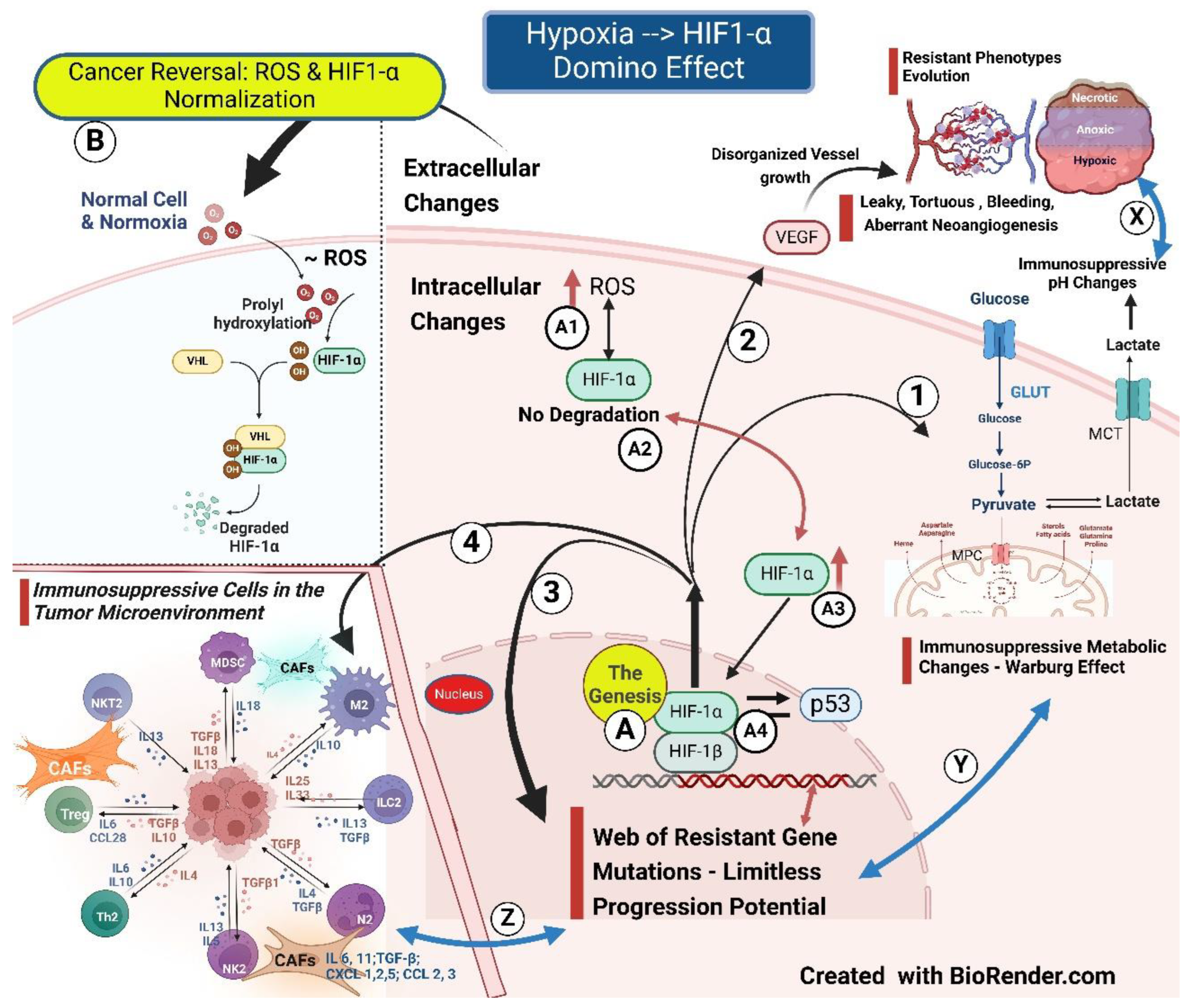
Figure 2.
Angiogenesis, Tumor (neo) Angiogenesis, Normalization, and Cyclicity strategy -Morphological Features of Cancer: A). The usual inner diameter of a cancer cell is about 20-30 µ depending on the site of origin. B). Oxygen and nutrient diffusion (and drugs) ceases beyond 100 µ (about 5 to 3 cell layers). Cancer cells become overtly hypoxic-anoxic resistant/dormant cell populations; later, some undergo necrosis. C). Leaky aberrant Angiogenesis with extravasation of hemopoietic cells. Evolution of Tumor angiogenesis, induced cyclicity, and extended vascular normalization: 1). Normal Angiogenesis leads to well-formed new tube-like blood vessels with stalk and tip cells covered with pericytes close in contact. 2). Tumor angiogenesis is elongated and distorted; weak, displaced pericytes; weak or absent tip cells leading to dysfunctional leaky aberrant Angiogenesis. 3). Within hours of starting antiangiogenics (Day 0), the normalization process of tumor angiogenesis begins. There will be improved oxygenation, reduced pH, increased drug delivery, immune cell infiltration, and tumor sensitivity to therapies. 4). From Day 0 to Day 28, induction of Vascular normalization. 5) Around day 28, the vascular trimming phase begins with increased pericytes and the reemergence of Hypoxia. 6). On stoppage of Antiangiogenics at this stage (Day 28), normalization of vasculature begins till the reestablishment of aberrant nature by another 4 weeks (as observed in patients taking drug holidays because of toxicity). 7). At this stage, restarting antiangiogenics repeats the normalization process (Day +0), lasting until Day +28 [Goel et al., Reference 11] 8). This cyclicity can be repeated to have an extended normalization phenomenon. Created in BioRender. Swamy, K. (2024) https://BioRender.com/y20r006.
Figure 2.
Angiogenesis, Tumor (neo) Angiogenesis, Normalization, and Cyclicity strategy -Morphological Features of Cancer: A). The usual inner diameter of a cancer cell is about 20-30 µ depending on the site of origin. B). Oxygen and nutrient diffusion (and drugs) ceases beyond 100 µ (about 5 to 3 cell layers). Cancer cells become overtly hypoxic-anoxic resistant/dormant cell populations; later, some undergo necrosis. C). Leaky aberrant Angiogenesis with extravasation of hemopoietic cells. Evolution of Tumor angiogenesis, induced cyclicity, and extended vascular normalization: 1). Normal Angiogenesis leads to well-formed new tube-like blood vessels with stalk and tip cells covered with pericytes close in contact. 2). Tumor angiogenesis is elongated and distorted; weak, displaced pericytes; weak or absent tip cells leading to dysfunctional leaky aberrant Angiogenesis. 3). Within hours of starting antiangiogenics (Day 0), the normalization process of tumor angiogenesis begins. There will be improved oxygenation, reduced pH, increased drug delivery, immune cell infiltration, and tumor sensitivity to therapies. 4). From Day 0 to Day 28, induction of Vascular normalization. 5) Around day 28, the vascular trimming phase begins with increased pericytes and the reemergence of Hypoxia. 6). On stoppage of Antiangiogenics at this stage (Day 28), normalization of vasculature begins till the reestablishment of aberrant nature by another 4 weeks (as observed in patients taking drug holidays because of toxicity). 7). At this stage, restarting antiangiogenics repeats the normalization process (Day +0), lasting until Day +28 [Goel et al., Reference 11] 8). This cyclicity can be repeated to have an extended normalization phenomenon. Created in BioRender. Swamy, K. (2024) https://BioRender.com/y20r006.
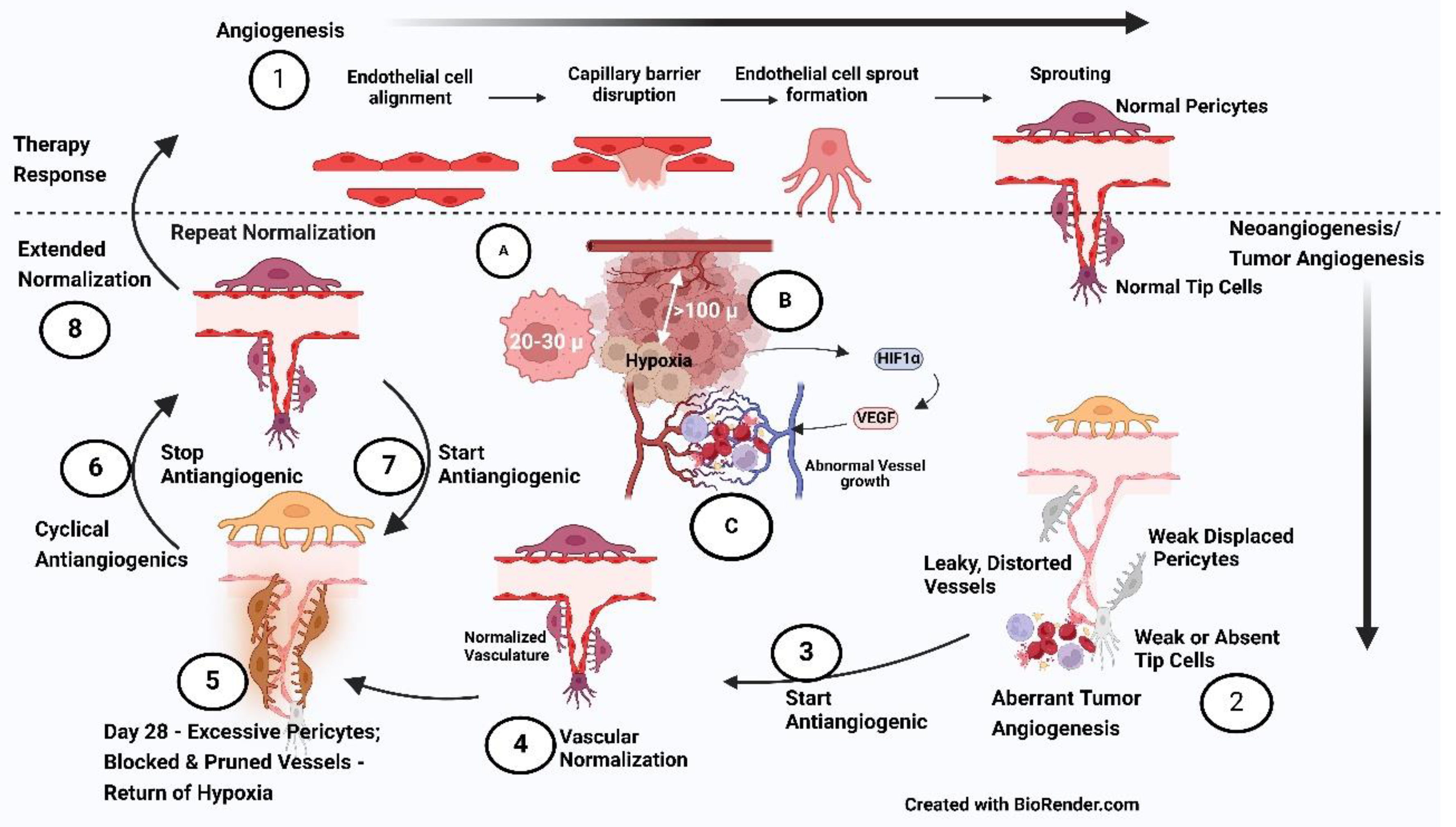
Figure 3.
Genomic/Phenotypic Evolutions - A. Hypoxia is the Spider that weaves a web of mutational cell types. B. Therefore, cancer tissue mass has diverse Spatial heterogeneity requiring combinations of curative methods. Clonogenic Cancer stem cells can proliferate and evolve into different subclones, contributing to cancer heterogeneity with varied resistant phenotypes. C. Two major types of cells are one with targetable mutations/receptors and the other without. D. Meanwhile, metastatic driver mutations manifest, leading to distant metastases whose response may vary from the primary lesions. E. There are also Temporal mutations over time, with or without therapy interventions, with increasing complexity and resistant phenotypes (Adaptive/Acquired resistance). F. These mutations are critically essential with the law of diminishing returns vis a vis cure and beg the question of preventability with an initial effective strategy. Created in https://BioRender.com.
Figure 3.
Genomic/Phenotypic Evolutions - A. Hypoxia is the Spider that weaves a web of mutational cell types. B. Therefore, cancer tissue mass has diverse Spatial heterogeneity requiring combinations of curative methods. Clonogenic Cancer stem cells can proliferate and evolve into different subclones, contributing to cancer heterogeneity with varied resistant phenotypes. C. Two major types of cells are one with targetable mutations/receptors and the other without. D. Meanwhile, metastatic driver mutations manifest, leading to distant metastases whose response may vary from the primary lesions. E. There are also Temporal mutations over time, with or without therapy interventions, with increasing complexity and resistant phenotypes (Adaptive/Acquired resistance). F. These mutations are critically essential with the law of diminishing returns vis a vis cure and beg the question of preventability with an initial effective strategy. Created in https://BioRender.com.
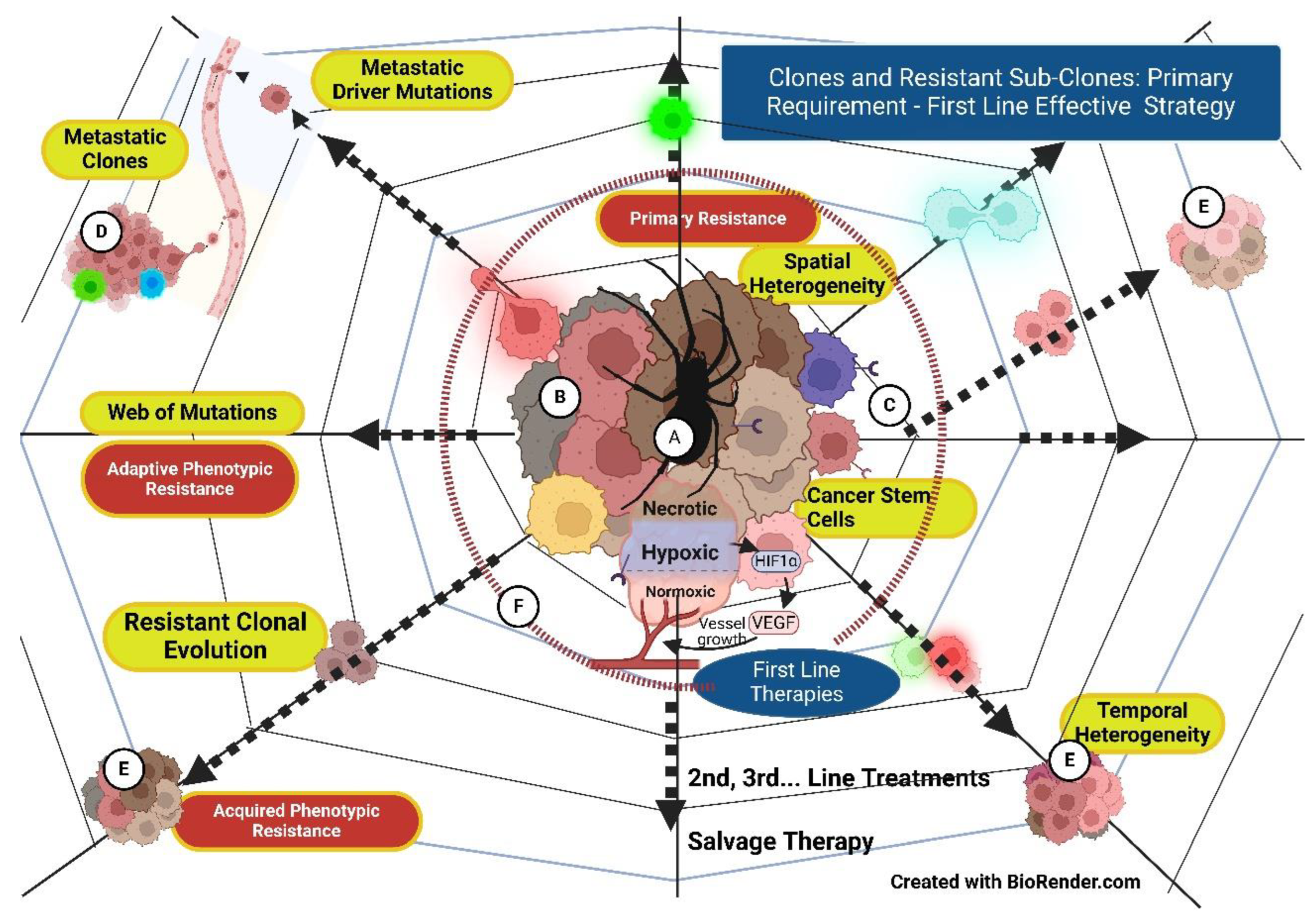
Figure 4.
Physical TME Barriers - However effective a therapy is, it may be unable to reach the cancer cell microenvironment (CCME) due to physical barriers called Physical TME, leading to treatment failure. Even though radiation can traverse through these barriers, its cancer cell kill effects can be limited by hypoxic resistance since radiation requires oxygen to prevent its DNA damage repair. 1. Drug diffusion barrier – One reason for the failure of CAR-T cell therapy in solid tumors is its inability to find its way into CCME. Even the nanoparticles (and the tagged drugs) with better diffusion ability will be unable to reach CCME due to the combined effects of aberrant Angiogenesis and physical barriers. 2. The other effects and barriers are Hypoxia, reduced nutrient supply, increased metabolic stress, and drug efflux with depleted concentration. 3 & 4. Anticancer immune cell trafficking into CCME is affected by immune cell trapping and immune exhaustion. 5. The wall of the necrotic areas within the cancer mass will be the sanctuary for the dormant/most resistant clonogenic cancer cells. Created in BioRender. Swamy, K. (2024) https://BioRender.com/e35m137.
Figure 4.
Physical TME Barriers - However effective a therapy is, it may be unable to reach the cancer cell microenvironment (CCME) due to physical barriers called Physical TME, leading to treatment failure. Even though radiation can traverse through these barriers, its cancer cell kill effects can be limited by hypoxic resistance since radiation requires oxygen to prevent its DNA damage repair. 1. Drug diffusion barrier – One reason for the failure of CAR-T cell therapy in solid tumors is its inability to find its way into CCME. Even the nanoparticles (and the tagged drugs) with better diffusion ability will be unable to reach CCME due to the combined effects of aberrant Angiogenesis and physical barriers. 2. The other effects and barriers are Hypoxia, reduced nutrient supply, increased metabolic stress, and drug efflux with depleted concentration. 3 & 4. Anticancer immune cell trafficking into CCME is affected by immune cell trapping and immune exhaustion. 5. The wall of the necrotic areas within the cancer mass will be the sanctuary for the dormant/most resistant clonogenic cancer cells. Created in BioRender. Swamy, K. (2024) https://BioRender.com/e35m137.
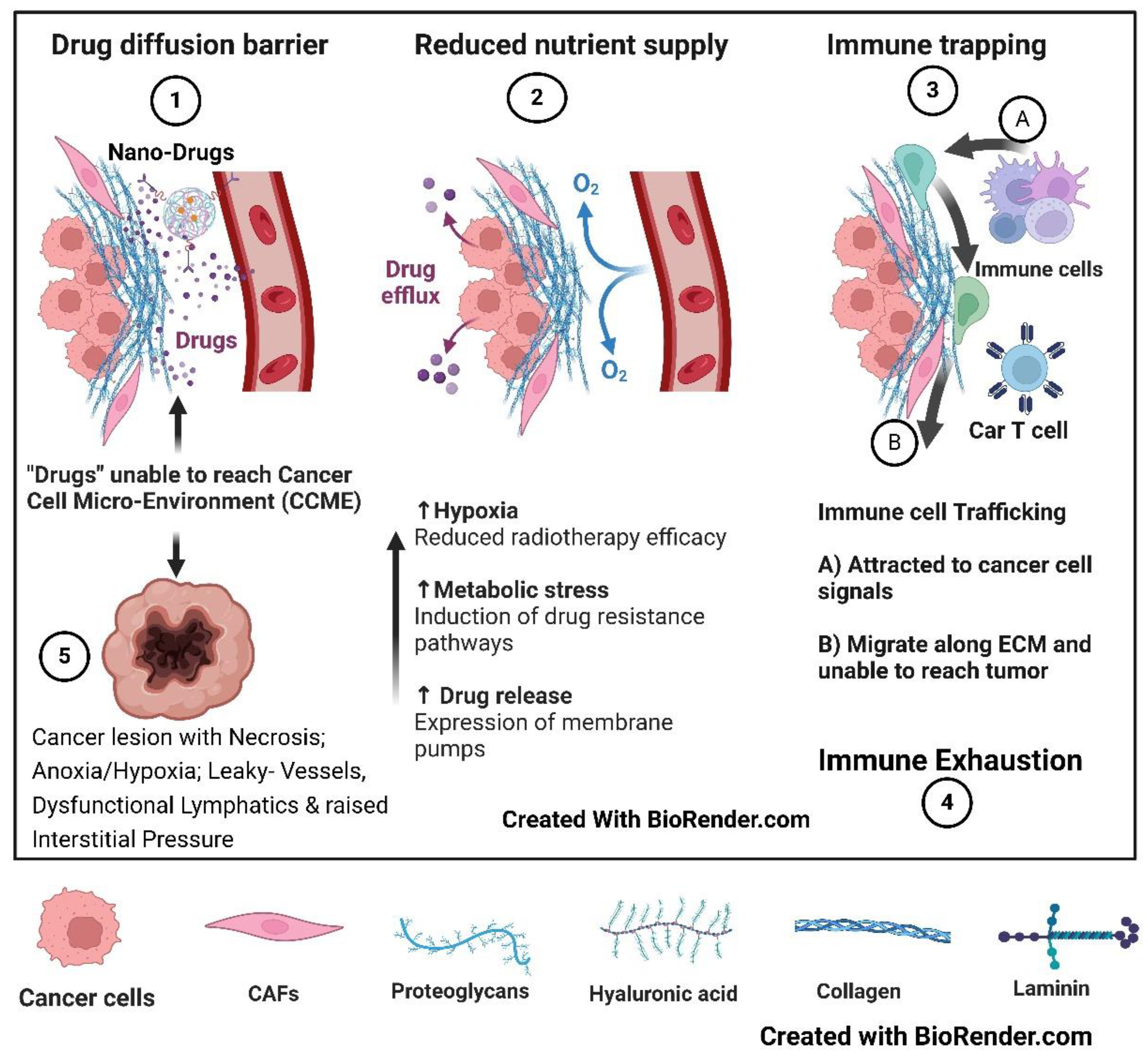
Figure 5.
Web of Therapy Options: The “Web” of available therapy options, each independently effective with innumerable combination possibilities. Cancer cure has always been about combinations of treatments that are effective independently, Timing, and sequencing. Additionally, periodically, newer targets are added.
Figure 5.
Web of Therapy Options: The “Web” of available therapy options, each independently effective with innumerable combination possibilities. Cancer cure has always been about combinations of treatments that are effective independently, Timing, and sequencing. Additionally, periodically, newer targets are added.
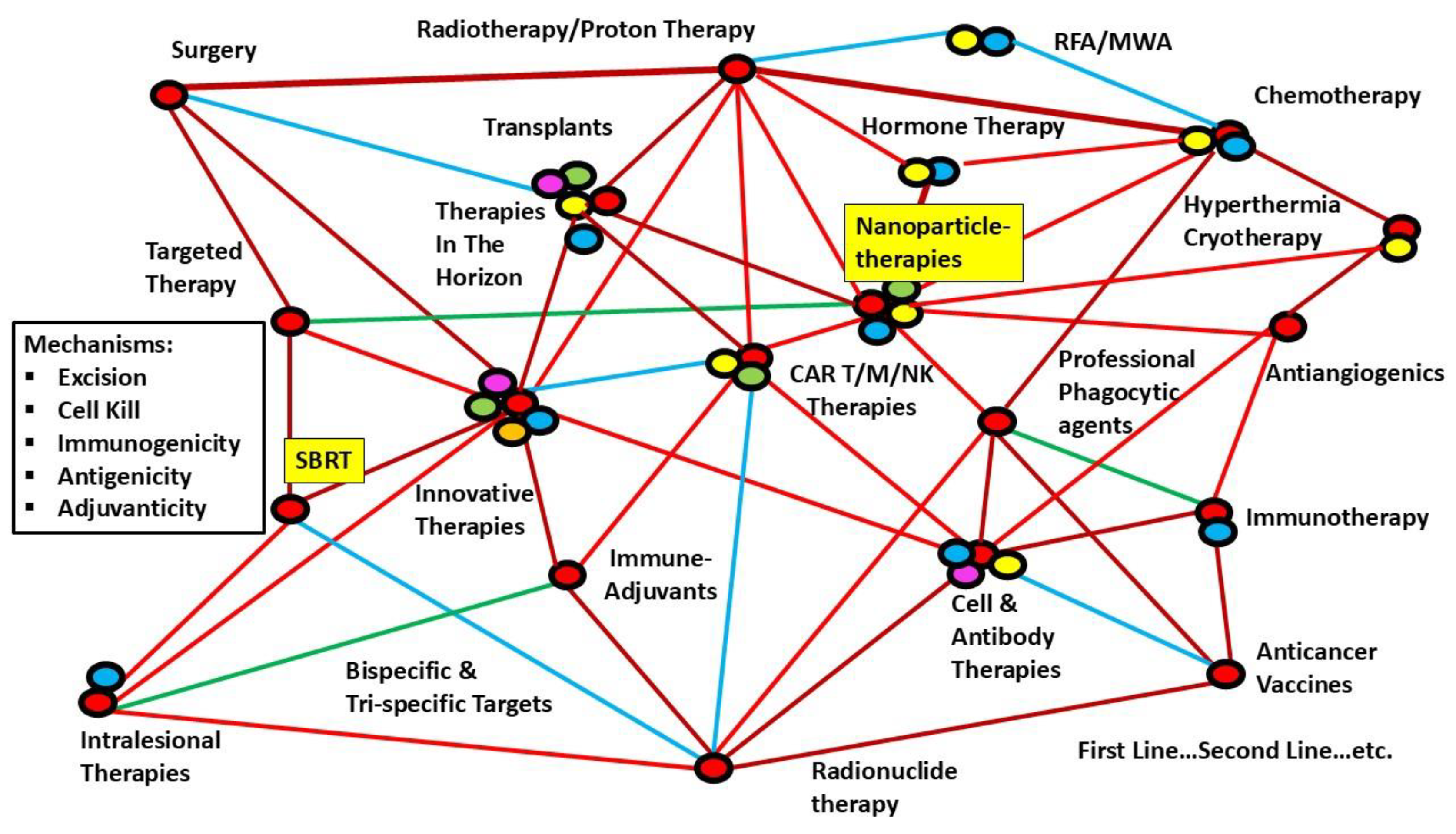
Figure 6.
Actionable Steps - Available therapies should be classified into one of the categories of vascular normalization, vascular promotion, immune promotion, or consolidation when Cancer becomes subclinical. Once the primary mechanism of action of the intervention modalities is decided, the particular approach can be “slotted” into one of the categories, and trials can be conducted in a stepwise flow, as shown in the Figure. Improving immune cells’ memory pool and suppleness of Extracellular Matrix (ECM) also needs to be considered for a particular treatment regarding the Timing and sequence in a combination. CT/RT = Chemotherapy/Radiotherapy; SBRT = Stereotactic radiotherapy. Created in BioRender. Swamy, K. (2024) https://BioRender.com/y21r646.
Figure 6.
Actionable Steps - Available therapies should be classified into one of the categories of vascular normalization, vascular promotion, immune promotion, or consolidation when Cancer becomes subclinical. Once the primary mechanism of action of the intervention modalities is decided, the particular approach can be “slotted” into one of the categories, and trials can be conducted in a stepwise flow, as shown in the Figure. Improving immune cells’ memory pool and suppleness of Extracellular Matrix (ECM) also needs to be considered for a particular treatment regarding the Timing and sequence in a combination. CT/RT = Chemotherapy/Radiotherapy; SBRT = Stereotactic radiotherapy. Created in BioRender. Swamy, K. (2024) https://BioRender.com/y21r646.
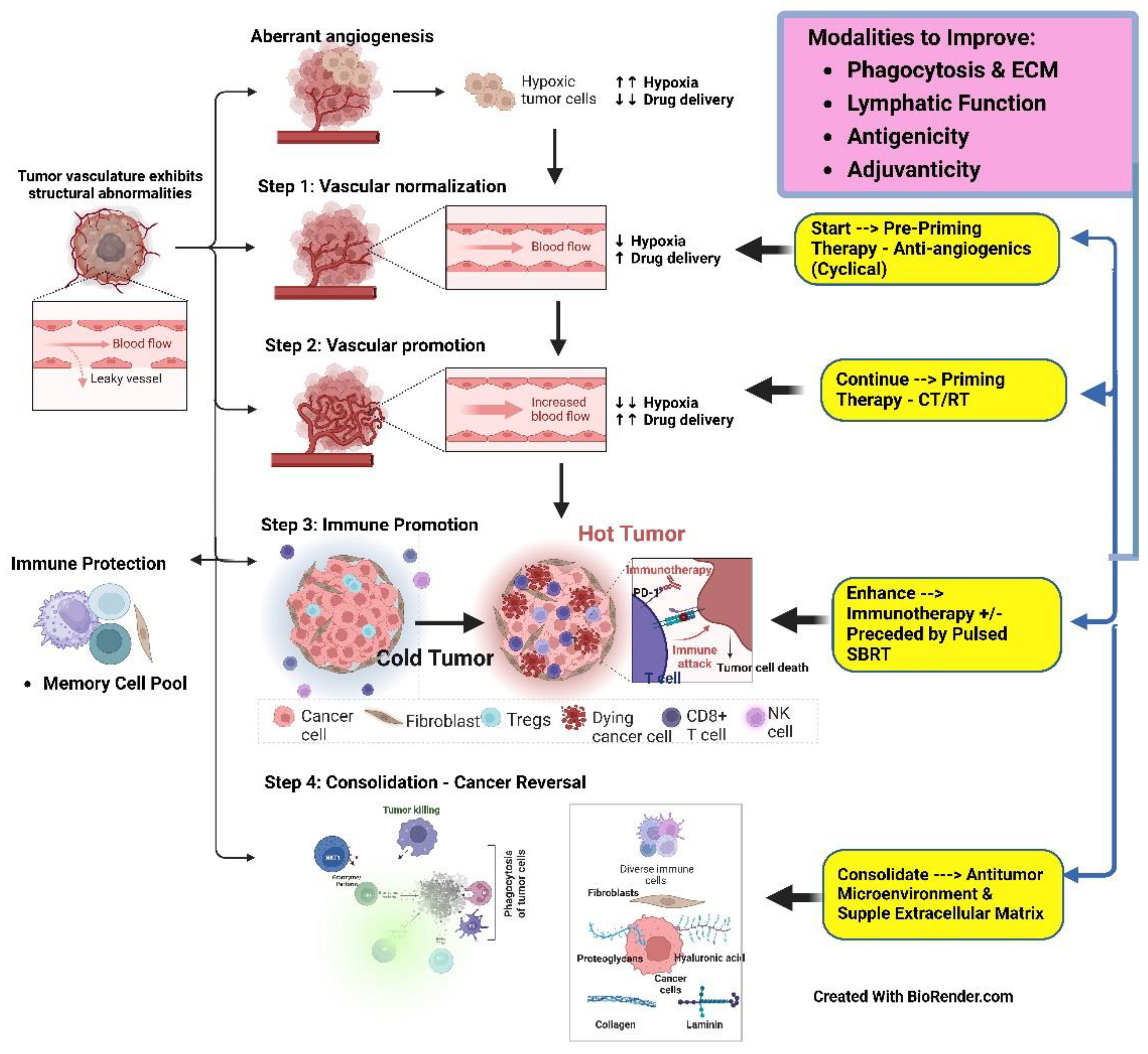
Figure 7.
Bridging the Foundational and Actionable Concepts - This requires classifying the treatment modality based on its primary mechanism of action and systematic adoption (as discussed in Figure 6) in Multi-Arm Multi-Stage (MAMS) combination therapy clinical trials [References Wason J. et al., 72; Serra A et al., 73]. The results set up the foundational-actionable cycle and fast resolution of the web of therapy options. Created in https://BioRender.com.
Figure 7.
Bridging the Foundational and Actionable Concepts - This requires classifying the treatment modality based on its primary mechanism of action and systematic adoption (as discussed in Figure 6) in Multi-Arm Multi-Stage (MAMS) combination therapy clinical trials [References Wason J. et al., 72; Serra A et al., 73]. The results set up the foundational-actionable cycle and fast resolution of the web of therapy options. Created in https://BioRender.com.
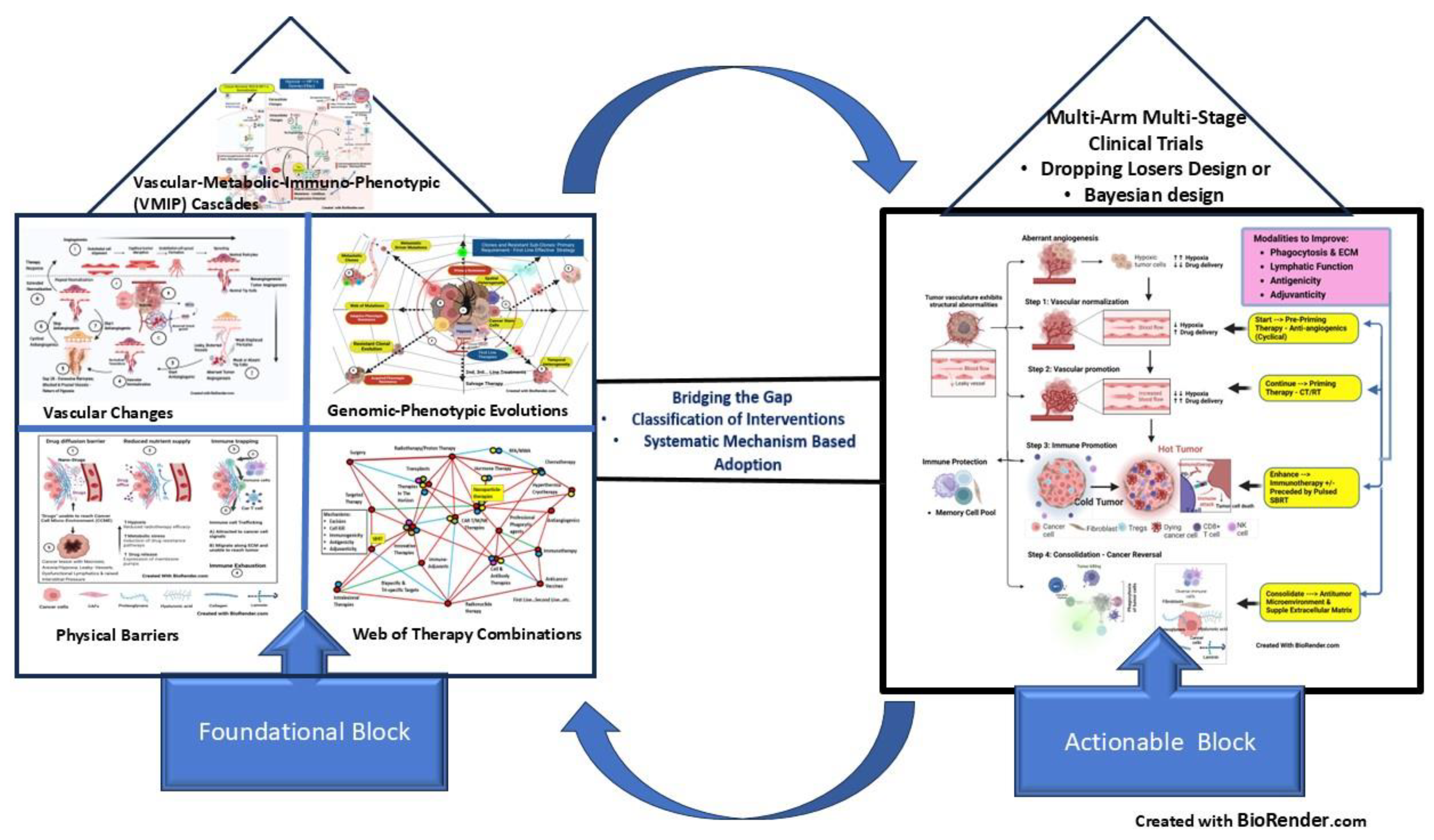
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



