
Submitted:
14 November 2024
Posted:
18 November 2024
You are already at the latest version
Abstract
Rubber compounds based on SBR and NBR were filled with constant amount of carbon black – 25 phr and calcium lignosulfonate – 30 phr. Glycerol as cheap and environmentally friendly softener was added into the rubber compounds in concentration scale from 0 to 20 phr to plasticize the rubber matrix and the biopolymer and to improve the adhesion between the components of rubber compounds. The results revealed that the addition of glycerol resulted in the decrease of rubber compounds viscosity. Based on dynamical-mechanical analysis it can be concluded that glycerol softened lignosufonate and lowered its glass transition temperature. The softened lignosufonate was better distributed and dispersed within the rubber matrices, which was clearly demonstrated by performing scanning electron microscopy. Glycerol contributed to better adhesion and compatibility between the rubber and the biopolymer, which was subsequently reflected in improvement of tensile behavior of vulcanizates. Due to compatibility in polarity among rubber matrix, biopolymer and plasticizer, higher tensile strength, and higher enhancement of tensile strength in dependence on glycerol content exhibited composites based on NBR. The main point of the work was to show that improvement of the composites properties can be reached by simple addition of suitable plasticizer into the rubber formulations with applied biopolymer without additional cost and time-consuming treatment procedures.
Keywords:
calcium lignosulfonate
; carbon black
; plasticizer
; composites
; cross-linking
; morphology
; properties
1. Introduction
Current trend in today's modern society is narrowly linked with utilization of biodegradable and environmentally friendly polymer materials. However, the production of 100% bio-based materials as a substitute for petroleum-based products is sometimes not a highly economically and technically viable solution. On the other hand, a combination of petroleum and bio-based sources has currently provided a better solution to develop a cost-effective, environmentally friendly products with a wide range of applications with a specific view to the pursuit of the European Green Deal [1,2,3,4]. The main aim to use natural-based sources in polymers is to mitigate the negative impact on the environment and to reduce the carbon footprint. A lot of natural based sources have been tested as potential fillers or components for polymer composites as starch, cellulose, lignins, lignosulfonates and their derivates [5,6,7,8,9].
Lignin is natural polymer, which does not occur in isolated form, but rather it is part of plant and wood cell walls along with cellulose and hemicellulose. Lignin is one of the most abundant biopolymers in the world (second after cellulose) and plays an important role in plant organism, where it controls cell wall structure, its rigidity, water permeability or pathogen attacks [10]. It has amorphous, three-dimensional highly branched aromatic structure. Phenylpropane units including p-hydroxyphenyl (H), syringyl (S) and guaiacyl (G) units connected by carbon-carbon (C–C) bonds and ether (C–O) bonds are the main building structures of lignin. Lignin can be extracted from plants, crops, and trees through a variety of extraction and delignification processes. Based on the selected method, lignin with different chemical composition and properties can be obtained. The most common methods to deliver lignin from natural sources are sulfur-based processes, Kraft pulping (alkali lignin or kraft lignin) and Sulfite pulping (lignosulfonates) [10,11,12,13]. Sulfur free processes include Soda and Organosolv procedures, which are of lower commercial importance [14,15]. Depending on the pulping process, the isolated lignins (technical lignins) differ in their composition, chemical structure, molecular weight, properties, purity, and solubility [16].
The sulfite pulping process is the main source of commercially available lignins, which are generally called lignosulfonates. Lignosulfonates are water-soluble polyaromatic polyelectrolytes having high molecular weight with broad distribution. They contain variety of functional groups as carboxylic groups, phenolic hydroxyl groups as well as sulfur containing moieties [17,18]. They are widely used as dispersants, emulsifiers, antistatic agents, or in the electronics sector [13,19].
Nowadays, only negligible amount (only 2-3%) of the derived technical lignins are commercially utilized. However, lignin exhibits very interesting characteristics, including good chemical and mechanical properties [20], low specific weight, antimicrobial [21] and antioxidant properties [22], excellent thermal and mechanical stability [23] arising from its aromatic structure and high carbon content. It is a low-cost and bio-renewable material. Lignin is a source of many chemical substances as well as is it highly used as a filler or component in polymer compounds, considering its ecological and environmental friendliness, availability, bio-degradability, reinforcing ability, thermo-oxidative stability or flame retardancy [24,25,26,27]. It also exhibits good viscoelastic characteristics, rheological and adhesive properties [28,29]. Outlining that and considering growing concern over protection of the environment and reducing industrial pollution, the effort to utilize lignin has considerably increased over last decades.
Lignin contains a large number of hydroxyl, methoxyl, carboxyl and carbonyl functional groups, due to which strong intra- and intermolecular bonds are formed between lignin macromolecules. In addition, the amorphous heterogeneous nature and hyperbranched structure hinders its compatibility and miscibility with most polymers. On the other hand, polar functional groups provide space for its surface modification with the aim to improve adhesion and compatibility between the biopolymer and the polymer matrix on their interface. Good adhesion and bonding between the polymer and the filler on the interfacial region are the basic condition to fabricate polymer compounds and composites with good processing characteristics, as well as suitable physical-mechanical and utility properties.
There have been applied and described several physical and chemical methods and procedures to modify lignin or polymers to enhance the interactions between both components, which provided very good physical-mechanical characteristics [19,30,31,32,33,34,35,36,37,38,39,40]. Though, a lot of these procedures are time and cost consuming or require additional equipment.
In this work, SBR and NBR were used as polymer matrices. SBR is non-polar general-purpose elastomer consisting of butadiene and styrene structural units and it is the most widely used in rubber-related technological applications. NBR is copolymer of butadiene and acrylonitrile and as a specialty polar rubber is mainly used in applications, where good resistance to oils and non-polar solvents is required. Both type rubbers are non-crystallizing and thus exhibit low tensile characteristics. Therefore, active fillers must be applied to increase physical-mechanical properties of the final products. Typically, carbon black is added into the rubber formulations to achieve the desired properties, mainly to improve tensile strength, hardness, modulus, or abrasion resistance. In addition, calcium lignosulfonate as biopolymer component was incorporated into rubber compounds as green alternative to carbon black, which is petroleum-based product. Since biopolymer fillers like lignins are, unless they are suitably modified, inactive fillers and usually do not contribute to the improvement of physical-mechanical properties of polymer materials, the combination of carbon-based fillers with biopolymers seems to be good way to fabricate rubber composites with good physical-mechanical and utility properties.
Plasticizer are usually used to improve polymer compounds process-ability, as they reduce viscosity, increase dispersion and distribution of fillers and additives. Thus, they help to prepare homogenous rubber compounds. They also increase rubber chains flexibility and lower glass transition temperature. Glycerol as cheap, commercially available, non-toxic and environmentally friendly softener was used for rubber formulations filled with carbon black and calcium lignosulfonate. It is a low molecular weight liquid and was chosen for its polar character, like lignosulfonate. It has been shown in our previous works [41,42] that glycerol contributed to better distribution and dispersion of the biopolymer within the rubber matrices and enhanced the adhesion between the components of rubber compounds. As a result, process-ability as well as tensile behavior of final materials were improved.
2. Experimental
2.1. Materials
Acrylonitrile-butadiene rubber NBR, type SKN 3345 (acrylonitrile content 31-35 %) was supplied from Sibur International, Russia. Cold emulsion polymerization produced styrene-butadiene rubber SBR, type Kralex 1502 (styrene content 23.5 %) was supplied from Synthos Kralupy, Czech Republic. NBR based rubber batch filled with 25 phr of carbon black (CB, type N330) as well as SBR based rubber batch filled with 25 phr of the same carbon black type were fabricated in Vipo, a.s. Partizánske, Slovakia. Calcium lignosulfonate having trade name Borrement CA120 was provided by Borregaard Deutschland GmbH, Germany. The average molecular weight of lignosulfonate was 24 000 g.mol-1 with specific surface area 3.9 m2.g-1. The elemental analysis revealed the presence of hydrogen (5.35 wt.%), carbon (46.63 wt.%), nitrogen (0.14 wt.%), sulfur (5.62 wt.%) and hydroxyl groups (1.56 wt.%) in its structure. Lignosulfonate was applied into rubber formulations in constant amount – 30 phr. Glycerol (86 % solution, supplied from Sigma-aldrich, USA) was added into rubber formulations as plasticizer from 5 to 20 phr. For cross-linking of rubber compounds, sulfur-based curing system consisting of 3 phr zinc oxide and 2 phr stearic acid (Slovlak, Košeca, Slovakia) as activators, 1.5 phr accelerator N-cyclohexyl-2-benzothiazole sulfenamide CBS (Duslo, Šaľa, Slovakia) and 3 phr sulfur (Siarkopol, Tarnobrzeg, Poland) was used.
2.2. Methods
2.2.1. Preparation and Curing of Rubber Compounds
Rubber compounds based on NBR or SBR, respectively were first pre-compounded with 25 phr of carbon black in industrial kneading machine Buzuluk (Buzuluk a.s., Komarov, Czech Republic). The following fabrication of rubber compounds proceeded in lab-scale conditions with utilization of laboratory kneading machine Brabender (Brabender GmbH & Co. KG, Duisburg, Germany).
The temperature of compounding was set up to 90 °C with rotor speed 55 rpm. The overall mixing process took 10 min. First, rubber-based batch was plasticated for 1 min, then zinc oxide and stearic acid were added with next 1 min compounding. Subsequently, biopolymer was incorporated. The amount of lignosulfonate was kept on constant dosage – 30 phr in each rubber compound. The kneading process continued for 2 min, and the glycerol was introduced. The rubber compounds were compounded for next 2 min. Then, they were taken out from the mixing chamber and cooled down in two-roll mill. In the second step, which took for 4 min at 90 °C and 55 rpm, sulfur and accelerator CBS were applied. In the final stage, the materials were homogenized and sheeted in two-roll mill. The composites based on NBR were designated as NBR/CB-L30 while the formulations based on SBR were marked as SBR/CB-L30.
The curing process of the fabricated composites was performed at 170 ºC and pressure of 15 MPa in a hydraulic press Fontijne (Fontijne, Vlaardingen, Holland) following their optimum cure time. After curing, thin sheets with dimensions 15 x 15 cm and thickness 2 mm were obtained.
2.2.2. Determination of Curing Characteristics
Curing characteristics were determined from corresponding curing isotherms, which were investigated in oscillatory rheometer MDR 2000 (Alpha Technologies, Akron, Ohio, USA).
The investigated curing parameters were:
ML (dN.m) - minimum torque
MH (dN.m) - maximum torque
∆M (dN.m) - torque difference, the difference between MH and ML
tc90 (min) - optimum curing time
ts1 (min) - scorch time
R (dN.m.min-1) – curing rate, defined as:
Mc90 (dN.m) – torque at tc90
Ms1 (dN.m) – torque at ts1
2.2.3. Determination of Cross-Link Density
The cross-link density ν was determined based on equilibrium swelling of composites in xylene. The weighted dried samples were placed into xylene in which they swelled within time at a laboratory temperature. The weight of samples was measured every hour until the equilibrium swelling was reached. The Flory-Rehner equation modified by Krause [43] was then used to calculate the cross-link density based upon the equilibrium swelling state:
ν - cross-link density (mol/cm3)
Vr0 - volume fraction of rubber in equilibrium swelling sample of vulcanizate in absence of fillers
Vr - volume fraction of rubber in equilibrium swelling sample of filled composite
VS - molar volume of the solvent
- Huggins interaction parameter (for NBR-xylene0.5316, SBR-xylene0.3908
2.2.4. Rheological Measurements
Dynamic viscosity was investigated using RPA 2000 (Alpha Technologies, Akron, Ohio, USA). The samples were analyzed under strain amplitude from 0.15 to 700 % at a constant frequency of 0.2 Hz and temperature 90 ºC.
2.2.5. Investigation of Physical-Mechanical Characteristics
The tensile properties were evaluated by using Zwick Roell/Z 2.5 appliance (Zwick GmbH & Co. KG, Ulm, Germany). The cross-head speed of the measuring device was set up to 500 mm.min-1 and the tests were carried out in compliance with the valid technical standards. Dumbbell-shaped test specimens (thickness 2 mm, length 80 mm, width 6.4 mm) were used for measurements. The hardness was measured by using durometer and was expressed in Shore A.
2.2.6. Microscopic Analysis
The surface morphology and microstructure were observed using scanning electron microscope JEOL JSM-7500F (Jeol Ltd., Tokyo, Japan). The samples were first cooled down in liquid nitrogen under glass transition temperature and then fractured into small fragments with surface area of 3 x 2 mm. The fractured surface was covered with a thin layer of gold and put into the microscope. The source of electrons is cold cathode UHV field emission gun, the accelerate voltage ranges from 0.1 kV to 30 kV and the resolution is 1.0 nm at 15 kV and 1.4 nm at 1 kV. SEM images are captured by CCD-Camera EDS (Oxford INCA X-ACT).
2.2.7. Determination of Dynamical-Mechanical Properties
Dynamical-mechanical performances were obtained by using a dynamical-mechanical analyzer DMTA MkIII, fy Rheometric Scientific. The samples were analyzed in tensile mode at a frequency 10 Hz, amplitude of dynamic deformation 64 μm and static force 0.2 N in temperature range from -60 ºC to 80 ºC.
3. Results and Discussion
3.1. Curing Process and Cross-Link Density
The influence of plasticizer glycerol on curing process was assessed based on the curing characteristics, which were determined from the corresponding curing isotherms (Figure 1 and Figure 2). It becomes apparent form them that the highest difference between maximum and minimum torque ΔM (ΔM = MH - ML) exhibited reference, glycerol free composites based on both type rubbers. With increasing content of glycerol, the differences between maximum and minimum torque became smaller as also shown from Figure 3. The decrease in torque difference with increase of glycerol content clearly points out to its strong plasticizing effect on rubber formulations. Molecules of glycerol enter intermolecular space and disrupt intra- and intermolecular physical forces and entanglements between rubber chains. This leads to the reduction of internal friction, to the increase of distance between rubber chains and to the increase of the chain segments mobility. Those factors result in the decrease of rubber compounds viscosity (see next chapter), which is very closely related to the decrease of torque difference on curing isotherms. Another factor that is closely connected with the difference between maximum and minimum torque is the cross-link density. In general, the lower the cross-link density, the lower the torque difference. The results obtained from experimental determination of cross-link density (Figure 4) confirmed certain correlation with the torque difference (Figure 3). When compared to the reference NBR based composite, the cross-link density first slightly increased by application of 5 phr glycerol. Then, the decrease in cross-linking degree of NBR based composites was recorded with next increase in glycerol content. The cross-link density of materials based on SBR did not change very much up to 10 phr of glycerol, then it dropped down at high plasticizer content. The cross-link density of composites based on NBR was about three times higher when compared to the equivalents based on SBR, which was reflected in higher torque difference of NBR based rubber formulations. Outlining that, it can be stated that the decrease in torque difference with increase of glycerol content is caused by the decrease in viscosity of rubber compounds in line with the decrease in their cross-link density.
In comparison with the reference, glycerol free sample based on NBR, the application of 5 phr plasticizer caused a slight decrease in scorch time, but then the scorch time almost did not change with next increase in glycerol content (Figure 5). On the other hand, the presence of plasticizer in NBR based rubber compounds resulted in prolongation of the optimum cure time (Figure 6). The longest time needed for optimum cross-linking required the rubber compound with 5 phr of glycerol. By next increase in glycerol content, the optimum cure time decreased and was found to be independent on plasticizer content. The ts1 and tc90 for the sample based on SBR with 5 phr of glycerol were almost the same as for the SBR based reference. Then, both curing characteristics showed decreasing trend with next increase in glycerol content. As seen in Figure 7, the highest curing rate R demonstrated the reference sample based on NBR. The application of glycerol caused a significant decrease in curing rate, which then seemed to be independent on plasticizer content. Similarly, the presence of glycerol resulted in the decrease in curing rate of rubber compounds based on SBR. The curing rate of SBR based composites was lower in comparison with the equivalents based on NBR. The biggest difference in R was recorded between reference samples. As shown, the curing rate of the reference sample based on NBR was about four times higher. The curing rate characterizes not only time required to achieve curing optimum, but also accounts transformation degree of uncured rubber compounds into vulcanizates. Thus, it might be stated that glycerol applied in rubber formulations decelerates the curing process, which seems to be more pronounced for composites based on NBR. Very similar dependences were obtained by calculating the maximum curing rate. From Figure 8 it becomes obvious that higher maximum curing rate exhibited rubber compounds based on NBR. The added glycerol negatively influenced the maximum curing rate for both types of composites. The possible explanation for decrease in curing kinetics can be the polarity of the plasticizer, due to which it might dilute or absorb polar curing reagents and thus making them ineffective during vulcanization [44]. The lower the amount of the curing additives, the slower the curing process. This could also provide explanation for the decrease in cross-link density of composites, mainly those with higher glycerol content. Negative effect on curing kinetics can also spring from acidic character of the biopolymer filler (pH lower than 5). It is generally known that acidic substances have negative effect on sulfur vulcanization and decelerate the curing process [45].
3.2. Rheological Study
The dependences of dynamic complex viscosity η* on shear rate are presented in Figure 9 and Figure 10. Looking at them one can see similar dependences of complex viscosity for both rubber formulations not only on shear rate but also on the amount of glycerol. As seen, the highest dynamic viscosity exhibited the reference compounds without plasticizer in all shear rate range. The higher the amount of glycerol, the lower the viscosities. The lowest viscosities were found to have NBR as well as SBR based composites with maximum glycerol content. The biggest differences in viscosities in dependence of glycerol content were recorded at lower shear rates. The higher the shear rate, the lower the viscosities. And the differences between the viscosities of rubber compounds became less visible, too. It also becomes apparent that the dynamic complex viscosities for both types of composites were very similar. The results obtained from rheological measurements clearly demonstrated that glycerol has strong plasticizing effect on rubber compounds and reduces their viscosities.
3.3. Physical-Mechanical Properties
The experimentally determined physical-mechanical characteristics of composites are graphically illustrated in Figure 11, Figure 12, Figure 13 and Figure 14. It becomes apparent that the dependences of modulus M300 (Figure 11) are in very close correlation with the dependences of cross-link density (Figure 4). Higher cross-link density of composites based on NBR was reflected in their higher modulus. Also, upon initial slight increase at 5 phr of glycerol, the modulus showed a decreasing tendency with next increase in glycerol content, following the cross-link density. Very precise correlation between both parameters can also be observed for systems based on SBR.
Higher cross-linking degree of composites based on NBR was also responsible for their higher hardness. The influence of glycerol on hardness for both composite types was almost not observed with exception of the sample based on NBR with maximum plasticizer content, in which small decline of the property was recorded (Figure 12). On the other hand, the higher the cross-link density, the lower the mobility and elasticity of rubber chains, thus resulting in lower elongation at break of the composites based on NBR when compared to the equivalents based on SBR. Another point contributing to differences in elongation at break is the structure of rubbers. NBR contains roughly 33 % of acrylonitrile structural units, which are thermoplastics elements of the rubber. On the other hand, SBR contains around 25 % of styrene units as thermoplastics element. Thus, it becomes apparent that SBR consists of higher amount of butadiene units, which are responsible for elastic properties of the rubber. As also shown in Figure 13, the elongation at break for both composite types showed increasing trend with increasing content of glycerol just up to 15 phr of glycerol. The increase of elongation at break can be attributed to the plasticizing effect of glycerol on rubber compounds, weakening of physical interactions between rubber chain segments and increase of their mobility and elasticity. This subsequently leads to the increase of elongation at break.
In general, it can be said that the application of glycerol resulted to the increase of tensile strength for both types of composites, although the effect of plasticizer on the tensile behavior was different (Figure 14). When comparing reference samples without glycerol, higher tensile strength exhibited that based on SBR. By addition of 5 phr plasticizer, the tensile strength decreased. Then, the positive effect of glycerol on tensile strength of SBR based formulations was recorded with next increase in plasticizer content. At maximum glycerol content, the tensile strength increased in more than 3 MPa when compared to reference (from about 16.5 MPa for the reference up to nearly 19.5 MPa for the composite with maximum plasticizer content). On the other hand, the tensile strength of composites based on NBR showed continuous increasing tendency with increasing content of glycerol up to 15 phr. The increase of tensile strength presented more than 5 MPa by increase of plasticizer content from 0 to 15 MPa (from around 15 MPa for the reference up to above 20 MPa for the composite with 15 phr of glycerol).
3.4. Morphology
The morphology of composites was observed on their surface structure by employing Scanning Electron Microscopy (SEM). To better distinguish the dispersion and distribution of calcium lignosulfonate, after fracturing the composites under the glass transition temperature, the uncovered surfaces were washed in boiling water for 2 hours. As calcium lignosulfonate is water soluble, it was extracted from the surface unveiling holes or cavities, which were occupied by the biopolymer filler before its extraction by hot water. SEM images of composites based on SBR are presented in Figure 15, while SEM images of those based on NBR are demonstrated in Figure 16. It becomes apparent from SEM images of both NBR and SBR based reference samples that lignosulfonate forms agglomerates and was poorly dispersed within the rubber matrices (Figure 15 and Figure 16A). The composite based on SBR with 5 phr of glycerol showed inhomogeneous structure still pointing out to poor dispersion of the biopolymer filler (Figure 15B). By next increase in glycerol content, better dispersion of calcium lignosulfonate was observed. As shown in Figure 15C–E, lignosulfonate formed smaller domains, which were more homogenously distributed within the rubber matrix. Similarly, the application of glycerol resulted in the formation of small biopolymer domains in composites based on NBR, which were more uniformly distributed within the matrix (Figure 16B-E). Glycerol is hydrophilic, low molecular substance that acts by softening effect on lignosulfonate, leads to its plasticization and lowering of its viscosity. Simultaneously glycerol has plasticizing effect on rubber matrix. As a result, the viscosity of rubber formulations decreased with increase in glycerol content (Figure 9 and Figure 10). The viscosity of the filler and the matrix became closer, which led to better miscibility and compatibility between both components during the mixing process. This resulted in better distribution and dispersion of the filler within the rubber matrix and better homogeneity and adhesion between the biopolymer and the rubber matrix on their interface. As outlined, lignosulfonate formed small soft filler like-domains. The soft domains with higher flexibility deform much more easily, when composites are exposed to deformation strains, similarly to the rubber systems containing reinforcing fillers [46,47]. This is in high contrast to rigid agglomerates of fillers, which act as stress concentrators upon deformation stretching. The outlined facts are responsible for enhancement of tensile behavior of composites. As shown in Figure 14, higher tensile strength demonstrated composites based on NBR. As calcium lignosulfonate and glycerol are polar materials, their compatibility with polar rubbers as NBR is higher when compared to non-polar elastomers, like SBR. This was confirmed by SEM analysis.
3.5. Dynamic-Mechanical Properties
The dynamic-mechanical measurements were conducted to evaluate the influence of plasticizer on viscoelastic properties of composites in dependence on temperature. The temperature dependences of tan δ for materials based on NBR with different glycerol content are graphically illustrated in Figure 17. The specific values of storage modulus G', loss modulus G'' and loss factor tan δ at -20, 0, 20 a 60 °C as well as glass transition temperature Tg are summarized in Table 1 and Table 2. It becomes apparent from Table 1 that the highest G' as well as G'' were exhibited at -20 °C. Storage and loss modulus of composites at 0 °C were significantly lower. With next increase in temperature, the moduli showed decreasing trend. It can also be stated that no significant changes of moduli were recorded in dependence on glycerol content at 0 and 20 °C, even in comparison with the reference, glycerol free sample. Storage as well as loss modulus at -20 °C passed over a maximum at 10 phr of glycerol, while G' modulus of composites at 60 °C showed decreasing trend with increasing content of glycerol. At 20 phr of glycerol, the storage modulus decreased to less than half of the equivalent modulus value of the reference composite at a given temperature. As seen in Table 2, no significant changes of tan δ in dependence on glycerol content were recorded at 0 and 20 °C. On the other hand, the increase of loss factor with the increase of glycerol content was observed at -20°C and 60 °C. The increase of loss factor at higher temperatures can also be seen from Figure 17, but its dependence on glycerol content seems to be different. The first peak maximum corresponds to glass transition temperature Tg of composites. As it becomes apparent from Figure 17 and Table 2, there was almost no change of Tg in dependence on glycerol content as it moved only from roughly -4.5 to -5.5 °C. As shown in Figure 17, there was almost no change in loss factor of the reference sample and vulcanizate with 5 phr of glycerol at higher temperatures. The composite with 10 phr of glycerol shows an increasing trend of tan δ with increase in temperature. The composites with 15 and 20 phr of glycerol reached the second maximum (tan δ = 0.28 at around 70 °C or tan δ = 0.31 at about 40 °C for the systems with 15 or 20 phr of the plasticizer, respectively).
The values of storage G' and loss G'' modulus for composites based on SBR at different temperatures are summarized in Table 3 and Table 4. As seen, the highest G' as well as G'' were found to have composites at -20 °C. With increase in temperature, both moduli showed decreasing trend. The influence of glycerol amount on moduli seems to be dependent on temperature. From -20 °C to 20 °C, G' passed over a maximum at 15 phr of plasticizer. On the other hand, storage modulus was found to decrease with increasing content of glycerol at 60 °C. The loss modulus did not change very much in dependence on glycerol content. At 60 °C it passed over a maximum at a loading of 15 phr. Similarly to G' and G'', composites exhibited the highest tan δ at -20 °C. The higher the temperature, the lower the loss factor. Tan δ seems to be independent on glycerol content at -20 °C and 0 °C, while the increase of loss factor was recorded at 20 °C and 60 °C. At 60 °C, more than twofold increase of tan δ was recorded for the composite with maximum glycerol content in comparison with the reference. From temperature dependences of loss factor (Figure 18) it becomes apparent that the first peak corresponding to glass transition temperature does not change with temperature very much. As shown in Table 4, the Tg moved around -25 °C. The temperature behavior of tan δ at higher temperatures was very similar as in the previous case. Almost no change in loss factor was recorded for the reference sample and composite with 5 phr of glycerol. Small gradual increase at about 60 °C was recorded for the composite with 10 phr of the plasticizer. The composite with 15 phr of glycerol reached the second peak at 75 °C (tan δ = 0.29), while the sample with 20 phr obtained the second maximum at 55 °C (tan δ = 0.29).
The achieved results suggest that the composite systems show two transition temperatures. The first peak maximum corresponds to glass transition temperature of composites and was strongly influenced by the type of rubber matrix. It becomes apparent that Tg for both composite types almost did not change in dependence on glycerol content. Lower Tg were found to have vulcanizates based on SBR, which is a logical reflection of higher amount of rubbery butadiene units in SBR structure. The second peak corresponds to glass transition temperature of lignosulfonate and it can be stated that the higher was the amount of glycerol, the lower was Tg of the biopolymer. This points out to strong plasticizing effect of glycerol on lignosulfonate. As suggested, plasticized lignosulfonate formed softer domains of lover rigidity and higher deformation ability. It is very likely that softened lignosulfonate can behave more as a polymer than as a filler. Thus, it might be stated that two polymer phases are formed when lignosulfonate is plasticized with higher amount of glycerol. Also, based upon the achieved results it becomes apparent that glycerol acts by plasticizing effect more on lignosulfonate rather than on rubber matrices, as evidence of the lowering of Tg of the biopopolymer.
4. Conclusions
In this work, calcium lignosulfonate in constant amount was added into carbon black rubber batches based on NBR and SBR. Glycerol was applied into rubber compounds to improve process-ability and properties of rubber compounds.
The results demonstrated that glycerol influenced the course of curing isotherms and decelerated the curing kinetics. This was reflected in the decrease of cross-linking degree, which was more pronounced for composites with higher amount of glycerol. The higher the amount of glycerol in rubber compounds, the lower their viscosities. This points out to plasticizing effect of glycerol on rubber formulations. The dynamic-mechanical analysis revealed that two transition regions occurred on loss factor - temperature dependences for composites with higher glycerol content, suggesting the formation of two polymer phases. The first peak that represents the glass transition temperature of composites is strongly dependent on the type of rubber matrix. Though, it was not influenced by the amount of glycerol. The second peak relates with glass transition temperature of lignosulfonate and it was evident that the higher was the amount of glycerol, the lower was Tg of the biopolymer. This points out to higher plasticizing effect of glycerol on lignosulfonate than on rubber matrices. The softened lignosulfonate was then better dispersed and distributed within the rubber formulations. Glycerol also contributed to better compatibility and adhesion between the biopolymer and rubber matrices on their interface, resulting in the improvement of tensile characteristics of composites. On the other hand, the dependencies of modulus and hardness were in closer correlations with the dependencies of cross-link density.
Acknowledgments
This work was supported by the Slovak Research and Development Agency under the contract No. APVV-19-0091 and APVV-22-0011.
References
- European Green Deal. https://commission.europa.eu/strategy-and-policy/priorities-2019-2024/european-green-deal_en (2019).
- Jacobs, B.; Yao, Y.; Van Nieuwenhove, I.; Sharma, D.; Graulus, G.J.; Bernaerts, K.; Verberckmoes, A. Sustainable lignin modifications and processing methods: green chemistry as the way forward. Green Chem. 2023, 25, 2042–2086. [Google Scholar] [CrossRef]
- Thomas, S.K.; Parameswaranpillai, J.; Krishnasamy, S.; Sabura Begum, P.M.; Nandi, D.; Siengchin, S.; Jacob George, J.; Hameed, N.; Salim, N.V.; Sienkiewicz, N. A comprehensive review on cellulose, chitin, and starch as fillers in natural rubber biocomposites. Carbohydr Polym Technol Appl. 2021, 2, 100095. [Google Scholar] [CrossRef]
- Sternberg, J.; Sequerth, O.; Pilla, S. Green chemistry design in polymers derived from lignin: review and perspective. Prog Polym Sci. 2021, 113, 101344. [Google Scholar] [CrossRef]
- Temesgen, S.; Rennert, M.; Tesfaye, T.; Grosmann, L.; Kuehnert, I.; Smolka, N.; Nase, M. Thermal, morphological, and structural characterization of starch-based bio-polymers for melt spinnability. e-Polymers. 2024, 24, 20240025. [Google Scholar] [CrossRef]
- Sakakibara, K.; Tsujii, Y. Visualization of fibrillated cellulose in polymer composites using a fluorescent-labeled polymer dispersant. ACS Sustainable Chem Eng. 2023, 11, 6332–6342. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Yang, D.; Wang, H.; Liu, W.; Li, Z. Multilayer two-dimensional lignin/ZnO composites with excellent anti-UV aging properties for polymer films. Green Chem Eng. 2022, 3, 338–348. [Google Scholar] [CrossRef]
- Kim, S.; Chung, H. Convenient cross-linking control of lignin-based polymers influencing structure–property relationships. ACS Sustainable Chem Eng. 2023, 11, 1709–1719. [Google Scholar] [CrossRef]
- Ridho, M.R.; Agustiany, E.A.; Rahmi Dn, M. Lignin as green filler in polymer composites: development methods, characteristics, and potential applications. Adv Mater Sci Eng. 2022, Article ID 1363481, 33p. [Google Scholar] [CrossRef]
- Araujo, T.R.; Bresolin, D.; de Oliveira, D.; Sayer, C.; de Araújo, P.H.P.; de Oliveira, J.V. Conventional lignin functionalization for polyurethane applications and a future vision in the use of enzymes as an alternative method. Eur Polym J. 2023, 188, 111934. [Google Scholar] [CrossRef]
- Henriksson, G.; Germgård, U.; Lindström, M.E. A review on chemical mechanisms of kraft pulping. Nord Pulp Paper Res J. 2024, 39, 297–311. [Google Scholar] [CrossRef]
- Fabbri, F.; Bischof, S.; Mayr, S.; Gritsch, S.; Bartolome, M.J.; Schwaiger, N.; Guebitz, G.M.; Weiss, R. The biomodified lignin platform: A review. Polymers. 2023, 15, 1694. [Google Scholar] [CrossRef] [PubMed]
- Yu, O.; Kim, K.H. Lignin to materials: A focused review on recent novel lignin applications. Appl Sci. 2020, 10, 4626. [Google Scholar] [CrossRef]
- Schulze, P.; Leschinsky, M.; Seidel-Morgenstern, A.; Lorenz, H. Continuous separation of lignin from organosolv pulping liquors: combined lignin particle formation and solvent recovery. Ind Eng Chem Res. 2019, 58, 3797–3810. [Google Scholar] [CrossRef]
- Guadix-Montero, S.; Sankar, M. Review on catalytic cleavage of C–C inter-unit linkages in lignin model compounds: towards lignin depolymerisation. Top Catal. 2018, 61, 183–198. [Google Scholar] [CrossRef]
- Vasile, C.; Baican, M. Lignins as promising renewable biopolymers and bioactive compounds for high-performance materials. Polymers. 2023, 15, 3177. [Google Scholar] [CrossRef]
- Shorey, R.; Salaghi, A.; Fatehi, P.; Mekonnen, T.H. Valorization of lignin for advanced material applications: a review. RSC Sustainability. 2024, 2, 804. [Google Scholar] [CrossRef]
- Gonçalves, S.; Ferra, J.; Paiva, N.; Martins, J.; Carvalho, L.H.; Magalhães, F.D. Lignosulphonates as an alternative to non-renewable binders in wood-based materials. Polymers. 2021, 13, 4196. [Google Scholar] [CrossRef]
- Libretti, C.; Correa, L.S.; Meier, M.A.R. From waste to resource: advancements in sustainable lignin modification. Green Chem. 2024, 26, 4358. [Google Scholar] [CrossRef]
- Wysocka, K.; Szymona, K.; McDonald, A.G.; Mamiński, M. Characterization of thermal and mechanical properties of lignosulfonate- and hydrolyzed lignosulfonate-based polyurethane foams. BioRes. 2016, 11, 7355–7364. [Google Scholar] [CrossRef]
- Sugiarto, S.; Leow, Y.; Li Tan, C.; Wang, G.; Kai, D. How far is lignin from being a biomedical material? Bioact Mater. 2022, 8, 71–94. [Google Scholar] [CrossRef]
- Xu, P.; Lin, Q.; Fang, L. Study on the mechanical properties of loess improved by lignosulfonate and its mechanism analysis and prospects. Appl Sci. 2022, 12, 9843. [Google Scholar] [CrossRef]
- Vera, M.; Bischof, S.; Rivas, B.L.; Weber, H.; Mahler, A.K.; Kozich, M.; Guebitz, G.M.; Nyanhongo, G.S. Biosynthesis of highly flexible lignosulfonate–starch based materials. Eur Polym J. 2023, 198, 112392. [Google Scholar] [CrossRef]
- Dai, P.; Liang, M.; Ma, X.; Luo, Y.; He, M.; Gu, X.; Gu, Q.; Hussain, I.; Luo, Z. Highly efficient, environmentally friendly lignin-based flame retardant used in epoxy resin. ACS Omega. 2020, 5, 32084–32093. [Google Scholar] [CrossRef] [PubMed]
- Komisarz, K.; Majka, T.M.; Pielichowski, K. Chemical transformation of lignosulfonates to lignosulfonamides with improved thermal characteristics. Fibers. 2022, 10, 20. [Google Scholar] [CrossRef]
- Jedrzejczyk, M.A.; den Bosch, S.V.; Van Aelst, J.; Van Aelst, K.; Kouris, P.D.; Moalin, M.; Haenen, G.R.M.M.; Boot, M.D.; Hensen, E.J.M.; Lagrain, B.; Sels, B.F.; Bernaerts, K.V. Lignin-based additives for improved thermo-oxidative stability of biolubricants. ACS Sustainable Chem Eng. 2021, 9, 12548–12559. [Google Scholar] [CrossRef]
- Ye, D.Z.; Jiang, L.; Hu, X.Q.; Zhang, M.H.; Zhang, X. Lignosulfonate as reinforcement in polyvinyl alcohol film: Mechanical properties and interaction analysis. Int J Biol Macromol. 2015, 83, 209–215. [Google Scholar] [CrossRef]
- Cai, M.; Zhao, X.; Han, X.; Du, P.; Su, Y.; Chen, C. Effect of thermal oxygen aging mode on rheological properties and compatibility of lignin-modified asphalt binder by dynamic shear rheometer. Polymers. 2022, 14, 3572. [Google Scholar] [CrossRef]
- Mili, M.; Hasmi, S.A.R.; Ather, M.; Hada, V.; Markandeya, N.; Kamble, S.; Mohapatra, M.; Rathore, S.K.S.; Srivastava, A.K.; Verma, S. Novel lignin as natural-biodegradable binder for various sectors-A review. J Appl Polym Sci. 2022, 139, e51951. [Google Scholar] [CrossRef]
- Bahl, K.; Jana, S.C. Surface modification of lignosulfonates for reinforcement of styrene–butadiene rubber compounds. J Appl Polym Sci. 2014, 131, 40123. [Google Scholar] [CrossRef]
- Shorey, R.; Gupta, A.; Mekonnen, T.H. Hydrophobic modification of lignin for rubber composites. Ind Crops Prod. 2021, 174, 114189. [Google Scholar] [CrossRef]
- Mohamad Aini, N.A.; Othman, N.; Hussin, M.H.; Sahakaro, K.; Hayeemasae, N. Hydroxymethylation-modified lignin and its effectiveness as a filler in rubber composites. Processes. 2019, 7, 315. [Google Scholar] [CrossRef]
- Koskinen, J.; Kemppainen, N.; Sarlin, E. Lignin dispersion in polybutadiene rubber (BR) with different mixing parameters. Prog Rubber Plast Recycl Technol. 2024. [CrossRef]
- Ferruti, F.; Carnevale, M.; Giannini, L.; Guerra, S.; Tadiello, L.; Orlandi, M.; Zoia, L. Mechanochemical methacrylation of lignin for biobased reinforcing filler in rubber compounds. ACS Sustain Chem Eng. 2024, 12, 14028–14037. [Google Scholar] [CrossRef]
- Li, M.; Zhu, L.; Xiao, H.; Shen, T.; Tan, Z.; Zhuang, W.; Xi, Y.; Ji, X.; Zhu, C.; Ying, H. Design of a lignin-based versatile bioreinforcement for high-performance natural rubber composites. ACS Sustain Chem Eng. 2022, 10, 8031–8042. [Google Scholar] [CrossRef]
- Atifi, S.; Miao, C.; Hamad, W.Y. Surface modification of lignin for applications in polypropylene blends. J Appl Polym Sci. 2017, 134, 45103. [Google Scholar] [CrossRef]
- Komisarz, K.; Majka, T.M.; Pielichowski, K. Chemical and physical modification of lignin for green polymeric composite materials. Materials. 2023, 16, 16. [Google Scholar] [CrossRef]
- Hait, S.; Kumar, L.; Ijaradar, J.; Ghosh, A.K.; De, D.; Chanda, J.; Ghosh, P.; Gupta, S.D.; Mukhopadhyay, R.; Wiesner, S.; Heinrich, G.; Das, A. Unlocking the potential of lignin: Towards a sustainable solution for tire rubber compound reinforcement. J Clean Prod. 2024, 470, 143274. [Google Scholar] [CrossRef]
- Adibi, A.; Jubinville, D.; Chen, G.; Mekonnen, T.H. In-situ surface grafting of lignin onto an epoxidized natural rubber matrix: A masterbatch filler for reinforcing rubber composites. React Funct Polym. 2024, 197, 105856. [Google Scholar] [CrossRef]
- Kun, D.; Pukánszky, B. Polymer/lignin blends: Interactions, properties, applications. Eur Polym J. 2017, 93, 618–641. [Google Scholar] [CrossRef]
- Kruželák, J.; Hložeková, K.; Kvasničáková, A.; Džuganová, M.; Chodák, I.; Hudec, I. Application of plasticizer glycerol in lignosulfonate-filled rubber compounds based on SBR and NBR. Materials. 2023, 16, 635. [Google Scholar] [CrossRef]
- Kruželák, J.; Hložeková, K.; Kvasničáková, A.; Džuganová, M.; Hronkovič, J.; Preťo, J.; Hudec, I. Calcium-lignosulfonate-filled rubber compounds based on NBR with enhanced physical–mechanical characteristics. Polymers. 2022, 14, 5356. [Google Scholar] [CrossRef] [PubMed]
- Kraus, G. Swelling of filler-reinforced vulcanizates. J Appl Polym Sci, 1963, 7, 861–871. [Google Scholar] [CrossRef]
- Alam, M.N.; Kumar, V.; Potiyaraj, P.; Lee, D.J.; Choi, J. Mutual dispersion of graphite–silica binary fillers and its effects on curing, mechanical, and aging properties of natural rubber composites. Polym Bull. 2022, 79, 2707–2724. [Google Scholar] [CrossRef]
- Ansarifar, A. Highly Efficient Methods for Sulfur Vulcanization Techniques, Results and Implications. Balboa Press, United Kongdom, 2022.
- Maciejewska, M.; Krzywania-Kaliszewska, A.; Zaborski, M. Ionic liquids applied to improve the dispersion of calcium oxide nanoparticles in the hydrogenated acrylonitrile-butadiene elastomer. Am J Mater Sci. 2013, 3, 63–69. [Google Scholar]
- Kruželák, J.; Sýkora, R.; Hudec, I. Vulcanization of rubber compounds with peroxide curing systems. Rubber Chem Technol. 2017, 90, 60–88. [Google Scholar] [CrossRef]
Figure 1.
Vulcanization isotherms of composites based on NBR.
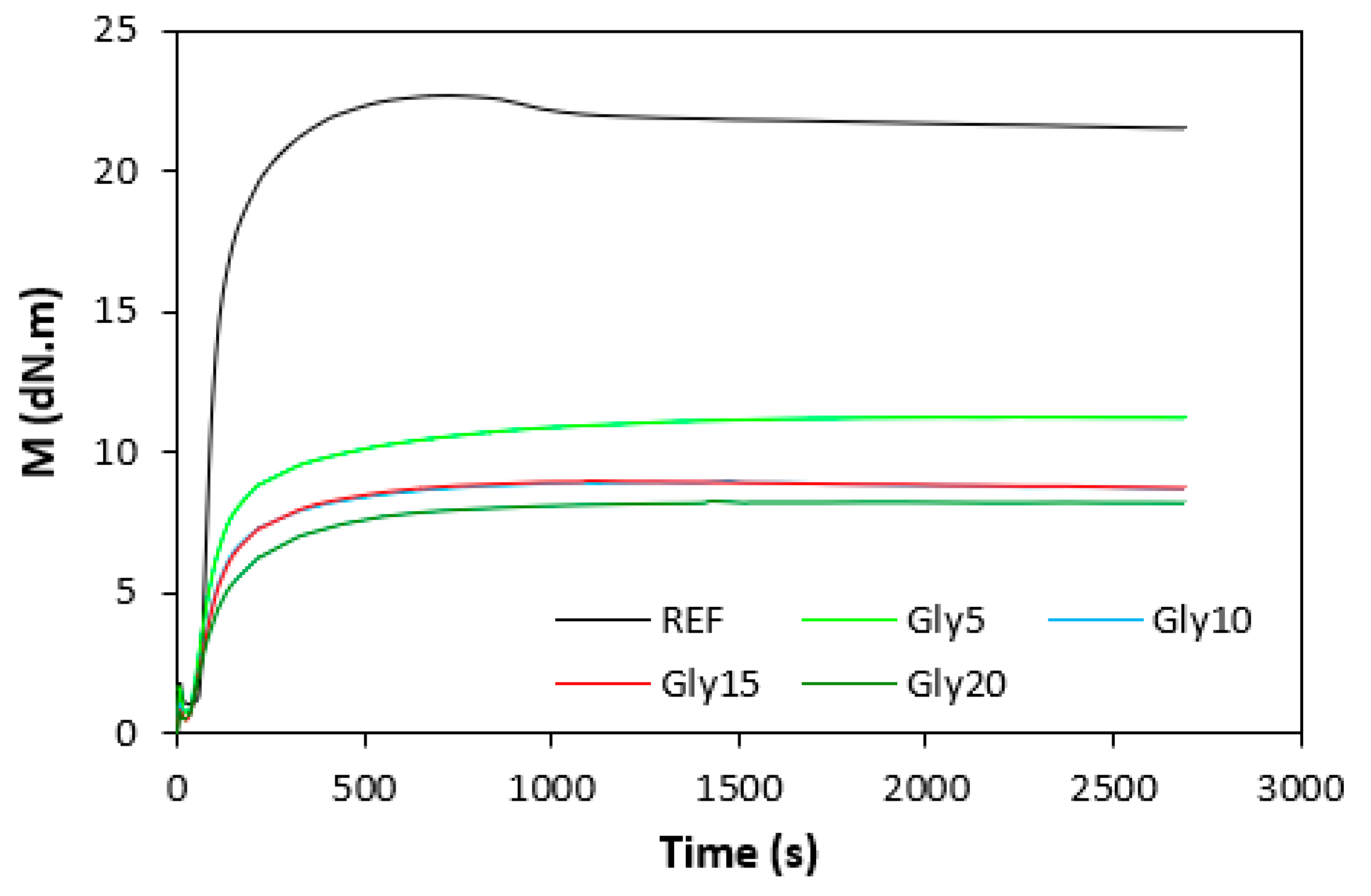
Figure 2.
Vulcanization isotherms of composites based on SBR.
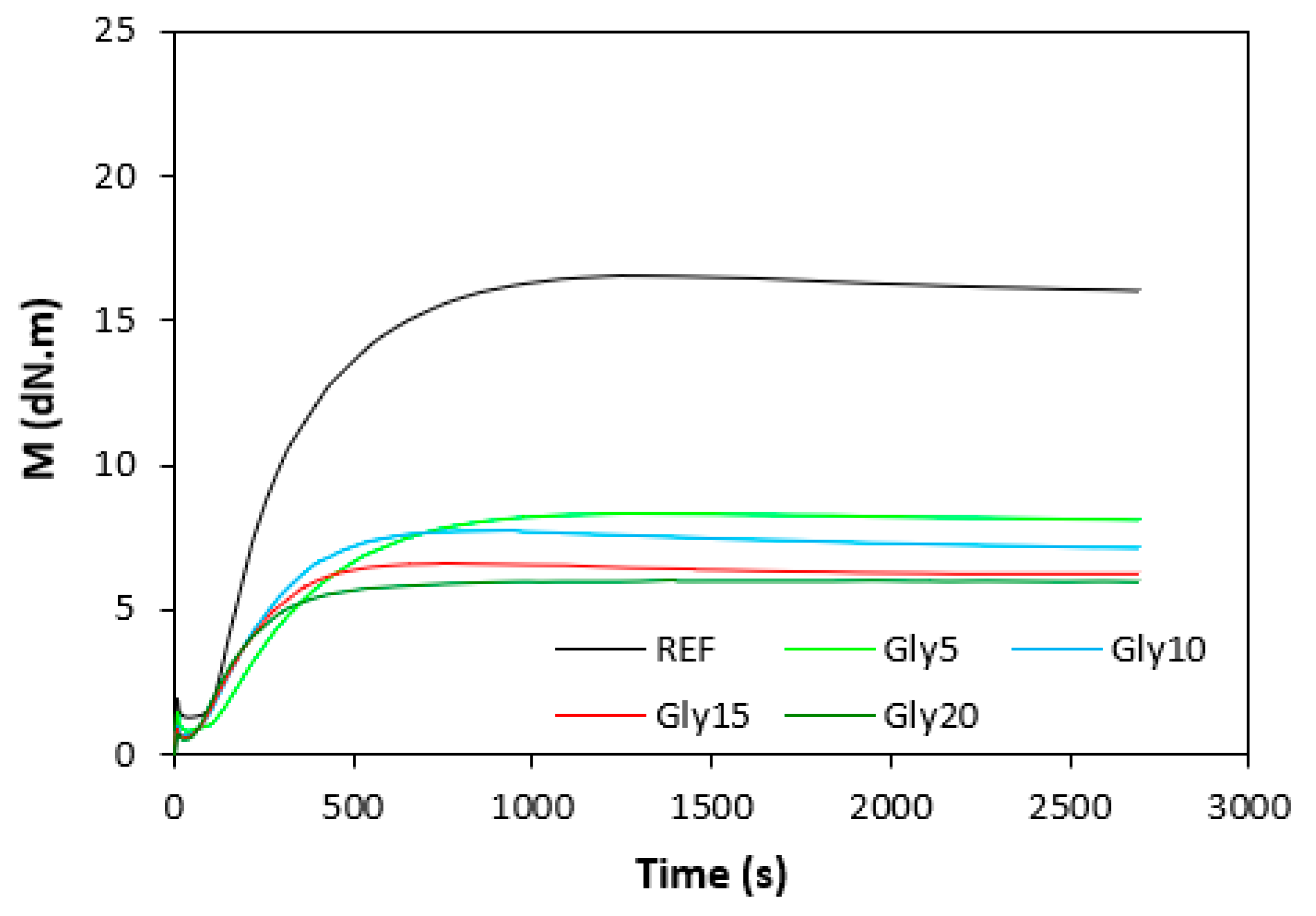
Figure 3.
Influence of glycerol content on torque difference ΔM of composites.
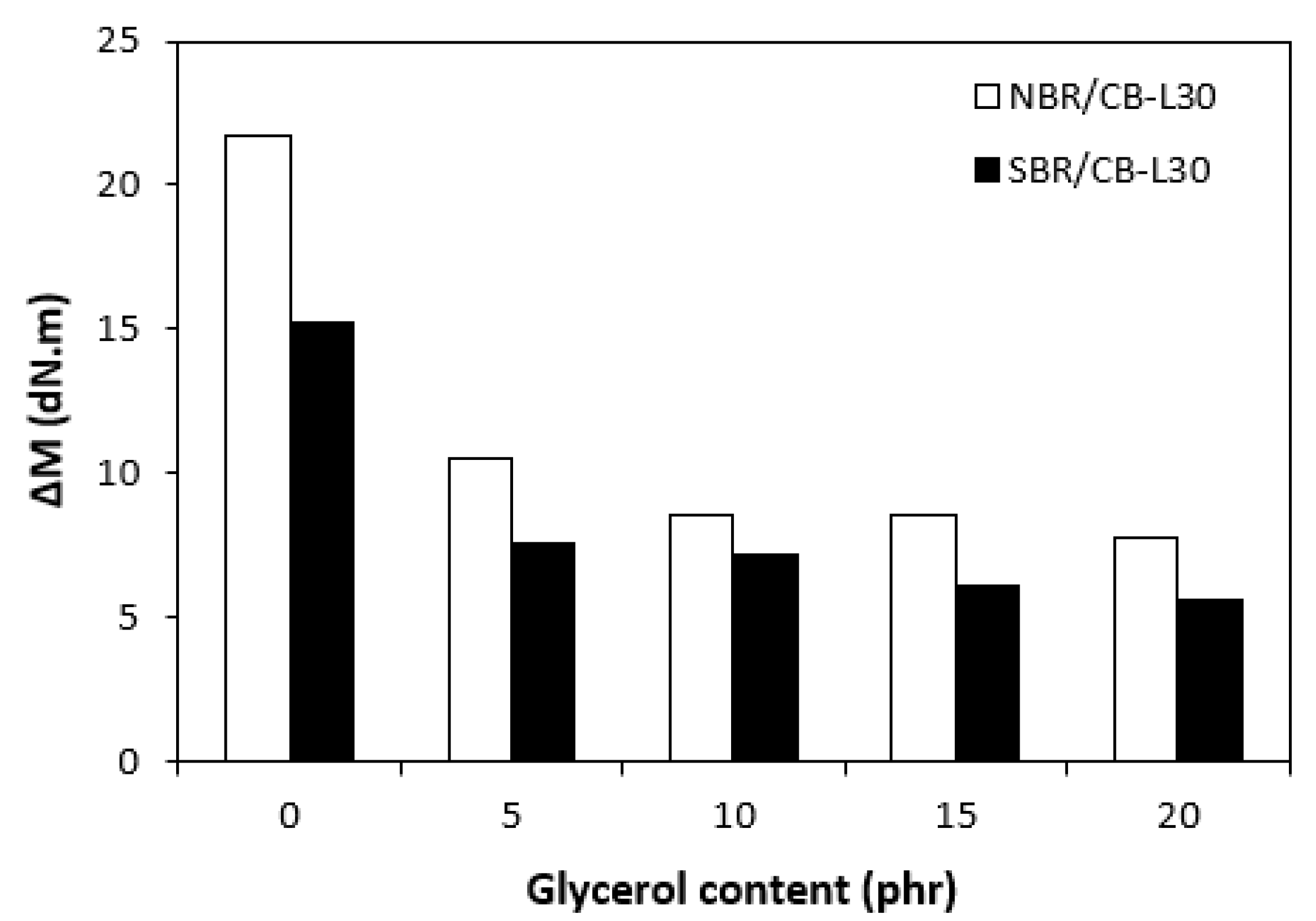
Figure 4.
Influence of glycerol content on cross-link density υ of composites.
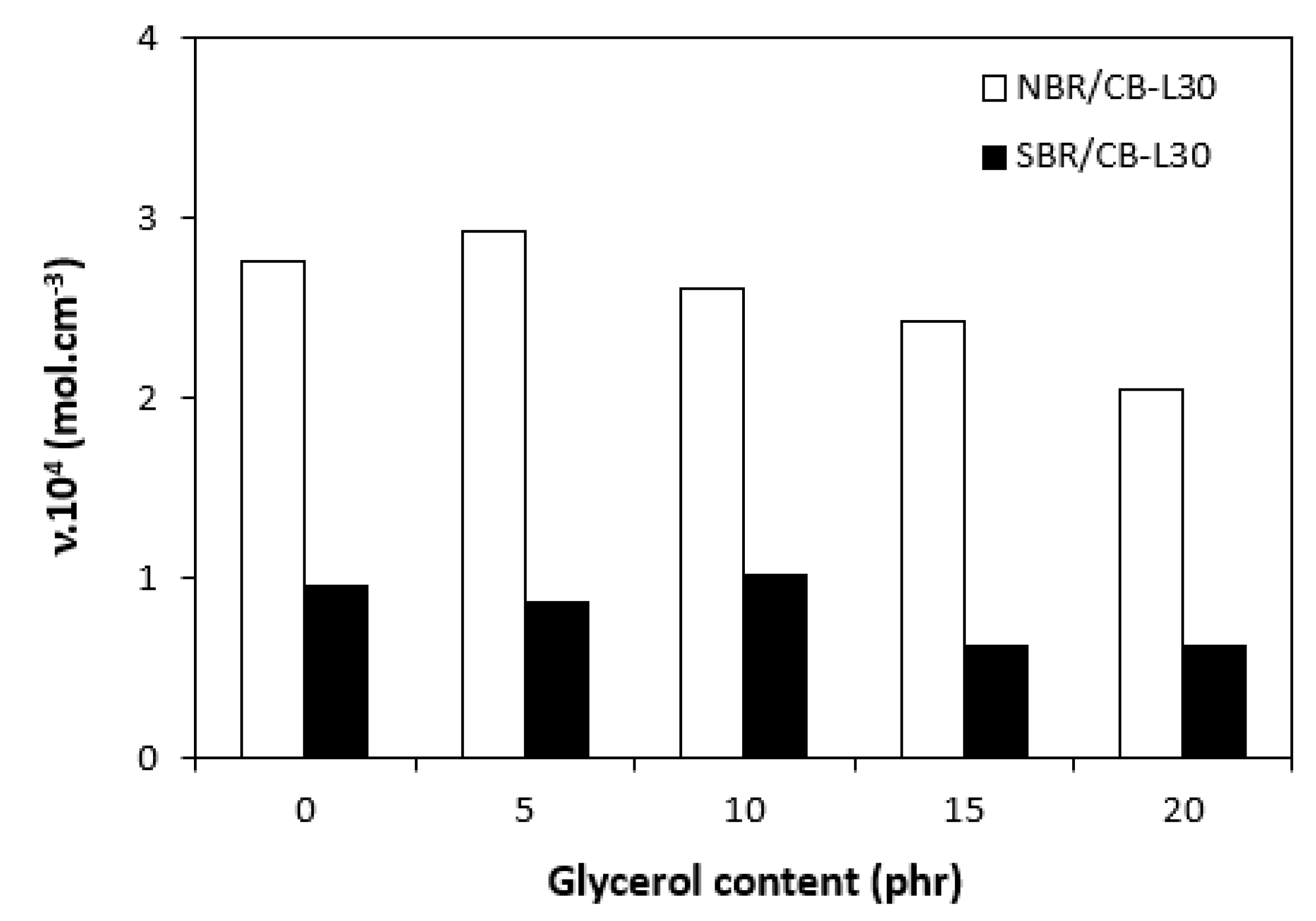
Figure 5.
Influence of glycerol content on scorch time ts1 of composites.
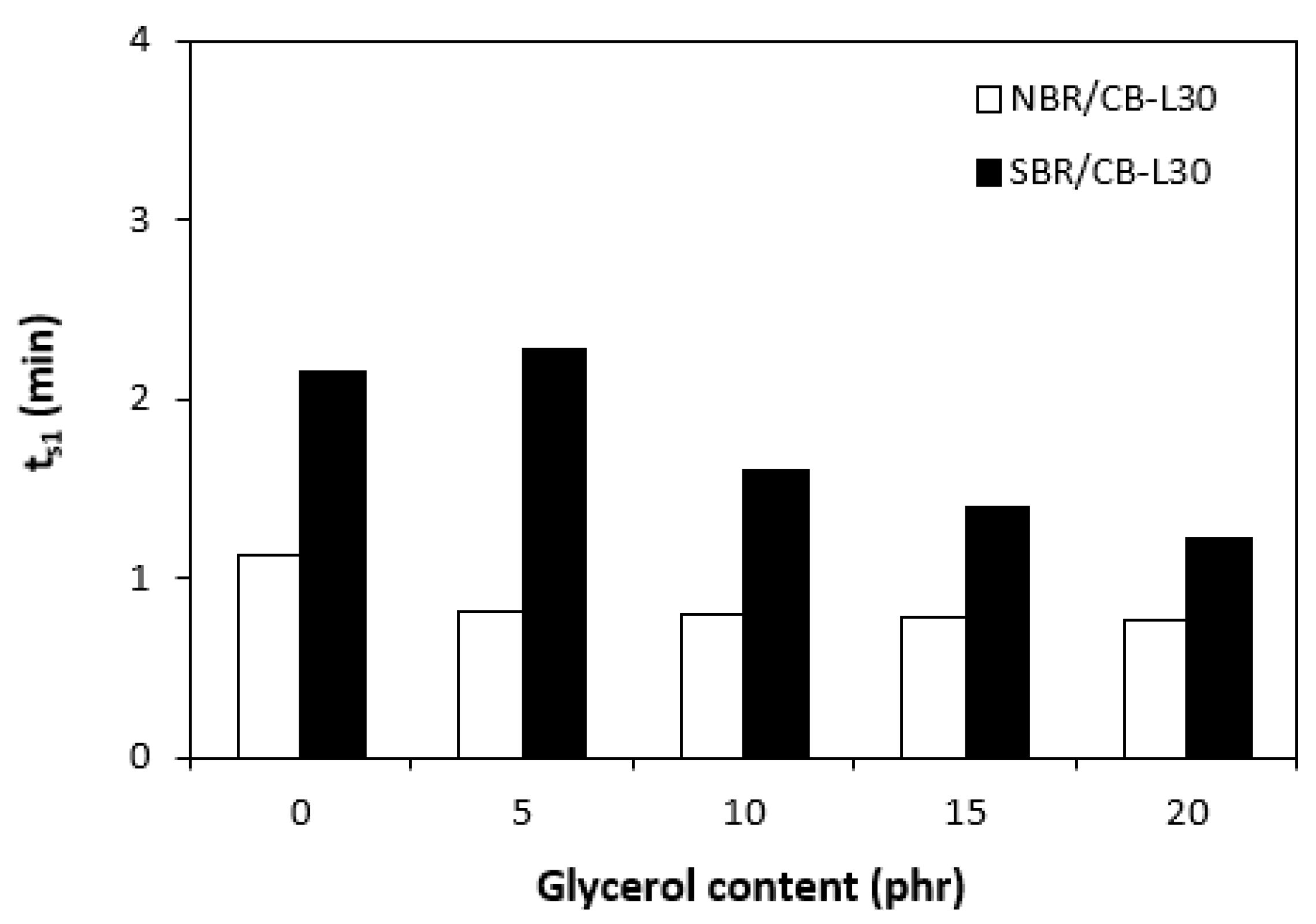
Figure 6.
Influence of glycerol content on optimum cure time tc90 of composites.
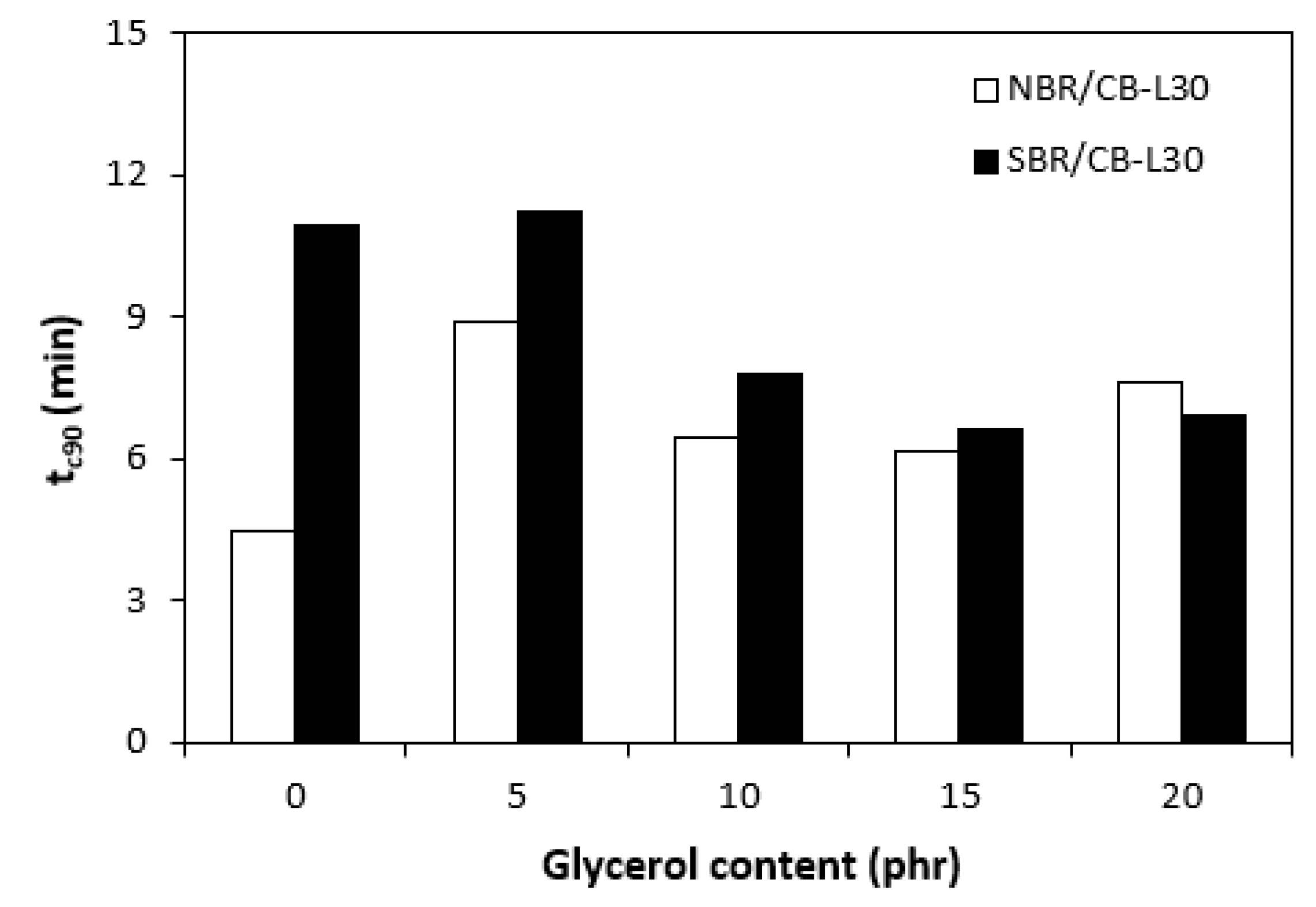
Figure 7.
Influence of glycerol content on curing rate R of composites.
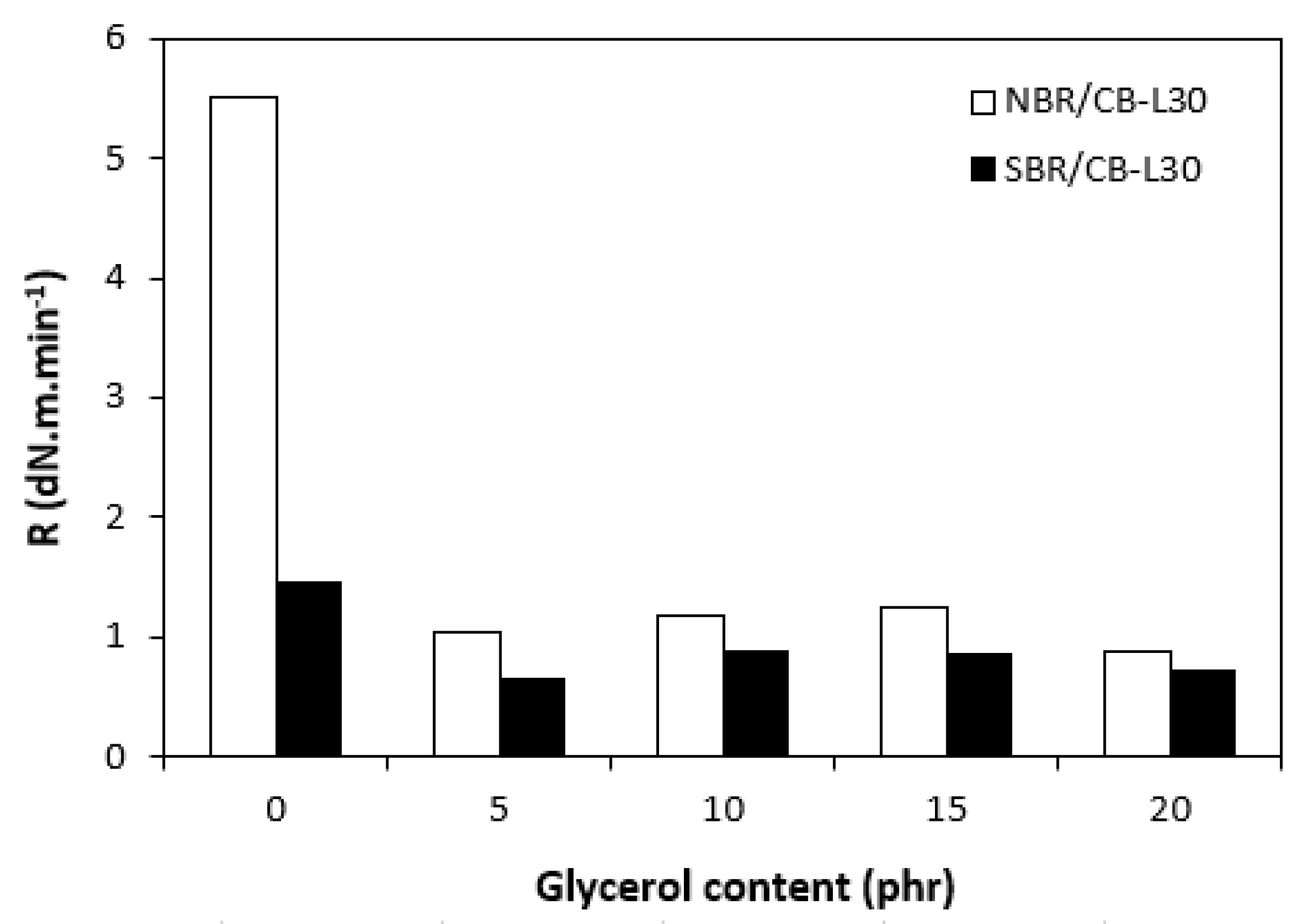
Figure 8.
Influence of glycerol content on maximum curing rate of composites.
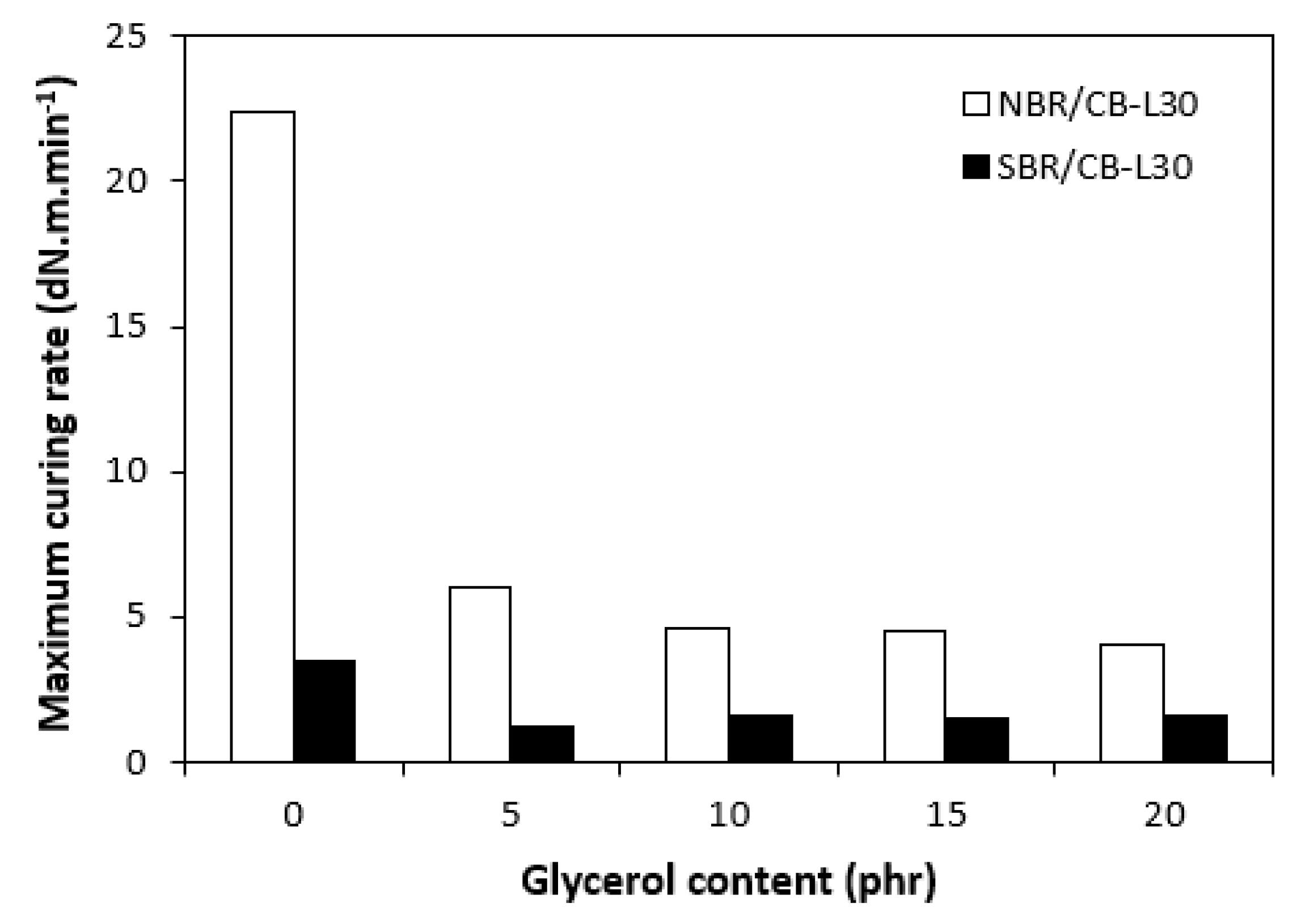
Figure 9.
Dependence of dynamic complex viscosity η* of composites based on NBR on shear rate.
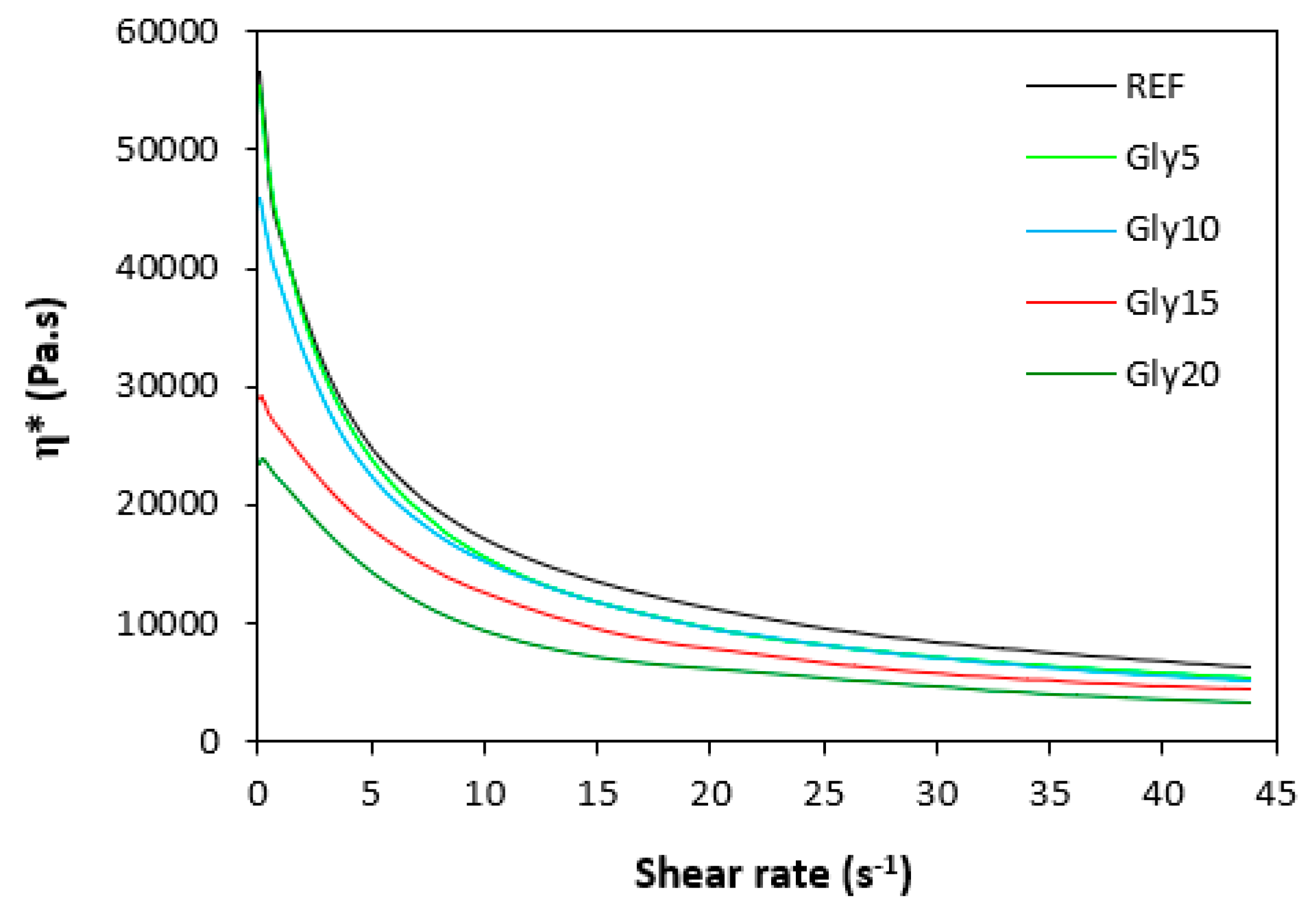
Figure 10.
Dependence of dynamic complex viscosity η* of composites based on SBR on shear rate.
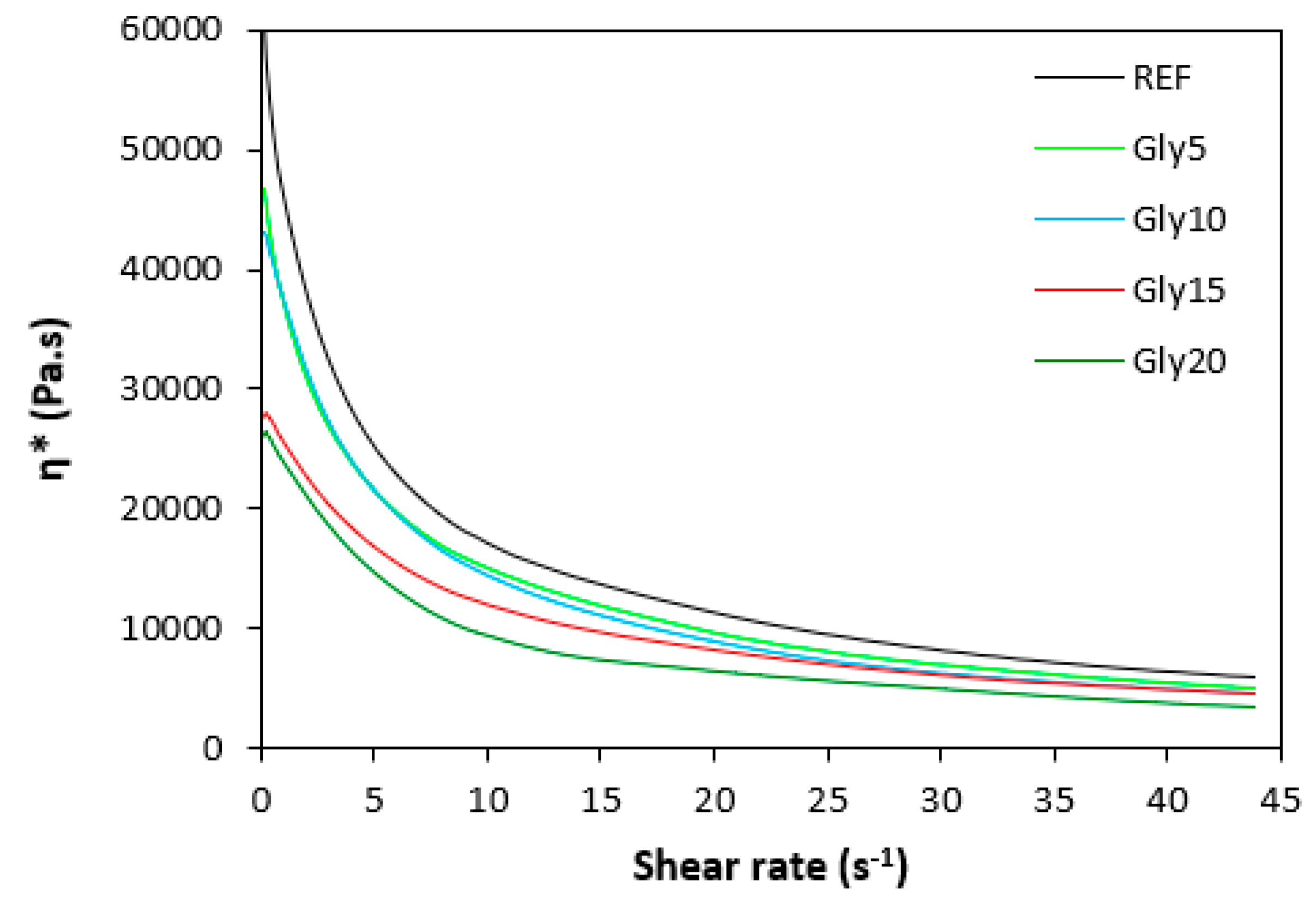
Figure 11.
Influence of glycerol content on modulus M300 of composites.

Figure 12.
Influence of glycerol content on hardness of composites.
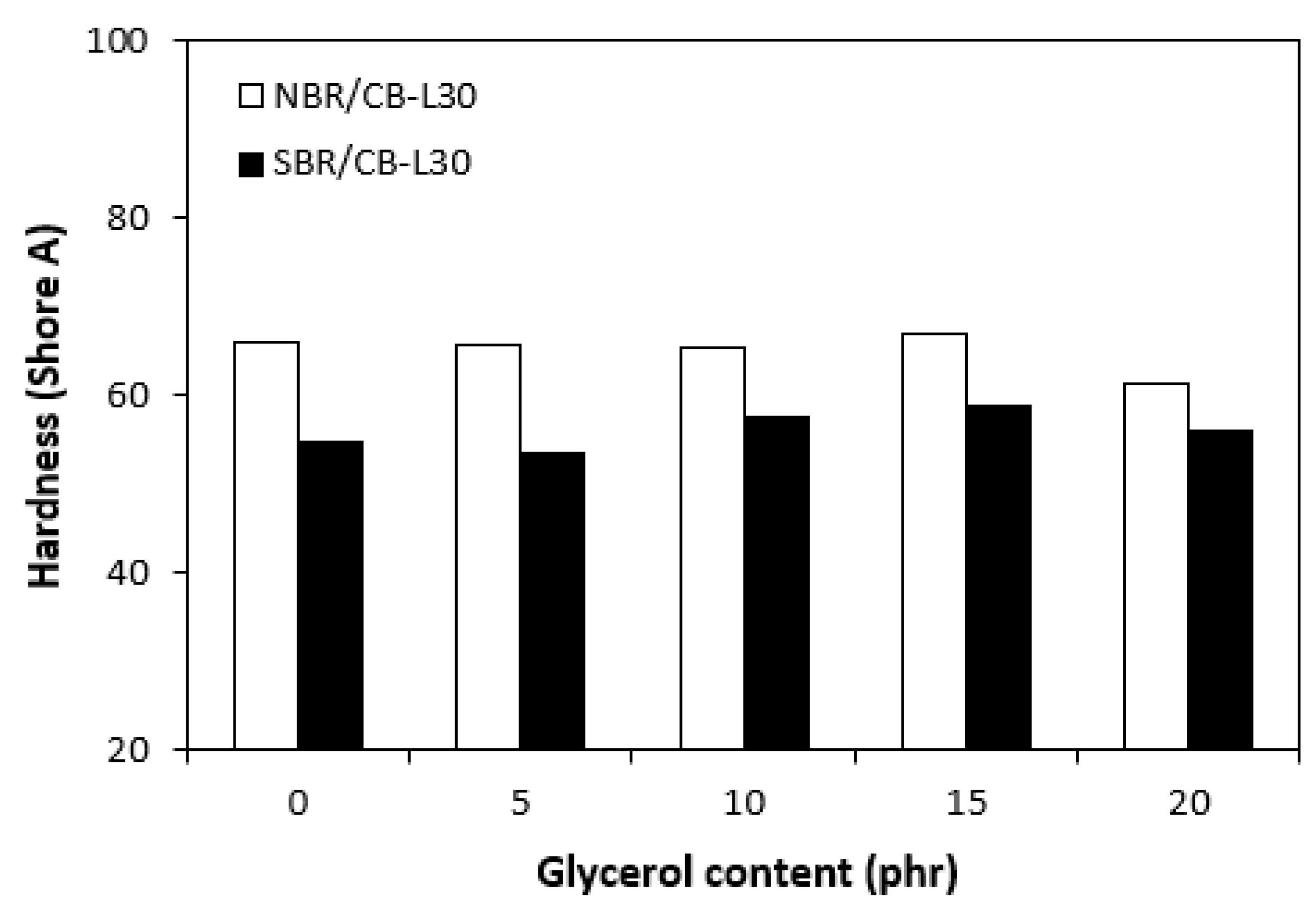
Figure 13.
Influence of glycerol content on elongation at break of composites.
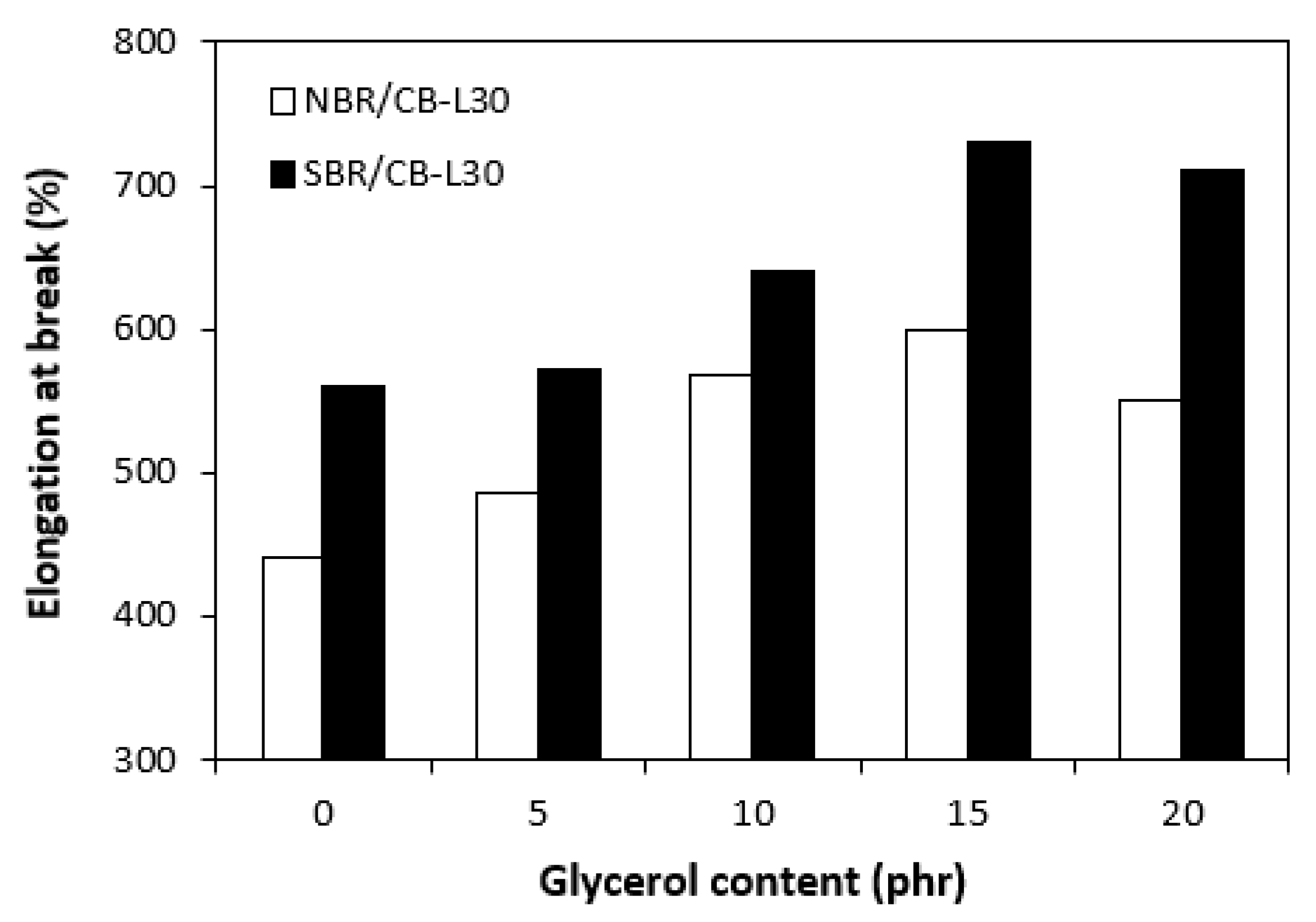
Figure 14.
Influence of glycerol content on tensile strength of composites.
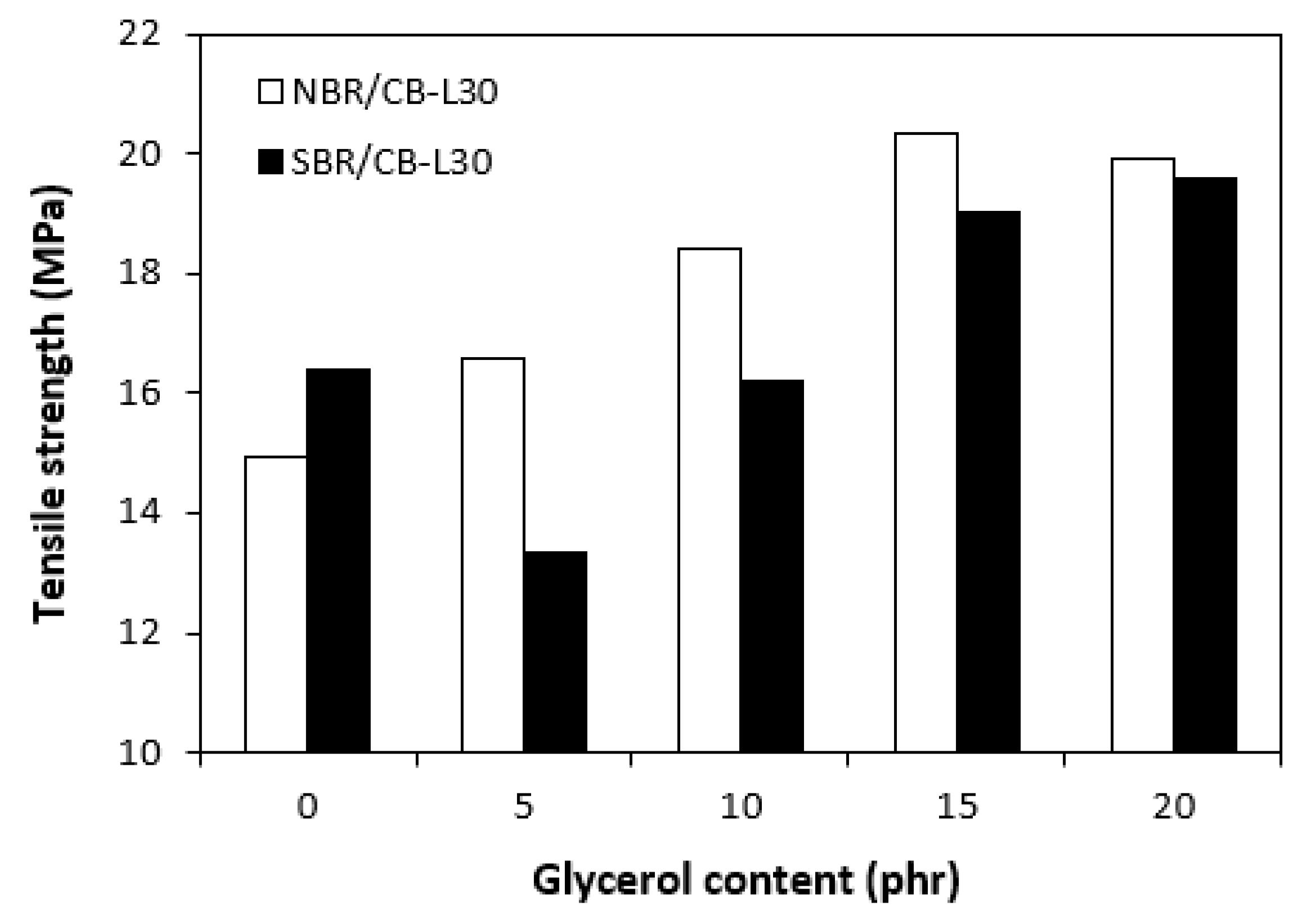
Figure 15.
SEM images of composites based on SBR: (A) reference sample without glycerol, (B) composite with 5 phr of glycerol, (C) composite with 10 phr of glycerol, (D) with 15 composite phr of glycerol, (E) composite with 20 phr of glycerol, .
Figure 15.
SEM images of composites based on SBR: (A) reference sample without glycerol, (B) composite with 5 phr of glycerol, (C) composite with 10 phr of glycerol, (D) with 15 composite phr of glycerol, (E) composite with 20 phr of glycerol, .
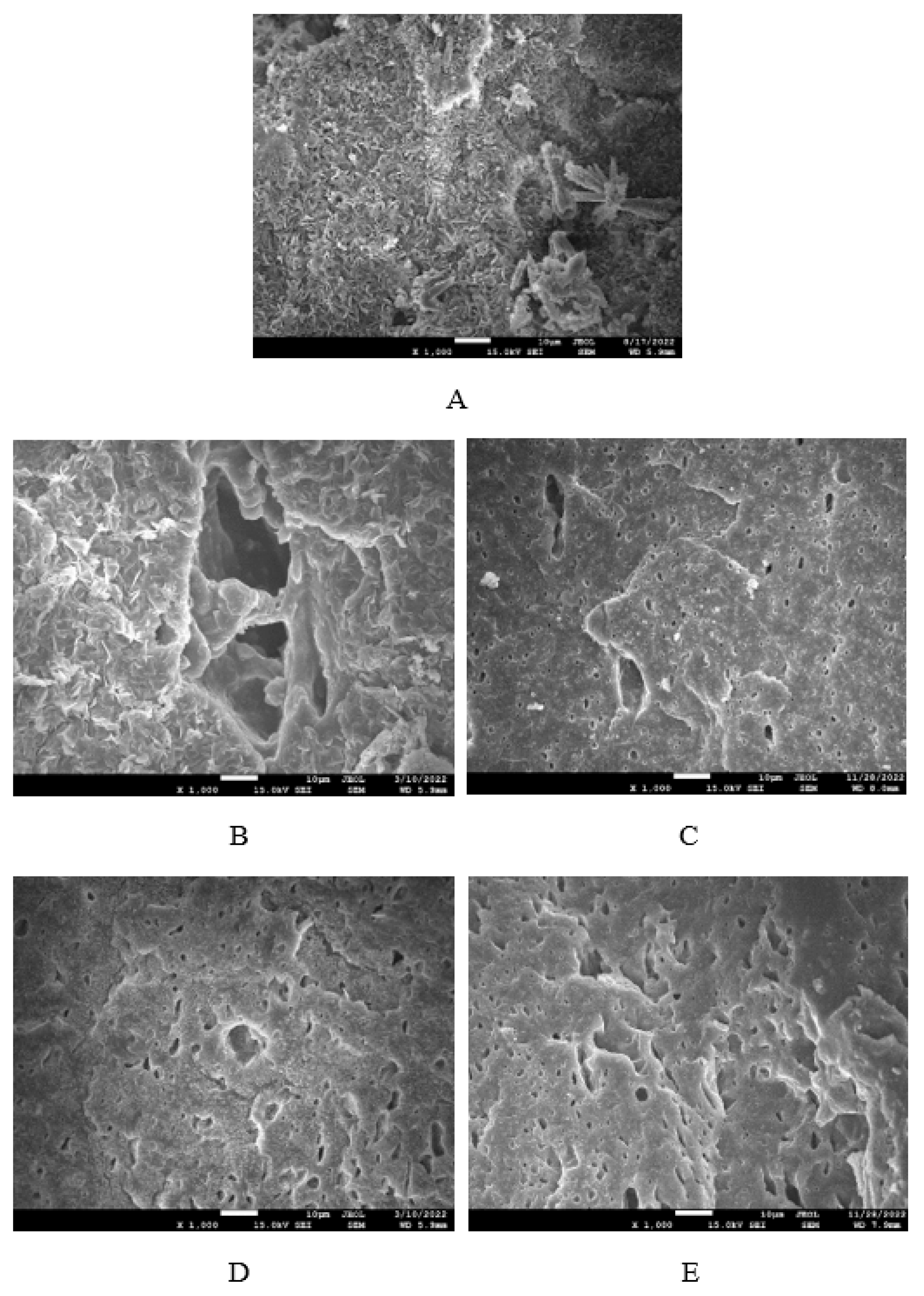
Figure 16.
SEM images of composites based on NBR: (A) reference sample without glycerol, (B) composite with 5 phr of glycerol, (C) composite with 10 phr of glycerol, (D) composite with 15 phr of glycerol, (E) composite with 20 phr of glycerol, .
Figure 16.
SEM images of composites based on NBR: (A) reference sample without glycerol, (B) composite with 5 phr of glycerol, (C) composite with 10 phr of glycerol, (D) composite with 15 phr of glycerol, (E) composite with 20 phr of glycerol, .
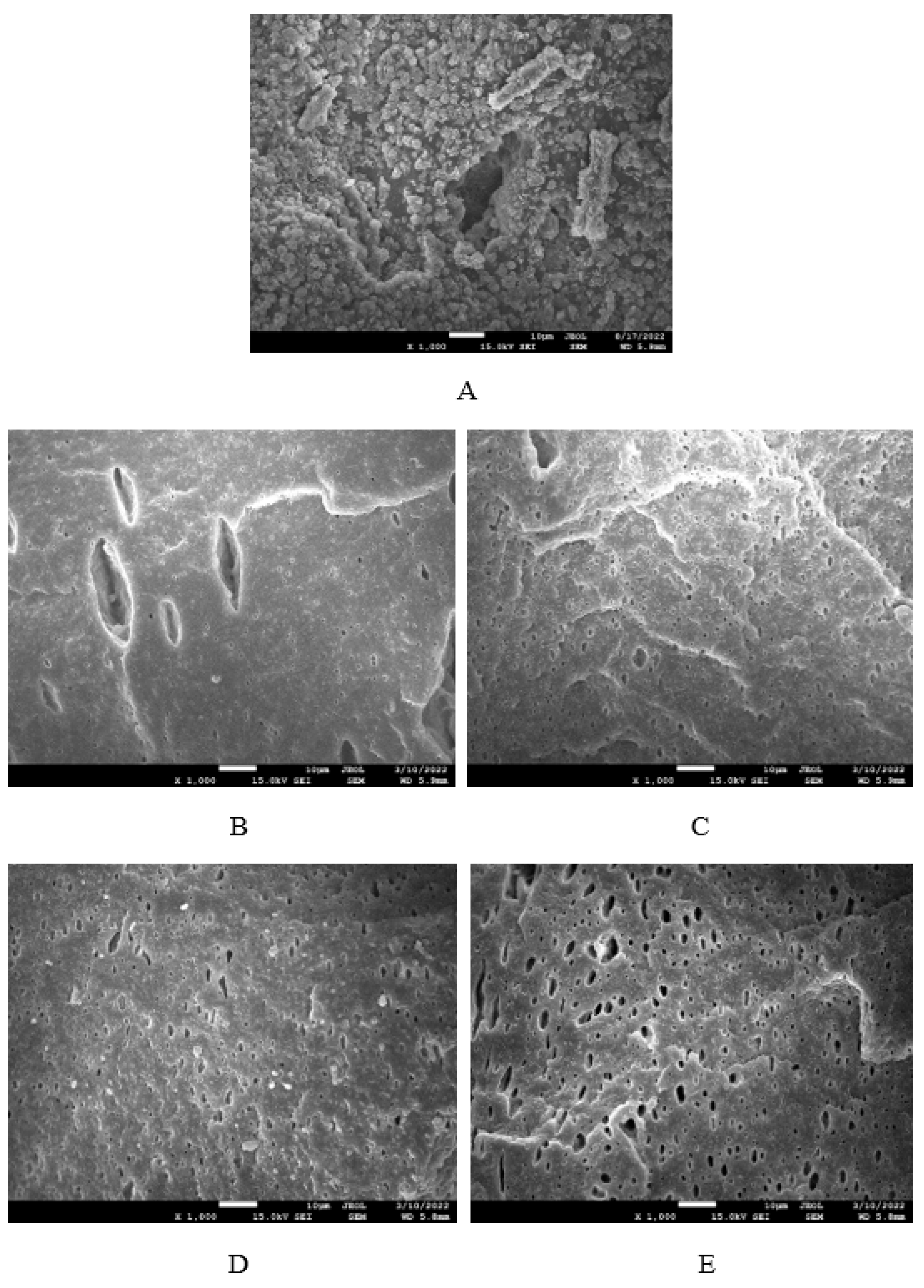
Figure 17.
Temperature dependences of loss factor tan δ for composites based on NBR.
Figure 18.
Temperature dependences of loss factor tan δ for composites based on SBR.
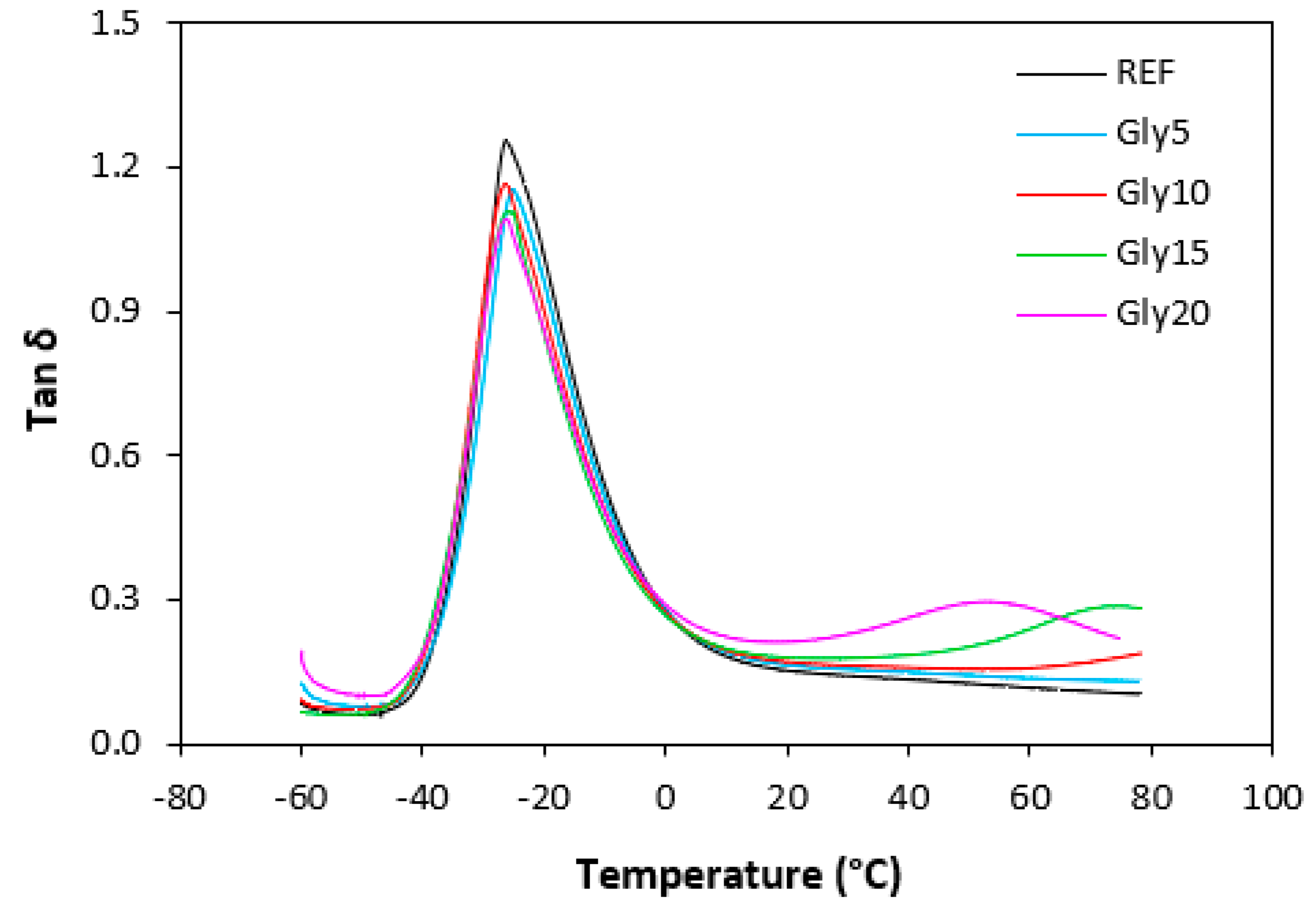

Table 1.
Storage G' and loss G'' modulus of composites based on NBR.
| Glycerol (phr) | G' (MPa) (-20 °C) |
G' (MPa) (0 °C) |
G' (MPa) (20 °C) |
G' (MPa) (60 °C) |
G" (MPa) (-20 °C) |
G" (MPa) (0 °C) |
G" (MPa) (20 °C) |
G" (MPa) (60 °C) |
|---|---|---|---|---|---|---|---|---|
| 0 | 3430 | 46.64 | 16.64 | 10.75 | 296 | 47.49 | 4.7 | 1.29 |
| 5 | 3227 | 45.62 | 15.68 | 9.83 | 343 | 47.37 | 4.47 | 1.34 |
| 10 | 3988 | 48.01 | 16.65 | 9.41 | 444 | 49.68 | 5.09 | 1.70 |
| 15 | 3137 | 43.84 | 16.59 | 7.66 | 434 | 39.86 | 4.71 | 2.00 |
| 20 | 2410 | 41.42 | 14.52 | 4.94 | 383 | 35.97 | 4.89 | 1.29 |
Table 2.
Glass transition temperature Tg and loss factor tan δ of composites based on NBR.
| Glycerol (phr) | Tg (°C) | tan δ (-20 °C) |
tan δ (0 °C) |
tan δ (20 °C) |
tan δ (60 °C) |
|---|---|---|---|---|---|
| 0 | -4.4 | 0.09 | 1.02 | 0.28 | 0.12 |
| 5 | -4.4 | 0.11 | 1.04 | 0.29 | 0.14 |
| 10 | -3.8 | 0.11 | 1.03 | 0.31 | 0.18 |
| 15 | -5.5 | 0.14 | 0.91 | 0.28 | 0.26 |
| 20 | -5.6 | 0.16 | 0.87 | 0.34 | 0.26 |
Table 3.
Storage G' and loss G'' modulus of composites based on SBR.
| Glycerol (phr) | G' (MPa) (-20 °C) |
G' (MPa) (0 °C) |
G' (MPa) (20 °C) |
G' (MPa) (60 °C) |
G" (MPa) (-20 °C) |
G" (MPa) (0 °C) |
G" (MPa) (20 °C) |
G" (MPa) (60 °C) |
|---|---|---|---|---|---|---|---|---|
| 0 | 36.57 | 13.75 | 10.63 | 8.18 | 37.05 | 3.81 | 1.63 | 0.96 |
| 5 | 42.53 | 15.91 | 12.13 | 8.79 | 41.01 | 4.47 | 2.01 | 1.22 |
| 10 | 44.61 | 17.31 | 12.85 | 8.55 | 40.09 | 4.75 | 2.21 | 1.36 |
| 15 | 45.62 | 18.29 | 13.31 | 7.2 | 39.09 | 4.94 | 2.42 | 1.74 |
| 20 | 43.67 | 17.19 | 11.67 | 4.62 | 37.45 | 4.98 | 2.48 | 1.31 |
Table 4.
Glass transition temperature Tg and loss factor tan δ of composites based on SBR.
| Glycerol (phr) |
Tg (°C) | tan δ (-20 °C) |
tan δ (0 °C) |
tan δ (20 °C) |
tan δ (60 °C) |
|---|---|---|---|---|---|
| 0 | -26.3 | 1.01 | 0.28 | 0.15 | 0.12 |
| 5 | -25.1 | 0.96 | 0.28 | 0.17 | 0.14 |
| 10 | -26.3 | 0.90 | 0.27 | 0.17 | 0.16 |
| 15 | -24.9 | 0.86 | 0.27 | 0.18 | 0.24 |
| 20 | -26.3 | 0.86 | 0.29 | 0.21 | 0.28 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



