
Submitted:
16 October 2024
Posted:
17 October 2024
You are already at the latest version
Abstract
Cancer remains a primary cause of mortality, with over 18.1 million new cases and 9.6 million deaths globally in 2018. Chemotherapy, which utilizes a spectrum of cytotoxic drugs targeting rapidly dividing cancer cells, is a predominant treatment modality. However, the tendency of chemotherapeutics to induce drug resistance and exhibit non-specific cytotoxicity necessitates the development of new anticancer agents with heightened efficacy and minimized toxicity. In recent years, the discovery of safe and effective antibacterial/antiviral agents has also been a hot spot in medicinal chemistry. This paper comprehensively reviews the synthesis, anticancer/ antibacterial/antiviral activity, and structure-activity relationships of natural 1,4-naphthoquinones and their derivatives. It highlights their potential as efficient and low-toxicity anti-tumor and anti-infectious drug candidates.
Keywords:
1
; 4-Naphthoquinones
; Anticancer
; Antimicrobial
; Antiviral
; Structure-activity relationships
; Natural products
1. Introduction
Cancer is a leading cause of death worldwide, posing a severe threat to human health. World Health Organization (WHO) data from 2018 indicates a staggering 18 million new cases, leading to approximately 9.5 million fatalities globally. China, the most populous nation in the world, reported 4.28 million new cases in the same year. This figure represents 23.8% of the global cancer incidence and contributes to 30.1% of cancer-related deaths globally [1]. Lung, breast, and colorectal cancers are the most prevalent and account for a third of incidence and mortality rates in the world. The primary treatment strategy in clinical settings includes radiotherapy, surgery, and chemotherapy, each playing a vital role in combating these formidable diseases [2].
Rooted in the principles of cancer biology and molecular genetics, chemotherapy is considered a cornerstone of cancer treatment and employs a wide range of drugs known for their cytotoxic effects. These drugs primarily, though not exclusively, target the rapidly dividing cancer cells [3]. However, its effectiveness is often overshadowed by significant limitations. Many anticancer drugs lack specificity, leading to collateral damage to healthy cells and causing severe toxic side effects, for example, skin and hair changes, nausea, blood clotting issues, fatigue, immune suppression, and potential damage to vital organs, with some effects causing long-term harm [4].
A critical challenge in chemotherapy is drug resistance, which accounts for approximately 90% of treatment failures due to cancer's ability to evade and metastasize [5]. This has steered the scientific community towards developing targeted anticancer therapies that promise higher efficacy and lower toxicity.
The issue of treatment resistance extends beyond cancer. Infectious diseases, notably caused by bacteria and viruses, also pose a global health threat. The COVID-19 pandemic, triggered by the SARS-CoV-2 virus, illustrates this issue significantly, which caused over 591 million cases and 6.43 million deaths by August 2022. Annually, bacterial infections lead to approximately 700,000 deaths, with projections suggesting a rise to 10 million by 2050, surpassing cancer fatalities [6]. The growing challenge of multidrug resistance (MDR) infections has been recognized by the World Health Organization as one of the top ten global health threats, emphasizing the urgent need for novel therapeutic strategies [7].
Malignant tumors present a significant threat to human health, with increasing cancer incidence underscoring the urgency of effective cancer prevention and treatment. In the United States, approximately 1,958,310 new cancer cases are projected for 2023. Looking ahead, the total number of cancer cases in the U.S. is expected to reach 2.3 million by 2025, driven by an aging population. Despite chemotherapy being the primary treatment modality, its extensive cytotoxic effects, poor drug selectivity, and tendency to foster drug resistance pose significant challenges. Consequently, developing highly effective, less toxic anti-tumor drugs has become a focal point in contemporary anti-tumor drug research. Addressing these issues is crucial for improving patient outcomes and reducing the global cancer burden.
2. Natural 1,4-naphthoquinones and Synthetic Studies
The exploration of lead compounds from natural products, followed by their structural modification, forms the cornerstone of drug research and development. Quinone compounds, a notable class of natural organic substances, have substantial medicinal value. Clinically, several drugs employed in solid tumor treatments, such as erythromycin, adriamycin, mitomycin, and mitoxantrone, incorporate quinone structures [8,9,10]. Moreover, naphthoquinones, prevalent in plants, fungi, and some animals, exhibit a broad spectrum of pharmacological properties [11].
Among various naphthoquinones, natural 1,4-naphthoquinones, which are naphthol analogs with a conjugated diketone group on the naphthalene structure, are particularly significant. The most crucial subset within this family is 1,4-naphthoquinone, widely utilized as an Active Pharmaceutical Ingredient (API) in the industry. These derivatives display diverse biological activities, including antifungal, antibacterial, antiplatelet, antiallergic, antiinflammatory, antithrombotic, antiviral, anti-tumor effects, and activities related to lipoxygenase inhibition, free radical scavenging, and antiradical properties [12,13,14,15].
Notable natural 1,4-naphthoquinone products include lawsone (7, Figure 1), juglone (5), shikonin (9), alkannin (10), vitamin K (1), plumbagin (6), and lapachol (8) [16,17]. Vitamin K analogs (1), characterized by a methyl group on the quinone ring (1,4-naphthoquinone), encompass several natural products (11, 12). Juglone and plumbagin share homologous structures, and similarly, the structures of lawsone and lapachol are closely related and can be discussed in conjunction. The naturally occurring naphazarin compounds, such as shikonin and alkannin, are a pair of enantiomers with significant relevance.
2.1. Synthesis of Natural 1,4-naphthoquinones
2.1.1. Synthesis of Vitamin K and Its Analogs
Vitamin K is an essential class of disubstituted 1,4-naphthoquinone derivatives, plays a crucial role in blood coagulation and is essential for synthesizing coagulation factors in the liver. There are two naturally occurring forms: Vitamin K1 (Phylloquinone), an oil-soluble vitamin derived from alfalfa, and Vitamin K2 (Menatetrenone), a crystalline form obtained from fermented fish. Vitamin K1 is abundant in plants and is known as phylloquinone due to its phytane side chain [18,19]. Vitamin K2, the biologically active form of Vitamin K, is subdivided into various subtypes like MK-4 and MK-7, differentiated by the number of isoprene units at the 3-position, the two most studied subtypes [20].
Following the isolation of Vitamin K1 (11, Figure 2) from alfalfa leaves, initial synthesis approaches involved Friedel-Crafts acylation using phytobromine or chlorophyll alcohol and 2-methyl-1,4-naphthoquinone. While presenting a straightforward strategy, these methods result in low yield [21,22]. Subsequent optimizations by Chenard [23] and Lipshutz [24] significantly improved yields. Chenard's method involved coupling protected naphthoquinone cuprate (13) with phytyl bromine, while Lipshutz's approach used chloromethylated 2-methyl-1,4-naphthoquinone (15) coupled under nickel chloride and triphenylphosphine catalytic conditions.
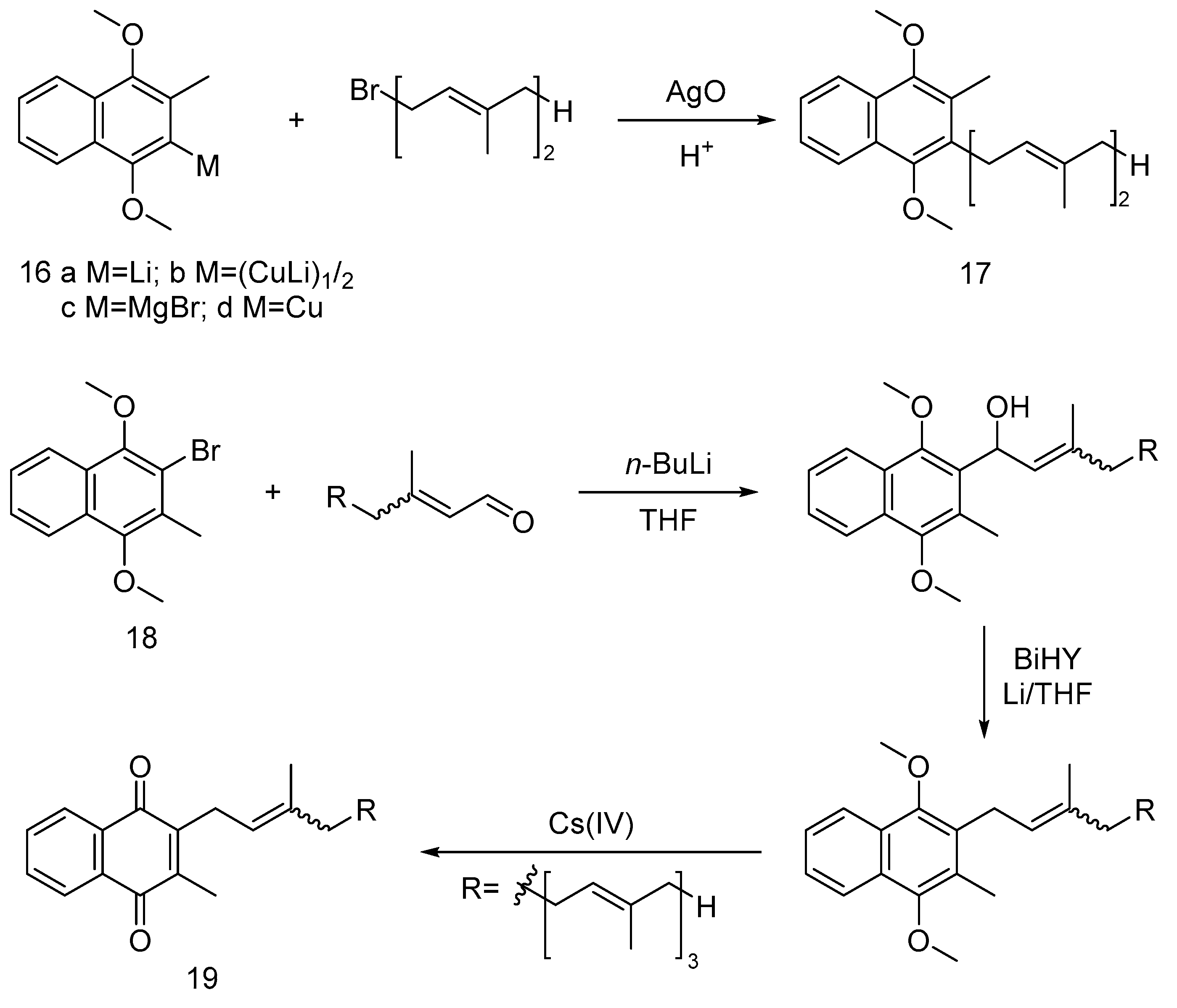
For the synthesis of vitamin MK-2 (17), Snyder et al. [25] reported a method using 2-metal-3-methyl-1,4-dimethoxynaphthalene (16) and aldehydes or chloroalkanes, with the Grignard reagent (16c) proving most effective for high-yield MK-2 synthesis up to 80% and 99% retention of E-configuration. Garcia [26] described a synthesis route for MK-4 (19), involving 2-bromo-3-methyl-1,4-dimethoxynaphthalene (18) and allyl aldehyde coupling using n-butyllithium to deliver corresponding alcohol, followed by Birch reduction to retain the conformation of desired intermediate and finally ceric ammonium nitrate (IV) oxidation to obtain vitamin MK-4.
Figure 4.
The synthetic route of vitamin MK-4.
Vitamin MK-7(30, Figure 5), a subtype of Vitamin K2, has distinguished high biological activity and better bioavailability than other Vitamin K forms. It features minimal intestinal absorption and is retained in the serum for up to three days. Such efficiency allows small daily doses to suffice in supporting Vitamin K-dependent enzymes and proteins. MK-7 also plays a role in calcium metabolism, contributing to bone formation and influencing the health of arterial blood vessel walls, unlike Vitamin K1.
The synthesis of Vitamin MK-7, based on its molecular structure, can be achieved from menadione or its protected derivatives. In a notable advancement, Krajewski et al. [27] developed a multistep synthesis process, which has been the subject of a U.S. patent application. The synthesis begins with farnesol (21, Figure 5), which undergoes acetylation and then oxidizes its hydroxyl group in the presence of tin dioxide. The resulting product is then esterified using phosphorus tribromide, leading to intermediate 23. This is followed by a substitution reaction with brominated farnesol to form intermediate 24, and then the substitution of benzenesulfonyl and acetyl groups yielded intermediate 26. The next step involves bromination to produce key intermediate 27. Concurrently, diethylmethanaphthoquinone (20) undergoes a substitution reaction to form intermediate 28. These intermediates then undergo a nucleophilic substitution to produce intermediate 29. The final synthesis stages include dephenylsulfonylation and oxidation, culminating in the production of Vitamin MK-7.
2.1.2. Synthesis of Juglone
Juglone (5, Figure 6), scientifically denoted as 5-hydroxy-1,4-naphthoquinone, is an intrinsic dye compound naturally found in Lawsonia inermis leaves and widely distributed across the roots, husks, fruits and bark of the walnut family [28,29]. However, due to its low content and extraction efficiency in P. nutans, chemical synthesis is the predominant source of juglone [30].
The early synthesis of juglone was achieved through a Friedel-Crafts acylation and cyclization reaction utilizing 1,4-dimethoxybenzene (31) as the foundational substrate, forming an intermediate (32). Khalafy et al. [31] pioneered juglone synthesis, yielding the intermediate 5,8-dihydroxy-1-tetrahydronaphthalenone (32), which was subsequently oxidized using DDQ to yield juglone. Notably, this method offers relatively high yields; however, its dependence on toxic solvents, such as benzene, renders it useless for industrial-scale production.
Subsequent methodologies harnessed juglone's quinone structure, focusing on the oxidation of naphthalene and naphthol as critical intermediates. Mamchur et al. [32] employed naphthalene (33) as the primary substrate, initially sulfonation to form a salt, followed by alkalinization to obtain a phenol sodium salt and ultimately subjecting it to oxidation with dichromate to yield juglone. However, it is a straightforward synthetic strategy. The main disadvantages are low yields and the use of heavy metals like chromium, which presents environmental contamination concerns.
Mingxiao Zhang et al. [33] introduced an innovative one-pot synthesis method for juglone. This approach synthesized the peracetic acid from acetic anhydride and hydrogen peroxide, eliminating the need for sulfuric acid. Subsequently, the authors incrementally added a methanolic solution of 1,5-dihydroxynaphthalene (34) and stirred at 50 °C for 4 hours to yield juglone. The main advantages of this method are ease of operation, high yields, cost-effectiveness, readily accessible raw materials, and eliminating the necessity for toxic solvents.
Hashemi et al. [34] embraced an environmentally conscientious synthetic technique, solid-phase microwave oxidation, for juglone synthesis. The method entailed mixing 1,8-dihydroxynaphthalene (35), periodate, and kaolinite clay in a mortar, followed by microwave irradiation for 50 seconds, resulting in a commendable 91% yield of juglone. This approach has a brief reaction duration, high yield, and negligible environmental impact.
2.1.3. Synthesis of Lawsone
Lawsone, also known as 2-hydroxy-1,4-naphthoquinone, is a reddish-orange dye naturally occurring in the leaves of Lawsonia inermis and the flowers of water hyacinth. Its historical use as a hair and skin dye dates back over 5000 years [35]. Lawsone has the remarkable ability to form a Michael addition reaction with the protein keratin in the skin and hair, resulting in long-lasting colouration until natural shedding occurs. Furthermore, it exhibits strong UV-absorbing properties, making aqueous extracts effective in preventing skin tanning. Due to its susceptibility to biodegradation during extraction, contemporary production primarily relies on chemical synthesis [27,36,37].
Fieser et al. [38] accomplished the synthesis of lawsone (7, Figure 7) through the closure of the phenyl derivative's ring (36) via a cyclization reaction. This reaction can occur under alkaline conditions in atmospheric air or catalyzed by p-nitrophenyl and copper sulfate. Inoue et al. [39] employed a multistep process to convert 4-chlorobutyryl compounds (37) into lawsone. Initially, naphthol (38, 39) was oxidized to 2-hydroxy-1,4-naphthoquinone using potassium superoxide (KO2) in a toluene solution of 18-crown-6 [40] or a tetrahydrofuran solution at -10 ℃ [41], delivering 34% yield. Subsequently, 4-amino-1,3-naphthalenediphenol (40) underwent oxidation to deliver lawsone under HCl or NaOH conditions at 45°C for an hour [42]. 3,4-dihydroxy-1-naphthoic acid (41) was hydrolyzed under alkaline KOH conditions in the presence of air to yield the target product [43]. Further oxidation of 3,4-dimethoxy-1-naphthol (42) was carried out in aqueous NaOH and potassium ferricyanide to afford lawsone (65% yield) and a dimer product [44]. Subsequently, 2-allyl-1,4-naphthoquinone (43) underwent oxidation using KOH and H2O2, yielding a lawsone. Finally, the hydrolysis of 4-allyloxy-1,2-naphthalenedione (44) under alkaline conditions resulted in the formation of lawsone and allyl alcohol.
2.1.4. Synthesis of Shikonin and Alkannin
Red-root gromwell, belonging to the Comfrey family, encompasses two distinct species: Lithospermum erythrorhizon and Arnebia euchroma. It is predominantly found in regions spanning China, Korea, and Japan [45]. Within this botanical genus, shikonin (9, Figure 8) and alkannin (10) emerge as a pair of enantiomeric isomers distinguished by their naphthazarin structure. Shikonin serves as the primary active constituent of Lithospermum erythrorhizon, whereas alkannin takes precedence in Arnebia euchroma. While conventional methods for obtaining shikonin have traditionally involved plant tissue extraction or cellular tissue cultivation, sustained research efforts have now made it feasible to achieve high enantiomeric excess (ee) values of shikonin, alkannin, and their derivatives through chemical synthesis [46,47].
Most synthetic approaches to shikonin rely on 1,4,5,8-tetramethoxynaphthaldehyde as a key starting material. Terada et al. [48] made a pioneering contribution by synthesizing racemic shikonin, commencing with 1,4,5,8-tetramethoxynaphthaldehyde (45) as the initial substrate. The method involves a nucleophilic addition reaction of a glycol-protected Grignard reagent to the aldehyde, followed by subsequent hydrolysis, ketone oxidation, and, ultimately, demethylation in the presence of AgO/AgNO3 to yield racemic shikonin. Subsequently, Moiseenkov et al. [49] employed a similar strategy using 1,4,5,8-tetramethoxynaphthaldehyde (45) as the starting material. This method encompassed epoxidation followed by ring opening through reaction with organic lithium reagents, forming 1,4,5,8-tetramethoxy shikonin (46). The final step involved racemic shikonin synthesis through oxidation with ceric ammonium nitrate and demethylation with AgO/AgNO3.
Nicolaou et al. [50] have meticulously documented a sophisticated total synthesis strategy aimed at elucidating the absolute conformation of shikonin (9, Figure 9) and alkannin (10). The method commences with the strategic utilization of commercially available 2,3-dichloronaphthazarin (47) as the primary starting material. This strategic approach involves the initial protection of the hydroxyl group, concomitant with the simultaneous bromination of the naphthazarin ring, culminating in the formation of intermediate 48. Subsequently, introducing the shikonin side chain at the brominated position yields the aryl ketone intermediate 49. This intermediate is of paramount significance, as it is an exceptionally well-suited substrate for numerous asymmetric reducing agents, rendering it the optimal choice for synthesizing both enantiomers. The reduction step, meticulously orchestrated through utilizing DIP-Cl, a chiral reducing agent, leads to forming intermediates 50 (S-configuration) and 51 (R-configuration), respectively. These intermediates are further subjected to oxidation and the subsequent removal of formaldehyde, ultimately revealing the absolute configurations of alkannin and shikonin.
This groundbreaking methodology stands out on several fronts. Firstly, it harnesses naphthazarin's economic and abundant resources as the primary starting material, contributing to its overall feasibility and accessibility. Secondly, it ingeniously generates an intermediate tailored to synthesize alkannin and shikonin with exceptionally high enantiomeric excess (ee) values with a method that is convenient to operate in the industry, attesting to its precision and efficiency. Lastly, the protective strategy demonstrates remarkable ingenuity, facilitating a straightforward, one-step deprotection process under mild conditions, underscoring its practicality and elegance.
In parallel, Rubing Wang et al. [51] have undertaken a study of utmost relevance, successfully yielding R and S configuration intermediates through the strategic introduction of S-1-phenylethylamine at the chiral carbon. Subsequent multistep hydrolysis and reduction reactions culminated in acquiring shikonin and alkannin with extraordinarily high ee values, underscoring their exemplary craftsmanship and commitment to scientific rigour.
In this section, we introduced several typical naphthoquinone synthesis routes, most of which were multistep protocols. However, the commonplace among them was the construction of the naphthoquinone moiety. Apparently, the most convenient strategy to synthesize naphthoquinone was the oxidation of the naphthalene, which provides insight into the oxidation reagent. Standard oxidation reagents, including cerium ammonium nitrate (CAN), Ag2O, periodic acid, oxygen, KO2 and H2O2, could oxidize the hydroxyl naphthalene or methoxy naphthalene to naphthoquinone. Currently, the most widely used reagent for oxidation is CAN, which is relatively optimal since the condition is mild and the cost is relatively low. However, because of the existent substitutions on the naphthoquinone moiety, the oxidation strategy encountered regioselectivity problems. Refers to Figure 10, the oxidation could react with both the 6’ site and 2’ site of the naphthoquinone ring to give a pair of enantiomers. In that case, figuring out a novel oxidation strategy to improve the regioselectivity could enormously improve naphthoquinone synthesis.
2.2. Biological Activity of 1,4-naphthoquinone Derivatives
1,4-Naphthoquinone derivatives induce cytotoxicity through mechanisms like redox cycling, arylation, DNA strand breakage, free radical generation, and quinone methylation. These compounds, featuring quinone structures, inhibit specific proteins, resulting in antibacterial, antiviral, and anti-tumor effects. Agents like adriamycin, mitoxantrone, and compounds like lawsone, β-lapachol, and shikonin primarily inhibit topoisomerase I for anti-tumor activity [51,52,53].
Anticancer activity
DNA topoisomerases control DNA's topological structure by precisely breaking and rejoining DNA strands. They fall into two major classes: topoisomerase I and topoisomerase II. Mammalian topoisomerase II is a crucial target in clinical tumor therapy.
The tumor suppressor gene p53 responds to a myriad of both intracellular and extracellular cues, such as growth factor deprivation, hypoxia, radiation, chemical deficiencies, and nucleotide synthesis aberrations. Upon activation, p53 predominantly instigates apoptosis through transcriptional regulation, eliminating cells with severely damaged DNA. Alternatively, it can initiate growth arrest, facilitating DNA repair and preventing tumor formation [54,55].
The c-Jun N-terminal kinase (JNK), a member of the mitogen-activated protein kinase family, is pivotal in the signal transduction pathway responsible for conveying extracellular signals into intracellular responses. It is crucial in various physiological processes, including cell growth, differentiation, and apoptosis [56]. JNK can also induce apoptosis in response to different stresses, primarily by altering gene expression and initiating the phosphorylation of specific substrates. Furthermore, JNK activation leads to the phosphorylation of p53 and its stabilization, achieved by disrupting its binding to MDM2. Numerous studies have established that JNK activation is essential for stress-induced mitochondrial cytochrome release and apoptosis through the mitochondrial caspase-9 pathway. Moreover, phosphorylation of JNK enhances the pro-apoptotic activity of multiple factors [57].
Vitamin K2 serves a critical function by facilitating the carboxylation of primary osteocalcin secreted by osteocytes, thereby promoting the deposition of calcium ions from the bloodstream into bone tissue. It is pivotal in the blood coagulation cascade and bone formation [58]. Recent research by Elikan Mishima et al. [59] has unveiled vitamin K's potential as a ferroptosis inhibitor, suggesting its application in treating organ injuries, therapy-resistant cancers, Parkinson's disease, and Alzheimer's disease. However, due to its limited absorption, substantial and frequent doses of the synthetic MK-4 type of vitamin K2 must meet the average body requirements. Additionally, a recent study has highlighted the substantial anticancer effects of vitamin K2, showing its synergistic potential when combined with various anticancer agents [60]. Furthermore, research into various vitamin K analogs with different substituents is ongoing, contributing to developing new anticancer agents.
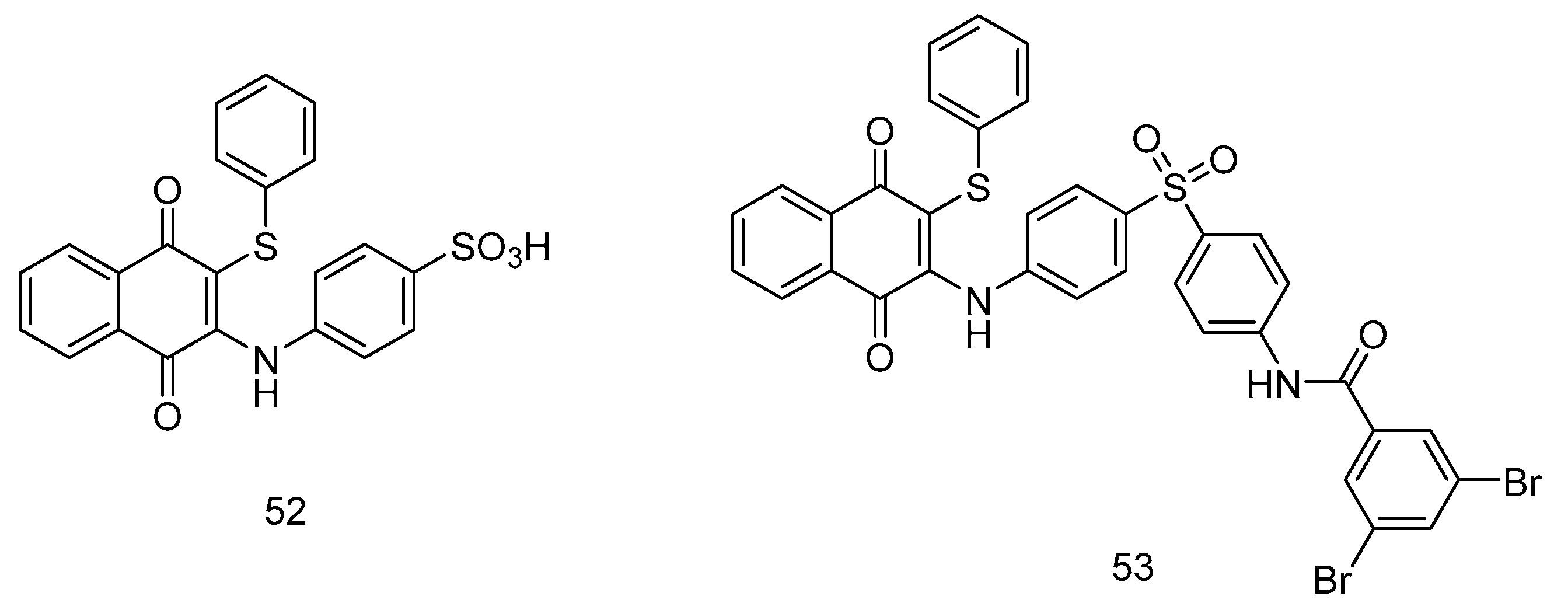
Yoo et al. [61] introduced phenylaminophenylthiosulfone compounds, incorporating an acyl group at the 3-position and a benzenethiol group at the 2-position of the naphthoquinone core scaffold, resulting in the synthesis of compounds 52-53 (Fig 10a). These compounds underwent rigorous in-vitro cytotoxicity assessments using three distinct cell lines: MCF-7, A549 and HeLa. Furthermore, compound 53 (IC50 = 40 μM) demonstrated robust cytotoxic effects against HeLa, comparable to doxorubicin (IC50 =30 μM). An in-depth structure-activity relationship analysis underscored the critical role of the 3-position substitution for anti-tumor selectivity, while the 2-position benzenethiol group substitution significantly enhanced cytotoxic activity.
Figure 10.
1,4-naphthoquinone derivatives as anticancer agents.
In a pioneering study, Wellington et al. [62] synthesized a novel series of vitamin VK3 analogs, innovatively incorporating sulfur-containing side chains at either the 2- or 3- position of the naphthoquinone moiety. This modification was geared towards enhancing anticancer efficacy. The research team rigorously tested these compounds against a diverse six-cell line panel to evaluate their anticancer activities. The findings were substantial. Compounds 54-55 (Figure 11) exhibited a remarkable inhibitory effect on cancer cells, surpassing the effectiveness of traditional vitamin VK3, as indicated by their lower GI50 values. Crucially, these compounds demonstrated heightened selectivity for cancer cells over normal human cells (specifically WI-38 cells), a significant advancement in targeted cancer therapy. This selectivity reduces potential collateral damage to healthy cells, a critical consideration in chemotherapy. A significant aspect of Wellington et al. research was the exploration of structure-activity relationships within these compounds. Introducing a fluorinated phenyl group at the 1,4-naphthoquinone sulfide moieties significantly boosted their anticancer activity.
Additionally, the study shed light on the potent anti-tumor properties of the 1,4-naphthoquinone sulfide dimer. In a comparative analysis of compounds 55a and 55b, a critical observation was made: the fluorine atom positioned at the 4-position was instrumental in enhancing the anticancer activities of these compounds. This insight underscores fluorination's impact in elevating anticancer agents' efficacy.
Wellington et al. research represents a significant stride in anticancer drug development. It exemplifies the potential of structural modifications in drug design and sets a precedent for future research in developing more effective and selective anticancer therapies.
Yu-Pu Juang et al. [63] conceived and synthesized a batch of juglone derivatives as the cell surface PDI (protein disulfide isomerase) inhibitor and evaluated their anticancer activity. The cytotoxicity experiments showed that compound 56 (Figure 11) exhibited distinct activity against several cell lines, including MDA-MB-231 (IC50 = 6.96 ± 0.47 μM), RPMI8226 (IC50 = 0.57 ± 0.01 μM) and EAhy926 (IC50 = 7.27 ± 0.08 μM). Additionally, compound 57 also showed remarkable cytotoxicity activity against the tested cell lines, especially RPMI8226 (IC50 = 0.66 ± 0.08).
Gokemen et al., in their significant research study [64], embarked on the synthesis of a series of N-substituted and S,S-substituted naphthoquinone derivatives (Figure 12), aiming to explore their potential as cytotoxic agents against various cancer cell lines (A-549, DU145, HCT-116, and MDA-MB-231 cancer cells), representing a diverse set of neoplastic conditions.
Suresh Awale et al. conceived and synthesized a series of plumbagin derivatives, among which compound 60 showed a distinct anticancer activity against PANC-1 (IC50 = 47.2 μM in DMEM; IC50 = 0.11 μM in NDM). In vivo, the anti-tumor evaluation indicated that the plumbagin derivative 60 possessed potent inhibition activity against pancreatic tumor growth. With the treatment of compound 60, at the dosage of 50 μg/day and 250 μg/day, the weight of the tumor was suppressed (205.8 and 107.6 mg relatively), compared with the control group 465.4 mg.
The results of their study were notable. Compounds 58a and 59, as illustrated in Figure 13, demonstrated considerable cytotoxic activity across the examined cell lines. These findings are significant as they suggest a broad-spectrum efficacy of these compounds against different types of cancer cells. In addition, compound 57b exhibited specific inhibitory effects against MDA-MB-231 cells. Furthermore, compound 58c showed a distinct inhibition profile, effectively targeting MDA-MB-231 and DU-145 cells.
Khoshneviszadeh et al. [65] synthesized a novel series of aminonaphthoquinone compounds, distinctively integrating 1,2,3-triazole hybrids at the 2-position of the naphthoquinone ring. The primary aim was to evaluate the cytotoxic activity of these compounds against three human cancer cell lines: MCF-7 (breast cancer), HT-29 (colorectal cancer), and MOLT-4 (acute lymphoblastic leukemia).
Among the synthesized compounds, 59c (Figure 13) emerged as the most effective, demonstrating potent anticancer activity across all three cell lines. This efficacy is quantified by its IC50 values, 10.4 μM for MCF-7, 6.8 μM for HT-29, and 8.4 μM for MOLT-4. These values suggest a notable capacity of compound 59c to inhibit cancer cell proliferation at relatively low concentrations.
A critical aspect of the study was SAR studies to enlighten the cytotoxicity effects of different compounds. It was observed that the presence of a halogen atom on the benzene ring notably increased the cytotoxic potency of the 1,2,3-triazole hybrids. This was particularly evident when comparing compound 61c with its counterparts 61a, 61b, 61e and 61d. Such findings underscore the impact of minor structural modifications on the biological activity of these compounds.
In general, this work contributes significantly to the field of anticancer drug design, particularly in highlighting the therapeutic potential of aminonaphthoquinone derivatives. The study demonstrates the capability of these compounds to target various cancer cell lines. It offers valuable insights into the role of structural elements, such as halogenation, in enhancing cytotoxic effects. This work paves the way for future explorations into more effective and targeted cancer therapies.
The study by Kirpotina et al. [66] ventures into the realm of cellular signaling, explicitly targeting the Cell division cycle 25 (Cdc25) and Mitogen-Activated Protein Kinase Kinase 7 (MKK7), both intimately linked to tumor development. The authors explored this by introducing a phenylamino side chain at the 2-position of the naphthoquinone ring, resulting in two structurally similar compounds, 62 and 63 (Figure 14). Their investigation further expanded to a series of naphthoquinone compounds (63a-i) with diverse substituent groups, evaluating their anticancer activities against nine tumor cell lines, inhibitory effects on MKK7 and MKK4, as well as Cdc25 A/B, complemented by molecular docking studies.
In their findings, compounds 63a and 63f-i stood out for their notably low IC50 values, indicating potent cytotoxicity. Among these, compound 63c emerged as a significant inhibitor of MKK7, showcasing an IC50 value as low as 0.23 μM. Compounds 63b, 63c, 63d, and 62a also demonstrated substantial inhibitory potential against both MKK4 and MKK7. In the context of Cdc25A and Cdc25B inhibition, compounds 63a, 63b, 63i, and 62b were particularly notable. The correlation between the inhibition of Cdc25A/B and the anticancer activity was evident, suggesting a potential pathway for therapeutic targeting. Conversely, the study observed no clear correlation between the anticancer activity and inhibition of MKK7.
This detailed investigation by Kirpotina et al. contributes significantly to our understanding of the structural-activity relationships, specifically in cancer signaling pathways. Their work underscores the potential of naphthoquinone derivatives in targeted cancer therapy, offering insights into the structural determinants of their biological efficacy. The lack of correlation between MKK7 inhibition and anticancer activity also opens new avenues for understanding the complex mechanisms of tumor suppression and developing more effective cancer treatments.
Olawode et al. [67] synthesized a series of naphthoquinone compounds, innovatively substituted with a 2-chloro-3-(thiazol-2-yl) amino group. Their research aimed to explore the compounds' multifaceted biological activities, including anticancer, antimycobacterial, and antimalarial effects. Notably, compounds 64a (IC50 = 1.8 µM), 64b (IC50 = 2.7 µM), 64c (IC50 = 1.5 µM) and 64d (IC50 = 0.004 µM) demonstrated significant cytotoxicity against SH-SY5Y neuroblastoma cells, as evidenced by their low IC50 values (Figure 15). The compound 64d reveals a significant cytotoxic activity that can contribute to the electronegative substitution at the phenyl group. However, these compounds exhibited minimal inhibition against HeLa cervical adenocarcinoma cells, indicating selective cytotoxicity towards SH-SY5Y cells. This selectivity is critical, as it suggests potential avenues for targeted neuroblastoma therapy, a vital aspect of personalized medicine in oncology.
Iryna Ivasechko et al. [68] got inspiration from thiazole and naphthoquinone compounds. They designed a series of naphthoquinone derivatives to determine the anticancer activity in vitro of condensed hybrid 4-thiazolidinone derivatives and their DNA interaction and general toxicity in vivo. A compound (65, Figure 15) with furan out of the condensed 4-thiazolidinone moiety showed relatively high anti-tumor activity, and the highest IC50 value was up to 0.6 μM. Furthermore, they also demonstrated that compound 63 could increase the percentage of cells in the G2/M phase of the cell cycle and reduce the synthesis of DNA and RNA in tumor cells.
Dyshlovoy and Pelageev et al. [69] approached naphthoquinone synthesis by introducing a 2-chloroethylthio group onto the naphthoquinone moiety. They tested the resulting compounds against advanced prostate cancer cell lines, uncovering that the compounds 65, 66 and 67 (Figure 16) exhibited potent inhibitory effects. Compounds 65 and 67a, in particular, showed remarkable activity and selectivity, with IC50 values ranging from 0.14 μM to 0.77 μM. The study provides valuable insights into the structural determinants of cytotoxicity, indicating that introducing the 2-chloroethylthio group enhances the compounds' anticancer potential. Moreover, the research demonstrated that compounds 65 and 67b exert their anticancer effects primarily through the induction of DNA damage and cysteinase-dependent apoptosis, offering a novel therapeutic mechanism against prostate cancer.
Ferraris et al. [70] explored a unique approach by dimerizing amino alcohol-substituted naphthoquinone compounds. Their investigation focused on the compounds' anticancer activities against Acute Myeloid Leukemia cell lines, including MOLM-14 and MV4-11. The study highlighted compounds 66a-e presenting a remarkable anticancer activity (Figure 17), along with 66d, 67a and 67b exhibiting pronounced cytotoxic effects. The findings also revealed that compounds 67a and 67b inhibited Indoleamine 2,3-dioxygenases (IDO), suggesting a mechanism that disrupts cancer cell proliferation, a significant advance in understanding the molecular pathways of these compounds.
Shikonin is a naturally occurring compound capable of excreting profound anti-tumor, antitelomerase and antiangiogenesis effects and is also responsible for DNA topoisomerase I/II inhibition [71,72]. A pair of enantiomers, identified as alkannin (Figure (X, XX) and shikonin (XX), were isolated and identified from the roots of two different plants: Alkanna tinctoria (L.) Tausch in Europe and Lithospermum erythrorhizon Sieb. et Zucc. in the Orient, respectively. These compounds demonstrated remarkable antiproliferative properties, making them attractive lead compounds in developing anticancer drugs. Nevertheless, alkannin/shikonin and their synthetic counterparts were not employed in clinical trials as anticancer medications. Their non-specific cytotoxicity in vitro and in vivo posed the biggest challenge. In literature, it has been demonstrated that ROS production and bioreductive alkylation are intimately linked to the mechanism of shikonin's anticancer activity and widespread toxicity. It has been demonstrated that the cytotoxicity caused by reactive oxygen species (ROS) and alkylation is non-specific, impacting a wide range of biological macromolecules that include key proteins and nucleic acids in cellular biology, thus not only destroying cancer cells but also impacting healthy cells. Therefore, the structural modification of shikonin derivatives is a highly desirable strategy for medicinal chemists. Yang et al. [73] modified shikonin, a compound known for its anti-tumor properties, by introducing furoic acid on its hydroxyl group side chain. This modification resulted in a novel derivative 70 (Figure 18) exhibiting enhanced topoisomerase II inhibitory activity and substantial cytotoxicity (ave. IC50 value of 7.75 µM) against a range of cancer cells, including drug-resistant variants. This derivative also showed effectiveness in vivo against several human carcinomas. Complementing this, Durchschein et al. [17] developed new shikonin derivatives, discovering that compound 71 displayed robust inhibitory activity against melanoma cells (WM164 and MUG-Me12) and induced apoptosis in a caspase-dependent manner without causing cell membrane damage or cell cycle arrest, highlighting a distinct mechanism of action.
Hongyan Lin et al. designed and synthesized a series of shikonin derivatives to determine their prospect as anticancer agents. They found that compound 72 (Figure 18) showed remarkable cytotoxicity activity against several cell lines, including H1975 (IC50 = 1.51 ± 0.42 μM), H1299 (IC50 = 5.48 ± 1.63 μM) and HCC827 (IC50 = 5.19 ± 1.10 μM).
Shore et al. [74] focused on 7-methyl juglone derivatives from Euclea natalensis, a plant traditionally used in African cancer treatment. These compounds demonstrated significant toxicity against various human cancer cell lines (MCF-7, HeLa, SNO and DU145), such as a significantly active compound 73, IC50 values of 5.3 µM and 6.8 µM (Figure 19) and 74e IC50 values of 10.1 µM and 9.3 µM against HeLa and DU145 respectively. The SAR studies reveal that the hydroxyl groups at the 2- and 5-positions play a crucial role in their cytotoxic effects. This study contributes to understanding naturally derived naphthoquinones and their potential in cancer therapy.
In recent studies, we synthesized several naphthoquinone oximes, including both shikonin and alkannin oximes [75,76,77,78]. We assessed their antiproliferative properties against certain cancer cell lines and the normal ones. Most of the prepared naphthoquinone oximes were shown to be strong anticancer agents against the tested cancer cell lines based on the results of biological evaluations. The alkannin oxime (DMAKO-05, Figure 19a) with an isovaleryl group as the side chain showed highly potent antiproliferative activity against the human leukemia K562 cells and human breast cancer MCF-7 cells (IC50 value of 0.7 μM and 7.5 μM) [75]. Exchange of the aliphatic isobutyl group of DMAKO-05 with an aromatic ring will further potentiate the anticancer activity since compound 75 (Figure 19a) was a five-fold more potent anticancer agent towards MCF-7 cells (IC50=1.5 μM), as compared with the lead compound. In addition, all of the produced oximes were identified as a group of selective anticancer agents with an IC50 value greater than 50 μM for the standard HSF cell line. Cui et al. focused on a kind of shikonin derivative named DMAKO-20 (75, Figure 19a), which possessed a remarkable anti-tumor activity against several cancer cell lines, including HCT-15 (IC50 = 0.63 ± 0.10 μM), HCT-116 (IC50 = 1.08 ± 0.21 μM) and K562 (IC50 = 0.4 ± 0.1 μM) [76]. Notably, DMAKO-20 was non-toxic to normal human cells (VEC and HSF). In-vivo experiments indicated that the treatment of DMAKO-20 could prominently diminish the tumor volume. Upon the dosage at 10 mg/kg, the average volume of HCT-15 xenografts (305.8 ± 25.8 mm) was much smaller than that of the control group (59.3% reduction) after 14 days’ treatment. The mechanistic studies demonstrated that the quinone derivative DMAKO-20 underwent tumor-specific CYP1B1-catalyzed oxidation to produce active naphthoquinone mono-oximes and nitric oxide, which exhibited synergistic anticancer effects.
Figure 19.
a: 1,4-naphthoquinone oximes as anticancer agents. b: The binding mode between DMAKO-20 and CYP1B1
Figure 19.
a: 1,4-naphthoquinone oximes as anticancer agents. b: The binding mode between DMAKO-20 and CYP1B1
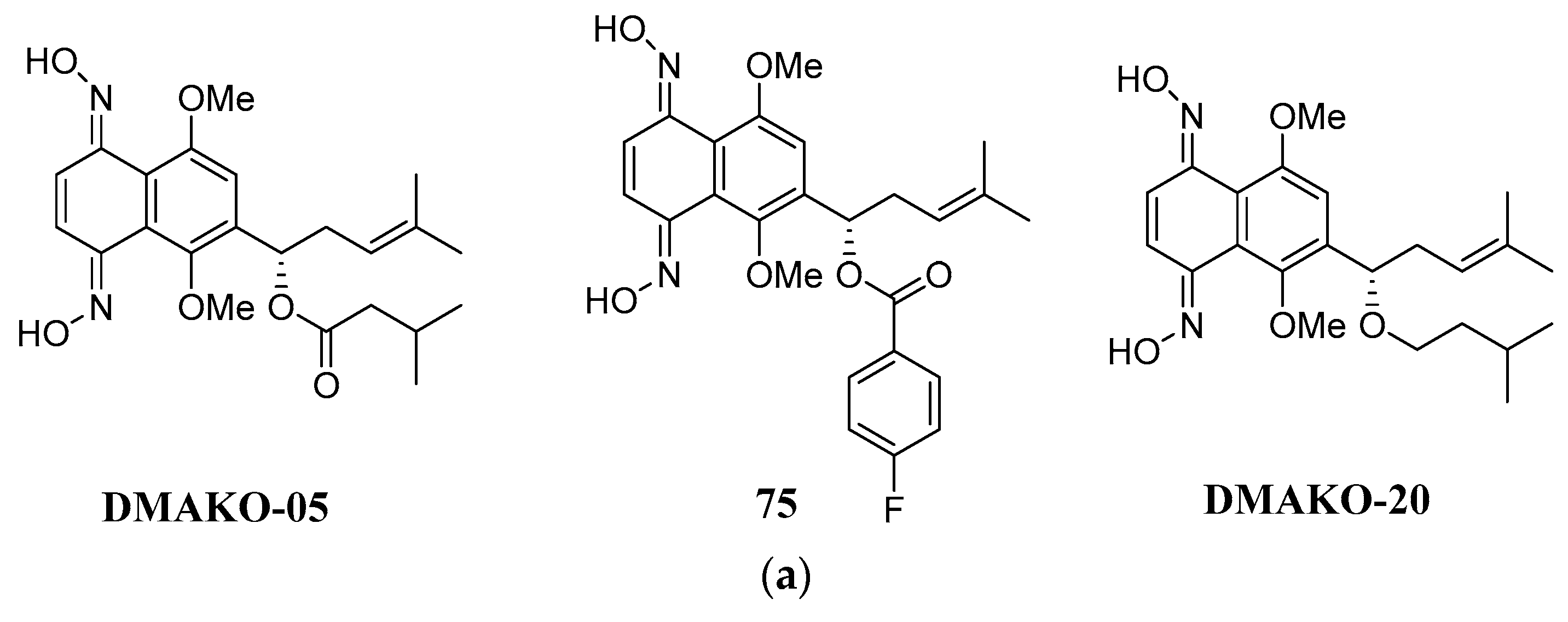
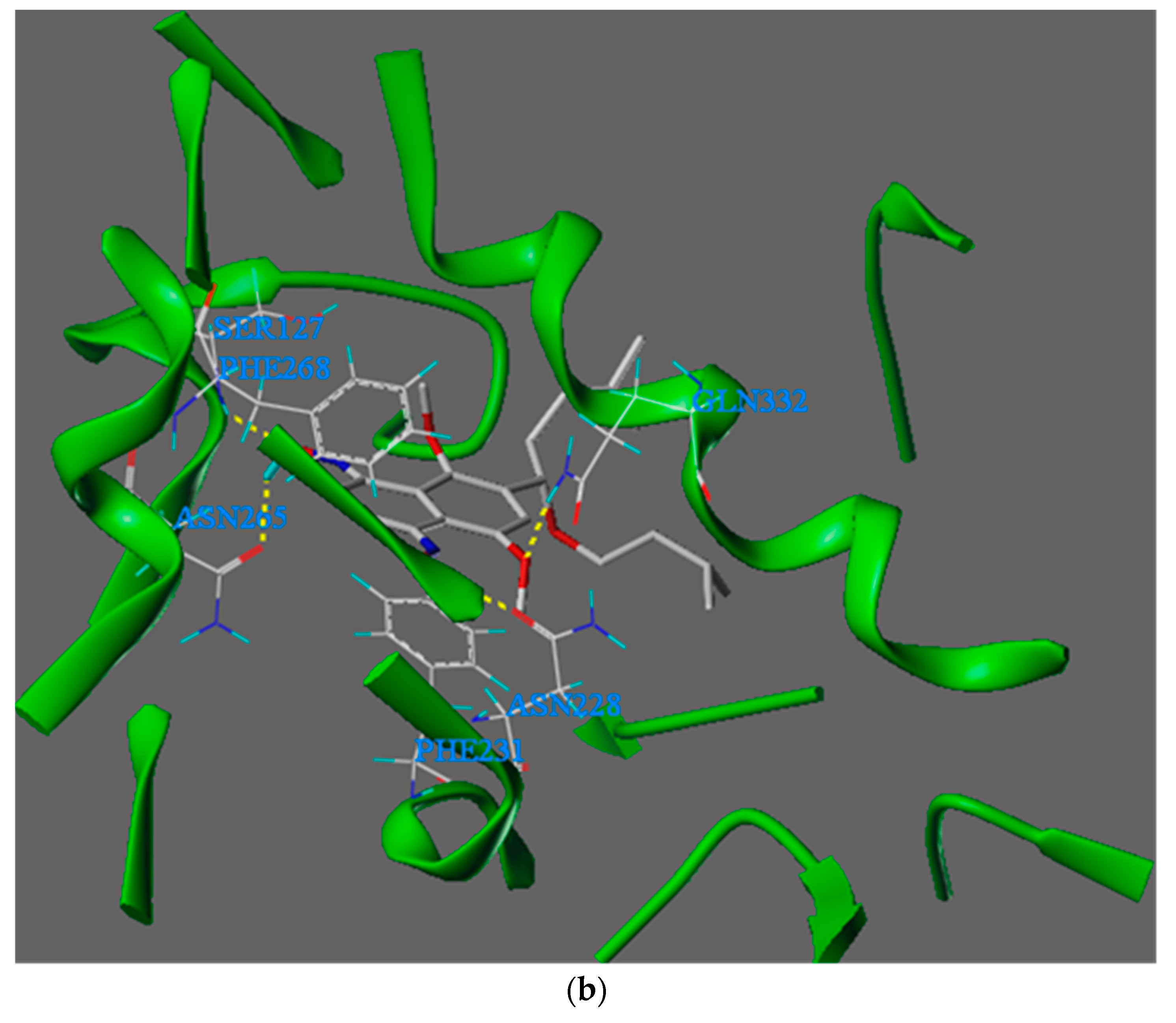

Table 1.
In vitro anticancer activity of DMAKO-20 against a panel of human cancer cell lines.
| IC50 (μM) | |||||
| DMAKO-20 | MCF-7 | MDA-MB-231 | DU145 | 22RV1 | K562 |
| 11.4±2.2 | 9.1±1.2 | 15.3±1.7 | 20.4±2.5 | 0.4±0.1 | |
| SKOV-3 | HepG2 | PANC | A549 | MGC-803 | |
| 20.6±1.6 | 2.2±0.5 | 2.3±0.4 | 16.8±0.9 | 2.33±0.89 | |
| HCT-15 | HCT-116 | VEC∆ | HSF∆ | HL-7702∆ | |
| 0.63±0.10 | 1.08±0.21 | 61.9±2.5 | 75.9±2.1 | 66.5±3.9 | |
∆: the cell lines in bold characters represent normal cell lines.
Molecular docking is based on the "lock and key principle" of the interaction between ligands and receptors, simulating the interactions between small molecule ligands and biological macromolecule receptors. The interaction between ligand and receptor is a process of molecular recognition, mainly including electrostatic interaction, hydrophobic interaction, hydrogen bonding interaction, van der Waals interaction and so on. The binding mode and affinity between the ligand and corresponding receptor could be predicted through calculation, thereby conducting virtual screening of drugs.
To illustrate the binding interaction between quinone oxime DMAKO-20 and CYP1B1, we docked DMAKO-20 into the active site of CYP1B1 (PDB ID: 3PM0). Their binding mode is shown in Figure 19b.
Figure 19b shows that the compound DMAKO-20 can bind tightly to the CYP1B1 enzyme. Therefore, it has a strong binding affinity (docking score of -9.7 KJ/mol). The oxygen atom at the 1 position on the oxime structure of the DMAKO-20 naphthalene ring forms a hydrogen bond with Ser127, and the distance is 2.56 Å. The hydrogen atom forms a hydrogen bond with the carbonyl oxygen atom of the Asn265 side chain, and the distance is 1.79 Å; the hydrogen atom Asn228 in the oxime structure at position 4 forms a hydrogen bond, and the distance is 2.04 Å; the oxygen atom in the methoxy structure at position 5 forms a hydrogen bond with Gln332, and the distance is 2.63 Å. The naphthalene ring part in the DMAKO-20 molecule overlaps to a large extent with the benzene ring part directly below Phe231 and above Phe268, and π-π stacking interactions occur, which is beneficial to improving the binding affinity; in addition, the 6-position of the target molecule is relatively small. The long, flexible hydrophobic side chain partially avoids the influence of steric hindrance and is deeply inserted into the hydrophobic pocket of the CYP1B1 enzyme. This hydrophobic interaction also improves the binding affinity of the compound to the active site.
Thakore et al. [79] introduced amino and phenyl groups to the lawsone scaffoldin their research. Subsequently, they assessed the anticancer activities of these modified compounds against the HepG2 human liver carcinoma cell line. Among the compounds investigated, precisely compounds 76-79 (Figure 20) exhibited significant cytotoxicity against HepG2 cells, with IC50 values falling within the range of 1.68 to 11.01 μM. Notably, an analysis of structure-activity relationships revealed that incorporating octyl and hydroxyphenyl groups into the side chain substantially augmented the anti-tumor activity of these compounds.
Interestingly, it was observed that the introduction of heterocyclic groups at the N-position, as seen in compounds 77a, 77 and 77c, did not contribute significantly to the cytotoxic activity when compared to the nitrophenyl group in these naphthoquinone compounds. Moreover, the presence of hydrocarbon groups at the N-position and aryl groups at the 1-position of the side chain appeared to enhance the compounds' anti-tumor efficacy.
In a related study, Araújo et al. [80] synthesized a series of hydrazone compounds derived from lapachol, with naphthoquinone as the core structure. The researchers then conducted an extensive investigation into the anticancer properties of these synthesized compounds across a panel of human cancer cell lines, including human promyelocytic leukemia (HL-60), human colorectal carcinoma (HCT-16), and human prostate adenocarcinoma (PC-3). Among the compounds evaluated, specifically compounds 80b, 81a, and 81b (Figure 21), demonstrated notable cytotoxic activity against the tested cancer cell lines. Notably, compounds 81a and 81b exhibited the highest potency in their anticancer effects.
In addition to assessing their anticancer potential, the researchers also examined the selectivity of these synthetic compounds by testing them on a non-malignant murine fibroblast cell line (L929). The results of this selectivity evaluation highlighted compound 81a as the most promising, particularly in its selectivity against HL-60 cells.
Khan and Said et al. [81] conducted an isolation study involving a series of furonaphthoquinone compounds 82 and 83 (Figure 22) extracted from the Amycolatopsis sp. strain to evaluate their antimicrobial properties. Notably, compounds 78 and 79 demonstrated potent and selective antibacterial activity, particularly against Gram-positive bacteria, compared to the positive controls of Ampicillin and Kanamycin. Compounds 82 and 83 exhibited significantly more inhibition against methicillin-resistant Staphylococcus aureus (MRSA), with MIC values ranging from 4 to 16 μg/ml. However, their antimicrobial activity against the other six tested Gram-positive bacterial strains was comparatively less pronounced. Notably, both compounds 82 and 83 showed limited inhibition against Gram-negative bacteria.
In a separate study, Nural et al. [82] synthesized a series of compounds 84a-c (Figure 23) by introducing triazole hybrid groups at the 2-position of naphthoquinone. Their research aimed to assess the synthesized compound's antimicrobial activity against different microbial strains and bacterial strains such as E. coli, B. cereus, S. aureus, P. aeruginosa, E. hirae, L. pneumophila subsp: pneumophila, and fungal strains including C. albicans and C. tropicalis.
Comparatively, when assessed against the positive control ampicillin, compounds 84a-c exhibited moderate inhibition against the tested bacterial strains. Notably, compounds 84a and 84b exhibited potent inhibition against specific microbial strains, with MIC values ranging from 4 to 16 μg/ml. Furthermore, compound 84b demonstrated moderate inhibition against the evaluated fungal strains compared to the positive antifungal drug.
3. Antimicrobial Activity
Balachandran et al. [83] isolated a novel naphthoquinone derivative bluemomycin 85 (Figure 24) from the Gram-positive filamentous bacterium Streptomyces sp. Their research focused on evaluating the antimicrobial activity of bluemomycin against various bacterial strains. Notably, bluemomycin displayed remarkable inhibitory effects, particularly against Gram-negative bacteria. This included strains such as M. luteus, E. aerogenes, P. aeruginosa, S. typhimurium, S. flexneri, K. pneumoniae (ESBL-3894), K. pneumoniae (ESBL-75,799), and E. coli (ESBL-3984), with MIC values consistently around 12.5 μg/ml. Additionally, bluemomycin exhibited potent antifungal activity against A. flavus, with a MIC value of 6.25 μg/ml.
Supratman et al. [84] investigated naphthoquinone compounds 86-88 (Figure 24) obtained from F. napiforme. Their investigation highlighted the substantial inhibitory potential of compounds 86-88 against S. aureus NBRC 13276 and P. aeruginosa ATCC 15442, with MIC values consistently below 12.5 μg/ml. However, these compounds exhibited limited activity against A. clavatus F 318a and C. albicans ATCC 2019. Notably, the introduction of hydroxy substitution at the 6-position of naphthoquinone was observed to enhance their antimicrobial activity.
López-López et al. [85] synthesized naphthoquinone derivatives 89a-e and 90 (Figure 25) enriched with amino acids in their side chains. Their antimicrobial assays revealed that these compounds displayed moderate inhibition against a spectrum of Gram-positive (E. faecalis and S. aureus) and Gram-negative bacteria (E. coli and P. aeruginosa), with MIC values consistently below 25 μg/ml. Of particular interest, compound 86 exhibited potent inhibition against a clinical isolate of E. coli, showcasing a MIC value of 24.7 μg/ml. Remarkably, this strain demonstrated substantial resistance to several conventional drugs, including cephalosporins.
In another study, Song et al. [86] engaged in the synthesis of lawsone derivatives 91a and 91b (Figure 26) and assessed their efficacy against multidrug-resistant strains, including methicillin-resistant S. aureus (MRSA), methicillin-sensitive S. aureus (MSSA), and vancomycin-intermediate S. aureus (VISA). Their findings indicated that compounds 91a and 91b exhibited moderate inhibition against the tested strains. Significantly, 91a demonstrated robust antibacterial activity against MRSA strains, with MIC values ranging from 1.25 to 2.5 μg/ml, a potency comparable to that of positive drugs like vancomycin and daptomycin (MIC = 1 μg/mL) and suppression of other tested strains (MIC value 2.5 μg/mL). Moreover, 91a was noted for generating reactive oxygen species (ROS) and chelating intracellular iron, contributing to its antibacterial activity by disrupting bacterial cell membranes upon entering the cytoplasm.
Sabutski et al. [87] synthesized Naphthoquinone–Sugar Tetracyclic Conjugates, with compounds 92 and 93a-d (Figure 27) exhibiting potent inhibition (MIC = 6.25 μM) against the S. aureus ATCC 21027 cell line. Additionally, 93a-c showed moderate antimicrobial activity, highlighting the potential activity enhancement through heterocyclic structures with mercapto-sugars.
Kim et al. [88] conducted a comprehensive investigation into pyrimidinone-fused naphthoquinone derivatives' synthesis and antibacterial activity, offering valuable insights into their potential as effective agents against drug-resistant oral bacteria strains. Their meticulous study systematically assessed a range of compounds 94a-c and 94e-h (Figure 41), revealing their impressive inhibitory capabilities across a diverse spectrum of bacterial strains, as evidenced by MIC ranging from 1.56 μg/mL to 12.5 μg/mL. This wide-ranging activity underscores the versatility of these compounds. Notably, the study observed moderate yet substantial antibacterial effects exhibited by compounds 94d and 94h against specific bacterial strains. The standout observation was the remarkable inhibition of P. gingivalis by compound 94d, signifying its potency against this oral bacterial strain. Furthermore, compared to the positive control oxytetracycline, these active compounds consistently demonstrated superior inhibitory properties, extending their effectiveness to critical bacterial strains relevant to oral health, including E. faecalis, S. epidermidis, S. mutans and P. gingivalis. The collective findings presented in this study accentuate the promise of pyrimidinone-fused naphthoquinone derivatives as potential therapeutic agents in the battle against drug-resistant oral bacteria strains, offering compelling prospects for future research and clinical applications in oral health management.
Gemili et al. [89] ventured into the synthesis of 2-iminothiazole substituted naphthoquinone compounds 95a-c (Figure 28). These compounds exhibited remarkable antibacterial activity against M. tuberculosis H37Rv, with MIC values consistently below 7.81, 3.9 and 1.96 μg/ml respectively, compared to positive control Isoniazid (MIC value = 0.98 μg/mL) and Ethambutol (MIC value = 1.96 μg/mL).
Erasmus et al. [90] synthesized a series of 2-amino-3-triazol substituted naphthoquinone derivatives 96 and 97a-e (Figure 29). These compounds 96 and 97 b displayed moderate antimycobacterial activity against Mtb, while the rest of compound demonstrating potent inhibition, as their MIC values were all below or equal to 4.1 μM. Notably, the introduction of electro-positive groups enhances the inhibitory activity against Mtb.
The antimicrobial activity of 1,4-naphthoquinone derivatives is often contingent upon their capacity to accept one or two electrons, forming anionic or double anionic radicals. The presence of electron-donating or electron-withdrawing substituents in 1,4-naphthoquinone determines the production of anionic radicals and the redox properties of the compounds, including the generation of reactive oxygen species such as hydrogen peroxide and superoxide, which can damage cells. Consequently, nitrogen and sulfur atoms in the side chain of naphthoquinone play a pivotal role in enhancing the antimicrobial activity of these compounds [91].
Tandon et al. [92] embarked on synthesizing a series of compounds enriched with nitrogen and sulfur atoms, strategically positioned at the 2- or 3-position of the side chain of 1,4-naphthoquinone and incorporated within the ring structure. Their comprehensive investigation thoroughly assessed these compounds' antimicrobial potential against a diverse array of pathogens, spanning C. albicans, C. neoformans, S. schenckii, T. mentagraphytes, C. parapsilosis, and A. fumigatus.
Among the synthesized compounds, compound 98a (Figure 30) emerged as a standout, demonstrating remarkable inhibitory activity with a minimum inhibitory concentration (MIC) of 12.5 μg/mL against C. albicans. Impressively, this level of inhibition surpassed that of the positive drug Miconazole, which exhibited an MIC of 25 μg/mL against the same pathogen. Furthermore, compound 98a displayed comparable inhibitory efficacy against C. neoformans (MIC = 12.5 μg/mL) and A. fumigatus (MIC = 12.5 μg/mL) when benchmarked against Miconazole.
The study also unveiled the notable performance of compounds 98b and 98c, both exhibiting comparable inhibitory activity against C. neoformans (MIC = 12.5 μg/mL) and C. albicans (MIC = 25 μg/mL) in comparison to the positive drug Miconazole. Compound 94c exhibited an intriguingly higher inhibitory potential against S. schenckii than the antifungal drug Nystatin, with an MIC value of 13.2 μg/mL.
These compelling findings underscore the promise of these meticulously designed compounds as potent candidates in antimicrobial agents, holding substantial potential for advancing therapeutic approaches against a spectrum of clinically relevant fungal pathogens.
Tandon et al. [93] reported that a series of aromatic, sulfur-containing 1,4-naphthoquinone derivatives were synthesized and assessed for their antibacterial and antifungal activities. Among these compounds, 99b, 100a, and 100b (Figure 31) demonstrated robust antifungal effects and notable antibacterial activity against S. aureus and K. pneumoniae. Intriguingly, compound 99b exhibited exceptional inhibitory potency against S. schenckii (MIC = 1.56 μg/mL), surpassing the performance of clinical antifungal drugs such as Fluconazole MIC = 2.0 μg/mL and Miconazole MIC=12.5 μg/mL. Furthermore, when compared to the positive drug Amphotericin-B, compound 99b consistently exhibited significant inhibitory activity against a spectrum of fungal species, including C. albicans MIC=1.56 μg/mL, C. neoformans MIC=0.78 μg/mL, T. mentagrophytes MIC=1.56 μg/mL, and A. fumigatus MIC=3.12 μg/mL. It is worth noting that compound 99b is currently undergoing further investigation for toxicological evaluation as a potential lead drug candidate.
Additionally, compound 100b displayed comparable inhibitory activity against C. neoformans MIC=12.5 μg/mL, aligning closely with the performance of the positive drug Miconazole. Conversely, compound 100a exhibited superior inhibition against C. albicans MIC=12.5 μg/mL compared to Miconazole MIC=25 μg/mL, while the activity of compound 100c closely mirrored that of the positive drug.
4. Antiviral Activity
In the realm of medicinal chemistry, Gonzaga, Daniel T. G., et al. [94] made significant strides in antiviral research by synthesizing a series of bis-naphthoquinone compounds, focusing on their activity against the Zika virus. They identified several high-activity compounds 101 and 102a-d (Figure 32). These compounds were found to exhibit potent antiviral properties, with EI50 values at or below 1.38 µM, notably surpassing the efficacy of the standard antiviral agent ribavirin, which has an EI50 value of 2.98 µM.
In a related study, our lab [95] explored the antiviral potential of juglone derivatives against SARS-CoV-2. We discovered that compounds 103-106 (Figure 33) showed remarkable inhibitory effects on SARS-CoV-2 Mpro. This enzyme is vital for the virus’s replication and transcription within host cells. The tested compounds demonstrated IC50 values ranging from 72.07 to 220.9 nM. Compared to the positive control shikonin, with an IC50 value of 15.75 ± 8.22 µM, compounds 104-106 exhibited more robust activity against SARS-CoV-2 Mpro. Compound 106 notably emerged as the most potent inhibitor, with an IC50 value of 72.07 nM.
Additionally, compound 105 significantly suppressed the proliferation of SARS-CoV-2 Mpro in vitro (EC50 = 4.55 µM). The molecular docking assays revealed that these compounds interact effectively with the enzyme's active site, primarily through hydrogen bonding with adjacent amino residues. Our study also provided insights into structure-activity relationships, suggesting that the incorporation of an acyl group or benzyl group enhances antiviral efficacy. In contrast, adding a methyl group, methoxy group, or the annulation of the naphthoquinone core with a phenyl group decreases the inhibition activities.
Naphthoquinone derivatives are the subject of considerable interest to the scientific community throughout the globe, particularly concerning their impact on virus replication and immune response modulation [96]. For instance, Sendl et al. [97] isolated two novel 1,4-naphthoquinone-active compounds, Rhinacanthin-C 107 (Figure 34) and Rhinacanthin-D 108 from Rhinacanthus nasutus. These compounds exhibited significant antiviral activity against cytomegalovirus (CMV), with EC50 values of 0.02 μg/mL and 0.22 μg/mL, respectively. Alcides et al. [98] synthesized derivatives 109a and 109b using lapachol (tobacco genus) as a lead compound and found them to be significantly effective against Herpes Simplex Virus-2, with IC50 values of 180 ng/mL and 25 ng/mL, respectively. This highlights the potential for developing a broader antiviral spectrum through chemical modifications and animal models of viral infection studies.
Additionally, HIV-1 reverse transcriptase inhibitors are of remarkable interest to medicinal chemists, such as the aryl-substituted naphthoquinone compounds synthesized by Ilina et al. [99], showed potent inhibition against four reverse transcriptase M(215), M(67), and M(67, 70, 215) in HIV-1 recombinant wild-type and mutant forms. Compounds 110, 111a and 111b (Figure 34) were particularly effective, indicating that 1,4-naphthoquinone derivatives are potent inhibitors of HIV-1 reverse transcriptase. Furthermore, some compounds inhibit azidothymidine-resistant wild-type and mutant reverse transcriptase. Indicates that 1,4-naphthoquinone derivatives may offer an alternative mechanism of action to existing drugs like nevirapine.
Singh et al. [100] synthesized 2-substituted-1,4-naphthoquinone derivatives with sulfur atoms in their side chains, demonstrating strong inhibitory activity against influenza-A and HSV-1 viruses with compounds 112 and 113 (Figure 35). Similarly, Yadav et al. synthesized compound 114 from 1,4-naphthoquinone, showing notable activity against type II poliovirus.
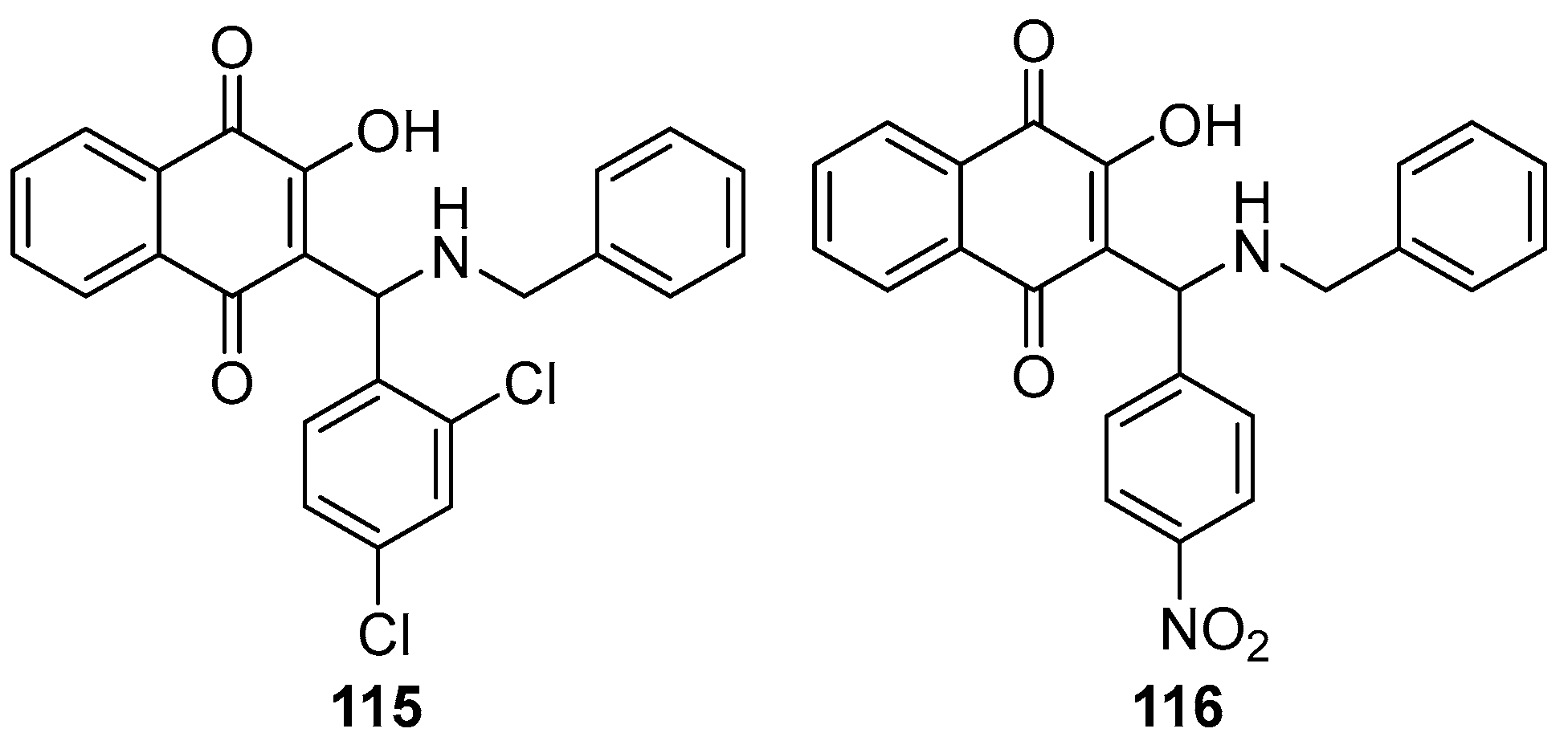
Behl et al. synthesized a series of 2-aminomethyl-3-hydroxy-1,4-naphthoquinones derivatives. Subsequently, they encapsulated these quinones in liposomes for antiviral testing against HSV-1. The study revealed that these aminomethyl naphthoquinones derivatives, when enclosed within phosphatidylcholine liposomes, effectively inhibited HSV-1 replication. Notably, derivatives featuring a benzyl group (115, see Figure 37) and a nitrobenzene moiety (116) demonstrated selective index values over nine times greater than those of acyclovir, used as the positive control. The EC50 values for compounds 115 and 116 were 0.56 ± 0.02 µM and 0.36 ± 0.04 µM, respectively, indicating a potency almost four and nine times higher than acyclovir, which showed an EC50 of 3.16 ± 0.09 µM under identical experimental conditions.
Figure 36.
2-aminomethyl-3-hydroxy-1,4-naphthoquinones derivatives as antiviral agents.
Ngoc et al. conducted a study to evaluate the antiviral efficacy of rhinacanthins C (117), D (118), N (119)(Figure 37), extracted from the roots of Rhinacanthus Nasutus – a medicinal plant of the Acanthaceae family traditionally used in treating herpes virus infections [101]. The in vitro experiments demonstrated that all four compounds effectively inhibited the influenza virus A/PR/8/34 (H1N1). The mean IC50 values were recorded as 0.30 µM for rhinacanthin C (117), 0.95 µM for rhinacanthin D (118), and 1.95 µM for rhinacanthin N (119).
Furthermore, Lisseth Cetina-Montejo and colleagues successfully isolated several novel naphthoquinones from the stem bark of Diospyros anisandra, with a specific focus on zeylanone epoxide (ZEP) (120, see Figure 38) for antiviral activity assessment [102]. The team conducted experiments by infecting MDCK cells with strains H1N1, H1N1-H275Y, and H3N2, followed by treatment with ZEP. Virus titers were quantified using a plaque assay. The research revealed mean IC50 values of 0.65 µM for H1N1, 2.77 µM for H1N1-H275Y, and 1.6 µM for H3N2.
Zima and colleagues focused on the isolation of natural naphthoquinones, specifically lapachol (8), mompain (121), and quambalarine (122) (Figure 39) from the Quambalaria Cyanescens fungus [103]. Their investigation into the antiviral properties of these compounds against H1N1 revealed varying levels of efficacy. Lapachol exhibited moderate inhibitory activity with an IC50 value of 19 μM. In contrast, mompain and quambalarine demonstrated significant potency as sub-micromolar inhibitors, with IC50 values of 0.43 μM and 0.29 μM, respectively.
Garima and colleagues embarked on a project to synthesize a series of compounds, subsequently testing their efficacy against the neuraminidase (NA) of the H5N1 virus [104]. Among these synthesized compounds, compound 123a (Figure 40) and compound 123b emerged as the most potent in terms of antiviral activity. Specifically, compound 123a exhibited an IC50 of 29 ± 0.9 μM, while compound 123b demonstrated an IC50 of 26.5 ± 0.7 μM.
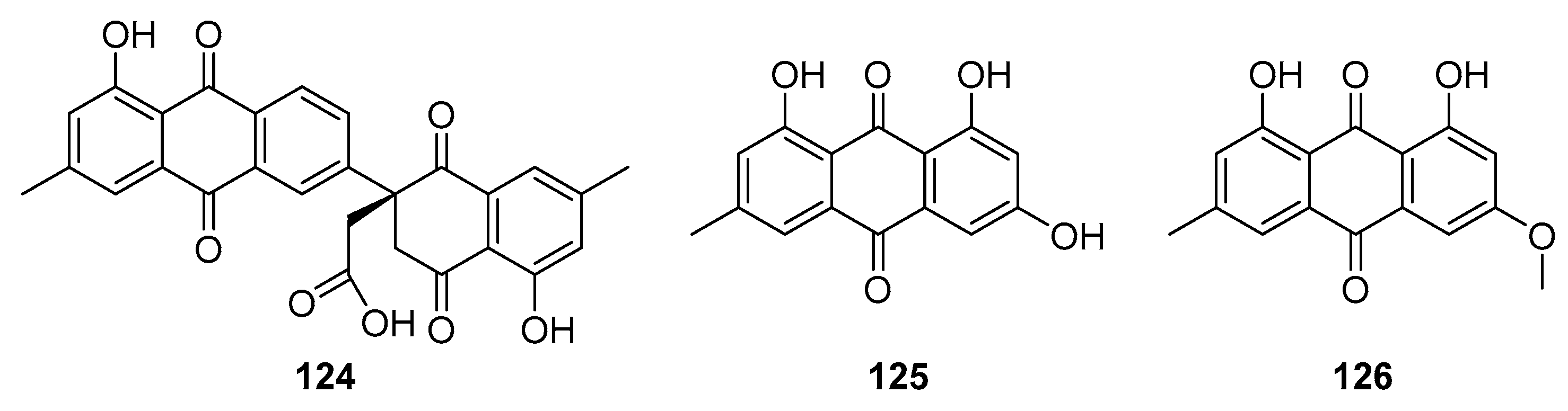
Xu et al. successfully isolated three anthraquinone analogues from the roots of P. Odoratum [105]. Utilizing NMR, MS, and CD techniques, they identified several novel natural naphthoquinones, notably compound 124 (see Figure 41). Additionally, compounds emodin (125) and physcion (126) were also detected in the extracts. The team then proceeded to evaluate the antiviral efficacy of these compounds against the influenza A virus (H1N1). Their findings indicated IC50 values of 11.4 μM for compound 124, 11.0 μM for emodin (125), and notably lower at 2.3 μM for physcion (126).
Figure 41.
Natural anthraquinone analogs as antiviral agents.
Miscellaneous
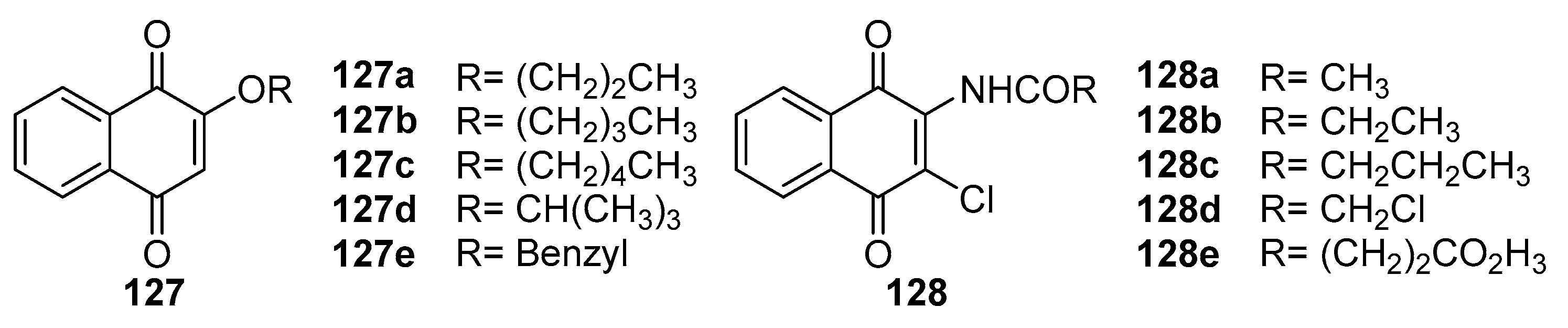
Lien et al. [106] synthesized a series of 2-substituted aliphatic and aromatic 1,4-naphthoquinone derivatives. These derivatives effectively inhibited the formation of neutrophil superoxide anion, demonstrating substantial anti-inflammatory activity. Notably, compounds 127a and 127b (Figure 42) achieved IC50 values of 0.6 μM, exhibiting inhibition intensity 25 times greater than the positive control, trifluoperazine. Additionally, these compounds showed promising antiplatelet agglutination activity. Mass cells release various inflammatory regulatory factors during immune responses, which is crucial in the passive skin allergic response (PCA). In a related study, Huang et al. [107] synthesized amide-substituted 1,4-naphthoquinone derivatives, assessing their antiallergic activity. Compounds 128a-128c significantly inhibited mast cell growth and degranulation at 0.3-1 μg/mL concentrations, thus showing notable antiallergic potential. Other biological activities of 1,4-naphthoquinones, such as antithrombotic, free radical scavenging, and antiradical activities, have been reported [35].
Figure 42.
The chemical structure of 1,4-naphthoquinone derivatives.
5. Conclusions and Further Perspectives
The exploration and structural modification of lead compounds derived from natural products is a leading approach in new drug development. Natural 1,4-naphthoquinone derivatives, with their diverse biological activities, including anti-tumor, antibacterial, antiviral, and anti-inflammatory properties, have garnered increasing attention. However, the extensive cytotoxic effects of 1,4-naphthoquinone have limited its clinical application.
From a chemical perspective, the molecular mechanisms underlying naphthoquinone's anticancer activity and pervasive toxicity are closely associated with generating reactive oxygen species (ROS) and bio-reductive alkylation. The damage caused by ROS and alkylation, often non-specific, can adversely affect various biological macromolecules, such as nucleic acids and proteins, impacting cancer and normal cells.
Shikonin, a bioactive component of the traditional Chinese medicine Lithospermum erythrorhizon, has well-documented anti-tumor effects. However, neither shikonin nor its synthetic derivatives have seen clinical use as anticancer drugs, largely due to their non-specific cytotoxicity. Recent studies have shifted focus to modifying the naphthoquinone scaffold of alkannin and shikonin to minimize cytotoxicity caused by ROS generation and alkylation. Innovations like shikonin oximes, which act as tumor-specific CYP1-activated prodrugs, have emerged as potent, selective anticancer agents, both in vivo and in vitro. This successful prodrug strategy offers new perspectives on mitigating the non-selective cytotoxicity of 1,4-naphthoquinones, highlighting their potential as promising anticancer drug candidates.
Reasonable structural modifications of 1,4-naphthoquinone can lead to compounds with enhanced activity and reduced toxicity. Current modifications mainly involve introducing various substituents (halogen, methyl, methoxy, hydroxyl, etc.) at strategic positions on the quinone ring or modifying the ring itself. While these alterations often increase activity, they can also intensify toxicity. Therefore, improving the selective anti-tumor efficacy of quinones while reducing their cytotoxicity remains a crucial goal in rational structural modification.
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. , Cancer statistics, 2020. CA: A Cancer Journal for Clinicians 2020, 70. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. , Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Katzung, B.G.; Masters, S.B.; Trevor, A.J. , Basic and Clinical Pharmacology, 11th Edition. Acta Agronomica Sinica 2009, 36–42. [Google Scholar]
- Baig, M. , Side Effects of Chemotherapy. 2018.
- Mishra, A.P.; Salehi, B.; Sharifi-Rad, M.; Pezzani, R.; Kobarfard, F.; Sharifi-Rad, J.; Nigam, M. , Programmed Cell Death, from a Cancer Perspective: An Overview. Molecular Diagnosis & Therapy 2018, 22, 281–295. [Google Scholar]
- Wang, Y.; Alenazy, R.; Gu, X.; Polyak, S.W.; Zhang, P.; Sykes, M.J.; Zhang, N.; Venter, H.; Ma, S. , Design and structural optimization of novel 2H-benzo[h]chromene derivatives that target AcrB and reverse bacterial multidrug resistance. European Journal of Medicinal Chemistry 2021, 213, 113049. [Google Scholar] [CrossRef]
- Wahab, S.; Khan, T.; Adil, M.; Khan, A. , Mechanistic aspects of plant-based silver nanoparticles against multi-drug resistant bacteria. Heliyon 2021, 7, e07448. [Google Scholar] [CrossRef]
- Verma, P.R. , Anti-Cancer Activities of 1,4-Naphthoquinones: A QSAR Study. Anti-Cancer Agents in Medicinal Chemistry 2006, 6, 489–499. [Google Scholar] [CrossRef]
- Shikov, A.N.; Pozharitskaya, O.N.; Krishtopina, A.S.; Makarov, V.G. , Naphthoquinone pigments from sea urchins: Chemistry and pharmacology. Phytochemistry Reviews 2018, 17, 509–534. [Google Scholar] [CrossRef]
- Soldi, R.; Horrigan, S.K.; Cholody, M.W.; Padia, J.; Sorna, V.; Bearss, J.; Gilcrease, G.; Bhalla, K.; Verma, A.; Vankayalapati, H.; Sharma, S. , Design, Synthesis, and Biological Evaluation of a Series of Anthracene-9,10-dione Dioxime β-Catenin Pathway Inhibitors. Journal of Medicinal Chemistry 2015, 58, 5854–5862. [Google Scholar] [CrossRef]
- Kishore, N.; Binneman, B.; Mahapatra, A.; van de Venter, M.; du Plessis-Stoman, D.; Boukes, G.; Houghton, P.; Marion Meyer, J.J.; Lall, N. , Cytotoxicity of synthesized 1,4-naphthoquinone analogues on selected human cancer cell lines. Bioorganic & Medicinal Chemistry 2014, 22, 5013–5019. [Google Scholar]
- Song, G.-Y.; Kim, Y.; You, Y.-J.; Cho, H.; Kim, S.-H.; Sok, D.-E.; Ahn, B.-Z. , Naphthazarin Derivatives (VI): Synthesis, Inhibitory Effect on DNA Topoisomerase-I and Antiproliferative Activity of 2- or 6-(1-Oxyiminoalkyl)-5,8-dimethoxy-1,4-naphthoquinones. Archiv der Pharmazie 2000, 333, 87–92. [Google Scholar] [CrossRef]
- Durchschein, C.; Hufner, A.; Rinner, B.; Stallinger, A.; Deutsch, A.; Lohberger, B.; Bauer, R.; Kretschmer, N. In Molecules, 2018; Vol. 23.
- V. P. Papageorgiou, A.N.A., V. F. Samanidou, I. N. Papadoyannis, Recent Advances in Chemistry, Biology and Biotechnology of Alkannins and Shikonins. Current Organic Chemistry 2006, 10, 2123–2142. [Google Scholar] [CrossRef]
- Tandon, V.K.; Kumar, S. , Recent development on naphthoquinone derivatives and their therapeutic applications as anticancer agents. Expert Opinion on Therapeutic Patents 2013, 23, 1087–1108. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-H.; Lo, C.-Y.; Gwo, Z.-H.; Lin, H.-J.; Chen, L.-G.; Kuo, C.-D.; Wu, J.-Y. In Molecules, 2015; Vol. 20, pp 11994-12015.
- Pingaew, R.; Prachayasittikul, V.; Worachartcheewan, A.; Nantasenamat, C.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. , Novel 1,4-naphthoquinone-based sulfonamides: Synthesis, QSAR, anticancer and antimalarial studies. European Journal of Medicinal Chemistry 2015, 103, 446–459. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, C.; Schurgers, L.J. , A COMPREHENSIVE REVIEW OF VITAMIN K AND VITAMIN K ANTAGONISTS. Hematology/Oncology Clinics of North America 2000, 14, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Ansell; Jack; Hirsh; Poller; Leon; Bussey; Henry; Jacobson; Alan; Hylek, The Pharmacology and Management of the Vitamin K Antagonists. CHEST, 2004.
- Pucaj, K.; Rasmussen, H.; Møller, M.; Preston, T. , Safety and toxicological evaluation of a synthetic vitamin K2, menaquinone-7. Toxicology Mechanisms and Methods, 2011. [Google Scholar]
- Almquist, H.J.; Klose, A.A. , SYNTHETIC AND NATURAL ANTIHEMORRHAGIC COMPOUNDS. Journal of the American Chemical Society 1939, 61, 2557–2558. [Google Scholar] [CrossRef]
- Fieser, L.F.; Campbell, W.P.; Fry, E.M.; Gates, M.D. , SYNTHETIC APPROACH TO VITAMIN K1. Journal of the American Chemical Society 1939, 61, 2559–2559. [Google Scholar] [CrossRef]
- Bertrand; L.; Chenard; Michael; J.; Manning; Peter; W.; Raynolds; John, Organocopper chemistry of quinone bisketals. Application to the synthesis of isoprenoid quinone systems. Journal of Organic Chemistry, 1980.
- Lipshutz, B.H.; Kim, S.-k.; Mollard, P.; Stevens, K.L. , An expeditious route to CoQn, vitamins K1 and K2, and related allylated para-quinones utilizing Ni(0) catalysis. Tetrahedron 1998, 54, 1241–1253. [Google Scholar] [CrossRef]
- Snyder, C.D.; Rapoport, H. , Synthesis of menaquinones. Journal of the American Chemical Society 1974, 96, 8046–8054. [Google Scholar] [CrossRef]
- Garcias, X.; Ballester, P.; Capo, M.; Saa, J.M. , (2)DELTA-STEREOCONTROLLED ENTRY TO (E)-PRENYL OR (Z)-PRENYL AROMATICS AND AND QUINONES - SYNTHESIS OF MENAQUINONE-4. The Journal of Organic Chemistry 1994, 59, 5093–5096. [Google Scholar] [CrossRef]
- Krajewski, K.; Kutner, A.; Dzikowska, J.; Gutowska, R.; Napiórkowski, M.; Winiarski, J.; Kubiszewski, M.; Jedynak, L.; Morzycki, J.; Witkowski, S. , Process for preparation of NK-7 type of vitamin K2. 2015.
- Strugstad, M.P.; Despotovski, S. , A summary of extraction, properties, and potential uses of juglone: A literature review. Journal of Ecosystems and Management, 2012. [Google Scholar]
- Inbaraj, J.J.; Chignell, C.F. , Cytotoxic Action of Juglone and Plumbagin: A Mechanistic Study Using HaCaT Keratinocytes. Chemical Research in Toxicology 2004, 17, 55. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.-h.; Cui, Q.; Zhang, Q.-j.; Li, S.-s. , An Efficient Multigram Synthesis of Juglone Methyl Ether. Journal of Chemical Research 2015, 39, 553–554. [Google Scholar] [CrossRef]
- Khalafy, J.; Bruce, J.M. , OXIDATIVE DEHYDROGENATION OF 1-TETRALONES: SYNTHESIS OF JUGLONE, NAPHTHAZARIN, AND α-HYDROXYANTHRAQUINONES. 2002.
- Mamchur, A.V.; Gorbas, L.F.; Galstyan, G.A. , Use of Ozone in Synthesis of 5-Hydroxy-1,4-naphthoquinone. Russian Journal of Applied Chemistry 2001, 74, 1405–1407. [Google Scholar] [CrossRef]
- Zhang, M.a.X.L. , One-pot synthesis of pecan quinone from acetic anhydride, H2O2 and 1,5-dihydroxynaphthol. Chinese Journal of Applied Chemistry 2007, 24, 3. [Google Scholar]
- Hashemi, M.; Akhbari, M. , Efficient Solvent-Free Oxidation of Phenols to p-Quinones with Iodic Acid on the Surface of K10 Montmorillonite. Russian Journal of Organic Chemistry 2005, 41, 935–936. [Google Scholar] [CrossRef]
- Dweck, A.C. , Natural ingredients for colouring and styling. International Journal of Cosmetic Science 2002, 24, 287–302. [Google Scholar] [CrossRef]
- Hamama, W.S.; Hassanien, A.E.-D.E.; Zoorob, H.H. , Advanced Routes in Synthesis and Reactions of Lawsone Molecules (2-Hydroxynaphthalene-1,4-dione). Journal of Heterocyclic Chemistry 2017, 54, 2155–2196. [Google Scholar] [CrossRef]
- Anaissi-Afonso, L.; Oramas-Royo, S.; Ayra-Plasencia, J.; Martín-Rodríguez, P.; García-Luis, J.; Lorenzo-Castrillejo, I.; Fernández-Pérez, L.; Estévez-Braun, A.; Machín, F. , Lawsone, Juglone, and β-Lapachone Derivatives with Enhanced Mitochondrial-Based Toxicity. ACS Chemical Biology 2018, 13, 1950–1957. [Google Scholar] [CrossRef]
- Fieser, L.F.; Berliner, E. , Naphthoquinone antimalarials; general survey. Journal of the American Chemical Society 1948, 70, 3151–3155. [Google Scholar] [CrossRef]
- Inoue, K.; Ueda, S.; Nayeshiro, H.; Moritome, N.; Inouye, H. , Biosynthesis of naphthoqinones and anthraquinones in streptocarpus dunnii cell cultures. Phytochemistry 1984, 23, 312–318. [Google Scholar] [CrossRef]
- Lissel, M. , Über die reaktion von α- und β -tetralon mit kaliumsuperoxid. Tetrahedron Letters 1984, 25, 2213–2214. [Google Scholar] [CrossRef]
- De Min, M.; Croux, S.; Tournaire, C.; Hocquaux, M.; Jackquet, B.; Oliveros, E.; Maurette, M.-T. , Réactivité du superoxyde de Potassium en phase hétérogéne: Oxydation de naphtalénedios en naphtoquinones hydroxylées. Tetrahedron 1992, 48, 1869–1882. [Google Scholar] [CrossRef]
- Yan, Y.; Kang, E.-H.; Yang, K.-E.; Tong, S.-L.; Fang, C.-G.; Liu, S.-J.; Xiao, F.-S. , High activity in selective catalytic oxidation of naphthol to 2-hydroxy-1,4-naphthoquinone by molecular oxygen under air pressure over recycled iron porphyrin catalysts. Catalysis Communications 2004, 5, 387–390. [Google Scholar] [CrossRef]
- Barange, D.K.; Kavala, V.; Raju, B.R.; Kuo, C.-W.; Tseng, C.; Tu, Y.-C.; Yao, C.-F. , Facile and highly efficient method for the C-alkylation of 2-hydroxy-1,4-naphthoquinone to nitroalkenes under catalyst-free ‘on water’ conditions. Tetrahedron Letters 2009, 50, 5116–5119. [Google Scholar] [CrossRef]
- Laatsch, H. , Dimere Naphthochinone, VIII. Synthese 3,3′-dihydroxylierter 2,2′-Bi-1,4-naphthochinone. Liebigs Annalen der Chemie 1983, 1983, 1886–1900. [Google Scholar] [CrossRef]
- Papageorgiou, V.; Assimopoulou, A.; Ballis, A. Alkannins and Shikonins: A New Class of Wound Healing Agents. CMC 2008, 15, 3248–3267. [Google Scholar] [CrossRef]
- Papageorgiou, V.P.; Assimopoulou, A.N.; Couladouros, E.A.; Hepworth, D.; Nicolaou, K.C. , The Chemistry and Biology of Alkannin, Shikonin, and Related Naphthazarin Natural Products. Angewandte Chemie International Edition 1999, 38, 270–301. [Google Scholar] [CrossRef]
- Gong, K.; Li, W. , Shikonin, a Chinese plant-derived naphthoquinone, induces apoptosis in hepatocellular carcinoma cells through reactive oxygen species: A potential new treatment for hepatocellular carcinoma. Free Radical Biology and Medicine 2011, 51, 2259–2271. [Google Scholar] [CrossRef]
- Terada, A.; et al. , Total synthesis of shikalkin [(±)-shikonin]. Journal of the Chemical Society Chemical Communications, 1983. [Google Scholar]
- Moiseenkov, A.M.; et al. , Total synthesis of shikalkin. Proceedings of the USSR Academy of Sciences 1987, 295, 614–617. [Google Scholar]
- Nicolaou, K.C.; Hepworth, D. , Concise and Efficient Total Syntheses of Alkannin and Shikonin. Angewandte Chemie International Edition 1998, 37, 839–841. [Google Scholar] [CrossRef]
- Wang, R.; Zhou, S.; Jiang, H.; Zheng, X.; Zhou, W.; Li, S. , An Efficient Multigram Synthesis of Alkannin and Shikonin. European Journal of Organic Chemistry 2012, 2012, 1373–1379. [Google Scholar] [CrossRef]
- D'Arpa, P.; Liu, L.F. , Topoisomerase-targeting antitumor drugs. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 1989, 989, 163–177. [Google Scholar] [CrossRef]
- Prachayasittikul, V.; Pingaew, R.; Worachartcheewan, A.; Nantasenamat, C.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. , Synthesis, anticancer activity and QSAR study of 1,4-naphthoquinone derivatives. European Journal of Medicinal Chemistry 2014, 84, 247–263. [Google Scholar] [CrossRef]
- Harris, S.L.; Levine, A.J. , The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef]
- Tang, J.C.; Zhao, J.; Long, F.; Chen, J.Y.; Mu, B.; Jiang, Z.; Ren, Y.; Yang, J. , Efficacy of Shikonin against Esophageal Cancer Cells and its possible mechanisms in vitro and in vivo. J Cancer 2018, 9, 32–40. [Google Scholar] [CrossRef]
- Sanclemente, M.; et al. , c-RAF Ablation Induces Regression of Advanced Kras/Trp53 Mutant Lung Adenocarcinomas by a Mechanism Independent of MAPK Signaling. Cancer Cell 2018, 217. [Google Scholar] [CrossRef]
- Hsu, Y.-L.; Cho, C.-Y.; Kuo, P.-L.; Huang, Y.-T.; Lin, C.-C. , Plumbagin (5-Hydroxy-2-methyl-1,4-naphthoquinone) Induces Apoptosis and Cell Cycle Arrest in A549 Cells through p53 Accumulation via c-Jun NH<sub>2</sub>-Terminal Kinase-Mediated Phosphorylation at Serine 15 in Vitro and in Vivo. Journal of Pharmacology and Experimental Therapeutics 2006, 318, 484. [Google Scholar] [CrossRef]
- Tabb, M.M.; Sun, A.; Zhou, C.; Grün, F.; Errandi, J.; Romero, K.; Pham, H.; Inoue, S.; Mallick, S.; Lin, M.; Forman, B.M.; Blumberg, B. , Vitamin K2 Regulation of Bone Homeostasis Is Mediated by the Steroid and Xenobiotic Receptor SXR*. Journal of Biological Chemistry 2003, 278, 43919–43927. [Google Scholar] [CrossRef]
- Mishima, E.; Ito, J.; Wu, Z.; Nakamura, T.; Wahida, A.; Doll, S.; Tonnus, W.; Nepachalovich, P.; Eggenhofer, E.; Aldrovandi, M.; Henkelmann, B.; Yamada, K.-i.; Wanninger, J.; Zilka, O.; Sato, E.; Feederle, R.; Hass, D.; Maida, A.; Mourão, A.S.D.; Linkermann, A.; Geissler, E.K.; Nakagawa, K.; Abe, T.; Fedorova, M.; Proneth, B.; Pratt, D.A.; Conrad, M. , A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature 2022, 608, 778–783. [Google Scholar] [CrossRef]
- Jiang, Y.a.Z.R. , Research progress of antitumor effect of vitamin K2. The Practical Journal of Cancer, 2015. [Google Scholar]
- Ravichandiran, P.; Subramaniyan, S.A.; Kim, S.-Y.; Kim, J.-S.; Park, B.-H.; Shim, K.S.; Yoo, D.J. , Synthesis and Anticancer Evaluation of 1,4-Naphthoquinone Derivatives Containing a Phenylaminosulfanyl Moiety. ChemMedChem 2019, 14, 532–544. [Google Scholar] [CrossRef]
- Wellington, K.W.; Hlatshwayo, V.; Kolesnikova, N.I.; Saha, S.T.; Kaur, M.; Motadi, L.R. , Anticancer activities of vitamin K3 analogues. Investigational New Drugs 2020, 38, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Juang, Y.-P.; Tsai, J.-Y.; Gu, W.-L.; Hsu, H.-C.; Lin, C.-L.; Wu, C.-C.; Liang, P.-H. , Discovery of 5-Hydroxy-1,4-naphthoquinone (Juglone) Derivatives as Dual Effective Agents Targeting Platelet-Cancer Interplay through Protein Disulfide Isomerase Inhibition. Journal of Medicinal Chemistry 2024, 67, 3626–3642. [Google Scholar] [CrossRef] [PubMed]
- Gokmen, Z.; Onan, M.E.; Deniz, N.G.; Karakas, D.; Ulukaya, E. , Synthesis and investigation of cytotoxicity of new N- and S,S-substituted-1,4-naphthoquinone (1,4-NQ) derivatives on selected cancer lines. Synthetic Communications 2019, 49, 3008–3016. [Google Scholar]
- Gholampour, M.; Ranjbar, S.; Edraki, N.; Mohabbati, M.; Firuzi, O.; Khoshneviszadeh, M. , Click chemistry-assisted synthesis of novel aminonaphthoquinone-1,2,3-triazole hybrids and investigation of their cytotoxicity and cancer cell cycle alterations. Bioorganic Chemistry 2019, 88, 102967. [Google Scholar] [CrossRef]
- Schepetkin, I.A.; Karpenko, A.S.; Khlebnikov, A.I.; Shibinska, M.O.; Levandovskiy, I.A.; Kirpotina, L.N.; Danilenko, N.V.; Quinn, M.T. , Synthesis, anticancer activity, and molecular modeling of 1,4-naphthoquinones that inhibit MKK7 and Cdc25. European Journal of Medicinal Chemistry 2019, 183, 111719. [Google Scholar] [CrossRef]
- Olawode, E.O.; Tandlich, R.; Prinsloo, E.; Isaacs, M.; Hoppe, H.; Seldon, R.; Warner, D.F.; Steenkamp, V.; Kaye, P.T. , Synthesis and biological evaluation of 2-chloro-3-[(thiazol-2-yl)amino]-1,4-naphthoquinones. Bioorganic & Medicinal Chemistry Letters 2019, 29, 1572–1575. [Google Scholar]
- Ivasechko, I.; Lozynskyi, A.; Senkiv, J.; Roszczenko, P.; Kozak, Y.; Finiuk, N.; Klyuchivska, O.; Kashchak, N.; Manko, N.; Maslyak, Z.; Lesyk, D.; Karkhut, A.; Polovkovych, S.; Czarnomysy, R.; Szewczyk, O.; Kozytskiy, A.; Karpenko, O.; Khyluk, D.; Gzella, A.; Bielawski, K.; Bielawska, A.; Dzubak, P.; Gurska, S.; Hajduch, M.; Stoika, R.; Lesyk, R. , Molecular design, synthesis and anticancer activity of new thiopyrano[2,3-d]thiazoles based on 5-hydroxy-1,4-naphthoquinone (juglone). European Journal of Medicinal Chemistry 2023, 252, 115304. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Pelageev, D.N.; Jakob, L.S.; Borisova, K.L.; Hauschild, J.; Busenbender, T.; Kaune, M.; Khmelevskaya, E.A.; Graefen, M.; Bokemeyer, C.; Anufriev, V.P.; von Amsberg, G. In Pharmaceuticals, 2021; Vol. 14.
- Ferraris, D.; Lapidus, R.; Truong, P.; Bollino, D.; Carter-Cooper, B.; Lee, M.; Chang, E.; LaRossa-Garcia, M.; Dash, S.; Gartenhaus, R.; Choi, Y.E.; Kipe, O.; Lam, V.; Mason, K.; Palmer, R.; Williams, E.; Ambulos, N.; Kamangar, F.; Zhang, Y.; Kapadia, B.; Jing, Y.; Emadi, A. , Pre-Clinical Activity of Amino-Alcohol Dimeric Naphthoquinones as Potential Therapeutics for Acute Myeloid Leukemia. Anti-Cancer Agents in Medicinal Chemistry 2022, 22, 239–253. [Google Scholar] [CrossRef]
- Yu, X.; Xiao-Long, H.; Chun-Lian, W.U. , The Research Progress of Cell Apoptosis Induced by Shikonin and Signal Pathway of Apoptosis. Pharmaceutical Biotechnology, 2016. [Google Scholar]
- Zhang, X.; Cui, J.-H.; Meng, Q.-Q.; Li, S.-S.; Zhou, W.; Xiao, S. , Advance in Anti-tumor Mechanisms of Shikonin, Alkannin and their Derivatives. Mini-Reviews in Medicinal Chemistry 2018, 18, 164–172. [Google Scholar] [CrossRef]
- Yang, F.; Chen, Y.; Duan, W.; Zhang, C.; Zhu, H.; Ding, J. , SH-7, a new synthesized shikonin derivative, exerting its potent antitumor activities as a topoisomerase inhibitor. International Journal of Cancer 2006, 119, 1184–1193. [Google Scholar] [CrossRef]
- Kamo, S.; Kuramochi, K.; Tsubaki, K. , Recent topics in total syntheses of natural dimeric naphthoquinone derivatives. Tetrahedron Letters 2018, 59, 224–230. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, X.; Song, H.; Zhou, S.; Li, S. , Synthesis and evaluation of novel alkannin and shikonin oxime derivatives as potent antitumor agents. Bioorganic & Medicinal Chemistry Letters 2014, 24, 4304–4307. [Google Scholar]
- Cui, J.; Zhang, X.; Huang, G.; Zhang, Q.; Dong, J.; Sun, G.; Meng, Q.; Li, S. , DMAKO-20 as a New Multitarget Anticancer Prodrug Activated by the Tumor Specific CYP1B1 Enzyme. Molecular Pharmaceutics 2019, 16, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Zhao, H.-R.; Meng, Q.-Q.; Zhang, Q.-J.; Dong, J.-Y.; Zhu, B.-q.; Li, S.-S. , Synthesis and biological evaluation of sulfur-containing shikonin oxime derivatives as potential antineoplastic agents. European Journal of Medicinal Chemistry 2018, 143, 166–181. [Google Scholar] [CrossRef]
- Huang, G.; Dong, J.-Y.; Zhang, Q.-J.; Meng, Q.-Q.; Zhao, H.-R.; Zhu, B.-Q.; Li, S.-S. , Discovery and synthesis of sulfur-containing 6-substituted 5,8-dimethoxy-1,4-naphthoquinone oxime derivatives as new and potential anti-MDR cancer agents. European Journal of Medicinal Chemistry 2019, 165, 160–171. [Google Scholar] [CrossRef]
- Nariya, P.; Shukla, F.; Vyas, H.; Devkar, R.; Thakore, S. , Synthesis and characterization of Mannich bases of lawsone and their anticancer activity. Synthetic Communications 2020, 50, 1724–1735. [Google Scholar] [CrossRef]
- Guimaráes, G.D.; de Assis Gonsalves, A.; Rolim, A.L.; Araújo, C.E.; dos Anjos Santos, L.V.; Silva, F.S.M.; de Cássia Evangelista de Oliveira, F.; da Costa, P.M.; Pessoa, C.; Fonseca Goulart, O.M.; Silva, L.T.; Santos, C.D.; Araújo, R.M.C. , Naphthoquinone-based Hydrazone Hybrids: Synthesis and Potent Activity Against Cancer Cell Lines. Medicinal Chemistry 2021, 17, 945–955. [Google Scholar] [CrossRef]
- Khan, A.; Said, M.S.; Borade, B.R.; Gonnade, R.; Barvkar, V.; Kontham, R.; Dastager, S.G. , Enceleamycins A–C, Furo-Naphthoquinones from Amycolatopsis sp. MCC0218: Isolation, Structure Elucidation, and Antimicrobial Activity. Journal of Natural Products 2022, 85, 1267–1273. [Google Scholar] [CrossRef]
- Nural, Y.; Ozdemir, S.; Doluca, O.; Demir, B.; Yalcin, M.S.; Atabey, H.; Kanat, B.; Erat, S.; Sari, H.; Seferoglu, Z. , Synthesis, biological properties, and acid dissociation constant of novel naphthoquinone–triazole hybrids. Bioorganic Chemistry 2020, 105, 104441. [Google Scholar] [CrossRef]
- Balachandran, C.; Al-Dhabi, N.A.; Duraipandiyan, V.; Ignacimuthu, S. , Bluemomycin, a new naphthoquinone derivative from Streptomyces sp. with antimicrobial and cytotoxic properties. Biotechnology Letters 2021, 43, 1005–1018. [Google Scholar] [CrossRef]
- Supratman, U.; Hirai, N.; Sato, S.; Watanabe, K.; Malik, A.; Annas, S.; Harneti, D.; Maharani, R.; Koseki, T.; Shiono, Y. , New naphthoquinone derivatives from Fusarium napiforme of a mangrove plant. Natural Product Research 2021, 35, 1406–1412. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.G.T.; Miranda, F.F.; Ferreira, V.F.; Freitas, C.C.; Rabello, R.F.; Carballido, J.M.; Corrêa, L.C.D. , Synthesis and antimicrobial evaluation of 3-hydrazino-naphthoquinones as analogs of lapachol. Journal of the Brazilian Chemical Society 2001, 12, 791–803. [Google Scholar] [CrossRef]
- Song, R.; Yu, B.; Friedrich, D.; Li, J.; Shen, H.; Krautscheid, H.; Huang, S.D.; Kim, M.-H. , Naphthoquinone-derivative as a synthetic compound to overcome the antibiotic resistance of methicillin-resistant S. aureus. Communications Biology 2020, 3, 529. [Google Scholar] [CrossRef] [PubMed]
- Sabutski, Y.E.; Menchinskaya, E.S.; Shevchenko, L.S.; Chingizova, E.A.; Chingizov, A.R.; Popov, R.S.; Denisenko, V.A.; Mikhailov, V.V.; Aminin, D.L.; Polonik, S.G. In Molecules, 2020; Vol. 25.
- Kim, K.; Kim, D.; Lee, H.; Lee, T.H.; Kim, K.-Y.; Kim, H. In Biomedicines, 2020; Vol. 8.
- Gemili, M.; Nural, Y.; Keleş, E.; Aydıner, B.; Seferoğlu, N.; Ülger, M.; Şahin, E.; Erat, S.; Seferoğlu, Z. , Novel highly functionalized 1,4-naphthoquinone 2-iminothiazole hybrids: Synthesis, photophysical properties, crystal structure, DFT studies, and anti(myco)bacterial/antifungal activity. Journal of Molecular Structure 2019, 1196, 536–546. [Google Scholar] [CrossRef]
- Erasmus, C.; Aucamp, J.; Smit, F.J.; Seldon, R.; Jordaan, A.; Warner, D.F.; N'Da, D.D. , Synthesis and comparison of in vitro dual anti-infective activities of novel naphthoquinone hybrids and atovaquone. Bioorganic Chemistry 2021, 114, 105118. [Google Scholar] [CrossRef]
- Tandon, V.K.; Maurya, H.K.; Yadav, D.B.; Tripathi, A.; Kumar, M.; Shukla, P.K. , Naphtho[2,3-b][1,4]-thiazine-5,10-diones and 3-substituted-1,4-dioxo-1,4-dihydronaphthalen-2-yl-thioalkanoate derivatives: Synthesis and biological evaluation as potential antibacterial and antifungal agents. Bioorganic & Medicinal Chemistry Letters 2006, 16, 5883–5887. [Google Scholar]
- Tandon, V.K.; Yadav, D.B.; Singh, R.V.; Chaturvedi, A.K.; Shukla, P.K. , Synthesis and biological evaluation of novel (l)-α-amino acid methyl ester, heteroalkyl, and aryl substituted 1,4-naphthoquinone derivatives as antifungal and antibacterial agents. Bioorganic & Medicinal Chemistry Letters 2005, 15, 5324–5328. [Google Scholar]
- Tandon, V.K.; Maurya, H.K.; Mishra, N.N.; Shukla, P.K. , Design, synthesis and biological evaluation of novel nitrogen and sulfur containing hetero-1,4-naphthoquinones as potent antifungal and antibacterial agents. European Journal of Medicinal Chemistry 2009, 44, 3130–3137. [Google Scholar] [CrossRef]
- Gonzaga, D.T.G.; Gomes, R.S.P.; Marra, R.K.F.; Silva, F.C.d.; Gomes, M.W.L.; Ferreira, D.F.; Santos, R.M.A.; Pinto, A.M.V.; Ratcliffe, N.A.; Cirne-Santos, C.C.; Barros, C.S.; Ferreira, V.F.; Paixão, I.C.N.P. , Inhibition of Zika Virus Replication by Synthetic Bis-Naphthoquinones. Journal of the Brazilian Chemical Society 2019, 30. [Google Scholar] [CrossRef]
- Cui, J.; Jia, J. , Discovery of juglone and its derivatives as potent SARS-CoV-2 main proteinase inhibitors. European Journal of Medicinal Chemistry 2021, 113789. [Google Scholar] [CrossRef]
- Fu, Z.; Huang, B.; Tang, J.; Liu, S.; Liu, M.; Ye, Y.; Liu, Z.; Xiong, Y.; Zhu, W.; Cao, D. , The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nature Communications.
- Sendl, A.; Chen, J.L.; Jolad, S.D.; Stoddart, C.; Rozhon, E.; Kernan, M.; Nanakorn, W.; Balick, M. , Two new naphthoquinones with antiviral activity from Rhinacanthus nasutus. Journal of Natural Products 1996, 59, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.J.M.D.; Buarque, C.D.; Brito, F.V.; Aurelian, L.; Costa, P.R.R. , Synthesis and preliminary pharmacological evaluation of new (+/-) 1,4-naphthoquinones structurally related to lapachol. Bioorganic & Medicinal Chemistry 2002, 10, 2731–2738. [Google Scholar]
- Ilina, T.V.; Semenova, E.A.; Pronyaeva, T.R.; Pokrovskii, A.G.; Tolstikov, G.A. , Inhibition of HIV-1 reverse transcriptase by aryl-substituted naphto- and anthraquinones. Doklady Biochemistry & Biophysics 2002, 382, 56. [Google Scholar]
- A, V.K.T.; B, R.B.C.; C, R.V.S.; B, S.R.; A, D.B.Y. , Design, synthesis and evaluation of novel 1,4-naphthoquinone derivatives as antifungal and anticancer agents. Bioorganic & Medicinal Chemistry Letters 2004, 14, 1079–1083. [Google Scholar]
- Ngoc, T.M.; Phuong, N.T.T.; Khoi, N.M.; Park, S.J.; Kwak, H.J.; Nhiem, N.X.; Trang, B.T.T.; Tai, B.H.; Song, J.H.; Ko, H.J. , A new naphthoquinone analogue and antiviral constituents from the root of Rhinacanthus nasutus. Natural Product Research 2018, 1. [Google Scholar] [CrossRef]
- Cetina-Montejo, L.; Ayora-Talavera, G.; Borges-Argáez, R. , Zeylanone epoxide isolated from Diospyros anisandra stem bark inhibits influenza virus in vitro. Archives of Virology, 2019. [Google Scholar]
- Zima, V.; Radilová, K.; Koíek, M.; Albiana, C.B.; Machara, A. , Unraveling the Anti-Influenza Effect of Flavonoids: Experimental Validation of Luteolin and its Congeners as Potent Influenza Endonuclease Inhibitors. European Journal of Medicinal Chemistry 2020, 112754. [Google Scholar] [CrossRef]
- Tisseh, Z.N.; Bazgir, A. , An efficient, clean synthesis of 3,3′-(arylmethylene)bis(2-hydroxynaphthalene-1,4-dione) derivatives. dyes\&\pigments 2009, 83, 258–261. [Google Scholar]
- Pang, X.; Zhao, J.Y.; Liu, N.; Chen, M.H.; Ma, B.P. , Anthraquinone analogues with inhibitory activities against influenza a virus from Polygonatum odoratum. Journal of Asian Natural Products Research 2020, 1–7. [Google Scholar] [CrossRef]
- Lien, J.C.; Huang, L.J.; Teng, C.M.; Wang, J.P.; Kuo, S.C. , Synthesis of 2-alkoxy 1,4-naphthoquinone derivatives as antiplatelet, antiinflammatory, and antiallergic agents. Chemical & Pharmaceutical Bulletin 2010, 33, 105–105. [Google Scholar]
- Lien, J.C.; Huang, L.J.; Wang, J.P.; Teng, C.M.; Lee, K.H.; Kuo, S.C. , Synthesis and antiplatelet, antiinflammatory and antiallergic activities of 2,3-disubstituted 1,4-naphthoquinones. Cheminform 1996, 27, 1181–1187. [Google Scholar] [CrossRef]
Figure 1.
The chemical structures of 1,4-naphthoquinone derivatives.
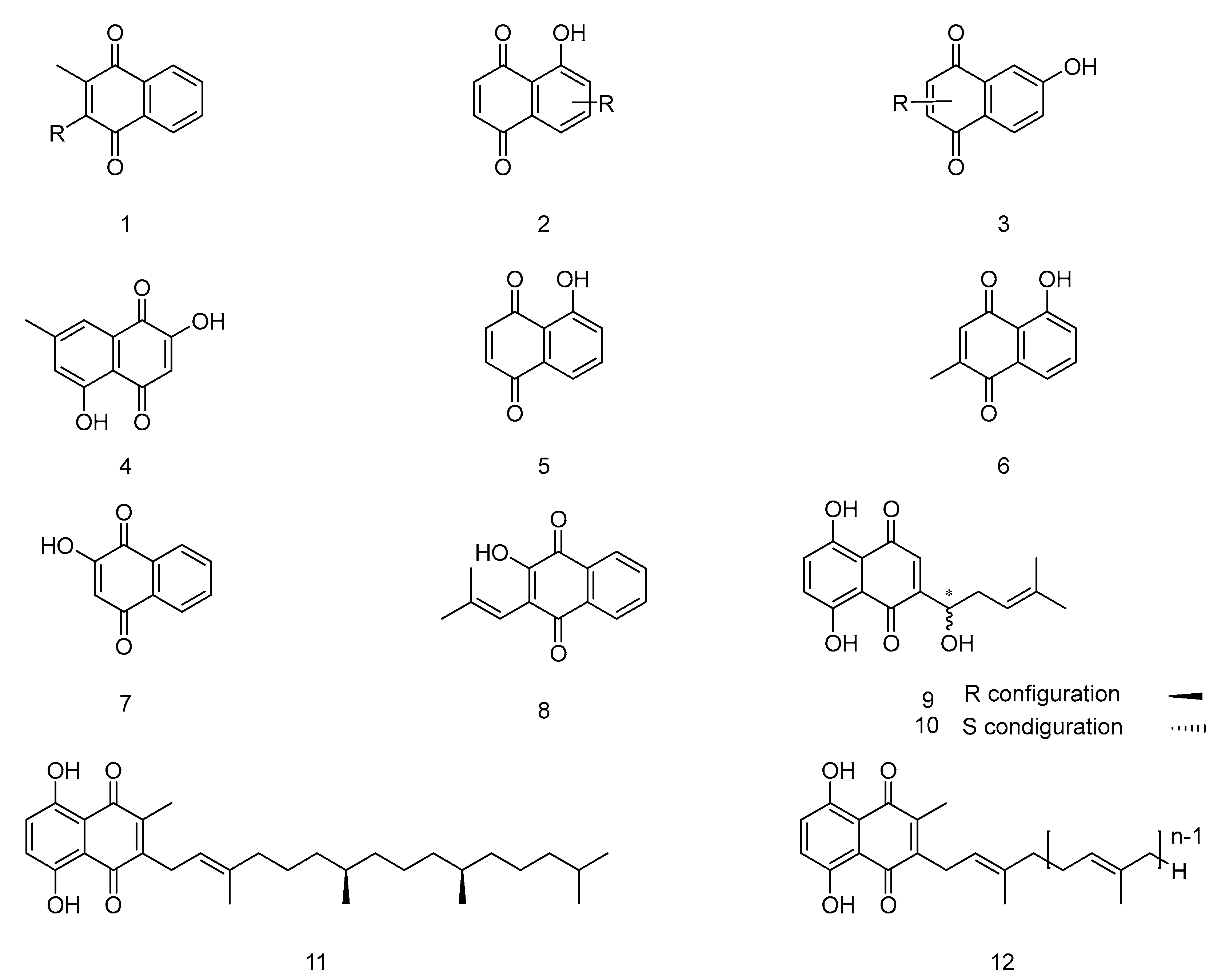
Figure 2.
The chemical structure of Vitamin K.
Figure 3.
The synthetic routes of Vitamin K1. Reagents and conditions: a) Phytyl bromide, -60 ℃, 30 min, H3O+; b) 0.5 % (NiCl2, PPh3, n-BuLi), THF, -78℃, 15 min.
Figure 3.
The synthetic routes of Vitamin K1. Reagents and conditions: a) Phytyl bromide, -60 ℃, 30 min, H3O+; b) 0.5 % (NiCl2, PPh3, n-BuLi), THF, -78℃, 15 min.
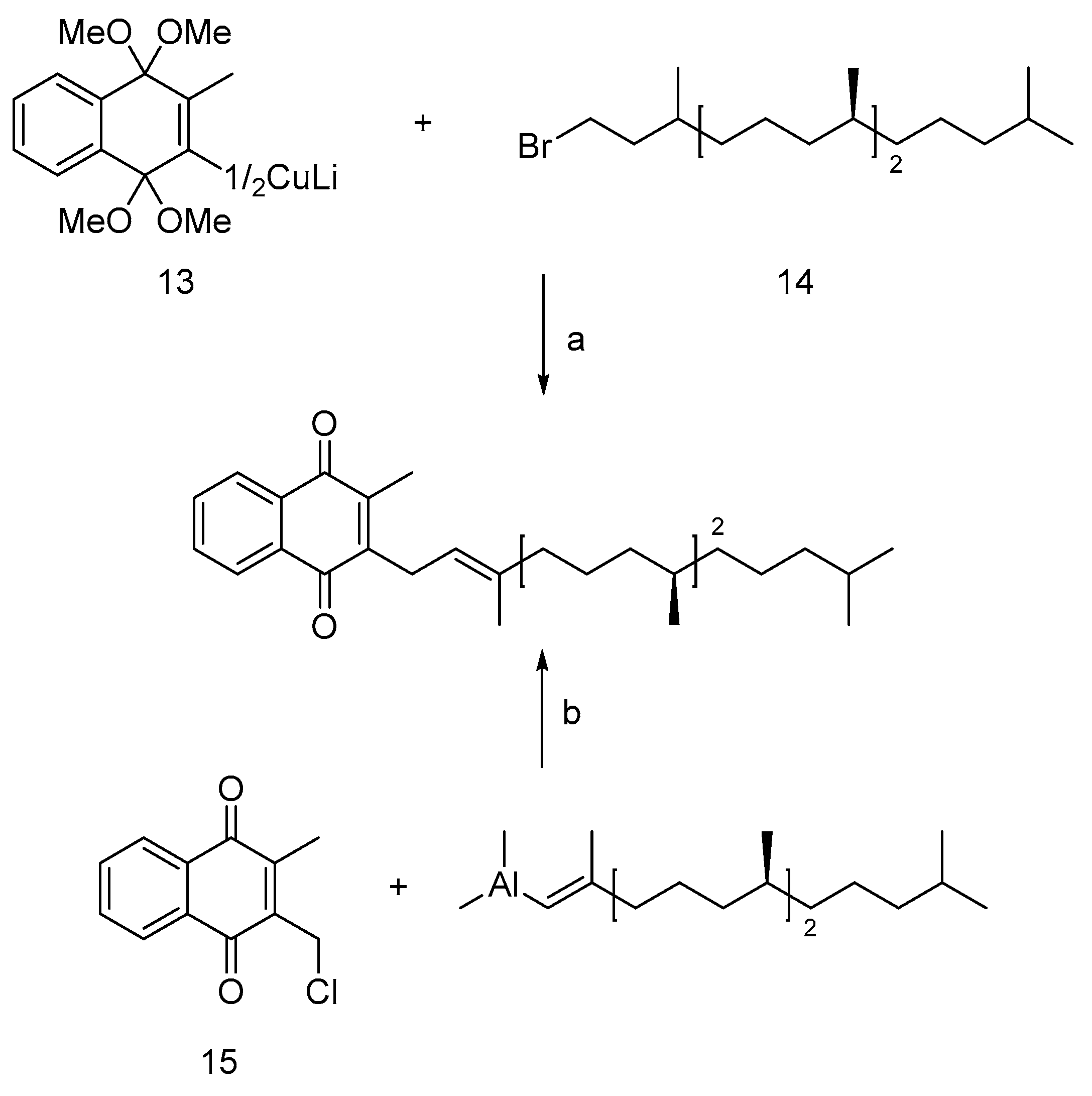
Figure 5.
The synthetic route of Vitamin MK-7. Conditions: a) Ac2O, Py, SeO2, tBuO2H, Salicylic acid; b) PBr3, PhSO2Na; c) tBuOK, Farnesyl bromide; d) LiEt3BH, Pd(dppe)Cl2; e) OH; f) PBr3, CH2Cl2; g) SnCl4, CH2Cl2, 0 ℃; h) NaHMDS, THF, -20 ℃; i) CAN, CH3CN/CH2Cl2.
Figure 5.
The synthetic route of Vitamin MK-7. Conditions: a) Ac2O, Py, SeO2, tBuO2H, Salicylic acid; b) PBr3, PhSO2Na; c) tBuOK, Farnesyl bromide; d) LiEt3BH, Pd(dppe)Cl2; e) OH; f) PBr3, CH2Cl2; g) SnCl4, CH2Cl2, 0 ℃; h) NaHMDS, THF, -20 ℃; i) CAN, CH3CN/CH2Cl2.
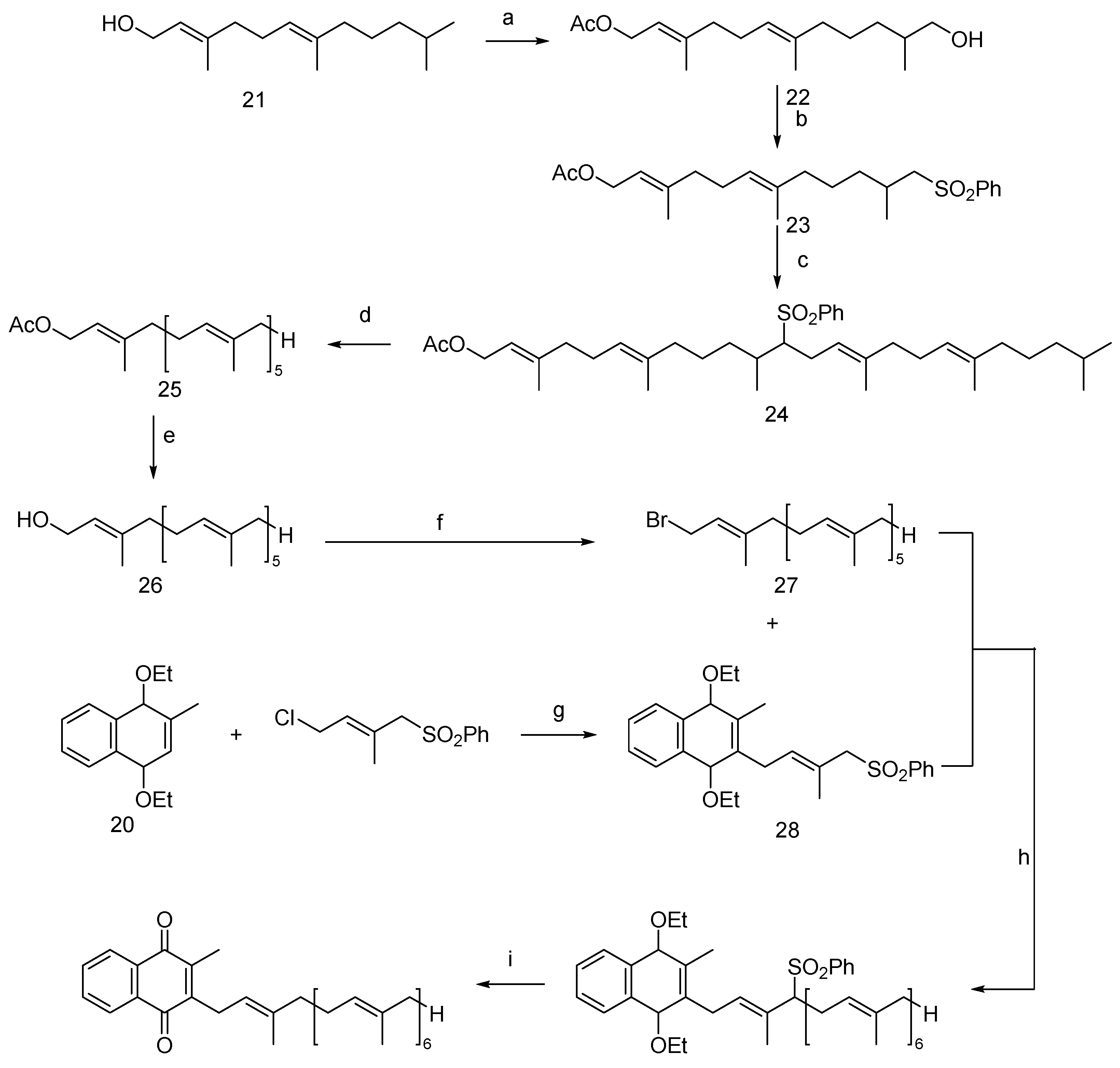
Figure 6.
The synthetic routes of juglone. Conditions: a) Succinic anhydride, PhNO2, AlCl3, Et3SiH, CF3COOH, HBr, reflux; b) DDQ, Benzene, reflux; c) H2SO4, SO3, 55 ℃; d) NaOH, 330 ℃, H2SO4, Na2Cr2O7, H2O, 15 ℃; e) Ac2O, H2O2, MeOH, 50 ℃; f) HIO4, Microwave, 50 ℃.
Figure 6.
The synthetic routes of juglone. Conditions: a) Succinic anhydride, PhNO2, AlCl3, Et3SiH, CF3COOH, HBr, reflux; b) DDQ, Benzene, reflux; c) H2SO4, SO3, 55 ℃; d) NaOH, 330 ℃, H2SO4, Na2Cr2O7, H2O, 15 ℃; e) Ac2O, H2O2, MeOH, 50 ℃; f) HIO4, Microwave, 50 ℃.
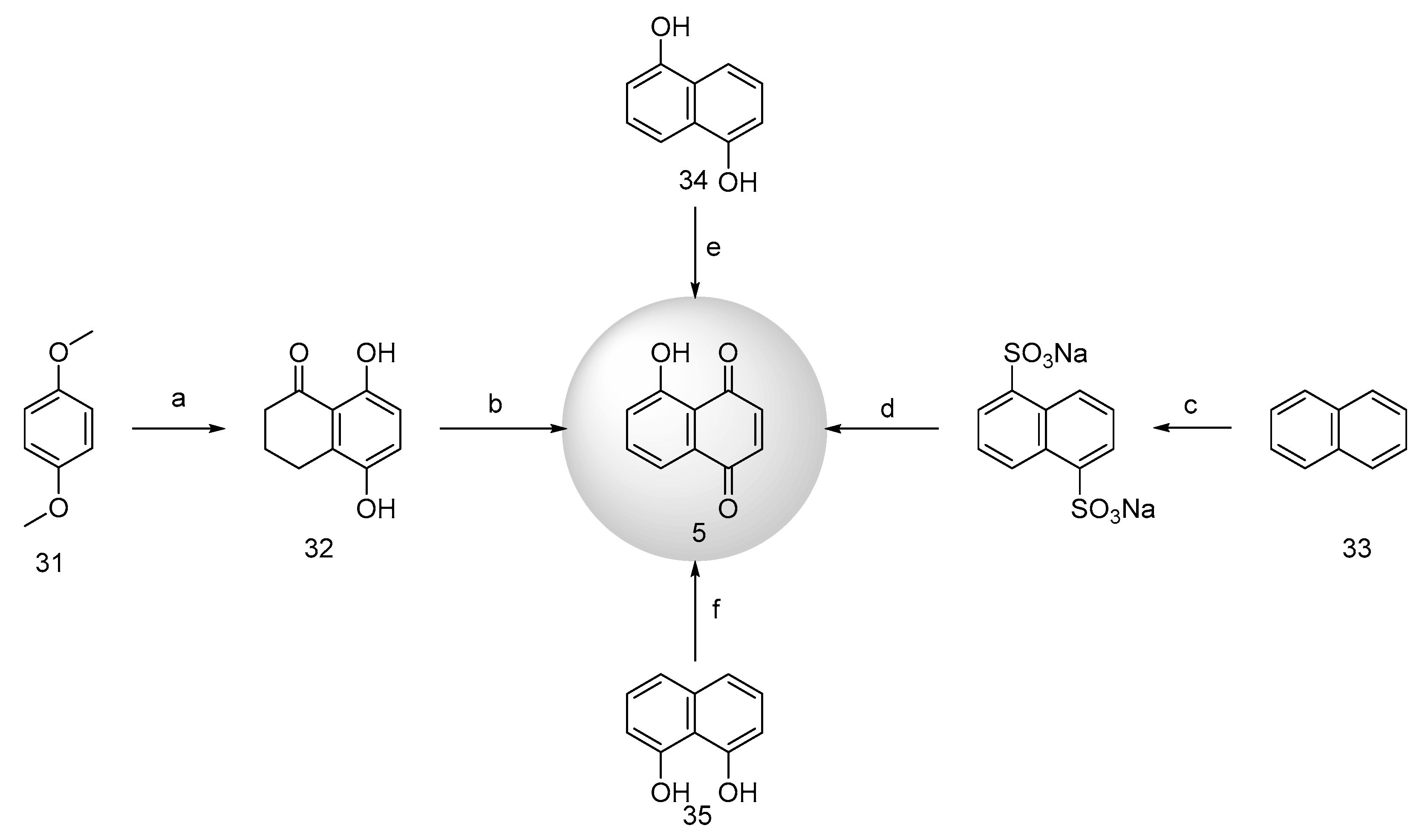
Figure 7.
The synthetic routes of 2-hydroxy-1,4-naphthoquinone. Conditions: a) p-Nitrotoluene, CuSO4 or NaOH, air; b) HCl, NaOH, AlCl3, O2, PdCl2, MeOH, sod. tetrahydroborate, CS2, CH2Cl2; c) KO2, 18-crown-6, toluene; d) Crown-ether/pyridine or KO2/THF, -10 ℃; e) [O], H2O, HCl or aq. KOH, air; f) Prussian red, H2O, NaOH; g) H2O2, KOH; h) Basic hydrolysis.
Figure 7.
The synthetic routes of 2-hydroxy-1,4-naphthoquinone. Conditions: a) p-Nitrotoluene, CuSO4 or NaOH, air; b) HCl, NaOH, AlCl3, O2, PdCl2, MeOH, sod. tetrahydroborate, CS2, CH2Cl2; c) KO2, 18-crown-6, toluene; d) Crown-ether/pyridine or KO2/THF, -10 ℃; e) [O], H2O, HCl or aq. KOH, air; f) Prussian red, H2O, NaOH; g) H2O2, KOH; h) Basic hydrolysis.
Figure 8.
The synthetic route of racemic Shikonin. Conditions: a) TsOH, CH3COCH3, MgMeI, THF; b) CAN, CH3CN/CH2Cl2, AgO, HNO3; c) AcOH, Py, SOCl2, NaOH, CH3COOH; d) Me2SCH2; e) Isopropyl thin lithium; f) CAN, CH3CN/CH2Cl2; g) AgO, AgNO3.
Figure 8.
The synthetic route of racemic Shikonin. Conditions: a) TsOH, CH3COCH3, MgMeI, THF; b) CAN, CH3CN/CH2Cl2, AgO, HNO3; c) AcOH, Py, SOCl2, NaOH, CH3COOH; d) Me2SCH2; e) Isopropyl thin lithium; f) CAN, CH3CN/CH2Cl2; g) AgO, AgNO3.
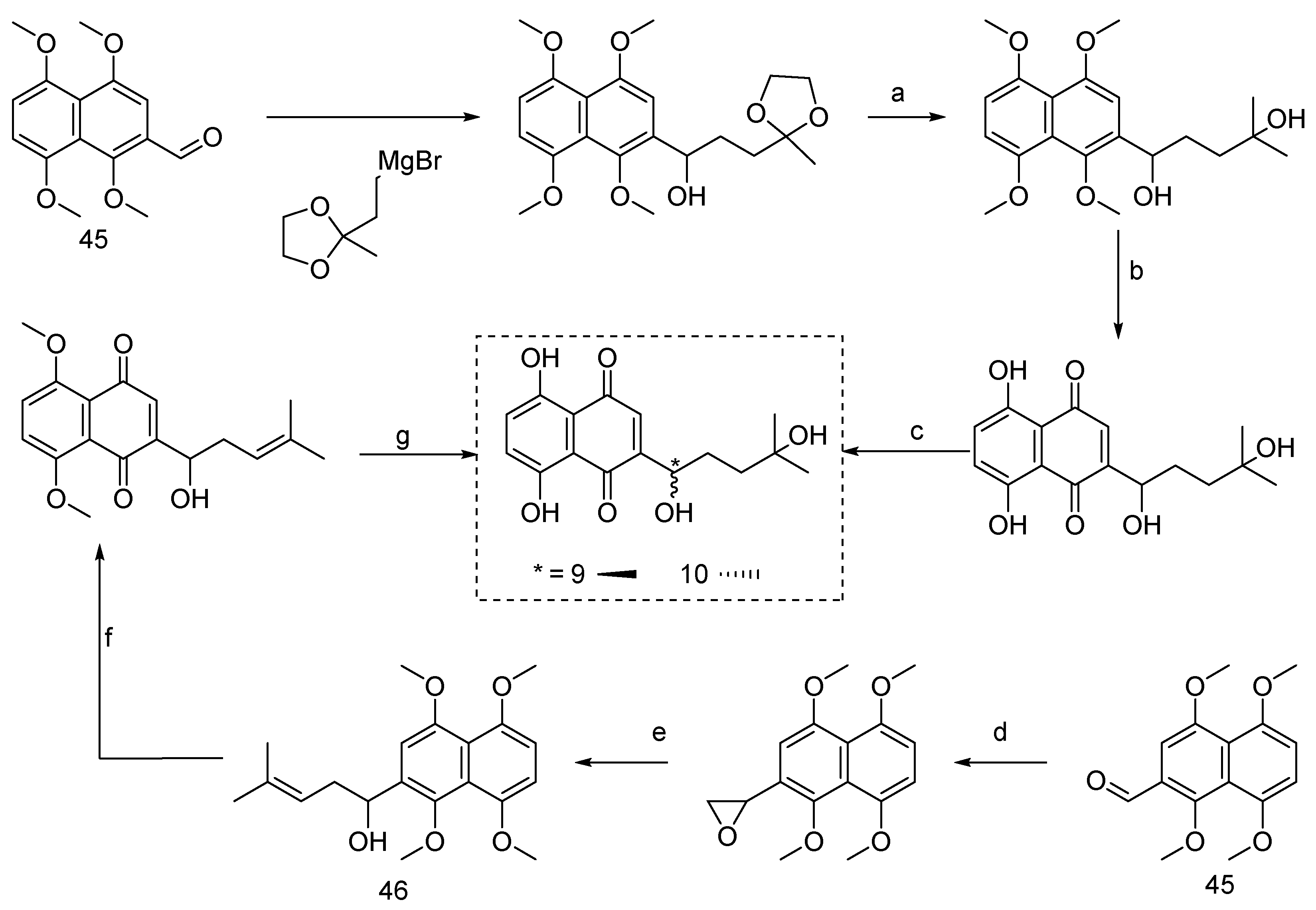
Figure 9.
The synthetic routes of Shikonin and Alkannin. Conditions: a) SnCl2, HCl, BrCH2Cl, K2CO3, DMF, NBS, CHCl3; b) tBuLi, THF; c) (+)-DIP-Cl, THF; d) (-)-DIP-Cl, THF.
Figure 9.
The synthetic routes of Shikonin and Alkannin. Conditions: a) SnCl2, HCl, BrCH2Cl, K2CO3, DMF, NBS, CHCl3; b) tBuLi, THF; c) (+)-DIP-Cl, THF; d) (-)-DIP-Cl, THF.
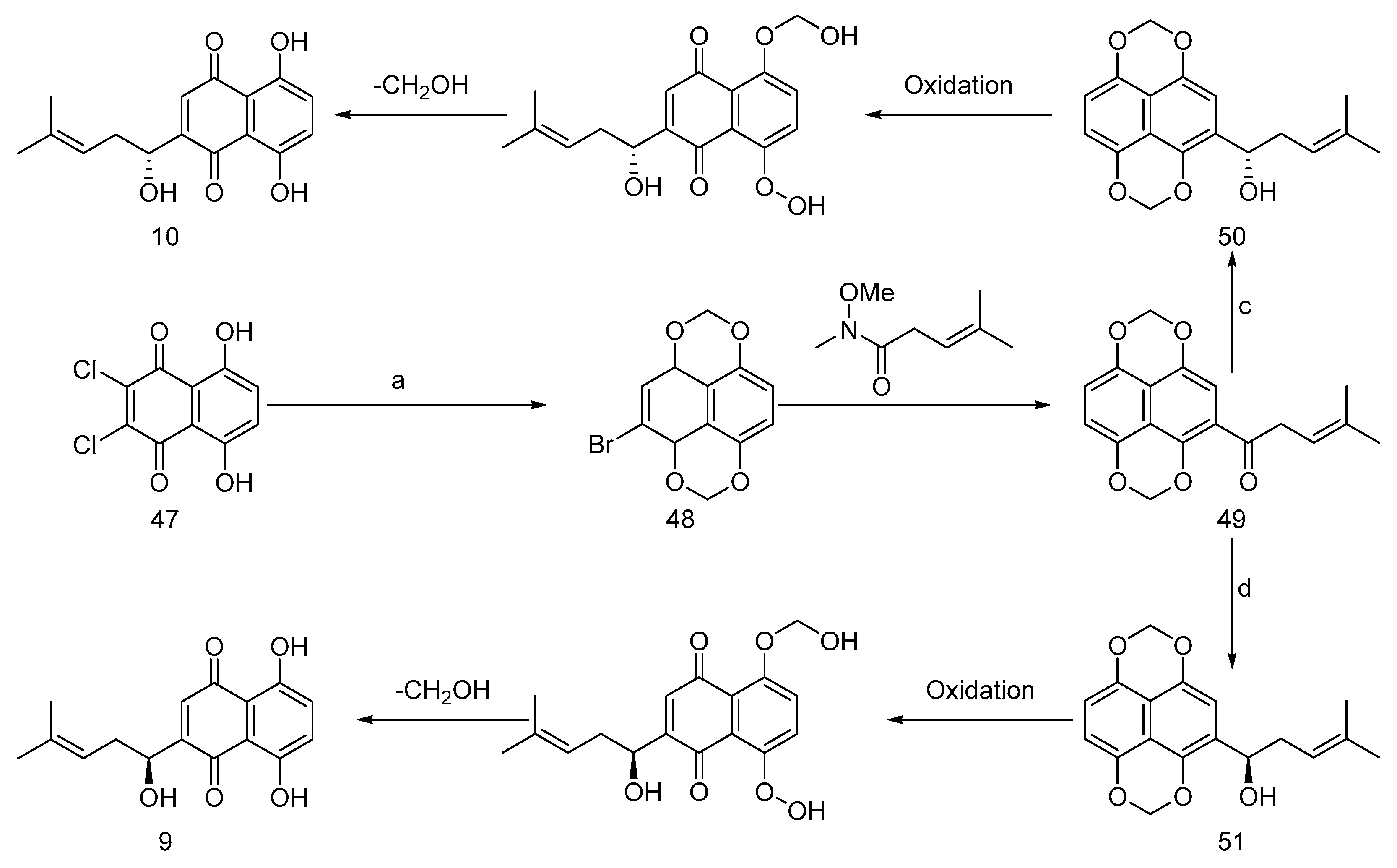
Figure 10.
Oxidation strategy to synthesize naphthoquinone.
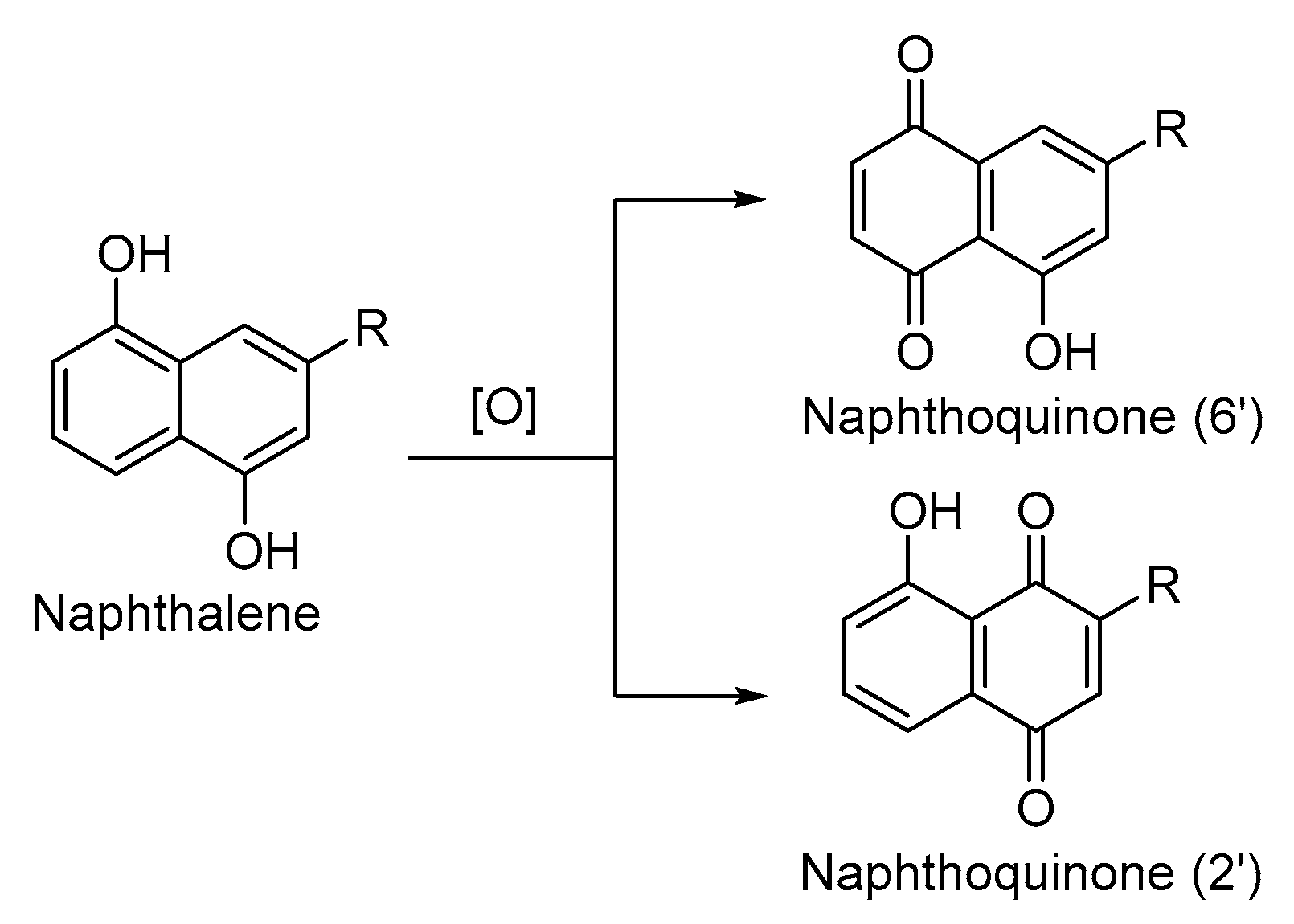
Figure 11.
Sulfur-contained 1,4-naphthoquinone derivatives as anticancer agents.
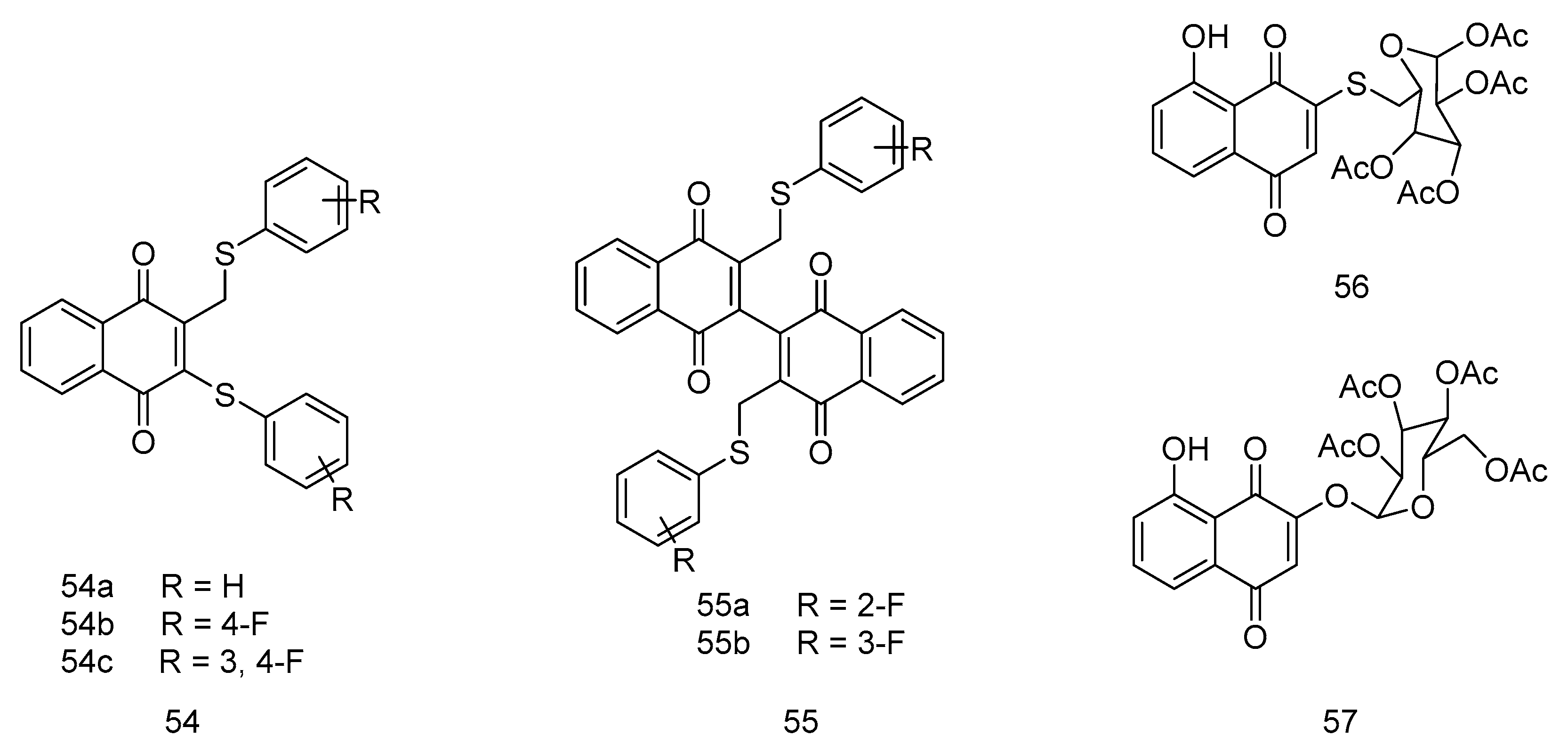
Figure 12.
1,4-naphthoquinone derivatives as anticancer agents.
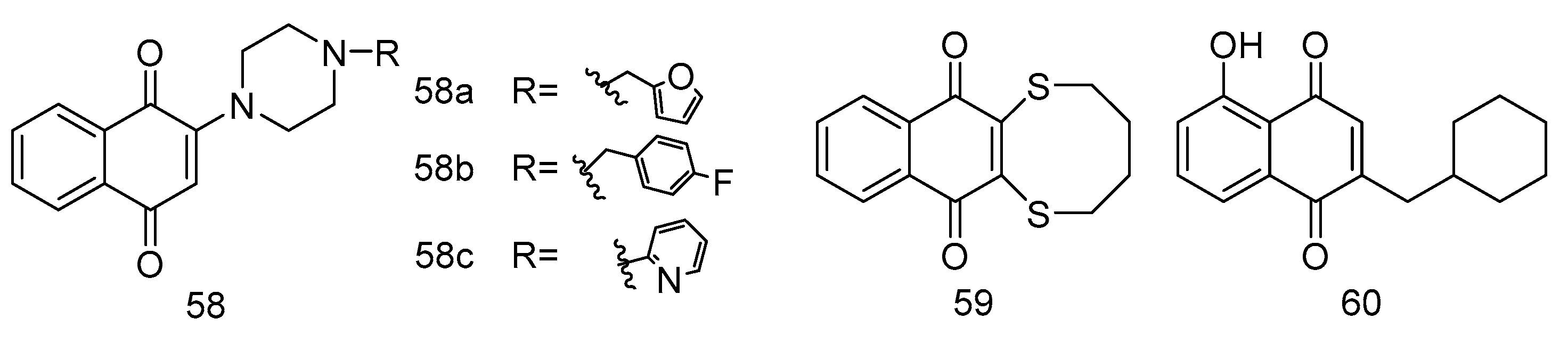
Figure 13.
1,4-naphthoquinone derivatives as anticancer agents.
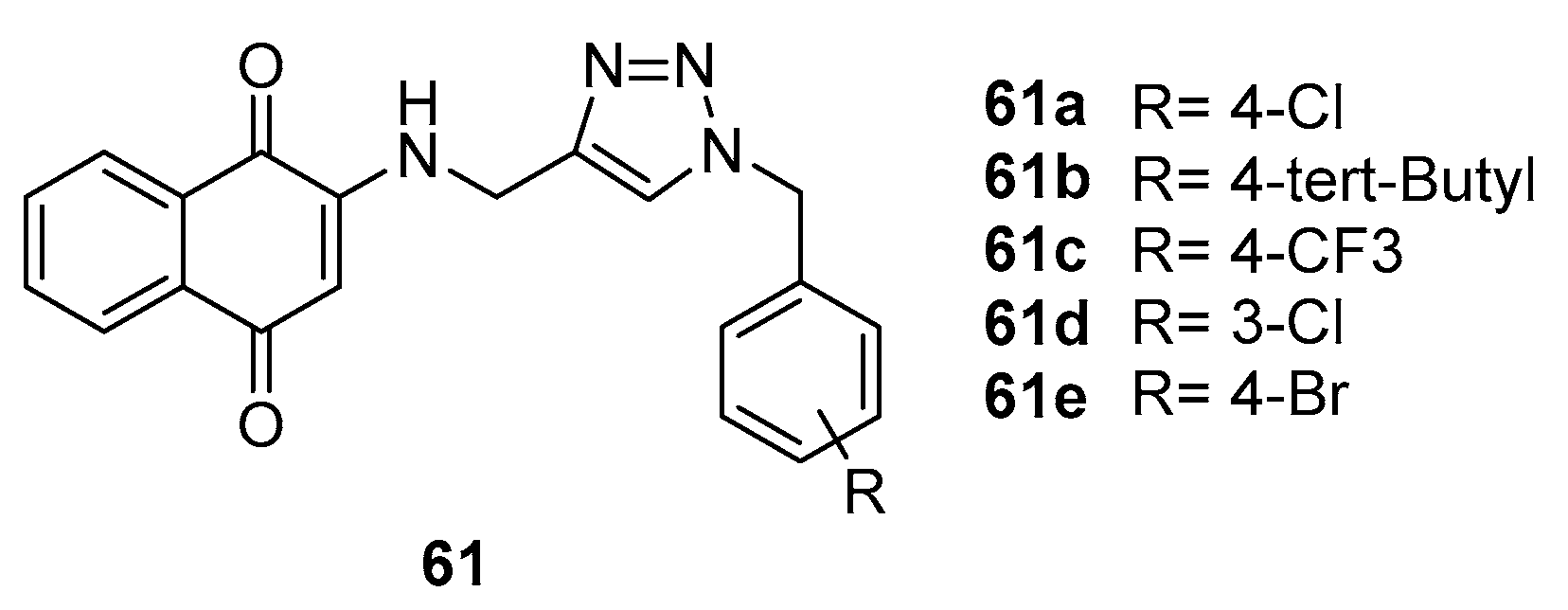
Figure 14.
1,4-naphthoquinone derivatives as anticancer agents.
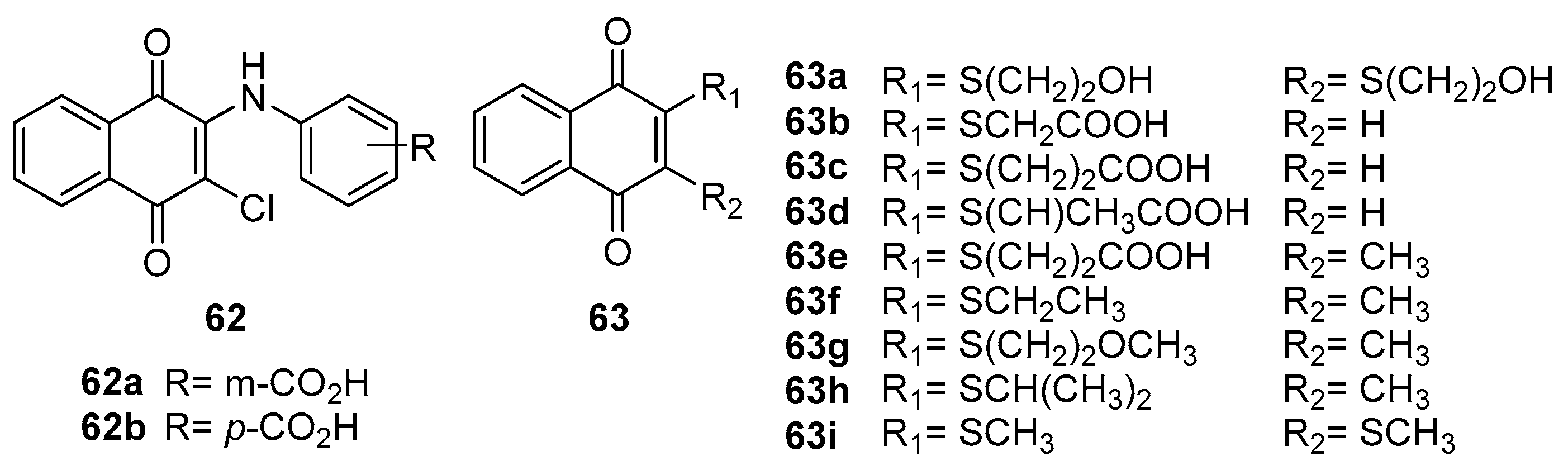
Figure 15.
1,4-naphthoquinone derivatives as anticancer agents.
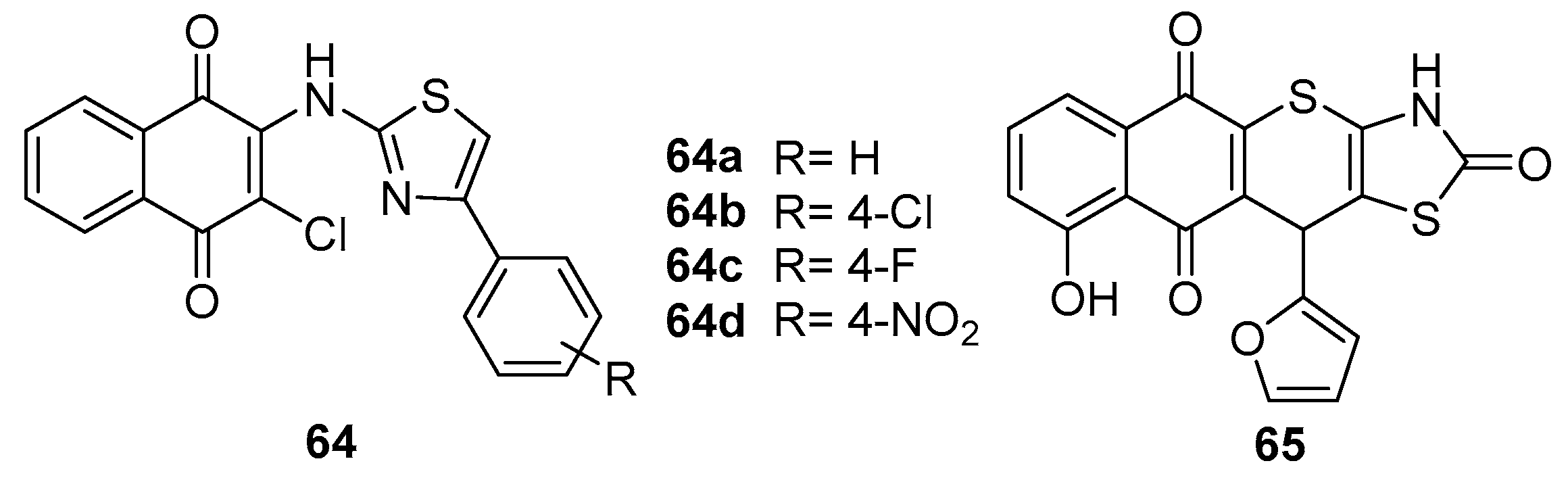
Figure 16.
2-chloroethylthio substituted 1,4-naphthoquinone derivatives as anticancer agents.
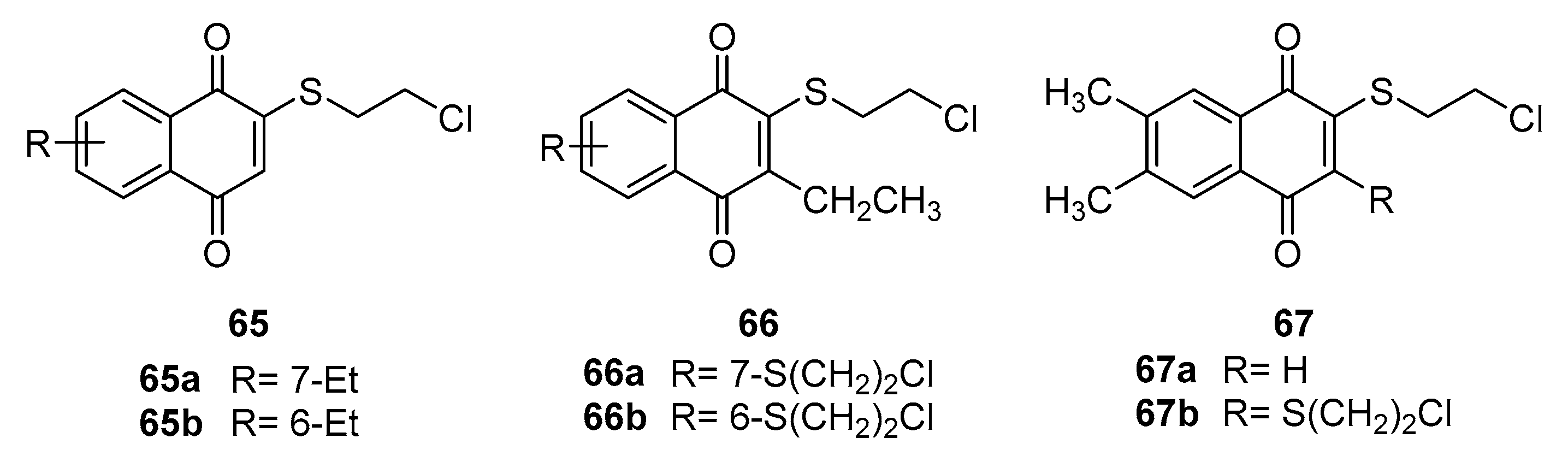
Figure 17.
Naphthoquinone dimers as anticancer agents.
Figure 18.
Shikonin derivatives as anticancer agents.
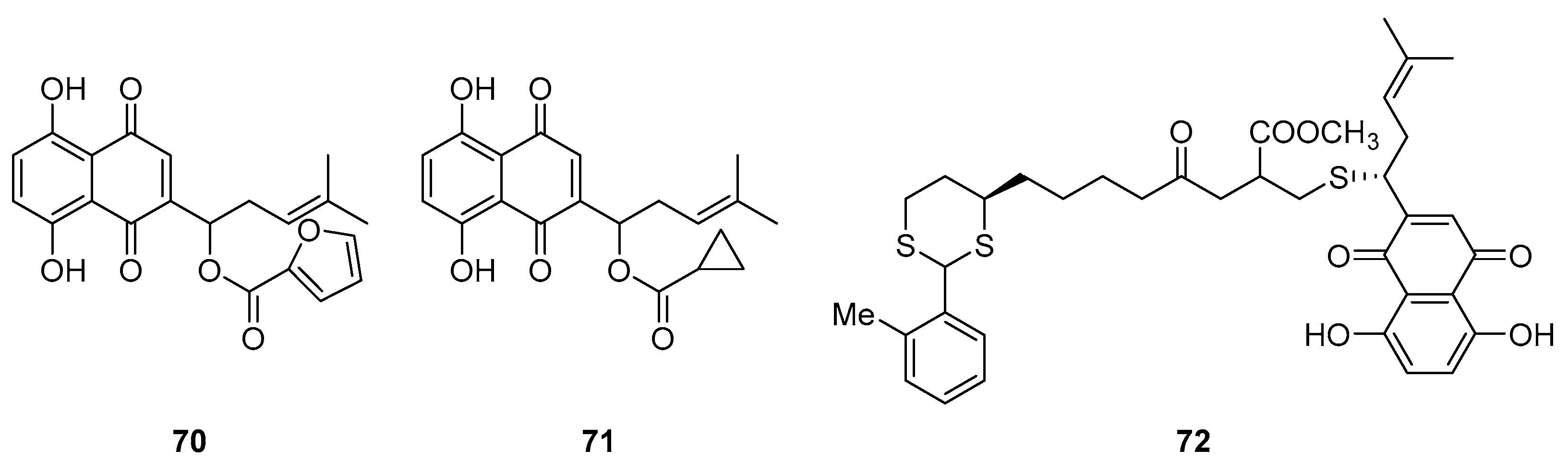
Figure 19.
1,4-naphthoquinone derivatives as anticancer agents.
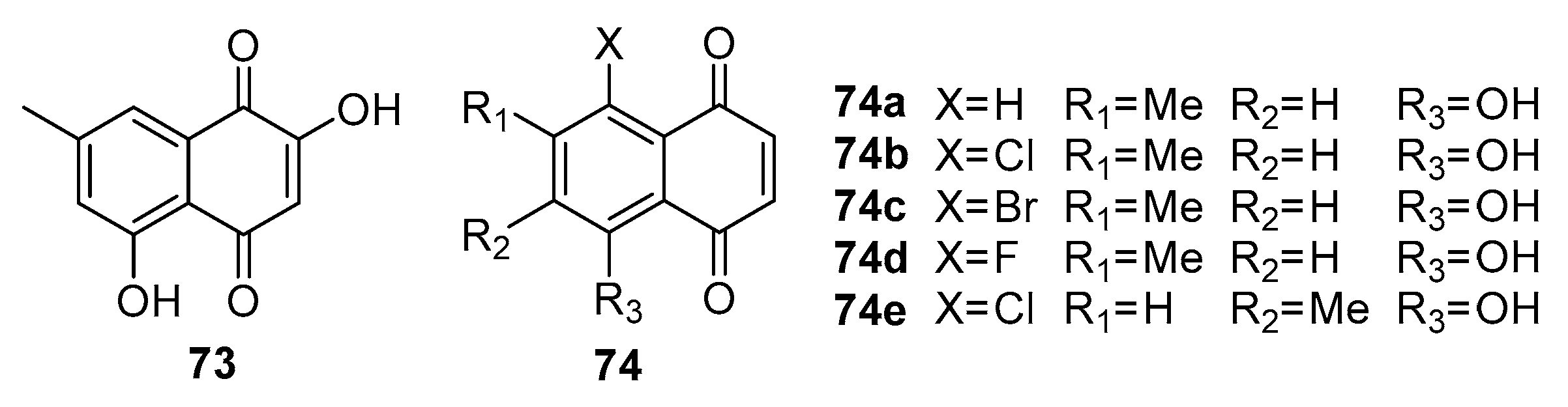
Figure 20.
Lawsone derivatives as anticancer agents.
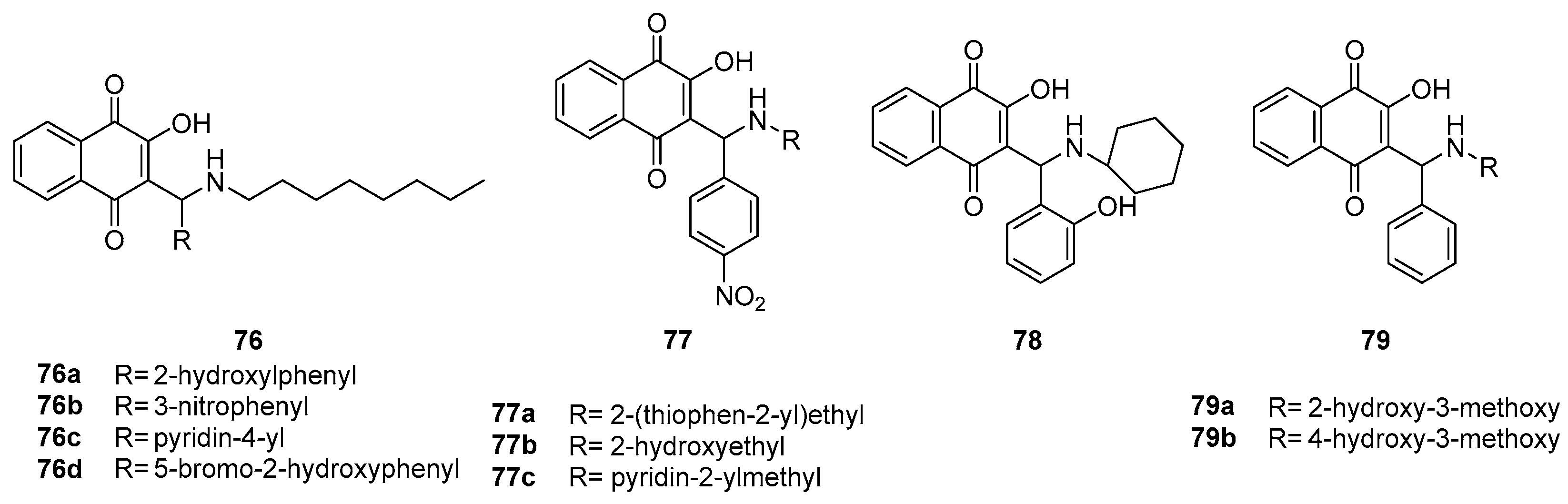
Figure 21.
Lapachol derivatives as anticancer agents.
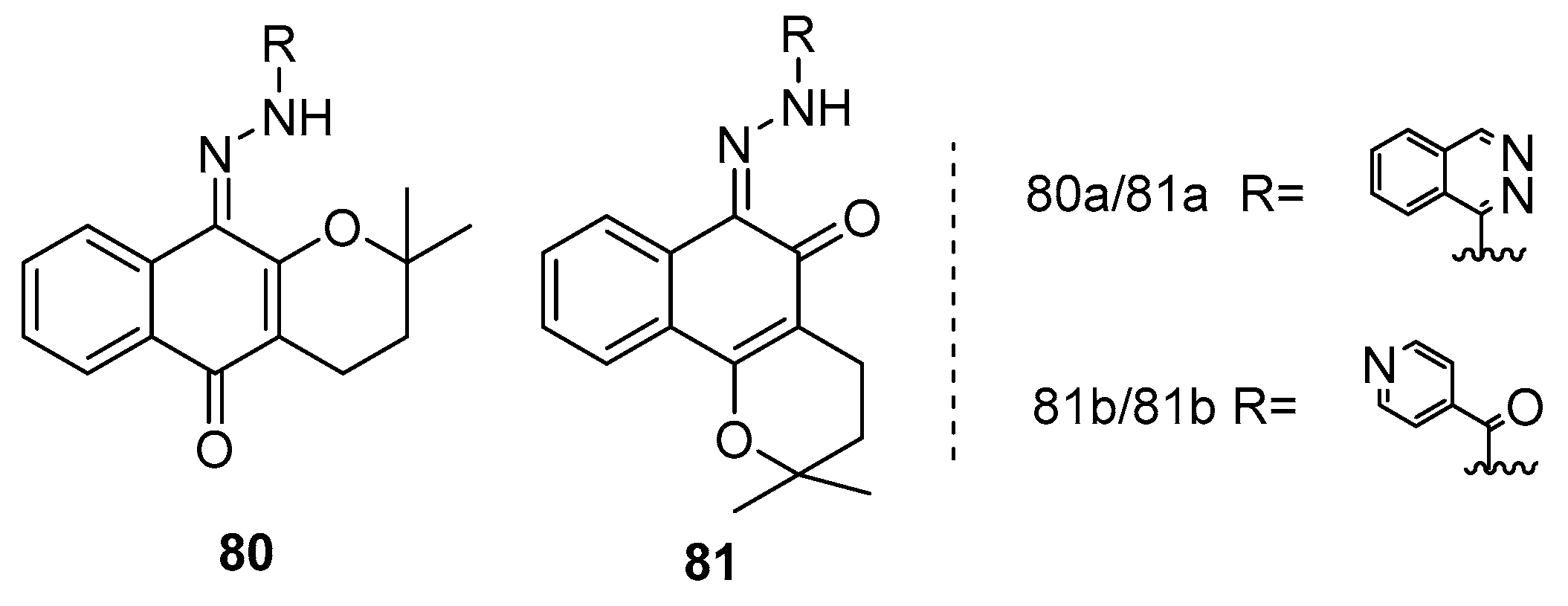
Figure 22.
Furo-naphthoquinones as antimicrobial agents.
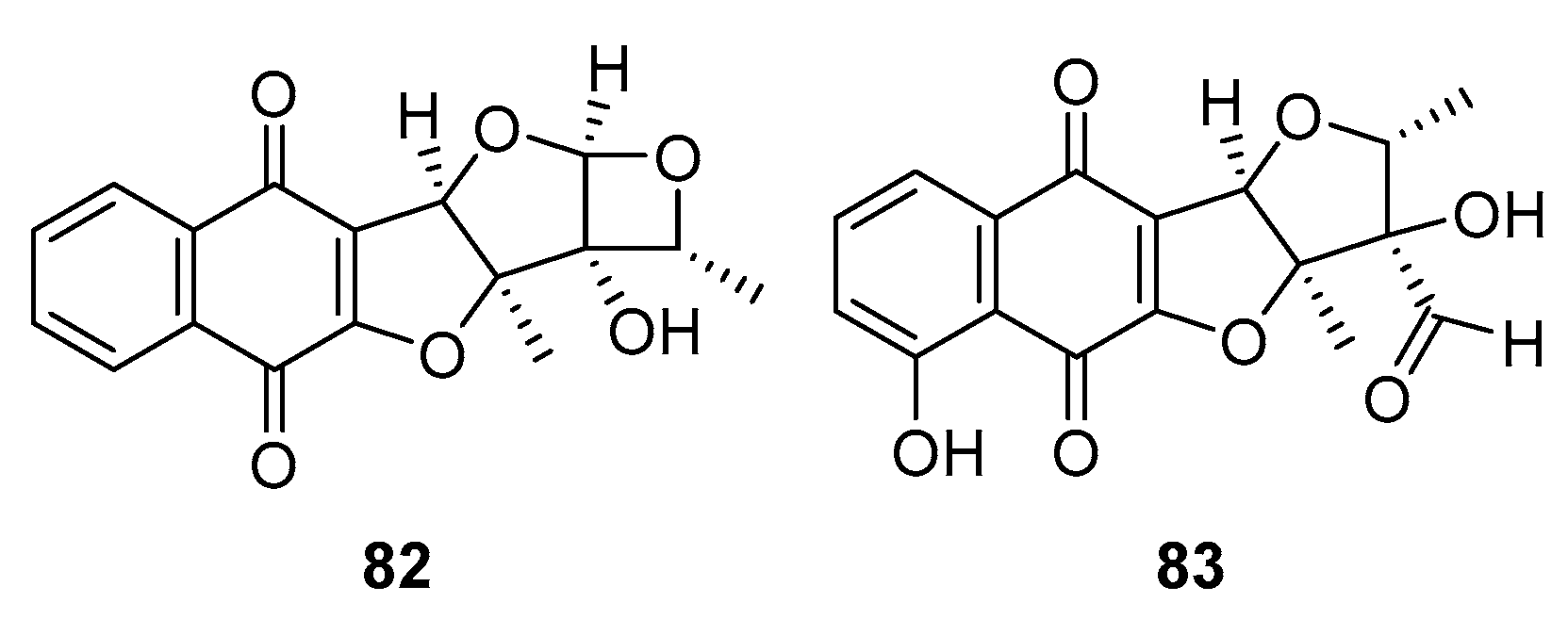
Figure 23.
1,4-naphthoquinone derivatives as antimicrobial agents.
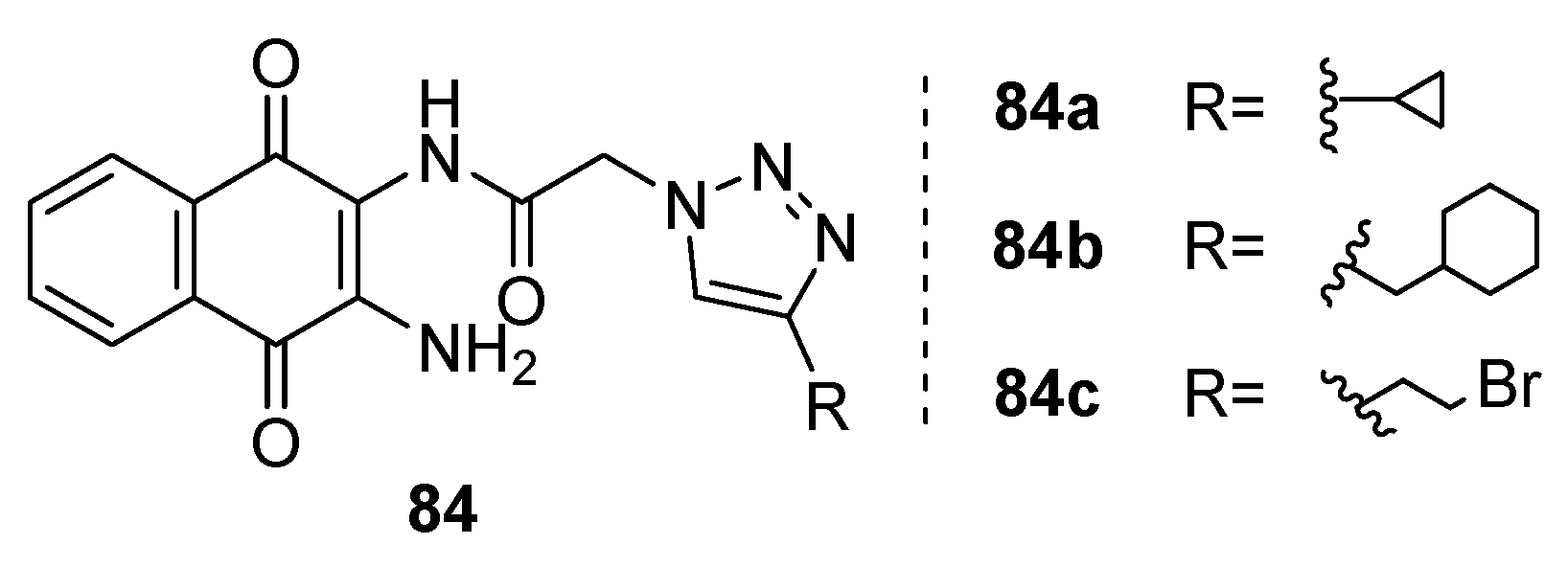
Figure 24.
Fusarium napiforme as antimicrobial agents.
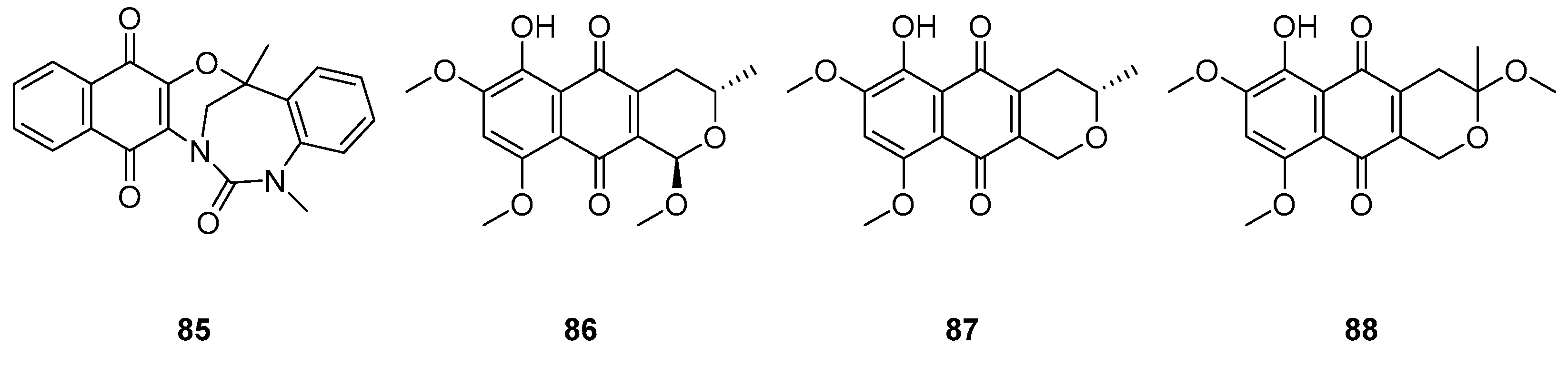
Figure 25.
1,4-naphthoquinone derivatives as antimicrobial agents.
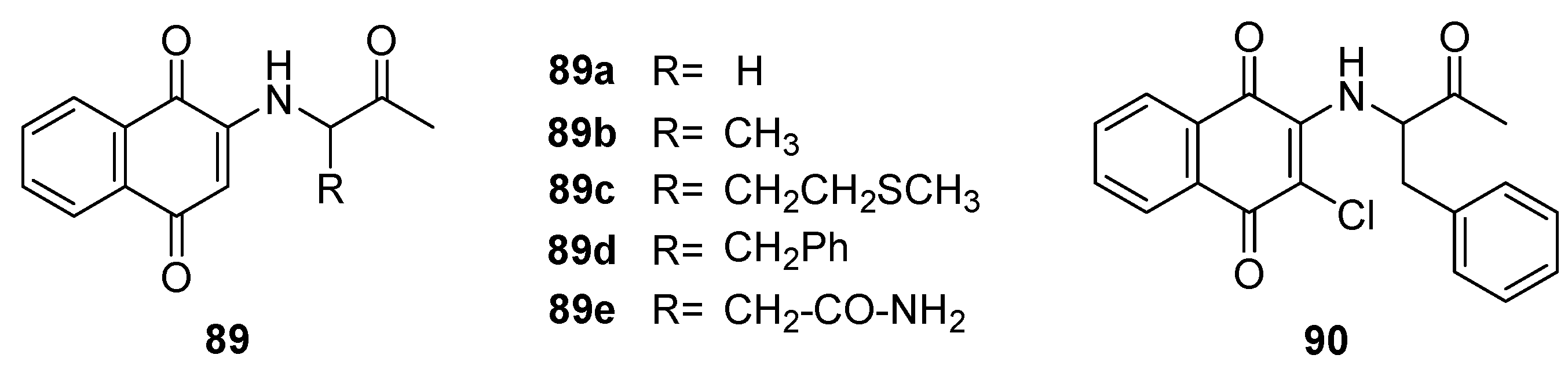
Figure 26.
Lawsone derivatives as antimicrobial agents.
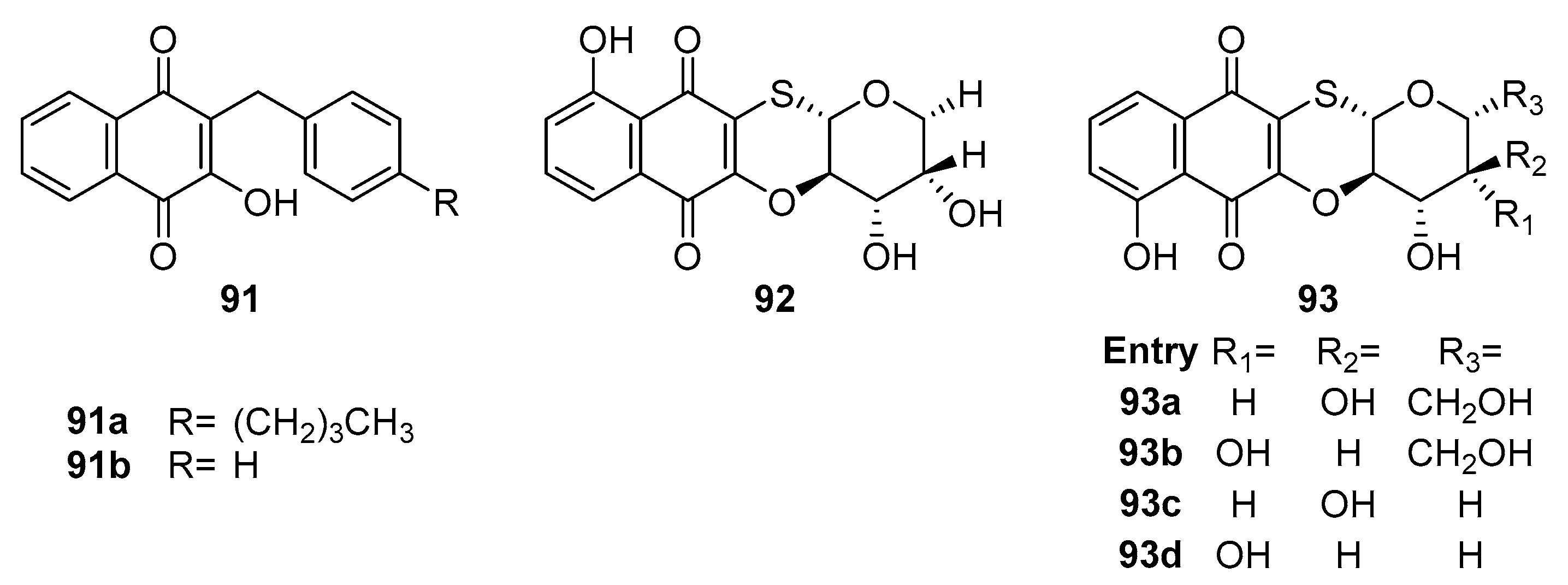
Figure 27.
Pyrimidinone-fused naphthoquinone derivatives as antimicrobial agents.
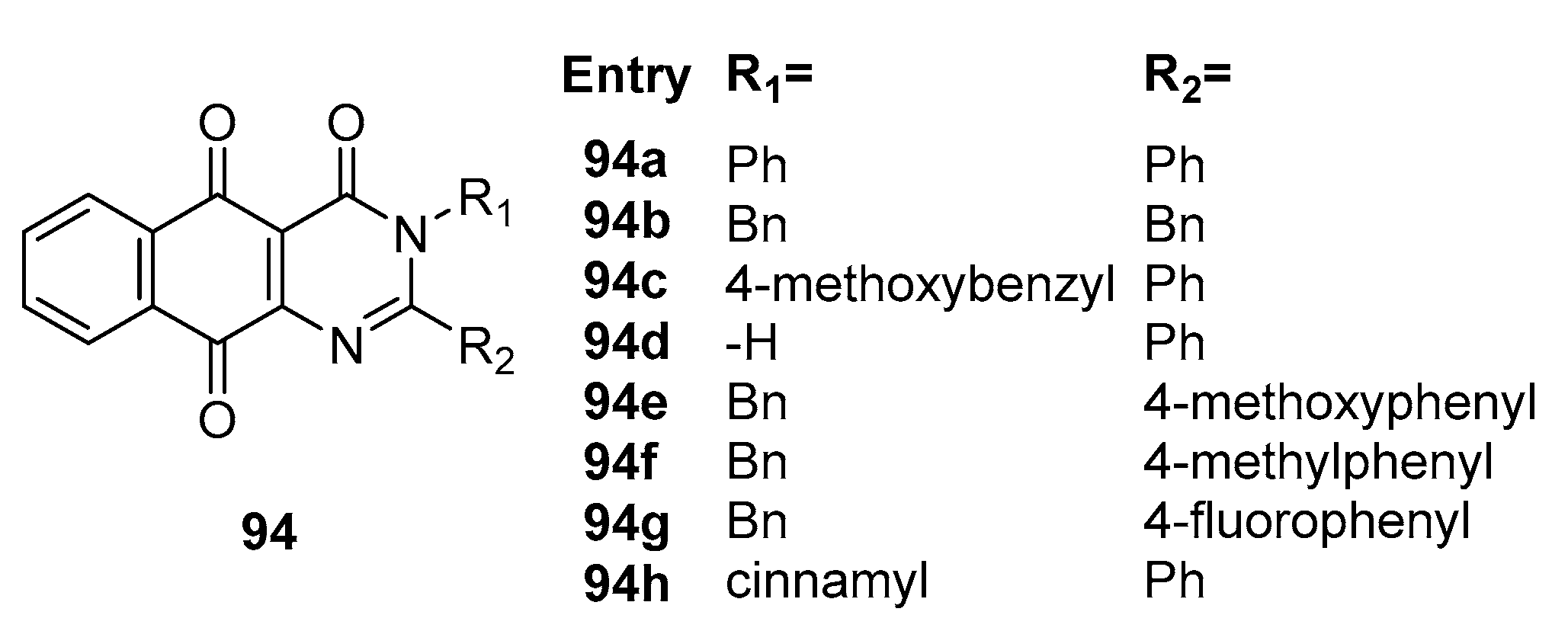
Figure 28.
2-iminothiazole substituted naphthoquinone compounds as antimicrobial agents.
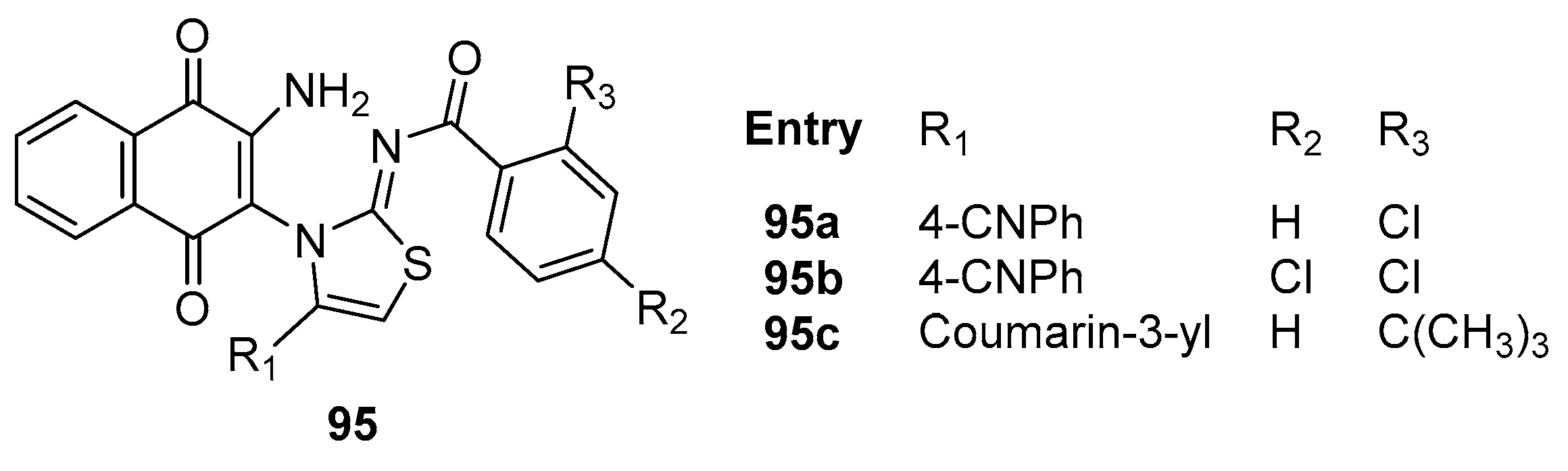
Figure 29.
2-amino-3-triazol substituted naphthoquinone compounds as antimicrobial agents.
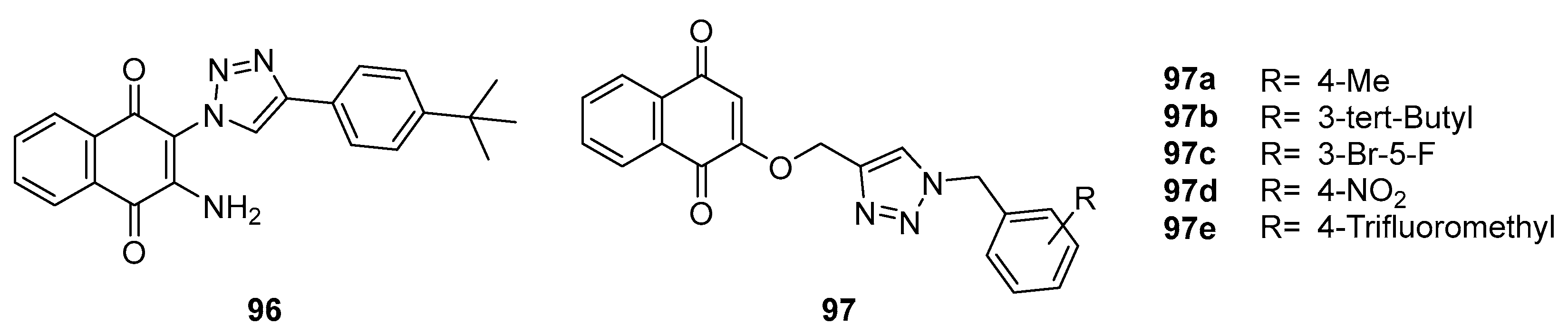
Figure 30.
1,4-naphthoquinone derivatives as antimicrobial agents.
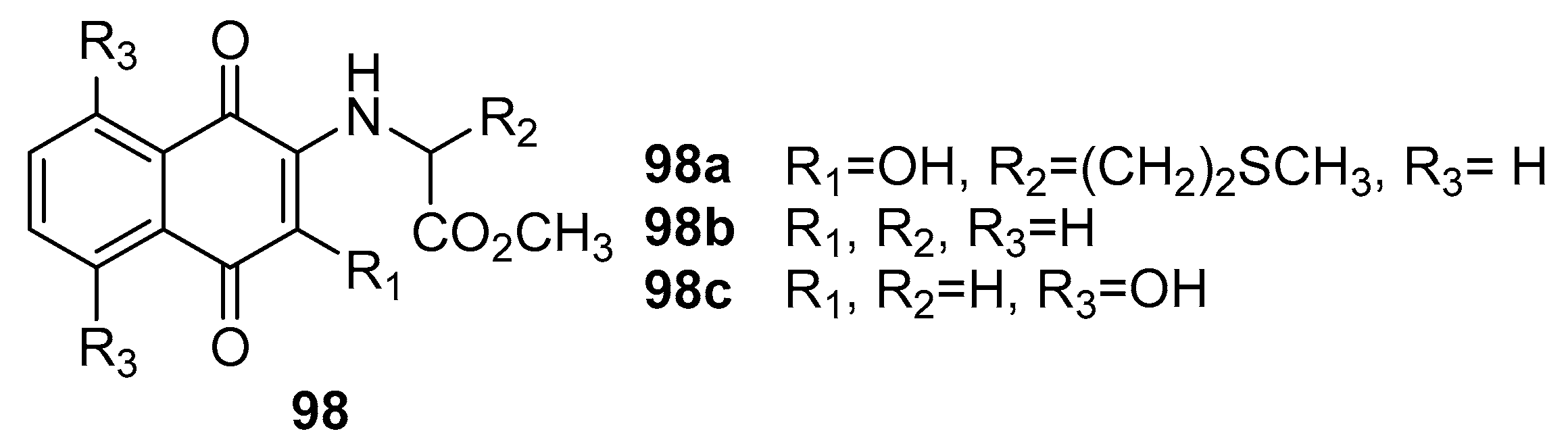
Figure 31.
1,4-naphthoquinone derivatives as antimicrobial agents.
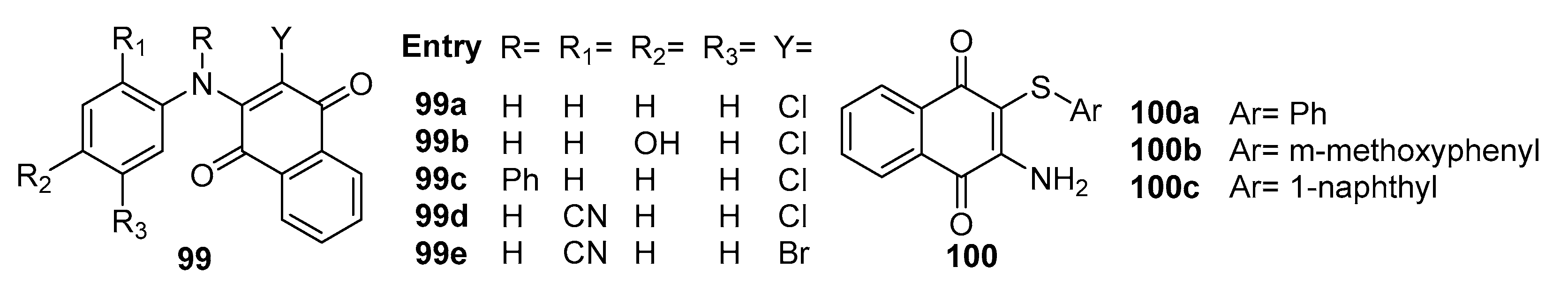
Figure 32.
Bis-naphthoquinone compounds as antiviral agents.
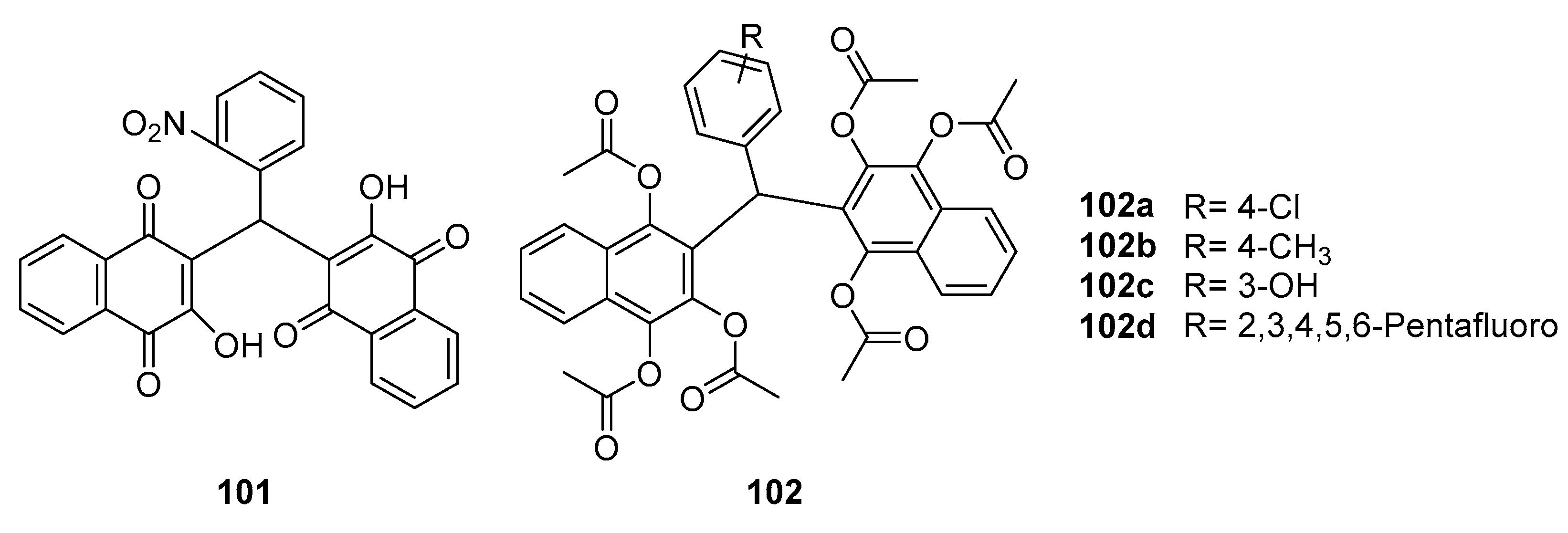
Figure 33.
Juglone derivatives as antiviral agents.
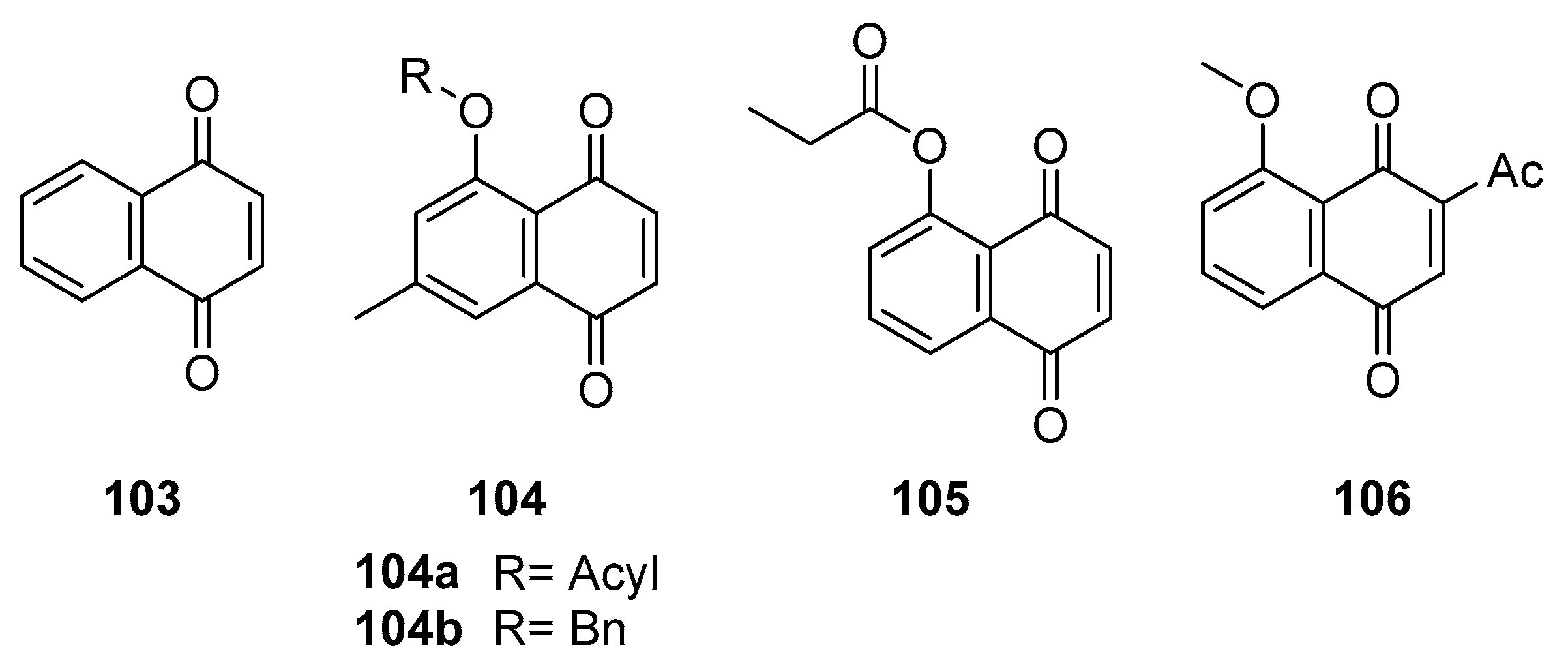
Figure 34.
1,4-naphthoquinone derivatives as antiviral agents.
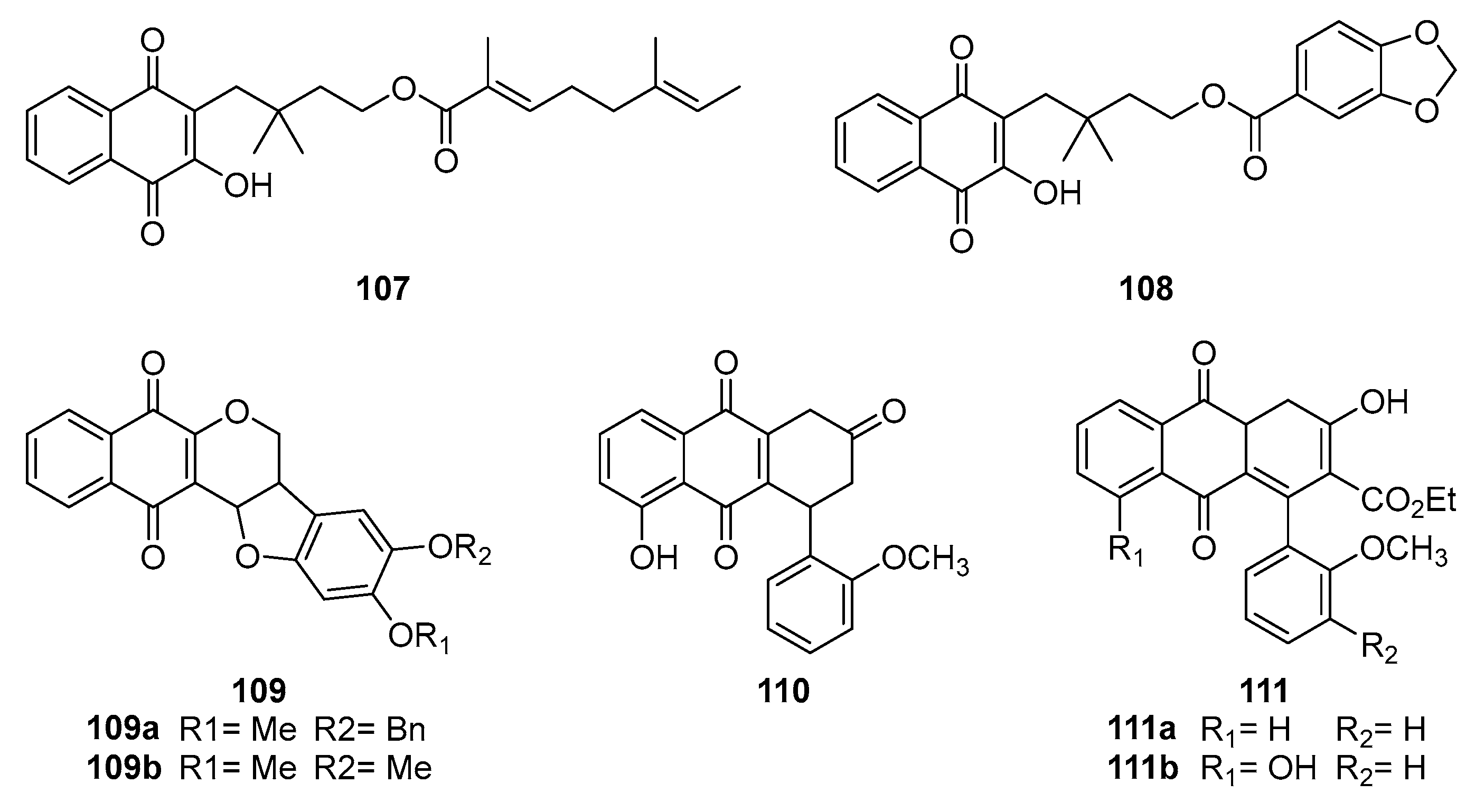
Figure 35.
1,4-naphthoquinone derivatives as antiviral agents.
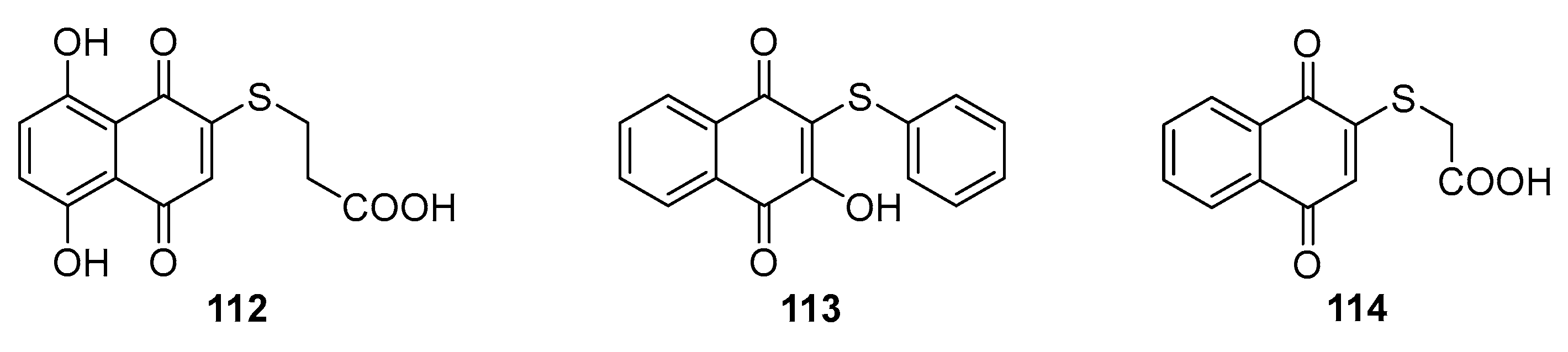
Figure 37.
1,4-naphthoquinones derivatives as antiviral agents.
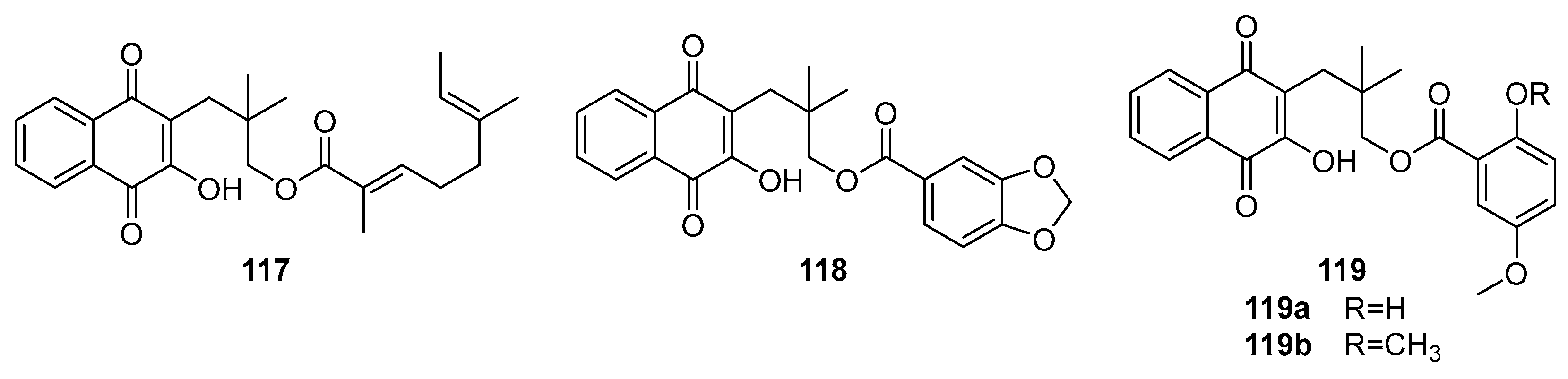
Figure 38.
1,4-naphthoquinones derivatives as antiviral agents.
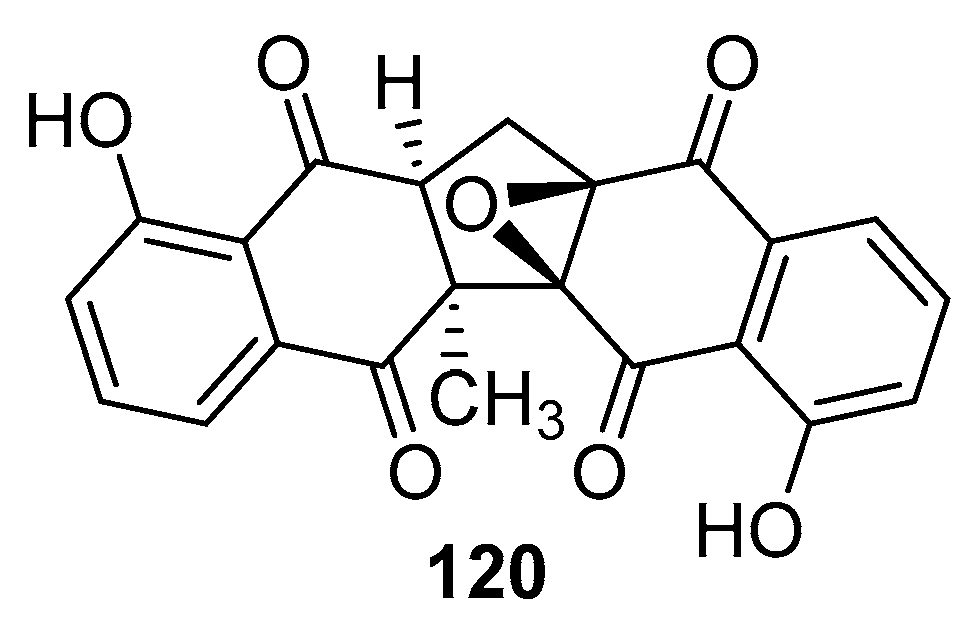
Figure 39.
Natural naphthoquinone compounds as antiviral agents.
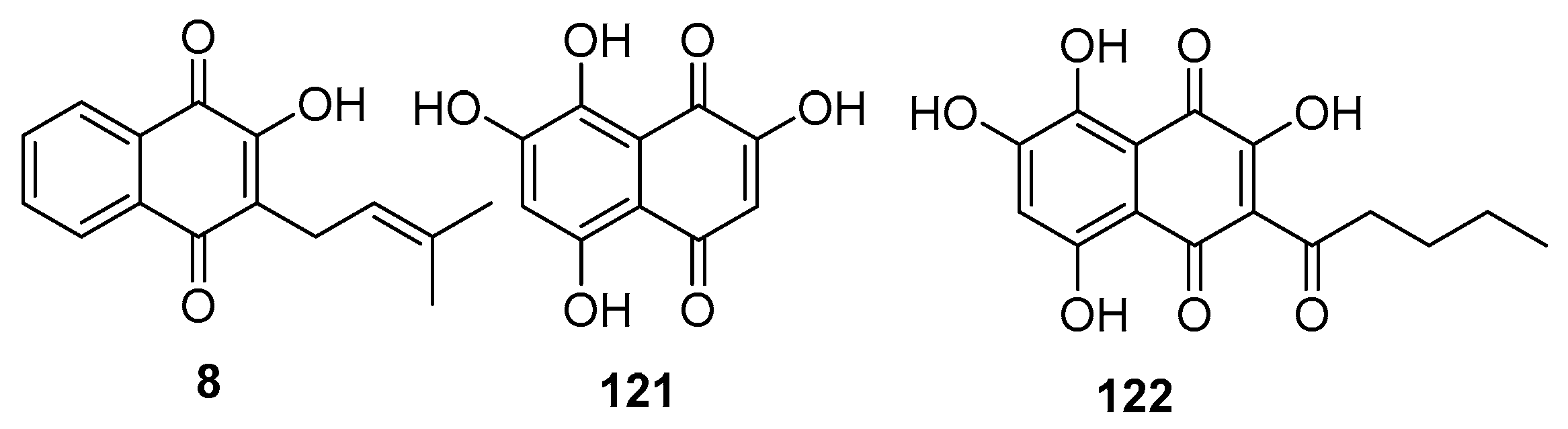
Figure 40.
1,4-Naphthaquinone compounds as antiviral agents.
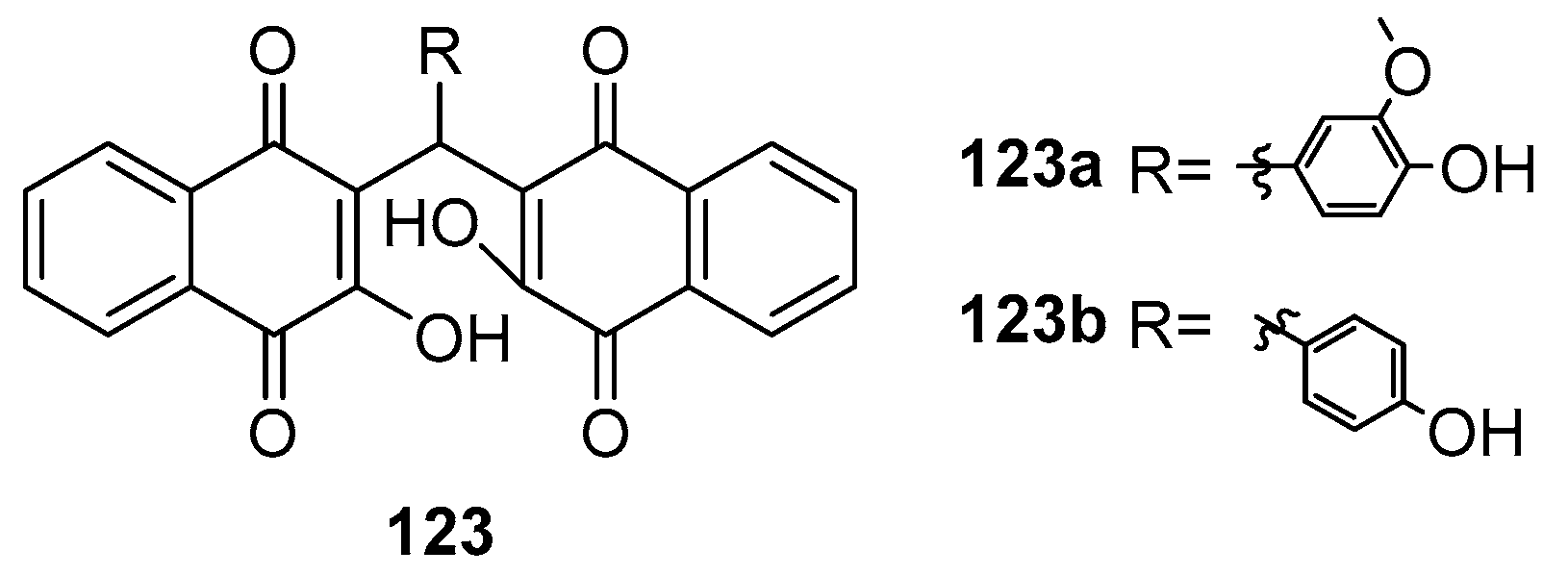
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



