Submitted:
01 October 2024
Posted:
01 October 2024
You are already at the latest version
Abstract
This review aims to describe the role of Limbic System-Associated Membrane Protein (LSAMP) in normal and pathophysiology and its potential implications in oncogenesis. We have summarized research articles reporting the role of LSAMP in the development of a variety of malignancies such as clear cell renal cell carcinoma, prostatic adenocarcinoma, lung adenocarcinoma, osteosarcoma, neuroblastoma, acute myeloid leukemia, and epithelial ovarian cancer. We also examine current understandings of how defects in LSAMP gene function may contribute to oncogenesis. Finally, this review discuss-es the implications of future LSAMP research and clinical applications.
Recent Findings LSAMP has been originally described as a surface adhesion glycoprotein expressed on cortical and subcortical neuronal somas and dendrites during the development of the limbic system. It is categorized as part of the IgLON immunoglobulin superfamily of cell-adhesion molecules and is involved in regulating neurite outgrowth and neural synapse generation. LSAMP is both aberrantly expressed and implicated in the development of neuropsychiatric disorders due to its role in the formation of specific neuronal connections within the brain. Additionally, LSAMP has been shown to support brain plasticity via the formation of neuronal synapses and is involved in modulating the hypothalamus in anxiogenic environments. In murine studies, loss of LSAMP expression was associated with decreased sensitivity to amphetamine, increased sensitivity to benzodiazepines, increased hyperactivity in new environments, abnormal social behavior, decreased aggressive behavior, and decreased anxiety. Findings have suggest-ed that LSAMP plays a role in attuning serotonergic activity as well as GABA activity. Given its importance to limbic system development, LSAMP has also been studied in the context of suicide.
In malignancies, LSAMP plays a significant role as a putative tumor suppressor, the loss of which leads to more aggressive phenotypes and mortality from metastatic disease. Loss of the LSAMP gene facilitates epithelial-mesenchymal transition, or EMT, where epithelial cells lose adhesion and gain the motile properties associated with mesenchymal cells. Additionally, LSAMP and the function of the RTK pathway have been implicated in tumorigenesis through the modulation of RTK expression in cell membranes and the activation of second messenger pathways and β-catenin.
Keywords:
Limbic System-Associated Membrane Protein (LSAMP)
; epithelial-mesenchymal transition (EMT)
; cell adhesion
; RTK pathway
1. Introduction
Prostate cancer is a common malignancy and one of the leading causes of cancer deaths [1]. Prostate cancer diagnosis rates and diagnoses with advanced disease are increasing. Prostate cancer mortality among patients of African ancestry is 2- to 4 -times higher than in patients of other ancestries [2]. The mortality of prostate cancer is largely due to inherited factors, and ancestry-associated germline mutations have been found to lead to more aggressive cancer and early disease progression [3]. In somatic contexts, genomic structural variations have been linked to the development of metastatic castration-resistant prostate cancer [4]. A growing body of evidence points to how the LSAMP gene functions in prostate cancer, as well as other cancers, and the role of LSAMP in tumor suppression.
2. History of LSAMP Research
Limbic system-associated membrane protein (LSAMP) is a surface glycoprotein expressed on cortical and subcortical neuronal somas and dendrites during the development of the limbic system [5]. LSAMP is categorized as part of the immunoglobulin-like family of cell-adhesion molecules IgLON, which also includes opioid- binding cell adhesion molecule OBCAM, neurotrimin, neuronal growth regulator 1, and IgLON5 [6]. LSAMP is involved with other members of the IgLON family in regulating neurite outgrowth and neural synapse generation [7].
LSAMP is aberrantly expressed in and is implicated in the development of neuropsychiatric disorders, especially schizophrenia, due to its normal function in the formation of specific neuronal connections within the brain [7]. Additionally, LSAMP has been shown to support brain plasticity via the formation of neuronal synapses, especially in the hippocampus, and is involved in modulating the hypothalamus in stressful or novel environments [5]. Studies in mice with wild- type, heterozygous, and homozygous loss of LSAMP expression have demonstrated that LSAMP loss results in decreased sensitivity to amphetamine with increased serotonin release and subsequently blunted serotonin turnover, as well as decreased serotonin at baseline compared with wild- type mice [8]. Loss of LSAMP in mice also resulted in hyperactivity in new environments, less aggressive behavior, less anxiety, and less whisker trimming, a marker of social dominance [7, 8]. Loss of LSAMP in mice increased the anxiolytic and hypnotic effects of benzodiazepines [7]. These findings suggest that LSAMP modulates both serotonergic activity via presynaptic serotonergic neurons in the brainstem, and GABA activity via modulation of GABAA receptor subunits [7, 8]. Lastly, LSAMP alterations have an association with suicide, which was investigated due to genomic structural differences in the limbic system among suicide victims [9]. Thus, the function of LSAMP as a cell-adhesion protein is important to understanding neural development in the limbic system [9, 10]. Beyond its many roles in the limbic system, LSAMP has been suggested to function as a tumor suppressor in early studies [11-13].
In malignancies, LSAMP may play a significant role as a tumor suppressor, the loss of which leads to more aggressive phenotypes and mortality from metastatic disease. Loss of LSAMP precipitates epithelial-mesenchymal transition (EMT), where epithelial cells lose adhesion and gain the motile properties associated with mesenchymal cells [6]. This transition rids cells of apical-basal polarity and allows them to break down extracellular matrix, lose adhesion, and migrate away from the basement membrane and into vasculature where they continue their migration [14]. The process of EMT permits formation of new structures in embryonal development, but also allows for metastasis when it occurs in the setting of oncogenesis [6]. The EMT that occurs in oncogenesis displays some differences from normal physiologic EMT, including in the EMT markers that cancer cells display, and that the cells themselves can be epithelial-like, less mesenchymal, often with incomplete transition [14]. The process of EMT in oncogenesis may imply that only a few cells, having reverted to a mesenchymal stem cell-like state, are required for tumor metastasis and invasion in a distant organ, consistent with the cancer stem cell phenotype [6]. These characteristics of LSAMP defects are evident in reports on clear cell renal cell carcinoma, lung adenocarcinoma, prostatic adenocarcinoma, osteosarcoma, neuroblastoma, acute myeloid leukemia, and epithelial ovarian cancer (Table 1).
LSAMP defects have been implicated in multiple cancers. These malignancies have poor outcomes affecting large numbers of patients. For example, renal cell carcinoma is typically discovered incidentally on imaging and incidence increases yearly, with over 81,610 new diagnoses and 14,390 deaths estimated for 2024 [1]. Familial RCC occurring due to inherited genetic mutations encompasses 2-8% of tumors, but up to 16% of advanced RCC demonstrate mutations in cancer-associated genes [15]. Lung cancer remains the leading cause of cancer death in the U.S. across genders, and while incidence is declining annually, only 1 in 4 patients will survive to five years [1]. Osteosarcoma is an aggressive cancer and the most common primary malignancy of bone, with a 65-66% 5-year survival in patients younger than 20 years [1]. Brain cancer is the leading cause of cancer death in children younger than 20 years, and 18% of patients with neuroblastoma will die within five years [1]. Acute myeloid leukemia will see an estimated 20,800 new cases and 11,220 deaths in 2024, and is the second most common leukemia in adults and children, with mortality and risk of invasive disease increasing with age [1]. Ovarian cancer is the leading cause of death from gynecologic cancer, where approximately 50% of patients present with disease that has already metastasized, and less than half of patients survive to five years [16]. Given the heavy mortality load of cancers of every type, identifying specific factors causing tumor aggression and decreased survival is crucial.
3. Renal Cell Carcinoma
Renal cell carcinoma comprises most primary renal neoplasms and develops sporadically from tubular epithelium in the renal cortex. RCC can arise in a nonhereditary fashion or can occur as part of an inherited syndrome such as Von Hippel-Lindau syndrome, Birt-Hogge-Dube syndrome, and tuberous sclerosis. Clear cell is the most common subtype of RCC and develops from the proximal renal tubule along with papillary RCC, while chromophobe RCC develops from the collecting tubule [17].
A three-hit model of oncogenesis has been described in renal cell carcinoma, similar to the two-hit model, where an initial genetic deactivation followed by subsequent epigenetic changes deactivating the second allele increases an individual’s susceptibility to developing cancer [17]. For individuals without an inherited mutation, two hits of genetic mutation, deletion, or silencing through epigenetic modification would be required to lead to tumorigenesis. For familial syndromes such as VHL, the initial inherited mutation of the VHL gene provides the first hit [18]. Inheritance of clear cell renal cell carcinoma (CCRCC) is primarily due to mutations in VHL, but chromosomal translocations on chromosome 3 have also been identified in both VHL-related RCC and non-VHL hereditary RCC [17, 18]. In the three-hit model, chromosomal translocation on chromosome 3 provides the first hit, loss of VHL on the derivative chromosome 3 provides the second hit, and a mutation of the second allele of VHL provides the third hit [17]. However, many of the hereditary forms of RCC which displayed a translocation and subsequent loss of the derivate chromosome 3 allele did not have VHL mutations [18]. The translocations implicated in the development of these hereditary forms of RCC span multiple different genes, all of which are located on chromosome 3 [18]. The literature reports eight chromosomal translocation families, which as in VHL are all linked to chromosome 3, suggesting that these translocation families might contribute to the development of CCRCC in ways similar to VHL [18]. One of the genes located on chromosome 3 involved in a translocation implicated in hereditary CCRCC, is LSAMP [18].
In CCRCC, the LSAMP gene located on at 3q13.3 has been identified as part of a translocation associated with the development of CCRCC. LSAMP spans the chromosome 3 involved in a t(1;3) (q32.1;q13.3) translocation. This t(1;3) translocation increases predisposition to loss of the derivative chromosome 3, which causes the loss of one of the copies of a tumor suppressor gene, or the “second hit” in the three-hit model [18]. Further genetic or epigenetic alterations to a gene such as LSAMP leads to CCRCC tumor development [18]. In an analysis of nine CCRCC cell lines, 53 sporadic tumors, and four familial tumors, no mutations in LSAMP itself were discovered, but downregulation of LSAMP expression was observed in nearly all the tumor samples [18]. Epigenetic silencing due to LSAMP promoter methylation was discovered in 78% of CCRCC cell lines, 26% of sporadic tumors, and 100% of familial tumors, and expression of LSAMP increased following application of DNA methyl transferase inhibitors [18]. Additionally, loss of heterozygosity of the LSAMP locus was found to be significantly associated with tumor development and occurred independently of LSAMP promotor methylation [18].
In RCC cell line models lacking LSAMP expression, activations or re-expression of LSAMP resulted in the suppression of tumor growth. Plasmids expressing green fluorescent protein EGFP-LSAMP were introduced to two RCC cell lines containing methylated LSAMP promotors, and these cells were allowed to proliferate along with control cells expressing green fluorescent protein EGFP alone. While cells expressing EGFP alone grew like non-transfected cells, EGFP-LSAMP transfected cells showed significant reduction in cell growth, and these cells did not undergo apoptosis [18].
4. Prostatic Adenocarcinoma
The disparity in prostate cancer mortality between American men of African Ancestry (AA) and those of Caucasian Ancestry (CA) has been highlighted in numerous studies. Although to a lesser extent, prostate cancer disparities are also apparent in equal-access healthcare settings arguing for the examination of biologic and genetic factors. While the presence of commonly observed ERG and PTEN genomic defects in prostate tumor genomes has been highlighted in previous studies, these studies were conducted primarily on datasets of tumors from patients of CA [3]. The inactivation of LSAMP has been understood as a cancer genomic event via deletion involving the LSAMP locus at chromosome 3q13.31 that occurs at significantly higher rates in tumors of patients of AA than CA [3]. Deletions involving the LSAMP locus were reported in 27% of AA compared with 13% of CA tumors in tumor copy number variation data, and in 26% of AA compared with 7% of CA patients by Fluorescence in situ Hybridization Assays (FISH) on tumor tissue microarrays [3]. Further, the association of LSAMP deletion with rapid disease progression suggests that detecting LSAMP deletions may serve as a predictor of disease progression in patients of AA [3]. The chromosomal mapping of prostate tumor DNA breakpoints in a recent report pinpointed the LSAMP locus and its association with localized and metastatic castration-resistant prostate cancers [4].
The LSAMP homologue, OPCML, a tumor suppressor and cell adhesion protein and member of the IgLON family, regulates downstream receptor tyrosine kinases affecting the extracellular signal-regulated kinase (ERK), as well as β-catenin through the Akt signaling pathway [19]. In prostate cancer cell culture model experiments the absence of LSAMP affected RTKs and activated both the ERK and Akt pathways and downstream β-catenin through RAS signaling [20]. This is one of the canonical pathways in receptor tyrosine kinase to RAS-to-RAF activity that affects cell adhesion in cancer context [21]. Parallel to RAS activation, Akt can be activated by focal adhesion kinase FAK in response to integrin signals [22, 23]. Taken together, LSAMP deletion and subsequent alteration of integrins may promote loss-of-adhesion of the cell to the basement membrane in known pro-metastatic pathways associated with disease progression (Figure 1). Thus, normal expression of LSAMP may act as a stabilizer of cell-to-basement membrane adhesion.
5. Lung Adenocarcinoma
Lung cancer remains the primary cause of cancer death worldwide and lung cancer mortality is marked by aggressive metastasis, which occurs due to the EMT mechanism and subsequent mesenchymal-epithelial transition, or MET, once the cancer cells reach their new organ [14]. LSAMP alterations have been reported in the context of lung cancer progression [6]. In an LSAMP gene expression study, a small set of lung adenocarcinoma samples was compared with non-malignant control samples suggesting that LSAMP was expressed at lower levels in the cancer tissue [6]. LSAMP, more so than any other member of the IgLON family, was associated with cancer aggressiveness where lower levels of LSAMP expression resulted in increased nodal metastasis and more advanced tumor stage [6]. Survival analyses demonstrated that decreased expression of LSAMP was associated with shorter survival time [6]. Other members of the IgLON family did not significantly affect survival in this study. Hypermethylation, copy number variation, and microRNA post-translational mechanisms were also investigated as possible regulatory mechanisms of LSAMP expression in lung adenocarcinoma. Copy number variation and microRNA appeared to be unlikely contributors to LSAMP regulation, but methylation of the LSAMP promotor resulted in decreased copies of LSAMP mRNA, increased nodal metastasis, and more advanced tumor stages [6]. The role of LSAMP in the epithelial mesenchymal transition EMT was also investigated. In cell culture models, markers of EMT were found to be modified in response to silencing of LSAMP via short hairpin RNA [6]. Expression of epithelial marker E-cadherin decreased, while mesenchymal markers such as N-cadherin, vimentin, alpha-smooth muscle actin, and EMT-associated zinc finger proteins Snail and Slug, increased [6].
6. Osteosarcoma
Osteosarcoma is an aggressive malignancy and a common primary bone tumor. Osteosarcomas are most frequently found in children, with a smaller spike seen in ages over 50 years, and they are associated with poor prognosis due to their high grade at diagnosis and frequent metastases [11]. Osteosarcomas develop at sites of bone growth, typically at the metaphysis, and most commonly around the knee at the proximal tibia, proximal humerus, or distal femur [11].
LSAMP has been investigated for its tumor-suppressive role in osteosarcoma and was reported in several studies. A dataset of 36 samples of human osteosarcoma tumors and 20 osteosarcoma cell lines was analyzed for DNA copy number changes, from which gains or losses of chromosomal regions were assessed [11]. The chromosomal locus 3q.13.31, harboring LSAMP, had a deletion rate of 56% in the tumor samples [11]. Another study on a dataset of 49 tumor biopsy tissues and DNA samples also identified locus 3q13.31 as the most common deletion in osteosarcoma, and analysis of 43 tumor biopsies with the addition of 5 osteosarcoma cells lines demonstrated decreased LSAMP expression in over 64% of cases [13]. A subsequent study further investigating LSAMP on chromosomal region 3q13.31 found reduced copy number of LSAMP in 59% of osteosarcoma samples and reduced expression of LSAMP in 60% of samples [24]. Analysis of a database of single-cell osteosarcoma messenger RNA expression data confirmed aberrant transcription levels in osteoblasts and in carcinoma-associated fibroblasts, significantly more than in other cell types [25]. Methylation of the LSAMP promoter region was suggested as the epigenetic mechanism contributing to decreased expression of LSAMP in osteosarcoma, as in RCC and lung adenocarcinoma. From a total number of tumor samples and cell lines that showed either partial or full methylation of the LSAMP promoter, 60% had subsequent decreased expression of LSAMP [11].
LSAMP expression has also been shown to affect tumor growth and proliferation in osteosarcoma. Decreased expression of LSAMP via small interfering RNA resulted in enhanced proliferation of osteoblast cells compared with controls [13]. When comparing cell lines with and without LSAMP expression, the presence of LSAMP was significantly associated with a 15-20% reduction in subsequent cell growth [24]. Further, LSAMP expression in mouse osteosarcoma models resulted in significant delay in tumorigenesis. In these osteosarcoma models, survival analyses showed association of decreased LSAMP mRNA expression with decreased survival [11]. Mechanistically, suppression of LSAMP by siRNA increased transcription of cyclin A2 and cyclin B1, pointing to a role of LSAMP in affecting the cell cycle [13].
7. Neuroblastoma
Neuroblastoma is a primarily pediatric neuroendocrine malignancy that develops from neural crest precursor cells. Structural alterations in LSAMP at chromosomal region 3q13.31 have been found in whole genome sequencing studies of relapsed and high-risk neuroblastoma [26]. Heterozygous rearrangements in 3q13.31 at the region of LSAMP were present in 17% of tumor samples, while 43% of neuroblastoma cell lines displayed segmental chromosomal alterations in LSAMP [26]. When LSAMP expression was silenced via short hairpin RNA, significantly increased cell growth was noted in neuroblastoma cell lines [26]. Following transfection with LSAMP in cell lines with heterozygous and homozygous LSAMP deletions, a significant decrease in growth was observed [26]. For a dataset of 498 neuroblastoma tumors, decreased LSAMP expression was significantly associated with decreased event-free survival and overall survival [26]. In neuroblastoma, LSAMP, as in RCC and osteosarcoma, appears to function as a tumor suppressor associating with increased survival.
8. Acute Myeloid Leukemia
LSAMP defects have been identified in acute myeloid leukemia (AML), specifically in the cytogenetic category of AML associated with either t(8;21) or inv(16) referred to as core-binding factor-AML, or CBF-AML [27]. CBF-AML has a good prognosis in both adult and pediatric populations, but most adult patients, and up to 20% of pediatric patients will eventually experience relapse [27]. Of 39 bone marrow samples of CBF-AML that relapsed, matched with samples taken at diagnosis, deletion of chromosomal region 3q13.31 that includes LSAMP was the most common copy number alteration, occurring in 12% [27]. The larger deletions associated with relapses were all shown to include deletion of LSAMP, and the minimally deleted region associated with relapse samples was found to contain exons of LSAMP [27].
9. Ovarian Epithelial Carcinoma
Ovarian epithelial cancer is the most common type of ovarian cancers and is a leading cause of ovarian cancer mortality [28]. It is not well understood where ovarian epithelial cancers originate, but an increasing number of reports suggests it originates in the epithelial lining of the uterine tubes [29]. Physiologically normal ovarian epithelium is a simple squamous to simple cuboidal epithelium, but unlike true epithelial tissue, exhibits attributes of mesenchymal cells [28, 30]. Ovarian epithelium displays more fragile adherence to the basement membrane than in typical epithelium, minimal expression of E-cadherin except in metaplasia and other instances where the epithelium becomes more columnar, and characteristics that support repair of the ovarian surface following follicular rupture, including contractile capacity, motility, and proliferative capability [30]. These attributes give ovarian epithelium similarities to the process of EMT underlying the role of LSAMP defects in tumorigenesis and indicate that ovarian epithelium is not well-differentiated and prone to malignant transformation [28, 30].
LSAMP has been linked to ovarian cancer, similar to the IgLON family member OBCAM, where the pathophysiology of the disease is better understood. To some extent, LSAMP expression was found to be reduced in all types of epithelial ovarian cancers but to a statistically significant level it was linked to the endometrioid subtype [28]. Analysis of cancer genomic copy number variations in serous ovarian cancer identified focal losses of LSAMP in malignant, but not in normal tissues [31]. In studies on OBCAM, the presence of OBCAM was shown to have a tumor suppressive effect via extracellular binding to receptor tyrosine kinases and subsequent attenuation of downstream pathways, specifically, inhibition of phosphorylation of ERK1, ERK2, and Akt [32]. Introduction of OBCAM in ovarian cancer cell culture models resulted in significantly decreased growth [32]. In mice, intraperitoneal injections of OBCAM-expressing malignant cells resulted in significantly decreased rate of tumorigenesis, decreased development of ascites, and decreased peritoneal extension of ovarian cancer cell lines, compared with controls [32]. These data are consistent with findings showing that in prostate cancer cell modulation of the receptor tyrosine kinase pathways is a potential mechanism by which LSAMP enacts its tumor suppressive activity [20].
Interestingly, high levels of LSAMP expression were seen in moderately-differentiated and well-differentiated ovarian cancers but were associated with decreased overall survival. Thus, measuring the levels of LSAMP expression may help prognosing a poor outcome beyond the morphological differentiation status [28]. The reason for this is not well understood at this time but it is possibly related to the role of LSAMP in the physiologically normal ovary, and that normal ovarian epithelium is not well-differentiated compared to other epithelial tissues.
10. Discussion
LSAMP is a cell surface adhesion molecule originally known for its role in the limbic system and in the development of neuropsychiatric disorders. LSAMP was shown to have tumor suppressive activity in a variety of cancers, first in renal cell carcinoma where the LSAMP gene was shown to play a part in the three-hit model, similar to VHL, for hereditary clear cell RCC [17, 18]. In RCC, LSAMP was shown to suppress growth in vitro and that suppression was reversed with a DNA methylation inhibitor [18]. Studies of ovarian epithelial carcinoma reported that LSAMP was reduced in tumor samples and was a prognostic factor, associated with decreased survival [28]. Osteosarcoma studies also linked LSAMP to worsened survival and found that heterologous expression of LSAMP delayed tumor growth in vivo [24]. In neuroblastoma, LSAMP suppressed growth in vitro and was again associated with worsened survival [26]. Studies in AML found decreased expression of LSAMP in relapsed cases [27]. Prostatic adenocarcinoma studies linked LSAMP aberration with disease progression, found more frequently in AA patients [3]. Across disease stages, breakpoints in the LSAMP region have emerged among structural variation patterns including metastatic castration-resistant prostate cancers [4]. It has been suggested that LSAMP functions by downregulating RTKs and downstream oncogenic signaling [20]. There are various mechanisms of LSAMP downregulation in several cancers, by genomic rearrangements, most frequently deletions, epigenetic silencing, or potentially post-transcriptional or post-translational inactivation, each of which may be related to predisposition to disease progression. Current data unanimously support the notion that downregulation of LSAMP is associated with oncogenesis in multiple cancer types, as well as worse outcomes for patients. Notably, detection of LSAMP loss has potential to detect clinically significant malignancies. Additionally, the molecular mechanism by which LSAMP functions as a tumor suppressor gene by modulating known oncogenic pathways may provide new opportunities for treating patients with LSAMP associated malignancies.
The goal of our review was to summarize published studies that investigate the role of LSAMP deletion or decreased expression in tumorigenesis, clinical and biological changes associated with LSAMP gene defects (Table 2). Emerging data underscore that LSAMP losses in the setting of cancer can increase expression of receptor tyrosine kinases on the surface of the cell, leading to increased activation of tumorigenic ERK and Akt pathways and increased activation of β-catenin. Akt is known to activate β-catenin via phosphorylation while deactivating GSK3, which inhibits the activity of β-catenin at baseline in the resting cell [33]. ERK is activated via the RAS/MAPK pathway, and then goes on to activate downstream pathways involved in cell proliferation, growth, and survival, including transcription factors and C-MYC as reported in AML [34].
The mechanism of action of LSAMP is likely context dependent. The Drosophila analog of IgLON, or Amalgam (Ama), was studied in a glia cell model and was found to function as a modulator of the receptor tyrosine kinase pathway [35]. However, Ama in glial cells was shown to act indirectly to increase receptor tyrosine kinase activity and downstream ERK signaling [35]. A knockdown of LSAMP in this context was subsequently performed, and in the same vein as Ama, increased expression of RTK pathway inhibitor SPROUTY2 was observed [35]. Effectively, the drosophila Ama, and LSAMP, worked in this model as an inhibitor of the inhibitor. Moreover, in astroglia cells, expression of other members of the IgLON family increased FGF signaling [35]. This contrasts with the currently understood action of LSAMP in other human adenocarcinoma models and the proposed suppressor role of LSAMP among other human malignancies where LSAMP expression downregulates the RTK pathway and suppresses tumor growth. The differences between the mechanism of drosophila Ama versus human LSAMP suggests a context dependent interaction with downstream RTK signaling.
A clinically important question that begun to emerge from reports on LSAMP is the potential association between LSAMP deletion in malignancies and ancestry. A study conducted on analyses of 435 prostate cancer patients established a significant association between defects in the LSAMP gene and tumors of African ancestry [3]. In general, variations across ancestry have been established in some, but not all, types of cancer. In neuroblastoma no association between ancestry and 5-year cause-specific survival was found in a study of 2119 pediatric neuroblastoma patients (age less than 18 years at diagnosis) [36]. Malignancies discussed in this review that have known associations with patient ancestry include renal cell carcinoma, lung adenocarcinoma, osteosarcoma, and AML. While Chen et al. did not stratify their results by ancestry, data show decreased survival rates in AA patients for every subtype of renal cell carcinoma [1]. Alaiwi et al. report that of 1,829 patients with RCC who were provided germline genetic testing, pathogenic or likely pathogenic variants in the FH gene were more common in AA patients, while the CHEK2 gene was more common in non-African patients [37]. The study authors note that of the 1,829 patient sample size, only 99 were of African descent, highlighting a need for larger studies of minority populations to lend more power to genetic associations [37].
Strong associations with ancestry can be seen in multiple genes implicated in lung adenocarcinoma. A study on the genomic sequencing data of 6,238 lung adenocarcinoma patients included CA, AA, American Indian or Alaska Native (AI/AN), Asian, and other descents (Native Hawaiian/Pacific Islander, Hispanic/Latino, and interracial or multiracial) [38]. The distribution of ancestry in the adenocarcinoma study, as in RCC, encompassed mostly CA patients (84.89% CA, 8.64% Asian, 4.81% AA, 0.16% AI/AN, and 1.50% Other) [38]. The data demonstrated that point mutations in EGFR were three times higher in Asian patients than in others, and mutations in KRAS were significantly higher in CA, followed by AA, Other, AI/AN, and lastly Asian. The study did not evaluate for ancestry associations with LSAMP, while Chang et al. found decreased LSAMP expression in cell lines and tumor samples but did not correlate their findings with ancestry.
Studies on osteosarcoma, AML, and ovarian cancer have reported significant differences between ancestry but did not correlate findings with changes in LSAMP. A sample of 537 osteosarcoma tumors demonstrated a 15% higher incidence among AA patients and an 11% higher incidence among patients of Hispanic ancestry (HA) [39]. In a sample of 814 pediatric patients (age less than 21 years at diagnosis) with acute myeloid leukemia, AA and HA were more likely to have AML with t(8;21) [40]. Compared with CA and HA, AA patients had significantly higher risk of events, higher risk of death, and higher rates of early death within fifty days of diagnosis [40]. Among patients with CBF-AML, AA patients had the lowest event free survival and the highest rates of early death [40]. Interestingly, the authors report that despite the favorable prognosis of CBF-AML, outcomes for African American patients with CBF-AML were similar to those of AML with higher risk cytogenetics [40]. In a study of 91,625 epithelial ovarian cancer patients, African American and Japanese American patients had a lower risk of developing cancer after adjusting for multiple variables [41]. The authors found no associations of endothelial ovarian cancer with age at menarche, obesity, smoking status, or diabetes among any ancestries [41]. However, the study only investigated associations with hormonal factors, and did not investigate genetic variations across ancestry. Genetic explanations for these findings in osteosarcoma, AML, and ovarian cancer remain unclear. In summary, discerning associations between ancestry and LSAMP-driven malignancies appears to be an exciting future direction.
11. Conclusions
Limitations of this review include variation in methodology across the included publications. The studies reviewed focused on different endpoints associated with LSAMP’s involvement in oncogenesis, including nodal metastasis, tumor stage, tumor aggressiveness, or rates of cell line growth. Additionally, differences by ancestry have been found in most cancers, but studies vary in investigating correlations between genomic changes and ancestry. Cancers such as RCC, prostatic adenocarcinoma, lung adenocarcinoma, and AML have defined genetic risk factors for the disease, and cancers such as RCC, prostatic adenocarcinoma, osteosarcoma, AML, and ovarian have significant differences in disease outcome between ancestries. RCC, prostatic adenocarcinoma, AML, and ovarian cancer have stratified genetic risk factors by ancestry, but other cancers have not investigated the role of LSAMP or other significant factors in the context of the ancestries of patients. Clinical applications of LSAMP’s function to diagnostics, prognostics, and therapies will require continued investigation. Further research needs to be done to determine how integrin signaling contributes to adherence in the presence and absence of LSAMP. Broader implications of these findings include development of treatment therapies targeted to tumors harboring defective LSAMP that will allow more informed risk and perhaps therapeutic stratification of patients.
12. Future Directions
The clinical application of LSAMP has potential in diagnostics, prognostics, risk stratification, and development of treatment therapies. Further research will determine how LSAMP influences cell adherence in the presence of RTK and integrin signaling. Additionally, investigating associations between ancestry and LSAMP-driven malignancies appears to be an exciting future direction.
References
- Siegel, R.L., A. N. Giaquinto, and A. Jemal, Cancer statistics 2024, CA Cancer J Clin 2024, 74, 12–49. [Google Scholar]
- Schaeffer, E.M. , et al. , Prostate Cancer, Version 4.2023, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2023, 21, 1067–1096. [Google Scholar] [PubMed]
- Petrovics, G. , et al. , A novel genomic alteration of LSAMP associates with aggressive prostate cancer in African American men. EBioMedicine 2015, 2, 1957–1964. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M. , et al. JCI Insight 2022, 7. [Google Scholar]
- Heinla, I. , et al. , Gene expression patterns and environmental enrichment-induced effects in the hippocampi of mice suggest importance of Lsamp in plasticity. Front Neurosci 2015, 9, 205. [Google Scholar] [CrossRef]
- Chang, C.Y. , et al. J Pers Med 2021, 11. [Google Scholar]
- Bregin, A. , et al. , Expression and impact of Lsamp neural adhesion molecule in the serotonergic neurotransmission system. Pharmacol Biochem Behav 2020, 198, 173017. [Google Scholar] [CrossRef]
- Innos, J. , et al. , Lsamp⁻/⁻ mice display lower sensitivity to amphetamine and have elevated 5-HT turnover. Biochem Biophys Res Commun 2013, 430, 413–418. [Google Scholar]
- Must, A. , et al. , Association of limbic system-associated membrane protein (LSAMP) to male completed suicide. BMC Med Genet 2008, 9, 34. [Google Scholar] [CrossRef]
- Sokolowski, M., J. Wasserman, and D. Wasserman, Polygenic associations of neurodevelopmental genes in suicide attempt. Mol Psychiatry 2016, 21, 1381–1390. [Google Scholar] [CrossRef]
- Kresse, S.H. , et al. , LSAMP, a novel candidate tumor suppressor gene in human osteosarcomas, identified by array comparative genomic hybridization. Genes Chromosomes Cancer 2009, 48, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.C. , et al. , Identification of chromosomal aberrations associated with disease progression and a novel 3q13.31 deletion involving LSAMP gene in osteosarcoma. Int J Oncol 2009, 35, 775–788. [Google Scholar] [CrossRef]
- Pasic, I. , et al. , Recurrent focal copy-number changes and loss of heterozygosity implicate two noncoding RNAs and one tumor suppressor gene at chromosome 3q13.31 in osteosarcoma. Cancer Res 2010, 70, 160–171. [Google Scholar] [CrossRef]
- Banyard, J. and D. R. Bielenberg, The role of EMT and MET in cancer dissemination. Connect Tissue Res 2015, 56, 403–413. [Google Scholar] [CrossRef]
- Bratslavsky, G. , et al. , Genetic risk assessment for hereditary renal cell carcinoma: Clinical consensus statement. Cancer 2021, 127, 3957–3966. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.K. , et al. , NCCN Guidelines® Insights: Ovarian Cancer, Version 3.2022. Journal of the National Comprehensive Cancer Network : JNCCN 2022, 20, 972–980. [Google Scholar]
- Bodmer, D. , et al. , Understanding familial and non-familial renal cell cancer. Hum Mol Genet 2002, 11, 2489–2498. [Google Scholar] [CrossRef]
- Chen, J. , et al. , The t(1;3) breakpoint-spanning genes <em>LSAMP</em> and <em>NORE1</em> are involved in clear cell renal cell carcinomas. Cancer Cell 2003, 4, 405–413. [Google Scholar]
- Antony, J. , et al. Cancer Gene Ther 2021, 28, 18–26. [Google Scholar] [CrossRef]
- Babcock, K. , et al., Abstract 5305, Reexpression of LSAMP, a gene frequently deleted in African American prostate cancers, suppresses tumor growth and β-catenin activity. Cancer Research 2019, 79 (13_Supplement), 5305–5305. [Google Scholar] [CrossRef]
- Götz, R. , Inter-cellular adhesion disruption and the RAS/RAF and beta-catenin signalling in lung cancer progression. Cancer Cell Int 2008, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Velling, T. , et al. , beta1-Integrins induce phosphorylation of Akt on serine 473 independently of focal adhesion kinase and Src family kinases. EMBO Rep 2004, 5, 901–905. [Google Scholar] [PubMed]
- Pavlakis, E. , et al. , Mutant p53-ENTPD5 control of the calnexin/calreticulin cycle: a druggable target for inhibiting integrin-α5-driven metastasis. J Exp Clin Cancer Res 2023, 42, 203. [Google Scholar] [CrossRef]
- Barøy, T. , et al. , Reexpression of LSAMP inhibits tumor growth in a preclinical osteosarcoma model. Mol Cancer 2014, 13, 93. [Google Scholar] [CrossRef]
- Feleke, M. , et al. , Single-cell RNA-seq identification of four differentially expressed survival-related genes by a TARGET: Osteosarcoma database analysis. Exp Biol Med (Maywood) 2022, 247, 921–930. [Google Scholar] [CrossRef]
- Martinez-Monleon, A. , et al. 31 chromosomal rearrangement indicates LSAMP as a tumor suppressor gene in neuroblastoma. Int J Oncol 2023, 62. [Google Scholar]
- Kühn, M.W.M. , et al. , High-resolution genomic profiling of adult and pediatric core-binding factor acute myeloid leukemia reveals new recurrent genomic alterations. Blood 2012, 119, e67–e75. [Google Scholar] [CrossRef] [PubMed]
- Ntougkos, E. , et al. , The IgLON family in epithelial ovarian cancer: expression profiles and clinicopathologic correlates. Clin Cancer Res 2005, 11, 5764–5768. [Google Scholar] [CrossRef]
- Nik, N.N. , et al. , Origin and pathogenesis of pelvic (ovarian, tubal, and primary peritoneal) serous carcinoma. Annu Rev Pathol 2014, 9, 27–45. [Google Scholar] [CrossRef]
- Auersperg, N. , et al. , Ovarian surface epithelium: biology, endocrinology, and pathology. Endocr Rev 2001, 22, 255–288. [Google Scholar]
- Zhang, L. , et al. , Identification of recurrent focal copy number variations and their putative targeted driver genes in ovarian cancer. BMC Bioinformatics 2016, 17, 222. [Google Scholar]
- McKie, A.B. , et al. , The OPCML tumor suppressor functions as a cell surface repressor-adaptor, negatively regulating receptor tyrosine kinases in epithelial ovarian cancer. Cancer discovery 2012, 2, 156–171. [Google Scholar] [CrossRef] [PubMed]
- Fang, D. , et al. , Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem 2007, 282, 11221–11229. [Google Scholar] [PubMed]
- Papa, S., P. M. Choy, and C. Bubici, The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene 2019, 38, 2223–2240. [Google Scholar] [CrossRef]
- Ariss, M.M. , et al. J Cell Sci 2020, 133. [Google Scholar]
- Farouk, F.S. , et al. , The Association between Race and Survival among Pediatric Patients with Neuroblastoma in the US between 1973 and 2015. Int J Environ Res Public Health 2020, 17. [Google Scholar] [CrossRef]
- Abou Alaiwi, S. , et al. , Trans-ethnic variation in germline variants of patients with renal cell carcinoma. Cell Rep 2021, 34, 108926. [Google Scholar] [CrossRef]
- Shi, H. , et al. , Genomic landscape of lung adenocarcinomas in different races. Front Oncol 2022, 12, 946625. [Google Scholar] [CrossRef]
- Zhang, C. , et al. , Common genetic variation and risk of osteosarcoma in a multi-ethnic pediatric and adolescent population. Bone 2020, 130, 115070. [Google Scholar] [CrossRef]
- Conneely, S.E. , et al. , Association of race and ethnicity with clinical phenotype, genetics, and survival in pediatric acute myeloid leukemia. Blood Adv 2021, 5, 4992–5001. [Google Scholar] [CrossRef]
- Sarink, D. , et al. , Racial/Ethnic Differences in Ovarian Cancer Risk: Results from the Multiethnic Cohort Study. Cancer Epidemiol Biomarkers Prev 2020, 29, 2019–2025. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The contents of this publication are the sole responsibility of the author(s) and do not necessarily reflect the views, opinions, or policies of Uniformed Services University of the Health Sciences (USUHS), the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., the Department of Defense (DoD) or the Departments of the Army, Navy, or Air Force. Mention of trade names, commercial products, or organizations does not imply endorsement by the U.S. Government. |
Figure 1.
LSAMP in prostate cancer. The loss of LSAMP in the cell membrane upregulates RTK signaling and downstream ERK and Akt pathways. Deletion in LSAMP result in the loss of adhesion to normal extracellular matrix elements resulting in the detachment of a nest of tumor cells from the primary tumor site and attachment to lymphatic or vascular endothelium where the LSAMP-deleted cancer cells spread to distant sites, such as lymph node and bone. Abbreviations: ERK, extracellular signal-regulated kinase; FAK, focal adhesion kinase; MEK, mitogen-activated protein kinase; RTK, receptor tyrosine kinase.
Figure 1.
LSAMP in prostate cancer. The loss of LSAMP in the cell membrane upregulates RTK signaling and downstream ERK and Akt pathways. Deletion in LSAMP result in the loss of adhesion to normal extracellular matrix elements resulting in the detachment of a nest of tumor cells from the primary tumor site and attachment to lymphatic or vascular endothelium where the LSAMP-deleted cancer cells spread to distant sites, such as lymph node and bone. Abbreviations: ERK, extracellular signal-regulated kinase; FAK, focal adhesion kinase; MEK, mitogen-activated protein kinase; RTK, receptor tyrosine kinase.
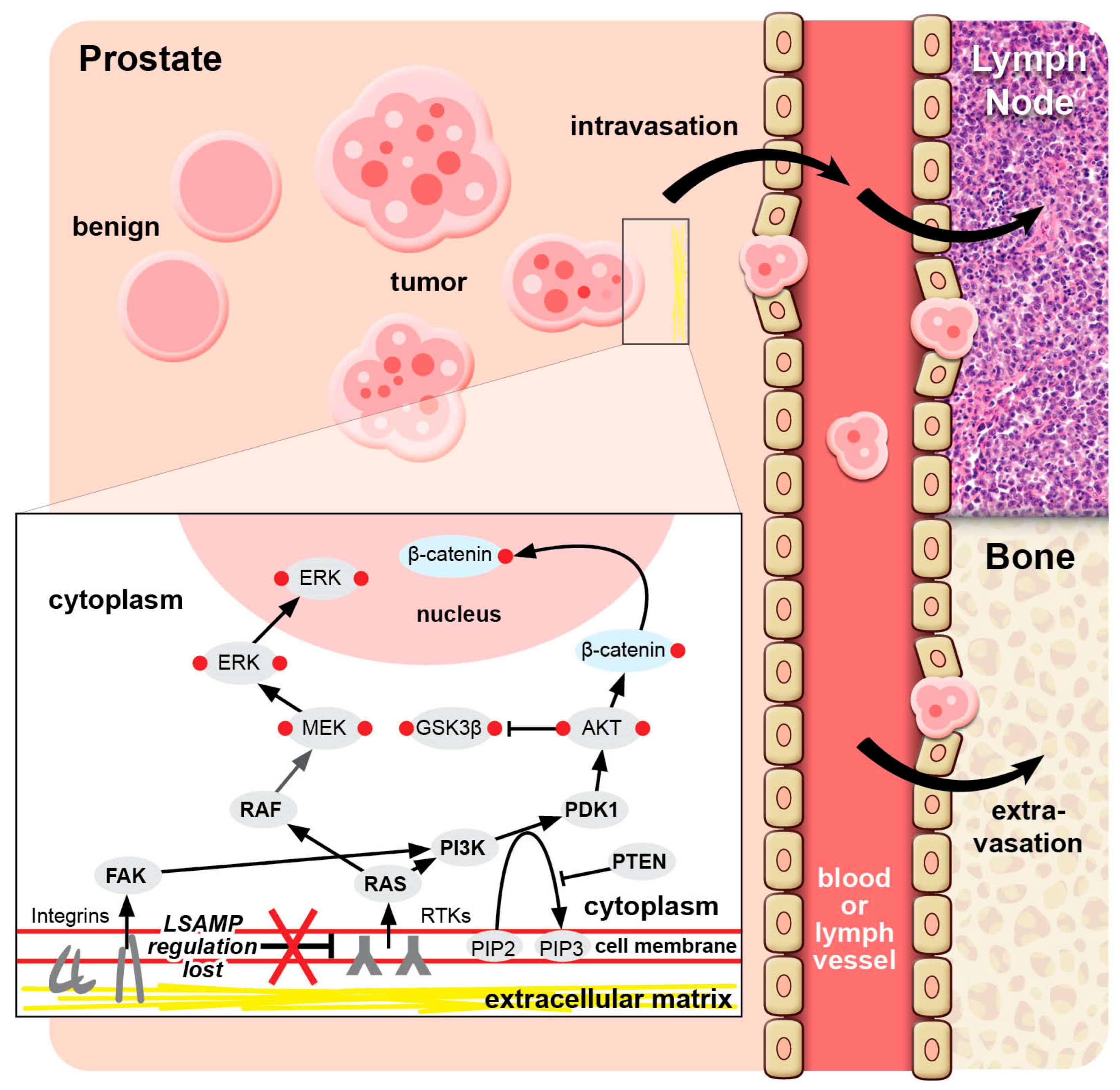

Table 1.
Studies investigating LSAMP in malignancies.
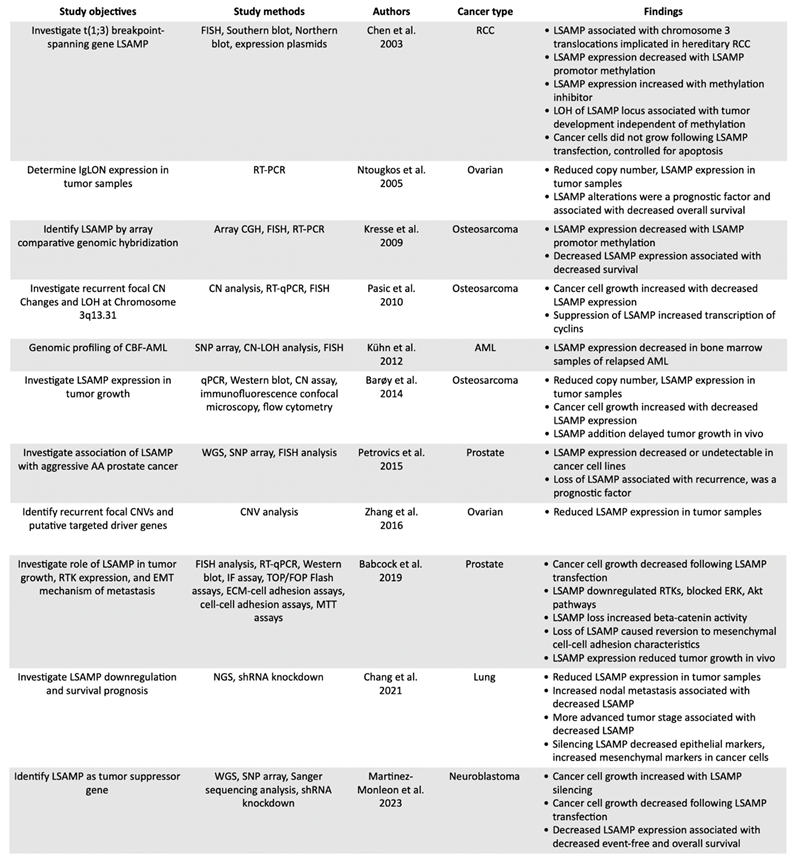 |
Table 2.
Changes associated with the LSAMP gene in cancers.
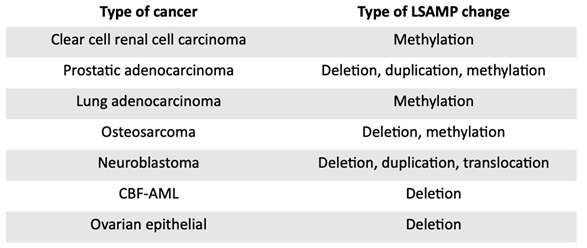 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



