
Submitted:
05 September 2024
Posted:
06 September 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The innate and adaptive immune systems collaborate to detect SARS-CoV-2 infection, minimize the viral spread, and kill infected cells, ultimately leading to the resolution of the infection. The adaptive immune system develops a memory of previous encounters with the virus, providing enhanced responses when rechallenged by the same pathogen. Such immunological memory is the basis of vaccine function. Here, we review the current knowledge on the immune response to SARS-CoV-2 infection and vaccination, focusing on the pivotal role of T cells in establishing protective immunity against the virus. After providing an overview of the immune response to SARS-CoV-2 infection, we describe the main features of SARS-CoV-2-specific CD4+ and CD8+ T cells, including cross-reactive T cells, generated in patients with different degrees of COVID-19 severity, and of Spike-specific CD4+ and CD8+ T cells induced by vaccines. Finally, we discuss T cell responses to SARS-CoV-2 variants and hybrid immunity and conclude by highlighting possible strategies to improve the efficacy of COVID-19 vaccination.
Keywords:
SARS-CoV-2 infection
; COVID-19 vaccine
; human T cells
; immunological memory
; antigen-specific T cells
; cross-reactive T cells
; hybrid immunity
; resident memory T cells
; vaccine improvement
1. Introduction
The T cell-mediated immunity is key to controlling intracellular pathogens, such as viruses. CD8+ T cells directly kill infected cells whereas CD4+ T cells provide “help” through cytokine production and optimize durable and effective CD8+ T cell and humoral responses [1].
Naïve T cells are activated in secondary lymphoid organs upon interacting with antigen-loaded dendritic cells (DCs). DCs present antigens to T cells in the form of short peptides loaded on the major histocompatibility complex (MHC), stimulate the cognate T cell receptor (TCR), provide co-stimulatory signals, and produce polarizing cytokines and metabolites that modulate the effector function and homing capacity of activated T cells. Upon stimulation, activated naïve T cells start to proliferate and differentiate into effector T cells that migrate to peripheral tissues where they exert their protective function [2]. After clearance of the pathogen, most effector T cells die by apoptosis and only a small fraction of them persist as central memory, effector memory, and resident memory T cells that will provide an enhanced systemic or local immune protection to the host when re-exposed to the same antigen [3]. The immunological memory is a peculiar property of the adaptive immune system and underlies vaccine-induced protective immunity.
Here, we aim to review the current knowledge of the immune response to SARS-CoV-2 infection and vaccination, revolving around T cells and cellular immunity. First, we will give an overview of the immune response to SARS-CoV-2 infection, including a brief description of the role of innate immunity in controlling the virus. We will then focus on the role of T cells, highlighting their capacity to provide immune protection in the absence of humoral immune responses. We will describe the different features of SARS-CoV-2-specific CD4+ and CD8+ T cells generated in patients with mild and severe COVID-19, and compare them with Spike-specific CD4+ and CD8+ T cells induced by vaccination. Finally, we will highlight some open questions and future perspectives in the field, discussing possible strategies to improve the efficacy of COVID-19 vaccines.
We are aware and would like to remind the readers that the large majority of published studies on the topic have been conducted on the peripheral blood of patients with COVID-19 and vaccinated individuals, which may convey a partial view of the immune response to SARS-CoV-2 infection and vaccination since most of immune cells are not in circulation [4]. Nonetheless, by being relatively easy to perform and minimally invasive, investigating the immune responses in peripheral blood has allowed longitudinal sampling to monitor the evolution of the effector response and the generation of immunological memory against SARS-CoV-2. Moreover, by being performed on large cohorts of individuals, it has enabled the scientific community to identify correlates of protective immunity, generating fundamental knowledge that has been instrumental in tackling the COVID-19 pandemic.
2. Overview of the Immune Response to SARS-CoV-2 Infection
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causing the Coronavirus Disease 2019 (COVID-19) is a single-stranded positive-sense RNA virus belonging to the Sarbecovirus subgroup of the Betacoronavirus genus, which also includes the SARS-CoV-1 and MERS viruses that have caused epidemic and pandemic outbreak of diseases in the last 20 years [5].
SARS-CoV-2 infection occurs without symptoms or with mild ones in most individuals and is usually resolved in 10-20 days [6]. When symptomatic it results in an influenza-like illness that can eventually progress to interstitial pneumonia, acute respiratory distress syndrome, and death. The most common COVID-19 symptoms include fever, dry cough, and shortness of breath, followed by fatigue, myalgias, headache, rhinorrhea, anosmia, ageusia, diarrhea, nausea, and vomiting [7,8,9].
The optimal immune response to SARS-CoV-2 requires innate and adaptive immunity to function coordinately (Figure 1). The proper setup of an innate immune response mediated by type-I and type-III interferon (IFN) is critical for establishing an effective antiviral adaptive immune response [10,11]. The innate immune system can detect the presence of RNA viruses, such as SARS-CoV-2, through two classes of pattern recognition receptors: Toll-like receptors (TLR3 and TLR7/8) and RIG-I-like receptors (RIG-I and MDA5) [12]. The two pathways converge into the activation of IRF3 and IRF7 transcription factors that cooperate with the nuclear factor κB (NF-κB) to trigger the expression of type-I and type-III IFNs. In turn, type-I and type-III IFNs bind their cognate receptors IFNAR1/IFNAR2 and IFNRL1/IL10R2 on infected cells and antigen-presenting cells (APCs) and initiate a signaling cascade that induces the STAT1/STAT2/IRF9-driven activation of several interferon-stimulated genes (ISGs), which interfere with the viral life cycle [13], and pro-inflammatory cytokines production. In addition, type-I IFN can directly act on T cells to support clonal expansion and memory formation in response to viral infections [14,15].
SARS-CoV-2 can inhibit or delay type-I/III IFN-mediated immune response by antagonizing type-I/III IFN production and the downstream signaling through different mechanisms mediated, sometimes redundantly, by several structural and non-structural viral proteins [16,17]. Therefore, temporally delayed and lower levels of type-I or type-III IFNs are detected in the lungs or the peripheral blood of patients with severe COVID-19 compared with other respiratory infections [18,19]. The suppression of the type-I/III response results in higher viral replication, delayed activation of the adaptive immune system, excessive inflammation, and tissue damage [20], which underlie the systemic inflammatory syndrome that characterizes severe COVID-19 cases [21]. Notably, genetic mutations and autoantibodies that interfere with IFN pathways have been detected, respectively, in about 3-5% of younger adults and 15-20% of patients over 70 years old with critical COVID-19 pneumonia [22,23,24], indicating that the IFN-mediated immune response is impaired both by virus-dependent and host-dependent mechanisms in a sizeable proportion of patients with severe disease.
Despite the important role of innate immunity, the best control of SARS-CoV-2 infection and protection from severe COVID-19 is achieved by a coordinated adaptive immune response made by virus-specific neutralizing antibodies (nAbs) and CD4+ and CD8+ T cells [25]. Indeed, the early induction of SARS-CoV-2-specific T cells and nAbs positively correlates with a better clinical outcome in patients with COVID-19 [26,27] (Figure 1). Antibodies recognize viral epitopes in their native conformation, can block the binding of the viral Spike protein to the ACE2 receptor on human cells preventing infection, and can promote immune effector function by binding to complement and Fc receptors [28]. Antibodies act on the extracellular virus and are most effective when present before the beginning of an infection. On the contrary, T cells cannot recognize a virus until cells are infected as they need viral epitopes to be processed and presented by MHC molecules on APCs. However, T cells can recognize epitopes derived from any viral protein thus broadening the repertoire of targetable viral structures [29]. CD8+ T cells identify and directly kill infected cells, and are critical in eliminating the virus in many viral infections [30]. CD4+ T cells contribute to antiviral immunity by, at least, three distinct mechanisms involving follicular helper T (TFH) cells, TH1 cells, and cytotoxic (CD4+-CTL) cells. TFH cells help the humoral response sustaining the affinity maturation of B cells and the generation of durable antibody responses [31]. TH1 cells produce IFN-γ and other cytokines that activate cell-intrinsic antiviral responses in infected cells and phagocytes and promote the recruitment of effector cells at the site of infection. They have been shown to contribute to immune protection against influenza [32] and SARS-CoV-1 [33] infections. Finally, CD4+-CTL cells bear a cytotoxic activity similar to CD8+ T cells and can directly kill MHC-II-expressing infected cells. They have been detected in several viral infections and associated with protection in patients infected by influenza [32] and Dengue [34] viruses. Notably, MHC-II-expression has been widely detected in the inflamed lung epithelial and endothelial cells of patients with lethal COVID-19 [35], suggesting that CD4+-CTL cells may cooperate with CD8+ T cells in killing SARS-CoV-2 infected cells.
Important information about the contribution of the different arms of the immune system in controlling SARS-CoV-2 infection comes from large studies performed on individuals with various types of inborn errors of immunity (IEI). These studies showed that IEI patients typically experience prolonged viral shedding and have higher mortality rates compared to the age-matched general population [36,37,38]. However, most IEI patients (90-95%) can resolve the infection, often with mild or moderate symptoms, highlighting redundancies and compensatory mechanisms in the human immune system for host defense against SARS-CoV-2 [36,37,38]. The largest group of patients with IEI analyzed so far was made by individuals affected by deficiency in antibody production, such as common variable immunodeficiency and hypogammaglobulinemia, indicating that SARS-CoV-2 infection can be controlled by the immune system in the absence of nAbs production, likely by T cells. Similarly, therapeutic depletion of B cells by rituximab in patients with multiple sclerosis is associated with an increased risk of developing a severe disease requiring hospitalization but does not significantly correlate with higher mortality rates [39,40,41].
Investigating the opposite scenario, namely the ability of B cells and antibodies to control SARS-CoV-2 infection without T cells, is more difficult because a complete lack of T cells is incompatible with life. However, evidence about the important role of T cells in the resolution of SARS-CoV-2 infection emerges from various clinical settings. In patients with Acquired Immune Deficiency Syndrome (AIDS) having an active SARS-CoV-2 infection, CD4+ T cell lymphopenia is associated with poorer outcomes [42,43], even when nAbs are produced. Among patients with hematologic cancer, the ones with defective CD4+ and CD8+ T cell responses had the highest mortality, regardless of the presence of B cell responses [44]. On the contrary, in patients having compromised humoral immunity due to the disease or therapy, the presence of SARS-CoV-2-specific CD8+ T cells was associated with improved survival [44]. This is consistent with the observation that CD8+ T cells can control viral loads upon rechallenge with SARS-CoV-2 in convalescent rhesus macaques with waning antibody titers [45]. The ability of T cells to control SARS-CoV-2 infection in the absence of humoral response is corroborated by the identification of SARS-CoV-2-specific T cells in the absence of seroconversion in asymptomatic individuals exposed to the virus and in some patients with paucisymptomatic COVID-19 [46,47]. Notably, we and others have reported cases of patients with COVID-19 who failed to mount a T cell response, measured either by T cell clonal expansion [48] or antigen-specific stimulation [25], and succumbed to the disease despite the production of SARS-CoV-2-specific antibodies, further supporting the important role for T cells in resolving SARS-CoV-2 infection.
Altogether, these data demonstrate that the best protection against SARS-CoV-2 infection is provided by integrated and coordinated innate and adaptive immune responses and indicate a superior capacity of T cells to control the virus.
3. Qualitative and Quantitative Alterations of T Cell Populations in Patients with COVID-19
One of the main clinical features of patients with COVID-19 is T cell lymphopenia correlating with the severity of the disease. A transient lymphopenia occurring during an acute infection before the peak in the T cell response is a characteristic common to many severe viral infections, can be induced by type-I IFN signaling, and may be useful to create the space for a robust virus-specific T cell response [49]. However, in patients with severe COVID-19, the T cell lymphopenia can persist weeks after infection or symptoms onset [50]. An increased CD4+/CD8+ T cell ratio has been reported in patients with severe COVID-19 [51,52], suggesting that SARS-CoV-2 infection might preferentially impair CD8+ T cells, especially from the effector memory population [48]. Initially, it was speculated that the stronger reduction of circulating T cells in patients with severe COVID-19 could result from increased recruitment of T cells at the site of infection. However, the observation that in bronchoalveolar lavage (BAL) fluid, the number of T cells was higher in patients with moderate disease than in those with severe disease [53] argues against this hypothesis. Presumably, a temporally dysregulated type-I IFN response, the inability to mount a virus-specific T cell response, the increased apoptosis of T cells, or a combination of these factors, may better explain the stronger lymphopenia in patients with severe disease.
Defects in type 1 immune responses [51] and skewing toward type 2 immunity [48,54] have been associated with COVID-19 severity, similar to what was observed in fatal SARS-CoV-1 infections [55] and in an experimental model of influenza infection [56], indicating that an inappropriate immune response to the virus may cause delayed viral clearance and disease deterioration.
The impaired ability to mount an effective antiviral T cell-mediated immune response underlies the uncoordinated adaptive immune response in the elderly [25] that, together with a higher rate of comorbidities, renders age the major risk factor in developing severe COVID-19 (Figure 2). In addition to the mentioned alteration in type-I IFN signaling due to the production of auto-antibodies and a decline in the antigen presentation potential of APCs [57], the capacity of setting up an adaptive immune response to SARS-CoV-2 in older adults is dampened by the age-related decrease in the repertoire of naïve T cells [58], which causes a contraction in the pool of T cells able to react to a new pathogen (Figure 2). The reduction of naïve T cells is stronger among CD8+ than CD4+ T cells, possibly because CD8+ T cells are more susceptible to the homeostatic proliferation-induced differentiation [59], and can be exacerbated by common chronic infections [60] that leads to the expansion of terminally differentiated and exhausted T cells. Both terminally differentiated CD8+ T cells and memory-like CD8+ T cells differentiated in response to cytokines include innate-like cytotoxic cells that can be activated in the absence of TCR stimulation [61,62]. Early activation of these bystander-activated cytotoxic T cells can cooperate in viral clearance [63], but their prolonged complement-mediated activation can also contribute to the excessive inflammation and tissue damage characterizing severe COVID-19 [64].
Together, these data indicate that prolonged T cell lymphopenia and maladapted CD4+ and CD8+ T cell responses are associated with severe COVID-19 and that these conditions, for different reasons, occur more frequently in the elders who are, indeed, at higher risk of clinical deterioration upon SARS-CoV-2 infection.
4. SARS-CoV-2 T Cell Antigens and Immunodominant Epitopes
CD4+ and CD8+ T cells activated by SARS-CoV-2 infection recognize a broad range of viral antigens derived from structural and non-structural proteins. A meta-analysis of 25 studies identified 1,434 non-redundant epitopes recognized by T cells, 399 of which have been defined as immunodominant (110 CD4+ and 289 CD8+ T cell epitopes) [65]. These data are constantly updated by the scientific community and, as of September 2024, the Immune Epitope Database [66] has cataloged over 3,700 records. The most immunodominant CD4+ T cell epitopes show a high HLA-II binding promiscuity, defined as the capacity to bind multiple HLA allelic variants [67]. Immunodominant epitopes mainly derive from the Spike (S), Nucleoprotein (N), and Membrane (M) structural proteins, but also from non-structural proteins such as nsp3, nsp12, and ORF3a [65,67]. CD4+ and CD8+ T cell responses to S, N, M, and nsp3 proteins are highly coordinated, meaning that CD4+ and CD8+ T cells specific to these proteins are simultaneously generated in most individuals [67]. It has been conservatively estimated that each person recognizes on average 19 different CD4+ and 17 CD8+ T cell epitopes [67], but none of the identified epitopes elicit T cell responses in 100% of the donor tested.
Looking at the distribution of the immunodominant epitopes from S and N proteins, it emerged that the epitopes recognized by CD4+ and CD8+ T cells possess distinct features. CD4+ T cell epitopes tend to be concentrated in discrete regions, while CD8+ T cell epitopes are more dispersed throughout the sequence of the antigens. Immunodominant CD4+ T cell epitopes in the S protein are enriched in the N-terminal domain of the S1 subunit, the C-terminal (aa 686-816) region and the fusion protein region in the S2 subunit, and a narrow conserved region (aa 346-365) of the receptor binding domain (RBD) [65,68]. Similarly, CD4+ T cell epitopes in the N protein are localized in the N-terminal and the C-terminal domains with little contribution from the linker region and protein tails. On the contrary, CD4+ T cell immunogenic regions are distributed along the entire M protein [65]. Interestingly, CD4+ T cell immunodominant regions identified in S and N proteins show a limited overlap with the immunodominant linear regions targeted by antibody responses [65], indicating complementarity between the humoral and cellular immunity, and corroborating the correlation between a coordinated adaptive immune response and a favorable clinical outcome.
Several studies have identified SARS-CoV-2-reactive T cells in about 50% of unexposed individuals [69,70,71,72]. These T cells cross-react with conserved epitopes from endemic coronaviruses (NL63, OC43, HKU1, and 229E) and have low-avidity T cell receptors (TCRs) [73,74]. M-specific CD4+ T cells and S-specific CD8+ T cells can also originate from cross-reactive cytomegalovirus-specific T cells [75], extending the cross-reactivity to other virus types. Despite cross-reactive T cells have been associated with less severe disease and enhanced immune responses [76,77,78], their protective role is still debated [79]. Moreover, although there is evidence for the recall of pre-existing cross-reactive memory T cells during SARS-CoV-2 infection, the majority of SARS-CoV-2-reactive T cells recognize new epitopes [67,68] through a heterogeneous repertoire of high-avidity TCRs [73,80,81].
These data indicate that T cell responses to SARS-CoV-2 are broad, multi-antigenic, and complementary to humoral responses.
5. Kinetics of T Cell Responses to SARS-CoV-2 Infection and Phenotype of SARS-CoV-2-Specific T Cells
As mentioned before, the early induction of SARS-CoV-2-specific T cells positively correlates with a better clinical outcome in patients with COVID-19 [26], and their protective function is corroborated by studies in non-human primates [45]. The identification and quantification of SARS-CoV-2-specific T cells are more complex than the measurement of specific Abs. They can be performed by different immune assays based either on the ex-vivo identification through tetramers and multimers staining or on the in-vitro stimulation with peptide pools, representing the whole SARS-CoV-2 peptidome or just selected epitopes, followed by monitoring of cytokine production, cell proliferation, or the expression of activation-induced markers (AIMs) on the cell surface. Both methods have advantages and limitations. Detection by tetramers and multimers binding does not require any T cell stimulation and the identification of antigen-specific T cells is independent of the upregulation of specific markers or the production of effector molecules. This technique is precise, but does not provide information on T cell functionality (unless cells are somehow stimulated) and usually allows the detection of a limited repertoire of antigen-specific T cells. Also, it requires knowledge of patients' HLA haplotypes, although this issue may be partially overcome by using complex libraries [82,83]. On the contrary, the in-vitro stimulation with peptide pools, or other sources of specific antigens, enables the monitoring of antigen-specific T cell effector function and is usually more sensitive. However, the high sensitivity may be paralleled by a lower specificity, due to the activation of low-affinity T cells that are not relevant for the in-vivo response to the virus, and to the detection of bystander-activated T cells if the antigenic stimulation is too long (e.g., > 24 hours). Moreover, it is possible that recently-in-vivo-activated T cells, such as effector cells during acute infection, may not properly respond to the in-vitro stimulation or that the in-vitro stimulation may alter the original phenotype of T cells.
Despite the mentioned limitations, the results produced by many studies describe defined patterns of induction, expansion, and contraction of the cellular immune response during and after SARS-CoV-2 infection (Figure 3). The timescale of the described immune responses may carry some unavoidable variability since the timing of infection cannot be precisely determined in humans and results are commonly reported as “time after symptoms onset” or “time after positive test”. However, such variability should be limited to a few days and affect the punctual definition only of the very early events of the immune response, since the median incubation period of SARS-CoV-2 infection has been estimated to be 5 days and the great majority (> 97%) of individuals develop symptoms within 11 days from viral infection [84]. Notably, these epidemiologic data agree with a study performing a controlled SARS-CoV-2 challenge in healthy volunteers [85]. SARS-CoV-2-specific T cells can be detected already 3-5 days after symptoms onset and expand in the following 10-20 days [25,26,86,87]. Delayed and weaker induction of SARS-CoV-2-specific T cells is observed in patients with severe COVID-19 [25,88], associated with a misfiring of the immune response, characterized by exaggerated and persistent activation of some components of the innate immunity and the lack of type-1 adaptive immune response [54].
SARS-CoV-2-specific CD4+ T cells are detected in nearly 100% of infected people and mainly produce TH1-associated cytokines, such as IFN-γ and TNF-α, although also TH17-associated cytokines have been detected [25,86,89,90]. CD4+ T cells from asymptomatic patients or patients with mild disease produce higher amounts of IFN-γ and IL-2 than those from patients with severe COVID-19, indicating a higher functionality and proliferative capacity [89,91]. Clonally expanded CD4+-CTL cells have been identified in hospitalized patients [92], but a clear correlation between this T cell subset and immune protection or disease severity is still missing. Circulating SARS-CoV-2-reactive TFH (cTFH) cells, specific for the S, N, and M proteins, are also found upon infection and correlate with nAbs production [93,94,95]. Notably, post-mortem examination of thoracic lymph nodes and spleen highlighted a strong reduction of BCL-6+ germinal center B cells that was associated with an early block of BCL-6+ TFH differentiation [96], demonstrating the relevance of TFH cells in supporting the humoral immune response to SARS-CoV-2 infection. Interestingly, the S-specific cTFH cells have been reported to be more abundant than S-specific TH1 cells, while the cTFH/TH1 ratio was inverted for the N-specific CD4+ T cells [89], suggesting a specialization of effector responses to different viral antigens.
SARS-CoV-2-specific CD8+ T cells are identified in about 70% of infected individuals and their induction strongly and significantly correlates with a better clinical outcome in patients with COVID-19 [25]. SARS-CoV-2-specific CD8+ T cells in patients with acute infection have mainly an effector memory phenotype (CD45RA– CCR7–) and produce high levels of effector molecules, such as granzyme B, perforin, and IFN-γ [89,97,98]. Patients with mild COVID-19 have a higher frequency of global and SARS-CoV-2-specific CD8+ memory precursor effector T cells compared with those with severe disease [48,97], suggesting an impaired or delayed generation of memory CD8+ T cells in patients with severe COVID-19.
Two-three weeks after symptoms onset the frequency of circulating SARS-CoV-2-specific T cells starts declining [99] (Figure 3). The kinetics of the contraction phase differs, at least in part, between CD4+ and CD8+ T cells: CD8+ T cells are progressively reduced starting about 1 month after infection, while the frequency of CD4+ T cells is more stable at least until 2 months [25,100]. The reasons underlying the different kinetics of contraction are still debated. They might result from the higher tendency of CD8+ T cells to reside in peripheral tissues than in the circulation and from persisting antigenic depots in dendritic cells within lymph nodes that sustain CD4+ T cell activation and proliferation. Nevertheless, more recent studies analyzing the presence of memory T cells within 8 months from the infection calculated a comparable half-life for CD4+ (t1/2 94-207 days) and CD8+ (t1/2 125-196 days) T cells [101,102], indicating that the different kinetics are limited to the first part of the contraction phase. Interestingly, a longitudinal study investigating the presence of SARS-CoV-2-specific CD4+ T cells at 6 to 15 months after infection calculated a t1/2 of 377 days, indicating a flattening of the contraction phase and suggesting the establishment of virus-specific long-term memory T cells.
Data collected in the first months after resolution of SARS-CoV-2 infection indicate that SARS-CoV-2-specific CD4+ T cells are mainly central memory (TCM) and effector memory (TEM) T cells, with a trend of TCM increasing and TEM decreasing over time [101,102]. On the contrary, SARS-CoV-2-specific CD8+ T cells are mainly effector memory (TEM) and effector memory CD45RA+ (TEMRA) T cells, with an increasing accumulation of TEMRA over time [98,101,102]. Nonetheless, it has been reported that the majority of SARS-CoV-2-specific CD8+ T cells one year after infection express the transcription factor cell factor 1 (TCF-1) but not the thymocyte selection-associated high-mobility group box (TOX) [98], suggesting they are endowed with self-renewal capacity and not terminally differentiated or exhausted. Moreover, a small population of SARS-CoV-2-specific CD4+ and CD8+ stem-cell memory T cells (TSCM) have been detected post-infection, up to 10 months after symptoms onset, hinting at the formation of long-lasting virus-specific memory cells [103]. These data are consistent with the observation made in the context of SARS-CoV-1 infection where virus-specific memory T cells are detected over 10 years post-infection [90,104].
The investigation of SARS-CoV-2-specific T cells has been primarily performed in peripheral blood. However, T cells showing expression of activation markers or clonal expansion have been identified in the lungs of patients with COVID-19 [53,105,106], suggesting the establishment of T cell-mediated responses in the infected tissues. Although studies investigating the antigen specificity of tissue-infiltrating T cells during the acute infection are still scarce, SARS-CoV-2-specific CD4+ and CD8+ T cells have been found in the bone marrow, spleen, lymph nodes, lung, and nasal mucosa of COVID-19 patients up to 6 months after infection [107,108,109]. The generation of SARS-CoV-2-specific and cross-reactive resident memory T (TRM) cells in the upper and lower airways [107,110] may contribute to the protection against the disease upon reinfection by rapidly recognizing the virus and providing an alarm function also in case of failure or delay of the innate immune response [111,112]. Indeed, a longitudinal study of BAL fluid from 273 patients with severe pneumonia showed an association between the presence of alveolar T cells targeting structural SARS-CoV-2 proteins and a better clinical outcome in unvaccinated patients [113].
These data demonstrate that virus-specific CD4+ and CD8+ effector T cells are induced in response to SARS-CoV-2 infection and contribute to the resolution of the infection. Infected individuals develop memory T cells that can be detected up to 15 months after infection and are predicted to last for years. SARS-CoV-2-specific memory T cells include circulating and tissue-resident T cells (Figure 4) that can provide multiple layers of enhanced protection against a severe disease upon reinfection.
6. SARS-CoV-2-Specific T Cell Responses to COVID-19 Vaccines
The development of vaccines against SARS-CoV-2 has been the breakthrough for mitigating the severe illness and hospitalization associated with COVID-19. The objective of vaccines is to stimulate an immune response against a pathogen without causing the pathogen-associated disease to train the immune system to face the same pathogen in the context of a natural infection. Four different types of COVID-19 vaccines have been developed and approved. They use as antigen source the inactivated whole virus (e.g., CoronaVac by Sinovac, BBIBP-CorV by Sinopharm, and BBV125 COVAXIN by Bharat Biotech) or just the S protein, which can be delivered in the form of messenger RNA (e.g., BNT162b2 by Pfizer-BioNTech and mRNA-1273 by Moderna), adenoviral vector (e.g., ChAdOx1-S by Oxford/AstraZeneca, Ad26.COV2.S by Janssen, and Gam-COVID-Vac by Gamaleya), or recombinant protein (e.g., NVX-CoV2373 by Novavax). According to the results from phase-III clinical trials, the protection against the wild-type SARS-CoV-2-induced disease ranges from 94-95% of two doses of mRNA-based vaccines [114,115] to 67-74% of one or two doses of adenoviral vector-based vaccines [116,117]. The efficacy of the other vaccines lies in between: it was about 89% for the protein-based vaccine [118] and 78-83% for those using inactivated viral particles [119,120].
The primary output of vaccine effectiveness is the production of nAbs that can potentially prevent the infection, providing a sterilizing immunity. Therefore, the presence and abundance of S-specific nAbs provide the strongest correlate of protection from subsequent infection with the same viral strain and disease development [121,122]. However, studies performed in relevant animal models demonstrate that antigen-specific T cells induced by vaccination contribute to the efficacy of vaccines [123,124]. They can extend the duration of vaccine-induced protective immunity after circulating antibodies start waning and broaden the protection against antibody-escaping virus variants.
S-specific CD4+ T cells are found in nearly all human subjects receiving two doses of mRNA COVID-19 vaccines and memory CD4+ T cells are maintained up to 6 months after the second dose [125,126,127,128]. The frequencies of S-specific CD4+ memory T cells elicited by the two doses of mRNA COVID-19 vaccines and their distribution among the TCM, TEM, and TEMRA subsets are comparable with those induced by natural infection, and similar are also the patterns of contraction and estimated half-lives [127,128,129,130]. Interestingly, vaccine-induced CD4+ memory T cells include a fraction of TSCM cells that may support establishing long-term memory [129]. Looking at the effector function of S-specific CD4+ T cells, they are mainly cTFH cells and TH1 cells producing IFN-γ, TNF-α, and IL-2 [131]. As for the natural infection, the frequency of vaccine-induced cTFH cells positively correlates with nAb titers [125,132]. Notably, the frequency of vaccine-induced cTFH and TH1 cells after the first immunization correlate with the abundance of nAbs and frequency of S-specific CD8+ T cells following the second dose of the vaccine, highlighting the role of rapidly stimulated CD4+ T cells in coordinating the immune response to the second vaccine dose, especially in individuals who did not experience previous SARS-CoV-2 infection [130]. Moreover, although the frequency of S-specific cTFH cells peaks one week after the second immunization, S-specific TFH cells in lymph nodes persist at least for 6 months [133] and their impairment is strictly associated with compromised germinal center reactions and nAbs production, as shown in immunocompromised individuals undergoing kidney transplantation [134]. S-specific polyfunctional TH1 cells and cTFH cells are comparably induced by adenoviral vector-based and recombinant protein-based vaccines [128,135], although it is difficult to assess a quantitative side-by-side comparison, also due to different immunization schedules [128].
S-specific CD8+ T cells are induced in about 70-90% of subjects receiving two doses of mRNA vaccines, but memory CD8+ T cells are maintained only in 40-65% of individuals at 6 months after the second dose [125,126,127,128]. Among individuals with detectable CD8+ memory T cells, the magnitude of memory is lower compared to CD4+ T cells [136,137]. Nonetheless, vaccine-induced CD8+ T cells are polyfunctional, able to produce different effector molecules, such as IFN-γ, TNF-α, and granzyme B, and, like S-specific CD4+ T cells, their distribution among T cell subsets mirrored the one observed in a natural infection, with a prevalence of TEM and TEMRA cells [128,129,131,137]. Notably, a vaccine based on adjuvanted recombinant S protein elicits a very low frequency of S-specific CD8+ memory T cells compared with mRNA vaccines [128] with minimal effects on the protection from the disease measured during clinical trials, indicating a superior role for CD4+ T cells in establishing the vaccine-induced protective immunity.
The knowledge about generating S-specific TRM cells upon vaccination is still inadequate. However, one study reported the absence of vaccine-induced S-specific T cells in the BAL fluid of vaccinated subjects, despite their detection in peripheral blood [138]. Similarly, a second study showed a very limited induction of S-specific TRM cells in lung biopsies following mRNA vaccination compared to infection [139].
These data demonstrate that COVID-19 vaccines trigger the development of S-specific CD4+ memory T cells and, often, CD8+ memory T cells. In particular, S-specific CD4+ T cells have a pivotal role in orchestrating the humoral and cellular responses to the vaccine. Moreover, vaccine-induced S-specific CD4+ and CD8+ memory T cells have a functional phenotype similar to that observed in response to natural infection and are predicted to be long-lived. On the contrary, COVID-19 vaccines seem unable to elicit the differentiation of S-specific TRM cells (Figure 4).
7. T Cell Responses to SARS-CoV-2 Variants and Hybrid Immunity
Despite having evolved an RNA proofreading mechanism acting during replication [140], SARS-CoV-2 has accumulated many mutations over time. Most of these mutations do not modify the amino acid sequence of viral proteins or do not provide any evolutionary benefit. However, some mutations that provide survival advantages and improved viral "fitness" have spread worldwide and are defined as variants of concern (VoCs). The major VoCs include Alpha (B.1.17 lineage), Beta (B.1.351), Delta (B.1.617), and Omicron (B.1.1.529). In particular, Delta and Omicron VoCs are characterized by significantly higher transmissibility and infectivity. The presence of multiple mutations in the S protein, especially in the RBD domain, allows these VoCs to escape, at least in part, the immune protection mediated by nAbs generated in response to previous infection and vaccination [141,142]. Moreover, Omicron can escape the neutralizing activity of most of, but not all, the therapeutic monoclonal antibodies currently available for clinical use [143].
The ability of VoCs to escape the control of vaccine-induced nAbs has raised concerns. However, several pieces of evidence show that vaccine-induced T cells can recognize all the SARS-CoV-2 variants of concern, including Omicron, largely preserving the protection against severe COVID-19 [126,127,131,144,145]. This is possible because S-specific CD4+ and CD8+ T cells recognize a median of 10-11 Spike epitopes in each person, and the great majority of these epitopes are conserved across VoCs [126].
The emergence of antibody-escaping VoCs and the drop in nAb titers 6-8 months after vaccination induced many countries to implement the third dose of vaccine (booster) at about 6 months from the second dose, especially to protect fragile subjects. The booster dose of mRNA vaccine promptly restores S-specific antibody titers and elicits potent neutralization across different VoCs, including Omicron, at least for three months after vaccination [146,147,148]. The enhanced functionality of the induced nAbs derives from the re-expansion of pre-existing memory B cell clones and the stimulation of new B cell responses with increased potency and breadth [149]. Evidence from other vaccines with a 3-dose schedule indicates that durable Ab responses are triggered after the third dose [136], but evaluating the durability of the protective immunity induced by the COVID-19 vaccination booster will require more time and additional studies. S-specific T cells are also rapidly re-stimulated after the booster vaccine [150], even in a good proportion (> 50%) of patients with compromised immune responses secondary to different diseases or therapies [151,152]. However, data about T cells are largely limited to the measurement of IFN-γ production and additional information on their functional phenotype is still missing.
The vaccination of people who had been previously infected by SARS-CoV-2 and the infection of vaccinated people by SARS-CoV-2 VoCs have provided additional insights on T cell responses to SARS-CoV-2 following repeated exposures as well as on hybrid immunity, intended as a combined immune response to natural infection and a vaccine. S- and RBD-specific memory B cell frequencies substantially increase and have higher somatic hypermutation and affinity maturation in hybrid immunity than after vaccination [127,153,154]. Consequently, nAbs titers and breadth of neutralization of VoCs are significantly improved in hybrid immune responses compared with the vaccination or the infection alone [136]. On the contrary, there are only modest differences in the frequency of circulating S-specific CD4+ and CD8+ T cells and their IFN-γ production capacity between hybrid immunity and vaccination [130,155,156,157]. However, data on the quality of T cell responses elicited by hybrid immunity are still scattered. A study reported that, although the order in the type of exposure (infection or vaccination first) affects the distribution between S- and non-S-specific T cell responses, there is no evidence of major alterations in the TCR repertoire of epitope-specific CD8+ T cells upon repeated exposure [158]. The same study showed that repeated stimulations lead to a shift of SARS-CoV-2-specific CD8+ memory T cells toward TEMRA cells, but these cells are not exhausted [158]. Another report showed, instead, that vaccination can induce a repertoire of S-specific CD4+ T cell clones that substantially diverges from the one previously activated by the infection [159], thus further broadening the antigen-specific T cell response against the virus. Similarly, SARS-CoV-2 breakthrough infections have been reported to enhance the magnitude, breadth, and repertoire of T cell responses [160]. Another possible advantage of hybrid immunity compared to vaccination alone is the generation of TRM cells [161] observed after SARS-CoV-2 infection but not upon vaccination (Figure 4). However, further investigation of vaccine-induced T cell tissutal immunity is required to clarify this topic.
Together, these data indicate that memory T cells induced by infection and vaccination can protect against SARS-CoV-2 antibody-escaping variants thanks to a polyclonal response that can recognize multiple conserved epitopes.
8. Conclusions and Future Perspectives
The optimal protection against SARS-CoV-2 infection requires all the components of the innate and adaptive immune system to function coordinately. When this happens, SARS-CoV-2 infection is resolved in a few days without or with mild symptoms. On the contrary, an impairment or delay in activating one of these components, due to virus-dependent or host-dependent factors, results in an uncoordinated response that can lead to severe disease. Upon infection, the adaptive immune system develops a memory of SARS-CoV-2 that generates enhanced immune responses and protection in case of re-exposure to the same virus. COVID-19 vaccines are designed to mimic the SARS-CoV-2 infection without disease to train the adaptive immune system to develop an immune memory that protects us when we are naturally exposed to the virus.
The immunological memory is mediated by four different arms of the adaptive immune system: antibodies, memory B cells, and memory CD4+ and CD8+ T cells. Antibodies are the only component that can prevent infection. Their continuous production is sustained by long-lived plasma cells, but it is clear that circulating SARS-CoV-2 nAbs levels decline some months after infection or vaccination. Moreover, the emergence of viral variants has made it evident that reaching herd immunity and completely blocking the circulation of the virus are targets currently not achievable. Nonetheless, memory T cells can efficiently protect us from severe disease and, in part, from symptomatic infection. SARS-CoV-2-specific memory T cells can recognize several conserved viral epitopes and protect against SARS-CoV-2 variants that breach the antibody barrier. Therefore, the efficacy of vaccines in establishing protective immunity should be routinely evaluated not only based on the capacity to elicit the production of nAbs, as currently done but also on the ability to generate antigen-specific memory T cells. However, T cells can recognize antigens only in the context of MHC presentation, making the ex-vivo identification and characterization of antigen-specific T cells difficult and hindering the development of high-throughput screening platforms. This aspect will require technical and technological improvements, which will be critical not only for clinical applications but also for the fundamental investigation aimed at increasing our knowledge of the mechanisms guiding the development of human memory T cells.
Approved COVID-19 vaccines have been developed extremely quickly thanks to the knowledge acquired in the last decades and have proven to be highly effective in protecting against severe COVID-19, but not from reinfection, also due to the emergence of viral variants. Currently, pharmaceutical companies are updating COVID-19 vaccines with the Spike sequence of the emerging SARS-CoV-2 variants, but they are evolving too fast to keep pace [162]. The development of a pan-coronavirus vaccine would overcome this issue and have a great epidemiologic and economic value [163]: about fifteen of these vaccine candidates are in development [164,165,166]. They have different degrees of target breadth, ranging from SARS-CoV-2 variants to the whole coronavirus genus but they are still far from reaching clinical use [165,166].
In the meantime, there is room for improving approved vaccines. First, the observation that circulating nAbs titers decline a few months after vaccination suggests that current vaccines cannot efficiently trigger the differentiation of long-lived plasma cells [167]. Second, approved vaccines, different from natural infection, fail to generate resident memory T and B cells and mucosal Abs. TRM cells can provide a rapid reaction of the immune system when the virus infects peripheral tissues, such as the nasopharyngeal mucosa and the lungs, thus preventing viral spreading from the site of entry and symptomatic disease. The differentiation of vaccine-induced TRM cells may depend on the route of immunization [124,168,169], the kind of antigen [170], or the use of specific adjuvants [171,172]. The “prime-and-pull” vaccination strategy has proven to generate both systemic and local memory T cells [173] and was effective in reducing Herpes simplex virus type-2 recurrent infections in preclinical studies [174]. In this setting, a conventional parenteral vaccination that elicits a systemic T cell response (prime) is followed by the local application of the antigen or a chemoattractant (pull) to establish a pool of TRM cells [173,175]. Interestingly, multiple parenteral immunizations can also induce the differentiation of TRM cells [176], although with lower efficiency compared with the local antigen administration [177], suggesting that the third or the fourth dose of current vaccines may generate S-specific TRM cells to a certain extent. A “prime-and-pull” strategy could be recapitulated for COVID-19 vaccines by combining the current schedules of intramuscular vaccination with a boost of a locally administered vaccine, such as through the nasal route. Intranasal COVID-19 vaccines are under development [178] and have shown promising results in animal models [179,180,181]. Upon completion of clinical trials, they may represent an additional weapon in the fight against COVID-19.
Another element that may be improved to ameliorate vaccine efficacy is the adjuvant. Adjuvants are vaccine components that stimulate the immune system to enhance the magnitude, breadth, and durability of the vaccine-induced immune response when these signals are not provided by the antigen, namely in all vaccines except for those made of live attenuated pathogens [182]. At the moment there are very few vaccine adjuvants approved for clinical use and, while they have been extensively tested for their capacity to enhance Abs production, the knowledge of their effect on memory T cell differentiation is surprisingly scarce [182]. Developing vaccine adjuvants that can induce the generation of long-lived memory T cells with the proper polarization and homing capacity will certainly improve the efficacy of vaccines.
To conclude, the COVID-19 pandemic has greatly challenged humanity with huge social and economic costs and the loss of millions of lives. At the same time, it has boosted unprecedented cooperation in the scientific community and has led to the rapid development of therapeutic and preventive strategies to tackle the emergency. Extraordinary progress has been quickly made in disentangling and understanding the immune response to SARS-CoV-2 infection and vaccination, and this knowledge will be precious to guide the development of improved vaccines against SARS-CoV-2 and other diseases.
Funding
This work has been supported by a Ricerca Finalizzata Giovani Ricercatori grant from the Italian Ministry of Health (grant number GR-2021-12374097).
Acknowledgments
As of September 2024, a PubMed search of the “(SARS-CoV-2 OR COVID-19) AND (T cells OR T lymphocytes)” terms returned more than 5,400 results. Therefore, reviewing all the literature on this topic is virtually impossible. I thank all the researchers who have dedicated their time to seriously investigate the immune response to SARS-CoV-2 infection and COVID-19 vaccines and apologize to those not mentioned in this review. Figure 1, Figure 3, and Figure 4 have been prepared with the help of Biorender.com.
Conflicts of Interest
The author declare no conflicts of interest.
References
- Sallusto, F. Heterogeneity of Human CD4(+) T Cells Against Microbes. Annu Rev Immunol 2016, 34, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Notarbartolo, S.; Abrignani, S. Human T lymphocytes at tumor sites. Semin Immunopathol 2022, 44, 883–901. [Google Scholar] [CrossRef] [PubMed]
- Farber, D.L.; Yudanin, N.A.; Restifo, N.P. Human memory T cells: Generation, compartmentalization and homeostasis. Nat Rev Immunol 2014, 14, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Ganusov, V.V.; De Boer, R.J. Do most lymphocytes in humans really reside in the gut? Trends Immunol 2007, 28, 514–518. [Google Scholar] [CrossRef]
- Singh, D.; Yi, S.V. On the origin and evolution of SARS-CoV-2. Exp Mol Med 2021, 53, 537–547. [Google Scholar] [CrossRef]
- Cevik, M.; Tate, M.; Lloyd, O.; Maraolo, A.E.; Schafers, J.; Ho, A. SARS-CoV-2, SARS-CoV, and MERS-CoV viral load dynamics, duration of viral shedding, and infectiousness: A systematic review and meta-analysis. Lancet Microbe 2021, 2, e13–e22. [Google Scholar] [CrossRef]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Lechien, J.R.; Chiesa-Estomba, C.M.; Place, S.; Van Laethem, Y.; Cabaraux, P.; Mat, Q.; Huet, K.; Plzak, J.; Horoi, M.; Hans, S.; et al. Clinical and epidemiological characteristics of 1420 European patients with mild-to-moderate coronavirus disease 2019. J Intern Med 2020, 288, 335–344. [Google Scholar] [CrossRef]
- Sette, A.; Crotty, S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 2021, 184, 861–880. [Google Scholar] [CrossRef]
- Manfrini, N.; Notarbartolo, S.; Grifantini, R.; Pesce, E. SARS-CoV-2: A Glance at the Innate Immune Response Elicited by Infection and Vaccination. Antibodies (Basel) 2024, 13. [Google Scholar] [CrossRef]
- Kasuga, Y.; Zhu, B.; Jang, K.J.; Yoo, J.S. Innate immune sensing of coronavirus and viral evasion strategies. Exp Mol Med 2021, 53, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu Rev Immunol 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Kolumam, G.A.; Thomas, S.; Thompson, L.J.; Sprent, J.; Murali-Krishna, K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med 2005, 202, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Tough, D.F.; Borrow, P.; Sprent, J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science 1996, 272, 1947–1950. [Google Scholar] [CrossRef]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.Y. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep 2020, 33, 108234. [Google Scholar] [CrossRef]
- Lee, J.H.; Koepke, L.; Kirchhoff, F.; Sparrer, K.M.J. Interferon antagonists encoded by SARS-CoV-2 at a glance. Med Microbiol Immunol 2022. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Pere, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Katze, M.G.; Fornek, J.L.; Palermo, R.E.; Walters, K.A.; Korth, M.J. Innate immune modulation by RNA viruses: Emerging insights from functional genomics. Nat Rev Immunol 2008, 8, 644–654. [Google Scholar] [CrossRef]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [PubMed]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Beziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bastard, P.; Effort, C.H.G.; Cobat, A.; Casanova, J.L. Human genetic and immunological determinants of critical COVID-19 pneumonia. Nature 2022, 603, 587–598. [Google Scholar] [CrossRef]
- Rydyznski Moderbacher, C.; Ramirez, S.I.; Dan, J.M.; Grifoni, A.; Hastie, K.M.; Weiskopf, D.; Belanger, S.; Abbott, R.K.; Kim, C.; Choi, J.; et al. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell 2020, 183, 996–1012.e19. [Google Scholar] [CrossRef]
- Tan, A.T.; Linster, M.; Tan, C.W.; Le Bert, N.; Chia, W.N.; Kunasegaran, K.; Zhuang, Y.; Tham, C.Y.L.; Chia, A.; Smith, G.J.D.; et al. Early induction of functional SARS-CoV-2-specific T cells associates with rapid viral clearance and mild disease in COVID-19 patients. Cell Rep 2021, 34, 108728. [Google Scholar] [CrossRef]
- Lucas, C.; Klein, J.; Sundaram, M.E.; Liu, F.; Wong, P.; Silva, J.; Mao, T.; Oh, J.E.; Mohanty, S.; Huang, J.; et al. Delayed production of neutralizing antibodies correlates with fatal COVID-19. Nat Med 2021, 27, 1178–1186. [Google Scholar] [CrossRef]
- Gruell, H.; Vanshylla, K.; Weber, T.; Barnes, C.O.; Kreer, C.; Klein, F. Antibody-mediated neutralization of SARS-CoV-2. Immunity 2022, 55, 925–944. [Google Scholar] [CrossRef] [PubMed]
- Vardhana, S.; Baldo, L.; Morice, W.G., 2nd; Wherry, E.J. Understanding T cell responses to COVID-19 is essential for informing public health strategies. Sci Immunol 2022, 7, eabo1303. [Google Scholar] [CrossRef]
- Chang, J.T.; Wherry, E.J.; Goldrath, A.W. Molecular regulation of effector and memory T cell differentiation. Nat Immunol 2014, 15, 1104–1115. [Google Scholar] [CrossRef]
- Crotty, S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 2019, 50, 1132–1148. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, T.M.; Li, C.K.; Chui, C.S.; Huang, A.K.; Perkins, M.; Liebner, J.C.; Lambkin-Williams, R.; Gilbert, A.; Oxford, J.; Nicholas, B.; et al. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat Med 2012, 18, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhao, J.; Mangalam, A.K.; Channappanavar, R.; Fett, C.; Meyerholz, D.K.; Agnihothram, S.; Baric, R.S.; David, C.S.; Perlman, S. Airway Memory CD4(+) T Cells Mediate Protective Immunity against Emerging Respiratory Coronaviruses. Immunity 2016, 44, 1379–1391. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, D.; Bangs, D.J.; Sidney, J.; Kolla, R.V.; De Silva, A.D.; de Silva, A.M.; Crotty, S.; Peters, B.; Sette, A. Dengue virus infection elicits highly polarized CX3CR1+ cytotoxic CD4+ T cells associated with protective immunity. Proc Natl Acad Sci U S A 2015, 112, E4256–E4263. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, N.; Boucau, J.; Kuo, H.H.; Perugino, C.; Mahajan, V.S.; Farmer, J.R.; Liu, H.; Diefenbach, T.J.; Piechocka-Trocha, A.; Lefteri, K.; et al. Expansion of Cytotoxic CD4+ T cells in the lungs in severe COVID-19. medRxiv 2021. [Google Scholar] [CrossRef]
- Meyts, I.; Bucciol, G.; Quinti, I.; Neven, B.; Fischer, A.; Seoane, E.; Lopez-Granados, E.; Gianelli, C.; Robles-Marhuenda, A.; Jeandel, P.Y.; et al. Coronavirus disease 2019 in patients with inborn errors of immunity: An international study. J Allergy Clin Immunol 2021, 147, 520–531. [Google Scholar] [CrossRef]
- Goudouris, E.S.; Pinto-Mariz, F.; Mendonca, L.O.; Aranda, C.S.; Guimaraes, R.R.; Kokron, C.; Barros, M.T.; Anisio, F.; Alonso, M.L.O.; Marcelino, F.; et al. Outcome of SARS-CoV-2 Infection in 121 Patients with Inborn Errors of Immunity: A Cross-Sectional Study. J Clin Immunol 2021, 41, 1479–1489. [Google Scholar] [CrossRef]
- Giardino, G.; Milito, C.; Lougaris, V.; Punziano, A.; Carrabba, M.; Cinetto, F.; Scarpa, R.; Dellepiane, R.M.; Ricci, S.; Rivalta, B.; et al. The Impact of SARS-CoV-2 Infection in Patients with Inborn Errors of Immunity: The Experience of the Italian Primary Immunodeficiencies Network (IPINet). J Clin Immunol 2022. [Google Scholar] [CrossRef]
- Sormani, M.P.; De Rossi, N.; Schiavetti, I.; Carmisciano, L.; Cordioli, C.; Moiola, L.; Radaelli, M.; Immovilli, P.; Capobianco, M.; Trojano, M.; et al. Disease-Modifying Therapies and Coronavirus Disease 2019 Severity in Multiple Sclerosis. Ann Neurol 2021, 89, 780–789. [Google Scholar] [CrossRef]
- Langer-Gould, A.; Smith, J.B.; Li, B.H.; Group, K.M.S. Multiple sclerosis, rituximab, and COVID-19. Ann Clin Transl Neurol 2021, 8, 938–943. [Google Scholar] [CrossRef]
- McKay, K.A.; Piehl, F.; Englund, S.; He, A.; Langer-Gould, A.; Hillert, J.; Frisell, T. Rituximab Infusion Timing, Cumulative Dose, and Hospitalization for COVID-19 in Persons With Multiple Sclerosis in Sweden. JAMA Netw Open 2021, 4, e2136697. [Google Scholar] [CrossRef] [PubMed]
- Bhaskaran, K.; Rentsch, C.T.; MacKenna, B.; Schultze, A.; Mehrkar, A.; Bates, C.J.; Eggo, R.M.; Morton, C.E.; Bacon, S.C.J.; Inglesby, P.; et al. HIV infection and COVID-19 death: A population-based cohort analysis of UK primary care data and linked national death registrations within the OpenSAFELY platform. Lancet HIV 2021, 8, e24–e32. [Google Scholar] [CrossRef]
- Geretti, A.M.; Stockdale, A.J.; Kelly, S.H.; Cevik, M.; Collins, S.; Waters, L.; Villa, G.; Docherty, A.; Harrison, E.M.; Turtle, L.; et al. Outcomes of Coronavirus Disease 2019 (COVID-19) Related Hospitalization Among People With Human Immunodeficiency Virus (HIV) in the ISARIC World Health Organization (WHO) Clinical Characterization Protocol (UK): A Prospective Observational Study. Clin Infect Dis 2021, 73, e2095–e2106. [Google Scholar] [CrossRef] [PubMed]
- Bange, E.M.; Han, N.A.; Wileyto, P.; Kim, J.Y.; Gouma, S.; Robinson, J.; Greenplate, A.R.; Hwee, M.A.; Porterfield, F.; Owoyemi, O.; et al. CD8(+) T cells contribute to survival in patients with COVID-19 and hematologic cancer. Nat Med 2021, 27, 1280–1289. [Google Scholar] [CrossRef]
- McMahan, K.; Yu, J.; Mercado, N.B.; Loos, C.; Tostanoski, L.H.; Chandrashekar, A.; Liu, J.; Peter, L.; Atyeo, C.; Zhu, A.; et al. Correlates of protection against SARS-CoV-2 in rhesus macaques. Nature 2021, 590, 630–634. [Google Scholar] [CrossRef]
- Swadling, L.; Diniz, M.O.; Schmidt, N.M.; Amin, O.E.; Chandran, A.; Shaw, E.; Pade, C.; Gibbons, J.M.; Le Bert, N.; Tan, A.T.; et al. Pre-existing polymerase-specific T cells expand in abortive seronegative SARS-CoV-2. Nature 2022, 601, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Shomuradova, A.S.; Vagida, M.S.; Sheetikov, S.A.; Zornikova, K.V.; Kiryukhin, D.; Titov, A.; Peshkova, I.O.; Khmelevskaya, A.; Dianov, D.V.; Malasheva, M.; et al. SARS-CoV-2 Epitopes Are Recognized by a Public and Diverse Repertoire of Human T Cell Receptors. Immunity 2020, 53, 1245–1257.e5. [Google Scholar] [CrossRef]
- Notarbartolo, S.; Ranzani, V.; Bandera, A.; Gruarin, P.; Bevilacqua, V.; Putignano, A.R.; Gobbini, A.; Galeota, E.; Manara, C.; Bombaci, M.; et al. Integrated longitudinal immunophenotypic, transcriptional and repertoire analyses delineate immune responses in COVID-19 patients. Sci Immunol 2021, 6. [Google Scholar] [CrossRef]
- Welsh, R.M.; Selin, L.K.; Szomolanyi-Tsuda, E. Immunological memory to viral infections. Annu Rev Immunol 2004, 22, 711–743. [Google Scholar] [CrossRef]
- Tian, Y.; Carpp, L.N.; Miller, H.E.R.; Zager, M.; Newell, E.W.; Gottardo, R. Single-cell immunology of SARS-CoV-2 infection. Nat Biotechnol 2022, 40, 30–41. [Google Scholar] [CrossRef]
- Mathew, D.; Giles, J.R.; Baxter, A.E.; Greenplate, A.R.; Wu, J.E.; Alanio, C.; Oldridge, D.A.; Kuri-Cervantes, L.; Pampena, M.B.; D'Andrea, K.; et al. Deep immune profiling of COVID-19 patients reveals patient heterogeneity and distinct immunotypes with implications for therapeutic interventions. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mazzoni, A.; Salvati, L.; Maggi, L.; Capone, M.; Vanni, A.; Spinicci, M.; Mencarini, J.; Caporale, R.; Peruzzi, B.; Antonelli, A.; et al. Impaired immune cell cytotoxicity in severe COVID-19 is IL-6 dependent. J Clin Invest 2020, 130, 4694–4703. [Google Scholar] [CrossRef]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med 2020, 26, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef]
- Li, C.K.; Wu, H.; Yan, H.; Ma, S.; Wang, L.; Zhang, M.; Tang, X.; Temperton, N.J.; Weiss, R.A.; Brenchley, J.M.; et al. T cell responses to whole SARS coronavirus in humans. J Immunol 2008, 181, 5490–5500. [Google Scholar] [CrossRef]
- Graham, M.B.; Braciale, V.L.; Braciale, T.J. Influenza virus-specific CD4+ T helper type 2 T lymphocytes do not promote recovery from experimental virus infection. J Exp Med 1994, 180, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Bartleson, J.M.; Radenkovic, D.; Covarrubias, A.J.; Furman, D.; Winer, D.A.; Verdin, E. SARS-CoV-2, COVID-19 and the Ageing Immune System. Nat Aging 2021, 1, 769–782. [Google Scholar] [CrossRef]
- Britanova, O.V.; Putintseva, E.V.; Shugay, M.; Merzlyak, E.M.; Turchaninova, M.A.; Staroverov, D.B.; Bolotin, D.A.; Lukyanov, S.; Bogdanova, E.A.; Mamedov, I.Z.; et al. Age-related decrease in TCR repertoire diversity measured with deep and normalized sequence profiling. J Immunol 2014, 192, 2689–2698. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Weyand, C.M. Mechanisms underlying T cell ageing. Nat Rev Immunol 2019, 19, 573–583. [Google Scholar] [CrossRef]
- Nicoli, F.; Clave, E.; Wanke, K.; von Braun, A.; Bondet, V.; Alanio, C.; Douay, C.; Baque, M.; Lependu, C.; Marconi, P.; et al. Primary immune responses are negatively impacted by persistent herpesvirus infections in older people: Results from an observational study on healthy subjects and a vaccination trial on subjects aged more than 70 years old. EBioMedicine 2022, 76, 103852. [Google Scholar] [CrossRef]
- Kim, T.S.; Shin, E.C. The activation of bystander CD8(+) T cells and their roles in viral infection. Exp Mol Med 2019, 51, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sottile, R.; Panjwani, M.K.; Lau, C.M.; Daniyan, A.F.; Tanaka, K.; Barker, J.N.; Brentjens, R.J.; Sun, J.C.; Le Luduec, J.B.; Hsu, K.C. Human cytomegalovirus expands a CD8(+) T cell population with loss of BCL11B expression and gain of NK cell identity. Sci Immunol 2021, 6, eabe6968. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, L.; Mescia, F.; Turner, L.; Hanson, A.L.; Kotagiri, P.; Dunmore, B.J.; Ruffieux, H.; De Sa, A.; Huhn, O.; Morgan, M.D.; et al. Longitudinal analysis reveals that delayed bystander CD8+ T cell activation and early immune pathology distinguish severe COVID-19 from mild disease. Immunity 2021, 54, 1257–1275.e8. [Google Scholar] [CrossRef] [PubMed]
- Georg, P.; Astaburuaga-Garcia, R.; Bonaguro, L.; Brumhard, S.; Michalick, L.; Lippert, L.J.; Kostevc, T.; Gabel, C.; Schneider, M.; Streitz, M.; et al. Complement activation induces excessive T cell cytotoxicity in severe COVID-19. Cell 2022, 185, 493–512.e25. [Google Scholar] [CrossRef]
- Grifoni, A.; Sidney, J.; Vita, R.; Peters, B.; Crotty, S.; Weiskopf, D.; Sette, A. SARS-CoV-2 human T cell epitopes: Adaptive immune response against COVID-19. Cell Host Microbe 2021, 29, 1076–1092. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res 2019, 47, D339–D343. [Google Scholar] [CrossRef]
- Tarke, A.; Sidney, J.; Kidd, C.K.; Dan, J.M.; Ramirez, S.I.; Yu, E.D.; Mateus, J.; da Silva Antunes, R.; Moore, E.; Rubiro, P.; et al. Comprehensive analysis of T cell immunodominance and immunoprevalence of SARS-CoV-2 epitopes in COVID-19 cases. Cell Rep Med 2021, 2, 100204. [Google Scholar] [CrossRef]
- Low, J.S.; Vaqueirinho, D.; Mele, F.; Foglierini, M.; Jerak, J.; Perotti, M.; Jarrossay, D.; Jovic, S.; Perez, L.; Cacciatore, R.; et al. Clonal analysis of immunodominance and cross-reactivity of the CD4 T cell response to SARS-CoV-2. Science 2021, 372, 1336–1341. [Google Scholar] [CrossRef]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e15. [Google Scholar] [CrossRef]
- Braun, J.; Loyal, L.; Frentsch, M.; Wendisch, D.; Georg, P.; Kurth, F.; Hippenstiel, S.; Dingeldey, M.; Kruse, B.; Fauchere, F.; et al. SARS-CoV-2-reactive T cells in healthy donors and patients with COVID-19. Nature 2020, 587, 270–274. [Google Scholar] [CrossRef]
- Mateus, J.; Grifoni, A.; Tarke, A.; Sidney, J.; Ramirez, S.I.; Dan, J.M.; Burger, Z.C.; Rawlings, S.A.; Smith, D.M.; Phillips, E.; et al. Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science 2020, 370, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Nelde, A.; Bilich, T.; Heitmann, J.S.; Maringer, Y.; Salih, H.R.; Roerden, M.; Lubke, M.; Bauer, J.; Rieth, J.; Wacker, M.; et al. SARS-CoV-2-derived peptides define heterologous and COVID-19-induced T cell recognition. Nat Immunol 2021, 22, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Bacher, P.; Rosati, E.; Esser, D.; Martini, G.R.; Saggau, C.; Schiminsky, E.; Dargvainiene, J.; Schroder, I.; Wieters, I.; Khodamoradi, Y.; et al. Low-Avidity CD4(+) T Cell Responses to SARS-CoV-2 in Unexposed Individuals and Humans with Severe COVID-19. Immunity 2020, 53, 1258–1271.e5. [Google Scholar] [CrossRef] [PubMed]
- Dykema, A.G.; Zhang, B.; Woldemeskel, B.A.; Garliss, C.C.; Cheung, L.S.; Choudhury, D.; Zhang, J.; Aparicio, L.; Bom, S.; Rashid, R.; et al. Functional characterization of CD4+ T cell receptors crossreactive for SARS-CoV-2 and endemic coronaviruses. J Clin Invest 2021, 131. [Google Scholar] [CrossRef]
- Pothast, C.R.; Dijkland, R.C.; Thaler, M.; Hagedoorn, R.S.; Kester, M.G.D.; Wouters, A.K.; Hiemstra, P.S.; van Hemert, M.J.; Gras, S.; Falkenburg, J.H.F.; et al. SARS-CoV-2-specific CD4(+) and CD8(+) T cell responses can originate from cross-reactive CMV-specific T cells. Elife 2022, 11. [Google Scholar] [CrossRef]
- Sagar, M.; Reifler, K.; Rossi, M.; Miller, N.S.; Sinha, P.; White, L.F.; Mizgerd, J.P. Recent endemic coronavirus infection is associated with less-severe COVID-19. J Clin Invest 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Loyal, L.; Braun, J.; Henze, L.; Kruse, B.; Dingeldey, M.; Reimer, U.; Kern, F.; Schwarz, T.; Mangold, M.; Unger, C.; et al. Cross-reactive CD4(+) T cells enhance SARS-CoV-2 immune responses upon infection and vaccination. Science 2021, 374, eabh1823. [Google Scholar] [CrossRef]
- Mallajosyula, V.; Ganjavi, C.; Chakraborty, S.; McSween, A.M.; Pavlovitch-Bedzyk, A.J.; Wilhelmy, J.; Nau, A.; Manohar, M.; Nadeau, K.C.; Davis, M.M. CD8(+) T cells specific for conserved coronavirus epitopes correlate with milder disease in COVID-19 patients. Sci Immunol 2021, 6. [Google Scholar] [CrossRef]
- Moss, P. The T cell immune response against SARS-CoV-2. Nat Immunol 2022, 23, 186–193. [Google Scholar] [CrossRef]
- Peng, Y.; Felce, S.L.; Dong, D.; Penkava, F.; Mentzer, A.J.; Yao, X.; Liu, G.; Yin, Z.; Chen, J.L.; Lu, Y.; et al. An immunodominant NP105-113-B*07:02 cytotoxic T cell response controls viral replication and is associated with less severe COVID-19 disease. Nat Immunol 2022, 23, 50–61. [Google Scholar] [CrossRef]
- Nguyen, T.H.O.; Rowntree, L.C.; Petersen, J.; Chua, B.Y.; Hensen, L.; Kedzierski, L.; van de Sandt, C.E.; Chaurasia, P.; Tan, H.X.; Habel, J.R.; et al. CD8(+) T cells specific for an immunodominant SARS-CoV-2 nucleocapsid epitope display high naive precursor frequency and TCR promiscuity. Immunity 2021, 54, 1066–1082.e5. [Google Scholar] [CrossRef] [PubMed]
- Kared, H.; Redd, A.D.; Bloch, E.M.; Bonny, T.S.; Sumatoh, H.; Kairi, F.; Carbajo, D.; Abel, B.; Newell, E.W.; Bettinotti, M.P.; et al. SARS-CoV-2-specific CD8+ T cell responses in convalescent COVID-19 individuals. J Clin Invest 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.K.; Hersby, D.S.; Tamhane, T.; Povlsen, H.R.; Amaya Hernandez, S.P.; Nielsen, M.; Gang, A.O.; Hadrup, S.R. SARS-CoV-2 genome-wide T cell epitope mapping reveals immunodominance and substantial CD8(+) T cell activation in COVID-19 patients. Sci Immunol 2021, 6. [Google Scholar] [CrossRef]
- Lauer, S.A.; Grantz, K.H.; Bi, Q.; Jones, F.K.; Zheng, Q.; Meredith, H.R.; Azman, A.S.; Reich, N.G.; Lessler, J. The Incubation Period of Coronavirus Disease 2019 (COVID-19) From Publicly Reported Confirmed Cases: Estimation and Application. Ann Intern Med 2020, 172, 577–582. [Google Scholar] [CrossRef]
- Killingley, B.; Mann, A.J.; Kalinova, M.; Boyers, A.; Goonawardane, N.; Zhou, J.; Lindsell, K.; Hare, S.S.; Brown, J.; Frise, R.; et al. Safety, tolerability and viral kinetics during SARS-CoV-2 human challenge in young adults. Nat Med 2022, 28, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, D.; Schmitz, K.S.; Raadsen, M.P.; Grifoni, A.; Okba, N.M.A.; Endeman, H.; van den Akker, J.P.C.; Molenkamp, R.; Koopmans, M.P.G.; van Gorp, E.C.M.; et al. Phenotype and kinetics of SARS-CoV-2-specific T cells in COVID-19 patients with acute respiratory distress syndrome. Sci Immunol 2020, 5. [Google Scholar] [CrossRef]
- Lindeboom, R.G.H.; Worlock, K.B.; Dratva, L.M.; Yoshida, M.; Scobie, D.; Wagstaffe, H.R.; Richardson, L.; Wilbrey-Clark, A.; Barnes, J.L.; Kretschmer, L.; et al. Human SARS-CoV-2 challenge uncovers local and systemic response dynamics. Nature 2024, 631, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; To, K.K.; Wong, Y.C.; Liu, L.; Zhou, B.; Li, X.; Huang, H.; Mo, Y.; Luk, T.Y.; Lau, T.T.; et al. Acute SARS-CoV-2 Infection Impairs Dendritic Cell and T Cell Responses. Immunity 2020, 53, 864–877.e5. [Google Scholar] [CrossRef]
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Stralin, K.; Gorin, J.B.; Olsson, A.; Llewellyn-Lacey, S.; Kamal, H.; Bogdanovic, G.; Muschiol, S.; et al. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell 2020, 183, 158–168.e14. [Google Scholar] [CrossRef]
- Le Bert, N.; Tan, A.T.; Kunasegaran, K.; Tham, C.Y.L.; Hafezi, M.; Chia, A.; Chng, M.H.Y.; Lin, M.; Tan, N.; Linster, M.; et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature 2020, 584, 457–462. [Google Scholar] [CrossRef]
- Le Bert, N.; Clapham, H.E.; Tan, A.T.; Chia, W.N.; Tham, C.Y.L.; Lim, J.M.; Kunasegaran, K.; Tan, L.W.L.; Dutertre, C.A.; Shankar, N.; et al. Highly functional virus-specific cellular immune response in asymptomatic SARS-CoV-2 infection. J Exp Med 2021, 218. [Google Scholar] [CrossRef] [PubMed]
- Meckiff, B.J.; Ramirez-Suastegui, C.; Fajardo, V.; Chee, S.J.; Kusnadi, A.; Simon, H.; Eschweiler, S.; Grifoni, A.; Pelosi, E.; Weiskopf, D.; et al. Imbalance of Regulatory and Cytotoxic SARS-CoV-2-Reactive CD4(+) T Cells in COVID-19. Cell 2020, 183, 1340–1353.e16. [Google Scholar] [CrossRef]
- van der Ploeg, K.; Kirosingh, A.S.; Mori, D.A.M.; Chakraborty, S.; Hu, Z.; Sievers, B.L.; Jacobson, K.B.; Bonilla, H.; Parsonnet, J.; Andrews, J.R.; et al. TNF-alpha(+) CD4(+) T cells dominate the SARS-CoV-2 specific T cell response in COVID-19 outpatients and are associated with durable antibodies. Cell Rep Med 2022, 3, 100640. [Google Scholar] [CrossRef]
- Boppana, S.; Qin, K.; Files, J.K.; Russell, R.M.; Stoltz, R.; Bibollet-Ruche, F.; Bansal, A.; Erdmann, N.; Hahn, B.H.; Goepfert, P.A. SARS-CoV-2-specific circulating T follicular helper cells correlate with neutralizing antibodies and increase during early convalescence. PLoS Pathog 2021, 17, e1009761. [Google Scholar] [CrossRef] [PubMed]
- Koutsakos, M.; Rowntree, L.C.; Hensen, L.; Chua, B.Y.; van de Sandt, C.E.; Habel, J.R.; Zhang, W.; Jia, X.; Kedzierski, L.; Ashhurst, T.M.; et al. Integrated immune dynamics define correlates of COVID-19 severity and antibody responses. Cell Rep Med 2021, 2, 100208. [Google Scholar] [CrossRef]
- Kaneko, N.; Kuo, H.H.; Boucau, J.; Farmer, J.R.; Allard-Chamard, H.; Mahajan, V.S.; Piechocka-Trocha, A.; Lefteri, K.; Osborn, M.; Bals, J.; et al. The Loss of Bcl-6 Expressing T Follicular Helper Cells and the Absence of Germinal Centers in COVID-19. SSRN 2020, 3652322. [Google Scholar] [CrossRef] [PubMed]
- Rha, M.S.; Jeong, H.W.; Ko, J.H.; Choi, S.J.; Seo, I.H.; Lee, J.S.; Sa, M.; Kim, A.R.; Joo, E.J.; Ahn, J.Y.; et al. PD-1-Expressing SARS-CoV-2-Specific CD8(+) T Cells Are Not Exhausted, but Functional in Patients with COVID-19. Immunity 2021, 54, 44–52.e3. [Google Scholar] [CrossRef]
- Adamo, S.; Michler, J.; Zurbuchen, Y.; Cervia, C.; Taeschler, P.; Raeber, M.E.; Baghai Sain, S.; Nilsson, J.; Moor, A.E.; Boyman, O. Signature of long-lived memory CD8(+) T cells in acute SARS-CoV-2 infection. Nature 2022, 602, 148–155. [Google Scholar] [CrossRef]
- Minervina, A.A.; Komech, E.A.; Titov, A.; Bensouda Koraichi, M.; Rosati, E.; Mamedov, I.Z.; Franke, A.; Efimov, G.A.; Chudakov, D.M.; Mora, T.; et al. Longitudinal high-throughput TCR repertoire profiling reveals the dynamics of T-cell memory formation after mild COVID-19 infection. Elife 2021, 10. [Google Scholar] [CrossRef]
- Ferretti, A.P.; Kula, T.; Wang, Y.; Nguyen, D.M.V.; Weinheimer, A.; Dunlap, G.S.; Xu, Q.; Nabilsi, N.; Perullo, C.R.; Cristofaro, A.W.; et al. Unbiased Screens Show CD8(+) T Cells of COVID-19 Patients Recognize Shared Epitopes in SARS-CoV-2 that Largely Reside outside the Spike Protein. Immunity 2020, 53, 1095–1107.e3. [Google Scholar] [CrossRef]
- Dan, J.M.; Mateus, J.; Kato, Y.; Hastie, K.M.; Yu, E.D.; Faliti, C.E.; Grifoni, A.; Ramirez, S.I.; Haupt, S.; Frazier, A.; et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science 2021, 371. [Google Scholar] [CrossRef] [PubMed]
- Cohen, K.W.; Linderman, S.L.; Moodie, Z.; Czartoski, J.; Lai, L.; Mantus, G.; Norwood, C.; Nyhoff, L.E.; Edara, V.V.; Floyd, K.; et al. Longitudinal analysis shows durable and broad immune memory after SARS-CoV-2 infection with persisting antibody responses and memory B and T cells. Cell Rep Med 2021, 2, 100354. [Google Scholar] [CrossRef]
- Jung, J.H.; Rha, M.S.; Sa, M.; Choi, H.K.; Jeon, J.H.; Seok, H.; Park, D.W.; Park, S.H.; Jeong, H.W.; Choi, W.S.; et al. SARS-CoV-2-specific T cell memory is sustained in COVID-19 convalescent patients for 10 months with successful development of stem cell-like memory T cells. Nat Commun 2021, 12, 4043. [Google Scholar] [CrossRef] [PubMed]
- Ng, O.W.; Chia, A.; Tan, A.T.; Jadi, R.S.; Leong, H.N.; Bertoletti, A.; Tan, Y.J. Memory T cell responses targeting the SARS coronavirus persist up to 11 years post-infection. Vaccine 2016, 34, 2008–2014. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.A.; Morales-Nebreda, L.; Markov, N.S.; Swaminathan, S.; Querrey, M.; Guzman, E.R.; Abbott, D.A.; Donnelly, H.K.; Donayre, A.; Goldberg, I.A.; et al. Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature 2021, 590, 635–641. [Google Scholar] [CrossRef]
- Szabo, P.A.; Dogra, P.; Gray, J.I.; Wells, S.B.; Connors, T.J.; Weisberg, S.P.; Krupska, I.; Matsumoto, R.; Poon, M.M.L.; Idzikowski, E.; et al. Longitudinal profiling of respiratory and systemic immune responses reveals myeloid cell-driven lung inflammation in severe COVID-19. Immunity 2021, 54, 797–814.e6. [Google Scholar] [CrossRef] [PubMed]
- Poon, M.M.L.; Rybkina, K.; Kato, Y.; Kubota, M.; Matsumoto, R.; Bloom, N.I.; Zhang, Z.; Hastie, K.M.; Grifoni, A.; Weiskopf, D.; et al. SARS-CoV-2 infection generates tissue-localized immunological memory in humans. Sci Immunol 2021, 6, eabl9105. [Google Scholar] [CrossRef]
- Roukens, A.H.E.; Pothast, C.R.; Konig, M.; Huisman, W.; Dalebout, T.; Tak, T.; Azimi, S.; Kruize, Y.; Hagedoorn, R.S.; Zlei, M.; et al. Prolonged activation of nasal immune cell populations and development of tissue-resident SARS-CoV-2-specific CD8(+) T cell responses following COVID-19. Nat Immunol 2022, 23, 23–32. [Google Scholar] [CrossRef]
- Ramirez, S.I.; Faraji, F.; Hills, L.B.; Lopez, P.G.; Goodwin, B.; Stacey, H.D.; Sutton, H.J.; Hastie, K.M.; Saphire, E.O.; Kim, H.J.; et al. Immunological memory diversity in the human upper airway. Nature 2024, 632, 630–636. [Google Scholar] [CrossRef]
- Niessl, J.; Sekine, T.; Lange, J.; Konya, V.; Forkel, M.; Maric, J.; Rao, A.; Mazzurana, L.; Kokkinou, E.; Weigel, W.; et al. Identification of resident memory CD8(+) T cells with functional specificity for SARS-CoV-2 in unexposed oropharyngeal lymphoid tissue. Sci Immunol 2021, 6, eabk0894. [Google Scholar] [CrossRef]
- Schenkel, J.M.; Fraser, K.A.; Vezys, V.; Masopust, D. Sensing and alarm function of resident memory CD8(+) T cells. Nat Immunol 2013, 14, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Fraser, K.A.; Beura, L.K.; Pauken, K.E.; Vezys, V.; Masopust, D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 2014, 346, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Markov, N.S.; Ren, Z.; Senkow, K.J.; Grant, R.A.; Gao, C.A.; Malsin, E.S.; Sichizya, L.; Kihshen, H.; Helmin, K.A.; Jovisic, M.; et al. Distinctive evolution of alveolar T cell responses is associated with clinical outcomes in unvaccinated patients with SARS-CoV-2 pneumonia. Nat Immunol 2024, 25, 1607–1622. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N Engl J Med 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N Engl J Med 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Falsey, A.R.; Sobieszczyk, M.E.; Hirsch, I.; Sproule, S.; Robb, M.L.; Corey, L.; Neuzil, K.M.; Hahn, W.; Hunt, J.; Mulligan, M.J.; et al. Phase 3 Safety and Efficacy of AZD1222 (ChAdOx1 nCoV-19) Covid-19 Vaccine. N Engl J Med 2021, 385, 2348–2360. [Google Scholar] [CrossRef]
- Sadoff, J.; Gray, G.; Vandebosch, A.; Cardenas, V.; Shukarev, G.; Grinsztejn, B.; Goepfert, P.A.; Truyers, C.; Fennema, H.; Spiessens, B.; et al. Safety and Efficacy of Single-Dose Ad26.COV2.S Vaccine against Covid-19. N Engl J Med 2021, 384, 2187–2201. [Google Scholar] [CrossRef]
- Heath, P.T.; Galiza, E.P.; Baxter, D.N.; Boffito, M.; Browne, D.; Burns, F.; Chadwick, D.R.; Clark, R.; Cosgrove, C.; Galloway, J.; et al. Safety and Efficacy of NVX-CoV2373 Covid-19 Vaccine. N Engl J Med 2021, 385, 1172–1183. [Google Scholar] [CrossRef]
- Tanriover, M.D.; Doganay, H.L.; Akova, M.; Guner, H.R.; Azap, A.; Akhan, S.; Kose, S.; Erdinc, F.S.; Akalin, E.H.; Tabak, O.F.; et al. Efficacy and safety of an inactivated whole-virion SARS-CoV-2 vaccine (CoronaVac): Interim results of a double-blind, randomised, placebo-controlled, phase 3 trial in Turkey. Lancet 2021, 398, 213–222. [Google Scholar] [CrossRef]
- Al Kaabi, N.; Zhang, Y.; Xia, S.; Yang, Y.; Al Qahtani, M.M.; Abdulrazzaq, N.; Al Nusair, M.; Hassany, M.; Jawad, J.S.; Abdalla, J.; et al. Effect of 2 Inactivated SARS-CoV-2 Vaccines on Symptomatic COVID-19 Infection in Adults: A Randomized Clinical Trial. JAMA 2021, 326, 35–45. [Google Scholar] [CrossRef]
- Goldblatt, D.; Alter, G.; Crotty, S.; Plotkin, S.A. Correlates of protection against SARS-CoV-2 infection and COVID-19 disease. Immunol Rev 2022. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.S.; Nason, M.C.; Flach, B.; Gagne, M.; O'Connell, S.; Johnston, T.S.; Shah, S.N.; Edara, V.V.; Floyd, K.; Lai, L.; et al. Immune correlates of protection by mRNA-1273 vaccine against SARS-CoV-2 in nonhuman primates. Science 2021, 373, eabj0299. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, A.; Yu, J.; McMahan, K.; Jacob-Dolan, C.; Liu, J.; He, X.; Hope, D.; Anioke, T.; Barrett, J.; Chung, B.; et al. Vaccine protection against the SARS-CoV-2 Omicron variant in macaques. Cell 2022, 185, 1549–1555.e11. [Google Scholar] [CrossRef] [PubMed]
- Kingstad-Bakke, B.; Lee, W.; Chandrasekar, S.S.; Gasper, D.J.; Salas-Quinchucua, C.; Cleven, T.; Sullivan, J.A.; Talaat, A.; Osorio, J.E.; Suresh, M. Vaccine-induced systemic and mucosal T cell immunity to SARS-CoV-2 viral variants. Proc Natl Acad Sci U S A 2022, 119, e2118312119. [Google Scholar] [CrossRef]
- Mateus, J.; Dan, J.M.; Zhang, Z.; Rydyznski Moderbacher, C.; Lammers, M.; Goodwin, B.; Sette, A.; Crotty, S.; Weiskopf, D. Low-dose mRNA-1273 COVID-19 vaccine generates durable memory enhanced by cross-reactive T cells. Science 2021, 374, eabj9853. [Google Scholar] [CrossRef]
- Tarke, A.; Coelho, C.H.; Zhang, Z.; Dan, J.M.; Yu, E.D.; Methot, N.; Bloom, N.I.; Goodwin, B.; Phillips, E.; Mallal, S.; et al. SARS-CoV-2 vaccination induces immunological T cell memory able to cross-recognize variants from Alpha to Omicron. Cell 2022, 185, 847–859.e11. [Google Scholar] [CrossRef]
- Goel, R.R.; Painter, M.M.; Apostolidis, S.A.; Mathew, D.; Meng, W.; Rosenfeld, A.M.; Lundgreen, K.A.; Reynaldi, A.; Khoury, D.S.; Pattekar, A.; et al. mRNA vaccines induce durable immune memory to SARS-CoV-2 and variants of concern. Science 2021, 374, abm0829. [Google Scholar] [CrossRef]
- Zhang, Z.; Mateus, J.; Coelho, C.H.; Dan, J.M.; Moderbacher, C.R.; Galvez, R.I.; Cortes, F.H.; Grifoni, A.; Tarke, A.; Chang, J.; et al. Humoral and cellular immune memory to four COVID-19 vaccines. Cell 2022, 185, 2434–2451.e17. [Google Scholar] [CrossRef]
- Guerrera, G.; Picozza, M.; D'Orso, S.; Placido, R.; Pirronello, M.; Verdiani, A.; Termine, A.; Fabrizio, C.; Giannessi, F.; Sambucci, M.; et al. BNT162b2 vaccination induces durable SARS-CoV-2-specific T cells with a stem cell memory phenotype. Sci Immunol 2021, 6, eabl5344. [Google Scholar] [CrossRef]
- Painter, M.M.; Mathew, D.; Goel, R.R.; Apostolidis, S.A.; Pattekar, A.; Kuthuru, O.; Baxter, A.E.; Herati, R.S.; Oldridge, D.A.; Gouma, S.; et al. Rapid induction of antigen-specific CD4(+) T cells is associated with coordinated humoral and cellular immunity to SARS-CoV-2 mRNA vaccination. Immunity 2021, 54, 2133–2142.e3. [Google Scholar] [CrossRef]
- Gao, Y.; Cai, C.; Grifoni, A.; Muller, T.R.; Niessl, J.; Olofsson, A.; Humbert, M.; Hansson, L.; Osterborg, A.; Bergman, P.; et al. Ancestral SARS-CoV-2-specific T cells cross-recognize the Omicron variant. Nat Med 2022, 28, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Tauzin, A.; Nayrac, M.; Benlarbi, M.; Gong, S.Y.; Gasser, R.; Beaudoin-Bussieres, G.; Brassard, N.; Laumaea, A.; Vezina, D.; Prevost, J.; et al. A single dose of the SARS-CoV-2 vaccine BNT162b2 elicits Fc-mediated antibody effector functions and T cell responses. Cell Host Microbe 2021, 29, 1137–1150.e6. [Google Scholar] [CrossRef] [PubMed]
- Mudd, P.A.; Minervina, A.A.; Pogorelyy, M.V.; Turner, J.S.; Kim, W.; Kalaidina, E.; Petersen, J.; Schmitz, A.J.; Lei, T.; Haile, A.; et al. SARS-CoV-2 mRNA vaccination elicits a robust and persistent T follicular helper cell response in humans. Cell 2022, 185, 603–613.e15. [Google Scholar] [CrossRef] [PubMed]
- Lederer, K.; Bettini, E.; Parvathaneni, K.; Painter, M.M.; Agarwal, D.; Lundgreen, K.A.; Weirick, M.; Muralidharan, K.; Castano, D.; Goel, R.R.; et al. Germinal center responses to SARS-CoV-2 mRNA vaccines in healthy and immunocompromised individuals. Cell 2022, 185, 1008–1024.e15. [Google Scholar] [CrossRef]
- Ewer, K.J.; Barrett, J.R.; Belij-Rammerstorfer, S.; Sharpe, H.; Makinson, R.; Morter, R.; Flaxman, A.; Wright, D.; Bellamy, D.; Bittaye, M.; et al. T cell and antibody responses induced by a single dose of ChAdOx1 nCoV-19 (AZD1222) vaccine in a phase 1/2 clinical trial. Nat Med 2021, 27, 270–278. [Google Scholar] [CrossRef]
- Sette, A.; Crotty, S. Immunological memory to SARS-CoV-2 infection and COVID-19 vaccines. Immunol Rev 2022. [Google Scholar] [CrossRef]
- Oberhardt, V.; Luxenburger, H.; Kemming, J.; Schulien, I.; Ciminski, K.; Giese, S.; Csernalabics, B.; Lang-Meli, J.; Janowska, I.; Staniek, J.; et al. Rapid and stable mobilization of CD8(+) T cells by SARS-CoV-2 mRNA vaccine. Nature 2021, 597, 268–273. [Google Scholar] [CrossRef]
- Tang, J.; Zeng, C.; Cox, T.M.; Li, C.; Son, Y.M.; Cheon, I.S.; Wu, Y.; Behl, S.; Taylor, J.J.; Chakraborty, R.; et al. Respiratory mucosal immunity against SARS-CoV-2 following mRNA vaccination. Sci Immunol 2022, eadd4853. [Google Scholar] [CrossRef]
- Pieren, D.K.J.; Kuguel, S.G.; Rosado, J.; Robles, A.G.; Rey-Cano, J.; Mancebo, C.; Esperalba, J.; Falco, V.; Buzon, M.J.; Genesca, M. Limited induction of polyfunctional lung-resident memory T cells against SARS-CoV-2 by mRNA vaccination compared to infection. Nat Commun 2023, 14, 1887. [Google Scholar] [CrossRef]
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.L. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol Cell 2020, 79, 710–727. [Google Scholar] [CrossRef]
- Planas, D.; Saunders, N.; Maes, P.; Guivel-Benhassine, F.; Planchais, C.; Buchrieser, J.; Bolland, W.H.; Porrot, F.; Staropoli, I.; Lemoine, F.; et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature 2022, 602, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef] [PubMed]
- VanBlargan, L.A.; Errico, J.M.; Halfmann, P.J.; Zost, S.J.; Crowe, J.E., Jr.; Purcell, L.A.; Kawaoka, Y.; Corti, D.; Fremont, D.H.; Diamond, M.S. An infectious SARS-CoV-2 B.1.1.529 Omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat Med 2022, 28, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Keeton, R.; Tincho, M.B.; Ngomti, A.; Baguma, R.; Benede, N.; Suzuki, A.; Khan, K.; Cele, S.; Bernstein, M.; Karim, F.; et al. T cell responses to SARS-CoV-2 spike cross-recognize Omicron. Nature 2022, 603, 488–492. [Google Scholar] [CrossRef]
- Liu, J.; Chandrashekar, A.; Sellers, D.; Barrett, J.; Jacob-Dolan, C.; Lifton, M.; McMahan, K.; Sciacca, M.; VanWyk, H.; Wu, C.; et al. Vaccines elicit highly conserved cellular immunity to SARS-CoV-2 Omicron. Nature 2022, 603, 493–496. [Google Scholar] [CrossRef]
- Falsey, A.R.; Frenck, R.W., Jr.; Walsh, E.E.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Bailey, R.; Swanson, K.A.; Xu, X.; et al. SARS-CoV-2 Neutralization with BNT162b2 Vaccine Dose 3. N Engl J Med 2021, 385, 1627–1629. [Google Scholar] [CrossRef]
- Garcia-Beltran, W.F.; St Denis, K.J.; Hoelzemer, A.; Lam, E.C.; Nitido, A.D.; Sheehan, M.L.; Berrios, C.; Ofoman, O.; Chang, C.C.; Hauser, B.M.; et al. mRNA-based COVID-19 vaccine boosters induce neutralizing immunity against SARS-CoV-2 Omicron variant. Cell 2022, 185, 457–466.e4. [Google Scholar] [CrossRef]
- Chu, L.; Vrbicky, K.; Montefiori, D.; Huang, W.; Nestorova, B.; Chang, Y.; Carfi, A.; Edwards, D.K.; Oestreicher, J.; Legault, H.; et al. Immune response to SARS-CoV-2 after a booster of mRNA-1273: An open-label phase 2 trial. Nat Med 2022, 28, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Muecksch, F.; Wang, Z.; Cho, A.; Gaebler, C.; Ben Tanfous, T.; DaSilva, J.; Bednarski, E.; Ramos, V.; Zong, S.; Johnson, B.; et al. Increased memory B cell potency and breadth after a SARS-CoV-2 mRNA boost. Nature 2022, 607, 128–134. [Google Scholar] [CrossRef]
- Wragg, K.M.; Lee, W.S.; Koutsakos, M.; Tan, H.X.; Amarasena, T.; Reynaldi, A.; Gare, G.; Konstandopoulos, P.; Field, K.R.; Esterbauer, R.; et al. Establishment and recall of SARS-CoV-2 spike epitope-specific CD4(+) T cell memory. Nat Immunol 2022, 23, 768–780. [Google Scholar] [CrossRef]
- Corradini, P.; Agrati, C.; Apolone, G.; Mantovani, A.; Giannarelli, D.; Marasco, V.; Bordoni, V.; Sacchi, A.; Matusali, G.; Salvarani, C.; et al. Humoral and T-cell immune response after three doses of mRNA SARS-CoV-2 vaccines in fragile patients: The Italian VAX4FRAIL study. Clin Infect Dis 2022. [Google Scholar] [CrossRef]
- Lim, S.H.; Stuart, B.; Joseph-Pietras, D.; Johnson, M.; Campbell, N.; Kelly, A.; Jeffrey, D.; Turaj, A.H.; Rolfvondenbaumen, K.; Galloway, C.; et al. Immune responses against SARS-CoV-2 variants after two and three doses of vaccine in B-cell malignancies: UK PROSECO study. Nat Cancer 2022, 3, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Muecksch, F.; Schaefer-Babajew, D.; Finkin, S.; Viant, C.; Gaebler, C.; Hoffmann, H.H.; Barnes, C.O.; Cipolla, M.; Ramos, V.; et al. Naturally enhanced neutralizing breadth against SARS-CoV-2 one year after infection. Nature 2021, 595, 426–431. [Google Scholar] [CrossRef]
- Sokal, A.; Barba-Spaeth, G.; Fernandez, I.; Broketa, M.; Azzaoui, I.; de La Selle, A.; Vandenberghe, A.; Fourati, S.; Roeser, A.; Meola, A.; et al. mRNA vaccination of naive and COVID-19-recovered individuals elicits potent memory B cells that recognize SARS-CoV-2 variants. Immunity 2021, 54, 2893–2907.e5. [Google Scholar] [CrossRef]
- Stamatatos, L.; Czartoski, J.; Wan, Y.H.; Homad, L.J.; Rubin, V.; Glantz, H.; Neradilek, M.; Seydoux, E.; Jennewein, M.F.; MacCamy, A.J.; et al. mRNA vaccination boosts cross-variant neutralizing antibodies elicited by SARS-CoV-2 infection. Science 2021. [Google Scholar] [CrossRef]
- Naranbhai, V.; Nathan, A.; Kaseke, C.; Berrios, C.; Khatri, A.; Choi, S.; Getz, M.A.; Tano-Menka, R.; Ofoman, O.; Gayton, A.; et al. T cell reactivity to the SARS-CoV-2 Omicron variant is preserved in most but not all individuals. Cell 2022, 185, 1041–1051.e6. [Google Scholar] [CrossRef] [PubMed]
- Rodda, L.B.; Morawski, P.A.; Pruner, K.B.; Fahning, M.L.; Howard, C.A.; Franko, N.; Logue, J.; Eggenberger, J.; Stokes, C.; Golez, I.; et al. Imprinted SARS-CoV-2-specific memory lymphocytes define hybrid immunity. Cell 2022, 185, 1588–1601.e14. [Google Scholar] [CrossRef]
- Minervina, A.A.; Pogorelyy, M.V.; Kirk, A.M.; Crawford, J.C.; Allen, E.K.; Chou, C.H.; Mettelman, R.C.; Allison, K.J.; Lin, C.Y.; Brice, D.C.; et al. SARS-CoV-2 antigen exposure history shapes phenotypes and specificity of memory CD8(+) T cells. Nat Immunol 2022, 23, 781–790. [Google Scholar] [CrossRef]
- Dykema, A.G.; Zhang, B.; Woldemeskel, B.A.; Garliss, C.C.; Rashid, R.; Westlake, T.; Zhang, L.; Zhang, J.; Cheung, L.S.; Caushi, J.X.; et al. SARS-CoV-2 vaccination diversifies the CD4+ spike-reactive T cell repertoire in patients with prior SARS-CoV-2 infection. EBioMedicine 2022, 80, 104048. [Google Scholar] [CrossRef] [PubMed]
- Tarke, A.; Zhang, Y.; Methot, N.; Narowski, T.M.; Phillips, E.; Mallal, S.; Frazier, A.; Filaci, G.; Weiskopf, D.; Dan, J.M.; et al. Targets and cross-reactivity of human T cell recognition of common cold coronaviruses. Cell Rep Med 2023, 4, 101088. [Google Scholar] [CrossRef]
- Neidleman, J.; Luo, X.; McGregor, M.; Xie, G.; Murray, V.; Greene, W.C.; Lee, S.A.; Roan, N.R. mRNA vaccine-induced T cells respond identically to SARS-CoV-2 variants of concern but differ in longevity and homing properties depending on prior infection status. Elife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. Fast-evolving COVID variants complicate vaccine updates. Nature 2022, 607, 18–19. [Google Scholar] [CrossRef]
- Bartsch, S.M.; O'Shea, K.J.; John, D.C.; Strych, U.; Bottazzi, M.E.; Martinez, M.F.; Ciciriello, A.; Chin, K.L.; Weatherwax, C.; Velmurugan, K.; et al. The potential epidemiologic, clinical, and economic value of a universal coronavirus vaccine: A modelling study. EClinicalMedicine 2024, 68, 102369. [Google Scholar] [CrossRef]
- Cohen, J. Making broader coronavirus vaccines is a struggle. Science 2022, 377, 566–567. [Google Scholar] [CrossRef]
- Dolgin, E. Pan-coronavirus vaccine pipeline takes form. Nat Rev Drug Discov 2022, 21, 324–326. [Google Scholar] [CrossRef] [PubMed]
- Cankat, S.; Demael, M.U.; Swadling, L. In search of a pan-coronavirus vaccine: Next-generation vaccine design and immune mechanisms. Cell Mol Immunol 2024, 21, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.C.; Hentenaar, I.T.; Morrison-Porter, A.; Solano, D.; Haddad, N.S.; Castrillon, C.; Lamothe, P.A.; Andrews, J.; Roberts, D.; Lonial, S.; et al. The Majority of SARS-CoV-2 Plasma Cells are Excluded from the Bone Marrow Long-Lived Compartment 33 Months after mRNA Vaccination. medRxiv 2024. [Google Scholar] [CrossRef]
- Liu, L.; Fuhlbrigge, R.C.; Karibian, K.; Tian, T.; Kupper, T.S. Dynamic programming of CD8+ T cell trafficking after live viral immunization. Immunity 2006, 25, 511–520. [Google Scholar] [CrossRef]
- Louis, L.; Clark, M.; Wise, M.C.; Glennie, N.; Wong, A.; Broderick, K.; Uzonna, J.; Weiner, D.B.; Scott, P. Intradermal Synthetic DNA Vaccination Generates Leishmania-Specific T Cells in the Skin and Protection against Leishmania major. Infect Immun 2019, 87. [Google Scholar] [CrossRef]
- Zens, K.D.; Chen, J.K.; Farber, D.L. Vaccine-generated lung tissue-resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight 2016, 1. [Google Scholar] [CrossRef]
- Marinaik, C.B.; Kingstad-Bakke, B.; Lee, W.; Hatta, M.; Sonsalla, M.; Larsen, A.; Neldner, B.; Gasper, D.J.; Kedl, R.M.; Kawaoka, Y.; et al. Programming Multifaceted Pulmonary T Cell Immunity by Combination Adjuvants. Cell Rep Med 2020, 1, 100095. [Google Scholar] [CrossRef] [PubMed]
- Lapuente, D.; Storcksdieck Genannt Bonsmann, M.; Maaske, A.; Stab, V.; Heinecke, V.; Watzstedt, K.; Hess, R.; Westendorf, A.M.; Bayer, W.; Ehrhardt, C.; et al. IL-1beta as mucosal vaccine adjuvant: The specific induction of tissue-resident memory T cells improves the heterosubtypic immunity against influenza A viruses. Mucosal Immunol 2018, 11, 1265–1278. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Iwasaki, A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature 2012, 491, 463–467. [Google Scholar] [CrossRef]
- Bernstein, D.I.; Cardin, R.D.; Bravo, F.J.; Awasthi, S.; Lu, P.; Pullum, D.A.; Dixon, D.A.; Iwasaki, A.; Friedman, H.M. Successful application of prime and pull strategy for a therapeutic HSV vaccine. NPJ Vaccines 2019, 4, 33. [Google Scholar] [CrossRef]
- Hobbs, S.J.; Nolz, J.C. Targeted Expansion of Tissue-Resident CD8(+) T Cells to Boost Cellular Immunity in the Skin. Cell Rep 2019, 29, 2990–2997.e2. [Google Scholar] [CrossRef]
- Van Braeckel-Budimir, N.; Varga, S.M.; Badovinac, V.P.; Harty, J.T. Repeated Antigen Exposure Extends the Durability of Influenza-Specific Lung-Resident Memory CD8(+) T Cells and Heterosubtypic Immunity. Cell Rep 2018, 24, 3374–3382.e3. [Google Scholar] [CrossRef]
- Davies, B.; Prier, J.E.; Jones, C.M.; Gebhardt, T.; Carbone, F.R.; Mackay, L.K. Cutting Edge: Tissue-Resident Memory T Cells Generated by Multiple Immunizations or Localized Deposition Provide Enhanced Immunity. J Immunol 2017, 198, 2233–2237. [Google Scholar] [CrossRef] [PubMed]
- Alu, A.; Chen, L.; Lei, H.; Wei, Y.; Tian, X.; Wei, X. Intranasal COVID-19 vaccines: From bench to bed. EBioMedicine 2022, 76, 103841. [Google Scholar] [CrossRef]
- Adler, J.M.; Martin Vidal, R.; Langner, C.; Vladimirova, D.; Abdelgawad, A.; Kunecova, D.; Lin, X.; Nouailles, G.; Voss, A.; Kunder, S.; et al. An intranasal live-attenuated SARS-CoV-2 vaccine limits virus transmission. Nat Commun 2024, 15, 995. [Google Scholar] [CrossRef]
- Warner, B.M.; Yates, J.G.E.; Vendramelli, R.; Truong, T.; Meilleur, C.; Chan, L.; Leacy, A.; Pham, P.H.; Pei, Y.; Susta, L.; et al. Intranasal vaccination with an NDV-vectored SARS-CoV-2 vaccine protects against Delta and Omicron challenges. NPJ Vaccines 2024, 9, 90. [Google Scholar] [CrossRef]
- Jearanaiwitayakul, T.; Sunintaboon, P.; Kittiayuwat, A.; Limthongkul, J.; Wathanaphol, J.; Janhirun, Y.; Lerdsamran, H.; Wiriyarat, W.; Ubol, S. Intranasal immunization with the bivalent SARS-CoV-2 vaccine effectively protects mice from nasal infection and completely inhibits disease development. Vaccine 2024, 42, 3664–3673. [Google Scholar] [CrossRef] [PubMed]
- Pulendran, B.; P, S.A.; O'Hagan, D.T. Emerging concepts in the science of vaccine adjuvants. Nat Rev Drug Discov 2021, 20, 454–475. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Coordinated innate and adaptive immune responses to SARS-CoV-2 infection correlate with better clinical outcomes. Protective immunity to SARS-CoV-2 infection requires the prompt stimulation of the innate immune system that, through the activation of type I/III IFN pathways, limits viral replication and triggers the timely induction of adaptive immune responses. In turn, SARS-CoV-2-specific TH1 and TFH CD4+ T cells optimize the effector function of cytotoxic CD8+ T cells and the production of potent neutralizing Abs, leading to the resolution of the infection in about two weeks. On the contrary, the defective activation of type I/III IFN responses results in unrestrained viral replication, delayed activation of the adaptive immune response, and sustained late production of proinflammatory cytokines. The impaired induction of SARS-CoV-2-specific cytotoxic CD8+ T cells and the wrong polarization of CD4+ T cells results in a reduced capacity to control the infection, while excessive inflammation causes extensive tissue damage.
Figure 1.
Coordinated innate and adaptive immune responses to SARS-CoV-2 infection correlate with better clinical outcomes. Protective immunity to SARS-CoV-2 infection requires the prompt stimulation of the innate immune system that, through the activation of type I/III IFN pathways, limits viral replication and triggers the timely induction of adaptive immune responses. In turn, SARS-CoV-2-specific TH1 and TFH CD4+ T cells optimize the effector function of cytotoxic CD8+ T cells and the production of potent neutralizing Abs, leading to the resolution of the infection in about two weeks. On the contrary, the defective activation of type I/III IFN responses results in unrestrained viral replication, delayed activation of the adaptive immune response, and sustained late production of proinflammatory cytokines. The impaired induction of SARS-CoV-2-specific cytotoxic CD8+ T cells and the wrong polarization of CD4+ T cells results in a reduced capacity to control the infection, while excessive inflammation causes extensive tissue damage.
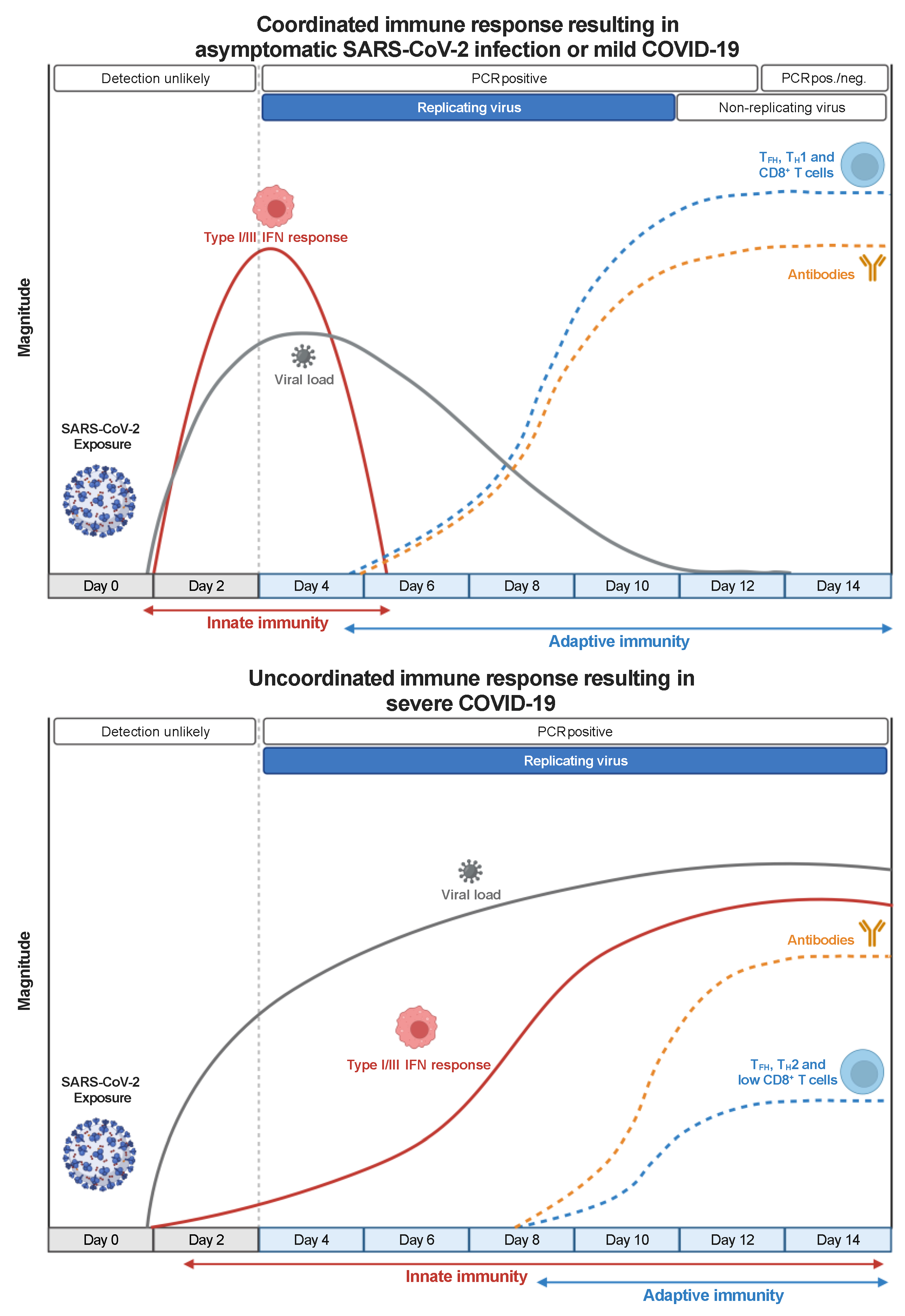
Figure 2.
Correlates of increased severe COVID-19 risk with aging. The risk of developing severe COVID-19 is significantly higher in the elderly. Several factors contribute to the increased risk, including an elevated incidence of co-morbidities, a reduced repertoire of naïve T cells, alterations in type-I/III signaling, and a higher basal inflammatory level. These factors lead to the uncoordinated innate and adaptive immune responses underlying severe COVID-19.
Figure 2.
Correlates of increased severe COVID-19 risk with aging. The risk of developing severe COVID-19 is significantly higher in the elderly. Several factors contribute to the increased risk, including an elevated incidence of co-morbidities, a reduced repertoire of naïve T cells, alterations in type-I/III signaling, and a higher basal inflammatory level. These factors lead to the uncoordinated innate and adaptive immune responses underlying severe COVID-19.
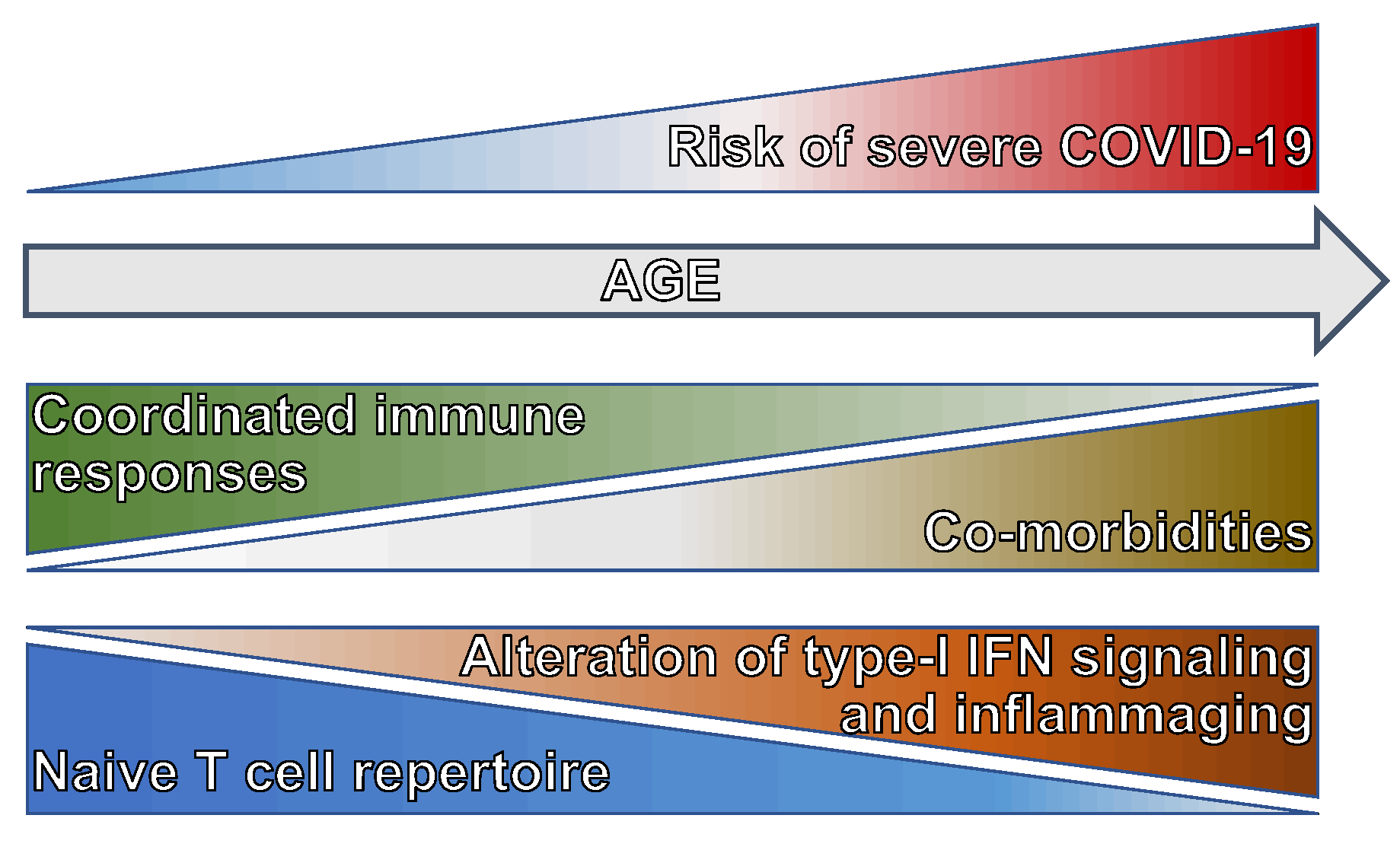
Figure 3.
Kinetics of CD4+ and CD8+ T cell responses to SARS-CoV-2 infection and vaccination. SARS-CoV-2-specific T cells are detected as early as 3-5 days after symptoms onset and expand in the following 2-3 weeks. Then, their frequencies start to decline with kinetics that, in the first two months, are slightly faster for CD8+ T cells than CD4+ T cells. SARS-CoV-2-specific memory CD4+ and CD8+ T cells are detectable up to 12-15 months after infection or vaccination and predicted to be long-lived.
Figure 3.
Kinetics of CD4+ and CD8+ T cell responses to SARS-CoV-2 infection and vaccination. SARS-CoV-2-specific T cells are detected as early as 3-5 days after symptoms onset and expand in the following 2-3 weeks. Then, their frequencies start to decline with kinetics that, in the first two months, are slightly faster for CD8+ T cells than CD4+ T cells. SARS-CoV-2-specific memory CD4+ and CD8+ T cells are detectable up to 12-15 months after infection or vaccination and predicted to be long-lived.
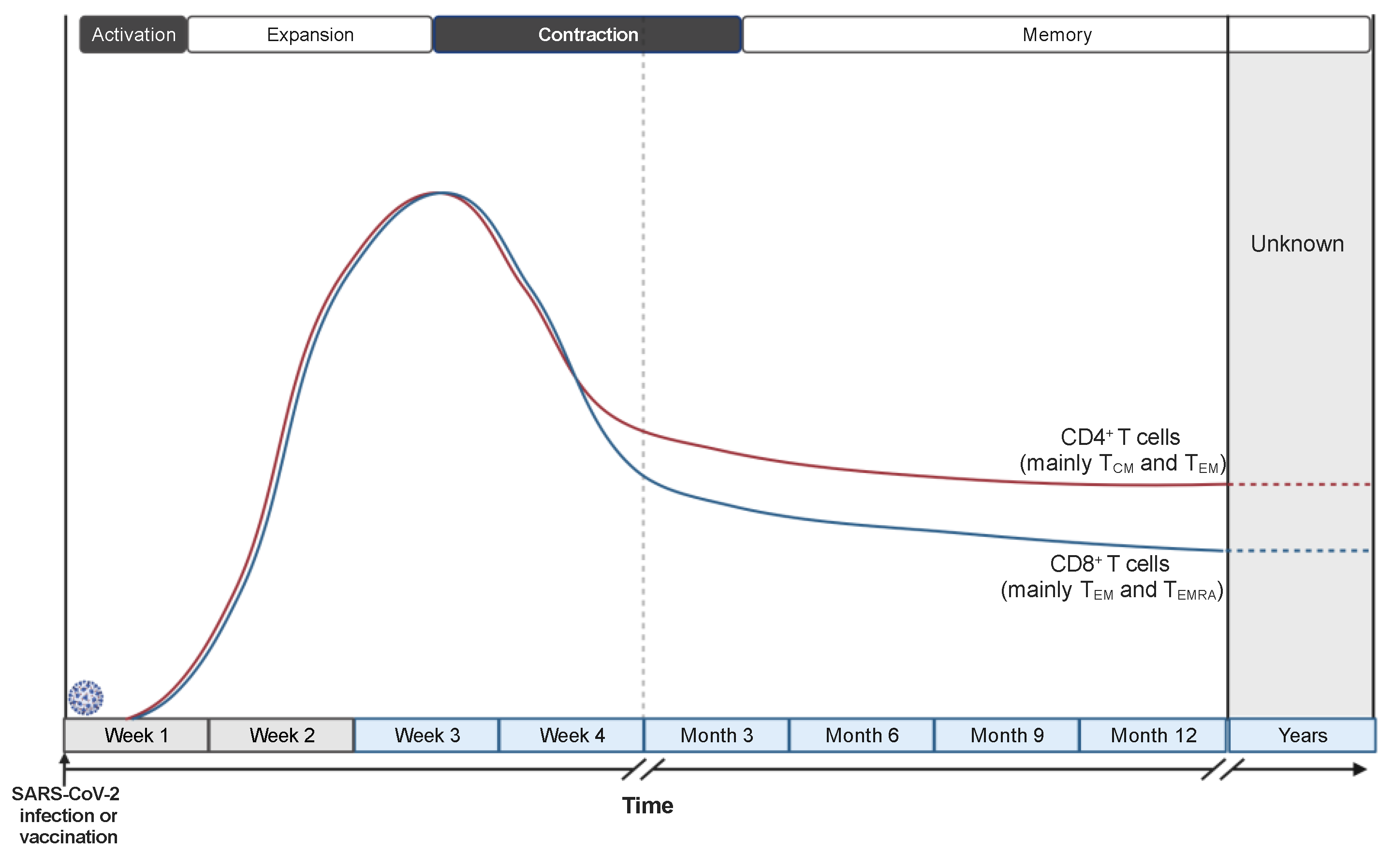
Figure 4.
Protective immunological memory induced by SARS-CoV-2 infection and vaccination. SARS-CoV-2 infection and COVID-19 vaccines elicit the production of circulating neutralizing and non-neutralizing Abs and induce the differentiation of memory B, CD4+ T cells, and CD8+ T cells that can significantly protect against severe COVID-19, hospitalization, and death. The natural infection also stimulates the differentiation of mucosal neutralizing Abs and CD4+ and CD8+ TRM cells that provide local protection at the site of viral entry and avoid virus spread. Neutralizing Abs are the only component of the adaptive immune system that can prevent infection and generate sterilizing immunity.
Figure 4.
Protective immunological memory induced by SARS-CoV-2 infection and vaccination. SARS-CoV-2 infection and COVID-19 vaccines elicit the production of circulating neutralizing and non-neutralizing Abs and induce the differentiation of memory B, CD4+ T cells, and CD8+ T cells that can significantly protect against severe COVID-19, hospitalization, and death. The natural infection also stimulates the differentiation of mucosal neutralizing Abs and CD4+ and CD8+ TRM cells that provide local protection at the site of viral entry and avoid virus spread. Neutralizing Abs are the only component of the adaptive immune system that can prevent infection and generate sterilizing immunity.
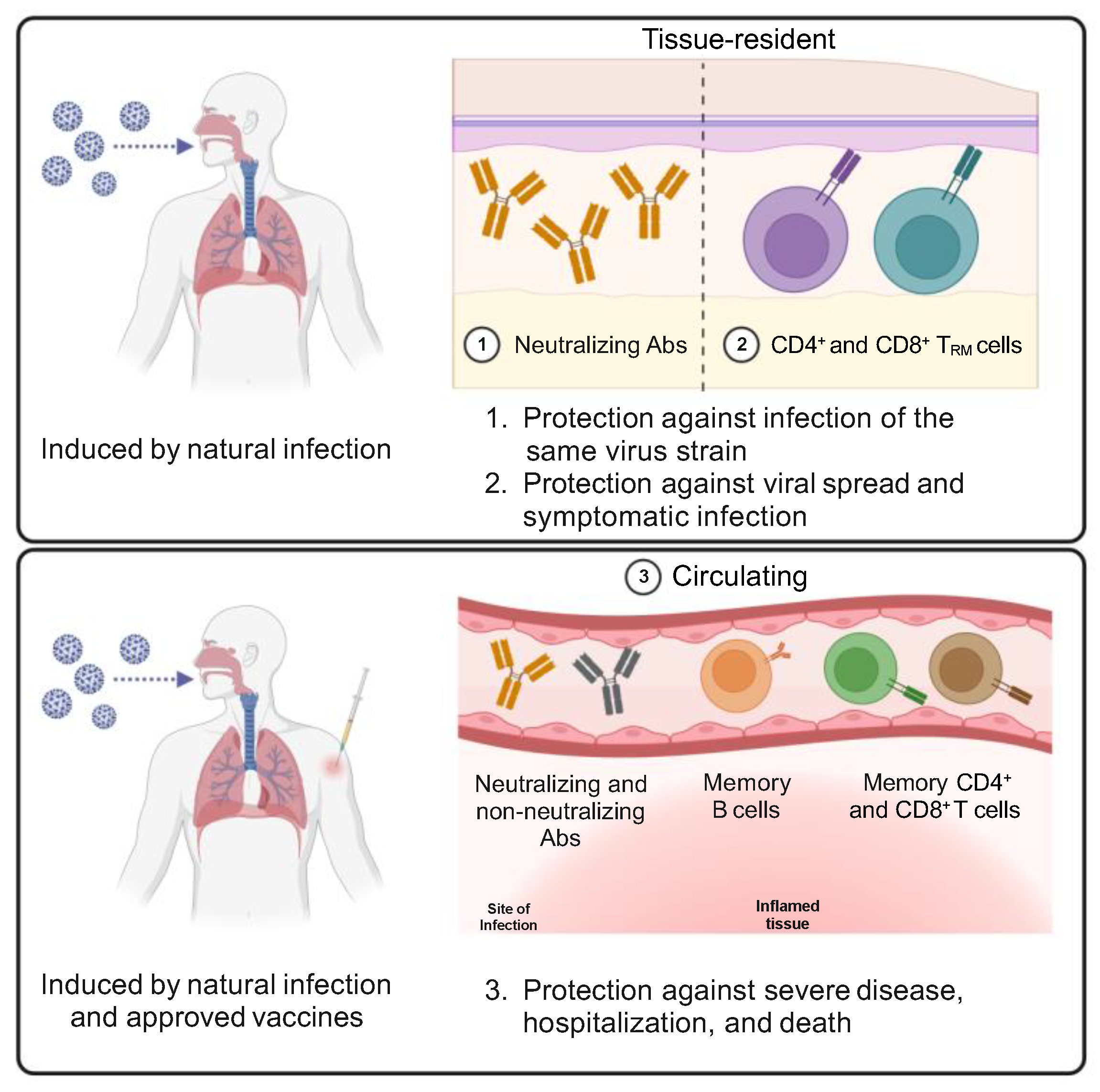
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



