
Submitted:
28 June 2024
Posted:
28 June 2024
You are already at the latest version
Abstract
Evans' Syndrome (ES) is a rare autoimmune disorder characterized by the simultaneous occurrence of immune thrombocytopenia (ITP) and autoimmune hemolytic anemia (AIHA). Thrombotic complications in ES patients are uncommon, particularly involving Buerger's Disease (BD). We report a case of a 49-year-old male with ES and a history of diabetes and heavy smoking, presenting with a necrotic wound on his right great toe. Diagnostic evaluations revealed severe stenosis and thrombosis in the lower limb arteries, diagnosed as BD. The patient underwent successful popliteal-tibioperoneal artery bypass surgery and subsequent disarticulation and revision of the distal phalanx, followed by the application of an acellular dermal matrix (ADM) to promote healing. Post-surgery, the patient showed significant improvement in blood flow and complete epithelialization without complications. This case highlights the importance of a multidisciplinary approach in managing complex wounds in ES patients, suggesting potential treatment pathways for future cases involving BD
Keywords:
Evans' syndrome
; Thromboangiitis Obliterans
; Thrombosis
; Autoimmune Disease
; Case Reports
1. Introduction
Evans syndrome (ES), first described by Evans in 1951, is defined as the simultaneous or sequential occurrence of immune thrombocytopenia (ITP) and warm autoimmune hemolytic anemia (AIHA) [1]. Studies performed over the past 40 years have revealed that ES has an annual incidence of only 1.8 cases per million people and a prevalence of 21.3 cases per million [2]. Because of its rarity, comparative clinical trials focusing on treatment methodologies are limited, and most current treatment recommendations are derived from those established for ITP and AIHA. Additionally, studies examining the involvement of plastic surgery departments in treating wounds in patients with ES are rare [3,4].
Here, we report a novel case of a patient typically managed for ES who concurrently developed Buerger’s disease (BD), also known as thromboangiitis obliterans. BD is a rare inflammatory condition primarily affecting the blood vessels of the arms and legs. This condition is characterized by inflammation and clotting in small- and medium-sized vessels, which reduces blood flow to affected areas [5]. Reduced blood flow can lead to pain, numbness, and swelling within angiosomes; patients may exhibit a bluish appearance at the extremities because of impaired circulation. Skin necrosis or ulceration can occur in severe cases [6,7].
Although a direct correlation between ES and thrombogenic diseases such as BD has not been conclusively established, recent studies indicated that autoimmune hemolytic anemia, a component of ES, is associated with a heightened risk of venous thromboembolism [8]. This case report reviews the association between ES and thrombotic complications, emphasizing the clinical diversity and complexity of ES. Our case of successful management of BD using a multidisciplinary approach demonstrates the importance of tailored management strategies and treatment of associated complications in ES.
2. Case Presentation
2.1. Initial Clinical Findings
A 49-year-old male, diagnosed with ES 3 years prior at our institution, presented with an open wound in his right great toe. Initial examination revealed symptoms of blue toe syndrome along with partial exposure of the distal phalanx (Figure 1).
2.2. Medical and Treatment History
The patient was a smoker with a 20-pack-year history and had been under continuous medical supervision for ES. The patient has a history of diabetes mellitus; however, no other significant conditions were identified. In 2018, the patient presented to our emergency department with persistent epistaxis, gingival bleeding, and petechial hemorrhage. These symptoms prompted his admission to the hematology-oncology department, where he began treatment with steroid pulse therapy. Multiple interventions were implemented following disease progression, characterized by a continuous decline in the platelet count. These interventions included repeated doses of intravenous immunoglobulin and platelet transfusions, ultimately necessitating a splenectomy in 2019 (Table 1).
The patient was admitted to the plastic surgery department for treatment of a wound on the right great toe (highlighted in red).
2.3. Diagnostic Evaluation
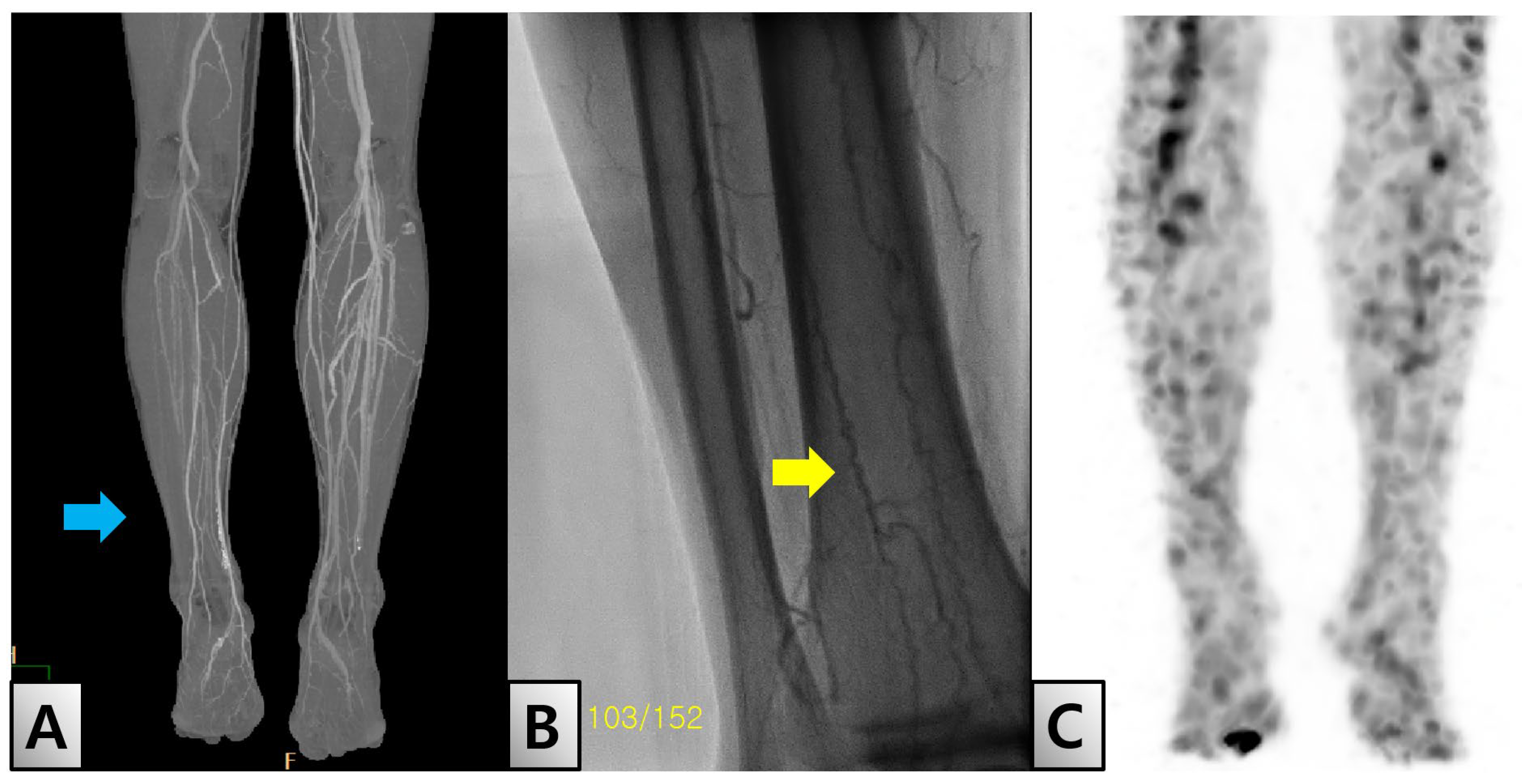
In April 2022, the patient returned to our facility one week after experiencing direct trauma to his toe, which led to necrosis of the distal phalanx. Diagnostic evaluations revealed an ankle-brachial index (ABI) of 0.85 on the right side and 1.3 on the left side, suggesting potential vascular occlusion. Further assessment using transcutaneous oxygen pressure showed measurements of 15 and 19 mmHg on the right and left sides, respectively. Detailed imaging studies, including lower-extremity angio-computed tomography (CT), identified thrombosis in both the popliteal and distal anterior tibial arteries (Figure. 2-A). The imaging results, along with the characteristic corkscrew pattern observed on subsequent angiography, led to a diagnosis of BD (Figure. 2-B). Additionally, white blood cell single-photon emission CT imaging showed uptake in the distal phalanx consistent with osteomyelitis (Figure. 2-C).
Figure 2.
Radiologic findings. (A) Lower extremity computed tomography shows severe stenosis in the right popliteal artery (blue arrow). (B) Lower extremity angiography shows a ‘corkscrew appearance’ (yellow arrow), which is characteristic of Buerger’s disease. (C) White blood cell single-photon emission computed tomography showed osteomyelitis in the right great toe.
Figure 2.
Radiologic findings. (A) Lower extremity computed tomography shows severe stenosis in the right popliteal artery (blue arrow). (B) Lower extremity angiography shows a ‘corkscrew appearance’ (yellow arrow), which is characteristic of Buerger’s disease. (C) White blood cell single-photon emission computed tomography showed osteomyelitis in the right great toe.
2.4. Intervention and Outcomes at Vascular Surgery Department
Twelve days after admission, an interdisciplinary approach involving both the cardiology and vascular surgery teams was employed to address the patient’s severe vascular complications. The initial attempt at angioplasty was hindered by pronounced stenosis of the popliteal artery. Consequently, a decision was made to perform a right popliteal-tibioperoneal artery bypass using the great saphenous vein to restore adequate blood flow. Postoperative imaging using follow-up CT showed significant improvements in popliteal flow. Furthermore, ABI measurements showed marked improvement, with readings of 0.97 on the right and 1.25 on the left, indicating successful vascular restoration. Three days after bypass surgery, the great toe displayed mild induration but remained stable and well-demarcated, indicating that the surgical intervention was successful (Figure. 3).
Figure 3.
Post-bypass surgery status of the right great toe. Three days following bypass surgery, the right great toe displayed mild induration but remained stable and well-demarcated, indicating the initial success of the surgical intervention. (A) Plantar view. (B) Dorsal view. (C) Anterior view.
Figure 3.
Post-bypass surgery status of the right great toe. Three days following bypass surgery, the right great toe displayed mild induration but remained stable and well-demarcated, indicating the initial success of the surgical intervention. (A) Plantar view. (B) Dorsal view. (C) Anterior view.

2.5. Surgical Procedures at Plastic Surgery Department
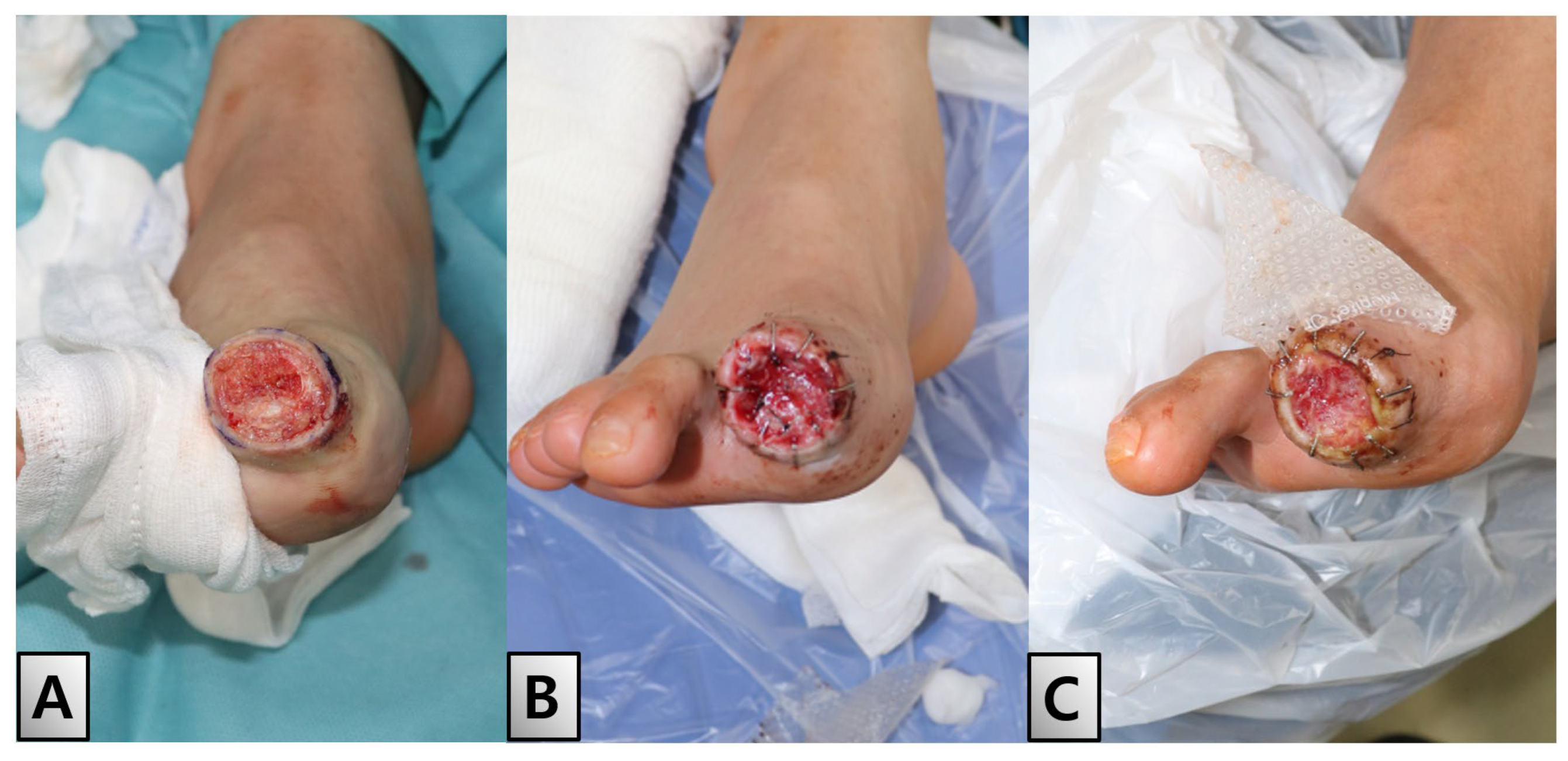
On day 21 of hospitalization, disarticulation and revision of the distal phalanx was performed under local anesthesia to address ongoing issues in the demarcated area. Despite the progression of tip necrosis, debridement revealed a healthy raw surface (Figure. 4-A). This favorable outcome enabled application of an acellular dermal matrix (SureDerm®, HansBiomed Corp, Seoul, Korea), which was used to promote healing and tissue regeneration at the injury site (Figure. 4-B, C).
Figure 4.
Post-disarticulation and healing process. On day 21 of hospitalization, a disarticulation and revision of the distal phalanx was performed under local anesthesia. Despite necrosis, debridement revealed a healthy raw surface, allowing for the application of an acellular dermal matrix (SureDerm®, HansBiomed Corp, Seoul, Korea). (A) Post-debridement view. (B) Application of the dermal matrix. (C) Healing with the well-taken matrix in place.
Figure 4.
Post-disarticulation and healing process. On day 21 of hospitalization, a disarticulation and revision of the distal phalanx was performed under local anesthesia. Despite necrosis, debridement revealed a healthy raw surface, allowing for the application of an acellular dermal matrix (SureDerm®, HansBiomed Corp, Seoul, Korea). (A) Post-debridement view. (B) Application of the dermal matrix. (C) Healing with the well-taken matrix in place.
2.6. Discharge and Follow-Up
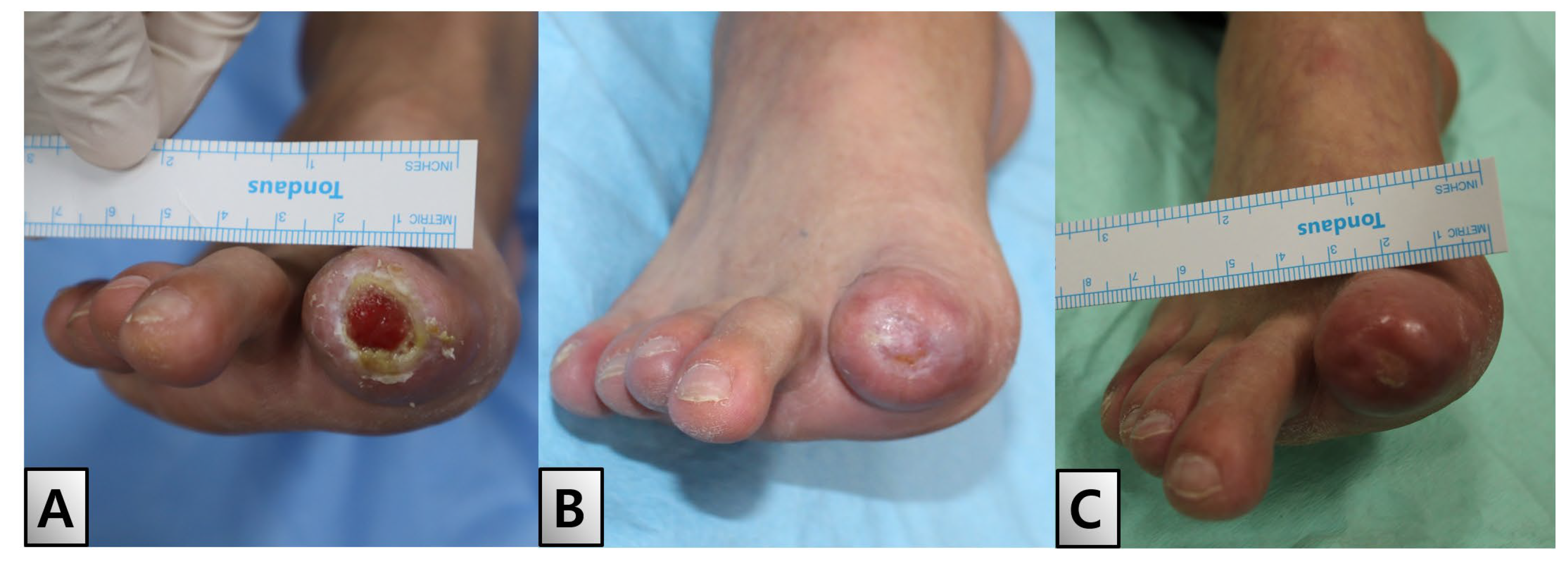
The patient was discharged after two weeks of dressing changes, during which initial epithelialization commenced from a 20% margin (Figure. 5-A). Outpatient follow-up visits were scheduled biweekly to monitor the patient’s progress. Full epithelialization was achieved within two weeks of discharge No complications or adverse effects were observed during the six-month follow-up period (Figure. 5-B, C).
Figure 5.
Discharge and follow-up. (A) Patient was discharged after two weeks, during which initial epithelialization commenced from a 20% margin. Outpatient follow-up visits were scheduled bi-weekly to monitor progress. (B) Full epithelialization achieved two weeks after discharge. (C) Condition six months post-discharge. Throughout the follow-up period, no complications or adverse effects were reported.
Figure 5.
Discharge and follow-up. (A) Patient was discharged after two weeks, during which initial epithelialization commenced from a 20% margin. Outpatient follow-up visits were scheduled bi-weekly to monitor progress. (B) Full epithelialization achieved two weeks after discharge. (C) Condition six months post-discharge. Throughout the follow-up period, no complications or adverse effects were reported.
2.7. Informed Consent
The patient provided written informed consent for the publication and use of his images.
3. Discussion
ES is a rare and complex autoimmune disorder characterized by the simultaneous or sequential occurrence of warm AIHA and ITP [1,9]. This condition often presents alongside autoimmune neutropenia, contributing to a variety of clinical symptoms due to anemia, thrombocytopenia, and leukopenia. Anemia associated with ES typically results from warm IgG autoantibodies, specifically IgA, but not cold agglutinins. Autoimmune neutropenia can also manifest as a symptom of ES, occurring in 15% adults and 20% children [9].
ES symptoms can resemble those of leukemia and lymphoma, necessitating a differential diagnosis to exclude these conditions. Hemolysis can lead to jaundice, dark brown urine, pallour, weakness, fatigue, and dyspnea. Thrombocytopenia may cause increased bruising and petechiae, which appear as small vessels bleeding into the skin, making blood vessels appear more prominent. Additionally, patients may exhibit bleeding tendencies such as epistaxis and menorrhagia. A low neutrophil count increases the risk of fever, oral ulcers, and bacterial infections [4].
The diagnosis of ES is based on characteristic symptoms, patient history, thorough clinical evaluation, and various specialized tests. Although there is no definitive test for ES, it is often diagnosed by excluding other potential conditions [3,4]. Specifically, ES can be diagnosed in the presence of both AIHA (positive direct Coombs test) and ITP in the same patient, regardless of whether these conditions occur simultaneously or sequentially [3,9]. In this case, the diagnosis was also based on symptoms and signs, with a positive direct Coombs test.
First-line treatments for ES typically involve steroids such as prednisone and prednisolone. In severe cases of immune thrombocytopenic purpura, intravenous immunoglobulin may be administered as life-saving therapy. Second-line treatments for refractory ES include rituximab, mycophenolate mofetil, cyclosporine, vincristine, azathioprine, sirolimus, and thrombopoietin receptor agonists [3,9]. The patient in this case was also treated with blood and platelet transfusions and primarily treated with steroids. Our therapeutic approach was consistent with those reported in the literature, starting with intravenous steroids and later including splenectomy and intravenous immunoglobulin when persistent thrombocytopenia and steroid resistance were observed. Steroids primarily act by removing macrophages, which destroy red blood cells and platelets. Concentrated IgG from human plasma donors blocks Fcγ receptors on macrophages, although this treatment remains controversial [10] Splenectomy is effective for treating thrombocytopenia; however, the potential side effects must be considered [11].
Reports of thrombotic complications associated with ES are rare. Neurological complications, such as cerebral thrombosis or hemorrhage, can occur, presenting with severe headaches, language difficulties, or paralysis [4]. Pulmonary embolism should be suspected in patients showing respiratory symptoms, such as difficulty breathing and chest pain. Thrombotic complications can be a serious issue in patients with ES because of decreased platelet counts and anemia, which can reduce the blood clotting ability [4]. Therefore, accurate diagnosis and treatment of thrombotic complications in patients with ES are crucial. Diagnostic tests for thrombotic complications in patients with ES may include blood tests such as D-dimer and coagulation studies, whereas imaging studies such as ultrasound and CT can help specify the location and severity of thrombi. Anticoagulation therapy and thrombolytic agents may be considered to prevent and manage thrombotic complications [4].
Because of the very low prevalence of ES, research on the association between ES and thrombotic complications is lacking. However, previous studies reported an increased likelihood of thrombosis as a complication of ITP and AIHA [12]. To our knowledge, no cases of lower-limb thrombotic complications in patients with ES have been reported to date. In our case, BD was accompanied by severe stenosis of the popliteal artery as observed in imaging (Figure. 2-A). BD primarily involves inflammation and thrombus formation in the small-to medium-sized vessels of the lower limbs, which can lead to narrowing and occlusion of the vessels, cutting off of the blood supply to related tissues, and symptoms such as pain and blue toe syndrome with tissue damage [5]. Typical corkscrew patterns were observed on angiography (Figure. 2-B). Therefore, appropriate radiological assessment and intervention planning considering BD are necessary in cases of lower-limb ulcers in patients with ES.
Our patient was previously treated for ES and had undergone splenectomy and steroid therapy; he also had a history of heavy smoking and diabetes. The patient showed improved blood flow and verified recovery of the AB) following vascular surgery and bypass surgery. Thus, a multidisciplinary approach should be considered for managing patients with ES and associated BD. The patient achieved successful epithelialization following debridement of the necrotic tissue, including the distal phalanx. During this process, an acellular dermal matrix was applied to promote healing, minimize damage to the surrounding tissues, and facilitate secondary intention healing. This method also provides a scaffold that supports cellular infiltration and tissue regeneration. The patient recovered completely without signs of infection or other complications, underscoring the effectiveness of this approach in managing complex wounds associated with ES and BD.
The primary limitation of this study is its reliance on a single patient. Thus, the reproducibility and generalizability of our results must be further analyzed before universal guidelines are recommended. However, based on the successful recovery of this patient, we suggest directions for treatment and diagnostic plans for future cases of ES with BD.
BD is a potential complication of EA, underscoring the need for precise diagnostic strategies and tailored treatment plans for patients with EA and highlighting the critical role of a multidisciplinary approach in managing the complexities of this disorder.
Author Contributions
Conceptualization, S.-Y.K.; methodology, S.-Y.K.; formal analysis, H.-J.C.; data curation, J.-Y.B.; writing—original draft preparation, H.-J.N.; writing—review and editing, H.-J.C.; visualization, H.-J.C.; supervision, H.-J.C.; project administration, J.-Y.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Institutional Review Board Statement
The study was approved for exemption by the Institutional Review Board of Soonchunhyang University Hospital (IRB exemption No. 2024-05).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on reasonable request from the corresponding author.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (grant number 2020R1A2C1100891) and Soonchunhyang Research Fund.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Evans RS,; Takahashi K,; Duane RT,; Payne R,; Liu C. Primary thrombocytopenic purpura and acquired hemolytic anemia: Evidence for a common etiology. Arch Intern Med 1951, 87, 48–65. [CrossRef] [PubMed]
- Hansen DL,; Möller S,; Andersen K,; Gaist D,; Frederiksen H. Evans syndrome in adults - incidence, prevalence, and survival in a nationwide cohort. Am J Hematol. 2019, 94, 1081–1090. [Google Scholar] [CrossRef]
- Audia S,; Grienay N,; Mounier M,; Michel M,; Bonnotte B. Evans’ syndrome: from diagnosis to treatment. J Clin Med 2020, 9, 3851. [Google Scholar] [CrossRef] [PubMed]
- Fattizzo B,; Michel M,; Giannotta JA, et al. Evans syndrome in adults: an observational multicenter study. Evans syndrome in adults: an observational multicenter study 2021, 5, 5468–5478. [Google Scholar]
- Olin JW,; Shih A. Thromboangiitis obliterans (Buerger’s disease). Curr Opin Rheumatol 2006, 18, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Cooper LT,; Tse TS,; Mikhail MA,; McBane RD,; Stanson AW,; Ballman KV. Long-term survival and amputation risk in thromboangiitis obliterans (Buerger’s disease). J Am Coll Cardiol 2004, 44, 2410–2411. [CrossRef] [PubMed]
- Le Joncour A,; Soudet S,; Dupont A, et al. French Buerger’s Network. Long-term outcome and prognostic factors of complications in thromboangiitis obliterans (Buerger’s disease): a multicenter study of 224 patients. J Am Heart Assoc 2018, 7, e010677. [Google Scholar] [CrossRef] [PubMed]
- Audia S,; Bach B,; Samson M, et al. Venous thromboembolic events during warm autoimmune hemolytic anemia. PLoS One. 2018, 13, e0207218. [Google Scholar]
- Michel M,; Chanet V,; Dechartres A, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood 2009, 114, 3167–3172. [Google Scholar] [CrossRef] [PubMed]
- Schwab I,; Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol. 2013, 13(3), 176–189. [Google Scholar] [CrossRef] [PubMed]
- Zanella A,; Barcellini W. Treatment of autoimmune hemolytic anemias. Haematologica. 2014, 99(10), 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Capecchi M,; Ciavarella A,; Artoni A,; Abbattista M,; Martinelli I. Thrombotic complications in patients with immune-mediated hemolysis. J Clin Med 2021, 10, 1764. [CrossRef] [PubMed]
Figure 1.
Initial findings. The entire right foot was chilled, and the right great toe had a bluish color change with a 2 × 1.5 cm-sized eschar formation on the toe tip, which had bone exposed depth. (A) Plantar view. (B) Dorsal view. (C) Anterior view.
Figure 1.
Initial findings. The entire right foot was chilled, and the right great toe had a bluish color change with a 2 × 1.5 cm-sized eschar formation on the toe tip, which had bone exposed depth. (A) Plantar view. (B) Dorsal view. (C) Anterior view.
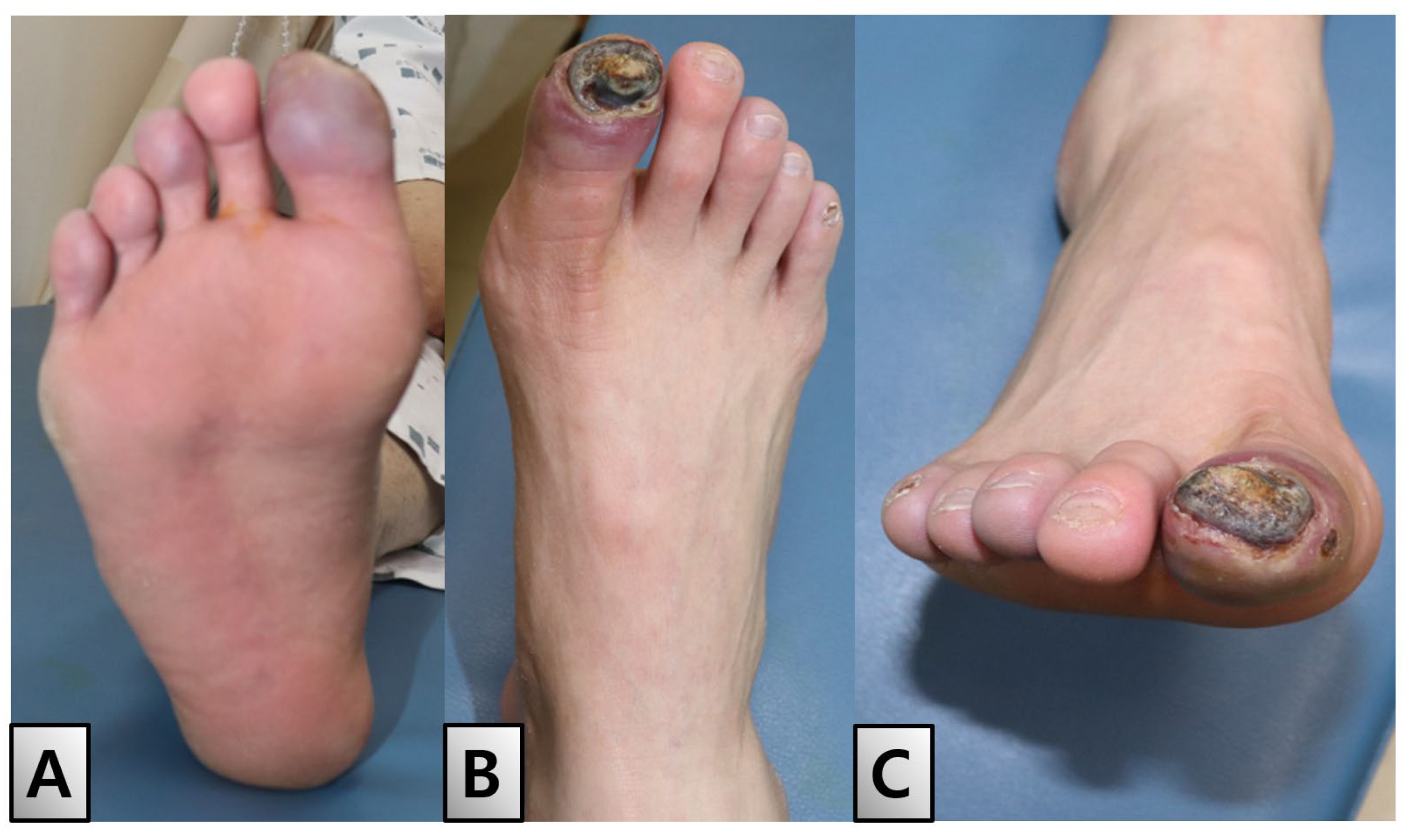

Table 1.
Comprehensive medical timeline of a patient with Evans’ syndrome.
| Date | Event | Details |
|---|---|---|
| Underlying Disease | Diagnosed with diabetes mellitus | Well controlled. |
| 2018.02.11 | Visit Emergency Room first time First Diagnosis of Evans’ Syndrome | Presented with epistaxis, gingival bleeding, and petechial hemorrhages.Antibody screening test: positive. Direct Coombs test: 4 positive. |
| 2018-2019 | Continuous Treatment | Multiple interventions including repeated doses of intravenous immunoglobulin and platelet transfusions. |
| 2019.08.03–2019.08.13 | Admission for Laparoscopic Splenectomy | Surgery performed to manage Evans’ Syndrome effectively. |
| 2022.04.18 | Admission by Plastic Surgery Department | For treatment of Right great toe wound. |
| 2022.04.21 | Lower Extremity CT | Focal thrombus identified in right popliteal artery and distal ATA. |
| 2022.05.04 | Right Femoral Arteriography | Severe stenosis in right popliteal artery; chronic total occlusions in right ATA, right posterior tibial artery (PTA), and right peroneal arteryCorkscrew findings suggestive of Buerger’s Disease. |
| 2022.05.12 | Right Popliteal-tibioperoneal artery bypass surgery with GSV. | Surgical intervention to bypass identified thrombus, using GSV. |
Abbreviations: CT; computed tomography, ATA; anterior tibial artery, PTA; posterior tibial artery, GSV; great saphenous vein.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



