
Submitted:
24 June 2024
Posted:
25 June 2024
You are already at the latest version
Abstract
Lirianthe delavayi has valuable ornamental and medicinal value, is an important economic tree, and has a long history of cultivation. As important regulatory molecules, miRNAs are involved in important physiological processes, such as the plant stress response, organ morphogenesis, hormone secretion, and signaling. In this study, based on miRNA sequencing analysis of wild and cultivated Lirianthe delavayi, we explored the differences in posttranscriptional regulation during the cultivation of Lirianthe delavayi from the aspect of miRNA molecular regulation. The results revealed the following: 1. A total of 365 miRNAs were sequenced and identified, and their first bases had obvious uracil (U) base preferences. Among them, 242 miRNAs were classified into 32 miRNA families; 2. Analysis of miRNA expression differences revealed that 39 miRNAs had significant expression differences between wild and cultivated populations; 3. Bioinformatics target gene prediction and target gene enrichment analysis of significantly differentially expressed miRNAs revealed that the target genes were significantly enriched in plant‒pathogen interactions, sphingolipid metabolism, defense responses, and ADP binding. 4. The plant‒pathogen interaction pathway involves seven differentially expressed miRNAs and 30 miRNA‒target gene regulatory pairs, which are involved mainly in pathogen signature molecule recognition and binding, signal transduction, and the regulation of disease resistance molecules or gene expression; Ath-MIR169g-p3_2ss7AG19GA, ath-MIR169g-p5_2ss7AG19GA, and ath-miR408-3p_L-3 are expressed only in wild population, and it is hypothesized that the domestication process from wild to cultivation species may be accompanied by the loss of disease resistance-related miRNAs. This study provides a reference for exploring the posttranscriptional-level regulatory mechanisms of adaptive changes in Lirianthe delavayi from wild to cultivated.
Keywords:
Lirianthe delavayi
; microRNA
; plant-pathogen interaction pathway
; miRNA-loss
1. Introduction
Lirianthe delavayi belongs to the Lirianthe family of Magnoliaceae, a dicotyledonous plant class of angiosperms. This species has a luxuriant crown, large turquoise leaves, and large, milky-white flowers with an aromatic odor, making it a valuable ornamental tree for gardens [1]. The bark and flowers can be used as medicine; the bark has the effect of strengthening the spleen and is used for treating dyspepsia, chronic gastritis, abdominal pain, diarrhea, etc. The flowers have the effect of relieving coughs and are used for treating rhinitis, sinusitis, bronchitis, coughing, etc. [2]. Due to its valuable ornamental and medicinal value, Lirianthe delavayi has a long history of cultivation in Yunnan Province as an economically important tree [3]. In the process of artificial cultivation, excellent traits such as unique flower color and strong flower fragrance have become the key and direction of plant selection [4], and cultivated populations produce some adaptive changes to adapt to the cultivation environment [5]; however, the related molecular regulatory mechanisms need to be further explored [6,7].
MicroRNAs (miRNAs) are a class of noncoding, endogenous, single-stranded small RNAs with regulatory functions that are found in eukaryotes and have a length of approximately 18-25 nt [8]. Plant miRNAs usually pair completely or nearly completely with target genes (target mRNAs), causing the cutting of target genes and thus regulating gene expression [8,10], thereby exerting a negative regulatory role at the posttranscriptional level [10]. As important regulatory molecules, miRNAs are involved in a series of important processes in plant life, including the plant stress response, organ morphogenesis, hormone secretion, and signal transduction [12,13,14,15]. The systematic identification of miRNAs in specific species is valuable for understanding their biological properties and regulatory networks. The expression of miRNAs in important economic crops, such as tomato [16], tobacco [17,18], olive [19], cabbage [20], tea tree [21], ginkgo [22], and Apocynum venetum [23], has been reported, and these miRNAs have been shown to play important roles in the regulation and improvement of important traits. Despite the increasing number of miRNA reports, information related to miRNAs in Lirianthe delavayi is still lacking.
The study of plant adaptation involves complex gene regulatory mechanisms [23], and the study of gene regulation can help to explore the effects of habitat differences (artificial versus natural environments) on plant biological processes and metabolic pathways during the cultivation of Lirianthe delavayi. Many studies have shown that miRNAs respond significantly to environmental stressors such as high salinity, drought and low temperature [23]. Many studies have demonstrated that some miRNAs, such as miR156, miR166, miR398, miR482, and miR858, are effective at promoting the development of miRNAs by targeting factors such as SQUAMOSA promoter-binding proteins [25], HD-zip [26], Cu-Zn superoxide dismutase [27], NBS-LRR [28], R2 R3-MYB [29], transcription factors and other functional genes involved in the environmental stress response. These miRNAs are thought to play critical roles in plant hormone regulation, signal transduction, plant‒pathogen interactions, and metabolic pathways [23]. Ma Yanming has conducted systematic identification and analysis of miRNAs in cultivated and wild adzpea and found that the differential expression of miRNAs is mainly related to disease resistance and stress resistance pathways [30]. In addition, our group reported in previous research that the abundance of pathogenic bacteria in cultivated Lirianthe delavayi increased compared with that in wild Lirianthe delavayi [31], suggesting that miRNA, an important regulatory molecule at the posttranscriptional level, may play an important regulatory role in the interaction between plants and pathogens. In recent years, the continuous updating and iteration of sequencing platforms have made large-scale analyses of miRNAs in nonmodel plants a reality [32]. In 2016, Xiaojiao Han et al. revealed the miRNA regulatory network under cadmium stress in southeastern Sedum through miRNA sequencing analyses [33]. Tan Jinhua et al. used a combination of miRNA and mRNA sequencing technologies to systematically identify and analyze miRNAs and mRNAs in Apocynum venetum under methyl jasmonate stress, revealing that the key regulatory pathways were mainly involved in biological processes such as redox homeostasis and phytohormone signaling [23]. Therefore, it is necessary and important to explore the potential posttranscriptional regulatory mechanisms of Lirianthe delavayi during cultivation via miRNA sequencing analysis and to identify the key functional pathways and regulatory networks involved in plant adaptation to different habitats to gain an in-depth understanding of the growth and developmental regulatory mechanisms of Lirianthe delavayi.
In this study, we used miRNA high-throughput sequencing technology and bioinformatics methods to systematically identify and analyze miRNAs in Lirianthe delavayi and screened out miRNAs that were differentially expressed in cultivated Lirianthe delavayi and wild Lirianthe delavayi. Then, the biological functions and key metabolic pathways of the target genes of the differentially expressed miRNAs were explored. Under the plant‒pathogen interaction pathway, the gene regulatory network and target gene functions of the differentially expressed miRNAs were explored, which provides a reference for understanding the molecular mechanism involved in adapting to different habitats between wild and cultivated Lirianthe delavayi.
2. Materials and Methods
2.1. Experimental Material
Samples were collected from wild and cultivated populations of Lirianthe delavayi (N:25°14 ′12.39″, E103°11′ 52.40″) in Majie town, Yiliang County. Four samples were collected, for a total of 8 samples. Leaf tissue from each sample was taken from the plant during the same period of the growing season, and after collection, it was quickly frozen in liquid nitrogen and stored at -80 degrees Celsius for further use.
2.2. Library Construction and Sequencing
The above eight samples were sent to Hangzhou Lianchuan Biotechnology Co., Ltd., the RNA was extracted, and after passing the quality inspection, the small RNA sequencing library was prepared by using the TruSeq Small RNA Sample Prep Kit (Illumina, San Diego, USA). After library preparation was completed, the constructed library was sequenced using an Illumina HiSeq 2000/2500, and the sequencing read length was single-ended 50 bp (SE50).
2.3. miRNA Identification
The raw sequencing data were counted to obtain the unique sequences of the sequencing data and the corresponding number of copies of each unique sequence. Raw sequencing data were first removed from 3′ adapters, sequences with sequencing base lengths less than 18 nt were removed, and sequences were filtered out if they had 80% A or C or G or T, only A, C and no G, T, or only G, T and no A, C, or were sequential nucleotide dimers, trimers, or sequential nucleotide dimers. Moreover, we compared and filtered the measured sequences from the mRNA, RFam (including rRNA, tRNA, snRNA, snoRNA, etc.) and Repbase (without miRNA) databases, called the filtered data valid data, and carried out further miRNA comparison, identification and prediction analyses on the valid data. Since there is no reported genome for Camellia sinensis, we used the Camellia sinensis genome from the TPIA database as the reference genome (downloaded at http://tpia.teaplant.org/web/Download/Genomic_data/CSS_ChrLev_20200506_Genome.fas.gz) and used ACGT101-miR to perform miRNA identification on valid data, including comparisons with miRNA precursors and mature bodies in the miRBase database, comparisons with the reference genome, and predictions of miRN precursors in the genome based on the principle of the 11 hairpin structure to identify possible miRNAs. miRNAs were identified and analyzed via the miRBase database and reference genome for possible miRNAs.
2.4. Differential Expression Analysis
The identification of miRNAs was performed using ACGT101-miR, and the expression level of miRNAs in each sample was calculated. In measuring expression, the normal value was used as a measure of miRNA expression [33], and thus, the correlation of miRNA expression in samples within and between groups and the differences in miRNA expression were assessed. Differential miRNAs were screened using the t test with a threshold of P<=0.05. Differential miRNAs were visualized and analyzed using an online tool (https://www.omicstudio.cn/tool. ).
2.5. miRNA Target Gene Prediction and Enrichment Analysis
For miRNA target gene prediction, transcriptome data of Camellia sinensis were used as a target background library, and psRobot (1.2) software was used for prediction with a score <2.5 as a threshold [34].
The enrichment analysis of target genes included two main steps: GO functional enrichment analysis and KEGG pathway functional enrichment analysis, which can determine the most important biochemical metabolic pathways and biological functions in which target genes are involved [30]. First, all target genes of differentially expressed miRNAs corresponded to each GO function or each KEGG pathway, and the number of genes was counted. Then, a hypergeometric test was applied to calculate the p value, and the specific research method used was described in existing reports [36].
2.6. miRNA-Gene-KEGG Network Regulation and Target Gene Function Analysis
Pathways that were significantly enriched and of interest were selected for analysis. In this study, the plant‒pathogen interaction pathway was selected for analysis, and target genes enriched in this pathway and their corresponding miRNAs were mapped and analyzed using Cytoscape 3.10.1. Based on the KEGG database, the target genes were projected under the plant‒pathogen interaction pathway (ko04626) in Arabidopsis thaliana to determine the position of the target genes under the pathway.
3. Results
3.1. Identification of miRNAs of Lirianthe Delavayi
To explore the regulatory mechanism of miRNA-mediated posttranscriptional regulation in cultivated and wild populations of Lirianthe delavayi, we established 8 miRNA libraries and conducted high-throughput miRNA sequencing. The original sequence data login number was PRJNA1109302. The 3′ adapters and garbage sequences were removed, and sequences with base lengths ranging from 18 to 25 nt were retained. A total of 365 miRNAs were identified, including 13 novel miRNAs (Supplementary Table S1). Analysis of the first base of all the identified miRNAs showed that their first bases had an obvious uracil (U) base preference, and this was the highest percentage of miRNAs with a sequence length of 21 bp (61.49%) (Figure 1).
3.1.1. miRNA Family Analysis of Lirianthe delavayi
The family analysis of the 365 miRNAs identified in Lirianthe delavayi revealed that 242 miRNAs were classified into 32 families. The MIR482 family had the most family members, with 32 family members, followed by the MIR167 family, with 20 family members, and a majority of the families had 1 to 3 family members (Attached Table 2). A comparison of the distribution of miRNA family members in Lirianthe delavayi with the miRNA families of other species included in the miRBase database revealed that 19 species, such as Lirianthe delavayi, Arabidopsis thaliana and potato, were clustered into one team, and the rest of the species were clustered into another group, with similar numbers and distributions of their members in each miRNA family. The number and distribution of 21 miRNA families, such as MIR390 and MIR482, were similar across species, and their number and distribution were greater than those of other families (Figure 2A).
Among the 365 miRNAs identified, 29 could not only be compared with the pre-miRNAs of the reference species Camellia sinensis but also with precursors that could be further compared with the Camellia sinensis genome. Phylogenetic analysis of these 29 miRNAs revealed that the miRNAs of the same family were basically clustered together, with five main miRNA families, namely, MIR171, MIR156, MIR319, MIR167, and MIR166, and the same family exhibiting more similar sequence information (Figure 2B).
3.1.2. Chromosomal Localization of Identified miRNAs in Lirianthe delavayi
Chromosomal localization analysis of miRNAs was performed using the MG2C online program. The miRNA precursors were distributed on all 15 chromosomes (Figure 3). Many miRNAs are distributed on chromosomes 14, 2, and 3, with 35, 26, and 26 miRNA precursors, respectively. The lowest number of miRNAs was distributed on chromosome 9, with 8. The most differentially expressed miRNAs were distributed on chromosome 6, with 3, and the rest of the chromosomes were distributed with 1 to 2 differentially expressed miRNAs. The novel miRNAs were distributed on chromosomes 5, 6, 7, 9, 10, 11, 12, 14, and 15, and chromosome 14 had the highest number of novel miRNAs, with 4 (Figure 3).
3.2. Differential Expression Analysis of miRNAs in Lirianthe delavayi
Samples of wild and cultivated Lirianthe delavayi were sequenced, and 365 miRNAs were identified and predicted. After the normalization of miRNA expression, a t test was used to screen the differentially expressed miRNAs (DEMs) using the threshold of P<=0.05, and 39 miRNAs were screened for significant differences in expression levels (Supplementary Table S1). Among them, 11 miRNAs were upregulated and 28 miRNAs were downregulated in the wild-type group (YW group) (Figure 4A and Figure 4B). For the 39 differentially expressed miRNAs, heatmap clustering analysis was performed, and the differentially expressed miRNAs were similarly expressed within their respective population (wild and cultivated) groups, with good homogeneity and significant differences between groups (Figure 5).
3.3. Prediction and Functional Analysis of Target Genes of DEMs in L. delavayi
3.3.1. Target Gene Prediction of DEMs
Target gene prediction of significantly differentially expressed miRNAs (DEMs) was performed using psRobot (v1.2) [35], and target genes were predicted using a plant-based target penalty scoring strategy (with a default threshold of score ≤ 2.5), resulting in a total of 841 target genes targeted by 39 differentially expressed miRNAs from wild and cultivated populations of Lirianthe delavayi (Supplementary Table S2).
3.3.2. KEGG Enrichment Analysis
In organisms, different genes coordinate with each other to exercise their biological functions, and pathway-based enrichment analysis can identify the main biological functions exercised by miRNA‒mRNA relationship pairs. The 841 target genes of 39 differentially expressed miRNAs were annotated and enriched in KEGG pathway units, 292 target genes were finally annotated and enriched in 79 pathway units, and the pathways with significant enrichment were screened out by applying a hypergeometric test (Figure 6). The first two pathways were significantly enriched: Plant‒pathogen interaction and Sphingolipid metabolism.
3.3.3. GO Enrichment Analysis
GO enrichment analysis can help us understand the main functions and biological processes associated with target genes. The basic unit of GO is the term (term, node), and each term corresponds to an attribute. First, 841 target genes were mapped to each term in the Gene Ontology database, among which 772 target genes were mapped to 989 terms. The number of genes in each term was calculated, and a hypergeometric test was applied to determine the significantly enriched GO terms. The first three significantly enriched terms were extrinsic component of plasma membrane, defense response and ADP binding (Figure 7).
3.4. Analysis of miRNAs and Target Genes Involved in the Plant‒Pathogen Interaction Pathway
3.4.1. Analysis of DEMs in the Plant‒Pathogen Interaction Pathway
We explored plant‒pathogen interactions for the pathways that were most highly enriched and of interest to us, including ath-miR168a-5p, ath-MIR169g-p3_2ss7AG19GA, ath-MIR169g-p5_2ss7AG19GA, ath-miR408-3p_L-3, gra-miR482d_R-2, ptc-miR530a_L+1_1ss21TG, and stu-miR482a-3p_R-2_1ss8TC, which are 7 significantly differentially expressed miRNAs (Table 1). The cultivated populations showed upregulation of ath-miR168a-5p and downregulation of the remaining six miRNAs relative to the wild populations, and three miRNAs, ath-MIR169g-p3, ath-MIR169g-p5, and ath-miR408-3p, were not expressed in cultivated Lirianthe delavayi.
3.4.2. Analysis of DEMs and Target Gene Function and Regulation in the Plant‒Pathogen Interaction Pathway
The seven miRNAs and their target genes involved in the plant‒pathogen interaction pathway were analyzed, and the following neural network diagram was drawn (Figure 8). From the network diagram, we can see the pairing of the 7 miRNAs with the target genes, and a total of 30 miRNA‒target gene regulatory pairs were obtained (Figure 8, Supplementary Table S3). A miRNA can regulate multiple target genes, and the same target gene may be targeted by multiple miRNAs, such as CERK1, At4g27190, and other target genes regulated by multiple miRNAs. In the plant‒pathogen pathway, ath-miR168a-5p only targets RAR1, and ath-miR408-3p_L-3 only targets RPP13 (Figure 8).
Based on the KEGG database, the target genes involved in the plant‒pathogen interaction pathway were projected onto the Arabidopsis pathway map (Figure 9) to explore the specific life activities in which the target genes are involved. CERK1, CNGCs, CDPK (CPK26), RPM1 (RPM1, RPP13), RPS2 (RPS2, At4g27190, SUMM2, At5g63020, At4g27220, At1g50180, At1g61180), RAR1, WRKY33, and other target genes in the plant‒pathogen interaction pathway are shown at specific locations (Figure 9). Ptc-miR530a_L+1_1ss21TG targets CNGC, which mainly serves as a carrier of calcium ions across the cell membrane, transporting them from the extracellular membrane to the intracellular cytoplasm; ath-MIR169g-p3_2ss7AG19GA and ath-MIR169g-p5_2ss7AG19GA target and regulate CERK1, a target gene that acts as a pattern recognition receptor molecule on the cell surface and stimulates an immune response by recognizing characteristic molecules (PAMPs) of pathogenic bacteria; ath-MIR169g-p3_2ss7AG19GA and ath-MIR169g-p5_2ss7AG19GA also target and regulate the plant calcium-dependent protein kinase CDPK, which is capable of calcium-signal activation and modulates downstream immune responses by binding to substrates and amplifying pathogen-stimulated signals; ath-MIR169g-p5_2ss7AG19GA targets and regulates WRKY33, which, as a transcriptional regulator, receives MAPK signals and participates in the regulation of the expression of resistance genes, thus contributing to resistance to pathogen attack; and RPM1 and RPS2, which are plant disease resistance genes.
4. Discussion
4.1. Identification and Survey of miRNAs of L. delavayi
In this study, miRNA sequencing was carried out on cultivated and wild Lirianthe delavayi, and 365 miRNAs were identified, including 13 novel miRNAs whose first bases had obvious uracil (U) base preferences, which was consistent with the findings of Fairview azalea [36]. These miRNAs were categorized into 32 families, and the MIR482 family had the greatest number of family members with 32 family members, followed by the MIR167 family with 20 family members, and most families had 1~3 family members. The number and distribution of members of Lirianthe delavayi in each miRNA family were similar to those in 19 other species, including Arabidopsis thaliana, and the number distribution pattern in each miRNA family was most similar to that in potato (Figure 2A). The number and distribution of miRNA members in 21 miRNA families, such as MIR390 and MIR482, were similar in each species, and the number of members was greater than those in other families (Figure 2A).
The miRNAs of Lirianthe delavayi were found on all chromosomes; chromosome 14 had the highest number of miRNAs (35), and chromosome 9 had the lowest number of miRNAs (8) (Figure 3). This phenomenon is the same as the lowest number of miRNAs distributed on chromosome 9 of Apocynum venetum [23]. The novel miRNAs were distributed on chromosomes 5, 6, 7, 9, 10, 11, 12, 14, and 15, and chromosome 14 had the greatest number of novel miRNAs (4) (Figure 3).
4.1. Major Regulatory Pathways of DEMs during the Cultivation of L. delavayi
Thirty-nine miRNAs that were significantly differentially expressed in wild and cultivated Lirianthe delavayi were screened from the 365 miRNAs that were initially identified, and 841 target genes of these 39 miRNAs were predicted. The target gene enrichment results showed that the target genes regulated by the differentially expressed miRNAs were mainly involved in the regulation of plant‒pathogen interactions, sphingolipid metabolism, defense responses, and ADP binding. Plant sphingolipids and their metabolites are active lipid molecules with signal transduction roles that not only regulate cell growth, differentiation and proliferation but also play important roles in plant disease prevention and resistance [38]. Plant defense responses are a series of coping strategies adopted by plants when faced with external threats, such as pest and disease infestation, changes in the physical environment, or competition with other plants, and these responses help plants enhance their adaptability, competitiveness, and resistance to disease [39,40]. ADP binding plays diverse roles in organisms, ranging from energy transfer to kinase activity regulation, which maintain normal cellular function and organismal homeostasis [41,42,43]. This implies that wild and cultivated populations of L. delavayi may have adapted to their environment during cultivation by influencing these important biological processes.
4.1. Regulation of DEMs and Their Target Genes in the Plant‒Pathogen Interaction Pathway
We performed functional enrichment analysis of target genes for DEMs in wild and cultivated L. delavayi and found that the target genes were significantly enriched in the plant‒pathogen interaction pathway associated with disease and stress resistance. Using this pathway, we obtained a total of 30 miRNA‒target gene regulatory pairs (Figure 8), which were mainly involved in pathogen signature molecule binding and recognition, signal transduction, and regulation of the expression of disease resistance molecules or genes (Figure 9). Ptc-miR530a_L+1_1ss21TG targets the calcium ion carrier CNGC to regulate the through-mode transport of calcium ions, thereby participating in calcium signaling [44]. Ath-MIR169g-p3_2ss7AG19GA regulates the plant calcium-dependent protein kinase CDPK, which amplifies pathogen-stimulated signals to drive downstream immune responses [45], and this miRNA also regulates the transcriptional regulator WRKY33, which is involved in the regulation of resistance gene expression [46]. In addition, ath-MIR169g-p3_2ss7AG19GA also targets CERK1, which recognizes characteristic molecules (PAMPs) of pathogens on the cell surface, thereby triggering an immune response [47]. RPM1 and RPS2 are regulated as plant disease resistance genes by the majority of the candidate miRNAs (Figure 8), and their encoded proteins are both capable of interacting with RIN4 proteins, recognizing effector molecules (PAMPs) released by pathogens and triggering a series of signal transduction processes that activate plant body defense response mechanisms, including the expression of disease resistance genes, the production of disease resistance substances, and cell apoptosis [48,49,50]. Ath-miR168a-5p targets the RAR1 protein-encoding gene, which is able to bind to the proteins of the two disease resistance genes mentioned above, enhancing their disease resistance activity and transducing the disease resistance signals they produce, triggering a defense response in the plant [51,52].
Previous studies have shown that many species have undergone considerable genome evolution during domestication and that the transition from wild to cultivated species is accompanied not only by the loss of disease resistance genes [53] but also by the loss of disease-resistant miRNAs [30]. Under the plant‒pathogen interaction pathway, ath-MIR169g-p3_2ss7AG19GA, ath-MIR169g-p5_2ss7AG19GA, and ath-miR408-3p_L-3 were expressed only in the wild population and not in the cultivated Lirianthe delavayi population, and we speculated that these three miRNAs might be missing during the cultivation process, which is in accordance with a previous report [30] (Ma Yenming, 2016). Plant domestication has long been oriented toward artificially selected superior traits, either to increase nutrient content, improve edible palatability, or obtain better ornamental qualities, and this process is often accompanied by a significant reduction in genetic diversity [54], a decrease in nucleotide diversity, and the loss of a large number of genotypes [55]. In addition, the intensive use of anthelmintic and antimicrobial drugs during domestication creates a favorable growing environment for cultivated plants, and favorable conditions reduce the need for plant resistance, coupled with the effects of reduced genetic diversity [56,57], which ultimately results in the loss of resistance genes and resistance miRNAs [58,59] (Gepts, 2004; Purugganan et al., 2009). The target gene functions of missing miRNAs were explored, and ath-MIR169g-p3_2ss7AG19GA and ath-MIR169g-p5_2ss7AG19GA are homologous miRNAs that regulate mostly the same target genes, such as transcriptional regulators (WRKY23, WRKY24, WRKY33, and WRKY35), the pattern recognition receptor molecule CERK1, the plant calcium-dependent protein kinase CPK26, and RPS2 (SUMM2 and At4g27190). These target genes mainly play roles in recognizing pathogen signature proteins [47], signaling [45], and regulating the transcription of disease resistance genes [46]. Ath-miR408-3p_L-3 targets RPP13, which mainly recognizes and binds antibodies that trigger defense responses [60].
5. Conclusions
The study of miRNAs can help to explore the adaptation mechanism of Lirianthe delavayi in the face of habitat differences (artificial versus natural environment) during cultivation. Through miRNA sequencing analysis of wild and cultivated Lirianthe delavayi, 365 miRNAs were identified, including 13 novel miRNAs, of which 39 miRNAs were significantly differentially expressed. The target genes of the significantly differentially expressed miRNAs were significantly enriched in plant‒pathogen interactions, sphingolipid metabolism, defense responses, and ADP binding for these biological functions, suggesting that wild and cultivated populations of Lirianthe delavayi may adapt to the environment during cultivation by influencing these important biological processes. The plant‒pathogen interaction pathway involves seven differentially expressed miRNAs and 30 miRNA‒target gene regulatory pairs, which are mainly involved in pathogen signature molecule recognition and binding, signal transduction, and regulation of the expression of disease-resistant molecules or genes. In addition, three significant differential miRNAs (ath-MIR169g-p3_2ss7AG19GA, ath-MIR169g-p5_2ss7AG19GA, and ath-miR408-3p_L-3) were expressed only in wild Lirianthe delavayi, and it was hypothesized that the domestication process from wild to cultivated species might be accompanied by the loss of disease resistance-related miRNAs. This study provides a reference for exploring the posttranscriptional regulatory mechanism of adaptive changes in Lirianthe delavayi during domestication.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1:Status of all identified mRNAs in L. delavayi; Table S2: miRNA family data; Table S3: Expression and differential miRNAs; Table S4: Target prediction and gene enrichment; Table S5: DEMs and Target genes in Plant-pathogen interaction.
Author Contributions
Conception, T.X.; methodology, T.X.; software, T.-X.Z.; formal analysis, T.-X.Z.; data curation, investigation, T.-X.Z., L.Y., J.Y., Y.L., P.-Z.Z., and T.X.; writing—original draft preparation, T.-X.Z.; writing—review and editing, T.-X.Z.,T.X. ; supervision and funding acquisition, T.X. All authors have read and agreed to the published version of the manuscript.
Funding
This research was financially supported by the Specific Funds 2024 of Forestry and Grassland Bureau of Yunnan province for the Research of the Investigation and Detection of Two Extremely Small Populations of Magnoliaceae Plants and Collection of Germplasm Resources (Grant No. ) to T.X. The funding sources had no role in the design of the study, the collection and analysis of the data, the decision to publish, or the preparation of the manuscript.
Data availability statement
The raw miRNA sequencing data were deposited in the GenBank Short Read Archive (SRA) database under accession number PRJNA1109302.and accession number(s) can be found below: https://www. ncbi.nlm.nih.gov/sra/PRJNA1109302.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Chinese Botanical Society; Chinese Academy of Sciences. China flora. Science Press: China, 1996; 30 volumes, Part I.112.
- National compilation group of Chinese herbal medicine. National Compilation of Chinese Herbal Medicine, People’s Medical Publishing House: Beijing, China, 1996.
- Zhu, R.-J. A good tree species for courtyard greening--Lirianthe delavayi. Yunnan Forestry 1985, 01, 27. [Google Scholar]
- Xia, B.; He, T.-T. Application of Southern Breeding Technology in the Development of Garden Ornamental Plant Resources. Molecular Plant Breeding 2024, 22, 3811–3816. [Google Scholar]
- Huang, Y. Adaptation of plants to the environment and utilization of environmental resources. China Resources Comprehensive Utilization 2018, 36, 89–91. [Google Scholar]
- Feng, X.; Chen, L.-Q. Research progress on the regulation of lotus plant type. Journal of Southwest Forestry University (Natural Science) 2024, 44, 216–220. [Google Scholar]
- Chen, W.; Zhou, Y.-Y.; LUO, P.; Cui, Y.-Y. Molecular regulation of flower repetalisation in angiosperms. Journal of Botany 2024, 59, 257–277. [Google Scholar]
- Zhang, L.; Chao, J.-T.; Cui, M.-M.; Chen, Y.-Q.; Zong, P.; Sun, Y.-H. Bioinformatics prediction of aubergine microRNAs with their target genes. Hereditas. 2011, 33, 116–124. [Google Scholar]
- Llave, C. Cleavage of Scarecrow-like mRNA Targets Directed by a Class of Arabidopsis miRNA. Science 2002, 297, 2053. [Google Scholar] [CrossRef]
- Kasschau, K.D.; Xie, Z.; Allen, E.; Llave, C.; Chapman, E.J.; Krizan, K.A.; Carrington, J.C. P1/HC-Pro, a Viral Suppressor of RNA Silencing, Interferes with Arabidopsis Development and miRNA Function. Developmental Cell 2003, 4, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-M. MicroRNA biogenesis and function in plants. FEBS Lett 2005, 579, 5923–5931. [Google Scholar] [CrossRef]
- Zhang, B.H.; Pan, X.P.; Anderson, T.A. Identification of 188 conserved maize microRNAs and their targets. FEBS Lett 2006, 580, 3753–3762. [Google Scholar] [CrossRef]
- Song, X.; Li, Y.; Cao, X.; Qi, Y. MicroRNAs and their regulatory roles in plant-environment interactions. Annu. Rev. Plant Biol. 2019, 70, 489–525. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Wang, X.; Zhou, Y.; Tan, J.; Zhou, Y.; Gao, F. Small RNA Sequencing Revealed that miR4415, a Legume-Specific miRNA, was Involved in the Cold Acclimation of. Frontiers in genetics 2022, 13, 870446. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.-J.; Li, C.-H.; Han, X.-L.; Shen, F.-F. Identification of conserved microRNAs and their target genes in tomato (Lycopersicon esculentum). Gene 2008, 414, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Lang, Q.-L.; Jin, C.-Z.; Lai, L.-Y.; Feng, J.-L.; Chen, S.-N.; Chen, J.-H. Tobacco microRNAs prediction and their expression infected with Cucumber mosaic virus and Potato virus X. Mol Biol Rep 2010, 38, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Frazier, T.P.; Xie, F.L.; Freistaedter, A.; Burklew, C.E.; Zhang, B.H. Identification and characterization of microRNAs and their target genes in bobacco (Nicotiana tabacum). Plant 2010, 232, 1289–1308. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.; Guan, Q.; Shen, C.; Feng, X.; Zhang, Y.; Chen, Y.; Wu, R. Integrated sRNA-seq and RNA-seq analysis reveals the regulatory roles of miRNAs in the low-temperature responses of Canarium album. Horticulturae 2022, 8, 667. [Google Scholar] [CrossRef]
- Sajad, S.; Dai, Q.; Yang, J.; Song, J. Identification of miRNAs involved in male fertility and pollen development in Brassica oleracea var. capitata L. by High-Throughput Sequencing. Horticulturae 2023, 9, 515. [Google Scholar] [CrossRef]
- Cao, D.; Li, J.; Ma, L.; Liu, Y.; Huang, J.; Jin, X. Genome-Wide identification of selenium-responsive microRNAs in tea Plant (Camellia sinensis LO Kuntze)[J]. Horticulturae 2023, 9, 1278. [Google Scholar] [CrossRef]
- Lu, J.; Mao, X.; Xu, Y.; Liu, S.; Wang, L. MicroRNA identification and integrated network analyses for age-dependent flavonoid biosynthesis in Ginkgo biloba. Forests 2023, 14, 1706. [Google Scholar] [CrossRef]
- Tan, J.-H.; Han, X.-W.; Liu, Q.; Dorjee, T.; Zhou, Y.J.; Sun, H.-G.; Gao, F. Joint Analysis of Small RNA and mRNA Sequencing Unveils miRNA-Mediated Regulatory Network in Response to Methyl Jasmonate in Apocynum venetum L. Horticulturae 2024, 10, 173. [Google Scholar] [CrossRef]
- Evans, L.M.; Slavov, G.T.; Rodgers-Melnick, E.; Martin, J.; Ranjan, P.; Muchero, W.; Brunner, A.M.; Schackwitz, W.; Gunter, L.; Chen, J.G. Population genomics of Populus trichocarpa identifies signatures of selection and adaptive trait associations. Nature Genetics 2014, 46, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Stief, A.; Altmann, S.; Hoffmann, K.; Pant, B.D.; Scheible, W.R.; Baurle, I. Arabidopsis miR156 Regulates Tolerance to Recurring Environmental Stress through SPL Transcription Factors. Plant Cell 2014, 26, 1792. [Google Scholar] [CrossRef] [PubMed]
- Williams, L. ; Regulation of Arabidopsis shoot apical meristem and lateral organ formation by microRNA miR166g and its AtHD-ZIP target genes. Development 2005, 132. [Google Scholar]
- Val-Torregrosa, B.; Mireia, B.; Segundo, B.S. Crosstalk between Nutrient Signaling Pathways and Immune Responses in Rice. Agriculture 2021, 11. [Google Scholar]
- Liao, L.; Xie, B.; Guan, P.; Jiang, N.; Cui, J. New insight into the molecular mechanism of miR482/2118 during plant resistance to pathogens. Front Plant Sci 2022, 13, 1026762. [Google Scholar] [CrossRef] [PubMed]
- Sumbur, B.; Gao, F.; Liu, Q.; Feng, D.; Bing, J.; Dorjee, T.; Zhou, Y. The Characterization of R2R3-MYB Genes in Ammopiptanthusnanus uncovers that the miR858-AnaMYB87 Module mediates the accumulation of anthocyanin under osmotic stress. Biomolecules 2023, 13, 1721. [Google Scholar] [CrossRef] [PubMed]
- Ma Y.-M. wild and cultivated adzuki bean mirnas genome-wide identification and comparative analysis Beijing agronomy courtyard 2016.
- Wu, N.-N.; Zeng, Z.-Y.; Xu, Q.-B.; Zhang, H.-B.; Xu, T. Artificial Cultivation Changes Foliar Endophytic Funga Community of the Ornamental Plant Lirianthe delavayi. Microorganisms 2023. [Google Scholar] [CrossRef] [PubMed]
- Mutz, K.O.; Heilkenbrinker, A.; L€onne, M.; Walter, J.G.; Stahl, F. Transcriptome analysis using next-generation sequencing. Curr Opin Biotech 2013, 24, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yin, H.; Song, X.; Zhang, Y.; Liu, M.; Sang, J.; Jiang, J.; Li, J .; Zhuo, R. Integration of small RNAs, degradome and transcriptome sequencing in hyperaccumulator Sedum alfredii uncovers a complex regulatory network and provides insights into cadmium phytoremediation. Plant Biotechnology Journal 2016.
- Xiang, L.; Muhammad, S.; Jinwen, W.; Lan, W.; Xiangdong, L.; Yonggen, L. Comparative Small RNA Analysis of Pollen Development in Autotetraploid and Diploid Rice. Molecular Sciences 2016, 17, 499. [Google Scholar]
- Wu, H.-J.; Ma, Yi.-Ke.; Tong, C.; Meng, W.; Xiu-Jie, W. PsRobot: a web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res 2012, 40, 22–8. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; et al. KEGG for linking genomes to life and the environment[J]. Nucleic Acids Res 2008, D480–484. [Google Scholar] [CrossRef] [PubMed]
- Fang, B.; Huang, Z.; Sun, Y.; Zhang, W.; Yu, J.; Zhang, J.; Dong, H.; Wang, S. Small RNA sequencing provides insights into molecular mechanism of flower development in Rhododendron pulchrum Sweet. Scientific reports 2023, 13, 17912. [Google Scholar] [CrossRef] [PubMed]
- Michaelson; Louise, V.; Napier; Johnathan; A.; Molino; Diana; Faure; Jean-Denis. Plant sphingolipids: Their importance in cellular organization and adaptation. Biochimica et Biophysica Acta (BBA) - Molecular & Cell Biology of Lipids 2016, 1861, 1329–1335.
- Chen, Y.-Y.; Lan, J.-X.; Wang, S.-Y. Research progress on early signal events of plant defense response. Acta PhytoPhysiologica Sinica 2023, 59, 829–838. [Google Scholar]
- Lu, Y.X.; Yang, Q.L.; Kang, H.W. ; Endosymbiont Buchnera assists aphids in suppressing host plant defense responses. Journal of plant protection 2024, 51, 69–77. [Google Scholar]
- Debabrata, D.; Kumar, V.A.; Gaurab, G. Amino acid substitution in the conserved motifs of a hypothetical R-protein in sesame imparts a significant effect on ADP binding position and hydrogen bond interaction. Physiological and Molecular Plant Pathology 2021, 113101588. [Google Scholar]
- Zhang, Q.; Liu, X.; Liu, H.; Zhang, B.; Yang, H.; Mi, K.; Guddat, L.W.; Rao, Z. Conformational Changes in a Macrolide Antibiotic Binding Protein From Mycobacterium smegmatis Upon ADP Binding. Frontiers in Microbiology 2021, 12780954–780954. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.; Eastlund, A.; Fischer, C.; Jardine, P. Kinetics of ATP/ADP Binding to the gp16 ATPase. Biophysical journal 2022, 1211909–1918. [Google Scholar] [CrossRef]
- Wang, Y.; Hou, H.S. Structure and function of cyclic nucleotide gated channel (CNGC) gene family in plants. Plant Physiology Communications 2007, 1, 1–8. [Google Scholar]
- Ma, H.; Wang, S.; Zhou, Y. Research progress of calcium-dependent protein kinases in plants. Journal of Nanjing Agricultural University 2017, 40, 565–572. [Google Scholar]
- Tao, H.; Miao, H.; Chen, L.; Wang, M.; Xia, C.; Zeng, W.; Sun, B.; Zhang, F.; Zhang, S.; Li, C. WRKY33-mediated indolic glucosinolate metabolic pathway confers resistance against Alternaria brassicicola in Arabidopsis and Brassica crops. Journal of Integrative Plant Biology 2022, 64, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wang, E.; Liu, J. CERK1, more than a coreceptor in plant–microbe interactions. New Phytologist, 2022.
- Alam, M.; Tahir, J.; Siddiqui, A.; Magzoub, M.; Shahzad-Ul-Hussan, S.; Mackey, D.; Afzal, A.J. RIN4 homologs from important crop species differentially regulate the Arabidopsis NB-LRR immune receptor, RPS2. Plant cell reports 2021, 40, 2341–2356. [Google Scholar] [CrossRef] [PubMed]
- Minsoo Y., J M M,A H E R.A conserved glutamate residue in RPM1-INTERACTING PROTEIN4 is ADP-ribosylated by the Pseudomonas effector AvrRpm2 to activate RPM1-mediated plant resistance.[J]. The Plant cell,2022, 34(12):.
- Prachumporn, N.; Anis, A.; Suthathip, K. Phosphorylation of CAD1, PLDdelta, NDT1, RPM1 Proteins Induce Resistance in Tomatoes Infected by Ralstonia solanacearum. Plants 2022, 11, 726–726. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Ohnishi, K.; Hikichi, Y.; Kiba, A. Molecular chaperons and cochaperons, Hsp90, RAR1, and SGT1 negatively regulate bacterial wilt disease caused by Ralstonia solanacearum in Nicotiana benthamiana. Plant signaling behavior, 2015, 10, 970410. [Google Scholar] [CrossRef] [PubMed]
- Hao; Qunqun; Wu; Jiajie; Pei; Hongcui; Lv; Bo; Fu; Daolin. The HSP90-RAR1-SGT1-based protein interactome in barley and stripe rust. Physiological and Molecular Plant Pathology 2015, 9111–19.
- Zhou, Z.; et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat Biotechnol 2015, 33, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Lozano, R.; Kim, J.H.; Bae, D.N.; Jeong, S.C. The patterns of deleterious mutations during the domestication of soybean. Nat Commun 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Thudi, M.; Roorkiwal, M.; et al. Resequencing of 429 chickpea accessions from 45 countries provides insights into genome diversity, domestication and agronomic traits. Nat Genet 2019, 51, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.I.; Bi, I.V.; Schroeder, S.G.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S. The effects of artificial selection on the maize genome. Science 2005, 308, 1310–1314. [Google Scholar] [CrossRef]
- Glauser, G.; Marti, G.; Villard, N.; Doyen, G.A.; Wolfender, J.-L.; Turlings, T.C.J.; Erb, M. Induction and detoxification ofmaize 1,4-benzoxazin-3-ones by insect herbivores. PlantJournal 2011, 68, 901–911. [Google Scholar]
- Gepts, P. Crop domestication as a long-term selection experiment. Plant Breeding Reviews 2004, 24, 1–41. [Google Scholar]
- Purugganan, M.D.; Fuller, D.Q. The nature of selection during plant domestication. Nature 2009, 457, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Bittner·Eddy, P.D.; Crute, L.R.; Holub, E.B.; Beynon, J.L. RPP13 is a simple locus in Arabidopsis thaliana for alleles that specify downy mildew resistance to different avirulence determinants in Peronospora parasitica. Plant 2000, 21, 177–188. [Google Scholar]
Figure 1.
Proportion of first base classes of miRNAs of different lengths.
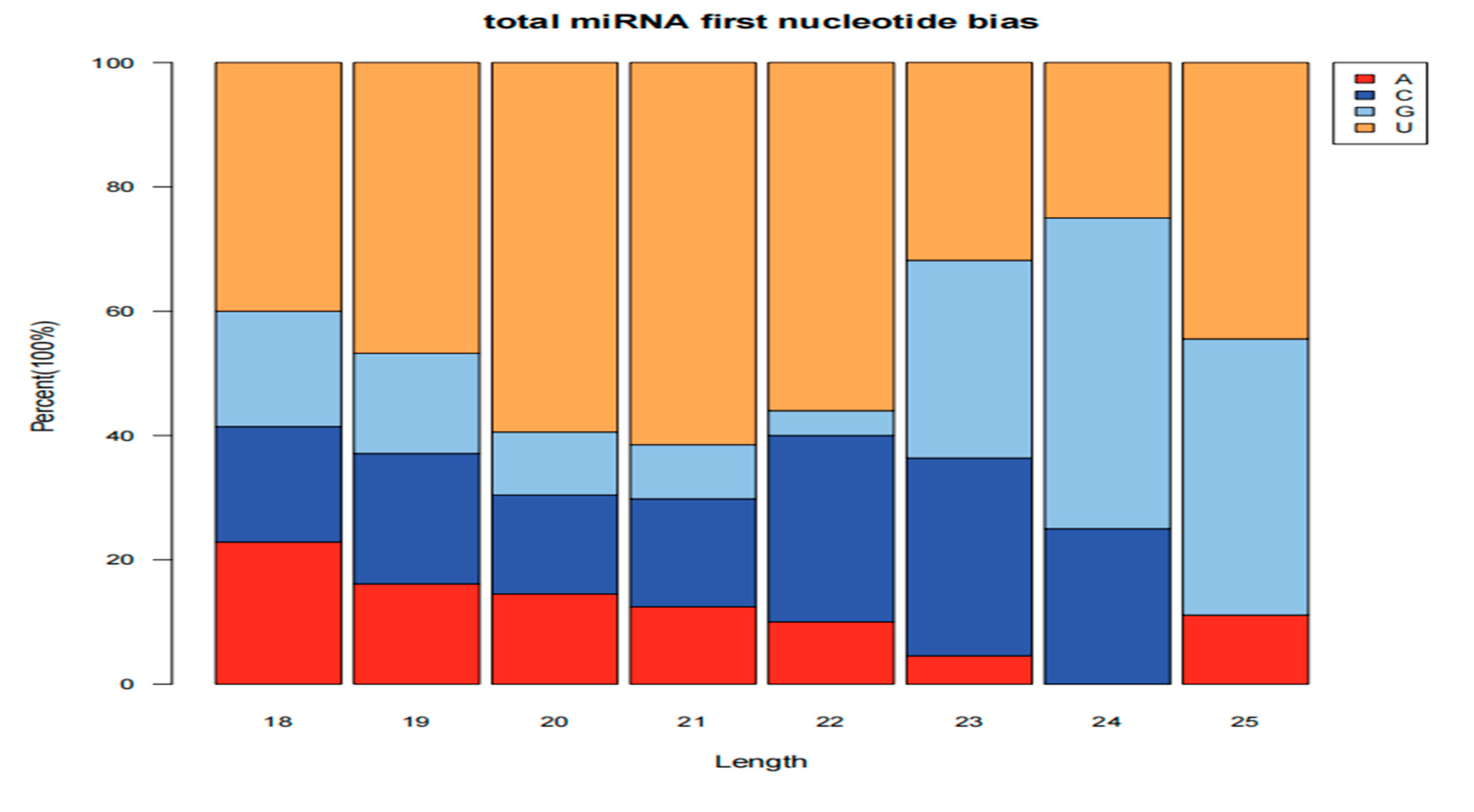
Figure 2.
(A) Comparison of the number of miRNA family members in Lirianthe delavayi and miRBase database species. (B) Phylogenetic analysis was performed on 29 miRNAs that could be compared to the reference species precursors (pre-miRNAs) and whose precursors could be compared to the reference genome. miRNAs of the same family and clustered together are marked with the same color.
Figure 2.
(A) Comparison of the number of miRNA family members in Lirianthe delavayi and miRBase database species. (B) Phylogenetic analysis was performed on 29 miRNAs that could be compared to the reference species precursors (pre-miRNAs) and whose precursors could be compared to the reference genome. miRNAs of the same family and clustered together are marked with the same color.
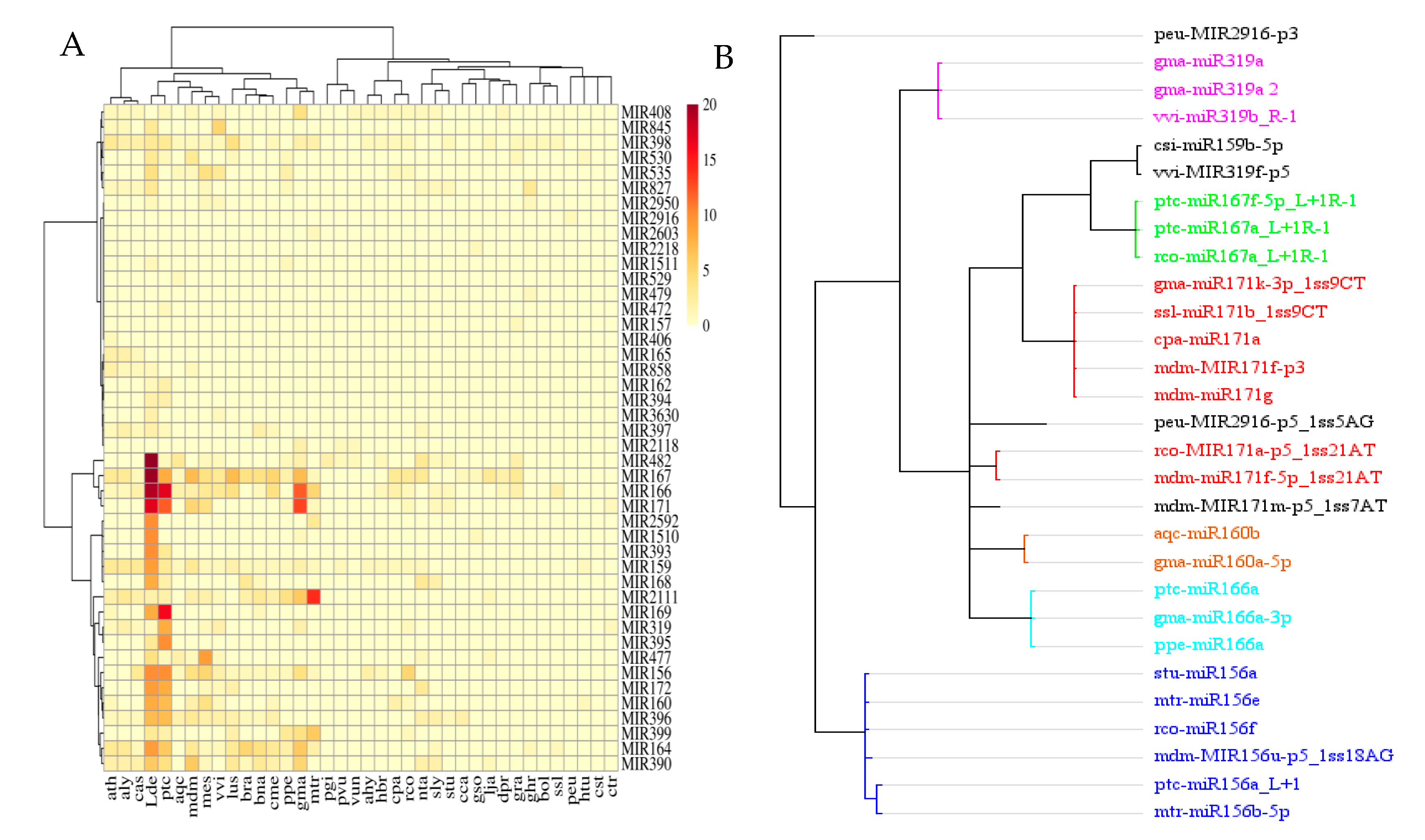
Figure 3.
miRNA chromosome localization map based on the MG2C online program; red indicates differentially expressed miRNAs.
Figure 3.
miRNA chromosome localization map based on the MG2C online program; red indicates differentially expressed miRNAs.
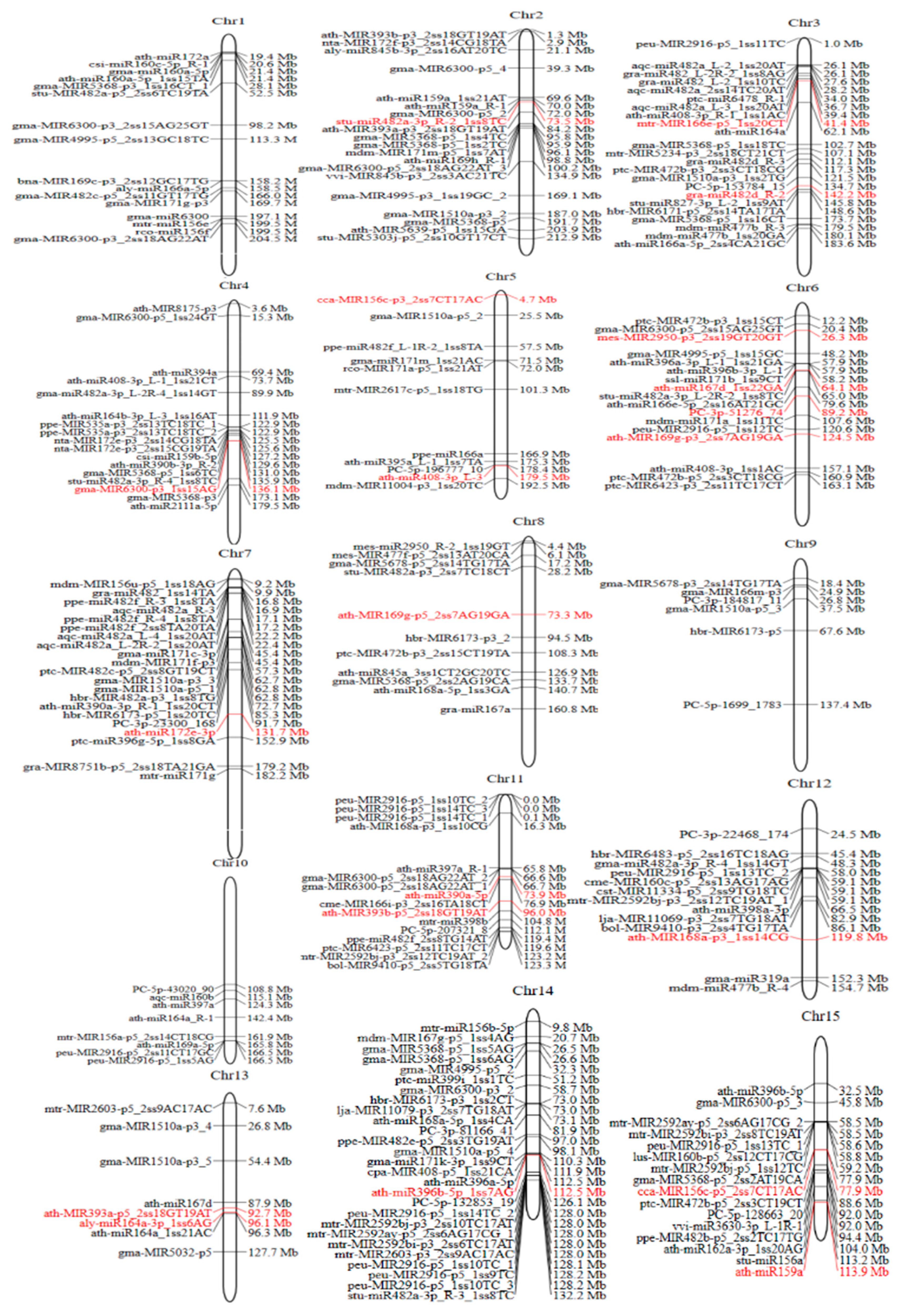
Figure 4.
(A) T test of differentially expressed miRNAs. (B) Volcano plot of differentially expressed miRNAs; red indicates significant and upregulated differences, blue indicates significant and downregulated differences, and gray indicates nonsignificant differences.
Figure 4.
(A) T test of differentially expressed miRNAs. (B) Volcano plot of differentially expressed miRNAs; red indicates significant and upregulated differences, blue indicates significant and downregulated differences, and gray indicates nonsignificant differences.
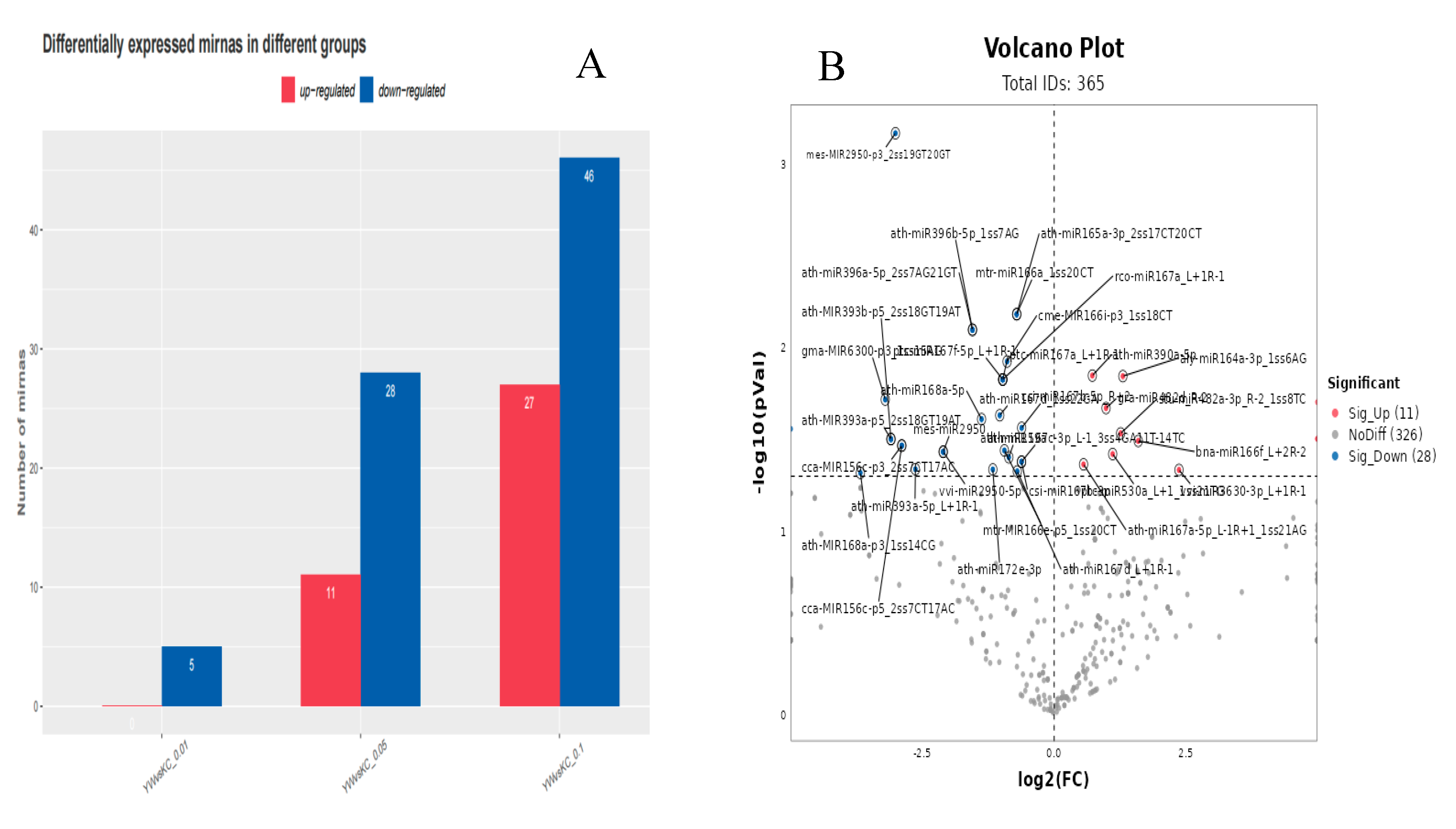
Figure 5.
Heatmap of significantly different miRNA expression profiles of wild and cultivated Lirianthe delavayi.
Figure 5.
Heatmap of significantly different miRNA expression profiles of wild and cultivated Lirianthe delavayi.
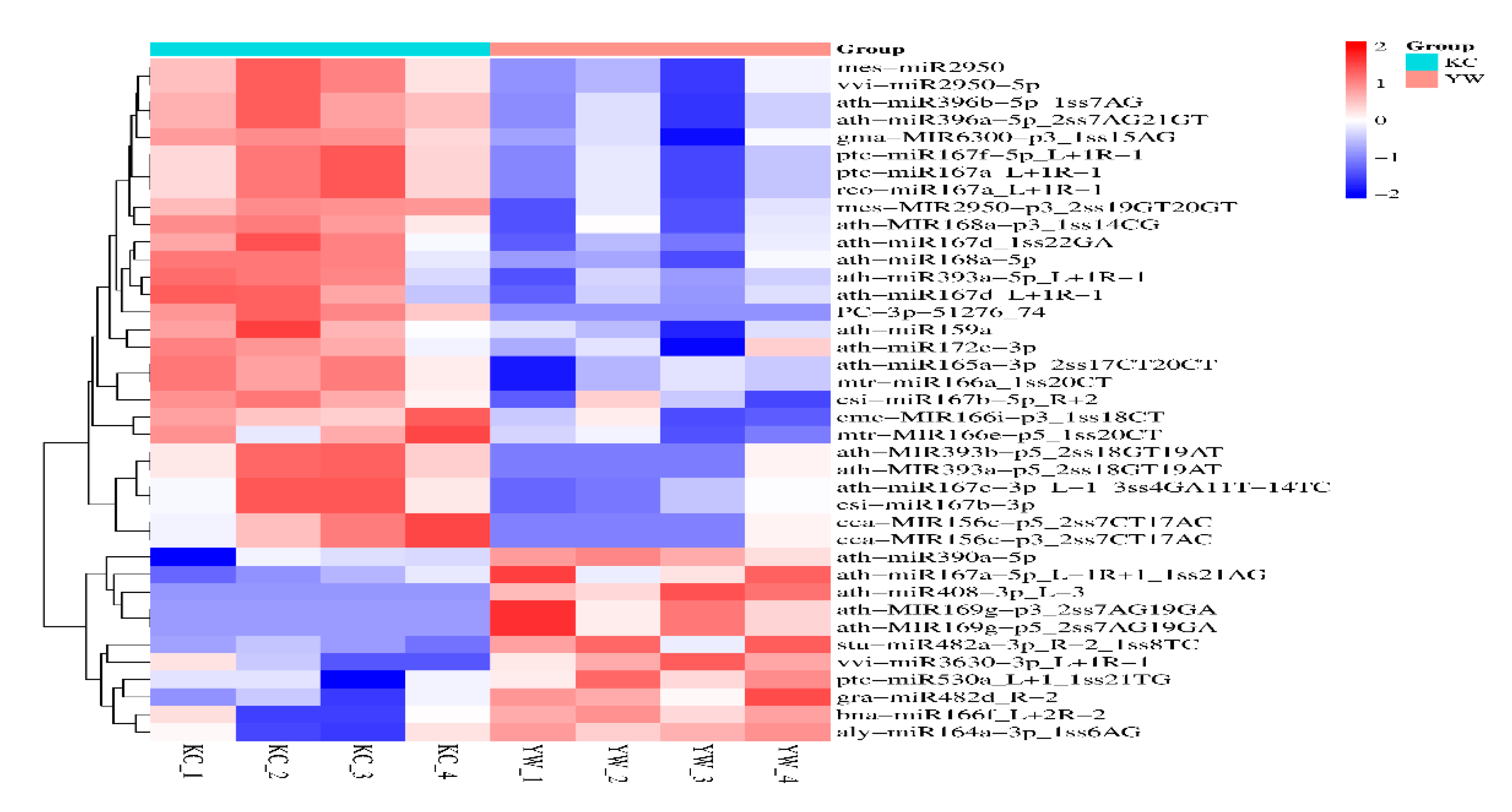
Figure 6.
miRNA KEGG enrichment bubble map of wild and cultivated Lirianthe delavayi.
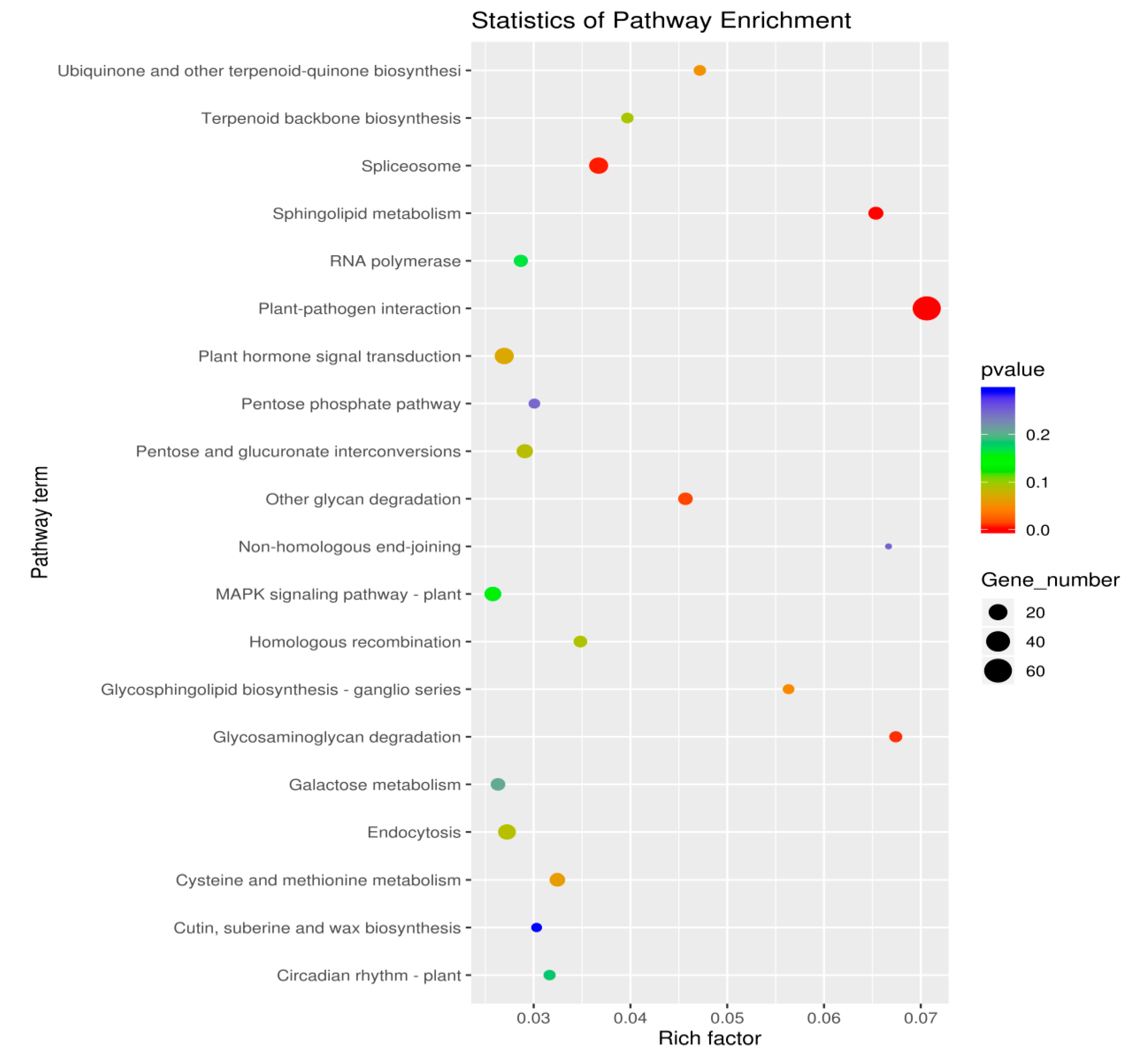
Figure 7.
GO analysis of significantly differentially expressed miRNAs in wild and cultivated Lirianthe delavayi.
Figure 7.
GO analysis of significantly differentially expressed miRNAs in wild and cultivated Lirianthe delavayi.
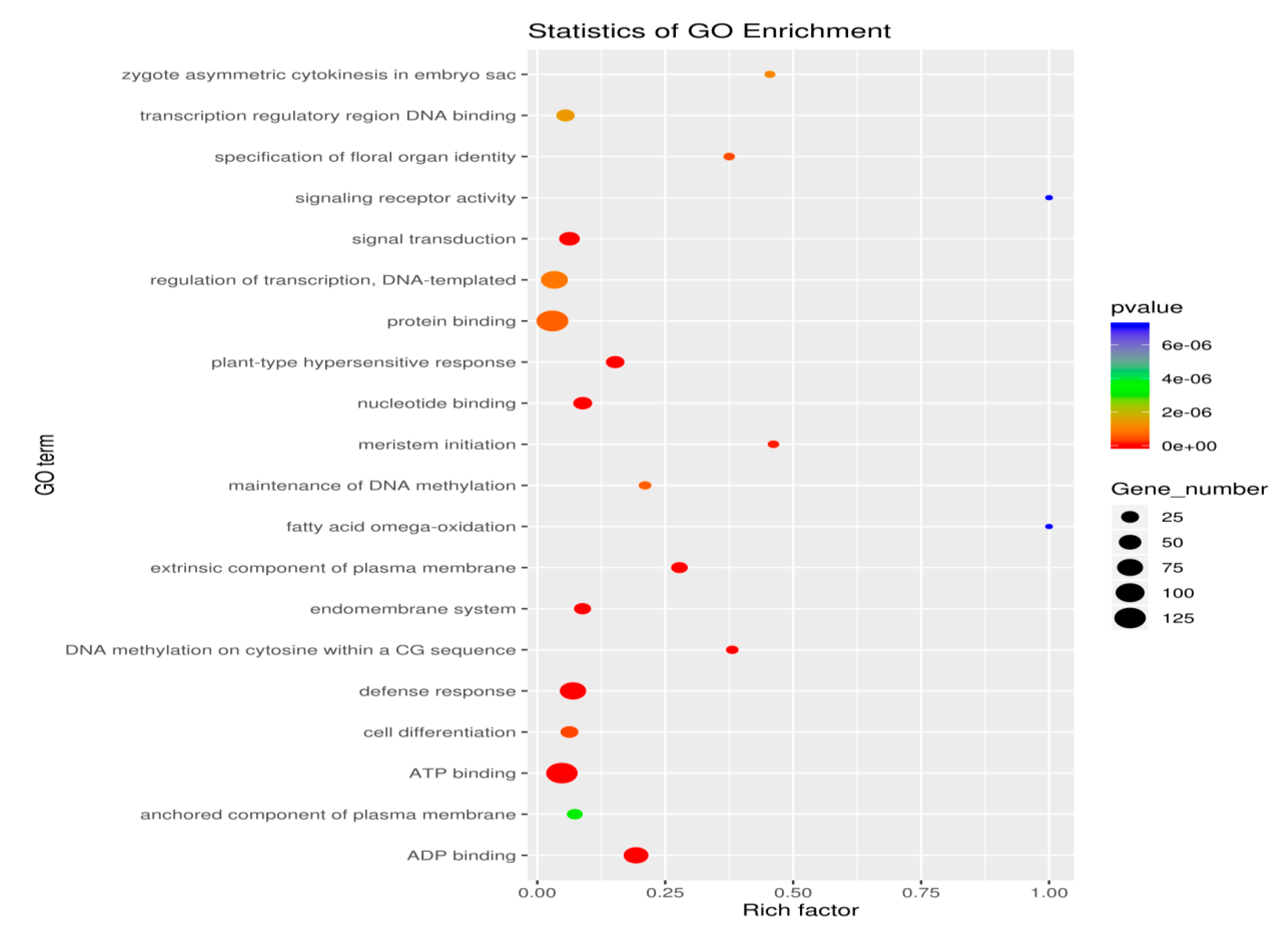
Figure 8.
miRNA-gene-KEGG network diagram.
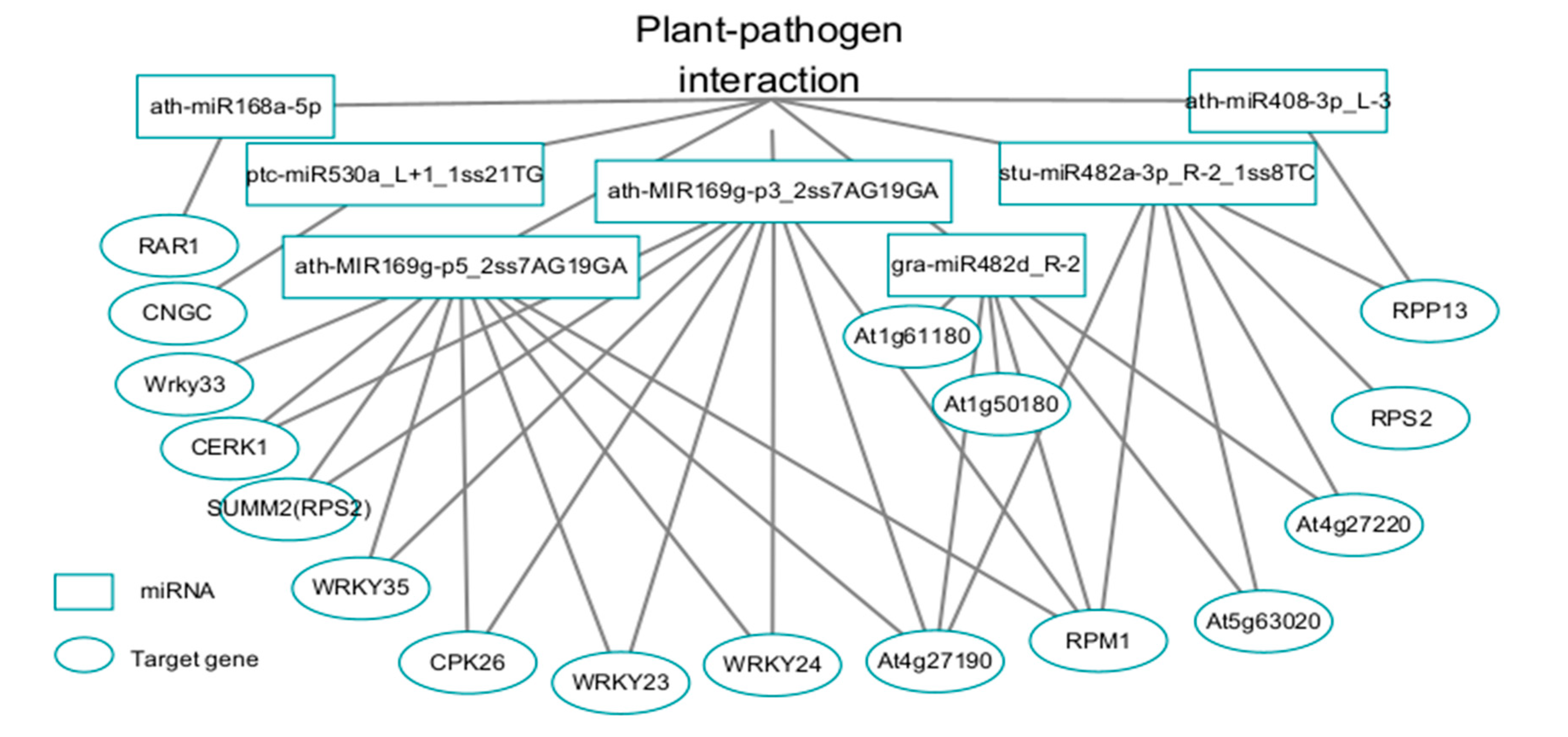
Figure 9.
Location and functional analysis of the miRNA target gene pathway network.
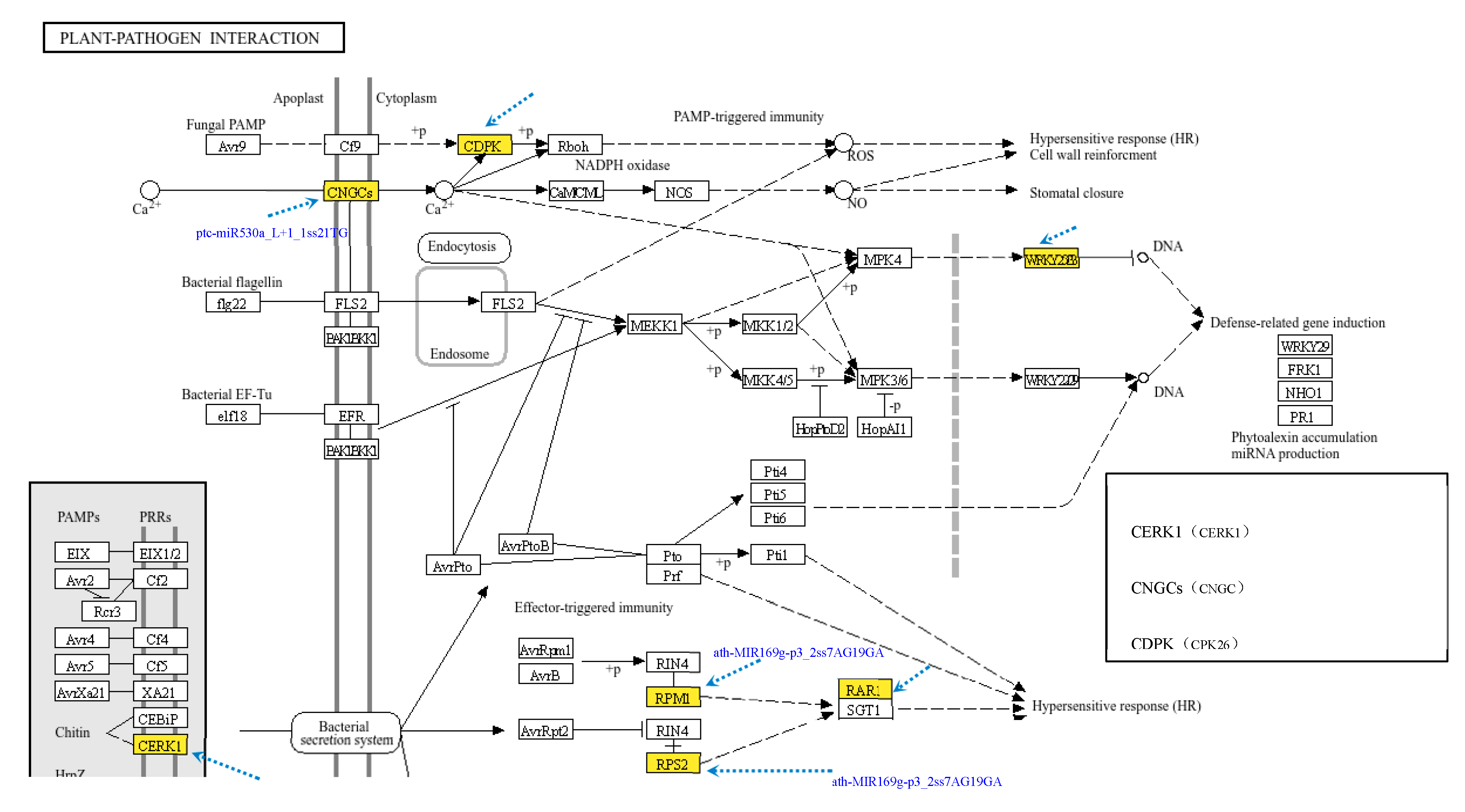

Table 1.
differentially expressed miRNAs in Plant‒pathogen interaction.
| miR_name | miR_seq | up/down | fold_change (KC/YW) |
|---|---|---|---|
| ath-miR168a-5p | TCGCTTGGTGCAGGTCGGGAA | up | 1.38 |
| ath-MIR169g-p3_2ss7AG19GA | CATGATGATGATGATTACA | down | -inf |
| ath-MIR169g-p5_2ss7AG19GA | CATGATGATGATGATTACA | down | -inf |
| ath-miR408-3p_L-3 | CACTGCCTCTTCCCTGGC | down | -inf |
| stu-miR482a-3p_R-2_1ss8TC | TTTCCAACTCCACCCATTCC | down | -2.40 |
| gra-miR482d_R-2 | TTTCCTATGCCCCCCATTCC | down | -1.98 |
| ptc-miR530a_L+1_1ss21TG | CTGCATTTGCACCTGCACCTG | down | -2.16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



