
Submitted:
25 May 2024
Posted:
27 May 2024
You are already at the latest version
Abstract
Retinitis pigmentosa (RP) is a heterogeneous inherited retinal disorder. Mutations in KIZ cause autosomal recessive (AR) RP. We aimed to characterize the genotype, expression pattern, and phenotype in a large cohort of KIZ cases. Sanger and whole exome sequencing were used to identify KIZ variants. Medical records were reviewed and analyzed. Thirty one patients with biallelic KIZ mutations were identified: 28 homozygous for c.226C>T (p.R76*), 2 compound heterozygous for p.R76* and c.3G>A (p.M1?), and one homozygous for c.247C>T (p.R83*). c.226C>T is a founder mutation among patients of Jewish descent. Clinical parameters were less severe in KIZ compared to DHDDS and FAM161A cases. RT-PCR analysis in fibroblast cells revealed the presence of four different transcripts in both WT and mutant samples with a lower percentage of the WT transcript in patients. Sequence analysis identified an exonic sequence enhancer (ESE) that includes the c.226 position which is affected by the mutation. KIZ mutations are an uncommon cause of IRD worldwide, but are not rare among Ashkenazi Jews. Our data indicate that p.R76* affect an ESE which in turn results in pronounced skipping of exon 3. Therefore, RNA-based therapies might show low efficacy since the mutant transcripts is spliced.
Keywords:
Clinical manifestation
; Exonic sequence enhancer
; Gene expression
; Retinal disease
1. Introduction
Inherited retinal diseases (IRDs) are heterogenous group of ocular disorders characterized by degeneration and dysfunction of photoreceptor cells leading to vision impairment. There are more than 50 major types of IRDs among which retinitis pigmentosa (RP- OMIM 268000) is considered one of the most genetically and clinically heterogeneous diseases in humans [1]. Population-based analysis indicated that 1 in 4000 individuals are affected by RP worldwide [2]. RP is characterized by significant variability among patients, with many exhibiting the classic symptoms such as impaired dark adaptation and night blindness during adolescence and loss of mid-peripheral visual field during young adulthood, both attributed to rod dysfunction. As the disease progresses, patients suffer loss of far peripheral vision, eventually developing tunnel vision, and ultimately experiencing compromise of the central vision due to cone impairment in the later stages of life [2].
The ocular phenotype of RP follows Mendelian pattern of inheritance where 15-25% are autosomal dominant, 35-50% are autosomal recessive (AR), and 7-15% are X-linked [3]. Pathogenic variants in over 60 genes are responsible for RP [1]. A unique group of genes that are associated with IRDs are those encoding proteins that are involved in the morphogenesis and function of the photoreceptor sensory cilia, a microtubule-based organelle coordinating various cellular processes [4,5]. Pathogenic variants in cilia-related genes result in retinal ciliopathies including Leber congenital amaurosis (LCA), RP, macular degeneration, cone-dystrophy, and cone-rod dystrophy [6]. Among these genes, KIZ was reported in 2014 to cause ARRP due to biallelic pathogenic variants [7]. To date only seven additional publications reported biallelic KIZ mutations in IRD patients (Table S1) [8,9,10,11,12,13,14,15,16]. The most common reported KIZ pathogenic variant is c.226C>T (p.R76*), a nonsense mutation leading to a premature termination codon (PTC) in exon 3. This variant has been reported in 18 patients with biallelic mutations, among whom seven subjects were previously included in a comprehensive analysis of IRD cases reported by us [12]. The current study will focus on further investigating this particular variant. For eight additional patients, clinical analysis was reported [8,14,17], and is largely in-line with the phenotypic spectrum of ARRP. The KIZ gene encodes the centrosomal protein kizuna that is expressed in the primary cilia and functions by stabilizing as well as strengthening the pericentriolar region prior to spindle formation, thus playing a vital role in cell cycle progression [18]. The human KIZ gene shows high expression levels in the retina while lower levels were detected in the retinal pigment epithelium (RPE), fibroblast cell-lines and white blood cells [17]. In addition, transcriptomic analysis revealed the presence of the mouse ortholog (Plk1s1) in rod photoreceptors and reduced Plkls1 expression was evident in rd1 mouse model exhibiting photoreceptor degeneration [7,19].
The current study includes clinical information on 31 RP patients harbouring biallelic KIZ mutations, the most common of which is c.226C>T (p.R76*) identified in 58 of the 62 (93.5%) mutated alleles. In addition, through gene expression analysis conducted on skin fibroblasts of WT individuals and patients carrying this mutation (c.226C>T), we provide evidence suggesting a dual mutational mechanism that potentially impacts the efficiency of RNA-based treatment modalities.
2. Materials and Methods
Please see Supplemental Methods for full description of the methods used in this study.
2.1. Recruitment and Clinical Analyses
Participants were recruited from Hadassah Medical Center, Tel Aviv Medical Center, and Rambam Medical Center, Israel. Ethical approval was obtained at individual Institutional Review Boards. The tenets of the Declaration of Helsinki were followed. Participants provided written informed consent after receiving an explanation about the study and its possible consequences before donating the blood sample. Ophthalmic evaluation was performed as previously described [20] and included a full ophthalmological examination, Goldmann perimetry (using Humphrey or Octopus systems), full field electroretinography (FFERG) according to the ISCEV standard (using LKC or Diagnosys systems), color vision testing using the Ishihara 38-panel and Farnsworth–Munsell D-15 tests, and color, autofluorescence and OCT imaging using Optos Silverstone, Optos California, TOPCON, Eidon and Heidelberg Spectralis systems.. The clinical assessment of KIZ patients encompassed visual acuity (VA) testing (25 cases), refraction (12), visual fields (10), funduscopy (15), optical coherence tomography (OCT- 12), electroretinography (ERG- 14), and electro-oculography (EOG- 6).
2.2. Statistical Analysis
Principal component analysis (PCA) of clinical parameters was performed using RStudio. PCA is a dimensionality reduction method that can translate n variables into n orthogonal linear combinations of those variables. Those linear combinations preserve the total variance. The first linear combination, PC1, is the linear combination with the greatest sample variance among all n possible linear combinations. The second principal component (PC2) is defined as the linear combination that accounts for a maximal proportion of the remaining variance subject to being uncorrelated with the first principal component and so on. In the current study, we analyzed the following seven variables by PCA: disease onset age, age of presentation, rod response, cone flicker amplitude, a-wave cone-rod response, b-wave cone-rod response, and slope of best-corrected visual acuity (BCVA) with age.
2.3. Genetic Analyses
DNA was extracted by the Maxwell DNA purification kit (Promega). Whole exome sequencing (WES) was performed on genomic DNA samples at Pronto Diagnostics Inc. (using Agilent SureSelect Human All Exon V4 kit), Variantyx Ltd. (using Agilent SureSelect v6, on NovaSeq, Illumina) and 3billion (using IDT xGen Exome Research Panel V2). Alignment of NGS fastq files was performed using the Galaxy platform (https://galaxyproject.org). Reads were initially aligned to hg19 using HISAT2 aiming to identify the transcripts present in the analysis, followed by Bowtie2 alignment to a custom-made fasta reference file containing the different transcripts. BAM files were viewed by IGV and the number of reads representing each transcript was tabulated. Variant files were annotated using ANNOVAR according to the dbSNP database as well as using the Genoox platform (https://franklin.genoox.com/) with the following filtering steps: (1) Variants in known IRD genes that are located within homozygous regions were analyzed prior to any other analysis; (2) All variants in known IRD genes (based on RetNet https://sph.uth.edu/retnet/) were analyzed; (3) Variant type: Missense, nonsense, splice-site, stop-loss, insertions and deletions in the coding region were included; (4) Variants with minor allele frequency (MAF) greater than 1.0% in the gnomAD database (https://gnomad.broadinstitute.org) were excluded; (5) Prediction of the possible effect of each variant was analyzed by 3 prediction online programs [SIFT (http://sift.jcvi.org/), MutationTaster (http://www.muta- tiontaster.org/) and PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/). All suspected pathogenic variants were validated by Sanger sequencing of PCR products. cDNA was synthesized using the qPCRBIO cDNA Synthesis Kit (PCRBIOSYSTEMS). RT-PCR products were treated by ExoSAP and underwent library preparation for NGS based on commercial recommendations (Illumina, San Diego, California).
2.4. Extraction of RNA and cDNA Synthesis
Total RNA was isolated from confluent fibroblast cell cultures established from each sample using RNeasy mini kit (Qiagen, Hilden, Germany). Following RNA isolation, cDNA was synthesized using qScript™cDNA synthesis kit (Quantabio, Beverly, MA). Total RNA was also isolated from mouse and sheep retinal tissues by manual Trizol based RNA extraction method followed by DNase treatment (Thermofisher Scientific, Waltham, MA) and cDNA was synthesized as previously mentioned.
2.5. RT-PCR and Next Generation Sequencing (NGS)
The primers amplifying exons 2 to 5 were designed using Primer3 (https://bioinfo.ut.ee/primer3-0.4.0/) to perform RT-PCT: Forward primer- 5’- TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG aaagaagagattggacctgg-3’, Reverse primer- 3’- GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG ggctatcttttgagcatagc-5’. Further, the designed primer sequences were linked to universal adapter sequence to facilitate indexing of each sample before NGS. RT-PCR was carried out using 2X PCRBIO Taq mix red (PCR Biosystems, London). The products were loaded on an agarose gel to visualise the expression pattern of KIZ. In addition, gel densitometry analysis was carried out using ImageJ software. Further, the RT-PCR products were treated by ExoSAP followed by either Sanger sequencing or library preparation for NGS that was based on commercial recommendations (Illumina, San Diego, California). Single-reads (1x150) were sequenced using Illumina NextSeq500 with an average of >100,000 reads per sample. Following sequencing, the NGS fastq files were aligned using Usegalaxy (https://galaxyproject.org). The files were uploaded to the program and initially groomed using Fastq Groomer followed by Bowtie2 alignment to a custom-made fasta reference file containing the different transcripts. The generated BAM files were viewed by IGV and the number of reads representing each transcript was tabulated. Further, the percentage of reads corresponding to each transcript was calculated.
2.6. Exonic Splicing Enhancer Sequence (ESE) Analysis
Putative ESE sequences in exon 3 of the control sequence were identified using the online program ESEfinder 3.0 (http://krainer01.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi).21 Each predicted ESE sequence was recognized by a Serine/Arginine-rich splicing factor (SRSF) and the tool provides a score for each suspected ESE. Based on the inbuilt algorithm, the thresholds obtained for SRSF1, SRSF2, SRSF5 and SRSF6 are 1.956, 2.38, 2.67 and 2.676 respectively. Predicted sequences holding a threshold higher than the above mentioned values are suggested to be a potential ESE sequence. The present study has included only the highest scored ESE sequence including the c.226 position. In addition, the mutant sequence was also fed into the program to determine if the mutation had an effect on the potential ESE by projecting a score below the threshold
3. Results
3.1. Clinical Evaluation of KIZ Patient
Our cohort of IRD cases within the Israeli and Palestinian populations comprises 839 families affected by RP (with 1163 recruited patients). Using various genetic analysis tools (such as Sanger sequencing of founder mutations, whole exome sequencing- WES, and mutation panels), we identified 30 families with RP (Table 1 and Figure S1), including 31 RP patients harboring biallelic pathogenic variants in KIZ. In addition, we have identified three other siblings affected by RP who have not yet been recruited to this study. The most common mutation was c.226C>T (p.R76*) that we identified homozygously in 27 families (28 patients) and heterozygously in two families (2 patients) in trans with c.3G>A (p.M1?). Both mutations have been previously reported (Table S1). In addition, we identified one index case who was homozygous for a novel variant, c.247C>T (p.R83*).
With an objective to study the retinal clinical features associated with KIZ-related RP, we collected clinical data from 25 patients (Table S2). No consistent involvement of organs other than the eye was identified in this cohort, suggesting that KIZ is associated predominantly with nonsyndromic RP.
The VA metrics collected in 26 KIZ subjects showed that most patients maintain relatively good vision through the 6th decade of life. Up to the age of 50 years, all patients but one had VA>0.3 and two patients above the age of 60 years had VA>0.6 (Table S2 and Figure 1A). All patients exhibited unremarkable anterior segments and any cataractous changes were mild: notably, the youngest patient with cataract was 29 years old. In our cohort, only two patients underwent cataract surgery, at the ages of 44 and 56 years. Data on refractive error was available for 12 patients with variable values ranging from myopic to hyperopic correction along with variable astigmatism (Table S2). Among our cohort, the visual fields (n=10) were affected relatively early in the course of the disease, and visual field testing showed progression from multiple scotomas to severely restricted, tunnel vision typical for advanced RP. Fundus findings ranged from very mild to typical RP-like appearance (Figure 2). Seven patients had minimal funduscopic changes which included subtle attenuation of the retinal vessels and fine mottling of the RPE. On fundus autofluorescence (FAF) of these eyes, hypoautofluorescent spots and patchas surrounding the arcades along with a hyperautofluorescent ring in the macular area were noted. One of the patients (MOL1605-1 in Figure 2) had a pigmented lesion, which appeared to be not related to the rest of the findings, and the position close to the optic nerve seemed to not affect the VA as well as the retinal architecture. Eight other patients exhibited retinal findings consistent with RP, including bone spicule-like pigmentation, vessels attenuation and waxy pallor of the optic disc. These changes were more easily discernible on FAF imaging accompanied by a hyperautofluorescent ring in the macular region. OCT was performed on 12 patients demonstrating a reduction in the outer nuclear layer (ONL) thickness and a small island of ellipsoid zone (EZ) preservation in the fovea (Figure 2). Six patients presented with an epiretinal membrane (ERM) which did not appear to affect their vision. Two patients had cystoid macular edema (CME) with good response to carbonic anhydrase inhibitors.
Full-field ERG (ffERG) was performed in 14 patients (Figure 1B–D), revealing a pattern consistent with rod-cone dystrophy. Initially, at the first examination most patients exhibited measurable amplitudes which remained recordable even at the age of 75 years. Despite the persistence of measurable amplitudes, there were observable alterations in the ERG responses. Specifically, the cone flicker responses exhibited relatively preserved amplitudes but delayed implicit times (Figure 1B), while the rod responses were either reduced or completely extinguished (Figure 1C-D), consistent with the diagnosis of rod-cone dystrophy in these patients.
To derive insight on the similarity between KIZ-related RP and other prevalent variants, we performed comparative analysis. Pairwise t-test analysis comparing VA and ERG responses of patients with KIZ mutations to previously described cases harbouring mutations in DHDDS, FAM161A, or MAK [21,22], revealed that KIZ patients have on average higher cone and mixed ERG responses while the MAK group showed higher age of onset and better VA compared to the other three groups (Figure S2). We subsequently performed a principal component analysis (PCA) on the following clinical variables: disease onset age, age of presentation, rod response, cone flicker amplitude, a-wave and b-wave cone-rod responses, and slope of best-corrected VA (BCVA) with age. The sample variances and variable coefficient of PC1-7 are showed in Tables S3 and S4. The analysis revealed that PC1 accounts for 67.3% of the variability whereas PC2 explains only 12.9% of the remaining variability. The subsequent principal components (PC3-PC7) accounted for variability ranging between 0.3% to 8.9%. Therefore, we used PC1 values as a measure of disease severity to compare between four gene-based groups (Figure 3). There were discernible differences in the PCA1 scores between 2 groups; patients with mutations in DHDDS and FAM161A exhibited lower PCA1 scores, indicative of a more severe retinal disease compared to patients with mutations in KIZ and MAK. EOG was performed in 6 patients, showing a reduced Arden ratio to values that were lower than those expected by the amplitudes of the ffERG in five out of the six cases (Figure S3).
3.2. Characterization of the Expression Pattern of KIZ in Patient-Derived Skin Fibroblasts
The KIZ gene generates one major transcript encompassing all coding exons, alongside six minor transcripts primarily characterized by the exclusion of specific exons (Table S5). RT-PCR analysis of RNA isolated from primary fibroblast cell cultures of four control subjects and three patients homozygous for KIZ-c.226C>T revealed the presence of four different products (Figure 4A,B). Aiming to identify the corresponding KIZ transcripts, we performed both Sanger sequencing and NGS-based sequencing that revealed the following: Transcript 1- the full-length canonical transcript that includes all exons that are flanked by the designed set of primer (exons 2-5), Transcript 2- skipping of exon 3 and inclusion of an alternative exon within intron 3, Transcript 3- skipping of exon 3, and Transcript 4- skipping of both exons 3 and 4. We subsequently used gel densitometry analysis as well as NGS analysis to measure the intensity of each band and the coverage of each transcript as a measure of expression level (Figure 4C,D). Surprisingly, both analyses revealed lower expression of Transcript 1 in patients compared to controls. On the other hand, a higher expression level was evident for transcripts 3 and 4 (Table S5 and Figure 4C,D).
Hence, pronounced skipping of exon 3 which carries the pathogenic nonsense variant was evident in patients compared to healthy controls. This observation indicates that the variant may disrupt the normal splicing mechanism. It should be noted here that transcripts 2-4, in which exon 3 is skipped, are out of frame.
3.3. Identification of Exonic Splicing Enhancer (ESE) Sequences in KIZ-Exon 3
ESEs are discrete purine-rich sequences of 6-8 bps located within an exon. These short sequences facilitate exon definition by assisting in the recruitment of splicing factors to the adjacent intron. Changes in the ESE sequence due to mutations may affect the splicing mechanism [23]. We hypothesized that the KIZ nonsense variant might have disrupted a potential ESE leading to pronounced exon skipping in patients.
The analysis of exon 3 sequence using ESEfinder revealed a 7bp sequence that is recognized by Serine/Arginine-rich Splicing Factor 5 (SRSF5) and includes the c.226th position (Figure 4E,F), with a higher score for the control sequence (ATACTCG- 4.411) compared to the mutant sequence (ATACTTG- 2.694). The data indicates that the nonsense variant in KIZ might affect this ESE sequence, resulting in pronounced skipping of exon 3, as identified in primary fibroblasts of patients.
3.4. Characterization of KIZ Expression in Normal Mice and Sheep Retina
Aiming to characterize the expression of KIZ in the retina, we performed RT-PCR analysis on RNA isolated from normal mice (n=6) and sheep retina (n=1). We observed only two different transcripts: Transcript 1 corresponds to the full-length canonical transcript and Transcript 2 in which exon 4 is skipped (Figure S4A). NGS analysis revealed a similar expression level of both transcripts (Figure S4B,C). In addition, a small proportion of transcripts (only 0.10%) in which exon 3 is skipped was observed in the mouse retina by NGS (Figure S4D). Therefore, the expression pattern obtained in the mouse and sheep retina is very different from the one obtained in human fibroblast cells.
3.5. Analysis of Cilia Generation and Length in Controls and Patients
To examine the functional effect of the KIZ-c.226C>T mutation on the cilia, we immunostained the primary fibroblast cells from one control subjects and two patients targeting pericentrin and acetylated-α-tubulin. We found that 97.4±3.6% and 96.9±0.9% of cells (n=100 in each sample) were ciliated in controls and patients, respectively. In addition, mean ciliary length was measured to be 4.6±0.9 µm and 4.3±0.1 µm in control and patients, respectively (Figure S5). No significant difference was evident in these parameters.
4. Discussion
KIZ mutations are an uncommon cause of IRDs on a global scale, but they are notably more prevalent among individuals of Ashkenazi Jewish descent, with a carrier rate of 1 in 79 individuals for the p.R76* mutation according to the gnomAD database. This prevalence accounts for the occurrence of the disease in non-consanguineous families. In a previous study, we estimated that 4,090 individuals worldwide harbour biallelic KIZ mutations, with the majority attributable to the p.R76* variant that is particularly prevalent among individuals of Latino descent [24]. With the inclusion of the current study, a total of 51 biallelic KIZ cases associated with ARRP have been reported in the scientific literature, the majority of whom (31 cases) residing in Israel. Therefore, the current study represents, to the best of our knowledge, the largest cohort of KIZ-related ARRP cases reported to date.
All subjects with biallelic KIZ mutations were diagnosed with RP but displayed clinical variability in the age of onset and disease progression, similar to the variability reported for other ARRP genes [21,22]. Compared to RP associated with DHDDS or MAK mutations, patients with KIZ-related IRD presented with milder clinical features, although early-onset severe RP was noted in previous studies [25]. The retinal manifestations of KIZ patients are often subtle and may be missed on routine clinical examination, hence it should be noted that all patients exhibited retinal changes that were readily identifiable on fundus autofluorescence.
Our data suggest that individuals with RP exhibiting either mild or typical retinal abnormalities along with visual field defects, displayed a pattern of rod dysfunction exceeding cone dysfunction on a ffERG. This characteristic feature was accompanied by an EOG Arden ratio below the expected level. Hence, patients who belong to Ashkenazi or North-African Jewish ancestry, may potentially carry biallelic KIZ mutation(s) as the underlying cause of their disease.
The genetic analysis conducted in this study revealed two unexpected findings. First, we identified four transcripts generated by KIZ in WT primary fibroblast cells, three of which are out-of-frame due to exon skipping. In one of these transcripts, exon 3, harbouring the c.226C>T nonsense variant, is skipped. Interestingly, skipping of exon 3 was found to be enhanced in homozygous patients. This led us to hypothesize not only that c.226C>T introduce a nonsense mutation but might also affect a potential ESE sequence that is recognized by a SRSF. We therefore predict that the c.226C>T variant acts in a dual mutational mechanism, causing extra skipping of exon 3 (creating a frameshift) and introducing a PTC in normally spliced transcript. Variants that affect ESEs are difficult to identify and only one such variant, c.624G>A in BEST1, that is predicted to be a silent variant, has been reported thus far [26]. The effect of this variant on ESEs was studied by sequence analysis and the effect on splicing was investigated using a splicing assay. Since KIZ is expressed in fibroblasts, we examined the natural expression pattern of KIZ in WT and mutant primary cells. Such dual mechanisms of mutations might have important implication for developing mutation-specific therapeutic modalities. Translation readthrough is currently being studied and developed as a potential therapy for nonsense mutations that cause a variety of inherited diseases, either by readthrough drugs such as ataluren [27,28,29] or by suppressor tRNAs [30,31]. Both methods rely on a substantial amount of available transcripts, however two mechanisms might limit this amount- the nonsense mediated mRNA decay (NMD) that degrades transcripts bearing a PTC [32] and ESE-affecting nonsense mutations, such as KIZ-c.226C>T, leading to aberrant splicing. While NMD does not seem to affect KIZ transcripts in fibroblasts (and potentially in the retina), however, the effect of c.226C>T on splicing might limit the number of transcripts for readthrough drugs. RNA editing, which is another mutation-specific therapy that is based on the adenosine deaminases acting on RNA (ADAR) enzyme, is usually performed at the pre-mRNA stage and is therefore less affected by NMD, however editing levels reported thus far are for this mutation are relatively low and need to be improved before considered as a potential therapy [33]. As such, the most suitable therapeutical approach for KIZ is currently gene augmentation therapy, which has not yet been developed for this particular gene.
One of the primary functional assays used to assess the efficacy of readthrough for nonsense mutations in ciliary proteins involves observing the ability of starved mutant fibroblast cells to develop cilia both before and after undergoing readthrough treatment [34]. This approach has been well-established for other retinal proteins that are localized to the photoreceptor cilia, such as USH2A [29], RPGR[35], and FAM161A [34]. Although KIZ is a known ciliary protein [7], we could not determine any effect of the c.226C>T variant on ciliary growth of mutant fibroblast cells, in agreement with previous results reported for other KIZ mutations [19]. Therefore, ciliary growth cannot serve as a functional assay for assessing KIZ recovery.
In conclusion, we present the largest cohort of biallelic KIZ cases reported to date. The 31 individuals we recruited were all diagnosed with ARRP of varying severity, which on average was slightly greater compared to cases associated with MAK mutations, but milder than the phenotype observed in cases linked to DHDDS and FAM161A mutations. The predominantly identified mutation is c.226C>T that acts as a nonsense mutation as well as an ESE-affecting mutation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org; Figure S1: Pedigrees of KIZ-related families; Figure S2: Comparison of clinical parameters by two-sided t-test; Figure S3: EOG versus ERG data; Figure S4: RT-PCR analysis of normal mice and sheep retina; Figure S5: Ciliary analysis of c.226C>T fibroblasts; Table S1: Reported mutations in KIZ; Table S2: Clinical information of patients will biallelic KIZ pathogenic variants; Table S3: The seven principal components sample variances obtained in this study; Table S4: The variable coefficient in each of the seven principal component; Table S5: KIZ transcript information.
Author Contributions
Conceptualization, Y.S., E.B., and D.S.; methodology, Y.S., A.R., A.O., P.G., H.O.H., A.S., M.R., and S.D.; writing—original draft preparation, Y.S. and D.S; writing—review and editing, A.R., A.O. P.G., H.O.H., A.S., S.K., M.R., R.O., S.D., H.N., R.L., S.S., D.Z., T.B.Y., E.B. funding acquisition, D.S., E.B., T.B.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Israel Science Foundation, grant number 1778/20, the Israeli Ministry of Health, grant number 3-14995, and the foundation fighting blindness (FFB), grant number BR-GE-0518-0734.
Institutional Review Board Statement
Ethical approval was obtained from Hadassah University Medical Center Institutional Review Board (approval code 21-03.08.07, approved on 3 August 2007). The tenets of the Declaration of Helsinki were followed. Participants provided written informed consent after receiving an explanation about the study and its possible consequences and before donating blood samples.
Informed Consent Statement
nformed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data of this study are presented within the article and its Supplementary Materials. The authors are willing to share materials, data sets, and protocols used in the acquisition of data presented in this publication with other researchers upon request (contact Dror Sharon, E-mail: dror.sharon1@mail.huji.ac.il).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Schneider, N.; Sundaresan, Y.; Gopalakrishnan, P.; Beryozkin, A.; Hanany, M.; Levanon, E.Y.; Banin, E.; Ben-Aroya, S.; Sharon, D. Inherited Retinal Diseases: Linking Genes, Disease-Causing Variants, and Relevant Therapeutic Modalities. Prog Retin Eye Res 2021, 101029. [Google Scholar] [CrossRef] [PubMed]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis Pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef] [PubMed]
- Ayuso, C.; Millan, J.M. Retinitis Pigmentosa and Allied Conditions Today: A Paradigm of Translational Research. Genome Med 2010, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Welby, E.; Li, T.; Swaroop, A. Retinal Disease in Ciliopathies: Recent Advances with a Focus on Stem Cell-Based Therapies. Transl Sci Rare Dis 2019, 4, 97–115. [Google Scholar] [CrossRef] [PubMed]
- Tatour, Y.; Ben-Yosef, T. Syndromic Inherited Retinal Diseases: Genetic, Clinical and Diagnostic Aspects. Diagnostics 2020, 10, 779. [Google Scholar] [CrossRef]
- Chen, H.Y.; Kelley, R.A.; Li, T.; Swaroop, A. Primary Cilia Biogenesis and Associated Retinal Ciliopathies. Semin Cell Dev Biol 2021, 110, 70–88. [Google Scholar] [CrossRef] [PubMed]
- El Shamieh, S.; Neuillé, M.; Terray, A.; Orhan, E.; Condroyer, C.; Démontant, V.; Michiels, C.; Antonio, A.; Boyard, F.; Lancelot, M.-E.; et al. Whole-Exome Sequencing Identifies KIZ as a Ciliary Gene Associated with Autosomal-Recessive Rod-Cone Dystrophy. Am J Hum Genet 2014, 94, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, K.; Duncan, J.L.; Biswas, P.; Soto-Hermida, A.; Matsui, H.; Jakubosky, D.; Suk, J.; Telenti, A.; Frazer, K.A.; Ayyagari, R. Whole Genome Sequencing Revealed Mutations in Two Independent Genes as the Underlying Cause of Retinal Degeneration in an Ashkenazi Jewish Pedigree. Genes (Basel) 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- El Shamieh, S.; Méjécase, C.; Bertelli, M.; Terray, A.; Michiels, C.; Condroyer, C.; Fouquet, S.; Sadoun, M.; Clérin, E.; Liu, B.; et al. Further Insights into the Ciliary Gene and Protein KIZ and Its Murine Ortholog PLK1S1 Mutated in Rod-Cone Dystrophy. Genes (Basel) 2017, 8. [Google Scholar] [CrossRef]
- Méjécase, C.; Kozak, I.; Moosajee, M. The Genetic Landscape of Inherited Eye Disorders in 74 Consecutive Families from the United Arab Emirates. Am J Med Genet C Semin Med Genet 2020, 184, 762–772. [Google Scholar] [CrossRef]
- Jauregui, R.; Chan, L.; Oh, J.K.; Cho, A.; Sparrow, J.R.; Tsang, S.H. Disease Asymmetry and Hyperautofluorescent Ring Shape in Retinitis Pigmentosa Patients. Sci Rep 2020, 10, 3364. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Ben-Yosef, T.; Goldenberg- Cohen, N.; Pras, E.; Gradstein, L.; Soudry, S.; Mezer, E.; Zur, D.; Abbasi, A.H.; Zeitz, C.; et al. A Nation-wide Genetic Analysis of Inherited Retinal Diseases in Israel as Assessed by the Israeli Inherited Retinal Disease Consortium (IIRDC). Hum Mutat 2019, 41, 140–149. [Google Scholar] [CrossRef]
- Weisschuh, N.; Obermaier, C.D.; Battke, F.; Bernd, A.; Kuehlewein, L.; Nasser, F.; Zobor, D.; Zrenner, E.; Weber, E.; Wissinger, B.; et al. Genetic Architecture of Inherited Retinal Degeneration in Germany: A Large Cohort Study from a Single Diagnostic Center over a 9-Year Period. Hum Mutat 2020, 41, 1514–1527. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Xu, C.L.; Breazzano, M.P.; Tanaka, A.J.; Ryu, J.; Levi, S.R.; Yao, K.; Sparrow, J.R.; Tsang, S.H. Progressive RPE Atrophy and Photoreceptor Death in KIZ-Associated Autosomal Recessive Retinitis Pigmentosa. Ophthalmic Genet 2020, 41, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Villafuerte-de la Cruz, R.A.; Garza-Garza, L.A.; Garza-Leon, M.; Rodriguez-De la Torre, C.; Parra-Bernal, C.; Vazquez-Camas, I.; Ramos-Gonzalez, D.; Rangel-Padilla, A.; Espino Barros-Palau, A.; Nava-García, J.; et al. Spectrum of Variants Associated with Inherited Retinal Dystrophies in Northeast Mexico. BMC Ophthalmol 2024, 24, 60. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Coussa, R.G.; DeBenedictis, M.J.M.; Traboulsi, E.I. Retinal Dystrophy Associated with a Kizuna (KIZ) Mutation and a Predominantly Macular Phenotype. Ophthalmic Genet 2019, 40, 455–460. [Google Scholar] [CrossRef] [PubMed]
- El Shamieh, S.; Neuillé, M.; Terray, A.; Orhan, E.; Condroyer, C.; Démontant, V.; Michiels, C.; Antonio, A.; Boyard, F.; Lancelot, M.-E.; et al. Whole-Exome Sequencing Identifies KIZ as a Ciliary Gene Associated with Autosomal-Recessive Rod-Cone Dystrophy. Am J Hum Genet 2014, 94, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Oshimori, N.; Ohsugi, M.; Yamamoto, T. The Plk1 Target Kizuna Stabilizes Mitotic Centrosomes to Ensure Spindle Bipolarity. Nat Cell Biol 2006, 8, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- El Shamieh, S.; Méjécase, C.; Bertelli, M.; Terray, A.; Michiels, C.; Condroyer, C.; Fouquet, S.; Sadoun, M.; Clérin, E.; Liu, B.; et al. Further Insights into the Ciliary Gene and Protein KIZ and Its Murine Ortholog PLK1S1 Mutated in Rod-Cone Dystrophy. Genes (Basel) 2017, 8. [Google Scholar] [CrossRef]
- Beryozkin, A.; Khateb, S.; Idrobo-Robalino, C.A.; Khan, M.I.; Cremers, F.P.M.; Obolensky, A.; Hanany, M.; Mezer, E.; Chowers, I.; Newman, H.; et al. Unique Combination of Clinical Features in a Large Cohort of 100 Patients with Retinitis Pigmentosa Caused by FAM161A Mutations. Sci Rep 2020, 10. [Google Scholar] [CrossRef]
- Kimchi, A.; Khateb, S.; Wen, R.; Guan, Z.; Obolensky, A.; Beryozkin, A.; Kurtzman, S.; Blumenfeld, A.; Pras, E.; Jacobson, S.G.; et al. Nonsyndromic Retinitis Pigmentosa in the Ashkenazi Jewish Population. Genetic and Clinical Aspects. Ophthalmology 2018, 125, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Beryozkin, A.; Khateb, S.; Idrobo-Robalino, C.; Khan, M.; Cremers, F.; Obolensky, A.; Hanany, M.; Mezer, E.; Chowers, I.; Newman, H.; et al. Unique Combination of Clinical Features in a Large Cohort of 100 Patients with Retinitis Pigmentosa Caused by FAM161A Mutations. Sci Rep 2020, 10, 15156. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, Y.; Banin, E.; Sharon, D. Exonic Variants That Affect Splicing - An Opportunity for “Hidden” Mutations Causing Inherited Retinal Diseases. Adv Exp Med Biol 2023, 1415, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Hanany, M.; Rivolta, C.; Sharon, D. Worldwide Carrier Frequency and Genetic Prevalence of Autosomal Recessive Inherited Retinal Diseases. Proceedings of the National Academy of Sciences 2020, 117, 2710–2716. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Xu, C.L.; Breazzano, M.P.; Tanaka, A.J.; Ryu, J.; Levi, S.R.; Yao, K.; Sparrow, J.R.; Tsang, S.H. Progressive RPE Atrophy and Photoreceptor Death in KIZ-Associated Autosomal Recessive Retinitis Pigmentosa. Ophthalmic Genet 2020, 41, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Kramer, F.; Mohr, N.; Kellner, U.; Rudolph, G.; Weber, B.H. Ten Novel Mutations in VMD2 Associated with Best Macular Dystrophy (BMD). Hum Mutat 2003, 22, 418. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Flanigan, K.M.; Wong, B.; Bonnemann, C.; Sampson, J.; Sweeney, H.L.; Reha, A.; Northcutt, V.J.; Elfring, G.; Barth, J.; et al. Phase 2a Study of Ataluren-Mediated Dystrophin Production in Patients with Nonsense Mutation Duchenne Muscular Dystrophy. PLoS One 2013, 8, e81302. [Google Scholar] [CrossRef] [PubMed]
- Wilschanski, M.; Miller, L.L.; Shoseyov, D.; Blau, H.; Rivlin, J.; Aviram, M.; Cohen, M.; Armoni, S.; Yaakov, Y.; Pugatsch, T.; et al. Chronic Ataluren (PTC124) Treatment of Nonsense Mutation Cystic Fibrosis. Eur Respir J 2011, 38, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Samanta, A.; Stingl, K.; Kohl, S.; Nagel-Wolfrum, K.; Ries, J.; Linnert, J. Ataluren for the Treatment of Usher Syndrome 2A Caused by Nonsense Mutations. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Mendonca, C.A.; Yukselen, O.; Muneeruddin, K.; Ren, L.; Liang, J.; Zhou, C.; Xie, J.; Li, J.; et al. AAV-Delivered Suppressor TRNA Overcomes a Nonsense Mutation in Mice. Nature 2022, 604, 343–348. [Google Scholar] [CrossRef]
- Albers, S.; Allen, E.C.; Bharti, N.; Davyt, M.; Joshi, D.; Perez-Garcia, C.G.; Santos, L.; Mukthavaram, R.; Delgado-Toscano, M.A.; Molina, B.; et al. Engineered TRNAs Suppress Nonsense Mutations in Cells and in Vivo. Nature 2023, 618, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Kulozik, A.E. A Perfect Message: RNA Surveillance and Nonsense-Mediated Decay. Cell 1999, 96, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Schneider, N.; Steinberg, R.; Ben-David, A.; Valensi, J.; David-Kadoch, G.; Rosenwasser, Z.; Banin, E.; Levanon, E.Y.; Sharon, D.; Ben-Aroya, S. A Pipeline for Identifying Guide RNA Sequences That Promote RNA Editing of Nonsense Mutations That Cause Inherited Retinal Diseases. Mol Ther Nucleic Acids 2024, 35, 102130. [Google Scholar] [CrossRef] [PubMed]
- Beryozkin, A.; Samanta, A.; Gopalakrishnan, P.; Khateb, S.; Banin, E.; Sharon, D.; Nagel-Wolfrum, K. Translational Read-Through Drugs (TRIDs) Are Able to Restore Protein Expression and Ciliogenesis in Fibroblasts of Patients with Retinitis Pigmentosa Caused by a Premature Termination Codon in FAM161A. Int J Mol Sci 2022, 23, 3541. [Google Scholar] [CrossRef]
- Vössing, C.; Owczarek-Lipska, M.; Nagel-Wolfrum, K.; Reiff, C.; Jüschke, C.; Neidhardt, J. Translational Read-through Therapy of Rpgr Nonsense Mutations. Int J Mol Sci 2020, 21, 1–16. [Google Scholar] [CrossRef]
Figure 1.
Graphs showing the decline of visual acuity (A) and ERG responses (B-D) versus age (years) in KIZ patients. A- Snellen visual acuity. B. Cone photoreceptor function as measured in response to a white 30-Hz flicker stimulus versus age (lower limit of normal cone 30-Hz flicker ERG amplitudes, 60 uV). C. Dark-adapted rod response. D. Dark-adapted a-wave. Each point represents the average of both eyes versus age. Data from cases with multiple data points are connected with a line. Data were collected from patients with biallelic mutations in KIZ (green), MAK (red), DHDDS (grey), and FAM161A (yellow).
Figure 1.
Graphs showing the decline of visual acuity (A) and ERG responses (B-D) versus age (years) in KIZ patients. A- Snellen visual acuity. B. Cone photoreceptor function as measured in response to a white 30-Hz flicker stimulus versus age (lower limit of normal cone 30-Hz flicker ERG amplitudes, 60 uV). C. Dark-adapted rod response. D. Dark-adapted a-wave. Each point represents the average of both eyes versus age. Data from cases with multiple data points are connected with a line. Data were collected from patients with biallelic mutations in KIZ (green), MAK (red), DHDDS (grey), and FAM161A (yellow).
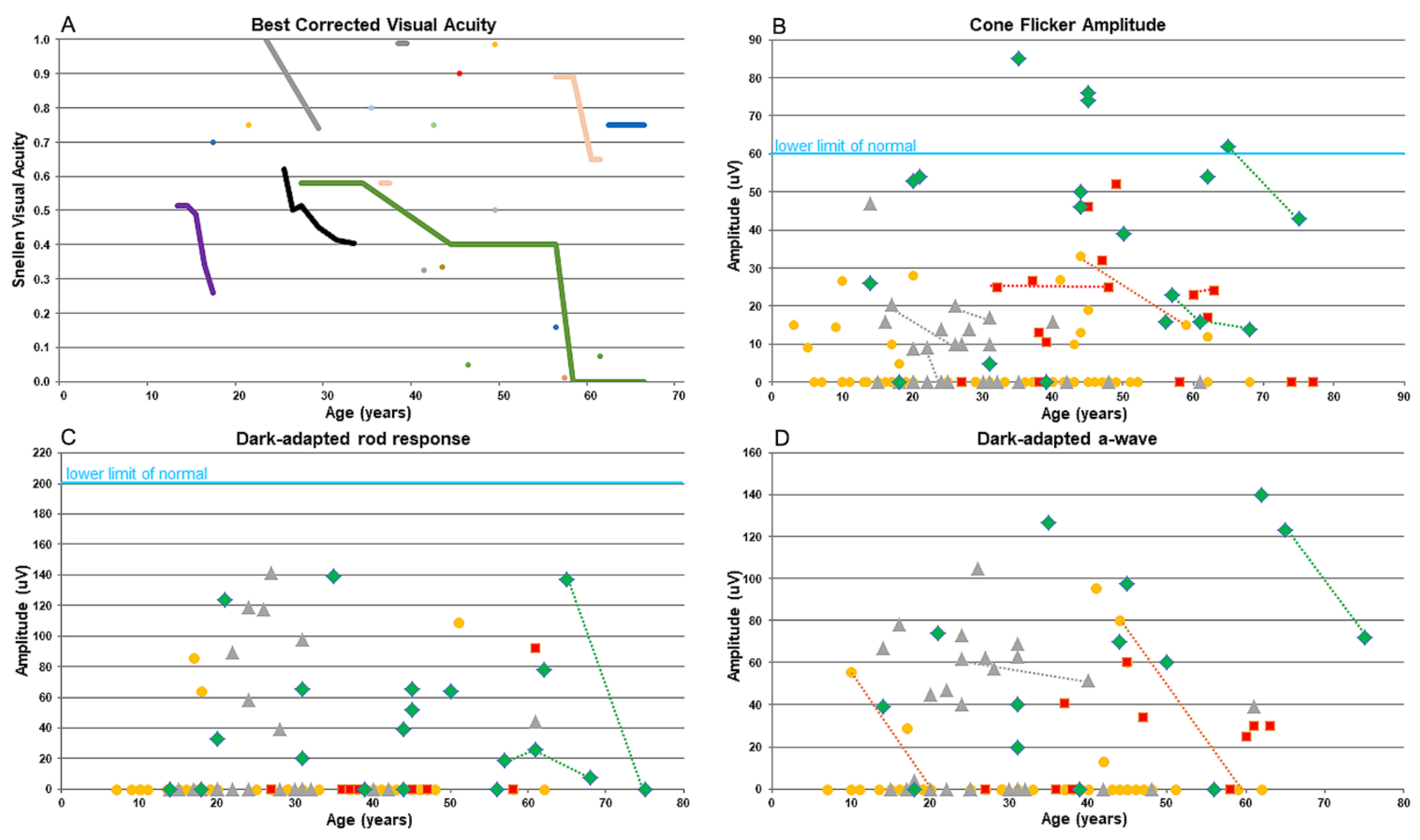
Figure 2.
Ultra-wide field pseudocolor, fundus autofluorescence (FAF), infra-red (IR), and OCT imaging in patients with retinitis pigmentosa (RP) resulting from KIZ mutations at different ages. See detailed description in the Supp info.
Figure 2.
Ultra-wide field pseudocolor, fundus autofluorescence (FAF), infra-red (IR), and OCT imaging in patients with retinitis pigmentosa (RP) resulting from KIZ mutations at different ages. See detailed description in the Supp info.
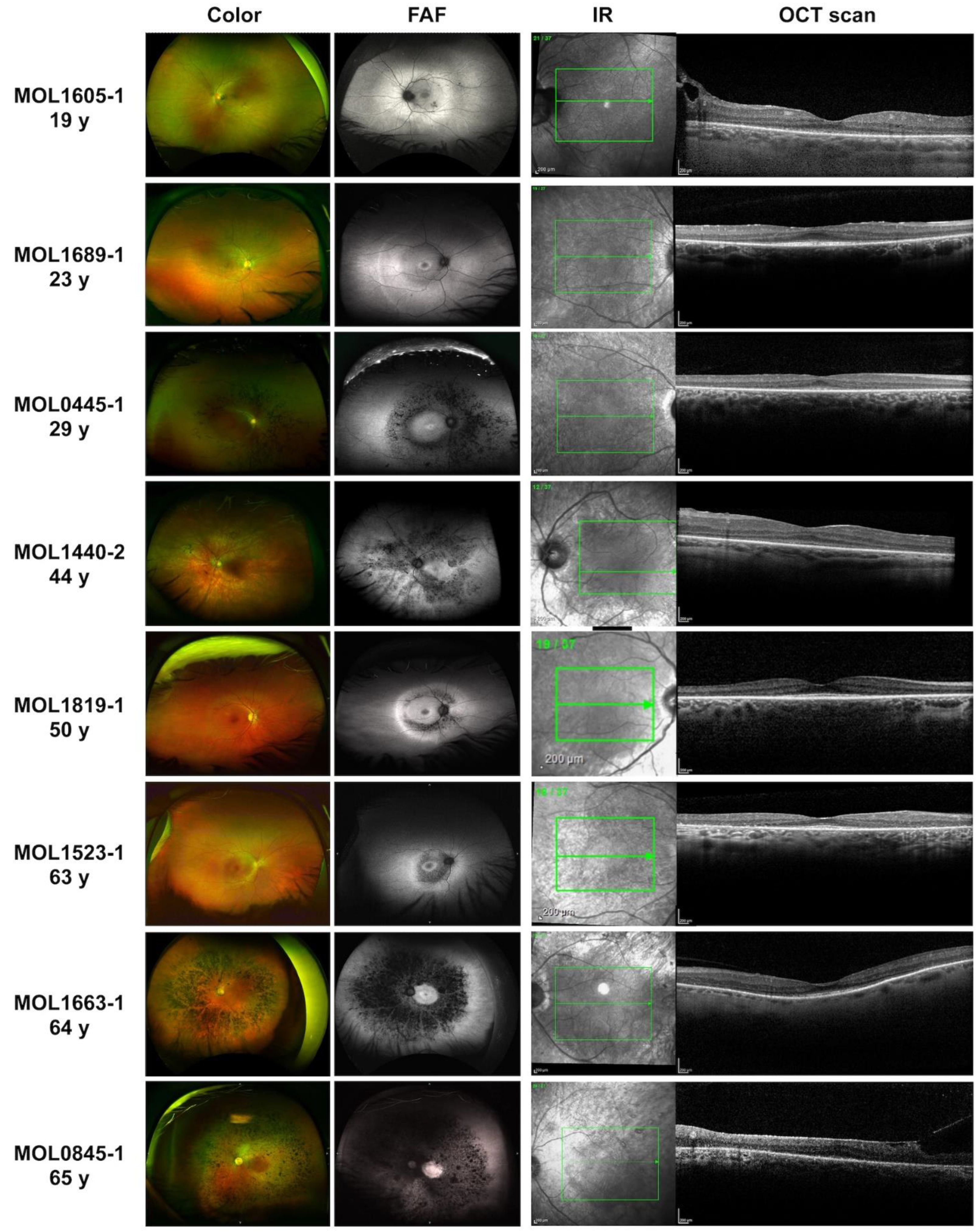
Figure 3.
PCA1 analysis of clinical parameters collected from patient harbouring mutations in four IRD-causing genes. Since in all PC1 variables low levels reflect a more severe disease, and since their coefficient are positive scalars (see column 1 in Table S4), one can refer to high patient scores as indicating a more severe disease. Post hoc analysis comparing each paired of genes, showing a significant difference between MAK and DHDDS (p-value = 0.017), KIZ and FAM161A (p-value = 0.018), MAK and FAM161A (p-value = 0.002).
Figure 3.
PCA1 analysis of clinical parameters collected from patient harbouring mutations in four IRD-causing genes. Since in all PC1 variables low levels reflect a more severe disease, and since their coefficient are positive scalars (see column 1 in Table S4), one can refer to high patient scores as indicating a more severe disease. Post hoc analysis comparing each paired of genes, showing a significant difference between MAK and DHDDS (p-value = 0.017), KIZ and FAM161A (p-value = 0.018), MAK and FAM161A (p-value = 0.002).
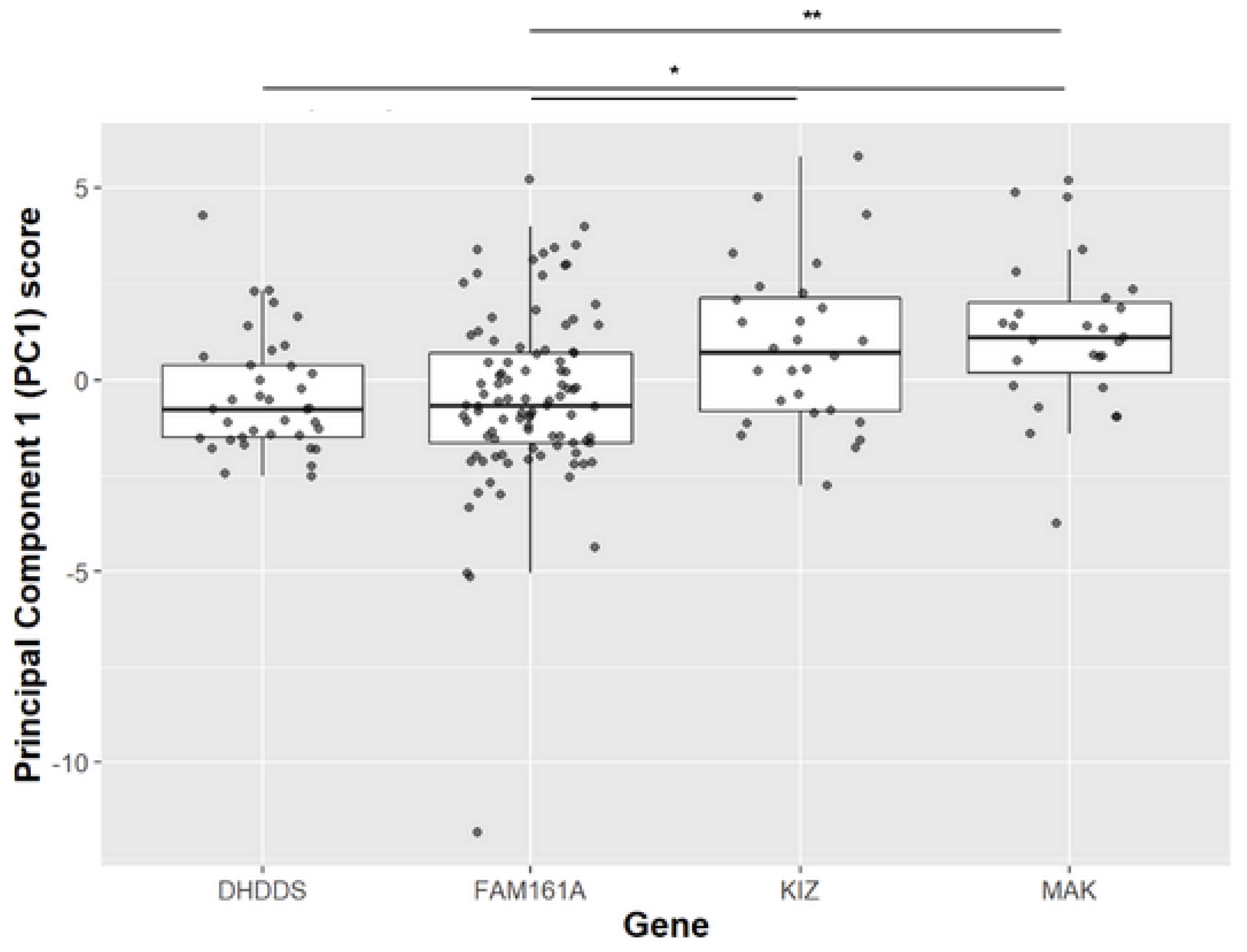
Figure 4.
Expression of KIZ in human fibroblast cells. A- Schematic structure of the human KIZ gene indicating exons that are relevant for the current analysis. B- RT-PCR agarose gel analysis of primary fibroblast cells from four controls (C1-C4) and three KIZ patients (P1-P3). Following NGS and Sanger sequencing of the PCR products, the identity of each fragment was determined as schematically depicted on the right. C,D- Gel densitometry (C) and NGS-based analysis (D) of transcripts 1, 3, and 4. Although Transcript 2 was evident in both Sanger and NGS analyses, its expression level was extremely low in all samples and therefore it was excluded from this analysis. E,F- Exonic sequence enhancer (ESE) analysis of WT (E) and the c.226C>T mutant allele (F) of KIZ Exon 3. The binding site and score of the SRSF5 protein are shown above the sequence.
Figure 4.
Expression of KIZ in human fibroblast cells. A- Schematic structure of the human KIZ gene indicating exons that are relevant for the current analysis. B- RT-PCR agarose gel analysis of primary fibroblast cells from four controls (C1-C4) and three KIZ patients (P1-P3). Following NGS and Sanger sequencing of the PCR products, the identity of each fragment was determined as schematically depicted on the right. C,D- Gel densitometry (C) and NGS-based analysis (D) of transcripts 1, 3, and 4. Although Transcript 2 was evident in both Sanger and NGS analyses, its expression level was extremely low in all samples and therefore it was excluded from this analysis. E,F- Exonic sequence enhancer (ESE) analysis of WT (E) and the c.226C>T mutant allele (F) of KIZ Exon 3. The binding site and score of the SRSF5 protein are shown above the sequence.
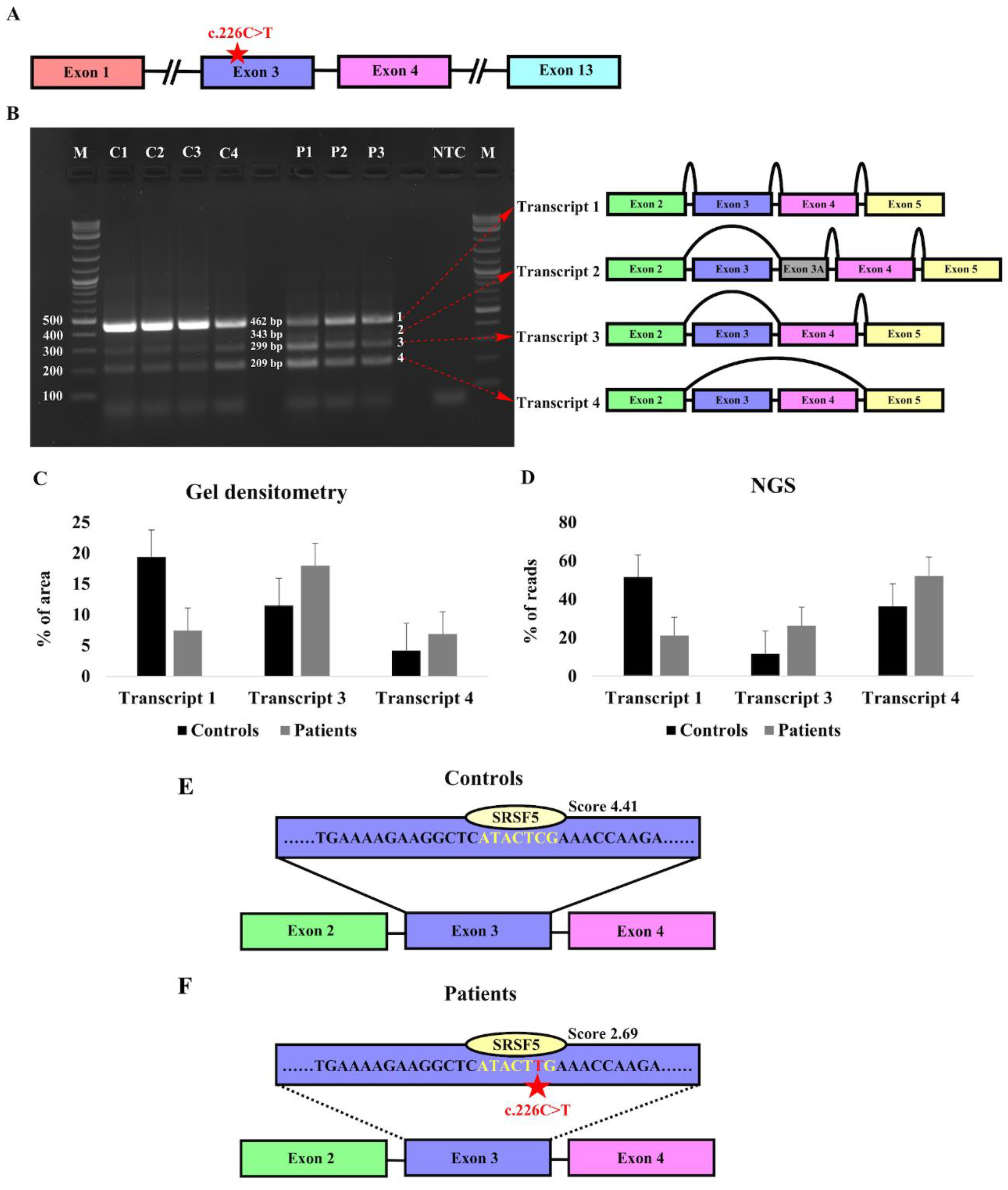

Table 1.
Families with biallelic KIZ mutations.
| Family Number | Origin | Consanguinity | Mutation 1 | Mutation 2 | # affected recruited | # affected- not recruited |
|---|---|---|---|---|---|---|
| MOL0289 | Turkish Jew | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| MOL0336 | NAJ | 2:3 | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| MOL0445 | NAJ | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| MOL0588 | ASH | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 1 |
| MOL0610 | ASH | 3:3 | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| MOL0845 | ASH | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| MOL1015 | ASH/Turkish Jew | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| MOL1156 | ASH | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| MOL1236 | AM | 2:2 | c.247C>T, p.R83* | c.247C>T, p.R83* | 1 | 0 |
| MOL1329 | ASH | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| MOL1440 | ASH | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 2 | 0 |
| MOL1523 | ASH | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| MOL1605 | ASH | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 1 |
| MOL1621 | ASH/Iraq | No | c.226C>T, p.R76* | c.3G>A, p.M1? | 1 | 1 |
| MOL1663 | ASH | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| MOL1684 | ASH | 3:3 | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| MOL1689 | ASH | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| MOL1720 | ASH | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| MOL1819 | NAJ | No | c.226C>T, p.R76* | c.3G>A, p.M1? | 1 | 0 |
| MOL2026 | NAJ | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| TB240 | ASH | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| TB244 | ASH | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| TB338 | ASH | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| TB675 | ASH | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| TB711 | ASH | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| TB736 | ASH | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| TB928 | ASH | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| TB980 | NAJ | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| TB1044 | ASH | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
| TB1212 | ASH | No | c.226C>T, p.R76* | c.226C>T, p.R76* | 1 | 0 |
NAJ- North-African Jews, ASH- Ashkenazi Jews, AM- Arab Muslim
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



