Submitted:
27 March 2024
Posted:
28 March 2024
You are already at the latest version
Abstract
Immune-cell reprogramming driven by mitochondria-derived reactive electrophilic immunometabolites (mt-REMs—e.g., fumarate, itaconate) is an emerging phenomenon of major biomedical importance. Despite their localized production, mt-REMs elicit significantly-large local and global footprints within and across cells, through mechanisms involving electrophile signaling. Burgeoning efforts are being put into profiling mt-REMs’ potential protein-targets and phenotypic mapping of their multifaceted inflammatory behaviors. Yet, precision indexing of mt-REMs’ first-responders with spatiotemporal intelligence and locale-specific function assignments remains elusive. Highlighting the latest advances and overarching challenges, this perspective aims to stimulate thoughts and spur interdisciplinary innovations to address these unmet chemical-biotechnological needs at therapeutic immuno-signaling frontiers.
Keywords:
Immunometabolite
; Mitochondria-derived metabolite
; Itaconate
; Aconitate
; Fumarate
; Reactive metabolite
; Electrophile signaling
; Protein profiling
1. Introduction
The human body harbors thousands of endogenously-generated metabolites that exhibit divergent bioactivities and pathway functions. The origins of these metabolites are also diverse, stemming from varied pathophysiological processes [1]. Some of these metabolites—broadly coined as reactive electrophilic or oxidant species (RES/ROS) [2]—manifest innate reactivity [3,4] (Figure 1a). This reactivity inevitably translates to their long-appreciated damaging roles when available in excess: e.g., when cell-autonomous regulation goes awry, or following RES/ROS bulk administration in high dosage, and/or over prolonged duration. Conversely, under controlled/physiological conditions, RES/ROS constitute essential context-specific signaling mediators for cell decision-making [4]. The latest chemical-biology innovations have cast light on the capability of individual RES to drive non-enzyme-assisted posttranslational protein modifications (nE-PTMs), even at low ligand occupancy, analogous to canonical enzymatic PTMs, e.g., phosphorylation [5]. Such RES-driven gain-of-function (GOF) or dominant loss-of-function (dLOF) signaling events open a nature-inspired in-road toward precision covalent-drug candidates [6,7] (Figure 1b,c). This perspective primarily focuses on a class of RES of mitochondria origin—here termed ‘mitochondrial-derived reactive electrophilic metabolites (mt-REMs) (Figure 1a)—that has gained attention from myriad research communities: inflammation [8], infectious disease [9], cancer immunology [10,11], etc.
Thoughtful integrations of metabolic and immunological research programs have led to the burgeoning appreciation of immunometabolic-regulatory powers of mt-REMs, and their therapeutic potential. The generation of mt-REMs is intricately linked to the tricarboxylic acid (TCA) cycle [12], which involves a series of enzymatic reactions that produce key mt-REMs or their precursors (Figure 2a). Activity of some of these enzymes—and levels of associated mt-REMs—directly correlates with clinical response to immune checkpoint blockade (ICB) therapies [10]. However, because of the nature of the tools deployed and inherent complexities as we discussed herein, we currently have little clue, as to how these individual mt-REMs modulate specific pathways to effect such profound changes. Our current understanding of locale-/context-specific protein responders that engage with specific mt-REMs and trigger functional GOF/dLOF nE-PTM signaling (Figure 1c) also remains limited. These fundamental knowledge gaps need to be filled.
Our perspective first undertakes an in-depth examination of itaconate (Figure 1a)—arguably one of the most popular mt-REM with extensive broad-spectrum immunomodulatory operations [13,14]. We discuss the biosynthesis, feedback regulation, and functional ramifications of itaconate on representative targets/pathways. Next, we analyze other notable mt-REMs (Figure 1a) and associated immune-relevant mechanisms-of-action (MoAs). Subsequently, we describe chemical-biological methods to profile mt-REMs’ prospective cellular targets. Finally, we envision how the emerging electrophile-function-guided proximity-mapping and signaling-interrogation tools—when accompanied by innovative adaptations and careful considerations—hold exciting prospects toward deciphering the local signaling activities of mt-REMs that influence global decision-making. While summarizing new knowledge and advances in each section, key limitations are also emphasized with the aim to stimulate thought and discussion and spur new innovations.
2. mt-REMs and Their Immunometabolic Signaling
2.1. Itaconate
2.1.1. Biosynthesis, Metabolic Transformations, and ‘Self-Modulation’
Itaconate is produced by the decarboxylation of cis-aconitate in a reaction catalyzed by cis-aconitate decarboxylase (ACOD1, also known as IRG1 and CAD), a mitochondrial matrix enzyme encoded in immune-responsive gene 1 (irg1) (Figure 2b). Cis-aconitate is a transitory intermediate within the TCA cycle, and thus not typically featured within the latter’s 8 different steps (Figure 2a). Cis-aconitate can be released during the rate-limiting step [15] of aconitase-catalyzed isomerization that involves two-step dehydration – rehydration sequence, converting citrate to iso-citrate, via cis-aconitate (Figure 2b). Citrate is derived from the first committed step of the TCA cycle: citrate-synthase (CS)-catalyzed condensation of acetyl-CoA and oxaloacetate (Figure 2a,b).
The so-produced itaconate can complete its metabolic cycle as follows: transformation into itaconyl-coenzyme A (CoA); subsequent conversion to citramalyl-CoA, which is catabolized by citramalyl-CoA lyase (CLYBL), yielding pyruvate and acetyl-CoA; and acetyl-CoA can re-enter the TCA cycle (Figure 2b) [16]. Recent reports further indicate the intracellular isomerization potential of itaconate to structural isomers, mesaconate, and likely, citraconate (Figure 1a) [17]. [Section 2.2 discusses these isomers]. Interestingly, the effects of itaconate—generated within the mitochondrial matrix—extend beyond its site of production. SLC25A11—dicarboxylate, citrate, and alpha-oxoglutarate solute-carrier protein on the inner mitochondrial membrane, is implicated in itaconate’s export out of mitochondrion (Figure 3). [18] Export into the extracellular space is thought to be mediated by ATP-binding cassette (ABC) transporter G2 (ABCG2) [19]—a member of the ABC transporter superfamily that exports numerous exo(endo)genous agents, e.g., cytotoxic drugs, toxins, and hormones (Figure 3). ABCG2 is a promising target in combating multidrug resistance in chemotherapy [20]. Nonetheless, the genetic necessity and sufficiency of transporters in mt-REMs’ inter-compartmental and cell-cell signaling roles remain to be validated.
IRG1 is inducibly expressed in innate-immune cells of myeloid lineage under pro-inflammatory conditions, particularly in macrophages, and to a reduced extent in dendritic cells and neutrophils. Highly conserved across different taxa, from marine invertebrates such as mussels [21], to mammalian macrophages, irg1 is an essential gene for the host’s innate-immune response. IRG1 expression is significantly (up to ~200-fold) upregulated in activated macrophages [13]. Toll-like receptor (TLR) signaling operative in several types of immune cells, and signaling actions driven by cytokines (protein-based factors involved in immune signaling), are among the pathways that drive IRG1 upregulation. Additionally, bacterial antigen lipopolysaccharide (LPS) can stimulate TLR4 on the surface of macrophages, which initiates a signaling cascade that activates various transcription factors and upregulates immune response genes, including irg1. Furthermore, oxidative stress-associated upregulation of ROS supports IRG1 induction [7]. RAW264.7 mouse macrophage-like cells following LPS-induced immune activation, harbor up to ~8 mM intracellular itaconate, from sub-micromolar concentrations in basal, non-activated, macrophages [22,23]. Such millimolar concentrations are also the dosage range within which itaconate exerts antimicrobial efficacies [24].
Interestingly, accumulating (pre)clinical studies indicate IRG1’s oncogene-like behaviors in many types of cancers. IRG1-knockout (KO) positively improves clinical outcomes of ICB-based anticancer immunotherapies. For instance, IRG1-KO reverses the undesirable immunosuppression by tumor-associated macrophages (TAMs) [25] and myeloid-derived immunosuppressor cells (MDSCs) [26] within the tumor microenvironment. Furthermore, therapeutic impacts against tumors are amplified when IRG1-KO chimeric antigen receptor (CAR)-macrophages are used alongside ICB therapies, such as anti-CD47 or anti-PD1 antibodies [27]. Given the clinical significance of IRG1 activity that highlights the importance of itaconate in regulating immune behaviors, one may expect that itaconate-mimicking small-molecule modulators are a promising gateway toward anticancer-drug development. But such endeavors presently face significant challenges. One major difficulty is due to the growing inventory of itaconate-regulated proteins/pathways, yet with largely poorly-resolved context-specific understanding.
Critically, itaconate’s ability to engage with a wide repertoire of targets enables the cell to ‘self-modulate’ itaconate’s levels, without absolute reliance on IRG1-expression/activity levels. Even within the mitochondrion, itaconate acts as a reversible competitive substrate-mimic inhibitor of succinate dehydrogenase (SDH) [28,29] (Figure 2d), downstream of cis-aconitate production in the TCA cycle (Figure 2a). This feedback inhibitory event reduces available local itaconate pools inside the mitochondrion, and slows down TCA cycle flux. The latter aspect may further reduce IRG1-dependent itaconate biosynthesis due to reduced production of cis-aconitate as a result of reduced TCA cycle flux. In certain contexts, such as in hypoxia where reverse TCA flux occurs via reductive carboxylation of α-oxoglutarate to isocitrate [at the mitochondrial-resident isoform 2 of isocitrate dehydrogenase (IDH2) [30], Figure 2a], IRG1-depleted macrophages manifests augmented IDH2 activity. Since isocitrate to citrate reversible isomerization via cis-aconitate can operate bidirectionally (Figure 2b), this reverse TCA flux affords an additional context-specific avenue to IRG1-catalyzed itaconate production. On the other hand, itaconate can also inhibit purified IDH2-enzyme (and its cytosolic variant, IDH1) [31], although other findings insinuate the lack of direct inhibition [32]. Likely, contexts, timing, and locale, are key in deconstructing stimulatory versus inhibitory behaviors.
2.1.2. Immunomodulatory Activities
With its unique potential to bridge the gap between metabolic processes and immune responses in activated macrophages, phenotypic ramifications of itaconate are of paramount importance. Activated macrophages are often simplified as M1 (classically-activated) and M2 (alternatively-activated) subtypes [33]. M1-macrophages—typically induced by LPS and IFN-γ—are associated with pro-inflammatory responses and essential for host defense against pathogens. M2-macrophages—induced by, e.g., IL-4 and IL-13—perform anti-inflammatory activities: e.g., tissue repair, and inflammation resolution. In tumor microenvironment, M2-macrophages are typically tumor-promoting as a result of their immunosuppressive behaviors hampering cytotoxic T-cell priming [34,35]. However, M1/M2-categorization is oversimplified, as macrophages can display mixed or transitional phenotypes with both pro- and anti-inflammatory characteristics depending on environmental cues/contexts [36]. Nonetheless, itaconate production is primarily associated with classically-activated M1-macrophages linking mitochondrial metabolism to pro-inflammatory immune response [37]. However, the so-generated itaconate can spur both pro- and anti-inflammatory behaviors in yet-poorly-understood context-specific manners (Figure 3). Conversely, IRG1 expression and itaconate production are less pronounced in alternatively-activated M2-macrophages [10].
Itaconate also plays critical roles in regulating the production and secretion of cytokines and chemokines (i.e., chemotactic cytokines involved in cell migration) secreted from macrophages [13] (Figure 3). Whether this involves direct interaction between itaconate and cytokines/chemokines, remains unresolved. Cytokines and chemokines can either act locally, or systemically through paracrine and endocrine signaling. In M1-macrophages, itaconate downregulates the secretion of IL-6 and IL-12 pro-inflammatory cytokines, while upregulating the production of CXCL10-chemokine, which drives trafficking of numerous innate and adaptive immune-cell types [14]. As such, itaconate influences both proximal and distal immune responses. However, cell-type-specific ramifications of itaconate’s bioactivities, and underlying mediators that support its contextual bioactivities and help shape the observed phenotypes, remain largely intractable (Figure 3). Additionally, given the postulated proclivity of itaconate to interconvert with its isomers (Figure 1a) (Section 2.2), conventional genetics/biochemical tools are unable to paint a comprehensive picture of itaconate’s mechanistic impacts. New technologies—particularly those capable of spatiotemporally-controlled perturbation with mt-REMs—are thus necessary.
2.1.3. Mechanistic Pathway Actions
Besides the reversible inhibition of SDH [38] (Figure 2d), much of the itaconate-driven phenotypic outcomes are linked to covalent irreversible adduction of nucleophilic residues within specific proteins. However, following uncontrolled flooding of cells/animals with mt-REMs, indiscriminate adduction of proteomes across many cell types typically ensues. Teasing apart specific signaling events influencing pathway-flux from target engagements non-attributable to phenotypes thus remains a formidable challenge. Alkylation of target proteins may or may not alter the protein/pathway function/activity. Thus, careful data interpretation and validations are necessary to link targets captured from profiling studies, to actionable signaling outputs. Below, we discuss a few select examples of pathway-level mechanistic behaviors of itaconate.
(a) KEAP1/NRF2
KEAP1 is a key negative regulator of NRF2, a transcription factor that drives the antioxidant response (AR) (Figure 4b). AR modulation is critical for electrophilic-stress defense: covalent KEAP1-modification by a range of RES—spanning from endogenous electrophiles to exogenous xenobiotics—disrupts KEAP1-NRF2 association, leading to NRF2/AR-upregulation [39]. Several clinically-used drugs—e.g., Tecfidera, Vumerity, Bafiertam (Figure 1b) that treat relapsing-remitting multiple sclerosis (RRMS [40]), and omaveloxolone (Figure 1b), recently approved for MS and Friedrich’s ataxia [41]—covalently label KEAP1, upregulating NRF2/AR as part of their therapeutic programs. Although KEAP1/NRF2-signaling is present in all cells, significant activation of this axis occurs in M1-macrophages under oxidative/electrophilic stress [42], or upon exposure to RES, including itaconate and its derivatives [39] (Figure 4a). Following bolus treatment of 4-octyl itaconate (4-OI, Figure 4a, a putative ‘mimic’ of itaconate with enhanced cell-permeablity, but also enhanced reactivity and potentially modified target spectra [14]) in HEK293T cells, a broad-spectrum/non-residue-specific covalent modification of KEAP1 was mapped at C151, C257, C273, C288, C297, and K615. As expected for the canonical KEAP1/NRF2/AR-signaling cascade, this process activates NRF2 (Figure 4b), upregulating a battery of cytoprotective/detoxification AR-genes, including hmox1, and those involved in glutathione-(GSH)-biosynthesis [43]. In the NRF2-KO mouse bone marrow-derived macrophages (BMDMs), the anti-inflammatory effects of 4-OI—including the reduction of LPS-induced IL-1β—are significantly reduced, compared to wild-type (WT) BMDMs [18,44]. Unfortunately, given the known sensitivity of KEAP1/NRF2/AR to numerous other RES [39], such as lipid-derived electrophiles (LDEs), whose generation is also upregulated during inflammation/stress [4], a direct causal link between these observations to 4-OI’s (and itaconate’s) target engagement with KEAP1 cannot be drawn definitively. Multi-site engagement of 4-OI to KEAP1 renders mutagenesis-based validations of on-target specificity on KEAP1 challenging. Furthermore, even in IRG1-KO/KD (knockout/knockdown) systems, the capability of itaconate to ‘self-modulate’ through additional routes (Section 2.1.1), makes it difficult to pinpoint functionally/genetically-sufficient mechanisms of mt-REM signaling following bolus treatment.
(b) STING
Activation of STING—an ER-mitochondrial-membrane-associated transmembrane protein—is a crucial innate-immune response to various types of cytosolic DNA, including pathogenic DNA and damaged nuclear/mitochondrial DNA from the host. STING’s binding to cGAMP and subsequent recruitment of TBK1, which phosphorylates STING, is intricately linked to initiating cell-death pathways [45], e.g., ferroptosis, pyroptosis, and necroptosis (Figure 4c). Metabolomics studies reveal a surge in itaconate levels upon DMXAA (a mouse STING agonist) treatment of RAW264.7 cells and mouse primary BMDMs [46]. The measured elevation of itaconate; an upregulation of IRG1; and associated decrease in STING-phosphorylation, constitute initial evidence to indicate the inhibitory role of itaconate in STING-signaling (Figure 4c,h).
Further explorations unveil that putative itaconate mimic, 4-OI, significantly curtails STING-phosphorylation and pathway activation [46]. In HEK293T cells overexpressing STING, 4-OI treatment results in STING-alkylation at multiple cysteine sites (148; and 65, 71, 88, and 147 in human and the mouse STING-ortholog, respectively). Notably, C148 (147 in mouse) is a key functional site for cGAMP-binding-induced STING-phosphorylation, obligatory for STING’s function. Thus, C148(147)-specific STING-alkylation is thought to suppress STING’s function. Importantly, the fact that C148(147) is a site essential for STING’s basal function makes it unable to use mutagenesis to link site-specific labeling to 4-OI-induced nE-PTM-signaling via STING. This reported STING-inhibition suppresses the production of inflammatory factors (e.g., type I interferons) and pro-inflammatory cytokines (e.g., TNF-α and IL-6). This action is particularly relevant in the mice sepsis model, in which intraperitoneal injection of itaconate or 4-OI significantly suppresses STING-phosphorylation.
An independent finding pinpoints NRF2 acting as a significant counter-regulator of STING [44] (Figure 4c,h). NRF2/AR-upregulation dampens STING-expression in KEAP1-KD A549 cells, and increases susceptibility to DNA-virus infection. RNA-Seq and qPCR analysis of NRF2-KD cells against controls suggests that NRF2 activation downregulates tmem173-mRNA, the gene encoding STING. The result is supported by the lower tmem173-mRNA levels detected in sulforaphane (a natural RES-based inducer of KEAP1/NRF2/AR)-treated cells and KEAP1-KD cells. Furthermore, results from ChIP-seq and experiments utilizing NRF2 inhibitor ML385 (which disrupts NRF2-DNA binding) indicate that the mechanism via which activated NRF2 downregulates tmem173-transcripts is not through genomic interaction, but likely through post-transcriptional regulation [44]. This is backed up by a slower decay of tmem173 mRNA in NRF2-KD A549 cells (against WT groups), following actinomycin-D-induced mRNA-synthesis inhibition, implying that NRF2 reduces sting-mRNA stability. The study underlines potential itaconate-mediated crosstalk between KEAP1/NRF2/AR and STING-signaling.
(c) ATF3/IκBζ
ATF3 is an anti-inflammatory transcription factor and a negative regulator of IκBζ, a key pro-inflammatory member of the IκB-protein family. ATF3-IκBζ axis regulates cytokine production and is implicated in mitochondrial-stress response [47] (Figure 4d). Dysregulation of ATF3-IκBζ-pathway leads to imbalanced immune responses, such as in autoimmune diseases, cancer, and infections, by either promoting excessive inflammation or failing to adequately manage pathogenic threats [48]. An inference of itaconate’s ability to modulate ATF3-IκBζ-axis is made using dimethyl itaconate [47] (DI, Figure 4a, another putative mimic of itaconate of increased cell-permeability, and strongly-elevated reactivity; subsequent studies implicate its metabolically-defunct behaviors [49]). In mouse primary BMDMs, DI treatment upregulates ATF3, thereby inhibiting IκBζ-driven inflammatory response. NRF2-KO BMDMs cannot suppress DI-induced IκBζ- and IL6-downregulation, indicating that this itaconate/ATF3/IκBζ axis is NRF2-independent. Critically, ATF3-KO in BMDMs restores IκBζ-protein expression and DI-induced pro-inflammatory IL-6 upregulation, demonstrating on-target pathway sufficiency in ATF3/DI-target engagement (Figure 4d,h). ATF3-upregulation observed in WT BMDMs cells is lost in IRG1-KO cells, indicating IRG1-dependence in ATF3-activation. Mice model with psoriasis, induced by TLR7/8-agonist imiquimod (IMQ), further supports the regulatory impacts of DI on ATF3-IκBζ-signaling. Mice treated with DI exhibit significantly-reduced psoriasis-like skin alterations and a decrease in the mRNA-expression of IMQ-inducible IκBζ-driven genes [47].
(d) JAK1/STATs
JAK/STAT-axis is a classical membrane-to-nucleus signal transduction, essential for cytokine-mediated immune signaling [50]. >40% of cytokines in human can stimulate this pathway. Binding of cytokines/growth factors to cell-surface cytokine-receptors activates JAK protein-tyrosine kinase by phosphorylation. Activated JAK subsequently phosphorylates STAT transcription factors, resulting in STATs’ nuclear translocation and transcriptional activation (Figure 4e,f). Itaconate (Figure 1a) and 4-OI (Figure 4a) inhibit JAK1-phosphorylation, suppressing downstream STAT1-mediated M1-macrophage polarization and STAT6-mediated M2-polarization. Following bolus 4-OI administration in HEK293T cells, JAK1 is modified at several cysteines (716, 817, 944, and 1131) (Figure 4h) [51]. By inhibiting cytokine-induced JAK1-activation, 4-OI is proposed to offer therapeutic benefits for conditions characterized by hyperactive inflammatory responses [52]. However, an integrated examination of multifactorial pathway effects reported for 4-OI/DI/itaconate is necessary, in order to pinpoint specific disease contexts that can offer the most discriminatory immunotherapeutic outcomes. Differences in models and biological conditions/contexts deployed across this and many other studies collectively make it challenging to draw concrete comparisons and conclusions.
(e) TFEB
Transcription factor EB (TFEB) is a key regulator of lysosomal biogenesis and autophagy, processes essential for metabolic-stress response [53]. TFEB is regulated by mTOR, which phosphorylates TFEB at S211. This phosphorylation event retains TFEB in the cytoplasm where it interacts with 14-3-3 proteins, preventing TFEB’s nuclear translocation and transcriptional activation (Figure 4g). Endogenous itaconate directly alkylates TFEB at C212 in LPS-stimulated monocyte-like THP-1 cells [54]. Immunofluorescence analysis of 4-OI-treated PMA (phorbol 12-myristate 13-acetate)-differentiated THP1 cells, or iBMDMs (immortalized BMDMs), shows that itaconylated-TFEB can undergo nuclear translocation. This alkylation counteracts the mTOR-mediated TFEB-phosphorylation, disrupting TFEB’s interaction with 14-3-3 proteins and eliciting TFEB nuclear translocation/transcription (Figure 4h). Chromatin immunoprecipitation assays further validate itaconate’s role in this cascade: IRG1-deficiency diminishes TFEB’s binding to the promoters of lysosomal biogenesis genes; this effect is reversed by 4-OI treatment. Functionally, this process is crucial for the antibacterial capacity of iBMDMs and THP-1 cells to S. typhimurium infection. Additionally, mice with a genetically-engineered itaconate-sensing-resistant-but-otherwise-functional TFEB-mutant (C270S, corresponding to C212S in human; which can still be phosphorylated by mTOR) exhibit increased vulnerability to S. typhimurium infection. Conversely, administering 4-OI can curtail inflammation [54].
2.2. Mesaconate and Citraconate
The cis/trans-geometrical isomers, mesaconate and citraconate, constitute structural isomers of itaconate (Figure 1a). Enzymatic processes, as well as specific biological contexts that lead to their formation, are not well characterized. Limited evidence hints that they can also be formed from itaconate or other TCA-cycle intermediates through non-enzymatic interconversions [17,55], under specific conditions dependent on pH, temperature, and the presence of specific ions or cofactors. Their potential for spontaneous isomerization post cell lysis, during sample preparation prior to metabolite-ID, using MS- or NMR-based analyses, could further complicate these outcomes. Like itaconate, mesaconate and citraconate also exhibit immunomodulatory, antioxidative, and antiviral properties [17].
When introduced to PMA-differentiated THP1 monocytes at supraphysiological concentrations (25 mM, 6 h), all three isomers impose distinct effects on metabolism [55]. Clearly, interpretation needs caution due to poorly-understood inherent differences in cell-uptake efficiency, potential spontaneous or enzymatic interconversions within the cell, alongside endogenous biosynthesis and/or differences in target spectra (see Section 2.1.1). For instance, the lack of cellular response from one isomer treatment could be due to its reduced permeation, bioavailability, etc., in reaching a certain target/locale in sufficient dosage. In PMA-differentiated THP1 and A549 cells infected with influenza-A virus, all three isomers were found to alter amino-acid metabolism and modulate cytokine/chemokine release, diminishing interferon signaling, oxidative stress, and viral particle release.
When treating Raw264.7 cells or LPS-stimulated BMDMs with itaconate or mesaconate, both metabolites exhibit similar effects in reducing glycolytic activity; yet, itaconate uniquely suppresses the TCA-cycle and cellular respiration [17]. Despite these metabolic differences, both similarly modulate immune responses in pro-inflammatory macrophages, notably downregulating IL-6 and IL-12 secretion, while upregulating CXCL10 pro-inflammatory chemokine production [17]. Among the three isomers assayed at 10-25 mM dosage range in HaCaT keratinocytes, citraconate is reportedly the strongest activator of NRF2 [55]. Although all three isomers influence the KEAP1/NRF2/AR-pathway, more pronounced impacts of citraconate on NRF2 and its downstream genes, such as akr1b10, and in modulating oxidative stress in macrophages, are recorded. However, the results require further validations to unambiguously rule out the effects not being caused by inherent differences in permeability, metabolic lability, and bioavailability. Nonetheless, available data thus far implicate citraconate as a unique innate inhibitor of IRG1′s itaconate biosynthesis, likely through competitive binding. Citraconate thus marks itself as the first naturally-occurring IRG1 inhibitor.
2.3. Fumarate
Fumarate—another emerging important mt-REM—is an intermediate within the classical 8-step TCA-cycle, where the enzyme SDH catalyzes its biosynthesis from succinate (Figure 2a). Fumarate’s inflammatory behaviors and therapeutic potentials have gained widespread attention over the recent years [56,57]. Indeed, several block-buster electrophilic drugs derived from fumarate—e.g., Tecfidera (DMF), Vumerity, and Bafiertam (Figure 1b)—have recently appeared in the treatment of autoimmune diseases [40]. Another derivative of fumarate, tepilamide fumarate (Figure 1b), which is used in treating psoriasis, has also recently completed the Phase II clinical trial [58]. Fumarate is coined as an oncometabolite, beyond immunometabolite, as its accumulation is associated with tumor evolution [10,59]. For instance, mutation in fumarase-encoding fh gene—responsible for converting fumarate to (S)-malate, the ensuing step in the TCA cycle (Figure 2a)—is linked to hereditary leiomyomatosis and renal cell cancer (HLRCC) syndrome [60]. Ongoing (pre)clinical studies are also aimed at repurposing DMF to diseases beyond MS, e.g., cutaneous T cell lymphoma (CTCL) (Phase II) [61], obstructive sleep apnea (Phase II) [62], intracranial unruptured aneurysms [63] (Phase IV, ongoing), cardiomyopathies (myocardial infarction, preclinical) [64], and other neurodegenerative diseases (Parkinson’s disease and Alzheimer’s disease, preclinical) [65,66,67,68,69]. DMF has also been used in combination with other therapies in disease treatment, such as with alteplase for treating acute ischemic stroke [70] (Phase I/II), and with temozolomide and radiation therapy for treating glioblastoma multiforme (Phase I) [71].
As with all RES that also feature broad-spectrum cytotoxicity, dissecting the on-target MoA of fumarate (or DMF/related analogs) poses significant challenges [40]. Although clinical MoA of DMF at the point of the drug approval, was described as covalent adduction of KEAP1, eliciting NRF2/AR-upregulation, a subsequent landmark study documents that DMF-induced therapeutic outcomes are maintained in NRF2-KO mice [72], ruling out the NRF2/AR-pathway as a genetically-sufficient/-necessary MoA. Many succeeding publications focused on the discovery of novel targets and pathways, distinct from KEAP1/NRF2/AR cascade, that can better explain the DMF MoA. However, similar genetic sufficiency tests via KD/KO of the discovered targets—in unison with fumarate/DMF-bolus dosing approaches—did not identify functionally-sufficient players. These interesting seminal studies have been reviewed in detail [40].
Intriguingly, given that NRF2/AR is a central cytoprotective pathway required for cell survival/proliferation [43], the clinical cell-killing action of DMF—particularly immune cells—appears conflicting. To resolve this pharmaceutically-relevant dichotomy, our laboratory set out to harness an in-house-developed precision electrophile-signaling interrogation technology (T-REX) [73,74,75]. T-REX can on-demand enact electrophile modification of a specific electrophile-sensor protein in vivo, in an otherwise largely-unperturbed cell/animal. [Section 4 delineates underlying technical concepts]. Leveraging T-REX to deliver specific reactive metabolites to KEAP1 led us to uncover a novel electrophile-induced selective loss of innate-immune cells, driven by KEAP1/Wdr1-mediated mitochondrial-targeted apoptotic signaling of conserved importance from human, mice, to fish [76] (Figure 5).
This pathway discovery was exclusively enabled by T-REX. However, should this pathway be a genetically- and functionally-sufficient MoA of DMF unique to innate immune cells, this model should hold true under pharmacological (whole-cell/-animal DMF) treatment. Consistent with this posit, Wdr1-depletion in live zebrafish and primary mouse BMDMs ablates the drug (DMF)-induced innate-immune cell depletion, otherwise observed in wild-type controls, following administration of DMF in non-cytotoxic amounts (25-50 μM, 4-6 h) [76]. This novel pathway/MoA is selectively chemotype-specific: beyond DMF, short-chain endogenous LDEs (e.g., 4-hydroxynonenal, Figure 1a) promote this cell-type-specific behavior, whereas longer-chain LDEs (e.g., 4-hxdroxydodecenal, Figure 1a) do not. This aspect is intriguing since all compounds studied are able to covalently engage with KEAP1 (and can promote NRF2). The necessity of KEAP1/Wdr1-axis also likely explains why NRF2-KO mice still support DMF-induced response [72] (vide supra). DMF tags KEAP1 at multiple sites (C151, 273, 288). Fortunately, because the triple-Cys-mutant, KEAP1(C151S,C273W,C288E), is able to maintain wild-type KEAP1′s canonical function [77] and is only defective of DMF-electrophile-sensing property, mutagenesis approach remains a viable validation path [76]. Being able to draw a precise relationship between specific reactive-ligand chemotype, specific target (and binding site), and specific pathway operative in select locale/cell-types—while sparing other cell-types and maintaining overall cell/animal growth/viability—promises to enable gateways to translational benefits.
3. Chemical Biology Approaches to Study mt-REMs
3.1. Protein Target-ID
(a) Indirect target profiling following bulk treatment with designated mt-REMs
First reported in early 2000′s, competitive activity-based protein profiling (ABPP) and its variants constitute a venerable high-throughput means to quantitative rank potential protein-targets of a given small-molecule ligand of interest (LOI) [78] (Figure 6a). The hit identification in ABPP relies upon an indirect readout through the use of proxy-electrophile probes, typically iodoacetamide-alkyne (IAA, Figure 6b). The proteins that have lost IAA-engagement in cell lysates (derived from LOI-treated cell/animal samples), compared to those from non-LOI-treated conrol specimens, are scored as hits. IAA-signal is analyzed following enrichment-based proteomics workflow. ABPP has been applied to immune-relevant instances, where LOI is an Immunometabolite, e.g., fumarate [79], or related drugs, e.g., DMF [40]. For instance, ABPP target-ID of fumarate in cell-based HLRCC models reveals the subtle nuances of fumarate’s reactivity with its targets, influenced by physicochemical factors, e.g., pH (see also Section 2.3) [80].
Furthermore, ABPP has been innovatively adapted to high-throughput identification of compounds from LOI-libraries [81]. This platform variant was recently applied to ID LOIs that can modulate T-cell activation [82]. Another interesting recent study captures potential targets of itaconate using ABPP-based workflow Involving a new carbohydrate-based cysteine-reactive blocking reagent, 3,4,6-O-Ac3ManNAz (Figure 6b) [83]. 260 potential protein-cysteines reactive to itaconate were identified in Raw264.7 cells. Further investigations reveal itaconate’s modification of glycolytic enzymes, e.g., ALDOA, GAPDH, and LDHA, resulting in inhibition of glycolysis—a process significantly upregulated in M1-macrophages. Glycolysis inhibition attenuates inflammatory responses in LPS-stimulated Raw264.7 cells.
Beyond protein-cysteines, ABPP has been applied to screen potentially-ligandable protein-lysines in primary human peripheral blood mononuclear cells (PBMCs), following LPS stimulation [84]. Pentynoic acid sulfotetrafluorophenyl (STP) ester is used as an indirect lysine-reactive proxy-electrophile probe (Figure 6b). Despite these advances in immune-relevant target-profiling, general limitations underpinning ABPP-based approaches [5] extend to these scenarios. The intrinsic impediments associated with compartment-specific and/or low-occupancy target-ID; indirect nature of the readout (where target-capture by proxy-electrophlic probes is performed in cell-lysate) constitute some of the key unresolved challenges.
(b) Direct target profiling
The direct capture of mt-REM-modified targets typically requires care in MS-workflow and sample processing, due to the lability of the resulting adducts (especially for cysteine-based adducts, via retro-Michael addition [4,85], Figure 7a). To date, strategies used to directly map mt-REM-targets can be subcategorized into antibody-based approaches (Figure 7b), and those leveraging Clickable mt-REM analogs (Figure 7c). The latter suffers from the requirement to flood the cells with (modified) mt-REMs, but the former is limited by the lack of chemotype-specific antibodies to individual mt-REMs.
Within the first approach (Figure 7b), a study using an anti-succinyl lysine antibody reports an increase in succinylated proteins (referred to as fumarate-modified proteins) following LPS-stimulation of mouse primary BMDMs [86]. Subsequent immunoprecipitation-based enrichment proteomics target-ID shows MDH, GAPDH, GC1, LDHA, and TAL, among others, as potential fumarate targets (although whether these modifications are functionally relevant requires further investigations). Another study takes advantage of the promiscuity of the anti-succinyl-lysine antibody in potential target-ID of itaconatylated proteins [87]. Exogenous supplementation of itaconate (10 mM) to LPS-activated macrophages, results in upregulation of itaconyl-CoA levels, as analyzed by selected reaction monitoring based LC-MS/MS. Itaconylation of several proteins, e.g., GAPDH, ENO1, PKM2 LDHA, NPM1, H2B1B, and SHMT2, are reported, although functional studies remain limited. Notably, to mitigate the use of anti-succinyl lysine antibody in identifying itaconylated proteomes, the authors thoughtfully attempt to produce anti-itaconyl lysine antibody but insufficient avidity to-date hampers applications.
In the second approach (Figure 7c), installing Clickable handles (typically an alkyne) into mt-REMs, in tandem with additional structural modifications aimed at enhancing cell permeability, bioavailability, and/or metabolic stability of the resulting modified analogs, are typically pursued. Accordingly alkyne-functionalized probes are rapidly emerging, although limited nature of these analogs in mimicking itaconate is now well-reported [14,49]. Alkyne-integrated version of 4-OI (ITalk) (Figure 7d) is a recently-reported Clickable probe used in the direct profiling of potential targets. Modification-site-ID is inferred from an aniline-derived probe that traps the remnant protein-carbonyls [88]. This strategy of direct capture side-steps the limitations surrounding indirect target-ID [5]; however, care is required since carbonyl-functions could result from several endogenous RES-modifications of proteins, e.g., LDEs, whose extent could differ between treated and non-treated samples.
Critically, under the contexts studied, whole-cell ITalk/4-OI treatment recapitulates the anti-inflammatory behaviors observed following itaconate stimulation. Downstream investigations uncover RIPK3-kinase activation—via RIPK3(S232)-phosphorylation—of relevance in necroptotic cell-death that is inducible by both itaconate and 4-OI. Although proteomics site-ID maps ITalk-alkylation to 3 cysteines of RIPK3, mutagenesis studies show that this signaling functionally requires RIPK3(C360)-specific alkylation [89]. Although the use of structural analogs has advanced our understanding of mt-REMs’ prospective target spectra, it is crucial to recognize the potential pitfalls. First, analogs may not fully recapitulate the natural behavior of mt-REMs. Second, such significant structural modifications with extended alkyl chain, for instance, will likely impact target-binding efficacies, as thoughtfully described by the developers of ITalk. These differences could further alter subcellular distribution, permeability, metabolic vulnerability, as well as intrinsic electrophilicity, among others, leading to different targets captured beyond inducing distinct biological effects. Rigorous validation experiments—e.g., ablation of measured outputs in knock-in (KI) Cys-mutant cell lines—are essential to accurately decipher the MoA.
(c) Function-guided proximity mapping by precision localized electrophile delivery
Clearly, both profiling approaches above have significantly advanced the field. However, they both rely upon bulk administration of reactive metabolites to cells/organisms, affording limited spatiotemporal resolution among others. Low-occupancy electrophile sensors and locale-specific kinetically-previleged responders known to drive GOF/dLOF nE-PTM signaling often escape identification. Caution should be exercised when different RES-chemotypes are compared using bulk administration, since outputs can be biased by differences in cellular uptake, diffusion, bioavailability, and the extent of metabolic vulnerability of each reactive entity. These issues are magnified when functional impacts are studied in complex live animals, with active metabolism.
The development of biocompatible photocaged-electrophiles—and the concept of precision localized electrophile delivery and associated REX-technologies—offers a complementary approach that addresses the above limitations (Figure 8a). REX-technologies—applicable in various living models (cells, worms, fish) [73]—leverage the Halo-protein, functionally-expressible in cell/organ-specific and subcellular/ organelle-specific manner. Following administration of bioinert and cell/animal-permeable REX-probes, and wash-out periods, the REX-probe irreversibly binds Halo in 1:1 stoichiometry. Light exposure at a preordained time enables rapid (t1/2 < 1 min) electrophile liberation. The approach mimics how nature builds up endogenous electrophiles locally with spatiotemporal contexts. The limited quantity of electrophile transiently made available within the designated Halo locale, is competed by native electrophile-sensor proteins proximal to Halo. Coupling this electrophile function-guided proximity-mapping platform (Localis-REX) to enrichment-proteomics workflows [90] enables quantitative target-ID/ranking among kinetically-privileged local responders. A recent example of Localis-REX demonstrates nucleus- vs. mitochondrial outermembrane-specific precision localized delivery of lipid-derived signaling electrophiles (LDEs) [91].
Using this setup, the apporach pinpoints locale-specific LDE-responsivity of proteins in locales that are not their primary resident [91]. For instance, primarily nuclear-resident kinase, CDK9, is uniquely responsive to an LDE only when the reactive metabolite is delivered in cytoplasm. Such low-abundant electrophile-sensor proteins performing ‘minority sensing’ in non-canonical locales are not easily identified using profiling tools delineated above that rely upon bulk administration [92]. Notably, locale specificity in delivery is intrinsically linked to reactive compounds’ diffusion distances. Unlike synthetic radicals and carbenes deployed in emerging innovative proximity-mapping tools [93,94,95], LDEs are subjected to endogenous metabolic clearance/transformations: e.g., glutathione-conjugation, GST-mediated detoxification/degradation, that have been reviewed [5]. Diffusion distances of LDEs are thus context-dependent. Nonetheless, Localis-REX profiling with Halo restricted to the cell nucleus, shows 62% canonical nuclear residents, supporting that LDE released from Localis-REX is largely localized. Interestingly, several of the remaining hits—canonically not nucleus-associated proteins—are also functionally validated to have nucleus-specific LDE-sensing ability [91].
As with all proteomics target-mapping approaches, mechanistic studies are pivotal in comprehensive understanding of the role of newly-captured first-responders. Gratifyingly, unlike all other proteomics profiling approaches that are end-point assays, i.e., once targets are captured, different types of functional assays are required for validations, Localis-REX is not an end-point omics assay. This is because it can be performed in tandem with a sister technology, T-REX (discussed in Section 4) that enables both target validation and on-target signaling interrogations. This tandem Localis-REX–T-REX mechanistic tool unmasks how minority reactive-metabolite sensing capability, choreographed by the cytosolic pool of CDK9, downregulates nuclear-specific RNA-Pol-II-mediated transcriptional regulation [91]. The nuclear pool of CDK9 is unable to sense LDEs, as its sensing cysteine site is blocked by the nuclear-resident binder, Cyclin T1 [91].
Localis-REX thus offers complementary solutions to existing chemoproteomics-profiling and proximity-mapping omics strategies. Nonetheless, it is worth noting that the approach requires meticulous design and engineering. Photocaged REX-probes necessitate novel biocompatible chemistries tailored to a specific reactive-electrophile class, beyond general requirements: non-toxicity, permeability, metabolic-stability, photouncaging efficiency, etc. Implementation of biocompatible chemical-biology/chemical-genetics workflows requires de-novo developments, particularly in complex live animals. Indeed, Halo-functionality in vivo, was not established prior to REX-technologies. REX-probe Halo-binding specificity in vivo must also be optimized, while uncompromising animal viability/morphology/development. Considerations into potential photo-induced toxicities and off-target outputs should also be carefully made, and to that end, we have installed a suite of technical and biological controls that can unambiguously rule out potential interferences/misinterpretations that we have reviewed elsewhere (Figure 8e) [73,96]. Unfortunately, the published photocaged-electrophile probes can only cage enal/enones as reactive motifs. For instance, photocaged-4-oxononenal (ONE), unsurprisingly, shows broad-spectrum reactivity prior to photouncaging [97]. Innovative biocompatible means to adapt REX-technologies and enable Localis-REX mt-REM-guided proximity-mapping are currently ongoing in our laboratory.
3.2. Potential Biosensing of mt-REMs
Genetically-encoded biosensors (GEBs) are traditionally proven useful in monitoring and reporting various cellular small-molecule metabolites. These biosensors are categorized based on their architecture, either as integrated systems where the biosensor is part of the protein or DNA/RNA of interest, or functions independently. GEBs with high specificity enable real-time reporting of specific metabolite’s dynamics. However, GEBs’ stability and potential invasiveness, etc., need to be resolved/optimized case-by-case. Small-molecule-based sensors, conversely, offer alternative approaches that can complement GEBs in detecting specific metabolites [98,99] or ions [100] within the cell. But locale-specific control of small-molecule-based biosensors typically falls short compared to GEBs encodable with numerous localization-tags.
The deployment of biosensors for covalently-reactive metabolites, e.g., LDEs, mt-REMs, requires key considerations. Covalent adduction, and hence inability of GEB to report further once the preexisting biosensor molecules are ligand-bound, restrict wider applicability. Instantaneous reporting of ligand-binding/debinding events—unbiased by changes in microenvironments that could impact kon/koff/Kd of the binding/dissociation equilibrium—is also likely difficult to achieve. Conversely, small-molecule-based biosensors for mt-REMs with chemotype-specificity will be generally difficult to achieve, against numerous endogenous Michael-acceptor-based RES, e.g., LDEs [4]. Such sensors would also need to outcompete 5-10 mM cellular GSH [4]. Importantly, to what extent trapping and depletion of cellular metabolite pools in the process of ‘sensing’ a given metabolite, alters localized availability and results in artifactual reporting of non-physiological trafficking-dynamics, need to be carefully evaluated.
A fluorescent-protein-based GEB for itaconate, BioITA, has recently surfaced [101]. BioTA is derived from itaconate-binding domain (IBD) of a bacterial transcriptional regulatory protein, ItcR (Figure 7e). The real-time reporting of itaconate upregulation by fluorescence turn-on within the subcellular compartments of RAW264.7 cells is demonstrated following LPS-stimulation. The reduction of signal after the removal of itaconate confirms that the BioITA-itaconate binding is reversible; additionally, the signal can be restored by re-adding itaconate. The Kd of BioITA is 203 ± 18.8 µM, as measured by isothermal titration calorimetry (ITC).
3.3. Precision Interrogations into Reactive-Metabolite-Directed nE-PTM Signaling
In the broader realms of reactive-metabolite signaling biology, the capability to precisely trigger target-specific electrophilic modifications has opened a direct lens into precision electrophile-driven GOF/dLOF nE-PTM-signaling. This tool is termed T-REX (Figure 8b) [73,74,75]. Proven compatible with use in both cultured cells and live organisms, C. elegans and zebrafish (i.e., Z-REX, for T-REX in zebrafish, Figure 8c), T(Z)-REX is based on the central tenet of precision localized reactive-metabolite delivery, as in Localis-REX (Section 3.1c). The only technical difference between Localis-REX and T-REX is the use of Halo-fused POI. T(Z)-REX shepherds a specific reactive metabolite to a specific POI and assay the ensuing effects on POI’s quantitative responsivity (i.e., ligand-occupancy) to a given electrophile, and critically, signal-propagation mechanism. A suite of technical and functional controls [73] enables discrimination of on-target consequences against those potentially arising from off-targets/engagements with ~200,000 other unique protein-cysteines and other nucleophilic protein-residues [102,103]. Z-REX is recently adapted to organ-specific LDE-responsivity mapping in transgenic zebrafish. Heart-specific KEAP1-LDEylation upregulates the NRF2/AR-axis selectively in the heart, and not in other tissues/locales [75,76]. This experiment not only demonstrates the specificity of Z-REX but also documents its potential in studying non-invasively, precise signaling pathways within whole vertebrate animals with high spatiotemporal precision. As validated through this [75] and other Z-REX studies in fish [76,103,104], bolus LDE-dosing of the whole animal renders it nearly impossible to control when a given LDE reaches the heart-specific protein-target (and in what amount); or if it at all does prior to metabolic conversions to other reactive species/chemotypes, or encounters endogenous degradation/clearance pathways, notwithstanding potential artifactual readouts due to bolus-LDE-induced toxicity. As alluded to above, expanding the biocompatible chemical space of REX-tools to integrate a range of mt-REMs is ongoing.
4. Outlook
Over the coming years, functional amalgamation of precision chemical-biology tools with the growing armory of single-cell-Seq and spatial-omics technologies will likely catalyze the creation of next-generation precision immunotherapies. The efficacy of these therapies will be gauged by their specificity in reaching intended targets and outcomes, while minimizing harmful off-target consequences. Targeting the metabolic underpinnings of immune responses will doubtless prove instrumental. But such advancements hinge on our capacity to precisely decipher the biological effects of reactive immunometabolites, in defined biological contexts, locale, timescale, and critically, inform on chemically-actionable protein targets that together with specific mt-REMs orchestrate functional signaling responses. Such an understanding will enable strategic manipulation of metabolic pathways, potentially transforming prophylactic and therapeutic approaches across a spectrum of immune-system disorders. Indeed, the most recent years have witnessed growing inventories of quantitative spatial atlases cartographing altered (epi)genetics, (epi)transcriptomics signatures, as well as shifts in local proteome-abundance/canonical-PTMs in manifold immune-microenvironments. The ability to crown such spatial annotations with locale-specific chemical actionability remains one of the key challenges of this decade. Decoding the powerful immunometabolic roles of mt-REMs and associated nE-PTM-signaling modalities with spatiotemporal intelligence represents an ideal niche. In this context, precision immuno-chemical biology tools hold great promise to bring about unique and enabling solutions.
Author Contributions
Concept and writing: K-T.H., Y.A.; figures: K-T.H. All authors agree to the final version of the manuscript.
Acknowledgements
Novartis FreeNovation Grant; European Research Council (ERC) grant (Project no. 101043303) funded by the State Secretariat for Education, Research and Innovation, Switzerland (SERI), and Swiss Federal Institute of Technology Lausanne (EPFL) (to YA). Dr. Marcus Long, Department of Immunology, University of Lausanne, for critical review.
Conflicts of interest
There are no conflicts to declare.
References
- Baker, S.A.; Rutter, J. Metabolites as signalling molecules. Nature Reviews Molecular Cell Biology 2023, 24, 355–374. [Google Scholar] [CrossRef] [PubMed]
- Long, M.J.C.; Huang, K.T.; Aye, Y. The not so identical twins: (dis)similarities between reactive electrophile and oxidant sensing and signaling. Chem Soc Rev 2021, 50, 12269–12291. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; et al. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat Rev Mol Cell Biol 2022, 23, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Parvez, S.; Long, M.J.C.; Poganik, J.R.; Aye, Y. Redox Signaling by Reactive Electrophiles and Oxidants. Chem Rev 8798, 118, 8798–8888. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Long, M.J.C.; Aye, Y. Proteomics and Beyond: Cell Decision-Making Shaped by Reactive Electrophiles. Trends Biochem Sci 2019, 44, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Long, M.J.C.; Aye, Y. Privileged Electrophile Sensors: A Resource for Covalent Drug Development. Cell Chem Biol 2017, 24, 787–800. [Google Scholar] [CrossRef]
- Liu, X.; et al. Precision Targeting of pten-Null Triple-Negative Breast Tumors Guided by Electrophilic Metabolite Sensing. ACS Cent Sci 2020, 6, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; et al. The signaling pathways and therapeutic potential of itaconate to alleviate inflammation and oxidative stress in inflammatory diseases. Redox Biol 2022, 58, 102553. [Google Scholar] [CrossRef]
- Peace, C.G.; O’Neill, L.A. The role of itaconate in host defense and inflammation. J Clin Invest 2022, 132. [Google Scholar] [CrossRef]
- Chen, F.; Dowerg, B.; Cordes, T. The yin and yang of itaconate metabolism and its impact on the tumor microenvironment. Curr Opin Biotechnol 2023, 84, 102996. [Google Scholar] [CrossRef]
- Lau, A.N.; Heiden, M.G.V. Metabolism in the Tumor Microenvironment. Annual Review of Cancer Biology 2020, 4, 17–40. [Google Scholar] [CrossRef]
- Angajala, A.; et al. Diverse Roles of Mitochondria in Immune Responses: Novel Insights Into Immuno-Metabolism. Front Immunol 2018, 9, 1605. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Artyomov, M.N. Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat Rev Immunol 2019, 19, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zheng, W.; Kong, W.; Zeng, T. Itaconate: A Potent Macrophage Immunomodulator. Inflammation 2023, 46, 1177–1191. [Google Scholar] [CrossRef]
- Lauble, H.; Stout, C.D. Steric and conformational features of the aconitase mechanism. Proteins 1995, 22, 1–11. [Google Scholar] [CrossRef]
- Adler, J.; Wang, S.F.; Lardy, H.A. The metabolism of itaconic acid by liver mitochondria. J Biol Chem 1957, 229, 865–879. [Google Scholar] [CrossRef]
- He, W.; et al. Mesaconate is synthesized from itaconate and exerts immunomodulatory effects in macrophages. Nat Metab 2022, 4, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Chen, C. ABCG2 is an itaconate exporter that limits antibacterial innate immunity by alleviating TFEB-dependent lysosomal biogenesis. Cell Metab 2024. [Google Scholar] [CrossRef]
- Mo, W.; Zhang, J.T. Human ABCG2: structure, function, and its role in multidrug resistance. Int J Biochem Mol Biol 2012, 3, 1–27. [Google Scholar]
- Sendra, M.; Saco, A.; Rey-Campos, M.; Novoa, B.; Figueras, A. Immune-responsive gene 1 (IRG1) and dimethyl itaconate are involved in the mussel immune response. Fish Shellfish Immunol 2020, 106, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Strelko, C.L.; et al. Itaconic acid is a mammalian metabolite induced during macrophage activation. J Am Chem Soc 2011, 133, 16386–16389. [Google Scholar] [CrossRef]
- Michelucci, A.; et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci U S A 2013, 110, 7820–7825. [Google Scholar] [CrossRef] [PubMed]
- Duncan, D.; Lupien, A.; Behr, M.A.; Auclair, K. Effect of pH on the antimicrobial activity of the macrophage metabolite itaconate. Microbiology (Reading) 2021, 167. [Google Scholar] [CrossRef]
- Chen, Y.J.; et al. Targeting IRG1 reverses the immunosuppressive function of tumor-associated macrophages and enhances cancer immunotherapy. Sci Adv 2023, 9, eadg0654. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; et al. Myeloid-derived itaconate suppresses cytotoxic CD8(+) T cells and promotes tumour growth. Nat Metab 2022, 4, 1660–1673. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; et al. Metabolic Reprogramming via ACOD1 depletion enhances function of human induced pluripotent stem cell-derived CAR-macrophages in solid tumors. Nat Commun 2023, 14, 5778. [Google Scholar] [CrossRef]
- Lampropoulou, V.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab 2016, 24, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Cordes, T.; et al. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J Biol Chem 2016, 291, 14274–14284. [Google Scholar] [CrossRef]
- Filipp, F.V.; Scott, D.A.; Ronai, Z.e.A.; Osterman, A.L.; Smith, J.W. Reverse TCA cycle flux through isocitrate dehydrogenases 1 and 2 is required for lipogenesis in hypoxic melanoma cells. Pigment Cell & Melanoma Research 2012, 25, 375–383. [Google Scholar] [CrossRef]
- Aso, K.; et al. Itaconate ameliorates autoimmunity by modulating T cell imbalance via metabolic and epigenetic reprogramming. Nat Commun 2023, 14, 984. [Google Scholar] [CrossRef] [PubMed]
- Heinz, A.; et al. Itaconate controls its own synthesis via feedback-inhibition of reverse TCA cycle activity at IDH2. Biochim Biophys Acta Mol Basis Dis 2022, 1868, 166530. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef]
- Barry, S.T.; Gabrilovich, D.I.; Sansom, O.J.; Campbell, A.D.; Morton, J.P. Therapeutic targeting of tumour myeloid cells. Nat Rev Cancer 2023, 23, 216–237. [Google Scholar] [CrossRef]
- Binnewies, M.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Ruytinx, P.; Proost, P.; Van Damme, J.; Struyf, S. Chemokine-Induced Macrophage Polarization in Inflammatory Conditions. Front Immunol 2018, 9, 1930. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sanchez-Rodriguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front Immunol 2019, 10, 1462. [Google Scholar] [CrossRef]
- Cordes, T.; Metallo, C.M. Itaconate Alters Succinate and Coenzyme A Metabolism via Inhibition of Mitochondrial Complex II and Methylmalonyl-CoA Mutase. Metabolites 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Hakomaki, H.; Levonen, A.L. Electrophilic metabolites targeting the KEAP1/NRF2 partnership. Curr Opin Chem Biol 2024, 78, 102425. [Google Scholar] [CrossRef]
- Poganik, J.R.; Aye, Y. Electrophile Signaling and Emerging Immuno- and Neuro-modulatory Electrophilic Pharmaceuticals. Front Aging Neurosci 2020, 12, 1. [Google Scholar] [CrossRef]
- Dayalan Naidu, S.; Dinkova-Kostova, A.T. Omaveloxolone (Skyclarys(TM)) for patients with Friedreich’s ataxia. Trends Pharmacol Sci 2023, 44, 394–395. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Olagnier, D.; et al. Nrf2 negatively regulates STING indicating a link between antiviral sensing and metabolic reprogramming. Nat Commun 2018, 9, 3506. [Google Scholar] [CrossRef]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet 2019, 20, 657–674. [Google Scholar] [CrossRef]
- Li, W.; et al. 4-octyl itaconate as a metabolite derivative inhibits inflammation via alkylation of STING. Cell Rep 2023, 42, 112145. [Google Scholar] [CrossRef] [PubMed]
- Bambouskova, M.; et al. Electrophilic properties of itaconate and derivatives regulate the IkappaBzeta-ATF3 inflammatory axis. Nature 2018, 556, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; et al. The central inflammatory regulator IkappaBzeta: induction, regulation and physiological functions. Front Immunol 2023, 14, 1188253. [Google Scholar] [CrossRef] [PubMed]
- ElAzzouny, M.; et al. Dimethyl Itaconate Is Not Metabolized into Itaconate Intracellularly. J Biol Chem 2017, 292, 4766–4769. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther 2021, 6, 402. [Google Scholar] [CrossRef]
- Runtsch, M.C.; et al. Itaconate and itaconate derivatives target JAK1 to suppress alternative activation of macrophages. Cell Metab 2022, 34, 487–501 e488. [Google Scholar] [CrossRef] [PubMed]
- Vale, K. Targeting the JAK-STAT pathway in the treatment of ‘Th2-high’ severe asthma. Future Med Chem 2016, 8, 405–419. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; et al. From the regulatory mechanism of TFEB to its therapeutic implications. Cell Death Discov 2024, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; et al. Itaconate is a lysosomal inducer that promotes antibacterial innate immunity. Mol Cell 2022, 82, 2844–2857 e2810. [Google Scholar] [CrossRef]
- Chen, F.; et al. Citraconate inhibits ACOD1 (IRG1) catalysis, reduces interferon responses and oxidative stress, and modulates inflammation and cell metabolism. Nat Metab 2022, 4, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Onuora, S. Mitochondrial fumarate implicated in inflammation. Nature Reviews Rheumatology 2023, 19, 257–257. [Google Scholar] [CrossRef] [PubMed]
- Seton-Rogers, S. Functions of fumarate. Nature Reviews Cancer 2016, 16, 617–617. [Google Scholar] [CrossRef] [PubMed]
- Mrowietz, U.; et al. Tepilamide Fumarate (PPC-06) Extended Release Tablets in Patients with Moderate-to-Severe Plaque Psoriasis: Safety and Efficacy Results from the Randomized, Double-blind, Placebo-controlled AFFIRM Study. J Clin Aesthet Dermatol 2022, 15, 53–58. [Google Scholar] [PubMed]
- Sullivan Lucas, B.; et al. The Proto-oncometabolite Fumarate Binds Glutathione to Amplify ROS-Dependent Signaling. Molecular Cell 2013, 51, 236–248. [Google Scholar] [CrossRef]
- Menko, F.H.; et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer 2014, 13, 637–644. [Google Scholar] [CrossRef]
- Nicolay, J.P.; et al. Dimethyl fumarate treatment in relapsed and refractory cutaneous T-cell lymphoma: a multicenter phase 2 study. Blood 2023, 142, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Braley, T.J.; et al. A randomized, subject and rater-blinded, placebo-controlled trial of dimethyl fumarate for obstructive sleep apnea. Sleep 2018, 41. [Google Scholar] [CrossRef]
- Pascale, C.L.; et al. Treatment with dimethyl fumarate reduces the formation and rupture of intracranial aneurysms: Role of Nrf2 activation. J Cereb Blood Flow Metab 2020, 40, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Meili-Butz, S.; et al. Dimethyl fumarate, a small molecule drug for psoriasis, inhibits Nuclear Factor-kappaB and reduces myocardial infarct size in rats. Eur J Pharmacol 2008, 586, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Campolo, M.; et al. The Neuroprotective Effect of Dimethyl Fumarate in an MPTP-Mouse Model of Parkinson’s Disease: Involvement of Reactive Oxygen Species/Nuclear Factor-kappaB/Nuclear Transcription Factor Related to NF-E2. Antioxid Redox Signal 2017, 27, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; et al. Repurposing the NRF2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson’s Disease. Antioxid Redox Signal 2016, 25, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; et al. Dimethyl fumarate attenuates 6-OHDA-induced neurotoxicity in SH-SY5Y cells and in animal model of Parkinson’s disease by enhancing Nrf2 activity. Neuroscience 2015, 286, 131–140. [Google Scholar] [CrossRef]
- Majkutewicz, I.; et al. Dimethyl fumarate attenuates intracerebroventricular streptozotocin-induced spatial memory impairment and hippocampal neurodegeneration in rats. Behav Brain Res 2016, 308, 24–37. [Google Scholar] [CrossRef]
- Majkutewicz, I.; et al. Age-dependent effects of dimethyl fumarate on cognitive and neuropathological features in the streptozotocin-induced rat model of Alzheimer’s disease. Brain Res 2018, 1686, 19–33. [Google Scholar] [CrossRef]
- Zhu, Z.; et al. Combination of the Immune Modulator Fingolimod With Alteplase in Acute Ischemic Stroke: A Pilot Trial. Circulation 2015, 132, 1104–1112. [Google Scholar] [CrossRef]
- Shafer, D.; et al. Phase I trial of dimethyl fumarate, temozolomide, and radiation therapy in glioblastoma. Neurooncol Adv 2020, 2, vdz052. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Topphoff, U.; et al. Dimethyl fumarate treatment induces adaptive and innate immune modulation independent of Nrf2. Proc Natl Acad Sci U S A 2016, 113, 4777–4782. [Google Scholar] [CrossRef]
- Long, M.J.C.; Rogg, C.; Aye, Y. An Oculus to Profile and Probe Target Engagement In Vivo: How T-REX Was Born and Its Evolution into G-REX. Acc Chem Res 2020. [Google Scholar] [CrossRef] [PubMed]
- Parvez, S.; et al. T-REX on-demand redox targeting in live cells. Nat Protoc 2016, 11, 2328–2356. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.T.; et al. Z-REX: shepherding reactive electrophiles to specific proteins expressed tissue specifically or ubiquitously, and recording the resultant functional electrophile-induced redox responses in larval fish. Nat Protoc 2023, 18, 1379–1415. [Google Scholar] [CrossRef]
- Poganik, J.R.; et al. Wdr1 and cofilin are necessary mediators of immune-cell-specific apoptosis triggered by Tecfidera. Nat Commun 2021, 12, 5736. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Tong, K.I.; Yamamoto, M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic Biol Med 2004, 36, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Cravatt, B.F.; Wright, A.T.; Kozarich, J.W. Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu Rev Biochem 2008, 77, 383–414. [Google Scholar] [CrossRef]
- Blewett, M.M.; et al. Chemical proteomic map of dimethyl fumarate-sensitive cysteines in primary human T cells. Sci Signal 2016, 9, rs10. [Google Scholar] [CrossRef]
- Kulkarni, R.A.; et al. A chemoproteomic portrait of the oncometabolite fumarate. Nat Chem Biol 2019, 15, 391–400. [Google Scholar] [CrossRef]
- Desai, H.S.; Yan, T.; Backus, K.M. SP3-FAIMS-Enabled High-Throughput Quantitative Profiling of the Cysteinome. Curr Protoc 2022, 2, e492. [Google Scholar] [CrossRef]
- Vinogradova, E.V.; et al. An Activity-Guided Map of Electrophile-Cysteine Interactions in Primary Human T Cells. Cell 2020, 182, 1009–1026 e1029. [Google Scholar] [CrossRef]
- Qin, W.; et al. S-glycosylation-based cysteine profiling reveals regulation of glycolysis by itaconate. Nat Chem Biol 2019, 15, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Abbasov, M.E.; et al. A proteome-wide atlas of lysine-reactive chemistry. Nat Chem 2021, 13, 1081–1092. [Google Scholar] [CrossRef]
- Poganik, J.R.; Long, M.J.C.; Aye, Y. Getting the Message? Native Reactive Electrophiles Pass Two Out of Three Thresholds to be Bona Fide Signaling Mediators. Bioessays 2018, 40, e1700240. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; et al. Discovery of Itaconate-Mediated Lysine Acylation. J Am Chem Soc 2023, 145, 12673–12681. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; et al. Quantitative Profiling of Protein Carbonylations in Ferroptosis by an Aniline-Derived Probe. J Am Chem Soc 2018, 140, 4712–4720. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; et al. Chemoproteomic Profiling of Itaconation by Bioorthogonal Probes in Inflammatory Macrophages. J Am Chem Soc 2020, 142, 10894–10898. [Google Scholar] [CrossRef]
- Long, M.J.C.; Liu, J.; Aye, Y. Finding a vocation for validation: taking proteomics beyond association and location. RSC Chem Biol 2023, 4, 110–120. [Google Scholar] [CrossRef]
- Zhao, Y.; et al. Function-guided proximity mapping unveils electrophilic-metabolite sensing by proteins not present in their canonical locales. Proc Natl Acad Sci U S A 2022, 119. [Google Scholar] [CrossRef] [PubMed]
- Long, M.J.C.; Miranda Herrera, P.A.; Aye, Y. Hitting the Bullseye: Endogenous Electrophiles Show Remarkable Nuance in Signaling Regulation. Chemical Research in Toxicology 2022, 35, 1636–1648. [Google Scholar] [CrossRef]
- Cho, K.F.; et al. Proximity labeling in mammalian cells with TurboID and split-TurboID. Nature Protocols 2020, 15, 3971–3999. [Google Scholar] [CrossRef]
- Hung, V.; et al. Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nature Protocols 2016, 11, 456–475. [Google Scholar] [CrossRef] [PubMed]
- Geri, J.B.; et al. Microenvironment mapping via Dexter energy transfer on immune cells. Science 2020, 367, 1091–1097. [Google Scholar] [CrossRef]
- Long, M.J.C.; Assari, M.; Aye, Y. Hiding in Plain Sight: The Issue of Hidden Variables. ACS Chem Biol 2022, 17, 1285–1292. [Google Scholar] [CrossRef]
- Lin, H.Y.; Haegele, J.A.; Disare, M.T.; Lin, Q.; Aye, Y. A generalizable platform for interrogating target- and signal-specific consequences of electrophilic modifications in redox-dependent cell signaling. J Am Chem Soc 2015, 137, 6232–6244. [Google Scholar] [CrossRef] [PubMed]
- Jaffrey, S.R. RNA-Based Fluorescent Biosensors for Detecting Metabolites in vitro and in Living Cells. Adv Pharmacol 2018, 82, 187–203. [Google Scholar] [CrossRef]
- Ohata, J.; Bruemmer, K.J.; Chang, C.J. Activity-Based Sensing Methods for Monitoring the Reactive Carbon Species Carbon Monoxide and Formaldehyde in Living Systems. Accounts of Chemical Research 2019, 52, 2841–2848. [Google Scholar] [CrossRef]
- Chyan, W.; Zhang, D.Y.; Lippard, S.J.; Radford, R.J. Reaction-based fluorescent sensor for investigating mobile Zn2+ in mitochondria of healthy versus cancerous prostate cells. Proc Natl Acad Sci U S A 2014, 111, 143–148. [Google Scholar] [CrossRef]
- Sun, P.; et al. A genetically encoded fluorescent biosensor for detecting itaconate with subcellular resolution in living macrophages. Nat Commun 2022, 13, 6562. [Google Scholar] [CrossRef] [PubMed]
- Long, M.J.; et al. beta-TrCP1 Is a Vacillatory Regulator of Wnt Signaling. Cell Chem Biol 2017, 24, 944–957 e947. [Google Scholar] [CrossRef]
- Long, M.J.; et al. Akt3 is a privileged first responder in isozyme-specific electrophile response. Nat Chem Biol 2017, 13, 333–338. [Google Scholar] [CrossRef]
- Van Hall-Beauvais, A.; et al. Z-REX uncovers a bifurcation in function of Keap1 paralogs. Elife 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; et al. Crystal structure of cis-aconitate decarboxylase reveals the impact of naturally occurring human mutations on itaconate synthesis. Proc Natl Acad Sci U S A 2019, 116, 20644–20654. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Maklashina, E.; Cecchini, G.; Iverson, T.M. The roles of SDHAF2 and dicarboxylate in covalent flavinylation of SDHA, the human complex II flavoprotein. Proc Natl Acad Sci U S A 2020, 117, 23548–23556. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Reactive metabolites (ROS/RES) and covalent drugs. (a,b) Chemical structures of representative (a) endogenous ROS and RES, including LDEs and mt-REMs; (b) fumarate-derived drugs (top row): all approved except tepilamide fumarate that completed Phase-II in the treatment of psoriasis. Michael-acceptor-based electrophilic motifs within each RES/drug are highlighted in gray. (c) RES can functionally regulate their target proteins through gain-of-function (GOF) or dominant loss-of-function (dLOF) mechanisms, eliciting an amplified signaling output, from substoichiometric electrophile ligand occupancy. In this context, RES-directed nE-PTMs are akin to GOF/dLOF signal-propagation mechanisms in canonical enzymatic PTMs such as phosphorylation.
Figure 1.
Reactive metabolites (ROS/RES) and covalent drugs. (a,b) Chemical structures of representative (a) endogenous ROS and RES, including LDEs and mt-REMs; (b) fumarate-derived drugs (top row): all approved except tepilamide fumarate that completed Phase-II in the treatment of psoriasis. Michael-acceptor-based electrophilic motifs within each RES/drug are highlighted in gray. (c) RES can functionally regulate their target proteins through gain-of-function (GOF) or dominant loss-of-function (dLOF) mechanisms, eliciting an amplified signaling output, from substoichiometric electrophile ligand occupancy. In this context, RES-directed nE-PTMs are akin to GOF/dLOF signal-propagation mechanisms in canonical enzymatic PTMs such as phosphorylation.
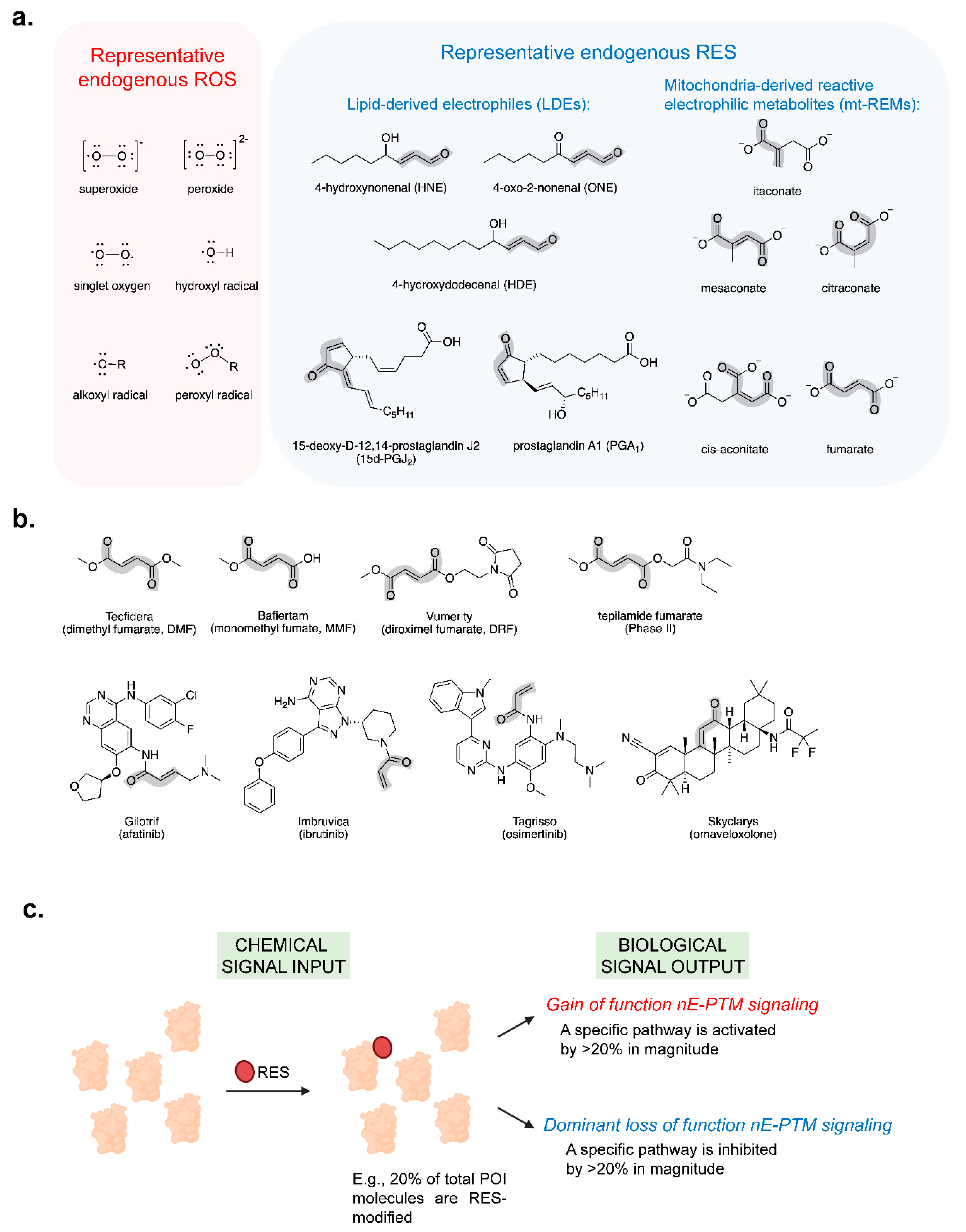
Figure 2.
Key enzymes involved in itaconate and fumarate biosynthesis and metabolic transformations. (a) TCA cycle. (b) IRG1-enzymatic conversion of cis-aconitate—an intermediate within the ACO2-catalyzed reversible conversion of citrate to/from isocitrate (green box)—to itaconate (blue box), which can further be metabolized to acetyl-CoA and pyruvate, and re-enter the TCA cycle in (Figure 2a). (c) Crystal structure of human IRG1 (PDB: 6R6U, left panel) and predicted cis-aconitate binding pocket featuring H-bonding interactions of cis-aconitate with proximal amino-acid residues (right panel). The putative active site was determined through comparison with the crystal structure of Agrobacterium tumefaciens iminodisuccinate epimerase (PDB 2HP3), electron density analysis of human IRG1 (PDB: 6R6U), and enzymatic activity tests of IRG1 active-site mutants [105]. (d) Crystal structure of porcine SDH flavoprotein subunit (SDHA) bound to oxaloacetate (as opposed to native substrate and product, succinate and fumarate) and flavin adenine dinucleotide (FAD) (PDB: 3SFD), along with surrounding active-site residues [106]. PDH: pyruvate dehydrogenase; CS: citrate synthase; ACO2: Mitochondrial aconitase; IDH2/3: mitochondrial isocitrate dehydrogenase; OGDH: oxoglutarate dehydrogenase; SCS: succinyl coenzyme A synthetase; SDH: succinate dehydrogenase; FH: fumarase; MDH2: mitochondrial malate dehydrogenase; IRG1: cis-aconitate decarboxylase; AUH: methylglutaconyl-CoA hydratase; CLYBL: mitochondrial citramalyl-CoA lyase.
Figure 2.
Key enzymes involved in itaconate and fumarate biosynthesis and metabolic transformations. (a) TCA cycle. (b) IRG1-enzymatic conversion of cis-aconitate—an intermediate within the ACO2-catalyzed reversible conversion of citrate to/from isocitrate (green box)—to itaconate (blue box), which can further be metabolized to acetyl-CoA and pyruvate, and re-enter the TCA cycle in (Figure 2a). (c) Crystal structure of human IRG1 (PDB: 6R6U, left panel) and predicted cis-aconitate binding pocket featuring H-bonding interactions of cis-aconitate with proximal amino-acid residues (right panel). The putative active site was determined through comparison with the crystal structure of Agrobacterium tumefaciens iminodisuccinate epimerase (PDB 2HP3), electron density analysis of human IRG1 (PDB: 6R6U), and enzymatic activity tests of IRG1 active-site mutants [105]. (d) Crystal structure of porcine SDH flavoprotein subunit (SDHA) bound to oxaloacetate (as opposed to native substrate and product, succinate and fumarate) and flavin adenine dinucleotide (FAD) (PDB: 3SFD), along with surrounding active-site residues [106]. PDH: pyruvate dehydrogenase; CS: citrate synthase; ACO2: Mitochondrial aconitase; IDH2/3: mitochondrial isocitrate dehydrogenase; OGDH: oxoglutarate dehydrogenase; SCS: succinyl coenzyme A synthetase; SDH: succinate dehydrogenase; FH: fumarase; MDH2: mitochondrial malate dehydrogenase; IRG1: cis-aconitate decarboxylase; AUH: methylglutaconyl-CoA hydratase; CLYBL: mitochondrial citramalyl-CoA lyase.
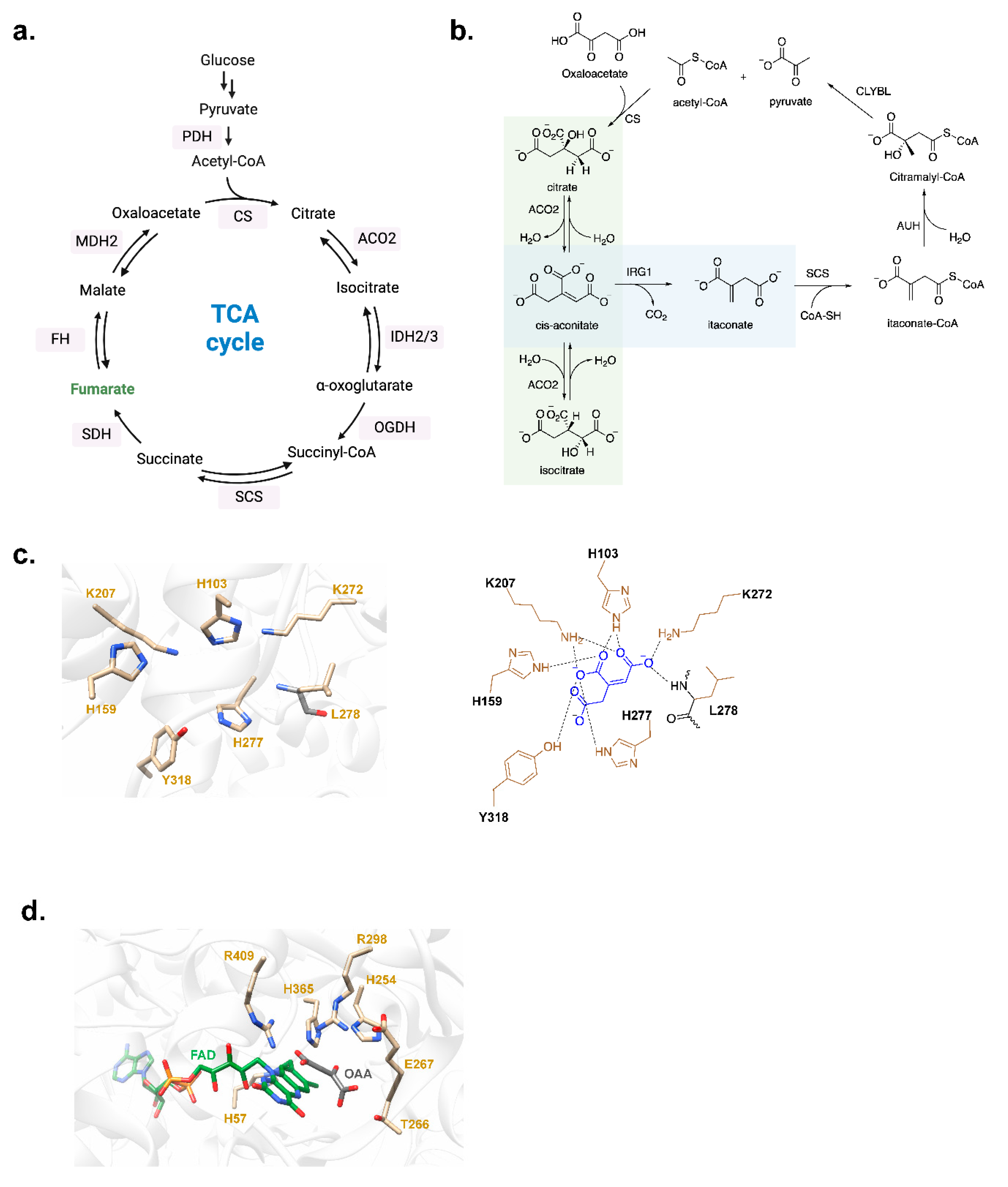
Figure 3.
Regulatory roles of itaconate. Inflammatory stimuli (cytokines or pathogenic molecules, such as LPS) bind to TLRs and promote the expression of pro-inflammatory cytokines as well as IRG1. The latter upregulates itaconate production. Itaconate is thought to be exported to the cytosol via SLC25A11, and subsequently, to the extracellular space by ABCG2. Itaconate can label myriad cellular proteins, and some of these target engagements are thought to modulate several processes, such as antioxidant response, pathogen clearance, leukocyte recruitment, and secretion of anti-inflammatory cytokines (see also Figure 4h). Both the target spectra of itaconate and associated signaling networks, including phenotypes implicated to be affected by itaconate, are rapidly expanding and evolving.
Figure 3.
Regulatory roles of itaconate. Inflammatory stimuli (cytokines or pathogenic molecules, such as LPS) bind to TLRs and promote the expression of pro-inflammatory cytokines as well as IRG1. The latter upregulates itaconate production. Itaconate is thought to be exported to the cytosol via SLC25A11, and subsequently, to the extracellular space by ABCG2. Itaconate can label myriad cellular proteins, and some of these target engagements are thought to modulate several processes, such as antioxidant response, pathogen clearance, leukocyte recruitment, and secretion of anti-inflammatory cytokines (see also Figure 4h). Both the target spectra of itaconate and associated signaling networks, including phenotypes implicated to be affected by itaconate, are rapidly expanding and evolving.
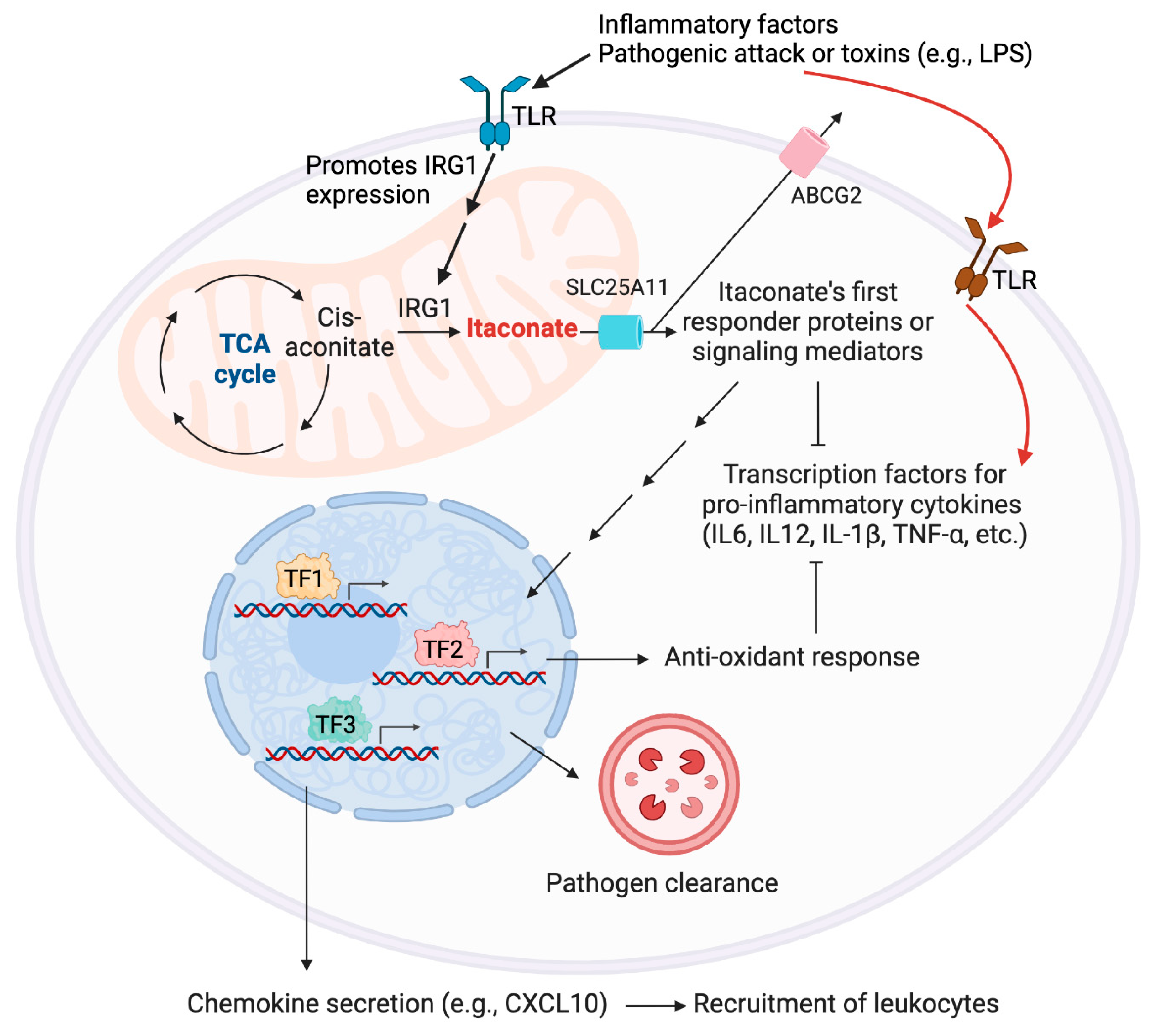
Figure 4.
Representative pathways reportedly regulated by itaconate (and its derivatives). (a) Chemical structures of Itaconate derivatives: DMI and 4-OI. (b) KEAP1/NRF2/AR axis: KEAP1 is a cytosolic protein and a negative regulator of NRF2. RES modification of KEAP1 hampers NRF2 degradation. The accumulated cytosolic NRF2 translocates to the nucleus and drives the expression of antioxidant response (AR) genes. (c) The formation of 2′3′-cGAMP from cytosolic/damaged DNA through cCAS activates STING on ER, through the recruitment of TBK1 and subsequent phosphorylation of STING and IRF3. The phosphorylated IRF3 translocates to the nucleus and drives the expression of several interferons and pro-inflammatory genes. (d) ATF3/IκBζ axis: ATF3 promotes eIF2α phosphorylation, which suppresses eIF2α-initiated translation. IκBζ, which is downstream of the eIF2α-initiated translation process, promotes the expression of pro-inflammatory genes. The detailed mechanisms regarding which proteins mediated among these processes (connected by double arrows) remain largely unclear. (e) JAK1/STAT1 axis: pro-inflammatory interferon promotes JAK1 phosphorylation, which subsequently phosphorylates STAT1. Phosphorylated STAT1 translocates to the nucleus and drives the expression of several pro-inflammatory genes. (f) JAK1/STAT6 axis: anti-inflammatory interferon promotes JAK1 phosphorylation, although through different receptors, resulting in STAT6 phosphorylation. Phosphorylated STAT6 upregulates the expression of several anti-inflammatory genes. (g) Phosphorylation of TFEB by mTOR prevents TFEB’s translocation from cytosol to the nucleus through the interaction between p-TFEB and 14-3-3 protein. The process suppresses TFEB-regulated lysosomal genes. (h) A representative set of pathways reported to be associated with itaconate-mediated immune response.
Figure 4.
Representative pathways reportedly regulated by itaconate (and its derivatives). (a) Chemical structures of Itaconate derivatives: DMI and 4-OI. (b) KEAP1/NRF2/AR axis: KEAP1 is a cytosolic protein and a negative regulator of NRF2. RES modification of KEAP1 hampers NRF2 degradation. The accumulated cytosolic NRF2 translocates to the nucleus and drives the expression of antioxidant response (AR) genes. (c) The formation of 2′3′-cGAMP from cytosolic/damaged DNA through cCAS activates STING on ER, through the recruitment of TBK1 and subsequent phosphorylation of STING and IRF3. The phosphorylated IRF3 translocates to the nucleus and drives the expression of several interferons and pro-inflammatory genes. (d) ATF3/IκBζ axis: ATF3 promotes eIF2α phosphorylation, which suppresses eIF2α-initiated translation. IκBζ, which is downstream of the eIF2α-initiated translation process, promotes the expression of pro-inflammatory genes. The detailed mechanisms regarding which proteins mediated among these processes (connected by double arrows) remain largely unclear. (e) JAK1/STAT1 axis: pro-inflammatory interferon promotes JAK1 phosphorylation, which subsequently phosphorylates STAT1. Phosphorylated STAT1 translocates to the nucleus and drives the expression of several pro-inflammatory genes. (f) JAK1/STAT6 axis: anti-inflammatory interferon promotes JAK1 phosphorylation, although through different receptors, resulting in STAT6 phosphorylation. Phosphorylated STAT6 upregulates the expression of several anti-inflammatory genes. (g) Phosphorylation of TFEB by mTOR prevents TFEB’s translocation from cytosol to the nucleus through the interaction between p-TFEB and 14-3-3 protein. The process suppresses TFEB-regulated lysosomal genes. (h) A representative set of pathways reported to be associated with itaconate-mediated immune response.
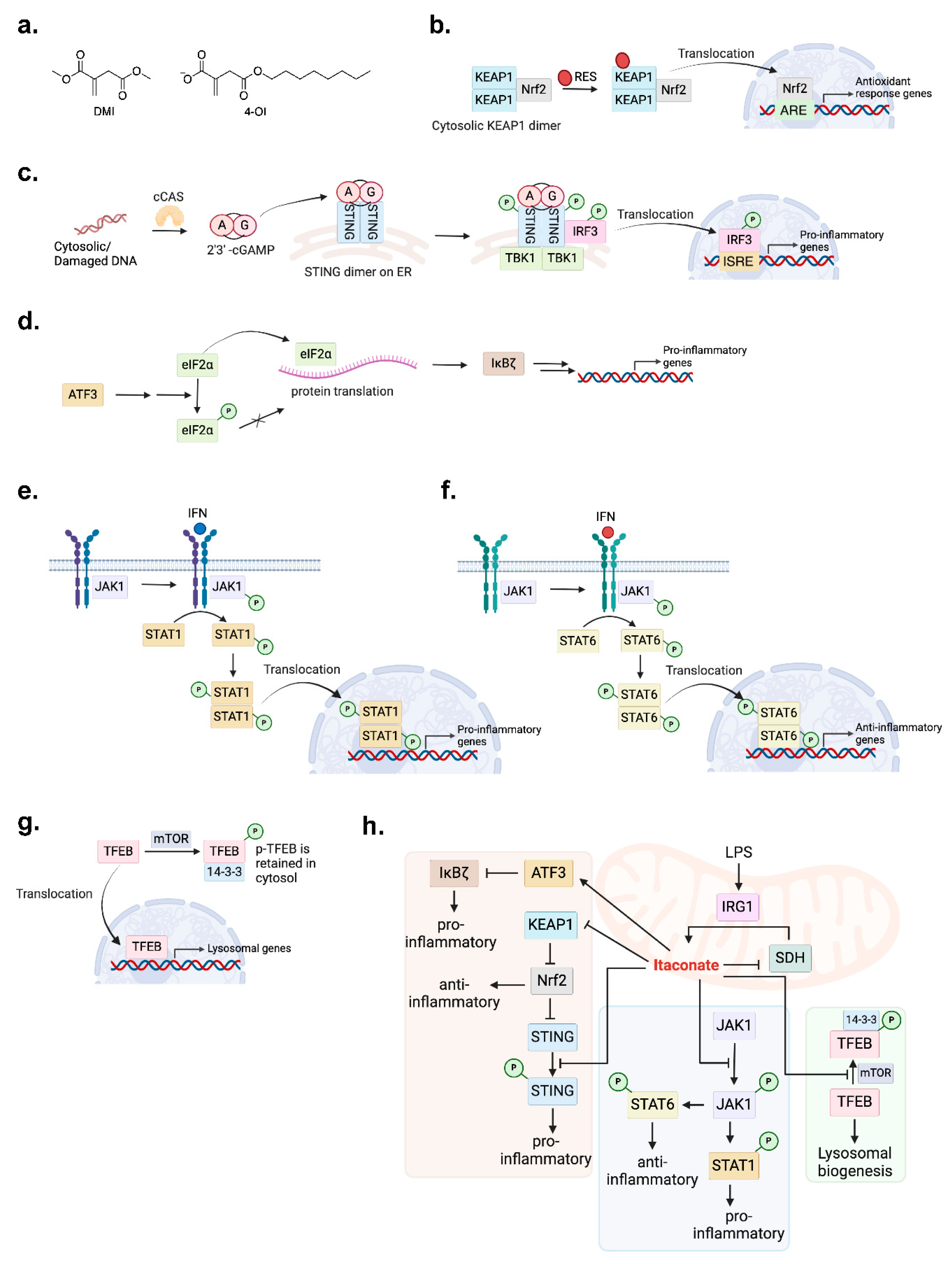
Figure 5.
DMF/HNE-modification of Keap1 induces mitochondria-targeted apoptosis selectively in neutrophils and macrophages. (a) DMF or HNE modification on KEAP1 interrupts KEAP1-WDR1 interaction. The dissociated WDR1 associates with cofilin and actin, which promotes cytochrome C release from Bax channel on the mitochondria. This event activates caspase 9 and subsequently caspase 3. The activated Caspase 3 initiates cell death in neutrophils/macrophages. Cell-type specificity is conferred by increased abundance of Wdr1 in innate immune cells. (b) T-REX-mediated KEAP1-specific HNEylation induces neutrophil and macrophage loss, against untreated controls, in Tg(lyz:TagRFP) and Tg(mpeg1:eGFP) larval zebrafish, respectively reporting neutrophil and macrophage cells [76]. (Figure adapted from published data [76]; the authors own the copyrights). lyz is a neutrophil-specific promoter in larval zebrafish, while mpeg1 is a macrophage-specific promoter. Scale bar, 500 µm in all images.
Figure 5.
DMF/HNE-modification of Keap1 induces mitochondria-targeted apoptosis selectively in neutrophils and macrophages. (a) DMF or HNE modification on KEAP1 interrupts KEAP1-WDR1 interaction. The dissociated WDR1 associates with cofilin and actin, which promotes cytochrome C release from Bax channel on the mitochondria. This event activates caspase 9 and subsequently caspase 3. The activated Caspase 3 initiates cell death in neutrophils/macrophages. Cell-type specificity is conferred by increased abundance of Wdr1 in innate immune cells. (b) T-REX-mediated KEAP1-specific HNEylation induces neutrophil and macrophage loss, against untreated controls, in Tg(lyz:TagRFP) and Tg(mpeg1:eGFP) larval zebrafish, respectively reporting neutrophil and macrophage cells [76]. (Figure adapted from published data [76]; the authors own the copyrights). lyz is a neutrophil-specific promoter in larval zebrafish, while mpeg1 is a macrophage-specific promoter. Scale bar, 500 µm in all images.
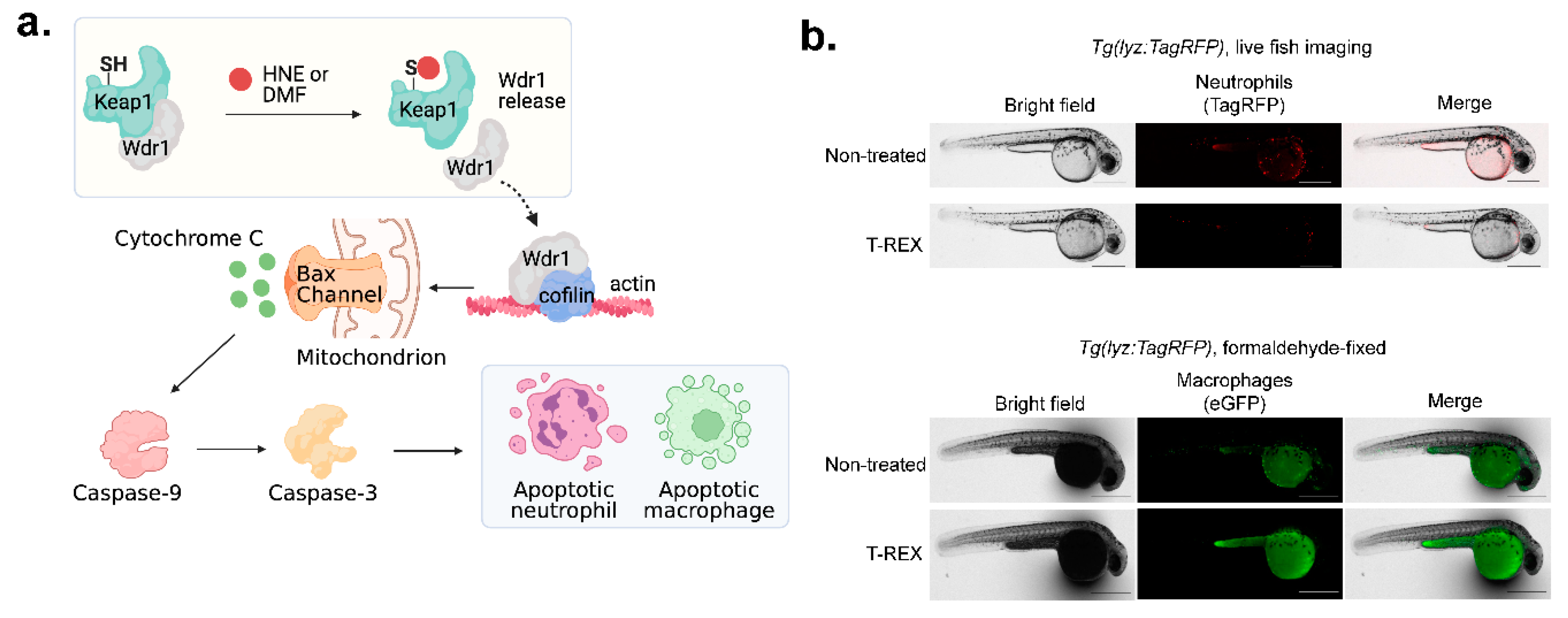
Figure 6.
Indirect target profiling. (a) Competitive isoTOP-ABPP workflow. Cells are treated with mt-REM of interest (or DMSO), harvested, and lysed. The lysate is subsequently treated with isotopically-labeled (typically, alkyne- or azide-functionalized) Cys- or Lys-reactive proxy-electrophile probe (see also in Figure 6b). This proxy-electrophile probe is assumed to react with all the remaining unreacted Cys or Lys. Subsequent enrichment and digest LC-MS/MS target-ID workflow identifies protein targets that have lost proxy-electrophile probe signal in the mt-REM-treated group, compared to DMSO-treated group. These proteins are scored as the targets of mt-REM. (b) Chemical structures of cysteine-reactive (the first two on the left) and lysine-reactive proxy-electrophile probes used in ABPP- and analogous indirect target-ID approaches.
Figure 6.
Indirect target profiling. (a) Competitive isoTOP-ABPP workflow. Cells are treated with mt-REM of interest (or DMSO), harvested, and lysed. The lysate is subsequently treated with isotopically-labeled (typically, alkyne- or azide-functionalized) Cys- or Lys-reactive proxy-electrophile probe (see also in Figure 6b). This proxy-electrophile probe is assumed to react with all the remaining unreacted Cys or Lys. Subsequent enrichment and digest LC-MS/MS target-ID workflow identifies protein targets that have lost proxy-electrophile probe signal in the mt-REM-treated group, compared to DMSO-treated group. These proteins are scored as the targets of mt-REM. (b) Chemical structures of cysteine-reactive (the first two on the left) and lysine-reactive proxy-electrophile probes used in ABPP- and analogous indirect target-ID approaches.
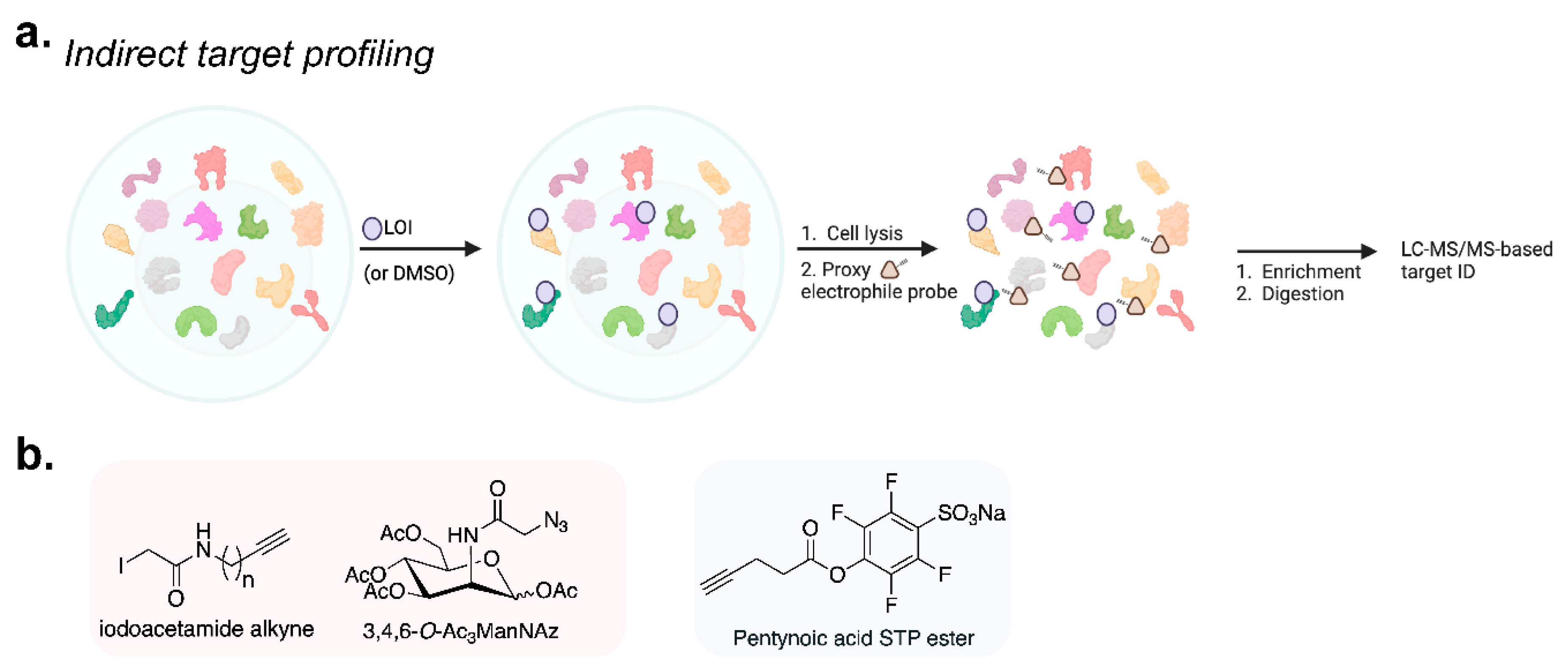
Figure 7.
Direct target profiling and itaconate biosensor bioITA. (a) Protein cysteines are modified by reactive electrophiles through Michael addition; the adducts so formed are labile, typically via retro-Michael addition process (mechanistic arrows for reversal are omitted for clarity). (b) Principle of antibody-based direct target capture: endogenous mt-REM-modified proteins are enriched by resin coated with antibody specific to the designated mt-REM. After washing away unbound proteins, mt-REM-modified proteins are eluted off the resin, and characterized by methods such as digest LC-MS/MS. (c) Principle of Clickable-ligand-based direct target capture: mt-REM analogs functionalized with Clickable moieties (exemplified here by alkyne) are added to cell lysates, cells, or animals. Post-treatment (and lysis where applicable), Click reaction with biotin-azide (or other purification tags), facilitates affinity enrichment. Subsequent digest LC-MS/MS analysis maps targets that are covalently bound to the mt-REM analogs deployed. (d) Chemical structures of ITalk. (e) BioITA biosensor: conformational change in BioITA protein upon non-covalent binding with itaconate induces a fluorescent signal. IBD: itaconate-binding domain; cpGFP: circularly permuted green fluorescent protein; CENP-B: centromere protein B.
Figure 7.
Direct target profiling and itaconate biosensor bioITA. (a) Protein cysteines are modified by reactive electrophiles through Michael addition; the adducts so formed are labile, typically via retro-Michael addition process (mechanistic arrows for reversal are omitted for clarity). (b) Principle of antibody-based direct target capture: endogenous mt-REM-modified proteins are enriched by resin coated with antibody specific to the designated mt-REM. After washing away unbound proteins, mt-REM-modified proteins are eluted off the resin, and characterized by methods such as digest LC-MS/MS. (c) Principle of Clickable-ligand-based direct target capture: mt-REM analogs functionalized with Clickable moieties (exemplified here by alkyne) are added to cell lysates, cells, or animals. Post-treatment (and lysis where applicable), Click reaction with biotin-azide (or other purification tags), facilitates affinity enrichment. Subsequent digest LC-MS/MS analysis maps targets that are covalently bound to the mt-REM analogs deployed. (d) Chemical structures of ITalk. (e) BioITA biosensor: conformational change in BioITA protein upon non-covalent binding with itaconate induces a fluorescent signal. IBD: itaconate-binding domain; cpGFP: circularly permuted green fluorescent protein; CENP-B: centromere protein B.
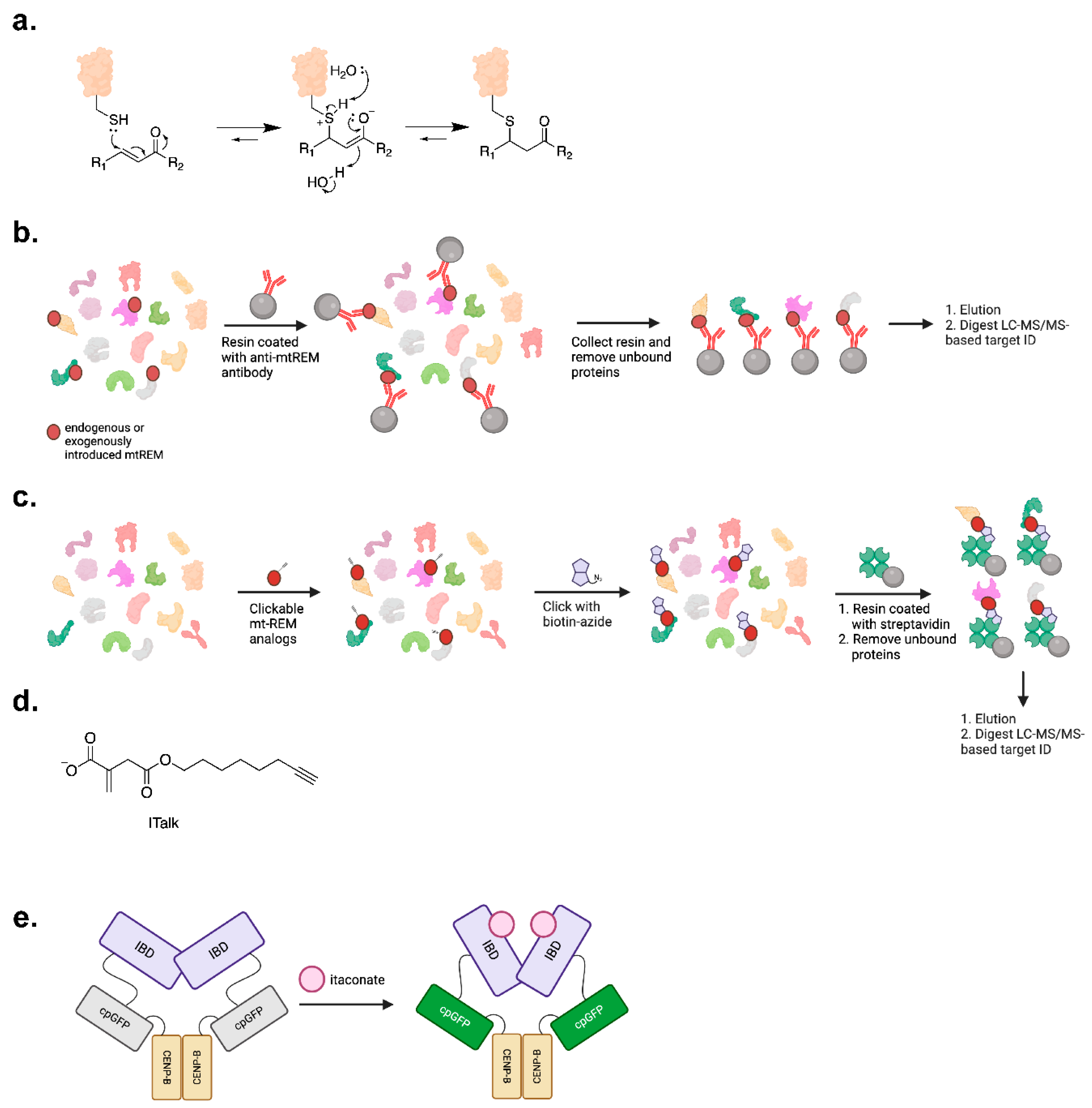
Figure 8.
Tandem Localis-REX—T-REX enables LDE functional target identification and target/ligand-pair-specific signaling interrogation in living systems. (a) Localis REX. Cells/fish/worms expressing Halo protein at a specific subcellular locale, organelle, or tissue/organ/cell-type, are treated with Halo-targetable (Ht) photocaged alkyne-functionalized electrophile probe (non-alkyne-functionalized probe is used as the control in Localis-REX; or for T-REX-based signaling investigations). Illumination (1-3 min, 5 mW/cm [2], 365 nm), rapidly liberates the designated electrophile with spatiotemporal precision. Electrophile-responsive proteins within the locale of Halo are enriched via Click coupling to biotin azide and streptavidin-pulldown. Digest LC-MS/MS proteomics afford a direct function-guided proximity mapping of localized first responders. Localis-REX can be coupled with SILAC, TMT-labeling, or LFQ (label-free quantification) for quantitative target-ID. (b) Technically, T-REX differs from Localis-REX only in that Halo-fused POI (protein of interest) is deployed in place of Halo. T-REX can be used for both upstream target engagement investigations such as quantification of ligand occupancy, quantitative ranking among best first responders, in cells, fish, and worms; and for monitoring downstream functional consequences triggered by on-target electrophile labeling in living subjects with high spatiotemporal resolution. (c) Images of Tg(myl7:DsRed-P2A-Halo-TEV-Keap1-2xHA, cry:mRFP1, gstp1:eGFP) fish treated with DMSO (vehicle/control), light-illumination alone, 0.3 μM Ht-PreHNE probe alone, or Z-REX (0.3 μM Ht-PreHNE + illumination). After the treatment, fish were fixed with 4% formaldehyde solution, and stained with goat anti-GFP and then anti-goat-Alexa647 to visualize AR reported by gstp1:eGFP transcriptional activation. The region of the heart was defined by DsRed signal, whose expression, alongside Halo-TEV-Keap1-HA, is driven by the cardiomyocyte-specific promoter, myl7. Z-REX-treated groups exhibit increased AR compared to all 3 negative control groups (DMSO, light, or Ht-PreHNE alone). mRFP1, driven by the eye-specific promoter, cry, and located in the fish eye, is used as a transgenesis marker (image not shown). Scale bar, 500 µm in all images [75]. (Figure adapted from published data [75]; the authors own the copyrights). (d) Chemical structure of Halo-targetable photocage LDE probe, Ht-PreLDE (left). The probe can form a stable covalent bond with Halo protein through a hexyl chloride component (indicated in red). The structure of released LDE (tangerine) is on the right. (e) A suite of negative controls in REX technologies ensures the validity of the measured signaling outputs. This includes technical controls such as treating with light alone, probe alone, and DMSO alone, to rule out any potential confounding effects induced by these individual components. Additionally, biological controls, in which either Halo and POI are expressed separately (e.g., by introducing a P2A linker) instead of as a fusion construct, or a mutant version of POI (which cannot sense electrophile but remains functional) is used in a Halo fusion construct; in these scenarios, signaling outputs are not be observed, should the measured effects be genuine consequences of target-ligand engagement in cells/animals. Likewise, no labeling of POI by the electrophile should occur in these control groups.
Figure 8.
Tandem Localis-REX—T-REX enables LDE functional target identification and target/ligand-pair-specific signaling interrogation in living systems. (a) Localis REX. Cells/fish/worms expressing Halo protein at a specific subcellular locale, organelle, or tissue/organ/cell-type, are treated with Halo-targetable (Ht) photocaged alkyne-functionalized electrophile probe (non-alkyne-functionalized probe is used as the control in Localis-REX; or for T-REX-based signaling investigations). Illumination (1-3 min, 5 mW/cm [2], 365 nm), rapidly liberates the designated electrophile with spatiotemporal precision. Electrophile-responsive proteins within the locale of Halo are enriched via Click coupling to biotin azide and streptavidin-pulldown. Digest LC-MS/MS proteomics afford a direct function-guided proximity mapping of localized first responders. Localis-REX can be coupled with SILAC, TMT-labeling, or LFQ (label-free quantification) for quantitative target-ID. (b) Technically, T-REX differs from Localis-REX only in that Halo-fused POI (protein of interest) is deployed in place of Halo. T-REX can be used for both upstream target engagement investigations such as quantification of ligand occupancy, quantitative ranking among best first responders, in cells, fish, and worms; and for monitoring downstream functional consequences triggered by on-target electrophile labeling in living subjects with high spatiotemporal resolution. (c) Images of Tg(myl7:DsRed-P2A-Halo-TEV-Keap1-2xHA, cry:mRFP1, gstp1:eGFP) fish treated with DMSO (vehicle/control), light-illumination alone, 0.3 μM Ht-PreHNE probe alone, or Z-REX (0.3 μM Ht-PreHNE + illumination). After the treatment, fish were fixed with 4% formaldehyde solution, and stained with goat anti-GFP and then anti-goat-Alexa647 to visualize AR reported by gstp1:eGFP transcriptional activation. The region of the heart was defined by DsRed signal, whose expression, alongside Halo-TEV-Keap1-HA, is driven by the cardiomyocyte-specific promoter, myl7. Z-REX-treated groups exhibit increased AR compared to all 3 negative control groups (DMSO, light, or Ht-PreHNE alone). mRFP1, driven by the eye-specific promoter, cry, and located in the fish eye, is used as a transgenesis marker (image not shown). Scale bar, 500 µm in all images [75]. (Figure adapted from published data [75]; the authors own the copyrights). (d) Chemical structure of Halo-targetable photocage LDE probe, Ht-PreLDE (left). The probe can form a stable covalent bond with Halo protein through a hexyl chloride component (indicated in red). The structure of released LDE (tangerine) is on the right. (e) A suite of negative controls in REX technologies ensures the validity of the measured signaling outputs. This includes technical controls such as treating with light alone, probe alone, and DMSO alone, to rule out any potential confounding effects induced by these individual components. Additionally, biological controls, in which either Halo and POI are expressed separately (e.g., by introducing a P2A linker) instead of as a fusion construct, or a mutant version of POI (which cannot sense electrophile but remains functional) is used in a Halo fusion construct; in these scenarios, signaling outputs are not be observed, should the measured effects be genuine consequences of target-ligand engagement in cells/animals. Likewise, no labeling of POI by the electrophile should occur in these control groups.
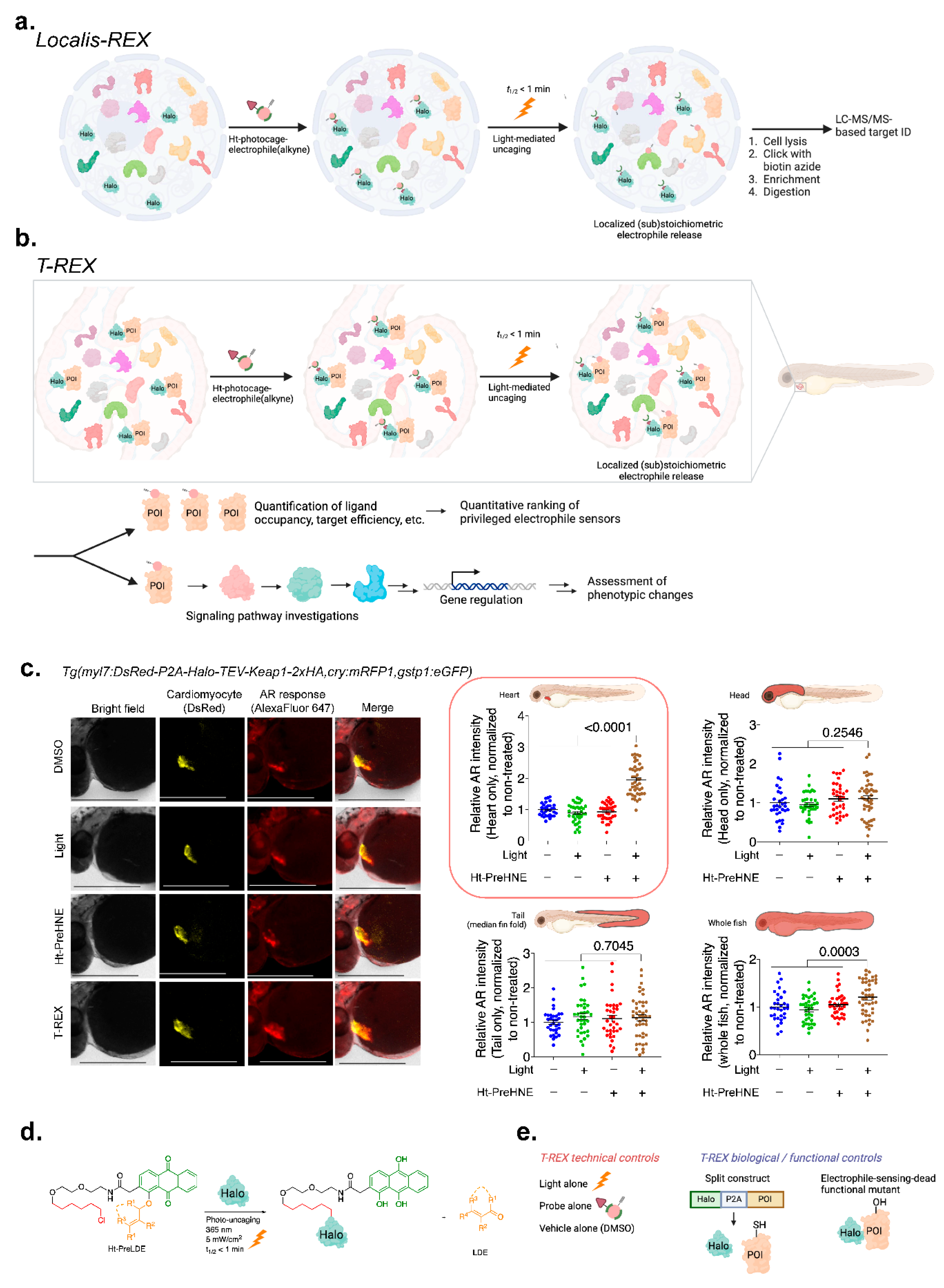
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



